Submitted:
04 September 2025
Posted:
05 September 2025
You are already at the latest version
Abstract
Heart failure (HF) has become an emerging problem, especially in regions where life expectancy is increasing. Despite its prevalence, the mechanisms behind HF development are not well understood, which is reflected in the lack of curative therapies. Mitochondria, autophagy, and sirtuins form a crucial triad involved in HF pathogenesis, interconnected by oxidative stress. Identifying a common pathway involving these three components could be valuable in developing new treatment strategies. Since HF is the end result of several cardiovascular diseases, this review highlights the main HF precursors and explores the roles of mitochondrial quality control (mtQC), autophagy, and sirtuins in HF development. Dysfunctional mitochondria may play a key role by enhancing oxidative stress and influencing autophagy and sirtuins, both of which have antioxidant properties. The dual nature of autophagy—its pro-life and pro-death roles—may contribute to different outcomes in HF related to oxidative stress. As mtQC, autophagy, and sirtuins may interact, we present data on their mutual dependencies in HF. However, the specificity of these interactions remains unclear and warrants further research, which could lead to identifying new therapeutic targets. In conclusion, the interplay between mtQC, autophagy, and sirtuins may be important in HF pathogenesis and could be leveraged in developing HF treatments.
Keywords:
Heart failure
; sirtuins
; autophagy
; oxidative stress
; mitochondria
1. Introduction
Heart failure (HF) is an emerging medical and social problem worldwide, especially in regions where life expectancy has significantly increased in recent years [1]. Heart failure was identified as a new epidemic in 1997 [2]. Its prevalence is estimated at 64 million worldwide, with roughly one million new cases reported each year in the US [3]. The incidence and prevalence of HF differ among various populations [4]. For example, the incidence of HF in White populations has not risen since the 1960s. However, the increase in hospitalizations highlights improved survival rates after a HF diagnosis, leading to more individuals with HF needing repeated hospital stays [5,6].
Heart failure is the final stage of most heart syndromes. Therefore, known risk factors for heart diseases—including hypertension, diabetes, lack of physical activity, hyperlipidemia, and smoking—are associated with the development of HF [7]. Traditionally, HF is classified into HF with preserved left ventricular ejection fraction (HFpEF, diastolic HF) and HF with reduced ejection fraction (HFrEF, systolic HF). The former is much more challenging to manage than the latter, and it remains a topic of debate whether it should be regarded as a distinct entity within the EF spectrum [8]. Additionally, the term HF with mid-range EF was proposed by the American College of Cardiology, the American Heart Association, and the European Society of Cardiology [9]. Some other classifications of HF can be utilized (reviewed in [10]. Regardless of the ejection fraction, HF can progress from its chronic, compensated state—where symptoms are managed with treatment and the patient remains stable—to an acute, decompensated state, in which symptoms suddenly worsen and urgent medical care is often needed [11].
Although the exact mechanism of HF development is not fully understood, it involves a complex interplay of structural, functional, and molecular changes that gradually decrease the heart’s ability to pump blood effectively (reviewed in [10,12]). The most important aspects of HF pathogenesis include ventricular remodeling, hemodynamic overload, neurohormonal activation, inflammation, oxidative stress, metabolic disturbances, endothelial and microvascular dysfunction, along with some comorbidities such as hypertension, coronary artery disease, diabetes, and obesity. Therefore, HF pathogenesis involves multiple factors, as outlined in several recent reviews, e.g., [13]. However, this list of the most important aspects of HF pathogenesis suggests that molecular mechanisms may play a role in the overall development of HF in many ways.
According to recent global and regional guidelines, HF therapy should include a combination of pharmacological, non-pharmacological, and device-based strategies tailored to individual patient conditions and comorbidities. Guideline-Directed Medical Therapy (GDMT) for HFrEF involves angiotensin receptor-neprilysin inhibitors, beta-blockers, mineralocorticoid receptor antagonists, sodium-glucose cotransporter 2 (SGLT2) inhibitors (also used for HFpEF), and diuretics [14]. Besides SGLT2 inhibitors, medications for blood pressure control and managing comorbidities, especially atrial fibrillation, diabetes, and obesity, are used in HFrEF [15]. Non-pharmacological HF management involves lifestyle changes to prevent heart diseases and patient education for early detection of decompensation [16]. Device therapy for HF includes implantable cardioverter-defibrillators, cardiac resynchronization therapy, and remote monitoring devices [17]. Advanced therapies for HF include mechanical circulatory support, heart transplantation, and gene therapy [18,19]. However, all these therapies are either in early stages or not curative, despite being foundational. Worsening heart failure is a clear indication that new therapeutic targets and strategies are highly needed. The challenges in HF therapy mainly result from incomplete knowledge of the mechanisms of HF pathogenesis, which are driven by molecular events.
Oxidative stress is involved in the development of many diseases, but the direct cause-and-effect relationship between stress and disease, as well as the pathways through which oxidative stress impacts physiological processes, are often unclear. In many cases, mitochondria are closely linked to oxidative stress. Conversely, dysfunctional mitochondria are reported to occur in HF [20]. Sirtuins (SIRTs), a class of histone deacetylases located in the nucleus/nucleolus, mitochondria and cytosol, are involved in mitochondrial quality control and antioxidant defense. They are reported to play a role in HF pathogenesis [20]. The products of oxidative stress, reactive oxygen and nitrogen species (RONS), damage cellular components, and autophagy is a main mechanism responsible for removing these damaged and often toxic cellular components [21]. Therefore, similar to oxidative stress, autophagy is involved in the development of many diseases, including HF [22]. Therefore, mitochondria, sirtuins, and autophagy form a crucial triad in HF pathogenesis, and identifying pathways shared by these three components in HF may lead to the development of new therapeutic targets.
To take preventive action against HF, it is essential to recognize pre-HF states and prodromal stages of this disease, which may also be present in its preclinical stage. Several precursors to HF have been identified (Figure 1). However, not all these precursors are specific to HF, so further assessment is needed to determine HF risk. Additionally, since HF is often the endpoint of other cardiovascular diseases, a prior diagnosis of these diseases increases the specificity of HF precursors. Clinically, cardiac hypertrophy is the most commonly used precursor for HF.
In this narrative/hypothesis review, we highlight molecular mechanisms that may be involved in HF pathogenesis, with a particular emphasis on oxidative stress, mitochondrial homeostasis, and autophagy. Sirtuins are presented as important players in both mitochondria and autophagy. Common pathways among these three elements are identified and proposed as new potential targets for HF prevention and treatment.
2. Energy Metabolism, Oxidative Stress and Mitochondrial Dysfunction in Heart Failure
2.1. Energy Metabolism
Under normal conditions, the heart uses energy mainly generated from fatty acids oxidation (FAO) in mitochondria, while glucose constitutes 20-30% of energy substrates (Figure 2) [23].
Under normal conditions, glycolysis is of minor significance in energy production in the heart, but in certain circumstances, such as ischemia, physical overload, HF, or stress, it becomes more important [24]. Generally, the heart shows metabolic flexibility that allows it to adjust to fuel availability by changing its substrate preference [25]. It was suggested that HF can be linked to a metabolic shift in the heart from primarily using fatty acids to relying more on glucose for energy production [26]. This shift toward glycolysis in HF is the primary aspect of metabolic heart remodeling in HF and is attributed to dysfunctional mitochondria [27]. However, the relationship between fatty acid and glucose utilization in HF is reported with conflicting results, which is not surprising given the variety of HF clinical presentations. Recently, it has been noted that these conflicting results are mainly due to methodological differences, and direct measurements consistently show that FAO is active or increased in HFpEF hearts. In contrast, glucose oxidation becomes impaired [28]. Therefore, the relationship between fatty acids and glucose utilization, and between oxidative phosphorylation (OXPHOS) and glycolysis, may be crucially connected to HF occurrence and severity [29]. Importantly, ATP levels in a failing heart are maintained until the final stage, despite early signs of a mismatch between energy supply and demand [30,31]. Additionally, ketone bodies can be used as energy sources in both healthy and failing hearts [32]. In all these relationships, mitochondria are essential.
2.2. Oxidative Stress
Impaired mitochondria are directly linked to oxidative stress, a state where RONS production surpasses their neutralization. It may result from external factors, such as radiation and certain chemicals, or be induced internally due to disturbances in metabolic processes, especially those related to energy production. Overproduction of RONS can damage cellular molecules, including proteins, lipids, and nucleic acids. To defend against oxidative stress, cells have developed an antioxidant system with three main components: antioxidant enzymes, DNA repair proteins, and low-molecular-weight antioxidants, which collectively maintain RONS at a normal level. Dysfunction in any of these antioxidant elements may cause pathological events. Cells constantly produce RONS as byproducts of many physiological processes, and RONS production by mitochondrial complexes occurs during their normal operation. However, damage to the complexes that form the mitochondrial electron transport chain (ETC) can lead to RONS overproduction, potentially damaging ETC proteins and the genes that encode them. This can create a “vicious cycle” resulting in cell death [33]. During their normal operation, complexes I and III of the ETC leak electrons that can lead to mitochondrial superoxide production [34]. This superoxide is dismutated by superoxide dismutase 2 (SOD2) into another RONS, hydrogen peroxide, which is then neutralized by peroxiredoxin (PRX) and glutathione peroxidase (GPX) [35]. Damage caused by mitochondrial RONS is considered a key mechanism in HF development, as demonstrated in several studies on failing hearts in HF patients and animal models [20]. RONS scavenging in mitochondria was shown to be beneficial in HF animal models [36,37,38]. Reduced nicotinamide adenine dinucleotide (NADH) plays an important role in the functioning of the PRX and GPX systems in mitochondria, and its supply to this organelle requires two enzymes: isocitrate dehydrogenase 2 (IDH2) and nicotinamide nucleotide transhydrogenase (NNT), whose impairment has been shown to contribute to HF development [39]. However, increased NTN activation in the mouse failing heart under oxidative stress was also observed [40]. This effect might disconnect NADPH from ATP production pathways and hinder energy metabolism in the HF heart during oxidative stress.
A failing heart requires more energy, and stimulation of mitochondrial ETC is linked to increased oxidative stress due to more RONS leakage from overactive ETC complexes. However, because of increased protein acetylation, ATP synthase is inhibited in HF [41]. This results not only in decreased energy production but also in increased RONS generation due to impaired electron flow in the ETC. Animal studies showed that the mitochondrial antioxidant system, including SOD2, was compromised in HF [42]. Human studies show decreased SOD2 activity in HF or no changes [43,44]. Moreover, studies on myocardial tissue homogenates from the left ventricular wall of hearts with end-stage failure caused by dilated or ischemic cardiomyopathy showed increased levels of catalase, the primary enzyme responsible for breaking down hydrogen peroxide, at both the mRNA and protein levels [45].
In summary, several studies report the involvement of oxidative stress in HF pathogenesis, and much evidence suggests that stress may be both a cause and a consequence of the disease. However, the exact mechanisms behind this involvement remain unclear, as does its source in HF. Consistent reports indicate that impaired mitochondria may be a primary source of RONS in HF. Human and animal studies suggest that hyperacetylation of proteins essential for mitochondrial homeostasis may contribute to oxidative stress in HF. Additionally, Ca2+ overload and impaired removal of damaged mitochondria through mitophagy can increase RONS levels. Mitochondrial damage, if not repaired, may lead to more severe damage in a “vicious cycle” or “RONS-induced RONS release.” These issues are discussed in more detail in the next section.
2.3. Mitochondrial Dysfunction
Oxidative stress can be both a cause and a result of mitochondrial impairment. Therefore, maintaining mitochondrial homeostasis is essential for HF development. This is managed through mitochondrial quality control (mtQC), a comprehensive system of cellular processes that protect mitochondrial integrity, function, and adaptability based on physiological needs and stress [46]. This system maintains mitochondria’s effectiveness in energy production while reducing damage from reactive oxygen species and other stress factors. The main components of mtQC are mitochondrial biogenesis, mitochondrial dynamics (fusion and fission), and mitophagy. Additionally, the maintenance of mitochondrial DNA (mtDNA), mitochondrial proteostasis, and unfolded protein response also contribute to mtQC [47]. Several studies report mtQC impairments in HF.
Peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1A, PGC-1α) is the key regulator of mtQC [48]. PGC-1α is highly expressed in the heart, promoting fatty acid oxidation and mitochondrial respiration [49]. Although PGC-1α lacks intrinsic enzymatic activity and a DNA binding domain, it plays a crucial role in mitochondrial biogenesis through the interaction with transcription factors nuclear respiratory factor 1 and 2 (NRF1/NRF2) and mitochondrial transcription factor A (TFAM) [50]. Additionally, PGC-1α is crucial for mitochondrial dynamics and mitophagy.
Studies on HF patients and animal models of this disease have shown inconsistent results, with some showing decreases and others showing no effect (reviewed in [51]). However, a 2018 study showed that the level of PGC-1α after transverse aortic constriction (TAC), a primary animal model for cardiac hypertrophy and heart failure, fluctuated over 5-14 days during HF progression [51]. This effect may, at least in part, explain discrepancies between the results obtained in different studies and suggests that research on PGC-1a in HF should instead focus on its time dependence rather than “static” measurements. Animal studies showed that mice with a double PGC-1α knockout exhibited more pronounced cardiac dysfunction and a higher mortality rate under stress conditions, like TAC, than their normal counterparts [52,53]. Conversely, overexpression of PGC-1α promoted the development of HF [54,55].
PTEN-induced kinase 1 (PINK1) is an important factor in mtQC because it initiates mitophagy, a process for the selective removal of damaged mitochondria, and plays a key role in the development of cardiovascular disease [56]. It was observed that PINK1 was entirely localized to the mitochondria, and its levels decreased in the end stage of HF in humans [57]. PINK1−/− mice showed increased oxidative stress, fibrosis, cardiomyocyte apoptosis, mitochondrial dysfunction, and developed left ventricular failure, along with signs of cardiac hypertrophy by 2 months of age. These studies clearly demonstrate that PINK1 activity is essential for postnatal myocardial development, maintaining mitochondrial function, and redox balance in cardiomyocytes. Mitophagy was reported to be impaired in HF in several other studies (reviewed in [58]). This problem will be discussed in more detail later, focusing on the role of autophagy in HF.
The mitochondrial DNA is a double-stranded, short (16,569 base pairs), closed DNA molecule that occurs in several copies within the mitochondria. Although the proximity of the electron transport chain (ETC) makes mtDNA more exposed to external DNA-damaging agents than nuclear DNA, its maintenance is less efficient. The DNA damage response (DDR), the main pathway for DNA maintenance in mitochondria, is not fully understood but involves fewer subpathways and proteins than the nuclear DDR. A reduced number of mtDNA copies was observed in a mouse model of myocardial infarction and remodeling created by ligation of the left anterior descending coronary artery [59]. Also, there is a decrease in the transcripts of mtDNA-encoded genes, including subunits of complex I, complex III (cytochrome b), and 12S and 16S rRNA. These changes and related adverse phenotypic alterations are attributed to increased oxidative stress associated with HF as an end stage of myocardial infarction. N-glycosylase/DNA lyase (8-oxo-deoxyguanosine glycosylase, OGG1) is the primary mammalian enzyme responsible for repairing oxidative DNA damage. It has 8 isoforms, with 7 targeting mitochondria, and the OGG1-2a isoform being the most abundant. An increased migration of OGG1-2a to mitochondria was observed in the early stage of compensated cardiac hypertrophy induced by abdominal aortic constriction [60]. This helped maintain mtDNA integrity despite increased oxidative stress. Therefore, increased migration of OGG1-2a may be seen as a mechanism that supports cardiac function during the compensatory stage. A direct causal link between mtDNA damage and heart failure (HF) was shown in a study where transgenic mice with Tet-on inducible, cardiomyocyte-specific expression of a mutant uracil-DNA glycosylase 1 (mutUNG1) were created [61]. In addition to uracil, this mutated UNG1 variant also removes thymine from mtDNA, leading to transitional apyrimidinic sites that impair mtDNA functions. After inducing mutUNG1 in cardiac myocytes, the mice developed hypertrophic cardiomyopathy, which progressed to congestive heart failure and resulted in premature death within about two months. The affected hearts showed reduced mtDNA replication and transcription, suppressed mitochondrial respiration despite higher levels of PGC-1α, mitochondrial mass, and antioxidant enzymes, and impaired mitochondrial fission and fusion dynamics. These changes caused worsening myocardial contractility, serving as the mechanism behind heart failure.
