
Submitted:
01 October 2025
Posted:
02 October 2025
You are already at the latest version
Abstract
Hypertrophic cardiomyopathy (HCM) progressing to end-stage heart failure and heart transplantation (HT) is a rare clinical scenario with insufficiently explored genetic background. In this single-center retrospective cohort study we aimed to characterize the genetic spectrum, variants of HCM adverse remodeling, and aspects of molecular pathogenesis of this subgroup. The study included 14 patients (9 females), among whom 10 developed a dilated/hypokinetic phenotype and 4 a restrictive phenotype. In 13 pa-tients (93%), at least one pathogenic or likely pathogenic genetic variant was identified. Dilated remodeling/hypokinesis was associated with loss-of-function variants in LAMP2 (3) in females, ALPK3homo (1), MYH7 (1), MYBPC3 (1), heterozygous missense variant in TRIM63 (1), FLNCtv (1), TTNtv (2). For the latter two electrophoretic analysis of titin isoform composition and protein content in myocardial fragments from ex-planted hearts confirmed the functional significance of TTN gene variants. The restric-tive phenotype in the adult group was associated with carriage of multiple pathogenic sarcomere gene variants: MYL3homo (1), MYBPC3+TPM1 (1), a MYH7 converter domain variant (1), and in 1 child, with a TNNT2 variant. This fundings supports HCM pro-gressing to HT is characterized by a higher frequency of variants in non-sarcomeric genes and Danon disease compared to the general HCM cohort.
Keywords:
hypertrophic cardiomyopathy
; heart transplantation
; adverse remodeling
; dilated phenotype
; restrictive phenotype
; non-sarcomeric genetic variants
; titin
; filamin C
; truncating variants
; Danon disease
1. Introduction
Hypertrophic cardiomyopathy (HCM) is one of the most common inherited cardiac disorders [1]. It demonstrates pronounced phenotypic heterogeneity, encompassing variation in myocardial morphology and the degree of hypertrophy, as well as differences in the extent of interstitial and replacement fibrosis, the presence and severity of diastolic dysfunction, and incomplete penetrance [1]. Consequently, the risk of arrhythmic events and progression to heart failure varies considerably across patients, driving an ongoing search for robust high-risk markers, including those related to genetic etiology.
Adverse myocardial remodeling in HCM can evolve along two major trajectories. One is a dilated “burn-out” phase, characterized by declining contractility, chamber enlargement, and wall thinning. The other is a restrictive phenotype, marked by severe diastolic dysfunction and often accompanied by a small left-ventricular (LV) cavity and low stroke volume [2,3]. In such cases, the ejection fraction (EF) may remain preserved or only mildly reduced [2,3]. Registry data suggest that LV systolic dysfunction occurs in ~8% of HCM patients, while the restrictive phenotype accounts for ~1.3% [4,5]. According to the UNOS registry, patients with HCM represent ~2% of those undergoing heart transplantation (HT) for end-stage heart failure [6].
Although many studies support the role of genotype in determining prognosis in HCM, some evidence inconsistency is still there. [4,7,8]. This inconsistency likely reflects the heterogeneity of causal variants - acting through distinct deleterious mechanisms and with variable effect sizes. Its impact may be obscured when patients are classified simply as genotype-positive or genotype-negative, with acquired factors adding to the complexity of disease penetrance and expressivity [9], also major adverse outcomes remain relatively infrequent in the broader HCM population [4,6].
Therefore, we aimed to characterize the spectrum of genetic findings, clinical course, variants of adverse myocardial remodeling, and elements of molecular pathogenesis in patients with HCM who progressed to end-stage heart failure requiring heart transplantation - a patient group exhibiting one of the most severe clinical scenarios.
2. Materials and Methods
We analyzed patients managed between 2010 and 2025 at the Almazov National Medical Research Center (Saint Petersburg, Russia). The diagnosis of HCM as a cardiomyopathy phenotype (ESC 2023 criteria [1]) was based on imaging data, including echocardiography and cardiac magnetic resonance, and was subsequently corroborated by histological examination of explanted hearts. Patterns of myocardial remodeling were assessed, and patients were classified into dilated/hypokinetic or restrictive phenotypes. Statistical analyses and figure generation were performed using Stata 18.
Genetic testing was performed using next-generation sequencing with Sanger confirmation. The number of genes included in the panels (39, 108, or 172) varied according to the diagnostic strategy applied by the laboratory (detailed gene lists are provided in the Appendix). Testing could be carried out either before or after HT. Target enrichment was performed with SureSelect (Agilent Technologies, CA), and sequencing was conducted on an Illumina HiSeq platform with SBSv4 reagents (Illumina, San Diego, CA). Sequence alignment, processing, and annotation were performed against the hg38 human genome reference. All novel and previously reported variants with an allele frequency <0.01% were classified according to ACMG recommendations. For detailed analyses, we focused on pathogenic, likely pathogenic, and variants of uncertain significance in genes with high myocardial expression.
Electrophoretic separation of titin isoforms, including the NT isoform described in mammalian striated muscle [10], was performed in 2.3% large-pore polyacrylamide gels reinforced with agarose, following the method of Yakupova et al. [11] with modifications. In particular, during sample preparation for SDS-PAGE, heating was limited to ≤40 °C to preserve protein integrity [12]. Titin content was quantified by densitometry relative to myosin heavy chains, an established approach for evaluating changes in high-molecular-weight titin isoforms.
