Submitted:
16 September 2025
Posted:
17 September 2025
You are already at the latest version
Abstract
The Hallmarks of Cancer framework distilled the complexity of tumour biology into a shared set of capabilities, but in its static form it cannot capture where in a tumour these traits occur, when they arise, or how they reorganize during cancer initiation and progression. We propose Spatiotemporal Hallmark Ecosystems (SHEs) as a unifying lens that integrates spatial architecture, temporal dynamics, and hallmark biology into a single operational unit of tumour evolution. In this view, hallmarks are not fixed consequences of specific mutations, but emergent, context-dependent programs shaped by interactions between genetic drivers, tissue structure, and systemic cues. SHEs help provide insight into why identical mutations can yield divergent outcomes, why some premalignant states persist for years, and how transitions, whether toward invasion, metastasis, or resistance, reflect coordinated reconfiguration of multiple hallmarks. By reframing cancer as a set of evolving ecosystems rather than isolated traits, this perspective outlines a path toward more predictive, adaptive, and context-aware oncology.
Keywords:
tumour biology
; Cancer Hallmarks
; tumor evolution
; intratumoral heterogeneity
; tumour microenvironment
; spatial transcriptomics
; ecological models of cancer
; therapy resistance
; cancer progression
; biomarker design
Cancer as an Evolving Ecosystem of Hallmark Programs
Cancer is extraordinarily complex, with each tumour exhibiting a unique combination of genetic alterations and cellular behaviors. In a bid to make sense of the extensive inter-tumour diversity, researchers have therefore sought unifying principles. This led to the concept of Cancer Hallmarks: a set of fundamental capabilities and biological traits (e.g., sustained proliferation, immune evasion) commonly acquired by cells to facilitate malignant transformation and subsequent cancer progression [1]. By distilling molecular aberrations into distinct broad biological traits, the Cancer Hallmarks framework provided a roadmap to understand the “endless complexity” of tumour biology [2,3].
However, traditional depictions of the Cancer Hallmarks are static and homogeneous: they describe what a tumour must do, but not where in a tumour these functions occur, nor when during tumorigenesis and throughout the clinical disease course they arise. Critically, a tumour is not a homogeneous mass, but rather, an evolving ecosystem of diverse cells whose interactions and behaviors change over time and space.
In this Perspective, we treat Cancer Hallmarks as tumour-relevant phenotypes: observable traits that emerge from diverse underlying mechanisms. In this framework, whether a tumour achieves sustained proliferation through oncogenic mutations, copy number alterations, epigenetic deregulation, or transcriptional rewiring, what ultimately matters is the acquisition of the phenotype itself. This framing allows us to collapse molecular heterogeneity into a shared functional language that shifts emphasis from the origin of a trait to its consequence, enabling a phenotypic view of tumour behavior that is compatible with spatial and temporal analysis. Building on this logic, we propose a new conceptual framework, Spatiotemporal Hallmark Ecosystems (SHEs), that integrates spatial and temporal heterogeneity into a unified view of how hallmark traits emerge, interact, and evolve within tumours.
Temporal and spatial heterogeneity have long been recognized in cancer. Nowell’s 1976 model of clonal evolution emphasized the importance of sequential genomic events [4], while subsequent iterations of the Hallmarks framework acknowledged the role of the tumour microenvironment (TME) [5,6]. Furthermore, the most recent update to the Hallmarks of Cancer added dimensions such as phenotypic plasticity, non-mutational epigenetic reprogramming, and senescence [7]. Each of these is inherently spatiotemporal [7], for example, plasticity may manifest in spatially zonated stem-like niches [8], while epigenetic programs can shift rapidly in response to localized cues or therapy [9].
Technological advances now make it possible to chart these dimensions in unprecedented detail. Single-cell sequencing has revealed striking variability in gene expression among tumour cells [10], and spatial profiling methods can map tumour and TME phenotypes in situ [6]. With spatially resolved transcriptomics technologies now reaching single-cell resolution [11,12], and new approaches such as SPRINTER enabling clonal-level inference of hallmark activity, like proliferation, across a single tumour’s evolutionary branches [13], it is now increasingly feasible to comprehensively study these interactions in patient samples. Collectively, these innovations set the stage for a new perspective: viewing cancer hallmarks through the lens of tumour structure and progression [14].
While both spatial and temporal heterogeneity have been individually explored across diverse studies, the hallmark framework itself is yet to fully reflect this complexity. By explicitly considering where and when hallmark traits arise, SHEs offer a way to reconcile fragmented observations across modalities and build a mechanistic, testable model of tumour evolution. In the sections that follow, we examine how SHEs manifest in tissue architecture and disease progression, and how they can guide experimental design and clinical strategies.
The SHE framework builds on and integrates concepts from multiple schools of thought in cancer biology. Like the somatic mutation theory (SMT), it recognises mutation-driven potential; like the tissue organisation field theory (TOFT), it accounts for structural and microenvironmental constraints; like ecological models, it emphasises interactions, niche construction, and resource competition; and like critical transition (CT) theory, it focuses on tipping points between system states. It extends the Hallmarks of Cancer from a static list of capabilities to a dynamic, spatiotemporally organised unit of analysis. In doing so, SHEs introduce several advances: they explicitly integrate spatial, temporal, and trait dimensions into a single framework; treat hallmarks as context-dependent state variables that can be measured and modelled; and place transitions, including therapy-induced or exposure-driven reorganisations,at the centre of analysis. They also link physiology and pathology by capturing how normal cyclic or developmental programs can be hijacked and locked into malignant states, operate across biological scales from molecular to clinical, and are inherently compatible with the operational needs of spatial omics in translational research and trials.
The Spatial Organization of Hallmarks
Tumours are composed of mosaics of distinct cellular neighborhoods. A growing body of evidence indicates that the canonical Cancer Hallmarks are not uniformly active throughout a tumour mass. Instead, they are compartmentalized in space, reflecting interactions between cancer cells and the TME. Recent spatial transcriptomic studies across multiple tumour types have revealed that most hallmarks segregate by compartment, with some predominant in cancer cells, others in the surrounding cells of the TME [15,16]. In one comprehensive analysis of 91 tumours (spanning 10 cancer types), 7 of 13 hallmark programs were found to be consistently more active in the malignant epithelial compartment, while the remaining hallmarks were governed largely by TME cells [15]. This essentially binary partition of hallmark activity between tumour vs. stroma, which had been suspected, is now empirically confirmed across diverse cancers. Notably, these spatial patterns are non-random and remarkably structured, akin to an organized ecosystem rather than a haphazard cell mixture [17]. Therefore, cell identity and location emerge as key determinants of which hallmark processes are active in specific compartments of a tumour mass.
Surprising insights have surfaced from mapping hallmarks in situ. The hallmark “sustaining proliferative signaling” (intuitively associated with cancer cells themselves) was found to be more active in the TME at the tumour’s invasive margins [15,16]. This counterintuitive finding aligns with earlier reports that even pre-cancerous stromal environments can release signals driving adjacent epithelial cells to proliferate [18]. In other words, surrounding fibroblasts, immune cells, and other TME components can act as the engine of tumour cell proliferation, especially at the tumour edge, effectively sustaining growth signals for the incipient cancer [19]. Conversely, within the tumour core, cancer cells often activate programs for autonomy and survival that need less direct TME input, such as, resisting cell death, enabling replicative immortality, and accumulating genome instability [15]. These traits ensure survival and unlimited growth in the core, an area relatively sheltered from immune attack and rich in tumour cell density. At the tumour’s periphery, cancer cells instead upregulate hallmarks that facilitate invasion and adaptation, such as evading growth suppressors or undergoing non-mutational epigenetic reprogramming to increase plasticity, suggesting that cancer cells at the invasive front confront normal tissue boundaries and immune surveillance, necessitating them to be highly adaptable [15].
The TME plays an active role in shaping hallmark activity across space [20]. In many regions, immune and stromal cells express hallmark programs that complement those of nearby tumour cells. Cells from the TME at the tumour boundary often display Hallmarks such as “tumour-promoting inflammation” and “avoiding immune destruction” [15] whereas deeper regions of the TME (further from cancer cells) are enriched for hallmarks such as “inducing angiogenesis” and “activating invasion and metastasis”. This suggests that blood vessel formation and invasive behavior often originate in the outer regions of the stromal compartment, facilitating outward tumour expansion and access to vasculature. Thus, a clear spatial architecture of hallmarks emerges: the tumour core is optimized for unchecked cell-autonomous growth, the invasive front is a zone of intense tumour–TME interaction and adaptation, and the outer TME supports processes for dissemination and nutrient supply. The interdependence is striking: wherever a cancer cell hallmark is active, nearby TME cells often exhibit complementary hallmarks that together form a functional unit, akin to ecological niches within the tumour [17,21].
Importantly, this spatial complementarity can be quantified and carries predictive value. Hallmark programs in tumour and TME compartments have been shown to exhibit measurable spatial interdependence, as captured by spatial autocorrelation and cross-correlation metrics [15]. The spatial relationship between different Cancer Hallmarks (i.e., the configuration of their “Hallmark Ecosystem”) can impact sensitivity to neoadjuvant chemotherapy or immunotherapy in patients with bladder cancer, suggesting that coordinated spatial organization can reflect underlying ecosystem dynamics with clinical relevance [15,22].
Spatial context can also constrain the expansion and expression of hallmark traits in specific clones. In normal epithelium, clonal expansion has been shown to depend on the fitness of neighboring cells, a principle known as neighbor-constrained fitness (NCF). When multiple clones grow in proximity, their expansion can be limited by mechanical boundaries or local competition, preventing even strongly mutated clones from dominating [23]. This suggests that even hallmark programs driven by strong oncogenic mutations may be modulated, or even suppressed, by spatially structured competition or interactions with the TME. Consistent with this, recent work in mammary epithelium shows that most Brca1;Trp53 mutant clones are eliminated through local remodeling processes, and only a minority survive to expand [24]. These findings suggest that spatial constraints can act as a filter on clonal expansion and on whether and where hallmark programs emerge.
