Submitted:
01 October 2025
Posted:
02 October 2025
You are already at the latest version
Abstract
Multiple sclerosis (MS), an autoimmune chronic inflammatory disorder of the Central Nervous System (CNS), leading to demyelination of CNS neurons, with its pathology being related with various genetic and environmental factors, such as diet and obesity. Obesity has reached the dimensions of a global epidemic. It is due to excessive fat accumulation and hypertrophy of adipocytes and results in insufficient energy expenditure in combination with genetic, social and environmental factors. The aim of this review is to analyze at molecular level, the relationship of obesity, as a chronic inflammatory condition, with the pathophysiology of MS as a chronic autoimmune inflammatory disease. Particular emphasis is placed on the role of cytokines, adiponectin and oxidative stress (OS) in neuronal damage and the pathogenesis of MS, in order to understand the complex links between obesity and MS. Chronic inflammation and OS, which are implicated and promoted in obesity, are interconnected processes, causing a toxic state, which contributes to the development of CNS neuroinflammation and neuronal damage, resulting in neuronal demyelination and the onset of MS. Adipose tissue is a complex endocrine organ, in addition to being a lipid storage organ, that secretes cytokines and adipokines, which are involved in the regulation of hormones, metabolism, inflammation, and whole-body homeostasis. Obesity triggers a chronic low-grade inflammation causing insulin resistance (IR) and leptin resistance, disruption of the blood-brain barrier (BBB) and brain metabolism, infiltration of the CNS by immune cells, production of reactive oxygen species (ROS), and generation of oxidative stress (OS). Therefore, obesity affects MS through common underlying mechanisms and thus seems to be a modifiable risk factor for this disease, whilst compounds with multi-functional characteristics, and antioxidant and anti-inflammatory potential could be an additive tool towards the progression of MS and its promotion through obesity.
Keywords:
Obesity
; Multiple Sclerosis
; Oxidative Stress
; Inflammation
; Cytokines
; Adipokines
; Antioxidants
; Natural antioxidants
1. Introduction
Obesity is defined as the excessive accumulation of body fat with detrimental effects on human health [1]. It represents a state of chronic, low-grade inflammation that has been associated with a wide range of pathological conditions, such as metabolic syndrome (MetS) and insulin resistance (IR), as well as numerous diseases including type 2 diabetes mellitus (T2DM), increased blood pressure, cardiovascular diseases (CVDs), non-alcoholic fatty liver disease (NAFLD), kidney disorders, musculoskeletal disorders, infertility, psychological problems, specific types of cancer, and autoimmune diseases [2,3,4,5,6,7]. Dietary shifts observed in recent year, characterized by increased consumption of foods high in fats and sugars, have contributed significantly to the global obesity epidemic [4,8]. According to the World Health Organization (WHO), an estimated 35% of the global population is either overweight [Body Mass Index (BMI), 25–30 kg/m²] or obese (BMI > 30 kg/m²) [9].
Multiple sclerosis (MS) is a chronic, autoimmune, inflammatory disease of the central nervous system (CNS), involiving demyelination, the destruction of the myelin sheath surrounding nerve fibers and neurodegeneration, whilst these processes impair or block the conduction of nerve impulses along axons [10]. The clinical forms of MS include: (a) relapsing-remitting MS, (b) secondary progressive MS, and (c) primary progressive MS [11]. The disease primarily affects young adults, typically manifesting between the ages of 20 and 50, with a mean age of onset around 30 years [12]. However, MS can also develop in childhood or later in life, beyond the age of 60 [10]. Over the past decades, the global prevalence of MS has been rising, particularly among women [13,14,15,16].
MS is thought to arise from the complex interplay of genetic, environmental, and lifestyle factors, including diet and obesity [17,18,19]. Overweight and obese individuals are reported to have a significantly elevated risk of developing MS [20]. In women diagnosed with MS, obesity at the time of diagnosis has been associated with a relapsing disease course [21]. Moreover, in individuals with MS, a positive correlation has been observed between obesity and disability severity, as measured by higher scores on the Expanded Disability Status Scale (EDSS) [22]. Additionally, comorbid conditions such as T2DM, hypertension, hypercholesterolemia, and peripheral vascular disease appear to be independent risk factors for increased disability in MS patients [21]. There is also evidence linking obesity during childhood, adolescence, and early adulthood with an increased risk of MS development [19,23]. Furthermore, obesity may influence disease progression and response to treatment [10]. Notably, a correlation has been reported between the extent of disability and oxidative stress (OS) in MS patients [22].
The aim of the present study is to investigate at molecular level the potential pathophysiological link between obesity, as a chronic inflammatory condition, and the pathogenesis of MS, which is likewise a chronic inflammatory autoimmune disease of the CNS. This includes the analysis of mechanisms such as the secretion of pro-inflammatory adipokines and cytokines, inflammasome activation, gut microbiota alterations, and oxidative stress. Additionally, the study focuses on the complementary role of multi-functional antioxidant and anti-inflammatory compounds in the treatment of MS, particularly through the resolution of chronic inflammation and oxidative stress.
2. Pathophysiological Association Between Obesity and Multiple Sclerosis
2.1. The Role of Chronic Inflammation in the Pathophysiology of MS
Obesity and multiple sclerosis (MS) share common pathophysiological mechanisms that interconnect them, particularly through the promotion of chronic inflammation, alterations in adipokine and cytokine secretion, associations with oxidative stress, and dysregulation of both the inflammasome and the gut microbiota [24,25].
Obesity is a metabolic condition characterized by adipocyte hypertrophy (increase in cell size) and hyperplasia (increase in cell number) [7,26]. This expansion of adipose tissue promotes the secretion of various chemotactic factors into the bloodstream, such as monocyte chemoattractant proteins MCP-1, -2, -3, and -4; CXCL10 (interferon-gamma-inducible protein 10); eotaxin; and CCL5/RANTES (regulated upon activation, normal T-cell expressed and secreted), all of them facilitating the recruitment of circulating immune cells [7] (Figure 1). These monocytes are subsequently activated into M1 pro-inflammatory macrophages, replacing the M2 anti-inflammatory phenotype typically found in individuals with normal body weight [7]. M1 macrophages in adipose tissue secrete elevated levels of (a) pro-inflammatory cytokines such as IL-6, IL-1β, and TNF-α, and (b) pro-inflammatory adipokines including leptin, visfatin, resistin, and plasminogen activator inhibitor-1 (PAI-1). In obesity, the secretion of anti-inflammatory adipokines such as adiponectin and apelin, as well as anti-inflammatory cytokines like IL-10, is reduced [7]. Collectively, these inflammatory changes in adipose tissue contribute to the development of chronic, systemic, low-grade inflammation, which underlies metabolic syndrome (MetS), insulin resistance (IR), type 2 diabetes mellitus (T2DM), and other obesity-related conditions, including MS [7,27]. Obesity also promotes the recruitment and expansion of pro-inflammatory T helper 1 (Th1) and Th17 cells, as well as CD8+ neutrophilic T lymphocytes [28], while reducing anti-inflammatory Th2 and regulatory T (Treg) cells, which are essential for maintaining immune homeostasis [29]. Furthermore, there is a decrease in invariant natural killer T (iNKT) cells and type 2 innate lymphoid cells (ILC2), along with diminished expression of peroxisome proliferator-activated receptor gamma (PPAR-γ), a key regulator of adipose tissue homeostasis [30]. Chronic inflammation in adipose tissue leads to an altered adipokine profile marked by increased secretion of leptin, visfatin, resistin, and PAI-1, and decreased adiponectin, an adipokine with anti-inflammatory properties [7,27].
Similarly, early stages of MS are characterized by chronic inflammation within the CNS, which progresses to neurodegeneration in the later phases of the disease [31]. Chemokines such as MCP-1, -2, -3, and CCL5/RANTES play a critical role in MS pathophysiology [32,33]. The immune profile of MS mirrors that of obesity, with an increase in Th1 and Th17 cells and a corresponding decrease in Th2 and Treg lymphocytes [28]. The dominance of Th17 cells is particularly implicated in promoting autoimmune inflammation within the CNS, thereby contributing to MS pathogenesis [16]. Additionally, elevated levels of pro-inflammatory adipokines may exacerbate disease progression in obese MS patients [29]. The dysregulated secretion of pro-inflammatory cytokines by adipocytes and macrophages in obesity is also believed to contribute to MS development [16]. Moreover, excessive lipolysis in obesity, driven by macrophage apoptosis inhibitor (AIM) in an attempt to counteract disease progression, results in the release of large amounts of saturated fatty acids (SFAs) [16]. These SFAs activate toll-like receptor 4 (TLR-4), triggering CNS demyelination and inflammation through nuclear factor-kappa B (NF-κB) signaling, thus promoting MS pathogenesis [16,34]. Finally, obesity is associated with increased production of IgG autoantibodies, which are involved in the pathogenesis of several autoimmune diseases, including MS [16].
2.1.1. Anti-Inflammatory Adipokines in MS
2.1.1.1. Adiponectin
Adiponectin is an anti-inflammatory adipokine. It is a peptide with a molecular weight of 30 kDa, primarily secreted by white adipose tissue [7]. Its actions include: a) suppression of NF-κB activity, b) reduction of TNF-α and IL-6 secretion from macrophages, c) decreased production of reactive oxygen species (ROS), d) reduced glucose levels in tissues, e) increased insulin secretion, f) inhibition of hepatic gluconeogenesis, g) cardioprotection, h) increased nitric oxide (NO) synthesis in the vascular endothelium, and i) promotion of angiogenesis [7] (Figure 2). Serum adiponectin levels are decreased in obesity, insulin resistance, type 2 diabetes (T2D), dyslipidemia, and cardiovascular diseases (CVDs) [7]. Moreover, its synthesis is inhibited by proinflammatory cytokines such as TNF-α and IL-6 [7]. In animal models, the absence of adiponectin in mice with experimental autoimmune encephalomyelitis (EAE) was found to result in enhanced lymphocyte activation and increased disease severity [35].
Tehrani et al. reported elevated adiponectin levels in female patients with early-onset relapsing-remitting multiple sclerosis (RRMS) compared to healthy controls [36]. Similarly, Düzel et al. observed comparable findings [37]. Furthermore, higher serum adiponectin levels at the time of MS diagnosis—prior to any treatment—have been associated with an increased risk of disease progression and disability [38]. Signoriello et al. found elevated adiponectin concentrations in the cerebrospinal fluid (CSF) of patients with primary progressive MS (PPMS) compared to RRMS patients, with higher CSF adiponectin levels being correlated with greater Expanded Disability Status Scale (EDSS) scores at disease onset and more severe disease course after 4.5 years of follow-up [39]. Conversely, Musabak et al. reported lower adiponectin levels in MS patients [40], a finding that was also confirmed by Kraszula et al. [41]. Therefore, adiponectin may represent a valuable biomarker at the onset of MS, potentially aiding in predicting disease progression and severity [42].
2.1.1.2. Apelin
Apelin is a small molecular weight peptide classified among the anti-inflammatory adipokines and is primarily secreted by adipocytes [7]. Its main actions include a) promoting the differentiation and metabolic activity of brown adipocytes and inducing the browning of white adipose tissue, b) increasing glucose uptake by cells, c) enhancing insulin sensitivity, d) inhibiting lipogenesis, lipolysis, and fatty acid oxidation, e) promoting the synthesis of antioxidant enzymes, and f) reducing oxidative stress (OS) [7] (Figure 3). Serum levels of apelin have been found to be elevated in obesity, which may be attributed either to peripheral apelin resistance or to a compensatory mechanism aiming to counteract insulin resistance in peripheral tissues [7]. In in cellulo studies using N9 microglial cell lines, apelin suppressed LPS (lipopolysaccharide )-induced production of iNOS and IL-6, while upregulating IL-10 and arginase-1 [43].
Rasooli et al. reported significantly lower plasma apelin levels in MS patients compared to healthy controls, with a statistically significant negative correlation with Expanded Disability Status Scale (EDSS) scores and relapse rate [36]. Similarly, Tehrani et al. observed reduced apelin levels in the blood of women with very early-stage relapsing-remitting MS (RRMS), which were positively correlated with both EDSS scores and the number of relapses [36]. In contrast, Alpua et al. found elevated apelin levels in patients with RRMS compared to controls, though no correlation was found with disease severity or duration [44]. Apelin and its receptor, APJ, are widely expressed in the central nervous system (CNS), particularly in neurons and oligodendrocytes [45]. Activation of APJ has been shown to promote remyelination by modulating myelin-associated regulatory factors, especially in demyelinating conditions related to aging or experimental autoimmune encephalomyelitis (EAE) [46]. Furthermore, apelin appears to facilitate the differentiation of neural stem cells, suggesting a potential role in remyelination processes during the course of MS [47].
2.1.2. Proinflammatory Adipokines in Multiple Sclerosis (MS)
2.1.2.1. Leptin
Leptin is classified among the proinflammatory adipokines [48]. It is a peptide consisting of 167 amino acids with a molecular weight of 16 kDa [48]. Leptin is primarily secreted by white adipose tissue, and its circulating levels correlate directly with the mass of adipose tissue [49]. Its main functions include: (a) regulation of food intake and metabolism through communication with the central nervous system (CNS) via specific receptors [48], (b) upregulation of proinflammatory cytokines such as TNF-α and IL-6, and (c) promotion of the proliferation of proinflammatory Th1 and Th17 cells [50,51] (Figure 4). In obesity, leptin resistance occurs, resulting in elevated serum leptin levels [52], which may also be associated with alterations observed in the blood-brain barrier (BBB) of obese individuals [53]. Furthermore, increased leptin concentrations have been detected in the serum and cerebrospinal fluid (CSF) of patients with relapsing-remitting MS (RRMS) [54,55]. Ouyang et al. demonstrated in an experimental autoimmune encephalomyelitis (EAE) mouse model that removal of leptin receptors attenuated leukocyte infiltration into the CNS and reduced BBB disruption [56].
2.1.2.2. Visfatin
Visfatin is a proinflammatory adipokine with a molecular weight of 52 kDa, secreted by macrophages in visceral adipose tissue, bone marrow, skeletal muscles, liver, lungs, pancreas, heart, brain, and various other organs [7]. Visfatin upregulates the expression of chemokines CCL2, CXCL2 (macrophage inflammatory protein 2α, MIP2-α), and CXCL8 (IL-8), as well as adhesion molecules such as ICAM-1 (intercellular adhesion molecule 1) and VCAM-1 (vascular cell adhesion molecule 1), acting as a chemoattractant for monocytes and lymphocytes [57]. Moreover, it induces (a) the release of proinflammatory cytokines including IL-1β, IL-6, and TNF-α, (b) endothelial dysfunction via the NF-κB pathway, and (c) oxidative stress also through NF-κB signaling [7] (Figure 5). Elevated visfatin levels have been reported in obesity, type 2 diabetes mellitus (T2DM), metabolic syndrome (MetS), and cardiovascular diseases (CVDs), while weight reduction in obese patients has been shown to decrease visfatin concentrations [7]. In microglial cells, visfatin plays a facilitative role in promoting the production of proinflammatory mediators such as IL-1β, IL-6, inducible nitric oxide synthase (iNOS), nitric oxide (NO), and ROS in response to LPS stimulation [58]. In experimental autoimmune encephalomyelitis, pharmacological suppression of visfatin after the appearance of clinical symptoms leads to a marked reduction in neurological impairment and spinal cord demyelination [59]. Moreover, research by Emamgholipour et al. demonstrated that individuals with RRMS exhibited elevated visfatin concentrations compared to those with primary or secondary progressive MS (PPMS and SPMS), with visfatin levels showing a positive correlation with proinflammatory cytokines TNF-α and IL-1β [60].
2.1.2.3. Resistin
Resistin is a proinflammatory adipokine with a molecular weight of 12.5 kDa, primarily secreted by macrophages within adipose tissue [7]. Resistin (a) promotes the activation of proinflammatory cytokines such as IL-1β, IL-6, and TNF-α, (b) upregulates several adhesion molecules, and (c) enhances oxidative stress (OS) [7]. Some studies have reported that elevated serum resistin levels are associated with obesity, insulin resistance, and type 2 diabetes mellitus (T2DM), while other studies have not confirmed these findings [7]. Additionally, Hossein-Nezhad and colleagues found increased serum resistin levels in patients with multiple sclerosis (MS) compared to controls, along with elevated levels of IL-1β, TNF-α, and C-reactive protein (CRP) [61]. Moreover, in patients with relapsing-remitting MS (RRMS), higher serum resistin levels correlated with reduced regulatory T cell (Treg) activity [41] (Figure 6).
2.1.2.4. Plasminogen Activator Inhibitor-1 (PAI-1)
PAI-1 is a physiological inhibitor of plasminogen activators (tPAs) [7,62,63]. Its increased activity is associated with impaired fibrinolysis and consequently a higher risk of cardiovascular diseases (CVDs) [7]. PAI-1 is secreted by adipose tissue, fibroblasts, vascular endothelial cells, and immune cells, and elevated serum levels have been observed in obese individuals, correlating with insulin resistance (IR), metabolic syndrome (MetS), and atherosclerosis [7]. Increased PAI-1 concentrations have also been detected in the serum and cerebrospinal fluid (CSF) of patients with MS [64]. Furthermore, serum PAI-1 levels were higher in active MS, compared to stable disease, and were positively associated with neurological deterioration and disability [64].
2.1.2.5. Chemerin
Chemerin is a proinflammatory adipokine that regulates adipocyte differentiation and is associated with obesity and MetS [65]. It acts as a chemoattractant for plasmacytoid dendritic cells (pDCs) and macrophages activated via Toll-like receptor 9 (TLR9) and high-mobility group box 1 protein (HMGB1), promoting type I interferon production [66]. Chemerin binds to the CCRL2 receptor (C-C chemokine receptor-like 2), facilitating macrophage infiltration and inducing IR [67]. In the central nervous system (CNS), chemerin is expressed by endothelial cells in MS sections and meninges, while its receptor CMKLR1 (chemokine-like receptor 1) is primarily found on infiltrating lymphocytes, dendritic cells, and macrophages [68]. Tomalka-Kochanowska et al. reported elevated chemerin levels in MS patients, correlating with obesity and increased body weight [69], whereas Koskderelioglu et al. did not observe similar findings [70], highlighting the need for further clinical investigations.
2.1.2.6. Fatty Acid Binding Protein 4 (FABP-4)
FABP-4 is a proinflammatory adipokine produced by adipocytes, monocytes, and macrophages, with its expression upregulated by Toll-like receptor 2 (TLR2) stimulation [71]. FABP-4 levels are higher in obese patients compared to overweight or normal-weight individuals [73]. Deficiency of FABP-4 reduces the secretion of proinflammatory cytokines via suppression of the NF-κB pathway [73], whereas administration of recombinant FABP-4 promotes proinflammatory cytokine secretion through the p38/NF-κB pathway [74]. Moreover, FABP-4 transports free fatty acids (FAs), released from lipolysis of fat droplets in the bloodstream, to various organs including the CNS [75,76]. FABP-4 knockout mice exhibit reduced clinical symptoms in experimental autoimmune encephalomyelitis (EAE) and decreased production of proinflammatory cytokines by dendritic cells [77,78]. In pediatric MS patients, a positive correlation between FABP-4, leptin, and relapsing-remitting MS (RRMS) has been observed, suggesting a potential role of these adipokines in disease progression [74]. Furthermore, in adult MS patients, FABP-4 has been associated with increased disability independently of body mass index (BMI) [72]. Similarly, in women, higher serum FABP-4 levels correlated with elevated Expanded Disability Status Scale (EDSS) scores [72]. Additionally, reduced expression of miR-34a has been detected in peripheral blood mononuclear cells (PBMCs) from patients with RRMS [79].
2.1.3. Pro-inflammatory Cytokines in MS
2.1.3.1. TNF-α
TNF-α is a proinflammatory adipokine primarily produced by macrophages, T and B lymphocytes, adipocytes, vascular endothelial cells, astrocytes, neurons, and muscles [7,80,81]. TNF-α exerts its effects through two distinct receptors, TNF-R1 and TNF-R2, which differ in their chemical affinity for TNF-α [7,81]. TNFR1 is expressed in all cell types [82], whereas TNFR2 is mainly found in neurons, endothelial cells, and various immune cells [83]. Expression of TNFR1 induces pathogenic effects, while TNFR2 expression has protective roles [84]. Specifically, soluble TNF-α mainly acts via TNFR1, mediating cellular apoptosis including oligodendrocytes [85] and chronic inflammation, whereas membrane-bound TNF-α acts primarily through TNFR2, activating genes involved in cell survival and resolution of inflammation [81]. It has been observed that in obesity and insulin resistance (IR), TNF-α expression is increased, with levels decreasing upon weight loss [7]. Experimental evidence also shows that TNF-α treatment increases IR in adipocytes [7]. TNF-α actions include: (a) stimulating MCP-1 (monocyte chemoattractant protein-1) and IL-6 secretion from preadipocytes [86], (b) inhibiting adiponectin synthesis [87], (c) increasing release of free fatty acids (FFAs) from adipocytes [87], (d) activating NF-κB, leading to elevated expression of adhesion molecules on endothelial and vascular smooth muscle cells and promoting atherogenesis [87], (e) reducing insulin action on peripheral glucose uptake [7], (f) increasing lipolysis in adipocytes [7], and (g) promoting ROS and superoxide anion production, thereby causing OS [7] (Figure 7). TNF-α appears to be involved in MS activation [88,89]. Sharief and Henges reported increased TNF-α levels in active MS [90]. Moreover, elevated TNF-α levels in cerebrospinal fluid (CSF) correlated with MS severity and progression [90]. Single nucleotide polymorphisms (SNPs) in the TNFR1 gene (TNFRSF1A) have been linked to increased MS risk [91]. TNF-α is upregulated in mice with EAE, and TNF-α administration worsens disease progression in these mice [89,92,93]. Additionally, mice with EAE lacking TNFR1 are either fully resistant or develop milder disease [94,95]. Selective TNFR1 blockade improves EAE outcomes, while TNFR2 deficiency worsens EAE [96,97,98].
2.1.3.2. IL-6
Interleukin-6 (IL-6) is a proinflammatory cytokine that induces the acute phase response of inflammation [7,99,100]. It is secreted by various cell types, including adipocytes, endothelial cells, monocytes, T and B lymphocytes, fibroblasts, microglia, neurons, and pancreatic beta cells [7,99,100]. IL-6 signals either by binding to its alpha receptor IL-6Rα (classical signaling) or through binding to a soluble IL-6 receptor form (sIL-6R) [7,99]. IL-6 signaling via gp130 (glycoprotein 130) activates the JAK1/STAT3 pathway (janus kinase 1/signal transducer and activator of transcription 3), resulting in gene transcription [99]. IL-6 also activates the MAPK (mitogen-activated protein kinase) pathway, leading to transcription of additional genes [99]. Obese patients exhibit higher serum IL-6 levels, as do patients with chronic inflammatory diseases and abnormal blood lipid profiles [63,101]. Furthermore, one-third of circulating IL-6 is produced by adipose tissue [63,101]. The hypothalamus shows the highest expression of IL-6 receptors, suggesting IL-6’s possible involvement in appetite and food intake regulation [63,101]. IL-6 appears to contribute to inflammation linked to MS pathogenesis [99]. It also promotes Th17 lymphocyte development [99], while IL-6-deficient mice display resistance to EAE [102,103].
