
Submitted:
09 October 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
Background and Aims: Despite substantial statin-induced low-density lipoprotein cholesterol (LDL-C) lowering, residual cardiovascular risk remains a major clinical challenge. A mechanistic synthesis of preclinical and clinical evidence was undertaken to explain why inflammation and risk persist despite optimal lipid control. Methods: Literature from preclinical models, clinical trial data, mechanistic modeling, and biomarker trajectories was reviewed and integrated to construct a unified framework linking lipid lowering with persistent immunometabolic activity. Results: Long-term, high-dose statin exposure has been associated with paradoxical effects in arterial macrophages, including activation of the NOD-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome, impaired resolution pathways, and promotion of elevated blood glucose and insulin resistance, including via reduction of circulating glucagon-like peptide-1 (GLP-1) in a microbiota-dependent manner. These local effects may coexist with systemic anti-inflammatory benefits, creating a lower bound beyond which arterial inflammation does not regress. Adaptive immune feedback, Lipoprotein(a)-driven amplification, and vascular remodeling further contribute to inter-individual variability. Temporal biomarker evolution defines three mechanistic phases that may assist in stratifying patient response and guiding therapy design. Conclusions: Residual cardiovascular risk can be reframed as an unintended but potentially modifiable immunometabolic plateau. By integrating established lipid-lowering outcomes with emerging insights into inflammation and metabolism, this framework provides a testable model to support biomarker-driven precision strategies and the earlier adoption of complementary therapies, thereby improving outcomes.
Keywords:
statin paradox
; NLRP3 inflammasome
; protein prenylation
; lipoprotein(a)
; IL-1β
; insulin resistance
; residual risk
; cardiovascular disease
1. Introduction
Despite substantial [LDL-C]1 lowering [1], statin-treated cohorts still exhibit ~75% baseline relative risk that plateaus, in absolute terms, at ≈5–10% by year five [2,3,4]. Anti-inflammatory trials (CANTOS, LoDoCo2 [5,6]) confirm that arterial inflammation drives residual events beyond lipid control, yet despite comparable [LDL-C], anti-C-Reactive Protein (CRP) therapy responses vary, underpinning the effect that chronic high-dose statins induce a persistent inflammatory minimum.
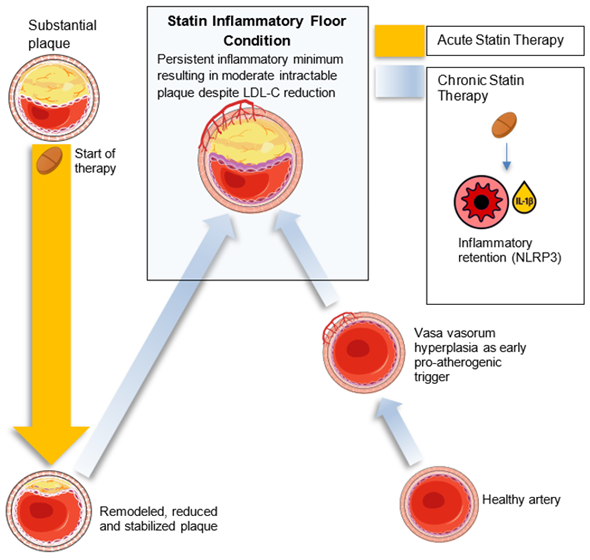
Figure 1.
Graphical Abstract – The Statin Floor Effect. Conceptual overview of the Statin Floor Effect. Long-term high-intensity statin therapy lowers LDL-C but establishes a localized arterial inflammatory floor through NLRP3 activation, adaptive immune modulation, and metabolic disruption. Vasa vasorum hyperplasia and diffuse intimal hyperplasia are depicted as early pro-atherogenic triggers that create a permissive substrate for lipid retention and inflammation. Together, these elements integrate molecular mechanisms, biomarker trajectories, and clinical observations to explain the plateau in cardiovascular risk reduction and highlight avenues for therapeutic innovation.
Figure 1.
Graphical Abstract – The Statin Floor Effect. Conceptual overview of the Statin Floor Effect. Long-term high-intensity statin therapy lowers LDL-C but establishes a localized arterial inflammatory floor through NLRP3 activation, adaptive immune modulation, and metabolic disruption. Vasa vasorum hyperplasia and diffuse intimal hyperplasia are depicted as early pro-atherogenic triggers that create a permissive substrate for lipid retention and inflammation. Together, these elements integrate molecular mechanisms, biomarker trajectories, and clinical observations to explain the plateau in cardiovascular risk reduction and highlight avenues for therapeutic innovation.
The Statin Floor Effect (SFE) is presented as a conceptual framework that integrates the temporal dynamics and pleiotropic complexity of statin therapy. Statin effects are not uniformly beneficial, and chronic, high-dose statin therapy can establish a persistent inflammatory minimum in arterial macrophages that limits further cardiovascular event risk reduction, despite continued systemic benefits. In constructing this model, the post-statin residual inflammatory floor has been explicitly distinguished from the mechanisms driving plaque initiation [7]. Multiple studies demonstrate that foam cells originate from monocyte-derived macrophages, and via Kruppel-like Factor 4-dependent phenotypic modulation and transdifferentiation of vascular smooth muscle cells [8,9]. Accordingly, the residual inflammatory floor encompasses contributions from both monocyte- and smooth muscle cell-derived foam cells within the atherosclerotic plaque.
In murine macrophages, statins trigger NLRP3 inflammasome activation [10], and intriguingly, context-dependent paradoxes between toll-like receptor (TLR) signaling and inflammasome engagement have been reported [11]. Mechanistic studies show that chronic statin exposure impairs protein prenylation and small GTPase function, offering a unified upstream pathway linking NLRP3 activation with insulin resistance [12]. In addition, recent translational work has demonstrated that statin exposure can aggravate insulin resistance through a gut microbiota–bile acid-GLP-1 axis, with reductions in circulating GLP-1 providing an additional route by which high-dose therapy reinforces a persistent inflammatory floor [13]. Direct confirmation of these interconnected processes in human atheroma, however, remains outstanding.
There is recent evidence also to suggest that an inflammatory floor may be preceded by structural vascular priming. Adventitial vasa vasorum hyperplasia and diffuse intimal hyperplasia, proposed as initiating events in atherosclerosis, may create a permissive microenvironment for lipid retention and immune activation, particularly in high-risk individuals. These early non-inflammatory structural changes may well interact with the mechanisms described in this framework and support the need for temporally stratified preventive approaches. In parallel, residual risk may also be sustained or amplified by LDL-independent pathways, including elevated [Lp(a)], [small dense LDL], and inflammatory cytokine axes such as interleukins (IL) IL-17/IL-23. These factors may converge on macrophage activation and inflammasome priming, reinforcing the localized inflammatory floor and contributing to inter-individual heterogeneity in treatment response. These elements collectively form a unified model for residual cardiovascular risk in the statin era, mechanistically grounded and clinically actionable. By locking arterial macrophages into a low-grade inflammatory floor, subtly deranging insulin signaling, and possibly influencing angiogenesis, chronic high-dose statins may limit full resolution of old lesions.
Although the framework centers on unintended immunometabolic adaptation, it also aligns with established observations that plaque burden alters local hemodynamics. Regions of disturbed flow, characterized by low turbulence or oscillatory shear stress, are known to promote endothelial dysfunction, leukocyte adhesion, and pro-inflammatory signaling [14]. These shear-sensitive pathways can be activated even in the absence of lipid level elevation and may synergize with residual inflammatory cues to sustain arterial lesion progression. Importantly, such flow disturbances persist or worsen in the presence of pre-existing plaques, creating a feed-forward loop that enhances plaque vulnerability and propagation [15].
Low shear stress has also been directly implicated in NLRP3 inflammasome activation in vascular endothelial cells, providing a mechanistic link between mechanical stress and innate immune priming [16]. These structural and mechanical factors may therefore act as physical amplifiers of the inflammatory floor, particularly in advanced atheroma, where statin effects on plaque architecture may reach their limits.
Although statins are not classified as antihypertensive agents, they may modestly influence blood pressure and vascular tone through pleiotropic effects [17]. Acute administration increases endothelial nitric oxide (NO) bioavailability and inhibits Ras homolog family member A/Rho-associated coiled-coil containing protein kinase (RhoA/ROCK) signaling, promoting vasodilation and transient reductions in peripheral resistance [18]. With chronic therapy, additional benefits emerge, including improved arterial compliance, reduced oxidative stress, and modulation of sympathetic tone and the renin–angiotensin–aldosterone system [19]. Meta-analyses of randomized controlled trials report small but statistically significant reductions in systolic (1–5 mmHg) and diastolic (0.5–3 mmHg) blood pressure with long-term statin use [17]. While insufficient to replace conventional antihypertensive therapy, these effects may contribute additively to vascular stabilization, particularly during early atheroma remodeling, when statin-mediated lipid lowering and inflammation resolution are most responsive. Moreover, computational modeling supports the interplay between hemodynamics and vascular biology. Recent mechano-chemo-biological models and multiscale simulations demonstrate that adventitial remodeling and vasa vasorum proliferation, hallmarks of the “outside-in” theory, can reinforce lesion development independent of luminal lipid burden [20,21]. These structural changes may serve as chronic reinforcers of localized inflammation, especially when coupled with sub-resolution plaque instability and altered flow dynamics. Paradoxically, chronic high-dose statin therapy has been shown to induce vascular endothelial growth factor and matrix metalloproteinase (MMP) expression in vessel wall cells, thereby potentiating adventitial vasa vasorum proliferation. These findings suggest that mechanical, structural, and immunometabolic pathways converge to establish and maintain the inflammatory floor observed in statin-treated patients.
Within this framework, mechanistic contributors are organized into primary pathways, centered on dose-dependent NLRP3 inflammasome activation in arterial macrophages, and secondary pathways that amplify or sustain the inflammatory floor. These include impaired protein prenylation, insulin resistance, adaptive immune modulation, and emerging factors such as red-blood-cell–derived extracellular vesicles, as well as parallel pathways involving oxidized lipoproteins, Lp(a), and IL-17/IL-23 signaling. This structure provides a scaffold for the mechanistic framework that follows.
2. Methods / Approach
Literature Search
This work was undertaken as a mechanistic narrative review, integrating preclinical studies, clinical trial findings, biomarker trajectories, and computational modeling relevant to statin pharmacology and residual cardiovascular risk. An integrative PubMed, Google Scholar, and Web of Science search (2000–September 2025) was conducted for terms covering statin pharmacology, residual cardiovascular risk, NLRP3 inflammasome priming, foam cell biology, protein prenylation, LDL-C, apoprotein B (ApoB), lipoprotein(a)/oxidized phospholipids (OxPL), and adaptive immunity. Peer-reviewed mechanistic studies, meta-analyses, and key reviews were prioritized, with additional references obtained by backward citation tracking to develop thematic clusters and identify testable predictions. The aim was to construct a unifying conceptual framework, the Statin Floor Effect, linking established lipid-lowering outcomes with emerging immunometabolic and vascular mechanisms. The focus was the development of a mechanistic framework; consequently, the work was not designed as a systematic review; PRISMA methodology and formal risk-of-bias assessment were therefore not applied.
Conceptual Framework Development
The model was built by first gathering and synthesizing key themes from the literature on long-term statin use. As the narrative was refined, mechanistic findings were organized into five core pathways:
- Inflammasome activation (NLRP3, IL-1β, IL-18)
- Disruption of protein prenylation (Ras-related C3 Botulinum Toxin Substrate [Rac1], geranylgeranylation)
- Insulin resistance acting as an inflammation amplifier
- Shifts in thymus-derived lymphocyte (T-cell) balance (T Helper 17 cell subtype [Th17] versus regulatory T-cells [Tregs])
- Immune priming driven by Lp(a) and oxidized phospholipids
These pathways were then aligned with clinical observations, such as the plateauing of major adverse cardiovascular events (MACE) reduction and persistent CRP elevation despite low [LDL-C], to generate clear, testable predictions. This process defined a unified framework for explaining residual cardiovascular risk in patients receiving statin therapy.
3. Mechanistic Framework
The mechanistic framework of the Statin Floor Effect is organized around a primary pathway, dose-dependent NLRP3 inflammasome activation in arterial macrophages, and a set of secondary pathways that amplify or sustain this inflammatory floor, including impaired protein prenylation, insulin resistance, and adaptive immune modulation. In addition, parallel pathways such as Lp(a)–oxidized phospholipids and IL-17/IL-23 signaling act as independent but convergent reinforcers. Against this backdrop, three paradoxes highlight the clinical and biological challenges that shape residual cardiovascular risk.