3. Results
3.1. General Clinical Characteristics
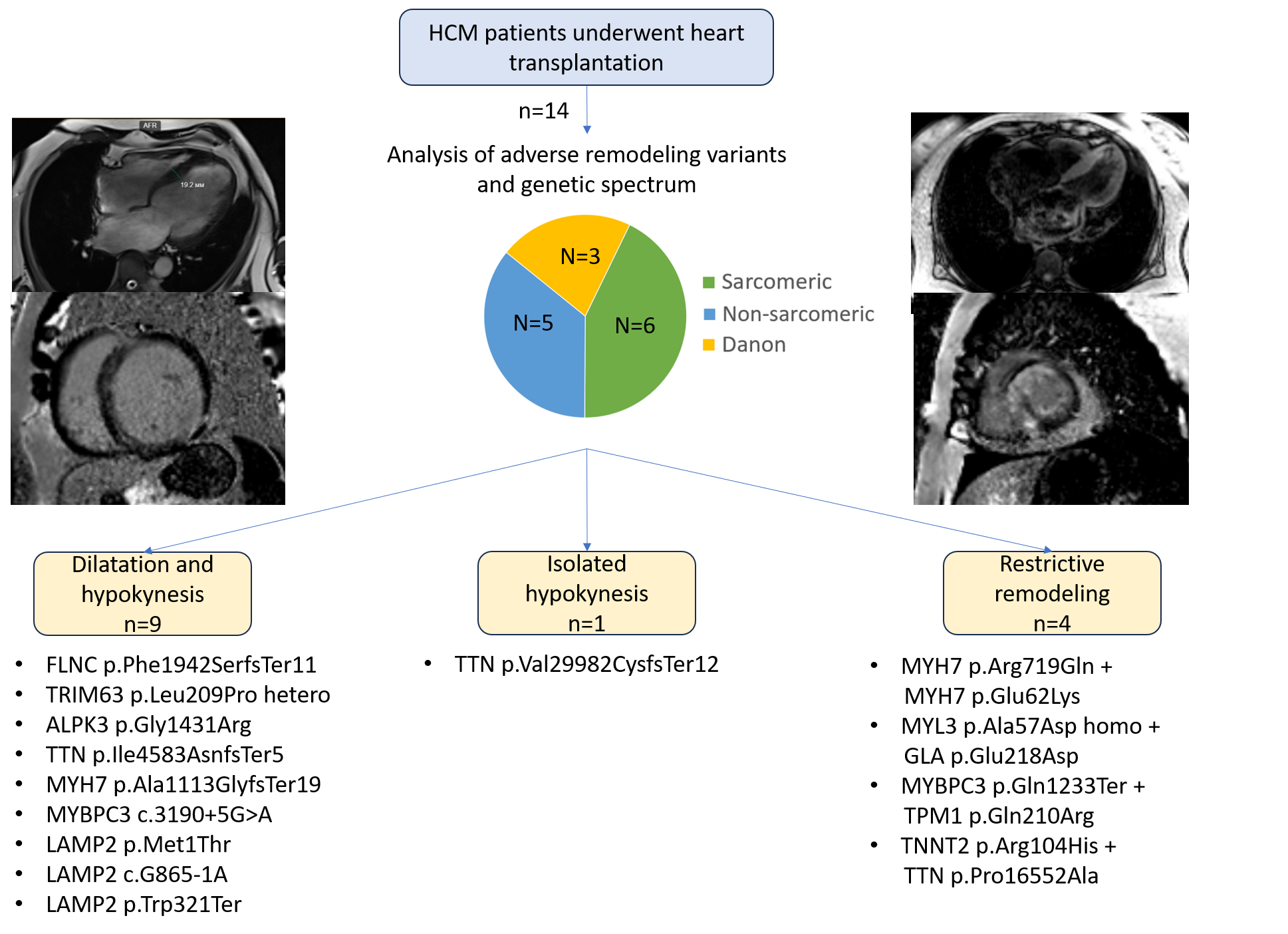
The study included 14 patients, 9 of them women (64%) (Table 1, Figure 1). The mean age at symptom onset was 30.6 ± 3.6 years, and the mean age at HT was 38.4 ± 3.5 years. Disease onset displayed three peaks: in childhood, at 20–30 years, and after 40 years (Figure 2).
The mean time from diagnosis to HT was 7.8 ± 1.3 years. A history of hypertension was present in 3 patients (21.4%), all diagnosed after the age of 40. Atrial fibrillation occurred in 9 patients (64.3%), and all patients had documented high-grade ventricular arrhythmias. None had concomitant coronary artery disease or diabetes.
In 9 patients (64.3%), adverse remodeling primarily manifested as dilation and hypokinesis; in one patient (7.1%), isolated hypokinesis occurred without marked dilation; and in 4 patients (28.6%), a restrictive phenotype was observed (Figure 1). In the dilated phenotype subgroup, the mean maximal LV wall thickness decreased from 18.6 ± 0.9 mm at disease onset to 12.7 ± 0.9 mm at the terminal stage (Table 1). In the restrictive subgroup, the respective values were 18.0 ± 1.6 mm and 17.0 ± 1.1 mm (Table 1).
No patient had evidence of LV outflow tract obstruction at presentation, nor had any undergone septal myectomy. Right ventricular (RV) hypertrophy was observed in 9 of 14 patients (64%). Increased trabeculation or criteria consistent with noncompaction myocardium were documented at certain stages of disease progression in 7 of 14 patients (50%) (Table 1). Clinical data, echocardiographic parameters, and remodeling dynamics are summarized in Table 1.
3.2. Genetic Spectrum and Association with Clinical Course and Adverse Remodeling Variants
Variants in sarcomere genes were identified in 6 patients: TNNT2, MYH7 (n=2), MYBPC3, MYBPC3+TPM1, and homozygous MYL3 (Figure 1, Table 2). Variants in non-sarcomeric or non-contractile genes were found in 5 patients: ALPK3homo, FLNC frameshift (n=2), TTN frameshift variants (n=2), and a TRIM63 missense variant. In 3 female-patients, genetic testing revealed Danon disease (LAMP2), which had not been clinically suspected prior to testing. In 13 of 14 patients (93%), at least one variant was classified as pathogenic or likely pathogenic (Table 2).
Adults transplanted before 35 years included all patients with Danon disease (P.8–P.10), one patient with a homozygous missense variant in ALPK3 (P.3), and both patients with TTN variants (P.4–P.5) (Figure 2). Patients transplanted after 45 years included P.6–P.7 and P.11–P.13, who carried variants in MYH7, MYL3, TRIM63, and FLNC (Figure 2). The pediatric subgroup was represented by P.14, who carried a likely pathogenic TNNT2 variant in combination with a TTN variant of uncertain significance, however, childhood-onset end-stage HCM is underrepresented in this study owing to the specifics of the pediatric HT-system in Russia.
The restrictive phenotype subgroup (P.11–P.14) was enriched for carriage of multiple variants in sarcomere genes (homozygous MYL3, MYBPC3+TPM1) (Picture 1). Of note, P.12 (female) carried homozygous MYL3 p.Ala57Asp together with a GLA variant of uncertain significance linked to Fabry disease; segregation data were unavailable to clarify pathogenicity. This subgroup also included a patient with the MYH7 converter-domain variant p.Arg719Gln.
The dilated phenotype subgroup (P.1–P.10) included all patients with Danon disease, the patient with a homozygous ALPK3 variant, carriers of in-frame deletions in TTN and FLNC, a heterozygous TRIM63 missense variant, and carriers of splice-site variants in MYH7 and MYBPC3 (Figure 1, Table 2). Dilated transformation in patients with ALPK3 and LAMP2 variants occurred against a background of histologically verified (Dallas criteria) chronic lymphocytic, virus-negative myocarditis (per endomyocardial biopsy or explanted heart specimens). Patient 9 received immunosuppressive therapy, which did not modify the avert progression to HT. Notably, none of the women with Danon disease showed evidence of multisystem involvement (skeletal muscle, liver, or nervous system).
In P.6 with the MYH7 splice-site+frameshift variant p.Ala1113GlyfsTer19 causing skipping of exon 27, segregation analysis enabled classification of the variant as likely pathogenic (Figure 3).
3.3. Titin Isoforms Electrophoretic Analysis.
To assess the functional consequences of truncating TTN variants in P.4 and P.5, electrophoretic analysis of titin isoform composition and content was performed in myocardial samples from the explanted hearts (Figure 4). In P.4 (p.Val29982CysfsTer12), there was a twofold reduction in intact full-length titin-1 (T1) molecules, spanning the sarcomere from the M-line to the Z-disk (sum of NT, N2BA, and N2B isoforms) - 48% in the LV and 58% in the RV compared with 100% in control. The largest titin isoform (NT) was completely absent on the electrophoretograms of both the RV and LV. These changes indicate enhanced titin proteolysis in the patient’s heart, as further evidenced by a marked increase in the ratio of proteolytic T2 fragments to intact T1 isoforms (T2/T1 ratio 42%/58% vs. 26%/74% in control) (Figure 4a.1–3).
In P.5 (p.Ile4583AsnfsTer5), the overall content of T1 isoforms was preserved in both ventricles; however, NT isoform abundance in the LV was reduced 1.5-fold (67% of control). In the myocardium of both ventricles, there was a more than twofold increase in the content of proteolytic T2 fragments of titin, which interact with myosin filaments in the A-band of the sarcomere. These findings suggest enhanced proteolysis of high-molecular-weight T1 isoforms and possibly increased titin turnover (Figure 4b.1-4).