2.1.3.3. IL-8
Interleukin-8 (IL-8) is a proinflammatory cytokine mainly produced by monocytes and macrophages [101]. Oxidized LDL (oxLDL) promotes IL-8 production and release from macrophages derived from human atherosclerotic plaques and foam cells [101]. IL-8 also induces matrix metalloproteinase-9 (MMP-9) release from neutrophils [104]. Elevated IL-8 levels are found in obese patients and those with T2DM and MetS [105,106]. IL-8 is responsible for BBB disruption and immune cell migration into the CNS, with lower IL-8 levels in blood serum and higher levels in CSF of MS patients compared to healthy controls [107]. Lund et al. demonstrated significantly higher serum IL-8 in untreated MS patients compared to controls, which significantly decreased following interferon-beta-1a therapy [108]. Neuteboom et al. found that elevated IL-8 during pregnancy correlated with increased risk of postpartum relapse [109].
2.1.3.4. IL-18
Interleukin-18 (IL-18), also known as interferon-gamma (IFN-γ) inducing factor, is produced by hematopoietic and non-hematopoietic cells such as monocytes, macrophages, keratinocytes, and mesenchymal cells [110]. IL-18 belongs to the IL-1 cytokine family, which comprises 11 cytokines that promote innate immune system activity [110]. IL-18 stimulates both innate and adaptive immunity [110] and is implicated in MS and EAE development [111]. Increased IL-18 expression has been found in serum and peripheral blood mononuclear cells (PBMCs) of MS patients and in brain and spinal cord tissues of mice with EAE [112,113]. IL-18 binds to the IL-18 receptor (IL-18R) to exert proinflammatory effects by activating NF-κB signaling and promoting Th1 differentiation, leading to IFN-γ induction. However, the precise mechanisms by which IL-18 regulates MS and EAE progression still remain unclear [111,114,115].
2.1.3.5. IL-1β
Interleukin-1 beta (IL-1β) is a proinflammatory cytokine secreted by M1 macrophages [7]. Its roles in obesity include (a) promoting ectopic fat accumulation, (b) increasing blood glucose levels, (c) disrupting insulin secretion, (d) causing insulin resistance, (e) inducing T2DM, (f) forming atherosclerotic plaques, (g) causing hepatic steatosis, (h) promoting liver cirrhosis, (i) suppressing PPARγ expression, (j) increasing cytokine and chemokine expression, and (k) inducing oxidative stress [7] (Figure 8). IL-1β mediates neuroinflammation by promoting innate immune responses during MS pathophysiology [116]. In EAE or MS progression, a characteristic feature is BBB and blood-spinal cord barrier (BSCB) leakage, leading to IL-1β release and subsequent neuroinflammation [117,118,119]. Mice deficient in IL-1β or IL-1 receptor (IL-1R) are resistant to EAE [120,121]. The exact mechanism of IL-1β’s role in EAE or MS is unclear, but IL-1β appears to promote differentiation of autoimmune CD4+ T cells into pathogenic Th1 and Th17 phenotypes, especially Th17 cells that express high IL-1R levels, thereby contributing to neuroinflammation and MS pathogenesis and clinical progression [119].
2.1.4. Anti-Inflammatory Cytokines in MS
2.1.4.1. IL-10
Interleukin-10 (IL-10) is an anti-inflammatory cytokine that plays a critical role in preventing inflammatory and autoimmune pathological conditions [122]. IL-10 is mainly produced by activated myeloid cells and lymphocytes, and to a lesser extent by other cells during inflammation [123]. IL-10 dimerizes and binds to the extracellular domain of two IL-10R1 subunits of the IL-10 receptor complex [123]. The IL-10 receptor complex is a tetramer consisting of two IL-10R1 subunits and two IL-10R2 subunits [123]. The IL-10R1 subunit is associated with JAK1 (Janus kinase 1), while the IL-10R2 subunit is associated with TYK2 (tyrosine kinase 2) [123]. Activation of the IL-10 receptor complex induces phosphorylation and activation of STAT1 (Signal Transducer and Activator of Transcription 1), STAT3, STAT5, and SOCS1/3 (Suppressor of Cytokine Signaling 1 and 3), ultimately leading to the inhibition of NF-κB-mediated signaling [123]. IL-10 suppresses the production of pro-inflammatory cytokines such as IL-1β, IL-6, IL-12, IL-18, and TNF-α, while promoting the production of other anti-inflammatory mediators, such as IL-1β receptor antagonist [123]. The anti-inflammatory effects of IL-10 have been demonstrated in experimental models of multiple sclerosis (MS), where it attenuates neuroinflammation [123,124]. In EAE (experimental autoimmune encephalomyelitis) mouse models, IL-10-deficient mice developed more severe disease compared to wild-type mice, whereas those overexpressing IL-10 were resistant to EAE [125,126].
2.2. Activation of the NLRP3 Inflammasome in Obesity and MS
Inflammasomes are large cytoplasmic multiprotein complexes that exert their effects through pattern recognition receptors (PRRs), promoting the secretion of mature pro-inflammatory cytokines IL-1β and IL-18 involved in inflammatory processes [25,127]. Inflammasomes consist of three components: (a) a sensor protein, such as the NOD-like receptor (NLR) family; (b) an adaptor protein (ASC or PYCARD, an apoptosis-associated protein containing a caspase recruitment domain); and (c) caspase-1 [128]. Toll-like receptors (TLRs) and NLRs are members of the PRR family, recognizing pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs), respectively [128]. PRR activation initiates inflammasome assembly and triggers NF-κB activation. The adaptor protein ASC links the sensor protein (NLR) with caspase-1 [128]. Activation of the NLRP3 inflammasome occurs when PAMPs or DAMPs bind to NLR receptors due to various metabolic abnormalities, including lysosomal disruption, release of mitochondrial DNA (mtDNA), ROS, and elevated intracellular calcium levels (Ca²⁺) [128]. Subsequent NLR oligomerization occurs via interactions of their PYD (pyrin domain) or CARD (caspase recruitment domain), followed by activation of caspase-1, which then leads to the production and secretion of mature IL-1β and IL-18 [128]. Activation of caspase-1 triggers pyroptosis programmed cell death [128].
NLRP3 inflammasomes are part of the NLR family and contain a leucine-rich repeat (LRR) domain, a nucleotide-binding site (NBS), and a pyrin domain-containing component (PDC-3) [128]. Esser et al. found increased expression of NLRP3 and IL-1β in macrophages infiltrating visceral fat of obese individuals with MetS compared to obese individuals without MetS [129]. In obesity, NLRP3 inflammasomes are activated by an excess of metabolic DAMPs such as ATP, glucose, fatty acids, ceramides, oxidized low-density lipoproteins (ox-LDL), crystallized uric acid, cholesterol crystals, and monosodium urate. Moreover, pro-inflammatory adipokines, like leptin, resistin, and TNF-α, contribute to NLRP3 activation, leading to caspase-1-mediated secretion of mature IL-1β and IL-18 [16,130]. Palmitic acid, a saturated fatty acid, activates the NLRP3-PYCARD inflammasome via mitochondrial ROS, AMPK (AMP-activated protein kinase), and the autophagy signaling cascade involving ULK1 (UNC-51-like kinase), resulting in caspase-1, IL-1β, and IL-18 production. This suggests a connection between high-fat diets and inflammation [131]. Conversely, oleic acid, an unsaturated fatty acid, inhibits palmitic acid’s effects by promoting AMPK activation and reducing endoplasmic stress (ER stress) [132]. Similarly, long-chain polyunsaturated fatty acids (PUFAs), such as omega-3s, inhibit caspase-1 activation through their receptor GPR120 (G-protein-coupled receptor 120). GPR120 binds to β-arrestin-2, forming a complex that interacts with and inhibits NLRP3 inflammasome activation [133]. In contrast, cholesterol crystals activate the NLRP3 inflammasome through lysosomal membrane rupture [134], and ox-LDL via NF-κB activation [135]. Hyperglycemia can also stimulate NLRP3 inflammasome activation in human adipose tissue through upregulation of TXNIP (thioredoxin-interacting protein), resulting in elevated IL-1β expression and contributing to the development of insulin resistance (IR) [136]. Increased glutamate levels during glucose deprivation and hypoxia induce endoplasmic reticulum stress (ER stress), elevated intracellular calcium (Ca²⁺), and upregulation of TXNIP expression [137]. Lipopolysaccharides (LPS) are potent PAMPs that act as endotoxins. Their levels increase in conditions like obesity and T2DM due to altered gut microbiota and increased intestinal permeability. LPS are taken up by macrophages and adipocytes and activate NLRP3 inflammasomes, leading to the production of pro-IL-1β via TLR4- and NF-κB-dependent pathways and the initiation of inflammatory responses [138]. Additionally, ATP, beyond its role as an intracellular energy carrier, is a major NLRP3 inflammasome activator via the NF-κB pathway [139]. NLRP3 inflammasomes are involved in the pathophysiology of MS [25]. Elevated expression levels of NLRP3 and IL-1β have been found in MS lesions [140,141], together with increased ASC, caspase-1, and IL-18 at the serum [140,141]. In EAE mouse models, elevated expression of NLRP3 mRNA and protein levels has been reported [111,142]. Additionally, Nlrp3⁻/⁻ mice showed reduction of Th1 and Th17 lymphocytes in the spinal cord and peripheral lymphoid tissues, as well as significantly milder EAE symptoms compared to wild-type mice [111,143]. Moreover, Yu et al. showed that mice lacking thymic stromal lymphopoietin (TSLP) (Tslpr⁻/⁻ mice) exhibited decreased NLRP3 expression and lower EAE scores [144].
2.3. Gut Microbiota Dysbiosis in Obesity and Multiple Sclerosis (MS)
Significant differences have been observed between the gut microbiota of obese individuals and that of people with normal body weight [16], which are attributed to alterations in the composition and function of the host microbiome [145]. The gut microbiota of obese individuals has the capacity to increase intestinal and blood–brain barrier (BBB) permeability [16,146,147]. Thus, in obese patients, lipopolysaccharides (LPS) from the outer membrane of Gram-negative bacteria, produced following fermentation, translocate across the intestinal mucosa and cross the BBB, acting on astrocytes and microglia [146,147,148,149,150], thereby altering the Treg/Th17 cell ratio in the central nervous system (CNS) and promoting neuroinflammation [151]. In obesity, there is an abundance of certain bacterial species (e.g., Archaea), along with a decrease or absence of other groups such as the Firmicutes and Bacteroidetes phyla [152]. The presence of certain bacteria in the gut microbiota of patients with MS, such as Fusobacteria, has been associated with an increased risk of disease relapse [153]. Therapeutic approaches in MS patients using probiotics and fecal microbiota transplantation aimed at modifying the gut microbial ecosystem have not yielded promising results to date [154]. Furthermore, it has been observed that MS patients exhibit low levels of propionic acid in both feces and blood [155]. Exogenous administration of propionic acid as adjunct therapy in MS was found to significantly reduce Th1 cell activity and enhance Treg cell function, resulting in less disability and fewer disease relapses [155].
3. Oxidative Stress (OS) in Obesity and Multiple Sclerosis
Oxidative stress (OS) in obesity arises through two main pathways. The first involves chronic inflammation in adipose tissue, where overproduction of proinflammatory cytokines, such as TNF-α, IL-6, and IL-1β, stimulates mitochondrial and peroxisomal oxidative phosphorylation, resulting in the generation of free radicals (FRs), mitochondrial DNA damage, ATP depletion, and lipotoxicity [7,60]. Chronic inflammation is further exacerbated by activation of PGE2 (prostaglandin E2), COX-2 (cyclooxygenase-2), and MAPK pathways [156]. Moreover, in obesity there is reduced expression of PPAR-γ by fatty acids and their metabolites. As a result, PPAR-γ fails to upregulate antioxidant genes and downregulate proinflammatory mediators [157]. The second pathway of OS involves the formation of NADPH oxidases, both through the pentose phosphate pathway signaling mechanism and the increased expression of NADPH oxidases in macrophages [156]. These enzymes catalyze the transfer of electrons from NADPH, reducing oxygen and generating the O2•-, which is subsequently reduced to H₂O₂ [156]. H₂O₂ is then broken down into water and oxygen by antioxidant enzymes such as catalase (CAT) and glutathione peroxidase (GPx). In cases of antioxidant enzyme deficiency, reactive oxygen species (ROS) accumulate [156,158]. Thus, ROS overproduction in obesity results from both NADPH oxidase activity and mitochondrial oxidative phosphorylation. Within mitochondria, molecular oxygen (O₂) is reduced to H₂O through the flavin mononucleotide and ubiquinone cycle, producing both superoxide anions (O2•-) and hydrogen peroxide (H₂O₂) [159]. The excessive generation of ROS leads to oxidative stress. In OS, increased levels of malondialdehyde (MDA), a biomarker of cellular damage and lipid peroxidation, are also observed [156].
Similarly, in MS, neuroinflammation activates the MAPK (p38 mitogen-activated protein kinase) signaling pathway in the nuclei of macrophages and dendritic cells, resulting in overproduction of proinflammatory cytokines such as TNF-α, IFN-γ, IL-1β, IL-12, IL-6, and IL-23 [160]. Moreover, activation of the transcription factor NF-κB in CNS cells, including T lymphocytes, macrophages, microglia, astrocytes, and oligodendrocytes, enhances neuroinflammation and drives MS pathogenesis [161]. Neuroinflammation also activates JNK signaling, leading to demyelination and neuronal apoptosis [162], and disrupts the PI3K/AKT (phosphatidylinositol 3-kinase/protein kinase B) pathway, contributing to MS exacerbation [163]. Neuroinflammation induces the overproduction of ROS and nitric oxide (NO) by glial cells and activates the arachidonic acid pathway in the CNS via COX and lipoxygenase (LOX), triggering OS [164,165]. OS can damage DNA, lipids, and proteins in neurons, leading to cell death [166]. It can also impair mitochondrial function and sodium-potassium pump activity, reducing ATP production, causing potassium accumulation, and inducing apoptosis [167]. Furthermore, ROS and nitric oxide synthase (NOS) overproduction in neural cells damages the myelin sheath and disrupts the integrity of the BBB [168,169,170].
Figure 9 schematically illustrates the interplay among obesity, chronic inflammation, oxidative stress (OS), and MS pathogenesis, highlighting the following: OS in obesity arises via two mechanisms. The first stems from chronic adipose tissue inflammation, which through excessive secretion of proinflammatory cytokines like TNF-α, IL-6, and IL-1β, triggers mitochondrial and peroxisomal oxidative phosphorylation, leading to ROS production. Chronic inflammation is further exacerbated by activation of PGE2, COX-2, and MAPK. In addition, obesity is associated with reduced expression of PPAR-γ by fatty acids and their metabolites, limiting the upregulation of antioxidant enzymes such as catalase, glutathione peroxidase, and superoxide dismutase. The second mechanism involves NADPH oxidase formation. These enzymes transfer electrons from NADPH to oxygen, generating superoxide (O2•-), which is then converted to hydrogen peroxide (H₂O₂). H₂O₂ is subsequently broken down into H₂O by antioxidant enzymes such as CAT and GPx. In the absence of sufficient antioxidant defenses, ROS accumulate. Thus, ROS overproduction in obesity stems from both NADPH oxidases and mitochondrial oxidative phosphorylation. The resulting oxidative stress (OS) is marked by elevated MDA levels and increased peroxidized lipid ratios, promoting chronic low-grade inflammation. OS also promotes further activation of PGE2, COX-2, and MAPK, reinforcing inflammation. Both chronic inflammation and OS contribute to the pathogenesis of MS.
4. Antioxidant Compounds in the Management of Multiple Sclerosis
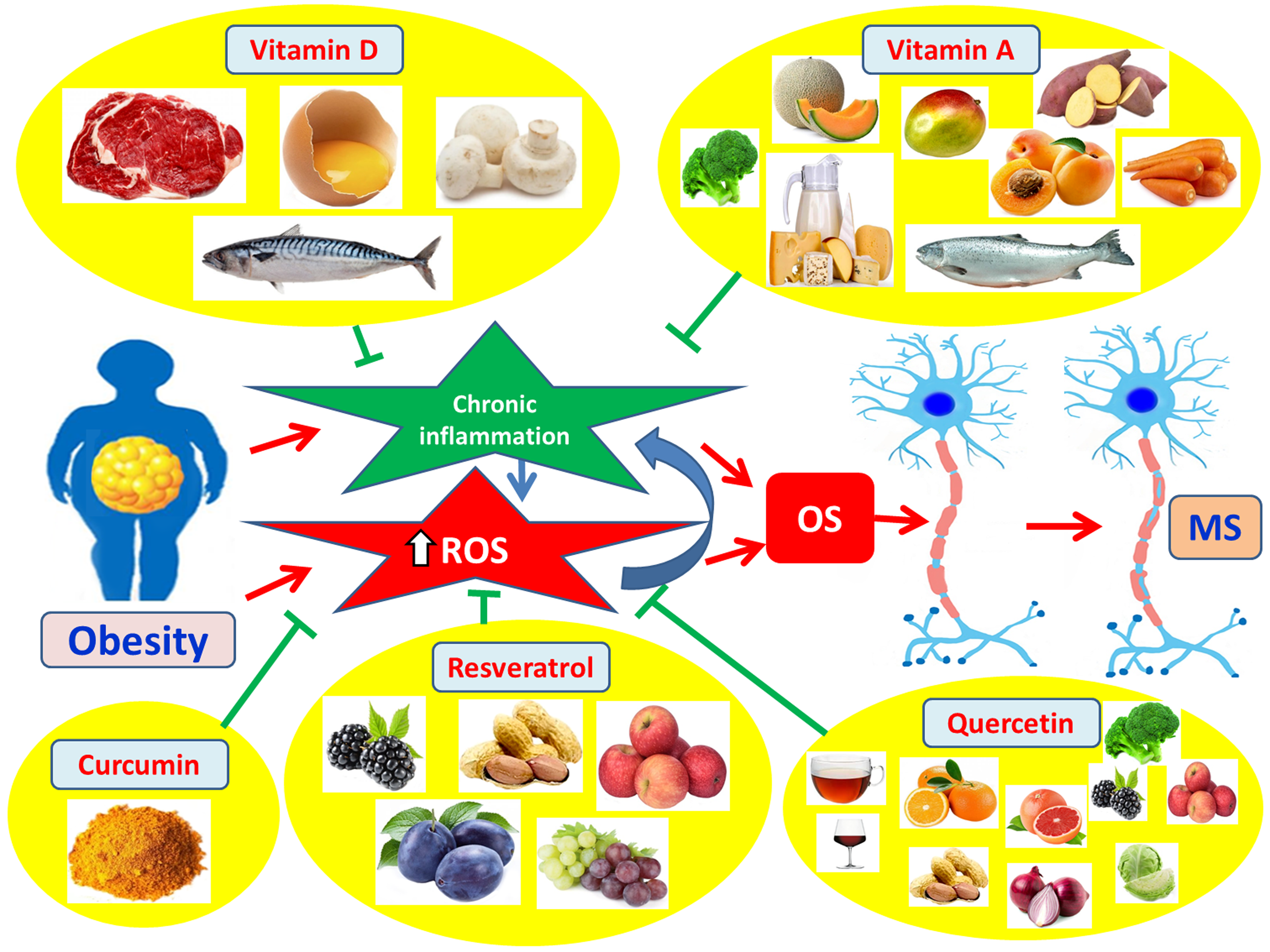
A key regulator of the antioxidant response is Nrf2 (nuclear factor erythroid 2-related factor 2) [171]. Under normal conditions, Nrf2 is bound to the Keap1 protein in the cytoplasm, which signals its degradation via the ubiquitin-proteasome system [171]. Under oxidative stress (OS), Nrf2 dissociates from Keap1 (Kelch-like ECH-associated protein 1) and translocates to the nucleus, where it binds to antioxidant response elements (AREs) and induces the transcription of target genes encoding antioxidant enzymes and detoxification proteins, thereby protecting cells from the damaging effects of reactive oxygen species (ROS) and maintaining redox homeostasis [172]. The Nrf2 signaling pathway is a promising target for activating antioxidant mechanisms in MS, as it regulates the expression of several antioxidant enzymes [173]. Moreover, the JNK and ERK signaling pathways can phosphorylate Nrf2 and promote its nuclear translocation [174,175]. There are two endogenous antioxidant defense systems, enzymatic antioxidants, such as glutathione peroxidase, catalase, superoxide dismutase (SOD), and paraoxonase 2 (PON2), and free radical scavenging molecules, such as alpha-tocopherol (vitamin E) and glutathione [164,176]. Some of the enzymes regulated by Nrf2 include superoxide dismutase (SOD), heme oxygenase-1 (HO-1), glutathione peroxidases (GPxs), and catalase [172]. Catalase is a first-line defense enzyme against ROS and OS [172]. Gray et al. found increased catalase expression in the gray matter of MS patients compared to controls, suggesting an attempt by the enzyme to neutralize oxidative stress [177]. Similarly, SOD acts as a primary defense against OS, and reduced SOD activity has been associated with uncontrolled ROS production [172]. Obradovic and colleagues reported elevated SOD activity in the serum of MS patients, confirming oxidative and inflammatory injury in MS [178]. In addition, the coenzyme CoQ10 may exert dose-dependent beneficial effects on improving OS and inflammation in MS [179]. Furthermore, several non-enzymatic compounds, such as polyunsaturated fatty acids, glutathione, N-acetylcysteine, alpha-lipoic acid, melatonin, L-carnitine, and various polyphenols (e.g., epigallocatechin, quercetin, curcumin, and resveratrol), have shown beneficial effects against obesity-related metabolic disorders [180] and may serve as adjunct therapies in MS [181]. Likewise, trace elements such as iron (Fe), zinc (Zn), manganese (Mn), and copper (Cu) exhibit beneficial effects on metabolic disorders related to obesity [179] and may play a supportive role in MS treatment [181]. Additionally, targeting inflammatory mediators represents an alternative therapeutic strategy for MS. However, the safety and efficacy of such interventions must be further studied and validated in future clinical trials. Anti-inflammatory compounds that interfere with inflammatory mediators include: a) IL-1R antagonists [7,182], b) recombinant anti-IL-1β antibodies [7], c) NF-κB inhibitors [7], d) anti-TNF-α agents [7], e) inflammasome-targeting compounds [127,183,184,185], and f) thiazolidinediones, which act as potent and selective activators of PPAR [7,183,184,185]. Finally, anti-obesity drugs with anti-inflammatory and antioxidant potential could prove useful as adjunct therapies in the management of MS [186].
