Submitted:
05 September 2025
Posted:
08 September 2025
You are already at the latest version
Abstract
Ferroptosis, an iron-dependent programmed cell death driven by lipid peroxidation, plays a critical role in autoimmune diseases such as rheumatoid arthritis, systemic lupus erythematosus, and psoriasis. This review systematically explores the interaction between ferroptosis and the immune system, highlighting its dynamic regulation of immune cell function (e.g., Treg cell stability, neutrophil activity) and inflammatory microenvironments via signaling pathways including JAK/STAT and NF-κB. Ferroptosis suppresses inflammation in rheumatoid arthritis by eliminating pro-inflammatory synoviocytes but exacerbates tissue damage in systemic lupus erythematosus through neutrophil ferroptosis. While ferroptosis inhibitors (e.g., Fer-1) and inducers (e.g., IKE) show promise in preclinical models, clinical translation faces challenges such as disease-specific mechanistic heterogeneity, insufficient drug selectivity, and complex metabolic interactions. Future research should integrate multi-omics, organoid models, and AI-driven predictions to develop precision-targeted strategies, offering novel therapeutic paradigms for autoimmune diseases.
Keywords:
ferroptosis
; autoimmune diseases
; JAK/STAT pathway
; NF-κB pathway
; lipid peroxidation
; precision therapy
1. Introduction
The immune system uses a variety of immune cells to eliminate senescent cells and immune complexes in the body and establish autoimmune tolerance to resist foreign invading pathogens, which is a defense mechanism of the human body.In certain instances, the disruption of autoimmune tolerance can lead to an aberrant immune response where the body’s immune system attacks its own tissues and cells, causing cellular or tissue damage and associated clinical manifestations, ultimately resulting in autoimmune diseases[1,2,3].
Approximately 20% of the global population is impacted by over 100 distinct types of autoimmune diseases. Among them, psoriasis (2%-4%), rheumatoid arthritis (0.5%-1%), Graves’ disease (0.5%), Crohn’s disease (0.2%-0.3%) and systemic involvement of systemic lupus erythematosus were the most common. As early as 1999, the World Health Organization listed autoimmune diseases as the third major threat to human health after cardiovascular diseases and cancer.After summarizing the different prevalence of AIDs in 11 countries, T Ngo found that the incidence of AIDs in women was higher than that in men[4]. In the past few decades, the overall incidence of AIDs has shown an upward trend, which has brought harm to people’s life and economy [5].At present, the research on AIDs is multi-faceted, involving a variety of pathways and microstates. Among them, ferroptoisi is a more popular research direction.Ferroptosis is a new type of programmed cell death different from apoptosis, which has the characteristics of immunogenic cell death.Ferroptosis can be caused by abnormal lipid accumulation in immune cells, which in turn can act on abnormal expression of immune cell function[6]. Ferroptosis has been proved to play a role in different diseases, and previous studies have also carried out experimental analysis and explanation of the mechanism of ferroptosis in the immune system from different angles.In this paper, a comprehensive review of multiple literatures was conducted to examine the role of ferroptosis in the immune system.
2. Overview and Important Components of Ferroptosis
2.1. Overview
The concept of ferroptosis is a form of programmed cell death (PCD) that is different from apoptosis and autophagy, which was first proposed by Dixon et al in 2012.Cellular features of ferroptosis include loss of membrane integrity, increased membrane density, mitochondrial shrinkage, and rupture of the mitochondrial outer membrane, but normal nuclear morphology[7].Current studies suggest that ferroptosis is related to fatal lipid peroxidation, and ferroptosis is also the result of imbalance of cellular metabolism and REDOX homeostasis. After literature review, we found that ferroptosis plays an important physiological role in the occurrence of neurodegenerative diseases, ischemic organ damage [8], gastrointestinal system diseases, tumors, and immune diseases.
2.2. Important Components and Pathways of Ferroptosis
GPX4 and system Xc- are key components in the occurrence of ferroptosis. GPX4 is one of the major antioxidoreductases in the glutathione reductant scavenging lipid peroxides products, which plays a role in protecting cells and tissues from free radical damage.GPX4 protects the integrity of cell membrane by resisting lipid peroxidation mainly through glutathione (GSH) [9] Studies have shown that GPX4 can reduce phospholipid peroxides (PLOOH) and reduce thymine hydrogen peroxide, cholesterol hydrogen peroxide and fatty acid hydrogen peroxide, so as to protect cells from oxidative damage to a certain extent.In addition, GPX4 also plays a role in regulating immune homeostasis and anti-tumor immunity.Genetics has shown that Treg cells play a crucial role in maintaining immune tolerance, and the loss of GPX4 leads to ferroptosis of Treg cells stimulated by T cell receptors, thereby disrupting immune system homeostasis[10]。Li et al. showed in clinical studies that the increase of CaMKIV/CREMα nuclear translocation will lead to the decrease of GPX4, and the inhibition of GPX4 expression will stimulate the increase of lipid reactive oxygen species, which will cause ferroptosis of neutrophils, and clinically presents as lupus-like lesions[11].
The cystine-glutamate antitransporter system Xc− consists of SLC3A2 and SLC7A11, which mediate the exchange of extracellular cystine and intracellular glutamate across the plasma membrane.System Xc- activity is usually positively correlated with SLC7A11 encoding.ATF3 binds to the SLC7A11 promoter to down-regulate SLC7A11 expression and inhibit system Xc-, which depletes intracellular GSH and promotes ferroptosis activator induced lipid peroxidation leading to cell death. Amino acid metabolism, iron accumulation and lipid peroxidation related to systems XC-GSH are the substrates for the occurrence of ferrodeath, which will be described in the next section.
3. Metabolism of Ferroptosis
3.1. Role of Amino Acid Metabolism in Ferroptosis
System Xc- is an antiporter responsible for the exchange of glutamate and cystine, playing an indispensable role in amino acid metabolism. On the extracellular side, it facilitates cystine uptake, while intracellularly, it is crucial for the synthesis of cysteine and GSH, thereby contributing to the cellular antioxidant defense system. In the 1950s, Harry Eagle’s research demonstrated that amino acid deprivation, particularly cysteine deficiency, resulted in cell death [12]. Additionally, his studies revealed that the endogenous synthesis of cysteine from methionine and glucose provided a protective effect against this cell death [13].Cell survival depends on cysteine, which is an essential cellular antioxidant.Cysteine is also a substrate of GSH [14], and cysteine can protect cells from oxidative stress damage by promoting GSH synthesis. GSH is not only the most abundant reductant in mammalian cells, but also a cofactor of many enzymes, which can reduce the accumulation of lipid peroxides through oxidation reduction, thereby inhibiting the occurrence of ferrodeath.In addition to cystine, the common amino acid metabolism also includes glutamic acid.Glutamine is the most abundant amino acid in blood and cell culture media, and its dependent performance is regulated by SLC7A11 in system XC-and has an impact on cancer [15]which once again proves the importance of amino acid metabolism for ferrodeath.
3.2. Role of Iron Metabolism in Ferroptosis
In addition to amino acid metabolism, the occurrence of ferroptosis is also related to iron metabolism. Ferroptosis is characterized by lipid peroxidation and iron accumulation, and its induction process is inseparable from the metabolic process of REDOX active iron.Iron metabolism is mainly regulated by the liver, which maintains systemic iron balance by producing and secreting factors. In mice fed a high-iron diet, liver ferroptoisi caused by iron overload occurred, which was mitigated by PPARα activation through Gpx4 and transferrin(TRF) [16]. Moreover, transferrin (Tf), ferritin, ferrimodulin and ferritransporter (FPN) also play a key role in the maintenance of systemic iron homeostasis. Fe3+ binds to the circulating transferrin (TF) in the blood and is transferred to the endosome of the cell through the transferrin receptor 1 (TfR1) on the cell membrane, where most of the iron can form ferritin and is stored, while the excess iron is transported outside the battery and converted into Fe3+, which maintains the iron balance in the cytoplasm through the ferritransport protein.Theoretically, almost all circulating iron needs to bind to TF/TRF, and iron-containing TF can further bind to the transferrin receptor TFRC, and then be converted into divalent iron ions by iron reductase in vivo and imported into the cytoplasm through transmembrane transporters.The key factor of iron export is SLC40A1. Studies have confirmed that the overexpression of SLC40A1 can improve ferroptosis, and the knockdown of SLC40A1 can promote ferroptosis [17], which also indirectly demonstrates the role of iron metabolism.
3.3. Lipid Metabolism - An Important Link in Ferroptosis
Lipid metabolism is an important link in ferroptosis, and the occurrence of ferroptosis is a death process driven by iron-dependent phospholipid peroxidation (PL), which is characterized by the accumulation of iron-dependent lethal lipid peroxide (LPO) [8].Reactive oxygen species (ROS) are by-products of aerobic metabolism that are constantly produced, converted and consumed in all living organisms.ROS can lead to DNA damage, genetic instability and cell death by enhancing cell proliferation and survival [18].It is well known that mammalian cells contain a certain level of PUFA-PL and bioactive iron. In the presence of bioactive iron, PUFA-PL can convert ROS into phospholipid peroxides (PLOOH) in an enzymatic or non-enzymatic manner.If PLOOH is not effectively neutralized, it will destroy the integrity of the plasma membrane and cause ferroptosis in vivo.There are many pathways to prevent lipid peroxidation, and the GPX4 pathway mentioned above is the most classic inhibitory mode, which can catalyze the reduction of toxic PLOOH to non-toxic Plol (PLOH) [19].Moreover, GPX4 is the only enzyme that directly reduces lipid hydroperoxides in biofilms, and it can convert GSH to oxidized glutathione disulfide to reduce LPO, thus maintaining cell REDOX homeostasis [20]. In addition, there are lipophilic free radical trapping antioxidants (RTA) that can terminate the propagation of PL peroxidation, thereby blocking ferrodeath caused by GPX4 deficiency [7]
In conclusion, the occurrence of ferroptosis is not a single factor, but a combination of factors. Because ferroptosis has multiple triggers, the link to the immune system is not unique.Next, we will describe the signaling pathways of ferroptosis and the immune system to further elaborate the relationship between them.
4. Ferroptosis and Signaling Pathways of the Immune System
4.1. The JAK/STAT Signaling Pathway
Janus kinase/signal transducer and activator of transcription (JAK/STAT) signaling pathway is considered to be one of the central communication nodes in cellular functions. Studies have found that JAK/STAT signaling pathway participates in the generation of more than 50 cytokines and growth factors, and plays an important role in immune regulation, which is closely related to various immune diseases[21]
Figure 1.
Mechanisms and signaling pathways of ferroptosis.
