
Submitted:
22 September 2025
Posted:
22 September 2025
You are already at the latest version
Abstract
Xenotransplantation using pig cells, tissues or organs may be associated with the trans-mission of porcine zoonotic or xenozoonotic microorganisms. Porcine endogenous retro-viruses (PERVs) pose a special risk for xenotransplantation as these viruses can infect human cells and are integrated in multiple copies in the genome of all pigs and therefore, they cannot be eliminated as other viruses can. To prevent PERV transmission to the re-cipient, several strategies have been developed: PERV-C-free animals, siRNA and genomic editing. Another strategy is the generation of vaccines based on neutralizing antibodies in order to protect the recipient. Here we cloned, produced and purified the recombinant transmembrane (p15E) and the surface envelope (gp70) protein of PERV to immunize rats. For the first time, an adjuvant type that is approved for human use was used. In all cases we obtained virus binding antibodies as shown in Western blot assays and neutralizing antibodies as shown in neutralization assays. The epitopes recognized by the antisera against p15E were determined using overlapping peptides. Two main epitopes were found in the sequence of p15E, one in the membrane proximal external region (MPER) and one in the fusion peptide proximal region (FPPR). The epitopes correspond to epitopes de-termined previously when immunizing different animal species with p15E of PERV. The epitope in the MPER is related by sequence and location to an epitope in the transmem-brane envelope protein of the human immunodeficiency virus-1 (HIV-1) recognized by a broadly neutralizing antibody from infected patients.
Keywords:
porcine endogenous retroviruses (PERVs)
; transmembrane envelope protein
; surface envelope protein
; neutralizing antibodies
; epitope mapping
; human immunodeficiency virus-1 (HIV-1)
1. Introduction
Xenotransplantation, using genetically modified pigs, offers a promising solution to the shortage of allogeneic donor organs. Preclinical trials have demonstrated long survival times in non-human primates transplanted with hearts and kidneys from genetically modified pigs [1]. Moreover, the first transplantations of pig organs into human patients and brain-dead individuals indicate that this emerging technology is steadily progressing toward clinical application. Genetically modified pigs are needed to prevent hyperacute, antibody-mediated and cellular rejection as well as to preserve coagulation [2]. Furthermore, it is essential to prevent transmission of potentially zoonotic or xenozoonotic porcine microorganisms to the recipient [3,4]. The transmission of the porcine cytomegalovirus/porcine roseolovirus (PCMV/PRV) to the first human patient who received a pig heart in Baltimore, which contributed to the death of the patient [5], indicates the importance of preventing such transmissions. Whereas PCMV/PRV and many other porcine viruses can be eliminated by selection of virus-negative animals, early weaning, colostrum deprivation, treatment with antiviral drugs or vaccines if available, porcine endogenous retroviruses (PERVs) cannot. The proviruses are integrated with up to 60 copies in the genome of all pigs.
PERV-A and PERV-B are present in all pigs, whereas PERV-C is not found in all pigs [6,7]. While PERV-A and PERV-B have been shown to infect human cells under in vitro conditions, PERV-C is an ecotropic virus that infects only pig cells. Recombination between PERV-C and PERV-A results in recombinant viruses with a high replication rate and the capacity to infect human cell [8]. To date, there is no evidence of transmission of these viruses in either preclinical studies or first human xenotransplantations (for review see [9]). Despite this, various strategies have been developed to prevent PERV transmissions. First of all, the selection of PERV-C-negative animals to prevent PERV-A/C recombination. This strategy was used when PERV-C-free Auckland Island pigs were selected [10]. Auckland Island pigs represent an inbred population of feral pigs isolated on the sub-Antarctic island for over 100 years. The animals have been maintained under pathogen-free conditions in New Zealand; they are virologically well characterized and have been used as donor sources in first clinical trials of porcine neonatal islet cell transplantation for the treatment of human diabetes patients. In the first clinical trials, no porcine viruses, including PERVs were transmitted to the human recipients. Auckland Island pig kidney cells (selected to be free of PERV-C) were imported from New Zealand, and founder animals were established by somatic cell nuclear transfer (SCNT) at the Ludwig Maximilian University of Munich (LMU) [11].
Another strategy is based on RNA interference. PERV-specific small hairpin (sh) RNA directed against sequences in the polymerase gene (pol) were expressed in pig cells in vitro and in transgenic animals in vivo and reduced the PERV expression [12,13,14]. Although gene editing by zinc finger nucleases (ZFN) did not succeed [15], using clustered regularly interspaced short palindromic repeats/CRISPR-associated 9 (CRISPR/Cas9) resulted in the inactivation of all PERV proviruses in vitro [16] and in vivo [17].
Another promising strategy is the generation of vaccines against PERVs in order to protect the recipient. Vaccines against the murine leukemia virus (MuLV) and the feline leukemia virus (FeLV), two gammaretroviruses closely related to PERV, were able to prevent leukemia in mice [18] and cats [19], respectively. 10µg of the surface envelope protein gp70 of the Friend-MuLV coupled to keyhole limpet hemocyanin by glutaraldehyde protected more than 90% of the mice from erythroleukemia [18]. Live and inactivated vaccines against FeLV were found efficacious in reducing clinical signs in cats [19].
In previous immunization studies using recombinant p15E and gp70 of PERV, binding and neutralizing antibodies were induced in different species [20,21,22,23,24]. In those studies, however, Freund’s adjuvant was applied, which is not suitable for use in humans. To extend these findings, we immunized rats with newly produced and purified recombinant p15E and gp70 in combination with a type of adjuvant approved for human use. AddaVax was employed, a squalene-based oil-in-water nano-emulsion similar to MF59 that has been licensed for use in adjuvanted influenza vaccines [25]. In addition, a modified neutralization assay and a novel method for epitope mapping were applied.
2. Materials and Methods
2.1. Rats, Normal Rat Sera and Cells
Wistar rats were maintained and immunized by the Davids Biotechnologie GmbH (Regensburg, Germany). Five rats were immunized with the transmembrane envelope protein p15E, five rats with the surface envelope protein gp70. Additional normal Wistar rat sera were obtained from the Institute for Animal Welfare, Animal Behavior and Laboratory Animal Science, Department of Veterinary Medicine, Free University Berlin.
Human embryonic kidneys 293T(293T) cells were cultured in Dulbecco’s modified eagle medium (DMEM) (Thermo Fisher Scientific, Waltham, Massachusetts, USA) supplemented with 10 % heat-inactivated fetal bovine serum (FBS, PAN-Biotech, Germany) Penicillin and streptomycin were not added to the culture medium.
2.2. Cloning, Expression and Purification of the Recombinant Proteins
Two antigens were prepared and used for screening assays and immunization: the recombinant transmembrane envelope protein p15E and recombinant surface envelope protein the gp70 (Figure 1).
The ectodomain sequence of p15E (amino acids 447-477, accession numberHQ688786) was cloned into the pCal-n vector (Stratagene Europe, Amsterdam, Netherlands) [20]. The recombinant plasmid was transferred to Escherichia coli DH5α (E. coli DH5α) competent cells. Single colonies were selected for sequencing, then the recombinant p15E was expressed in E. coli BL21 DE3 and induced with 0.2 mM isopropyl β-D-thiogalactopyranoside (IPTG) (Thermo Fisher Scientific, Waltham, MA, USA) in Luria-Bertani (LB) medium at OD600 of 0.6. After incubation for 3 h at 37 °C with shaking at 200 rpm, cells were harvested by centrifugation at 6000 x g for 30 min, at 4 °C using a Sorvall RC 6 Plus Superspeed Centrifuge (Thermo Fisher Scientific, Waltham, Massachusetts, USA). Cells were lysed by incubation in 50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 2 mM CaCl2 and 10 mM dithiothreitol (DTT), supplemented with 1 mg/mL lysozyme, 1000 U benzonase (Merck, Darmstadt, Germany) and cOmplete ethylenediaminetetraacetic acid (EDTA)-free protease inhibitor cocktail (Merck, Darmstadt, Germany) for 30 min at room temperature. Subsequently, the lysate was subjected to 20 s sonification cycles with 1 min cooling on ice. The suspension was diluted in a 1:1 ratio with binding buffer (50 mM Tris/HCl (pH 7.5), 150 mM NaCl, 2 mM CaCl2) and centrifuged at 50,000 x g for 20 min; supernatant was filtered through a 0.45 µm filter using. The protein was purified by calmodulin resin affinity chromatography using an ÄKTAprime plus system (both from Cytiva Life Sciences, Freiburg, Germany) with 5 mM DTT included in binding buffer and 2 mM DTT in the washing (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM CaCl2) and elution buffers (50 mM Tris/HCl, pH 7.5, 150 mM NaCl, 2 mM EGTA).
The entire sequence of the surface envelope protein gp70 of PERV-A (amino acids 34-146, accession number Q688785) and a short stretch of the ectodomain sequence of p15E were cloned into pET22b (+) vector (Merck, Darmstadt, Germany) [21]. The p15E sequence was included to make the antigen similar to the sequence of the recombinant gp70/p15E of FeLV successfully used as Leucogen to vaccinate cats [25]. The pET22b (+)-gp70 was transferred into E. coli DH5α competent cells. Confirmative sequencing was performed after single-colony selection. The recombinant protein was expressed in E. coli BL21 DE3 cells and induced with 0.1 mM IPTG in LB medium at an OD600 of 0.6. After incubation for 3 h at 37 °C with shaking at 200 rpm, cells were harvested as mentioned above. E. coli DH5α and BL21 strains were grown on LB agar or in LB medium with ampicillin (100 µg/mL) as the selective antibiotic [21]. Cells were lysed in phosphate buffered saline (PBS) (pH 7.4) with 10 mM DTT, supplemented with 1 mg/mL lysozyme by 20 s sonification cycles with 1 min intervals on ice. Cells were harvested by centrifugation at 22,000 x g for 30 min at 4 °C and the insoluble pellet was lysed by incubation in lysis buffer (pH 8.0) (100 mM NaH2PO4, 10 mM Tris/HCl 6 M GuHCl and 10 mM DTT, supplemented with 3 U/µL benzonase) overnight at 4 °C. The supernatant was filtered through a 0.45 µM filter. Purification was performed by Ni-NTA resin affinity chromatography (Cytiva Life Sciences, Freiburg, Germany) using an ÄKTAprime plus system with 5 mM DTT included in binding (pH 8.0), two washing (pH 6.3 and pH5.0) and elution buffers (pH 4.5) (100 mM NaH2PO4, 10 mM Tris/HCl, 8 M urea). Purified recombinant gp70 was concentrated to 1mg/mL using Vivaspin 15R Centrifugal Concentrator (Vivaproducts, Littleton, USA) and subsequently dialyzed against PBS using dialysis cassettes (15 ml, Thermo Fisher Scientific, Waltham, MA, USA). Recombinant proteins were characterized by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting.