4.1. Antioxidants as Complementary Therapy in MS
4.1.1. Vitamin D
Vitamin D reduces the number of activated autoreactive T lymphocytes in the CNS [187], which attack the myelin sheath and contribute to the pathogenesis of multiple sclerosis (MS) [188]. It also appears to inhibit the differentiation of dendritic cells [189,190], which are innate immune cells involved in MS pathogenesis [191]. Furthermore, vitamin D reduces the accumulation of macrophages in the CNS in experimental autoimmune encephalomyelitis (EAE) models, indicating potential neuroprotective effects [192,193,194,195]. In parallel, vitamin D enhances the activity of regulatory T cells (Tregs) [196], which suppress autoreactive T lymphocytes of the immune system [197]. Additionally, vitamin D induces a shift from Th1 toward Th2 cells [198], thereby promoting a shift in T-cell-derived cytokine production from pro-inflammatory Th1 cytokines, such as TNF-α, IFN-γ, and IL-2 [207], to anti-inflammatory Th2 cytokines, including IL-4, IL-5, and IL-10 [199]. Moreover, vitamin D reduces the production of nitric oxide (NO) [200] and suppresses the inducible nitrogen monoxide synthase (iNOS) pathway [201] in microglia, macrophages, and astrocytes [202]. This is particularly important, as NO contributes to BBB disruption, damage of oligodendrocytes and axons, and demyelination [203]. Overall, vitamin D plays a protective role in the onset of MS, reduces disease severity, and decreases the frequency of relapses [204,205] (Figure 10).
4.1.2. Vitamin A
Vitamin A, particularly through its active metabolite retinoic acid, enhances the integrity of the blood–brain barrier (BBB) [206], thereby restricting the passage of peripheral pro-inflammatory substances and immune cells into the brain parenchyma [181,207]. In addition, vitamin A exerts anti-inflammatory and antioxidant properties in the CNS [181,208]. It inhibits the secretion of pro-inflammatory cytokines such as IL-1, IL-12, TNF-α, and nitric oxide (NO) [181,209]. Vitamin A also increases the secretion of the anti-inflammatory cytokine IL-10 by B cells in MS and simultaneously enhances the production of antioxidant enzymes, thereby protecting the brain from oxidative stress [181,210]. Furthermore, it activates PPARs, modulating the phenotype of CNS macrophages, reducing neuroinflammation, neuronal oxidative stress, and axonal demyelination [181]. Additionally, vitamin A inhibits the transcription factor NF-κB and the activator protein 1 (AP-1), both involved in T cell activation, as well as STAT-1, which plays a role in neuroinflammation [181,211]. Vitamin A also promotes the differentiation of T cells into Th2 to produce anti-inflammatory cytokines [181,212]. Likewise, it increases the number of regulatory T cells (Tregs), which suppress autoreactive T lymphocytes of the immune system that attack the myelin sheath and contribute to MS pathogenesis [181,213]. At the same time, vitamin A inhibits the differentiation of CD4+ T cells into Th1 and Th17, thereby protecting against neuroinflammation [181,213] (Figure 11). Similarly, vitamin A suppresses the ability of CD4+ T cells to induce EAE and plays a protective role in the onset of MS [181,213].
4.1.3. Curcumin
Curcumin has the ability to cross the blood–brain barrier (BBB) and protect the brain from inflammatory damage [214,215]. It prevents the degradation of tight junction proteins of the BBB [216], thus preserving the barrier’s integrity and limiting the entry of peripheral immune cells and inflammatory mediators into the brain parenchyma [181]. Moreover, curcumin has been shown to promote remyelination of neurons in animal models of MS [217]. It also exerts anti-inflammatory and antioxidant properties in the CNS, promotes the clearance of ROS, and chelates metal ions such as manganese, iron, copper, and zinc [181,218,219]. Additionally, curcumin inhibits the expression of pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, IL-17, and TNF-α in the CNS, as well as the expression of COX-2, MCP-1, and macrophage inflammatory protein-1α (MIP-1α) [220,221]. Curcumin also inhibits the activation of NF-κB, thereby suppressing the pro-inflammatory responses involved in the pathogenesis of both MS and EAE [181]. Likewise, it inhibits the differentiation of Th17 cells, which play a key role in MS pathogenesis [222]. Furthermore, curcumin restores the function of Tregs [223], which suppress the autoreactive T lymphocytes that attack the myelin sheath and contribute to MS development [224]. Curcumin also activates Nrf2, resulting in protection against oxidative stress, mitochondrial dysfunction, neuroinflammation, and neurodegeneration in MS [218,225]. Additionally, curcumin exerts neuroprotective effects in neurodegenerative diseases by activating antioxidant systems, such as heat shock proteins 70 (Hsp70s), heme oxygenase-1 (HMOX-1), and thioredoxin, which is essential for maintaining mitochondrial ROS homeostasis [215,225,226] (Figure 12).
4.1.4. Resveratrol
Resveratrol enhances the integrity of the BBB, thereby limiting the infiltration of peripheral pro-inflammatory substances and immune cells into the brain parenchyma [227]. It also exerts anti-inflammatory and antioxidant effects in the CNS, reducing reactive oxygen species (ROS) and pro-inflammatory cytokines such as TNF-α, IL-1β, IL-9, IL-12, IL-17, IL-23, and interferon-gamma (IFN-γ) in the CNS [181,225]. Additionally, resveratrol inhibits the expression of MIP-1α [225] and suppresses Th17 helper T cells [228]. At the same time, it promotes the shift of Th1 cells toward Th2 cells, altering cytokine production by T lymphocytes from pro-inflammatory (e.g., TNF-α, IFN-γ, IL-2) to anti-inflammatory (e.g., IL-4, IL-5, IL-10) cytokines [228]. Moreover, resveratrol activates sirtuin 1 (SIRT1), a NAD⁺-dependent deacetylase, whose overexpression appears to exert neuroprotective effects [181,228,229]. Resveratrol also enhances remyelination of neurons in experimental autoimmune encephalomyelitis (EAE) models [225]. Due to its anti-inflammatory, antioxidant, and anti-apoptotic properties, it reduces neuronal damage and the severity of MS [225] (Figure 13).
4.1.5. Quercetin
Quercetin strengthens the BBB and limits the passage of peripheral pro-inflammatory substances and immune cells into the brain parenchyma [230]. It also demonstrates anti-inflammatory and antioxidant properties in the CNS by promoting ROS clearance, inhibiting the secretion of pro-inflammatory cytokines such as IL-1β, IL-12, and TNF-α, and chelating metal ions [181,230]. Furthermore, quercetin inhibits nitric oxide synthase activity in cells like macrophages and astrocytes and suppresses the proliferation of autoreactive T cells, which attack the myelin sheath and contribute to the pathogenesis of MS [230]. Similarly, it inhibits the differentiation of Th1 helper T cells [230]. Additionally, quercetin reduces IFN-γ production and inhibits calcium-mediated signaling in CNS cells, exerting neuroprotective effects [181]. It also inhibits xanthine oxidase, an enzyme implicated in axonal and myelin loss in EAE models [181]. Simultaneously, quercetin inhibits the phosphorylation of JAK2, tyrosine kinase 2 (TYK2), and signal transducer and activator of transcription 3 (STAT3), thus exhibiting anti-inflammatory and anti-apoptotic effects in the CNS [181] (Figure 14).
5. Conclusions
It is now well established that obesity induces neurodegenerative damage in the CNS, due to the chronic inflammation, insulin resistance (IR), and oxidative stress (OS) it causes. In the brains of obese individuals, elevated levels of ROS are produced as a result of persistent neuroinflammation. These high ROS levels act on brain mitochondria, impairing the production of ATP, which is essential for the functioning of neurons and glial cells. Moreover, elevated ROS levels cause damage to the phospholipid membranes of neural cells in the brain. Both chronic neuroinflammation and oxidative stress are involved in the pathophysiology of MS, and the two processes interact with and exacerbate one another. OS results from an imbalance between reactive species and antioxidant systems, favoring oxidative damage. It is also associated with inflammasome signaling, gut microbiota dysbiosis, cytokine-induced synaptic hyperexcitability, abnormal iron accumulation in the brain, and microglial activation, all of which contribute to neuronal injury. Oxidative stress biomarkers measured in serum or cerebrospinal fluid (CSF) may hold diagnostic and prognostic value, while antioxidant compounds may have potential clinical applications as adjunct therapies in MS. These antioxidant agents appear to exert a positive impact on disease progression, as do innovative therapeutic strategies that target inflammatory mediators by blocking the IL-1 receptor, inhibiting IL-1β or NF-κB, targeting TNF-α, or modulating inflammasomes and activating PPAR pathways. Furthermore, anti-obesity drugs with anti-inflammatory and antioxidant properties may serve as beneficial adjunct treatments for MS. More research is needed to assess the safety and efficacy of new antioxidant compounds as complementary therapies in MS, in two processes, obesity and MS, which, also as this study showed, appear to interact and share common pathophysiological mechanisms.
Author Contributions
The authors confirm their contribution to the paper as follows: study conception and study design: F.N.V., O.P., M.V., P.T.N.; literature review F.N.V., V.K.V.; draft manuscript: F.N.V., O.P., M.V., V.K.V., P.T.N.; editing and critical revision of the manuscript: F.N.V., O.P., M.V., V.K.V., P.T.N. All authors reviewed and approved the final version of the manuscript.
Funding
This review did not receive financial support.
Conflict of Interest
The authors declare no conflict of interest.
References
- Wen, X.; Zhang, B.; Wu, B.; et al. Signaling pathways in obesity: mechanisms and therapeutic interventions. Sig Transduct Target Ther 2022, 7, 298. [Google Scholar] [CrossRef] [PubMed]
- Overs, S.; Hughes, C.M.; Haselkorn, J.K.; Turner, A.P. Modifiable comorbidities and disability in multiple sclerosis. Curr Neurol Neurosci Rep 2012, 12, 610–617. [Google Scholar] [CrossRef]
- Beltrá-Sánchez, H.; Harhay, M.O.; Harhay, M.M.; McElligott, S. Prevalence trends of metabolic syndrome in the adult, U.S. population, 1999-2010. JACC 2013, 62, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Versini, Μ.; Jeandel, P.-Y.; Rosenthal, E.; Shoenfeld, Y. Obesity in autoimmune diseases: not a passive bystander. Autoimmunity Reviews 2014, 13, 981–1000. [Google Scholar] [CrossRef]
- Gremese, E.; Tolusso, B.; Gigante, M.R.; Ferraccioli, G. Obesity as a risk and severity factor in rheumatic diseases (autoimmune chronic inflammatory diseases). Front Immunol 2014, 5, 576. [Google Scholar] [CrossRef]
- Kwiat, V.R.; Reis, G.; Valera, I.C.; Parvatiyar, K.; Parvatiyar, M.S. Autoimmunity as a sequela to obesity and systemic inflammation. Front Physiol 2022, 13, 887702. [Google Scholar] [CrossRef]
- Varra, F.-N.; Varras, M.; Varra, V.-K.; Theodosis-Nobelos, P. Molecular and pathophysiological relationship between obesity and chronic inflammation in the manifestation of metabolic dysfunctions and their inflammation-mediating treatment options (Review). Mol Med Rep 2024, 29, 95. [Google Scholar] [CrossRef]
- LMoroni, I. Bianchi, and A. Lleo: Geoepidemiology, gender and autoimmune disease. Autoimmun Rev 2012, 11, A386–A392. [Google Scholar] [CrossRef]
- WHO, Overweight/obesity: overweight by country. Global Health Observatory Data Repository 2008–2013, 2016, [internet]. Available online: http://www.who.int/gho/ncd/risk factors/obesity text/en/.
- Guerrero-García, J.J.; Carrera-Quintanar, L.; López-Roa, R.I.; Márquez-Aguirre, A.L.; Rojas-Mayorquín, A.E.; Daniel Ortuño-Sahagún, D. Multiple Sclerosis and Obesity: Possible Roles of Adipokines. Mediators Inflamm 2016, 2016, 4036232. [Google Scholar] [CrossRef]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Thompson, A.J.; Wolinsky, J.S.; et al. Defining the clinical course of multiple sclerosis. Neurology 2013, 83, 278–286. [Google Scholar] [CrossRef]
- Milo, R.; Miller, A. Revised diagnostic criteria of multiple sclerosis. Autoimmun Rev 2014, 13, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Koch-Henriksen, N.; Sørensen, P.S. The changing demographic pattern of multiple sclerosis epidemiology. Lancet Neurol 2010, 9, 520–532. [Google Scholar] [CrossRef] [PubMed]
- Trojano, M.; Lucchese, G.; Graziano, G.; Taylor, B.V.; Simpson SJr Lepore, V.; et al. Geographical variations in sex ratio trends over time in multiple sclerosis. PloS One 2012, 7, e48078. [Google Scholar] [CrossRef]
- Melcon, M.O.; Correale, J.; Melcon, C.M. Is it time for a new global classification of multiple sclerosis? J Neurol Sci 2014, 344, 171–181. [Google Scholar] [CrossRef]
- Correale, J.; Marrodan, M. Multiple sclerosis and obesity: The role of adipokines. Front Immunol 2022, 13, 1038393. [Google Scholar] [CrossRef]
- Schwarz, S.; Leweling, H. Multiple sclerosis and nutrition. Mult Scler 2005, 11, 24–32. [Google Scholar] [CrossRef]
- Marck, C.H.; Neate, S.L.; Taylor, K.L.; Weiland, T.J.; Jelinek, G.A. Prevalence of comorbidities, overweight and obesity in an international sample of people withmultiple sclerosis and associations with modifiable lifestyle factors. PLoS ONE 2016, 11, e0148573. [Google Scholar] [CrossRef]
- Hedström, A.; Alfredsson, L.; Olsson, T. Environmental factors and their interactions with risk genotypes in MS susceptibility. Curr Opin Neurol 2016, 29, 293–298. [Google Scholar] [CrossRef]
- Mohammadi, M.; Mohammadi, A.; Habibzadeh, A.; Korkorian, R.; Mohamadi, M.; Shaygannejad, V.; Zabeti, A.; Mirmosayyeb, O. Abnormal body mass index is associated with risk of multiple sclerosis: A systematic review and meta-analysis. Obes Res Clin Pract 2024, 18, 311–321. [Google Scholar] [CrossRef]
- Marrie, R.A.; Horwitz, R.I.; Cutter, G.; Tyry, T.; Vollmer, T. Association between comorbidity clinical characteristics of MS. Acta Neurol Scand 2011, 124, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, S.R.; Colado Simão, A.N.; Kallaur, A.P.; et al. Disability in patients with multiple sclerosis: influence of insulin resistance, adiposity, and oxidative stress. Nutrition 2014, 30, 268–273. [Google Scholar] [CrossRef]
- Mokry, L.E.; Ross, S.; Timpson, N.J.; et al. Obesity and multiple sclerosis: a mendelian randomization study. PLoS Med 2016, 13, e1002053. [Google Scholar] [CrossRef]
- Alfredsson, L.; Olsson, T. Lifestyle and environmental factors in multiple sclerosis. Cold Spring Harb Perspect Med 2019, 9, a028944. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Yu, H.; Bu, Z.; Wen, L.; Yan, L.; Feng, J. Focus on the Role of the NLRP3 Inflammasome in multiple sclerosis: Pathogenesis, diagnosis, and therapeutics. Front Mol Neurosci 2022, 15, 894298. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.; Birk, R. Adipose tissue hyperplasia and hypertrophy in common and syndromic obesity-The case of BBS obesity. Nutrients 2023, 15, 3445. [Google Scholar] [CrossRef]
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am J Physiol Cell Physiol 2021, 320, C375–C391. [Google Scholar] [CrossRef]
- Balasa, R.; Maier, S.; Barcutean, L.; Stoian, A.; Motataianu, A. The direct deleterious effect of Th17 cells in the nervous system compartment in multiple sclerosis and experimental autoimmune encephalomyelitis: One possible link between neuroinflammation and neurodegeneration. Rev Romana Med Laborator 2020, 28. [Google Scholar] [CrossRef]
- Raud, B.; McGuire, P.J.; Jones, R.G.; Sparwasser, T.; Berod, L. Fatty acid metabolism in CD8+ T cell memory: Challenging current concepts. Immunol Rev 2018, 283, 213–231. [Google Scholar] [CrossRef]
- Varra, F.-N.; Varras, M.; Karikas, G.-A.; Theodosis-Nobelos, P. Mechanistic interrelation between Multiple Sclerosis and the factors related to obesity. Involvement of antioxidants. Pharmakeftiki, 2025, 37, 2–16. [Google Scholar]
- Giovannoni, G. The neurodegenerative prodrome in multiple sclerosis. Lancet Neurol 2017, 16, 413–414. [Google Scholar] [CrossRef]
- Jalonen, T.O.; Pulkkinen, K.; Ukkonen, M.; Saarela, M.; Elovaara, I. Differential intracellular expression of CCR5 and chemokines in multiple sclerosis subtypes. J Neurol 2002, 249, 576–583. [Google Scholar] [CrossRef] [PubMed]
- Cheng, W.; Chen, G. Chemokines and Chemokine Receptors in Multiple Sclerosis. Mediators Inflamm 2014, 2014, 659206. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of TLR4 signaling in hypothalamus: implications for the pathogenesis of obesity. J Neurosci 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Piccio, L.; Cantoni, C.; Henderson, J.G.; Hawiger, D.; Ramsbottom, M.; Mikesell, R.; Ryu, J.; Hsieh, C.S.; Cremasco, V.; Haynes, W.; et al. Lack of adiponectin leads to increased lymphocyte activation and increased disease severity in a mouse model of multiple sclerosis. Eur J Immunol 2013, 43, 2089–2100. [Google Scholar] [CrossRef]
- Tehrani, A.R.; Gholipour, S.; Sharifi, R.; Yadegari, S.; Abbasi-Kolli, M.; Masoudian, N. Plasma levels of CTRP-3, CTRP-9 and apelin in women with multiple sclerosis. J Neuroimmunol 2019, 333, 576968. [Google Scholar] [CrossRef]
- Düzel, B.; Tamam, Y.; Çoban, A.; Tüzün, E. Adipokines in multiple sclerosis patients with and without optic neuritis as the first clinical presentation. Immunol Investig 2018, 48, 190–197. [Google Scholar] [CrossRef]
- Signoriello, E.; Lus, G.; Polito, R.; Casertano, S.; Scudiero, O.; Coletta, M.; Monaco, M.L.; Rossi, F.; Nigro, E.; Daniele, A. Adiponectin profile at baseline is correlated to progression and severity of multiple sclerosis. Eur J Neurol 2019, 26, 348–355. [Google Scholar] [CrossRef]
- Signoriello, E.; Mallardo, M.; Nigro, E.; Polito, R.; Casertano, S.; Di Pietro, A.; Coletta, M.; Monaco, M.L.; Rossi, F.; Lus, G.; et al. Adiponectin in cerebrospinal fluid from patients affected by multiple sclerosis is correlated with the progression and severity of disease. Mol Neurobiol 2021, 58, 2663–2670, Erratum in 2021, 58, 2671. [Google Scholar] [PubMed]
- Musabak, U.H.; Demirkaya, S.; Genç, G.; Ilikci, R.S.; Odabasi, Z. Serum adiponectin, TNF-α, IL-12p70, and IL-13 levels in multiple sclerosis and the effects of different therapy regimens. Neuroimmunomodulation 2011, 18, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Kraszula, Ł.; Jasińska, A.; Eusebio, M.-O.; Kuna, P.; Głąbiński, A.; Pietruczuk, M. Evaluation of the relationship between leptin, resistin, adiponectin and natural regulatory T cells in relapsing-remitting multiple sclerosis. Neurol Neurochir Polska 2012, 46, 22–28. [Google Scholar] [CrossRef]
- Rijnsburger, M.; Djuric, N.; Mulder, I.A.; de Vries, H.E. Adipokines as immune cell modulators in multiple sclerosis. Int J Mol Sci 2021, 22, 10845. [Google Scholar] [CrossRef]
- Chen, Y.; Pitzer, A.L.; Li, X.; Li, P.L.; Wang, L.; Zhang, Y. Instigation of endothelial Nlrp3 inflammasome by adipokine visfatin promotes inter-endothelial junction disruption: role of HMGB1. J Cell Mol Med 2015, 19, 2715–2727. [Google Scholar] [CrossRef]
- Alpua, M.; Turkel, Y.; Dag, E.; Kisa, U. Apelin-13, A promising biomarker for multiple sclerosis? Ann Indian Acad Neurol 2018, 21, 126–129. [Google Scholar] [CrossRef]