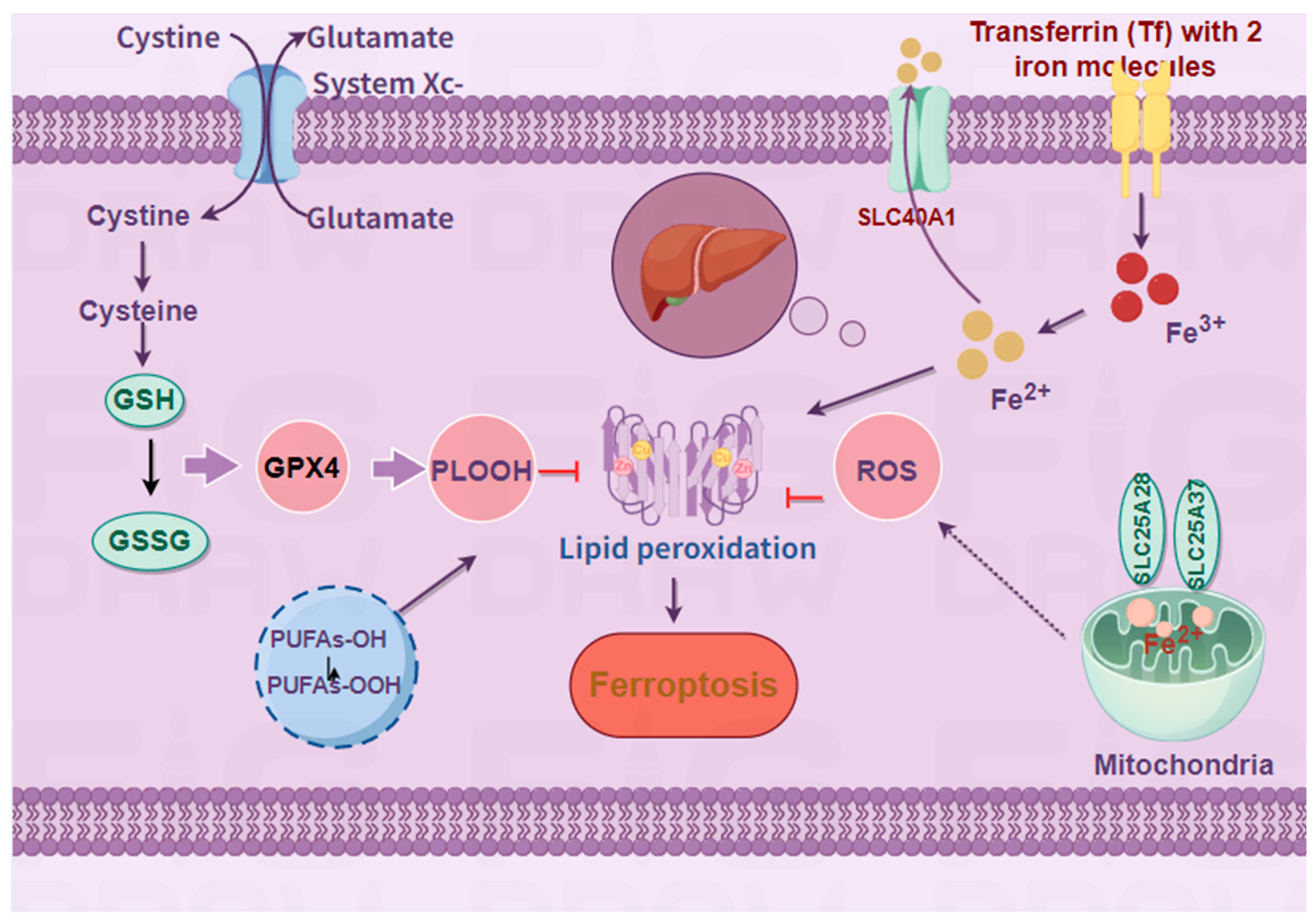
The occurrence of ferroptosis is mainly related to system Xc-, lipid peroxidation, and iron metabolism.System Xc-, as a cystine/glutamate antitransporter, can reduce GSH to GSSG through GSR and GPX4, affect lipid peroxidation, and also participate in the generation of PLOOH with polyunsaturated fatty acids.All aspects of iron metabolism, including iron absorption, storage and export, play an important role in ferroptosis.
4.1.1. JAK/STAT Signaling Pathway and Immune Response
STAT transcription factors are regulated by various cytokines and growth factors and are involved in the regulation of immune responses in the microenvironment [22].Myeloid-derived suppressor cells(MDSC) are derived from hematopoietic stem cells in bone marrow and have immunosuppressive properties of adaptive and innate immunity. A report in 2016 stated that VEGF, G-CSF, GM-CSF, Flt3L and other anti-inflammatory cytokines (such as interleukin-6) can activate STAT signaling and regulate the proliferation and activation of MDSC [23].In particular, VEGF and IL-6 can activate STAT3 on MDSC and accelerate the proliferation of MDSC [24].IFN-y is a key cytokine in anti-tumor host immunity, and blocking IFN-γ or destroying STAT1 will affect the inhibitory effect of MO-MDSCs[25].Natural killer (NK) cells are important immune cells in the body and participate in the occurrence of autoimmune diseases.Proliferation of NK cells depends on IL-15 induced by STAT1 in the JAK/STAT signaling pathway [26].In addition, STAT5 and STAT5b are important transcription factors for NK cell activation and proliferation, and the number of NK cells is significantly decreased in patients with STAT5b deficiency [27].T cells are also associated with immune responses, and the JAK/STAT pathway plays a key role in T cells, which are the cells that produce the largest number of cellular immunity among lymphocytes.T cells can differentiate into multiple effector subsets, among which Treg cells play a regulatory role and suppress potential pathological immune responses.Treg specific transcription factor is FoxP3[28], and its promoter binds to STAT5 to promote Treg differentiation [29]. Studies have shown that STAT3 and FoxP3 can also be used as transcription factors to regulate the biological function of Treg [30].
Activation of the JAK/STAT pathway has been found to promote the progression of various diseases, including various solid tumors [31], leukemia, inflammatory diseases, and immune diseases [32].In recent decades, JAK/STAT inhibitors have gained widespread application in the treatment of immune-related diseases. Tofacitinib, an orally administered small-molecule JAK inhibitor recently approved by the U.S. Food and Drug Administration (FDA), exerts its anti-inflammatory effects on inflammatory bowel diseases via the JAK/STAT signaling pathway, thereby demonstrating therapeutic efficacy [33].In addition, by nonlinear mixed-effect modeling of the aggregated data to characterize the population pharmacokinetics of tofacitinib in patients with ulcerative colitis (UC), 5 mg b.i.d. tofacitinib has better clinical efficacy in UC patients, while 10 mg b.i.d has additional clinical benefits [34].
The medicinal value of tofatinib was further substantiated in a randomized, double-blind, placebo-controlled trial conducted in patients with ankylosing spondylitis (AS). In this study, administration of tofatinib at doses of 5 mg and 10 mg twice daily demonstrated significantly superior efficacy compared to placebo at week 12, while its safety profile remained consistent with that observed in other indications[35]. Moreover, JAK inhibitors also have a good clinical effect on skin diseases caused by immune system disorders. For example, in the randomized, double-blind, placebo-controlled clinical trial of systemic lupus erythematosus (SLE), it was found that tofacitinib not only has a significant effect on SLE, but also improves cardiometabolic and immunological parameters associated with early onset atherosclerosis in SLE [36].In another randomized double-blind clinical trial, tofacitinib has also proved its efficacy and safety in treating atopic dermatitis in dogs [37]. Coincidentally, tofacitinib has also been used in the treatment of psoriasis, and has good efficacy in both cowhide and shingles [38].
4.1.2. JAK/STAT Signaling Pathway and Ferroptosis
IFN-γ, a key cytokine in anti-tumor host immunity mentioned above, is not only associated with the immune system, but also inhibits the transcription of Xc- in erastin or RSL3-induced cell death through the JAK/STAT signaling pathway, thereby increasing cell sensitivity to ferroptoisi activators [39].In addition, it has been found that IFN-γ interacts with transcription factors in the JAK/STAT signaling pathway to affect cell ferroptosis.IFN-γ can promote the binding of STAT1 to SLC7A11, a member of the XC-family of the system, thereby increasing lipid peroxidation in vivo and slowing the growth of xenograft tumors, while STAT1 deficiency can reverse this situation [40].This was further corroborated by YU, who demonstrated that the down-regulation of SLC7A11 by IFN-γ through the JAK/STAT signaling pathway enhances the sensitivity of adrenocortical cells to erastin-induced ferroptosis [41]. IFN-γ has also been found to activate the JAK/STAT pathway in hepatocellular carcinoma by down-regulating the mRNA and protein levels of SLC3A2 and SLC7A11 in system Xc- [39].As mentioned above, one of the typical characteristics of ferroptosis is iron accumulation, and the maintenance of iron homeostasis without causing iron accumulation depends on the action of ferriregulin.Studies have shown that the JAK/STAT signaling pathway is a clear mechanism to drive the expression of ferrimodulin [42], and some drugs can drive ferrimodulin through the JAK/STAT pathway to affect the occurrence of diseases, such as the regulation of hepatocellular carcinoma by dandelion polysaccharide [43], and the treatment of rheumatoid arthritis by Jinnophin[44].These studies have demonstrated the association between JAK/STAT signaling pathway and ferroptosis, suggesting a therapeutic approach for the future treatment of ferroptosis related diseases (Table 1).
4.2. NF-κB Signaling Pathway
4.2.1. NF-κB Signaling Pathway and Immune Response
NF-κB is a classical transcription factor, and its signaling pathways are activated by different mechanisms, which can be divided into typical pathway and atypical pathway.Activation of NF-κB signaling pathway is associated with apoptosis, viral replication, tumorigenesis, inflammation and various autoimmune diseases [45].The classical NF-κB pathway is activated by proinflammatory signals, Toll-like receptors (TLR) and lymphocyte receptors.TLR are pattern recognition receptors, which can recognize damage-related molecular patterns and activate the expression of related receptors and inflammatory genes, thus playing a protective role [46]. In addition, TLR is also crucial in the production of autoantibodies, and studies have found that the pathogenesis of autoimmune diseases is related to defective clearance of apoptotic cell debris [47].For example, in the mouse model, the nucleic acid-activated endosome TLR7 and TLR9 in the cytoplasm cannot be completely removed, leading to systemic lupus erythematosus [48].Activation of the NF-κB signaling pathway is also closely related to lymphoid tissue development and function.It not only makes the development of myeloid thymic epithelial cells more clear, but also promotes the maintenance and activation of mature lymphocytes[49], which is specifically manifested as mediating T and B cell responses.The differentiation of T cells into TH1 or TH2 subgroups is dependent on the contribution of NF-κB.Activation of naive T cells requires antigen specificity provided by activated APC and co-stimulatory signal transduction presented by TCR binding to MHC. The activated T cells then multiply rapidly and rely on NF-κB activity to prevent apoptosis and produce cytokines [50].The atypical pathway is mediated by NEMO and IKKβ independent IKKα dimer complexes.Recent studies have revealed that the non-classical NF-κB signaling pathway can regulate different aspects of immune function, and NIK is the core component of the non-classical NF-κB pathway[51].The first aspect pertains to the impact on lymphocytes, which constitute a critical component of the body’s immune response. The non-canonical NF-κB signaling pathway plays an essential role in mediating the proper development of secondary lymphoid organs[52]. Research has demonstrated that the loss of NIK function in mice with lymphatic dysplasia impairs lymph node development and results in splenic structural abnormalities. Furthermore, mice deficient in NIK exhibit severe defects in the development of primary lymphoid organs, specifically the thymus. The non-canonical NF-κB pathway facilitates dendritic cells (DCs) in recognizing infections and tissue damage via pattern recognition receptors, enabling their maturation into competent antigen-presenting cells. This process is pivotal for T cell generation and activation and serves as a bridge between innate and adaptive immunity [53].