Thus, electrophoretic analysis of titin isoform composition and content demonstrated the pathogenic impact of the identified TTNtv, manifested in particular by enhanced proteolytic degradation of the protein and by a reduction in the quantitative content of its isoforms.
4. Discussion
In this study we specifically examined the genetic spectrum in a cohort of HCM patients who underwent HT; to our knowledge, no prior publications have used a comparable design. Our findings may help delineate genetic subgroups at highest risk of progressing to end-stage heart failure despite contemporary guideline-directed medical therapy.
Variants in “non-sarcomeric or non-contractile” genes (ALPK3, TTN, FLNC, TRIM63) accounted for 35.7% of our cohort, a proportion substantially higher than in unselected HCM populations [13]. They are not classified as “classic” sarcomere HCM-associated genes, although some are also described as sarcomere-associated [14]. In addition, a considerable fraction (21.4%) of female patients were diagnosed with Danon disease (LAMP2). Variants in classic sarcomeric/contractile genes comprised 42.8%, and genotype-negative patients were virtually absent. The distribution of age at disease onset in our cohort showed three peaks, which is consistent with published data on age at onset in unselected HCM populations [15]. We identified patients carrying frameshift variants of TTN (n=2) and FLNC (n=1), resulting in truncating proteins, which are classically associated with dilated cardiomyopathy [16,17], whereas the association of TTNtv with HCM has been reported only sporadically [18]. Mechanisms underlying TTNtv-associated cardiomyopathy may include haploinsufficiency and aggregate formation due to impaired ubiquitin-mediated degradation [19], the amyloidogenic potential of titin has also been described [20]. Previously, we reported a patient with a mixed cardiomyopathy phenotype carrying an extended deletion in TTN, for which bioinformatic modeling showed unfolding of protein motifs and the formation of amyloid-like structures [21]. In the patient with the TTN variant p.Val29982CysfsTer12, results of SDS-gel electrophoresis followed by densitometric analysis revealed reduced titin isoform content in the myocardium of the explanted heart, thereby confirming the pathogenic effect of the identified variant and suggesting a possible contribution of titin haploinsufficiency to the phenotype. In contrast, in the patient with the TTN variant p.Ile4583AsnfsTer5, the predominant abnormality was not enhanced proteolysis but rather accelerated titin turnover, which suggests pathological aggregation of T2 fragments and shorter titin fragments.
No descriptions of frameshift or truncating FLNC variants associated with HCM were identified in the available literature. Two principal mechanisms have been proposed for FLNC-related cardiomyopathy: protein aggregation caused by non-truncating mutations, and haploinsufficiency due to truncating mutations [22]. For HCM, protein aggregation appears to be the more likely mechanism, but this mechanism is not typical for FLNCtv. In this regard, it may be assumed that in patients with FLNCtv, the development of an HCM phenotype is also possible - either due to protein aggregation or through other mechanisms - or that in our patient, FLNCtv acted as a trigger of dilated transformation, whereas the HCM phenotype arose from other, unidentified causes, such as an non-target genetic background or the influence of polygenic determinants in an older patient with a history of hypertension [9]. Confirmation of the first hypothesis would require advanced proteomic and structural studies. The second hypothesis is consistent with current evidence that HCM may develop as a polygenic disorder with additional contributions from acquired factors [9,23].
In female patients with Danon disease, beyond the well-described isolated cardiac involvement without significant multisystem manifestations, we emphasize the possible role of myocarditis in accelerating fibrosis and driving heart failure, potentially reflecting an autoimmune response to primary genetic injury. Such a mechanism may also be relevant in other genetic cardiomyopathies.
For ALPK3, homozygous loss-of-function variants have been linked to severe autosomal-recessive HCM with childhood onset [14]. More recently, heterozygous loss-of-function variants were shown to underlie autosomal-dominant adult-onset HCM with reduced penetrance, resembling genotype-negative disease [14,24]. In this study, we report a case of HCM associated with a homozygous ALPK3 missense variant identified at a young age, which may indicate a role of missense variants in the development of autosomal-recessive ALPK3 cardiomyopathy. The clinical course and genetic data of this patient we have described in detail previously [25].
For TRIM63, an autosomal-recessive inheritance model in HCM is now considered highly probable [14]. However, we cannot exclude a modifying contribution of heterozygous missense TRIM63 variants in patients with poorly controlled hypertension, as in our case.
Special attention should be paid to variants in classic HCM-associated sarcomeric genes and their combinations. One of our patients carried a splice-site variant in MYH7 leading to exon 27 skipping, which implies haploinsufficiency. This mechanism is atypical for MYH7-related HCM but more characteristic of dilated cardiomyopathy and LV noncompaction [26]. Another patient carried MYH7 p.Arg719Gln and p.Glu62Lys, located in the converter and SH3 domains, respectively. Variants in the MYH7 converter domain (amino acids 709–777) are well known to be associated with adverse prognosis. The p.Arg719Gln variant has also been previously reported as pathogenic, characterized by high penetrance, increased risk of sudden cardiac death, and absence of intraventricular obstruction [27,28]. Notably, atrial enlargement and atrial fibrillation have been described in a number of patients [29], features consistent with the restrictive phenotype observed in our patient. Nevertheless, it cannot be excluded that the second variant, p.Glu62Lys, also contributed to the development of restrictive hemodynamics.
For the MYL3 p.Ala57Asp variant, usually linked to autosomal-dominant HCM, an autosomal-recessive form has recently been described [30] without restrictive features. Thus, in our patient, the restrictive phenotype may have been influenced by concomitant carriage of a GLA variant of uncertain significance.
The limitations of our study include its retrospective cohort design, the use of different gene panels across patients (Table 1), and restriction to individuals who underwent HT rather than all patients with end-stage heart failure. Also, childhood-onset end-stage HCM is underrepresented in our cohort, as pediatric organ donation is not permitted in Russia, and size incompatibility often precludes transplantation from adult donors. This subgroup warrants focused study in the future.
5. Conclusions
The genetic basis of HCM progressing to HT is heterogeneous. In comparison with unselected HCM cohorts, our series was markedly enriched for non-sarcomeric variants. Among sarcomeric genes, carriage of multiple pathogenic variants (within a single gene or across genes) and MYH7 converter-domain variants appear to predispose to restrictive remodeling, whereas splice-site variants in MYH7 and MYBPC3 may lead to haploinsufficiency and dilated transformation. Danon disease accounted for a substantial proportion of cases, and its storage-disease nature may remain unrecognized until genetic testing, particularly in women. Potentially high-risk non-sarcomeric variants include homozygous ALPK3. Finally, truncating variants in sarcomere-associated genes not typically linked to HCM, such as TTN and FLNC, may also contribute to HCM or its dilated phase, likely through haploinsufficiency or protein aggregation.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/doi/s1, Figure S1: title; Table S1: title; Video S1: title.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, writing—original draft preparation, S.A., A.K.; methodology, A.K, I.V.; formal analysis, L.K.; investigation, A.B., L.G., L.M., A.G.; resources, A.S., M.B., O.M., P.F.; data curation, L.K., A.F., A.S., M.M., Yu.S., S.F., M.S.; writing—review and editing, A.K., I.M., A.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Russian Scientific Organization, grant number 25-15-00552.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of Almazov National Medical Research Center.
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study. Written informed consent has been obtained from the patients to publish this paper.
