
Submitted:
10 September 2025
Posted:
14 October 2025
You are already at the latest version
Abstract
Cancer stem cells and induced pluripotent stem cells (iPSCs) differ from normal tissue stem cells in their ability to maintain telomere length stability and permanent capacity for self-renewal. In normal stem cells these traits gradually decrease with age and/or continuous or accelerated cell division. An origin of cancer cells from committed cells is consistent with a similar mechanism of telomere maintenance in cancer and iPSCs. Telomere shortening/damage is believed to protect cells from malignant transformation but, paradoxically, telomere shortening has been observed repeatedly to precede most cancers and the absence of apoptotic factors like p53 may reveal this inherent favourable role of telomere damage in cancer development. Architectural changes in telomeric chromatin such as those underlying the telomere position effect and others support telomere maintenance of some tumors. Here, we propose that several signaling pathways in conjunction with telomere shortening/damage may result in release of Rap1 from telomeres and its subsequent interaction with the embryonic stem cell marker Zscan4 might support immortalization and malignant transformation of the target cell.
Keywords:
telomeres
; Rap1
; Zscan4
; PI3K/AKT pathway
Introduction
In the quest to understand the nature of cancer the point of departure has been provided by one of the most fundamental aspects of living organisms, the capacity for regeneration and self-renewal which constitutes also the hallmark of cancer. It was realized that this property is compartmentalized within organisms and linked to germinal structures or cells that have been endowed with these embryonic features that allow preservation of the capacity of regeneration. This idea led Conheim to the first proposal on the origin of cancer in embryonic remains that persist in adult life disconnected from their original sources and which later in life manifest themselves by growing in an aberrant fashion. Later, histological research established that specialized somatic cells in every tissue, initially known as reserve cells and later as normal stem cells, are responsible for replenishing the tissue cell population continually lost by wear and tear or shed after multiple rounds of proliferation and differentiation. The shared property of indefinite self-renewal between the so-called reserve cells and cancer cells with its double physiological and pathological face led to the assumption that cancer growth arises from a failure of tissue stem cells to respond to regulatory signals that maintain normal tissue architecture. In other words, cancer directly originates in tissue stem cells. The modern concept of the cancer stem cell assumes this hierarchical organization ascribing embryonic/stem features to these cells generally disregarding an origin in embryonic rests. However, this feature is often interpreted as precluding the origin of cancer in committed cells.
Regeneration potential is lost as soon as cells leave the stem cell compartment as first confirmed in the hematopoietic system. Even in the transition from long term hematopoietic stem cells (LT-HSC) to short-term hematopoietic stem cells (ST-HSC) there is some loss of regeneration potential. In the next stages, transit amplifying cells that expand by continuous cell division, lose the capacity of indefinite self-renewal (1) which is restricted to normal stem cells and cancer cells. A similar contention can apply to other tissues. Muscle satellite stem cells have been shown to lose indefinite self-renewal immediately after their first division.”Pax7+ Myf5- stem cells that undergo asymmetric self-renewal divisions on muscle fibres, give rise to a basal Pax7+ Myf5- stem cell daughter and an apical Pax7+ Myf5- satellite cell with a more restricted proliferative potential” (2)
Mammary epithelium exhibits identical behaviour. Mammospheres isolated by short-term stem markers (Lin-, CD24+, CD29hi) give rise to multilineage colonies in culture but over time these colonies lose their ability to self-renew and start to differentiate in vivo (3)
Moreover, regenerating potential is further decreased by cell differentiation which is linked to exit from the niche or stem cell compartment
The self-renewal potential of adult tissue stem cells is lower than that of embryonic stem cells (ESCs), cancer cells or induced pluripotent stem cells (iPSCs) (derive from fully differentiated somatic cells reprogrammed with Oct-3/4, Sox-2, c-Myc and Kfl4). In somatic adult stem cells, self-renewal is present throughout the life of the organism but lifelong regenerative potential decreases with age and with cell division. Thus, increased stem cell proliferation caused by different agents (the deletion of CKIs, oncogenes, mTORC1 activation, Foxo3a inactivation, Sirt1 deletion in HSCs) is rapidly followed by stem cell exhaustion as demonstrated by competitive repopulation assays. The limit to cell proliferation may be dictated by the concurrent shortening of telomere length which is linked to cell division and coordinated with telomerase expression which may counteract it. In tumorigenesis, telomere shortening’s role is a double edge sword. Sensing of telomere shortening by p53 results in senescence that protects from cancer development. Through senescence, the inherent tendency of telomere shortening to reactivate telomerase or other means of telomere maintenance (ALT), a pre-requisite for cancer development, would be abolished. (4).
The cancer stem cell needs to utilize a mechanism to preserve telomere length or it would be extinguished after some replication rounds. There are differences between iPSCs, ESCs and cancer cells on the one hand and normal stem cells on the other: telomeres of tumour cells are in general shorter than those of normal cells but their length is stabilized, there is no gradual reduction of length with continuous cell division which enables self-renewal. This suggests that a sudden change in telomerase control expression occurs at the moment of transformation. This pattern of telomerase expression of the cancer stem cell is reminiscent of that of reprogrammed fully differentiated cells such as iPSCs which suggests that the cancer stem cell also originates in post-stem cells. This concept is reinforced by the common observation that cancer stem cells usually possess shorter telomeres than normal counterparts which suggests that the cell of origin engaged the differentiation pathway and started the normal process of gradual telomere length reduction undergone by all normal post-stem cells. Of note, the proportion of stem and non-stem cells within different tumour types may vary widely as observed in leukaemia. A larger fraction of the non-stem population undergoing telomere erosion would make it difficult to notice telomere stabilization although this effect might not be discerned if the progeny were separated from the CSC by only a few replication rounds or it is endowed with the mechanism of telomere length preservation of the cancer stem cell
Normal Stem Cells and Oncogenic Pathways
In culture, stem cells are difficult to maintain in the undifferentiated state. Only when they remain anchored to their niche can they be kept in an undifferentiated state. Oncogenes such as Ras can decouple cell cycle control with adhesion to the niche leading to anchorage-independent growth which is one of the earliest events in transformation (5). At the same time, signalling pathways involved in oncogenesis trigger some degree of differentiation and exit from the stem cell compartment. In addition, most oncogenes disturb normal coordination between cell proliferation and cell differentiation which may potentiate telomere shortening/damage. Concomitantly, the DNA damage response in ageing and stress-induced senescence preferentially impacts on the telomere region (6). All these facts should make committed cells preferential targets of malignant transformation. Evidently, progenitors that retain telomerase expression can be targets of malignant transformation. However, they do not retain indefinite self-renewal which must be reacquired. This must coincide with a fundamental change in telomerase reactivation marked by telomere length stabilization.
In previous studies we argued that the role of oncogenes in disturbing tissue differentiation, developmental errors and other sources of DNA damage converge on inducing telomere erosion/damage which precedes and triggers malignant transformation, The process is reminiscent of a phenomenon observed in in vitro cell cultures known as crisis where sustained telomere length reduction leads to massive apoptosis and then to the emergence of cells with an immortal phenotype. In order to simplify the discussion, we used the term telomere erosion/shortening to refer to the undoubtedly complex disturbances that telomeres and the telomere complex may undergone as different routes of tumorigenesis may impact the telomere complex in addition to shortening.
In this study, we postulate that malignant transformation entails one or several reprogramming events that reactivate telomerase expression (or ALT). This takes place in the post-stem cell stage where telomerase (or ALT) is subject to a new form of control
Regarding the assumption that the process of transformation entails cell reprogramming and the gain of self-renewal ability by a committed cell, it can be expected that the resulting cancer stem cell would exhibit a phenotype partially overlapping with that of a normal stem cell. In fact, although cancer stem cells are defined and isolated by surface markers shared with normal stem cells, they practically always express differentiation markers that allow for assignment to a particular stage of a cell lineage. A widespread survey of CSCs identified in different cancer types confirms this presumption. The case of leukemia is particularly relevant as the origin of most of them can be traced to a block in differentiation which imparts to the leukemia stem cell the phenotype corresponding to the specific stage of the developmental block. (7).
The origin of malignant transformation in a committed cell could arise after aberrant proliferation which, in the presence of gradual undermining of telomerase activity would result in gradual telomere erosion. The implication is that telomerase (or ALT) should be reactivated during tumor progression, most likely as a late event that marks the acquisition of immortality.
The alternative theory of a direct stem cell of origin implies that tumors initiate from normal stem cells that have a higher mutation burden than somatic cells (disregarding that the differentiated progeny of a stem cell carries the same mutation burden) as a result of age and have not ceased to express telomerase implying that control of telomerase would not change hands.
In former papers we highlighted multiple observations from human clinic and tumor models (8,7,9) in favour of the telomere erosion hypothesis, i.e., the origin of cancer in committed cells. Howver, let us examine the opposite hypothesis of a direct origin of cancer in stem cells.
Presumably, cancers recapitulate phenotypic features of their cell of origin. Therefore, we will focus on cancers commonly occurring in highly proliferative tissues that develop at early differentiation stages as it is expected that they will contain more numerous targets for transformation in a stem cell stage. Consistent with these population being the target for tumorigenesis, the oncogenic pathways that drive these cancers consist of those deregulated pathways that signal physiological differentiation on the same cells placing them in a progenitor rather than stem cell stage.
The Wnt Pathway and Crosstalk Between the Wnt/βcatenin and AKT/mTOR Pathways
The Wnt pathway supports the self-renewal of stem and progenitor cells and is conspicuously expressed in highly regenerating tissues. Β-catenin, the central player in this pathway, is maintained at low level by GSK3β phosphorylation in a destruction complex composed of GSK3β, adenomatous polyposis coli (APC), Axin, CKI kinase and β-catenin. Signaling by Wnt ligands acting on cell surface receptors of the Frizzle family inactivates GSK3β by phosphorylation at Serine 9 and blocks β-catenin destruction. This results in the accumulation of β-catenin which is then translocated to the nucleus where in conjunction with TCF factors (mainly TCF4 in the intestine) it stimulates transcription of target genes expressed physiologically in the stem cell areas and proliferative compartment of different tissue organs
There is evidence of a cross-talk between the Wnt/β-catenin pathway and the PI3-AKT-mTOR pathway which might underlie some findings that will be discussed later. GSK3β is a critical node of both pathways. On the one hand, it has been shown that GSK3β inhibition and subsequent Wnt/β-catenin activation enhances HSC self-renewal. Concomitantly, GSK3β inhibition stimulates mTORC1 which promotes proliferation and differentiation. By simultaneously stimulating the Wnt pathway (enhancing of HSC self-renewal) with inhibitors of GSK3β CHIR99021 or lithium and inhibiting the mTORC1 pathway with rapamycin, Huang et al. (10) were able to maintain long-term multilineage haematopoiesis in cytokine free cultures treated with both inhibitors. These culture conditions allowed tri-lineage hematopoietic reconstitution and gave rise to mature myeloid cells, contrary to control cells cultured in a standard cytokine cocktail. Transplantation to secondary recipients confirmed the improved preservation of HSC function as assessed by a higher chimerism detected in cells treated with both inhibitors. This implies that Wnt signaling activated in most instances through GSK3β inhibition should drive some differentiation without the artificial inhibition of mTORC1. GSK3β is also inhibited by AKT ( 11) which, in turn, requires the activity of the PI3K/AKT pathway which is antagonized by PTEN. This is why the LT-HSC exhaustion caused by deregulated Wnt/β-catenin signaling is recapitulated by hyperactivation of the PI3K/AKT pathway as seen after PTEN deletion. Here, we want to stress that mTORC1 activation through GSK·β inhibition associated with Wnt/β-catenin activation stimulates growth and, like c-Myc, promotes exit from the stem cell compartment. Stimulation of mTORC1 that takes place through deregulated Wnt signaling leads to engagement in cell proliferation and differentiation (exit from the stem cell compartment) which leads to stem cell exhaustion that might likely impact on telomere shortening.
Before dealing with intestinal adenoma, it is important to remember that this lesion contains progenitor cells because Myc (an essential target of Wnt/βcatenine pathway) accelerates exit from the stem cell compartment and detachment from the niche. This entails loss of stem cell regenerating potential which is crucial for tumor maintenance that consequently must be obtained from other sources. This has been demonstrated in hematopoietic tissue. Quoting from Wilson et al. “the inability to down-regulate c-Myc resulted in increased differentiation at the expense of self-renewal. C-Myc transduced cells repressed expression of N-cadherin and other cell adhesion molecules such as LFA-1 receptor and αL and β2-integrins accompanied by detachment to the niche and premature differentiation. Functionally, however, repopulation experiments showed that the repopulating ability of c-Myc transduced KLS-HSC cells was severely impaired. However, in addition to increased exit from the stem cell compartment, Myc overexpression can impair normal differentiation.” (12). Similarly, p27 helps to maintain cells in an undifferentiated stage and it has been found that Skp2-dependent degradation of p27 is crucial for progression of adenomas to carcinomas (13)
Adenoma-Carcinoma Sequence
Van de Wetering et al. have extensively characterized the Wnt/β-catenin signature in the intestine. Downstream targets include CD44, BMP4, claudin-1, ENC1 and c-MYC, c-ETS2, EPHB2. Cyclin D1, a previously published TCF-4 target was not affected by dn-TCF-4. P21 was induced independently of p53 and marks the differentiated compartment, p15, p19, p16 not expressed or unaffected Only c-MYC overrode the effect of dnTCF-4 on cell growth. It was established that c-MYC represses p21 and c-MYC and MIZ-1 were bound to p21 promoter (14)
Activating mutations of the Wnt pathway are the only known genetic alterations in early premalignant lesions in the intestine, such as aberrant crypt foci and small polyps (15, 16). In the hereditary syndrome known as hereditary polyposis as well as in more than 85% of colorectal tumors the initiating mutation is a loss of function mutation of APC and the central oncogenic player is the nuclear accumulation of β-catenin and its binding to TCF molecules (mainly TCF4, in the intestine). Zhang et al. showed that hTERT is a target of the Wnt/β-catenin pathway. They also reported that silencing β-catenin suppressed telomerase activity which was mediated by the suppression of TCF-4 (17).
The immediate consequence of deregulated β-catenin stimulation is the formation of a polyp or adenoma which in time may transform into a fully malignant cancer. Although the rate of conversion from adenoma to cancer is not high it is believed that most colorectal cancers arise from adenomas Therefore, this lesion provides an opportunity to study the main steps involved in conversion to malignancy.
Telomerase activity has been frequently evaluated in colorectal adenomas and its incidence has varied in different studies from 0% to 100%. However, there is ample consensus that telomerase activity increases in parallel with the size of the polyp and malignant potential. A report by Tang (18) that examined telomerase activity in 31 adenomatous polyps and 22 paired cancer-normal mucosa from nonhereditary nonpolyposis colorectal cancer patients found a linear correlation between polyp progression and telomerase activity. The latter was detected in 18% of normal mucosa specimens, 16% of small (<1cm) polyps, 20% of intermediate (>2cm) polyps and 71% of large polyps and 96% of adenocarcinomas suggesting that progenitors that composed the bulk of adenomas preserve telomerase expression.
