
Submitted:
21 September 2025
Posted:
29 September 2025
You are already at the latest version
Abstract
Accessory breasts denote the formation of extra breast tissue along the milk line, being more prevalent among Black and Asian populations, affecting both genders. This study aimed to determine the genetic aetiology of accessory breasts in a multiplex family, where all female siblings presented with bilateral accessory breasts. The study also ascertained secondary findings (SFs) that may impact the health of the family members. Clinical data and saliva samples were obtained from all family members. Ultrasound and histopathology confirmed the diagnosis. Whole-exome sequencing was conducted on DNA samples obtained from the saliva, with variant calling conducted utilizing Sentieon workflow. Variant classification was based on American College of Medical Genetics and Genomics guidelines. After segregation analysis, 12 candidate genes emerged. Among these, PRSS50 and FANCC emerged as top candidates, being implicated in breast diseases. However, two variants in FANCC (c.360del; p.His120GlnfsTer24 and c.355_358del; p.Ser119IlefsTer24) were selected as the most probable causal variants because of the role of this gene in familial hereditary breast and ovarian cancer syndromes. The remaining ten genes were reported as potentially accounting for co-occurring conditions segregating with accessory breasts. Reported SFs involve TTR and RYR1. In conclusion, pathogenic variants in FANCC are causal for familial accessory breasts.
Keywords:
accessory breasts
; polymastia
; polythelia
; multiplex family
; whole exome sequencing
; FANCC gene
; secondary findings
; co-occurring conditions
1. Introduction
Accessory breast tissue is a rare anomaly characterized by extra breast tissue that persists after typical embryonic growth. Occurring in about 2% to 6% of the global human population, it is more prevalent in Black and Asian ancestries, with the Japanese being the most affected. Although it affects both sexes, males experience it the least [1,2]. Anomalous breast tissue refers to both supernumerary breasts and abnormal breast tissue [3,4]. Supernumerary breasts can be found along the milk line, which runs bilaterally and symmetrically from the front axillary lobes to the groin area and the inner portion of the thigh [5]. The armpit is the primary site where extra breast tissue is often detected, although it can also be observed in other body parts, such as the face, back of the neck, and the back and sides of the thigh [6].
Two sets of ectoderm-derived mammary ridges appear during the fourth week of human development [7]. These ridges, often called the “milk line,” extend bilaterally from the front axillary folds to the inguinal folds on the ventral side of the human body. Typically, these ridges disappear, except for the areas where the breasts form [7]. Axillary breast tissue contains most of the components found in normal breast tissue, including the parenchyma, areola, and nipple [8]. Kajava developed a classification system for additional breast tissue in 1915 [2,4]. Class 1, or polymastia, refers to a complete breast with glandular tissue, an areola, and a nipple. A supernumerary breast with only glandular tissue, a nipple, and no areola is classified as Class 2. Class 3 includes an areola with glandular tissue present. Class 4 supernumerary breasts exhibit only glandular tissue. A “pseudomamma,” which comprises only an areola and a nipple, is classified as Class 5. Class 6 is known as polythelia and consists entirely of a nipple, whereas Class 7, termed polythelia areolaris, consists solely of an areola. Class 8 contains only one hair patch, called “polythelial pilosis” [2]. The most common phenotype of accessory breast tissue observed is Class 4 fibroglandular tissue, which often occurs in the axilla.
Most instances of accessory breasts are sporadic, despite the potential for an autosomal dominant inheritance pattern with incomplete penetrance [6]. A person with the condition may not exhibit any symptoms and may be unaware of its development, regardless of where the extra breast tissue is located. Furthermore, accessory breast tissue can be noted during menstruation, pregnancy, or breastfeeding, as it responds to hormonal stimuli in the same manner as normal breast tissue [8].
The identification of supplementary breast tissue has significant treatment implications for each patient. If additional breast tissue is not recognised for what it is, a common variation could be misidentified as another medical defect. Adipose tissue tumours, lymphadenopathy, cutaneous cysts, arterial and venous abnormalities, and cancer are among the most frequently reported probable diagnoses in scientific investigations [9]. According to Goyal et al. (2008), several pathological conditions in normal breast tissue can also arise in accessory breast tissue. These conditions include benign processes such as fibrocystic changes, fibroepithelial lesions, mastitis, atypical ductal or lobular hyperplasia, and true malignancy [8].
The current study sought to identify the genetic basis of accessory breasts in a multiplex family from Ghana, utilising whole exome sequencing (WES). To the best of our knowledge, this study pioneers genetic studies on accessory breasts among the Ghanaian population.
2. Results
2.1. Clinical Characteristics of the Accessory Breast Phenotype in the Multiplex Family
Bilateral accessory breasts were observed in all three female siblings but not in their male siblings or the father (Figure 1A). However, the mother was deceased at the time of recruitment and was unavailable for sample collection and family history. The oldest sibling (27 years), Parity 1, noticed bilateral accessory breast at age of 13 in both axillae (2.5 cm in the right and 3 cm in the left) since age 13. During pregnancy, the masses increase in size; they decrease in size after lactation.
The second sibling (25 years) with Gravida and Parity 0 became aware of her bilateral accessory breast at age 12. The mass of the left accessory breast measured approximately 2 cm, while the right measured about 2.5 cm. The masses were occasionally painful, usually during her menstrual cycle, but subsided after. She noticed an enlargement in the accessory breasts at the age of 25. The mass measured roughly 4 × 4 cm in the left and right axillae.
The Proband (23 years) also had bilateral accessory breasts in the axilla. The mass was discovered at 14 years, measuring just 2 cm in the right axilla and 3 cm in the left. Upon physical examination at age 23, the mass was non-tender and measured about 4 cm in both axillae. Each mass was not attached to the overlying skin, showed no hyperpigmentation or palpable axillary lymph nodes. Routine laboratory analysis of the proband was normal. Ultrasound of the breast suggested bilateral accessory breast tissue (Figure 1B,C). The proband had excision biopsy of both accessory breasts under general anaesthesia (Figure 1D–F) for histopathological examination. The two tissue samples were submitted for histopathological analysis fixed in 10% buffered formalin. The larger nodule weighed 146.0 g and measured 10 × 7.0 × 5.0 cm. The smaller weighed 94.0 g and measured 8.0 × 6.0 × 0.4 cm. The cut surfaces of the tissues showed mostly yellow fat with focal areas of grey fibrous tissue, and the skin surfaces appearing normal. The microscopic anatomy of both tissue samples revealed breast lobules and ductal structures. Observation of fibrous tissue and biopsy showed no atypia. Sections of the nodule exhibited a thin, well-defined mass encased in a delicate fibrous capsule. The lesional area contained both epithelial and stromal components. The stroma comprised bland spindle cells, whereas the epithelial ducts showed no atypia. The overall morphology was characteristic of a fibroadenoma in accessory breast tissue.
2.2. Detection of Etiologic Variants for Accessory Breast Phenotype
The paired-end reads generated at a read depth of 100X had an average mean quality score of 35.85, suggesting a base calling accuracy of over 99%. More than 93% of the reads had a mean quality score above 30 (Supplementary Table S2). More than 99% of the unique reads aligned to the GRCh38 human reference genome in each sample. Furthermore, over 97% of the exons were sequenced with a coverage read depth of at least 20 reads, indicating uniformity and completeness of the coverage (Supplementary Table S3). About 40.4% of the variants were synonymous variants, while missense variants accounted for 35.6%. The remaining variants (frameshift, intron, upstream, downstream, splice region, etc.) accounted for the remaining 24%.
The ACMG guidelines [14] on variant classifications were used to prioritise the probable aetiological variant for the accessory breast phenotype in the family. The annotated rare variants with a MAF ≤ 0.01 were checked for pathogenicity using 11 variant effect predictors embedded in dbNSFP [18]. For missense variants, pathogenicity was established if at least six predictors indicated so. The tools employed included ClinPred, MetaRNN, BaysDel_addAF, REVEL, CADD, AlphaMissense, MutPred2, Polyphen-2, MutationAssessor, MutationTaster, and SIFT. Furthermore, SpliceAI was utilised to determine pathogenicity for splice site variants. CADD and REVEL scores were used to assess loss-of-function variants. These analyses revealed 129 probable pathogenic variants in the proband, comprising 70 missense variants and 59 loss-of-function (LoF) variants, with 9 being novel variants. Segregation analyses were subsequently conducted to ascertain variant(s) that segregated with the accessory breast phenotype in the family.
After analyses using the ACMG guidelines, variants in 12 genes were selected. However, variants in two candidate genes, PRSS50 and FANCC (Table 1), were prioritized as top candidates for the accessory breast phenotype in the family because these two genes have previously been implicated in breast diseases such as breast cancer. FANCC is a DNA repair protein that participates in post-replication repair and has a checkpoint function in the cell cycle. This gene has been associated with hereditary breast and ovarian diseases [28], making it the most probable candidate for the accessory breast phenotype in the family. Figure 2 depicts the bulk tissue expression patterns of FANCC and PRSS50 genes and their interactions with other genes. Both genes are expressed in the breasts. However, interactome analysis demonstrated that FANCC interacts with essential genes involved in inherited breast cancer syndromes such as BRCA1. These evidences support the FANCC variants as the most probable causative genetic factor for the familial accessory breast phenotype in the study family. As a quality control step, the FANCC variant from the WES analysis was confirmed using Sanger sequencing. The chromatograms from the Sanger sequencing are presented in Figure 3. This sequencing was carried out using both reverse and forward primer sets.