Data Availability Statement
Data is contained within the article or supplementary material
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACMG | American College of Medical Genetics |
| HCM | Hypertrophic cardiomyopathy |
| HT | Heart transplantation |
| LV | Left ventricle |
| RV | Right ventricle |
Appendix A
Target 172 Genes Panel
ABCC9, ACADVL, ACTA1, ACTC1, ACTN2, ACVR2B, AGK, AKAP9, ALPK3, ANK2, ANKRD1, ANO5, BAG3, BRAF, CACNA1C, CACNA2D1, CACNB2, CALM1, CALM2, CALM3, CALR3, CASQ2, CAV3, CBL, CDH2, CMYA5, CRELD1, CRYAB, CSRP3, CTNNA3, DES, DMD, DMPK, DNAAF1, DNAAF3, DPP6, DSC2, DSG2, DSP, DTNA, DYSF, EMD, EYA4, FHL1, FHL2, FHOD3, FKRP, FKTN, FLNA, FLNC, FXN, GAA, GATA4, GATA5, GATA6, GATAD1, GDF1, GJA5, GLA, GPD1L, HAND1, HCN4, HFE, HRAS, ILK, ISPD, JPH2, JUP, KCNA5, KCND3, KCNE1, KCNE2, KCNE3, KCNE5,KCNH2, CNJ2, KCNJ5, KCNJ8, KCNQ1, KRAS, LAMA4, LAMP2, LDB3, LEFTY2, LMNA, LMOD3, LRRC10, LZTR1, MAP2K1, MAP2K2, MIB1, MMP21, MRA, MYBPC3, MYBPHL, MYH6, MYH7, MYL2, MYL3, MYL4, MYLK2, MYOF, MYOM1,MYOT, MYOZ2, MYPN, NEBL, NEXN, NF1, NKX2-5, NKX2-6, NPPA, NRAS, NUP155, PDLIM3, PKD1L1, PKP2, PLEC, PLEKHM2, PLN, PPA2, PPP1CB, PRDM16, PRKAG2, PSEN1, PSEN2, PTPN11, RAF1, RANGRF,RBM20, RIT1,RRAS, RYR2, SALL4, SCN10A, SCN1B, SCN2B, SCN3B, SCN4B, SCN5A, SCNN1G, SDHA, SGCD, SHOC2, SLMAP, SNTA1, SOS1, SOS2, SPEG, SPRED1, SYNE1, SYNM, SYNPO2L, TAZ, TBX20, TBX5, TCAP, TECRL, TGFB3, TMEM43, TMPO, TNNC1, TNNI3, TNNI3K, TNNT2, TPM1, TRDN, TRPM4, TTN, TTR, VCL, ZIC3.
Target 108 genes panel:
ABCC9, ACTC1, ACTN2, AKAP9, ANK2, ANKRD1, BAG3, BRAF, CACNA1C, CACNA2D1, CACNB2, CALM1, CALR3, CASQ2, CAV3, CBL, CRYAB, CSRP3, DES, DMD, DMPK, DSC2, DSG2, DSP, DTNA, EMD, EYA4, FHL1, FHL2, FKTN, FXN, GAA, GLA, GPD1L, HCN4, HRAS, ILK, JPH2, JUP, KCND3, KCNE1, KCNE1L, KCNE2, KCNE3, KCNH2, KCNJ2, KCNJ5, KCNJ8, KCNQ1, KRAS, LAMA4, LAMP2, LDB3, LMNA, MAP2K1, AP2K2, MRPL3, MYBPC3, MYH6, MYH7, MYL2, MYL3, MYLK2, MYOM1, MYOZ2, MYPN, NEBL, NEXN, NF1, NOS1AP, NRAS, PDLIM3, PKP2, PLN, PRKAG2, SEN1, PSEN2, PTPN11, RAF1, RANGRF, RBM20, RYR2, SCN1B, SCN3B, SCN4B, SCN5A, SCO2, SDHA, SGCD, SHOC2, SLC25A3, SLMAP, SNTA1, SOS1, SPRED1, TAZ, TCAP, TGFB3, TMEM43, TMPO, TNNC1, TNNI3, TNNT2, TPM1, TRDN, TRPM4, TTN, VCL.
Target 39 genes panel:
ACTC1, ACTN2, ALPK3, BAG3, BRAF, CBL, CSRP3, FHL2, FHOD3, FLNC, FXN, GLA, HRAS, KRAS, LAMP2, LDB3, LZTR1, MAP2K1, MAP2K2, MYBPC3, MYH7, MYL2, MYL3, MYLK2, MYOM1, MYPN, NF1, NRAS, PRKAG2, PTPN11, RAF1, SHOC2, SOS1, TNNC1, TNNI3, TNNT2, TPM1, TRIM63, TTR.
References
- Arbelo E, Protonotarios A, Gimeno JR, Arbustini E, Arbelo E, Barriales-Villa R, et al. 2023 ESC Guidelines for the management of cardiomyopathies: Developed by the task force on the management of cardiomyopathies of the European Society of Cardiology (ESC). Eur Heart J. 2023;44(37):3503–626.
- Zampieri M, Salvi S, Fumagalli C, Argirò A, Zocchi C, Del Franco A, et al. Clinical scenarios of hypertrophic cardiomyopathy-related mortality: Relevance of age and stage of disease at presentation. Int J Cardiol [Internet]. 2023 Mar 1 [cited 2025 Sep 30];374:65–72. Available from: https://pubmed.ncbi.nlm.nih.gov/36621577/.
- Argirò A, Zampieri M, Marchi A, Cappelli F, Del Franco A, Mazzoni C, et al. Stage-specific therapy for hypertrophic cardiomyopathy. Eur Heart J Suppl [Internet]. 2023 May 1 [cited 2025 Sep 30];25(Suppl C):C155–61. Available from: https://pubmed.ncbi.nlm.nih.gov/37125313/.
- Marstrand P, Han L, Day SM, Olivotto I, Ashley EA, Michels M, et al. Hypertrophic Cardiomyopathy with Left Ventricular Systolic Dysfunction: Insights from the SHaRe Registry. Circulation [Internet]. 2020 Apr 28 [cited 2025 Jul 1];141(17):1371–83. Available from: https://pubmed.ncbi.nlm.nih.gov/32228044/.
- Li S, Wu B, Yin G, Song L, Jiang Y, Huang J, et al. MRI characteristics, prevalence, and outcomes of hypertrophic cardiomyopathy with restrictive phenotype. Radiol Cardiothorac Imaging [Internet]. 2020 Aug 1 [cited 2025 Jul 16];2(4):e190158. Available from: https://pubmed.ncbi.nlm.nih.gov/33778596/.
- Miklin DJ, DePasquale E. Transplantation Trends in End Stage Hypertrophic Cardiomyopathy Over 35 Years: A UNOS Registry Analysis. J Hear Lung Transplant [Internet]. 2023 Apr 1 [cited 2025 Apr 27];42(4):S240. Available from: https://www.jhltonline.org/action/showFullText?pii=S1053249823005843.
- Georgiopoulos G, Figliozzi S, Pateras K, Nicoli F, Bampatsias D, Beltrami M, et al. Comparison of Demographic, Clinical, Biochemical, and Imaging Findings in Hypertrophic Cardiomyopathy Prognosis: A Network Meta-Analysis. JACC Hear Fail [Internet]. 2023 Jan 1 [cited 2025 Jul 1];11(1):30–41. Available from: https://pubmed.ncbi.nlm.nih.gov/36599547/.