2.3. Immunization Schedule
Wistar rats (n=5 per group) were immunized with 300 µg of recombinant p15E or gp70 emulsified in AddaVax (InvivoGen, Toulouse, France) for the primary immunization, followed by two booster injections of the same dose emulsified in the same adjuvant on days 14 and 28. Preimmune sera were collected one day before immunization and immune sera were collected on days 14, 28, and 42 after the first immunization. The animal experiments were conducted by Davids Biotechnologie GmbH (Regensburg, Germany).
2.4. SDS-PAGE
Protein expressions and purifications were analyzed by SDS-PAGE, which was performed using a 17 % separating gel for p15E and a 12 % separating gel for gp70. 1 µg p15E and 100 nggp70 were mixed with 6 × SDS-PAGE loading buffer and denatured at 95 °C for 10 min in a metal heating block (Eppendorf Vertrieb Deutschland GmbH, Wesseling-Berzdorf, Germany). Proteins were separated by SDS-PAGE and stained by Imperial Protein Stain (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature and destained with double-distilled water (ddH2O) until background was clear.
2.5. Western Blotting
SDS-PAGE was performed as described above. The proteins were transferred onto 0.2 µm PVDF membranes (Carl Roth GmbH, Karlsruhe, Germany) using a semi-dry transfer system (Peqlab Biotechnologie GmbH, Erlangen, Germany) at a constant current of 0.13 A for 70 min. The blot was blocked with 5 % (w/v) non-fat dry milk (NFDM) powder in Tris-buffered saline with 0.05 % Tween-20 (TBST) for 1 h at room temperature. Membranes were then incubated with positive control sera (goat sera anti-p15E, #355 and anti-gp70, #62 [21]), rat anti-p15E (1:1000) or rat anti-gp70 (1:1000) overnight at 4 °C, followed by incubation with the corresponding horseradish peroxidase (HRP)-conjugated donkey anti-goat IgG antibody (1:20,000) (Merck, Darmstadt, Germany) and HRP-conjugated goat anti-rat IgG(H+L) secondary antibody (1:10,000) (Thermo Fisher Scientific, Waltham, MA, USA) for 1 h at room temperature. Membranes were washed three times for 10 min with TBST between each incubation step. Blots were developed with ECL Prime Western Blot Detection Reagent (Cytiva, USA) according to the manufacturer’s instructions.
2.6. In Vitro Neutralization Assays
293T cells cultured in DMEM supplemented with heat inactivated 10 % FBS at 37 °C in a 5 % CO2 incubator were used for neutralization assays. Rat anti-p15E and anti-gp70 sera were heat-inactivated at 56 °C for 30 min and serially diluted twofold in DMEM. 20 µL undiluted or serially diluted sera and 80 µL PERV supernatant (threshold cycle (Ct) <25) were mixed and incubated at 37 °C for 15 min. The mixture was added to 100 µL HEK 293T cells (5,000 cells/ 100 µL) in 96-well plates and incubated for 96 h. Lysis duplex real-time polymerase chain (real-time PCR) reaction detecting both, PERV-pol and human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was performed in situ. Direct PCR Lysis Reagent (Cell) (Viagen Biotech, Los Angeles, CAa, USA) and Nuclease-Free water were mixed at a 1:1 ratio and Proteinase K (Viagen Biotech, Los Angeles, CA, USA) was added at 40 µL per 1 mL of the resulting mixture. This mixture was applied to lyse cells in situ, adding 60 µL per well. The cell lysates were incubated overnight at 55 °C for lysis and wrapped in damp paper towels to prevent evaporation, followed by heat inactivation of proteinase K at 85 °C for 1.5 h. The resulting products were used directly for real-time PCR analysis.
2.7. Duplex Real-Time PCR for the Detection of Provirus
To determine the provirus in 293T cells, a duplex real-time PCR was performed. Genomic DNA was extracted using the method described above. Real-time PCR reactions were carried out using SensiFAST Probe No-ROX kit (Meridian Bioscience, Cincinnati, OH, USA), specific primers and probes (Table 1) on a qTOWER3G qPCR cycler (Analytik Jena, Jena, Germany). The cycling conditions used were as follows: initial denaturation at 95 °C for 5 min, followed by 45 amplification cycles of denaturation at 95 °C for 15 s, annealing at 62 °C for 30 s, and extension at 72 °C for 30 s.
2.8. Epitope Mapping
The ectodomain of PERV p15E (EPISLTLAVMLGLGVAAGVGTGTAALITGPQQLEKGLGEHAAMTEDLRLEESVSNLEESLTSLSEVVLQNRRGLDLLFLREGLCAALKEECCFYVDHSGAIRDSMSKLRERLERRRRREADQGWFENRSPWMTTLLSALTGPLVVLLLLLT) was synthesized as 74 peptides of 13-mers overlapping by 11 amino acids (JPT Peptide Technologies, Berlin, Germany) (Supplementary Table 1). N-terminal acetylation of the peptides makes the peptides more stable against N-terminal degradation, and the uncharged N-acetyl group corresponds more closely to the structure of the native antigen than a charged NH3+ group.
Peptides were covalently immobilized on the glass surface. An optimized hydrophilic linker moiety is inserted between the glass surface and the antigen derived peptide sequence to avoid false negatives caused by steric hindrances. For technical reasons all peptides contain a C-terminal glycine.
The profiling experiment was performed with a total of five rat serum samples diluted to 1:40, 1:200 and 1:1000 with blocking buffer and incubated for 1 hour at 30 °C on the Multiwell microarray slide. Each slide contained 21 individual mini-arrays (1 mini-array per sample dilution).
Subsequent to sample incubation, a secondary, fluorescently labeled anti-rat-IgG antibody, at 0.1 μg/ml, was added into the corresponding wells and left to react for 1 hour.
Additional control incubations applying the secondary antibody only (but no rat serum sample) were performed in parallel on the same microarray slide to assess false positive binding of the secondary antibody to peptides.
Serum samples were diluted in blocking buffer (Pierce International, Superblock TBS T20) and applied to microarrays for 1 h at 30 °C, then they were incubated with secondary antibody diluted in blocking buffer for 1 h at 30 °C. Secondary antibody used for detection: Anti-rat IgG (Jackson Immunoresearch, Ely, United Kingdom), label Cy5, applied concentration 0.1 μg/ml. Before each incubation step, microarrays were washed with washing buffer (50 mM TBS-buffer including 0.1 % Tween-20, pH 7.2). Finally, the microarrays were dried.
After washing and drying, the microarrays were scanned using a high-resolution fluorescence scanner (Axon Genepix Scanner 4300 SL50) at 635 nm to obtain fluorescence intensity profiles. Laser settings and applied resolution were identical for all measurements. The resulting images were analyzed und quantified using spot-recognition software GenePix (Molecular Devices). For each spot, the mean signal intensity was extracted (between 0 and 65535 arbitrary units).
For further data evaluation, the so called MMC2 values were determined. The MMC2 equals the mean value of all three instances on the microarray except when the coefficient of variation (CV) – standard-deviation divided by the mean value – is larger 0.5. In this case the mean of the two closest values (MC2) is assigned to MMC2.
3. Results
3.1. Production and Purification of the Antigens
To prepare the antigens required for immunization and testing, the DNA sequence of p15E or gp70 were cloned into vectors pCal-n or pET22b (+), respectively (Figure 1), then protein was expressed in E. coli and subsequently purified using calmodulin resin or Ni-NTA resin affinity chromatography under denatured conditions. Elution fractions showed a strongly enriched band at approximately 12 kDa and a single band at 54 kDa corresponding to the expected size of the recombinant p15E and gp70, respectively, (Figure 2). This indicates that the recombinant proteins were successfully expressed and purified.
3.2. Characterization of the Antigens
To verify the identity of the purified recombinant p15E and gp70, a Western blot was performed using goat anti-p15E (goat serum #355 [21]) and goat anti-gp70 (goat serum #62 [21]) antibodies. The blot showed single bands at 12 kDa and 54 kDa, corresponding to the expected molecular weights of p15E and gp70, respectively. These results confirm that the recombinant proteins were successfully expressed and purified without significant degradation (Figure 3).
3.3. Detection of Binding Antibodies
Five rats were immunized with the recombinant protein p15E, another five rats were immunized with the recombinant protein gp70 according to the immunization schedule (Figure 4).
To analyze whether specific antibodies against the recombinant proteins p15E or gp70 are present in the rat sera, Western blots were performed using control goat sera. The control sera were obtained previously by immunization of goats with recombinant p15E or gp70 [21]. In addition, to determine the titer of the specific antibodies in rat sera against antigens in the Western blot assay, rat sera were diluted at 1:100, 1:200, 1:500, 1:1000, and 1:10,000. The Western blot analysis showed single bands at 12kDa or 54kDa, which are the theoretical molecular weights of recombinant p15E or gp70. As expected, all rats produced specific antibodies against the recombinant protein p15E or gp70, and the titer of the rat sera against recombinant p15E or gp70 was at least 1:10,000 (Figure 5). This antibody titer is comparable with the titers found in previous immunizations [20,21].