Furthermore, the relationship between cell types and hallmark programs is inherently complex. A single hallmark such as angiogenesis, may involve diverse contributors, including fibroblasts, endothelial cells, and macrophages, each playing a distinct role in the overall process [19]. Conversely, a single cell type can engage in multiple hallmarks depending on its spatial position and local context. For instance, a macrophage at the tumour core may contribute to inflammation and immune evasion, while the same lineage at the periphery supports invasion or remodeling [25]. This functional plasticity, recently described in depth for fibroblasts [26] among others, means that hallmark signatures often reflect composite activity across heterogeneous populations. However, despite this cellular heterogeneity, hallmark programs tend to show consistent spatial organization, as they form regional patterns that become evident only when cells are analyzed in situ. Spatial transcriptomics thus reveals structured hallmark activity that may be obscured in bulk or dissociated single-cell profiles.
Beyond variation across cell types, spatial and single-cell data have also revealed [10,27] hallmark specialization at the clonal level, reflecting genetic divergence, environmental filtering, or both. In many cancers, no individual clone single-handedly executes all Cancer Hallmarks. Instead, different subclones tend to specialize in certain Hallmarks. In fact, as cancer clones within the same tumour evolve and phylogenetically diverge from each other at the genetic level, they tend to also diverge in hallmark activity profiles [22,28]. This, eventually, can lead to clone specialization, and segregation of functions within the tumour, with each clone exhibiting high activity of a particular subset of hallmarks and, therefore, phenotypic heterogeneity [29,30]. Importantly, this functional specialization can enhance overall tumour fitness. For example, if the activation of one Cancer Hallmark limits the resources available to the cell to activate a second Hallmark in the same cell (due to metabolic or regulatory trade-offs, for example), the tumour can maximize both Hallmarks if each clone maximizes a single hallmark programme [31]. This apparent specialization may reflect evolutionary pressures that favor complementary hallmark programs across subclones, as this would improve the overall fitness at the ecosystem level [32].
At the same time, tumours are not completely compartmentalized into one-hallmark clones. There may be cases of phenotypic convergence (distantly related clones displaying similar Hallmark profiles) and phenotypic divergence without genomic divergence (clones with the same genetic profile showing different Hallmark activation). The latter is especially intriguing: it implies that factors beyond DNA sequence (e.g., epigenetic states, local TME, stochastic variation) can lead two clones with the same genotype that follow different Hallmark paths [15,33]. These non-genetic influences can shape the tumour phenotype in a spatially localized manner, contributing to intratumoral heterogeneity that would not be discerned through sequencing approaches.
Taken together, these findings support the approach that tumours can be conceptualized as SHEs: dynamic, zonated communities in which distinct hallmark programs emerge, interact, and evolve [14]. Cancer and TME cells continuously interact and mutually influence each other’s hallmark behaviors, creating regional ecosystems within the tumour [34]. Appreciating this spatial architecture is not only theoretically important, but also has clinical relevance, since the location of a Hallmark with respect to others, can affect how a tumour develops, disseminates, and responds to therapy.
The Temporal Evolution of Hallmarks
The traditional Cancer Hallmark framework describes hallmark traits as intrinsic capabilities acquired in a relatively linear sequence. This view underestimates their temporal plasticity and the influence of dynamic selective pressures. Increasingly, tumour evolution studies have shown that hallmark traits fluctuate over time, shift in dominance, and arise in response to clonal competition, microenvironmental adaptation, and therapy [35].
Longitudinal studies like TRACERx in non-small cell lung cancer (NSCLC) [36,37,38,39,40,41,42,43], have shown that hallmark traits such as proliferation and immune evasion are unevenly expressed and evolve with disease progression. Genomic instability, often triggered by early TP53 loss or chromosomal missegregation, seeds clonal diversity that fuels the emergence of distinct phenotypic programs under selective pressure [10].
Foundational hallmarks such as genomic instability, unchecked proliferation, and replicative immortality often appear early in tumorigenesis, driven by canonical oncogenic mutations (e.g., in EGFR, BRAF, FGFR3) [44]. However, studies of normal tissues have shown that many of these alterations can persist for years without leading to malignancy, suggesting while additional cooperating mutations are typically required, even these are not sufficient on their own for hallmark activation. Instead, spatial and temporal context, including niche competition, immune surveillance, and tissue remodeling, likely determines whether these mutations give rise to malignant phenotypes. As selective pressures accumulate, some clones may gain additional capabilities such as angiogenesis, metabolic rewiring [45], or enhanced motility via EMT [46], enabling them to overcome local constraints and expand.
These filtering principles are already evident in normal tissue contexts, where clonal expansion and hallmark emergence are tightly regulated over time, even in the presence of oncogenic mutations. In the case of breast cancer, for example, recent in vivo lineage tracing in mammary epithelium [24] has shown that even cells harboring early oncogenic mutations (such as Brca1 and Trp53 loss) can expand widely across ductal networks without immediate transformation. Their fate is constrained by the tissue’s spatial architecture and by temporal cycles of remodeling, such as those driven by the oestrous cycle. These physiological cycles selectively eliminate most mutant clones, allowing only a few to survive and clonally expand, creating fields of cancer-prone tissue without overt malignancy. A similar principle has been observed in the esophageal epithelium, where NOTCH1 mutations, although frequent and positively selected in normal tissue [23], do not inevitably lead to cancer, underscoring how context and ecological constraints shape whether oncogenic alterations translate into hallmark activation. This highlights that hallmark emergence is not solely mutation-driven, but shaped by dynamic, tissue-level filters that operate over time (e.g., cyclical remodeling, spatial competition, or developmental timing) which collectively determine whether, when, and where hallmark programs are executed [14,24].
Importantly, the order of hallmark acquisition is not fixed. While proliferative traits are often dominant early, other hallmarks (e.g., immune evasion, metabolic adaptation, or angiogenesis) can arise at various stages, particularly in inflammation-prone or immunologically active tissues. Rather than being linear, hallmark evolution appears to follow a branching, context-specific trajectories shaped by both intrinsic genotype and extrinsic ecological pressures.
The temporal plasticity of Hallmark activation is further modulated by interactions between cancer cells and the surrounding TME [47]. Fibroblasts, myeloid cells, and lymphocytes dynamically respond to tumour-derived signals: remodeling the extracellular matrix, producing immunosuppressive cytokines, or inducing neovascularization. These effects are highly context-dependent: fibroblast subtypes can switch between pro- and anti-tumour phenotypes based on cytokine cues or therapeutic pressure, while chronic hypoxia selects for cells with increased HIF-1α activity, glycolysis, and autophagy [48], supporting survival under low-oxygen conditions. These responses in turn promote or suppress specific Hallmark programs in nearby cancer cells [49,50]. Thus, Cancer Hallmarks are not merely cell-autonomous features, but emergent properties of spatially and temporally evolving cell-to-cell interactions.
External interventions, particularly cancer therapies, can add potent layers of selective pressure. For example, in lung adenocarcinoma, EGFR inhibitors initially suppress proliferative hallmarks but select for subclones with resistance mutations (e.g., T790M or C797S), activation of bypass signaling (e.g., MET amplification), or histological transformation [51]. These adaptations often coincide with broader Hallmark remodeling, including reduced apoptotic priming, immune escape, and metabolic adaptation. Anti-angiogenic therapies limit vascular supply, leading to selection for clones capable of surviving in nutrient- and oxygen-poor environments through metabolic rewiring or induction of vasculogenic mimicry [52]. Likewise, immune checkpoint blockade creates a distinct set of selective pressures, particularly on adaptive hallmarks. Tumours under immune pressure may lose their antigen-presenting machinery [53], acquire specific mutations (e.g., in JAK1, JAK2, or B2M) [54], or upregulate immunosuppressive ligands such as PD-L1, LAG-3, or TIGIT [55]. These adaptations not only enhance immune evasion but might also influence other Cancer Hallmarks, such as reducing interferon-driven growth arrest or shifting metabolic profiles to outcompete infiltrating immune cells in the tumour niche.
Finally, insights from research autopsy programs such as the UK-wide PEACE [56] study (NCT03004755) have revealed how metastatic sites can impose distinct evolutionary constraints [57]. These programs provide unique access to end-stage tumours and metastatic sites, allowing to reconstruct hallmark evolution across space and time [58]. Autopsy-based studies have shown that metastases are not merely clonal offshoots of the primary tumour but often exhibit distinct hallmark activity adapted to the target organ. Brain metastasis, for example, frequently shows enhanced oxidative phosphorylation and immune suppression [59,60,61], while bone metastasis often favours survival and dormancy programs [62]. These findings illustrate how hallmark evolution is both temporally dynamic and spatially contingent, shaped by local niches throughout disease progression, and may even converge across genetically distinct subclones through parallel evolution, as tumours adapt to the same selective pressures within specific metastatic niches [63,64] [65].
Spatiotemporal Hallmark Ecosystems: A Conceptual Framework
The preceding sections explored how hallmark traits are shaped by the spatial structure of tumours and their temporal evolution across disease progression. Spatial analyses reveal compartmentalized, niche-specific programs that coordinate hallmark activity across cell types and tumour regions. Temporal studies, in turn, show how hallmark traits emerge, recede, or reconfigure under evolutionary and therapeutic pressures. But these are not separate processes: spatial organization and temporal change are deeply intertwined.
To fully capture how hallmark programs arise and persist, we propose the concept of SHEs: dynamic, multicellular systems in which hallmark traits emerge from context-dependent interactions across space and time [66]. Rather than existing as static traits confined to individual cells, hallmarks in this framework are ecosystem-level properties that evolve with tumour architecture and selective pressure (Figure 1).