- Bonaventura J, Rowin EJ, Chan RH, Chin MT, Puchnerova V, Polakova E, et al. Relationship Between Genotype Status and Clinical Outcome in Hypertrophic Cardiomyopathy. J Am Heart Assoc [Internet]. 2024 [cited 2025 Jul 1];13(10):e033565. Available from: https://pubmed.ncbi.nlm.nih.gov/38757491/.
- Harper AR, Goel A, Grace C, Thomson KL, Petersen SE, Xu X, et al. Common genetic variants and modifiable risk factors underpin hypertrophic cardiomyopathy susceptibility and expressivity. Nat Genet 2021 532 [Internet]. 2021 Jan 25 [cited 2022 Apr 23];53(2):135–42. Available from: https://www.nature.com/articles/s41588-020-00764-0.
- Vikhlyantsev IM, Podlubnaya ZA. New titin (connectin) isoforms and their functional role in striated muscles of mammals: facts and suppositions. Biochemistry (Mosc) [Internet]. 2012 [cited 2025 Sep 30];77(13):1515–35. Available from: https://pubmed.ncbi.nlm.nih.gov/23379526/.
- Yakupova EI, Abramicheva PA, Rogachevsky V V., Shishkova EA, Bocharnikov AD, Plotnikov EY, et al. Cardiac titin isoforms: Practice in interpreting results of electrophoretic analysis. Methods [Internet]. 2025 Apr 1 [cited 2025 Aug 25];236:17–25. Available from: https://pubmed.ncbi.nlm.nih.gov/39993454/.
- Vikhlyantsev IM, Podlubnaya ZA. Nuances of electrophoresis study of titin/connectin. Biophys Rev [Internet]. 2017 Jun 1 [cited 2025 Sep 30];9(3):189–99. Available from: https://pubmed.ncbi.nlm.nih.gov/28555301/.
- Hernandez SG, Romero L de la H, Fernández A, Peña-Peña ML, Mora-Ayestaran N, Basurte-Elorz MT, et al. Redefining the Genetic Architecture of Hypertrophic Cardiomyopathy: Role of Intermediate Effect Variants. Circulation [Internet]. 2025 Aug 13 [cited 2025 Sep 30];online ahead of print. Available from: /doi/pdf/10.1161/CIRCULATIONAHA.125.074529?download=true.
- Hespe S, Waddell A, Asatryan B, Owens E, Thaxton C, Adduru ML, et al. Genes Associated With Hypertrophic Cardiomyopathy: A Reappraisal by the ClinGen Hereditary Cardiovascular Disease Gene Curation Expert Panel. J Am Coll Cardiol [Internet]. 2025 Feb 25 [cited 2025 Jul 23];85(7):727–40. Available from: https://pubmed.ncbi.nlm.nih.gov/39971408/.
- Marston NA, Han L, Olivotto I, Day SM, Ashley EA, Michels M, et al. Clinical characteristics and outcomes in childhood-onset hypertrophic cardiomyopathy. Eur Heart J [Internet]. 2021 May 21 [cited 2025 Sep 30];42(20):1988. Available from: https://pmc.ncbi.nlm.nih.gov/articles/PMC8139852/.
- Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016 Dec 6;68(22):2440–51.
- J Verdonschot JA, Vanhoutte EK, F Claes GR, J M Helderman-van den Enden AT, J Hoeijmakers JG, E I Hellebrekers DM, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. | Stephane R B Heymans [Internet]. 2020 [cited 2023 Oct 12];41(6):1091–111. Available from: https://onlinelibrary.wiley.com/doi/10.1002/humu.24004.
- Sepp R, Hategan L, Csányi B, Borbás J, Tringer A, Pálinkás ED, et al. The Genetic Architecture of Hypertrophic Cardiomyopathy in Hungary: Analysis of 242 Patients with a Panel of 98 Genes. Diagnostics [Internet]. 2022 May 1 [cited 2025 Jul 28];12(5):1132. Available from: https://pubmed.ncbi.nlm.nih.gov/35626289/.
- Fomin A, Gärtner A, Cyganek L, Tiburcy M, Tuleta I, Wellers L, et al. Truncated titin proteins and titin haploinsufficiency are targets for functional recovery in human cardiomyopathy due to TTN mutations. Sci Transl Med [Internet]. 2021 Nov 3 [cited 2025 Jul 28];13(618):eabd3079. Available from: https://pubmed.ncbi.nlm.nih.gov/34731013/.
- Yakupova EI, Vikhlyantsev IM, Lobanov MY, Galzitskaya O V., Bobylev AG. Amyloid properties of titin. Biochem [Internet]. 2017 Dec 1 [cited 2025 Jul 28];82(13):1675–85. Available from: https://pubmed.ncbi.nlm.nih.gov/29523065/.
- Andreeva SE, Snetkov PP, Vakhrushev YA, Piankov IA, Yaznevich OO, Bortsova MA, et al. Molecular basis of amyloid deposition in myocardium: not only ATTR and AL. Case report. Transl Med. 2023 Mar 2;9(6):26–35.
- Song S, Shi A, Lian H, Hu S, Nie Y. Filamin C in cardiomyopathy: from physiological roles to DNA variants. Heart Fail Rev [Internet]. 2022 Jul 1 [cited 2025 Jul 28];27(4):1373–85. Available from: https://pubmed.ncbi.nlm.nih.gov/34535832/.
- Zheng SL, Jurgens SJ, McGurk KA, Xu X, Grace C, Theotokis PI, et al. Evaluation of polygenic scores for hypertrophic cardiomyopathy in the general population and across clinical settings. Nat Genet [Internet]. 2025 Mar 1 [cited 2025 Sep 30];57(3):563–71. Available from: https://www.nature.com/articles/s41588-025-02094-5.
- Ryu SW, Jeong WC, Hong GR, Cho JS, Lee SY, Kim H, et al. High prevalence of ALPK3 premature terminating variants in Korean hypertrophic cardiomyopathy patients. Front Cardiovasc Med [Internet]. 2024 [cited 2025 Jul 30];5(11):1424551. Available from: https://pubmed.ncbi.nlm.nih.gov/39036505/.
- Jorholt J, Formicheva Y, Vershinina T, Kiselev A, Muravyev A, Demchenko E, et al. Two New Cases of Hypertrophic Cardiomyopathy and Skeletal Muscle Features Associated with ALPK3 Homozygous and Compound Heterozygous Variants. Genes (Basel) [Internet]. 2020 Oct 1 [cited 2022 Jun 22];11(10):1–9. Available from: /pmc/articles/PMC7602582/.
- Myasnikov RP, Kulikova O V., Meshkov AN, Bukaeva AA, Kiseleva A V., Ershova AI, et al. A Splice Variant of the MYH7 Gene Is Causative in a Family with Isolated Left Ventricular Noncompaction Cardiomyopathy. Genes (Basel) [Internet]. 2022 Oct 1 [cited 2025 Aug 25];13(10):1750. Available from: https://pubmed.ncbi.nlm.nih.gov/36292635/.
- Consevage MW, Salada GC, Baylen BG, Ladda RL, Rogan PK. A new missense mutation, Arg719Gln, in the β-cardiac heavy chain myosin gene of patients with familial hypertrophic cardiomyopathy. Hum Mol Genet [Internet]. 1994 Jun [cited 2025 Jul 30];3(6):1025–6. Available from: https://pubmed.ncbi.nlm.nih.gov/7848441/.