2.3. Rare Genetic Variants for Co-Occurring Conditions in Individuals with Polymastia
Aside from PRSS50 and FANCC, numerous other aetiologic variants in 10 genes were found to segregate with the accessory breast phenotype in the family, even though these genes have not been previously linked to breast diseases. These other rare genetic variants could potentially explain co-occurring conditions in individuals with accessory breasts. These genes included SLC7A7, NDE1, DIP2B, ADGRG6, CHDH, OR2W1, ACKR2, C3orf62, OR4Q3, and MYO1H (Table 2). The pathogenicity of these variants was determined using the ACMG guidelines for variant classification mentioned earlier.
2.4. ACMG Secondary Finding Genes
The WES dataset was evaluated for secondary findings based on the ACMG list for reporting secondary findings in clinical exome and genome sequencing [29]. After variant prioritisation, no variant in the listed ACMG genes segregated perfectly with the accessory breast phenotype in the family. However, a heterozygous missense variant in the RYR1 gene was identified in the proband and the oldest affected female sibling (Table 3). The RYR1 variant is classified as likely benign or a variant of uncertain significance (VUS) in ClinVar. The oldest affected female sibling also had a heterozygous missense variant in the TTR gene (Table 3). The TTR variant observed in the current study is classified as pathogenic in ClinVar. According to the ACMG guidelines on secondary findings, RYR1 and TTR are associated with malignant hyperthermia and hereditary TTR amyloidosis, respectively. Both conditions are inherited in an autosomal dominant manner, as observed in those harbouring the variants [29]. Interestingly, no aetiologic variant in ACMG secondary findings genes was observed in the other affected female sibling with accessory breast, nor any of the male participants in the family.
3. Discussion
3.1. Clinical Presentations and Diagnosis of the Accessory Breasts
Accessory breast disease is an uncommon condition which is usually misdiagnosed as a result of its similarity to other medical conditions. Clinical recognition of this condition is necessary to ensure correct diagnosis and operative management. The genetic aetiology of accessory breasts remains largely undiscovered. This study aimed to determine the genetic aetiology of accessory breasts in a multiplex Ghanaian family of six individuals.
Accessory breast tissue responds to hormonal stimuli like normal breast tissue. It is primarily observed during menarche, pregnancy, or lactation. However, supplementary mammary tissue may not be evident in some individuals until pregnancy [7]. The oldest female sibling in the affected family reported that the supplementary accessory breasts were relatively small when they were first discovered. She noted that the masses increased in size during pregnancy and reduced in size after pregnancy. Hormones such as oestrogen, progesterone, and prolactin influence the growth of both supplementary and regular mammary tissue [30]. During pregnancy, increased oestrogen and progesterone levels produce anatomical and physiological changes in the breast tissue. These hormonal changes add to adipose tissue proliferation and ductal elongation. Post-pregnancy, oxytocin and prolactin levels increase, while progesterone levels reduce, resulting in a reduction in breast size, as noticed in the eldest affected sibling [31,32].
The second affected sibling became aware of her bilateral axillary breasts at the age of twelve. She reported that the masses were sometimes painful, particularly during her menstrual cycle, but resolved after menstruation. This condition, termed cyclical mastalgia, is also observed in typical breasts and is a common symptom among women. Previous studies have defined it as breast pain at the onset of menstruation [33]. The pain is sporadic and recurrent, corresponding in timing with the menstrual cycle. It may occur unilaterally or bilaterally and is associated with tenderness, heaviness, and swelling. The condition affects up to 70% to 80% of women during their lifetime [34]. A study [35] attributed breast pain during the luteal phase of menstruation to elevated serum oestrogen levels, lowered progesterone levels and hormonal imbalance. Additionally, sex hormones-binding protein (SHBP) has been connected to water retention in breast tissue, adding to breast pain [34]
The youngest affected sibling (proband) exhibited bilateral accessory breasts in the axilla, in line with studies on polymastia [2]. Clinical presentation varies, and mammography or ultrasound is recommended [36]. The current study made an initial ultrasound diagnosis and confirmed it with histopathology. Histopathological examination of excised tissue revealed predominantly yellow fat with focal fibrous tissue in line with normal breast composition [37]. Microscopic analysis recognised breast lobules, ductal structures, and fibrous tissue, with stromal engagement. Kajava (1915) classified accessory breast tissue into eight categories, with axillary fibroglandular tissues falling under class 4[2]. The histopathological observations in the proband align with those in the literature, revealing glandular epithelium, connective tissue stroma, and subcutaneous fat [38].
3.2. Genetic Aetiology of the Accessory Breast Phenotype in the Multiplex Family
Pathogenic variants in the PRSS50 and FANCC genes have been implicated in breast disease. Variants in PRSS50, encoding a proteolytic enzyme, have been observed in breast cancer [39]. It holds back the activin signalling, influencing cell regulation and proliferation by decreasing the expression of p27, also called CDKN1B [40]. It also activates NF-kappa B (NF-κB), a transcription factor in immune responses and tumorigenesis [41]. Overexpression of PRSS50 is associated with breast cancer invasion and malignancy [40], but it is not a top candidate for accessory breast aetiology [42]. FANCC is involved in DNA repair, cell cycle checkpoint regulation, and chromosomal stability. It also suppresses cytokine-induced apoptosis via STAT1 activation in response to cytokines and growth factors [43]. FANCC is regulated by TP53, which binds to its promoter to stimulate transcription [44]. Murine models of the Fancc gene display phenotypes such as hypersensitivity to DNA crosslinking agents, spontaneous chromosomal breakage, and loss of germ cells, which results in reduced fertility [45]. FANCC is involved in several pathways, including the Fanconi anaemia pathway, cytokine signalling, protein kinase R (PKR)-mediated signalling, and transcription regulation by TP53 [46]. Variants in FANCC are linked to hereditary breast and ovarian cancer syndromes, making it a key candidate gene for accessory breast tissue in the family studied. The current study discovered two pathogenic frameshift deletions: c.360del (p.His120GlnfsTer24, CADD score = 24.6) and c.355_358del (p.Ser119IlefsTer24, CADD score = 27.7), Table 1. The two variants segregated perfectly with the accessory breast phenotype within the family, further supporting the pathogenicity of these variants. These variants are classified as pathogenic in ClinVar and are associated with hereditary neoplastic syndromes, increasing susceptibility to benign and/or malignant neoplasms.
A previous study on actionable mutations in various non-BRCA cancer-associated genes found deletions in similar regions of the FANCC gene (c.355_360delTCTCATinsA) in Black/African American women with a familial history of breast cancer [47]. Pathogenic and likely pathogenic variants in FANCC were also reported in individuals with breast and ovarian cancer [28]. A study [48] in an Australian population examined the familial predisposition to breast and ovarian cancer in multiple multi-generational breast cancer families without BRCA1/2 mutations. Two protein-truncating variants in FANCC (c.535C>T, [p.Arg179*] and c.553C>T, [p.Arg185*]) were found in 15 BRCA1/2-negative families at high risk of breast cancer. This suggests a predisposing role for FANCC variants in breast cancer [48]. A similar study conducted in a Chinese cohort with familial breast and/or ovarian cancer that lacked BRCA1/2 mutations observed a deleterious variant (c.339G>A, W113X) in FANCC [49]. However, in a large-scale case-control study in a European population genotyped two truncating variants (p.R185X and p.R548X) of FANCC using microarray, as both variants are known to cause disease in the population. Although the variants were detected in approximately 50 individuals, there was no evidence of an association between these variants and the risk of breast cancer compared to BRCA1 and BRCA2 mutations [50].
