Submitted:
04 September 2025
Posted:
05 September 2025
You are already at the latest version
Abstract
L-asparaginase (L-ASNase) is a vital enzymatic drug widely used for treating acute lymphoblastic leukemia (ALL) and certain lymphomas. However, its clinical application is often limited by a short plasma half-life, pronounced immunogenicity, and systemic toxicities. To address these challenges, we recently developed conjugates of L-ASNase with cationic polymers, enhancing its cytostatic activity by increasing enzyme binding with cancer cells. The present study focuses on the development of liposomal formulations of E. coli L-asparaginase (EcA) and its conjugates with cationic polymers: the natural oligoamine spermine (spm) and a synthetic chitosan-polyethylenimine (Chit-PEI) copolymer. This approach aims to improve enzyme encapsulation efficiency and stability within liposomes. Various formulations—including EcA conjugates with polycations incorporated into 100 nm and 400 nm phosphatidylcholine/cardiolipin (PC/CL, 80/20) anionic liposomes—were synthesized as a delivery system of high enzyme load. Fourier Transform Infrared (FTIR) spectroscopy confirmed successful enzyme association with liposomal carriers by identifying characteristic changes in the vibrational bands corresponding to both protein and lipid components. In vitro release studies demonstrated that encapsulating EcA formulations in liposomes more than doubled their half-release time (T1/2), depending on the formulation. Cytotoxicity assays against Raji lymphoma cells revealed that liposomal formulations, particularly 100 nm EcA-spm liposomes, exhibited markedly superior anti-proliferative activity, reducing cell viability to 4.5%, compared to 35% for free EcA. Confocal Laser Scanning Microscopy (CLSM) provided clear visual and quantitative evidence that enhanced cellular internalization of the enzyme correlates directly with its cytostatic efficacy. Notably, formulations showing higher intracellular uptake produced greater cytotoxic effects, emphasizing that hydrolysis of asparagine inside cancer cells, rather than extracellularly, is critical for therapeutic success. Among all tested formulations, the EcA-spermine liposomal conjugate demonstrated the highest fluorescence intensity within cells providing enhanced cytotoxicity. These results strongly indicate that encapsulating cationically modified L-ASNase in liposomes is a highly promising strategy to improve targeted cellular delivery and prolonged enzymatic activity. This strategy holds significant potential for developing more effective and safer antileukemic therapies.
Keywords:
L-asparaginase
; liposomes
; drug delivery
; leukemia
; cationic polymers
; prolonged release
; cell viability
; targeted therapy
; Raji cells
1. Introduction
L-asparaginase (L-ASNase, EC 3.5.1.1) is an indispensable component of multi-agent chemotherapy protocols for acute lymphoblastic leukemia (ALL), the most common childhood cancer [1,2,3,4,5,6,7]. Its therapeutic mechanism is elegant yet effective: it catalyzes the hydrolysis of L-asparagine to L-aspartic acid and ammonia, thereby depleting the circulating pool of this amino acid. While normal cells can synthesize their own L-asparagine via the enzyme asparagine synthetase, many leukemic cells lack this capability. This metabolic defect renders them exquisitely sensitive to asparagine deprivation, leading to an inhibition of protein synthesis and subsequent apoptotic cell death.
Despite its critical role in modern oncology, the clinical utility of bacterial L-ASNases, particularly the one derived from Escherichia coli (EcA), is fraught with significant challenges [8,9,10,11,12,13,14,15]. A primary obstacle is its high immunogenicity. As a foreign protein, EcA can elicit a host immune response, leading to the formation of neutralizing antibodies that inactivate the enzyme and hypersensitivity reactions ranging from mild rashes to life-threatening anaphylaxis. Furthermore, EcA exhibits a short plasma half-life (typically < 24 hours), necessitating frequent administrations that increase the risk of immunogenic responses and patient discomfort. Other associated toxicities include pancreatitis, coagulopathy, and neurotoxicity.