A different study by Druliner (19) was aimed to determine whether telomere length and telomere maintenance mechanisms could distinguish cancer associated adenomas from those that are cancer-free. They classified adenomas into two groups: adenomas adjacent to cancer (residual adenomas contiguous with cancer) (CAP) and polyps not associated with cancer or cancer-free polyps (CFP). Further, they identified aggressive CAPs (if a stage I-II cancer recurred or presented a stage IV and aggressive CFPs (if there was a recurrence of the polyp. CAP polyps had shorter telomere length and higher telomerase reverse transcriptase (hTERT) expression compared to CFP polyps. The degree of difference in hTERT expression between the polyps and their normal colon tissue was significant for the CAP cases but not for the CFP cases and this could not be attributed to differences in the normal tissues. The aggressive CAPs exhibited the shortest range of telomere lengths with the polyp tissue exhibiting the shortest of telomere lengths. The researchers also measured telomere lengths of peripheral blood leucocytes (PBLs) and normal colon mucosa. After adjustment for age CAP patients avereged 91.7 bp longer PBL telomere length than CFP patients; the normal colon epithelium hosted significantly longer telomeres than PBL (we suggest that this indicates a relative loss of quiescence in normal stem cells which decreases their regenerative potential and paradoxically their resistance to cancer transformation.
Plentz (20) measured telomere fluorescence intensity in consecutive histological sections stained by hematoxilin-eosin from areas of high grade dysplasia and adenoma. Signal intensity was weaker at the earliest morphologically defined stage of carcinoma-high grade dysplasia with minimally invasive growth suggesting that malignant transformation starts in the cells with the shortest telomeres.
Our interpretation of these findings is that telomere shortening proceeds within adenomas in spite of a correlative increase in telomerase activity, a feedback loop insufficient to reverse telomere attrition, probably because differentiating cells lose gradually the capacity of their precursors stem cells for telomerase expression. At some point that marks the appearance of immortalization either telomerase re-expression or ALT will take place and telomere length becomes stabilized, a mark of malignant conversion. The precedent findings suggest that gradual telomere shortening (not necessarily a definitive length reduction but some level of telomere damage) is the trigger for re-expression of telomerase or ALT and telomere length stabilization.
The signalling pathway that connects progressive telomere shortening or damage to telomerase activation (or ALT) must be different from the mechanism underlying telomerase activation in stem cells. First, in cells entering the differentiation pathway, including progenitors, telomere maintenance is impaired compared to stem cells as suggested by an increased rate of telomere shortening in intestinal adenomas compared to stem cells. This is consistent with a negative impact of exit from the stem compartment and cell differentiation in the capability of telomerase to cope with erosion of telomeres (or impaired coupling of cell proliferation with differentiation caused by oncogenes). Later, a fundamental change in telomere maintenance can explain telomere stabilization. Up until this point the increased response in telomerase activity was unable to counteract continuous telomere shortening. The two faces of this process: accelerated telomere shortening and the appearance of a new mechanism of telomerase control which is permanent and coincides with immortalization strongly suggest that immortalization is facilitated by the process of telomere shortening preceding it and that it does not occur in the stem cell compartment. Instead, it occurs in a cell engaged in tissue turnover and some degree of differentiation before developing a new mechanism for telomere maintenance distinct from that of the stem cell. Both concepts are supported by recent discoveries on telomere reactivation in cancer (to be discussed). First, telomerase expression is controlled differently in malignant versus pre-malignant cells. Second, permanent reactivation of telomerase (or ALT) inherent to cell immortalization emerges simultaneously with other stem cell features, therefore in a non-stem cell. As stated above, interference of oncogenes with cell turnover appears to be a constant finding in tumour development. In a former paper we have shown that a true stem phenotype is extremely rare in cancer (7). Even tumours that reproduce phenotypic features of cells at early stages of development display features of premature differentiation (7). In the intestine, it has been irrefutably established that colon cancer initiating cells are included in the CD133 positive population. By limiting dilution analysis, it was calculated that there was one cancer initiating cell in 263 CD133+ cells, a >200-fold enrichment over the unfractionated population. The CD133- cells did not initiate tumour growth. (21). Prominin was detected throughout the lower half of the intestinal crypt and was not restricted to the rare stem cells sandwiched between Paneth cells. CD133 can be detected in Lgr+ stem cells but also on the more numerous transit amplifying compartment. Thus, it is highly likely that colon cancer initiating cells are early committed precursors rather than stem cells
Hoffmayer (22) showed that Wnt/β-catenin signalling regulates telomerase expression in the intestinal mucosa and mouse ES cells. They have revealed binding sites in the hTERT promoter and transcription start site for different downstream targets of the Wnt/TCF pathway. The complex arrangement of distinct targets of the Wnt/TCF pathway on the hTERT promoter enables for a flexible response of telomerase expression in different contexts or to the occurrence of mutations in these promoter binding sites. This could explain differences in the control in cancer versus adenoma even when minimal changes in the promoter have occurred. Thus, when colon carcinoma cell lines carrying tetracycline inducible constructs coding for dominant negative versions of TCF1 or TCF4 were subjected to tetracycline treatment, significant downregulation of TCF targets expressed in colon carcinomas were observed but hTERT transcript levels were comparable to that of the same tumors lacking the dominant negative constructs. As Ducrest et al. state, “ “these results quite strongly argue that TCF does not play a role, direct or indirect, in controlling hTERT expression in colon carcinomas” (23). In cancer transformation telomerase re-expression is under a new, different type of control relative to normal stem cells.
New discoveries are revealing the bewildering complexity of the mechanisms used by cancer cells to support telomerase reactivation. All of them appear to be specific of cancer cells and linked to the emergence of stemness. Cancer specific hTERT transcription may be due to hTert transcription being under the control of a mutated hTERT promoter or under the control of the wild type promoter which becomes responsible to changes in nearby altered chromatin architecture.
A report from Kim et al. (24) describes a new phenomenon that they named telomere position effect over long distance (TPE-OLD) that explains how telomere length is linked to telomerase expression. In cells with long telomeres chromatin folding governs the formation of a loop placing the sub telomeric region adjacent to the Tert locus. This is associated with repression of Tert expression. Conversely, telomere looping between the hTert locus and subtelomeric 5p which exists in normal cells is greatly reduced during in vitro aging leading to abrogation of Tert repression. These researchers point out that the conception that the hTert gene is not transcribed in normal telomerase silent cells is false. Expression of splice variants takes place but it does not result in full length RNA capable of producing telomerase activity. However, the amount of transcription is higher in the cells with shortest telomeres. They also observed higher level of active telomerase in two human fibroblast strains with short telomeres compared to the same cells with long telomeres. Chromatin looping affects the expression of genes located in the vicinity of Tert locus such as the CLPTM1L gene. CLPTM1L mRNA expression is detected in normal passaged BJ human fibroblasts but its expression increased with progressive telomere erosion. Expression of genes located at intermediate distance between CLPTM1l and telomer locus did not change. Active chromatin marks H3K4me3 and H3K9 acet were found increased across the hTert promoter of aged cells with short telomeres compared with young cells with long telomeres. Same active transcription marks were detected at the promoters of nearby genes such as CLPTM1L.
All these findings suggest that telomere shortening creates permissive conditions that may not be self-sufficient for the production of active telomerase but require cooperation with other factors such as oncogenes. With this idea in mind the researchers tested the knock down of p21 (CDKN1A t) which had previously been shown to de-repress hTERT expression. This resulted in increased number of transcripts including later exons 7/8 (coding for critical residues in the reverse transcriptase domain) and exons 15/16, which are responsive for putative active hTERT and total variants respectively. These variants increased with the knockdown of p21 in old but not young BL cells but they did not detect telomerase activity. However, this suggests that more drastic alteration or further shortening of telomeres could trigger telomerase re-expression
The role played by oncogenes or (epigenetic alterations mediated by oncogenes in cooperation with genomic changes in the proximity of the telomerase promoter) in the reactivation of Tert expression is exemplified by another mechanism of Tert regulation unveiled by Akincilar et al. (25). In CRC patient samples the authors found a significant correlation of JunD and β-catenin expression and hTERT transcription. Since JunD co-localizes with CTCF to regulate chromatin compactness, the authors compared of RNA-seq-data and ATAC-seq between high JunD and low JunD CRC cells and normal colon cells which identified 10 accessible chromatin regions co-enriched with JunD and CTCF that were not present in stem cells. Removal of a distal non-coding region located 140kb upstream of the WT promoter abrogated hTERT expression which demonstrates the influence of chromatin architecture on hTERT expression. Both JunD and CTCF were required for hTERT expression only in cells containing a wild type promoter and their deletion abrogated binding of Sp1 to the WT-hTERT promoter. Afterwards, circularized chromosome conformation capture (4C) sequencing identified a ,5 kb T-INT2 region (referred to as Tert interacting region 2) that regulates expression of WT-hTERT. It was proposed that JunD-CTCF binding relaxes and bends chromatin enabling the interaction between T-INT2 and the WT-hTERT
JunD upregulation of Tert in CRC cells also requires the recruitment of epigenetic factors, including CBP/p300 which are known to unwind chromatin in conjunction with other epigenetic modifiers like CTCF and Sp-1, to form bounds between distinct chromatin regions. A significant correlation was found in CRC patient samples between JunD and β-catenin expression although knocking-down of β-catenin with siRNA reduced hTert expression in HCT116 cells but it did not affect hTert expression in HT29 cells which have low basal β-catenin levels. The INT-2 region is operative in CRC cells where hTert is activated by oncogenic alterations but was not found in normal stem cells or induced pluripotent cells where 4 factors keep the WT-hTERT promoter active. On the other hand, INT-2 does not have a role on the regulation of mutated hTert promoters which are regulated by still another mechanism involving long-range chromatin interaction
Mutations of the hTERT promoter are common in cancer with higher incidence in glioblastoma, melanoma, urothelial, bladder and thyroid cancers and tend to affect older patients. Point mutations of the Tert promoter have been identified in two key positions, C250T and C228T in 19% of cancers. In general, these are single base mutations that create binding sites for members of the ETS transcription factor family (26). Regulation of mutant promoters appear to require long-range chromatin interaction between the mutant promoter and a genomic region located 260kb upstream. This genomic region called T-INT1 contains multiple binding motifs for GA-binding protein (GABP)α/β (members of the ETS transcription family). GABPA dimers on the proximal promoter have been shown to interact with GABP dimers located in a region 260 kb upstream of the TSS site of the hTERT gene to make a stable transcriptional hub (27). The MED12 subunit of the MED complex was identified as a mediator of that interaction. The same report identified other candidate genes for similar long-range interactions that require further validation. T-INT1 appears to be implicated exclusively in the regulation of mutant Tert promoters
Complementation Between Oncogenic Pathways Suggests Parallel Changes on Reprogramming of Stemness and Telomerase Control
As previously shown, adenomas display a crypt/adenoma TCF-dependent gene expression signature. SOX4, LGR5, AXIN2, cMYC and this signature together with tumour cell proliferation is abrogated in vitro by the inhibition of TCF function through the expression of dominant-negative TCF (dnTCF4). Another signalling pathway important for colorectal carcinomas (CCs) is Hedgehog (HH)-GLI. HH-GLI has been shown to be essential for proliferation and survival of primary colorectal carcinoma cell. In this pathway, signalling is normally triggered by secreted HH ligands, most often by Sonic Hedgehog (SHH), that inactivate the 12-transmembrane protein Patched1 (PTCH1). PTCH1 activity inhibits the function of the 7-transmembrane-G-couple-receptor-like protein Smoothened (SMOH) Upon PTCH1 inactivation by HH ligands, SMOH is free to signal intracellularly, involving several kinases and leading to the activation of Gli transcription factors. Gli factors have activator and repressor functions. Gli3 3 encodes the strongest repressor. In the absence of HH ligands GliR is dominant and Gli1 is not transcribed. Upon SMOH activation, the Gli code is switched so that Gli1 is transcribed and Gl3R repressed.
In a panel of tumors directly isolated and processed from patients, Varnat et al. (28) detected the Wnt/TCF signature (LGR5, SOX4, Axin2, cMYC, CD44) in CD133+ cells from early, non-metastatic TNM1,2 CCs compared to normal colon whereas this signature was downregulated on metastatic TNM 3,4 CD133+ and CD133- cells. Instead, the latter displayed elevated expression of the HH-GLI signature. Metastatic CCs also exhibited higher levels of Wnt/TCF inhibitors DKK1 and SFRP1 than non-metastatic CCs. Inhibition of a crypt/adenoma signature by dnTCF4 did not decrease the expression of the HH-Gli1 pathway activity markers and viceversa blockade of endogenous HH-Gli1 repressed Gli1 but it did not inhibit Wnt-TCF suggesting that, in vitro, Wnt-TCF and HH-Gli act in parallel. Analysis of cell lines and primary CCs revealed that exprewession of the Wnt/TCF signature was maintained in cells cultured in vitro whereas repression of this signature and enhanced expression of the HH-GLI and core ES- like signatures were detected in vivo.
In addition, a stem cell-like signature formed by NANOG, OCT4, SOX2, KLF4 and BMI1 similar to the reprogramming set involved in inducing iPS cells from differentiated cells was enriched in CD133+ versus CD133- especially when comparing metastatic with non-metastatic cases. The stem-like signature was dependent on the HH-GLI pathway but unaffected by blockade of endogenous Wnt/TCF signaling by dnTCF4.
The finding that β-catenin induced GLI1 activity in a GLI-binding site luciferase reporter assay independent of TCF function suggested a crosstalk between these pathways. KRASv12G, MEK and AKT also enhanced GLI activity on GLI-luciferase reporter assays. Exogenous Gli activity was also repressed by PTen and p53. Interestingly, MEK and AKT inhibitors repressed the epithelial-mesenchymal marker SNAIL1 only in metastatic CCs. On the other hand, β-catenin has been found to upregulate sonic hedhog (Shh) and Patched (PTCH1) as well as IGF signaling on the hair follicle (29).
Repression of the Wnt-TCF crypt/adenoma signature in patient samples and in xenografts versus in vitro culture indicated the possibility that Wnt-TCF signalling might be replaced by other signalling pathways during tumour progression ending in further reprogramming (immortalization emerging in cells within the same population through a secondary oncogenic pathway?).They made use of the two established CC cell lines previously used to demonstrate the key role of TCF function in vitro: Ls174T-dnTCFdox and DLD1-dnTCFdox. These clones display homogeneous dnTCF4 expression only upon doxycycline addition. Ls174T-dnTCFdox transplanted in vivo displayed TCF target inhibition to the same level as in vitro as compared with untreated cell controls but this did not have an impact on tumour growth: tumours resulting from Ls 174T-dnTCF4dox cells or parental Ls174T cells with or without dox treatment showed similar growth curves and tumour appearance. However, the same level of inhibition in vitro leads to complete growth arrest. This lack of tumour growth arrest in spite of inhibition of several targets of Wnt-TCF parallels results commented above (23) and suggests a critical change in tumour growth control probably associated with a separate control of telomerase expression. However, results with DLD1-dnTCF4dox were different. Treatment with dox repressed TCF targets as well as tumour growth both in vivo and in vitro. (telomerase expression control had not changed hands yet?)(28).