3.4. Detection of Neutralizing Antibodies
To detect neutralizing antibodies against PERV in the immune rat sera, a neutralization assay was established. This assay is based on PERV infection in human 293T cells measuring provirus integration into cellular DNA. For this, a PCR method called lysis duplex real-time PCR was used. Cells were lysed directly in the 96-well plate. Supernatant from 293T cells producing a PERV-A/C virus which was adapted to human cells by passaging on 293T cells [28] was pre-incubated with immune serum dilutions and added to uninfected 293T cells. The real-time PCR used human GAPDH for evaluation. Since GAPDH is present in all cells this value indicates the total amount of cells, whereas the value for PERVpol indicated the number of infected cells. Sera from five rats immunized with recombinant p15E and sera from five rats immunized with the recombinant gp70 showed neutralizing capacity (Figure 6). The higher the ct value of PERVpol, the lower the number of infected 293 cells, indicating a greater neutralizing capacity (Figure 6b, d). The neutralization capacity was dose-dependent. Identical ct values for GAPDH confirmed that the number of 293 cells was consistent across all tested wells and that the antiserum was not cytotoxic (Figure 6c, e). Cytotoxicity may cause misleading results, appearing as false-positive neutralizing activity. The neutralizing activity of the antisera against p15E was much weaker compared to the neutralizing activity of the antisera against gp70 (Figure 6).
3.5. Epitope Mapping
To identify the epitopes in PERV p15E recognized by the immune rat sera, an epitope mapping was performed using 74 overlapping 13-mer peptides (with 11 amino acids overlaps) spanning the ectodomain of p15E. The peptides immobilized on microarray slides were incubated with rat sera and with a secondary, fluorescently labeled anti-rat-IgG antibody. The microarrays were scanned using a high-resolution fluorescence scanner. Laser settings and applied resolution were identical for all performed measurements. The resulting images were analyzed and quantified using spot-recognition software. For each spot, the mean signal intensity was extracted (between 0 and 65535 arbitrary units) (Supplementary Table 1).
To visualize the results obtained and to compare binding regions across the individual incubations, a heatmap diagram (Figure 7) was computed showing fluorescence intensities in a color-coded manner from white (no binding) to red (strong binding).
Three of the five investigated rat sera (namely rat serum, 1, 2 and 5) showed a comparable binding profile indicating one putative epitope represented within peptide 12 and 13 (peptide 12 TAALITGPQQLEK, peptide 13 ALITGPQQLEKGL), indicating epitope ALITGPQQLEK (Figure 8). In addition to the peptides 11, 12 and 13, the incubation of rat serum 3 yielded significant signals in a region spanning peptides 55 to 63. Probably more than one antibody from the serum is responsible for the detected signal distribution (Figure 8). Rat serum 4 gave rise to several signals with intensities above 5000 more or less all over the peptide scan. The most prominent region with the strongest signals (above 20,000) is represented by peptide 55 and 56 (Figure 8). Therefore, three main epitopes were detected, ALITGPQQLEK, in the FPPR, as well as LERRRRE/LRERLERRRRERE and GWFEGWF/GWFEGWFNR in the MPER of p15E.
4. Discussion
Immunization of rats with recombinant PERV p15E and PERV gp70 induced neutralizing antibodies, indicating that these envelope proteins may serve as effective antigens in vaccines designed to protect against PERV infection.
These findings are consistent with previous studies in which goats, rats, mice, and guinea pigs were immunized with both proteins, despite several differences between those studies and the present immunization approach.
Previously, goats were immunized with 500 µg antigen, Wistar rats with 150 µg, Balb/c mice with 50 µg, and Guinea pigs with 200 µg antigen [20,21]. In this study, we increased the antigen dose to 300 µg in order to achieve improved immunization outcomes in Wistar rats. The immunization schedule was also different. In previous studies a second and third immunization was performed after 2 and 5 weeks, here booster immunizations were performed after 14 and 28 days (Figure 4). The main difference is the usage of a different adjuvant. In previous studies complete Freund´s adjuvant (CFA) was used for immunization and incomplete Freund´s adjuvant (IFA) for booster immunizations [20,21]. CFA is composed of mineral oil (paraffin oil and mannide monooleate) and inactivated and dried mycobacteria (M. tuberculosis). CFA contains trehalose 6,6’ dimycolate, which stimulates the macrophage inducible Ca2+-dependent lectin receptor (Mincle). Additionally, CFA has ligands for toll-like receptor 2 (TLR2), TLR4, and TLR9. IFA lacks the mycobacterial components. CFA cannot be used in humans because it causes severe side effects, it triggers strong and long-lasting inflammatory reactions, and can lead to abscesses, ulcerations, necrosis, and chronic granulomas at the injection site. Therefore, we tested AddaVax, which is a squalene-based oil-in-water nano-emulsion. Its formulation is similar to that of MF59 that has been licensed in Europe for adjuvanted flu vaccines [25]. Squalene is an oil more readily metabolized than the paraffin oil used in Freund’s adjuvants. This class of adjuvants is believed to act through recruitment and activation of antigen-presenting cells (APCs) and stimulation of cytokines and chemokines production by macrophages and granulocytes [25].
Immunization with AddaVax induced neutralizing antibodies, and in the case of sera against p15E, the antibodies recognized epitopes similar to those reported in previous studies using CFA/IFA (Figure 9). Previously, epitope mapping was carried out using a cellulose-adsorbed peptide spot library consisting of 15-mer peptides overlapping by 12 amino acids, with detection performed by chemiluminescence [20,21]. In the present study, a library of 74 peptides composed of 13-mers overlapping by 11 amino acids was employed. Shorter peptides are expected to allow more precise fine mapping of the epitopes. All peptides were covalently immobilized on a glass surface, and epitope detection was performed using a fluorescently labeled anti-rat IgG antibody in combination with a high-resolution fluorescence scanner. The epitopes identified by both approaches were highly similar (Figure 9).
It is of great interest that the main epitope in the MPER of p15E shows a sequence homology with an epitope in gp41 of HIV-1, which is recognized by a broadly neutralizing monoclonal antibody, 4E10, isolated from an AIDS patient [29] (Figure 10). 4E10 neutralizes diverse subtypes of HIV-1 (e.g., subtypes B, C, and E) [29]. Not surprisingly, similar epitopes were also found when cats were immunized with p15E of FeLV [30].
It is not only the partial sequence homology between the epitopes in the gammaretroviruses PERV and FeLV on the one hand and HIV-1 on the other hand, but also the localization of these epitopes in the MPER (Figure 11), what is important for the understanding of the function of this domain. In more recent studies with HIV-1 it has been demonstrated that the MPER is important for env-mediated fusion and virus infectivity [31,32]: mutations to Ala of three of five conserved Trp residues in this region are sufficient to abrogate syncytium formation [32]. In contrast to our success in inducing antibodies against the MPER of PERV (and FeLV), numerous attempts to induce antibodies of the type 2F5 and 4E10, broadly neutralizing HIV-1, failed [33,34,35]. Immunization experiment using the backbone of p15E of PERV in which the epitopes in the FPPR and MPER were substituted by the corresponding epitopes of 2F5 and 4E10 induced binding antibodies against the HIV epitopes, but despite the exact recognition of the 2F5 epitope, no or very weak neutralization of HIV-1NL4-3 by the immune sera was demonstrated [36].
In immunization studies using subunits of p15E, i.e., recombinant proteins corresponding to the N-terminal, the C-terminal helical region (NHR, CHR) and a p15E with a mutation in the Cys–Cys loop, no MPER-specific neutralizing antibodies were induced, indicating that the Cys-Cys loop is important [24]. However, when the animals were immunized with the FPPR/NHR subunit or the mutated p15E alone, novel neutralizing antibodies binding to the NHR were found. The epitope was IVTEDLQALEKS, and was localized in the NHR at a position where in gp41 of HIV-1 the neutralizing antibodies D5 and HK20 are binding [37,38,39]. Such antibodies were not detected in this immunization study.
Since no animal models are available to study the efficacy of PERV-neutralizing antibodies in vivo, we immunized cats with the corresponding p15E protein of FeLV. This approach induced neutralizing antibodies that bound to epitopes similar to those recognized in PERV p15E [30]. Upon challenge with infectious FeLV, the immunized cats were protected from leukemia [40], demonstrating that these antibodies are capable of neutralizing the virus in vivo.
Because immunization of different animal species with PERV p15E and gp70 consistently elicited neutralizing antibodies targeting identical epitopes critical for the infection process, it is reasonable to suggest that similar protective antibodies could also be induced in non-human primates and humans.
5. Conclusions
Immunization of rats with the recombinant transmembrane (p15E) and surface (gp70) envelope proteins of PERV induced both binding and neutralizing antibodies. The induction of such antibodies across multiple animal species as shown here and previously suggests that immunization of non-human primates and humans may similarly elicit protective responses. Immunization was carried out using an adjuvant comparable to MF59, which is licensed for human use. The neutralizing antibodies targeted epitopes within the FPPR and MPER, regions that play a critical role in the infection process. This study is also relevant for the development of vaccines against HIV-1, as the epitope in the MPER shares sequence and positional similarity with an epitope in the transmembrane envelope protein of HIV-1 that is recognized by a broadly neutralizing antibody from infected patients.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Figure S1: Overview of the overlapping peptides used for the epitope mapping and results of the binding experiments of five rat sera immunized with recombinant p15E at three different dilutions.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, J.D.; methodology, J.B., J.D., L.K.; validation, J.B., J.D., L.K.; formal analysis, J.B., J.D., L.K.; investigation, J.B., J.D., L.K.; resources, J.D.; data curation, J.B., J.D., L.K., B.K.; writing—original draft preparation, J.D., J.B.; writing—review and editing, J.B., J.D., L.K., B.K.; supervision, J.D.; project administration, J.D.; funding acquisition, J.D., B.K. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by Wacker Chemie AG, Munich, Germany, and the APC was funded by the Free University Berlin. J.B. was grateful for the support from the China Scholarship Council (CSC) (Grant No. 202206857010).