NIK serves as a central mediator in immune responses, and its abnormal activation or expression is closely associated with the onset of immune-related diseases. For instance, experiments conducted on aly mutant mice and NIK knockout mice revealed B cell deficiency due to disordered lymph nodes, Peyer’s patches, and spleen structures. Additionally, NIK mutations were identified in patients with combined immunodeficiency [54]. Furthermore, individuals with NIK mutations exhibited deficiencies in follicular helper cells, memory T cell populations, and natural killer cells[54]. These findings suggest that targeting NIK could offer novel therapeutic strategies for autoimmune diseases. Highly selective and potent small-molecule inhibitors of NIK have been shown to effectively treat experimental lupus in NZB/WF1 mice [55]. Moreover, NIK plays a role in promoting inflammatory activation of human endothelial cells in the synovial fluid of rheumatoid arthritis (RA) patients, and NIK inhibitors demonstrate promising efficacy in treating RA[56]. Consistent with this, the inflammatory activation of endothelial cells induced by synovial fluid from RA patients was significantly attenuated following NIK knockdown [57]. Collectively, these results indicate that targeting NIK represents a promising approach and methodology for the treatment of immune-related disorders (Table 1).
4.2.2. NF-κB Signaling Pathway and Ferroptosis
Numerous studies have shown that ferroptosis is closely related to NF-κB signaling pathway.In the classical NF-κB pathway, studies have found that miR-93-5p can promote apoptosis and ferroptosis of granulosa cells and improve polycystic ovarian syndrome (PCOS) by regulating the NF-κB signaling pathway [58].Dimethyl fumarate (DMF) alleviates neuroinflammation and ferroptosis in chronic cerebral hypoperfusion by mediating NF-κB signaling pathway, significantly improves cognitive impairment, and partially reverses hippocampal neuronal damage and loss [59].Heat shock protein beta-1 (HSPB1) binding to Ikβ-α and promoting its ubiquitin-mediated degradation can lead to the activation of NF-κB signal transduction, and inhibition of ferroptosis can up-regulate the expression of HSPB1, thereby promoting the resistance to breast cancer treatment drugs[60].Non-classical NF-κB activation has also been associated with ferroptosis.Specific knockout or inhibition of NIK prevented excessive lipid peroxidation in primary hepatocytes, thereby alleviating APAP-mediated hepatotoxicity in mice[61]. (Table 1)
5. Ferroptosis in Immune Diseases
5.1. Rheumatoid Arthritis (RA)
RA is a chronic progressive inflammatory disease. Abnormal proliferation of fibroblast-like synoviocytes (FLS) drives inflammatory signals leading to the appearance of RA.RA usually affects the knee joint and elbow joint, which may lead to joint and periarticular structure damage and systemic inflammation if not treated in time[62]. Moreover, RA is characterized by the infiltration of immune cells in the joints [63], and the main clinical symptoms are joint swelling, stiffness and pain, and even bone and joint deformation and loss of function in severe cases [64].According to the survey, the global incidence of RA is 0.5%-1%, and it mostly occurs in women aged 30-50 years[65]. RA is the strongest systemic immune system disease in autoimmune diseases, and it is difficult to treat clinically and has many complications [66].In recent years, ferroptosis has received widespread attention in the treatment of inflammatory arthritis. A bioinformatics analysis found that 34 potential ferroptosis related genes found in RA were mainly enriched in HIF-1 signaling pathway, FoxO signaling pathway, and ferroptoisi pathway[67].Most researchers hold two explanations for the role of ferroptosis in RA:
First, excessive iron accumulation can damage osteoblasts.As early as in 1996, Fritz conducted an experimental analysis of 86 synovium from patients with rheumatoid arthritis (RA) or osteoarthritis (OA) and found that iron deposits in RA synovium were significantly increased compared with OA patients[68].Iron overload is one of the characteristics of death, and the experimental evidence that iron overload is one of the culprits of osteoporosis and bone fracture, the mice was discovered in the iron overload in mice model of[63] bone balance is broken, and in vitro experiments have confirmed that iron overload can reduce the activity of osteoblast [69]. In addition, in vitro iron was found in a mouse model of hemophilia to lead to increased expression of the p53 binding protein mdm2, which may underlie the development of hemophilia synovitis [70].The above studies have proved that excessive iron in synovial fluid is positively correlated with the severity of RA, and iron deposition can also aggravate RA by inducing ferroptosis in macrophages. Therefore, the use of ferroptosis inhibitor (LPX-1) can alleviate the development of arthritis[71].(Table 2)
Second, lipid peroxidation can cause bone damage and aggravate immune disorders.Datta et al. measured the synovial fluid of RA patients by flow cytometry and found the existence of a large number of ROS [72] The results of another clinical experiment showed that the levels of GSH and GPX4 in the blood of were reduced [72]. GSH can inhibit the production of ROS and the occurrence of ferroptosis, and the decreased levels indicate the increase of ROS in patients.In addition, studies have confirmed that the tumor suppressor gene p53 is expressed in RA and FSL, and p53 can inhibit systemic Xc- by downregulating the expression of SLC7A11, resulting in a decrease in antioxidant capacity and ROS accumulation in the body [73].Similarly, Mateen et al. also detected ROS production and lipid peroxidation in the blood of RA patients[74], indicating that ROS can be used as a potential marker of RA disease progression.In addition, ROS is an important element in the ROS/TNF-α feedback pathway, and the production of TNF-α depends on the activation of NF-κB signaling pathway stimulated by ROS. Studies have shown that NF-κB signaling pathway can activate p38/JNK signaling pathway to accelerate the progression of RA [75].Other studies have shown that wasp venom (WV) can not only reduce the level of TNF-α by inactivating JAK/STAT signaling pathway, but also accumulate ROS to induce GPX4-mediated ferroptosis to treat RA[76].The treatment of RA by inducing ferroptoisi is not unique. FLS was significantly increased in a mouse model of collagen-induced arthritis, and the use of ferroptoisi inducer IKE could reduce the number of fibroblasts in the synovium of mice, thereby reducing inflammation and tissue damage[77].Glycine can reduce the expression of GPX4 and FTH1 by increasing the methylation of GPX4 promoter mediated by the concentration of S-adenosine methionine (SAM) and reducing the expression of FTH1 in RA and FLS, thereby enhancing ferroptosis and achieving the effect of RA treatment[78].Studies have found that targeted activation of Nrf2 can not only reduce ROS, but also inhibit the proliferation and migration of FSL, indicating that inhibition of ferroptosis can also improve RA. Similarly, inhibition of NCOA4-mediated iron phagocytosis can protect RA and FLS from ferroptosis in LPS-induced inflammation under hypoxia [79].(Table 2)
5.2. Systemic Lupus Erythematosus (SLE)
Systemic lupus erythematosus (SLE) is a typical autoimmune disease characterized by excessive activation of the immune system, resulting in lesions of autoantibodies and immune complexes, which may involve systemic tissues and organs [80]. The skin and mucosa are mainly involved, and butterfly erythema occurs, accompanied by nervous system involvement.Investigation shows that SLE is the most sex-different disease among autoimmune diseases, with a male to female incidence rate of 1:9, which is more common in young women [81].Currently, the management of SLE remains imperfect, characterized by high treatment costs and numerous sequelae, which impose significant psychological stress and economic burdens on patients.The pathogenesis of SLE is related to most immune cells, and the link between SLE and ferroptosis mainly involves iron overload and lipid peroxidation [82].
Neutrophils are the main immune cells in the circulation and are important in both innate and adaptive immunity [83].Neutrophil death in systemic lupus erythematosus (SLE) may serve as an autoantigen to induce interferon (IFN) production, thereby contributing to the pathogenesis of SLE [84].Li et al. demonstrated that GPX4-induced ferroptosis of neutrophils causes the emergence of autoimmune diseases [85].In addition, B cells also play an important role in maintaining immune homeostasis, and higher ROS levels affect the activation and differentiation process of B cells [11], while GPX4 is also essential in preventing ferroptosis of B cells [86].In addition, studies have found that iron in T cells of SLE patients is increased compared with normal people[87], and GSH level in T cells of SLE patients is lower, and the degree of GSH reduction is related to mitochondrial hyperpolarization and increased ROS [88].Therefore, SLE can be improved by mediating T cells, such as erucic acid inhibiting the effector function of T cells and improving the pregnancy response of SLE [89].
Kidney is one of the most severely damaged organs in SLE. Iron deposition and severe lipid peroxidation in the kidney have been observed in lupus-susceptible mouse models [90].Iron accumulation in the kidneys of lupus nephritis (LN) mice leads to albuminuria and transferrin uria[91], which can be prevented and alleviated by the ferroptosis inhibitor liproxstatin-2[92].In addition, the typical manifestation of SLE is skin lesions. Studies have found that the increase of skin iron content after ultraviolet B radiation (UVB) exposure leads to excessive accumulation of ROS and GSH depletion, leading to the death of immunogenic keratinocytes, thereby causing skin inflammation [93].(Table 2)
5.3. Psoriasis (PsO)
Psoriasis (PsO) is a common chronic autoimmune skin disease. The occurrence of psoriasis is related to the activation of abnormal infiltrating immune cells, excessive proliferation of keratinocytes and accumulation of inflammatory cytokines [94].PsO is the autoimmune disease with the highest incidence at present. According to statistics, about 125 million people worldwide suffer from psoriasis, and the incidence rate is as high as 2-4%[95].PsO clinically presents with localized or extensive erythema, papules, and desquamation, and even pruritus.According to different clinical manifestations, PsO is divided into four types, including plaque psoriasis, spotting psoriasis, erythrodermic psoriasis and pustular psoriasis, among which plaque psoriasis is the most common, accounting for 80-90% of the total incidence[96].Unlike other autoimmune diseases, while persistent inflammation in PsO is the primary cause, genetic and environmental factors also play important roles [95].PsO has a strong genetic susceptibility, with a prevalence of up to 17.7% in first-degree relatives of PsO, which is mainly related to alleles in the major histocompatibility complex genetic region in the short arm of chromosome 6 [97].In addition, obesity [98], smoking [99], and bacterial infection [100] were all positively associated with the development of PsO. These external causes can trigger immune inflammatory responses in genetically predisposed patients under certain conditions. The relationship between ferroptoisi and inflammatory diseases has been mentioned many times before, and PsO is no exception.