Further research suggests again that on the course of cancer progression the initial oncogenic pathways may intersect or overlap with new cancer-driving pathways. This process might entail a secondary event of reprogramming within an established cancer stem cell or within another cell from the tumour population. Findings by Asciutti et al. (30) again show the superimposition of distinct oncogenic pathways on carcinogenesis initiated by the Wnt/TCF pathway. They studied three gastric carcinomas: AGS that harbours a β-catenin activating mutation that prevents its proteasome degradation and MKN-28 and MKN-74, both containing inactivating mutations of APC. Downregulation of TCF signalling by dominant negative TCF4 (dnTCF4) led to reduced expression of TCF targets including c-Myc, BMP-4, Axin-2 and Lef-1 in all three tumours as well as inhibition of tumour cell proliferation in AGS. No inhibition of proliferation was observed in the other two cell lines in spite of a comparable response to dnTCF4 regarding downregulation of TCF targets including c-Myc inhibition and correlated increase in p21 level. Following overnight serum starvation ERK and AKT phosphorylation were detected in AGS and MKN-28 tumour lines while MKN-78 showed minimal if any AKT phosphorylation and very low ERK activation. AGS tumour growth could be inhibited by either dnTCF4 or c-Myc shRNA while neither of them affected MKN-28 tumour growth. Exposure to a MEK inhibitor or aPI3K inhibitor led to a marked inhibition of proliferation of MKN-28 tumour and slight reduction of AGS proliferation
The Epithelial-Mesenchymal Transition Appears to Entail a Secondary Stemness Reprograming Event and a New Change in the Control of Telomerase Reactivation
Superimposition of secondary oncogenic pathways during tumour progression goes hand and hand with some ominous signs such as invasion and metastasis. The onset of metastasis, is usually preceded by invasion of tumour cells into the surrounding soft tissues. The capacity for invasion is rooted on a physiological mechanism used during embryogenesis for tissue remodelling and amply used later in life by cancer cells, called epithelial-mesenchymal transition (EMT). A study by Medici et al. (31) on colon cancer and melanoma cell lines shows an intricate cooperation between the Wnt and TGF-β pathways in the establishment of EMT. Initially, TGFβ1 and TGFβ2 induction of Snail and Slug resulted in decreased level of E-cadherin with subsequent release of cytosolic β-catenin. Then, β-catenin-TCF4 upregulated TGF-β3 which promoted synthesis of LEF-1. As Snail and Slug are transcriptor repressors, their induction of TGFβ3 is indirect and mediated by β-catenin-TCF4 binding to the TGF-β3 promoter.
Loss of E-cadherin was accompanied by morphological changes characteristic of EMT. However, DN-LEF-1 prevented invasion associated with EMT.EMT Enhanced transcription of TGFβ3 results in upregulation of LEF-1 which after binding cytoplasmic β-catenin can enforce the acquisition of the mesenchymal phenotype and gene changes associated with EMT: increased vimentin, fibronectin, α-SMA and α-actinin expression.
The authors indicated the possible involvement of the MAPK pathway on the TGFβ1/2 upregulation of E-cadherin repressors Snail and Slug. As well as the possible role played in this and related processes by GSK3β, an important node in the bifurcation of this and other signalling pathways. TGFβ1, TGFβ2 and TGFβ3 all signal through PI3K (and downstream target AKT) which induces phosphorylation and inactivation of GSK3β, thereby releasing β-catenin from its destruction complex and creating a surplus of β-catenin which can associate with LEF-1 that was upregulated by TGFβ3 signalling.
The report by Medici et al. does not address the emergence of stem cell traits or the potential role of telomerase in EMT. This was addressed in a model of gastric cancer by Liu et al. (32)). Wnt-TCF supports self-renewal of gastric mucosa and its deregulation can initiate gastric cancer but EMT, appears to require, in addition, the participation of hTERT as well as the other factors described above. Liu et al. infected the GC cell line BGC-823 (and two more gastric cancer cell lines) with an hTERT retroviral vector and detected increased levels of mesenchymal markers vimentin and Snail and a reduction of E-cadherin. This led also to a significantly increased invasiveness of hTERT-BGC-823 cells compared with control pBabe-BGC-823 cells. Conversely, hTERT inhibition resulted in loss of these mesenchymal markers and diminished invasiveness. hTERT inhibition by siRNA substantially abolished TGFβ1-stimulated Snail and vimentin induction and conversely, hTERT overexpressing cells exhibited much higher levels of Snail and vimentin than control cells. hTERT and TGFβ1 affected β-catenin expression with overexpression of these factors leading to higher β-catenin levels and their inhibition correlating with slight but consistent reduction of β-catenin. In BGC-823 cells transfected with a dominant negative hTERT (mutation D869A) that loses a catalytic function or with C-terminal tagged hTERT that exhibits telomerase activity but is unable to elongate telomeres, increased expression of Snail and vimentin and activation of TCF/LEF reporter were detected.
After injection of hTERT-BGC-823 cells or of pBabe-BGC-823 control cells into the tail vein of nude mice, metastatic colonies were found in the lungs in both cases but those formed by hTERT-BGC-823 were larger and more numerous. hTERT-induced EMT was accompanied by the appearance of stem cell features such as dramatic enhancement of monosphere formation and overexpression of stem cell markers like OCT-4 and CD44, an established gastric CSC marker.
(32). These findings and the aforementioned study of Varnat et al. suggest the need for the simultaneous occurrence of reprogramming (as shown by the onset of stem cell traits) and inducers of EMT such as Snail and vimentin
Upregulation of TGF-β1-mediated upregulation of β-catenin by hTERT and the synergistic effect of TGF-β1 and hTERT in EMT induction demonstrates a positive feedback loop between these factors which may accelerate induction of EMT and acquisition of stemness traits. It is also possible that during cancer progression, interaction of hTERT with new partners leading to increased expression of hTERT, might induce further reprogramming events that could accelerate and worsen the course of the disease
Id1 and Epithelial-Mesenchymal Transition.
Ids are members of the helix-loop-helix (HLH) group of proteins which can form heterodimers with basic helix-loop-helix (bHLH) proteins. Lacking the basic domain necessary for DNA binding, Ids act as dominant negative transcription factors. In hematopoiesis Id1 preserves the undifferentiated state by antagonizing a differentiation step induced by E2A. Id1 null readily commit to myeloid differentiation consistent with de-repression of E2A as inducer of myeloid lineage (33) When bone marrow cells were transduced with an Id1 expressing MSCV retrovirus cells could divide for over 1 year in the presence of stem cell factor. Most cells were myeloblasts with some promyelocytes and myelocytes; these cells could be induced to differentiate with physiological inducers of differentiation. Transplantation of cultured cells to mice induced a myeloproliferative like-disease. However, secondary recipients did not develop leukaemia (7) suggesting that prevention of differentiation by Id1 does not induce transformation
The landscape afforded by in vivo experiments is somewhat different. One of the in vivo studies on Id1 took advantage of a murine mammary epithelial cell line, SCp2, wich can differentiate in vitro recapitulating the main aspects of development and lactation including proliferation and invasion of mammary during late pregnancy as reflected by growth arrest and formation of alveolar structures as well as milk proteins when serum is removed and cells are placed in contact with basement membrane components and given lactogenic hormones (insulin, prolactin and hydrocortisone) Upon constitutive Id1 expression these cells slowly invade the basement membrane and express a 120kd metalloproteinase (MMP) mimicking the capacity of normal mammary cells to secrete MMPs that degrade the ECM during involution. However, despite constitutive Id1 expression SCp2 cells do not grow in soft agar nor form tumors in nude mice. SCp2 cells transfected with the murine Id-1 cDNA driven by the mouse mammary tumor virus promoter (Cp2-Id-1 cells) formed alveolar structures but later (10 days) detached and migrated through the surrounding parenchyma. Among nine human breast tumors examined, only Id-1 expressing cells also expressed the 120-kDa gelatinase. A complete epithelial-mesenchymal transition was not observed as they still retained some epithelial markers. (34)
Expression of activated Ras in mammary epithelium of mice leads to formation of senescent foci containing disorganized epithelial cells. Mice that concurrently harbor deletion of the p19, p53 or p21 tumor suppressor develop rapidly mammary tumors suggesting that these tumor suppressors prevent tumorigenesis by inducing senescence. However, ectopic expression of Id1 co-expressed with activated Ras leads to rapid tumor development which indicates the capacity of Id1 to antagonize apoptosis induced by the tumor suppressor axis p19, p53, p21. In the same study conditional activation of hrasv12 in conjunction with constitutional activation of Id1 led to tumor formation but doxycycline inactivation of the hrasv12 promoter resulted in complete tumor regression within 40 days. This again suggests that Id1 alone, even if constitutively expressed cannot give rise to tumor development. It is true, however, as the same study informs, that overexpression of Id1 in the intestine leads to development of adenomas albeit with long latency and low penetrance. To our knowledge, there is only an example of tumor (leukemia) induced by Id1 alone. Its overexpression in the hematopoietic compartment leads to massive apoptosis, disruption of differentiation and leukemia (35). The finding of massive apoptosis induced by Id1 is surprising as it seems to contradict the anti-apoptotic function usually performed by Id1. The transgenic expression of Id1 in thymocytes was found to change the normal distribution of the thymocyte population at the DN and later stages in a way that is highly compatible with a hematopoietic developmental block, a situation which is a frequent cause of leukemia which suggests a totally different mechanism of tumorigenesis that must be imputed to the developmental block. (36).
In summary, Id1 does not meet the classical definition of an oncogene. No Id1 mutatios have been found although numerous reports have linked Id1 to induction of invasion and metastasis in human cancer through promotion of epithelial-mesenchymal transition (EMT). Two characteristic features of epithelial cells undergoing EMT are the adoption of a mesenchymal morphology and simultaneous expression of stem cell properties. This fact demonstrates the possibility of spontaneous reprogramming of committed cells to neoplastic stem cells. However, most information on EMT has been obtained with models involving ectopic expression of Id1 in immortalized cell lines although occasionally have involved committed cells. The main final downstream effectors of Id1 in EMT include negative regulators of E-cadherin such as Snail, Twist, fibronectin and vimentin, although various additional targets participate in this process in distinct tissues. Some examples follow
Breast
Mani et al. induced EMT by ectopic expression of Snail or Twist in cells isolated from non-tumorigenic, immortalized mammary epithelial cells (HMLE) sorted into enriched mammary stem cells (CD44hi CD24lo and committed CD44lo CD24hi. The mesenchymal-like cells generated by EMT acquired a CD44hi CD24lo expression pattern-precisely the same phenotype that has been ascribed to neoplastic mammary stem cells. Similarly, induction of an EMT in HMLEs by exposure to TGF-β1 (an inducer of Id-1) resulted in the appearance of mesenchymal-looking cells and the acquisition of the CD44hi CD24lo phenotype. (37). In the words of these authors: “In principle, the behaviour of the HMLE human mammary epithelial cells might be attributable to the introduced genes that were used previously to immortalize these cells, specifically hTERT, which encodes the catalytic subunit of human telomerase holoenzyme, as well as the SV40 early region. For example, these genes might facilitate the reprogramming of the HMLE cells initiated by EMT-inducing transcription factors such as Snail or Twist. However, we observe exact the same responses to Snail or Twist in non-immortalized primary mammary epithelial cells, which indicates that hTERT and SV40 antigens appear to have no effect on the induction of the EMT. In summary, the experiments indicate that committed cells are reprogrammed to stem cells when subjected to EMT.
Other experiments have revealed the wide diversity of downstream targets that may be involved in Id1-induced EMT. Gumireddy et al. (38) working also with breast cancer cell lines and clinical samples showed that Id1 interacts with TFAP2A which binds to the S100A9 promoter enhancing its activity. A consequence of this activation is suppression of RhoC. Therefore, Id1 by binding TFAP2A downregulates RhoC. RhoC appeared to be the main responsible for invasion and metastasis of breast cancer and its downregulation suppressed the migratory and invasive phenotype induced by Id1.
However, different experiments have incriminated the PI3K/AKT pathway as the backbone of EMT. Thus, In breast cell line MCF7 Lee et al. (39) showed that Id-1 activated the Akt pathway by inhibition of PTEN transcription through downregulation of p53. Akt was phosphorylated at Ser473 and glycogen synthase kinase at Ser9 with stabilization and nuclear localization of β-catenin, TCF/LEF, cyclin D1 and Akt-mediated p27 phosphorylation at Thr157 and translocation to cytoplasm. Fong et al. reported that Infection of a non-invasive breast cancer cell line with Id-1 rendered it invasive (40).
A study in lung cancer confirmed that induction of Id1 was mainly mediated by Akt activation. Id1-overexpressing H460 lung cancer cells had elevated expression of cyclin D1 and CDK4 as well as a decreased expression of CDK2, cdc2 and cyclin B1. The expression of phospho-p38MAPK (active form of p38MAPK) was increased in Id1-overexpressing H460 cells and repressed in Id1- knockdown H520 cells. However, when Id1-overexpressing H460 cells were treated with SB203580, an inhibitor of p38MAPK, or wortmannin, an inhibitor of PI3K/Akt, they found that only wortmannin had a significant effect on suppressing cell proliferation indicating that Akt was the main effector of Id1 expression. JNK, ERK and STAT3 activities were also analysed (41). Activation induced by Id-1 may extend a step further along the PI3K pathway affecting also the NFκB expression. Li (42) showed that in esophageal cancer cells, stable ectopic expression of Id-1 induced expression of pAKT, glycogen synthase kinase 3β and inhibitor of kappa B, as well as increased nuclear translocation of NFkB subunit p65 and NFkB binding activity. These effects were antagonized by treatment with the PI3K inhibitor LY294002. This experiment revealed the involvement of another downstream target of the pathway, NFκB which appears to be responsible for the apoptosis protection of cancer cells. Both, the PI3K inhibitor LY294002 or the NFkB inhibitor Bay 11-7082 increased the sensitivity of Id1 overexpressing esophageal cancer cells to TNF-α-induced apoptosis
The same conclusion can be derived from another study in prostate cancer cells. Id-1 induced nuclear translocation of NFκB along with upregulation of its downstream targets Bcl-xL and ICAM-1 which was accompanied by increased resistance to apoptosis induced by TNFα through inactivation of Bax and caspase 3 (43)
Liver
Direct origin of cancer from primary hepatocytes has been described associated or preceding EMT.. Cirrhotic livers provide a fertile soil for cancer development and some observations implicate fibroblastoid cells derived from hepatocytes as the cells of origin of hepatocarcinoma. TGF-β which is increased in chronic inflammation of the liver can promote this phenotypic change. Primary mouse hepatocytes have been observed to lose their epithelial phenotype in response to TGF-β accompanied by loss of E-cadherin and upregulation of Snail. EMT in the liver appears to be a complex process requiring inputs from several signaling pathways. In primary murine hepatocytes, EMT was associated to focal adhesion kinase (FAK)-Src-dependent activation of the PI3K/Akt and Erk1/2 pathways. On the other hand, EMT appears to be associated in human hepatocarcinoma with metastatic and highly invasive tumors. E-cadherin was reported to be strongly expressed at cell-to-cell contact in a group of non-metastatic HCC patients. Instead, reduced expression of E-cadherin and loss of cell-to-cell contact associates with nuclear translocation of β-catenin and predominates in the group of HCC patients with metastasis (44).