To address these shortcomings, several strategies have been pursued. The most clinically successful to date is PEGylation—the covalent attachment of polyethylene glycol (PEG) chains to the enzyme surface. This has led to marketed products like Oncaspar® (pegaspargase) [16,17,18,19,20,21], which offers a significantly longer half-life and reduced immunogenicity. However, PEGylation is not a universal solution; it can sometimes diminish the enzyme’s specific activity, and “silent inactivation” mediated by non-neutralizing anti-PEG antibodies may still occur. This has prompted the development of alternative delivery systems aimed at enhancing the therapeutic index of L-ASNase. Therefore, we focus on designing L-ASNase conjugates with polyamines, which provide an optimal pH environment, enzyme stabilization, and—most importantly—enhanced efficacy against cancer cells through interactions with the polyamine transport system (PTS) receptors on leukemic cells [22]. To realize the concept of a targeted therapeutic selectively acting on cancer cells, the internalization efficiency of the conjugates can be further improved by employing delivery platforms with increased permeability, such as liposomal formulations.
Liposomes, self-assembled vesicles composed of a lipid bilayer enclosing an aqueous core, represent a highly versatile and clinically validated platform for drug delivery [23,24,25,26,27,28,29]. By encapsulating L-ASNase, liposomes can effectively shield it from proteolytic enzymes and the host immune system, thereby prolonging its circulation time and mitigating immunogenic reactions. Moreover, the physicochemical properties of liposomes can be precisely tuned to control drug release and achieve targeted delivery. Cationic liposomes, for instance, can leverage the net negative charge of cancer cell membranes (due to an overabundance of anionic components like phosphatidylserine) to promote electrostatic interactions and enhance cellular uptake via endocytosis.
Early foundational work by Cruz et al. (1993) demonstrated that liposomal encapsulation of L-ASNase markedly improved enzymatic stability and reduced cellular toxicity, setting the stage for in vivo pharmacokinetic studies [30]. However, these initial formulations had moderate circulation times and suboptimal control over enzyme release, limiting therapeutic potential.
Based on this foundation, Jorge et al. [31], in their study from 1994, developed liposomal formulations of palmitoyl-L- ASNase with the aim of enhancing the loading of the enzyme into liposomes [31]. These formulations demonstrated a significant prolongation of the circulation half-life, while simultaneously minimizing acute toxicity, all while maintaining the antitumor efficacy. This approach addressed the issue of limited circulation time that had been observed in previous research. Despite these advances, immunogenicity and hypersensitivity concerns remained largely unresolved.
Gaspar et al. (1996) further advanced the field by demonstrating that liposomal encapsulation could prevent anaphylactic reactions and double survival in animal models, highlighting the immunoprotective role of liposomes [32]. While these results improved understanding of immune response mitigation, issues related to drug loading and dosing efficiency persisted.
More recently, De and Venkatesh (2012) delved into the realm of optimizing encapsulation efficacy and release kinetics within liposomal L-ASNase formulations, with a particular emphasis on charged liposomes supplemented with sterylamine or dicetyl phosphate. Their findings demonstrated enhanced enzyme retention, meticulously controlled release mechanisms, and enhanced cytotoxic effects against cancerous cells in vitro [33]. This refinement addressed earlier shortcomings related to enzyme leakage and inconsistent activity.
Finally, Guimarães et al. (2022) introduced next-generation PEG-grafted liposomes that significantly enhanced enzyme stability, reduced hypersensitivity, and exhibited superior cytotoxicity toward leukemic cells [25]. This approach marked a critical step toward improved targeting and immune evasion. Nonetheless, challenges related to low drug loading and active enzyme content still limit clinical translation.