3.3. Other Likely Pathogenic Variants Segregating with the Accessory Breasts
Accessory breasts can be observed as part of pleiotropic syndromes or as individual traits. Studies have reported co-occurring conditions in affected individuals. Based on this, we hypothesise that the variants in the 10 other genes could contribute to conditions that co-occur with accessory breast, but not necessarily the accessory breast phenotype in the family (Table 2). Six missense variants (occurring in SLC7A7, NDE1, DIP2B, ADGRG6, OR2W1, and CHDH), one stop-loss variant (in MYO1H), and three frameshift variants (observed in ACKR2, C3orf62, and OR4Q3) segregated perfectly with the accessory breast phenotype in all affected individuals. NDE1 is essential for brain function due to its role in cerebral cortex development [51]. Variants of NDE1 have been associated with neurodevelopmental disorders, particularly microcephaly-related diseases of cortical development [52]. Similarly, DIP2B plays a crucial role in neuronal cell growth, with variants in this gene linked to neurodevelopmental diseases [53]. ADGRG5 is a receptor protein vital for tissue and organ function, including the heart, ear, and sciatic nerve [52]. Variants of this gene have been associated with skeletal defects, intellectual disabilities, and lung disorders [54]. ACKR2 encodes a chemokine receptor, thus interacting with various inflammatory chemokines in the human body. Variants of this gene have been associated with multiple developmental disorders [55]. The protein encoded by MYO1H is pivotal to cellular defence mechanisms such as phagocytosis [56]. Variants of this gene have been linked to central hypoventilation syndromes [57]. C3orf62 is crucial for spermatogenesis and male fertility, although a recent study has associated a mutation in this gene with myofibroma [58]. OR4Q3 and OR2W1 encode olfactory receptors responsible for the sense of smell in humans. Although diseases associated with these genes have been reported less frequently, a recent study linked mutations in the OR4Q3 gene to glioblastoma [59]. Additionally, renal-urological and cardiovascular malformations can co-occur with accessory breast tissue [2]. The current study identified a variant in CHDH, a mitochondrial enzyme involved in choline metabolism [60]. Variants within this gene have been linked to metabolic disorders and, in severe cases, can lead to liver damage, as well as tumour prognosis [61]. Variants in SLC7A7 are associated with lysinuric protein intolerance. Patients with this condition display various clinical manifestations affecting multiple organs. Severe complications include growth retardation, along with lung and renal malformations [62].
3.4. Clinically Actionable Secondary Findings Observed in Family Members
In addition to ascertaining the primary genetic cause of accessory breasts, the current study also aimed to decipher gene variants linked to secondary findings according to ACMG guidelines [30]. A pathogenic missense variant (c.9355C>T, [p.Arg3119Cys]) in RYR1 was identified in the proband and the eldest affected female sibling. RYR1 encodes a ryanodine receptor in skeletal muscle and is associated with autosomal dominant malignant hyperthermia [30]. This condition is a musculoskeletal disorder that triggers a potentially fatal hypermetabolic response to stressors or anaesthetic agents. Studies have noted that the malignant hyperthermia phenotype varies significantly depending on the RYR1 variants. More severe phenotypes are observed in variants occurring at relatively conserved sites in the protein [63]. The current study also observed a pathogenic variant in TTR in the oldest female sibling affected by accessory breasts. Variants in TTR have been associated with autosomal dominant hereditary TTR amyloidosis [30]. A phenotype associated with the heart results from the accumulation of misfolded TTR protein in the organ, thereby impacting its function [64,65]. The TTR variant observed in the current study has been classified in ClinVar as pathogenic and is associated with congenital heart malformations.
3.5. Limitations of the Study
A limitation of this study is that the observation is limited to a single multiplex family, and more families are required to generalise these observations. The difficulty in distinguishing accessory breasts from other breast malformations may account for the underreporting of the condition, which explains the small sample size. Furthermore, additional functional genomics data on the implicated variants are needed to validate the role of the FANCC gene in the aetiology of accessory breasts in the affected multiplex family. However, in the current study, several strategies, including gene expression and interactome studies, were employed to decipher the role of FANCC in tissues associated with accessory breasts.
4. Materials and Methods
4.1. Study Population
The multiplex family is of Ghanaian descent and was recruited from the Kwame Nkrumah University of Science and Technology (KNUST) Hospital. The affected females presented with bilateral accessory breasts. The Institutional Review Board (IRB) at KNUST granted ethical approval for this cross-sectional study, with approval number CHRPE/AP/120/22. Written informed consent was obtained before subject recruitment, and the family was interviewed using a questionnaire.
The study recruited a family of six individuals: three males and three females. All three female siblings displayed phenotypes of bilateral accessory breasts. The mother was deceased at the time of subject recruitment and could not participate in the study. Thus, only a single multiplex family was recruited. The proband was the first to report to the study site and was clinically evaluated by qualified General Surgeons. Histopathological analysis was conducted on excision biopsies obtained from the bilateral accessory breasts of the proband. The proband then informed the study team of the two other siblings with the same condition, who were also subsequently recruited into the study.
4.2. Sample Collection, DNA Processing and Whole Exome Sequencing
Saliva samples were collected using the Oragene DNA Saliva Collection Kit (DNA Genotek, Canada). DNA processing was conducted at the Human Genetics and Genomics (HuGENE) Laboratory at KNUST. The workflow included DNA extraction and purification, quantification, and quality control checks such as XY genotyping for sex confirmation (Supplementary Figure S1) [10,11].
In summary, DNA extraction and purification from saliva samples followed the Oragene Saliva Protocol [10]. DNA quantification was performed using the Invitrogen QubitTM dsDNA BR Assay kit with the Qubit 4.0 Fluorometer (ThermoFisher Scientific, USA). A Nanodrop spectrophotometer was utilised to ascertain the DNA purity by detecting potential contaminants like protein, RNA, or phenol by determining 260/280 and 260/230 absorbance ratios. XY genotyping was carried out as a quality control step to confirm the sexes of study participants [10,11].
Genomic DNA samples were sent to Azenta Life Sciences (USA) for WES at a read depth of 100X (Supplementary Figure S2). In summary, DNA was re-quantified using the Qubit 4 Fluorometer, and the Twist Human Comprehensive Exome library was prepared per manufacturer guidelines. Fragmentation was performed using a Covaris S220, followed by end-repair, adenylation, and adapter ligation. Adapter-ligated fragments underwent PCR amplification, validation via Agilent Tapestation, and hybridization with biotinylated baits. Streptavidin-coated beads captured the hybrid DNA, which was cleaned, amplified, and indexed using Illumina primers.
Sequencing libraries were clustered onto multiple lanes of a flow cell and sequenced using the Illumina HiSeq with a 2x150 bp paired-end configuration. The HiSeq Control Software (HCS) handled image analysis and base calling, generating binary base call (BCL) files. These were converted to FastQ format and de-multiplexed using Illumina bcf2fastq v2.19, allowing one mismatch in index identification.
4.3. Bioinformatic Analysis of Whole Exome Sequencing Dataset
The bioinformatics analysis of the WES dataset included quality control checks with FastQC and Trimmomatic, variant calling with Sentieon pipeline (Supplementary Figure S3) and variant prioritisation workflows (Supplementary Figure S4)
The FastQ files obtained from the Illumina sequencing platform were utilised for bioinformatics analysis (Supplementary Figure S3). The raw reads underwent quality checks using FastQC 0.11.9 and were trimmed for low-quality bases and adapter sequences using Trimmomatic 0.39 [12]. The reads were aligned to the GRCh38 reference genome using BWA-MEM of Sentieon 202112.01[13]. After alignment, PCR and optical duplicates were identified, and BAM files were generated. Variant calling, including SNVs and indels, was conducted using Sentieon 202112.01 DNAscope. VCF files were normalised through left alignment of indels and splitting multiallelic sites into multiple sites using bcftools 1.13. Transcripts that overlapped were determined for every variant, and the effects of the variants on the transcripts were predicted utilising Ensembl Variant Effect Predictor (VEP) v104. The most severe consequence or impact was selected for every variant for downstream cohort analysis.
Variant prioritisation followed the ACMG guidelines [14]. Variants with an allele frequency 1%, and above were filtered out using databases such as the 1000 Genomes Project [15], Exome Variant Server, dbSNP and gnomAD [16,17]. Variants below this threshold were assessed for pathogenicity using 11 tools embedded in dbNSFP [18], including SIFT, Polyphen 2, Mutation Taster, Mutation Assessor, MetaRNN, REVEL, MutPred, BaysDel_addAF, ClinPred, CADD, and AlphaMissense (Supplementary Table S1). For missense variants, they were classified as pathogenic if at least six of these tools identified them as such. Splice AI evaluated splice variants. CADD was again used to assess loss-of-function variants. The functional impact of variants was further analysed using web-based bioinformatics tools to assess the effects of amino acid changes on protein function, along with expression and phenotypic data from animal models. The Genecard suite, including VarElect and Malacards, was used for phenotype- and expression-based variant prioritization [19]. Segregation analysis determined whether variants co-segregated with the phenotype in affected family members. Finally, pathogenicity was further validated using ClinVar, Online Mendelian Inheritance in Man (OMIM), the Alliance for Genomic Resources, and Human Genome Mutation Database (HGMD) [20,21,22].
4.4. Sanger Sequencing to Confirm Pathogenic Variants
Sanger Sequencing was carried out to confirm the variants from the WES data obtained from the Next-Generation Sequencing (NGS) platform. The procedure for Sanger sequencing has been published by our group [10]. In summary, one primer set was designed to cover the exonic region harbouring the c.360del and c.355_358del variants of the FANCC gene. The Primer3 software (https://primer3.ut.ee/) was used to design the primer set to amplify both variants by adding 500 bp upstream and downstream of the region harbouring the variants. The In Silico PCR platform from the UCSC genome browser was used to determine whether the primer sets annealed to the targeted genomic region. The forward and reverse primers used were 5-TGGCACATTCAGCATTAAACAT-3 and 5-TTGTTTCATAGAGACCACCCC-3, respectively, with an amplicon size of 271 base pairs. A total of 4 ng/μl of DNA in a 10 μl reaction volume was used for polymerase chain reaction (PCR). The PCR conditions have been published [10].