In cervical cancer the T-box transcription factor 3 (TBX3) promotes EMT and its expression correlates with Id1 mRNA and protein levels although it has not yet been confirmed whether TBX3 regulates Id1 directly. Silencing TBX3 reduced cell migration and invasion. However, TBX3 might act also through its ability to downregulate PTEN and suppression of p19-p53 pathway (45). In another study on cervical cancer, Id1-induced activation of the PI3K pathway could be followed, at least, to downstream target NFkBp65 (46).
Caveolin-1 is essential for the role of Id-1 in inducing EMT and cell survival in prostate cancer cells. Apparently, each Id1 and caveolin alone can induce EMT but they have a synergistic effect. In prostate cells, Id1 induced Akt (mediator of PI3K pathway) activation through promoting the binding between Cav-1 and PP2A. Cav-1 regulates signalling of membrane proteins such as epidermal growth factor receptor, Src and H-ras through direct physical interaction. Although Id1 and Cav-1 alone are each able to increase Akt phosphorylation they can act synergistically. Id1 promotes the inhibitory effect of Cav1 on PP2A but this does not result in lowering PP2A level. Id1 does not bind PP2A. Thus, it seems that Id1 promotes the binding activity between Cav-1 and PP2A which may lead to suppression of PP2A activity (47)
Glioblastoma
As first described in breast cancer, in glioblastoma, TGF-b has been shown to increase the CD44high cell population enriched for CICs through the induction of an epithelial-mesenchymal transition. The CD44high/Id1high population was enriched for GICs and generated tumors reproducing the heterogeneous cell population of the original tumor (48). Other experiments in non-transformed p16-p19 deficient astrocytes have revealed how the context can influence backbone signaling pathways Id1- mediated suppression of Cullin3 conferred stem cell like features and tumorigenicity to Ink4a/Arf-/- astrocytes. The authors identified GLI2 and DVL2 as Cullin3 interacting proteins which are involved in the SHH and Wnt pathways respectively and demonstrated that ID1-mediated suppression of Cullin3 may activate SHH and Wnt signaling by stabilizing GLI2 and DVL2 proteins, respectively. Id1 overexpression markedly increased DVL2 and GLI2 protein stability relative to VEC and ID1+Cullin3 overexpressing cells. However, qRT-PCR analysis revealed that Dvl2 and Gli2 mRNA levels were not significantly changed by ID1 overexpression which indicate that ID1 regulates DVL2 and GLI2 expression at a post-transcriptional level by suppressing Cullin3 expression (49). We will see again this phenomenon in tumorigenesis: the initial extracellular inducers of a tumorigenic signaling pathway cease to be necessary for stimulation of the pathway which becomes autonomous, that is, dependent only in the intracellular donstream targets.
Superimposition of the Wnt and Shh signaling pathways has also been demonstrated in the colon cancer line HCT116. Id1 maintained the stemness of CRC cells through activating the Wnt and Shh signaling pathways Combined inhibition of Wnt pathway with FH535 and Shh with HPI-1 showed greater inhibition than each single inhibitor. Concomitantly, there was significant reduced c-myc expression and c-myc downstream target PLAC8. Taken together, these data demonstrate that Id1 maintains the stemness of CRC cells via the Id1-c-Myc-PLAC8 axis through activating the Wnt/β-catenin and Shh signaling pathways (50).
A few reports have uncovered the participation of EGFR and MAPK in Id1 expression. In non-small-cell lung (NSCL) cancer, Pillai et al. (51) reported that signaling through the nicotine acetylcoline receptor or the EGF receptor upregulated Id1 in an Scr-dependent manner. Nicotine induced the expression of mesenchymal markers fibronectin and vimentin by downregulation of ZBP-89, a zinc finger repressor protein. Ectopic expression of a mutant Ras also resulted in increased Id1 level.
Despite the above reports, it is important to emphasize that EMT occurs as a physiological phenomenon in wound healing. In the early phase of wound healing EMT takes place as demonstrated by a change in keratinocyte morphology that shift to that of interstitial cells with the capacity to migrate and the typical molecular expression pattern of EMT: downregulation of E-cadherin and overexpression of vimentin and N-cadherin. This molecular expression pattern was amplified in the keratinocyte cell line HaCat by addition of TGF-β1. In addition, exogenous TGF-β1 accelerated cutaneous wound healing. It was shown that Foxo3a could revert these changes. Overexpression of Foxo3a suppressed the TGF-β1-induced EMT and the associated β-catenin activation. This key molecule of the Wnt pathway, β-catenin, was increased in cytoplasm and nucleus of HaCat cells by silencing Foxo3a expression and, conversely, the expression of β-catenin was decreased when infecting HaCat cells with lenti-flag- Foxo3a (52). Here a new player, FOXO, enters the game. There are reports showing that Foxo3a can suppress EMT in prostate cancer cells through inhibition of Wnt pathway. Foxo3a inhibits β-catenin both through direct binding and induction of miR-34 expression. β-catenin was shown to participate in Foxo3a-mediated suppression of EMT. In fact, decreased-E-cadherin and increased N-cadherin and fibronectin levels induced by Foxo3a KD and other features of EMT were reversed by silencing β-catenin expression (53). Another member of the Foxo family, FOXC1, has been shown to control stem cells of basal-like breast cancer via non-canonical activation of smoothened-independent Hedgehog signaling (54).
Evidently, EMT is a very complex process about which we have only fragmentary data, as illustrated in Figure 1
The above observations paint a picture of the crucial role of the crosstalk between the PI3K and Wnt/Shh oncogenic pathways. Some of the reports gathered have described activation of a downstream, converging molecule of these and other pathways, NF-kB, which is found to be involved in stemness and invasiveness. On the other hand, the common occurrence of stemness could be explained if oncogenic signaling extends to other downstream targets of the PI3K pathway such as the FOXO family and specially, Foxo3a, which are known to regulate stemness.
FOXOs and NF-kB Interplay (Figure 2)
The Forkhead box transcription factors family consist of 19 subclasses of Fox genes. They are involved in stemness, cell survival, redox homeostasis and stress resistance. In the absence of growth factor or insulin stimulation, Foxos reside in the nucleus where they activate transcription of cell cycle inhibitors such as p130, p27, p57 and p21 as well as proapptotic genes such as TRAIL, FasL and Bim. Foxo inactivation by PI3K/AKT signaling leads to its nuclear exclusion with enhanced cell proliferation, survival and stress sensitivity. (55). Loss of Foxo in the hematopoietic compartment results in an increase in HSC in cycle accompanied by exit of CD34- LSK cells and concomitant decreased p27 and p57. Stem cell quiescence depends on these cyclin dependent kinase inhibitors (CKIs). Ablation of CKIs leads to stem cell exhaustion (56). The transition from HSC to myeloid progenitos is accompanied by a 100-fold increase in ROS. This change appears to be regulated by a transcription program that is independent of Foxo. The high ROS level wich is appropriate for the functions of short- lived myeloid cells would be incompatible with HSC homeostasis. (57). Akt is the main kinase that regulates Foxos (Figure 2) although other kinases such as glucocorticoid inducible kinase (SGK), casein kinase (CK1), dual thyrosine phosphorylated regulated kinase 1 (DYRK1) and IKK may phosphorylate Foxos in specific residues and target many other protein substrates including GSK3β, p21 and p27. Phosphorylation of Foxos by JNK appears to be counterregulatory with respect to Akt (57, 58)
Figure 2.
P13K/ AKT signaling.
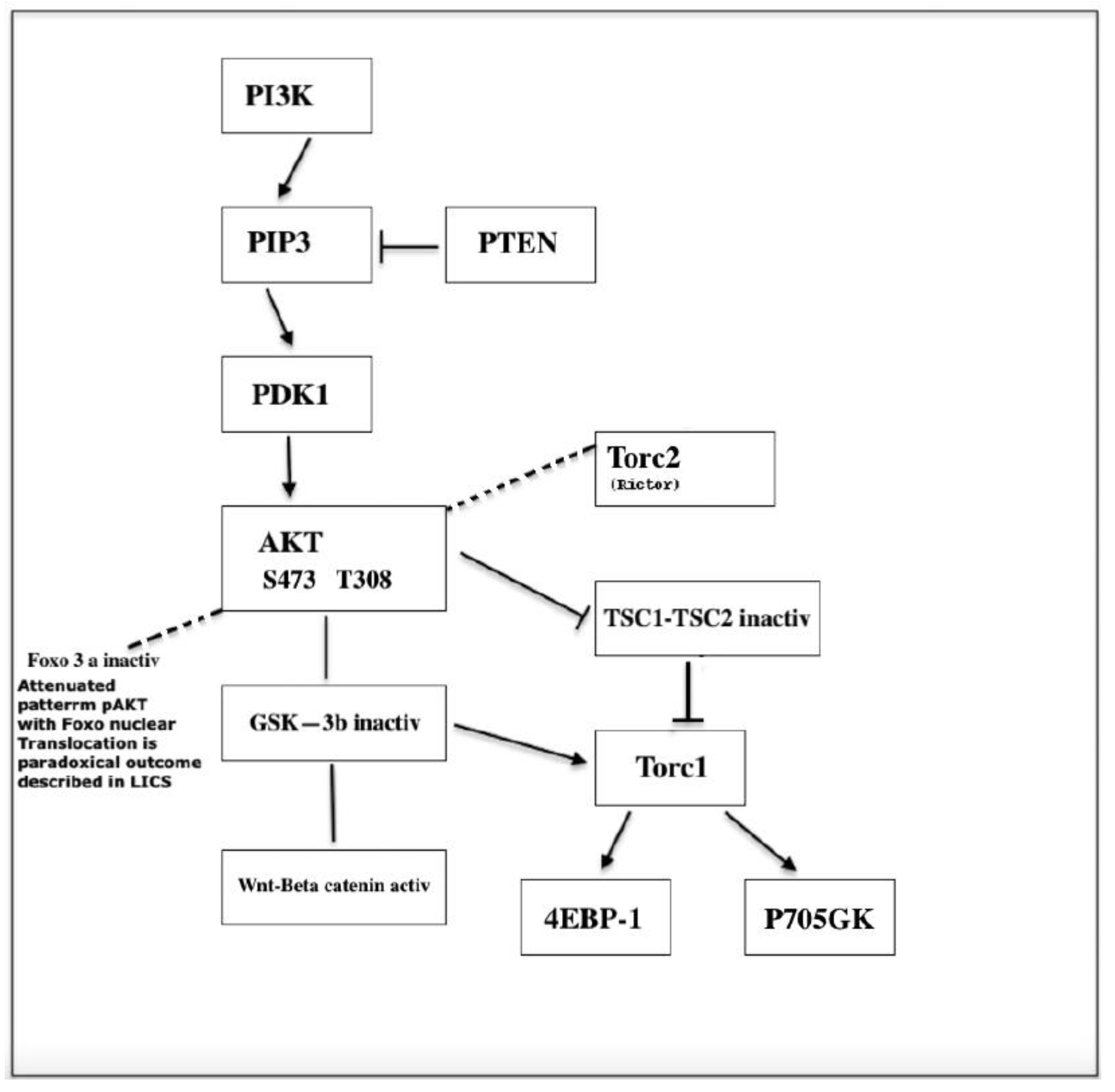
Studies on the role of PI3K pathway in tumorigenesis have revealed two apparently reversible functions of FOXOs associated with the activation and inactivation of AKT. Inactivation of Foxo3a which is always accompanied by cytoplasmic translocation of Foxo3a results in cell proliferation-differentiation. However, despite AKT signaling some cells show nuclear localization of Foxo3a and a pattern of attenuated AKT ( Figure 2). This reverse nuclear translocation opposite to the expected cytoplasmic translocation normally induced by phosphorylated AKT appears to be essential for the emergence of the cancer stem cell and maintenance of leukemia initiating cells whereas suppression of Foxo3a appears essential to maintain survival and proliferation. This double shutling of Foxo3a is observed in AML-AF9 leukemia where LICs have the GMP phenotype but contrary to the bulk tumor cells display an attenuated pattern of AKT activation. Sykes et al. ( 58) activated AKT in HSCs by Pten deletion or myr-AKT constructs transduced to bone marrow cells using the stem cell virus MSCV. Invariably, Pten deletion leads to HSC mobilization and HSC exhaustion followed by tumorigenesis.BM cells transduced with activated AKT (MSCV-IRES-GFP-myr construct) when injected to irradiated syngeneic recipients induce a myeloproliferative disease (MPD) that progresses to either AML or ALL. Despite the MPD, the proportion of GFP+ LSK cells were decreased compared to control, consistent with HSC mobilization and exhaustion of the HSC compartment. Paradoxically, Sykes et al. found that the same AKT construct induced suppression of growth of AML-AF9 leukemia. Suppression of growth was associated with myeloid maturation and myeloid maturation related cell death. Pten deletion, activates AKT and induce self-renewal and exhaustion of HSCs (likely accompanied by exit of the HSC compartment). In the AML-AF9 leukemia model, the expression of phosphorylated AKT followed by HSC exhaustion was accompanied by a conspicuous increased myeloid maturation and myeloid maturation related cell death (myeloid marker CD11b was higher on bone marrow cells injected with the stem cell virus MSCV (MSCV.IRES-GFP-myr-Akt). However, maturation related death induced by myr-Akt occurred in the presence of rapamycin, suggesting that Akt utilizes pathways other than mTOR for activation for myeloid maturation. (58). In fact, Foxo3a inactivation induces cell proliferation and differentiation of HSPCs as well as exhaustion of HSC as revealed in competitive repopulation assays. This has been observed in other tissue stem cells, in addition to HSCs. For instance, Foxo1 and Foxo 3 knockdown led to differentiation of intestinal stem cells into goblet cells and Paneth cells. Foxo 3 is higher in neural stem cells (NSCs) undergoing self-renewal and its ablation led to a decrease in NSC population accompanied by an increase in progenitors and the exhaustion of the NSC pool. (56). A parallelism on the effects of different targets of the PI3K pathway is well known, but a subtle difference may be revealed here: pAKT phosphorylates and inactivates different substrates such as TSC2 and GSK3-β. Interestingly, pAkt Ser473 is dispensable for TSC2 (which activates mTORC1) or GSK3-β inactivation but is required for Foxo3a inactivation (Figure 2). Thus, myeloid maturation and cell death might be attributed to the action of Foxo3a inactivation.. Apparently, growth inhibition in this system must be attributed entirely to maturation related cell death since pAKT only reduces tumor burden and induces apoptosis of the bulk of tumor cells but it does not affect the leukemia initiating cells (LICs). However, in the AML-AF9 leukemia LICs have the phenotype of GMPs but differ from GMPs by a pattern of attenuated Akt activity (reduced pAktThr308 and reduced pAkt Ser473) which is identical to that of normal HSCs and which seems essential to maintenance of self-renewal. In summary, AKT activation induced by MSCV virus improved leukemia by reducing tumor burden but did not have any effect on the AkT inactive stem cell. Since HSCs were pushed into cycle and engaged turnover by Foxo3a inactivation, the attenuated pattern of AKT expression which protects stem cells from exhaustion must have been gained after cells have been pushed out of the HSC stage by Foxo inactivation as indicates the GMP phenotype. The reversal to the AKT pattern associated to active Foxo3a must be concomitant with the triggering of immortalization and cancer transformation through unknown signaling. JNK was shown to be activated in response to Foxo deactivation but its activation resulted in further increased maturation related apoptosis. (58)
Assuming a process of tumorigenesis that involves Foxo deactivation (downstream target of PI3K pathway), which leads to differentiation, the emergence of the LIC after Akt-induced-Foxo inactivation implies an event of de-differentiation-reprogramming occurring in a cell previously engaged in pAkt-induced proliferation-differentiation. For instance, nuclear exclusion (inactivation) of Foxo3a is associated with the first step of hematopoietic differentiation as Foxo3a is present in the nucleus of freshly isolated Foxo3a+/+CD34- KSL cells (HSCs) but appears in the cytoplasm of freshly isolated Foxo3a+/+ CD34+ KSL cells (progenitors). Our proposal is that malignant transformation of a committed cell might be supported by a mechanism of telomerase reactivation more similar to that of iPSCs (triggered in a differentiated cell) rather than the telomere maintenance mechanism of the normal stem cell which is probably exhausted. Some Foxo family members such as FOXF2 can regulate stemness differentially in basal and luminal-like breast cells (59).