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Data is contained within the article or supplementary material.
Acknowledgments
We thank Hagen Richter and Lisa-Marie Eder (Wacker Chemie, Munich, Germany) for their valuable discussions and critical reading of the manuscript.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AIDS | Acquired immunodeficiency syndrome |
| CHR | C-terminal helical region |
| CRISPR/Cas9 | Clustered regularly interspaced short palindromic repeats /CRISPR-associated 9 |
| DMEM | Dulbecco’s modified eagle medium |
| FeLV | Feline leukemia virus |
| FPPR | Fusion peptide proximal region |
| LMU | Ludwig Maximilian University of Munich |
| MPER | Membrane proximal external region |
| MuLV | Murine leukemia virus |
| NHR | N-terminal helical region |
| PERV | Porcine endogenous retrovirus |
| PCMV/PRV | Porcine cytomegalovirus/porcine roseolovirus |
| SCNT | Somatic cell nuclear transfer |
| ZNF | Zinc finger nucleases |
References
- Cooper, D.K.C.; Pierson, R.N., 3rd. Milestones on the path to clinical pig organ xenotransplantation. Am J Transplant. 2023, 23, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Kurome, M.; Kessler, B.; Kemter, E.; Wolf, E. What Genetic Modifications of Source Pigs Are Essential and Sufficient for Cell, Tissue, and Organ Xenotransplantation? Transpl Int. 2024, 37, 13681. [Google Scholar] [CrossRef] [PubMed]
- Denner, j. Virus Safety of Xenotransplantation. Viruses. 2022, 14, 1926. [Google Scholar] [CrossRef]
- Fishman, J.A. Prevention of infection in xenotransplantation: Designated pathogen-free swine in the safety equation. Xenotransplantation. 2020, 27, e12595. [Google Scholar] [CrossRef]
- Griffith, B.P.; Grazioli, A.; Singh, A.K.; Tully, A.; Galindo, J.; Saharia, K.K.; Shah, A.; Strauss, E.R.; Odonkor, P.N.; Williams, B.; Silverman, H.J.; Burke, A.; Drachenberg, C.B.; Wells, C.L.; Dickfeld, T.; Hong, S.N.; Hicks AJ3rd Ananthram, M.; Gupta, A.; Christenson, R.H.; Tamburro, L.; Zhang, T.; Hershfeld, A.; Lewis, B.; Feller, E.D.; Kuravi, K.; Sorrells, L.; Morgand, E.; Mezine, F.; Goutaudier, V.; Rothblatt, M.; Lau, C.L.; Taylor, B.; Perrin, S.; Loupy, A.; Ayares, D.; Mohiuddin, M.M. Transplantation of a genetically modified porcine heart into a live human. Nat Med. 2025, 31, 589–598. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Patience, C.; Magre, S.; Weiss, R.A.; Banerjee, P.T.; Le Tissier, P.; Stoye, J.P. Host range and interference studies of three classes of pig endogenous retrovirus. J. Virol. 1998, 72, 9986–9991. [Google Scholar] [CrossRef]
- Denner, J.; Tönjes, R.R. Infection barriers to successful xenotransplantation focusing on porcine endogenous retroviruses. Clin Microbiol Rev 2012, 25, 318–343. [Google Scholar] [CrossRef] [PubMed]
- Bartosch, B.; Stefanidis, D.; Myers, R.; Weiss, R.; Patience, C.; Takeuchi, Y. Evidence and consequence of porcine endogenous retrovirus recombination. J. Virol. 2004, 78, 13880–13890. [Google Scholar] [CrossRef]
- Denner, J. Why was PERV not transmitted during preclinical and clinical xenotransplantation trials and after inoculation of animals? Retrovirology. 2018, 15, 28. [Google Scholar] [CrossRef]
- Fiebig, U.; Krüger, L.; Denner, J. Determination of the Copy Number of Porcine Endogenous Retroviruses (PERV) in Auckland Island Pigs Repeatedly Used for Clinical Xenotransplantation and Elimination of PERV-C. Microorganisms. 2024, 12, 98. [Google Scholar] [CrossRef]
- Lange, A.; Medugorac, I.; Ali, A.; Kessler, B.; Kurome, M.; Zakhartchenko, V.; Hammer, S.E.; Hauser, A.; Denner, J.; Dobenecker, B.; Wess, G.; Tan, P.L.J.; Garkavenko, O.; Reichart, B.; Wolf, E.; Kemter, E. Genetic diversity, growth and heart function of Auckland Island pigs, a potential source for organ xenotransplantation. Xenotransplantation. 2024, 31, e12858. [Google Scholar] [CrossRef]
- Dieckhoff, B.; Petersen, B.; Kues, W.A.; Kurth, R.; Niemann, H.; Denner, J. Knockdown of porcine endogenous retrovirus (PERV) expression by PERV-specific shRNA in transgenic pigs. Xenotransplantation. 2008, 15, 36–45. [Google Scholar] [CrossRef]
- Ramsoondar, J.; Vaught, T.; Ball, S.; Mendicino, M.; Monahan, J.; Jobst, P.; Vance, A.; Duncan, J.; Wells, K.; Ayares, D. Production of transgenic pigs that express porcine endogenous retrovirus small interfering RNAs. Xenotransplantation. 2009, 16, 164–80. [Google Scholar] [CrossRef] [PubMed]
- Semaan, M.; Kaulitz, D.; Petersen, B.; Niemann, H.; Denner, J. Long-term effects of PERV-specific RNA interference in transgenic pigs. Xenotransplantation 2012, 19, 112–121. [Google Scholar] [CrossRef]
- Semaan, M.; Ivanusic, D.; Denner, J. Cytotoxic Effects during Knock Out of Multiple Porcine Endogenous Retrovirus (PERV) Sequences in the Pig Genome by Zinc Finger Nucleases (ZFN). PLoS One. 2015, 10, e0122059. [Google Scholar] [CrossRef]
- Yang, L.; Güell, M.; Niu, D.; George, H.; Lesha, E.; Grishin, D.; Aach, J.; Shrock, E.; Xu, W.; Poci, J.; Cortazio, R.; Wilkinson, R.A.; Fishman, J.A.; Church, G. Genome-wide inactivation of porcine endogenous retroviruses (PERVs). Science. 2015, 350, 1101–4. [Google Scholar] [CrossRef]
- Niu, D.; Wei, H.J.; Lin, L.; George, H.; Wang, T.; Lee, I.H.; Zhao, H.Y.; Wang, Y.; Kan, Y.; Shrock, E.; Lesha, E.; Wang, G.; Luo, Y.; Qing, Y.; Jiao, D.; Zhao, H.; Zhou, X.; Wang, S.; Wei, H.; Güell, M.; Church, G.M.; Yang, L. Inactivation of porcine endogenous retrovirus in pigs using CRISPR-Cas9. Science. 2017, 357, 1303–1307. [Google Scholar] [CrossRef]
- Kleiser, C.; Schneider, J.; Bayer, H.; Hunsmann, G. Immunoprevention of Friend leukaemia virus-induced erythroleukaemia by vaccination with aggregated gp70. J Gen Virol 1986, 67 (Pt 9) Pt 9, 1901–1907. [Google Scholar] [CrossRef]
- Almeras, T.; Schreiber, P.; Fournel, S.; Martin, V.; Nicolas, C.S.; Fontaine, C.; et al. Comparative efficacy of the Leucofeligen™ FeLV/RCP and Purevax™ RCP FeLV vaccines against infection with circulating feline Calicivirus. BMC Vet Res, 2017; 13, 300. [Google Scholar]
- Fiebig, U.; Stephan, O.; Kurth, R.; Denner, J. Neutralizing antibodies against conserved domains of p15E of porcine endogenous retroviruses: basis for a vaccine for xenotransplantation? Virology 2003, 307, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Kaulitz, D.; Fiebig, U.; Eschricht, M.; Wurzbacher, C.; Kurth, R.; Denner, J. Generation of neutralising antibodies against porcine endogenous retroviruses (PERVs). Virology 2011, 411, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Denner, J.; Mihica, D.; Kaulitz, D.; Schmidt, C.M. Increased titers of neutralizing antibodies after immunization with both envelope proteins of the porcine endogenous retroviruses (PERVs). Virol. J. 2012, 9, 260. [Google Scholar] [CrossRef]
- Waechter, A.; Eschricht, M.; Denner, J. Neutralization of porcine endogenous retrovirus by antibodies against the membrane-proximal external region of the transmembrane envelope protein. J. Gen. Virol. 2013, 94, 643–651. [Google Scholar] [CrossRef]
- Waechter, A.; Denner, J. Novel neutralising antibodies targeting the N-terminal helical region of the transmembrane envelope protein p15E of the porcine endogenous retrovirus (PERV). Immunol. Res. 2014, 58, 9–19. [Google Scholar] [PubMed]