Importantly, despite these advances, none of these studies adequately addressed the crucial aspects of targeted drug release and selective penetration into cancer cells. The formulations largely relied on passive delivery mechanisms without specific tumor cell targeting or stimuli-responsive release strategies. Recognizing these unmet needs, we have turned our efforts toward developing conjugates and delivery systems that enable selective internalization by cancer cells—particularly through receptor-mediated mechanisms—and controlled, tumor-specific enzyme release. This focus aims to maximize therapeutic efficacy while minimizing off-target effects, thus overcoming a critical gap in previous liposomal L-ASNase research. Our work aims to fill this gap by designing systems that combine the pharmacokinetic advantages of liposomes with active targeting and controlled release features for improved leukemia therapy.
In this work, we hypothesized that a dual approach—combining the covalent modification of EcA with cationic agents and its subsequent encapsulation in liposomes—could synergistically enhance its anti-leukemic efficacy. We synthesized conjugates of L-asparaginase (EcA), known for its improved biopharmaceutical properties [3,34,35,36,37], with two polycationic carriers: the natural oligoamine spermine (EcA-spm) and a semi-synthetic cationic polymer, chitosan-polyethylenimine (EcA-Chit-PEI). The core rationale behind this approach is to increase the enzyme loading capacity within liposomal formulations. Due to their polycationic nature, these conjugates interact strongly with cardiolipin (CL), a diphosphatidyl lipid bearing two negative charges. This electrostatic interaction promotes the integration of conjugates into liposomal structures composed of phosphatidylcholine and cardiolipin (PC/CL). By manipulating the composition of these liposomes and exploring the impact of different polycations, we aim to determine whether a short or long-chain polycation is more effective in enhancing the properties of liposomal formulations. This process involves the formation of cardiolipin aggregates stabilized through polyamine-mediated cross-linking.
Beyond simply enhancing enzyme loading, conjugation with polyamines confers critical functional advantages during intracellular trafficking. Upon liposome-cell membrane fusion, the encapsulated enzyme is internalized via endocytosis, confining it initially within endosomes. For effective cytotoxic activity, EcA must efficiently escape from endosomes into the cytoplasm before lysosomal degradation can occur. So, the polyamine moieties play a pivotal protective role by facilitating endosomal escape, thereby accelerating enzyme release into the cytoplasm and preventing lysosomal digestion by lytic enzymes [38]. This enhanced endosomal escape mechanism extends the intracellular lifespan of EcA, allowing for sustained depletion of asparagine in cancer cells and enhancing therapeutic efficacy. PEI, for instance, is known for its rapid and efficient escape from endosomes, and the incorporation of polycations significantly enhances the intracellular bioavailability of the enzyme. However, pure PEI itself is not suitable due to its high cytotoxicity caused by its poisonous charge, which is why we are developing optimal formulations.
In summary, conjugating L-asparaginase with polyamines not only enhances liposomal loading through electrostatic clustering with cardiolipin but also promotes cellular uptake and endosomal escape, ultimately improving intracellular persistence and cytotoxic function against lymphoma cells. This dual advantage offers a promising strategy to overcome key limitations of current L-asparaginase delivery technologies.
2. Materials and Methods
2.1. Materials
L-asparaginase from E. coli (EcA) was purchased from Veropharm (Moscow, Russia). Phosphatidylcholine (PC) from soybean and cardiolipin (CL) from bovine heart were obtained from Avanti Polar Lipids (Alabaster, AL, USA). Spermine, chitosan (low molecular weight), and polyethylenimine (PEI, branched, avg. MW ~2 kDa) were sourced from Sigma-Aldrich. Eosin-5-isothiocyanate (EITC) and Bis-BODIPY™ FL C11-PC) (1,2-Bis-(4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Undecanoyl)-sn-Glycero-3-Phosphocholine) (BODIPY-lipid) were purchased from Thermo Fisher Scientific (Waltham, MA, USA). All other solvents and reagents were of analytical grade.