A previous report validated these findings and unveiled new aspects of the issue (60). BCR-ABL activates Akt signaling that suppress FOXO transcription factors, chiefly among them, in the hematopoietic system, Foxo3a. It was shown that Foxo3a has an essential role in the maintenance of CML LICs. Nuclear localization of Foxo3a and decreased Akt phosphorylation are enriched in the LIC population and as observed by Sykes et al. in the AML-AF9 model, low Akt phosphorylation in LICs correlated with mTORC1 inactivation. In order to prove the role of Foxo3a in the maintenance of LICs Naka et al. infected immature bone marrow cells from Foxo3a+/+ and Foxo3a-/- mice with a retrovirus carrying MSCV-BCR-ABL-IRES-GFP. The infected cells were transplanted into syngeneic recipients. Both recipient groups developed CML-like MPD demonstrating that FOXO3a is dispensable for the generation of CML-like disease. However, the in vitro colony forming ability of second BMT LICs was decreased by loss of Foxo3a. In a third BMT, mild CML-like disease developed in recipients within one month but Foxo3a deficiency prevented the propagation of CML cells in the peripheral blood and spleen. Foxo 3a+LICs also developed ALL as well as CML which was not observed in recipients of Foxo3a-. Thus, Foxo3a is essential for long-term maintenance of leukemia initiating potential whereas Foxo3a inactivation is a previous step in the initiation of leukemia. An important additional finding was that TGF-β1 signaling controls Akt activation in the nucleus of KLS+ cells but not in nucleus of KLS- (non-LICs) as indicated by phosphorylation of Smad2/3 restricted to nuclei of KLS+ cells. In view of the discussion in precedent paragraphs on the role of TGFβ on epithelial-mesenchymal transition, this finding raises the question of the involvement of EMT in these leukemia models. Moreover, the role of telomerase re-expression must be investigated as suggested by the report mentioned above by Liu et al. (32) where hTERT inhibition by siRNA substantially abolished TGFβ1-stimulated Snail and vimentin induction and conversely, hTERT overexpressing cells exhibited much higher levels of Snail and vimentin than control cells. Other outcomes of Foxo inactivation which are not usually contemplated may assist on the promotion of tumorigenesis such as the non-physiological type of myeloid differentiation induced by Foxo deactivation (57) as interference with hematopoiesis might by itself be a cause of tumor formation as numerous examples of hematopoietic developmental blocks indicate.
IκB Kinase (IKKβ, or IKKα to a lesser degree) may induce inactivation and cytoplasmic translocation of Foxo3a independent of pAKT at Ser 644 (61) (Figure 3) and both pAKT or IKK-induced nuclear inactivation of Foxo3a promotes cell proliferation and resistance to apoptosis. Therefore, it is believed that these molecular changes have an important role in the promotion of tumorigenesis. However, deepening the puzzle, the correlation between positive pAKT and IKK and cytoplasmic localization of Foxo3a may occur in normal tissues as well as in tumors (62). Nevertheless, the contribution of Foxo3a to leukemic transformation is indisputable as demonstrated by impaired leukemogenesis in Rictor-deleted mice (7) that cannot phosphorylate Akt Ser473 as illustrated in Figure 2 and are, therefore, unable to inactivate Foxo, suggesting Foxo`s role in tumorigenesis requires both inactivation in the initial phase within progenitors and its activation in LICs
The NF-κB Transcription Factors and Oncogenesis.
The NF-kB family of transcription factors plays a pivotal role in a multitude of process such as inflammation, cell proliferation, survival, adaptive and innate immunity, EMT and stemness Here we will discuss its role in cancer as one of the final downstream targets of the PI3K pathway
The NF-κB family consists of five members: RelA/p65, RelB, c-Rel, NFκB1/p50 and NF-κB2/p52. P50 and p52 lack a transcactivation domain and are generated by processing from p105 and p100 respectively
NF-κB signaling is primary enacted by the IKK complex (IkKα, IKKβ and IKKγ/Nemo) which phosphorylates inactive IκB proteins located in the cytosol resulting in the nuclear translocation of NF-κB factors and the release and degradation of IκB proteins (Figure 3). Signal transmission to NF-kB transcription targets is channelled according to two different routes designated as the canonical and the alternate pathways.
In the canonical pathway, IKKβ subunit directly phosphorylates NF-kB-associated IκBα (mainly the p50/p65/RelA which is the most abundant dimer) leading to IκBα degradation and p50 /p65 translocation to the nucleus where it regulates transcription of target genes.
The alternate pathway relies on the IKKα homodimer activation in a manner dependent on NF-kB-inducing kinase (NIK). Activated IKKα phosphorylates p100 yielding p52. Then p52, RelB dimer translocates into the nucleus to activate its target genes. Numerous translational modifications modulate the activity of IKK and IκB proteins. The canonical pathway can be activated by a plethora of stimuli, antigen/MHC, cytokines, LPS, IL-1β, TNF-α. Activators of the non-canonical pathway include the TNFR superfamily, CD40 B-cell activating factor (BAFF), TNF-like weak inducer of apoptosis (TWEAK) etc. Both pathways can interact at several levels (63)
Evidently, NF-kB activation and Foxo3a deactivation, are two possible outcomes of IKK activation that can contribute to promoting tumorigenesis
NF-κB can be Directly Activated Through DNA Damage Signaling
Induction of DNA damage has revealed the existence of an ATM-NEMO-IKK signaling pathway.
Grosjean-Raillard et al. (64) chose 4 AML cell lines to study DNA damage-induced NF-kB activation. increasing reactive oxygen species in KG1 AML cell line which did not exhibit NFkB activation resulted in phosphorylated ATM and phosphorylation of the IKK complex.It was shown that a complex of ATM and NEMO exited the nucleus to phosphorylate cytosolic IKK and p65 was translocated to nucleus. Conversely, ATM inhibition abolished IKK phosphorylation which was followed by apoptosis. The other three AML cell lines displayed constitutive NFkB activation and one of them P39 exhibited also strong constitutive ATM phosphorylation. In this latter line, ATM inhibition abolished IKK phosphorylation and p65 nuclear translocation. Next these authors compared ATM-NEMO-IKK signaling between CD34+ myeloblasts (hematopoietic progenitors) from healthy controls and patients with high-risk myelodysplastic syndrome (MDS) or acute myeloid leukemia (AML) (high-risk MDS differs from low-risk MDS in that apoptosis is widespread in the second and very rare in the former). Less than 1% of myeloblasts from healthy donors stain positive for phosphorylated ATM whereas a substantial fraction of myeloblasts from either high-risk MDS or AML patients exhibit positive pATM staining. In both, AML MDS myeloblasts pATM inhibition caused inhibition of NFkB (loss of nuclear p65) coupled to inactivation of the IKK complex and redistribution of NEMO from the nucleus to the cytoplasm
NFκB is constitutively activated in the mostf primary AML samples and high-risk myelodysplastic syndrome (MDS) but is not activated in normal HSCs (65) (66). IKK can be phosphorylated and activated by signaling through the PI3K/AKT or ERK/MAPK pathways which leads to Foxo3a inactivation and phosphorylation at S644 (and potentially NF-kB activation). Foxo3a can also be phosphorylated by Akt onT32, S253 and S315 and by ERK on S294, S344 and S425. Each of these phosphorylation events has been reported to stimulate FOXO3a nuclear exclusion, ubiquitination and proteasomal degradation
Reversion of signaling by inhibition of PI3K or AKT has been shown to result in redistribution of Foxo3a from the cytoplasm to the nucleus in numerous models but this was not observed in a study (66) involving primary AML human samples and a leukemic cell line. In a substantial fraction of the samples Foxo3a was excluded from the nucleus (nuclear localization was detected only in 7,5% of AML samples) in spite of Akt phosphorylation on S473 and thus of Foxo3a on T32 and S253. However, treatment with IC87114 which abolished these phosphorylation events did not result in Foxo nuclear translocation. Similarly, suppression of phosphorylation at ERK1/2 by a specific MEK inhibitor failed to induce Foxo nuclear translocation. In contrast, the anti-NEMO peptide that specifically inhibits IKK activity was found to induce Foxo3a nuclear localization in leukemic cells. Furthermore, an IKK-insensitive Foxo3a protein mutated at S644 translocated into the nucleus and activated the transcription of the Fas-L and p21Cip genes. This, in turn, inhibited leukemic cell proliferation and induced apoptosis. These, and the preceding observations may be interpreted to mean that in AML, IKK signaling takes control and replaces former signaling events. Whether this takeover of PI3K signaling by IKK is a general feature or is restricted to AML cells is unknown. The latter option would suggest a direct role of DNA damage in the origin of AML
Sequential Activation of the AKT/ IKK/NF-κB Pathway and Its Role in Cancer Metastasis and Angiogenesis
NF-kB activation via sequential activation of PI3K, AKT, and IKKβ is plainly shown in a report by Agarwal et al. (67). They studied two cell lines (SW480 and RKO) out of five CRC cell lines that displayed constitutive activation of AKT and both NF-κB and β-catenin dependent transcription. Inhibition of transcription of NF-kB targets was demonstrated by WT PTEN, KD AKT or KD IKKβ as well as by siRNA targeting of AKT and IKKα.Either WT PTEN, KD AKT or KD IKKα (a factor of the alternate pathway) inhibited both NF-κB and β-catenin transcription whereas KD IKKβ while inhibiting NF-κB did not have any effect on β-catenin expression in RKO and SW480). It is likely that Foxo3a deactivation would respond in the same way to the silencing of each of these members of the PI3K pathway. However, unfortunately Foxos were not examined in this report
An array-based gene expression analysis of the cell lines with constitutive expression of NF-kB and β-catenin following inhibition by KD IKKα or WT PETEN showed reduced expression of multiple genes implicated in angiogenesis and metastasis. Some of these genes are dependent on either NF-kB or β-catenin or both. IL-8, μPA or VEGF-C are dependent on both. Elevated expression of mRNA for these genes is observed in a substantial fraction of colorectal tumors compared to matched normal mucosa. (two-fold increase in IL-8 expression in about 85% of colon and 75% of rectal tumors whereas μPA was elevated in75% of colon and 79% of rectal tumors)
Other reports suggest, however, a more complex interplay between the intermediate targets of the pathway as some cooperation between intermediate targets rather than a strict linear sequence of activation, could be at play. Nevertheless, activation culminates in the final activation of NF-k via IKK (68).
Thus, returning to the dilemma raised above regarding AML, the independent signaling of IKK from previous steps of the PI3 pathway, suggests that an ATM-NEMO-IKK is exclusively responsible for NF-kB activation in AML, which emphasizes the primary role of DNA damage in AML. Similar reliance of Foxo regulation on IKK independently from other members of the PI3K pathway observed also in AM by Chapuis et al. above (66) ) raised the question of the extent of the coupled and simultaneous regulation of Foxo and NF-kB in tumorigenesis. Synergy between Foxos and NF-kB may blur attribution of effects to any of them in particular.
Tumor invasion through NF-kB activation has been associated with both the canonical and non-canonical pathways. In addition, it can be mediated through sequential activation of the pathway or single activation of a target at the converging downstream end such as any of IKK proteins. This route appears to be available to scaffold proteins, a class of proteins that regulate a signaling node in order to determine the choice of a downstream target. This has been shown for the scaffold protein connector enhancer of KSR (CNK) which facilitates Raf activation in the Ras-Raf-MAPK/ERK kinase (MEK)-ERK. CNK1 can regulate the ERK pathway and the JNK pathway. However, it was shown that CNK1 mediates human breast and cervical cancer by cooperation with the non-canonical NF-kB pathway, independently of ERK and JNK pathways (69)
To complicate matters further, positive feedback between FOXC1 and NF-kB has been described in the basal type of breast cancer. (BLBC). EGFR upregulates FOXC1 expression in BLBC and prostate cancer cells through Ras/ERK and PI3K pathways. Overexpression of the p65 subunit of NF-kB in MDA-MB-468 cell line (but not inMDA-MB-231 cell line with much lower level of EGFR expression) markedly increased FOXC1 promoter activity. A positive feedback regulatory loop has been demonstrated in BLBC between FOXC1 and NF-kB that may explain why both proteins are highly specific of BLBC. FOXC1 requires NF-kB for regulation of cell migration and invasion (70 (71) as it is seen in many other tumor types. As it has also been reported that FOXC1 controls stemness of basal-like breast cancer via Hh signaling, further work is necessary to elucidate the signaling sequence. (72)
A Collateral Note on the ATM-NEMO-NF-kB Pathway
A final comment raised by these observations concern an unsuspected parallelism in the oncogenic mechanisms of AML and BRCA-induced breast cancer. Frequent DNA damage is thought to be caused by homology directed repair, or homologous recombination (HR), a mechanism linked to the S phase of the cell cycle and therefore prevalent in highly proliferating cells such as hematopoietic progenitors and regenerating mammary cells. The BRCA tumor suppressor associated with breast cancer participates in homology-directed DNA repair and is involved in DNA repair in epithelial cells of the regenerating mammary gland. Thus, the underlying mechanism of breast cancer promotion by BRCA appears to be the same or closely related to that of AML cells. Enhanced cell proliferation-associated DNA damage would trigger the ATM-NEMO-IKK pathway and NF-kB could be the final target for tumorigenesis in both diseases.