Increased mitochondrial damage and impaired mtQC, leading to the release of mtDNA into circulation, may contribute to cardiac inflammation, as it can trigger activation of circulating immune cells [62]. As inducers of the NLRP3 inflammasome disrupted normal mitochondrial homeostasis, which decreased the levels of the coenzyme NAD+ and inactivated the NAD+-dependent SIRT2 [63]. This effect caused the buildup of acetylated α-tubulin, which helped dynein-dependent mitochondrial transport and the positioning of NLRP3 adaptor on mitochondria relative to NLRP3 on the endoplasmic reticulum. Since HF is linked with NAD+/NADH redox imbalance, overactivation of NLRP3 might be another factor, besides mtDNA release, that connects mitochondrial dysfunction with inflammation in HF.
The involvement of mitochondria in HF pathogenesis extends beyond mtQC in general and PGC-1α specifically. The acetylation of mitochondrial proteins, mainly at lysine residues and primarily facilitated by acyl coenzymes A (acyl-CoAs), is crucial not only for mtQC but also for enzyme activity, including SOD2, metabolic adaptation, and aging [64]. Recently, it was shown that the levels of sirtuin 3 (SIRT3), a key regulator that removes acetyl groups from lysine residues, and nicotinamide adenine dinucleotide (NAD+) decreased in HF in dogs, contributing to the state of hyperacetylation [65]. Hyperacetylation of mitochondrial proteins was also observed in other studies involving HF patients and animal models [66,67,68]. The mechanism behind the increased levels of acetylation of mitochondrial proteins in HF is not completely known, but the two main reasons for that may be increased levels of acyl-CoAs and/or increased activity of SIRT3, observed in failing heart [20]. The activity of each sirtuin depends on NAD+ accessibility, and decreased levels of NAD+ and reduced NAD+/NADH ratios have been observed in hearts with mitochondrial dysfunction [69]. The NAD+/NADH redox imbalance promotes activation of NACHT, LRR, and PYD domains-containing protein 3 (NLRP3) inflammasome, contributing to a vicious cycle and shifting mitochondria from energy producers to cell death initiators [20].
Another mitochondria-related hallmark of HF is the deregulation of Ca2+ homeostasis [70]. Impaired Ca2+ reuptake by the sarcoplasmic reticulum (SR) in a failing heart results in decreased cytosolic Ca2+ transients. Since mitochondria are located near the SR, it is believed that, under pathological conditions, the SR might act as a Ca2+ sink. This Ca2+ overload then leads to mitochondrial dysfunction [20]. As the Ca2+ ions decrease, PHPPP declines. Reduced mitophagy regulates many mitochondrial enzymes and other proteins, including those involved in oxidative defense, OXPHOS, cardiomyocyte death, and the mitochondrial permeability transition pore (mtPTP) opening, which are important in HF pathogenesis [71]. The mitochondrial Ca2+ uniporter (MCU) and mitochondrial Na+/Ca2+ exchanger (NCLX) are two main components involved in Ca2+ transport within mitochondria [72]. However, studies on the involvement of both transporters in HF pathogenesis have yielded inconsistent results, and overall, the mechanism of how Ca2+ overload causes damage in mitochondria remains unclear and requires further investigation (reviewed in [20]).
Mitochondria interact with other organelles such as the endoplasmic reticulum, lysosomes, ribosomes, lipid droplets, and the nucleus, and this interaction contributes to cardiac homeostasis. Its dysfunction may be involved in HF pathogenesis (reviewed in [73]).
In summary, mitochondrial dysfunction in HF heart, including defective mtQC, may lead to oxidative stress, increased mtDNA damage, NLRP-mediated inflammation, overload with Ca2+, disturbed NAD+/NADH redox homeostasis, impaired fusion/fission with the involvement of declined mitophagy (Figure 3). It should be stressed that the mutual dependence in the categories cause-effect between these effects related to dysfunctional mitochondria, oxidative stress and HF is not completely clear and requires further research.
Despite a clear connection between HF pathogenesis and mitochondrial dysfunction, there is no established therapeutic strategy targeting mitochondrial issues in HF, but Clinical.Trials.gov lists some ongoing or completed trials aiming to restore mitochondrial health, improve energy production, and reduce oxidative stress in cardiomyocytes in HF (https://clinicaltrials.gov/search?cond=Heart%20Failure&term=mitochondria&intr=treatment. Accessed August 30, 2025).
3. Sirtuins in Heart Failure
Sirtuins (silent information regulators, SIRTs) are a family of NAD+-dependent class III histone deacetylases and adenosine diphosphate ribosyl transferases, comprising seven members, SIRT1-7 in mammals [74]. They are localized in the nucleus, nucleolus, cytoplasm, and mitochondria, and most of them have dual localizations. They perform many functions, but their primary role is to regulate gene expression through the deacetylation of chromatin proteins and transcription factors (reviewed in [75]). Other functions are linked to their roles in antioxidant defense, DDR, autophagy, apoptosis, inflammation, metabolism, aging, and brain function, but these are mostly driven by their gene expression-regulating activities. Besides physiological roles, sirtuins are also involved in the development of many disorders, including cancer and neurodegenerative diseases [76,77]. However, in both of these syndromes and other pathologies, sirtuins can have both beneficial and adverse effects.
Some works cited in this section relate directly to syndromes that are precursors or associates of HF, including cardiac hypertrophy, ischemia-reperfusion damage, and fibrosis. It is justified that in most, if not all, cases, HF is linked to at least one of these symptoms.
Sirtuins play a key role in cardiac development mainly by regulating the expression of genes crucial for this process, and for example, SIRT1 deficiency has been linked to congenital heart defects [78]. Sirtuins are involved in heart development from the very beginning, as they participate in the differentiation of cardiac progenitor cells [79]. Additionally, heart morphogenesis involves sirtuins, which regulate key signaling pathways such as wingless-type MMTV integration site family (WNT), neurogenic locus notch homolog protein (NOTCH), and forkhead box protein O (FOXO) [80,81]. Sirtuins’ antioxidant properties enable them to protect the developing heart against oxidative stress [82]. Sirtuins regulate energy metabolism during heart development, especially by promoting mitochondrial biogenesis and fatty acid oxidation FAO), which are essential for the energy needs of the growing heart [83]. These properties of sirtuins will be discussed in more detail later, especially regarding mitochondrial functions and autophagy.
In general, SIRTs are seen to play a beneficial and important role in repressing HFpEF, but notable exceptions to this rule were observed [84]. Exercise training and caloric restriction have been shown to improve the quality of life in patients with HFpEF [85]. Two clinical trials demonstrating that a well-structured 4-week exercise training–caloric restriction program resulted in reductions in systolic blood pressure and increased EF suggest that these effects were partly due to increased NAD+ levels and enhanced SIRT1 activity [86,87]. Furthermore, an increase in antioxidant capacity was observed in one of these studies. SIRT1 was shown to protect against the harmful effects caused by cardiac remodeling in a mouse model of HFpEF by regulating lipid metabolism and inflammation [88].
There are issues with clearly classifying SIRT2 as either a protective or harmful factor for cardiac function [89]. Also in HF, the activity of SIRT2 was reported to have both beneficial and adverse effects. It was demonstrated that the hearts of the SIRT2-/- mice showed improved cardiac function after ischemia-reperfusion (I/R) and pressure overload (PO), suggesting that SIRT2 had maladaptive effects in the heart in response to stress [90]. Similar outcomes were observed in mice lacking SIRT2 specifically in cardiomyocytes. Further research showed that SIRT2 affects the levels and activity of NRF2 within cells, leading to a reduction in antioxidant enzyme expression. Deleting NRF2 from the hearts of SIRT2-/- mice eliminated their protection after PO. Additionally, giving a SIRT2 inhibitor to mouse hearts decreased cardiac size and lowered hypertrophy caused by PO. Therefore, SIRT2 seems to have harmful effects on the heart and influences its response to injury and the development of cardiac hypertrophy, which is a step toward HF. Conversely, SIRT2 was downregulated in hypertrophic hearts from mice [91]. SIRT2-/- mice showed cardiac hypertrophy and fibrosis, along with decreased cardiac ejection fraction and fractional shortening. Conversely, overexpressing SIRT2 specifically in the heart protected against hypertrophy and fibrosis caused by age and angiotensin II (ANGII), while also restoring cardiac function. Mechanistically, SIRT2 helps maintain AMP-activated protein kinase (AMPK) activity in hypertrophic hearts both in vivo, whether due to aging or ANGII, and in cardiomyocytes in vitro. The liver kinase B1 (LKB1), a key kinase upstream of AMPK, was identified as a direct target of SIRT2. Consequently, SIRT2 promotes AMPK activation by deacetylating LKB1. Loss of SIRT2 reduces AMPK activity, increasing susceptibility to aging-related and ANGII–induced cardiac hypertrophy. Elevated plasma SIRT2 levels positively correlate with heart failure (HF) occurrence in patients after an acute myocardial infarction [92]. A lower level of SIRT2 was observed during pathological heart hypertrophy in mice [93]. Animals lacking SIRT2 developed natural heart issues with hypertrophy, remodeling, fibrosis, and dysfunction as they aged. Young SIRT2-deficient mice also showed more severe hypertrophy when exposed to stimuli. Conversely, increasing SIRT2 levels decreased agonist-induced cardiac hypertrophy in cardiomyocytes through a cell-intrinsic process. Mechanistically, SIRT2 interacted with and deacetylated the NFATc2 transcription factor. The absence of SIRT2 stabilized NFATc2 and increased its presence in the nucleus, boosting its transcription activity. Therefore, inhibiting NFAT can restore heart functions in SIRT2-deficient mice.
The downregulation of SIRT3 was shown to promote fibrotic effects in a mouse model of HFpEF, linked to hyperacetylation of mitochondrial proteins, which leads to increased production of interleukins IL1B and IL18 and enhanced assembly of NLRP3 on hyperacetylated mitochondria [94]. Elevated β-hydroxybutyrate levels decreased NLPR3 formation and triggered proinflammatory cytokine-driven mitochondrial dysfunction and fibrosis. Additionally, β-hydroxybutyrate lowered the acetyl-CoA pool and mitochondrial acetylation, partly through activating citrate synthase and inhibiting fatty acid uptake. A disruption in mitochondrial fatty acid oxidation associated with hyperacetylation of key enzymes in this pathway was shown in a mouse HFpEF model [95]. Down-regulation of SIRT3 and a shortage of NAD+ contribute to the observed hyperacetylation of mitochondrial proteins. Reduced expression of genes involved in NAD+ synthesis was confirmed in cardiac tissue from HFpEF patients. HFpEF mice supplemented with nicotinamide riboside, a NAD+ precursor or an activator of NAD+ synthesis, showed improvements in mitochondrial functions and a reversal of the HFpEF phenotype. Therefore, HFpEF is linked to mitochondrial dysfunction in the myocardium and NAD+ deficiency, which may be related to SIRT3 downregulation. It was demonstrated that supplementation with nitrite and metformin, which activate the skeletal muscle SIRT3-5’-AMPK pathway, improved glucose uptake and metabolism in a mouse model of HFpEF [96]. In similar research, SIRT3 deficiency in skeletal muscle increased the secretion of lysyl oxidase homolog 2 and β2-microglobulin, contributing to pulmonary vascular remodeling in pulmonary hypertension caused by HFpEF [96]. A decrease in indole-3-propionic acid, which activates the nicotinamide N-methyl transferase (NNMT)-SIRT3 axis, was observed in HFpEF patients [97]. These studies were further confirmed by nicotinamide supplementation, NNMT knockdown and overexpression in a mouse model oh HFpEF.
SIRT4 is mainly located in mitochondria, but it can also be found in the nucleus, cytoplasm, centrosomes, and microtubules [98]. Although SIRT4 is highly expressed in the heart, its roles in cardiac development and disease are not fully understood. SIRT4 may affect cardiac processes such as ischemia-reperfusion and remodeling differently than other sirtuins [99]. It was shown that SIRT4 overexpression in mice with angiotensin II infusion promoted the progression from compensated to decompensated cardiac hypertrophy [100]. Recently, it was demonstrated that overexpressing SIRT4 specifically in cardiomyocytes accelerated heart failure development in response to pressure overload, primarily through a mitochondrial RONS-mediated increase in profibrotic transcriptional signaling [101]. These results suggest that SIRT4 could be a therapeutic target to inhibit in individuals with cardiac SIRT4 overexpression to prevent the development or progression of HF.
Fibrosis in HF was examined in a different study, this time involving SIRT5 [102]. In this study, a mouse strain overexpressing SIRT5 (SIRT5OE) was subjected to chronic pressure overload via TAC. Compared to controls, SIRT5OE mice were protected against the effects of TAC, including left ventricular dilation and reduced ejection fraction. Transcriptomic analysis showed that SIRT5 suppresses key heart failure (HF) consequences: the metabolic shift from fatty acid oxidation to glycolysis, immune activation, and fibrotic signaling pathways. Oxidative stress in mouse cardiomyocytes led to SIRT5 downregulation [103]. SIRT5 knockdown led to decreased cell viability and increased numbers of apoptotic cells and caspase 3/7 activity. A direct interaction between Bcl-2-like protein 1 (B2CL1) and SIRT5 was observed. Therefore, SIRT5 may negatively regulate oxidative stress-induced apoptosis in cardiomyocytes and could be considered in preventive and therapeutic strategies for oxidative stress-mediated cardiac injury, such as in HF. Ischemia-reperfusion injury (IRI) is often a precursor to HI. A cardioprotective role of mitochondrial SIRT5 in a mouse model of cardiac IR injury has been reported [104]. Downregulation of SIRT5 in IR-injured hearts was associated with increased protein lactylation and mitochondrial dysfunction. Conversely, elevating SIRT5 levels reduced mitochondrial damage and eased cardiac injury. SIRT5 interacts with the mitochondrial protein adenine nucleotide translocase 2 (ANT2), inhibiting its lactylation and strengthening its interaction with another mitochondrial protein, voltage-dependent anion-channel 1 (VDAC1). Lactylation-resistant ANT2 forms a more effective complex with VDAC1, improving cardiac function after injury. In conclusion, SIRT5 promotes the interaction between mitochondrial proteins ANT2 and VDAC1 to preserve mitochondrial homeostasis and support heart function in IRI.
Endothelial SIRT6 plays a role in transporting fatty acids across the endothelial barrier by suppressing peroxisome proliferator-activated receptor gamma (PPARγ, encoded by the PPARG gene) [105]. This mechanism was shown to be crucial for mitigating experimental HFpEF by restoring endothelial SIRT6 function, as evidenced by the deacetylation of histone H3K9 around the PPARG promoter. In similar research involving diabetic HF patients, a decrease in SIRT6 was observed in cardiac tissues [106]. Research also showed that restoring endothelial SIRT6 expression in a mouse model of HFpEF and diabetes—induced by a long-term high-fat diet combined with low-dose streptozocin—led to improvements in diastolic dysfunction and reduced cardiac lipid accumulation. SIRT6 decreased endothelial fatty acid uptake, which was linked to the suppression of endothelial PPARγ expression through SIRT6-dependent deacetylation of histone H3 near the PPARγ gene promoter. This indicates that decreased endothelial SIRT6 expression connects diabetes to HFpEF by influencing fatty acid transport across the endothelial barrier mediated by PPARγ. Additionally, SIRT6, telomerase reverse transcriptase (TERT), and telomere repeat binding factor 1 (TRF1) were downregulated in TAC mice [107]. Overexpression of SIRT6 increased TERT and TRF1 levels and improved survival in mice after TAC. Further analysis revealed that SIRT6 overexpression reduced TAC-induced heart dysfunction and cardiac inflammation, leading to less cardiac fibrosis and smaller infarcts.