- Ott, G.; Radhakrishnan, R.; Fang, J.H.; Hora, M. The adjuvant MF59: a 10-year perspective. In Vaccine Adjuvants: Preparation Methods and Research Protocols, Methods in Molecular Medicine; O’Hagan, D.T., Ed.; Humana Press: Totowa, NJ, USA, 2000; Volume 42, pp. 211–228. [Google Scholar]
- Marciani, D.J.; Kensil, C.R.; Beltz, G.A.; Hung, C.H.; Cronier, J.; Aubert, A. Genetically-engineered subunit vaccine against feline leukaemia virus: protective immune response in cats. Vaccine 1991, 9, 89–96. [Google Scholar] [CrossRef]
- Behrendt, R.; Fiebig, U.; Norley, S.; Gürtler, L.; Kurth, R.; Denner, J. A neutralization assay for HIV-2 based on measurement of provirus integration by duplex real-time PCR. J. Virol. Methods 2009, 159, 40–46. [Google Scholar] [CrossRef]
- Denner, J.; Specke, V.; Thiesen, U.; Karlas, A.; Kurth, R. Genetic alterations of the long terminal repeat of an ecotropic porcine endogenous retrovirus during passage in human cells. Virology 2003, 314, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Zwick, M.B.; Labrijn, A.F.; Wang, M.; Spenlehauer, C.; Saphire, E.O.; Binley, J.M.; et al. Broadly neutralizing antibodies targeted to the membrane-proximal external region of human immunodeficiency virus type 1 glycoprotein gp41. J. Virol. 2001, 75, 10892–10905. [Google Scholar] [CrossRef]
- Langhammer, S.; Hübner, J.; Kurth, R.; Denner, J. Antibodies neutralizing feline leukaemia virus (FeLV) in cats immunized with the transmembrane envelope protein p15E. Immunology 2006, 117, 229–237. [Google Scholar]
- Munoz-Barroso, I.; Salzwedel, K.; Hunter, E.; Blumenthal, R. Role of the membrane-proximal domain in the initial stages of human immunodeficiency virus type 1 envelope glycoprotein-mediated membrane fusion. J. Virol. 1999, 73, 6089–6092. [Google Scholar]
- Salzwedel, K.; West, J.T.; Hunter, E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 gp41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 1999, 73, 2469–2480. [Google Scholar] [CrossRef] [PubMed]
- Coëffier, E.; Clément, J.M.; Cussac, V.; Khodaei-Boorane, N.; Jehanno, M.; Rojas, M.; Dridi, A.; Latour, M.; El Habib, R.; Barré-Sinoussi, F.; Hofnung, M.; Leclerc, C. Antigenicity and immunogenicity of the HIV-1 gp41 epitope ELDKWA inserted into permissive sites of the MalE protein. Vaccine 2000, 19, 684–693. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Broder, C.C.; Long, D.; Lee, S.A.; Peterson, J.; Chakrabarti, S.; Doms, R.W.; Moss, B. Native oligomeric human immunodeficiency virus type 1 envelope glycoprotein elicits diverse monoclonal antibody reactivities. J. Virol. 1994, 68, 3015–3026. [Google Scholar] [CrossRef] [PubMed]
- Muster, T.; Guinea, R.; Trkola, A.; Purtscher, M.; Klima, A.; Steindl, F.; Palese, P. Cross-neutralizing activity against divergent human immunodeficiency virus type 1 isolates induced by the gp41 sequence ELDKWAS. J. Virol. 1994, 68, 4031–4034. [Google Scholar] [CrossRef]
- Strasz, N.; Morozov, V.A.; Kreutzberger, J.; Keller, M.; Eschricht, M.; Denner, J. Immunization with hybrid proteins containing the membrane proximal external region of HIV-1. AIDS Res. Hum. Retroviruses 2014, 30, 498–508. [Google Scholar] [CrossRef]
- Gustchina, E.; Li, M.; Louis, J.M.; Anderson, D.E.; Lloyd, J.; Frisch, C.; Bewley, C.A.; Gustchina, A.; Wlodawer, A.; Clore, G.M. Structural basis of HIV-1 neutralization by affinity matured Fabs directed against the internal trimeric coiled-coil of gp41. PLoS Pathog. 2010, 6, e1001182. [Google Scholar] [CrossRef]
- Miller, M.D.; Geleziunas, R.; Bianchi, E.; Lennard, S.; Hrin, R.; Zhang, H.; Lu, M.; An, Z.; Ingallinella, P.; Finotto, M.; Mattu, M.; Finnefrock, A.C.; Bramhill, D.; Cook, J.; Eckert, D.M.; Hampton, R.; Patel, M.; Jarantow, S.; Joyce, J.; Ciliberto, G.; Cortese, R.; Lu, P.; Strohl, W.; Schleif, W.; McElhaugh, M.; Lane, S.; Lloyd, C.; Lowe, D.; Osbourn, J.; Vaughan, T.; Emini, E.; Barbato, G.; Kim, P.S.; Hazuda, D.J.; Shiver, J.W.; Pessi, A. A human monoclonal antibody neutralizes diverse HIV-1 isolates by binding a critical gp41 epitope. Proc. Natl. Acad. Sci. USA 2005, 102, 14759–14764. [Google Scholar] [CrossRef]
- Sabin, C.; Corti, D.; Buzon, V.; Seaman, M.S.; Lutje Hulsik, D.; Hinz, A.; Vanzetta, F.; Agatic, G.; Silacci, C.; Mainetti, L.; Scarlatti, G.; Sallusto, F.; Weiss, R.; Lanzavecchia, A.; Weissenhorn, W. Crystal structure and size-dependent neutralization properties of HK20, a human monoclonal antibody binding to the highly conserved heptad repeat 1 of gp41. PLoS Pathog. 2010, 6, e1001195. [Google Scholar] [CrossRef]
- Langhammer, S.; Hübner, J.; Jarrett, O.; Kurth, R.; Denner, J. Immunization with the transmembrane protein of a retrovirus, feline leukemia virus: absence of antigenemia following challenge. Antivir. Res. 2011, 89, 119–123. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic presentation of the recombinant envelope proteins used for immunization. (a), recombinant gp70 protein with pelB leader sequence, partial sequence of p15E and His-tag; (b), recombinant p15E protein with calmodulin binding protein (CBP). PCS, protease cleavage site, numbering according accession number AJ133817.
Figure 1.
Schematic presentation of the recombinant envelope proteins used for immunization. (a), recombinant gp70 protein with pelB leader sequence, partial sequence of p15E and His-tag; (b), recombinant p15E protein with calmodulin binding protein (CBP). PCS, protease cleavage site, numbering according accession number AJ133817.

Figure 2.
(a) Elution profile of the calmodulin binding protein (CBP) affinity chromatography of recombinant p15E. (b) Coomassie blue staining of SDS-PAGE of fractions of the elution process L, protein ladder, 1, lysed cells, 2, supernatant, 3, flow through, 4, wash fraction, 5-9, elution fractions, i.e., recombinant p15E (12 kDa) protein purified by affinity chromatography. (c) Elution profile of the His tag affinity chromatography of recombinant gp70. (d) Coomassie blue staining of the SDS-PAGE of fractions of the elution process. L, protein ladder, 1, lysed cells, 2, soluble fraction, 3, insoluble fraction, 4, 5, cleared lysate filtered by 0.45µM filter, 6, flow through, 7, wash fraction, pH 6.3, 8 wash fraction at pH 5.0, 9, 10, elution fractions at pH 4.5, i.e., recombinant gp70 (54 kDa) purified by affinity chromatography.
Figure 2.
(a) Elution profile of the calmodulin binding protein (CBP) affinity chromatography of recombinant p15E. (b) Coomassie blue staining of SDS-PAGE of fractions of the elution process L, protein ladder, 1, lysed cells, 2, supernatant, 3, flow through, 4, wash fraction, 5-9, elution fractions, i.e., recombinant p15E (12 kDa) protein purified by affinity chromatography. (c) Elution profile of the His tag affinity chromatography of recombinant gp70. (d) Coomassie blue staining of the SDS-PAGE of fractions of the elution process. L, protein ladder, 1, lysed cells, 2, soluble fraction, 3, insoluble fraction, 4, 5, cleared lysate filtered by 0.45µM filter, 6, flow through, 7, wash fraction, pH 6.3, 8 wash fraction at pH 5.0, 9, 10, elution fractions at pH 4.5, i.e., recombinant gp70 (54 kDa) purified by affinity chromatography.
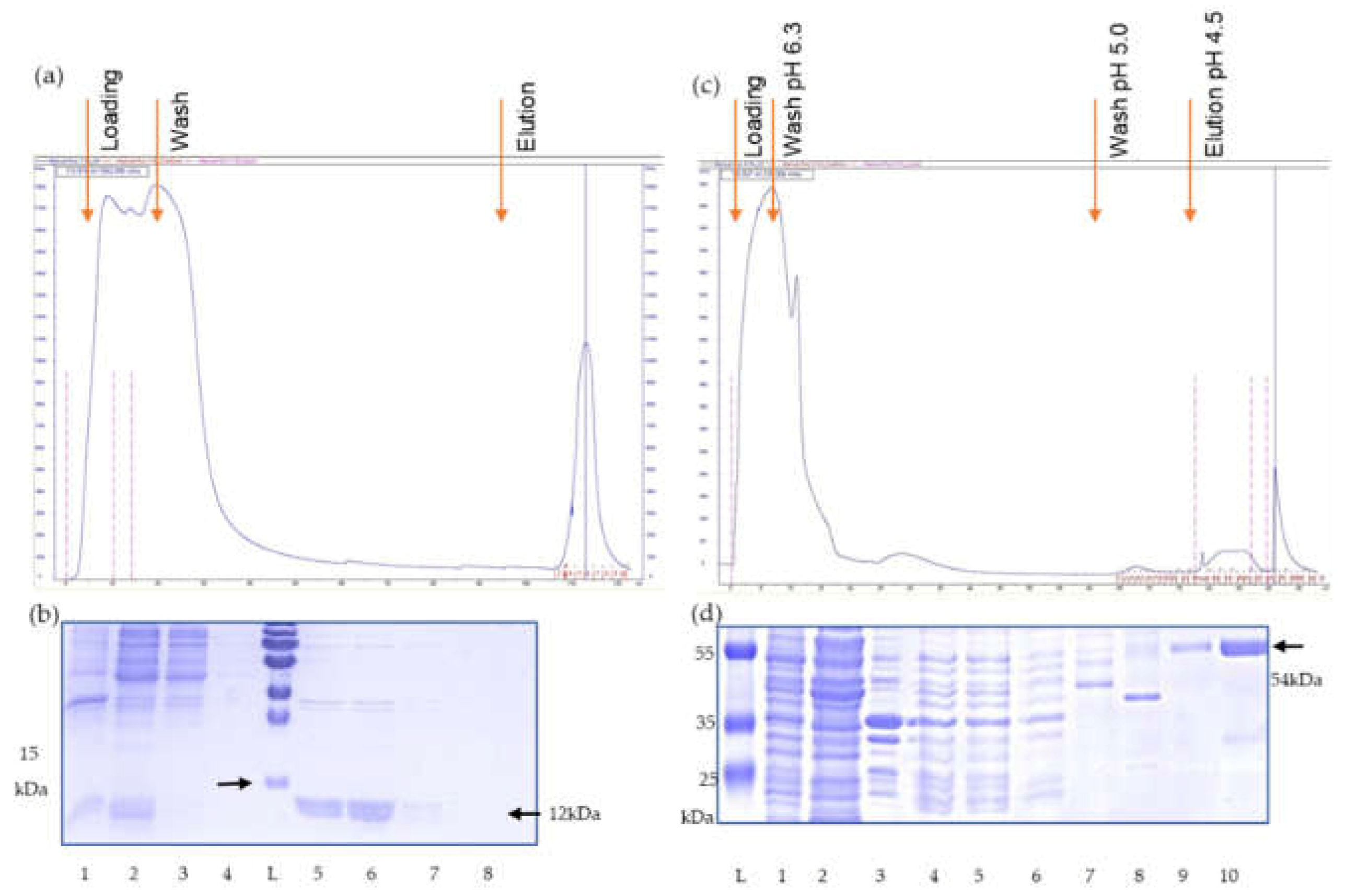
Figure 3.