Studies have shown that the occurrence of skin diseases is mainly related to oxidative stress in the skin microenvironment, and the anti-oxidative imbalance in the external environment is the key to the pathogenesis of skin diseases[101].Compared with healthy skin, skin cells of PsO patients have increased iron content and down-regulated GPX4 expression [102].However, GPX4 was significantly decreased in keratinocytes of PsO patients, while lipid peroxidation was enhanced [103]. Moreover, gene database analysis shows that genes related to ferroptosis in psoriasis are involved in the regulation of immune microenvironment [104]. For example, acyl-coa synthetase long-chain family member 4 (ACSL4) can enhance inflammatory response by promoting lipid peroxidation and activating ferroptosis[105].Therefore, regulation of ferroptosis is a new pathway for the treatment of psoriasis.Fer-1, as a ferroptosis inhibitor, can effectively treat psoriasis by blocking Erastin-induced production of lipid ROS and thereby inhibiting ferroptosis [106].In a mouse model of IMQ-induced psoriasiform dermatitis, application of Fer-1 significantly improved skin thickness and dyskeratosis [104].(Table 2)
5.4. Inflammatory Bowel Disease (IBD)
Inflammatory bowel disease (IBD) is a chronic inflammatory disorder of the gastrointestinal tract, clinically categorized into Crohn’s disease (CD) and ulcerative colitis (UC). CD typically manifests as widespread transmural inflammation of the gastrointestinal tract, whereas UC primarily affects the left side of the colon [107]. The most common early symptom of IBD is bloody diarrhea, with over 90% of UC patients reporting rectal bleeding [108]. Other frequent intestinal symptoms include abdominal pain, tenesmus, fecal incontinence, and vomiting [109], and in severe cases, it can lead to arthritis, liver dysfunction, and skin lesions[107]. Research has found that the pathogenesis of IBD is driven by the interaction of genetic and environmental factors. Environmental exposures such as diet, smoking, medication, and family history can disrupt the immune system [110], leading to abnormal responses of the gut microbiota [111]. Numerous studies have demonstrated the involvement of various pro-inflammatory cytokines, such as Th17, IL-1β, and IFN-γ. In UC, IL-1β has been shown to promote the development of intestinal inflammation [112]. In CD, IL-17 produced by Th17 is considered a crucial inflammatory factor in its pathogenesis, capable of activating STAT3 to induce a strong inflammatory response [113], and the inhibition of IL-17A can reduce the occurrence of inflammation [114].
Ferroptosis is a form of cell death, which plays a pivotal role in epithelial renewal, tissue homeostasis and chronic inflammation of intestinal epithelial cells [115].Intestinal epithelial cells are highly selective barriers between the intestinal lumen and cells in the lower layer of the immune system, and stress in the endoplasmic reticulum can promote the development of chronic intestinal inflammation by down-regulating tissue homeostasis[116]. Iron exposure to intestinal epithelial cells can easily trigger endoplasmic reticulum stress [117].In fact, reports of chronic inflammation of the gastrointestinal tract caused by iron exposure are not single. A Japanese study showed that either excessive dietary iron intake or oral iron treatment aggravated UC [118], and Carrier’s study also demonstrated that excessive iron aggravated intestinal inflammation [119].Iron overload will increase the disease activity of IBD, which is due to the damage of iron overload to the intestinal antioxidant defense system, resulting in increased ROS and oxidative stress response. In addition, the ROS production detected in the colonic mucosa of IBD patients is also a strong demonstration [120].Therefore, we reasoned that inhibition of the onset of ferroptosis could improve IBD.Curculigoside (CUR) is a natural ingredient with antioxidant and anti-inflammatory effects. The use of CUR in a mouse model of colitis can significantly up-regulate the active expression of GPX4 and reduce the occurrence of ferroptosis in UC[121].We know that GPX4 is an important antioxidant enzyme, and Dong found that by upregulating the expression of GPX4 in UC, it can significantly inhibit ferroptosis, thereby improving UC symptoms[122]. In the male Wistar rat model, the use of iron chelating agents (including maltol and kojic acid) can effectively reduce the inflammatory index [123].These data indicate that ferroptosis inhibitors have a significant effect on IBD.(Table 2)
5.5. Multiple Sclerosis (MS)
Multiple sclerosis (MS) is an autoimmune disease mediated by T cells, with common clinical manifestations of movement disorders, focal demyelination in the brain stem and spinal cord, which is the main cause of disability in young people [124]. In recent years, the prevalence of MS has shown an increasing trend [125], with its incidence rising as latitude increases[126]. Consequently, the etiology of MS is believed to be associated with environmental factors and geographical location [146]. Furthermore, exposure to ultraviolet B (UVB) radiation, Epstein-Barr (EB) virus infection, obesity, and smoking have been identified as potential contributors that may exacerbate the condition of MS [127].GPX4 is broadly expressed in neurons and glial cells, where it plays a critical role in protecting these cells from oxidative stress [128]. Both GPX4 mRNA and protein levels are reduced, while lipid peroxidation is elevated in the brains of MS patients[127]. Additionally, MRI imaging has revealed an increased iron concentration in gray matter structures, providing evidence that ferroptosis is implicated in the pathogenesis of MS [129].At present, the pathogenesis of ferroptosis in MS is not clear, and some researchers believe that oxidative stress caused by iron accumulation is one of the causes. Iron accumulation promotes neurodegeneration through proinflammatory mechanisms and mitochondrial dysfunction[130], and can also lead to impaired cerebral venous drainage [131].
The aforementioned studies indicate that ferroptosis inhibitors hold potential as therapeutic agents for MS. Fer-1, a commonly utilized ferroptosis inhibitor, has been shown to effectively prevent Cuprizone-induced loss of oligodendrocytes and myelin in demyelinated mice, thereby alleviating symptoms associated with MS [132].Dimethyl fumarate (DMF), a drug approved by the FDA for the treatment of multiple sclerosis (MS), exerts its therapeutic effects by mitigating oxidative stress damage through the NRF2/NF-κB signaling pathway, thereby demonstrating significant anti-inflammatory and antioxidant properties [59,133]. Additionally, DMF has been shown to ameliorate chronic cerebral insufficiency, markedly elevate glutathione (GSH) levels, reduce iron expression, and alleviate hippocampal neuronal damage in rat models. Another FDA-approved agent, Desferrione (DFP), serves as a potent inhibitor of ferroptosis and is utilized in the treatment of iron overload disorders [134]. In a lysophospholipid-induced mouse model of focal demyelination in the optic nerve, DFP effectively attenuates the proliferation of microglia and astrocytes, as well as the associated myelin loss[135].(Table 2)
5.6. Type I Diabetes
Type 1 Diabetes Mellitus (T1DM)is also a chronic autoimmune disease characterized by pancreatic β-cell damage.According to statistical analysis of epidemiological data, T1DM is more common in adults [136], and its incidence is related to diet and living habits[137]. Researchs have demonstrated that diets rich in meat and protein are associated with an increased risk of developing T1DM [138,139]. This correlation is attributed to the propensity of such diets to induce hypercholesterolemia and obesity. Hypercholesterolemia, in turn, exacerbates oxidative stress, leading to the apoptosis of pancreatic β-cells [140]. Furthermore, obesity triggers a state of low-grade inflammation, wherein infiltrating macrophages release pro-inflammatory cytokines, thereby intensifying β-cell autoimmunity [141].The pro-inflammatory cytokine IFN-γ has been shown to directly impair the function and viability of β-cells in cyclophosphamide-induced autoimmune diabetic mice[142]. The secretion of proinflammatory cytokines is closely associated with the activation of autoreactive T cells and the generation of ROS [143]. Moreover, lipid metabolism plays a significant role in the pathogenesis of T1DM. Free fatty acids (FFAs), which are crucial components of lipids, have been implicated in this process. Lipidomic analysis of serum FFAs in infants and young children revealed that T1DM significantly impacts the activity of lipid elongases [144].Research has demonstrated that elevated levels of FFAs not only diminish peripheral insulin sensitivity but also contribute to β-cell dysfunction and apoptosis [145].Long-term supplementation with ω-3 polyunsaturated fatty acids in children who have a genetic predisposition to T1DM during early stages of life can significantly reduce the likelihood of developing islet autoimmune diseases [146]. ω-3 polyunsaturated fatty acids and their bioactive derivatives have been shown to effectively suppress the inflammatory and immune responses associated with T1DM by modulating the NF-κB signaling pathway [147].Studies have demonstrated that NaHS can effectively inhibit the release of pro-inflammatory cytokines and alleviate depression-like and anxiety-like behaviors induced by T1DM. The underlying mechanism is associated with reducing iron accumulation and oxidative stress, while increasing the expression of GPX4 and SLC7A11, thereby significantly mitigating ferroptosis in mouse models[148]. Furthermore, berberine (BBR) has been identified as a GPX4-targeting agent that effectively inhibits ferroptosis in pancreatic beta cells[149] In addition, human umbilical cord mesenchymal stem cells (HUCMSCs) have been shown to significantly increase iron content and ROS levels in the penile tissue, leading to a notable improvement in erectile dysfunction in diabetic rats. Concurrently, HUCMSCs were found to markedly downregulate the expression of key lipid metabolism genes [150]. These findings collectively suggest a strong association between T1DM and ferroptosis, and further indicate that the inhibition of ferroptosis can ameliorate T1DM-related complications.(Table 2)
6. Conclusion
Ferroptosis, an iron-dependent form of programmed cell death driven by lipid peroxidation, plays a dual role in autoimmune diseases: it can suppress inflammation by eliminating hyperactivated immune cells (e.g., fibroblast-like synoviocytes in RA while exacerbating tissue damage through iron overload and lipid peroxidation (e.g., neutrophil ferroptosis in systemic lupus erythematosus (SLE). Its regulatory network involves cross-talk between signaling pathways (e.g., JAK/STAT, NF-κB) and metabolism of amino acids, iron, and lipids. For instance, IFN-γ downregulates system Xc⁻ via JAK/STAT to enhance ferroptosis sensitivity in cancer cells, whereas the NF-κB pathway influences macrophage iron metabolism through ferritinophagy. This tissue-specific dynamic regulation provides novel therapeutic targets but also poses challenges, including disease-specific mechanistic heterogeneity (e.g., protective ferroptosis in RA vs. pathogenic ferroptosis in SLE, insufficient drug selectivity, and complex metabolic interactions.
Future research must integrate interdisciplinary approaches, such as spatial transcriptomics to map ferroptosis in situ, AI-driven drug design, and humanized organoid models for translational validation. Key directions include developing tissue-targeted ferroptosis modulators (e.g., liposome-encapsulated GPX4 agonists), exploring combination therapies (e.g., ferroptosis inhibitors with anti-TNF-α monoclonal antibodies), deciphering crosstalk between ferroptosis and other cell death modalities (e.g., pyroptosis), and investigating metabolic reprogramming effects on ferroptosis susceptibility. These advancements will shift treatment strategies from “one-size-fits-all” approaches to precision regulation of the immune-metabolic-ferroptosis axis, opening new frontiers for autoimmune disease therapy.
Author Contributions
Conceptualization: Xiaokang Jia; data curation: Ziman He; funding acquisition: Xiaokang Jia;investigation: Ziman He; methodology: Xiaokang Jia; supervision:Xiaokang Jia; writing—original draft: Xiaokang Jia; writing—review and editing: all authors. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by National Natural Science Foundation of China Youth Program (82405525), and Hainan Provincial Natural Science Foundation Youth Program (825QN312).