Several prior studies have proposed ecological models of cancer, drawing parallels between tumours and ecosystems to highlight processes such as cellular competition, niche construction, and resource limitation [21,67,68,69,70,71,72,73]. These frameworks have enriched our understanding of tumour biology by emphasizing non–cell-autonomous behaviors and the role of the TME. What distinguishes SHEs is the explicit framing of hallmark traits as emergent ecosystem-level properties whose implementation depends on spatial context and temporal dynamics.
SHEs are governed by recursive feedback loops. Oncogenic mutations may reshape the local microenvironment (e.g., by promoting angiogenesis or altering immune cell recruitment) which in turn changes the selective pressures acting on nearby clones. A clone’s survival and contribution to hallmark programs thus depends not only on its genotype, but on its ability to engage with and shape its niche. This mutual dependency creates ecological filters that determine which hallmark configurations persist.
This perspective also reframes key transitions in cancer progression (Box 1). For example, the transformation of follicular lymphoma into diffuse large B-cell lymphoma is not merely driven by new mutations but represents a systems-level reorganization of the tumour ecosystem in the lymphoid tissue [74]. Malignant B cells become more proliferative and immune-evasive, while stromal and immune cell networks simultaneously shift to permit unchecked outgrowth [75,76]. In this view, transformation marks a tipping point in the coordination and stabilization of hallmark programs. It is an ecological threshold rather than a purely molecular switch.
Although the primary unit of analysis in SHEs is the local tumour microenvironment, we recognize that hallmark stabilization and ecosystem transitions are shaped by external, host-level forces [3]. Age, metabolic status, systemic inflammation, immune surveillance, and circadian rhythms impose time-varying constraints on how tumours evolve, respond to therapy, or remain dormant. These systemic cues interact with local ecosystems, modulating the likelihood of hallmark acquisition and ecological reorganization. Rather than treating these as separate models, we view host-level inputs as temporally dynamic modulators of local SHE states.
By mapping how hallmark traits are implemented, coordinated, and stabilized over time and space, SHEs provide a scaffold to interpret diverse data types, from spatial transcriptomics to longitudinal genomics, and to identify which tumours are poised for transition, resistance, or dissemination. Importantly, this framing shifts focus away from isolated markers toward ecosystem architectures, offering a more holistic, dynamic model for both basic research and translational application.
Box 1 - Comparative interpretations: traditional models vs the SHE framework
The Spatiotemporal Hallmark Ecosystem (SHE) framework offers a complementary perspective on well-known cancer phenomena by integrating spatial organization, temporal dynamics, and ecological interactions. Below, we contrast traditional molecular interpretations with SHE-based insights to highlight how this framework can generate novel hypotheses and refine mechanistic understanding.
- Oncogenic mutations in normal tissue
Traditional view: Mutations accumulate silently until a second hit enables transformation.
SHE interpretation: Mutant clones exist in ecosystems that suppress hallmark activation—through spatial constraints, immune surveillance, or niche competition. Transformation requires ecological permissiveness, not just additional mutations.
- Intratumoral heterogeneity
Traditional view: Genetic diversification produces subclones with different phenotypes.
SHE interpretation: Functional traits may be distributed across clones due to ecological trade-offs. Heterogeneity is not only genetic but reflects adaptive specialization and division of labor across the tumour ecosystem.
- Therapeutic resistance
Traditional view: Resistance emerges from pre-existing or acquired mutations in individual clones.
SHE interpretation: Resistance is a property of spatially resilient ecosystems. Even if a resistant clone is eliminated, the niche may reconstitute resistance through ecological buffering and plasticity.
- Transformation events
Traditional view: Transformation results from accumulation of new driver mutations.
SHE interpretation: Transformation is an ecosystem tipping point—where hallmark programs become co-activated and stabilized across the spatial network of tumour and stromal cells.
- Metastatic colonization
Traditional view: Successful metastasis requires genetic adaptation to new tissue environments.
SHE interpretation: Metastasis requires re-establishing a functional hallmark-supporting ecosystem. Failure to do so explains dormancy or non-viable micrometastases, even when disseminated clones are viable.
These comparisons do not reject classical models but extend them. The SHE framework adds a layer of ecological logic, explaining not just the what of cancer progression, but the where, when, and how these traits are implemented across dynamic multicellular systems.
Current Technical and Conceptual Limitations
Efforts to map cancer hallmarks in space and time are advancing rapidly, but remain constrained by critical technical and conceptual challenges. Chief among them is the disconnect between spatial and temporal modalities: spatial studies rely predominantly on transcriptomic data, while temporal analyses are often grounded in genomic sequencing. As a result, we are trying to reconstruct spatiotemporal ecosystems using data that are fragmented both in technology and biological focus, with one capturing mRNA expression in place, and the other tracking DNA-based clonal dynamics over time, but usually without spatial resolution.
Not all hallmarks are equally measurable under these conditions. Traits like proliferation or immune activation are relatively easy to infer from gene expression, especially when cell-type-specific signatures are available. Others such as genome instability, senescence, or epigenetic reprogramming, are far less tractable, especially using transcriptomics alone. Some hallmarks are best conceptualized as being mutation-driven (e.g., genome instability), while others are downstream products of transcriptional or signaling programs. But even for expression-driven hallmarks, inference is limited by platform-specific biases, gene panel constraints, and cross-study variability.
Additional complexity arises from the fact that hallmark programs do not map cleanly to specific genes or cell types. Many genes participate in multiple hallmarks, and many cell types express different hallmark traits depending on their spatial context. This many-to-many mapping complicates interpretation and risks conflating distinct biological processes. Without integrated, multimodal approaches that can link genotype, phenotype, and context, we remain limited in our ability to accurately resolve hallmark dynamics.
Despite these limitations, the framework of Spatiotemporal Hallmark Ecosystems remains useful, precisely because it accommodates these uncertainties. By treating hallmarks as emergent, ecosystem-level properties [67] rather than binary traits, we gain a conceptual structure that can evolve alongside the technologies used to study it. The following sections examine how spatial and temporal data, even in their current form, can already begin to illuminate the organization and evolution of hallmark traits in cancer.
Charting Hallmarks in Motion: Implications for Cancer Research
The SHE framework provides a unifying lens through which to reinterpret long-standing observations and unresolved paradoxes in cancer biology [77,78]. Spatial transcriptomics, single-cell genomics, lineage tracing, and longitudinal sampling have each illuminated fragments of hallmark behavior. However, without a coherent model, these insights remain siloed. SHEs offer that model by treating hallmark traits not as isolated, static features, but as emergent, ecosystem-level properties shaped by spatial context and temporal dynamics.
This reframing invites researchers to move beyond binary presence/absence descriptions of hallmark traits, toward probing their dependencies, hierarchies, and vulnerabilities. For example, identifying which combinations of hallmark programs co-occur within a spatial niche, or which traits emerge only after a specific ecological or mutational shift, becomes central to understanding progression. SHEs shift the analytical focus from whether a hallmark is present to why, where, and under what conditions it becomes dominant or dispensable. Importantly, this framework helps reinterpret several foundational concepts in cancer biology through a new, spatiotemporal lens.
It may explain, for instance, a persistent paradox: why oncogenic mutations are frequently found in normal tissues without leading to malignancy [23,79,80]. From a SHE perspective, this reflects ecosystems in which pro-tumorigenic traits fail to stabilize. Spatial compartmentalization, immune surveillance, ecological competition, or lack of enabling signals may prevent the emergence of fully functional hallmark programs; thus positive selection of oncogenic mutations is observed yet only limited clonal expansions occur. Studying these abortive ecosystems could reveal early constraints on transformation and suggest new avenues for cancer prevention.
The SHE model also challenges the traditional dichotomy between driver and passenger mutations [81,82]. A mutation may promote hallmark expression in one spatial or temporal context but become irrelevant, or even detrimental, as the ecosystem evolves [83]. Conversely, a mutation previously labeled as a passenger may become functionally important if the context shifts. In this view, driver status is not a binary label but a context-dependent state, shaped by both microenvironment and timing. This is consistent with recent reevaluations of the promoter model of cancer initiation.
Intratumoral heterogeneity, long viewed as an analytical challenge, becomes in this framework a necessary substrate for plasticity [84]. Diverse clones and cell states are not simply noise; they enable hallmark programs to be flexibly distributed, buffered, or reconfigured over time. The resilience of a tumour may depend not only on which hallmarks it expresses, but on how adaptively it can rewire their implementation across spatial compartments and different phases of tumour evolution, from early lesion development through progression, therapy response, and late-stage disease.
SHEs may also help reinterpret metastasis. Successful colonization of a secondary site likely requires more than just dissemination of aggressive clones. It demands the stepwise reconstruction of a hallmark-supporting ecosystem under new constraints [85]. This process may unfold slowly and stochastically, which may help explain why many micrometastases fail to grow, and why clinical metastases can often emerge years after initial seeding.
Methodologically, these insights call for integrated, multimodal studies. Spatial transcriptomics alone cannot capture how mutations reshape tumour niches [28,86]. Lineage tracing alone misses the phenotypic transitions that precede selection [87]. Cohorts that combine spatial proteogenomics, in situ clonal tracking, and temporally resolved sampling [37,88,89,90] are essential to reconstruct hallmark trajectories. The SHE framework provides a conceptual map to interpret these complex datasets.
Certain transitions, such as FL to tFL [91], NMIBC to MIBC [92], PanIN to PDAC [20,93], or DCIS to invasive breast cancer [94,95], offer tractable models for studying SHE dynamics. These systems retain some spatial structure while undergoing evolutionary and ecological reprogramming, making them ideal for identifying which configurations mark irreversible transformation, or resistance to it (Box 2).
By shifting focus from isolated mutations to the architecture and dynamics of hallmark ecosystems, SHEs offer a new logic for experimental design. They invite research questions that probe not just which traits are present, but how and where they are coordinated, and under what pressures they change.