Clinical Problem and Paradoxes
The Residual Risk Paradox
- Early epidemiological work, in particular the Framingham Study and its stress-defense analyses, established the baseline event rates and risk factors still used to benchmark modern therapies [24]
The Statin Pleiotropy Paradox
Statins exhibit systemic anti-inflammatory effects [25,26] but demonstrate diminishing MACE benefit at higher doses [3,26], increase diabetes risk [27] and appear to produce dose-dependent Fourier Transform Infrared spectroscopy markers of oxidative stress consistent with NLRP3 priming [28]; interindividual response variability underscores mechanistic heterogeneity [29].
The Temporal Paradox
Event rates plateau at ≈5–10% by year five [2,30,31]; CRP reductions may not predict long-term resolution [32,33]; in type 2 diabetes cohorts, statin therapy duration, more than dose or [LDL-C] achieved, best predicts risk reduction [34]; and secondary prevention patients still exhibit residual events after years of treatment [25].
The Enhanced Mechanistic Model
Core Framework Statement
| The statin floor effect emerges when chronic, high-intensity statin therapy creates a net pro-inflammatory state in arterial macrophages despite systemic anti-inflammatory benefits, establishing a localized inflammatory minimum that limits further cardiovascular risk reduction. |
Temporal Stratification Model
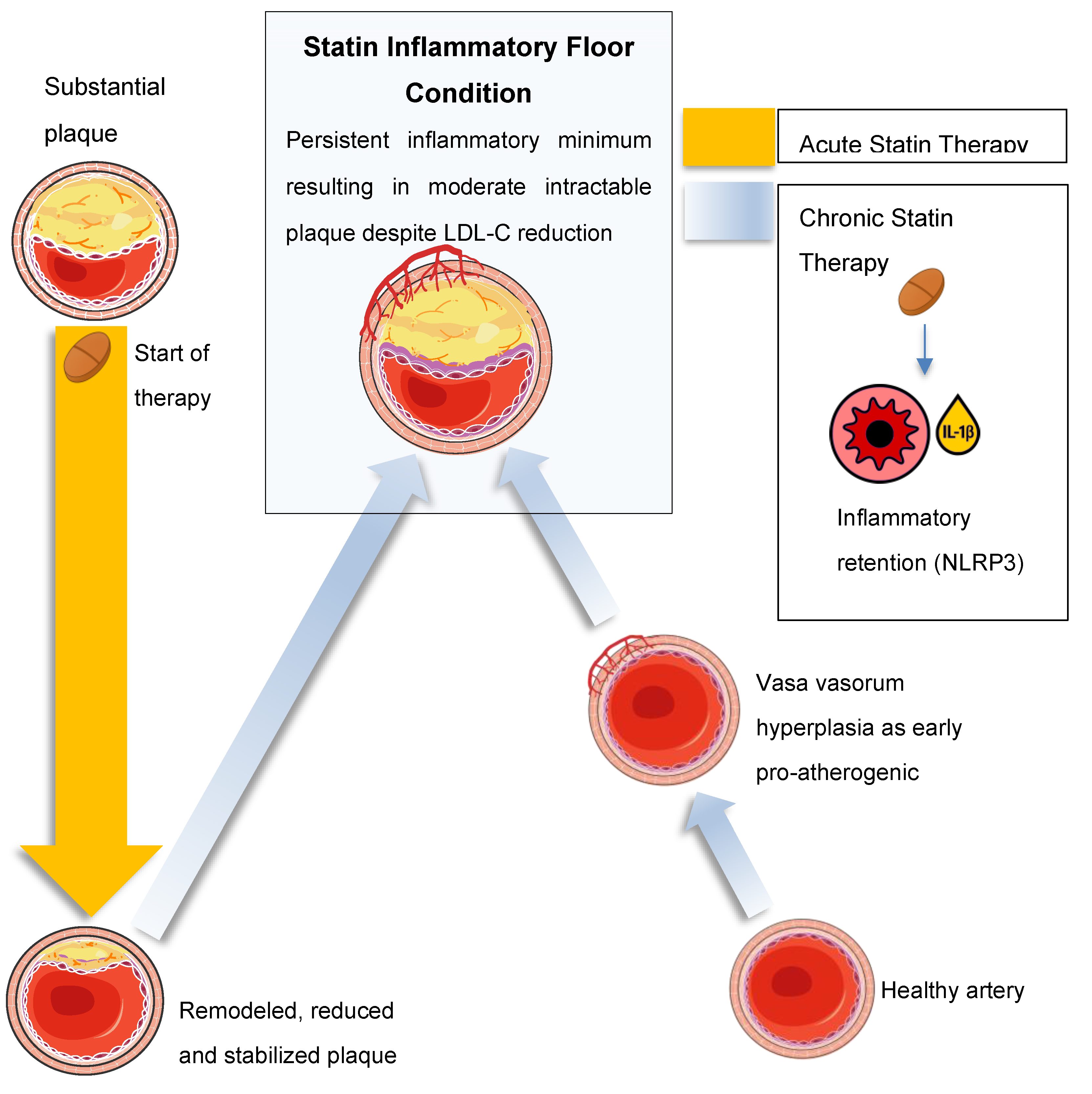
Three overlapping phases of statin-induced mechanistic shifts are conceptualized (Figure 2), each building on the last:
- Phase 1 (0–6 months): rapid [LDL-C] lowering with predominant anti-inflammatory benefit
- Phase 2 (6–24 months): competing beneficial and detrimental effects as mevalonate pathway disruption accumulates
- Phase 3 (>24 months): a persistent inflammatory floor limits further MACE reduction despite maintained [LDL-C] suppression
Figure 2.
Mechanistic details of each temporal phase of the SFE.Phase 1: Statin-mediated 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibition increases endothelial NO and inhibits RhoA/ROCK, stabilizing plaque and suppressing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)–driven cytokines[17,19,35]. Rapid [LDL-C] reduction drives foam cell apoptosis and efferocytosis [36,37,38]. Phase 2: Depletion of mevalonate-derived isoprenoids (GGPP, farnesyl pyrophosphate) impairs prenylation of small GTPases, resulting in NLRP3 inflammasome–dependent caspase-1 activation and IL-1β secretion—effects that are reversible with geranylgeraniol supplementation in both macrophages and adipocytes[10,39]. Concurrent mitochondrial stress fosters insulin resistance (↑ HOMA-IR) [10,27], dampening resolution pathways. Phase 3: Adaptive immune feedback (Th17 expansion, IL-17/IL-23 signaling) [40,41,42,43,44] and Lp(a)-derived OxPLs [45,46,47], potentially driven up by statins, reinforce NLRP3 activation. Adventitial vasa vasorum hyperplasia primes new LDL retention [48], cementing a self-sustaining inflammatory minimum that caps further MACE reduction.
Figure 2.
Mechanistic details of each temporal phase of the SFE.Phase 1: Statin-mediated 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibition increases endothelial NO and inhibits RhoA/ROCK, stabilizing plaque and suppressing nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)–driven cytokines[17,19,35]. Rapid [LDL-C] reduction drives foam cell apoptosis and efferocytosis [36,37,38]. Phase 2: Depletion of mevalonate-derived isoprenoids (GGPP, farnesyl pyrophosphate) impairs prenylation of small GTPases, resulting in NLRP3 inflammasome–dependent caspase-1 activation and IL-1β secretion—effects that are reversible with geranylgeraniol supplementation in both macrophages and adipocytes[10,39]. Concurrent mitochondrial stress fosters insulin resistance (↑ HOMA-IR) [10,27], dampening resolution pathways. Phase 3: Adaptive immune feedback (Th17 expansion, IL-17/IL-23 signaling) [40,41,42,43,44] and Lp(a)-derived OxPLs [45,46,47], potentially driven up by statins, reinforce NLRP3 activation. Adventitial vasa vasorum hyperplasia primes new LDL retention [48], cementing a self-sustaining inflammatory minimum that caps further MACE reduction.
Primary Pathway: Dose-Dependent NLRP3 Activation
- Statins inhibit the mevalonate pathway, reducing geranylgeranylation and impairing Rac1 regulation to augment NLRP3 activity2 [49,50], with mathematical and spectroscopic modeling confirming dose-dependent stress signatures consistent with inflammasome priming [28]. Moreover, statins modestly elevate Lp(a) levels [51], whose oxidized phospholipid cargo directly triggers NLRP3 activation, thereby offering a unified upstream pathway linking NLRP3 activation with insulin resistance and reinforcing the inflammatory floor despite optimal LDL-C levels3 [45,46].
- Statin-mediated menaquinone-4 (MK-4) depletion may impair inflammasome regulation via reduced SXR activation (vide infra: Section 4 - Therapeutic Potential subsection).
- Statins also inhibit GTPase prenylation, suppressing Th17 differentiation [56,57] and expanding Tregs [58,59], though these adaptive immune effects vary among individuals [60] and sustain a pro-inflammatory milieu4. In contrast, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors (FOURIER) and bempedoic acid (CLEAR Outcomes) show continuously diverging event-reduction curves at 2–3 years without the early plateau seen in statin trials [31,61].
Additional translational evidence strengthens this link between statins, mitochondrial dysfunction, and inflammasome priming. In a controlled study of overweight adults, 56 days of high-dose atorvastatin reduced insulin sensitivity in most participants and selectively inhibited mitochondrial complex IV activity at clinically relevant tissue concentrations. Mitochondrial calcium retention capacity was also diminished, a change that predisposes to permeability transition and ROS-driven NLRP3 activation. These findings provide direct human support for the notion that chronic statin therapy establishes a pro-inflammatory floor not only through prenylation-dependent Rac1 dysregulation but also by converging on mitochondrial stress pathways [62].
Recent evidence further reinforces the role of the NLRP3 inflammasome as a persistent driver of residual cardiovascular risk. Mo et al. [63] reviewed accumulating preclinical and clinical data showing that NLRP3 activation in macrophages, smooth muscle cells, endothelial cells, and cardiomyocytes contributes to lesion progression, fibrosis, and maladaptive remodeling even when lipid levels are reduced. Importantly, elevated IL-1β and IL-18 remain associated with adverse cardiovascular outcomes despite [LDL-C] lowering, consistent with the inflammatory plateau described in the SFE framework. The review also highlights that NLRP3 activation depends on multiple signals, including oxidative stress, mitochondrial dysfunction, and ionic flux, that are not addressed by statin therapy. This mechanistic independence from cholesterol burden provides a clear rationale for adjunctive therapeutic strategies, such as direct NLRP3 inhibition, which have shown benefit in preclinical models and are entering translational development.