The development of a novel, highly effective liposomal L-asparaginase platform was made with Liposome Extruder LiposoFast LF-50, 5-50 ml, 50-1000 nm, (Avestin China, distributer DIA-M, Russia, Moscow)
2.2. Preparation and Labeling of Formulations
2.2.1. L-ASNase Covalent Conjugation
Cationic conjugates were synthesized by reacting EcA with spermine or a pre-synthesized chitosan-PEI copolymer at a 3/1 (w/w) enzyme-to-polymer ratio in phosphate-buffered saline (PBS, pH 7.4) using EDC/NHS chemistry as earlier described [3,22,36,39]. Purification was carried out using centrifuge filters with a cut-off weight of 100 kDa (5,000g, three times for 7 minutes).
2.2.2. Fluorescent Labeling of L-ASNase
For CLSM studies, EcA was labeled with EITC (EcA-eosin) by incubation at a 1:10 molar ratio in PBS (pH 7.4) for 2 hours at room temperature. Unconjugated dye was removed by dialysis (50 kDa cut-off). To obtain fluorescently labeled conjugates, the labeled enzyme was modified as described in paragraph 2.2.1 above.
2.2.3. Liposome Preparation
Liposomes were prepared by the thin-film hydration method. A mixture of PC and CL (80/20 mass ratio) was dissolved in EtOH, and the solvent was evaporated to form a thin lipid film. For fluorescent liposomes, 0.1 mol% of BODIPY-lipid was included in the initial mixture. The film was hydrated with a PBS solution containing either native EcA or its conjugates to achieve a final enzyme/lipid ratio of 30/70 (w/w). The resulting multilamellar vesicles were extruded 5 times through polycarbonate membranes with pore sizes of 100 nm or 400 nm using a mini-extruder (Avanti Polar Lipids) to produce unilamellar vesicles of defined size.
2.3. FTIR Spectroscopy
Fourier Transform Infrared (FTIR) spectra were recorded on Bruker TensorFlow 27 device (Bruker, Ettlingen, Germany) and MICRAN-3 FTIR microscope (Simex, Novosibirsk, Russia) equipped with an Attenuated Total Reflectance (ATR) accessory. A small aliquot of each sample (EcA, conjugates, EcA-eosin, empty liposomes, EcA-eosin loaded liposomes, etc) was deposited on the ATR crystal and dried. Spectra were collected in the 4000–900 cm⁻¹ range with a resolution of 2 cm⁻¹ and averaged over 50-70 scans.
2.4. In Vitro Release Study
The release kinetics of L-ASNase were evaluated using a dialysis method against a PBS buffer (pH 7.4). 1 mL of each formulation was placed in a dialysis bag (Spectra/Por, 300 kDa MWCO) and submerged in 50 mL of release medium at 37°C with gentle stirring. At predetermined time intervals (0, 15, 30, 60, 90, 180, 240, 360, 480, 600 min), 0.2 mL aliquots were withdrawn from the external medium and replaced with fresh buffer. The concentration of released L-ASNase was quantified by measuring A290 and Trp-fluorescence (λex = 290 nm, λemi = 350 nm) on SpectraMax M5 (Molecular Devices, CA, USA). The cumulative release percentage was calculated and plotted against time.
2.5. Cell Culture and Viability Assay
Human Burkitt’s lymphoma Raji cells (ATCC® CCL-86™) were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin at 37°C in a 5% CO₂ humidified atmosphere.
For the viability assay, cells were seeded in 96-well plates at a density of 2 × 10⁴ cells/well and incubated for 24 hours. The medium was then replaced with fresh medium containing various concentrations of L-ASNase formulations (from 0.1 to 30 U/mL). After 72 hours of incubation, cell viability was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. The absorbance was measured at 570 nm, and viability was expressed as a percentage relative to untreated control cells.