SIRT7 has this unique feature among other SIRTs: it can be localized in the nucleolus [108]. Although there is no direct evidence of the specific role that this unique localization of SIRT7 may have in cardiac function, it cannot be ruled out, as it is linked to protein synthesis, stress responses, and chromatin dynamics [109]. An increased expression of SIRT7 was observed in cardiomyocyte-specific SIRT7-knockout mice subjected to pressure overload induced by TAC [110]. These animals also showed an increase in heart weight relative to tibial length and exhibited reduced cardiac contractile function compared to controls. A direct interaction between SIRT7 and the transcription factor GATA-4 was identified, and GATA4 knockdown lessened the severity of phenylephrine-induced cardiac hypertrophy. SIRT7 deacetylated GATA4 in cardiomyocytes, affecting its transcriptional activity. This suggests that SIRT7 deficiency in cardiomyocytes promotes hypertrophy under pressure overload, and SIRT7’s antihypertrophic effects are mediated through its interaction with and promotion of GATA4 deacetylation. In another study, SIRT7-deficient animals showed a reduction in lifespan and developed heart hypertrophy and inflammatory cardiomyopathy [111]. Hearts of SIRT7 mutants showed widespread fibrosis. The study also demonstrated that SIRT7 interacts with the cellular tumor antigen p53 and deacetylates it in vitro, resulting in hyperacetylation of p53 in vivo and an increased rate of apoptosis in the mutant mice’s myocardium. Primary cardiomyocytes lacking SIRT7 exhibited higher baseline apoptosis and were less resistant to oxidative and genotoxic stress, indicating that SIRT7 is crucial for regulating stress responses and cell death in the heart.
Major effects of SIRTs related to HF are summarized in Table 1. The selection of these effects presented in this section is subjective and not exhaustive. More information can be found elsewhere, e.g., [75,82,112]. In summary, SIRTs are important players in HF pathogenesis, and most studies suggest their beneficial potential and protective effects against HF. However, due to incomplete knowledge of the basic mechanisms of SIRTs homeostasis, further research is necessary to determine their potential in HF, especially regarding their mutual interactions.
4. Autophagy in Heart Failure
Autophagy is a process in which cells remove damaged, dysfunctional, or unnecessary material, as well as invaders and their remnants, through a central molecular pathway essential for maintaining cellular and organismal homeostasis [113]. Autophagy occurs as either degradative autophagy or secretory autophagy. In degradative autophagy, materials to be removed are broken down inside the cell and may then be recycled. In secretory autophagy, the cargo is exported out of the cell. Research on secretory autophagy is still in the early stages of understanding its basic mechanisms, and its involvement in HF has not been definitively confirmed [114]. Therefore, we will concentrate exclusively on degradative autophagy.
Degradative autophagy is categorized into three types: macroautophagy (commonly called autophagy), microautophagy, and chaperone-mediated autophagy (CMA) (Figure 4). The primary components of the autophagy machinery include sequestosome 1 (SQSTM1/p62), optineurin (OPTN), ubiquilin 2, nibrin 1, WD repeat and FYVE domain-containing 3, calcium-binding and coiled-coil domain 2, mechanistic target of rapamycin (MTOR), and huntingtin [113]. A key feature of degradative autophagy is the formation of autolysosomes through the fusion of autophagosomes with lysosomes, a process that involves SNARE proteins such as syntaxin 17, SNAP29, and vesicle-associated membrane proteins 7 and 8 [115]. This system directs cargo through specific receptors or adaptors that recognize degradation signals on cargo proteins and bind to LC3 and γ-aminobutyric acid receptor-associated protein on autophagosomes [116].
Autophagy is strongly linked to regulating cardiac structure and function across different physiological and pathological conditions, including heart failure and its precursors (reviewed in [117]). The role of autophagy in HF development is particularly significant because of the rising prevalence of HF and the decrease of autophagy with age [118]. However, our understanding of how impaired autophagic flux leads to cardiac dysfunction is still limited. Usually, the harmful effects of disrupted autophagy in the heart are seen as the buildup of damaged proteins and organelles that are toxic to cardiomyocytes, but it is unclear why and how this happens in HF.
Rapamycin is used to stimulate autophagy, while chloroquine suppresses it [119]. It was observed that ANGII-infused mice showed increased co-localization of LC3 puncta with vimentin [120]. Furthermore, treating cardiac fibroblasts with rapamycin increased type I collagen (COLI) and decreased fibronectin (FN) after ANGII stimulation. Similarly, inhibiting autophagy with chloroquine or knocking down ATG5 worsened ANGII-induced buildup of COLI and FN. Additionally, rapamycin improved cardiac fibrosis and dysfunction in mice infused with ANGII, while chloroquine worsened both fibrosis and dysfunction and also impaired heart function. This suggests that autophagy may play a protective role in reducing extracellular matrix buildup in the heart. In similar studies, activating the AMPK/MTOR/ULK1 pathway to stimulate autophagy decreased collagen deposits and myocardial fibrosis in mice [121].
Hypertrophic cardiomyocytes, typical of HF and its prodrome, accumulate misfolded proteins and damaged organelles [122]. These potentially toxic cellular debris are removed by the combined action of the ubiquitin-proteasome system and autophagy. Therefore, impaired autophagy may be both a cause and a consequence of cardiac hypertrophy. Several hypertrophy-related signaling pathways overlap with those involved in autophagy regulation [123]. The Ca2+/calcineurin signaling pathway was shown to activate the transcription of hypertrophy-related genes and inhibit autophagy in cardiomyocytes, involving AMPK [124,125]. MTOR, a critical autophagy protein, is, along with glycogen synthase kinase-3 (GSK3), a downstream target that underpins the involvement of the PI3K/AKT pathway in cardiac hypertrophy [126]. Cardiac autophagy and mitophagy might be controlled by mitogen-activated protein kinases (MAPKs), which also play a key role in hypertrophic signaling [127,128]. Therefore, several signaling pathways connect cardiac hypertrophy with autophagy, but more research is needed to establish a clear cause-and-effect relationship between these processes. Deletion of the ATG5 gene in the mouse heart led to cardiac hypertrophy and functional problems, which resulted in HF and ultimately animal death [129]. This confirms that autophagy may decrease during cardiac hypertrophy. In the final stage of HF, the myocardium becomes acutely decompensated, which is linked to the overproduction of misfolded proteins, damaged organelles, and other RONS-related products [130]. This may trigger an excessive autophagic response, potentially damaging normal proteins and organelles, which can lead to apoptosis and loss of cardiomyocytes [131]. Therefore, autophagy may have both beneficial and harmful effects in HF, reflecting its pro-life and pro-death properties [132].
A combined in vivo and in vitro study was conducted to examine the role of epigenetic modifications in cardiac fibrosis [133]. The involvement of miR-17-5p and BCL2/adenovirus E1B 19 kDa protein-interacting protein 3 (BNIP3) in modulating mitophagy and alleviating pathological cardiac fibrosis was studied in the TAC mice model of myocardial fibrosis and in cardiac fibroblasts treated with ANGII. Lower levels of myocardial miR-17-5p were associated with decreased left ventricular systolic function and increased collagen buildup in heart tissue. In vitro, treatment with angiotensin II led to reduced expression of miR-17-5p, increased BNIP3 levels, and excessive mitophagy, as shown by higher RONS levels, decreased ATP production, and increased markers of fibrosis. Additionally, overexpression of miR-17-5p directly bound to the 3’-untranslated region of BNIP3, significantly reducing its expression, which helped restore mitochondrial balance and decrease collagen synthesis. Conversely, overexpressing BNIP3 negated the anti-fibrotic and mitochondrial-protective effects of miR-17-5p. Therefore, the miR-17-5p/BNIP3 signaling pathway regulates mitophagy in cardiac fibroblasts and is essential for fibrotic remodeling. As a result, it may serve as a target for therapeutic strategies aimed at reducing cardiac fibrosis and delaying heart failure progression. In another epigenetic-related study, it was observed that the miRNA-212/132 family influenced cardiac hypertrophy and autophagy in cardiomyocytes [134]. MiR-212/132 null mice were resistant to pressure-overload-induced heart failure (HF), while cardiomyocyte-specific overexpression of the miR-212/132 family caused pathological cardiac hypertrophy, HF, and death. Both miR-212 and miR-132 directly suppress the anti-hypertrophic and pro-autophagic transcription factor FOXO3. Overexpressing these miRNAs led to excessive activation of the pro-hypertrophic calcineurin/NFAT signaling pathway and impaired autophagy during starvation. Consequently, inhibiting miR-132 with antagomir injections could potentially restore normal cardiac hypertrophy and reduce HF, offering a promising therapeutic strategy for this condition.
The importance of the FOXO3-BNIP3 pathway in mitochondria-related heart failure development was confirmed in another study showing a FOXO3-driven increase of BNIP3 in normal and phenylephrine (PE)-stressed cardiomyocytes from a rat HFpEF model [135]. This effect was linked to increased mitochondrial Ca2+, which led to reduced mitochondrial membrane potential, mitochondrial fragmentation, and cell death. A cardiotropic adeno-associated virus serotype 9 encoding dominant-negative FOXO3 (AAV9.dn-FX3) was used for gene delivery in the HFpEF model. While dn-FX3 lessened the rise in BNIP3 levels and its effects in PE-stressed ACM, delivering AAV9.dn-FX3a in an experimental HFpEF model decreased BNIP3 expression. This treatment reversed harmful left ventricular remodeling and improved both systolic and especially diastolic function, with additional benefits in mitochondrial structure and activity. Furthermore, FOXO3a promotes the expression of harmful genes related to mitochondrial apoptosis, autophagy, and cardiac atrophy. Therefore, FOXO3 may connect HF, mitochondria, and autophagy.
Oxidative stress triggers autophagy, and ANGII increases cardiac mitochondrial RONS production, leading to a decline in mitochondrial membrane potential in cardiomyocytes and resulting in greater oxidative damage to cardiac mitochondrial proteins and mtDNA deletions [136]. These harmful effects of ANGII on mitochondria were associated with an increase in the number of autophagosomes and amplified mitochondrial biogenesis, which may help replace damaged mitochondria and restore energy production. This study also highlighted the key role of RONS produced by mitochondria, as shown by experiments with mice overexpressing catalase targeted to mitochondria, compared to those overexpressing peroxisomal-targeted catalase. The mice with mitochondrial-targeted catalase, but not those with peroxisomal-targeted catalase, were resistant to cardiac hypertrophy, fibrosis, mitochondrial damage, and heart failure.
Ventricular remodeling plays a key role in the development and progression of HF. [137]. It is connected to oxidative stress, ER stress, and a weakened ubiquitin-proteasome system, making autophagy a vital process for reducing the effects of remodeling. In ventricular remodeling, autophagy can be both helpful and harmful, acting as a protective mechanism in early stages but potentially leading to cell death and heart failure if it becomes uncontrolled [138].
Impaired autophagy in mice caused by knocking ATG3, a key autophagy initiator, led to NAD+ deficiency in the heart due to increased NAD+ clearance [139]. This effect was caused by NNMT induction resulting from the activation of the SQSTM1-nuclear factor kappa-light-chain-enhancer of activated B cells (NFKB) signaling pathway.
Inflammation, a key aspect of HF pathogenesis, may influence autophagy and vice versa [140]. Several studies suggest that the interaction between autophagy and inflammation may be important in HF development [141]. Additionally, in this context, autophagy has two sides, as it can either promote or suppress NLRP3 activation [142,143]. Solute carrier family 26 member 4 (SLC26A4) might serve as an intermediate between inflammation, autophagy, and cardiac hypertrophy [144]. It was shown that SLC26A4 activated autophagy and NLRP3 inflammasome in a phenylephrine-induced cardiomyocyte hypertrophy in vitro model [143]. Furthermore, SLC26A4 activated NLRP3 inflammasome in vivo in TAC rats. Therefore, targeting the expression of SLC26A4 to regulate the activation of autophagy and the NLRP3 inflammasome pathway may be a perspective therapeutic target for cardiac hypertrophy and HF. In another study, it was demonstrated that mtDNA that escapes from autophagy resulted in toll-like receptor 9 (TLR9)-mediated inflammatory responses in cardiomyocytes and induced myocarditis and dilated cardiomyopathy [145]. Therefore, impaired mitophagy could be a molecular mechanism responsible for chronic inflammation in failing hearts.
Several substances, including those used in traditional Chinese medicine, were tested as potential therapies targeting autophagy in HF, yielding promising results (reviewed in [22]). However, ClinicalTrials.gov lists only two clinical trials related to autophagy/mitophagy in HF, with one completed and the other in the recruiting phase, but neither directly aims to target autophagy in HF (https://clinicaltrials.gov/search?cond=heart%20failure&term=Mitophagy, accessed August 21, 2025).
In summary, many studies suggest the beneficial effect of effective autophagy in HF and its precursors, starting with cardiac hypertrophy. However, overactive autophagy may be harmful to heart health. It is important to recognize that autophagy levels—normal, underactive, or overactive—can change depending on the stage of HF, especially during the acute decompensated phase, where high autophagy activity may cause damage to normal cellular components. Therefore, whether autophagy is a friend or foe in HF depends on the cellular context, similar to other syndromes.
5. Interplay between Mitochondrial Quality Control, Sirtuins, and Autophagy in Heart Failure
The common factor among mtQC, SIRTs, autophagy, and HF is oxidative stress. It usually results from impaired mitochondria caused by faulty mtQC and is linked to excessive production of RONS, which damage cellular components that are targeted for autophagy. Sirtuins, due to their antioxidant properties, can reduce oxidative stress. However, SIRTs, especially SIRTs3-5 located in the mitochondrial matrix, regulate mitochondrial health independently of oxidative stress. Additionally, SIRTs play a role in autophagy regulation. Mitochondrial dysfunction, which can be influenced by SIRTs and autophagy, leads to an energy deficit in the heart, contributing to HF and its precursor syndromes.
As mentioned, impaired autophagy in mice caused by knocking out ATG3, a key initiator of autophagy, led to NAD+ deficiency in hearts due to increased NAD+ clearance [117]. Although the status of sirtuins was not examined in that study, it can be speculated that they might be impaired due to the lack of their cofactor, highlighting the importance of sirtuin-autophagy interaction for cardiac health.
Acute cardiovascular injury, such as that seen in myocardial infarction, can lead to acute HF [146]. An increased expression of SIRT7 was observed in mice in response to injury caused by myocardial infarction, especially at the active wound healing site [147]. SIRT7-deficient mice showed increased susceptibility to cardiac rupture after myocardial infarction and experienced delayed wound healing after skin injury. They also demonstrated less fibrosis, reduced fibroblast differentiation, and fewer inflammatory cells in the infarct’s border zone. In laboratory experiments, cardiac fibroblasts lacking SIRT7 expressed lower levels of fibrosis-related genes and had decreased transforming growth factor receptor I (TGFBR1) protein. The absence of SIRT7 induced autophagy in cardiac fibroblasts, and inhibiting autophagy prevented the reduction of TGFBR1. This indicates that SIRT7 may play a vital role in tissue repair by interacting with autophagy and TGFBR1.