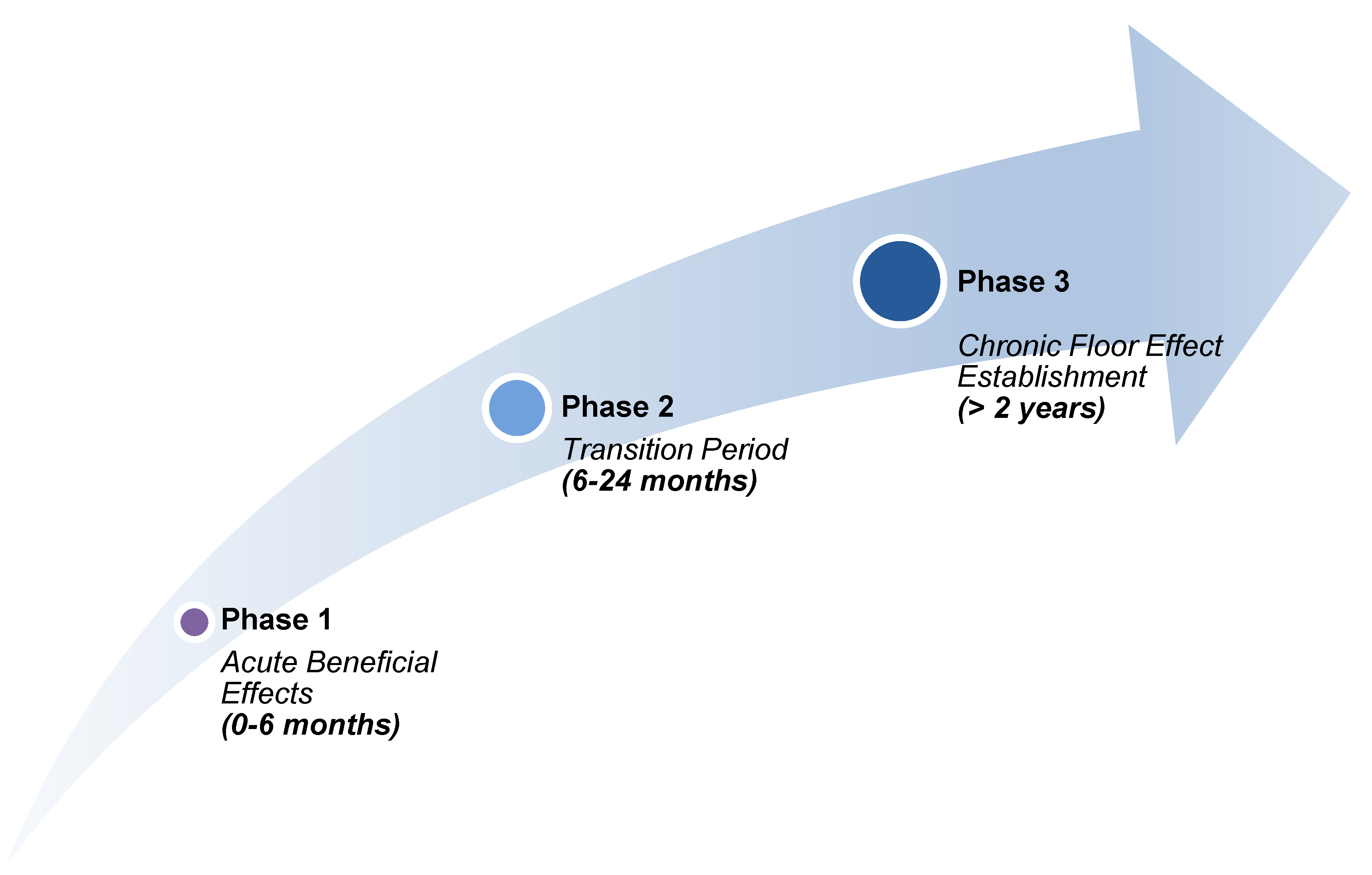
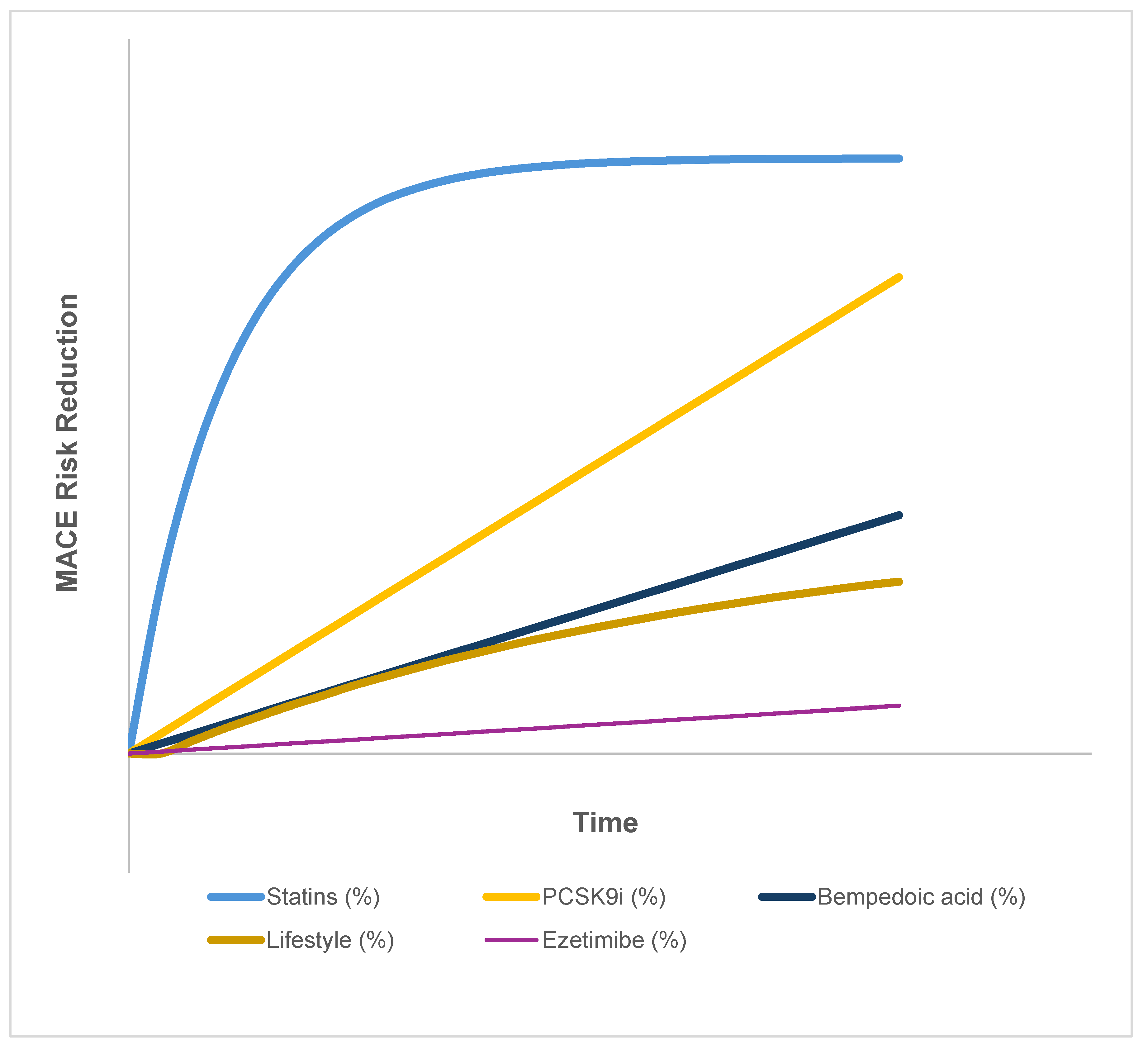
These findings indicate that while statins provide a rapid early reduction in risk, their long-term benefit plateaus in a way not observed with newer agents or lifestyle interventions. This pattern is summarized in Table 1 and illustrated in the hypothetical trajectories of Figure 3, which emphasize the differing temporal dynamics across therapeutic classes.

Table 1.
Temporal MACE Reduction Profiles by Therapy Class.
| Therapy Class | Trial(s) | Time for Initial Risk Reduction | Temporal Pattern | Plateau Observed | Notes |
|---|---|---|---|---|---|
| Statins (moderate–high dose) | CTT meta-analysis [2,3,26] | Within 3–6 months | Rapid early ↓, then plateau | Yes | Clear early benefit, but ~75% of events still occur despite [LDL-C] <1.4 mmol/L |
| PCSK9 inhibitors | FOURIER, ODYSSEY, ORION OUTCOMES [31,64,65] | 12–24 months | Progressive over time | No | Model-informed analyses calibrated to data show ongoing divergence of events over 2–3+ years, consistent with trial observations [66] |
| Ezetimibe | IMPROVE-IT [67] | ~24 months | Delayed, modest effect | Mild | Additive to statins; risk reduction ~6% over 6 years |
| Bempedoic acid | CLEAR Outcomes [61] | ≥24 months | Linear, steady | No | Benefit emerges gradually, consistent with an early plateau in statin curves, and raises the question of a statin-linked inflammatory floor |
| Lifestyle interventions | PREDIMED, PURE, observational cohorts [68,69,70] | 2–5 years | Slow, cumulative | Not applicable | Heterogeneous effects: within 2–5 year studies, no clear plateau is observed, but longer-term data are limited |
Statins demonstrate an early, steep reduction in major adverse cardiovascular events (MACE) that plateaus within 3–6 months, leaving ~75% of residual risk despite optimal [LDL-C] suppression. In contrast, non-statin agents and lifestyle interventions display more gradual or sustained trajectories without the early plateau, highlighting mechanistic differences that may relate to inflammasome activation, mitochondrial dysfunction, and other statin-linked adaptive processes.
Figure 3.
Hypothetical MACE Reduction Risk Trajectories Illustrative risk reduction curves are shown over time for statins, PCSK9 inhibitors, bempedoic acid, ezetimibe, and lifestyle interventions. Statins demonstrate an early, steep reduction in major adverse cardiovascular events (MACE) that plateaus over time, whereas non-statin therapies and lifestyle measures exhibit more gradual, additive, or linear trajectories. The figure highlights the relative differences in temporal dynamics between lipid-lowering and adjunctive approaches.
Figure 3.
Hypothetical MACE Reduction Risk Trajectories Illustrative risk reduction curves are shown over time for statins, PCSK9 inhibitors, bempedoic acid, ezetimibe, and lifestyle interventions. Statins demonstrate an early, steep reduction in major adverse cardiovascular events (MACE) that plateaus over time, whereas non-statin therapies and lifestyle measures exhibit more gradual, additive, or linear trajectories. The figure highlights the relative differences in temporal dynamics between lipid-lowering and adjunctive approaches.
Secondary Pathways5
- Statin-induced NLRP3 activation → systemic IL-1β elevation
- IL-1β → worsens insulin sensitivity [71]
- Insulin resistance → further chronic inflammation [72]
- Human and translational studies show statins can aggravate insulin resistance by reducing circulating GLP-1 via a Clostridium–Ursodeoxycholic acid (UDCA)–bile-acid axis; UDCA supplementation restores GLP-1 and improves glycemia [13]
- Red blood cell (RBC)-derived extracellular vesicles (EV) in diabetes: Glycated or fragile RBCs release vesicles enriched in arginase-1, which are taken up by endothelial cells to suppress nitric oxide bioavailability and increase reactive oxygen species, driving endothelial dysfunction and inflammation [73]. In the statin context, where diabetes risk is elevated, such RBC-EV-mediated insults may synergize with impaired metabolic resilience to sustain a permissive vascular milieu for lipid retention and inflammasome activation.
- Recent reviews underscore that statins, along with other cardiovascular agents, can directly impair insulin secretion, β-cell survival, and adipocyte signaling, producing drug-induced hyperglycemia and new-onset diabetes [74]. These glycemic perturbations not only reinforce insulin resistance but also create a permissive metabolic milieu for persistent inflammasome activation, amplifying the inflammatory floor effect despite optimal LDL-C lowering.
- A comprehensive mechanistic review [75] consolidates evidence from trials, observational cohorts, and molecular studies showing that chronic statin therapy can precipitate new-onset type 2 diabetes via multiple convergent mechanisms: impaired Ca²⁺ signaling in pancreatic β-cells, downregulation of GLUT4 in adipocytes, disrupted insulin signaling in peripheral tissues, and microRNA/epigenetic modulation. These pathways may amplify the residual inflammatory floor by increasing metabolic stress, promoting oxidative flux and inflammasome priming, reinforcing the SFE construct beyond lipid regulation.
- Metabolic dysfunction amplifies pro-inflammatory cytokine production [77]
- A self-reinforcing cycle maintains the inflammatory floor [77]
Tissue-Specific Context Dependency
Additional Amplification by Remnant Lipoproteins
Beyond oxidized phospholipids and [Lp(a)], residual cardiovascular risk is also propagated by triglyceride-enriched remnant lipoproteins, including very-low-density lipoprotein (VLDL) remnants and intermediate-density lipoproteins (IDL). Their cholesterol cargo is now recognized as approximately twice as atherogenic as LDL-C. These particles penetrate the arterial wall, are avidly taken up by macrophages without prior oxidative modification and amplify endothelial inflammation and foam cell persistence. Their shared structural backbone positions ApoB as a unifying biomarker of atherogenic burden across LDL, Lp(a), and remnants, consistent with an emerging consensus that residual risk persists even when [LDL-C] is well controlled [79]. Novel adjunctive approaches targeting ApoB or remnant metabolism, such as angiopoietin-like protein 3 (ANGPTL3) or apolipoprotein C3 (ApoC3) inhibitors, evinacumab, or icosapent ethyl, provide a mechanistic rationale for therapies designed to bypass the statin floor.
Parallel Amplification Pathways
Having outlined the core mechanistic pathways, including NLRP3 inflammasome activation, protein prenylation disruption, adaptive immune-macrophage crosstalk, and Lp(a) amplification, the following section examines the histological mapping and clinical correlates that substantiate each facet of the SFE (see Figure 4).