(a) Western blot analysis of recombinant p15E (12 kDa) protein. Goat anti-p15E serum (1:1000) was used as primary antibody, donkey anti-goat IgG antibody (1:20,000) as secondary antibody; (b) Western blot analysis of recombinant gp70 (54 kDa) protein. Goat anti-gp70 serum (1:1000) was used as primary antibody, donkey anti-goat IgG antibody (1:20,000) as secondary antibody.
Figure 3.
(a) Western blot analysis of recombinant p15E (12 kDa) protein. Goat anti-p15E serum (1:1000) was used as primary antibody, donkey anti-goat IgG antibody (1:20,000) as secondary antibody; (b) Western blot analysis of recombinant gp70 (54 kDa) protein. Goat anti-gp70 serum (1:1000) was used as primary antibody, donkey anti-goat IgG antibody (1:20,000) as secondary antibody.
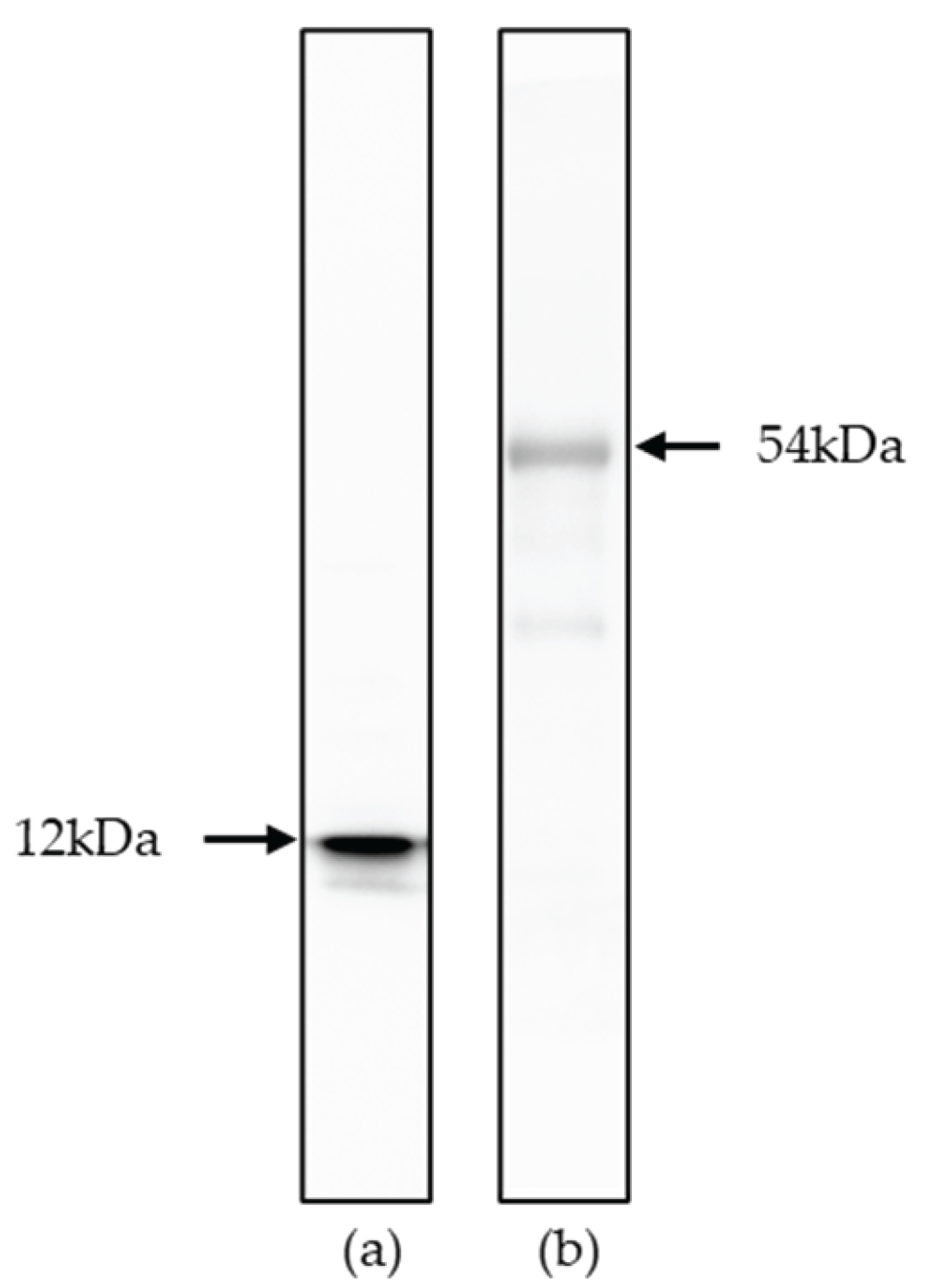
Figure 4.
Scheme of immunization and sample collection. Wistar rats were immunized with 300 µg purified p15E or gp70 protein mixed with the adjuvant AddaVax per rat for the primary immunization and followed by two boosts. Preimmune sera were collected before the immunization. On day 42 final sera were collected.
Figure 4.
Scheme of immunization and sample collection. Wistar rats were immunized with 300 µg purified p15E or gp70 protein mixed with the adjuvant AddaVax per rat for the primary immunization and followed by two boosts. Preimmune sera were collected before the immunization. On day 42 final sera were collected.
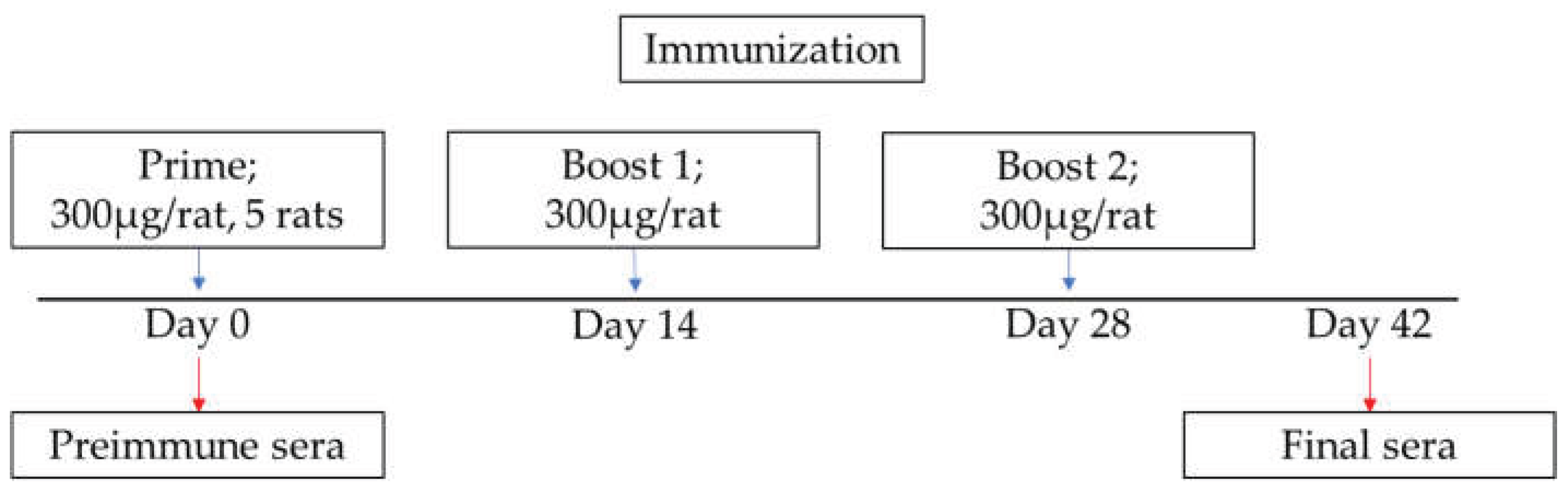
Figure 5.