- Huang X, Song L, Ma AQ, Gao J, Zheng W, Zhou X, et al. A malignant phenotype of hypertrophic cardiomyopathy caused by Arg719Gln cardiac beta-myosin heavy-chain mutation in a Chinese family. Clin Chim Acta [Internet]. 2001 Aug 20 [cited 2025 Jul 30];310(2):131–9. Available from: https://www.sciencedirect.com/science/article/abs/pii/S0009898101005381.
- Golubenko M V., Pavlyukova EN, Salakhov RR, Makeeva OA, Puzyrev K V., Glotov OS, et al. A New Leu714Arg Variant in the Converter Domain of MYH7 is Associated with a Severe Form of Familial Hypertrophic Cardiomyopathy. Front Biosci - Sch [Internet]. 2024 [cited 2025 Aug 29];16(1):1. Available from: https://pubmed.ncbi.nlm.nih.gov/38538344/.
- Osborn DPS, Emrahi L, Clayton J, Tabrizi MT, Wan AYB, Maroofian R, et al. Autosomal recessive cardiomyopathy and sudden cardiac death associated with variants in MYL3. Genet Med [Internet]. 2021 Apr 1 [cited 2025 Jul 30];23(4):787–92. Available from: https://www.sciencedirect.com/science/article/pii/S1098360021024473?ref=pdf_download&fr=RR-2&rr=964d1fdc4e046132.
Figure 1.
Study design and results. Central pie chart reflects proportion of sarcomeric, non-sarcomeric variants and patients with Danon disease. MRI pictures on the left side represent dilated remodeling of patient 2 with TRIM63 variant, on the right side – restrictive remodeling of patient 11 with MYH7 variant. HCM – hypertrophic cardiomyopathy, MRI – magnetic resonance imaging.
Figure 1.
Study design and results. Central pie chart reflects proportion of sarcomeric, non-sarcomeric variants and patients with Danon disease. MRI pictures on the left side represent dilated remodeling of patient 2 with TRIM63 variant, on the right side – restrictive remodeling of patient 11 with MYH7 variant. HCM – hypertrophic cardiomyopathy, MRI – magnetic resonance imaging.
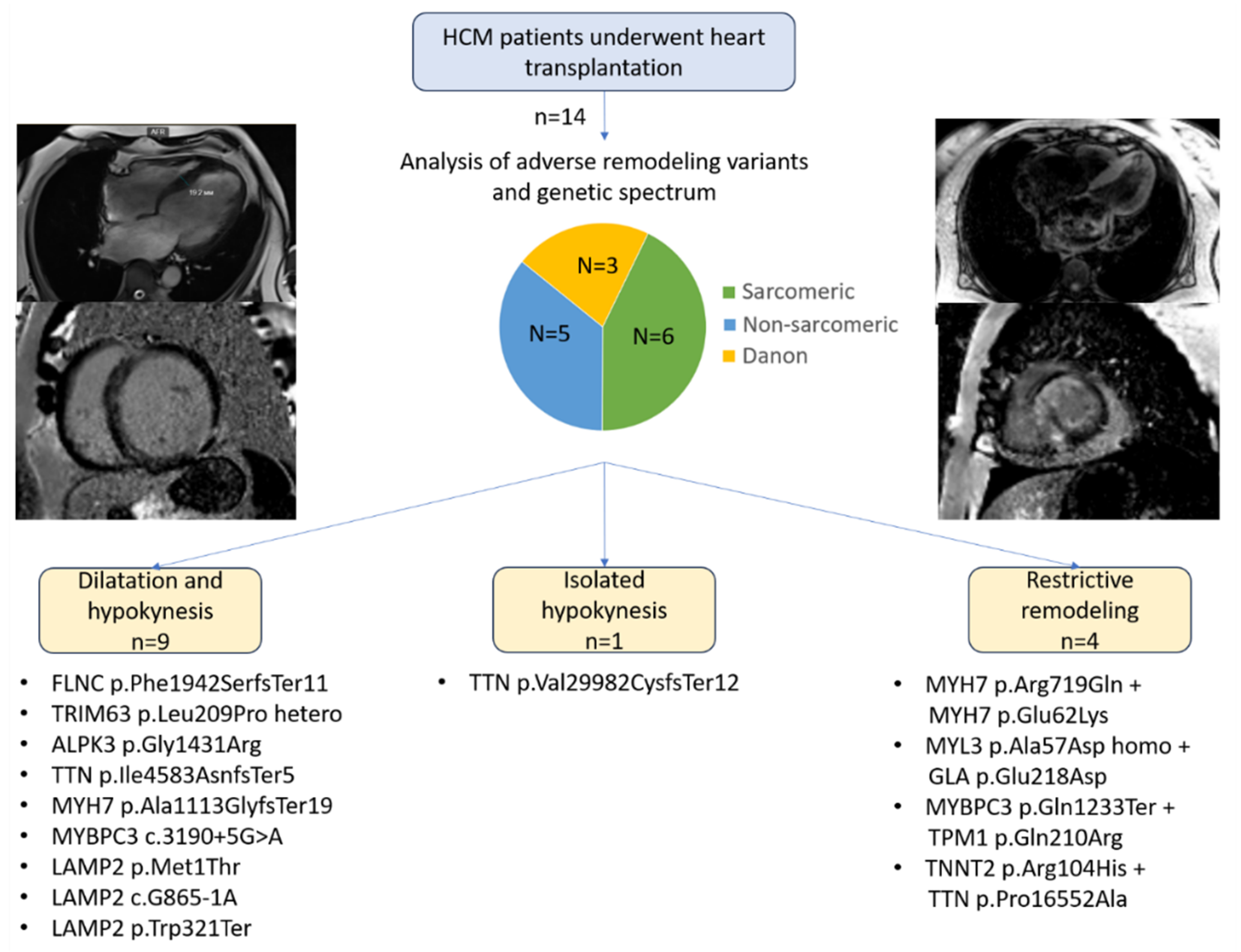
Figure 2.
Age at debut and heart transplantation and underlying genes. HT – heart transplantation.