Multifaceted Involvement of NF-κB in Tumorigenesis
NF-kB seems to be involved in all stages of tumorigenesis from initiation due to its role in chronic inflammation to induction of the invasive phenotype, EMT and stemness (73), (74). Curiously, no oncogenic mutations of IKKs have been found. In the regulation of EMT and stemness, NF-kB seems to act at a common converging node of signaling pathways such as PI3K/AKT as suggested by its cooperation with FOXOS or the ability of a super-repressor form of IκBα or genetic silencing of RelA to suppress Ras-mediated tumorigenesis (75). It is difficult to envisage how such a vast array of functions could be mediated by a factor or restricted group of factors. Interestingly, a protein that interacts with some of the downstream effectors of these pathways has also been implicated in invasiveness and especially in stemness, Rap1, which could be fundamental for immortalization
Possible Downstream Effectors of Telomerase Reactivation in Transformation
In a former paper (7) we hypothesized that Rap1, a protein that associates indirectly with telomeres through interaction with the sheltering complex protein TRF2, might be involved in coupling of telomere damage with oncogenic signaling. More recently, we have become aware of some previous publications that strongly suggest the implication of Rap1 in transformation. The E6 protein of high-risk papilloma HPV16 and HPV18 binds several proteins in addition to p53. One of them is E6TP1 (E6-targeted protein, a Rap GTPase-activating protein that negatively regulates mitogenic signalling mediated by Rap. The ability of E6 mutants to bind and degrade E6TP1 (Figure 3) correlates better than the ability to degrade p53 with E6-induced immortalization of mammary epithelial cells (MECs). The correlation of E6TP1 degradation with immortalization of MECs exceeds that of mutated p53. From a panel of E6 mutants all of those capable of immortalizing MECs were able to induce telomerase activity.. One mutant, previously reported not to activate telomerase in a transient expression system was shown to induce telomerase activity after analysis of immortalized MECs (76). Now, E6TP1 shows high sequence homology with Rap1 GAP which negatively regulates mitogenic signaling by Rap1. Presumably E6 can also degrade Rap GAP (Figure 3) and it is, therefore likely, that other molecules that degrade RapGAP could also stimulate Rap mitogenic activity and recapitulate E6-induced telomerase activity and immortalization of MECs.
Interplay Between Telomere Shortening and Rap1. (Figure 3)
Rap1 associates to telomeres through TRF2 as it does not directly bind DNA. Rap1 functions at telomeres have been difficult to elucidate. Its best characterized function is the regulation of homology directed repair, a pathway which creates telomere sister chromatide exchange unless it is inhibited by Rap1.
It has been described that Rap1 loss may increase telomere shortening and conversely, decrease telomere length increases Rap1 shuttling and binding to non-telomeric genomic sites. This may facilitate either Rap1 interaction with other DNA binding proteins such as NF-kB and FOXOs or Rap1 shuttling to the nuclear or cytoplasmic matrix where this interaction could take place (77). The finding by Blanco et al. that TRF2 overexpression in the absence of telomerase activity leads to critically short telomeres and increased epithelial carcinogenesis suggests that carcinogenesis could be related to disruption of TRF2-Rap1 interaction with release of non-telomeric Rap1 (78)
As Rap1 has been shown to form a complex with IKKs (Figure 3) and phosphorylate p65, thereby rendering NF-kB transcriptionally competent, it is possible that Rap1 is the final effector of many oncogenic signaling pathways described above. Furthermore, there are NF-kB binding sites in the Rap1 promoter. This and other findings suggest the existence of a feedback loop between these factors. (79) (80) (81) .
Interaction of Rap1 and Zscan4. (Figure 3)
Lee et al. have described an interaction of Rap1 with the newly identified embryonic stem cell marker Zinc finger and SCAN domain containing 4 gene (Zscan4) (Figure 3) which has a key function in genomic stability by regulating telomere elongation. These authors analysed two cell lines, MCF7 and SaOS2 (epithelial/telomerase+ and mesenchymal/telomerase--, respectively) and found that Zscan4 was present in cancer cells from both cell lines. Rap1 bound to the C-terminal domain of Zscan (82). Transfection of cancer cells with Rap1 siRNA resulted in significantly less expression of Zscan4 whereas overexpression of Rap1 gave rise to greater Zscan4 expression (82). It is plausible that Zscan4 could replace reprogramming factors that convert somatic committed cells into pluripotent stem cells triggering the final step of transformation. In this way, neoplastic transformation would require a surplus of non-telomeric Rap1 facilitated by repression of Rap1 GTPase (RapGAP) or by telomere shortening/damage
The interleukin-1 receptor accessory protein (IL1RAP) is a coreceptor of interleukin-1 receptor type1. IL1RAP expression is higher on the GMP population compared with other progenitor populations and with HSCs. It is not expressed on normal human HSCs but is highly expressed on primitive CD34+ CD38- CML cells. This fact suggests again that leukemic blast cells are derived from progenitors rather than directly from HSCs and that IL1 RAP upregulation might be related to increased non-telomeric RAP ((83)
Some reports consistent with the role of RAP1 as a converging node of several oncogenic pathways follow:
Niola et al. ((84) working on neural gliomas have shown that Id proteins repress Rap1GAP with concurrent activation of Rap (Figure 3). Active Rap1 through Id-mediated repression of Rap GAP leads to the emergence of stemness features such as stem cell marker Nestin and neurosphere generation as well as adhesion to the endothelial niche. Conversely, Id depletion determines the immediate release from the niche and expression of differentiation markers. Multipotent NSCs in the ventricular zone of the brain are anchored to the ventricular surface by an apical membrane. Neural progenitors acquire RapGAP expression as soon as they lose contact with the ventricular surface. Id deletion is associated with Rap1GAP expression and disruption of adhesion to the niche. Interestingly, these authors showed that the ID-RAP1 axis is especially relevant for the mesenchymal type of high-grade glioma (85)
Rap1 GAP has been found lost or downregulated in various types of tumors and with high frequency in thyroid tumors. Deficiency correlates with increased cell migration and invasiveness (86). In oropharyngeal squamous carcinoma active GTP-bound Rap1 was upregulated compared to normal or immortalized keratinocytes. Inactivation of Rap1 by Rap1GAP resulted in strong tumor inhibition (87). Rap1GAP was identified as a tumor suppressor also in pancreatic cancer (88). The effects of tetratricopeptide repeat domain 17 (TTC17) downregulation on invasiveness and metastasis of breast cancer were shown to be linked to activation of Rap1/CDC42 pathway (89)
Further Comments
In this essay we have focus especially on oncogenic signaling cascades that initiate in normal progenitor cells. Regenerating ability of early progenitors is maintained by stem cell associated telomerase. Nevertheless, sustained proliferation is usually followed by exhaustion of the regenerative potential. This is at loggerheads with the long-lasting, persisting proliferative ability observed in fully differentiated cells induced to de-differentiate by the four factors (iPSCs) or in cancer cells. Thus, we hypothesize that the process of cancer involves de-differentiation of a post-stem cell that resorts to a different type of telomerase reactivation or a distinct mechanism of telomere maintenance than the one that characterizes normal stem cells.
The GSK 3 node appears to be crucial in regulation/deregulation of growth in tissues endowed with high proliferation ability such as intestine or blood. Within chromosomal translocation associated leukaemia those involving MLL fusion proteins characteristically affect cells from very early differentiation stage showing immature features and high proliferating activity. MLL fusion proteins activate an HSC-self renewal program apparently initiated by HoxA9 and Meis1 genes. These genes are, under physiological conditions, only expressed in c-Kit+, Thylo Linlo/- Sca1+ rhodamine 123lo HSCs and their expression is rapidly downregulated in more differentiated multipotential common lymphoid and myeloid progenitors. However, these fusion proteins cause persistent expression of Hoxa7, Hoxa9 and Meis1 in cells at later stages. Interestingly, Hoxa9 and Meis are upregulated in advance of the full self-renewal signature. In vitro growth of MLL-AF9 transduced human cord blood cells was shown to be strictly dependent on the Flt3 ligand which indicates that the cancer stem cell and its normal precursor expressed FLT3 which marks them as located downstream of the HSC (7). Several facts indicate the involvement of the GSK3 node in oncogenesis in these and other immature leukemias. For instance, a reversible myelomonocytic block in MLL-ENL leukemia was shown to be dependent on c-myc. A dominant negative myc mutant prevented the block caused by MLL-ENL and precluded transformation (7). MLL transformed cells were shown to be highly dependent on GSK3 by culturing MLL-transduced myeloid progenitors with a GSK3 inhibitor which reduced their clonogenic potential and proliferation in contrast to cells immortalized by other fusion proteins that did not show adverse effects (90). However, translocation products from AML-ETO, PML-RARα and PLZF-RARα can activate Wnt signaling through induction of plakoglobin (γ-catenin) which blocks β-catenin degradation and activates TCF, LEF and their target c-Myc, thus enhancing cell proliferation and preservation of immature features (7)
Distinct Oncogenes Impact on Cells at Different Developmental Stages
Differentiation markers of tumours can be taken as an indication of the origin of cancer from a specific developmental stage of the corresponding cell lineage. In the mammary gland, keratin6 and Sca-1 are preferentially expressed by mammary stem or progenitor cells even if they are not restricted to these compartments. As in the intestine and other tissues, the Wnt pathway physiologically activates cells in the early developmental stages. Experiments on transgenic mice have shown (91) that overexpression of Wnt-1 or its downstream effectors β-catenin or c-Myc under the control of the MMTV promoter, resulted in development of hyperplastic mammary glands composed mainly of progenitor cells and mammary tumours derived from these cells. The early markers Keratin 6 and Sca-1were detected in a great number of hyperplastic ducts as well as in mammary tumours of Wnt-1 transgenics but were not increased in premalignant ducts or tumours from MMTV-Neu, MMTV-Hras or MMTV-PyMT transgenic mice. IH staining demonstrated that Wnt-1 induced tumours were composed of luminal and myoepithelial cells (both cell types appear to be malignant in Wnt-1 induced tumours whereas MMTV-β-catenin and MMTV-c-Myc showed an hyperplastic myoepithelial component and malignant luminal cells. In contrast, myoepitelial hyperplasia was absent from MMTV-Neu, MMTV-Hras and MMTV.PyMT induced tumours. The coexistence of myoepithelial and luminal cells in Wnt-1 induced tumours suggest transformation from a common progenitor. Some of them arose in Pten heterozygous MMTV-Wnt-1 transgenic mice and the wild type Pten locus was missing in in both luminal and myoepitehelial cells. Implication of Pten pathway in tumours induced by Wnt pathway may be facilitated by the crosstalk commented before between these pathways and the role played by the GSK3β node. The hyperplastic myoepithelial component not yet transformed in MMTV-β-catenin and MMTV-c-Myc may correspond to the adenomatous phase observed in intestinal adenomas that will ultimately give rise to adenocarcinomas. The authors`s interpretation is that deregulated Wnt signalling causes excessive proliferation and arrest cells are early mammary development consistent with our own interpretation above of a partial block in intestinal Wnt deregulated signalling. Other studies (92) have shown that the Neu oncogene induced tumors have a uniform luminal histology, require CDK4 and cyclin D1 for transformation and reflect the Erb2 subtype of human cancer. In contrast, Wnt-1 driven tumours exhibit features in common with the human basal subtype that falls within the group of triple negative (TN) tumors (lack ER and PR expression and Her2 gene amplification). Id1 and Id3 expression, reflective of an immature stage, is seen in tumor cells of Wnt-1 driven tumors but not in Neu-induced tumor cells.
The phenotypic difference in tumors arising in MMTV-Wnt1 on the one hand and MMTV-β-catenin and MMTV-c-myc suggest a cell of origin in progenitors that in the case of MMTV-Wnt1 belongs to an earlier developmental stage. In these tumors, the more immature component is already malignant whereas in the other two tumor types is adenomatous. Interestingly, immaturity is associated with the appearance of PTEN loss of heterozygosity, which was not detected in the other transgenics. Pathway deregulation in Wnt1 transgenics occurs at an earlier step than in the other two, a step that may involve the GSK3-β node and a wider range of downstream targets including Notch activation that is responsible for a more undifferentiated phenotype.
A wider stratification of the developmental stages of cancer origin is described in a paper by Lim et al.(93). These authors prepared cell suspensions from reduction mammoplasties from normal females or BRCA mutation carriers. After discarding hematopoietic and endothelial cells, they fractionated the resultant Lin- population into four distinct subsets with the help of two antibodies against CD49f (α6 integrin) and EpCAM (also referred to as CD326 (against epithelial cell adhesion molecule). EpCAM was predominantly expressed on luminal cells whereas CD49f marked basal cells. More specifically, the CD49fhi EpCAM- subpopulation expressed the basal lineage markers p63, cytikeratin 14 and vimentin but did not express the estrogen receptor or progesterone receptor. In contrast, the CD49f- EpCAM+ and CD49f+ and CD49f+ EpCAM+ subpopulations expressed luminal markers including cytokeratin 8/18, cytokeratin 19, GATA-3 (a regulator of luminal differentiation) and Mucin-1 (MUC-1). The CD49f- EpCAM+ fraction should contain mature luminal cells since cells negative for estrogen receptor and progesterone receptor are much les represented in this fraction. Only the CD49f+ EpCAM+ subset contained a high proportion of cells positive for CD133 (prominin-1), a progenitor marker in diverse tissues. The CD49f- EpCAM-fraction comprised mainly stromal fibroblasts. Only the CD49fhi EpCAM- subset (also referred to as MaSC-enriched population) showed mammary regenerating capacity. This latter population was substantially reduced in BRCA carriers whereas the luminal progenitor subpopulation (CD49f+ EpCAM+) was increased in them relative to normal breast tissue. Gene expression signatures derived from microarray studies disclose the association of luminal progenitor signature with basal cancers and that of the mature luminal signature with luminal A and B subtypes. The MaSC signature was more concordant with the normal-like and the newly tentatively defined claudin-low subtypes. We postulate that these latter subtypes arise from progenitors at earlier stage than basal cells. Their cell of origin is difficult to establish as might be reflective of a heterogeneous population including basal cells and myoepithelial cells. Furthermore, they do not have a clear counterpart in clinical classifications although they could be grouped with the poor outcome groups (LumB, basal-like and HER2+/ER- all of which might arise from early developmental stages.