As cited in earlier sections, myocardial IRI is directly connected to HF [148]. Briefly, Ca2+ overload, peroxidation, and inflammation are key factors in the development of myocardial IRI. The interaction among these factors leads to the activation of cardiomyocyte cell death pathways, including autophagy [148]. Therefore, targeting autophagy and other forms of programmed cell death may be an effective strategy to reduce IRI and, consequently, HF. Chloramphenicol succinate, an autophagy stimulant, administered to pigs that experienced coronary artery occlusion and reperfusion, resulted in a significant decrease in infarct size and increased expression of LC3 and another autophagy-related protein, BCN1, which also plays a role in regulating apoptosis [149,150]. Autophagy in cardiomyocytes is linked to SIRT1 through various pathways, including those involving ATG, LC3, and FOXOs (reviewed in [151,152]). Recent research has confirmed that SIRT1-mediated autophagy plays unique roles at various stages of myocardial I/R injury (reviewed in [151]). Therefore, targeting the mechanism of SIRT1-mediated autophagy with adjustments for a specific phase of IRI may be a therapeutic strategy for preventing HF development.
Hypoxic stress directly contributes to HF by causing myocardial damage and other effects related to HF, involving several signaling pathways, including HIF- and Na+/K+ ATPase [153]. Heart tissue samples from patients with cyanotic congenital heart disease showed increased autophagy, apoptosis, and higher SIRT1 levels compared to the noncyanotic control samples [152]. SIRT1 promoted autophagic flux and reduced apoptosis in hypoxic H9C2 cells. Additionally, SIRT1 stimulated AMPK, while the AMPK inhibitor Compound C blocked the activation of autophagy by SIRT1. SIRT1 protected hypoxic cardiomyocytes from apoptosis, involving inositol-requiring kinase enzyme 1α (IRE1α). The SIRT1 activator SRT1720 enhanced AMPK activity, suppressed IRE1α, increased autophagy, and decreased apoptosis in the heart tissues of normoxic mice compared to the hypoxic control group. Hypoxic mice treated with the SIRT1 inhibitor EX-527 showed opposite effects. Therefore, SIRT1 may promote autophagy through AMPK activation and reduce hypoxia-induced apoptosis via the IRE1α pathway, helping to protect cardiomyocytes from hypoxic stress and prevent HF development. This experiment exemplifies the regulation of cardiovascular autophagy by sirtuins. Initially identified for SIRT1, sirtuins can directly trigger autophagy by deacetylating autophagy-related genes or enhance autophagic flux by increasing the expression of genes that regulate autophagy [154].
Mitochondria may be linked to autophagy in HF through interactions with lysosomes, including the regulation of mitochondrial fission, modulation of lysosomal transport, and mediation of calcium signaling (reviewed in [73]).
Besides dysfunctional mtQC, protein quality control may also contribute to HF. It was demonstrated that impaired autophagic flux decreased the availability of NAD+, a cofactor of sirtuins, in cardiomyocytes [139]. NAD+ deficiency caused by impaired autophagy results from the induction of nicotinamide N-methyltransferase (NNMT), which methylates the NAD+ precursor nicotinamide (NAM) to produce N-methyl-nicotinamide (MeNAM). Administering nicotinamide mononucleotide (NMN) or inhibiting NNMT activity in autophagy-deficient hearts and cardiomyocytes restores NAD+ levels and enhances cardiac and mitochondrial function. Mechanistically, autophagic inhibition leads to the accumulation of SQSTM1, which activates NF-κB signaling and increases NNMT transcription. Therefore, this work suggests a novel mechanism showing how autophagic flux supports mitochondrial and cardiac health by mediating SQSTM1-NF-κB-NNMT signaling and regulating cellular NAD+ levels, resulting in changes in SIRTs activity.
In summary, the interaction between mtQC, autophagy, and sirtuins may contribute to HF development through many pathways. Dysfunctional mitochondria might be the key element in this interaction, as they are linked to increased oxidative stress, which can trigger autophagy to remove damaged parts of cardiocytes and activate SIRTs to reduce the stress. Many compounds can influence this process, but primarily, they are canonical proteins of mtQC, autophagy, and SIRTs. Further research is needed to clarify the details of their specific interactions in HF, which could help develop targeted therapeutic strategies based on this interaction.
6. Conclusions and Perspectives
Heart failure is the end stage of many cardiovascular diseases. Therefore, its mechanism of development shares aspects with these diseases, but it also has features that distinguish it from other heart syndromes. The common factor for most, if not all, of these aspects is oxidative stress, which is primarily related to dysfunctional mitochondria. Such mitochondria may be characterized by impaired mitophagy, which, in turn, reflects a compromised autophagic response of cardiomyocytes. Sirtuins are crucial regulators of cellular and organism-wide responses to oxidative stress. Consequently, oxidative stress, mitochondria, autophagy, and sirtuins may interact in the development of heart failure.
Currently, the literature broadly recognizes the protective role of autophagy in HF [155]. Autophagy removes damaged mitochondria that can increase oxidative stress through a vicious cycle, thereby reducing oxidative stress. This reduction is also aided by the antioxidative actions of SIRTs. Additionally, autophagy may lower the inflammatory response by degrading proinflammatory components, regulating inflammasomes, clearing pathogens and damage-associated molecular patterns, and modulating cytokine secretion [156]. Because of the important role of SIRTs in autophagy regulation, they might be involved in all these functions, providing a key element for HF mitigation through autophagy. Oxidative stress and inflammatory responses are critical aspects of ventricular remodeling, serving as hallmarks of HF. Conversely, excessive autophagy can accelerate this process, and further research should explore the involvement of SIRTs in this aspect of autophagy in HF development. Cardiomyocyte apoptosis, common in HF, may be driven by excessive autophagy and dysfunctional mitochondria, but SIRT3 controls mitochondrial outer membrane permeabilization, a vital step in the mitochondrial pathway of apoptosis [157]. Furthermore, SIRT1 was reported to have an anti-apoptotic role in hypoxic stress, which is characteristic of a significant portion of HF cases [158]. Therefore, further research into the mutual interaction and regulation of mitochondria, autophagy, and SIRTs is needed to develop potential therapeutic strategies for targeting HF.
The evolving nature of autophagy may differently affect HF and its precursors depending on the HF stage. Another consideration is the classification of HF into HFpEF and HFrEF. All these factors should be taken into account when planning HF treatment tailored to the molecular characteristics of the disease.
Although the involvement of mitochondrial dysfunctions in HF pathogenesis is documented, the relationship between RONS-induced mtDNA damage and its repair remains unclear based on current results, as the failing heart may compensate for oxidative stress effects by mobilizing DNA repair proteins to mitochondria [61]. Therefore, more research is necessary on DNA damage and repair in cardiomyocytes.
It is not surprising that sirtuins may interact with mtQC and autophagy, and this was not the purpose of this work to demonstrate that such an interaction occurs in HF. Instead, we aimed to show the mechanism underlying that interaction based on the roles of these three elements in HF pathogenesis.
As mentioned in the previous section, targeting SIRT1-related autophagy specifically during the phases of IRI might serve as a preventive and therapeutic strategy in HF. To achieve this, small-molecule drugs and miRNA regulators can be developed. Resveratrol, sevoflurane, quercetin, and melatonin could be considered during the ischemic stage, while coptisine, curcumin, berberine, and miRNA agomirs or antagomirs involved in regulating the SIRT1-autophagy axis may provide cardioprotective effects during reperfusion (reviewed in [151]).
In summary, mtQC, autophagy, and SIRTs may have significant potential in the development of HF and its precursors. However, further research on the molecular mechanisms underlying this potential is necessary to translate these mostly observational associations into effective treatments and prevention strategies for heart failure. The important role of sirtuins in HF development is suggested by several reports, but as they are relatively recently discovered molecules, more research is needed to fully understand their properties. Nevertheless, their role in regulating oxidative stress, mitochondria, and autophagy is well established, justifying their consideration in the context of heart failure pathogenesis and therapy.
Author Contributions
Conceptualization, J.K., and J.B.; writing—original draft preparation, J.K., J.D., M.D., E.P., and J.B.; writing—review and editing, J.B.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
None.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Savarese, G.; Becher, P.M.; Lund, L.H.; Seferovic, P.; Rosano, G.M.C.; Coats, A.J.S. Global burden of heart failure: a comprehensive and updated review of epidemiology. Cardiovasc Res 2023, 118, 3272–3287. [Google Scholar] [CrossRef]
- Braunwald, E. Shattuck lecture--cardiovascular medicine at the turn of the millennium: triumphs, concerns, and opportunities. The New England journal of medicine 1997, 337, 1360–1369. [Google Scholar] [CrossRef]
- Martin, S.S.; Aday, A.W.; Allen, N.B.; Almarzooq, Z.I.; Anderson, C.A.M.; Arora, P.; Avery, C.L.; Baker-Smith, C.M.; Bansal, N.; Beaton, A.Z.; et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association. Circulation 2025, 151, e41–e660. [Google Scholar] [CrossRef]
- Bozkurt, B.; Ahmad, T.; Alexander, K.; Baker, W.L.; Bosak, K.; Breathett, K.; Carter, S.; Drazner, M.H.; Dunlay, S.M.; Fonarow, G.C.; et al. HF STATS 2024: Heart Failure Epidemiology and Outcomes Statistics An Updated 2024 Report from the Heart Failure Society of America. Journal of Cardiac Failure 2025, 31, 66–116. [Google Scholar] [CrossRef]
- Levy, D.; Kenchaiah, S.; Larson, M.G.; Benjamin, E.J.; Kupka, M.J.; Ho, K.K.; Murabito, J.M.; Vasan, R.S. Long-term trends in the incidence of and survival with heart failure. The New England journal of medicine 2002, 347, 1397–1402. [Google Scholar] [CrossRef] [PubMed]
- Roger, V.L.; Weston, S.A.; Redfield, M.M.; Hellermann-Homan, J.P.; Killian, J.; Yawn, B.P.; Jacobsen, S.J. Trends in heart failure incidence and survival in a community-based population. Jama 2004, 292, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.A.; Zaccardi, F.; Squire, I.; Okhai, H.; Davies, M.; Huang, W.; Mamas, M.; Lam, C.S.P.; Khunti, K.; Kadam, U.T. Risk Factors for Heart Failure. Circulation: Heart Failure 2020, 13, e006472. [Google Scholar] [CrossRef] [PubMed]
- Francis, G.S.; Cogswell, R.; Thenappan, T. The heterogeneity of heart failure: will enhanced phenotyping be necessary for future clinical trial success? J Am Coll Cardiol 2014, 64, 1775–1776. [Google Scholar] [CrossRef]
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.; Coats, A.J.; Falk, V.; González-Juanatey, J.R.; Harjola, V.P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2016, 18, 891–975. [Google Scholar] [CrossRef]
- Schwinger, R.H.G. Pathophysiology of heart failure. Cardiovasc Diagn Ther 2021, 11, 263–276. [Google Scholar] [CrossRef]
- Zile, M.R.; Bennett, T.D.; St. John Sutton, M.; Cho, Y.K.; Adamson, P.B.; Aaron, M.F.; Aranda, J.M.; Abraham, W.T.; Smart, F.W.; Stevenson, L.W.; et al. Transition From Chronic Compensated to Acute Decompensated Heart Failure. Circulation 2008, 118, 1433–1441. [Google Scholar] [CrossRef]
- Mann, D.L.; Felker, G.M. Mechanisms and Models in Heart Failure. Circulation Research 2021, 128, 1435–1450. [Google Scholar] [CrossRef] [PubMed]
- Roman-Pepine, D.; Serban, A.M.; Capras, R.-D.; Cismaru, C.M.; Filip, A.G. A Comprehensive Review: Unraveling the Role of Inflammation in the Etiology of Heart Failure. Heart Failure Reviews 2025, 30, 931–954. [Google Scholar] [CrossRef]