Results of the Western blot analysis. (a) Titration of rat serum 1 against p15E using purified recombinant p15E protein as antigen. 1, goat serum against p15E, followed by dilutions of the rat serum 2, 1:100; 3, 1:200; 4, 1:500; 6, 1:1000; 6, 1:10,000. (b) Western blot analysis of 5 rat sera anti-p15E using purified recombinant p15E protein as antigen. 1, goat serum against p15E; 2, rat serum 1; 3, rat serum 2; 4, rat serum 3; 5, rat serum 4; 6, rat serum 5; -all at dilution 1:10,00. (c) Titration of rat serum 1 against gp70 using purified recombinant gp70 protein as antigen. 1, goat sera anti-gp70; followed by dilutions of the rat sera 2, 1:100; 3, 1:200; 4, 1:500; 5, 1:1000; 6, 1:10,000. (d) Western blot analysis of rat sera anti-gp70 using purified recombinant gp70 protein as antigen. 1, goat sera against gp70; 2, rat serum 1; 3, rat serum 2; 4, rats serum 3; 5, rat serum 4; 6, rat serum 5, all at dilution 1:1000.
Figure 5.
Results of the Western blot analysis. (a) Titration of rat serum 1 against p15E using purified recombinant p15E protein as antigen. 1, goat serum against p15E, followed by dilutions of the rat serum 2, 1:100; 3, 1:200; 4, 1:500; 6, 1:1000; 6, 1:10,000. (b) Western blot analysis of 5 rat sera anti-p15E using purified recombinant p15E protein as antigen. 1, goat serum against p15E; 2, rat serum 1; 3, rat serum 2; 4, rat serum 3; 5, rat serum 4; 6, rat serum 5; -all at dilution 1:10,00. (c) Titration of rat serum 1 against gp70 using purified recombinant gp70 protein as antigen. 1, goat sera anti-gp70; followed by dilutions of the rat sera 2, 1:100; 3, 1:200; 4, 1:500; 5, 1:1000; 6, 1:10,000. (d) Western blot analysis of rat sera anti-gp70 using purified recombinant gp70 protein as antigen. 1, goat sera against gp70; 2, rat serum 1; 3, rat serum 2; 4, rats serum 3; 5, rat serum 4; 6, rat serum 5, all at dilution 1:1000.

Figure 6.
Results of the neutralization assays. (a), Efficacy of the duplex real-time PCR used in the neutralization assay. A synthetic Gene block containing the sequences of the PERV pol region and of huGAPDH was in 10-fold dilution and a duplex real-time PCR was performed using primers and probes for PERV pol and huGAPDH; (b), the neutralization capacity of five rat sera against p15E, a preimmune serum and a control goat serum against p15E (serum #355) was measured detecting PERV pol using a duplex real-time PCR; (c), corresponding ct values of huGAPDH; (d), the neutralization capacity of five rat sera against gp70, a preimmune serum and goat anti-gp70 (serum #62) were measured detecting PERV pol by duplex real-time PCR; (e), corresponding ct values of huGAPDH.
Figure 6.
Results of the neutralization assays. (a), Efficacy of the duplex real-time PCR used in the neutralization assay. A synthetic Gene block containing the sequences of the PERV pol region and of huGAPDH was in 10-fold dilution and a duplex real-time PCR was performed using primers and probes for PERV pol and huGAPDH; (b), the neutralization capacity of five rat sera against p15E, a preimmune serum and a control goat serum against p15E (serum #355) was measured detecting PERV pol using a duplex real-time PCR; (c), corresponding ct values of huGAPDH; (d), the neutralization capacity of five rat sera against gp70, a preimmune serum and goat anti-gp70 (serum #62) were measured detecting PERV pol by duplex real-time PCR; (e), corresponding ct values of huGAPDH.
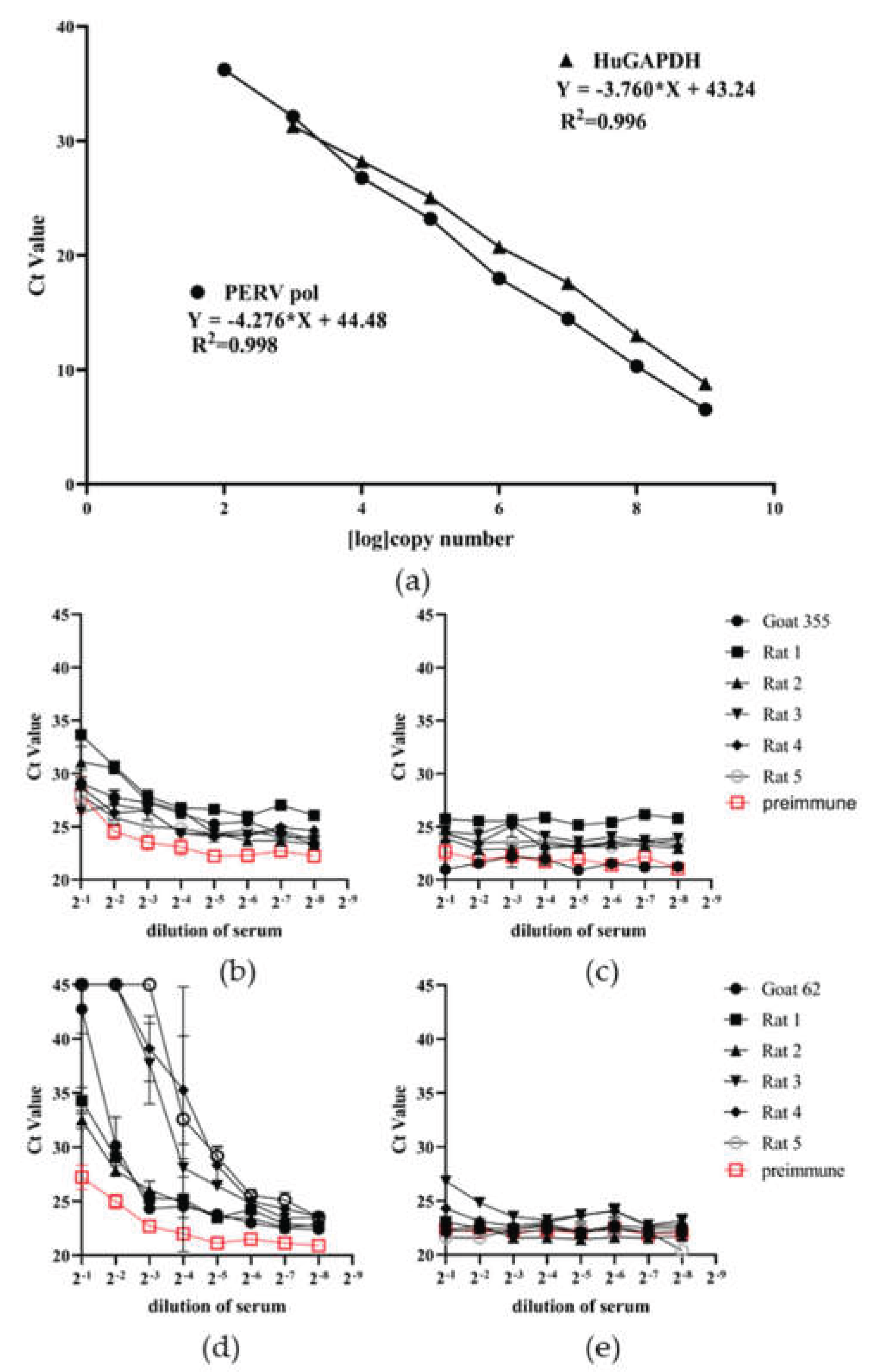
Figure 7.
Epitope mapping of five rat sera against p15E. Heatmap diagram showing all sample incubations; y-axis represents peptide sequences in the library (see Supplementary Table 1), x-axis specifies samples applied. The MMC2 values are shown color coded ranging from white (0 or low intensity) via yellow (middle intensity) to red (high intensity). A, rats serum 1; B, rat serum 2; C, rat serum 3; D, rat serum 4; E, rat serum 5. 1, serum dilution 1:40; 2, serum dilution 1:200; 3, serum dilution 1:1000. Strong signals were obtained with control spots containing the full-length rat IgG demonstrating a correct assay performance (not shown).
Figure 7.
Epitope mapping of five rat sera against p15E. Heatmap diagram showing all sample incubations; y-axis represents peptide sequences in the library (see Supplementary Table 1), x-axis specifies samples applied. The MMC2 values are shown color coded ranging from white (0 or low intensity) via yellow (middle intensity) to red (high intensity). A, rats serum 1; B, rat serum 2; C, rat serum 3; D, rat serum 4; E, rat serum 5. 1, serum dilution 1:40; 2, serum dilution 1:200; 3, serum dilution 1:1000. Strong signals were obtained with control spots containing the full-length rat IgG demonstrating a correct assay performance (not shown).