Figure 4.
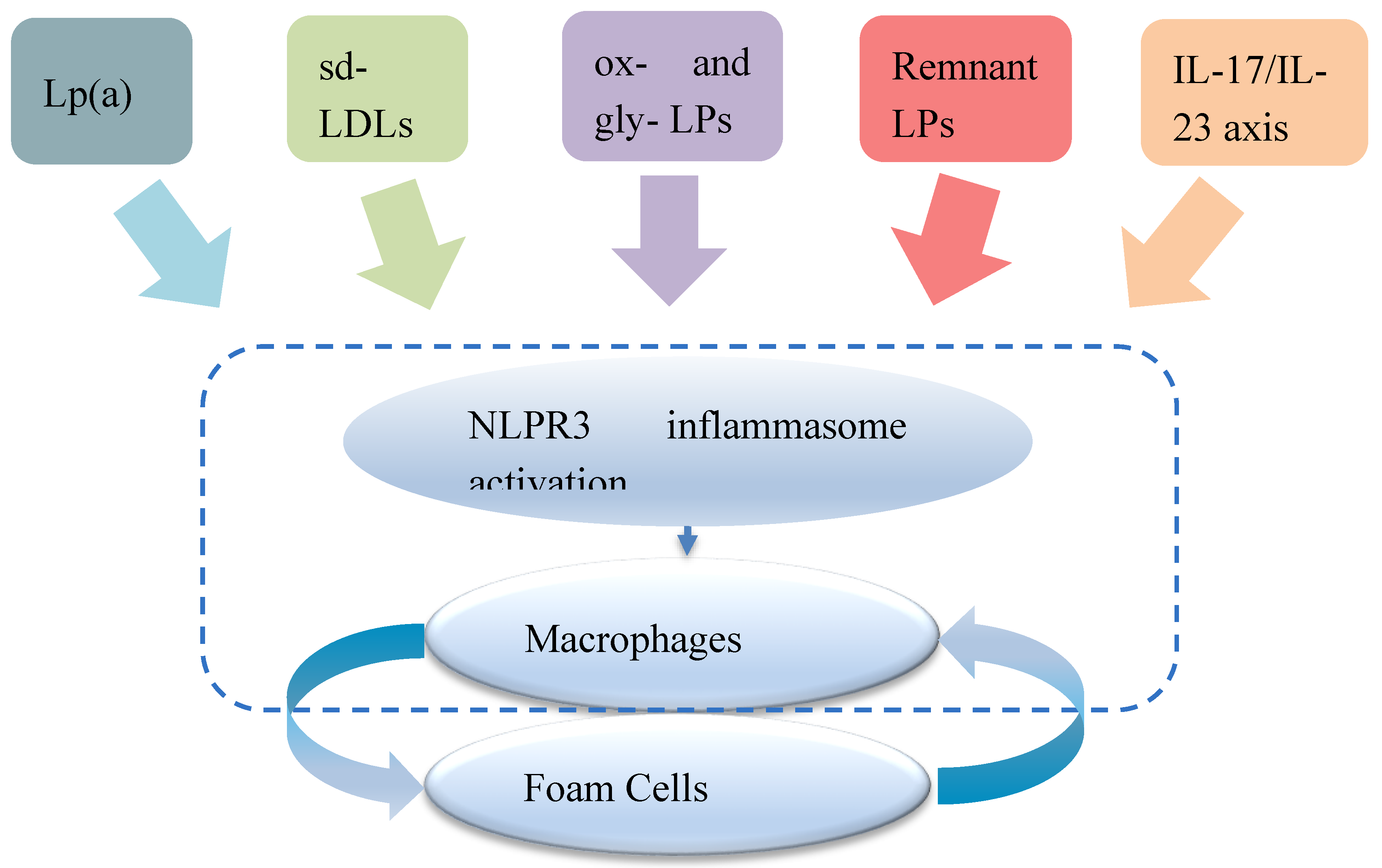
Parallel amplification pathways converging on the inflammatory floor Schematic illustrating distinct axes, lipoprotein(a), including oxidized phospholipids (Lp(a)–OxPL), oxidized and glycated lipoproteins (ox-LPs and gly-LPs), small dense low-density lipoprotein (sd-LDL), remnant lipoproteins (remnant LPs), and IL-17/IL-23 signaling, converging on activation NLRP3 inflammasome in arterial macrophages, thereby promoting foam cell persistence.Lp(a): Carries OxPLs that serve as damage-associated molecular patterns, directly triggering NLRP3 and IL-1β release in macrophages [45,46,51]; Apo(a) kringle repeat modulates OxPL burden [80,81] sd-LDL: Prone to oxidative modification and increased endothelial retention; engages lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) and cluster of differentiation 36 (CD36), generating reactive oxygen species and activating NF-κB [82,83,84] Remnant LPs: Cholesterol-rich remnants penetrate the intima, activate TLR2/4 on macrophages, and impair cholesterol efflux via ATP-binding cassette sub-family A member 1 (ABCA1) downregulation [85,86] IL-17/IL-23 axis: Vascular Th17 cells secrete IL-17A/IL-22, upregulating MMPs and downregulating IL-10 in macrophages, thereby sustaining foam cell survival [41,42,43,44].
Figure 4.
Parallel amplification pathways converging on the inflammatory floor Schematic illustrating distinct axes, lipoprotein(a), including oxidized phospholipids (Lp(a)–OxPL), oxidized and glycated lipoproteins (ox-LPs and gly-LPs), small dense low-density lipoprotein (sd-LDL), remnant lipoproteins (remnant LPs), and IL-17/IL-23 signaling, converging on activation NLRP3 inflammasome in arterial macrophages, thereby promoting foam cell persistence.Lp(a): Carries OxPLs that serve as damage-associated molecular patterns, directly triggering NLRP3 and IL-1β release in macrophages [45,46,51]; Apo(a) kringle repeat modulates OxPL burden [80,81] sd-LDL: Prone to oxidative modification and increased endothelial retention; engages lectin-like oxidized low-density lipoprotein receptor 1 (LOX-1) and cluster of differentiation 36 (CD36), generating reactive oxygen species and activating NF-κB [82,83,84] Remnant LPs: Cholesterol-rich remnants penetrate the intima, activate TLR2/4 on macrophages, and impair cholesterol efflux via ATP-binding cassette sub-family A member 1 (ABCA1) downregulation [85,86] IL-17/IL-23 axis: Vascular Th17 cells secrete IL-17A/IL-22, upregulating MMPs and downregulating IL-10 in macrophages, thereby sustaining foam cell survival [41,42,43,44].
Supporting Evidence
Molecular Mechanisms
- Histological mapping of human plaques confirms the persistence of macrophage-driven inflammation in morphologically stable lesions, reinforcing the concept of a durable, non-resolving inflammatory floor even in the absence of systemic flare markers [87].
- Disruption of Rac1 and protein prenylation alters macrophage phenotype [49]
- M2 macrophages retain pro-inflammatory characteristics under chronic statin exposure [88]
- Foam cells are maintained due to impaired mobility and IL-1β-mediated LDL uptake [54]
- Geranylgeraniol supplementation mitigates statin-induced inflammasome activation [89]
Clinical Correlates
- Dose-dependent diabetes risk with statin therapy; mechanistic human evidence indicates statin-associated GLP-1 reduction contributes to this phenotype [13]
- Colchicine reduces CV events by ~34% in statin-treated patients [6]
- Early vs late statin initiation shows different inflammatory profiles [32]
- Although statin therapy often reduces [CRP] to within the clinically acceptable range as measured by hs-CRP assays [91], concentrations may still exceed thresholds associated with arterial inflammation and cardiovascular risk. In other words, [CRP] falls, but not necessarily far enough to extinguish the inflammatory floor that sustains residual events. Meta-analytic evidence confirms that, even with optimal [LDL-C] reduction, many patients continue to experience cardiovascular events, often in association with persistent, albeit subclinical, elevations in inflammatory markers [23,32].
- PCSK9 inhibitors show diminishing returns in heavily statin-treated populations [92]
Biomarker Evolution Patterns
- Initial [CRP] reduction followed by plateau or slight increase, while IL-18 levels remain elevated and predictive of cardiovascular death in both stable and unstable angina [33]
- Persistent elevation of IL-18 in high-dose statin users [93]
- Divergence between systemic and arterial inflammatory markers [91]
- Elevated IL-1β and IL-18 remain predictive of adverse cardiovascular outcomes despite [LDL-C] lowering, consistent with persistent inflammasome activity [63]
The persistence of inflammatory signaling despite intensive [LDL-C] lowering indicates that the Statin Floor Effect represents a plausible molecular and cellular state, discernible through biomarker trajectories. This framework provides a rationale for therapeutic strategies that extend beyond statin intensification to address the immunometabolic mechanisms sustaining the floor.
4. Therapeutic Implications
The mechanistic framework suggests that residual cardiovascular risk persists because of a localized, statin-induced inflammatory floor. This recognition redirects therapeutic emphasis away from further intensification of statin monotherapy toward targeted combination approaches. The following predictions and implementation strategies are derived directly from the framework and highlight opportunities for earlier, more individualized interventions.
Refined Biomarker Strategy and Monitoring
Dynamic Biomarker Panel
A refined biomarker strategy is central to detecting and monitoring the inflammatory floor phenotype. Table 2 outlines a proposed dynamic panel that integrates inflammatory mediators, metabolic indices, and lipid-related markers to provide longitudinal resolution of residual risk.
Table 2.
Inflammatory and Metabolic Biomarkers Relevant to the Statin Floor Effect.
| Biomarker | Mechanistic Role | Interpretive Value | Preferred Monitoring |
|---|---|---|---|
| ApoB | Structural protein for all atherogenic lipoproteins | Direct measure of total atherogenic particle burden | Immunoassay; preferable to LDL-C in residual risk evaluation |
| IL-1β | Primary NLRP3 inflammasome effector cytokine | Direct marker of inflammasome activation and macrophage priming | Plasma assay (ELISA or multiplex) |
| IL-18 | Co-product of NLRP3 activation | Correlation with plaque activity; elevated in persistent risk | Plasma assay; potential for plaque-specific imaging correlation |
| IL-18/IL-10 Ratio | Balance between inflammation and resolution | Indicator of inflammatory floor and impaired resolution capacity | Calculated from serum cytokine panel |
| TNF-α | Promotes foam cell persistence, metabolic dysfunction | Marker of chronic plaque inflammation and macrophage dysfunction | Serum or plasma; longitudinal tracking preferred |
| CRP | Downstream IL-6–driven acute-phase reactant | Systemic surrogate for residual inflammatory risk | Standard clinical immunoassay |
| Adiponectin | Anti-inflammatory adipokine is suppressed in insulin resistance | Negative correlation of NLRP3 activation | ELISA or multiplex adipokine panel |
| HOMA-IR | Composite index of insulin resistance | Proxy for systemic metabolic reinforcement of NLRP3 | Derived from fasting insulin and glucose |
| GLP-1 | Incretin reduced by statins via microbiota–bile acid axis | Lower levels flag insulin-resistance amplification under statins; candidate for UDCA rescue | Fasting plasma GLP-1 (stabilized tubes); paired with HOMA-IR |
Functional Assays
- Ex vivo macrophage polarisation capacity
- LDL uptake studies
- Inflammasome activation assays
Precision Monitoring Strategy
- Genetic screening: 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMGCR), NLRP3, IL-1β, Rac1 variants
- Baseline phenotyping: Metabolic and inflammatory profile assessment
- Longitudinal tracking: Biomarker evolution over therapy duration
- Imaging correlation: Arterial inflammation persists despite systemic reduction8 [95]; advanced PET tracers such as ¹⁸F-NaF (detecting microcalcification) and ⁶⁸Ga-DOTATATE or ⁶⁸Ga-PentixaFor (targeting activated macrophages or C-X-C chemokine receptor type 4 (CXCR4) expression) provide tissue-specific insight into persistent plaque activity, helping distinguish inflammatory floor regions from systemic responses [91,96]
The mechanistic framework outlined above generates a set of predictions that can be evaluated in clinical cohorts and experimental systems. These predictions span the clinical, mechanistic, and interventional domains, linking biomarker trajectories with cellular behavior and therapeutic outcomes. By formalizing them, the framework provides a pathway from conceptual synthesis to empirical validation.