- Patel, J.; Rassekh, N.; Fonarow, G.C.; Deedwania, P.; Sheikh, F.H.; Ahmed, A.; Lam, P.H. Guideline-Directed Medical Therapy for the Treatment of Heart Failure with Reduced Ejection Fraction. Drugs 2023, 83, 747–759. [Google Scholar] [CrossRef]
- Lund, L.H.; Crespo-Leiro, M.G.; Laroche, C.; Zaliaduonyte, D.; Saad, A.M.; Fonseca, C.; Čelutkienė, J.; Zdravkovic, M.; Bielecka-Dabrowa, A.M.; Agostoni, P.; et al. Heart failure in Europe: Guideline-directed medical therapy use and decision making in chronic and acute, pre-existing and de novo, heart failure with reduced, mildly reduced, and preserved ejection fraction - the ESC EORP Heart Failure III Registry. Eur J Heart Fail 2024, 26, 2487–2501. [Google Scholar] [CrossRef]
- Jaarsma, T.; Hill, L.; Bayes-Genis, A.; La Rocca, H.B.; Castiello, T.; Čelutkienė, J.; Marques-Sule, E.; Plymen, C.M.; Piper, S.E.; Riegel, B.; et al. Self-care of heart failure patients: practical management recommendations from the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2021, 23, 157–174. [Google Scholar] [CrossRef]
- Mullens, W.; Dauw, J.; Gustafsson, F.; Mebazaa, A.; Steffel, J.; Witte, K.K.; Delgado, V.; Linde, C.; Vernooy, K.; Anker, S.D.; et al. Integration of implantable device therapy in patients with heart failure. A clinical consensus statement from the Heart Failure Association (HFA) and European Heart Rhythm Association (EHRA) of the European Society of Cardiology (ESC). European Journal of Heart Failure 2024, 26, 483–501. [Google Scholar] [CrossRef]
- Argiro, A.; Bui, Q.; Hong, K.N.; Ammirati, E.; Olivotto, I.; Adler, E. Applications of Gene Therapy in Cardiomyopathies. JACC: Heart Failure 2024, 12, 248–260. [Google Scholar] [CrossRef] [PubMed]
- Roussoulières, A.; Farrero, M.; Gustafsson, F.; Kittleson, M.; Munagala, M.; Stehlik, J. Heart Transplant and Durable Mechanical Circulatory Support for Specific Less-Common Cardiomyopathies. CJC Open 2025, 7, 813–820. [Google Scholar] [CrossRef]
- Zhou, B.; Tian, R. Mitochondrial dysfunction in pathophysiology of heart failure. The Journal of clinical investigation 2018, 128, 3716–3726. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J. Autophagy: from phenomenology to molecular understanding in less than a decade. Nature reviews. Molecular cell biology 2007, 8, 931–937. [Google Scholar] [CrossRef]
- Li, L.; Xi, R.; Gao, B.; Zeng, Y.; Ma, Q.; Gong, T.; Wang, J. Research progress of autophagy in heart failure. American journal of translational research 2024, 16, 1991–2000. [Google Scholar] [CrossRef]
- Ventura-Clapier, R.; Garnier, A.; Veksler, V. Energy metabolism in heart failure. J Physiol 2004, 555, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zou, Y.; Song, C.; Cao, K.; Cai, K.; Wu, Y.; Zhang, Z.; Geng, D.; Sun, W.; Ouyang, N.; et al. The role of glycolytic metabolic pathways in cardiovascular disease and potential therapeutic approaches. Basic Res Cardiol 2023, 118, 48. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz, S.C.; Purohit, S.; Tian, R. Cardiac Metabolism and its Interactions With Contraction, Growth, and Survival of Cardiomyocytes. Circulation Research 2013, 113, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ Res 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Chen, C.; Wang, J.; Zhu, X.; Hu, J.; Liu, C.; Liu, L. Energy metabolism and redox balance: How phytochemicals influence heart failure treatment. Biomedicine & Pharmacotherapy 2024, 171, 116136. [Google Scholar] [CrossRef]
- Shehadeh, L.A.; Robleto, E.; Lopaschuk, G.D. Cardiac energy substrate utilization in heart failure with preserved ejection fraction: reconciling conflicting evidence on fatty acid and glucose metabolism. Am J Physiol Heart Circ Physiol 2025, 328, H1267–H1295. [Google Scholar] [CrossRef]
- Carvalho, R.A. Chapter 12 - The glycolytic pathway to heart failure. In Glycolysis; Ferreira, R., Oliveira, P.F., Nogueira-Ferreira, R., Eds.; Academic Press, 2024; pp. 235–266. [Google Scholar]
- Smith, C.S.; Bottomley, P.A.; Schulman, S.P.; Gerstenblith, G.; Weiss, R.G. Altered Creatine Kinase Adenosine Triphosphate Kinetics in Failing Hypertrophied Human Myocardium. Circulation 2006, 114, 1151–1158. [Google Scholar] [CrossRef]
- Weiss, R.G.; Gerstenblith, G.; Bottomley, P.A. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proceedings of the National Academy of Sciences of the United States of America 2005, 102, 808–813. [Google Scholar] [CrossRef]
- Matsuura, T.R.; Puchalska, P.; Crawford, P.A.; Kelly, D.P. Ketones and the Heart: Metabolic Principles and Therapeutic Implications. Circ Res 2023, 132, 882–898. [Google Scholar] [CrossRef] [PubMed]
- Sb, H.; X, J.; Qh, Y.; Xr, Z.; Bb, Z.; Kh, W.; Xy, S.; Yt, C.; Xr, R.; Jf, M.; et al. The vicious circle between mitochondrial oxidative stress and dynamic abnormality mediates triethylene glycol dimethacrylate-induced preodontoblast apoptosis. Free Radical Biology and Medicine 2019, 134, 644–656. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z.B. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). International journal of molecular medicine 2019, 44, 3–15. [Google Scholar] [CrossRef]
- Hewitt, O.H.; Degnan, S.M. Antioxidant enzymes that target hydrogen peroxide are conserved across the animal kingdom, from sponges to mammals. Scientific reports 2023, 13, 2510. [Google Scholar] [CrossRef]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Experimental & molecular medicine 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Schwemmlein, J.; Maack, C.; Bertero, E. Mitochondria as Therapeutic Targets in Heart Failure. Current Heart Failure Reports 2022, 19, 27–37. [Google Scholar] [CrossRef]
- Teixeira, R.B.; Albro, J.H.; Sabra, M.; Abedin, T.; Tucker, A.N.; Sidharth, R.; Sellke, F.W.; Wipf, P.; Abid, M.R. Mitochondria-targeted ROS scavenger JP4-039 improves cardiac function in a post-myocardial infarction animal model and induces angiogenesis in vitro. PloS one 2025, 20, e0320703. [Google Scholar] [CrossRef]
- Ku, H.J.; Ahn, Y.; Lee, J.H.; Park, K.M.; Park, J.W. IDH2 deficiency promotes mitochondrial dysfunction and cardiac hypertrophy in mice. Free Radic Biol Med 2015, 80, 84–92. [Google Scholar] [CrossRef]
- Nickel, A.G.; von Hardenberg, A.; Hohl, M.; Löffler, Joachim R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metabolism 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Zhang, X.; Ji, R.; Liao, X.; Castillero, E.; Kennel, P.J.; Brunjes, D.L.; Franz, M.; Möbius-Winkler, S.; Drosatos, K.; George, I.; et al. MicroRNA-195 Regulates Metabolism in Failing Myocardium Via Alterations in Sirtuin 3 Expression and Mitochondrial Protein Acetylation. Circulation 2018, 137, 2052–2067. [Google Scholar] [CrossRef] [PubMed]
- Peoples, J.N.; Saraf, A.; Ghazal, N.; Pham, T.T.; Kwong, J.Q. Mitochondrial dysfunction and oxidative stress in heart disease. Experimental & molecular medicine 2019, 51, 1–13. [Google Scholar] [CrossRef]
- Borchi, E.; Bargelli, V.; Stillitano, F.; Giordano, C.; Sebastiani, M.; Nassi, P.A.; d’Amati, G.; Cerbai, E.; Nediani, C. Enhanced ROS production by NADPH oxidase is correlated to changes in antioxidant enzyme activity in human heart failure. Biochimica et biophysica acta 2010, 1802, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Sam, F.; Kerstetter, D.L.; Pimental, D.R.; Mulukutla, S.; Tabaee, A.; Bristow, M.R.; Colucci, W.S.; Sawyer, D.B. Increased Reactive Oxygen Species Production and Functional Alterations in Antioxidant Enzymes in Human Failing Myocardium. Journal of Cardiac Failure 2005, 11, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, S.; Bieligk, U.; Beulich, K.; Hasenfuss, G.; Prestle, J. Gene Expression of Antioxidative Enzymes in the Human Heart. Circulation 2000, 101, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Ng, M.Y.W.; Wai, T.; Simonsen, A. Quality control of the mitochondrion. Developmental cell 2021, 56, 881–905. [Google Scholar] [CrossRef]
- Held, N.M.; Houtkooper, R.H. Mitochondrial quality control pathways as determinants of metabolic health. Bioessays 2015, 37, 867–876. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Qian, L.; Zhu, Y.; Deng, C.; Liang, Z.; Chen, J.; Chen, Y.; Wang, X.; Liu, Y.; Tian, Y.; Yang, Y. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduction and Targeted Therapy 2024, 9, 50. [Google Scholar] [CrossRef]
- Abu Shelbayeh, O.; Arroum, T.; Morris, S.; Busch, K.B. PGC-1α Is a Master Regulator of Mitochondrial Lifecycle and ROS Stress Response. Antioxidants (Basel, Switzerland) 2023, 12. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Frontiers in cell and developmental biology 2022, Volume 10 - 2022. [Google Scholar] [CrossRef]
- Bhat, S.; Chin, A.; Shirakabe, A.; Ikeda, Y.; Ikeda, S.; Zhai, P.; Hsu, C.P.; Sayed, D.; Abdellatif, M.; Byun, J.; et al. Recruitment of RNA Polymerase II to Metabolic Gene Promoters Is Inhibited in the Failing Heart Possibly Through PGC-1α (Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α) Dysregulation. Circ Heart Fail 2019, 12, e005529. [Google Scholar] [CrossRef]
- Kärkkäinen, O.; Tuomainen, T.; Mutikainen, M.; Lehtonen, M.; Ruas, J.L.; Hanhineva, K.; Tavi, P. Heart specific PGC-1α deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc Res 2019, 115, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Lehman, J.J.; Kelly, D.P. Transcriptional activation of energy metabolic switches in the developing and hypertrophied heart. Clin Exp Pharmacol Physiol 2002, 29, 339–345. [Google Scholar] [CrossRef]
- Russell, L.K.; Mansfield, C.M.; Lehman, J.J.; Kovacs, A.; Courtois, M.; Saffitz, J.E.; Medeiros, D.M.; Valencik, M.L.; McDonald, J.A.; Kelly, D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ Res 2004, 94, 525–533. [Google Scholar] [CrossRef]
- Gwon, J.G.; Lee, S.M. Role of PTEN-Induced Protein Kinase 1 as a Mitochondrial Dysfunction Regulator in Cardiovascular Disease Pathogenesis. Vasc Specialist Int 2024, 40, 9. [Google Scholar] [CrossRef]
- Billia, F.; Hauck, L.; Konecny, F.; Rao, V.; Shen, J.; Mak, T.W. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proceedings of the National Academy of Sciences 2011, 108, 9572–9577. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, Y.; Bi, Y.; Zhao, Z.; Wang, S.; Lin, S.; Yang, Z.; Wang, X.; Mao, J. Emerging role of mitophagy in heart failure: from molecular mechanism to targeted therapy. Cell cycle (Georgetown, Tex.) 2023, 22, 906–918. [Google Scholar] [CrossRef]
- Ide, T.; Tsutsui, H.; Hayashidani, S.; Kang, D.; Suematsu, N.; Nakamura, K.; Utsumi, H.; Hamasaki, N.; Takeshita, A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res 2001, 88, 529–535. [Google Scholar] [CrossRef]
- Vela-Guajardo, J.E.; Pérez-Treviño, P.; Rivera-Álvarez, I.; González-Mondellini, F.A.; Altamirano, J.; García, N. The 8-oxo-deoxyguanosine glycosylase increases its migration to mitochondria in compensated cardiac hypertrophy. J Am Soc Hypertens 2017, 11, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Lauritzen, K.H.; Kleppa, L.; Aronsen, J.M.; Eide, L.; Carlsen, H.; Haugen Ø, P.; Sjaastad, I.; Klungland, A.; Rasmussen, L.J.; Attramadal, H.; et al. Impaired dynamics and function of mitochondria caused by mtDNA toxicity leads to heart failure. Am J Physiol Heart Circ Physiol 2015, 309, H434–H449. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, H.; Otsu, K. Mitochondrial DNA as an inflammatory mediator in cardiovascular diseases. The Biochemical journal 2018, 475, 839–852. [Google Scholar] [CrossRef]
- Misawa, T.; Takahama, M.; Kozaki, T.; Lee, H.; Zou, J.; Saitoh, T.; Akira, S. Microtubule-driven spatial arrangement of mitochondria promotes activation of the NLRP3 inflammasome. Nature Immunology 2013, 14, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Wagner, G.R.; Payne, R.M. Mitochondrial Acetylation and Diseases of Aging. Journal of aging research 2011, 2011, 234875. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.C.; Szekely, K.; Zhang, K.; Lanfear, D.E.; Sabbah, H.N. Evidence of Hyperacetylation of Mitochondrial Regulatory Proteins in Left Ventricular Myocardium of Dogs with Chronic Heart Failure. International journal of molecular sciences 2025, 26. [Google Scholar] [CrossRef]
- Castillo, E.C.; Morales, J.A.; Chapoy-Villanueva, H.; Silva-Platas, C.; Treviño-Saldaña, N.; Guerrero-Beltrán, C.E.; Bernal-Ramírez, J.; Torres-Quintanilla, A.; García, N.; Youker, K.; et al. Mitochondrial Hyperacetylation in the Failing Hearts of Obese Patients Mediated Partly by a Reduction in SIRT3: The Involvement of the Mitochondrial Permeability Transition Pore. Cell Physiol Biochem 2019, 53, 465–479. [Google Scholar] [CrossRef]