2.6. Confocal Laser Scanning Microscopy (CLSM)
Raji cells were seeded on poly-L-lysine coated coverslips in a 96-well plate. Cells were treated with fluorescently labeled formulations: EcA-eosin (red channel), BODIPY-liposomes (green channel), or the combined formulation. After a 4-hour incubation period, cells were washed three times with cold PBS to remove non-internalized formulations, fixed with 4% paraformaldehyde, and mounted on glass slides. Images were acquired using CLSM (Olympus FluoView FV1000). Eosin was excited at 488 nm and its emission collected at 560-590 nm. BODIPY was excited at 488 nm and its emission collected at 505-535 nm.
2.7. CLSM Image Quantification
To quantify the cellular uptake observed in CLSM images, the mean fluorescence intensity (MFI) per cell was calculated using ImageJ software. For each formulation, 10-15 distinct cells were randomly selected as regions of interest (ROIs). The MFI for both the red (Eosin-EcA) and green (BODIPY-Liposome) channels was measured within each ROI. A background region devoid of cells was also measured, and MFI of this background was used to normalize the cell-associated MFI by calculating the signal-to-background ratio (division). The average corrected MFI ratios and standard deviations were then calculated for each sample and plotted for comparison.
3. Results and Discussion
3.1. Design Rationale: Conjugate Synthesis and Liposomal Formulation
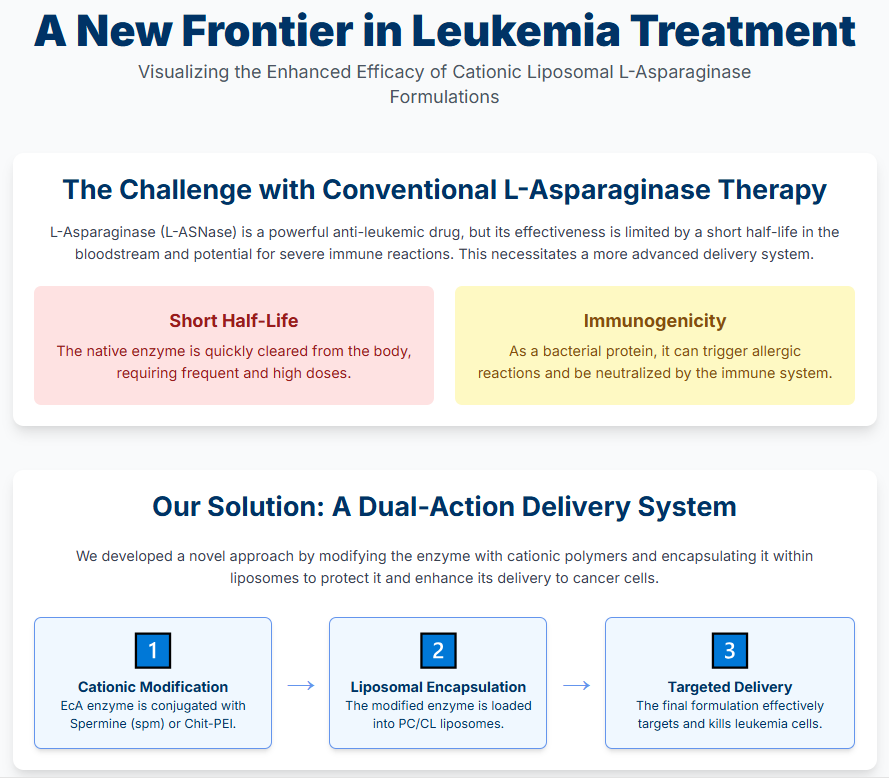
The strategic design of this study aimed to create robust delivery vehicles for L-ASNase. This was achieved through the conjugation of L-ASNase with functional molecules like spermine and a PEI-PEG copolymer, followed by their encapsulation within liposomes (Figure 1).