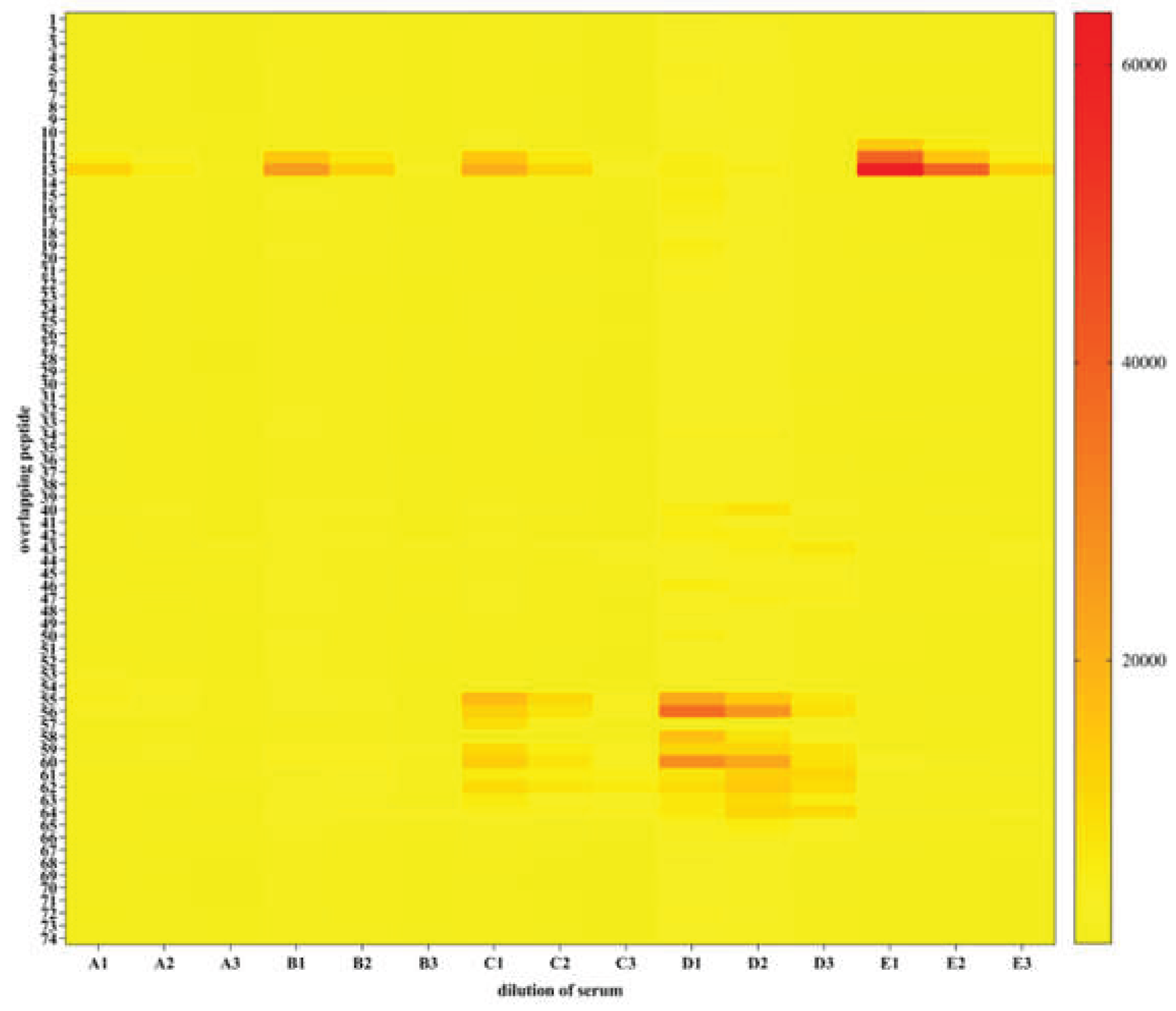
Figure 8.
Determination of the epitopes recognized by rat sera 1,2,3,4 and 5. The numbers of the rats and of the peptides are shown and the epitopes are framed.
Figure 8.
Determination of the epitopes recognized by rat sera 1,2,3,4 and 5. The numbers of the rats and of the peptides are shown and the epitopes are framed.

Figure 9.
Summary of epitopes recognized by five rat sera and one control goat serum (goat #355) against PERV p15E. The epitopes recognized by goat serum 355 were determined previously using another method, based on a cellulose-adsorbed peptide spot library of 15-mer peptides overlapping by 12 amino acids and a detection by chemiluminescence [21].
Figure 9.
Summary of epitopes recognized by five rat sera and one control goat serum (goat #355) against PERV p15E. The epitopes recognized by goat serum 355 were determined previously using another method, based on a cellulose-adsorbed peptide spot library of 15-mer peptides overlapping by 12 amino acids and a detection by chemiluminescence [21].

Figure 10.
A, Sequence comparison between the membrane proximal external region (MPER) of the transmembrane envelope protein PERV p15E and HIV-1 gp41. Identical amino acids are marked in red. The epitopes recognized by the immune serum from rat 3 and the epitopes recognized by the monoclonal antibodies 2F5 and 4E10, isolated from AIDS patients [29] are shown as blue bars. B, Sequence comparison of epitopes E1 and 2Eb of different retroviruses.
Figure 10.
A, Sequence comparison between the membrane proximal external region (MPER) of the transmembrane envelope protein PERV p15E and HIV-1 gp41. Identical amino acids are marked in red. The epitopes recognized by the immune serum from rat 3 and the epitopes recognized by the monoclonal antibodies 2F5 and 4E10, isolated from AIDS patients [29] are shown as blue bars. B, Sequence comparison of epitopes E1 and 2Eb of different retroviruses.

Figure 11.
Localization of the epitopes in the transmembrane envelope proteins p15E and gp41 of PERV and HIV-1, respectively, after interaction of the CHR and NHR during infection. FP, fusion peptide; NHR, N-terminal helical region; C-C loop, cysteine-cysteine loop; CHR, C-terminal helical region; MSD, membrane spanning domain.
Figure 11.
Localization of the epitopes in the transmembrane envelope proteins p15E and gp41 of PERV and HIV-1, respectively, after interaction of the CHR and NHR during infection. FP, fusion peptide; NHR, N-terminal helical region; C-C loop, cysteine-cysteine loop; CHR, C-terminal helical region; MSD, membrane spanning domain.
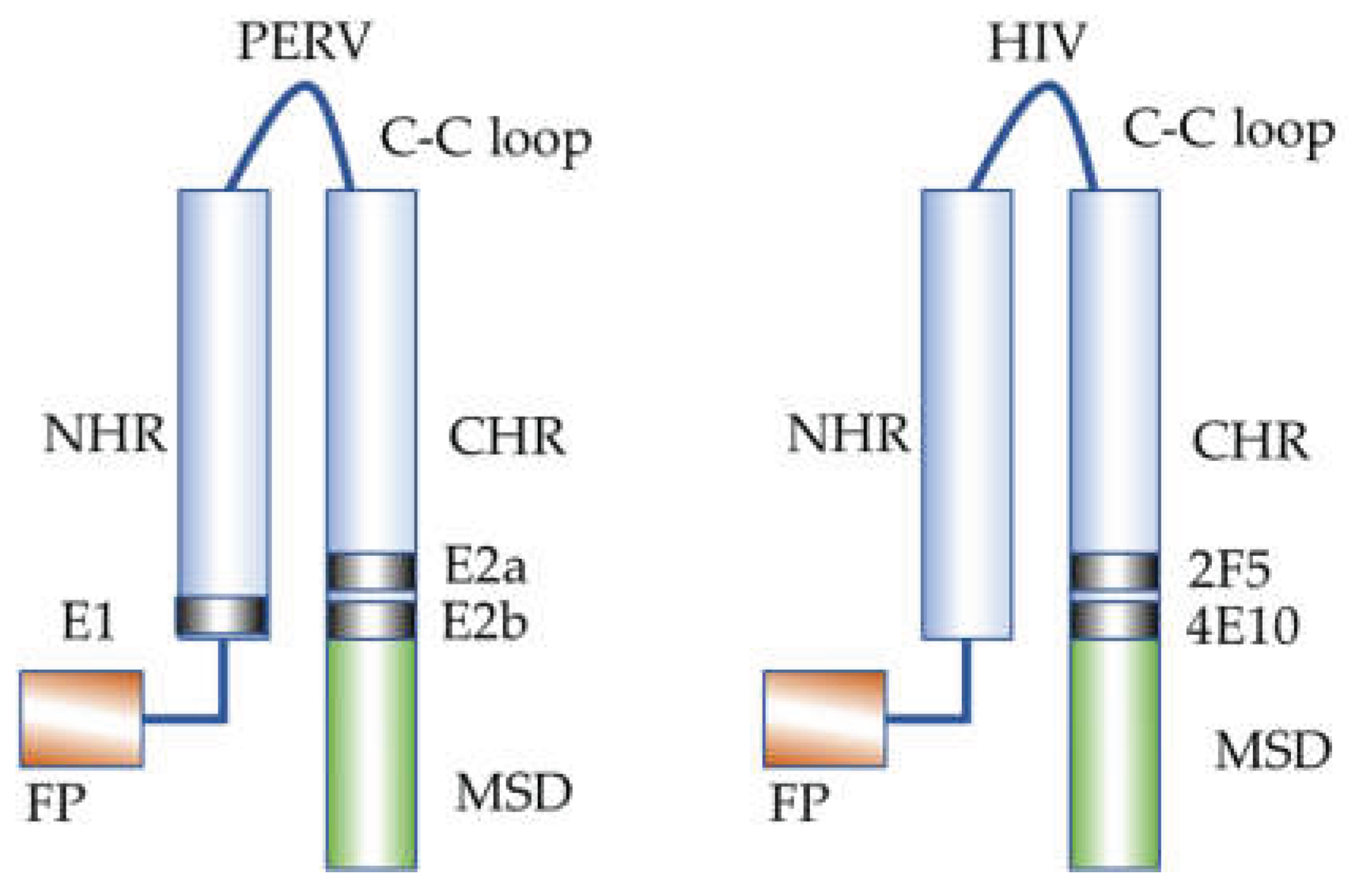

Table 1.
Primers and probes used for the duplex real-time PCR.
| Primer/probe | Sequence 5’-3’ | Direction | Location (nucleotide number) | Accession number | Reference |
|---|---|---|---|---|---|
| PERV pol for | CGACTGCCCCAAGGGTTCAA | + | 3568-3587 | GenBank HM159246 | Yang et al. [16] |
| PERV pol rev | TCTCTCCTGCAAATCTGGGCC | - | 3783-3803 | ||
| PERV pol probe | 6FAM-CACGTACTGGAGGAGGGTCACCTG-BHQ1 | + | 3655-3678 | ||
| GAPDH for | GGCGATGCTGGCGCTGAGTAC | + | 365-385 | GenBank AF261085 | Behrendt et al. [27] |
| GAPDH rev | TGGTTCACACCCATGACGA | - | 495-513 | ||
| GAPDH probe | HEX-CTTCACCACCATGGAGAAGGCTGGG-BHQ1 | + | 407-430 |
for, forward primer; rev, reverse primer
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