Testable Predictions
Clinical Predictions
- On maximal-dose statin therapy, IL-1β/IL-18 will fall initially in step with [LDL-C], but after 12–24 months will plateau or rebound, so that at long-term steady-state (≥2 years), patients have higher IL-1β/IL-18 despite [LDL-C] remaining low
- Carriers of HMGCR or NLRP3 variants show greater susceptibility to the inflammatory floor effect
- Arterial inflammation persists on PET imaging despite systemic normalization
- Statin-induced diabetics show higher arterial inflammatory markers independent of glycemic control
Mechanistic Predictions
- Statin-treated macrophages express M2 markers but retain IL-1β secretion9
- Geranylgeraniol supplementation suppresses IL-1β without altering [LDL-C]
- Early vs late statin exposure yields distinct epigenetic and inflammatory profiles
- Anti-inflammatory agents benefit genetically susceptible patients more than average
Intervention Predictions
- Early combination therapy prevents inflammatory floor effect establishment
- Metformin co-therapy lowers localized IL-1β levels and diabetes risk
- Biomarker-guided statin dosing improves risk/benefit balance
- Genetic profiling enables earlier combination treatment
- Adjunct UDCA will restore GLP-1 and attenuate elevated blood glucose and insulin-resistance amplification in patients on high-intensity statins [13]
Clinical Implementation Strategy
Translating these mechanistic insights into practice requires a structured approach to patient stratification and therapy sequencing. Figure 5 summarizes a proposed clinical implementation framework that links risk tiers to biomarker-guided monitoring and combination treatment strategies.
Figure 5.
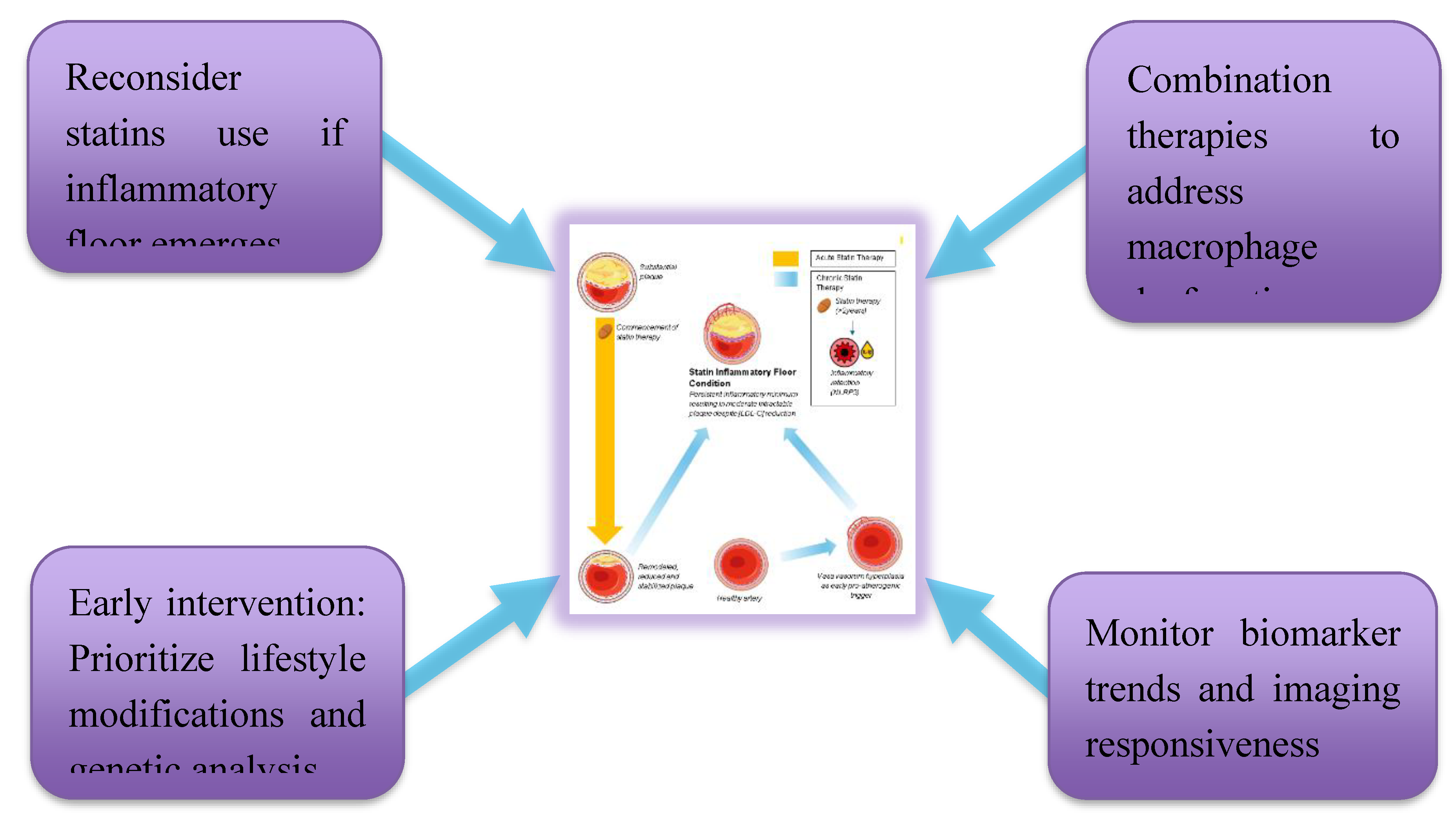
Proposed Clinical Implementation Strategies Framework linking early lifestyle modification, genetic risk assessment, and biomarker-guided monitoring to individualized treatment decisions. The figure emphasizes reconsideration of high-intensity statin therapy if an inflammatory floor phenotype emerges, the use of imaging and biomarker trends to guide precision intervention, and the role of combination therapies targeting macrophage dysfunction.
Figure 5.
Proposed Clinical Implementation Strategies Framework linking early lifestyle modification, genetic risk assessment, and biomarker-guided monitoring to individualized treatment decisions. The figure emphasizes reconsideration of high-intensity statin therapy if an inflammatory floor phenotype emerges, the use of imaging and biomarker trends to guide precision intervention, and the role of combination therapies targeting macrophage dysfunction.
Risk Stratification Approach
- Low Risk: Statin monotherapy, routine monitoring
- Moderate Risk: Enhanced biomarker tracking, conditional combination therapy
- High Risk: Immediate combination therapy and intensive monitoring
Precision Medicine Integration
- One off determination of [LP(a)]
- Genetic testing to identify susceptible variants
- Metabolic and inflammatory baseline profiling
- Individualized ApoB, LDL-C, and inflammatory targets
- Dynamic treatment adjustments
Therapeutic Sequencing
- Pre-Phase (Structural Priming and Vascular Neogenesis)In the SFE framework, adventitial vasa vasorum hyperplasia and diffuse intimal hyperplasia prime arteries for lipid retention and inflammation, Yang et al. showed in rabbits that adventitial vasa vasorum proliferation precedes intima–media thickening and predicts plaque development [102]. Following this, diffuse intimal hyperplasia (DIH), as described by Subbotin, may further predispose to lipid retention by increasing oxygen diffusion distance and creating localized hypoxia. This facilitates smooth muscle cell phenotypic modulation and extracellular matrix accumulation [103]. Together, adventitial vasa vasorum and DIH can create a permissive terrain upon which lipids accumulate and immune processes initiate. Identifying and interrupting these early, pre-lipid stages may offer an opportunity for pre-emptive vascular therapy in at-risk individuals.
- Phase 1 (0–6 months): Statin therapy initiation
- Phase 2 (6–24 months): Monitor for inflammatory floor effect
- Phase 3 (>2 years): Maintain combination therapy for at-risk individuals
Research Priorities and Study Design
Building on the predictions derived from the framework, specific research priorities can be defined to test, refine, and extend the model. These include longitudinal human studies, targeted intervention trials, mechanistic validation in preclinical systems, and integrative modeling approaches. Together, these strategies provide a roadmap for translating the SFE concept into evidence-based clinical practice.
Longitudinal Mechanistic Studies
- 5-year biomarker tracking studies
- Stratified cohorts by genetic risk
- Human arterial tissue sampling
- PET imaging to correlate markers with inflammation
Intervention Trials
- Early statin + anti-inflammatory randomized controlled trials
- Biomarker-guided versus standard care dosing
- Metformin combination trials
- Personalized therapy based on genomic risk
Mechanistic Validation
- Murine models to examine NLRP3–foam cell trajectory
- Ex vivo validation of macrophage phenotypes
- Prospective validation of biomarker panels
Temporal Stratification Mode Modeling Research
A multiscale hybrid model, combining statin pharmacokinetics, adherence, and plaque biomechanics, predicts plateaued regression and persistent plaque despite ongoing therapy, implying unmodeled resistance factors and, even without NLRP3 detail, supporting a residual inflammatory floor [104]. Several pharmacologic and systems-biology models demonstrate nonlinear, plateauing statin responses aligned with the framework; notably, Mylonas et al.’s fractional-calculus dose-response model identified thresholds where lipid-lowering plateaus and cellular toxicity/oxidative stress emerge, consistent with in vitro inflammasome priming [28]. Separately, Lei et al. modeled the growth of necrotic cores in atherosclerotic plaques under statin influence, showing that while progression slows, it rarely reverses completely, reinforcing the concept of partial resolution limitations central to the SFE construct [105]. While these models do not simulate NLRP3 activation or T-cell polarization explicitly, their combined outcomes support a multidimensional framework in which statin efficacy is modulated by nonlinear biology, treatment timing, and tissue-specific effects.
Clinical Implications and Future Directions
The framework also carries immediate clinical relevance and longer-term implications for cardiovascular medicine. By guiding risk stratification, informing therapeutic combinations, and shaping precision medicine approaches, it has the potential to reshape both patient care and research priorities. Future directions extend beyond statin therapy alone, toward broader immunometabolic strategies and innovative therapeutic development.
Immediate Clinical Applications
- Biomarker screening for residual inflammatory risk
- Early combination therapy consideration
- Avoid unnecessary statin up-titration in the inflammatory floor effect phenotype
- Better risk communication strategies
Long-Term Paradigm Shifts
- Broaden to immunometabolic cardiology
- Genetically informed prevention models
- Simultaneous targeting of lipid, metabolic, and immune pathways
- Integrated biomarker and imaging-based care models
- While NLRP3 remains the most extensively characterized inflammasome in atherosclerosis, other complexes such as absent in melanoma 2 and NOD-like receptor family CARD domain containing 4 may also contribute to vascular inflammation [53]. Recognizing the pivotal role of adaptive immunity in atherosclerosis, future paradigms may shift towards immunomodulatory therapies [50,106]. Their integration into the SFE framework awaits further mechanistic clarity.
Therapeutic Innovation Opportunities
- NLRP3-specific anti-inflammatory therapies
- Fixed-dose statin + anti-inflammatory combinations
- Genetic testing panels for cardiovascular risk
- Point-of-care inflammatory biomarker devices
- Adventitial vasa vasorum modulation: Early detection and targeting of proliferation, via ultrasound or contrast-enhanced imaging, may offer a window to prevent downstream plaque formation before intimal thickening or immune infiltration
- Structural-stage targeting: Therapeutic modulation of DIH and matrix remodeling, especially in metabolically susceptible individuals, may delay or prevent the creation of lipid-retentive terrain. This approach expands risk-reduction efforts upstream of lipid-lowering or anti-inflammatory therapies.
- Targeted modulation of Lp(a)-associated risk - is now achievable through gene-silencing therapies such as pelacarsen (antisense oligonucleotide), olpasiran and zerlasiran (small interfering RNAs, siRNAs), which have been shown to reduce [Lp(a)] by up to 90% in clinical trials. These agents provide precision strategies for genetically mediated [Lp(a)] elevation.
- Statins can variably raise [Lp(a)] in some individuals, though this effect is not universal and may not always be clinically significant
- Novel agents like muvalaplin (an oral inhibitor of Apo(a)-ApoB binding) offer non-injectable alternatives
- While colchicine does not reduce [Lp(a)], it may blunt downstream inflammation triggered by Lp(a)-bound OxPLs, offering potential therapeutic synergy in high-risk inflammatory phenotypes
Why This Framework Matters
Clinical Relevance
- Explains paradoxes in residual cardiovascular risk
- Enables identification of patients needing intensified therapy
- Enhances clinical efficiency via better risk stratification
- Supports enhanced cardiovascular disease risk reduction
Scientific Impact
- Postulates a unifying mechanistic model
- Encourages precision research designs
- Enhances biomarker development
- Sets the stage for future immunometabolic therapies
Therapeutic Potential
- RNA-silencing approaches: Recent preclinical studies demonstrate that hepatocyte-targeted siRNA and GalNAc-conjugated oligonucleotides against Farnesyl-diphosphate Farnesyltransferase 1 (FDFT1) achieve >70% knockdown of squalene synthase mRNA, reduce plasma [LDL-C] by 30–40%, and attenuate atherosclerotic lesion development in animal models. Early human Phase I data with GalNAc–FDFT1 siRNA suggest durable [LDL-C] lowering with quarterly subcutaneous dosing and a favorable safety profile [107,108].