The PCR products were run on a 2% agarose gel, along with size markers at 100 A and 200 V for 20 minutes to confirm the success of the PCR run. Ultraviolet (UV) light was used to view the gel electrophoretic product. Successfully amplified PCR products were sequenced using an ABI 3730XL DNA sequencer based on Sanger sequencing technology at Functional Biosciences, Madison, Wisconsin (https://functionalbio.com/).
Chromatograms from the Sanger sequencing platform were transferred to a Unix workstation, PHRED (http://www.phrap.org/phredphrapconsed.html, v.0.961028) utilised for base calling, PHRAP (http://www.phrap.org/, v.0.960731) employed to assemble bases, POLYPHRED (http://droog.gs.washington.edu/polyphred/, v. 0.970312) subsequently used to scan bases, and CONSED (http://www.phrap.org/consed/consed.html, v. 4.0) used for viewing reads. Reads were subsequently aligned to the GRCh38 human reference genome using the “Blat” function of UCSC Genome Browser (https://genome.ucsc.edu/).
4.5. Gene Expression and Interactome Analyses
The expression profiles of the two top candidate genes from bioinformatics analyses of WES datasets, FANCC and PRSS50, were ascertained using bulk tissues gene expression data in GTEX (https://www.gtexportal.org/home/gene/FANCC, 2024). The interactions of these two genes with other genes were deciphered employing STRING (https://string-db.org/cgi/network?taskId=bTGzi0bXr9XL&sessionId=bUkeU0Kv11w6, 2024).
5. Conclusions
Although accessory breast is a rare anomaly worldwide, this study recruited a multiplex family of six, with all three females exhibiting a bilateral accessory breast phenotype. Among the 12 candidate genes that perfectly segregated with the phenotype in all three affected females, the FANCC gene, a DNA repair protein, has been implicated in hereditary breast and/or ovarian disease, making it a top candidate gene underlying the aetiology of the accessory breast phenotype in the family. Pathogenic variants in other genes, such as SLC7A7 and CHDH, may predispose affected individuals to co-occurring conditions. Following ACMG guidelines on secondary findings, heterozygous variants in the RYR1 gene could potentially predispose the two females with the accessory breast phenotype to autosomal dominant malignant hyperthermia. A variant of the TTR gene may also predispose the eldest affected sibling to autosomal dominant hereditary TTR amyloidosis.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Figure S1: DNA processing workflow prior to sequencing; Figure S2: A detailed workflow of whole exome sequencing; Figure S3: The variant calling workflow depicting various processes involved in generating the VCF files; Figure S4: A detailed workflow of the variant prioritization process; Table S1: Predictive threshold of variant effect prediction tools; Table S2: Sample sequencing statistics of the multiplex family; Table S3: Summary statistics of alignment.
Author Contributions
Conceptualization, L.J.J.G. and S.M.; methodology, L.J.J.G., A.S.D., C.O.A., G.O.M., B.T., T.D.B., S.M. and A.B.; software, L.J.J.G., C.O.A., G.O.M. and B.T.; validation, A.S.D., C.O.A., G.O.M., B.T., T.D.B., F.K.N.A., I.K., L.K.B., S.M., A.A.A., A.B., P.D. and L.J.J.G.; formal analysis, A.S.D., C.O.A., G.O.M., B.T., T.D.B., S.M., A.B. and L.J.J.G.; investigation, A.S.D., S.M., A.B. and L.J.J.G.; resources, A.B. and L.J.J.G; data curation, L.J.J.G., A.S.D., C.O.A., G.O.M., B.T., T.D.B., S.M. and A.B.; writing—original draft preparation, A.S.D and L.J.J.G; writing—review and editing, A.S.D., C.O.A., G.O.M., B.T., T.D.B., F.K.N.A., I.K., L.K.B., S.M., A.A.A., A.B., P.D. and L.J.J.G.; visualization, A.S.D., C.O.A., G.O.M., B.T., T.D.B., F.K.N.A., I.K., L.K.B., S.M., A.A.A., A.B., P.D. and L.J.J.G.; supervision, S.M. and L.J.J.G; project administration, L.J.J.G; funding acquisition, L.J.J.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding, and the APC was funded by LJJG.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Institutional Review Board (or Ethics Committee) of KWAME NKRUMAH UNIVERSITY OF SCIENCE AND TECHNOLOGY, KUMASI, GHANA (protocol code CHRPE/AP/120/22 and 5th April 2022).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to privacy and ethical restrictions on the sharing of DNA sequence datasets.
Acknowledgments
We are grateful to the members of the multiplex family who consented and voluntarily participated in the study.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
ACMG American College of Medical Genetics and Genomics
BCL Binary base call
PKR Protein Kinase R
SHBP Sex hormone binding protein
SNVs Single nucleotide variations
WES Whole exome sequencing
TRH Thyrotropin releasing hormone
References
- Laor, T. , Collins, M.H., Emery, K.H., Donnelly, L.F., Bove, K.E. and Ballard, E.T. ‘MRI appearance of accessory breast tissue: A diagnostic consideration for an axillary mass in a peripubertal or pubertal girl. American Journal of Roentgenology 2004, 183, 1779–1781. [Google Scholar] [PubMed]
- Bellahsene-Bendib, S. , Achir, Y., Aiche, D. and Aimeur, C. ‘Polymastia: What you Should Know. From One Man Case Discovered by Chance. Clinics in Nursing 2023, 2. [Google Scholar]
- Rho, J.Y. , Juhng, S.K. and Yoon, K.J. ‘Carcinoma originating from aberrant breast tissue of the right upper anterior chest wall: a case report. Journal of Korean Medical Science 2001, 16, 519. [Google Scholar] [PubMed]
- Bakker, J.R. , Sataloff, D.M. and Haupt, H.M. ‘Breast cancer presenting in aberrant axillary breast tissue. Community Oncology 2005, 2, 117–120. [Google Scholar] [CrossRef]
- Hong, J.H. , Oh, M.J., Hur, J.Y. and Lee, J.K. ‘Accessory breast tissue presenting as a vulvar mass in an adolescent girl. Archives of Gynecology and Obstetrics 2009, 280, 317–320. [Google Scholar] [CrossRef]
- Youn, H.J. and Jung, S.H. ‘Accessory Breast Carcinoma. Breast Care 2009, 4, 104. [Google Scholar]
- Schoenwolf, G.C. , Bleyl, S.B., Brauer, P.R. and Francis-West, P.H. ‘Larsen’s Human Embriology. Larsen’s Human Embryology 2014, 5, 155–171. [Google Scholar]
- Goyal, S. , Puri, T., Gupta, R., Julka, P. and Rath, G. ‘Accessory breast tissue in axilla masquerading as breast cancer recurrence. Journal of Cancer Research and Therapeutics 2008, 4, 95–96. [Google Scholar]
- Sahu, S.K. , Husain, M. and Sachan, P.K. ‘Bilateral Accessory Breast. The Internet Journal of Surgery 2007, 17. [Google Scholar]
- Gowans, L.J.J. , Oseni, G., Mossey, P.A., Adeyemo, W.L., Eshete, M.A., Busch, T.D., Donkor, P., Obiri-Yeboah, S., Plange-Rhule, G., Oti, A.A., Owais, A., Olaitan, P.B., Aregbesola, B.S., Oginni, F.O., Bello, S.A., Audu, R., Onwuamah, C., Agbenorku, P., Ogunlewe, M.O., Abdur-Rahman, L.O., Marazita, M.L., Adeyemo, A.A., Murray, J.C. and Butali, A. ‘Novel GREM1 variations in Sub-Saharan African patients with cleft lip and/or cleft palate. Cleft Palate-Craniofacial Journal 2018, 55, 736–742. [Google Scholar]
- Alade, A. , Peter, T., Busch, T., Awotoye, W., Anand, D., Abimbola, O., Aladenika, E., Olujitan, M., Rysavy, O., Nguyen, P.F., Naicker, T., Mossey, P.A., Gowans, L.J.J., Eshete, M.A., Adeyemo, W.L., Zeng, E., Van Otterloo, E., O’Rorke, M., Adeyemo, A., Murray, J.C., Lachke, S.A., Romitti, P.A. and Butali, A. ‘Shared genetic risk between major orofacial cleft phenotypes in an African population. Genetic Epidemiology 2024, 48, 258–269. [Google Scholar] [PubMed]
- Bolger, A.M. , Lohse, M. and Usadel, B. ‘Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Feng, X. , Ping, J., Gao, S., Han, D., Song, W., Li, X., Tao, Y. and Wang, L. ‘Novel JAG1 variants leading to Alagille syndrome in two Chinese cases. Scientific Reports 2024, 14, 1812. [Google Scholar]
- Richards, S. , Aziz, N., Bale, S., Bick, D., Das, S., Gastier-Foster, J., Grody, W.W., Hegde, M., Lyon, E., Spector, E., Voelkerding, K. and Rehm, H.L. ‘Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine 2015, 17, 405–424. [Google Scholar]
- Johnston, J.J. and Biesecker, L.G. ‘Databases of genomic variation and phenotypes: existing resources and future needs. Human Molecular Genetics 2013, 22, R27. [Google Scholar] [CrossRef]
- Fairley, S. , Lowy-Gallego, E., Perry, E. and Flicek, P. ‘The International Genome Sample Resource (IGSR) collection of open human genomic variation resources. Nucleic Acids Research 2020, 48, D941–D947. [Google Scholar] [CrossRef]
- Gudmundsson, S. , Singer-Berk, M., Watts, N.A., Phu, W., Goodrich, J.K., Solomonson, M., Rehm, H.L., MacArthur, D.G. and O’Donnell-Luria, A. ‘Variant interpretation using population databases: Lessons from gnomAD. Human Mutation. 2022, 43, 1012–1030. [Google Scholar] [CrossRef]
- Liu, X. , Li, C., Mou, C., Dong, Y. and Tu, Y. ‘dbNSFP v4: a comprehensive database of transcript-specific functional predictions and annotations for human nonsynonymous and splice-site SNVs. Genome Medicine 2020, 12, 103. [Google Scholar] [CrossRef]
- Stelzer, G. , Plaschkes, I., Oz-Levi, D., Alkelai, A., Olender, T., Zimmerman, S., Twik, M., Belinky, F., Fishilevich, S., Nudel, R., Guan-Golan, Y., Warshawsky, D., Dahary, D., Kohn, A., Mazor, Y., Kaplan, S., Iny Stein, T., Baris, H.N., Rappaport, N., Safran, M. and Lancet, D. ‘VarElect: The phenotype-based variation prioritizer of the GeneCards Suite. BMC Genomics 2016, 17, 444. [Google Scholar]
- Stenson, P.D. , Ball, E. V., Mort, M., Phillips, A.D., Shaw, K. and Cooper, D.N. (2012) ‘The Human Gene Mutation Database (HGMD) and its exploitation in the fields of personalized genomics and molecular evolution. Current protocols in bioinformatics.