Data Availability
The original contributions presented in the study are included in the article, further inquiries can be directed to the corresponding authors.
Conflicts of Interest
The authors declare that they have no conflicts of interest.
References
- Y. Song, J. Li, and Y. Wu, “Evolving understanding of autoimmune mechanisms and new therapeutic strategies of autoimmune disorders,” (in eng), Signal Transduct Target Ther, vol. 9, no. 1, p. 263, Oct 4 2024. [CrossRef]
- C. Chen et al., “Mendelian randomization as a tool to gain insights into the mosaic causes of autoimmune diseases,” (in eng), Autoimmun Rev, vol. 21, no. 12, p. 103210, Dec 2022. [CrossRef]
- L. Wang, F. S. Wang, and M. E. Gershwin, “Human autoimmune diseases: a comprehensive update,” (in eng), J Intern Med, vol. 278, no. 4, pp. 369-95, Oct 2015. [CrossRef]
- S. T. Ngo, F. J. Steyn, and P. A. McCombe, “Gender differences in autoimmune disease,” (in eng), Front Neuroendocrinol, vol. 35, no. 3, pp. 347-69, Aug 2014. [CrossRef]
- F. W. Miller, “The increasing prevalence of autoimmunity and autoimmune diseases: an urgent call to action for improved understanding, diagnosis, treatment, and prevention,” (in eng), Curr Opin Immunol, vol. 80, p. 102266, Feb 2023. [CrossRef]
- D. M. Maahs, N. A. West, J. M. Lawrence, and E. J. Mayer-Davis, “Epidemiology of type 1 diabetes,” (in eng), Endocrinol Metab Clin North Am, vol. 39, no. 3, pp. 481-97, Sep 2010. [CrossRef]
- S. J. Dixon et al., “Ferroptosis: an iron-dependent form of nonapoptotic cell death,” (in eng), Cell, vol. 149, no. 5, pp. 1060-72, May 25 2012. [CrossRef]
- X. Jiang, B. R. Stockwell, and M. Conrad, “Ferroptosis: mechanisms, biology and role in disease,” (in eng), Nat Rev Mol Cell Biol, vol. 22, no. 4, pp. 266-282, Apr 2021. [CrossRef]
- Y. Xie, R. Kang, D. J. Klionsky, and D. Tang, “GPX4 in cell death, autophagy, and disease,” (in eng), Autophagy, vol. 19, no. 10, pp. 2621-2638, Oct 2023. [CrossRef]
- C. Xu et al., “The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity,” (in eng), Cell Rep, vol. 35, no. 11, p. 109235, Jun 15 2021. [CrossRef]
- P. Li et al., “Glutathione peroxidase 4-regulated neutrophil ferroptosis induces systemic autoimmunity,” (in eng), Nat Immunol, vol. 22, no. 9, pp. 1107-1117, Sep 2021. [CrossRef]
- H. Eagle, “Nutrition needs of mammalian cells in tissue culture,” (in eng), Science, vol. 122, no. 3168, pp. 501-14, Sep 16 1955. [CrossRef]
- H. Eagle, K. A. Piez, and V. I. Oyama, “The biosynthesis of cystine in human cell cultures,” (in eng), J Biol Chem, vol. 236, pp. 1425-8, May 1961.
- A. Meister, “Glutathione metabolism,” (in eng), Methods Enzymol, vol. 251, pp. 3-7, 1995. [CrossRef]
- L. A. Timmerman et al., “Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target,” (in eng), Cancer Cell, vol. 24, no. 4, pp. 450-65, Oct 14 2013. [CrossRef]
- G. Xing et al., “PPARα alleviates iron overload-induced ferroptosis in mouse liver,” (in eng), EMBO Rep, vol. 23, no. 8, p. e52280, Aug 3 2022. [CrossRef]
- N. Geng et al., “Knockdown of ferroportin accelerates erastin-induced ferroptosis in neuroblastoma cells,” (in eng), Eur Rev Med Pharmacol Sci, vol. 22, no. 12, pp. 3826-3836, Jun 2018. [CrossRef]
- J. N. Moloney and T. G. Cotter, “ROS signalling in the biology of cancer,” (in eng), Semin Cell Dev Biol, vol. 80, pp. 50-64, Aug 2018. [CrossRef]
- F. Ursini, M. Maiorino, M. Valente, L. Ferri, and C. Gregolin, “Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides,” (in eng), Biochim Biophys Acta, vol. 710, no. 2, pp. 197-211, Feb 15 1982. [CrossRef]
- R. Brigelius-Flohé and M. Maiorino, “Glutathione peroxidases,” (in eng), Biochim Biophys Acta, vol. 1830, no. 5, pp. 3289-303, May 2013. [CrossRef]
- X. Hu, J. Li, M. Fu, X. Zhao, and W. Wang, “The JAK/STAT signaling pathway: from bench to clinic,” (in eng), Signal Transduct Target Ther, vol. 6, no. 1, p. 402, Nov 26 2021. [CrossRef]
- Q. Hu et al., “JAK/STAT pathway: Extracellular signals, diseases, immunity, and therapeutic regimens,” (in eng), Front Bioeng Biotechnol, vol. 11, p. 1110765, 2023. [CrossRef]
- H. J. Ko and Y. J. Kim, “Signal transducer and activator of transcription proteins: regulators of myeloid-derived suppressor cell-mediated immunosuppression in cancer,” (in eng), Arch Pharm Res, vol. 39, no. 11, pp. 1597-1608, Nov 2016. [CrossRef]
- P. Jayaraman et al., “Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release,” (in eng), J Immunol, vol. 188, no. 11, pp. 5365-76, Jun 1 2012. [CrossRef]
- K. Movahedi et al., “Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity,” (in eng), Blood, vol. 111, no. 8, pp. 4233-44, Apr 15 2008. [CrossRef]
- B. Becknell and M. A. Caligiuri, “Interleukin-2, interleukin-15, and their roles in human natural killer cells,” (in eng), Adv Immunol, vol. 86, pp. 209-39, 2005. [CrossRef]
- A. Bernasconi et al., “Characterization of immunodeficiency in a patient with growth hormone insensitivity secondary to a novel STAT5b gene mutation,” (in eng), Pediatrics, vol. 118, no. 5, pp. e1584-92, Nov 2006. [CrossRef]
- L. Zhou, M. M. Chong, and D. R. Littman, “Plasticity of CD4+ T cell lineage differentiation,” (in eng), Immunity, vol. 30, no. 5, pp. 646-55, May 2009. [CrossRef]
- M. A. Burchill, J. Yang, C. Vogtenhuber, B. R. Blazar, and M. A. Farrar, “IL-2 receptor beta-dependent STAT5 activation is required for the development of Foxp3+ regulatory T cells,” (in eng), J Immunol, vol. 178, no. 1, pp. 280-90, Jan 1 2007. [CrossRef]
- D. M. Woods et al., “Decreased Suppression and Increased Phosphorylated STAT3 in Regulatory T Cells are Associated with Benefit from Adjuvant PD-1 Blockade in Resected Metastatic Melanoma,” (in eng), Clin Cancer Res, vol. 24, no. 24, pp. 6236-6247, Dec 15 2018. [CrossRef]
- D. Reddy, R. Kumavath, P. Ghosh, and D. Barh, “Lanatoside C Induces G2/M Cell Cycle Arrest and Suppresses Cancer Cell Growth by Attenuating MAPK, Wnt, JAK-STAT, and PI3K/AKT/mTOR Signaling Pathways,” (in eng), Biomolecules, vol. 9, no. 12, Nov 27 2019. [CrossRef]
- P. Xin et al., “The role of JAK/STAT signaling pathway and its inhibitors in diseases,” (in eng), Int Immunopharmacol, vol. 80, p. 106210, Mar 2020. [CrossRef]
- C. Nunes, L. Almeida, R. M. Barbosa, and J. Laranjinha, “Luteolin suppresses the JAK/STAT pathway in a cellular model of intestinal inflammation,” (in eng), Food Funct, vol. 8, no. 1, pp. 387-396, Jan 25 2017. [CrossRef]
- A. Mukherjee et al., “Exposure-Response Characterization of Tofacitinib Efficacy in Moderate to Severe Ulcerative Colitis: Results From Phase II and Phase III Induction and Maintenance Studies,” (in eng), Clin Pharmacol Ther, vol. 112, no. 1, pp. 90-100, Jul 2022. [CrossRef]
- Deodhar et al., “Tofacitinib for the treatment of ankylosing spondylitis: a phase III, randomised, double-blind, placebo-controlled study,” (in eng), Ann Rheum Dis, vol. 80, no. 8, pp. 1004-1013, Aug 2021. [CrossRef]
- S. A. Hasni et al., “Phase 1 double-blind randomized safety trial of the Janus kinase inhibitor tofacitinib in systemic lupus erythematosus,” (in eng), Nat Commun, vol. 12, no. 1, p. 3391, Jun 7 2021. [CrossRef]
- P. R. Little, V. L. King, K. R. Davis, S. B. Cosgrove, and M. R. Stegemann, “A blinded, randomized clinical trial comparing the efficacy and safety of oclacitinib and ciclosporin for the control of atopic dermatitis in client-owned dogs,” (in eng), Vet Dermatol, vol. 26, no. 1, pp. 23-30, e7-8, Feb 2015. [CrossRef]
- M. Abe et al., “Tofacitinib for the treatment of moderate to severe chronic plaque psoriasis in Japanese patients: Subgroup analyses from a randomized, placebo-controlled phase 3 trial,” (in eng), J Dermatol, vol. 44, no. 11, pp. 1228-1237, Nov 2017. [CrossRef]
- R. Kong, N. Wang, W. Han, W. Bao, and J. Lu, “IFNγ-mediated repression of system xc(-) drives vulnerability to induced ferroptosis in hepatocellular carcinoma cells,” (in eng), J Leukoc Biol, vol. 110, no. 2, pp. 301-314, Aug 2021. [CrossRef]
- W. Wang et al., “CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy,” (in eng), Nature, vol. 569, no. 7755, pp. 270-274, May 2019. [CrossRef]
- X. Yu, D. Zhu, B. Luo, W. Kou, Y. Cheng, and Y. Zhu, “IFNγ enhances ferroptosis by increasing JAK-STAT pathway activation to suppress SLCA711 expression in adrenocortical carcinoma,” (in eng), Oncol Rep, vol. 47, no. 5, May 2022. [CrossRef]
- K. V. Kowdley, E. M. Gochanour, V. Sundaram, R. A. Shah, and P. Handa, “Hepcidin Signaling in Health and Disease: Ironing Out the Details,” (in eng), Hepatol Commun, vol. 5, no. 5, pp. 723-735, May 2021. [CrossRef]