- Combination with Statins: Preclinical co-administration of low-dose statin plus FDFT1 siRNA yielded additive [LDL-C] lowering (~60-70% total) and greater atherosclerotic plaque regression than either alone, supporting a dual-mechanism strategy to overcome the inflammation floor of statin monotherapy
- Targeting the rescue of the impaired synthesis of MK-4: a form of vitamin K₂ that modulates inflammatory resolution and mitochondrial homeostasis - although direct clinical evidence is lacking, murine studies show that statins can suppress MK-4 formation in extrahepatic tissues. MK-4 deficiency impairs activation of matrix Gla protein (MGP), promoting microcalcification, and may enhance NLRP3 inflammasome activity by removing antioxidant and anti-NF-κB restraints [109]. Observational human data further link low circulating [MK-4] with greater coronary calcification, suggesting a plausible translational mechanism [110]. Thus, MK-4 depletion may represent a second direct molecular axis, alongside geranylgeranyl gyrophosphate / Rac1 disruption, through which statins promote a persistent inflammatory floor within atherosclerotic lesions. This again illustrates the paradoxical effect of statins in that they also enhance plaque stabilization through calcification.
- FDFT1 silencing: limits squalene-derived isoprenoids, reducing macrophage endoplasmic reticulum stress and NLRP3 activation, offering a novel strategy to target the inflammatory floor
- IL-10: may drive post-MI plaque regression and myocardial remodeling by enhancing M2 polarization, suppressing MMP-9, and boosting mitochondrial function, but its efficacy falls off above ~1 µg/mL, highlighting the need for controlled delivery [111]
- Eicosapentaenoic acid (EPA)-driven plaque reduction: a meta-analysis of 23 intravascular ultrasound trials showed each 1% decrease in percent atheroma volume was linked to a 19% lowering of MACE [112], and in statin-treated coronary artery disease patients, a higher ratio of EPA-derived mediators (18-hydroxyeicosapentaenoic acid + resolvin E1) to leukotriene B₄ strongly predicted actual plaque regression [113]
- New gene-editing approaches: such as VERVE-102, which offers durable, possibly lifelong PCSK9 silencing in humans, may further decouple [LDL-C] lowering from the immunometabolic disruptions observed with statins and thus represent an ideal platform for combination therapy targeting inflammatory floor mechanisms [114]
- Unique protein dysregulations: opportunities lie in their discovery by the application of proteomics mass spectrometry, paving the way to new therapies and mechanistic insights. Early spatially resolved proteomic studies of human plaques [115] (preprint) suggest that arterial PCSK9 secretion, ECM remodeling, and region-specific inflammatory pathways may be particularly tractable targets, underscoring the value of local proteomic mapping for future therapy development.
These therapeutic strategies and research priorities emphasize the SFE as a clinically relevant and experimentally tested construct, setting the stage for the conclusion of its greater significance.
5. Conclusions
The Statin Floor Effect framework integrates clinical and molecular observations to account for the early reduction in cardiovascular events with high-dose statin therapy and the subsequent plateau in benefit. Beyond [LDL-C] lowering, prolonged statin exposure may plausibly establish a localized arterial inflammatory state, driven by NLRP3 inflammasome activation, adaptive immune modulation, and depletion of metabolic cofactors, that constrains further risk reduction. Framed in this way, the construct offers both a coherent explanation for long-standing therapeutic paradoxes and a foundation for precision strategies aimed at overcoming residual cardiovascular risk.
Some aspects remain to be substantiated in larger human studies, but their inclusion highlights valuable avenues for mechanistic investigation and the identification of novel therapeutic targets. Clarifying the relative contribution of each mechanism will be essential to refining the model and informing intervention design.
While non-statin interventions often demonstrate extended, near-linear benefit within 2–5 year studies, their long-term trajectories may also converge toward asymptotic limits beyond typical follow-up durations. From a clinical perspective, the framework suggests that optimal risk reduction is likely to require combining lipid-lowering with interventions that ameliorate residual inflammation and metabolic dysregulation. Prospective studies incorporating serial biomarker assessments and carefully timed adjunct therapies will be essential to test the Statin Floor Effect and to determine whether addressing the “inflammatory floor” can extend and complement the benefits of statin treatment.
Declaration of Generative AI and AI-Assisted Technologies in the Writing Process
During the preparation of this work, the author used ChatGPT (OpenAI) to improve readability, refine section layouts, and offer discipline-specific terminology and editing suggestions. No figures or tables were produced by AI. After using this support, the author carefully reviewed, edited, and validated as needed, and assumes full responsibility for the content of the publication.
Author Contributions
E.J.P. conceived the framework, performed the literature review, and drafted and revised the manuscript.
Acknowledgements
The author thanks the University of Tasmania for providing subscription-journal access for retired staff members, which supported the literature review for this work. The author also gratefully acknowledges Dr Michael McCarthy, FRACGP, for sharing an unpublished discussion paper that resonated with independently developing ideas about persistent arterial inflammation in atherosclerosis. His framing of residual cardiovascular risk and his highlighting of Subbotin’s hyperplasia observations catalyzed a deeper exploration of statin therapy as a potential mechanistic contributor to that inflammatory persistence, ultimately refining the central framework presented here. Artery renders in the graphical abstract were adapted from Servier Medical Art (https://smart.servier.com/), licensed under CC BY 4.0 (https://creativecommons.org/licenses/by/4.0/), using Inkscape 1.4.2.
References
- Cannon Christopher, P.; Braunwald, E.; McCabe Carolyn, H.; Rader Daniel, J.; Rouleau Jean, L.; Belder, R.; etal., *!!! REPLACE !!!*. Intensive versus Moderate Lipid Lowering with Statins after Acute Coronary Syndromes. New England Journal of Medicine 2004, 350, 1495–1504. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.D.; Guyton, J.R.; Herrington, D.M.; McGowan, M.P.; Wenger, N.K.; Shear, C.; Shear C. Treating to New Targets (TNT) Study: Does lowering low-density lipoprotein cholesterol levels below currently recommended guidelines yield incremental clinical benefit? Am J Cardiol 2004, 93, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Cholesterol Treatment Trialists, C. Efficacy and safety of more intensive lowering of LDL cholesterol: A meta-analysis of data from 170,000 participants in 26 randomised trials. The Lancet 2010, 376, 1670–1681. [Google Scholar] [CrossRef]
- Navarese, E.P.; Robinson, J.G.; Kowalewski, M.; Kolodziejczak, M.; Andreotti, F.; Bliden, K.; etal., *!!! REPLACE !!!*. Association Between Baseline LDL-C Level and Total and Cardiovascular Mortality After LDL-C Lowering: A Systematic Review and Meta-analysis. Jama 2018, 319, 1566–1579. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ridker, P.M.; Everett, B.M.; Thuren, T. Antiinflammatory therapy with canakinumab for atherosclerotic disease. New England Journal of Medicine 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- Nidorf, S.M.; Fiolet, A.T.L.; Mosterd, A. Colchicine in patients with chronic coronary disease. New England Journal of Medicine 2020, 383, 1838–1847. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; etal., *!!! REPLACE !!!*. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; etal., *!!! REPLACE !!!*. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine 2015, 21, 628–637. [Google Scholar] [CrossRef]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; FrancisGA. Contribution of Intimal Smooth Muscle Cells to Cholesterol Accumulation and Macrophage-Like Cells in Human Atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef]
- Henriksbo, B.D.; Tamrakar, A.K.; Phulka, J.S.; Barra, N.G.; Schertzer, JD. Statins activate the NLRP3 inflammasome and impair insulin signaling via p38 and mTOR. American Journal of Physiology-Endocrinology and Metabolism 2020, 319, E110–E116. [Google Scholar] [CrossRef]
- Koushki, K.; Shahbaz, S.K.; Mashayekhi, K.; Sadeghi, M.; Zayeri, Z.D.; Taba, M.Y.; et al. Anti-inflammatory Action of Statins in Cardiovascular Disease: The Role of Inflammasome and Toll-Like Receptor Pathways. Clin Rev Allergy Immunol 2021, 60, 175–199. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sirtori, C.R.; Watts, G.F.; Corsini, A.; Ruscica, M. Understanding the molecular mechanisms of statin pleiotropic effects: The unresolved puzzle. Archives of Toxicology 2023, 97, 2861–2887. [Google Scholar] [CrossRef]
- She, J.; Tuerhongjiang, G.; Guo, M.; Liu, J.; Hao, X.; Guo, L.; et al. Statins aggravate insulin resistance through reduced blood glucagon-like peptide-1 levels in a microbiota-dependent manner. Cell Metabolism 2024, 36, 408–421.e5. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.J.; Chien, S. Effects of disturbed flow on vascular endothelium: Pathophysiological basis and clinical perspectives. Physiological Reviews 2011, 91, 327–387. [Google Scholar] [CrossRef]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef]
- Zhou, X.; Li, C.; Xu, W. Shear stress activates NLRP3 inflammasome to promote endothelial inflammation. Journal of Cellular Physiology 2020, 235, 491–500. [Google Scholar] [CrossRef]
- Ichihara, A.; Hayashi, M.; Koura, Y.; Tada, Y.; Kaneshiro, Y.; Saruta, T. Long-term effects of statins on arterial pressure and stiffness of hypertensives. Journal of Human Hypertension 2005, 19, 103–109. [Google Scholar] [CrossRef]
- Liao, J.K.; Seto, M.; Noma, K. Rho Kinase (ROCK) Inhibitors. Journal of Cardiovascular Pharmacology 2007, 50. [Google Scholar] [CrossRef]
- Liao, J.K.; Laufs, U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol 2005, 45, 89–118. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gierig, M.; Tragoudas, A.; Haverich, A.; Wriggers, P. Mechano-chemo-biological model of atherosclerosis formation based on the outside-in theory. Biomechanics and Modeling in Mechanobiology 2024, 23, 539–552. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, H.; Hou, Q.; Zhang, Y.; Qiao, A. Multiscale Modeling of Vascular Remodeling Induced by Wall Shear Stress. Front Physiol 2021, 12, 808999. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Sehayek, D.; Cole, J.; Björnson, E.; Wilkins, J.T.; Mortensen, M.B.; Dufresne, L.; et al. ApoB, LDL-C and non-HDL-C as Markers of Cardiovascular Risk. Journal of Clinical Lipidology. 2025. [CrossRef] [PubMed]
- Mortensen, MB. ApoB triumphs once more over LDL-C and non-HDL-C in risk prediction: Ready for guidelines? European Heart Journal 2024, 45, 2419–2421. [Google Scholar] [CrossRef] [PubMed]
- Abohelwa, M.; Kopel, J.; Shurmur, S.; Ansari, M.M.; Awasthi, Y.; Awasthi, S. The Framingham Study on Cardiovascular Disease Risk and Stress-Defenses: A Historical Review. Journal of Vascular Diseases 2023, 2, 122–164. [Google Scholar] [CrossRef]