- Hu, G.; Wang, Z.; Zhang, R.; Sun, W.; Chen, X. The Role of apelin/apelin receptor in energy metabolism and water homeostasis: A comprehensive narrative review. Front Physiol 2021, 12, 632886. [Google Scholar] [CrossRef]
- Ito, M.; Muramatsu, M.; Kato, Y.; Sharma, B. Age-dependent decline in remyelination capacity is mediated by apelin/APJ signaling. Nat Aging 2021, 1, 284–294. [Google Scholar] [CrossRef]
- Liu, Q.; Zhuo, S.; Wang, X.; Gu, C.; Guo, Q.; Li, X.; et al. Apelin alleviated neuroinflammation and promoted endogenous neural cells proliferation and differentiation after spinal cord injury in rats. J Neuroinflam 2022, 19, 160. [Google Scholar] [CrossRef]
- Dragano, N.R.; Haddad-Tovolli, R.; Velloso, L.A. Leptin, Neuroinflammation and Obesity. Front Horm Res 2017, 48, 84–96. [Google Scholar]
- Izquierdo, A.G.; Crujeiras, A.B.; Casanueva, F.F.; Carreira, M.C. Leptin, obesity, and leptin resistance: Where are we 25 years later? Nutrients 2019, 11, 2704. [Google Scholar] [CrossRef]
- Maurya, R.; Bhattacharya, P.; Dey, R.; Nakhasi, H.L. Leptin Functions in Infectious Diseases. Front Immunol 2018, 9, 2741. [Google Scholar] [CrossRef]
- Kiernan, K.; Nichols, A.G.; Alwarawrah, Y.; MacIver, N.J. Effects of T cell leptin signaling on systemic glucose tolerance and T cell responses in obesity. PLoS ONE 2023, 18, e0286470. [Google Scholar] [CrossRef]
- Liu, J.; Yang, X.; Yu, S.; Zheng, R. The leptin resistance. Adv Exp Med Biol 2018, 1090, 145–163. [Google Scholar]
- Pan, W.W.; Myers, M.G., Jr. Leptin and the maintenance of elevated body weight. Nat Rev Neurosci 2018, 19, 95–105. [Google Scholar] [CrossRef]
- Matarese, G.; Carrieri, P.B.; Montella, S.; De Rosa, V.; La Cava, A. Leptin as a metabolic link to multiple sclerosis. Nat Rev Neurol 2010, 6, 455–461. [Google Scholar] [CrossRef]
- Evangelopoulos, M.E.; Koutsis, G.; Markianos, M. Serum leptin levels in treatment-naive patients with clinically isolated syndrome or relapsing-remitting multiple sclerosis. Autoimmune Dis 2014, 2014, 486282. [Google Scholar] [CrossRef]
- Ouyang, S.; Hsuchou, H.; Kastin, A.J.; Mishra, P.K.; Wang, Y.; Pan, W. Leukocyte infiltration into spinal cord of EAE mice is attenuated by removal of endothelial leptin signaling. Brain Behav Immun 2014, 40, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.R.; Bae, Y.H.; Bae, S.K.; Choi, K.S.; Yoon, K.H.; Koo, T.H.; et al. Visfatin enhances ICAM-1 and VCAM-1 expression through ROS-dependent NF-kappaB activation in endothelial cells. Biochim Biophys Acta 2008, 1783, 886–895. [Google Scholar] [CrossRef] [PubMed]
- 58 Xu, Y.; Yu, L.; Liu, Y.; Tang, X.; Wang, X. Lipopolysaccharide-induced microglial neuroinflammation: Attenuation by FK866. Neurochem Res 2021, 46, 1291–1304. [Google Scholar] [CrossRef] [PubMed]
- Mariani, F.; Roncucci, L. Chemerin/chemR23 axis in inflammation onset and resolution. Inflammation Res 2015, 64, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Emamgholipour, S.; Eshaghi, S.M.; Hossein-nezhad, A.; Mirzaei, K.; Maghbooli, Z.; Sahraian, M.A. Adipocytokine profile, cytokine levels and foxp3 expression in multiple sclerosis: A possible link to susceptibility and clinical course of disease. PLoS ONE 2013, 8, e76555. [Google Scholar] [CrossRef]
- Hossein-Nezhad, A.; Varzaneh, F.N.; Mirzaei, K.; Emamgholipour, S.; Varzaneh, F.N.; Sahraian, M.A. A polymorphism in the resistin gene promoter and the risk of multiple sclerosis. Minerva Med. 2013, 104, 431–438. [Google Scholar]
- Bhatti, J.S.; Bhatti, G.K.; Reddy, P.H. Mitochondrial dysfunction and oxidative stress in metabolic disorders—A step towards mitochondria based therapeutic strategies. Biochim Biophys Acta-Mol Basis Dis 2016, 1863, 1066–1077. [Google Scholar] [CrossRef]
- Sethi, J.K.; Hotamisligil, G.S. Metabolic Messengers: Tumour necrosis factor. Nat Metab 2021, 3, 1302–1312. [Google Scholar] [CrossRef]
- Lebas, H.; Guérit, S.; Picot, A.; Boulay, A.C.; Fournier, A.; Vivien, D.; Salmon, M.C.; Docagne, F.; Bardou, I. PAI-1 production by reactive astrocytes drives tissue dysfibrinolysis in multiple sclerosis models. Cell Mol Life Sci 2022, 79, 323. [Google Scholar] [CrossRef]
- Bruzzone, S.; Fruscione, F.; Morando, S.; Ferrando, T.; Poggi, A.; Garuti, A.; et al. Catastrophic NAD+ depletion in activated Tlymphocytes through nampt inhibition reduces demyelination disability in, EAE. PloS One 2009, 4, e7897. [Google Scholar] [CrossRef]
- Ghosh, A.R.; Bhattacharya, R.; Bhattacharya, S.; Nargis, T.; Rahaman, O.; Duttagupta, P.; et al. Adipose recruitment and activation of plasmacytoid dendritic cells fuel metaflammation. Diabetes 2016, 65, 3440–3452. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.M.; Li, W.Q.; Huang, C.L.; Li, J.; Xie, W.H.; et al. Ccrl2 deficiency deteriorates obesity and insulin resistance through increasing adipose tissue macrophages infiltration. Genes Dis 2022, 9, 429–442. [Google Scholar] [CrossRef]
- Lande, R.; Gafa, V.; Serafini, B.; Giacomini, E.; Visconti, A.; Remoli, M.E.; et al. Plasmacytoid dendritic cells in multiple sclerosis: intracerebral recruitment and impaired maturation in response to interferon-beta. J Neuropathol Exp Neurol 2008, 67, 388–401. [Google Scholar] [CrossRef] [PubMed]
- Tomalka-Kochanowska, J.; Baranowska, B.; Wolinska-Witort, E.; Uchman, D.; Litwiniuk, A.; Martynska, L.; Kalisz, M.; Bik, W.; Kochanowski, J. Plasma chemerin levels in patients with multiple sclerosis. Neuro Endocrinol Lett 2014, 35, 218–223. [Google Scholar] [PubMed]
- Koskderelioglu, A.; Gedizlioglu, M.; Eskut, N.; Tamer, P.; Yalcin GBozkaya, G. Impact of chemerin, lipid profile, and insulin resistance on disease parameters in patients with multiple sclerosis. Neurol Sci 2021, 42, 2471–2479. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.J.; Thompson, B.R.; Sanders, M.A.; Bernlohr, D.A. Interaction of the adipocyte fatty acid-binding protein with the hormone-sensitive lipase: regulation by fatty acids and phosphorylation. J Biol Chem 2007, 282, 32424–32432. [Google Scholar] [CrossRef]
- Bove, R.; Healy, B.C.; Musallam, A.; Soltany, P.; Diaz-Cruz, C.; Sattarnezhad, N.; et al. Fatty acid binding protein-4 is associated with disability in multiple sclerosis. Mult Scler 2019, 25, 344–35. [Google Scholar] [CrossRef]
- Dou, H.X.; Wang, T.; Su, H.X.; Gao, D.D.; Xu, Y.C.; Li, Y.X.; et al. Exogenous FABP4 interferes with differentiation, promotes lipolysis and inflammation in adipocytes. Endocrine 2020, 67, 587–596. [Google Scholar] [CrossRef]
- Messina, S.; Vargas-Lowy, D.; Musallam, A.; Healy, B.C.; Kivisakk, P.; Gandhi, R.; et al. Increased leptin a-FABPlevels in relapsing progressive forms of MS. BMC Neurol 2013, 13, 172. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Saitoh, S.; Shimamoto, K.; Miura, T. Fatty Acid-Binding Protein 4 (FABP4): Pathophysiological insights and potent clinical biomarker of metabolic and cardiovascular diseases. Clin Med Insights Cardiol 2015, 8 (Suppl. S3), 23–33. [Google Scholar] [CrossRef]
- Furuhashi, M. Fatty acid-binding protein 4 in cardiovascular and metabolic diseases. J Atheroscler Thromb 2019, 26, 216–232. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, J.M.; Liu, Q.; Brittingham, K.C.; Liu, Y.; Gruentha, M.; Gorgun, C.Z.; et al. Deficiency of fatty acid-binding proteins in mice confers protection from development of experimental autoimmune encephalomyelitis. J Immunol 2007, 179, 313–321. [Google Scholar] [CrossRef]
- Jung, M.Y.; Kim, H.S.; Hong, H.J.; Youn, B.S.; Kim, T.S. Adiponectin induces dendritic cell activation via PLCg/JNK/NF-kB pathways, leading to Th1 and Th17 polarization. J Immunol 2012, 188, 2592–2601. [Google Scholar] [CrossRef] [PubMed]
- Pérez, M.M.G.; Eisele, S.J.G. MicroRNAs as a possible biomarker in the treatment of multiple sclerosis. IBRO Neurosci Rep 2022, 13, 492–499. [Google Scholar] [CrossRef]
- Ouchi, N.; Parker, J.L.; Lugus, J.J.; Walsh, K. Adipokines in inflammation and metabolic disease. Nat Rev Immunol 2011, 11, 85–97. [Google Scholar] [CrossRef]
- Brambilla, R.; Ashbaugh, J.J.; Magliozzi, R.; et al. Inhibition of soluble tumour necrosis factor is therapeutic in experimental autoimmune encephalomyelitis and promotes axon preservation and remyelination. Brain 2011, 134, 2736–2754. [Google Scholar] [CrossRef]
- Wajant, H.; Siegmund, D. TNFR1 and TNFR2 in the Control of the Life and Death Balance of Macrophages. Front Cell Dev Biol 2019, 7, 91. [Google Scholar] [CrossRef]
- Pegoretti, V.; Baron, W.; Laman, J.D.; Eisel, U.L.M. Selective Modulation of TNF–TNFRs Signaling: Insights for Multiple Sclerosis Treatment. Front Immunol 2018, 9, 925. [Google Scholar] [CrossRef]
- LProbert:, T.N.F.; its receptors in the, C.N.S. The essential, the desirable and the deleterious effects. Neuroscience 2015, 302, 2–22. [Google Scholar] [CrossRef]
- Hövelmeyer, N.; Hao, Z.; Kranidioti, K.; Kassiotis, G.; Buch, T.; Frommer, F.; von Hoch, L.; Kramer, D.; Minichiello, L.; Kollias, G.; Lassmann, H.; Waisman, A. Apoptosis of oligodendrocytes via Fas and TNF-R1 is a key event in the induction of experimental autoimmune encephalomyelitis. J Immunol 2005, 175, 5875–5884. [Google Scholar] [CrossRef]
- Sánchez-Muñoz, F.; García-Macedo, R.; Alarcón-Aguilar, F.; Cruz, M. Adipocinas, tejido adiposo y su relación con células del sistema inmune [Adipocitokines, adipose tissue and its relationship with immune system cells]. Gac Med Mex. 2005, 141, 505–512. (In Spanish) [Google Scholar]
- Lastra, G.; Manrique, C.M.; Hayden, M.R. The role of beta-cell dysfunction in the cardiometabolic syndrome. J Cardiometab Syndr 2006, 1, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Brück, W. The pathology of multiple sclerosis is the result of focal inflammatory demyelination with axonal damage. J Neurol 2005, 252 (Suppl. S5), v3–v9. [Google Scholar] [CrossRef]
- Fresegna, D.; Bullitta, S.; Musella, A.; Rizzo, F.R.; De Vito, F.; Guadalupi, L.; et al. Reexamining the role of TNF in MS pathogenesis and therapy. Cells 2020, 9, 2290. [Google Scholar] [CrossRef] [PubMed]
- Sharief, M.K.; Hentges, R. Association between tumor necrosis factor-alpha and disease progression in patients with multiple sclerosis. N Engl J Med 1991, 325, 467–472. [Google Scholar] [CrossRef]
- Gregory, A.P.; Dendrou, C.A.; Attfield, K.E.; Haghikia, A.; Xifara, D.K.; Butter, F.; et al. TNF receptor 1 genetic risk mirrors outcome of anti-TNF therapy in multiple sclerosis. Nature 2012, 488, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Maguire, A.D.; Bethea, J.R.; Kerr, B.J. TNFa in MS and its animal models: implications for chronic pain in the disease. Front Neurol 2021, 12, 780876. [Google Scholar] [CrossRef]
- van Oosten, B.W.; Barkhof, F.; Truyen, L.; Boringa, J.B.; Bertelsmann, F.W.; et al. Increased MRI activity and immune activation in two multiple sclerosis patients treated with the monoclonal antitumor necrosis factor antibody cA2. Neurology 1996, 47, 1531–1534. [Google Scholar] [CrossRef] [PubMed]
- Kassiotis, G.; Kollias, G. Uncoupling the proinflammatory from the immunosuppressive properties of tumor necrosis factor (TNF) at the p55 TNF receptor level: implications for pathogenesis and therapy of autoimmune demyelination. J Exp Med 2001, 193, 427–434. [Google Scholar] [CrossRef]
- Steeland, S.; Van Ryckeghem, S.; Van Imschoot, G.; et al. TNFR1 inhibition with a Nanobody protects against EAE development in mice. Sci Rep 2017, 7, 13646. [Google Scholar] [CrossRef]
- Nomura, T.; Abe, Y.; Kamada, H.; Shibata, H.; Kayamuro, H.; Inoue, M.; et al. Therapeutic effect of PEGylated TNFR1-selective antagonistic mutant TNF in experimental autoimmune encephalomyelitis mice. J Control Release 2011, 149, 8–14. [Google Scholar] [CrossRef]
- Williams, S.K.; Maier, O.; Fischer, R.; Fairless, R.; Hochmeister, S.; Stojic, A.; et al. Antibody-mediated inhibition of TNFR1 attenuates disease in a mouse model of multiple sclerosis. PLoS One 2014, 9, e90117. [Google Scholar] [CrossRef]
- Richter, F.; Williams, S.K.; John, K.; Huber, C.; Vaslin, C.; Zanker, H.; et al. The TNFR1 antagonist atrosimab is therapeutic in mouse models of acute and chronic inflammation. Front Immunol 2021, 12, 705485. [Google Scholar] [CrossRef]
- Ireland, S.J.; Monson, N.L.; Davis, L.S. Seeking Balance: Potentiation and Inhibition of Multiple Sclerosis Autoimmune Responses by IL-6 and IL-10. Cytokine 2015, 73, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Rothaug, M.; Becker-Pauly, C.; Rose-John, S. The role of interleukin-6 signaling in nervous tissue. Biochim Biophys Acta 2016, 1863, 1218–1227. [Google Scholar] [CrossRef] [PubMed]
- Jin, E.S.; Lee, M.H.; Murphy, R.E.; Malloy, C.R. Pentose phosphate pathway activity parallels lipogenesis but not antioxidant processes in rat liver. Am J Physiol-Endocrinol Metab 2017, 314, E543–E551. [Google Scholar] [CrossRef]
- Eugster, H.P.; et al. IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol 1998, 28, 2178–2187. [Google Scholar] [CrossRef]
- Okuda, Y.; et al. IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol 1998, 10, 703–708. [Google Scholar] [CrossRef]
- Chakrabarti, S.; Patel, K.D. Regulation of matrix metalloproteinase-9 release from IL-8-stimulated human neutrophils. J Leukoc Biol 2005, 78, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Straczkowski, M.; Dzienis-Straczkowska, S.; Stêpieñ, A.; Kowalska, I.; Szelachowska, M.; Kinalska, I. Plasma interleukin-8 concentrations are increased in obese subjects and related to fat mass and tumor necrosis factor-alpha system. J Clin Endocrinol Metab 2002, 87, 4602–4606. [Google Scholar] [CrossRef]
- Makiel, Κ.; Suder, A.; Targosz, A.; Maciejczyk, Μ.; Alon Haim, A. Exercise-Induced Alternations of Adiponectin, Interleukin-8 and Indicators of Carbohydrate. Metabolism in males with metabolic syndrome. Biomolecules 2023, 13, 852. [Google Scholar] [CrossRef]
- Matejčíkováa, Z.; Mareša, J.; Sládkováa, V.; Svrčinováa, T.; Vysloužilováa, J.; Zapletalováb, J.; Kaňovský, P. Cerebrospinal fluid and serum levels of interleukin-8 in patients with multiple sclerosis and its correlation with Q-albumin. Mult Scler Relat Disord 2017, 14, 12–15. [Google Scholar] [CrossRef]
- Lund, B.T.; Ashikian, N.; Ta, H.Q.; Chakryan, Y.; Manoukian, K.; Groshen, S.; Gilmore, W.; Cheema, G.S.; Stohl, W.; Burnett, M.E.; Ko, D.; Kachuck, N.J.; Weiner, L.P. 2004 Increased CXCL8 (IL-8) expression in Multiple Sclerosis. J. Neuroimmunol. 2004, 155, 161–171. [Google Scholar] [CrossRef] [PubMed]
- Neuteboom, R.F.; Verbraak, E.; Voerman, J.S.; van Meurs, M.; Steegers, E.A.; de Groot, C.J.; Laman, J.D.; Hintzen, R.Q. First trimester interleukin 8 levels are associated with postpartum relapse in multiple sclerosis. Mult Scler 2009, 15, 1356–1358. [Google Scholar] [CrossRef]
- Ihim, S.A.; Abubakar, S.D.; Zian, Z.; Sasaki, T.; Saffarioun, M.; Maleknia, S.; Azizi, G. Interleukin-18 cytokine in immunity, inflammation, and autoimmunity: Biological role in induction, regulation, and treatment. Front Immunol 2022, 13, 919973. [Google Scholar] [CrossRef] [PubMed]
- Gris, D.; Ye, Z.; Iocca, H.A.; Wen, H.; Craven, R.R.; Gris, P.; et al. NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immun 2010, 185, 974–981. [Google Scholar] [CrossRef]
- Nicoletti, F.; Di-Marco, R.; Mangano, K.; Patti, F.; Reggio, E.; Nicoletti, A.; et al. Increased serum levels of interleukin-18 in patients with multiple sclerosis. Neurology 2001, 57, 342–344. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Chen, S.D.; Miao, L.; Liu, Z.G.; Li, W.; Zhao, Z.X.; et al. Serum levels of interleukin (IL)-18, IL-23 and IL-17 in Chinese patients with multiple sclerosis. J Neuroimmunol 2012, 243, 56–60. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.D.; Takeda, K.; Akira, S.; Sarvetnick, N.; Ljunggren, H.G. IL-18 directs autoreactive T cells and promotes autodestruction in the central nervous system via induction of IFN-γ by NK cells. J Immun 2000, 165, 3099–3104. [Google Scholar] [CrossRef]
- Gutcher, I.; Urich, E.; Wolter, K.; Prinz, M.; Becher, B. Interleukin 18–independent engagement of interleukin 18 receptor-α is required for autoimmune inflammation. Nat Immunol 2006, 7, 946–953. [Google Scholar] [CrossRef]
- Lalor, S.J.; Dungan, L.S.; Sutton, C.E.; Basdeo, S.A.; Fletcher, J.M.; Mills, K.H. Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immun 2011, 186, 5738–5748. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; et al. The hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat Rev Immunol 2015, 15, 545–558. [Google Scholar] [CrossRef]
- Barclay, W.; Shinohara, M.L. Inflammasome activation inmultiple sclerosis and experimental autoimmune encephalomyelitis (EAE). Brain Pathology 2017, 27, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Bradstreet, T.R.; Schwarzkopf, E.A.; Jarjour, N.N.; Chou, C.; Archambault, A.S.; et al. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. The Journal of Experimental Medicine 2016, 213, 251. [Google Scholar] [CrossRef]
- Schiffenbauer, J.; Streit, W.J.; Butfiloski, E.; LaBow, M.; Edwards, C., III; Moldawer, L.L. The induction of EAE is only partially dependent on TNF receptor signaling but requires the IL-1 type I receptor. Clinical Immunology 2000, 95, 117–123. [Google Scholar] [CrossRef]
- Iyer SS, Cheng G: Role of Interleukin 10 Transcriptional Regulation in Inflammation and Autoimmune Disease. Crit Rev Immunol 2012, 32, 23–63. [CrossRef]
- Moore, K.W.; et al. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol 2001, 19, 683–765. [Google Scholar] [CrossRef]
- Bugbee, E.; Wang, A.A.; Gommerman, J.L. Under the influence: environmental factors as modulators of neuroinflammation through the IL-10/iL-10R axis. Front Immunol 2023, 14, 1188750. [Google Scholar] [CrossRef]
- Bettelli, E.; et al. IL-10 is critical in the regulation of autoimmune encephalomyelitis as demonstrated by studies of IL-10- and IL-4-deficient and transgenic mice. J Immunol 1998, 161, 3299–3306. [Google Scholar] [CrossRef]
- Samoilova, E.B.; Horton, J.L.; Chen, Y. Acceleration of experimental autoimmune encephalomyelitis in interleukin-10-deficient mice: roles of interleukin-10 in disease progression and recovery. Cell Immunol 1998, 188, 118–124. [Google Scholar] [CrossRef]
- Shao, S.; Chen, C.; Shi, G.; Zhou, Y.; Wei, Y.; Fan, N.; Yang, Y.; Wu, L.; Zhang, T. Therapeutic potential of the target on NLRP3 inflammasome in multiple sclerosis. Pharmacol Ther 2021, 227, 107880. [Google Scholar] [CrossRef] [PubMed]
- Govindarajan, V.; de Rivero Vaccari, J.P.; Keane, R.W. Role of inflammasomes in multiple sclerosis and their potential as therapeutic targets. J Neuroinflammation 2020, 17, 260. [Google Scholar] [CrossRef]
- Esser, N.; L’homme, L.; De Roover, A.; Kohnen, L.; Scheen, A.J.; Moutschen, M.; Piette, J.; Legrand-Poels, S.; Paquot, N. Obesity phenotype is related to nlrp3 inflammasome activity and immunological profile of visceral adipose tissue. Diabetologia 2013, 56, 2487–2497. [Google Scholar] [CrossRef] [PubMed]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The nlrp3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179. [Google Scholar] [CrossRef] [PubMed]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P. Fatty acid–induced nlrp3-asc inflammasome activation interferes with insulin signaling. Nat Immunol 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Zeng X, Zhu M, Liu X, Chen X, Yuan Y, Li L, Liu J, Lu Y, Cheng J, Chen, Y; et al. Oleic acid ameliorates palmitic acid induced hepatocellular lipotoxicity by inhibition of er stress and pyroptosis. Nutr Metab 2020, 17, 1–14.