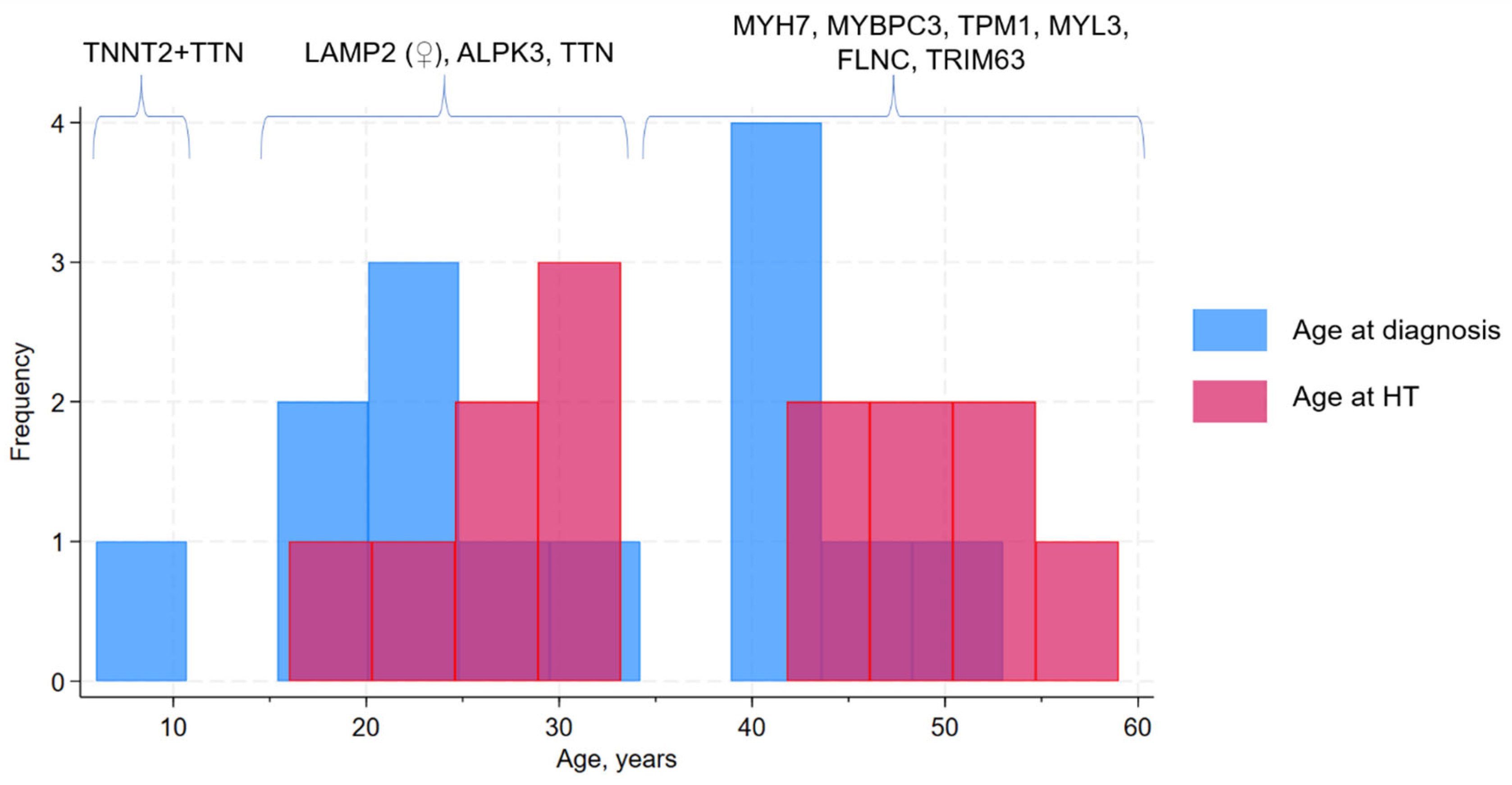
Figure 3.
Pedigree and segregation analysis of Patient 6 with the MYH7 p.Ala1113GlyfsTer19 variant. HCM – hypertrophic cardiomyopathy; HT – heart transplantation; SCD – sudden cardiac death. Black shading indicates presence of the HCM phenotype. “+/-” denotes heterozygous carriage of the variant; “-/-” denotes absence of the variant.
Figure 3.
Pedigree and segregation analysis of Patient 6 with the MYH7 p.Ala1113GlyfsTer19 variant. HCM – hypertrophic cardiomyopathy; HT – heart transplantation; SCD – sudden cardiac death. Black shading indicates presence of the HCM phenotype. “+/-” denotes heterozygous carriage of the variant; “-/-” denotes absence of the variant.
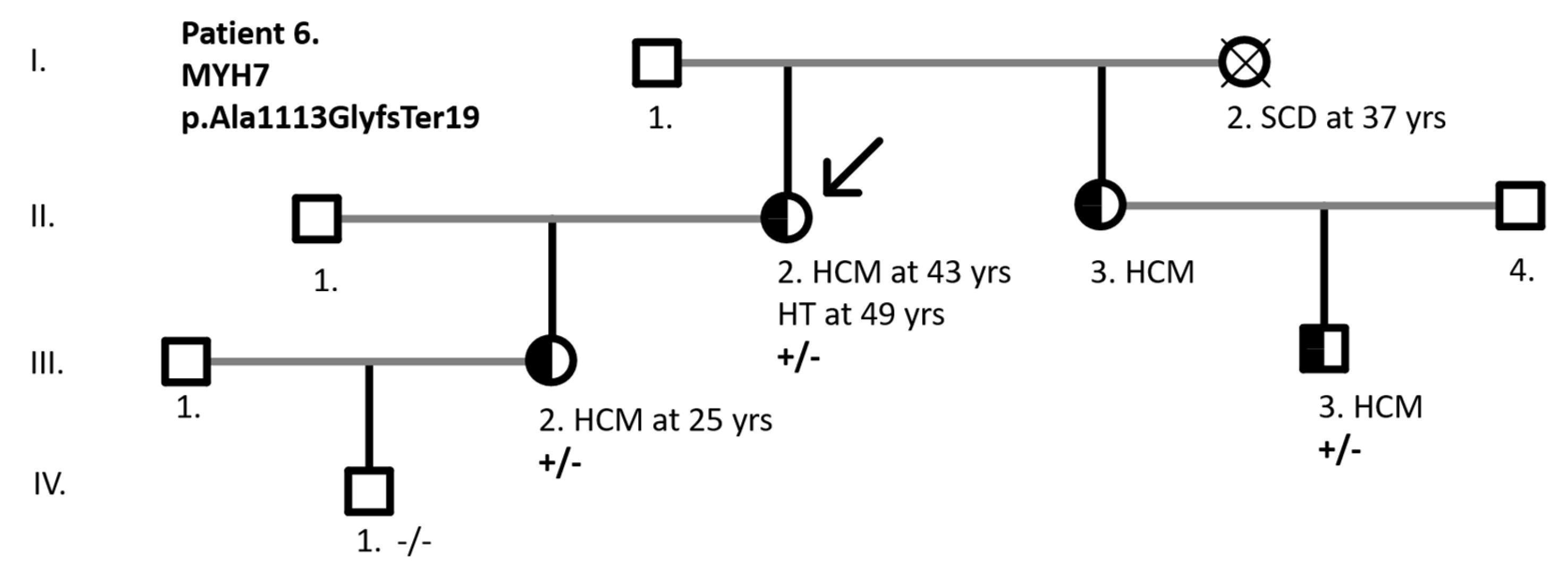
Figure 4.
SDS–PAGE of titin in patients with truncating TTN variants. (a) Patient with p.Val29982CysfsTer12: (1) RV, (2) LV, (3) rat heart (control). There is a twofold reduction in full-length T1 molecules, complete absence of the NT isoform, and an increased content of proteolytic T2 fragments compared with control. (b) Patient with p.Ile4583AsnfsTer5: (1, 2) control (human myocardium), (3) LV, (4) RV. In both ventricles, there is an approximately twofold increase in proteolytic T2 fragments compared with control, with preserved overall content of N2BA and N2B isoforms and a reduced NT isoform in the right ventricle. Notes: LV—left ventricle; RV—right ventricle; MyHC—myosin heavy chains.
Figure 4.
SDS–PAGE of titin in patients with truncating TTN variants. (a) Patient with p.Val29982CysfsTer12: (1) RV, (2) LV, (3) rat heart (control). There is a twofold reduction in full-length T1 molecules, complete absence of the NT isoform, and an increased content of proteolytic T2 fragments compared with control. (b) Patient with p.Ile4583AsnfsTer5: (1, 2) control (human myocardium), (3) LV, (4) RV. In both ventricles, there is an approximately twofold increase in proteolytic T2 fragments compared with control, with preserved overall content of N2BA and N2B isoforms and a reduced NT isoform in the right ventricle. Notes: LV—left ventricle; RV—right ventricle; MyHC—myosin heavy chains.