- F. Ren et al., “The Effects of Dandelion Polysaccharides on Iron Metabolism by Regulating Hepcidin via JAK/STAT Signaling Pathway,” (in eng), Oxid Med Cell Longev, vol. 2021, p. 7184760, 2021. [CrossRef]
- L. Yang et al., “Auranofin mitigates systemic iron overload and induces ferroptosis via distinct mechanisms,” (in eng), Signal Transduct Target Ther, vol. 5, no. 1, p. 138, Jul 31 2020. [CrossRef]
- S. C. Sun, “The non-canonical NF-κB pathway in immunity and inflammation,” (in eng), Nat Rev Immunol, vol. 17, no. 9, pp. 545-558, Sep 2017. [CrossRef]
- N. Jounai, K. Kobiyama, F. Takeshita, and K. J. Ishii, “Recognition of damage-associated molecular patterns related to nucleic acids during inflammation and vaccination,” (in eng), Front Cell Infect Microbiol, vol. 2, p. 168, 2012. [CrossRef]
- L. E. Muñoz, K. Lauber, M. Schiller, A. A. Manfredi, and M. Herrmann, “The role of defective clearance of apoptotic cells in systemic autoimmunity,” (in eng), Nat Rev Rheumatol, vol. 6, no. 5, pp. 280-9, May 2010. [CrossRef]
- A. N. Suthers and S. Sarantopoulos, “TLR7/TLR9- and B Cell Receptor-Signaling Crosstalk: Promotion of Potentially Dangerous B Cells,” (in eng), Front Immunol, vol. 8, p. 775, 2017. [CrossRef]
- M. Irla et al., “Autoantigen-specific interactions with CD4+ thymocytes control mature medullary thymic epithelial cell cellularity,” (in eng), Immunity, vol. 29, no. 3, pp. 451-63, Sep 19 2008. [CrossRef]
- M. S. Hayden and S. Ghosh, “NF-κB in immunobiology,” (in eng), Cell Res, vol. 21, no. 2, pp. 223-44, Feb 2011. [CrossRef]
- G. Xiao, E. W. Harhaj, and S. C. Sun, “NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100,” (in eng), Mol Cell, vol. 7, no. 2, pp. 401-9, Feb 2001. [CrossRef]
- S. C. Sun, “The noncanonical NF-κB pathway,” (in eng), Immunol Rev, vol. 246, no. 1, pp. 125-40, Mar 2012. [CrossRef]
- R. M. Steinman and H. Hemmi, “Dendritic cells: translating innate to adaptive immunity,” (in eng), Curr Top Microbiol Immunol, vol. 311, pp. 17-58, 2006. [CrossRef]
- K. L. Willmann et al., “Biallelic loss-of-function mutation in NIK causes a primary immunodeficiency with multifaceted aberrant lymphoid immunity,” (in eng), Nat Commun, vol. 5, p. 5360, Nov 19 2014. [CrossRef]
- H. D. Brightbill et al., “NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus,” (in eng), Nat Commun, vol. 9, no. 1, p. 179, Jan 12 2018. [CrossRef]
- A. R. Noort, P. P. Tak, and S. W. Tas, “Non-canonical NF-κB signaling in rheumatoid arthritis: Dr Jekyll and Mr Hyde?,” (in eng), Arthritis Res Ther, vol. 17, no. 1, p. 15, Jan 28 2015. [CrossRef]
- P. Kucharzewska et al., “NIK-IKK complex interaction controls NF-κB-dependent inflammatory activation of endothelium in response to LTβR ligation,” (in eng), J Cell Sci, vol. 132, no. 7, Apr 3 2019. [CrossRef]
- W. Tan et al., “MiR-93-5p promotes granulosa cell apoptosis and ferroptosis by the NF-kB signaling pathway in polycystic ovary syndrome,” (in eng), Front Immunol, vol. 13, p. 967151, 2022. [CrossRef]
- N. Yan, Z. Xu, C. Qu, and J. Zhang, “Dimethyl fumarate improves cognitive deficits in chronic cerebral hypoperfusion rats by alleviating inflammation, oxidative stress, and ferroptosis via NRF2/ARE/NF-κB signal pathway,” (in eng), Int Immunopharmacol, vol. 98, p. 107844, Sep 2021. [CrossRef]
- Y. Liang et al., “HSPB1 facilitates chemoresistance through inhibiting ferroptotic cancer cell death and regulating NF-κB signaling pathway in breast cancer,” (in eng), Cell Death Dis, vol. 14, no. 7, p. 434, Jul 15 2023. [CrossRef]
- X. Zhong et al., “Hepatic NF-κB-Inducing Kinase and Inhibitor of NF-κB Kinase Subunit α Promote Liver Oxidative Stress, Ferroptosis, and Liver Injury,” (in eng), Hepatol Commun, vol. 5, no. 10, pp. 1704-1720, Oct 2021. [CrossRef]
- J. J. Cush, “Rheumatoid Arthritis: Early Diagnosis and Treatment,” (in eng), Rheum Dis Clin North Am, vol. 48, no. 2, pp. 537-547, May 2022. [CrossRef]
- J. S. Smolen, D. Aletaha, and I. B. McInnes, “Rheumatoid arthritis,” (in eng), Lancet, vol. 388, no. 10055, pp. 2023-2038, Oct 22 2016. [CrossRef]
- W. Grassi, R. De Angelis, G. Lamanna, and C. Cervini, “The clinical features of rheumatoid arthritis,” (in eng), Eur J Radiol, vol. 27 Suppl 1, pp. S18-24, May 1998. [CrossRef]
- H. L. Wright, R. J. Moots, and S. W. Edwards, “The multifactorial role of neutrophils in rheumatoid arthritis,” (in eng), Nat Rev Rheumatol, vol. 10, no. 10, pp. 593-601, Oct 2014. [CrossRef]
- G. S. Firestein, “Evolving concepts of rheumatoid arthritis,” (in eng), Nature, vol. 423, no. 6937, pp. 356-61, May 15 2003. [CrossRef]
- X. Li, A. He, Y. Liu, Y. Huang, and X. Zhang, “Bioinformatics identification of ferroptosis-related genes and therapeutic drugs in rheumatoid arthritis,” (in eng), Front Med (Lausanne), vol. 10, p. 1192153, 2023. [CrossRef]
- P. Fritz, J. G. Saal, C. Wicherek, A. König, W. Laschner, and H. Rautenstrauch, “Quantitative photometrical assessment of iron deposits in synovial membranes in different joint diseases,” (in eng), Rheumatol Int, vol. 15, no. 5, pp. 211-6, 1996. [CrossRef]
- W. Xiao et al., “Iron overload increases osteoclastogenesis and aggravates the effects of ovariectomy on bone mass,” (in eng), J Endocrinol, vol. 226, no. 3, pp. 121-34, Sep 2015. [CrossRef]
- N. Hakobyan, T. Kazarian, A. A. Jabbar, K. J. Jabbar, and L. A. Valentino, “Pathobiology of hemophilic synovitis I: overexpression of mdm2 oncogene,” (in eng), Blood, vol. 104, no. 7, pp. 2060-4, Oct 1 2004. [CrossRef]
- Y. Liu et al., “Heterogeneous ferroptosis susceptibility of macrophages caused by focal iron overload exacerbates rheumatoid arthritis,” (in eng), Redox Biol, vol. 69, p. 103008, Feb 2024. [CrossRef]
- S. Datta, S. Kundu, P. Ghosh, S. De, A. Ghosh, and M. Chatterjee, “Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis,” (in eng), Clin Rheumatol, vol. 33, no. 11, pp. 1557-64, Nov 2014. [CrossRef]
- S. Z. Hassan, T. A. Gheita, S. A. Kenawy, A. T. Fahim, I. M. El-Sorougy, and M. S. Abdou, “Oxidative stress in systemic lupus erythematosus and rheumatoid arthritis patients: relationship to disease manifestations and activity,” (in eng), Int J Rheum Dis, vol. 14, no. 4, pp. 325-31, Oct 2011. [CrossRef]
- Y. Sun and H. S. Cheung, “p53, proto-oncogene and rheumatoid arthritis,” (in eng), Semin Arthritis Rheum, vol. 31, no. 5, pp. 299-310, Apr 2002. [CrossRef]
- S. Mateen, S. Moin, A. Q. Khan, A. Zafar, and N. Fatima, “Increased Reactive Oxygen Species Formation and Oxidative Stress in Rheumatoid Arthritis,” (in eng), PLoS One, vol. 11, no. 4, p. e0152925, 2016. [CrossRef]
- Z. Xie, H. Hou, D. Luo, R. An, Y. Zhao, and C. Qiu, “ROS-Dependent Lipid Peroxidation and Reliant Antioxidant Ferroptosis-Suppressor-Protein 1 in Rheumatoid Arthritis: a Covert Clue for Potential Therapy,” (in eng), Inflammation, vol. 44, no. 1, pp. 35-47, Feb 2021. [CrossRef]
- L. L. Ni et al., “The therapeutic effect of wasp venom (Vespa magnifica, Smith) and its effective part on rheumatoid arthritis fibroblast-like synoviocytes through modulating inflammation, redox homeostasis and ferroptosis,” (in eng), J Ethnopharmacol, vol. 317, p. 116700, Dec 5 2023. [CrossRef]
- J. Wu et al., “TNF antagonist sensitizes synovial fibroblasts to ferroptotic cell death in collagen-induced arthritis mouse models,” (in eng), Nat Commun, vol. 13, no. 1, p. 676, Feb 3 2022. [CrossRef]
- H. Ling et al., “Glycine increased ferroptosis via SAM-mediated GPX4 promoter methylation in rheumatoid arthritis,” (in eng), Rheumatology (Oxford), vol. 61, no. 11, pp. 4521-4534, Nov 2 2022. [CrossRef]
- Y. Zhang, G. Wang, T. Wang, W. Cao, L. Zhang, and X. Chen, “Nrf2-Keap1 pathway-mediated effects of resveratrol on oxidative stress and apoptosis in hydrogen peroxide-treated rheumatoid arthritis fibroblast-like synoviocytes,” (in eng), Ann N Y Acad Sci, vol. 1457, no. 1, pp. 166-178, Dec 2019. [CrossRef]
- Y. Wang et al., “Alleviated NCOA4-mediated ferritinophagy protected RA FLSs from ferroptosis in lipopolysaccharide-induced inflammation under hypoxia,” (in eng), Inflamm Res, vol. 73, no. 3, pp. 363-379, Mar 2024. [CrossRef]
- A. A. Justiz Vaillant, A. Goyal, and M. A. Varacallo, “Systemic Lupus Erythematosus,” in StatPearls. Treasure Island (FL) with ineligible companies. Disclosure: Amandeep Goyal declares no relevant financial relationships with ineligible companies. Disclosure: Matthew Varacallo declares no relevant financial relationships with ineligible companies.: StatPearls Publishing.
- Copyright © 2025, StatPearls Publishing LLC., 2025.