Box 2 - Illustrative examples where the SHE framework reframes cancer progression
These six transitions illustrate how Spatiotemporal Hallmark Ecosystems (SHEs) integrate genetic drivers, ecological context, and temporal change to explain oncogenesis, progression, and therapy response.
Follicular lymphoma → diffuse large B-cell lymphoma (FL→DLBCL): Gain-of-function EZH2 mutations alter germinal centre B-cell epigenetic programs, locking in proliferative and immune-evasive hallmarks. In the SHE view, these molecular changes reprogram the lymphoid niche—remodelling stromal and immune cell networks—which in turn stabilise the very hallmarks initiated by the mutation. Transformation marks an ecological tipping point where multiple hallmark programs co-activate across the tumour–stroma network.
Non-muscle-invasive → muscle-invasive bladder cancer (NMIBC→MIBC): Activating FGFR3 mutations are common in NMIBC, initiating proliferative hallmarks that are spatially constrained by immune-active and stromal compartments. Over time, the tumour and microenvironment co-evolve: immune surveillance is attenuated, stromal barriers are remodelled, and invasion-related hallmarks take hold. Progression reflects both the breakdown of these spatial constraints and the re-assembly of hallmarks into a more plastic, invasive ecosystem.
Brain metastasis dormancy → activation: Dormant micrometastases persist in the brain for years within niches that suppress proliferation and angiogenesis hallmarks. Activation follows microenvironmental changes—vascular remodelling, astrocyte reprogramming, or immune exclusion—that lift these constraints. SHE framing captures this as a shift from a low-complexity, stable hallmark configuration to a high-complexity, growth-permissive state, with spatial proximity of angiogenesis and immune evasion programs as a key enabler.
Ewing sarcoma emergence during puberty: The EWSR1–ETS fusion is an aberrant transcription factor that drives proliferative and migratory hallmarks, but these often remain latent in the pre-pubertal bone environment. Pubertal growth plate expansion and hormone-driven remodelling introduce angiogenic, proliferative, and matrix-remodelling cues. The oncogenic program and the transient, permissive niche synergise, locking in malignant hallmarks that would otherwise be switched off when development stabilises.
Microsatellite instability–high colorectal cancer under immune checkpoint blockade (MSI-H CRC + ICB): PD-1 blockade initially dismantles immune-evasion hallmarks in MSI-H CRC, driving tumour regression. Some tumours, however, reorganise their ecosystems: myeloid infiltration rises, T-cell exclusion zones emerge, and cancer cells adapt metabolically to inflamed niches. Resistance is thus an ecosystem-level phenomenon, where hallmarks are re-configured in space and time to restore growth despite continued therapy.
EGFR-mutant lung cancer under chronic pollution exposure: Chronic PM2.5 exposure induces repeated airway injury–repair cycles that activate inflammation, proliferation, and tissue-remodelling hallmarks. In EGFR-mutant lung epithelium, these environmental programs promote expansion of initiated clones without requiring new driver mutations. The SHE lens frames pollution as a chronic promoter that reshapes spatial niches, accelerating progression by stabilising hallmark configurations favourable to tumour growth.
From Maps to Models: Clinical Applications of SHEs
While Spatiotemporal Hallmark Ecosystems (SHEs) are not yet ready for translation into clinical use, they offer a powerful conceptual scaffold for understanding therapeutic response, resistance, and tumour evolution in the clinic. Many of the challenges faced in precision oncology (predicting heterogeneous responses, explaining relapse, or identifying optimal treatment windows) stem not from a lack of biomarkers, but from a failure to account for the spatial and temporal organization of hallmark programs. The SHE framework can help address this gap.
Most current biomarkers capture only limited biological context. Tissue biopsies and ctDNA assays (even the FDA-approved EGFR plasma test in lung cancer) typically provide a snapshot at a single time point, while classical tumour markers in GI, pancreatic, or ovarian cancers are tracked longitudinally but remain bulk, systemic readouts with little spatial resolution. These approaches assume that if a hallmark-associated mutation or transcript is detected, the corresponding phenotype is active. In reality, the activity of a hallmark depends on whether it is stabilized within a supportive ecosystem. A proliferation marker in a spatially constrained niche may not predict outgrowth; immune evasion genes may fail to matter if suppressive myeloid cells or exclusionary stroma are absent. SHEs recast biomarkers as context-dependent signals whose meaning comes from their spatial and temporal embedding.
This perspective also explains regional and temporal resistance. Therapy failure often emerges from niches where cancer, immune, and stromal cells interact to buffer against treatment selection. Rather than attributing resistance solely to clonal mutations, SHEs emphasize the role of local ecosystems, and point to strategies like disrupting niche architecture, modulating TME support, or timing therapy to catch transient vulnerabilities.
In the near term, SHEs could be used to reinterpret treatment response and resistance across existing datasets. Spatial analysis of pre- and post-treatment samples may reveal which hallmark programs collapse under therapy [6,96,97], which persist, and how their spatial coordination changes. Rather than asking whether a treatment reduced proliferation globally, clinicians could ask: which ecosystems were dismantled, and which reorganized to survive? This translational logic is summarized in Figure 2, which illustrates how multi-region sampling and multi-omics profiling can be used to map SHEs and connect them to clonal selection, dissemination, and therapy response.
Looking further ahead, the SHE model opens the door to spatiotemporal risk stratification. Instead of classifying tumours based only on mutational burden or immune infiltration, patients could be stratified by the architecture, plasticity, and coordination of their hallmark ecosystems [15]. For example, the proximity of immune-suppressive niches to proliferative zones may better predict checkpoint inhibitor failure than PD-L1 staining alone [22].
This vision aligns with emerging efforts to build digital twins of tumours [98,99,100,101,102]. These are computational models that simulate how tumours respond to therapy under different conditions. If populated with robust spatiotemporal inputs, such models could move beyond genomic personalization toward ecosystem modeling, predicting not just what mutations are present, but how hallmark traits will evolve under specific therapeutic pressures. SHEs provide the conceptual logic needed to design and validate such models, linking biological mechanism to clinical action [98].
As spatial and longitudinal profiling becomes routine in clinical trials, SHEs offer a principled way to interpret complex patient data, design adaptive treatment strategies, and anticipate tumour evolution. They recast precision oncology as a spatiotemporal challenge, where timing and location of intervention may matter as much as the molecular target itself.
Conclusions: A New Lens on Cancer Complexity
The Hallmarks of Cancer framework transformed oncology by distilling molecular chaos into a coherent set of capabilities. But today’s tools reveal not just what tumours are made of, but how they are organized and how they change. This calls for a new synthesis that integrates the classical Hallmarks with spatial and temporal dimensions.
Spatiotemporal Hallmark Ecosystems (SHEs) offer that synthesis. By framing hallmark traits as emergent, distributed, and context-dependent programs, SHEs reconcile contradictory observations: how identical mutations yield different outcomes, why some tumours remain indolent, and how resistance emerges. They reposition cancer not as a static entity but as an evolving system, structured by both internal dynamics and external pressures.
SHEs build on concepts of heterogeneity and the tumour microenvironment but go beyond them, shifting the focus from individual features to ecosystem-level coordination, emphasizing when, where, and how hallmark programs are implemented and stabilized. This reframing has practical consequences for biomarker design, therapeutic timing, and experimental modeling.
By integrating the mutational focus of SMT, the structural perspective of TOFT, the interaction-driven view of ecological models, the tipping-point logic of CT theory, and the trait-based lens of the Hallmarks, SHEs provide a unifying, operational framework for mapping and anticipating cancer evolution in both research and clinical settings.
As spatial and longitudinal profiling become more integrated into cancer research and care, SHEs encourage thinking in terms of ecosystems dynamics as well as mutations or markers. They connect molecular traits to phenotypic behavior and clinical outcomes through a dynamic, testable framework (Box 3).
Ultimately, tumours are not just collections of altered cells, but systems in motion, structured across time and space, shaped by interaction, and vulnerable at points of transition. By learning to map and anticipate these dynamics, we open the door to a more predictive, adaptive, and precise oncology, one the anticipates change rather than merely reacts to it.
Box 3 - Falsifiable hypotheses derived from the existence of SHEs
To transition from a conceptual model to a scientific theory, the Spatiotemporal Hallmark Ecosystem (SHE) framework must yield predictions that are falsifiable and empirically testable. Below are five core predictions that exemplify this principle:
- Prediction 1: Therapeutic resistance is an emergent property of the tumour ecosystem, not just a feature of individual clones: If resistance arises from the interaction of tumour cells with their microenvironment, then ablating a resistant clone alone should not lead to durable response unless its supporting ecosystem is also disrupted.
- Prediction 2: Subclonal hallmark specialization reflects ecological trade-offs rather than purely genetic divergence: In tumours with high subclonal diversity, hallmark programs (e.g., proliferation, immune evasion) should segregate across clones in a mutually exclusive fashion, even when those clones share a similar genetic background.
- Prediction 3: Transformation to malignancy requires the spatial coordination of multiple hallmark programs: Premalignant lesions exhibiting spatially co-localized hallmark traits should show higher progression rates than lesions in which these traits remain uncoordinated.
- Prediction 4: The degree of spatial complementarity between tumour and TME hallmark programs predicts treatment response: Tumours with high cross-compartment coordination (e.g., evading growth suppressors in tumour cells coupled with sustained proliferative signaling from the TME) should respond more favorably to therapies that target both compartments.
- Prediction 5: Phenotypic divergence among genetically identical clones arises from local niche conditions, and can be reversed: Isogenic clones located in different spatial niches should exhibit divergent hallmark activity, which should homogenize if their niche environment is disrupted (e.g., through fibroblast depletion or cytokine blockade).
These hypotheses provide a roadmap for converting the SHE framework into a research program guided by falsifiable, cross-platform experiments.