- Sabeel, S.; Motaung, B.; Nguyen, K.A.; Ozturk, M.; Mukasa, S.L.; Wolmarans, K.; et al. Impact of statins as immune-modulatory agents on inflammatory markers in adults with chronic diseases: A systematic review and meta-analysis. PLoS ONE 2025, 20, e0323749. [Google Scholar] [CrossRef]
- Ridker Paul, M.; Danielson, E.; Fonseca Francisco, A.H.; Genest, J.; Gotto Antonio, M.; Kastelein John, J.P.; et al. Rosuvastatin to Prevent Vascular Events in Men and Women with Elevated C-Reactive Protein. New England Journal of Medicine 2008, 359, 2195–2207. [Google Scholar] [CrossRef]
- Sattar, N.; Preiss, D.; Murray, HM. Statins and risk of incident diabetes: A collaborative meta-analysis of randomised statin trials. Lancet 2010, 375, 735–742. [Google Scholar] [CrossRef]
- Mylonas, E.; Mamareli, C.; Filippakis, M.; Mamarelis, I.; Anastassopoulou, J.; Theophanides, T. A Mathematical Model of Statin Anti-Hyperlipidemic Drug Reactivity and Diverse Concentrations of Risk Toxicity. Journal of Clinical Medicine 2025, 14. [Google Scholar] [CrossRef]
- Mega, J.L.; Stitziel, N.O.; Smith, JG. Genetic risk, coronary heart disease events, and the clinical benefit of statin therapy: An analysis of primary and secondary prevention trials. Lancet 2015, 385, 2264–2271. [Google Scholar] [CrossRef]
- Baigent, C.; Keech, A.; Kearney, PM. Efficacy and safety of cholesterol-lowering treatment: Prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005, 366, 1267–1278. [Google Scholar] [CrossRef]
- Sabatine Marc, S.; Giugliano Robert, P.; Keech Anthony, C.; Honarpour, N.; Wiviott Stephen, D.; Murphy Sabina, A.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. New England Journal of Medicine 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Sposito, A.C.; Carvalho, L.S.; Cintra, R.M.; Araújo, A.L.; Ono, A.H.; Andrade, J.M.; et al. Rebound inflammatory response during the acute phase of myocardial infarction after simvastatin withdrawal. Atherosclerosis 2009, 207, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Cannon, C.P.; Morrow, D.; Rifai, N.; Rose, L.M.; McCabe, C.H.; et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med 2005, 352, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Choi, J.; Kim, S.G.; Kim, NH. Relative contributions of statin intensity, achieved low-density lipoprotein cholesterol level, and statin therapy duration to cardiovascular risk reduction in patients with type 2 diabetes: Population based cohort study. Cardiovascular Diabetology 2022, 21, 28. [Google Scholar] [CrossRef] [PubMed]
- Laufs, U.; La Fata, V.; Plutzky, J.; Liao, JK. Upregulation of Endothelial Nitric Oxide Synthase by HMG CoA Reductase Inhibitors. Circulation 1998, 97, 1129–1135. [Google Scholar] [CrossRef]
- Feng, Q.; Jia, S.; Zhou, H.; Liu, S.; Li, Y.; Ding, J.; et al. An Atorvastatin/Ferrostatin-1 Codelivered Hybrid Exosome/Liposome System for Combinational Ferroptosis Inhibition, Inflammation Suppression, Efferocytosis Promotion, and Macrophage Reprogramming in Atherosclerosis Treatment. ACS Applied Materials & Interfaces 2025, 17, 36542–36556. [Google Scholar] [CrossRef]
- Tabas, I.; Seimon, T.; Timmins, J.; Li, G.; Lim, W. Macrophage apoptosis in advanced atherosclerosis. Ann N Y Acad Sci. 2009, 1173 (Suppl. 1), E40–E45. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yurdagul, A.; Jr Doran, A.C.; Cai, B.; Fredman, G.; Tabas, IA. Mechanisms and Consequences of Defective Efferocytosis in Atherosclerosis. Front Cardiovasc Med. 2017, 4, 86. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Henriksbo, B.D.; Tamrakar, A.K.; Xu, J.; Duggan, B.M.; Cavallari, J.F.; Phulka, J.; et al. Statins Promote Interleukin-1β–Dependent Adipocyte Insulin Resistance Through Lower Prenylation, Not Cholesterol. Diabetes 2019, 68, 1441–1448. [Google Scholar] [CrossRef]
- Gao, Q.; Jiang, Y.; Ma, T.; Zhu, F.; Gao, F.; Zhang, P.; et al. A critical function of Th17 proinflammatory cells in the development of atherosclerotic plaque in mice. The Journal of Immunology 2010, 185, 5820–5827. [Google Scholar] [CrossRef]
- Eid, R.E.; Rao, D.A.; Zhou, J. Interleukin-17 and related cytokines induce proinflammatory cytokines and activate human vascular smooth muscle cells. Arteriosclerosis, Thrombosis, and Vascular Biology 2009, 29, 490–497. [Google Scholar] [CrossRef]
- Hansson, G.K.; Hermansson, A. The immune system in atherosclerosis. Nature Immunology 2011, 12, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nature Reviews Immunology 2006, 6, 508–519. [Google Scholar] [CrossRef] [PubMed]
- Erbel, C.; Akhavanpoor, M.; Okuyucu, D.; Wangler, S.; Dietz, A.; Zhao, L.; et al. IL-17A influences essential functions of the monocyte/macrophage lineage and is involved in advanced murine and human atherosclerosis. J Immunol 2014, 193, 4344–4355. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Que, X.; Hung, M.Y.; Yeang, C.; Gonen, A.; Prohaska, T.A.; Sun, X.; et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018, 558, 301–306. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Pirro, M.; Simental-Mendía, L.E.; Bianconi, V.; Watts, G.F.; Banach, M.; Sahebkar, A. Effect of Statin Therapy on Arterial Wall Inflammation Based on 18F-FDG PET/CT: A Systematic Review and Meta-Analysis of Interventional Studies. J Clin Med. 2019, 8. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tsimikas, S.; Gordts, P.; Nora, C.; Yeang, C.; Witztum, JL. Statin therapy increases lipoprotein(a) levels. Eur Heart J 2020, 41, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Sedding, D.G.; Boyle, E.C.; Demandt, J.A.F.; Sluimer, J.C.; Dutzmann, J.; Haverich, A.; et al. Vasa Vasorum Angiogenesis: Key Player in the Initiation and Progression of Atherosclerosis and Potential Target for the Treatment of Cardiovascular Disease. Front Immunol 2018, 9, 706. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Healy, A.M.; Terkeltaub, R.A.; Moore, K.J.; Yvan-Charvet, L. Statins disrupt macrophage Rac1 regulation leading to increased calcification in atherosclerotic plaques. Arteriosclerosis, Thrombosis, and Vascular Biology 2020, 40, 714–726. [Google Scholar] [CrossRef]
- Grebe, A.; Hoss, F.; Latz, E. NLRP3 Inflammasome and the IL-1 Pathway in Atherosclerosis. Circulation Research 2018, 122, 1722–1740. [Google Scholar] [CrossRef]
- Tsimikas, S.; Fazio, S.; Ferdinand, KC. Lipoprotein(a): Novel target and emerging biomarker for cardiovascular risk. Journal of the American College of Cardiology 2017, 69, 692–711. [Google Scholar] [CrossRef]
- Zhang, X.; Qin, Y.; Wan, X.; Liu, H.; Lv, C.; Ruan, W.; et al. Rosuvastatin exerts anti-atherosclerotic effects by improving macrophage-related foam cell formation and polarization conversion via mediating autophagic activities. Journal of Translational Medicine 2021, 19, 62. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Kanneganti, TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 2016, 213, 617–629. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Peled, M.; Fisher, EA. Dynamic aspects of macrophage polarization during atherosclerosis progression and regression. Frontiers in Immunology 2014, 5, 579. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.W.; Huang, L.H.; Randolph, GJ. Cytokine circuits in cardiovascular disease. Immunity 2019, 50, 941–954. [Google Scholar] [CrossRef]
- Greenwood, J.; Steinman, L.; Zamvil, SS. Statin therapy and autoimmune disease: From protein prenylation to immunomodulation. Nat Rev Immunol 2006, 6, 358–370. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dehnavi, S.; Sohrabi, N.; Sadeghi, M.; Lansberg, P.; Banach, M.; Al-Rasadi, K.; et al. Statins and autoimmunity: State-of-the-art. Pharmacology & Therapeutics 2020, 214, 107614. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, J.; Peng, X.; Ramgolam, V.S.; Markovic-Plese, S. Simvastatin inhibits IL-17 secretion by targeting multiple IL-17-regulatory cytokines and by inhibiting the expression of IL-17 transcription factor RORC in CD4+ lymphocytes. J Immunol 2008, 180, 6988–6996. [Google Scholar] [CrossRef] [PubMed]
- Kwak, B.; Mulhaupt, F.; Myit, S.; Mach, F. Statins as a newly recognized type of immunomodulator. Nat Med 2000, 6, 1399–1402. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.; Zhang, K.; Li, J.; Dong, M.; Yang, J.; An, G.; et al. Statins Induce the Accumulation of Regulatory T Cells in Atherosclerotic Plaque. Molecular Medicine 2012, 18, 598–605. [Google Scholar] [CrossRef]
- Li, Y.; Gao, H.; Zhao, J.; Ma, L.; Hu, D. Safety and efficacy of bempedoic acid among patients with statin intolerance and those without: A meta-analysis and a systematic randomized controlled trial review. PLoS ONE 2024, 19, e0297854. [Google Scholar] [CrossRef] [PubMed]
- Ryan, T.E.; Torres, M.J.; Lin, C.T.; Clark, A.H.; Brophy, P.M.; Smith, C.A.; et al. High-dose atorvastatin therapy progressively decreases skeletal muscle mitochondrial respiratory capacity in humans. JCI Insight. 2024, 9. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mo, B.; Ding, Y.; Ji, Q. NLRP3 inflammasome in cardiovascular diseases: An update. Frontiers in Immunology 2025, 16. [Google Scholar] [CrossRef] [PubMed]
- Schwartz Gregory, G.; Steg, P.G.; Szarek, M.; Bhatt Deepak, L.; Bittner Vera, A.; Diaz, R.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. New England Journal of Medicine 2018, 379, 2097–2107. [Google Scholar] [CrossRef]
- Ray, K.K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. New England Journal of Medicine 2020, 382, 1507–1519. [Google Scholar] [CrossRef]
- Wang, Y.; Courcelles, E.; Peyronnet, E.; Porte, S.; Diatchenko, A.; Jacob, E.; et al. Credibility assessment of a mechanistic model of atherosclerosis to predict cardiovascular outcomes under lipid-lowering therapy. npj Digital Medicine 2025, 8, 171. [Google Scholar] [CrossRef]
- Cannon, C.P.; Blazing, M.A.; Giugliano, RP. Ezetimibe added to statin therapy after acute coronary syndromes. New England Journal of Medicine 2015, 372, 2387–2397. [Google Scholar] [CrossRef]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.-I.; Corella, D.; Arós, F.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. New England Journal of Medicine 2018, 378, e34. [Google Scholar] [CrossRef]
- Kariuki, J.K.; Imes, C.C.; Engberg, S.J.; Scott, P.W.; Klem, M.L.; Cortes, YI. Impact of lifestyle-based interventions on absolute cardiovascular disease risk: A systematic review and meta-analysis. JBI Evidence Synthesis 2024, 22, 4–65. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yusuf, S.; Rangarajan, S.; Teo, K.; Islam, S.; Li, W.; Liu, L.; et al. Cardiovascular Risk and Events in 17 Low-, Middle-, and High-Income Countries. New England Journal of Medicine 2014, 371, 818–827. [Google Scholar] [CrossRef]
- Corrao, G.; Ibrahim, B.; Nicotra, F.; Soranna, D.; Merlino, L.; Catapano, A.L.; et al. Statins and the risk of diabetes: Evidence from a large population-based cohort study. Diabetes Care 2014, 37, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Berbudi, A.; Khairani, S.; Tjahjadi, AI. Interplay Between Insulin Resistance and Immune Dysregulation in Type 2 Diabetes Mellitus: Implications for Therapeutic Interventions. Immunotargets Ther 2025, 14, 359–382. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Collado, A.; Humoud, R.; Kontidou, E.; Eldh, M.; Swaich, J.; Zhao, A.; et al. Erythrocyte-derived extracellular vesicles induce endothelial dysfunction through arginase-1 and oxidative stress in type 2 diabetes. J Clin Invest. 2025, 135. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Widiarti, W.; Saputra, P.B.T.; Savitri, C.G.; Putranto, J.N.E.; Alkaff, FF. The impact of cardiovascular drugs on hyperglycemia and diabetes: A review of 'unspoken' side effects. Hellenic J Cardiol 2025, 83, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Galicia-Garcia, U.; Jebari, S.; Larrea-Sebal, A.; Uribe, K.B.; Siddiqi, H.; Ostolaza, H.; et al. Statin Treatment-Induced Development of Type 2 Diabetes: From Clinical Evidence to Mechanistic Insights. Int J Mol Sci. 2020, 21. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jain, M.K.; Ridker, PM. Anti-inflammatory effects of statins: Clinical evidence and basic mechanisms. BMJ Open 2020, 10, e039034. [Google Scholar] [CrossRef]