- Amberger, J.S. , Bocchini, C.A., Scott, A.F. and Hamosh, A. ‘OMIM.org: leveraging knowledge across phenotype-gene relationships. Nucleic Acids Research 2019, 47, D1038–D1043. [Google Scholar] [CrossRef]
- Landrum, M.J. , Chitipiralla, S., Kaur, K., Brown, G., Chen, C., Hart, J., Hoffman, D., Jang, W., Liu, C., Maddipatla, Z., Maiti, R., Mitchell, J., Rezaie, T., Riley, G., Song, G., Yang, J., Ziyabari, L., Russette, A. and Kattman, B.L. ‘ClinVar: updates to support classifications of both germline and somatic variants. Nucleic acids research 2025, 53, D1313–D1321. [Google Scholar] [PubMed]
- Kendig, K.I. , Baheti, S., Bockol, M.A., Drucker, T.M., Hart, S.N., Heldenbrand, J.R., Hernaez, M., Hudson, M.E., Kalmbach, M.T., Klee, E.W. et al.. ‘Sentieon DNASeq Variant Calling Workflow Demonstrates Strong Computational Performance and Accuracy. Frontiers in Genetics 2019, 10, 736. [Google Scholar] [CrossRef] [PubMed]
- Mahmoud, M. , Huang, Y., Garimella, K., Audano, P. A., Wan, W., Prasad, N., Handsaker, R. E., Hall, S., Pionzio, A., Schatz, M. C., Talkowski, M. E., Eichler, E. E., Levy, S. E., and Sedlazeck, F. J. Utility of long-read sequencing for All of Us. Nature Communications 2024, 15, 837. [Google Scholar] [CrossRef] [PubMed]
- Wingett, S. W. , and Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Rappaport, N. , Twik, M., Plaschkes, I., Nudel, R., Stein, T. I., Levitt, J., Gershoni, M., Morrey, C. P., Safran, M., and Lancet, D. MalaCards: an amalgamated human disease compendium with diverse clinical and genetic annotation and structured search. Nucleic Acids Research 2017, 45, D877–D887. [Google Scholar] [CrossRef]
- Baldarelli, R.M. , Smith, C.L., Ringwald, M., Richardson, J.E., Bult, C.J., Anagnostopoulos, A., Begley, D.A., Bello, S.M., Christie, K., Finger, J.H., Hale, P., Hayamizu, T.F., Hill, D.P., et al. ‘Mouse Genome Informatics: an integrated knowledgebase system for the laboratory mouse. Genetics 2024, 227, iyae031. [Google Scholar]
- Susswein, L.R. , Marshall, M.L., Nusbaum, R., Vogel Postula, K.J., Weissman, S.M., Yackowski, L., Vaccari, E.M., Bissonnette, J., Booker, J.K., Cremona, M.L., Gibellini, F., Murphy, P.D., Pineda-Alvarez, D.E., Pollevick, G.D., Xu, Z., Richard, G., Bale, S., Klein, R.T., Hruska, K.S. and Chung, W.K. ‘Pathogenic and likely pathogenic variant prevalence among the first 10,000 patients referred for next-generation cancer panel testing. Genetics in Medicine 2016, 18, 823–832. [Google Scholar]
- Miller, D.T. , Lee, K., Abul-Husn, N.S., Amendola, L.M., Brothers, K., Chung, W.K., Gollob, M.H., Gordon, A.S., Harrison, S.M., Hershberger, R.E., Klein, T.E., Richards, C.S., Stewart, D.R. and Martin, C.L. ‘ACMG SF v3.1 list for reporting of secondary findings in clinical exome and genome sequencing: A policy statement of the American College of Medical Genetics and Genomics (ACMG)’. Genetics in Medicine. 2022, 24, 1407–1414. [Google Scholar]
- Lakkawar, N.J. , Maran, G., Srinivasan, S. and Rangaswamy, T. ‘Accessory breast tissue in the axilla in a puerperal woman- case study. Acta Medica Medianae 2010, 49, 45–48. [Google Scholar]
- Alex, A. , Bhandary, E. and McGuire, K.P. ‘Anatomy and physiology of the breast during pregnancy and lactation. in Advances in Experimental Medicine and Biology 2020, 1252, 3–7. [Google Scholar]
- Schock, H. , Zeleniuch-Jacquotte, A., Lundin, E., Grankvist, K., Lakso, H.Å., Idahl, A., Lehtinen, M., Surcel, H.M. and Fortner, R.T. ‘Hormone concentrations throughout uncomplicated pregnancies: A longitudinal study. BMC Pregnancy and Childbirth 2016, 16, 146. [Google Scholar]
- Jaiswal, G. and Thakur, G.S. ‘An alternative yogic approach for cyclical mastalgia—A narrative review. Journal of Family Medicine and Primary Care 2021, 10, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Sivarajah, R. , Welkie, J., Mack, J., Casas, R.S., Paulishak, M. and Chetlen, A.L. ‘A review of breast pain: Causes, imaging recommendations, and treatment. Journal of Breast Imaging. Oxford University Press 2020, 2, 101–111. [Google Scholar] [CrossRef] [PubMed]
- Eren, T. , Aslan, A., Ozemir, I.A., Baysal, H., Sagiroglu, J., Ekinci, O. and Alimoglu, O. ‘Factors effecting mastalgia. Breast Care 2016, 11, 188–193. [Google Scholar] [CrossRef]
- Farcy, D.A. , Rabinowitz, D. and Frank, M. ‘Ectopic glandular breast tissue in a lactating young woman. Journal of Emergency Medicine 2011, 41, 627–629. [Google Scholar] [CrossRef]
- Darlington, A.J. (2015) ‘Anatomy of the breast. In Digital Mammography: a holistic approach, pp. 3–10.
- Lopez, M.E. and Olutoye, O.O. (2017) ‘Breast embryology, anatomy, and physiology. in Endocrine Surgery in Children. Springer Berlin Heidelberg, pp. 365–376.