- L. Bowlus, “The role of iron in T cell development and autoimmunity,” (in eng), Autoimmun Rev, vol. 2, no. 2, pp. 73-8, Mar 2003. [CrossRef]
- Nathan, “Neutrophils and immunity: challenges and opportunities,” (in eng), Nat Rev Immunol, vol. 6, no. 3, pp. 173-82, Mar 2006. [CrossRef]
- G. S. Garcia-Romo et al., “Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus,” (in eng), Sci Transl Med, vol. 3, no. 73, p. 73ra20, Mar 9 2011. [CrossRef]
- H. Zhang, L. Wang, and Y. Chu, “Reactive oxygen species: The signal regulator of B cell,” (in eng), Free Radic Biol Med, vol. 142, pp. 16-22, Oct 2019. [CrossRef]
- J. Muri, H. Thut, G. W. Bornkamm, and M. Kopf, “B1 and Marginal Zone B Cells but Not Follicular B2 Cells Require Gpx4 to Prevent Lipid Peroxidation and Ferroptosis,” (in eng), Cell Rep, vol. 29, no. 9, pp. 2731-2744.e4, Nov 26 2019. [CrossRef]
- M. Zhao et al., “Downregulation of BDH2 modulates iron homeostasis and promotes DNA demethylation in CD4(+) T cells of systemic lupus erythematosus,” (in eng), Clin Immunol, vol. 187, pp. 113-121, Feb 2018. [CrossRef]
- A. Perl, P. Gergely, Jr., and K. Banki, “Mitochondrial dysfunction in T cells of patients with systemic lupus erythematosus,” (in eng), Int Rev Immunol, vol. 23, no. 3-4, pp. 293-313, May-Aug 2004. [CrossRef]
- Y. Chang et al., “Erucic acid improves the progress of pregnancy complicated with systemic lupus erythematosus by inhibiting the effector function of CD8(+) T cells,” (in eng), MedComm (2020), vol. 4, no. 5, p. e382, Oct 2023. [CrossRef]
- Q. Feng et al., “Broadening horizons: the multifaceted functions of ferroptosis in kidney diseases,” (in eng), Int J Biol Sci, vol. 19, no. 12, pp. 3726-3743, 2023. [CrossRef]
- S. Marks et al., “Renal iron accumulation occurs in lupus nephritis and iron chelation delays the onset of albuminuria,” (in eng), Sci Rep, vol. 7, no. 1, p. 12821, Oct 9 2017. [CrossRef]
- A. A. Alli et al., “Kidney tubular epithelial cell ferroptosis links glomerular injury to tubulointerstitial pathology in lupus nephritis,” (in eng), Clin Immunol, vol. 248, p. 109213, Mar 2023. [CrossRef]
- K. Vats et al., “Keratinocyte death by ferroptosis initiates skin inflammation after UVB exposure,” (in eng), Redox Biol, vol. 47, p. 102143, Nov 2021. [CrossRef]
- A. W. Armstrong and C. Read, “Pathophysiology, Clinical Presentation, and Treatment of Psoriasis: A Review,” (in eng), Jama, vol. 323, no. 19, pp. 1945-1960, May 19 2020. [CrossRef]
- X. Liang et al., “Interplay Between Skin Microbiota Dysbiosis and the Host Immune System in Psoriasis: Potential Pathogenesis,” (in eng), Front Immunol, vol. 12, p. 764384, 2021. [CrossRef]
- G. Schett, P. Rahman, C. Ritchlin, I. B. McInnes, D. Elewaut, and J. U. Scher, “Psoriatic arthritis from a mechanistic perspective,” (in eng), Nat Rev Rheumatol, vol. 18, no. 6, pp. 311-325, Jun 2022. [CrossRef]
- P. E. Stuart et al., “Genome-wide Association Analysis of Psoriatic Arthritis and Cutaneous Psoriasis Reveals Differences in Their Genetic Architecture,” (in eng), Am J Hum Genet, vol. 97, no. 6, pp. 816-36, Dec 3 2015. [CrossRef]
- V. M. Bhole et al., “Differences in body mass index among individuals with PsA, psoriasis, RA and the general population,” (in eng), Rheumatology (Oxford), vol. 51, no. 3, pp. 552-6, Mar 2012. [CrossRef]
- U. Gazel, G. Ayan, D. Solmaz, S. Akar, and S. Z. Aydin, “The impact of smoking on prevalence of psoriasis and psoriatic arthritis,” (in eng), Rheumatology (Oxford), vol. 59, no. 10, pp. 2695-2710, Oct 1 2020. [CrossRef]
- N. R. Telfer, R. J. Chalmers, K. Whale, and G. Colman, “The role of streptococcal infection in the initiation of guttate psoriasis,” (in eng), Arch Dermatol, vol. 128, no. 1, pp. 39-42, Jan 1992.
- O. Kızılyel, N. Akdeniz, M. S. Metin, and F. Elmas Ö, “Investigation of oxidant and antioxidant levels in patients with psoriasis,” (in eng), Turk J Med Sci, vol. 49, no. 4, pp. 1085-1088, Aug 8 2019. [CrossRef]
- Benhadou, D. Mintoff, and V. Del Marmol, “Psoriasis: Keratinocytes or Immune Cells - Which Is the Trigger?,” (in eng), Dermatology, vol. 235, no. 2, pp. 91-100, 2019. [CrossRef]
- Y. Shou, L. Yang, Y. Yang, and J. Xu, “Inhibition of keratinocyte ferroptosis suppresses psoriatic inflammation,” (in eng), Cell Death Dis, vol. 12, no. 11, p. 1009, Oct 27 2021. [CrossRef]
- M. N. Wu et al., “Genetic analysis of potential biomarkers and therapeutic targets in ferroptosis from psoriasis,” (in eng), Front Immunol, vol. 13, p. 1104462, 2022. [CrossRef]
- Miotto et al., “Insight into the mechanism of ferroptosis inhibition by ferrostatin-1,” (in eng), Redox Biol, vol. 28, p. 101328, Jan 2020. [CrossRef]
- M. Fumery, S. Singh, P. S. Dulai, C. Gower-Rousseau, L. Peyrin-Biroulet, and W. J. Sandborn, “Natural History of Adult Ulcerative Colitis in Population-based Cohorts: A Systematic Review,” (in eng), Clin Gastroenterol Hepatol, vol. 16, no. 3, pp. 343-356.e3, Mar 2018. [CrossRef]
- A. Dignass et al., “Second European evidence-based consensus on the diagnosis and management of ulcerative colitis part 1: definitions and diagnosis,” (in eng), J Crohns Colitis, vol. 6, no. 10, pp. 965-90, Dec 2012. [CrossRef]
- M. C. Dubinsky et al., “Impact of Bowel Urgency on Quality of Life and Clinical Outcomes in Patients With Ulcerative Colitis,” (in eng), Crohns Colitis 360, vol. 4, no. 3, p. otac016, Jul 2022. [CrossRef]
- E. V. Loftus, Jr., “Clinical epidemiology of inflammatory bowel disease: Incidence, prevalence, and environmental influences,” (in eng), Gastroenterology, vol. 126, no. 6, pp. 1504-17, May 2004. [CrossRef]
- J. T. Chang, “Pathophysiology of Inflammatory Bowel Diseases,” (in eng), N Engl J Med, vol. 383, no. 27, pp. 2652-2664, Dec 31 2020. [CrossRef]
- C. A. Dinarello, “Interleukin-1beta and the autoinflammatory diseases,” (in eng), N Engl J Med, vol. 360, no. 23, pp. 2467-70, Jun 4 2009. [CrossRef]
- M. Gu et al., “IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma,” (in eng), Mol Cancer, vol. 10, p. 150, Dec 15 2011. [CrossRef]
- M. M. Fort et al., “IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo,” (in eng), Immunity, vol. 15, no. 6, pp. 985-95, Dec 2001. [CrossRef]
- C. Günther, H. Neumann, M. F. Neurath, and C. Becker, “Apoptosis, necrosis and necroptosis: cell death regulation in the intestinal epithelium,” (in eng), Gut, vol. 62, no. 7, pp. 1062-71, Jul 2013. [CrossRef]
- T. Clavel and D. Haller, “Bacteria- and host-derived mechanisms to control intestinal epithelial cell homeostasis: implications for chronic inflammation,” (in eng), Inflamm Bowel Dis, vol. 13, no. 9, pp. 1153-64, Sep 2007. [CrossRef]
- T. Werner et al., “Depletion of luminal iron alters the gut microbiota and prevents Crohn’s disease-like ileitis,” (in eng), Gut, vol. 60, no. 3, pp. 325-33, Mar 2011. [CrossRef]
- Y. Kobayashi et al., “Association between dietary iron and zinc intake and development of ulcerative colitis: A case-control study in Japan,” (in eng), J Gastroenterol Hepatol, vol. 34, no. 10, pp. 1703-1710, Oct 2019. [CrossRef]
- J. C. Carrier, E. Aghdassi, K. Jeejeebhoy, and J. P. Allard, “Exacerbation of dextran sulfate sodium-induced colitis by dietary iron supplementation: role of NF-kappaB,” (in eng), Int J Colorectal Dis, vol. 21, no. 4, pp. 381-7, May 2006. [CrossRef]
- A. D. Millar, D. S. Rampton, and D. R. Blake, “Effects of iron and iron chelation in vitro on mucosal oxidant activity in ulcerative colitis,” (in eng), Aliment Pharmacol Ther, vol. 14, no. 9, pp. 1163-8, Sep 2000. [CrossRef]
- S. Wang, W. Liu, J. Wang, and X. Bai, “Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4,” (in eng), Life Sci, vol. 259, p. 118356, Oct 15 2020. [CrossRef]
- S. Dong et al., “Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway,” (in eng), Dig Liver Dis, vol. 53, no. 10, pp. 1276-1285, Oct 2021. [CrossRef]
- M. Minaiyan, E. Mostaghel, and P. Mahzouni, “Preventive Therapy of Experimental Colitis with Selected iron Chelators and Anti-oxidants,” (in eng), Int J Prev Med, vol. 3, no. Suppl 1, pp. S162-9, Mar 2012.