- Yan, Y.; Jiang, W.; Spinetti, T.; Tardivel, A.; Castillo, R.; Bourquin, C.; Guarda, G.; Tian, Z.; Tschopp, J.; Zhou, R.; et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of nlrp3 inflammasome activation. Immunity 2013, 38, 1154–1163. [Google Scholar] [CrossRef]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. Nlrp3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef]
- Sheedy, F.J.; Grebe, A.; Rayner, K.J.; Kalantari, P.; Ramkhelawon, B.; Carpenter, S.B.; Becker, C.E.; Ediriweera, H.N.; Mullick, A.E.; Golenbock, D.T.; et al. Cd36 coordinates nlrp3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol 2013, 14, 812–820. [Google Scholar] [CrossRef]
- Koenen, T.B.; Stienstra, R.; Van Tits, L.J.; De Graaf, J.; Stalenhoef, A.F.; Joosten, L.A.; Tack, C.J.; Netea, M.G. Hyperglycemia activates caspase-1 and TXNIP-mediated IL-1beta transcription in human adipose tissue. Diabetes 2011, 60, 517–524. [Google Scholar] [CrossRef]
- Yang, S.-J.; Han, A.R.; Kim, E.-A.; Yang, J.W.; Ahn, J.-Y.; Na, J.-M.; Cho, S.-W. Khg21834 attenuates glutamate-induced mitochondrial damage apoptosis nlrp3 inflammasome activation in sh-sy5y human neuroblastoma cells. Eur, J. Pharmacol. 2019, 856, 172412. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 2007, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, X.; Ghani, A.; Malik, A.; Wilder, T.; Colegio, O.R.; Flavell, R.A.; Cronstein, B.N.; Mehal, W.Z. Adenosine is required for sustained inflammasome activation via the a 2a receptor and the hif-1α pathway. Nat Commun 2013, 4, 1–9. [Google Scholar] [CrossRef]
- Keane, R.; Dietrich, W.; de Rivero Vaccari, J. Inflammasome proteins as biomarkers of multiple sclerosis. Front Neurol 2018, 9, 135. [Google Scholar] [CrossRef]
- Voet, S.; Mc Guire, C.; Hagemeyer, N.; Martens, A.; Schroeder, A.; Wieghofer, P.; et al. A 20 critically controls microglia activation and inhibits inflammasome-dependent neuroinflammation. Nat. Commun. 2018, 9, 2036. [Google Scholar] [CrossRef]
- Ke, P.; Shao, B.-Z.; Xu, Z.-Q.; Chen, X.-W.; Wei, W.; Liu, C. Activating a7 nicotinic acetylcholine receptor inhibits NLRP3 inflammasome through regulation of b-arrestin-1. CNS Neurosci Ther 2017, 23, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Williams, K.L.; Gunn, M.D.; Shinohara, M.L. NLRP3 inflammasome induces chemotactic immune cell migration to the CNSin experimental autoimmune encephalomyelitis. Proc Natl Acad Sci, U.S.A. 2012, 109, 10480–10485. [Google Scholar] [CrossRef]
- Yu, X.; Lv, J.; Wu, J.; Chen, Y.; Chen, F.; Wang, L. The autoimmune encephalitis-related cytokine TSLP in the brain primes neuroinflammation by activating the JAK2-NLRP3 axis. Clin Exp Immunol 2021, 207, 113–122. [Google Scholar] [CrossRef]
- Hörmannsperger, G.; Haller, D. Molecular crosstalk of probiotic bacteria with the intestinal immune system: clinical relevance in the context of inflammatory bowel disease. Int J Med Microbiol 2010, 300, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Lang, Y.; Shen, D.; Jin, T.; Zhu, J.; Cui, L. Gut microbiota in multiple sclerosis and experimental autoimmune encephalomyelitis: Current applications and future perspectives. Mediat Inflamm 2018, 2018, 8168717. [Google Scholar] [CrossRef]
- Forte, N.; Fernández-Rilo, A.C.; Palomba, L.; Di Marzo, V.; Cristino, L. Obesity Affects the Microbiota–Gut–Brain Axis and the Regulation Thereof by Endocannabinoids and Related Mediators. Int J Mol Sci 2020, 21, 1554. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2- driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef]
- Haase, S.; Haghikia, A.; Wilck, N.; Müller, D.N.; Linker, R.A. Impacts of microbiome metabolites on immune regulation and autoimmunity. Immunology 2018, 154, 230–238. [Google Scholar] [CrossRef]
- Dalile, B.; Van Oudenhove, L.; Vervliet, B.; Verbeke, K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol 2019, 16, 461–478. [Google Scholar] [CrossRef] [PubMed]
- Esplugues, E.; Huber, S.; Gagliani, N.; Hauser, A.E.; Town, T.; Wan, Y.Y.; et al. Control of TH17 cells occurs in the small intestine. Nature 2011, 475, 514–518. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann Neurol 2017, 81, 369–382. [Google Scholar] [CrossRef]
- Buscarinu, M.C.; Cerasoli, B.; Annibali, V.; Policano, C.; Lionetto, L.; Capi, M.; Mechelli, R.; Romano, S.; Fornasiero, A.; Mattei, G. Altered intestinal permeability in patients with relapsing-remitting multiple sclerosis: A pilot study. Mult Sclerosis 2017, 23, 442–446. [Google Scholar] [CrossRef]
- Bron, P.A.; Kleerebezem, M.; Brummer, R.J.; Cani, P.D.; Mercenier, A.; MacDonald, T.T.; Garcia-Rodenas, C.L.; Wells, J.M. Can probiotics modulate human disease by impacting intestinal barrier function? B J Nutr 2017, 117, 93–107. [Google Scholar] [CrossRef]
- Duscha, A.; Gisevius, B.; Hirschberg, S.; Yissachar, N.; Stangl, G.I.; Eilers, E.; Bader, V.; Haase, S.; Kaisler, J.; David, C.; et al. Propionic Acid Shapes the Multiple Sclerosis Disease Course by an Immunomodulatory Mechanism. Cell 2020, 180, 1067–1080.e16. [Google Scholar] [CrossRef] [PubMed]
- Naomi, R.; Teoh, S.H.; Embong, H.; Balan, S.S.; Othman, F.; Bahari, H.; Yazid, M.D. The Role of Oxidative Stress and Inflammation in Obesity and Its Impact on Cognitive Impairments—A Narrative Review. Antioxidants 2023, 12, 1071. [Google Scholar] [CrossRef] [PubMed]
- Muzio, G.; Barrera, G.; Pizzimenti, S. Peroxisome Proliferator-Activated Receptors (PPARs) and Oxidative Stress in Physiological Conditions and in Cancer. Antioxidants 2021, 10, 1734. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J Physiol 2003, 552, 335–344. [Google Scholar] [CrossRef]
- Tirichen, H.; Yaigoub, H.; Xu, W.; Wu, C.; Li, R.; Li, Y. Mitochondrial Reactive Oxygen Species and Their Contribution in Chronic Kidney Disease Progression Through Oxidative Stress. Front Physiol 2021, 12, 627837. [Google Scholar] [CrossRef]
- Krementsov, D.N.; Thornton, T.M.; Teuscher, C.; Rincon, M. The Emerging Role of p38 Mitogen-Activated Protein Kinase in Multiple Sclerosis and Its Models. Mol Cell Biol 2013, 33, 3728–3734. [Google Scholar] [CrossRef]
- Yue, Y.; Stone, S.; Lin, W. Role of nuclear factor κB in multiple sclerosis and experimental autoimmune encephalomyelitis. Neural Regen Res 2018, 13, 1507–1515. [Google Scholar]
- Singh, A.; Upadhayay, S.; Mehan, S. Understanding Abnormal c-JNK/p38MAPK Signaling Overactivation Involved in the Progression of Multiple Sclerosis: Possible Therapeutic Targets and Impact on Neurodegenerative Diseases. Neurotox Res 2021, 39, 1630–1650. [Google Scholar] [CrossRef]
- Chu, E.; Mychasiuk, R.; Hibbs, M.L.; Semple, B.D. Dysregulated phosphoinositide 3-kinase signaling in microglia: shaping chronic neuroinfammation. J Neuroinflammation 2021, 18, 276. [Google Scholar] [CrossRef]
- Ortiz, G.G.; et al. Immunology and oxidative stress in multiple sclerosis: clinical and basic approach. Clin Dev Immunol 2013, 2013, 708659. [Google Scholar]
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol 2016, 277, 58–67. [Google Scholar] [CrossRef]
- Tabassum, R.; Jeong, N.Y. Potential for therapeutic use of hydrogen sulfide in oxidative stress-induced neurodegenerative diseases. Int J Med Sci 2019, 16, 1386. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, J.M.; et al. Therapeutic potential of curcumin for multiple sclerosis. Neurol Sci 2018, 39, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Jadidi-Niaragh, F.; Mirshafiey, A. Th17 cell the new player of neuroinflammatory process in multiple sclerosis Scand. J. Immunol. 2011, 74, 1–13. [Google Scholar]
- Uttara, B.; et al. Oxidative stress and neurodegenerative diseases: a review of upstream and downstream antioxidant therapeutic options. Curr Neuropharmacol 2009, 7, 65–74. [Google Scholar] [CrossRef]
- Zha, Z.; et al. Potential utility of natural products against oxidative stress in animal models of multiple sclerosis. Antioxidants 2022, 11, 1495. [Google Scholar] [CrossRef]
- Tang, Z.; Hu, B.; Zang, F.; Wang, J.; Zhang, X.; Chen, H. Nrf2 drives oxidative stress-induced autophagy in nucleus pulposus cells via a Keap1/Nrf2/p62 feedback loop to protect intervertebral disc from degeneration. Cell Death & Disease 2019, 10, 510. [Google Scholar]
- Emamgholipour, S.; Hossein-Nezhad, A.; Sahraian, M.A.; Askarisadr, F.; Ansari, M. Expression and enzyme activity of MnSOD and catalase in peripheral blood mononuclear cells isolated from multiple sclerosis patients. Arch Med Lab Sci. 2015, 1, 23–28. [Google Scholar]
- Arredondo, F.; et al. After cellular internalization, quercetin causes Nrf2 nuclear translocation, increases glutathione levels, and prevents neuronal death against an oxidative insult. Free Radic Biol Med. 2010, 49, 738–747. [Google Scholar] [CrossRef]
- Lee, D.H.; Gold, R.; Linker, R.A. Mechanisms of oxidative damage in multiple sclerosis and neurodegenerative diseases: therapeutic modulation via fumaric acid esters. Int J Mol Sci 2012, 13, 11783–11803. [Google Scholar] [CrossRef]
- Hannan, M.A.; et al. Neuroprotection against oxidative stress: phytochemicals targeting TrkB signaling and the Nrf2-ARE antioxidant system. Front Mol Neurosci 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Lü, J.-M.; Lin, P.H.; Yao, Q.; Chen, C. Chemical and molecular mechanisms of antioxidants: experimental approaches and model systems. J Cell Mol Med 2009, 14, 840–860. [Google Scholar] [CrossRef]
- Gray, E.; Kemp, K.; Hares, K.; Redondo, J.; Rice, C.; Scolding, N.; Wilkins, A. Increased microglial catalase activity in multiple sclerosis grey matter. Brain Res 2014, 1559, 55–64. [Google Scholar] [CrossRef]
- Obradovic, D.; Andjelic, T.; Ninkovic, M.; Dejanovic, B.; Kotur-Stevuljevic, J. Superoxide dismutase (SOD), advanced oxidation protein products (AOPP), and disease-modifying treatment are related to better relapse recovery after corticosteroid treatment in multiple sclerosis. Neurol Sci 2021, 42, 3241–3247. [Google Scholar] [CrossRef]
- Salekzamani, S.; Pakkhesal, S.; Babaei, M.; Mirzaaghazadeh, E.; Mosaddeghi-Heris, R.; Talebi, M.; Sanaie, S.; Naseri, A. Coenzyme Q10 supplementation in multiple sclerosis; a systematic review. Multiple Sclerosis and Related Disorders 2024, 2024, 106212. [Google Scholar] [CrossRef] [PubMed]
- Varra, F.N.; Gkouzgos, S.; Varras, M.; Theodosis-Nobelos, P. Efficacy of antioxidant compounds in obesity and its associated comorbidities. Pharmakeftiki 2024, 36, 2–19. [Google Scholar]
- Theodosis-Nobelos, P.; Rekka, E.A. The Multiple Sclerosis Modulatory Potential of Natural Multi-Targeting Antioxidants. Molecules 2022, 27, 8402. [Google Scholar] [CrossRef]
- Luís, J.P.; Simões, C.J.V.; Brito, R.M.M. The Therapeutic Prospects of Targeting IL-1R1 for the Modulation of Neuroinflammation in Central Nervous System Disorders. Int J Mol Sci 2022, 23, 1731. [Google Scholar] [CrossRef]
- Sun, K.; Park, J.; Kim, M.; Scherer, P.E. Endotrophin, a multifaceted player in metabolic dysregulation and cancer progression, is a predictive biomarker for the response to PPAR agonist treatment. Diabetologia 2016, 60, 24–29. [Google Scholar] [CrossRef]
- Chuncha, T.; Thunapong, W.; Yasom, S.; Wanchai, K.; Eaimworawuthikul, S.; Metzler, G.; Lungkaphin, A.; Pongchaidecha, A.; Sirilun, S.; Chaiyasut, C.; et al. Decreased Microglial Activation Through Gut-brain Axis by Prebiotics Probiotics or Synbiotics Effectively Restored Cognitive Function in Obese-insulin Resistant Rats, J. Neuroinflammation 2018, 15, 11. [Google Scholar] [CrossRef]
- Long, X.; Zeng, X.; Tan, F.; Yi, R.; Pan, Y.; Zhou, X.; Mu, J.; Zhao, X. Lactobacillus plantarum KFY04 prevents obesity in mice through the PPAR pathway and alleviates oxidative damage and inflammation. Food Funct 2020, 11, 5460–5472. [Google Scholar] [CrossRef]
- Shirani, A.; Cross, A.H.; Stuve, O. Exploring the association between weight loss-inducing medications and multiple sclerosis: insights from the FDA adverse event reporting system database. Ther Adv Neurol Disord 2024, 17, 1–6. [Google Scholar]
- Sîrbe, C.; Rednic, S.; Grama, A.; Pop, T.L. An Update on the Effects of Vitamin D on the Immune System and Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 9784. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Du, S.; Zhao, L.; Jain, S.; Sahay, K.; Rizvanov, A.; et al. Autoreactive lymphocytes in multiple sclerosis: Pathogenesis and treatment target. Front Immunol 2022, 13, 996469. [Google Scholar] [CrossRef] [PubMed]
- Berer, A.; Stöckl, J.; Majdic, O.; Wagner, T.; Kollars, M.; Lechner, K.; et al. 1,25-Dihydroxyvitamin D3 inhibits dendritic cell differentiation and maturation in vitro. Exp. Hematol. 2000, 28, 575–583. [Google Scholar] [CrossRef]
- Barragan, M.; Good, M.; Jay KKolls, J.K. Regulation of Dendritic Cell Function by Vitamin D. Nutrients 2015, 7, 8127–8151. [Google Scholar] [CrossRef] [PubMed]
- Nuyts, A.H.; Lee, W.P.; Bashir-Dar, R.; Berneman, Z.N.; Cools, N. Dendritic cells in multiple sclerosis: key players in the immunopathogenesis, key players for new cellular immunotherapies? Mult Scler 2013, 19, 995–1002. [Google Scholar] [CrossRef]
- Nashold, F.E.; Miller, D.J.; Hayes, C.E. 1,25-dihydroxyvitamin D3 treatment decreases macrophage accumulation in the CNS of mice with experimental autoimmune encephalomyelitis. J Neuroimmunol 2000, 103, 171–177. [Google Scholar] [CrossRef]
- Koduah, P.; Paul, F.; Dörr, J.-M. Vitamin D in the prevention, prediction and treatment of neurodegenerative and neuroinflammatory diseases. EPMA J 2017, 8, 313–325. [Google Scholar] [CrossRef]
- Zorzella-Pezavento, S.F.G.; Mimura, L.A.N.; Fraga-Silva, T.F.C.; Ishikawa, L.L.W.; França, T.G.D.; Sartori, A. Experimental Autoimmune Encephalomyelitis Is Successfully Controlled by Epicutaneous Administration of MOG Plus Vitamin D Analog. Front. Immunol. 2017, 8, 1198. [Google Scholar] [CrossRef] [PubMed]
- Savran, Z.; Baltaci, S.B.; Aladag, T.; Mogulkoc, R.; Baltaci, A.K. Vitamin D and Neurodegenerative Diseases Such as Multiple Sclerosis (MS), Parkinson’s Disease (PD), Alzheimer’s Disease (AD), and Amyotrophic Lateral Sclerosis (ALS): A Review of Current Literature. Curr Nutr Rep 2025, 14, 77. [Google Scholar] [CrossRef]
- Farias, A.S.; Spagnol, G.S.; Bordeaux-Rego, P.; Oliveira, C.O.F.; Fontana, A.G.M.; de Paula, R.F.O.; et al. Vitamin D3 induces IDO+ tolerogenic DCs and enhances Treg, reducing the severity of EAE. CNS Neurosci Ther 2013, 19, 269–277. [Google Scholar] [CrossRef]
- Wan, Y.Y. Regulatory T cells: immune suppression and beyond. Cell Mol Immunol 2010, 7, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M. Vitamin D and the immune system: new perspectives on an old theme. Endocrinol Metab Clin North Am 2010, 39, 365–379. [Google Scholar] [CrossRef]
- Fenercioglu, A.K. The Anti-Inflammatory Roles of Vitamin D for Improving Human Health. Curr Issues Mol Biol 2024, 46, 13514–13525. [Google Scholar] [CrossRef]
- Mirarchi, A.; Albi, E.; Beccari, T.; Cataldo Arcuri, C. Microglia and Brain Disorders: The Role of Vitamin D and Its Receptor. Int J Mol Sci 2023, 24, 11892. [Google Scholar] [CrossRef] [PubMed]
- Dursun, E.; Gezen-Ak, D.; Yilmazer, S. The Influence of Vitamin D Treatment on the Inducible Nitric Oxide Synthase (INOS) Expression in Primary Hippocampal Neurons. Noro Psikiyatr Ars 2014, 51, 163–168. [Google Scholar] [CrossRef]
- Galoppin, M.; Kari, S.; Soldati, S.; Pal, A.; Rival, M.; Engelhardt, B.; et al. Full spectrum of vitamin D immunomodulation in multiple sclerosis: mechanisms and therapeutic implications. Brain Commun 2022, 4, fcac171. [Google Scholar] [CrossRef]
- Smith, K.J.; Lassmann, H. The role of nitric oxide in multiple sclerosis. Lancet Neurol 2002, 1, 232–241. [Google Scholar] [CrossRef]
- Sintzel, M.B.; Rametta, M.; Reder, A.T. Vitamin D and Multiple Sclerosis: A Comprehensive Review. Neurol Ther 2017, 7, 59–85. [Google Scholar] [CrossRef]
- Cassard, S.D.; Fitzgerald, K.C.; Qian, P.; Emrich, S.A.; Azevedo, C.J.; Goodman, A.D.; et al. High-dose vitamin D3 supplementation in relapsing-remitting multiple sclerosis: a randomised clinical trial. EclinicalMedicine 2023, 59, 101957. [Google Scholar] [CrossRef] [PubMed]
- Zlotnik, D.; Rabinski, T.; Halfon, A.; Anzi, S.; Plaschkes, I.; Benyamini, H.; et al. P450 oxidoreductase regulates barrier maturation by mediating retinoic acid metabolism in a model of the human BBB. Stem Cell Reports 2022, 17, 2050–2063. [Google Scholar] [CrossRef] [PubMed]
- Gryka-Marton, M.; Grabowska, A.D.; Szukiewicz, D. Breaking the Barrier: The Role of Proinflammatory Cytokines in BBB Dysfunction. Int. J Mole Sci 2015, 26, 3532. [Google Scholar] [CrossRef]
- Tschuck, J.; Nair, V.P.; Galhoz, A.; Zaratiegui, C.; Tai, H.-M.; Ciceri, G.; et al. Suppression of ferroptosis by vitamin A or radical-trapping antioxidants is essential for neuronal development. Nat Commun 2024, 15, 7611. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Drew, P.D. 9-Cis-retinoic acid suppresses inflammatory responses of microglia and astrocytes. J Neuroimmunol. 2005, 171, 135–144. [Google Scholar] [CrossRef]
- Wojtal, K.A.; Wolfram, L.; Frey-Wagner, I.; Lang, S.; Scharl, M.; Vavricka, S.R.; et al. The effects of vitamin A on cells of innate immunity in vitro. Toxicol in vitro 2013, 27, 1525–1532. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushali, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Dorosty-Motlagh, A.R.; Honarvar, M.N.; Sedighiyan, M.; Abdolahi, M. The molecular mechanisms of vitamin A deficiency in multiple sclerosis. J. Mol. Neurosci. 2016, 60, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Raverdeau, M.; Breen, C.J.; Misiak, A.; Mills, K.H. Retinoic acid suppresses IL-17productionandpathogenic activity of γδT-cells in CNS autoimmunity. Immunol. Cell Biol. 2016, 94, 763–773. [Google Scholar] [CrossRef] [PubMed]
- Miller, E.; Markiewicz, Ł.; Kabziński, J.; Odrobina, D.; Majsterek, I. Potential of redox therapies in neuro-degenerative disorders. Front. Biosci. 2017, 9, 214–234. [Google Scholar] [CrossRef]
- Dattilo, S.; Mancuso, C.; Koverech, G.; Di Mauro, P.; Ontario, M.L.; Petralia, C.C.; Petralia, A.; Maiolino, L.; Serra, A.; Calabrese, E.J.; et al. Heat shock proteins and hormesis in the diagnosis and treatment of neurodegenerative diseases. Immun. Ageing 2015, 12, 20. [Google Scholar] [CrossRef]
- Wu, S.; Guo, T.; Qi, W.; Li, Y.; Gu, J.; Liu, C.; et al. Curcumin ameliorates ischemic stroke injury in rats by protecting the integrity of the blood-brain barrier. Exp Ther Med 2021, 22, 783. [Google Scholar] [CrossRef]
- Motavaf, M.; Sadeghizadeh, M.; Babashah, S.; Zare, L.; Javan, M. Dendrosomal nanocurcumin promotes remyelination through induction of oligodendrogenesis in experimental demyelination animal model. J Tissue Eng Regen Med 2020, 14, 1449–1464. [Google Scholar] [CrossRef] [PubMed]
- Moldoveanu, C.-A.; Tomoaia-Cotisel, M.; Sevastre-Berghian, A.; Tomoaia, G.; Mocanu, A.; Pal-Racz, C.; Toma, V.-A.; Roman, I.; Ujica, M.-A.; Pop, L.-C. A Review on Current Aspects of Curcumin-Based Effects in Relation to Neurodegenerative, Neuroinflammatory and Cerebrovascular Diseases. Molecules 2025, 30, 43. [Google Scholar] [CrossRef]
- Smirnova, E.; Moniruzzaman, M.; Chin, S.; Sureshbabu, A.; Karthikeyan, A.; Do, K.; et al. A Review of the Role of Curcumin in Metal Induced Toxicity. Antioxidants (Basel) 2023, 12, 243. [Google Scholar] [CrossRef]
- Aggarwal, B.B.; Gupta, S.C.; Sung, B. Curcumin: an orally bioavailable blocker of TNF and other pro-inflammatory biomarkers. Br J Pharmacol 2013, 169, 1672–1692. [Google Scholar] [CrossRef]
- Yazdani, Y.; Zamani, A.R.N.; Majidi, Z.; Sharafkandi, Ν.; Alizadeh, S.; Amir MEMofrad, A.M.E.; et al. Curcumin and targeting of molecular and metabolic pathways in multiple sclerosis. Cell Biochem Funct 2023, 41, 779–787. [Google Scholar] [CrossRef]
- Zhao, G.; Liu, Y.; Yi, X.; Wang, Y.; Qiao, S.; Li, Z.; Ni, J.; Song, Z. Curcumin inhibiting Th17 cell differentiation by regulating the metabotropic glutamate receptor-4 expression on dendritic cells. Int Immunopharmacol 2017, 46, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Dolati, S.; Babaloo, Z.; Ayromlou, H.; Ahmadi, M.; Rikhtegar, R.; Rostamzadeh, D.; et al. Nanocurcumin improves regulatory T-cell frequency and function in patients with multiple sclerosis. J Neuroimmunol 2019, 327, 15–21. [Google Scholar] [CrossRef]
- Buc, M. Role of regulatory T cells in pathogenesis and biological therapy of multiple sclerosis. Mediators Inflamm 2013, 2013, 963748. [Google Scholar] [CrossRef]
- Miller, E.D.; Dziedzic, A.; Saluk-Bijak, J.; Bijak, M. A Review of Various Antioxidant Compounds and their Potential Utility as Complementary Therapy in Multiple Sclerosis. Nutrients 2019, 11, 1528. [Google Scholar] [CrossRef]
- Holzerova, E.; Danhauser, K.; Haack, T.B.; Kremer, L.S.; Melcher, M.; Ingold, I.; et al. Human thioredoxin 2 deficiency impairs mitochondrial redox homeostasis and causes early-onset neurodegeneration. Brain 2016, 139, 346–354. [Google Scholar] [CrossRef]
- Wang, D.; Li, S.-P.; Fu, J.-S.; Zhang, S.; Bai, L.; Guo, L. Resveratrol defends blood-brain barrier integrity in experimental autoimmune encephalomyelitis mice. J Neurophysiol 2016, 116, 2173–2179. [Google Scholar] [CrossRef]
- Shindler, K.S.; Ventura, E.; Dutt, M.; Elliott, P.; Fitzgerald, D.C.; Rostami, A. Oral resveratrol reduces neuronal damage in a model of multiple sclerosis. J. Neuroophthalmol. 2010, 30, 328–339. [Google Scholar] [CrossRef]
- Gianchecchi, E.; Fierabracci, A. Insights on the Effects of Resveratrol and Some of Its Derivatives in Cancer and Autoimmunity: A Molecule with a Dual Activity. Antioxidants 2020, 9, 91. [Google Scholar] [CrossRef] [PubMed]
- Javanbakht, P.; Yazdi, F.R.; Taghizadeh, F.; Khadivi, F.; Hamidabadi, H.G.; Kashani, I.R.; et al. Quercetin as a possible complementary therapy in multiple sclerosis: Anti-oxidative, anti-inflammatory and remyelination potential properties. Heliyon 2023, 9, e21741. [Google Scholar] [PubMed]
Figure 1.