- Grillon, J.M.; Johnson, K.R.; Kotlo, K.; Danziger, R.S. Non-histone lysine acetylated proteins in heart failure. Biochimica et biophysica acta 2012, 1822, 607–614. [Google Scholar] [CrossRef]
- Pei, Z.; Dong, M.; Meng, X.; Yao, W.; Guo, Y.; Wang, F. Effects of Nicotinamide Adenine Dinucleotide on Older Patients with Heart Failure. Rev Cardiovasc Med 2024, 25, 297. [Google Scholar] [CrossRef] [PubMed]
- Nasuhidehnavi, A.; Zarzycka, W.; Górecki, I.; Chiao, Y.A.; Lee, C.F. Emerging interactions between mitochondria and NAD+ metabolism in cardiometabolic diseases. Trends in Endocrinology & Metabolism 2025, 36, 176–190. [Google Scholar] [CrossRef]
- Johnson, E.; Albakri, J.S.; Allemailem, K.S.; Sultan, A.; Alwanian, W.M.; Alrumaihi, F.; Almansour, N.M.; Aldakheel, F.M.; Khalil, F.M.A.; Abduallah, A.M.; et al. Mitochondrial dysfunction and calcium homeostasis in heart failure: Exploring the interplay between oxidative stress and cardiac remodeling for future therapeutic innovations. Current Problems in Cardiology 2025, 50, 102968. [Google Scholar] [CrossRef]
- Modesti, L.; Danese, A.; Angela Maria Vitto, V.; Ramaccini, D.; Aguiari, G.; Gafà, R.; Lanza, G.; Giorgi, C.; Pinton, P. Mitochondrial Ca(2+) Signaling in Health, Disease and Therapy. Cells 2021, 10. [Google Scholar] [CrossRef]
- Pathak, T.; Trebak, M. Mitochondrial Ca(2+) signaling. Pharmacol Ther 2018, 192, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; He, P.; Liu, W.; Wu, H.; Wang, Z. Unraveling mitochondrial crosstalk: a new frontier in heart failure pathogenesis. Front Cardiovasc Med 2025, 12, 1641023. [Google Scholar] [CrossRef] [PubMed]
- North, B.J.; Verdin, E. Sirtuins: Sir2-related NAD-dependent protein deacetylases. Genome biology 2004, 5, 224. [Google Scholar] [CrossRef]
- Wu, Q.-J.; Zhang, T.-N.; Chen, H.-H.; Yu, X.-F.; Lv, J.-L.; Liu, Y.-Y.; Liu, Y.-S.; Zheng, G.; Zhao, J.-Q.; Wei, Y.-F.; et al. The sirtuin family in health and disease. Signal Transduction and Targeted Therapy 2022, 7, 402. [Google Scholar] [CrossRef]
- Gupta, R.; Ambasta, R.K.; Kumar, P. Multifaced role of protein deacetylase sirtuins in neurodegenerative disease. Neurosci Biobehav Rev 2022, 132, 976–997. [Google Scholar] [CrossRef]
- Yu, L.; Li, Y.; Song, S.; Zhang, Y.; Wang, Y.; Wang, H.; Yang, Z.; Wang, Y. The dual role of sirtuins in cancer: biological functions and implications. Frontiers in oncology 2024, 14, 1384928. [Google Scholar] [CrossRef] [PubMed]
- Ianni, A.; Yuan, X.; Bober, E.; Braun, T. Sirtuins in the Cardiovascular System: Potential Targets in Pediatric Cardiology. Pediatric Cardiology 2018, 39, 983–992. [Google Scholar] [CrossRef]
- Hsu, Y.C.; Wu, Y.T.; Tsai, C.L.; Wei, Y.H. Current understanding and future perspectives of the roles of sirtuins in the reprogramming and differentiation of pluripotent stem cells. Experimental biology and medicine (Maywood, N.J.) 2018, 243, 563–575. [Google Scholar] [CrossRef]
- Lipphardt, M.; Dihazi, H.; Müller, G.A.; Goligorsky, M.S. Fibrogenic Secretome of Sirtuin 1-Deficient Endothelial Cells: Wnt, Notch and Glycocalyx Rheostat. Frontiers in physiology 2018, 9, 1325. [Google Scholar] [CrossRef]
- Ronnebaum, S.M.; Patterson, C. The FoxO family in cardiac function and dysfunction. Annu Rev Physiol 2010, 72, 81–94. [Google Scholar] [CrossRef] [PubMed]
- Matsushima, S.; Sadoshima, J. The role of sirtuins in cardiac disease. Am J Physiol Heart Circ Physiol 2015, 309, H1375–H1389. [Google Scholar] [CrossRef]
- Liu, H.; Wang, S.; Wang, J.; Guo, X.; Song, Y.; Fu, K.; Gao, Z.; Liu, D.; He, W.; Yang, L.L. Energy metabolism in health and diseases. Signal Transduct Target Ther 2025, 10, 69. [Google Scholar] [CrossRef]
- Ding, Y.-N.; Wang, H.-Y.; Chen, X.-F.; Tang, X.; Chen, H.-Z. Roles of Sirtuins in Cardiovascular Diseases: Mechanisms and Therapeutics. Circulation Research 2025, 136, 524–550. [Google Scholar] [CrossRef] [PubMed]
- Kitzman, D.W.; Brubaker, P.; Morgan, T.; Haykowsky, M.; Hundley, G.; Kraus, W.E.; Eggebeen, J.; Nicklas, B.J. Effect of Caloric Restriction or Aerobic Exercise Training on Peak Oxygen Consumption and Quality of Life in Obese Older Patients With Heart Failure With Preserved Ejection Fraction: A Randomized Clinical Trial. Jama 2016, 315, 36–46. [Google Scholar] [CrossRef]
- Corbi, G.; Conti, V.; Troisi, J.; Colucci, A.; Manzo, V.; Di Pietro, P.; Calabrese, M.C.; Carrizzo, A.; Vecchione, C.; Ferrara, N.; et al. Cardiac Rehabilitation Increases SIRT1 Activity and β-Hydroxybutyrate Levels and Decreases Oxidative Stress in Patients with HF with Preserved Ejection Fraction. Oxidative medicine and cellular longevity 2019, 2019, 7049237. [Google Scholar] [CrossRef]
- Russomanno, G.; Corbi, G.; Manzo, V.; Ferrara, N.; Rengo, G.; Puca, A.A.; Latte, S.; Carrizzo, A.; Calabrese, M.C.; Andriantsitohaina, R.; et al. The anti-ageing molecule sirt1 mediates beneficial effects of cardiac rehabilitation. Immunity & ageing : I & A 2017, 14, 7. [Google Scholar] [CrossRef]
- Costantino, S.; Mengozzi, A.; Velagapudi, S.; Mohammed, S.A.; Gorica, E.; Akhmedov, A.; Mongelli, A.; Pugliese, N.R.; Masi, S.; Virdis, A.; et al. Treatment with recombinant Sirt1 rewires the cardiac lipidome and rescues diabetes-related metabolic cardiomyopathy. Cardiovasc Diabetol 2023, 22, 312. [Google Scholar] [CrossRef] [PubMed]
- Alhasaniah, A.H.; Alissa, M.; Elsaid, F.G.; Alsugoor, M.H.; AlQahtani, M.S.; Alessa, A.; Jambi, K.; Albakri, G.S.; Albaqami, F.M.K.; Bennett, E. The enigmatic role of SIRT2 in the cardiovascular system: Deciphering its protective and detrimental actions to unlock new avenues for therapeutic intervention. Curr Probl Cardiol 2025, 50, 102929. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chang, H.C.; Tatekoshi, Y.; Mahmoodzadeh, A.; Balibegloo, M.; Najafi, Z.; Wu, R.; Chen, C.; Sato, T.; Shapiro, J.; et al. SIRT2 inhibition protects against cardiac hypertrophy and ischemic injury. eLife 2023, 12. [Google Scholar] [CrossRef]
- Tang, X.; Chen, X.F.; Wang, N.Y.; Wang, X.M.; Liang, S.T.; Zheng, W.; Lu, Y.B.; Zhao, X.; Hao, D.L.; Zhang, Z.Q.; et al. SIRT2 Acts as a Cardioprotective Deacetylase in Pathological Cardiac Hypertrophy. Circulation 2017, 136, 2051–2067. [Google Scholar] [CrossRef]
- Zheng, M.; Du, X.; Zhao, L.; Sun, H.; Chen, M.; Yang, X. Elevated plasma Sirtuin2 level predicts heart failure after acute myocardial infarction. J Thorac Dis 2021, 13, 50–59. [Google Scholar] [CrossRef]
- Sarikhani, M.; Maity, S.; Mishra, S.; Jain, A.; Tamta, A.K.; Ravi, V.; Kondapalli, M.S.; Desingu, P.A.; Khan, D.; Kumar, S.; et al. SIRT2 deacetylase represses NFAT transcription factor to maintain cardiac homeostasis. Journal of Biological Chemistry 2018, 293, 5281–5294. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ Res 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Tong, D.; Schiattarella, G.G.; Jiang, N.; Altamirano, F.; Szweda, P.A.; Elnwasany, A.; Lee, D.I.; Yoo, H.; Kass, D.A.; Szweda, L.I.; et al. NAD(+) Repletion Reverses Heart Failure With Preserved Ejection Fraction. Circ Res 2021, 128, 1629–1641. [Google Scholar] [CrossRef] [PubMed]
- Jheng, J.R.; Bai, Y.; Noda, K.; Huot, J.R.; Cook, T.; Fisher, A.; Chen, Y.Y.; Goncharov, D.A.; Goncharova, E.A.; Simon, M.A.; et al. Skeletal Muscle SIRT3 Deficiency Contributes to Pulmonary Vascular Remodeling in Pulmonary Hypertension Due to Heart Failure With Preserved Ejection Fraction. Circulation 2024, 150, 867–883. [Google Scholar] [CrossRef]
- Wang, Y.C.; Koay, Y.C.; Pan, C.; Zhou, Z.; Tang, W.; Wilcox, J.; Li, X.S.; Zagouras, A.; Marques, F.; Allayee, H.; et al. Indole-3-Propionic Acid Protects Against Heart Failure With Preserved Ejection Fraction. Circ Res 2024, 134, 371–389. [Google Scholar] [CrossRef]
- Bergmann, L.; Lang, A.; Bross, C.; Altinoluk-Hambüchen, S.; Fey, I.; Overbeck, N.; Stefanski, A.; Wiek, C.; Kefalas, A.; Verhülsdonk, P.; et al. Subcellular localization and mitotic interactome analyses identify SIRT4 as a centrosomally localized and microtubule associated protein. bioRxiv 2020, 2020.2002.2017.940692. [Google Scholar] [CrossRef] [PubMed]
- Bugger, H.; Witt, C.N.; Bode, C. Mitochondrial sirtuins in the heart. Heart Failure Reviews 2016, 21, 519–528. [Google Scholar] [CrossRef]
- Luo, Y.X.; Tang, X.; An, X.Z.; Xie, X.M.; Chen, X.F.; Zhao, X.; Hao, D.L.; Chen, H.Z.; Liu, D.P. SIRT4 accelerates Ang II-induced pathological cardiac hypertrophy by inhibiting manganese superoxide dismutase activity. Eur Heart J 2017, 38, 1389–1398. [Google Scholar] [CrossRef]
- Byrne, N.J.; Koentges, C.; Khan, E.; Pfeil, K.; Sandulescu, R.; Bakshi, S.; Költgen, C.; Vosko, I.; Gollmer, J.; Rathner, T.; et al. Sirtuin 4 accelerates heart failure development by enhancing reactive oxygen species-mediated profibrotic transcriptional signaling. Journal of Molecular and Cellular Cardiology Plus 2025, 12, 100299. [Google Scholar] [CrossRef]
- Guo, A.H.; Baliira, R.; Skinner, M.E.; Kumar, S.; Andren, A.; Zhang, L.; Goldsmith, R.S.; Michan, S.; Davis, N.J.; Maccani, M.W.; et al. Sirtuin 5 levels are limiting in preserving cardiac function and suppressing fibrosis in response to pressure overload. Scientific reports 2022, 12, 12258. [Google Scholar] [CrossRef]
- Liu, B.; Che, W.; Zheng, C.; Liu, W.; Wen, J.; Fu, H.; Tang, K.; Zhang, J.; Xu, Y. SIRT5: A Safeguard Against Oxidative Stress-Induced Apoptosis in Cardiomyocytes. Cellular Physiology and Biochemistry 2013, 32, 1050–1059. [Google Scholar] [CrossRef]
- Li, S.; Shen, S.; Hong, Y.; Ding, K.; Chen, S.; Chen, J.; Wang, C.; Wen, Y.; Mo, G.; Yu, L.; et al. Sirt5 preserves cardiac function in ischemia-reperfusion injury by inhibiting ANT2 lactylation. bioRxiv 2024, 2024.2011.2025.625148. [Google Scholar] [CrossRef]
- Khan, D.; Ara, T.; Ravi, V.; Rajagopal, R.; Tandon, H.; Parvathy, J.; Gonzalez, E.A.; Asirvatham-Jeyaraj, N.; Krishna, S.; Mishra, S.; et al. SIRT6 transcriptionally regulates fatty acid transport by suppressing PPARγ. Cell reports 2021, 35, 109190. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, H.; Brooks, A.; Xu, S.; Luo, J.; Steiner, R.; Mickelsen, D.M.; Moravec, C.S.; Jeffrey, A.D.; Small, E.M.; et al. SIRT6 Mitigates Heart Failure With Preserved Ejection Fraction in Diabetes. Circ Res 2022, 131, 926–943. [Google Scholar] [CrossRef]
- Li, Y.; Meng, X.; Wang, W.; Liu, F.; Hao, Z.; Yang, Y.; Zhao, J.; Yin, W.; Xu, L.; Zhao, R.; et al. Cardioprotective Effects of SIRT6 in a Mouse Model of Transverse Aortic Constriction-Induced Heart Failure. Frontiers in physiology 2017, Volume 8 - 2017. [Google Scholar] [CrossRef]
- Kiran, S.; Chatterjee, N.; Singh, S.; Kaul, S.C.; Wadhwa, R.; Ramakrishna, G. Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. Febs j 2013, 280, 3451–3466. [Google Scholar] [CrossRef]
- Chen, Y.-F.; Qi, R.-Q.; Song, J.-W.; Wang, S.-Y.; Dong, Z.-J.; Chen, Y.-H.; Liu, Y.; Zhou, X.-Y.; Li, J.; Liu, X.-Y.; et al. Sirtuin 7 ameliorates cuproptosis, myocardial remodeling and heart dysfunction in hypertension through the modulation of YAP/ATP7A signaling. Apoptosis 2024, 29, 2161–2182. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, S.; Izumiya, Y.; Araki, S.; Nakamura, T.; Kimura, Y.; Hanatani, S.; Yamada, T.; Ishida, T.; Yamamoto, M.; Onoue, Y.; et al. Cardiomyocyte Sirt (Sirtuin) 7 Ameliorates Stress-Induced Cardiac Hypertrophy by Interacting With and Deacetylating GATA4. Hypertension 2020, 75, 98–108. [Google Scholar] [CrossRef]
- Vakhrusheva, O.; Smolka, C.; Gajawada, P.; Kostin, S.; Boettger, T.; Kubin, T.; Braun, T.; Bober, E. Sirt7 Increases Stress Resistance of Cardiomyocytes and Prevents Apoptosis and Inflammatory Cardiomyopathy in Mice. Circulation Research 2008, 102, 703–710. [Google Scholar] [CrossRef]
- Wang, Y.; Li, Y.; Ding, H.; Li, D.; Shen, W.; Zhang, X. The Current State of Research on Sirtuin-Mediated Autophagy in Cardiovascular Diseases. Journal of Cardiovascular Development and Disease 2023, 10, 382. [Google Scholar] [CrossRef]
- Aman, Y.; Schmauck-Medina, T.; Hansen, M.; Morimoto, R.I.; Simon, A.K.; Bjedov, I.; Palikaras, K.; Simonsen, A.; Johansen, T.; Tavernarakis, N.; et al. Autophagy in healthy aging and disease. Nature Aging 2021, 1, 634–650. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Peng, G.; Liu, H.; Wang, L.; Lu, R.; Li, L. Molecular mechanisms of secretory autophagy and its potential role in diseases. Life sciences 2024, 347, 122653. [Google Scholar] [CrossRef] [PubMed]