Acknowledgements
E.P-P. is supported by by the Spanish Ministry of Science (PID2024-159258OB-I00 and RYC2019-026415-I MICIU/AEI/10.13039/501100011033, “El FSE invierte en tu futuro”), Fundación FERO-ASEICA (BFERO2002.6). M.S. is supported by “la Caixa” Foundation (LCF/BQ/DR22/11950021). M.J-H has received funding from CRUK, NIH National Cancer Institute, IASLC International Lung Cancer Foundation, Lung Cancer Research Foundation, Rosetrees Trust, UKI NETs and NIHR. N.M. was a Sir Henry Dale Fellow, jointly funded by the Wellcome Trust and the Royal Society (211179/Z/18/Z), and also receives funding from a CRUK Programme Foundation Award (DRCPFA-Nov23/100003) and a CRUK Biomarker Project Award (CRCBPA-Mar24/100001), the Rosetrees Trust, the NIHR BRC at University College London Hospitals and the CRUK University College London Experimental Cancer Medicine Centre.
Conflicts of Interest
M.J-H. has consulted for Astex Pharmaceuticals, Pfizer and Achilles Therapeutics, and is a member of, the Achilles Therapeutics Scientific Advisory Board and Steering Committee, has received speaker honoraria from Pfizer, Astex Pharmaceuticals, Oslo Cancer Cluster, Bristol Myers Squibb, Genentech and GenesisCare. MJ-H is listed as a co-inventor on a European patent application relating to methods to detect lung cancer PCT/US2017/028013), this patent has been licensed to commercial entities and, under terms of employment, M.J.-H. is due a share of any revenue generated from such license(s), and is also listed as a co-inventor on the GB priority patent application (GB2400424.4) with title: Treatment and Prevention of Lung Cancer.
References
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100: 57–70.
- Weinberg, R.A. Coming Full Circle—From Endless Complexity to Simplicity and Back Again. Cell 2014, 157, 267–271. [Google Scholar] [CrossRef]
- Swanton, C.; Bernard, E.; Abbosh, C.; André, F.; Auwerx, J.; Balmain, A.; Bar-Sagi, D.; Bernards, R.; Bullman, S.; DeGregori, J.; et al. Embracing cancer complexity: Hallmarks of systemic disease. Cell 2024, 187, 1589–1616. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144: 646–674.
- Elhanani, O.; Ben-Uri, R.; Keren, L. Spatial profiling technologies illuminate the tumor microenvironment. Cancer Cell 2023, 41, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Burkhardt, D.B.; Juan, B.P.S.; Lock, J.G.; Krishnaswamy, S.; Chaffer, C.L. Mapping Phenotypic Plasticity upon the Cancer Cell State Landscape Using Manifold Learning. Cancer Discov. 2022, 12, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A.; Gaskell, E.; Bernstein, B.E. Epigenetic plasticity and the hallmarks of cancer. Science 2017, 357, eaal2380. [Google Scholar] [CrossRef]
- Gavish, A.; Tyler, M.; Greenwald, A.C.; Hoefflin, R.; Simkin, D.; Tschernichovsky, R.; Darnell, N.G.; Somech, E.; Barbolin, C.; Antman, T.; et al. Hallmarks of transcriptional intratumour heterogeneity across a thousand tumours. Nature 2023, 618, 598–606. [Google Scholar] [CrossRef]
- Oliveira MF de, Romero JP, Chung M, Williams SR, Gottscho AD, Gupta A, et al. High-definition spatial transcriptomic profiling of immune cell populations in colorectal cancer. Nat Genet. 2025;57: 1512–1523.
- Janesick, A.; Shelansky, R.; Gottscho, A.D.; Wagner, F.; Williams, S.R.; Rouault, M.; Beliakoff, G.; Morrison, C.A.; Oliveira, M.F.; Sicherman, J.T.; et al. High resolution mapping of the tumor microenvironment using integrated single-cell, spatial and in situ analysis. Nat. Commun. 2023, 14, 1–15. [Google Scholar] [CrossRef]
- Lucas, O.; Ward, S.; Zaidi, R.; Bunkum, A.; Frankell, A.M.; Moore, D.A.; Hill, M.S.; Liu, W.K.; Marinelli, D.; Lim, E.L.; et al. Characterizing the evolutionary dynamics of cancer proliferation in single-cell clones with SPRINTER. Nat. Genet. 2024, 57, 103–114. [Google Scholar] [CrossRef]
- Floor, S.L.; Dumont, J.E.; Maenhaut, C.; Raspe, E. Hallmarks of cancer: of all cancer cells, all the time? Trends Mol. Med. 2012, 18, 509–515. [Google Scholar] [CrossRef]
- Sibai, M.; Cervilla, S.; Grases, D.; Musulen, E.; Lazcano, R.; Mo, C.-K.; Davalos, V.; Fortian, A.; Bernat, A.; Romeo, M.; et al. The spatial landscape of cancer hallmarks reveals patterns of tumor ecological dynamics and drug sensitivity. Cell Rep. 2025, 44, 115229. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Cao, C.; Kumar, M.; Sinha, S.; Chanda, A.; McNeil, R.; Samuel, D.; Arora, R.K.; Matthews, T.W.; Chandarana, S.; et al. Spatial transcriptomics reveals distinct and conserved tumor core and edge architectures that predict survival and targeted therapy response. Nat. Commun. 2023, 14, 1–19. [Google Scholar] [CrossRef]
- Somarelli, J.A. The Hallmarks of Cancer as Ecologically Driven Phenotypes. Front. Ecol. Evol. 2021, 9. [Google Scholar] [CrossRef]
- Skrupskelyte G, Rojo Arias JE, Dang Y, Han S, Bejar MT, Colom B, et al. Pre-cancerous Niche Remodelling Dictates Nascent Tumour Survival. bioRxiv. 2024. p. 2024.07.04.602022. [CrossRef]
- Kang, J.; Lee, J.H.; Cha, H.; An, J.; Kwon, J.; Lee, S.; Kim, S.; Baykan, M.Y.; Kim, S.Y.; An, D.; et al. Systematic dissection of tumor-normal single-cell ecosystems across a thousand tumors of 30 cancer types. Nat. Commun. 2024, 15, 1–17. [Google Scholar] [CrossRef]
- Zhou, D.C.; Jayasinghe, R.G.; Chen, S.; Herndon, J.M.; Iglesia, M.D.; Navale, P.; Wendl, M.C.; Caravan, W.; Sato, K.; Storrs, E.; et al. Spatially restricted drivers and transitional cell populations cooperate with the microenvironment in untreated and chemo-resistant pancreatic cancer. Nat. Genet. 2022, 54, 1390–1405. [Google Scholar] [CrossRef]
- Bhattacharya, R.; Avdieiev, S.S.; Bukkuri, A.; Whelan, C.J.; Gatenby, R.A.; Tsai, K.Y.; Brown, J.S. The Hallmarks of Cancer as Eco-Evolutionary Processes. Cancer Discov. 2025, 15, 685–701. [Google Scholar] [CrossRef]
- Grande E, Sibai M, Andrada E, Grases D, Reig O, Escobosa M, et al. Spatial biomarkers of response to neoadjuvant therapy in muscle-invasive bladder cancer: the DUTRENEO trial. medRxiv. 2025. [CrossRef]
- Colom, B.; Alcolea, M.P.; Piedrafita, G.; Hall, M.W.J.; Wabik, A.; Dentro, S.C.; Fowler, J.C.; Herms, A.; King, C.; Ong, S.H.; et al. Spatial competition shapes the dynamic mutational landscape of normal esophageal epithelium. Nat. Genet. 2020, 52, 604–614. [Google Scholar] [CrossRef]
- Ciwinska, M.; Messal, H.A.; Hristova, H.R.; Lutz, C.; Bornes, L.; Chalkiadakis, T.; Harkes, R.; Langedijk, N.S.M.; Hutten, S.J.; Menezes, R.X.; et al. Mechanisms that clear mutations drive field cancerization in mammary tissue. Nature 2024, 633, 198–206. [Google Scholar] [CrossRef] [PubMed]
- Chu, X.; Tian, Y.; Lv, C. Decoding the spatiotemporal heterogeneity of tumor-associated macrophages. Mol. Cancer 2024, 23, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Sinjab, A.; Min, J.; Han, G.; Paradiso, F.; Zhang, Y.; Wang, R.; Pei, G.; Dai, Y.; Liu, Y.; et al. Conserved spatial subtypes and cellular neighborhoods of cancer-associated fibroblasts revealed by single-cell spatial multi-omics. Cancer Cell 2025, 43, 905–924.e6. [Google Scholar] [CrossRef]
- Tirosh, I.; Suva, M.L. Cancer cell states: Lessons from ten years of single-cell RNA-sequencing of human tumors. Cancer Cell 2024, 42, 1497–1506. [Google Scholar] [CrossRef]
- Lomakin, A.; Svedlund, J.; Strell, C.; Gataric, M.; Shmatko, A.; Rukhovich, G.; Park, J.S.; Ju, Y.S.; Dentro, S.; Kleshchevnikov, V.; et al. Spatial genomics maps the structure, nature and evolution of cancer clones. Nature 2022, 611, 594–602. [Google Scholar] [CrossRef]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: it takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Shi, H.; Williams, M.J.; Satas, G.; Weiner, A.C.; McPherson, A.; Shah, S.P. Allele-specific transcriptional effects of subclonal copy number alterations enable genotype-phenotype mapping in cancer cells. Nat. Commun. 2024, 15, 1–13. [Google Scholar] [CrossRef]
- Campbell, N.R.; Rao, A.; Hunter, M.V.; Sznurkowska, M.K.; Briker, L.; Zhang, M.; Baron, M.; Heilmann, S.; Deforet, M.; Kenny, C.; et al. Cooperation between melanoma cell states promotes metastasis through heterotypic cluster formation. Dev. Cell 2021, 56, 2808–2825.e10. [Google Scholar] [CrossRef] [PubMed]
- Adler, M.; Chavan, A.R.; Medzhitov, R. Tissue Biology: In Search of a New Paradigm. Annu. Rev. Cell Dev. Biol. 2023, 39, 67–89. [Google Scholar] [CrossRef]
- Roehrig, A.; Hirsch, T.Z.; Pire, A.; Morcrette, G.; Gupta, B.; Marcaillou, C.; Imbeaud, S.; Chardot, C.; Gonzales, E.; Jacquemin, E.; et al. Single-cell multiomics reveals the interplay of clonal evolution and cellular plasticity in hepatoblastoma. Nat. Commun. 2024, 15, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Dujon, A.M.; Aktipis, A.; Alix-Panabières, C.; Amend, S.R.; Boddy, A.M.; Brown, J.S.; Capp, J.; DeGregori, J.; Ewald, P.; Gatenby, R.; et al. Identifying key questions in the ecology and evolution of cancer. Evol. Appl. 2020, 14, 877–892. [Google Scholar] [CrossRef] [PubMed]
- Gourmet L, Ramazzoti D, Mallick P, Walker-Samuel S, Zapata L. The temporal evolution of cancer hallmarks. bioRxiv. 2024. [CrossRef]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Math, M.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Jamal-Hanjani, M.; Wilson, G.A.; McGranahan, N.; Birkbak, N.J.; Watkins, T.B.K.; Veeriah, S.; Shafi, S.; Johnson, D.H.; Mitter, R.; Rosenthal, R.; et al. Tracking the Evolution of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2017, 376, 2109–2121. [Google Scholar] [CrossRef]
- Gerstung M, Jolly C, Leshchiner I, Dentro SC, Gonzalez S, Rosebrock D, et al. The evolutionary history of 2,658 cancers. Nature. 2020;578: 122–128.