Schematic representation of the main mechanisms by which chronic inflammation contributes to the pathogenesis of multiple sclerosis in obesity. MCP-1, monocyte chemoattractant protein-1; CCL5-RANTES: C-C motif chemokine ligand 5 – regulated upon activation, normal T-cell expressed and secreted; Th1, T helper 1 cells; Th17, T helper 17 cells; Treg: regulatory T lymphocytes; PPAR-γ: peroxisome proliferator-activated receptor gamma; FAs: fatty acids; AIM: apoptosis inhibitor of macrophages; TLR-4, toll-like receptor 4; NF-κB: nuclear factor kappa B.
Figure 1.
Schematic representation of the main mechanisms by which chronic inflammation contributes to the pathogenesis of multiple sclerosis in obesity. MCP-1, monocyte chemoattractant protein-1; CCL5-RANTES: C-C motif chemokine ligand 5 – regulated upon activation, normal T-cell expressed and secreted; Th1, T helper 1 cells; Th17, T helper 17 cells; Treg: regulatory T lymphocytes; PPAR-γ: peroxisome proliferator-activated receptor gamma; FAs: fatty acids; AIM: apoptosis inhibitor of macrophages; TLR-4, toll-like receptor 4; NF-κB: nuclear factor kappa B.
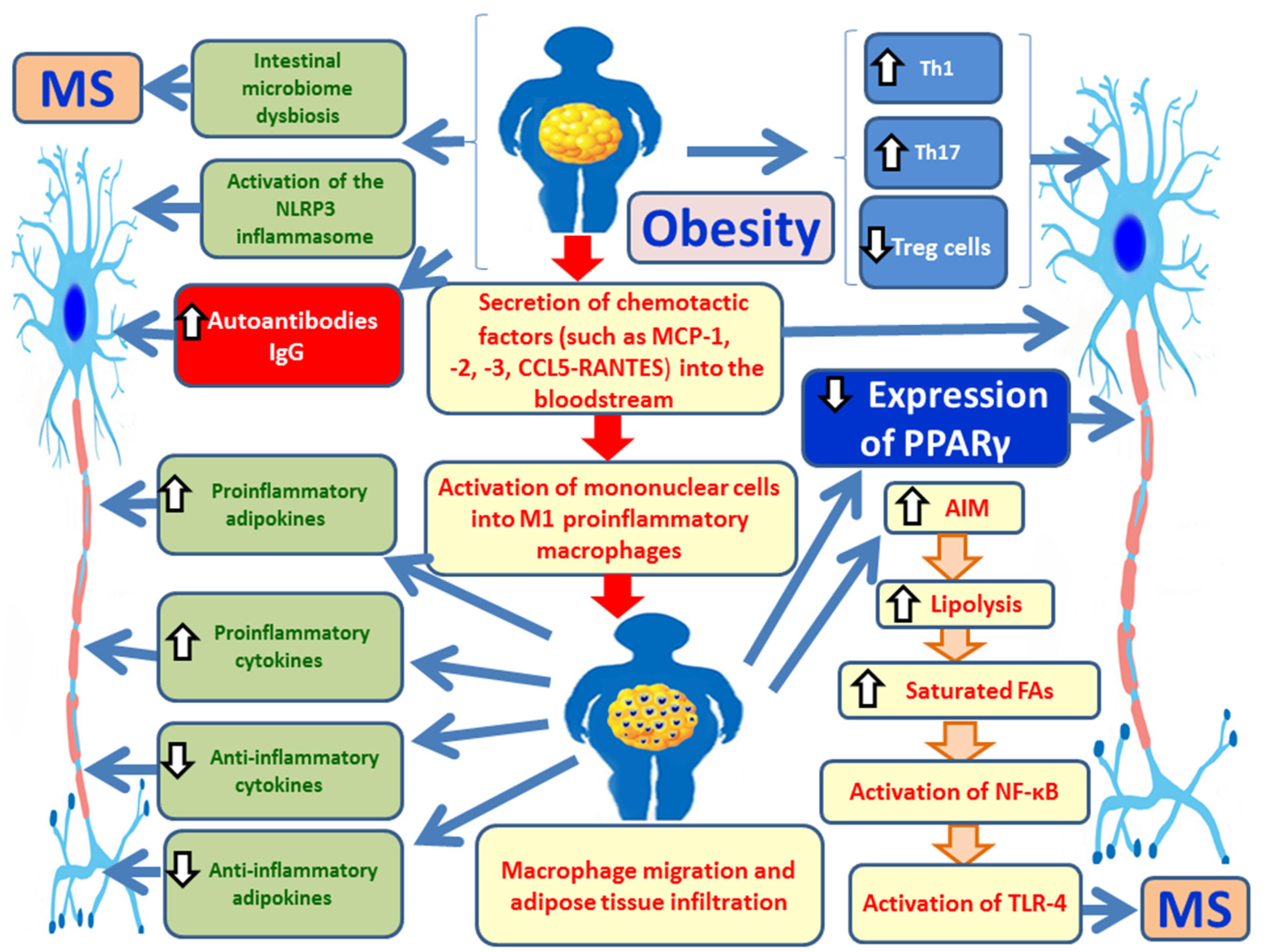
Figure 2.
Protective actions of adiponectin in obesity and its potential role as a biomarker for monitoring the progression and severity of multiple sclerosis (MS). ROS: reactive oxygen species; NF-κB: nuclear factor kappa B; IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; NO: nitric oxide; MS: multiple sclerosis.
Figure 2.
Protective actions of adiponectin in obesity and its potential role as a biomarker for monitoring the progression and severity of multiple sclerosis (MS). ROS: reactive oxygen species; NF-κB: nuclear factor kappa B; IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; NO: nitric oxide; MS: multiple sclerosis.
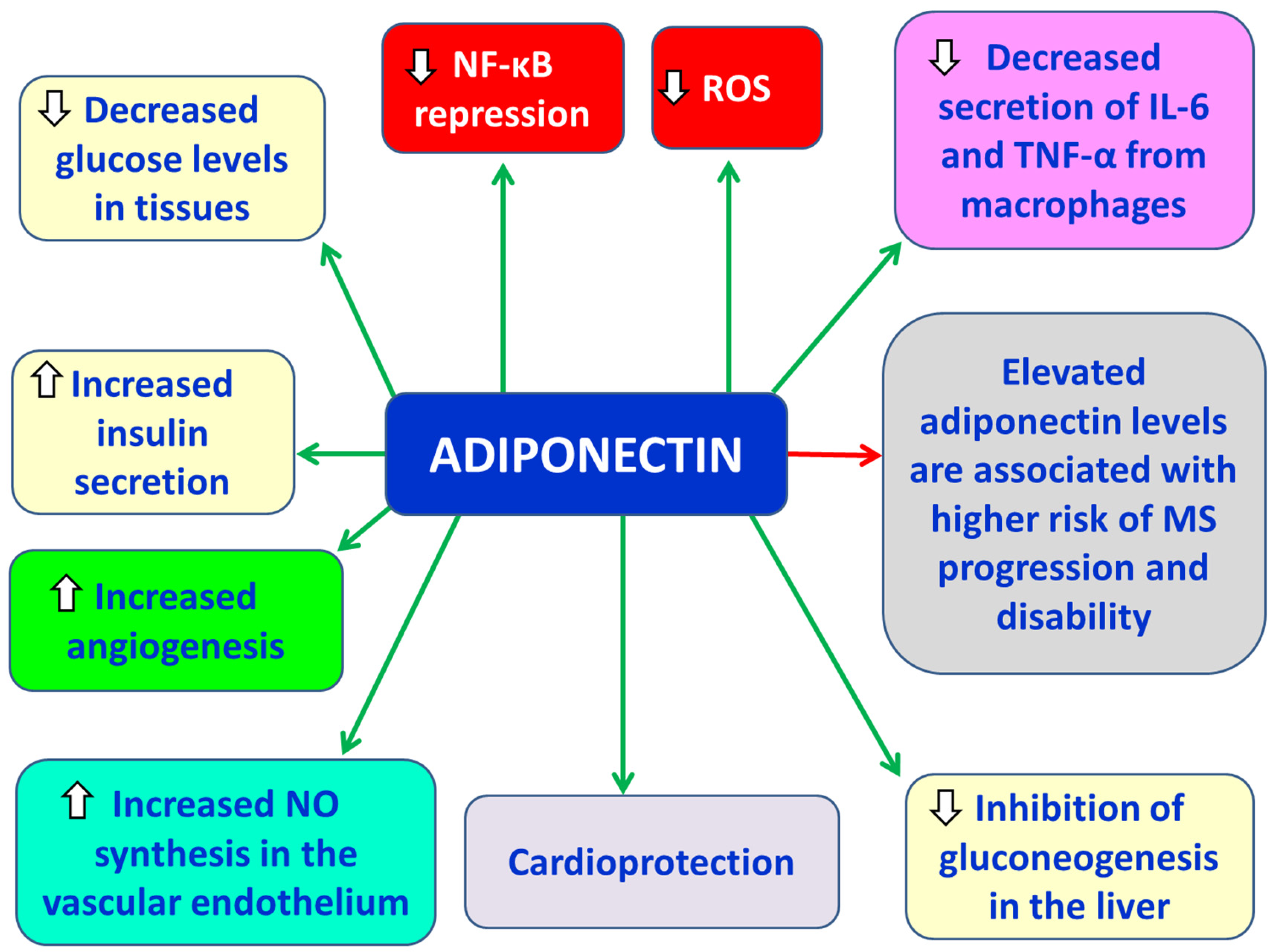
Figure 3.
Protective actions of apelin in obesity and its potency towards obesity-associated multiple sclerosis (MS). IL-6, interleukin-6; IL-10, interleukin-10; iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species; OS: oxidative stress; FAs: fatty acids; MS: multiple sclerosis.
Figure 3.
Protective actions of apelin in obesity and its potency towards obesity-associated multiple sclerosis (MS). IL-6, interleukin-6; IL-10, interleukin-10; iNOS: inducible nitric oxide synthase; ROS: reactive oxygen species; OS: oxidative stress; FAs: fatty acids; MS: multiple sclerosis.
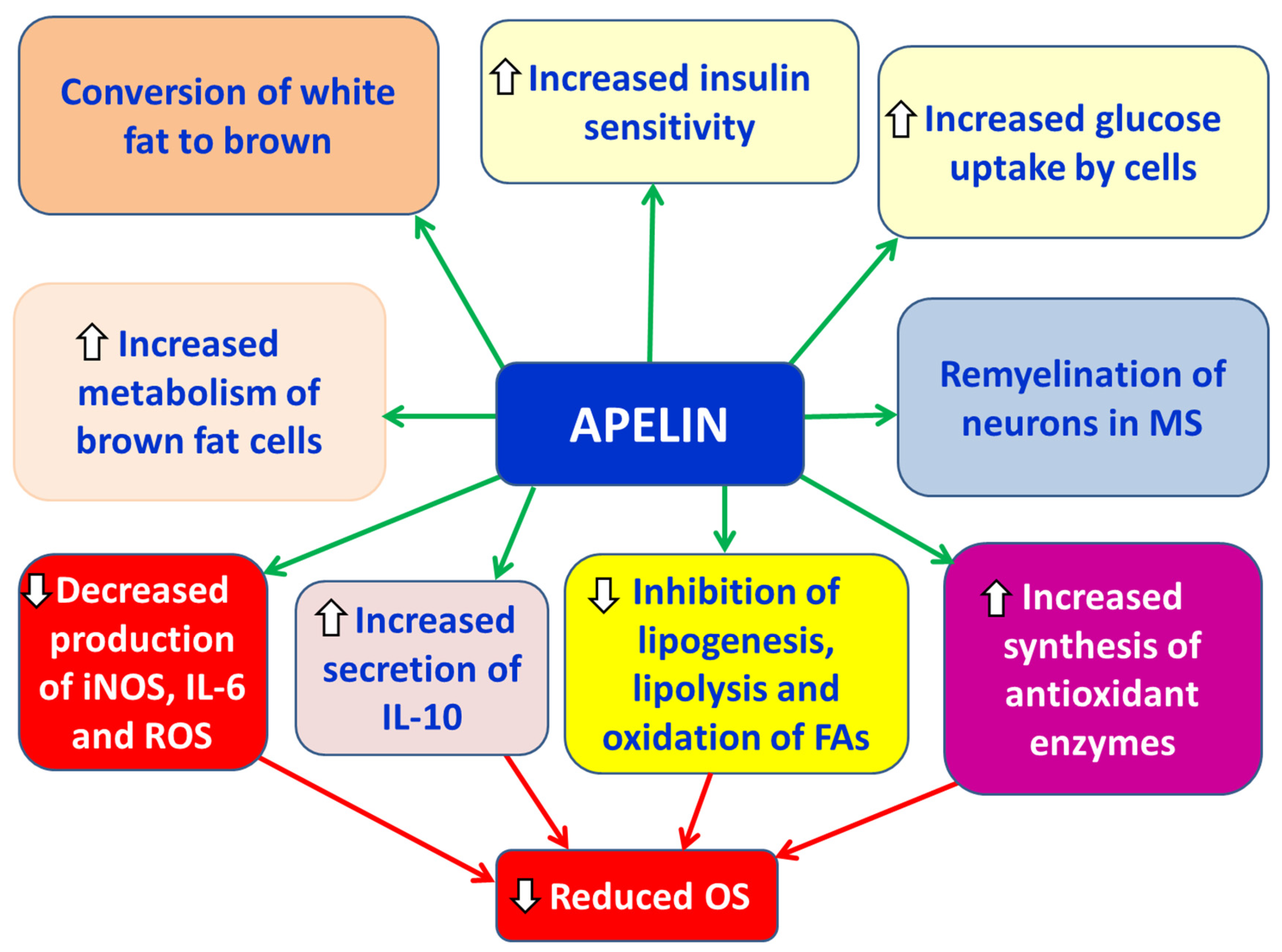
Figure 4.
Actions of leptin related to the pathophysiology of multiple sclerosis (MS). IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; Th1 lymphocytes: T helper 1 lymphocytes; Th17 lymphocytes: T helper 17 lymphocytes.
Figure 4.
Actions of leptin related to the pathophysiology of multiple sclerosis (MS). IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; Th1 lymphocytes: T helper 1 lymphocytes; Th17 lymphocytes: T helper 17 lymphocytes.
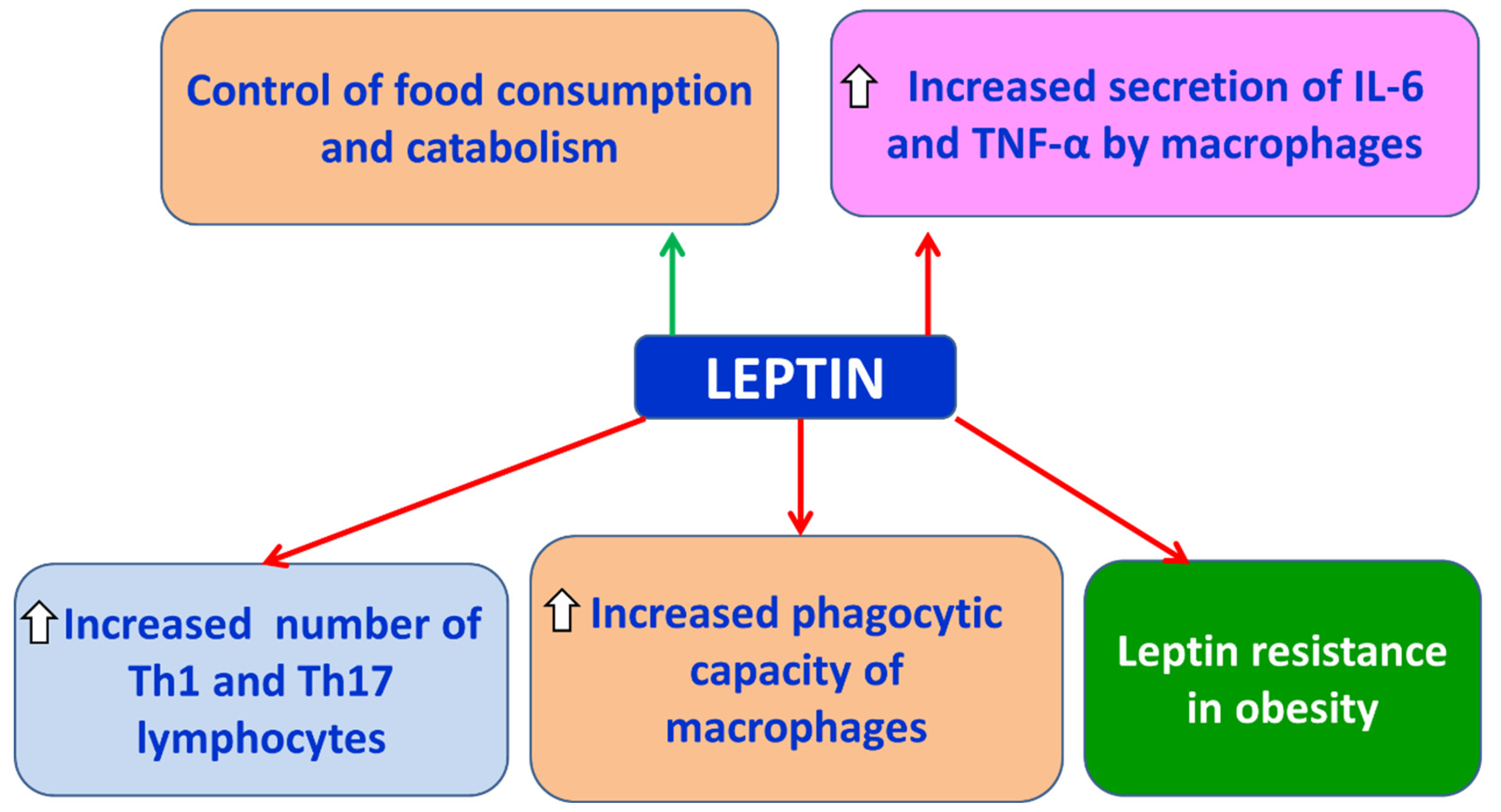
Figure 5.
Effects of visfatin on chronic inflammation and oxidative stress associated with obesity and multiple sclerosis (MS). CCL2, chemokine (C-C motif) ligand 2; CXCL2, chemokine (C-X-C motif) ligand 2; CXCL8, chemokine (C-X-C motif) ligand 8; T2DM: type 2 diabetes mellitus, MetS: metabolic syndrome; CVDs: cardiovascular diseases; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule-1; ROS: reactive oxygen species; NF-κB: ; MS: multiple sclerosis; IL-1β; IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; NOS: ; NO: nitric oxide: nitric oxide synthase.
Figure 5.
Effects of visfatin on chronic inflammation and oxidative stress associated with obesity and multiple sclerosis (MS). CCL2, chemokine (C-C motif) ligand 2; CXCL2, chemokine (C-X-C motif) ligand 2; CXCL8, chemokine (C-X-C motif) ligand 8; T2DM: type 2 diabetes mellitus, MetS: metabolic syndrome; CVDs: cardiovascular diseases; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule-1; ROS: reactive oxygen species; NF-κB: ; MS: multiple sclerosis; IL-1β; IL-6, interleukin-6; TNF-α: tumor necrosis factor-alpha; NOS: ; NO: nitric oxide: nitric oxide synthase.