- Jahn, R.; Cafiso, D.C.; Tamm, L.K. Mechanisms of SNARE proteins in membrane fusion. Nature Reviews Molecular Cell Biology 2024, 25, 101–118. [Google Scholar] [CrossRef] [PubMed]
- Stolz, A.; Ernst, A.; Dikic, I. Cargo recognition and trafficking in selective autophagy. Nature cell biology 2014, 16, 495–501. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Chiong, M.; Lavandero, S.; Klionsky, D.J.; Ren, J. Mitophagy in cardiovascular diseases: molecular mechanisms, pathogenesis, and treatment. Trends in molecular medicine 2022, 28, 836–849. [Google Scholar] [CrossRef]
- De Meyer, G.R.Y.; De Keulenaer, G.W.; Martinet, W. Role of autophagy in heart failure associated with aging. Heart Failure Reviews 2010, 15, 423–430. [Google Scholar] [CrossRef]
- Nalbandian, A.; Llewellyn, K.J.; Nguyen, C.; Yazdi, P.G.; Kimonis, V.E. Rapamycin and chloroquine: the in vitro and in vivo effects of autophagy-modifying drugs show promising results in valosin containing protein multisystem proteinopathy. PloS one 2015, 10, e0122888. [Google Scholar] [CrossRef]
- Liu, S.; Chen, S.; Li, M.; Zhang, B.; Shen, P.; Liu, P.; Zheng, D.; Chen, Y.; Jiang, J. Autophagy activation attenuates angiotensin II-induced cardiac fibrosis. Archives of biochemistry and biophysics 2016, 590, 37–47. [Google Scholar] [CrossRef]
- Wang, L.; Yuan, D.; Zheng, J.; Wu, X.; Wang, J.; Liu, X.; He, Y.; Zhang, C.; Liu, C.; Wang, T.; et al. Chikusetsu saponin IVa attenuates isoprenaline-induced myocardial fibrosis in mice through activation autophagy mediated by AMPK/mTOR/ULK1 signaling. Phytomedicine 2019, 58, 152764. [Google Scholar] [CrossRef]
- Li, Z.; Wang, J.; Yang, X. Functions of Autophagy in Pathological Cardiac Hypertrophy. International Journal of Biological Sciences 2015, 11, 672–678. [Google Scholar] [CrossRef]
- Sehrawat, A.; Mishra, J.; Mastana, S.S.; Navik, U.; Bhatti, G.K.; Reddy, P.H.; Bhatti, J.S. Dysregulated autophagy: A key player in the pathophysiology of type 2 diabetes and its complications. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2023, 1869, 166666. [Google Scholar] [CrossRef]
- He, H.; Liu, X.; Lv, L.; Liang, H.; Leng, B.; Zhao, D.; Zhang, Y.; Du, Z.; Chen, X.; Li, S.; et al. Calcineurin suppresses AMPK-dependent cytoprotective autophagy in cardiomyocytes under oxidative stress. Cell death & disease 2014, 5, e997. [Google Scholar] [CrossRef]
- Shaikh, S.; Troncoso, R.; Criollo, A.; Bravo-Sagua, R.; García, L.; Morselli, E.; Cifuentes, M.; Quest, A.F.; Hill, J.A.; Lavandero, S. Regulation of cardiomyocyte autophagy by calcium. American journal of physiology. Endocrinology and metabolism 2016, 310, E587–E596. [Google Scholar] [CrossRef]
- Sugden, P.; Fuller, S.; Weiss, S.; Clerk, A. Glycogen synthase kinase 3 (GSK3) in the heart: A point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. British journal of pharmacology 2008, 153 Suppl 1, S137–S153. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Jang, S.; Agostini, B. Crosstalk between mitogen-activated protein kinases and mitochondria in cardiac diseases: therapeutic perspectives. Pharmacol Ther 2014, 144, 202–225. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Zhou, X.; Yang, T.; Wang, L.; Feng, L.; Wang, Z.; Xu, J.; Jing, W.; Wang, T.; Su, H.; et al. The role of autophagy in cardiovascular disease: Cross-interference of signaling pathways and underlying therapeutic targets. Frontiers in Cardiovascular Medicine 2023, Volume 10 - 2023. [Google Scholar] [CrossRef]
- Ljubojević-Holzer, S.; Kraler, S.; Djalinac, N.; Abdellatif, M.; Voglhuber, J.; Schipke, J.; Schmidt, M.; Kling, K.M.; Franke, G.T.; Herbst, V.; et al. Loss of autophagy protein ATG5 impairs cardiac capacity in mice and humans through diminishing mitochondrial abundance and disrupting Ca2+ cycling. Cardiovasc Res 2022, 118, 1492–1505. [Google Scholar] [CrossRef]
- Mongirdienė, A.; Skrodenis, L.; Varoneckaitė, L.; Mierkytė, G.; Gerulis, J. Reactive Oxygen Species Induced Pathways in Heart Failure Pathogenesis and Potential Therapeutic Strategies. Biomedicines 2022, 10, 602. [Google Scholar] [CrossRef]
- Peng, Y.; Liao, B.; Zhou, Y.; Zeng, W. Ginsenoside Rb2 improves heart failure by down-regulating miR-216a-5p to promote autophagy and inhibit apoptosis and oxidative stress. J Appl Biomed 2023, 21, 180–192. [Google Scholar] [CrossRef]
- Denton, D.; Xu, T.; Kumar, S. Autophagy as a pro-death pathway. Immunol Cell Biol 2015, 93, 35–42. [Google Scholar] [CrossRef]
- Huang, D.; Wen, Q.; Su, Y.; Li, X. miR-17-5p Inhibits BNIP3-Mediated Mitochondrial Autophagy to Attenuate Pathological Cardiac Fibrosis. Balkan Med J 2025. [Google Scholar] [CrossRef]
- Ucar, A.; Gupta, S.K.; Fiedler, J.; Erikci, E.; Kardasinski, M.; Batkai, S.; Dangwal, S.; Kumarswamy, R.; Bang, C.; Holzmann, A.; et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun 2012, 3, 1078. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Kohlbrenner, E.; Gamb, S.I.; Guenzel, A.J.; Klaus, K.; Fayyaz, A.U.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. FOXO3a regulates BNIP3 and modulates mitochondrial calcium, dynamics, and function in cardiac stress. Am J Physiol Heart Circ Physiol 2016, 311, H1540–H1559. [Google Scholar] [CrossRef]
- Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress mediates induction of autophagy and hypertrophy in angiotensin-II treated mouse hearts. Autophagy 2011, 7, 917–918. [Google Scholar] [CrossRef]
- Mann, D.L. Basic mechanisms of left ventricular remodeling: the contribution of wall stress. Journal of Cardiac Failure 2004, 10, S202–S206. [Google Scholar] [CrossRef]
- Nishida, K.; Otsu, K. Autophagy during cardiac remodeling. Journal of Molecular and Cellular Cardiology 2016, 95, 11–18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Z.; Li, Q.; Trammell, S.A.; Schmidt, M.S.; Pires, K.M.; Cai, J.; Zhang, Y.; Kenny, H.; Boudina, S.; et al. Control of NAD<sup>+</sup> homeostasis by autophagic flux modulates mitochondrial and cardiac function. The EMBO journal 2024, 43, 362–390. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Tam, E.; Song, E.; Xu, A.; Sweeney, G. Crosstalk between myocardial autophagy and sterile inflammation in the development of heart failure. Autophagy Reports 2024, 3, 2320605. [Google Scholar] [CrossRef]
- Chiu, B.; Jantuan, E.; Shen, F.; Chiu, B.; Sergi, C. Autophagy-Inflammasome Interplay in Heart Failure: A Systematic Review on Basics, Pathways, and Therapeutic Perspectives. Ann Clin Lab Sci 2017, 47, 243–252. [Google Scholar] [PubMed]
- Pavillard, L.E.; Cañadas-Lozano, D.; Alcocer-Gómez, E.; Marín-Aguilar, F.; Pereira, S.; Robertson, A.A.B.; Muntané, J.; Ryffel, B.; Cooper, M.A.; Quiles, J.L.; et al. NLRP3-inflammasome inhibition prevents high fat and high sugar diets-induced heart damage through autophagy induction. Oncotarget 2017, 8, 99740–99756. [Google Scholar] [CrossRef]
- Tang, L.Q.; Wang, L.L.; Tang, Q.F.; Wang, W. SLC26A4 regulates autophagy and activates the NLRP3 inflammasome to mediate pathological cardiac hypertrophy. Scientific reports 2025, 15, 12511. [Google Scholar] [CrossRef]
- Tang, L.; Yu, X.; Zheng, Y.; Zhou, N. Inhibiting SLC26A4 reverses cardiac hypertrophy in H9C2 cells and in rats. PeerJ 2020, 8, e8253. [Google Scholar] [CrossRef]
- Oka, T.; Hikoso, S.; Yamaguchi, O.; Taneike, M.; Takeda, T.; Tamai, T.; Oyabu, J.; Murakawa, T.; Nakayama, H.; Nishida, K.; et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature 2012, 485, 251–255. [Google Scholar] [CrossRef] [PubMed]
- Jenča, D.; Melenovský, V.; Stehlik, J.; Staněk, V.; Kettner, J.; Kautzner, J.; Adámková, V.; Wohlfahrt, P. Heart failure after myocardial infarction: incidence and predictors. ESC Heart Fail 2021, 8, 222–237. [Google Scholar] [CrossRef] [PubMed]
- Araki, S.; Izumiya, Y.; Rokutanda, T.; Ianni, A.; Hanatani, S.; Kimura, Y.; Onoue, Y.; Senokuchi, T.; Yoshizawa, T.; Yasuda, O.; et al. Sirt7 Contributes to Myocardial Tissue Repair by Maintaining Transforming Growth Factor-β Signaling Pathway. Circulation 2015, 132, 1081–1093. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial ischemia/reperfusion: Translational pathophysiology of ischemic heart disease. Med 2024, 5, 10–31. [Google Scholar] [CrossRef] [PubMed]
- Kang, R.; Zeh, H.J.; Lotze, M.T.; Tang, D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 2011, 18, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Sala-Mercado, J.A.; Wider, J.; Undyala, V.V.; Jahania, S.; Yoo, W.; Mentzer, R.M., Jr.; Gottlieb, R.A.; Przyklenk, K. Profound cardioprotection with chloramphenicol succinate in the swine model of myocardial ischemia-reperfusion injury. Circulation 2010, 122, S179–S184. [Google Scholar] [CrossRef]
- Ding, X.; Zhu, C.; Wang, W.; Li, M.; Ma, C.; Gao, B. SIRT1 is a regulator of autophagy: Implications for the progression and treatment of myocardial ischemia-reperfusion. Pharmacological Research 2024, 199, 106957. [Google Scholar] [CrossRef]
- Luo, G.; Jian, Z.; Zhu, Y.; Zhu, Y.; Chen, B.; Ma, R.; Tang, F.; Xiao, Y. Sirt1 promotes autophagy and inhibits apoptosis to protect cardiomyocytes from hypoxic stress. International journal of molecular medicine 2019, 43, 2033–2043. [Google Scholar] [CrossRef] [PubMed]
- Baloglu, E. Hypoxic Stress-Dependent Regulation of Na,K-ATPase in Ischemic Heart Disease. International journal of molecular sciences 2023, 24. [Google Scholar] [CrossRef]
- Hariharan, N.; Maejima, Y.; Nakae, J.; Paik, J.; Depinho, R.A.; Sadoshima, J. Deacetylation of FoxO by Sirt1 Plays an Essential Role in Mediating Starvation-Induced Autophagy in Cardiac Myocytes. Circ Res 2010, 107, 1470–1482. [Google Scholar] [CrossRef] [PubMed]
- Ortega, M.A.; Fraile-Martinez, O.; de Leon-Oliva, D.; Boaru, D.L.; Lopez-Gonzalez, L.; García-Montero, C.; Alvarez-Mon, M.A.; Guijarro, L.G.; Torres-Carranza, D.; Saez, M.A.; et al. Autophagy in Its (Proper) Context: Molecular Basis, Biological Relevance, Pharmacological Modulation, and Lifestyle Medicine. Int J Biol Sci 2024, 20, 2532–2554. [Google Scholar] [CrossRef]
- Gan, T.; Qu, S.; Zhang, H.; Zhou, X.J. Modulation of the immunity and inflammation by autophagy. MedComm (2020) 2023, 4, e311. [Google Scholar] [CrossRef]
- Yapryntseva, M.A.; Maximchik, P.V.; Zhivotovsky, B.; Gogvadze, V. Mitochondrial sirtuin 3 and various cell death modalities. Frontiers in cell and developmental biology 2022, 10, 947357. [Google Scholar] [CrossRef]
- D’Onofrio, N.; Servillo, L.; Balestrieri, M.L. SIRT1 and SIRT6 Signaling Pathways in Cardiovascular Disease Protection. Antioxidants & redox signaling 2018, 28, 711–732. [Google Scholar] [CrossRef]
Figure 1.
Precursors of heart failure. These precursors partly overlap, as shown in this diagram. ER stands for endoplasmic reticulum; IRI stands for ischemia-reperfusion injury. Created in https://BioRender.com.
Figure 1.
Precursors of heart failure. These precursors partly overlap, as shown in this diagram. ER stands for endoplasmic reticulum; IRI stands for ischemia-reperfusion injury. Created in https://BioRender.com.
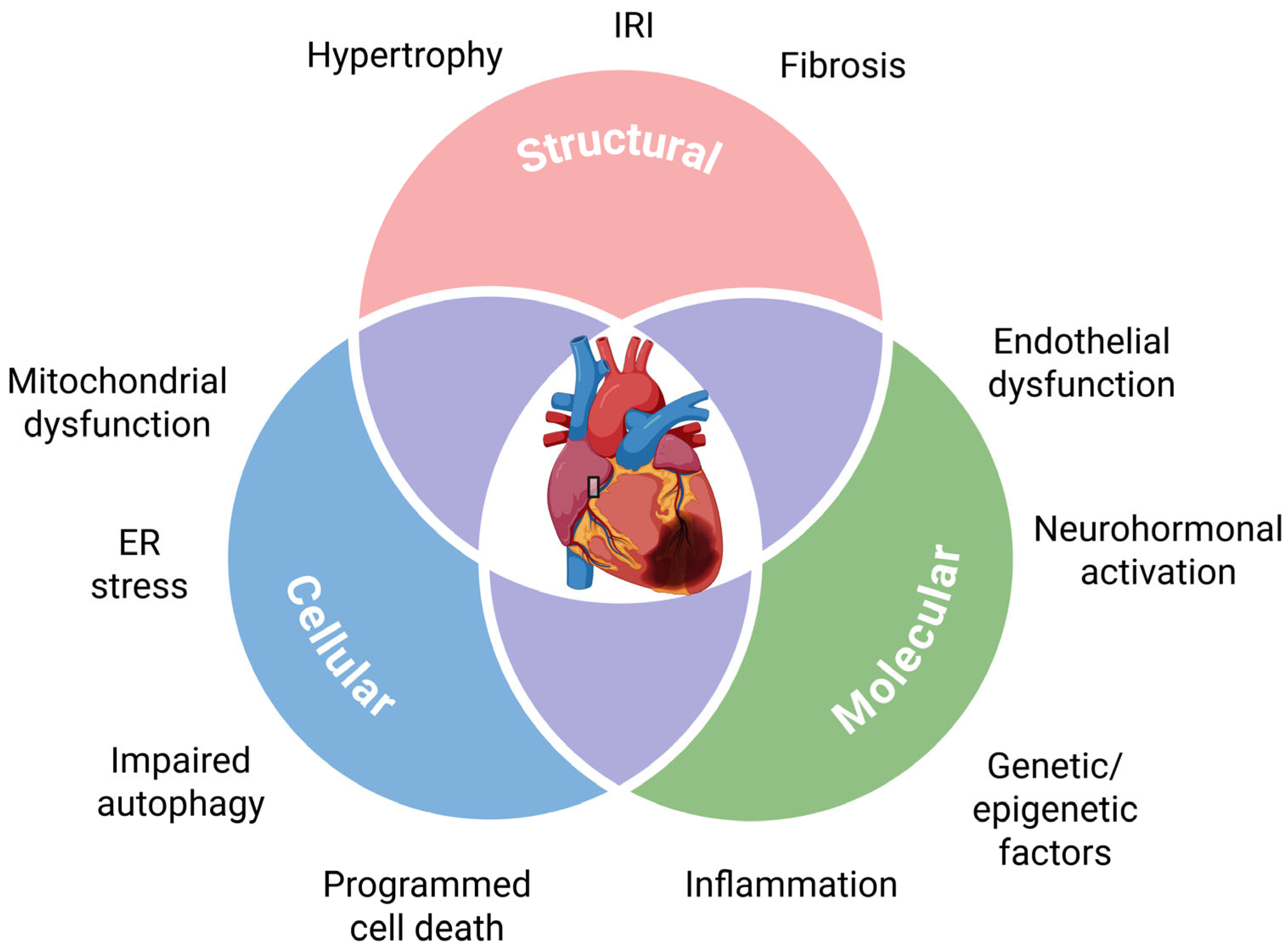
Figure 2.
Energy in the normal heart is primarily produced through oxidative phosphorylation in mitochondria, with a minor role for glycolysis in the cytosol. However, in a diseased heart, such as an ischemic or failing heart, these proportions vary as indicated. These changes can range widely depending on the specific disease and its clinical presentation. Significant differences exist in substrate utilization between diabetic and non-diabetic heart failure (HF), with the former relying more on fatty acid oxidation due to insulin resistance and impaired glucose uptake. Although only fatty acids and glucose are discussed here, ketone bodies, lactate, and other molecules can also serve as energy substrates. Created in https://BioRender.com.
Figure 2.
Energy in the normal heart is primarily produced through oxidative phosphorylation in mitochondria, with a minor role for glycolysis in the cytosol. However, in a diseased heart, such as an ischemic or failing heart, these proportions vary as indicated. These changes can range widely depending on the specific disease and its clinical presentation. Significant differences exist in substrate utilization between diabetic and non-diabetic heart failure (HF), with the former relying more on fatty acid oxidation due to insulin resistance and impaired glucose uptake. Although only fatty acids and glucose are discussed here, ketone bodies, lactate, and other molecules can also serve as energy substrates. Created in https://BioRender.com.
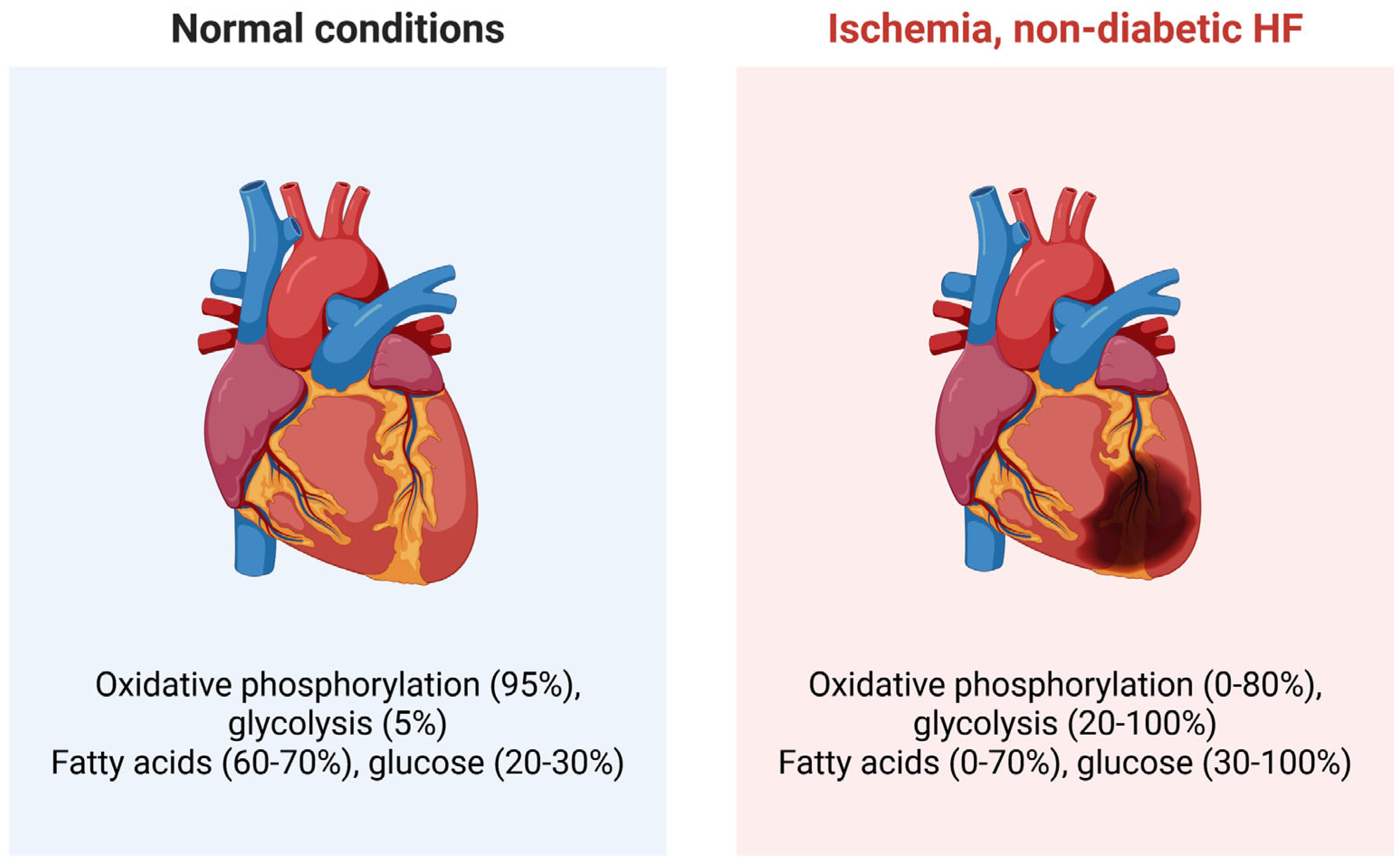
Figure 3.