- Youssef, H.M.K. , Radi, D.A. and Abd El-Azeem, M.A. ‘Expression of TSP50, SERCA2 and IL-8 in Colorectal Adenoma and Carcinoma: Correlation to Clinicopathological Factors. Pathology and Oncology Research 2021, 27, 1609990. [Google Scholar] [CrossRef]
- Song, Z.B. , Wu, P., Ni, J.S., Liu, T., Fan, C., Bao, Y.L., Wu, Y., Sun, L.G., Yu, C.L., Huang, Y.X. and Li, Y.X. ‘Testes-specific protease 50 promotes cell proliferation via inhibiting activin signaling. Oncogene 2017, 36, 5948–5957. [Google Scholar] [CrossRef]
- Wang, Y. , Xu, S., Wang, S., Song, Z., Zheng, L., Wang, G., Sun, Y. and Bao, Y. ‘miR-4709-3p Inhibits Cell Proliferation by Downregulating TSP50 Expression in Breast Cancer Cells. DNA and cell biology 2021, 40, 969–978. [Google Scholar] [CrossRef]
- Niu, C.X. , Li, J.W., Li, X.L., Zhang, L.L., Lang, Y., Song, Z.B., Yu, C.L., Yang, X.G., Zhao, H.F., Sun, J.L., Zheng, L.H., Wang, X., Sun, Y., Han, X.H., Wang, G.N. and Bao, Y.L. ‘PRSS50-mediated inhibition of MKP3/ERK signaling is crucial for meiotic progression and sperm quality. Zoological Research 2024, 45, 1037–1047. [Google Scholar]
- Pang, Q. , Christianson, T. A., Keeble, W., Diaz, J., Faulkner, G.R., Reifsteck, C., Olson, S. and Bagby, G.C. The Fanconi anemia complementation group C gene product: structural evidence of multifunctionality, Blood 2001, 98, 1392–1401. [Google Scholar]
- Bieging, K. T. , Mello, S. S., and Attardi, L. D. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews. Cancer 2014, 14, 359–370. [Google Scholar]
- Aubé, M. , Lafrance, M., Brodeur, I., Delisle, M.-C. and Carreau, M. Fanconi anemia genes are highly expressed in primitive CD34 + hematopoietic cells. BMC blood disorders 2003, 3, 1. [Google Scholar] [PubMed]
- Matthews, L. , Gopinath, G., Gillespie, M., Caudy, M., Croft, D., de Bono, B., D’Eustachio, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic acids research 2009, 37 suppl. 1, D619–D622. [Google Scholar] [CrossRef] [PubMed]
- Frey, M.K. , Sandler, G. , Sobolev, R., Kim, S.H., Chambers, R., Bassett, R.Y., Martineau, J., Sapra, K.J., Boyd, L., Curtin, J.P., Pothuri, B. and Blank, S. V. ‘Multigene panels in Ashkenazi Jewish patients yield high rates of actionable mutations in multiple non-BRCA cancer-associated genes. Gynecologic Oncology 2017, 146, 123–128. [Google Scholar]
- Thompson, E.R. , Doyle, M.A., Ryland, G.L., Rowley, S.M., Choong, D.Y.H., Tothill, R.W., Thorne, H., Barnes, D.R., Li, J., Ellul, J., Philip, G.K., Antill, Y.C., James, P.A., Trainer, A.H., Mitchell, G. and Campbell, I.G. ‘Exome Sequencing Identifies Rare Deleterious Mutations in DNA Repair Genes FANCC and BLM as Potential Breast Cancer Susceptibility Alleles. PLoS Genetics 2012, 8, e1002894. [Google Scholar]
- Pan, Z.W. , Wang, X.J., Chen, T., DIng, X.W., Jiang, X., Gao, Y., Mo, W.J., Huang, Y., Lou, C.J. and Cao, W.M. ‘Deleterious mutations in DNA repair gene FANCC exist in BRCA1/2-negative Chinese familial breast and/or ovarian cancer patients. Frontiers in Oncology 2019, 9, 169. [Google Scholar] [CrossRef]
- Dörk, T. , Peterlongo, P., Mannermaa, A., Bolla, M.K., Wang, Q., Dennis, J., Ahearn, T., Andrulis, I.L., Anton-Culver, H., Arndt, V., et al. ‘Two truncating variants in FANCC and breast cancer risk. Scientific Reports 2019, 9, 12524. [Google Scholar]
- Tsai, M.H. , Ke, H.C., Lin, W.C., Nian, F.S., Huang, C.W., Cheng, H.Y., Hsu, C.S., Granata, T., Chang, C.H., Castellotti, B., Lin, S.Y., Doniselli, F.M., Lu, C.J., Franceschetti, S., Ragona, F., Hou, P.S., Canafoglia, L., Tung, C.Y., Lee, M.H., Wang, W.J. and Tsai, J.W. ‘Novel lissencephaly-associated NDEL1 variant reveals distinct roles of NDE1 and NDEL1 in nucleokinesis and human cortical malformations. Acta Neuropathologica 2024, 147, 13. [Google Scholar]
- Soto-Perez, J. , Baumgartner, M. and Kanadia, R.N. ‘Role of NDE1 in the Development and Evolution of the Gyrified Cortex. Frontiers in Neuroscience 2020, 14, 617513. [Google Scholar] [CrossRef]
- Yao, M. , Pan, Y., Ren, T., Yang, C., Lei, Y., Xing, X., Zhang, L., Cui, X., Zheng, Y., Xing, L. and Wu, C. ‘Loss of Dip2b leads to abnormal neural differentiation from mESCs. Stem Cell Research & Therapy 2023, 14, 248. [Google Scholar]
- Musa, G. , Cazorla-Vázquez, S., van Amerongen, M.J., Stemmler, M.P., Eckstein, M., Hartmann, A., Braun, T., Brabletz, T. and Engel, F.B. ‘Gpr126 (Adgrg6) is expressed in cell types known to be exposed to mechanical stimuli. Annals of the New York Academy of Sciences 2019, 1456, 96–108. [Google Scholar] [CrossRef]
- Gowhari Shabgah, A. , Jadidi-Niaragh, F., Mohammadi, H., Ebrahimzadeh, F., Oveisee, M., Jahanara, A. and Gholizadeh Navashenaq, J. ‘The Role of Atypical Chemokine Receptor D6 (ACKR2) in Physiological and Pathological Conditions; Friend, Foe, or Both? Frontiers in Immunology 2022, 13, 861931. [Google Scholar] [CrossRef]
- Dalaie, K. , Yassaee, V.R., Behnaz, M., Yazdanian, M., Jafari, F. and Farimani, R.M. ‘Relationship of the rs10850110 and rs11611277 polymorphisms of the MYO1H gene with non-syndromic mandibular prognathism in the Iranian population. Dental and medical problems 2020, 57, 433–440. [Google Scholar] [CrossRef]
- Spielmann, M. , Hernandez-Miranda, L.R. , Ceccherini, I., Weese-Mayer, D.E., Kragesteen, B.K., Harabula, I., Krawitz, P., Birchmeier, C., Leonard, N. and Mundlos, S. ‘Mutations in MYO1H cause a recessive form of central hypoventilation with autonomic dysfunction. Journal of medical genetics 2017, 54, 754–761. [Google Scholar]
- Alerasool, N. , Leng, H., Lin, Z.Y., Gingras, A.C. and Taipale, M. ‘Identification and functional characterization of transcriptional activators in human cells. Molecular Cell 2022, 82, 677–695. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y. , Qi, P., Xiang, W., Yanhui, L., Yu, L. and Qing, M. ‘Multi-Omics Analysis Reveals Novel Subtypes and Driver Genes in Glioblastoma. Frontiers in Genetics 2020, 11, 565341. [Google Scholar] [CrossRef] [PubMed]
- Roci, I. , Watrous, J.D., Lagerborg, K.A., Jain, M. and Nilsson, R. ‘Mapping choline metabolites in normal and transformed cells. Metabolomics: Official journal of the Metabolomic Society 2020, 16, 125. [Google Scholar] [CrossRef] [PubMed]
- Li, Yifei, Shen, X. , Yang, X., Lian, F., Li, Yanping, Li, J., Huang, Y., Shen, W. and Liu, H. ‘CHDH, a key mitochondrial enzyme, plays a diagnostic role in metabolic disorders diseases and tumor progression. Frontiers in Genetics 2023, 14, 1240650. [Google Scholar] [CrossRef]
- Martinelli, D. , Schiff, M., Semeraro, M., Agolini, E., Novelli, A. and Dionisi-Vici, C. ‘CUGC for lysinuric protein intolerance (LPI). European Journal of Human Genetics 2020, 28, 1129–1134. [Google Scholar] [CrossRef]
- Carpenter, D. , Robinson, R.L., Quinnell, R.J., Ringrose, C., Hogg, M., Casson, F., Booms, P., Iles, D.E., Halsall, P.J., Steele, D.S., Shaw, M.A. and Hopkins, P.M. ‘Genetic variation in RYR1 and malignant hyperthermia phenotypes. British journal of anaesthesia 2009, 103, 538–548. [Google Scholar] [CrossRef]
- Spaccavento, A. , Rodríguez, M. del R., Meretta, A., Elissamburu, P., Carvelli, V., Gobbo, M., Rosa, D., Masoli, O., Conde, D. and Costabel, J.P. ‘Prevalence of transthyretin amyloid cardiomyopathy in patients admitted for acute heart failure. Current problems in cardiology 2024, 49, 102385. [Google Scholar] [CrossRef]
- Jacobson, D.R. , Alexander, A.A., Tagoe, C., Garvey, W.T., Williams, S.M., Tishkoff, S., Modiano, D., Sirima, S.B., Kalidi, I., Toure, A. and Buxbaum, J.N. ‘The prevalence and distribution of the amyloidogenic transthyretin (TTR) V122I allele in Africa. Molecular genetics & genomic medicine 2016, 4, 548–556. [Google Scholar]
Figure 1.