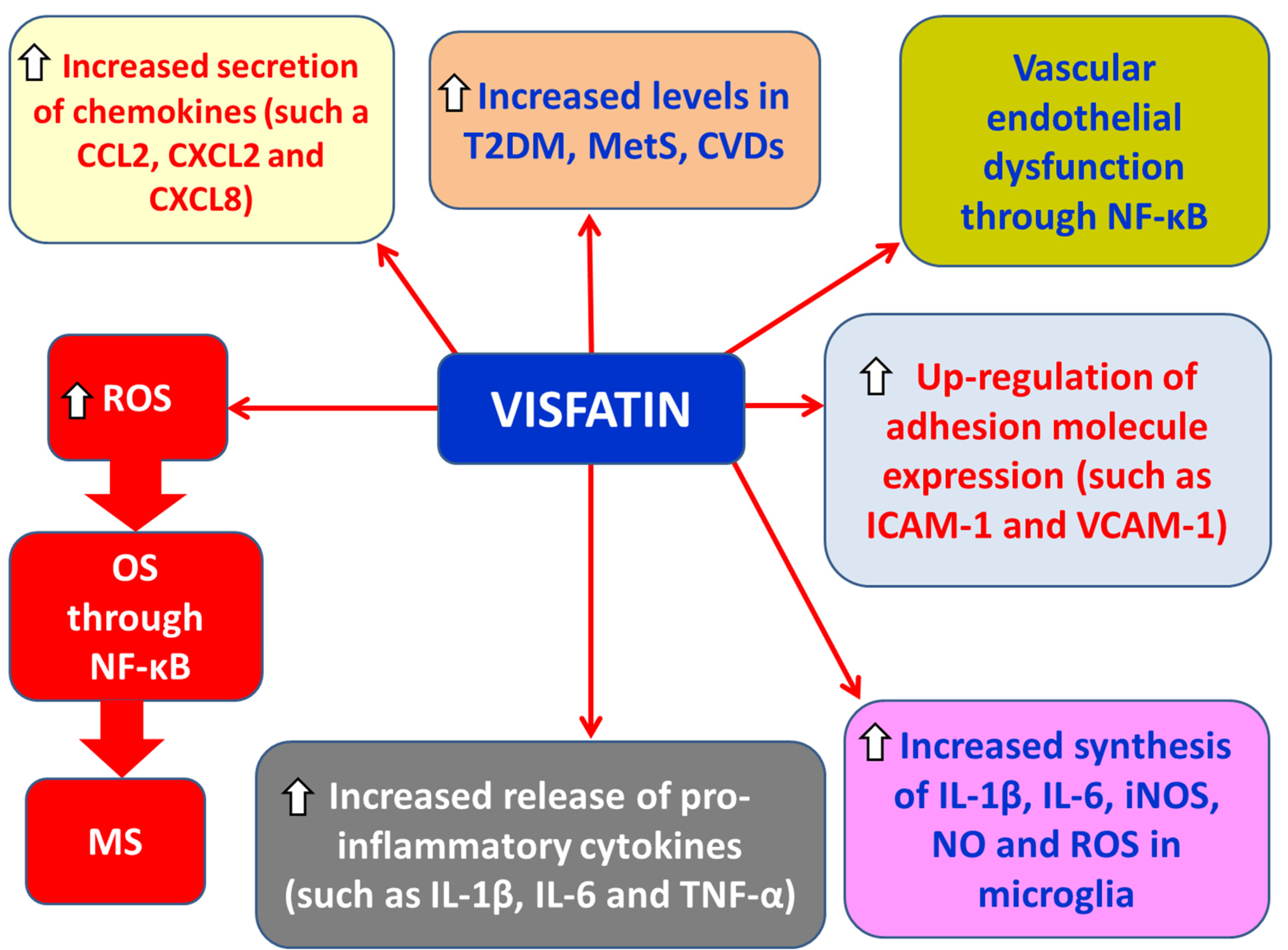
Figure 6.
Actions of resistin in chronic inflammation and oxidative stress associated with obesity and MS. IL-1β: interleukin-1β; TNF-α: tumor necrosis factor-alpha; ROS: reactive oxygen species; OS: oxidative stress; T2DM: type 2 diabetes mellitus; IR: insulin resistance; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule-1; MS: multiple sclerosis.
Figure 6.
Actions of resistin in chronic inflammation and oxidative stress associated with obesity and MS. IL-1β: interleukin-1β; TNF-α: tumor necrosis factor-alpha; ROS: reactive oxygen species; OS: oxidative stress; T2DM: type 2 diabetes mellitus; IR: insulin resistance; ICAM-1, intercellular adhesion molecule 1; VCAM-1, vascular cell adhesion molecule-1; MS: multiple sclerosis.
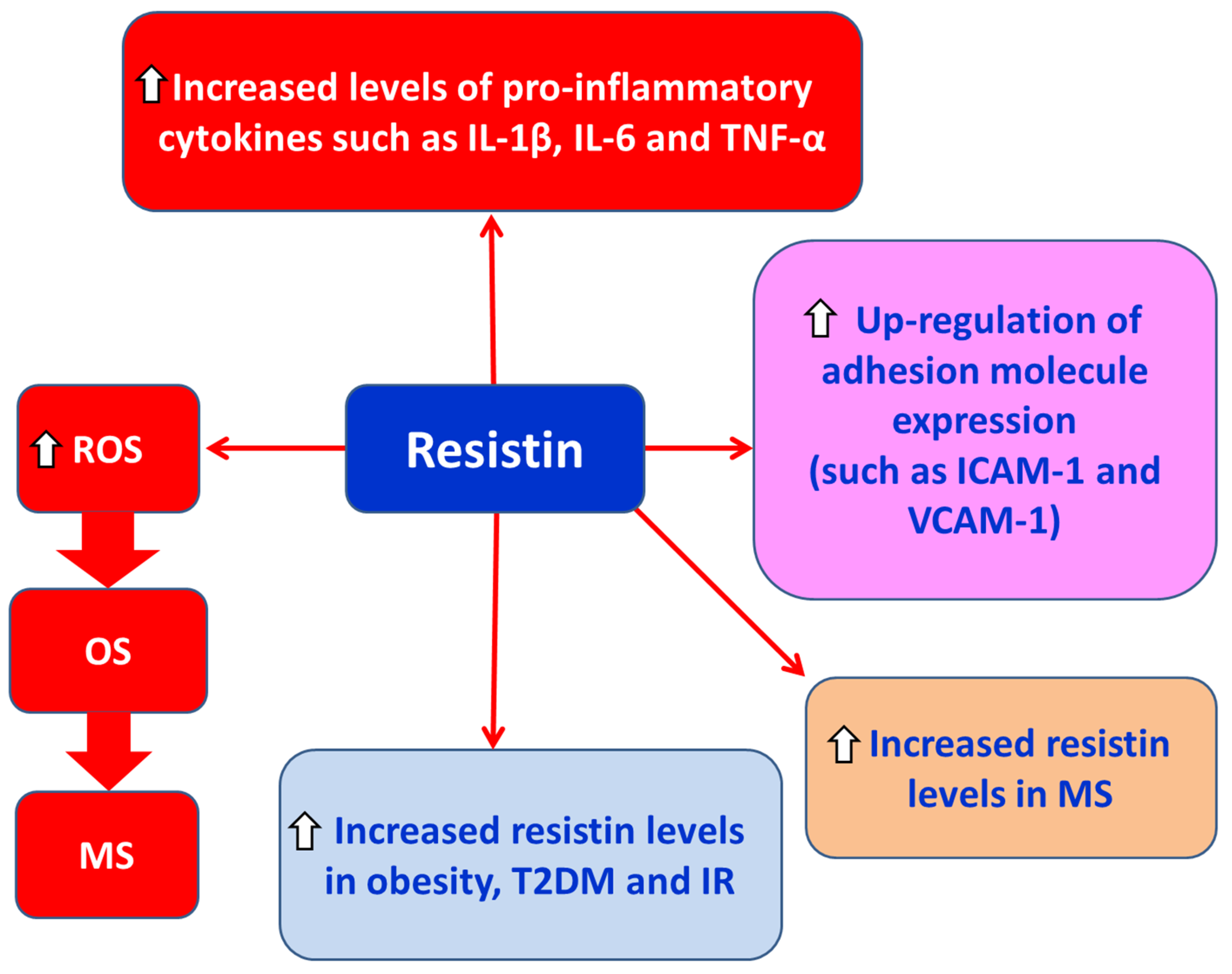
Figure 7.
Actions of TNF-α in chronic inflammation and oxidative stress associated with obesity and MS. ROS: reactive oxygen species; OS: oxidative stress; MS: multiple sclerosis; TNF-α: tumor necrosis factor-alpha; IR: insulin resistance; IL-6, interleukin-6; MCP-1, monocyte chemo-attractant protein-1; FAs: fatty acids.
Figure 7.
Actions of TNF-α in chronic inflammation and oxidative stress associated with obesity and MS. ROS: reactive oxygen species; OS: oxidative stress; MS: multiple sclerosis; TNF-α: tumor necrosis factor-alpha; IR: insulin resistance; IL-6, interleukin-6; MCP-1, monocyte chemo-attractant protein-1; FAs: fatty acids.
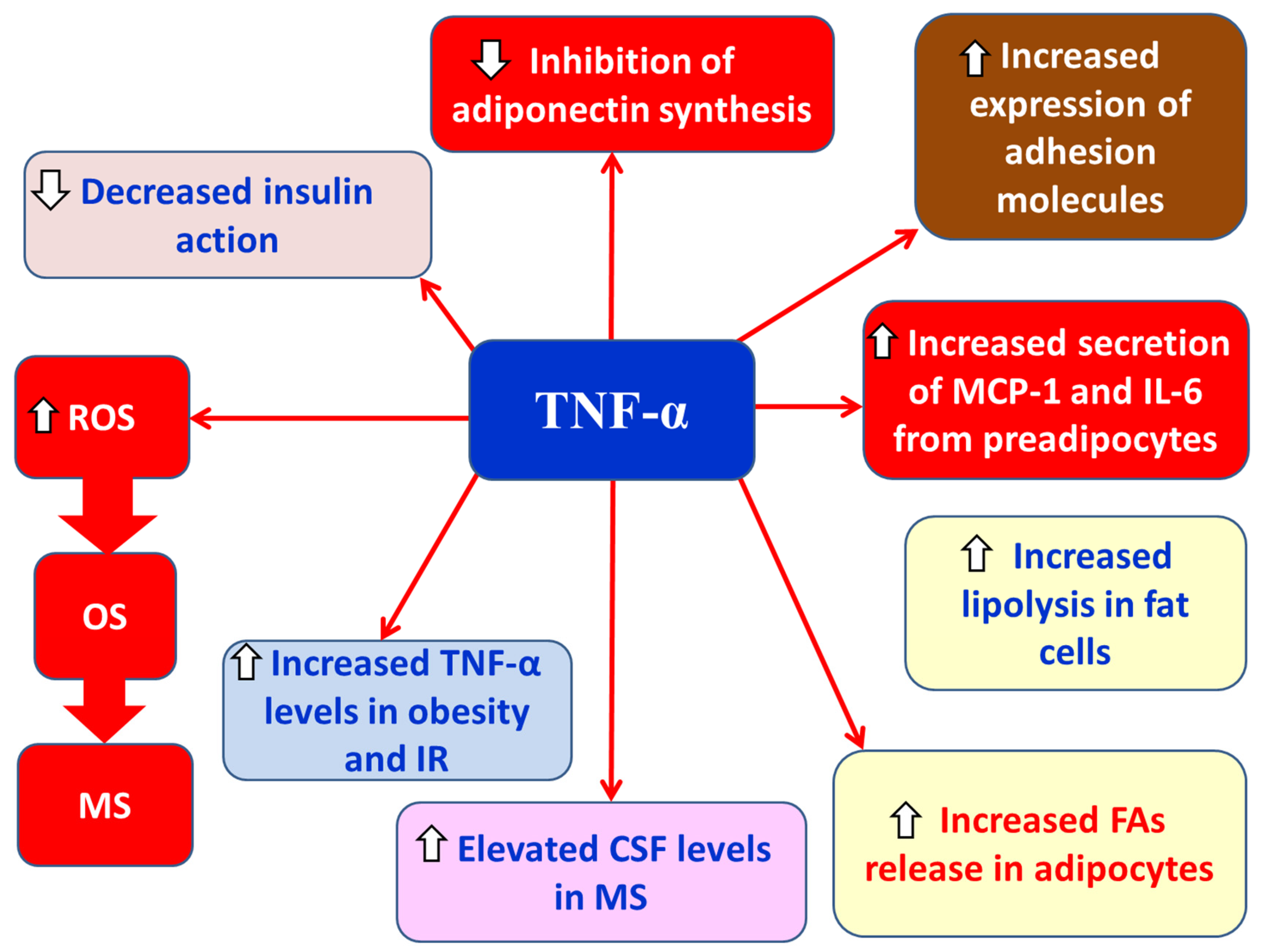
Figure 8.
Actions of IL-1β in chronic inflammation and OS associated with obesity and MS. ΙL-1β: interleukin-1β; ROS: reactive oxygen species; OS: oxidative stress; MS: multiple sclerosis; IR: insulin resistance; PPARγ: peroxisome proliferator-activated receptor gamma.
Figure 8.
Actions of IL-1β in chronic inflammation and OS associated with obesity and MS. ΙL-1β: interleukin-1β; ROS: reactive oxygen species; OS: oxidative stress; MS: multiple sclerosis; IR: insulin resistance; PPARγ: peroxisome proliferator-activated receptor gamma.
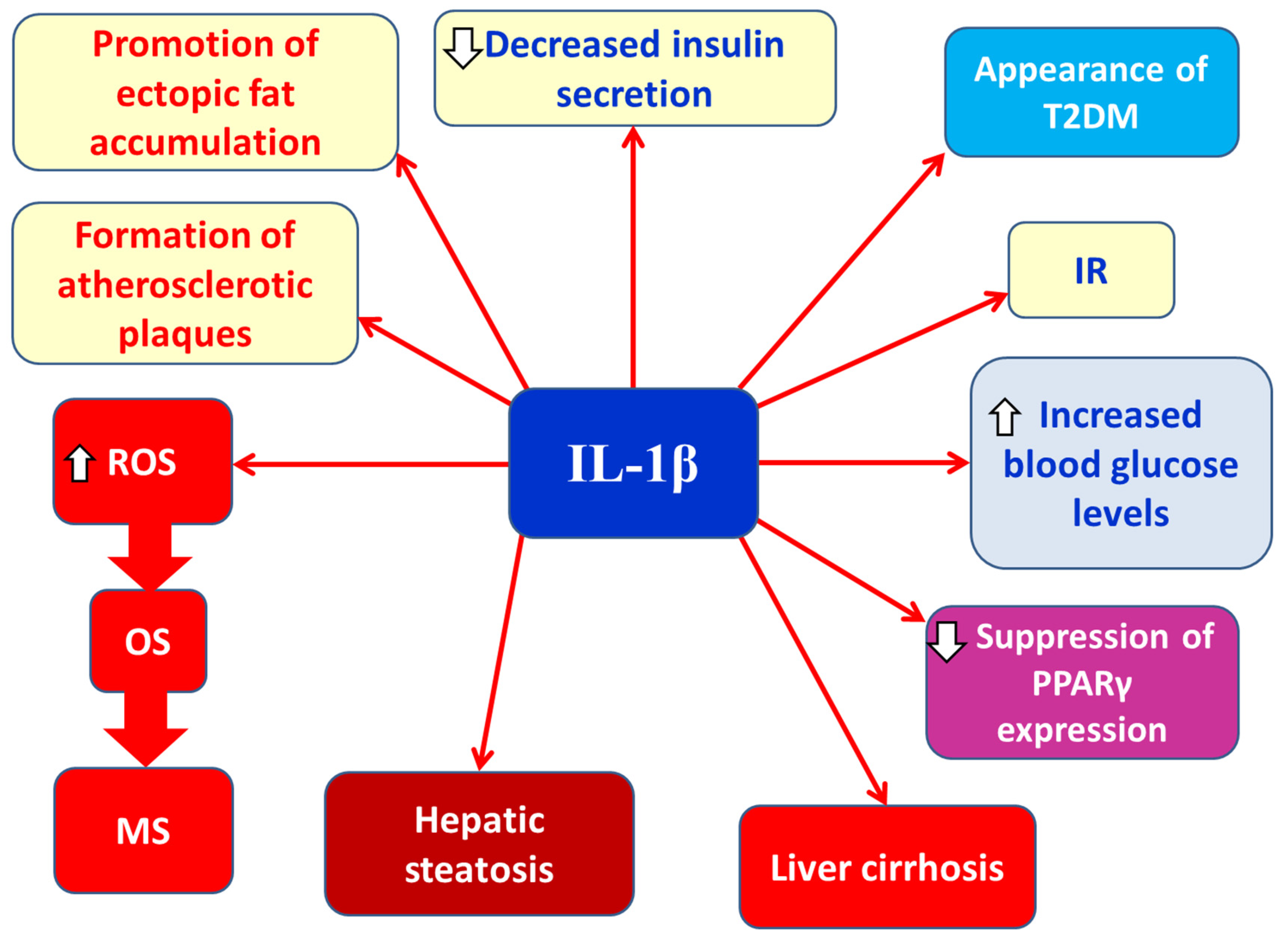
Figure 9.
Schematic representation of the interaction between obesity, chronic inflammation, and oxidative stress in the pathogenesis of multiple sclerosis (MS). ROS: reactive oxygen species; PGE2, prostaglandin E2; COX-2, cyclooxygenase-2; MAPK: mitogen-activated protein kinase; PPAR-γ: peroxisome proliferator-activated receptor gamma; NADPH: nicotinamide adenine dinucleotide phosphate; MDA: malondialdehyde; PKC: protein kinase C.
Figure 9.
Schematic representation of the interaction between obesity, chronic inflammation, and oxidative stress in the pathogenesis of multiple sclerosis (MS). ROS: reactive oxygen species; PGE2, prostaglandin E2; COX-2, cyclooxygenase-2; MAPK: mitogen-activated protein kinase; PPAR-γ: peroxisome proliferator-activated receptor gamma; NADPH: nicotinamide adenine dinucleotide phosphate; MDA: malondialdehyde; PKC: protein kinase C.
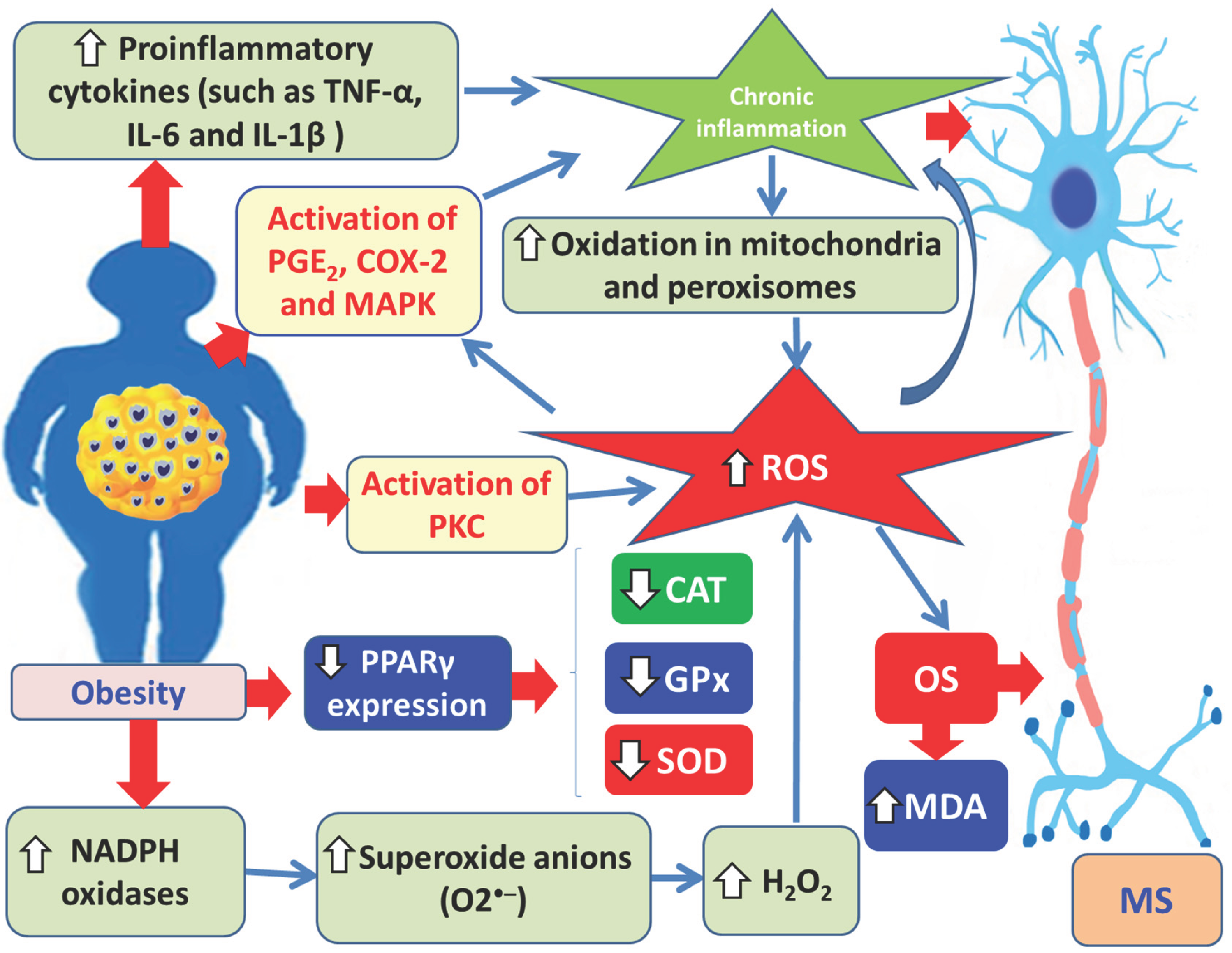
Figure 10.
Schematic representation of the beneficial effects of vitamin D against MS. CNS: central nervous system; Treg cell: regulatory T cell; Th1 cell: T helper 1 cell; Th2 cell: T helper 2 cell; NO: nitrogen oxide; iNOS: inducible nitric oxide synthase; MS: multiple sclerosis.
Figure 10.
Schematic representation of the beneficial effects of vitamin D against MS. CNS: central nervous system; Treg cell: regulatory T cell; Th1 cell: T helper 1 cell; Th2 cell: T helper 2 cell; NO: nitrogen oxide; iNOS: inducible nitric oxide synthase; MS: multiple sclerosis.