Mitochondrial dysfunction in heart failure. Two-headed arrows depict the mutual dependence between these dysfunctions and HF, indicating that, currently, only an association can be identified without a clear causal relationship. mtDNA, mitochondrial DNA; NLRP3, NACHT, LRR, and PYD domains-containing protein 3 (NLRP3); NAD+, nicotinamide adenine dinucleotide oxidized; NADH, nicotinamide adenine dinucleotide reduced. Created in https://BioRender.com.
Figure 3.
Mitochondrial dysfunction in heart failure. Two-headed arrows depict the mutual dependence between these dysfunctions and HF, indicating that, currently, only an association can be identified without a clear causal relationship. mtDNA, mitochondrial DNA; NLRP3, NACHT, LRR, and PYD domains-containing protein 3 (NLRP3); NAD+, nicotinamide adenine dinucleotide oxidized; NADH, nicotinamide adenine dinucleotide reduced. Created in https://BioRender.com.
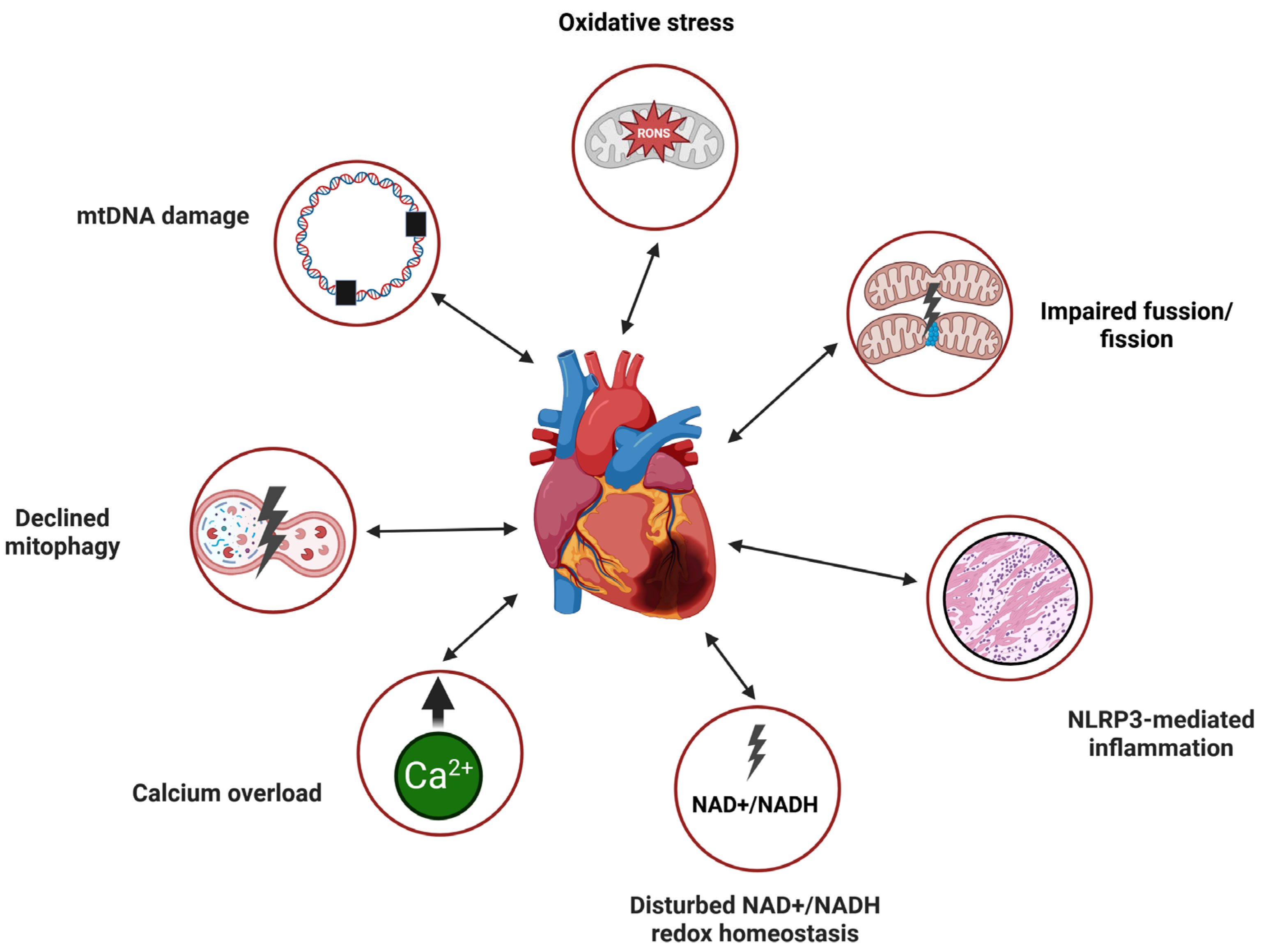
Figure 4.
Outline of autophagy pathways. Macroautophagy begins with the formation of the isolation membrane (phagophore), which expands to form an autophagosome that encloses the cargo. In macroautophagy, the autophagosome fuses with a lysosome to form an autolysosome, where lysosomal enzymes digest the cargo. In chaperone-mediated autophagy, the cargo contains a specific motif, such as the lysine-phenylalanine-glutamic acid-arginine-glutamine sequence, recognized by a lysosomal membrane protein with the help of a chaperone, exemplified here by heat shock cognate 71 kDa protein (HSC70). In microautophagy, the cargo is directly engulfed by a lysosome and broken down. Created in https://BioRender.com.
Figure 4.
Outline of autophagy pathways. Macroautophagy begins with the formation of the isolation membrane (phagophore), which expands to form an autophagosome that encloses the cargo. In macroautophagy, the autophagosome fuses with a lysosome to form an autolysosome, where lysosomal enzymes digest the cargo. In chaperone-mediated autophagy, the cargo contains a specific motif, such as the lysine-phenylalanine-glutamic acid-arginine-glutamine sequence, recognized by a lysosomal membrane protein with the help of a chaperone, exemplified here by heat shock cognate 71 kDa protein (HSC70). In microautophagy, the cargo is directly engulfed by a lysosome and broken down. Created in https://BioRender.com.
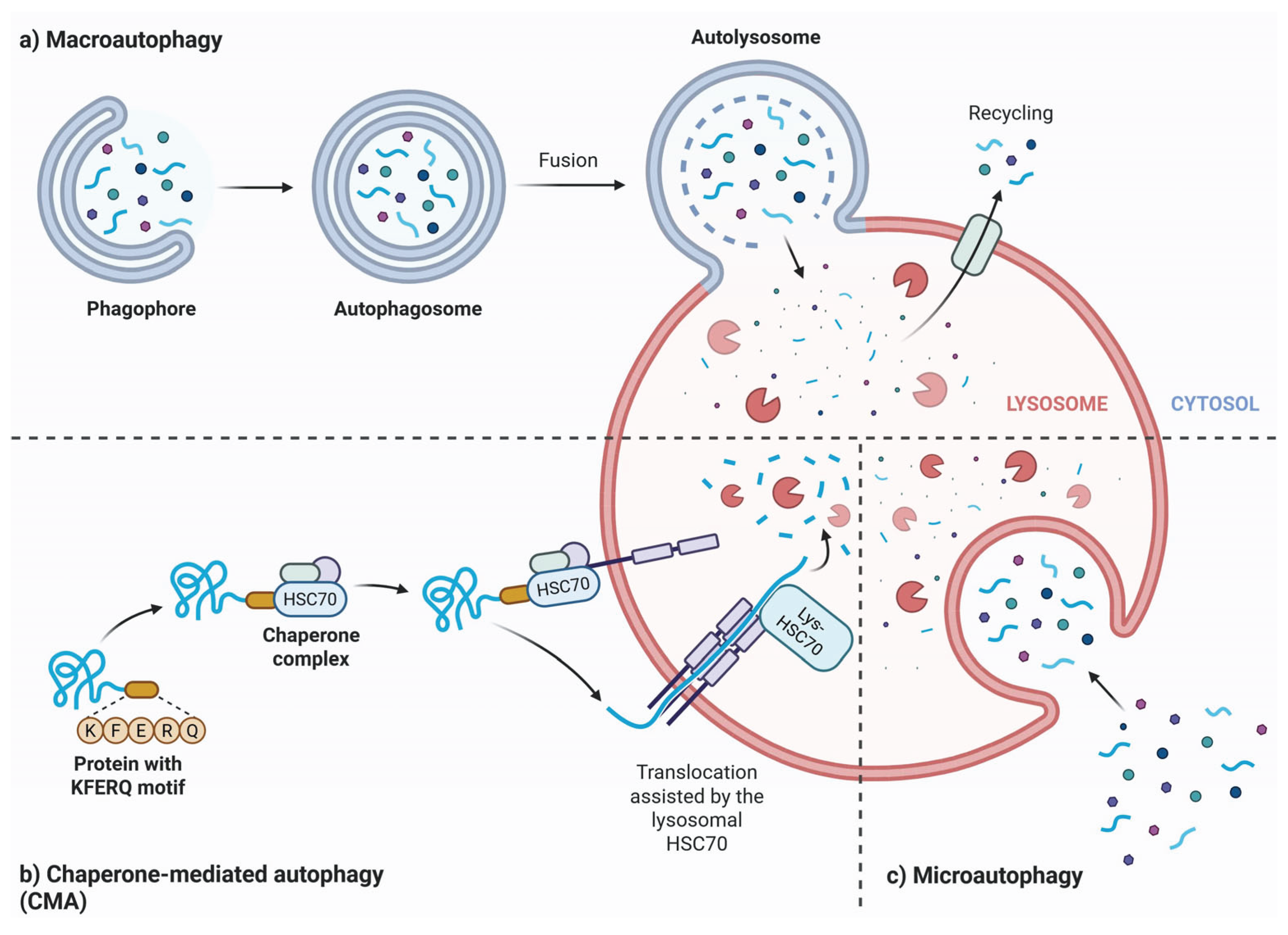

Table 1.
Some heart failure-related effects of sirtuins.
| Sirtuin | Subject | Effect | Mediators/mechanism | Reference |
|---|---|---|---|---|
| SIRT1a | HFpEF patients | Reduced systolic blood pressure, increased EF | Increased levels of SIRT1 and NAD+ in caloric restriction and exercise program; increased antioxidant capacity | [86,87] |
| Mouse model of HFpEF | Protection against harmful effects related to heart remodeling | Regulation of lipid metabolism and inflammation | [88] | |
| SIRT2 | SIRT2-/- mice and mice lacking SIRT1 in cardiomyocytes | Improved cardiac function after I/R and PO, reduced cardiac hypertrophy | Maladaptive effects in response to stress, decrease in antioxidant protection due to NRF2 inhibition | [90] |
| SIRT2-/- mice | Cardiac hypertrophy and fibrosis, reduced EF and fractional shortening | SIRT2-mediated AMPK activity through deacetylating LKB1 | [91] | |
| Mice with overexpressed SIRT2 | Protection against hypertrophy and fibrosis caused by age and ANGII | |||
| Patients after acute myocardial infarction | Positive correlation between SIRT2 and HF | [92] | ||
| SIRT2-/- mice | Heart hypertrophy, remodeling, fibrosis, and age-related dysfunction | SIRT2 interacted with and deacetylated the NFATc2 transcription factor | [93] | |
| SIRT3 | Mouse model of HFpEF | Fibrosis | hyperacetylation of mitochondrial proteins, resulting in enhanced production of interleukins IL1B and IL18 and increased assembly of NLRP3 | [94] |
| Mouse model of HFpEF | Impairment in mitochondrial fatty acid oxidation | Hyperacetylation of key enzymes of fatty acid oxidation, SIRT3 downregulation, NAD+ deficiency | [95] | |
| Mouse model of HFpEF | Improved glucose uptake and metabolism | Activation of the skeletal muscle SIRT3-5’-AMPK pathway | [96] | |
| Mouse model of HFpEF with SIRT3 deficiency | Pulmonary vascular remodeling | Increased secretion of lysyl oxidase homolog 2 and β2-microglobulin | [96] | |
| HFpEF patients | Heart remodeling | Reduction of indole-3-propionic acid, activating NNMT-SIRT3 axis | [97] | |
| SIRT4 | Mice with ANGII infusion | Progression from compensated to decompensated cardiac hypertrophy | SIRT4 overexpression | [100] |
| Mice with heart-specific SIRT4 overexpression | sped up heart failure development in response to pressure overload | Mitochondrial RONS-mediated increase in profibrotic transcriptional signaling | [101] | |
| SIRT5 | Mice with SIRT5 overexpression | Protection against TAC consequences | Suppression of metabolic switch from fatty acid oxidation to glycolysis, immune activation, and fibrotic signaling pathways | [102] |
| Mouse cardiomyocytes | Reduction in the cell viability, and an increase in the number of apoptotic cells and the caspase 3/7 activity | Direct interaction between B2CL1 and SIRT5 | [103] | |
| mouse model of cardiac IR injury | reduced mitochondrial damage and alleviated cardiac injury | Increasing SIRT5 levels reduced mitochondrial damage and alleviated cardiac injury through interaction with ANT2, inhibiting its lactylation and enhancing its interaction with VDAC1 | [104] | |
| SIRT6 | Mouse model of HFpEF | HF mitigation | restoring endothelial SIRT6 function and it was underlined by the deacetylation of histone H3K9 around the PPARG promoter | [105] |
| Diabetic HF patients | Decreased level of SIRT6 | [106] | ||
| Diabetic mouse model of HFpEF | Improvements in diastolic dysfunction and decreased cardiac lipid buildup | Suppression of endothelial PPARγ expression via SIRT6-dependent deacetylation of histone H3 near the PPARγ gene promoter. | [106] | |
| TAC mice | Mitigated TAC-induced heart dysfunction and decreased cardiac inflammation, resulting in reduced cardiac fibrosis and smaller infarcts | Overexpression of SIRT6 elevated TERT and TRF1 levels | [107] | |
| SIRT7 | TAC mice | Increase in heart weight relative to tibial length, and they demonstrated a reduced cardiac contractile function | Interaction between SIRT7 and the transcription factor GATA-4 was identified, and GATA4 knockdown lessened the severity of phenylephrine-induced cardiac hypertrophy. SIRT7 deacetylated GATA4 in cardiomyocytes, influencing its transcriptional activity. | [110] |
a All abbreviations are defined in the main text.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