An interesting finding that is concordant with findings commented above concerns the correlation of oncogenic pathways with distinct stages of presumed cell origin: PTEN LOH is an event correlating with basal type tumors and TP53 loss predominates in luminal tumors (94)
Other interesting aspect of secondary oncogenic pathways concerns the advent of some specific mutations that apparently endow tumor cells with further initiating capacity and are detected when tumors reach the clinical stage defined by metastasis. In breast cancer Id1 was identified as the only transcription factor within a set of genes associated with high risk of lung metastasis. A monospecific monoclonal antibody revealed positive staining for Id1 in 36% of TN breast cancers. This proportion being higher in the metaplastic subtype. Among non TN breast tumors only 1 out of 105 was positive for Id1 expression. Dependence of this secondary mutation on the primary oncogenic pathway may exist as Id1 expression was detected in 45% of lung metastasis in Er- Pr- cases and 0 out of 16 hormone receptor positive. Knock down of Id1 and Id3 either individually or in combination, from a cell line representative of the TN human subgroup of breast cancer resulted in partial or complete inhibition of tumor initiating capacity, respectively. Id1/Id3 knockdown suppressed lung metastatic potential of the selected subline. On the other hand, knockdown of Id1 and Id3 in a Wnt-driven tumor resulted in significant impairment of in vitro mammosphere generation as well as tumor initiation. The same knockdown did not cause alterations in Neu-induced tumors (mammosphere formation which is a measure of tumor initiating capacity is lower in Neu tumors but is not lowered by the knockdown of Id1 and Id3 (92)
Since oncogenes usually impact on physiological processes of differentiation along tissue turnover, a correlation of oncogenic signaling pathways and lineage developmental stage is expected. Evidently, the oncogenic signaling pathways reviewed here are but a fraction of those used by tumors which, in addition of the crosstalk between distinct pathways makes research on cancer such an unassailable task. Fortunately, it is likely that most of these pathways should converge on a few common targets essential to elicit immortalization. For instance, Ha-Ras or c-Neu can transactivate the peptidyl-prolyl isomerase Pin1 through E2F (95). In turn, Pin1 can stimulate NF-kB activity. It does so by inhibiting binding of p65 to IκB which results in the nuclear accumulation of p65/p50 (96)
Antonio Torres-Montaner
Acknowlegments: The author is indebted to Jorge Guio-Martinez and Marisa Peleato for their support in the initiation of this work
References
- Adolfsson J,Mansson R, Buza-Vidas, N. Hultquisty A. et al.Identification of Flt3+ lympho-myeloid stem cells lacking erythron-megakaryocytic potential: a revised road map for adult blood lineage commitment. Cell 2005, 121, 295–306. [Google Scholar] [CrossRef]
- Morrison SJ and Spradling, AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell 2008, 132, 598–611. [Google Scholar] [CrossRef]
- Wnt proteins are self-renewing factors for mammary stem cells and promote their long-term expansion in culture. Yi Arial Zeng and Roel Nusse. Cell Stem Cell 2010, 6, 568–577. [Google Scholar]
- Telomerase-deficient mice with short telomeres are resistant to skin tumorigenesis. Gonzalez-Suarez E, Samper E, Flores JM, Blasco M. Nature Genetics 2000, 26, 114–117. [Google Scholar]
- Ras signals to the cell cycle machinery via multiple pathways to induce anchorage-independent growth. J-J Yang, J-S Kang and RS Krauss. Mol Cell Biol 1998, 18, 2586–2595. [Google Scholar]
- Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. G. Hewitt, D. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. G. Hewitt, D. Jurk, FDM Marques, C. Correia-Melo et al. Nature communications 2012. [Google Scholar] [CrossRef]
- The telomere complex and the origin of the cancer stem cell. Torres-Montaner A. Biomarker research 2021. [CrossRef]
- Cancer origin in commited versus stem cells: hypothetical antineoplastic mechanism/s associated with stem cells. Critical reviews in oncology and hematology. Torres-Montaner A. 2011, 8, 209–224. [Google Scholar] [CrossRef]
- Interactions between the DNA damage response and the telomere complex in carcinogenesis. Torres-Montaner A. Current issues in molecular biology 2023, 45, 7582–7616. [Google Scholar] [CrossRef]
- Huang J, Nguyen-McCarty M, Hexner O, Danet-Desnoyes G, Klein PS. Maintenance of hematopoietic stem cells through regulation if Wnt and mTOR pathways. Nature medicine 2012, 18, 1778–1785. [Google Scholar] [CrossRef]
- Pivotal role for glycogen synthase kinase-3 in hematopoietic stem cell homeostasis in mice. J. Huang, Y. Zhang, A. Bersenev, W. Thimothy O`Brien et al. J. Clin. Invest. 2009, 119, 3519–3529. [Google Scholar] [CrossRef]
- C-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. A. Wilson et al. Genes & Development 2004, 18, 2747–2763.
- The role of Skp2 and its substrate CDKN1B (p27) in colorectal cancer. O.V. Bochis et al. J. Gastrointestin Liver Dis 2015, 24, 225–234. [CrossRef]
- The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. M van de Wetering, E. Sancho, C. Verweij, W. de Lau et al. Cell 2002, 111, 241–250. [Google Scholar]
- APC mutations occur early during rectal tumorigenesis. Powell S, Silz N, Beazer-Barclay, Bryan TM et al. 1992, 235-237. [CrossRef]
- Early genetic aberrations in patients with sporadic colorectal cancer. Druliner BR, Ruan X, Sicotte H, O`Brien D et al. Molecular carcinogénesis 2018, 57, 114–124. [CrossRef]
- Human telomerase reverse transcriptase (hTERT) is a novel target of the Wnt/β-catenin pathway in human cancer. Y. Zhang L Toh, P Lau and X Wang. The journal of biological chemistry 2012, 287, 32494–32511. [CrossRef]
- Close correlation between telomerase expression and adenomatous polyp progression in multistep colorectal carcinogenesis. Tang R, Cheng A-J, Wang J-Y and Wang T-Ch V. Cancer research 1998, 58, 4052–4054. [Google Scholar]
- Time lapse to colorectal cancer: telomere dynamics define the malignant potential of polyps. Druliner BR, Ruan X, Johnson R, Grill D et al. Clinical and Translational Gastroenterology 2016, 7, e188. [CrossRef]
- Telomere shortening of epithelial cells characterises the adenoma-carcinoma transition of human colorectal cancer. Plentz RR, Wiemann SU, Flemming P, Meier PN et al. Gut 2003, 52, 1304–1307.
- A human colon cancer cell capable of initiating tumor growth in immunodeficient mice. O`Brien CA, Pollet A, Gallinger S, Dick JE. Nature 2007. [CrossRef]
- Wnt/β-catenin signaling regulates telomerase in stem cells and cancer cells. Hoffmeyer K, Raggioli A, Rudloff S, Anton R et al. Science 2012, 336, 1549–1554.
- Regulation of the human telomerase reverse transcriptase gene. A-L Ducrest. H. Szutorisz, J. Lingner and M. Nabholz. Oncogene 2002, 21, 541–552. [Google Scholar]
- Regulation of the human telomerase gene TERT by telomere position effect-over long distances (TPE-OLD): implications for aging. W. Kim, AT. Regulation of the human telomerase gene TERT by telomere position effect-over long distances (TPE-OLD): implications for aging. W. Kim, AT. Ludlow, J. Min, JD Robin, C. Stadler, I. Mender, T-P Lai, N Zhang. WE Wright, JW Shaw. PLOS biology 2016. [Google Scholar] [CrossRef]
- Identification of mechanism of cancer-cell-specific reactivation of hTERT offers therapeutic opportunities for blocking telomerase specifically in human colorectal cancer. SC Akincilar, JY Heng Chua, QFeng Ng, CHT Chan,, Z. Eslami-S et al. Nucleic Acid Research 2023, 51, 1–16. [Google Scholar]
- Reactivation of telomerase in cancer. SC Akincilar, B. Unal, V. Tergaonkar. Cell. Mol. Life. Sci 2016, 73, 1659–1670. [Google Scholar]
- Genome-wide screens identify specific drivers of mutant hTERT promoters. R. Shanmugam, MB Ozturk, J-L Low, SC Akincilar et al. PNAS 2022, 119. [CrossRef]
- Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. F. Varnat, I. Siegl-Cachedenier, M. Malerba, P. Gervaz, A. Ruiz I Altaba. EMBO Mol. Med 2010, 2, 440–457. [Google Scholar] [CrossRef]
- Molecular conversations and the development of the hair follicle and basal cell carcinoma. PJ Harris, N. Takebe and S. Percy Ivy. Cancer Prev Res (Phila) 2010, 3, 1217–1221. [Google Scholar] [CrossRef]
- Diverse mechanisms of Wnt activation and effects of pathway inhibition on proliferation of human gastric carcinoma cells. S. Asciutti, G Akiri, L. Grumaloto, S. Vijayakumar and SA Aaronson. Oncogene 2011, 30, 956–966. [Google Scholar] [CrossRef]
- Snail and Slug promote epithelial-mesenchymal transition through β-catenin-T-cell factor-4-dependent expression of transforming growth factor-β-3. D. Medici, ED. Hay and BR. Olsen. Molecular Biology of the Cell 2008, 19, 4875–4887. [Google Scholar]
- Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Z. Liu, Q. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Z. Liu, Q. Li, K. Li, L. Chen, W. Li, M. Hou, T. Liu, J Yang et al. Oncogene 2012, 1-11. [CrossRef]
- Id1 restrains myeloid commitment, maintaining the slf-renewal capacity of hematopoietic stem cells. V. Jankovic, A. Ciarrochi, P. Boccuni, T. DeBlasio et al. PNAS 2007, 104, 1260–1265. [Google Scholar]
- A novel pathway for mammary epithelial cell invasion induced by the helix-loop-helix protein Id-1. Mol. Mol. Cell. Biol 1998, 4577–4588. [Google Scholar]
- Id1 cooperates with oncogenic Ras to induce metastatic mammary carcinoma by subversion of the cellular senescence response. A. Swarbrick, E. Roy, T. Allen and J. Michael Bishop. PNAS 2008, 105, 5402–5407. [Google Scholar]
- Massive apoptosis of thymocytes in T cell deficient Id1 transgenic mice, Kim D, Peng X-C, Sun X-H. Mol. Cell Biol 1999, 19, 8240–8253. [Google Scholar]
- The epithelial-mesenchymal transition generates cells with properties of stem cells. Mani SA, Guo W, Liao M-J, Ng E et al. Cell 2008, 133, 704–715.
- Id1 promotes breast cancer metastasis by S100A9 regulation. K. Gumireddy, A. Li, AV Kossenkov, KQ Cai et al.. Molecular cancer research 2014, 12, 1334–1343.
- Id-1 activates Akt-mediated Wnt signaling and p27kip1 phosphorylation through PTEN inhibition. JY Lee, M-B Kang, S-H Kang, T. Qian et al. Oncogene 2009, 824–831.
- Id-1 as a molecular target in therapy for breast cancer cell invasion and metastasis. S. Fong, Y. Itahana, T. Sumida, J. Singh et al. PNAS 2003, 100, 13543–13548. [Google Scholar]
- 41 ) Id1 promotes lung cancer cell proliferation and tumor growth through Akt-related pathway Yu-Jen Cheng, Jen-Wei Tsai, Kun-Chou Hsieh, Yu-Chi Yang, Yun-Ju Chen, Ming-Shyang Huang, Shyng-Shiou Yuan. Cancer Letters 2011, 307, 191–199. [CrossRef] [PubMed]
- Id-1 activation of PI3K/Akt/NFκB signaling pathway and its significance in promoting survival of esophageal cancer cells. B.Li, Y. Cheung, X Wang, SW Tsao et al. Carcinogenesis 2007, 28, 2313–2320.
- Id-1 expression promotes cell survival through activation of NF-κB signalling pathway in prostate cancer cells. M-T Ling, X. Wang, X-S Ouyang, K. Xu et al. Oncogene 2003, 22, 4498–4508. [Google Scholar]
- Epithelial to mesenchymal transition in hepatocellular carcinoma. W. Mikulits. Future Oncology 2009, 5, 1169–1179. [CrossRef] [PubMed]
- TBX3 promotes the epithelial mesenchymal transition of cervical cancer by upregulating ID1. H. Yang, Y. Sun, X. Jia, Y. Cai et al. Am J. Cancer Res 2023, 13, 4115–4124. [Google Scholar]
- Wu Z, Li J. Zhang Y et al. Synchronous coexpression of Id1 and nuclear NFkappaB p65 promotes cervical cancer progression and malignancy and is associated with a poor prognosis and chemosensitivity. Oncol. Rep 2019, 42, 2075–2086. [Google Scholar]
- 47 (Identification of a novel inhibitor of differentiation-1 (ID-1) binding partner, caveolin-1, and its role in epithelial-mesenchymal transition and resistance to apoptosis in prostate cancer cells. X. Zhang, M-T Ling, Q. Wang, C-K Lau, et al. J. Biol. Chem 2007, 282, 33284–33294. [Google Scholar]
- TGF-b Receptor Inhibitors Target the CD44high/Id1high Glioma-Initiating Cell Population in Human Glioblastoma Judit Anido, Andrea Saez-Borderıas, Alba Gonzalez-Junca, Laura Rodo’ Gerard Folch, 2010, 18, 655–668.
- The ID1-cullin3 axis regulates intracellular Shh and Wnt signaling in Glioblastoma stem cells. X. Jin, H-M Jeon, X.Jin, E-J Kim. J. Yin et al. Cell Reports 2016, 16, 1629–1641. [Google Scholar]
- Inhibitor of DNA binding I (Id I) mediates stemness of colorectal cancer cells through the IdI-c-myc-PLAC8 axis via the Wnt/β-catenin and Shh signaling pathways. Y.Sun, X. Lai, Y Yu, J. Li et al. Cancer Management and Research 2019, 11, 6855–6869. [Google Scholar]
- ID1 Facilitates the Growth and Metastasis of Non-Small Cell Lung Cancer in Response to Nicotinic Acetylcholine Receptor and Epidermal Growth Factor Receptor Signaling. Smitha Pillai, Wasia Rizwani, Xueli Li, Bhupendra Rawal, Sajitha Nair, Michael J. Schell, Gerold Bepler,† Eric Haura, Domenico Coppola, and Srikumar Chellappan. Molecular and cellular biology 2011, 31, 3052–3067. [Google Scholar] [CrossRef]
- FoxO3a depletion accelerates cutaneous wound healing by regulating epithelial-mesenchymal transition through β-catenin activation. T. Liu, J-Z Huang, Z-Y Lei, R-S Yan and D_l Fan. Molecular medicine reports 2020, 21, 1224–1232. [Google Scholar] [CrossRef]
- Foxo3a modulates WNT/β-catenin signaling and suppresses epithelial-to-mesenchymal transition in prostate cancer cells. Cell signaling 2015, 27, 510–518. [CrossRef]
- FOXC1 Activates smoothened-independent hedgehog signaling in basal-like breast cancer. Han B, Qu Y, Jin Y, Deng N et al. Cell reports 2015, 13, 1046–1058. [CrossRef]
- X. Zhang, N. Tang, TJ Hadden AK. Rishi. Akt, FoxO and regulation of apoptosis. Biochimica et Biophysica Acta 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- The Foxo signaling axis displays conjoined functions in redox homeostasis and stemness. R. Soh, A. Hardy, Ni zur Niede. Free Radic Biol Med 2021, 169, 224–237. [Google Scholar] [CrossRef]
- Foxo transcription factors and stem cell homeostasis: insights from the hematopoietic system. Z. Tothova and D. Far Gilliland. Cell stem cell 2007. [CrossRef]
- Akt/FOXO signaling enforces reversible differentiation blockade in myeloid leukemias. Sykes SM, Lane SW, Bullinger L, Kalaitzidis D et al. Cell 2011. [CrossRef]
- FOXF2 oppositely regulates stemness in luminal and basal-like breast cancer cells through the Wnt/beta-catenin pathway. Zhang X, Zhang R, Hou Ch, He R et al.. J.Biol. Chem 2022, 298, 102082.
- TGF-β-FOXO signaling maintains leukaemia initiating cells in chronic myeloid leukaemia. Naka K, Hoshii T, Muraguchi T, Tadokoro Y et al. Nature Letters, 2010.