Pedigree of the affected family and clinical presentations of the proband. A: Pedigree of the affected multiplex family. B and C: The proband, after an ultrasound examination, was diagnosed with bilateral accessory breasts. D to F: Excision biopsy of the accessory breast tissue in the proband for samples for histological examination.
Figure 1.
Pedigree of the affected family and clinical presentations of the proband. A: Pedigree of the affected multiplex family. B and C: The proband, after an ultrasound examination, was diagnosed with bilateral accessory breasts. D to F: Excision biopsy of the accessory breast tissue in the proband for samples for histological examination.
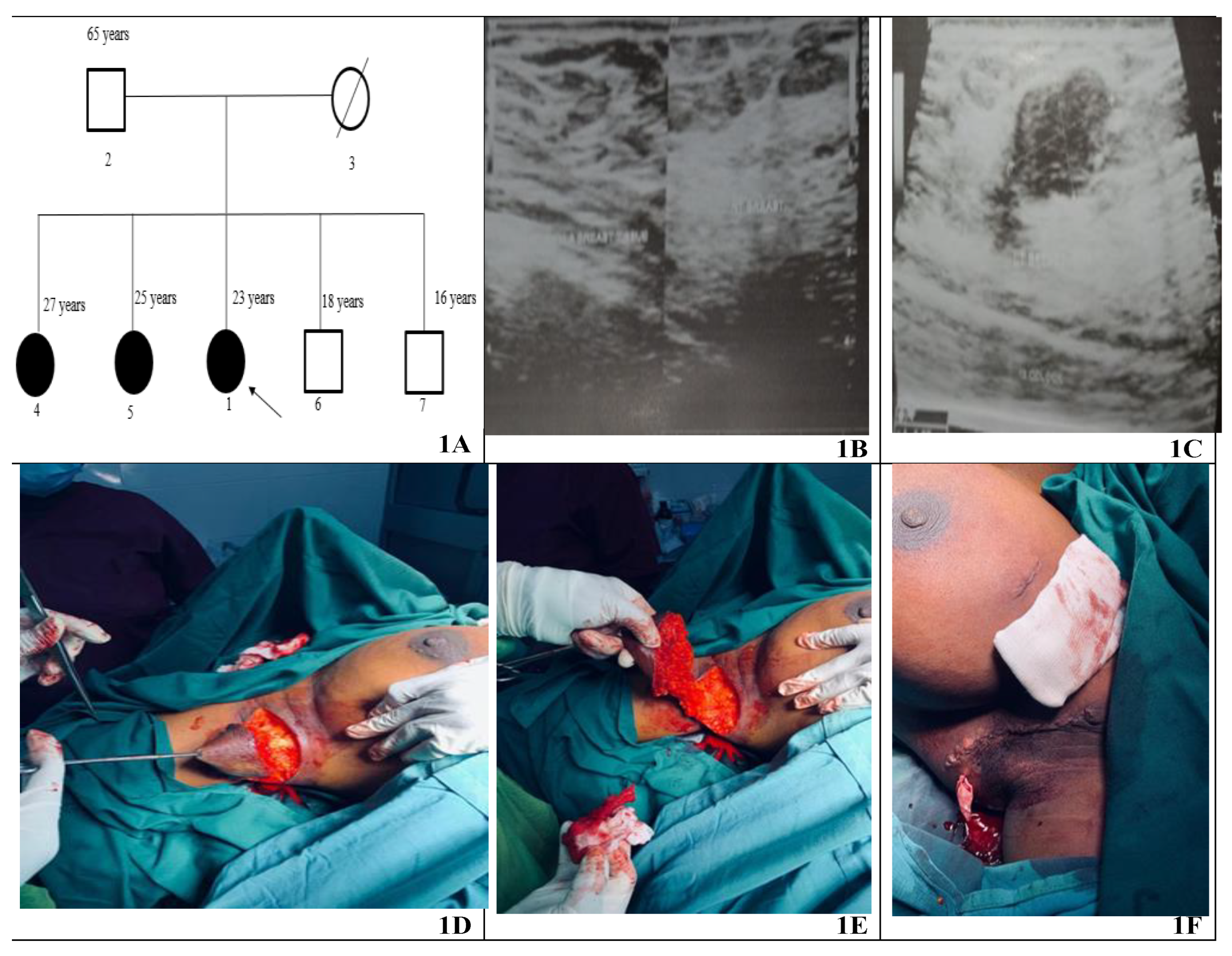
Figure 2.
Bulk tissue expression and interactome of the FANCC gene. A: Bulk tissue expression pattern of FANCC. The arrow indicates its expression in the mammary tissue (https://www.gtexportal.org/home/gene/FANCC, 2024). B: depicts the string interaction of the FANCC gene, showing its interaction with essential breast cancer-related genes like BRCA1 (https://string-db.org/cgi/network?taskId=bTGzi0bXr9XL&sessionId=bUkeU0Kv11w6, 2024). C: Bulk tissue expression pattern of PRSS50, demonstrating expression of the gene in the breast, indicated by an arrow. D: Interactome for PRSS50.
Figure 2.
Bulk tissue expression and interactome of the FANCC gene. A: Bulk tissue expression pattern of FANCC. The arrow indicates its expression in the mammary tissue (https://www.gtexportal.org/home/gene/FANCC, 2024). B: depicts the string interaction of the FANCC gene, showing its interaction with essential breast cancer-related genes like BRCA1 (https://string-db.org/cgi/network?taskId=bTGzi0bXr9XL&sessionId=bUkeU0Kv11w6, 2024). C: Bulk tissue expression pattern of PRSS50, demonstrating expression of the gene in the breast, indicated by an arrow. D: Interactome for PRSS50.
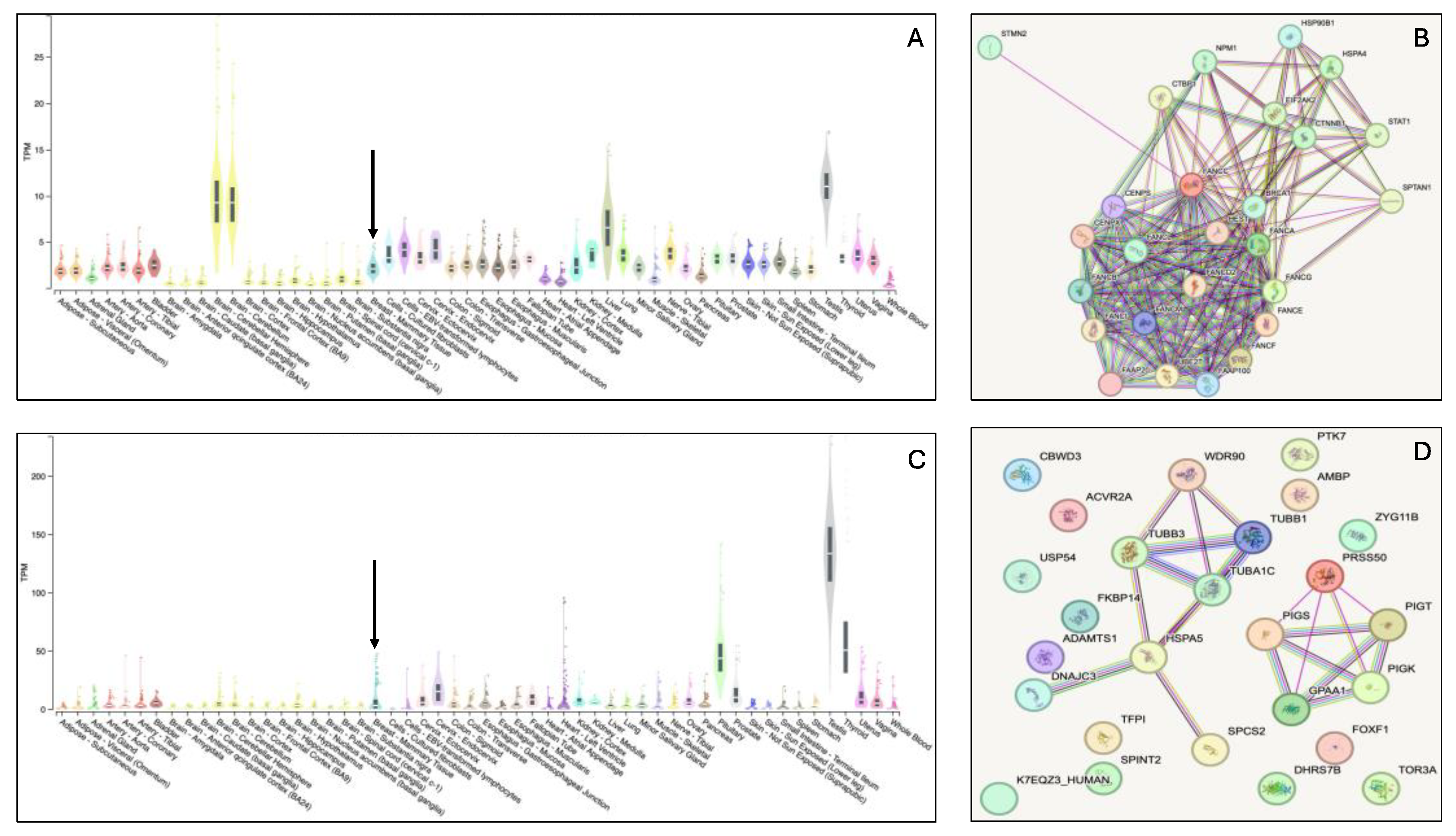
Figure 3.