- Dijkstra, K.K.; Vendramin, R.; Karagianni, D.; Witsen, M.; Gálvez-Cancino, F.; Hill, M.S.; Foster, K.A.; Barbè, V.; Angelova, M.; Hynds, R.E.; et al. Subclonal immune evasion in non-small cell lung cancer. Cancer Cell 2025. [Google Scholar] [CrossRef]
- Rosenthal, R.; Cadieux, E.L.; Salgado, R.; Bakir, M.A.; Moore, D.A.; Hiley, C.T.; Lund, T.; Tanić, M.; Reading, J.L.; Joshi, K.; et al. Neoantigen-directed immune escape in lung cancer evolution. Nature 2019, 567, 479–485. [Google Scholar] [CrossRef]
- Martínez-Ruiz, C.; Black, J.R.M.; Puttick, C.; Hill, M.S.; Demeulemeester, J.; Cadieux, E.L.; Thol, K.; Jones, T.P.; Veeriah, S.; Naceur-Lombardelli, C.; et al. Genomic–transcriptomic evolution in lung cancer and metastasis. Nature 2023, 616, 543–552. [Google Scholar] [CrossRef]
- Frankell, A.M.; Dietzen, M.; Al Bakir, M.; Lim, E.L.; Karasaki, T.; Ward, S.; Veeriah, S.; Colliver, E.; Huebner, A.; Bunkum, A.; et al. The evolution of lung cancer and impact of subclonal selection in TRACERx. Nature 2023, 616, 525–533. [Google Scholar] [CrossRef]
- Al Bakir, M.; Huebner, A.; Martínez-Ruiz, C.; Grigoriadis, K.; Watkins, T.B.K.; Pich, O.; Moore, D.A.; Veeriah, S.; Ward, S.; Laycock, J.; et al. The evolution of non-small cell lung cancer metastases in TRACERx. Nature 2023, 616, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.-T.; Zalmas, L.-P.; Bailey, C.; Black, J.R.M.; Martinez-Ruiz, C.; Pich, O.; Gimeno-Valiente, F.; Usaite, I.; Magness, A.; Thol, K.; et al. TRACERx analysis identifies a role for FAT1 in regulating chromosomal instability and whole-genome doubling via Hippo signalling. Nat. Cell Biol. 2024, 27, 154–168. [Google Scholar] [CrossRef]
- Arner, E.N.; Rathmell, J.C. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell 2023, 41, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2018, 20, 69–84. [Google Scholar] [CrossRef]
- Gourmet, L.; Sottoriva, A.; Walker-Samuel, S.; Secrier, M.; Zapata, L. Immune evasion impacts the landscape of driver genes during cancer evolution. Genome Biol. 2024, 25, 1–15. [Google Scholar] [CrossRef]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef]
- Winkler, J.; Abisoye-Ogunniyan, A.; Metcalf, K.J.; Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 2020, 11, 5120. [Google Scholar] [CrossRef]
- Greten, F.R.; Grivennikov, S.I. Inflammation and Cancer: Triggers, Mechanisms, and Consequences. Immunity 2019, 51, 27–41. [Google Scholar] [CrossRef]
- Leonetti, A.; Sharma, S.; Minari, R.; Perego, P.; Giovannetti, E.; Tiseo, M. Resistance mechanisms to osimertinib in EGFR-mutated non-small cell lung cancer. Br. J. Cancer 2019, 121, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-L.; Chen, H.-H.; Zheng, L.-L.; Sun, L.-P.; Shi, L. Angiogenic signaling pathways and anti-angiogenic therapy for cancer. Signal Transduct. Target. Ther. 2023, 8, 198. [Google Scholar] [CrossRef]
- Puttick, C.; Jones, T.P.; Leung, M.M.; Galvez-Cancino, F.; Liu, J.; Varas-Godoy, M.; Rowan, A.; Pich, O.; Martinez-Ruiz, C.; Bentham, R.; et al. MHC Hammer reveals genetic and non-genetic HLA disruption in cancer evolution. Nat. Genet. 2024, 56, 2121–2131. [Google Scholar] [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer Discov. 2017, 7, 188–201. [Google Scholar] [CrossRef] [PubMed]
- Joller, N.; Anderson, A.C.; Kuchroo, V.K. LAG-3, TIM-3, and TIGIT: Distinct functions in immune regulation. Immunity 2024, 57, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Hessey, S.; Fessas, P.; Zaccaria, S.; Jamal-Hanjani, M.; Swanton, C. Insights into the metastatic cascade through research autopsies. Trends Cancer 2023, 9, 490–502. [Google Scholar] [CrossRef]
- Fares, J.; Fares, M.Y.; Khachfe, H.H.; Salhab, H.A.; Fares, Y. Molecular principles of metastasis: a hallmark of cancer revisited. Signal Transduct. Target. Ther. 2020, 5, 28. [Google Scholar] [CrossRef]
- Geukens, T.; Maetens, M.; E Hooper, J.; Oesterreich, S.; Lee, A.V.; Miller, L.; Atkinson, J.M.; Rosenzweig, M.; Puhalla, S.; Thorne, H.; et al. Research autopsy programmes in oncology: shared experience from 14 centres across the world. J. Pathol. 2024, 263, 150–165. [Google Scholar] [CrossRef] [PubMed]
- Fischer, G.M.; Jalali, A.; Kircher, D.A.; Lee, W.-C.; McQuade, J.L.; Haydu, L.E.; Joon, A.Y.; Reuben, A.; de Macedo, M.P.; Carapeto, F.C.L.; et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019, 9, 628–645. [Google Scholar] [CrossRef]
- Karimi, E.; Yu, M.W.; Maritan, S.M.; Perus, L.J.M.; Rezanejad, M.; Sorin, M.; Dankner, M.; Fallah, P.; Doré, S.; Zuo, D.; et al. Single-cell spatial immune landscapes of primary and metastatic brain tumours. Nature 2023, 614, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.; Sheng, J.; Cui, J.Z.; Zhao, H.; Wong, S.T. Systems immunology insights into brain metastasis. Trends Immunol. 2024, 45, 903–916. [Google Scholar] [CrossRef]
- Croucher, P.I.; McDonald, M.M.; Martin, T.J. Bone metastasis: the importance of the neighbourhood. Nat. Rev. Cancer 2016, 16, 373–386. [Google Scholar] [CrossRef] [PubMed]
- Best, S.A.; Gubser, P.M.; Sethumadhavan, S.; Kersbergen, A.; Abril, Y.L.N.; Goldford, J.; Sellers, K.; Abeysekera, W.; Garnham, A.L.; McDonald, J.A.; et al. Glutaminase inhibition impairs CD8 T cell activation in STK11-/Lkb1-deficient lung cancer. Cell Metab. 2022, 34, 874–887.e6. [Google Scholar] [CrossRef]
- Zavitsanou, A.-M.; Pillai, R.; Hao, Y.; Wu, W.L.; Bartnicki, E.; Karakousi, T.; Rajalingam, S.; Herrera, A.; Karatza, A.; Rashidfarrokhi, A.; et al. KEAP1 mutation in lung adenocarcinoma promotes immune evasion and immunotherapy resistance. Cell Rep. 2023, 42, 113295–113295. [Google Scholar] [CrossRef]
- Gui, P.; Bivona, T.G. Evolution of metastasis: new tools and insights. Trends Cancer 2022, 8, 98–109. [Google Scholar] [CrossRef]
- Shi, Q.; Chen, Y.; Li, Y.; Qin, S.; Yang, Y.; Gao, Y.; Zhu, L.; Wang, D.; Zhang, Z. Cross-tissue multicellular coordination and its rewiring in cancer. Nature 2025, 643, 529–538. [Google Scholar] [CrossRef]
- Feunteun, J.; Ostyn, P.; Delaloge, S. Tumor cell malignancy: A complex trait built through reciprocal interactions between tumors and tissue-body system. iScience 2022, 25, 104217. [Google Scholar] [CrossRef]
- Zahir, N.; Sun, R.; Gallahan, D.; Gatenby, R.A.; Curtis, C. Characterizing the ecological and evolutionary dynamics of cancer. Nat. Genet. 2020, 52, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef]
- Noble, R.; Burri, D.; Le Sueur, C.; Lemant, J.; Viossat, Y.; Kather, J.N.; Beerenwinkel, N. Spatial structure governs the mode of tumour evolution. Nat. Ecol. Evol. 2021, 6, 207–217. [Google Scholar] [CrossRef]
- Khaliq, A.M.; Rajamohan, M.; Saeed, O.; Mansouri, K.; Adil, A.; Zhang, C.; Turk, A.; Carstens, J.L.; House, M.; Hayat, S.; et al. Spatial transcriptomic analysis of primary and metastatic pancreatic cancers highlights tumor microenvironmental heterogeneity. Nat. Genet. 2024, 56, 2455–2465. [Google Scholar] [CrossRef]
- Aguadé-Gorgorió, G.; Anderson, A.R.; Solé, R. Modeling tumors as complex ecosystems. iScience 2024, 27, 110699. [Google Scholar] [CrossRef]
- Ma, H.; Srivastava, S.; Ho, S.W.T.; Xu, C.; Lian, B.S.X.; Ong, X.; Tay, S.T.; Sheng, T.; Lum, H.Y.J.; Ghani, S.A.B.A.; et al. Spatially Resolved Tumor Ecosystems and Cell States in Gastric Adenocarcinoma Progression and Evolution. Cancer Discov. 2025, 15, 767–792. [Google Scholar] [CrossRef] [PubMed]
- Béguelin, W.; Teater, M.; Meydan, C.; Hoehn, K.B.; Phillip, J.M.; Soshnev, A.A.; Venturutti, L.; Rivas, M.A.; Calvo-Fernández, M.T.; Gutierrez, J.; et al. Mutant EZH2 Induces a Pre-malignant Lymphoma Niche by Reprogramming the Immune Response. Cancer Cell 2020, 37, 655–673.e11. [Google Scholar] [CrossRef]
- Mourcin F, Verdière L, Roulois D, Amin R, Lamaison C, Sibut V, et al. Follicular lymphoma triggers phenotypic and functional remodeling of the human lymphoid stromal cell landscape. Immunity. 2021;54: 1788–1806.e7.