The choice of carbodiimide chemistry for conjugating L-ASNase was based on its proven efficacy in forming stable amide linkages between carboxyl groups and amines. By activating the carboxyl functionalities of L-ASNase and reacting them with the amine groups of spermine or the PEI-PEG copolymer, stable bioconjugates (L-ASNase-spermine and L-ASNase-PEI-PEG) were synthesized. This covalent linkage is crucial for maintaining the structural integrity of the conjugate during formulation and administration. The inclusion of PEI-PEG is particularly strategic: PEI’s polycationic nature is known to facilitate cellular uptake and can potentially aid in endosomal escape (as discussed above), while PEGylation confers enhanced colloidal stability, increases circulation half-life by reducing immunogenicity and non-specific protein adsorption, and improves overall biocompatibility. Spermine, as natural polyamine, was included into the L-asparaginase conjugate due to its ability to enhance cellular targeting through polyamine transport system. Compared to synthetic polymers, spermine is considered a more “intelligent” and biocompatible cationic modifier, while still providing the essential positive charge required for effective interaction with negatively charged cancer cell membranes.
The preparation of liposomal formulations via extrusion method was applied to produce liposomes with controlled size and lamellarity. This method allows for the reproducible fabrication of liposomes, which is essential for consistent therapeutic outcomes. The initial formation of a thin lipid film ensures uniform distribution of phosphatidylcholine and cardiolipin, key components for creating stable, fluid lipid bilayers capable of encapsulating the enzyme conjugates. Subsequent hydration and extrusion through polycarbonate membranes of defined pore sizes (as illustrated in Figure 1) are critical steps for breaking down larger vesicle structures into smaller, unilamellar liposomes, typically in the nanometer range (100-400 nm). This controlled particle size is advantageous for achieving optimal biodistribution, potentially enhancing passive targeting to tumors via the enhanced permeability and retention (EPR) effect, and facilitating efficient interaction with target cells.
3.2. Physicochemical Characterization of Formulation
3.2.1. FTIR Spectroscopy
For CLSM studies, L-ASNase (EcA) was labeled with eosin isothiocyanate (EITC) to generate EcA-eosin conjugates by incubating the enzyme with EITC at a 1:10 molar ratio. This fluorescent labeling allowed direct visualization of the enzyme’s cellular uptake and intracellular distribution, providing critical insights into the interaction of the enzyme-loaded liposomes with target cells.
The successful incorporation of EcA within the liposomal structure was unequivocally confirmed by FTIR spectroscopy (Figure 2). Analysis of the spectra reveals a clear superposition of signals from individual components within the final formulation, evidencing the enzyme’s association with the lipid membrane. The spectrum of EcA-eosin incorporated in PC/CL liposomes (blue line) represents a composite profile combining key features of both lipids and protein. Prominent lipid bands include the intense symmetric and asymmetric ν(C-H) stretching vibrations of acyl chains between 2850 and 3000 cm⁻¹, as well as the characteristic ν(C=O) ester stretch of phosphatidylcholine at approximately 1738 cm⁻¹, which define the empty PC/CL liposome spectrum (green line).
Meanwhile, the final formulation’s spectrum displays hallmark protein absorptions consistent with EcA: the Amide I band (~1655 cm⁻¹), primarily from C=O stretching in the peptide backbone, and the Amide II band (~1545 cm⁻¹), arising from N-H bending and C-N stretching. These features align with the native EcA spectrum (black line). Importantly, the presence of the eosin fluorescent label is substantiated by distinctive aromatic ring vibrations at ~1400 cm⁻¹ (ν(C=C)) and ether stretches near ~1030 cm⁻¹ (ν(C-O)), clearly visible in the EcA-eosin spectrum (red line). The coexistence of these lipid, protein, and dye-specific bands within a single spectrum provides compelling evidence that EcA-eosin is physically associated with the liposomal matrix, confirming successful encapsulation and the formation of the intended drug-delivery system.
3.2.2. Loading Degree and Activity Studies
Following conjugation, any unbound or free enzyme was effectively separated from the liposomal one through dialysis and subsequently by HPLC methods, ensuring a high degree of purity for the conjugated forms. The entrapment efficiency and enzyme-to-lipid loading capacity of native EcA and its conjugates EcA-spm, EcA-PEI-g-PEG were evaluated in phosphatidylcholine/cardiolipin (PC/CL, 80/20 mol%) liposomes of two distinct sizes—100 nm and 400 nm (Table 1).