Chromatograms depicting the frameshift variants in the affected individuals. The Sanger sequencing was carried out using both forward and reverse primers. The chromatograms were generated from Sanger sequencing using the reverse primer set, except for C, where that of the forward primer is shown, because the reverse one was not very clear. The arrows indicate the position of the variants. Panels A, C and D are the chromatograms from the three affected females, whereas panels B, E and F are from the unaffected males in the family. A: 20222001_1, B: 20222001_2, C: 20222001_4, D: 20222001_5, E: 20222001_6, F: 20222001_7.
Figure 3.
Chromatograms depicting the frameshift variants in the affected individuals. The Sanger sequencing was carried out using both forward and reverse primers. The chromatograms were generated from Sanger sequencing using the reverse primer set, except for C, where that of the forward primer is shown, because the reverse one was not very clear. The arrows indicate the position of the variants. Panels A, C and D are the chromatograms from the three affected females, whereas panels B, E and F are from the unaffected males in the family. A: 20222001_1, B: 20222001_2, C: 20222001_4, D: 20222001_5, E: 20222001_6, F: 20222001_7.
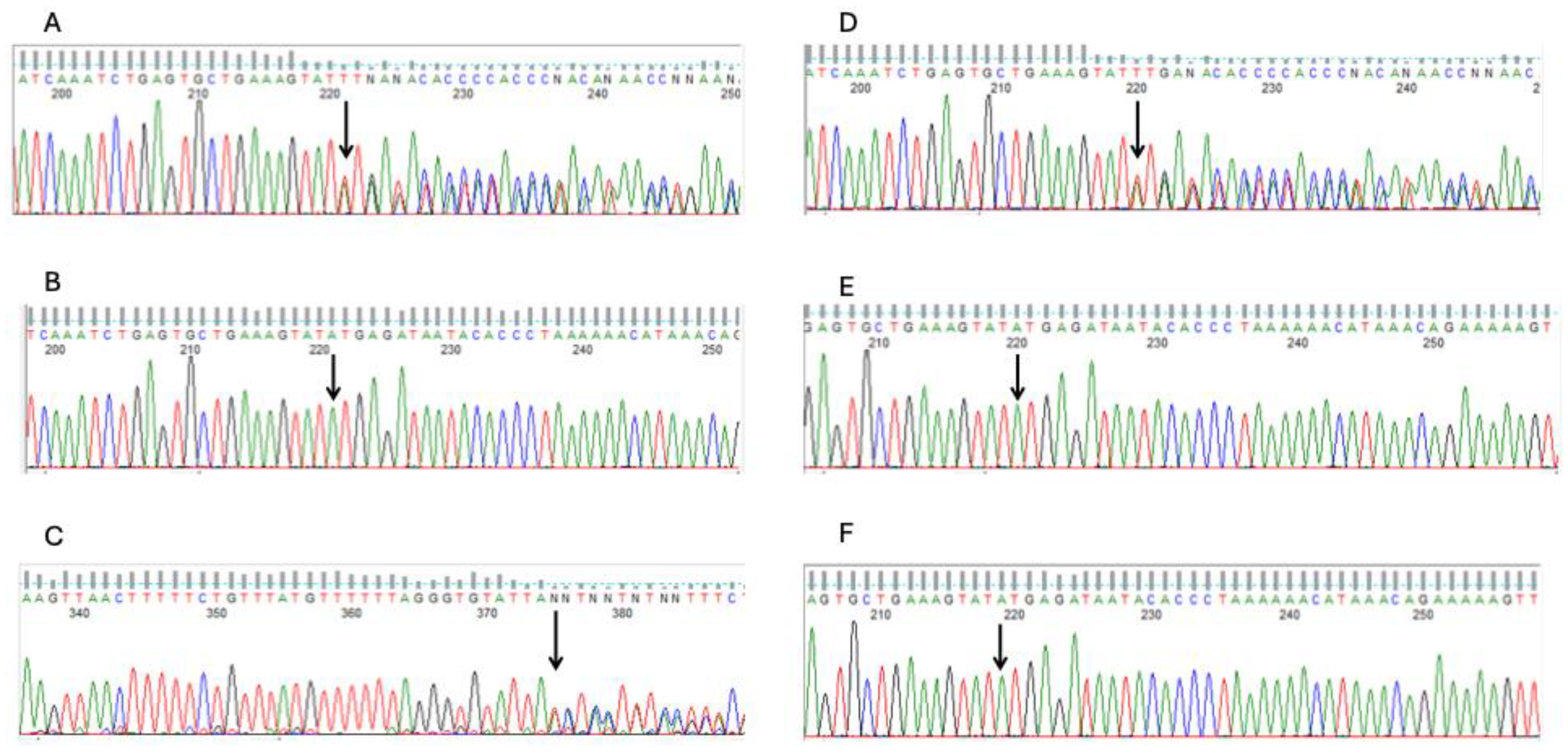

Table 1.
Rare genetic variants associated with the accessory breast phenotype in the family.
| Genomic coordinate | ETI | Gene Name | Ref# | Alt# | Genotype of affected individuals | HSVc | HSVp | NTP |
| Chr3:46714301 (rs145256818) | ENST00000460241 | PRSS50 | G | A | G/A | c.671C>T | p.Pro224Leu | 7 |
| Chr9:95172132 (rs766909460) | ENST00000289081 | FANCC | TA | T | TA/T | c.360del | p.His120GlnfsTer24 | 1 |
| Chr9:95172134 (rs750003253) | ENST00000289081 | FANCC | TGAGA | T | TGAGA/T | c.355_358del | p.Ser119IlefsTer24 | 1 |
All transcripts indicated for the genes are canonical transcripts. All variants were absent in unaffected members. ETI: Ensemble Transcript ID, Ref#: Reference allele, Alt#: Alternate allele, NTP: Number of tools predicting pathogenicity.
Table 2.
Rare genetic variants that may be responsible for co-occurring conditions.
| Genomic coordinate | ETI | Gene Name | Ref# | Alt# | Genotype of affected individuals | HSVc | HSVp | NTP |
| Chr14:22813019 (rs764284986) | ENST00000397532 | SLC7A7 | A | G | A/G | c.380T>C | p.Ile127Thr | 9 |
| Chr16:15687426 (Novel) |
ENST00000396355 | NDE1 | C | G | C/G | c.438C>G | p.Ile146Met | 9 |
| Chr12:50731373 (rs147225936) | ENST00000301180 | DIP2B | T | C | T/C | c.3646T>C | p.Tyr1216His | 8 |
| Chr6:142415804 (rs1204304460) | ENST00000367609 | ADGRG6 | G | A | G/A | c.2678G>A | p.Arg893Gln | 6 |
| Chr3:53823630 (rs765283936) | ENST00000315251 | CHDH | C | T | C/T | c.379G>A | p.Val127Ile | 6 |
| Chr3:42865498 (rs577087357) | ENST00000422265 | ACKR2 | ATGGCACC | A | ATGGCACC/A | c.1010_1016del | p.Pro337LeufsTer21 | 1 |
| Chr12:109447180 (rs200225794) | ENST00000310903 | MYO1H | T | C | T/C | c.3115T>C | p.Ter1039ArgextTer1 | 1 |
| Chr3:49276455 (rs868287641) | ENST00000343010 | C3orf62 | CCT | C | CCT/C | c.416_417del | p.Glu139GlyfsTer20 | 1 |
| Chr14:19747835 (Novel) |
ENST00000642117 | OR4Q3 | CATGAACCCCCAGCT | C | CATGAACCCCCAGCT/C | c.436_449del | p.Asn146ProfsTer32 | 1 |
| Chr6:29044316 (rs149813138) |
ENST00000377175 | OR2W1 | G | A | G/A | c.860C>T | p.Pro287Leu | 7 |
All transcripts indicated for the genes are canonical transcripts. All variants were absent in unaffected members. ETI: Ensemble Transcript ID, Ref#: Reference allele, Alt#: Alternate allele, NTP: Number of tools predicting pathogenicity.
Table 3.
Variants observed in ACMG secondary findings genes.
| Genomic coordinate | ETI | Gene Name | Ref allele | Alt allele | Genotype | HSVc | HSVp | NTP |
| Chr19:38512366 (rs61739911) |
ENST00000359596 |
RYR1 | C | T | C/T | c.9355C>T |
p.Arg3119Cys |
7 |
| Chr18:31595139 (rs1555631393) |
ENST00000237014 | TTR | G | A | G/A | c.220G>A | p.Glu74Lys | 11 |
Transcript indicated for the gene is a canonical transcript. ETI: Ensemble Transcript ID, NTP: Number of tools predicting pathogenicity.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