- De Nardo, D.; Latz, E. NLRP3 inflammasomes link inflammation and metabolic disease. Trends Immunol 2011, 32, 373–379. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Ramsey, L.B.; Johnson, JA. Influence of SLCO1B1 genotype on statin therapy. Genetics in Medicine 2021, 23, 1420–1430. [Google Scholar] [CrossRef]
- Toth, P.P.; Banach, M. It is time to address the contribution of cholesterol in all apoB-containing lipoproteins to atherosclerotic cardiovascular disease. Eur Heart J Open 2024, 4, oeae057. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- El-Menyar, A.; Khan, N.A.; Al Mahmeed, W.; Al Suwaidi, J.; Al-Thani, H. Cardiovascular Implications of Lipoprotein(a) and its Genetic Variants: A Critical Review From the Middle East. JACC: Asia 2025, 5, 847–864. [Google Scholar] [CrossRef]
- Tsimikas, S.; Clopton, P.; Brilakis, E.S.; Marcovina, S.M.; Khera, A.; Miller, E.R.; et al. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: Results from the Dallas Heart Study. Circulation 2009, 119, 1711–1719. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Jiang, H.; Zhou, Y.; Nabavi, S.M.; Sahebkar, A.; Little, P.J.; Xu, S.; et al. Mechanisms of Oxidized LDL-Mediated Endothelial Dysfunction and Its Consequences for the Development of Atherosclerosis. Frontiers in Cardiovascular Medicine 2022, 9. [Google Scholar] [CrossRef] [PubMed]
- Mitra, S.; Goyal, T.; Mehta, JL. Oxidized LDL, LOX-1 and Atherosclerosis. Cardiovascular Drugs and Therapy 2011, 25, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Tian, K.; Xu, Y.; Sahebkar, A.; Xu, S. CD36 in Atherosclerosis: Pathophysiological Mechanisms and Therapeutic Implications. Current Atherosclerosis Reports 2020, 22, 59. [Google Scholar] [CrossRef] [PubMed]
- Ginsberg, H.N.; Packard, C.J.; Chapman, M.J.; Borén, J.; Aguilar-Salinas, C.A.; Averna, M.; et al. Triglyceride-rich lipoproteins and their remnants: Metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies—A consensus statement from the European Atherosclerosis Society. European Heart Journal 2021, 42, 4791–4806. [Google Scholar] [CrossRef]
- Tall, A.R.; Yvan-Charvet, L. Cholesterol, inflammation and innate immunity. Nature reviews Immunology 2015, 15, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Kolodgie, F.D.; Yahagi, K.; Mori, H.; Romero, M.E.; Finn, A.V.; Virmani, R. A Comprehensive Histological Analysis of Human Atherosclerotic Plaques: Implications for Understanding Residual Cardiovascular Risk. Am J Pathol 2023, 193, 1501–1517. [Google Scholar] [CrossRef]
- Sadeghi, M.; Khayati, S.; Dehnavi, S.; Almahmeed, W.; Sukhorukov, V.N.; Sahebkar, A. Regulatory impact of statins on macrophage polarization: Mechanistic and therapeutic implications. Journal of Pharmacy and Pharmacology 2024, 76, 763–775. [Google Scholar] [CrossRef]
- Tan, B.; Chin, KY. Potential role of geranylgeraniol in managing statin-associated muscle symptoms: A COVID-19 related perspective. Front Physiol 2023, 14, 1246589. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Dimmitt, S.; Stampfer, H.; Martin, J.; Kennedy, M. Sufficient Statin Dose? Harms Increase More Than Benefits With Higher Doses. Heart, Lung and Circulation 2023, 32, S402. [Google Scholar] [CrossRef]
- Tarkin, J.M.; Joshi, F.R.; Evans, N.R.; Chowdhury, M.M.; Figg, N.L.; Shah, A.V.; et al. Detection of Atherosclerotic Inflammation by (68)Ga-DOTATATE PET Compared to [(18)F]FDG PET Imaging. J Am Coll Cardiol 2017, 69, 1774–1791. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Blankenberg, S.; Tiret, L.; Bickel, C.; Peetz, D.; Cambien, F.; Meyer, J.; et al. Interleukin-18 is a strong predictor of cardiovascular death in stable and unstable angina. Circulation 2002, 106, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Rogers, A.J.; Guan, J.; Trtchounian, A.; Hunninghake, G.M.; Kaimal, R.; Desai, M.; et al. Association of Elevated Plasma Interleukin-18 Level With Increased Mortality in a Clinical Trial of Statin Treatment for Acute Respiratory Distress Syndrome. Crit Care Med 2019, 47, 1089–1096. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Wang, X.; Kaiser, H.; Kvist-Hansen, A.; McCauley, B.D.; Skov, L.; Hansen, P.R.; et al. IL-17 Pathway Members as Potential Biomarkers of Effective Systemic Treatment and Cardiovascular Disease in Patients with Moderate-to-Severe Psoriasis. Int J Mol Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Joshi, N.V.; Vesey, A.T.; Williams, MC. 18F-fluoride positron emission tomography for identification of ruptured and high-risk coronary atherosclerotic plaques: A prospective clinical trial. Lancet 2014, 383, 705–713. [Google Scholar] [CrossRef]
- Lapa, C.; Reiter, T.; Li, X. Imaging of CXCR4 expression in atherosclerotic plaques with the novel PET tracer 68Ga-pentixafor: Comparison with 18F-FDG and histopathology. Journal of Nuclear Medicine 2015, 56, 751–756. [Google Scholar] [CrossRef]
- null, n. Final Report of a Trial of Intensive versus Standard Blood-Pressure Control. New England Journal of Medicine 2021, 384, 1921–1930. [Google Scholar] [CrossRef]
- Williams, B.; Mancia, G.; Spiering, W.; Agabiti Rosei, E.; Azizi, M.; Burnier, M.; et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology (ESC) and the European Society of Hypertension (ESH). European Heart Journal 2018, 39, 3021–3104. [Google Scholar] [CrossRef]
- McEvoy, J.W.; McCarthy, C.P.; Bruno, R.M.; Brouwers, S.; Canavan, M.D.; Ceconi, C.; et al. 2024 ESC Guidelines for the management of elevated blood pressure and hypertension: Developed by the task force on the management of elevated blood pressure and hypertension of the European Society of Cardiology (ESC) and endorsed by the European Society of Endocrinology (ESE) and the European Stroke Organisation (ESO). European Heart Journal 2024, 45, 3912–4018. [Google Scholar] [CrossRef]
- Gisterå, A.; Hansson, GK. The immunology of atherosclerosis. Nat Rev Nephrol 2017, 13, 368–380. [Google Scholar] [CrossRef] [PubMed]
- Taleb, S.; Tedgui, A.; Mallat, Z. IL-17 and Th17 cells in atherosclerosis: Subtle and contextual roles. Arterioscler Thromb Vasc Biol 2015, 35, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gao, P.; Li, Q.; Wang, T.; Guo, S.; Zhang, J.; et al. Arterial Adventitial Vasa Vasorum Hyperplasia involved in Atherosclerotic Plaque Formation in a Rabbit Model. Ultrasound Med Biol 2024, 50, 1273–1279. [Google Scholar] [CrossRef] [PubMed]
- Subbotin, VM. Excessive intimal hyperplasia in human coronary arteries before intimal lipid depositions is the initiation of coronary atherosclerosis and constitutes a therapeutic target. Drug Discov Today 2016, 21, 1578–1595. [Google Scholar] [CrossRef] [PubMed]
- Pichardo-Almarza, C.; Diaz-Zuccarini, V. Understanding the Effect of Statins and Patient Adherence in Atherosclerosis via a Quantitative Systems Pharmacology Model Using a Novel, Hybrid, and Multi-Scale Approach. Front Pharmacol 2017, 8, 635. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lei, W.; Hu, J.; Xie, Y.; Liu, C.; Chen, X. Mathematical modelling of the effects of statins on the growth of necrotic core in atherosclerotic plaque. Math Model Nat Phenom. 2023, 18. [Google Scholar] [CrossRef]
- Ouyang XZ, Liu. Regulatory T cells and macrophages in atherosclerosis: From mechanisms to clinical significance. Frontiers in Immunology 2024, 15. [Google Scholar] [CrossRef]
- Chen, X.; Li, Y.; Kumar, S. Hepatocyte-targeted siRNA silencing of FDFT1 reduces cholesterol synthesis and plasma LDL in preclinical models. Molecular Therapy 2018, 26, 2211–2221. [Google Scholar] [CrossRef]
- Wang, Z.; Patel, R.; Singh, A. GalNAc-conjugated FDFT1 siRNA lowers LDL cholesterol and attenuates atherosclerosis in mouse models. Journal of Lipid Research 2019, 60, 581–590. [Google Scholar] [CrossRef]
- Tan, J. Li Y. Revisiting the interconnection between lipids and vitamin K metabolism: Insights from recent research and potential therapeutic implications: A review. Nutrition & Metabolism 2024, 21, 6. [Google Scholar] [CrossRef]
- Torii, S.; Ikari, Y.; Tanabe, K.; Kakuta, T.; Hatori, M.; Shioi, A.; et al. Plasma phylloquinone, menaquinone-4 and menaquinone-7 levels and coronary artery calcification. Journal of Nutritional Science 2016, 5, e48. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, J.J.; Rizk, R.; Li, X.; Lee, B.G.; Matthies, M.L.; et al. Macrophages overexpressing interleukin-10 target and prevent atherosclerosis: Regression of plaque formation and reduction in necrotic core. Bioengineering & Translational Medicine 2024, 9, e10717. [Google Scholar] [CrossRef]
- Bhatt Deepak, L.; Libby, P.; Mason, RP. Emerging Pathways of Action of Eicosapentaenoic Acid (EPA). JACC: Basic to Translational Science 2025, 10, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Welty, F.K.; Schulte, F.; Alfaddagh, A.; Elajami, T.K.; Bistrian, B.R.; Hardt, M. Regression of human coronary artery plaque is associated with a high ratio of (18-hydroxy-eicosapentaenoic acid + resolvin E1) to leukotriene B(4). Faseb j 2021, 35, e21448. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Flight, P.; Rohde, E.; Lee, R.; Mazzola, A.M.; Platt, C.; Mizoguchi, T.; et al. † VERVE-102, a clinical stage in vivo base editing medicine, leads to potent and precise inactivation of PCSK9 in preclinical studies. Journal of Clinical Lipidology. 2025, 19 (Suppl. 3), e68. [Google Scholar] [CrossRef]
- Sinha, A.; Sachs, N.; Kratz, E.; Pauli, J.; Steigerwald, S.; Albrecht, V.; et al. Spatially resolved proteomic signatures of atherosclerotic carotid artery disease. medRxiv. 2025. [Google Scholar] [CrossRef]
| 1 | Square-bracket notation (e.g., [LDL-C]) denotes molar concentration throughout. |
| 2 | This mechanism is supported by murine and cell culture studies, full validated in human arterial macrophages is yet to occur |
| 3 | In vitro models support OxPL–NLRP3 activation, the in vivo contribution of Lp(a) to this pathway is yet to be quantified |
| 4 | The role of adaptive immune–macrophage crosstalk in this context has not yet been fully mapped, however, emerging data suggest that insulin resistance may modulate T-cell polarization and indirectly reinforce pro-inflammatory macrophage phenotypes. |
| 5 | While statins have clear cardiovascular benefit, the exacerbation of insulin resistance and potential diabetogenic effects (as summarized by Galicia-Garcia et al. [75]) warrants that glycemic trajectories (e.g. HbA1c, insulin signaling markers) be included as candidate biomarkers in future tests of the SFE framework. |
| 6 | Meta-analytic evidence links statin therapy to new-onset diabetes, with heightened risk in patients with pre-existing metabolic dysfunction—supporting a systemic feedback loop |
| 7 | While IL-1β–induced insulin resistance is supported by human and animal data, the bidirectional loop involving macrophages remains under active investigation |
| 8 | [CRP] reflects measured concentrations via hs-CRP assay, and there is a distinction between clinical targets and immunologic sufficiency |
| 9 | Bentzon et al. have extensively characterized macrophage dynamics in plaque progression and regression models, forming a foundational framework for interpreting phenotypic transitions in atherosclerosis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