- Laurent, C.; Dietrich, S.; Tarte, K. Cell cross talk within the lymphoma tumor microenvironment: follicular lymphoma as a paradigm. Blood 2024, 143, 1080–1090. [Google Scholar] [CrossRef]
- Laplane, L.; Maley, C.C. The evolutionary theory of cancer: challenges and potential solutions. Nat. Rev. Cancer 2024, 24, 718–733. [Google Scholar] [CrossRef]
- Huang, S.; Soto, A.M.; Sonnenschein, C. The end of the genetic paradigm of cancer. PLOS Biol. 2025, 23, e3003052. [Google Scholar] [CrossRef]
- Lawson, A.R.J.; Abascal, F.; Coorens, T.H.H.; Hooks, Y.; O’nEill, L.; Latimer, C.; Raine, K.; Sanders, M.A.; Warren, A.Y.; Mahbubani, K.T.A.; et al. Extensive heterogeneity in somatic mutation and selection in the human bladder. Science 2020, 370, 75–82. [Google Scholar] [CrossRef]
- Wijewardhane, N.; Dressler, L.; Ciccarelli, F.D. Normal Somatic Mutations in Cancer Transformation. Cancer Cell 2021, 39, 125–129. [Google Scholar] [CrossRef]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385.E18. [Google Scholar] [CrossRef]
- Li, Y.; Porta-Pardo, E.; Tokheim, C.; Bailey, M.H.; Yaron, T.M.; Stathias, V.; Geffen, Y.; Imbach, K.J.; Cao, S.; Anand, S.; et al. Pan-cancer proteogenomics connects oncogenic drivers to functional states. Cell 2023, 186, 3921–3944.e25. [Google Scholar] [CrossRef] [PubMed]
- Porta-Pardo, E.; Valencia, A.; Godzik, A. Understanding oncogenicity of cancer driver genes and mutations in the cancer genomics era. FEBS Lett. 2020, 594, 4233–4246. [Google Scholar] [CrossRef] [PubMed]
- Barkley, D.; Rao, A.; Pour, M.; França, G.S.; Yanai, I. Cancer cell states and emergent properties of the dynamic tumor system. Genome Res. 2021, 31, 1719–1727. [Google Scholar] [CrossRef]
- Massagué, J.; Ganesh, K. Metastasis-Initiating Cells and Ecosystems. Cancer Discov. 2021, 11, 971–994. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; Bonnardel, J.; Haest, B.; Vanderborght, B.; Wagner, C.; Remmerie, A.; Bujko, A.; Martens, L.; Thoné, T.; Browaeys, R.; et al. Spatial proteogenomics reveals distinct and evolutionarily conserved hepatic macrophage niches. Cell 2022, 185, 379–396.e38. [Google Scholar] [CrossRef]
- Bhang, H.-E.C.; Ruddy, D.A.; Krishnamurthy Radhakrishna, V.; Caushi, J.X.; Zhao, R.; Hims, M.M.; Singh, A.P.; Kao, I.; Rakiec, D.; Shaw, P.; et al. Studying clonal dynamics in response to cancer therapy using high-complexity barcoding. Nat. Med. 2015, 21, 440–448. [Google Scholar] [CrossRef]
- Gerlinger, M.; Horswell, S.; Larkin, J.; Rowan, A.J.; Salm, M.P.; Varela, I.; Fisher, R.; McGranahan, N.; Matthews, N.; Santos, C.R.; et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 2014, 46, 225–233. [Google Scholar] [CrossRef]
- de Bruin, E.C.; McGranahan, N.; Mitter, R.; Salm, M.; Wedge, D.C.; Yates, L.; Jamal-Hanjani, M.; Shafi, S.; Murugaesu, N.; Rowan, A.J.; et al. Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 2014, 346, 251–256. [Google Scholar] [CrossRef]
- Yates, L.R.; Gerstung, M.; Knappskog, S.; Desmedt, C.; Gundem, G.; Van Loo, P.; Aas, T.; Alexandrov, L.B.; Larsimont, D.; Davies, H.; et al. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat. Med. 2015, 21, 751–759. [Google Scholar] [CrossRef]
- Carbone A, Roulland S, Gloghini A, Younes A, von Keudell G, López-Guillermo A, et al. Follicular lymphoma. Nat Rev Dis Primers. 2019;5: 83.
- Dyrskjøt L, Hansel DE, Efstathiou JA, Knowles MA, Galsky MD, Teoh J, et al. Bladder cancer. Nat Rev Dis Primers. 2023;9: 58.
- Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: Advances and challenges. Cell. 2023;186: 1729–1754.
- Risom, T.; Glass, D.R.; Averbukh, I.; Liu, C.C.; Baranski, A.; Kagel, A.; McCaffrey, E.F.; Greenwald, N.F.; Rivero-Gutiérrez, B.; Strand, S.H.; et al. Transition to invasive breast cancer is associated with progressive changes in the structure and composition of tumor stroma. Cell 2022, 185, 299–310.e18. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, B.; Luo, M.; Huang, J.; Zhang, K.; Zheng, S.; Zhang, S.; Zhou, J. Progression from ductal carcinoma in situ to invasive breast cancer: molecular features and clinical significance. Signal Transduct. Target. Ther. 2024, 9, 1–28. [Google Scholar] [CrossRef]
- Chen, J.; Larsson, L.; Swarbrick, A.; Lundeberg, J. Spatial landscapes of cancers: insights and opportunities. Nat. Rev. Clin. Oncol. 2024, 21, 660–674. [Google Scholar] [CrossRef] [PubMed]
- Gong, D.; Arbesfeld-Qiu, J.M.; Perrault, E.; Bae, J.W.; Hwang, W.L. Spatial oncology: Translating contextual biology to the clinic. Cancer Cell 2024, 42, 1653–1675. [Google Scholar] [CrossRef]
- Johnson, J.A.; Bergman, D.R.; Rocha, H.L.; Zhou, D.L.; Cramer, E.; Mclean, I.C.; Dance, Y.W.; Booth, M.; Nicholas, Z.; Lopez-Vidal, T.; et al. Human interpretable grammar encodes multicellular systems biology models to democratize virtual cell laboratories. Cell 2025, 188, 4711–4733.e37. [Google Scholar] [CrossRef]
- Metzcar, J.; Wang, Y.; Heiland, R.; Macklin, P. A Review of Cell-Based Computational Modeling in Cancer Biology. JCO Clin. Cancer Informatics 2019, 3, 1–13. [Google Scholar] [CrossRef]
- Rocha, H.L.; Aguilar, B.; Getz, M.; Shmulevich, I.; Macklin, P. A multiscale model of immune surveillance in micrometastases gives insights on cancer patient digital twins. npj Syst. Biol. Appl. 2024, 10, 1–12. [Google Scholar] [CrossRef]
- Rocha, H.L.; Godet, I.; Kurtoglu, F.; Metzcar, J.; Konstantinopoulos, K.; Bhoyar, S.; Gilkes, D.M.; Macklin, P. A persistent invasive phenotype in post-hypoxic tumor cells is revealed by fate mapping and computational modeling. iScience 2021, 24, 102935. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Boussard, T.; Macklin, P.; Greenspan, E.J.; Gryshuk, A.L.; Stahlberg, E.; Syeda-Mahmood, T.; Shmulevich, I. Digital twins for predictive oncology will be a paradigm shift for precision cancer care. Nat. Med. 2021, 27, 2065–2066. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Spatiotemporal Hallmark Ecosystems. A schematic slope representation of cancer evolution, illustrating how hallmark programs emerge, stabilize, and reorganize across stages. Early lesions (emergence) are marked by genome instability, proliferative signaling, and inflammatory cues, but remain constrained by tissue context. During progression, additional hallmarks such as resistance to cell death, immune evasion, and replicative immortality allow expansion. Invasion is enabled by angiogenesis and phenotypic plasticity, while metastasis requires reassembly of hallmark programs in distant niches. Together, this highlights that cancer progression is not a linear sequence of traits but a dynamic reconfiguration of hallmark ecosystems across space and time.