- R. Dobson and G. Giovannoni, “Multiple sclerosis - a review,” (in eng), Eur J Neurol, vol. 26, no. 1, pp. 27-40, Jan 2019. [CrossRef]
- P. Browne et al., “Atlas of Multiple Sclerosis 2013: A growing global problem with widespread inequity,” (in eng), Neurology, vol. 83, no. 11, pp. 1022-4, Sep 9 2014. [CrossRef]
- N. Koch-Henriksen and P. S. Sørensen, “The changing demographic pattern of multiple sclerosis epidemiology,” (in eng), Lancet Neurol, vol. 9, no. 5, pp. 520-32, May 2010. [CrossRef]
- A. Ascherio, “Environmental factors in multiple sclerosis,” (in eng), Expert Rev Neurother, vol. 13, no. 12 Suppl, pp. 3-9, Dec 2013. [CrossRef]
- B. R. Cardoso, D. J. Hare, A. I. Bush, and B. R. Roberts, “Glutathione peroxidase 4: a new player in neurodegeneration?,” (in eng), Mol Psychiatry, vol. 22, no. 3, pp. 328-335, Mar 2017. [CrossRef]
- M. Neema et al., “Deep gray matter involvement on brain MRI scans is associated with clinical progression in multiple sclerosis,” (in eng), J Neuroimaging, vol. 19, no. 1, pp. 3-8, Jan 2009. [CrossRef]
- X. Zhang, N. Surguladze, B. Slagle-Webb, A. Cozzi, and J. R. Connor, “Cellular iron status influences the functional relationship between microglia and oligodendrocytes,” (in eng), Glia, vol. 54, no. 8, pp. 795-804, Dec 2006. [CrossRef]
- A. V. Singh and P. Zamboni, “Anomalous venous blood flow and iron deposition in multiple sclerosis,” (in eng), J Cereb Blood Flow Metab, vol. 29, no. 12, pp. 1867-78, Dec 2009. [CrossRef]
- P. Jhelum et al., “Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination,” (in eng), J Neurosci, vol. 40, no. 48, pp. 9327-9341, Nov 25 2020. [CrossRef]
- Cattani-Cavalieri et al., “Dimethyl Fumarate Attenuates Lung Inflammation and Oxidative Stress Induced by Chronic Exposure to Diesel Exhaust Particles in Mice,” (in eng), Int J Mol Sci, vol. 21, no. 24, Dec 18 2020. [CrossRef]
- L. Kwiatkowski et al., “Deferiprone vs deferoxamine for transfusional iron overload in SCD and other anemias: a randomized, open-label noninferiority study,” (in eng), Blood Adv, vol. 6, no. 4, pp. 1243-1254, Feb 22 2022. [CrossRef]
- A. Rayatpour, F. Foolad, M. Heibatollahi, K. Khajeh, and M. Javan, “Ferroptosis inhibition by deferiprone, attenuates myelin damage and promotes neuroprotection in demyelinated optic nerve,” (in eng), Sci Rep, vol. 12, no. 1, p. 19630, Nov 16 2022. [CrossRef]
- R. D. Leslie et al., “Adult-Onset Type 1 Diabetes: Current Understanding and Challenges,” (in eng), Diabetes Care, vol. 44, no. 11, pp. 2449-2456, Nov 2021. [CrossRef]
- Ogrotis, T. Koufakis, and K. Kotsa, “Changes in the Global Epidemiology of Type 1 Diabetes in an Evolving Landscape of Environmental Factors: Causes, Challenges, and Opportunities,” (in eng), Medicina (Kaunas), vol. 59, no. 4, Mar 28 2023. [CrossRef]
- S. Muntoni et al., “High meat consumption is associated with type 1 diabetes mellitus in a Sardinian case-control study,” (in eng), Acta Diabetol, vol. 50, no. 5, pp. 713-9, Oct 2013. [CrossRef]
- E. Syrjälä et al., “A Joint Modeling Approach for Childhood Meat, Fish and Egg Consumption and the Risk of Advanced Islet Autoimmunity,” (in eng), Sci Rep, vol. 9, no. 1, p. 7760, May 23 2019. [CrossRef]
- M. Cnop, J. C. Hannaert, A. Y. Grupping, and D. G. Pipeleers, “Low density lipoprotein can cause death of islet beta-cells by its cellular uptake and oxidative modification,” (in eng), Endocrinology, vol. 143, no. 9, pp. 3449-53, Sep 2002. [CrossRef]
- R. Buzzetti, S. Zampetti, and P. Pozzilli, “Impact of obesity on the increasing incidence of type 1 diabetes,” (in eng), Diabetes Obes Metab, vol. 22, no. 7, pp. 1009-1013, Jul 2020. [CrossRef]
- L. Campbell, T. W. Kay, L. Oxbrow, and L. C. Harrison, “Essential role for interferon-gamma and interleukin-6 in autoimmune insulin-dependent diabetes in NOD/Wehi mice,” (in eng), J Clin Invest, vol. 87, no. 2, pp. 739-42, Feb 1991. [CrossRef]
- E. Padgett, K. A. Broniowska, P. A. Hansen, J. A. Corbett, and H. M. Tse, “The role of reactive oxygen species and proinflammatory cytokines in type 1 diabetes pathogenesis,” (in eng), Ann N Y Acad Sci, vol. 1281, no. 1, pp. 16-35, Apr 2013. [CrossRef]
- A. I. S. Sobczak, S. J. Pitt, T. K. Smith, R. A. Ajjan, and A. J. Stewart, “Lipidomic profiling of plasma free fatty acids in type-1 diabetes highlights specific changes in lipid metabolism,” (in eng), Biochim Biophys Acta Mol Cell Biol Lipids, vol. 1866, no. 1, p. 158823, Jan 2021. [CrossRef]
- Y. Lee, H. Hirose, M. Ohneda, J. H. Johnson, J. D. McGarry, and R. H. Unger, “Beta-cell lipotoxicity in the pathogenesis of non-insulin-dependent diabetes mellitus of obese rats: impairment in adipocyte-beta-cell relationships,” (in eng), Proc Natl Acad Sci U S A, vol. 91, no. 23, pp. 10878-82, Nov 8 1994. [CrossRef]
- M. Norris et al., “Omega-3 polyunsaturated fatty acid intake and islet autoimmunity in children at increased risk for type 1 diabetes,” (in eng), Jama, vol. 298, no. 12, pp. 1420-8, Sep 26 2007. [CrossRef]
- Zhang, Y. Xiao, J. Hu, S. Liu, Z. Zhou, and L. Xie, “Lipid metabolism in type 1 diabetes mellitus: Pathogenetic and therapeutic implications,” (in eng), Front Immunol, vol. 13, p. 999108, 2022. [CrossRef]
- Y. Wang et al., “Hydrogen sulfide alleviates the anxiety-like and depressive-like behaviors of type 1 diabetic mice via inhibiting inflammation and ferroptosis,” (in eng), Life Sci, vol. 278, p. 119551, Aug 1 2021. [CrossRef]
- Bao, Y. Jin, J. Han, W. Wang, L. Qian, and W. Wu, “Berberine Regulates GPX4 to Inhibit Ferroptosis of Islet β Cells,” (in eng), Planta Med, vol. 89, no. 3, pp. 254-261, Mar 2023. [CrossRef]
- H. Feng et al., “Human umbilical cord mesenchymal stem cells ameliorate erectile dysfunction in rats with diabetes mellitus through the attenuation of ferroptosis,” (in eng), Stem Cell Res Ther, vol. 13, no. 1, p. 450, Sep 5 2022. [CrossRef]

Table 1.
The role of different signaling pathways on the disease.
| Pathway | Inhibitor | Experimental Methods |
Experimenta l Subject |
Experimental results |
|
| JAK/STAT | Tofacitinib | A nonlinear mixed-effects model |
Patients with ulcerative colitis (UC) |
Tofacitinib 5 mg b.i.d. has good clinical efficacy in UC patients, while 10 mg b.i.d has additional clinical benefits |
[34] |
| JAK/STAT | Tofacitinib | Randomized, double-blind, placebo-controlle d clinical trial |
Patients with ankylosing spondylitis (AS) |
Tofacitinib at a dose of 5mg and10mg twice daily will show superior efficacy over placebo at week 12, with a safety profile consistent with other indications |
[35] |
| JAK/STAT | Tofacitinib | Randomized, double-blind, placebo-controlle d clinical trial |
Patients with systemic lupus erythematos us (SLE) |
Improve SLE early-onset atherosclerosis related cardiac metabolism and immunological parameters |
[36] |
| JAK/STAT | Tofacitinib | Randomized, double-blind, placebo-controlle d clinical trial |
Patients with canine atopic dermatitis |
Good curative effects |
[37] |
| JAK/STAT | Tofacitinib | Randomized, double-blind, placebo-controlle d clinical trial |
Patients with psoriasis |
Both cow skin and shingles have good curative effect |
[38] |
| NF-κB | A small-m olecule NI K inhibitor |
Basic experiment | NZB/WF1 mice |
Good curative effects |
[55] |
| NF-κB | A small-m olecule NI K inhibitor |
Basic experiment | Pathological extracts from RA patients |
Good curative effects |
[56] |
| NF-κB | A small-m olecule NI K inhibitor |
Basic experiment | Pathological extracts from patients with endodermati tis |
Good curative effects |
[57] |
Table 2.
Effect of different ferroptosis treatment routes on the disease.
| Disease | Approach | Mechanism | Effect | Reference |
| RA | By inhibiting the occurrence of ferroptosis | Mice with induced arthritis were treated with ferroptosis inhibitor (LPX-1)) |
Effectively relieve joint swelling and synovial hyperplasia in mice, inhibit inflammation |
[74] |
| RA | By inhibiting the occurrence of ferroptosis |
Targeted activation of Nrf2 reduces ROS |
Effectively inhibit the proliferation and migration of FSL |
[80] |
| RA | By inhibiting the occurrence of ferroptosis |
FLS isolated from RA patients were treated with LPS and ferroptosis inducers and ferroptosis inhibitors, respectively |
Ferroptosis inhibitors can inhibit NCOA4-mediated iron phagocytosis to protect FLS |
[81] |
| RA | By inhibiting the occurrence of ferroptosis | Wasp venom (WV) accumulates ROS to induce GPX4-mediated ferroptosis |
Ferroptosis inducers are effective in RA treatment |
[77] |
| RA | By inducing the occurrence of ferroptosis |
For collagen-induced arthritis mice exhibiting a significant increase in fibroblast-like synoviocytes (FLS), the ferroptosis inducer IKE was administered. |
Ferroptosis inducer IKE can reduce inflammation and tissue damage by reducing the number of fibroblasts in mouse synovium |
[77] |
| RA | By inducing the occurrence of ferroptosis | Glycine was used in the CIA mouse model and the effect was evaluated |
Glycine promotes ferroptosis by increasing theconcentration of S-adenosylmethionine (SAM) to treat RA |
[79] |
| SLE | By inhibiting the occurrence of ferroptosis |
Erucic acid was used to suppress T cells in patients with SLE | Erucic acid regulates the immune response of pathogenic T cells and improves pregnancy response in SLE |
[90] |
| SLE | By inhibiting the occurrence of ferroptosis |
Erucic acid was used to suppress T cells in patients with SLE |
The ferroptosis inhibitor Liproxstatin-2 can reverse the serum induced ferroptosis in proximal renal tubular epithelial cells of LN patients and improve LN symptoms |
[81,93] |
| PsO | By inhibiting the occurrence of ferroptosis |
The ferroptosis inhibitor Fer-1 was administered |
Fer-1 inhibits lipid peroxidation to block the inflammatory response |
[106] |
| PsO | By inhibiting the occurrence of ferroptosis |
Fer-1 was applied to mice with IMQ-induced psoriasis-like dermatitis |
Fer-1 improved the increase of skin thickness and dyskeratosis in mice |
[104] |
| IBD | By inhibiting the occurrence of ferroptosis |
Use of CUR in a mouse model of colitis |
Significantly upregulated GPX4 expression and decreased UC ferroptosis |
[121] |
| IBD | By inhibiting the occurrence of ferroptosis |
The expression of Furin protease was measured in UC |
Significantly upregulated GPX4 expression and decreased UC ferroptosis |
[122] |
| IBD | By inhibiting the occurrence of ferroptosis |
Use of iron chelators (including maltol and kojic acid) in a male |
Effectively reduce inflammation index |
[123] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