Table 1.
Clinical data and trends of echocardiographic data reflecting adverse remodeling in 14 patients with HCM underwent heart transplantation. Patients with dilated remodeling demonstrated progressive decrease of EF, thinning of the left ventricle wall and enlargement of left ventricle (iEDV) and atrium (iLAV) over the time, while patients with restrictive remodelling had preserved or mildly reduced EF even at the terminal stage of the disease and severe atria enlargement with relatively preserved left ventricle volume (hallmark of restrictive phenotype). AF – atrial fibrillation, EF – ejection fraction, HCM – hypertrophic cardiomyopathy, HT – heart transplantation, iLAV – indexed left atrium volume, iEDV – indexed end-diastolic volume, LV – left ventricle, NT-proBNP - N-terminal pro-Brain Natriuretic Peptide, RV – right ventricle.
Table 1.
Clinical data and trends of echocardiographic data reflecting adverse remodeling in 14 patients with HCM underwent heart transplantation. Patients with dilated remodeling demonstrated progressive decrease of EF, thinning of the left ventricle wall and enlargement of left ventricle (iEDV) and atrium (iLAV) over the time, while patients with restrictive remodelling had preserved or mildly reduced EF even at the terminal stage of the disease and severe atria enlargement with relatively preserved left ventricle volume (hallmark of restrictive phenotype). AF – atrial fibrillation, EF – ejection fraction, HCM – hypertrophic cardiomyopathy, HT – heart transplantation, iLAV – indexed left atrium volume, iEDV – indexed end-diastolic volume, LV – left ventricle, NT-proBNP - N-terminal pro-Brain Natriuretic Peptide, RV – right ventricle.
| Mean (SD) | Dilated remodeling (n=10) | Restrictive remodeling (n=4) | Overall (n=14) | |
| EF, % | at debut | 48.3 (4.7) | 61.8 (5.6) | 52.1 (4.0) |
| at terminal stage | 22.3 (2.2) | 44.5 (2.) | 28.6 (3.2) | |
| Wall thicknes, mm | at debut | 18.6 (0.9) | 18 (1.60) | 18.5 (0.8) |
| at terminal stage | 12.7 (0.9) | 17 (1.1) | 13.9 (0.9) | |
| iEDV, ml/m3 | at debut | 90.1 (11.2) | 51.0 (7.2) | 78.9 (9.5) |
| at terminal stage | 140.8 (17.1) | 49.0 (10.0) | 114.6 (16.) | |
| iLAV, ml/m3 | at debut | 55.7 (6.0) | 56.3 (7.4) | 55.9 (4.6) |
| at terminal stage | 70.9 (6.7) | 90.8 (8.6) | 76.6 (5.8) | |
| RV hypertrophy | 5 (50%) | 4 (100%) | 9 (64%) | |
| LV hypertrabeculation | 6 (60%) | 1 (25%) | 7 (50%) | |
| Female sex | 6 (60%) | 3 (75) | 9 (64%) | |
| AF | 6 (60%) | 3 (75%) | 9 (64%) | |
| Hypertension | 3 (30%) | 0 | 3 (21%) | |
| Septal myoectomy | 0 | 0 | 0 | |
| Coronary artery disease | 0 | 0 | 0 | |
| Diabetes | 0 | 0 | 0 | |
| Liver fibrosis | 0 | 2 (50%) | 2 (14%) | |
| Precapillary pulmonary hypertension | 2 (20%) | 2 (75%) | 4 (29%) | |
| NT-proBNP, ng/ml | 6509.2 (1682.0) | 4444.5 (741.0) | 5873.9 (1193.2) | |
| Time from debut to HT | 6.9 (0.8) | 10 (4.2) | 7.8 (1.3) | |
| Age at debut | 31.1 (4.2) | 29.5 (8.3) | 30.6 (3.6) | |
| Age at HT | 38.0 (4.1) | 39.5 (8.0) | 38.4 (3.5) | |
Table 2.
Characteristics of the genetic variants identified in the study patients. ACMG – American College of Medical Genetics; LP – likely pathogenic; MAF – minor allele frequency; P – pathogenic; VUS – variant of uncertain significance.
Table 2.
Characteristics of the genetic variants identified in the study patients. ACMG – American College of Medical Genetics; LP – likely pathogenic; MAF – minor allele frequency; P – pathogenic; VUS – variant of uncertain significance.
| Patient | Gene | GRCh38 position, cDNA | Amino acid substitution | Variant type | Rs, MAF% | Zygosity | ACMG pathogentity | Gene panel |
| 1 | FLNC | 7:128851605, c.5823delC | p.Phe1942SerfsTer11 | Inframe deletion | - | hetero | LP | 39 |
| 2 | TRIM63 | Chr 1:26058595, c.T626C | p.Leu209Pro | missense | rs1553145730 0,00007 |
hetero | VUS | 39 |
| 3 | ALPK3 | Chr15:8486279, c.G4291A | p.Gly1431Arg | Missense | rs750258262 0,001 |
homo | LP | 172 |
| 4 | TTN | Chr2: 178552954-GACAdel: c.89943 89946del | p.Val29982CysfsTer12 | Inframe deletion | - | hetero | LP | 172 |
| Chr2: 178616815: c.G48074A | p.Ser16025Asn | Missense | rs727504720 0,00007 |
hetero | VUS | |||
| 5 | TTN | Chr2: 178748649: c.13748 13751delTTTA | p.Ile4583AsnfsTer5 | Frameshift | rs1460696675 0,002 |
hetero | LP | 172 |
| 6 | MYH7 | Chr14:23420234insC c.3337dupl |
p.Ala1113GlyfsTer19 | Splice-region+ frameshift | hetero | LP | 108 | |
| 7 | MYBPC3 | Сhr11: 47333552: c.3190+5G>A | - | Splice-region | rs587782958 0,0006 |
hetero | P | 39 |
| 8 | LAMP2 | ChrX: 20469168: c.T2C | p.Met1Thr | Start-loss | - | hetero | LP | 39 |
| 9 | LAMP2 | ChrX: 120442663: c.G865-1A | - | Splice-region | rs397516752 | hetero | P | 172 |
| 10 | LAMP2 | СhrX: 120441861: c.G962A | p.Trp321Ter | Nonsense | rs1060502306 | hetero | P | 39 |
| 11 | MYH7 | Сhr14: 23425970: c.G2156A | p.Arg719Gln | Missense | rs121913641 | hetero | P | 39 |
| Сhr14: 23433549: c.G184A | p.Glu62Lys | Missense | rs727504416 0,0005 |
hetero | VUS | |||
| 12 | MYL3 | Chr3: 46860813: c.C170A | p.Ala57Asp | Missense | rs139794067 0,01 |
homo | LP | 39 |
| GLA | ChrX: 101398932: c.A654T | p.Glu218Asp | Missense | - | hetero | VUS | ||
| 13 | MYBPC3 | Chr11: 47332189: c.C3697T | p.Gln1233Ter | Nonsense | rs397516037 0,0008 |
hetero | P | 172 |
| TPM1 | Chr15:63061778: c.A629G | p.Gln210Arg | Missense | rs777139450 | hetero | LP | ||
| 14 | TNNT2 | Chr1:201365291: c.G311GA | p.Arg104His | Missense | rs397516457 | hetero | P | 172? |
| TTN | Chr2:178613067: c.C49654G | p.Pro16552Ala | Missense | - | hetero | VUS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