Figure 1.
Spatiotemporal Hallmark Ecosystems. A schematic slope representation of cancer evolution, illustrating how hallmark programs emerge, stabilize, and reorganize across stages. Early lesions (emergence) are marked by genome instability, proliferative signaling, and inflammatory cues, but remain constrained by tissue context. During progression, additional hallmarks such as resistance to cell death, immune evasion, and replicative immortality allow expansion. Invasion is enabled by angiogenesis and phenotypic plasticity, while metastasis requires reassembly of hallmark programs in distant niches. Together, this highlights that cancer progression is not a linear sequence of traits but a dynamic reconfiguration of hallmark ecosystems across space and time.
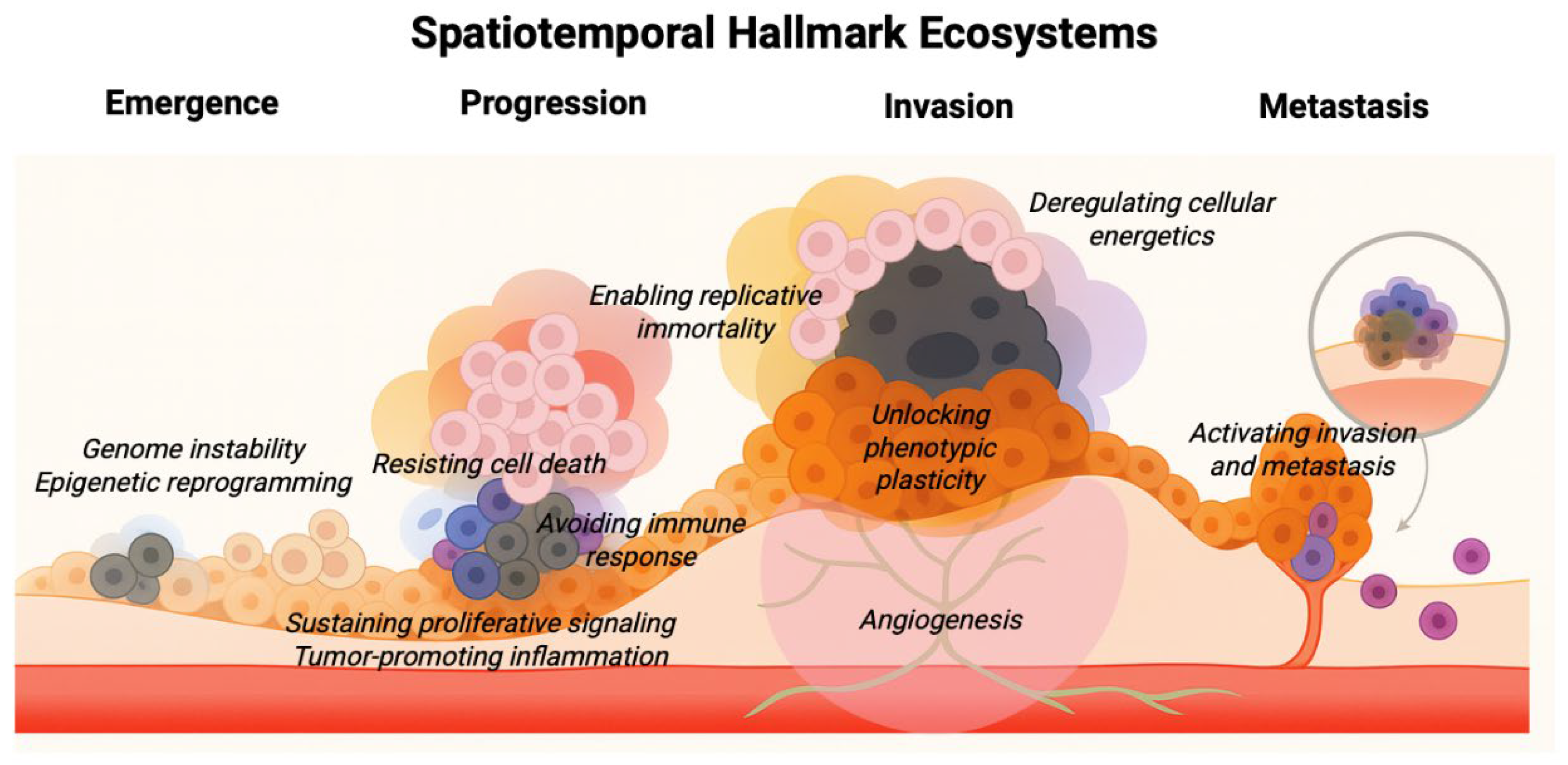
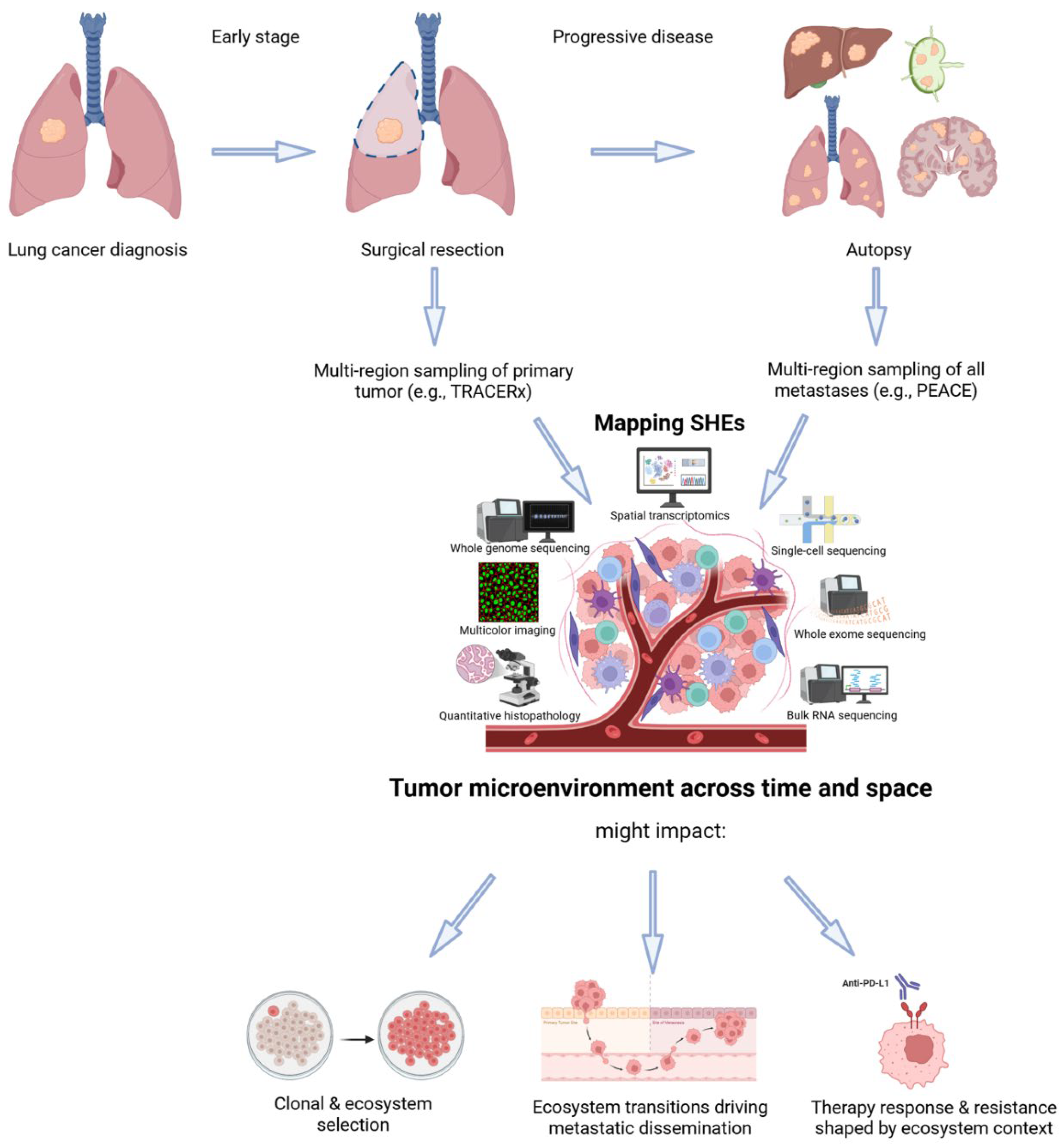
Figure 2.
Mapping Spatiotemporal Hallmark Ecosystems in clinical samples. Longitudinal and multi-region sampling strategies, such as those used in TRACERx and PEACE, enable the reconstruction of tumour ecosystems across space and time. Integration of spatial transcriptomics, single-cell profiling, bulk sequencing, and multiplex imaging allows mapping of SHEs in patient tumours. These approaches reveal how hallmarks stabilize or reorganize within ecosystems, with direct implications for clonal and ecosystem selection, metastatic dissemination, and therapy response or resistance.
Figure 2.
Mapping Spatiotemporal Hallmark Ecosystems in clinical samples. Longitudinal and multi-region sampling strategies, such as those used in TRACERx and PEACE, enable the reconstruction of tumour ecosystems across space and time. Integration of spatial transcriptomics, single-cell profiling, bulk sequencing, and multiplex imaging allows mapping of SHEs in patient tumours. These approaches reveal how hallmarks stabilize or reorganize within ecosystems, with direct implications for clonal and ecosystem selection, metastatic dissemination, and therapy response or resistance.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