- Dynamic of FoxO transcription factors. H. Huang & D.J. Tindall. J.cell science 2007, 120, 2479–2487. [CrossRef]
- IκB kinase promotes tumorigenesis through inhibition of Forkhead Foxo3a. M C-T Hu, D-F Lee, W. Xia, LS Golman et al. Cell 2004, 117, 225–237.
- NF-κB in biology and targeted therapy: new insights and translational implications. Guo Q, Jin Y, ChenX, Ye X et al. . Signal transduction and targeted therapy 2024, 9, 53. [Google Scholar]
- ATM mediates constitutive NF-κB activation in high-risk myelodysplastic syndrome and acute myeloid leukemia. Grosjean-Raillard J, Tailler M, Ades L, Perfettini J-L et al. Oncogene 2009, 28, 1099–1109.
- Warner JK, Wang JCY Hope KI, Jin L et al. Concepts of human leukemic development. Oncogene 2004, 23,, 7164.7177); Testa U. Leukemia stem cells Ann Hematol 2011, 90, 245–271.
- IkB Kinase overcomes PI3K/Akt and ERK/MAPK to control FOXO3a activity in acute myeloid leukemia. . Chapuis N, Park S, Leotoing L, Tamburini J et al. Blood 2010, 116, 4240–4250.
- The AKT/ IkB kinase pathway promotes angiogenic/metastatic gene expression in colorectal cancer by activating nuclear factor-κB and β-catenin. Agarwal A, Das K, Lerner N, Sathe S et al. Oncogene 2005, 24, 1921–1031. [Google Scholar]
- Akt-dependent regulation of NF-kB is controlled by mTOR and Raptor in association with IKK. Dan HC, Cooper MJ, Cogswell PC, Duncan JA et al. Genes & development 2008, 22, 1490–1500.
- CNK1 promotes invasion of cancer cells through NF-kB-dependent signaling. Molecular cancer research 2010, 8, 395–406. [CrossRef]
- Identification of EGF-NF-kB-FOXC1 signaling axis in basal-like breast cancer. Chung S, Jin Y, Han B, Qu Y et al. Cell Communication and Signaling 2017, 15-22. [CrossRef]
- FOXC1 regulates the functions of human basal-like breast cancer cells by activating NF-kB signaling. Wang J, Ray PS, Sim M-S, Zhen ZOU X et al. Oncogene 2012, 31, 4798–4802. [CrossRef]
- FOXC! Activates smoothened-independent hedgehog signaling in basal-like breast cancer. Han B, Qu Y, Jin Y, Deng N et al. Cell reports 2015, 13, 1046–1058. [CrossRef]
- NF-kB signaling in neoplastic transition from epithelial to mesenchymal phenotype. A. Oh, M. Pardo, A. Rodriguez, C. Yu et al. Cell Communication and Signaling 2023, 21, 291. [Google Scholar]
- NF-B, stem cells and breast cancer: the links get stronger. K. Shostak and A. Chariot. Breast cancer rsearch 2011, 13, 214–220.
- NF-κB and cancer: a paradigm of Yin-Yang. G. Xiao and J. Fu. Am J Cancer Res 2011, 1, 192–221. [Google Scholar]
- Human papillomavirus type 16 E6-induced degradation of E6TP1 correlates with its ability to immortalize human mammary epithelial cells. Q. Gao, L. Singh, A. Kumar, S. Srinivasan, DE. Wazer and V. Band. Journal of Virology 2001, 75, 4459–4466. [Google Scholar] [CrossRef]
- A genetic interaction between RAP1 and telomerase reveals an unanticipated role for RAP1 in telomere maintenance. P. Martinez, G. Gomez-Lopez, DG Pisano JM Flores and MA Blasco. Aging Cell 2016, 15, 1113–1125. [Google Scholar]
- Telomerase abrogation dramatically accelerates TRF2-induced epithelial carcinogenesis. Blanco R, Muñoz P, Flores JM, Klatt P and Blasco MA. Genes and development 2007, 21, 206–220. [Google Scholar]
- Crabbe L and Karlsseder, J. Mammalian Rap1 widens its impact. Nature cell biology 2010, 12, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Telomeres and inflammation Ghosh AS and Tergaonkar, V. 2010; 9.
- Telomere-independent Rap1 is an IKK adaptor and regulates NF-κB-dependent gene expression. Teo H, Ghosh S, Luesch H, Ghosh A et al. Nature cell biology 2010, 12. [CrossRef]
- Zscan4 interacts directly with human Rap1 in cancer cells regardless of telomerase status. Lee K & Gollahon S. Cancer Biolgy and Therapy 2014, 15, 1094–1105. [Google Scholar]
- IL1RAP antibodies block IL-1-induced expansion of candidate CML stem cells and mediate cell killing in xenograft models. Blood 2016, 128, 2683–2693. [CrossRef]
- Id proteins synchronize stemness and anchorage to the niche of neural stem cells. F. Niola, X. Zhao, D. Singh, A. Castano et al. Nat. Cell Biol 2012, 14, 477–487. [Google Scholar] [CrossRef]
- Mesenchymal high-grade glioma is maintained by the ID-RAP1 axis. F. Niola, X. Zhao, D. Singh, R. Sullivan, A. Castano et al. J. Clin. Invest. 2013, 123, 405–417. [Google Scholar]
- Downregulation of Rap1 GAP through epigenetic silencing and loss of heterozygosity promotes invasion and progression of thyroid tumors. H Zuo, M. Gandhi, MM Edreira, D. Hochbaum et al. Cancer Research 2010, 70, 1389–1397. [Google Scholar]
- Rap1GAP inhibits tumor growth in oropharyngeal squamous cell carcinoma. Zhang Z., Mitra RS, Henson BS, Datta NS et al. American Journal of pathology 2006, 168, 585–596. [Google Scholar] [CrossRef]
- Identification of a putative tumor suppressor gene Rap1GAP in pancreatic cancer. Zhang L, Chenwei L., Mahmood R, van Golen K., et al. Cancer Research 2006, 66, 898–906. [CrossRef]
- Loss of TTC17 promotes breast cancer metastasis through Rap1/CDC42 signaling and sensitizes it to rapamycin and paclitaxel. Zhang J, Guo F, Li Ch, Wang Y et al. Cell & Bioscience. [CrossRef]
- Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Z. Wang, KS Smith, M. Murphy, O. Piloto et al. Nature 2008, 455, 1205–1209. [Google Scholar] [CrossRef]
- Evidence that transgenes encoding components of the Wnt signaling pathway preferentially induce mammary cancers from progenitor cells. Y Li, B. Welm, K. Podsypanina, S. Huang, M. Chamorro et al. PNAS 2003, 100, 15853–15858.
- Id genes mediate tumor reinitiation during breast cancer lung metastasis. GP. Gupta et al. PNAS 104,19506-19511.
- Lim, E.; et al. et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA mutation carriers. Nature Medicine 2009, 15, 907–913. [Google Scholar]
- Martins, FC; et al. Evolutionary pathways in BRCA-1 associated breast tumors. Cancer Discovery 2012, 2, 503–511. [Google Scholar] [CrossRef]
- PIN1 is an E2F target gene essential for Neu/Ras-induced ransformation of mammary epithelial cells. Ryo A, Liou Y-Ch, Wulf G, Nakamura M et al. Molecular and cellular. Biology 2002, 22, 5281–5295. [Google Scholar]
- Regulation of NF-kB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Ryo A, Suizu F, Yoshida Y, Perrem K et al. Molecular cell 2003, 12, 1413–1426. [Google Scholar]
Figure 1.
Some circuity involved in increased cell migration, invasion and metastasis.
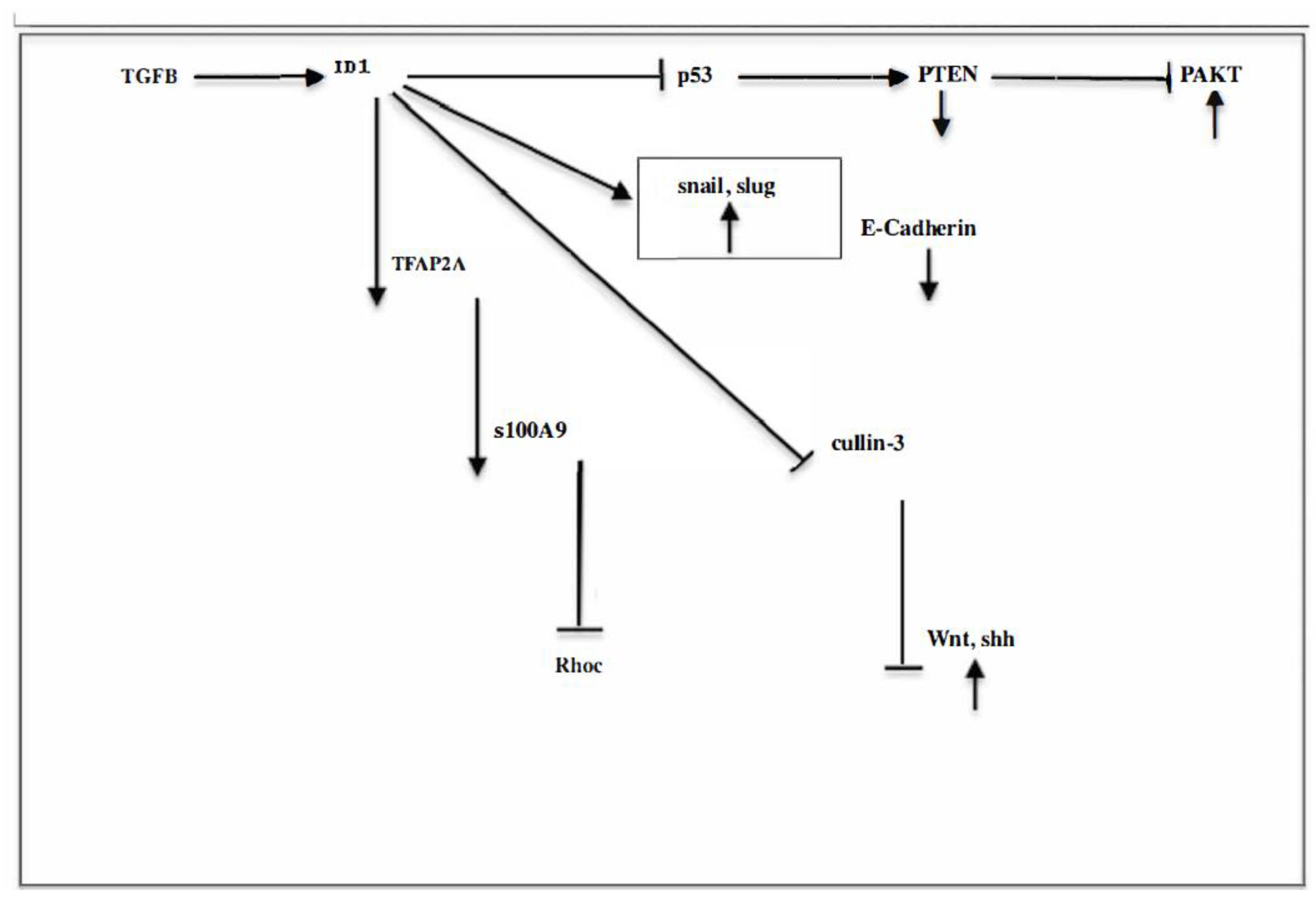
Figure 3.
Interaction RAP • Z scan4 • Telomere damage.
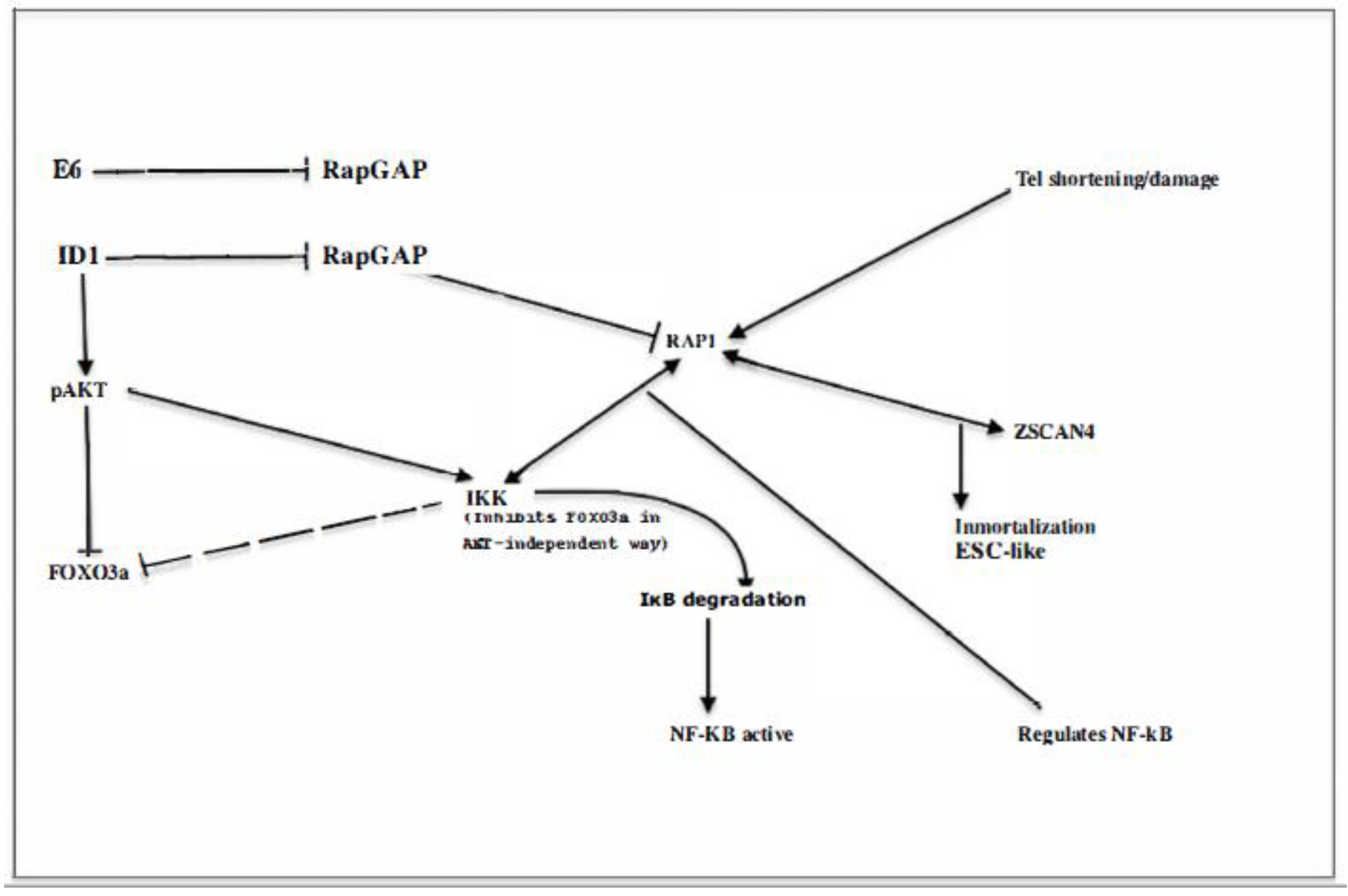
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