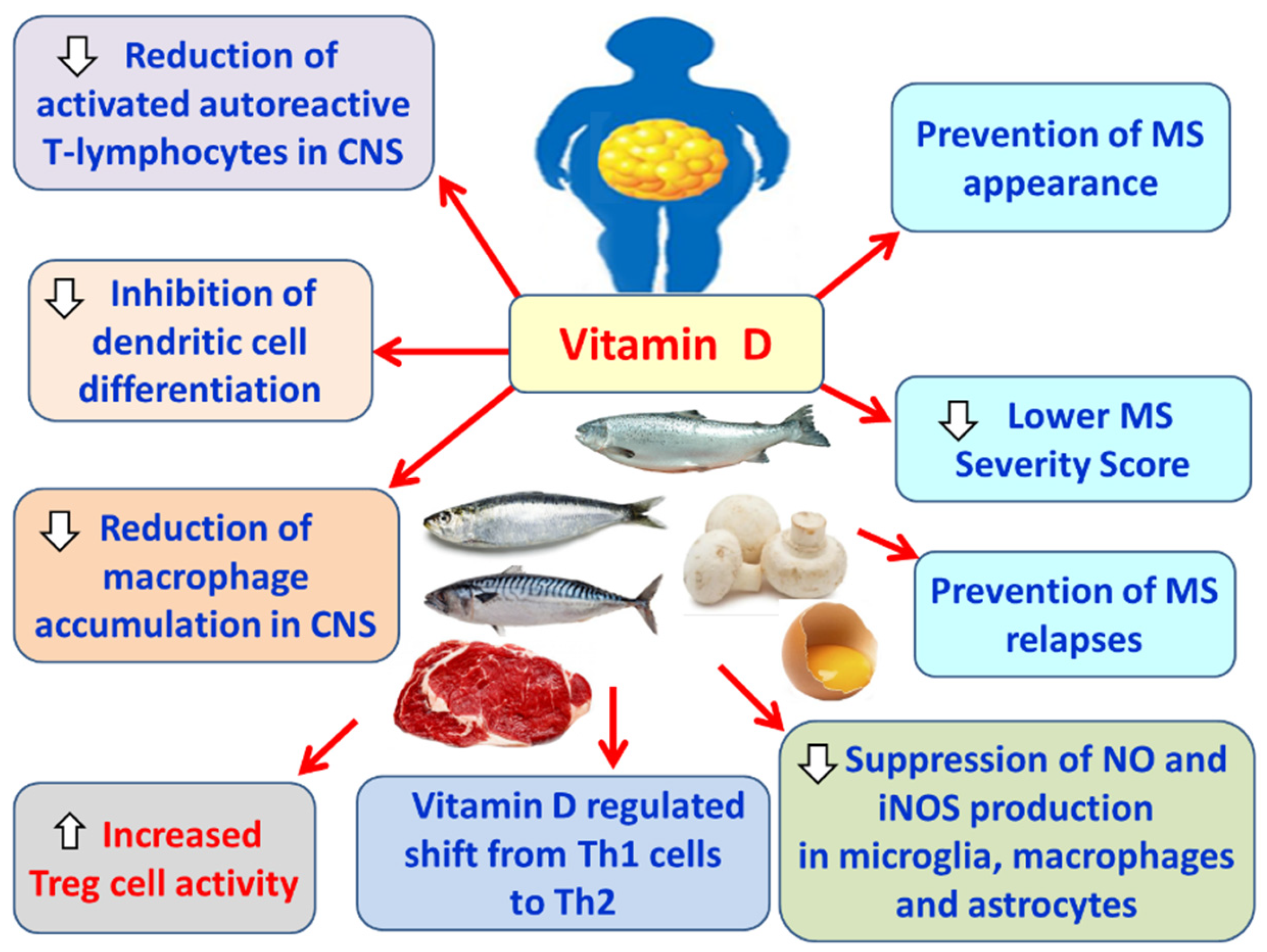
Figure 11.
Schematic representation of the beneficial effects of vitamin A against multiple sclerosis (MS). PPARs: peroxisome proliferation-activated receptor; NF-κB: nuclear factor-κΒ; STAT-1, signal trasducer and activator of transcription 1; AP-1, activator protein 1; CD4+ T lymphocytes: also known as helper T cells; Th1 cell: T helper 1 cell; Th17 cell: T helper 17 cell; CNS: central nervous system; BBB: blood-brain barrier; IL-1, interleukin-1; IL-12, interleukin ; TNF-α: tumor necrosis factor-alpha; STAT3, signal transducer and activator of transcription 3; MS: multiple sclerosis.
Figure 11.
Schematic representation of the beneficial effects of vitamin A against multiple sclerosis (MS). PPARs: peroxisome proliferation-activated receptor; NF-κB: nuclear factor-κΒ; STAT-1, signal trasducer and activator of transcription 1; AP-1, activator protein 1; CD4+ T lymphocytes: also known as helper T cells; Th1 cell: T helper 1 cell; Th17 cell: T helper 17 cell; CNS: central nervous system; BBB: blood-brain barrier; IL-1, interleukin-1; IL-12, interleukin ; TNF-α: tumor necrosis factor-alpha; STAT3, signal transducer and activator of transcription 3; MS: multiple sclerosis.
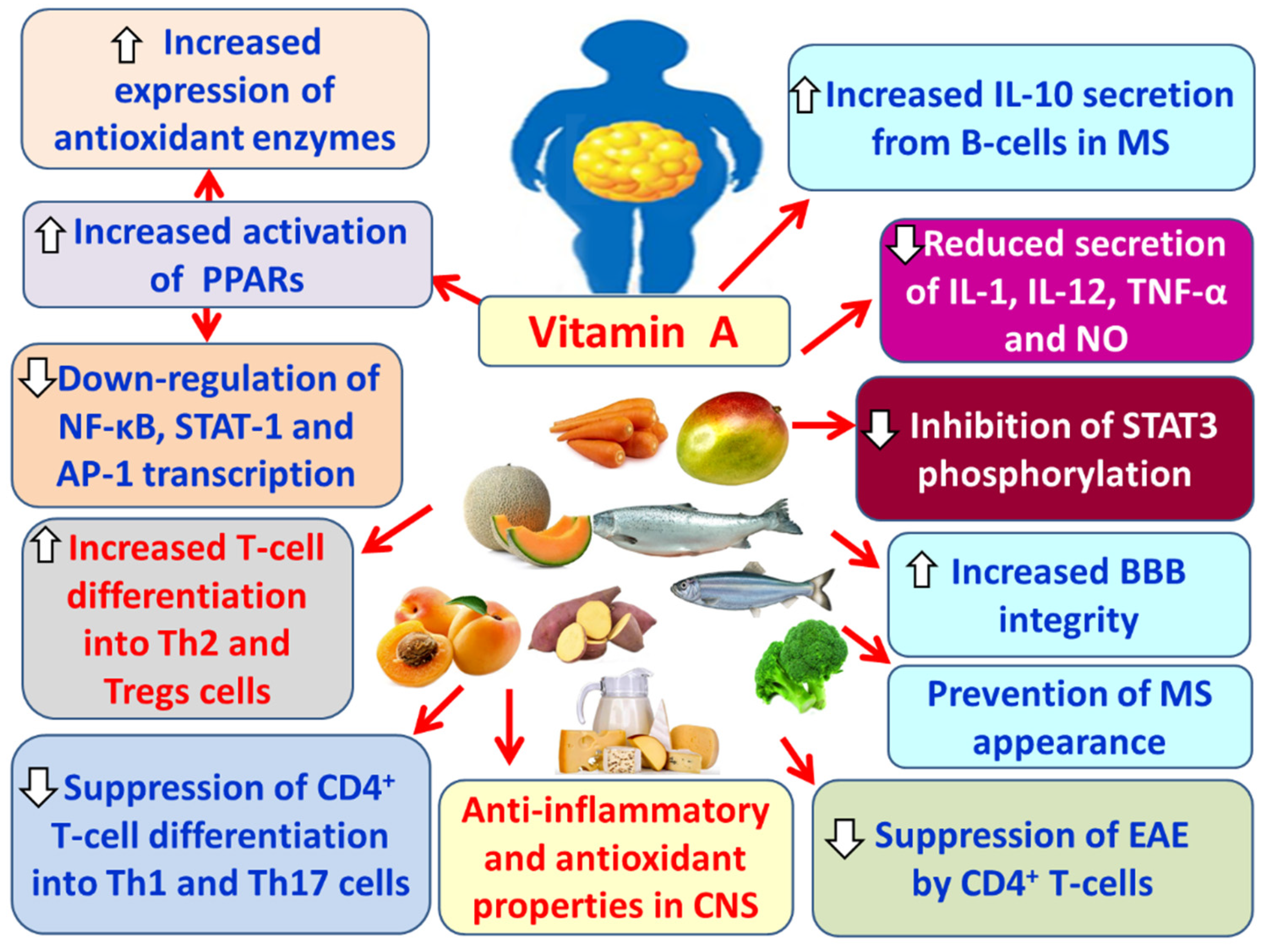
Figure 12.
Schematic representation of the beneficial effects of curcumin against MS. Nrf2, nuclear factor erythroid 2-related factor 2; HMOX-1, heme oxygenase 1, Hsp70s: heat shock proteins 70; Mn2+: manganous cation; Fe2+: ferrous cation; Cu2+: copper cation; Zn2+: zinc cation; NF-κB: nuclear factor-κB; CNS: central nervous system; COX-2, cyclooxygenase-2; MCP-1, monocyte chemoattractant protein 1; MIP-1α: macrophage inflammatory protein 1 alpha; IL-1β: interleukin 1β; IL-6, interleukin 6; IL-8, interleukin 8; IL-17; TNF-α: tumor necrosis factor alpha; Th17, T helper 17 cells; BBB: blood-brain barrier.
Figure 12.
Schematic representation of the beneficial effects of curcumin against MS. Nrf2, nuclear factor erythroid 2-related factor 2; HMOX-1, heme oxygenase 1, Hsp70s: heat shock proteins 70; Mn2+: manganous cation; Fe2+: ferrous cation; Cu2+: copper cation; Zn2+: zinc cation; NF-κB: nuclear factor-κB; CNS: central nervous system; COX-2, cyclooxygenase-2; MCP-1, monocyte chemoattractant protein 1; MIP-1α: macrophage inflammatory protein 1 alpha; IL-1β: interleukin 1β; IL-6, interleukin 6; IL-8, interleukin 8; IL-17; TNF-α: tumor necrosis factor alpha; Th17, T helper 17 cells; BBB: blood-brain barrier.
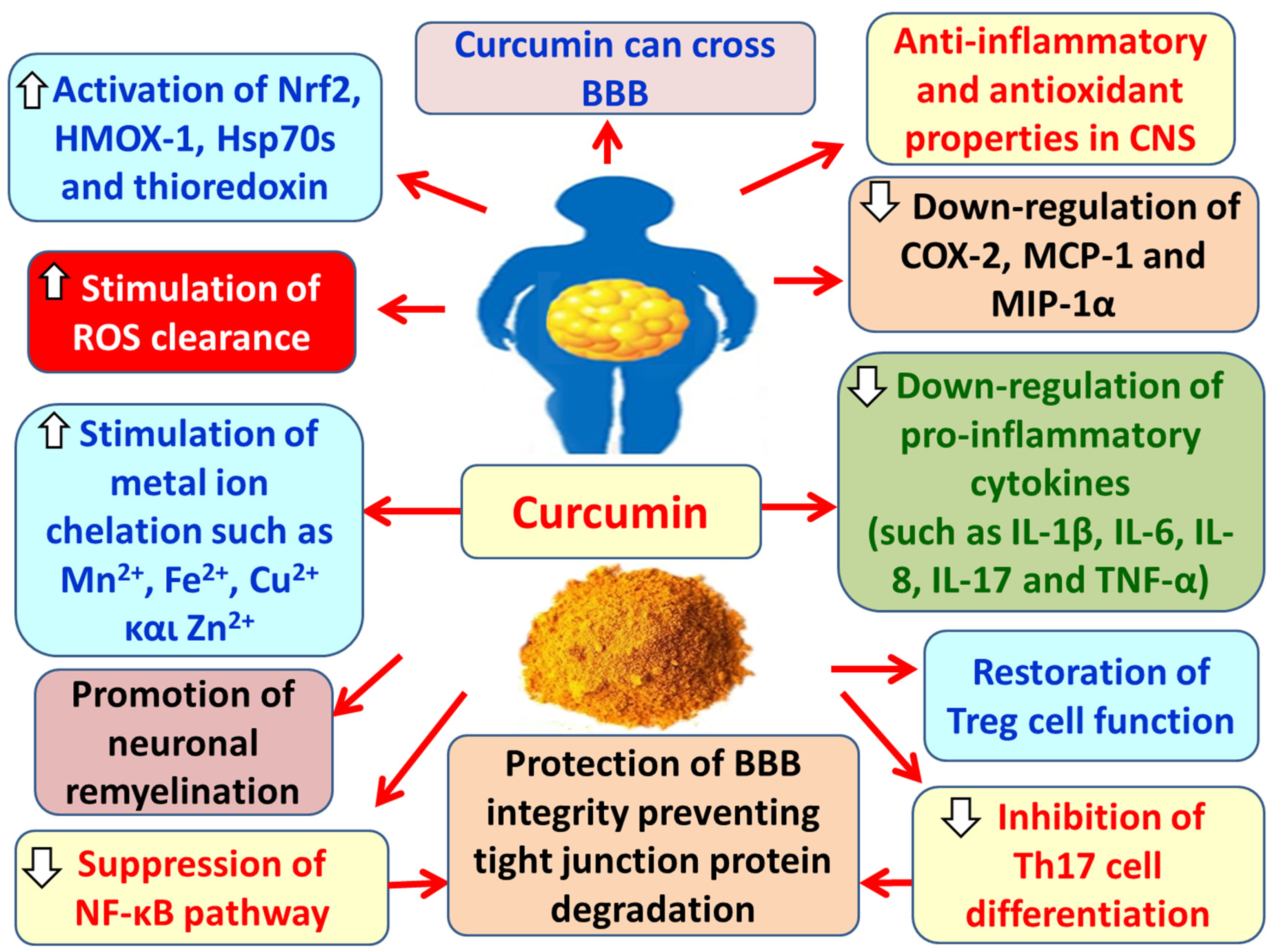
Figure 13.
Schematic representation of the beneficial effects of resveratrol against MS. MS: multiple sclerosis; SIRT1, surtuin 1; Th1 cells: T helper 1 cells; Th17 cells: T helper 17 cells; CNS: central nervous system; TNF-α: tumor necrosis factor-alpha; ΙL-1β: interleukin 1; IL-9, interleukin 9; IL-12, interleukin 12; IL-17, interleukin 17; IL-23, interleukin 23; INF-γ: interferon gamma; ROS: reactive oxygen species; BBB: blood-brain barrier.
Figure 13.
Schematic representation of the beneficial effects of resveratrol against MS. MS: multiple sclerosis; SIRT1, surtuin 1; Th1 cells: T helper 1 cells; Th17 cells: T helper 17 cells; CNS: central nervous system; TNF-α: tumor necrosis factor-alpha; ΙL-1β: interleukin 1; IL-9, interleukin 9; IL-12, interleukin 12; IL-17, interleukin 17; IL-23, interleukin 23; INF-γ: interferon gamma; ROS: reactive oxygen species; BBB: blood-brain barrier.
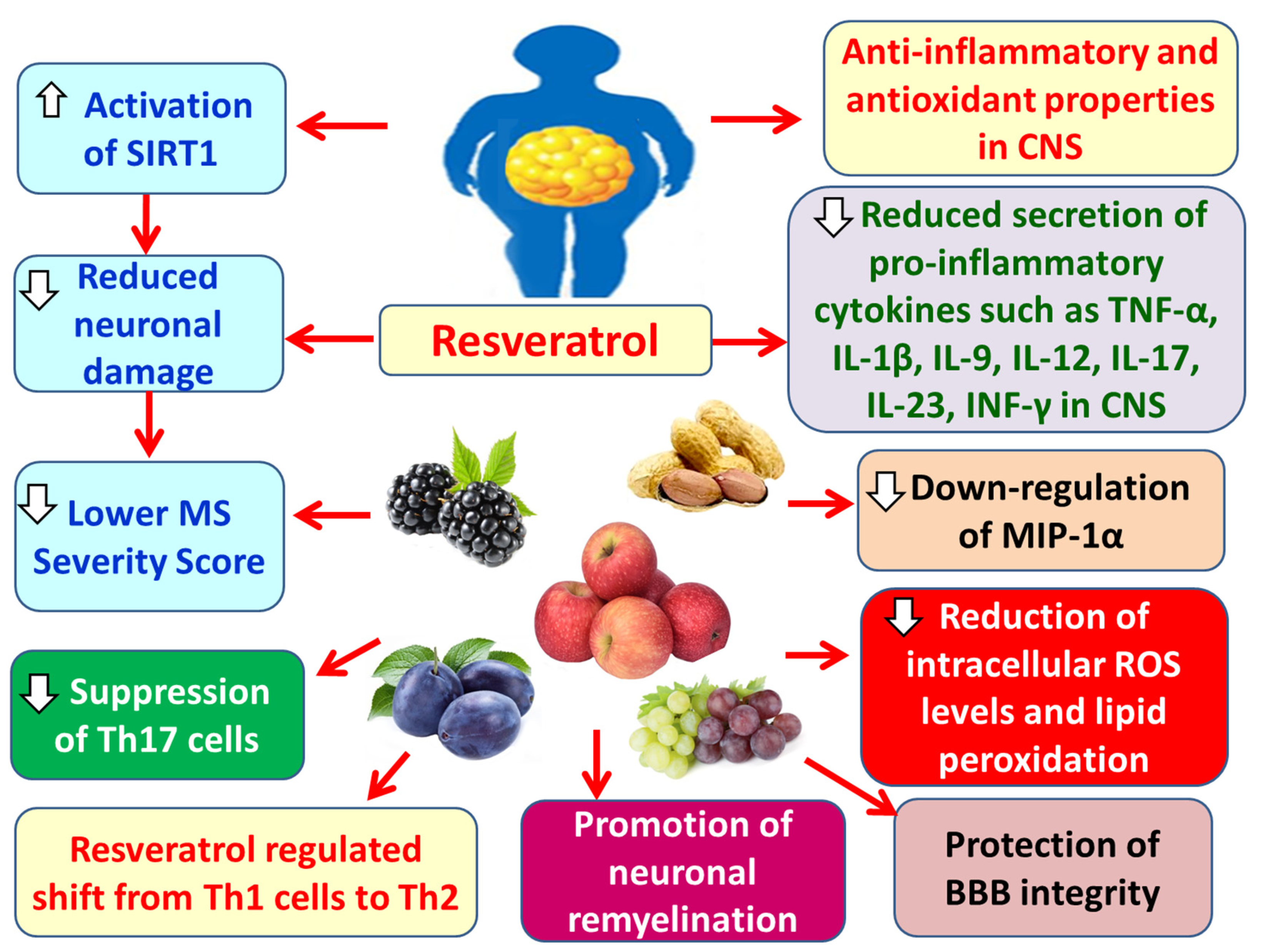
Figure 14.
Schematic representation of the beneficial effects of quercetin against MS. MS: multiple sclerosis; Th1 cells: T helper 1 cells; CNS: central nervous system; JKA2, Janus kinase 2; TYK2, tyrosine kinase 2; STAT3, signal transducer and activator of transcription 3; BBB: blood-brain barrier; IL-1β: interleukin 1β; IL-12, interleukin 12, TNF-α: tumor necrosis factor-alpha; ΙΝF-γ: interferon gamma.
Figure 14.
Schematic representation of the beneficial effects of quercetin against MS. MS: multiple sclerosis; Th1 cells: T helper 1 cells; CNS: central nervous system; JKA2, Janus kinase 2; TYK2, tyrosine kinase 2; STAT3, signal transducer and activator of transcription 3; BBB: blood-brain barrier; IL-1β: interleukin 1β; IL-12, interleukin 12, TNF-α: tumor necrosis factor-alpha; ΙΝF-γ: interferon gamma.
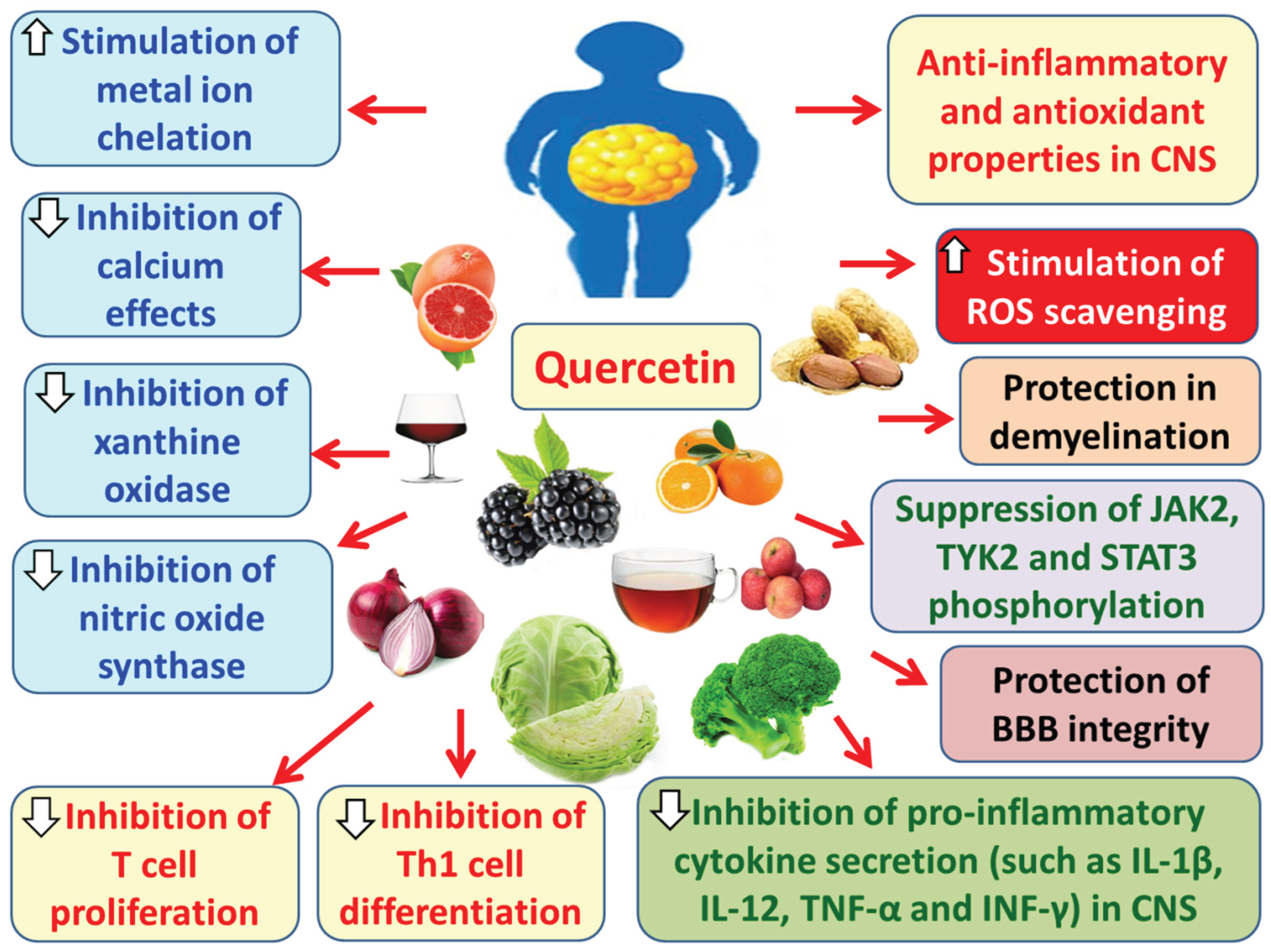
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