In 100 nm liposomes, native EcA showed a moderate entrapment efficiency of 63 ± 2%, reflecting typical encapsulation challenges for unmodified enzymes. Fotr conjugated enzyme, the entrapment efficiency significantly increased: EcA-spm reached 89 ± 1%, and EcA-PEI-g-PEG achieved 82 ± 2%. These considerable enhancements can be attributed to the introduction of polycationic groups on the enzyme surface, which foster stronger electrostatic interactions with the negatively charged cardiolipin, thereby promoting more effective incorporation within the liposomal bilayer or aqueous core.
For larger liposomes (400 nm diameter), entrapment efficiencies improved further across all formulations, with native EcA at 70 ± 5%, EcA-spm nearly complete at 97 ± 1%, and EcA-PEI-g-PEG at 94 ± 1%. The increased internal volume and surface area of the larger liposomes likely facilitate a more favorable microenvironment for enzyme encapsulation, particularly when enhanced by polycationic conjugation.
Correspondingly, the term that better reflects this parameter is the enzyme loading capacity of the carrier, expressed as the ratio of L-ASNase to lipid (µg enzyme per µmol lipid). This parameter exhibited a similar trend across formulations. In 100 nm liposomes, enzyme loadings were measured at 85 ± 3 µg/µmol for native EcA, 120 ± 2 µg/µmol for EcA-spm, and 111 ± 3 µg/µmol for EcA-PEI-g-PEG conjugates. Notably, these values significantly increased in 400 nm liposomes, reaching 95 ± 7 µg/µmol (EcA), 131 ± 2 µg/µmol (EcA-spm), and 127 ± 2 µg/µmol (EcA-PEI-g-PEG).
The pronounced increase in enzyme loading per unit lipid for the conjugated formulations clearly indicates improved payload capacity and entrapment efficiency. This demonstrates the successful achievement of the formulation goals. Importantly, the short-chain natural spermine conjugate sufficiently enhances loading capacity without restricting the enzyme’s conformational mobility, which is critical for maintaining activity and ensuring efficient encapsulation within the liposomal matrix. In contrast, longer-chain synthetic polymers, while effective, may impose steric constraints that limit enzyme dynamics and loading efficiency.
In summary, the results illustrate that conjugation of EcA with polycationic moieties significantly improves its incorporation into PC/CL liposomes, particularly favoring higher entrapment efficiencies and enzyme loading. Larger liposome size synergistically enhances these parameters, suggesting that both chemical modification and physical formulation variables are critical levers to optimize liposomal delivery systems for L-ASNase.
The enzymatic activity of native L-ASNase, its conjugates (L-ASNase-spermine and L-ASNase-PEI-PEG), and their respective liposomal formulations was evaluated using Circular Dichroism (CD) spectroscopy (Figure 3, Table 1). The CD signal at 210 nm corresponds to changes associated with the conversion of L-asparagine (Asn) to L-aspartate (Asp), allowing real-time monitoring of enzymatic activity [40]. Figure 3 show kinetic curves illustrating the substrate hydrolysis. The initial CD values for all samples start similarly (~36 mdeg). From these curves, it is clear that all formulations retain enzymatic activity to varying degrees, with differences attributable to conjugation and encapsulation effects. Notably, while native L-ASNase demonstrates the highest initial reaction rate, the conjugated and liposomal forms maintain substantial activity, indicating that neither chemical modification nor incorporation into liposomes abolishes catalytic function. This figure serves as a crucial baseline for the subsequent analysis and comparison of enzymatic efficiencies across different formulations. Understanding these activity profiles allows us to evaluate how modifications and delivery methods influence enzyme performance, stability, and potential therapeutic effectiveness, guiding further optimization of the delivery system.
Activities comparison of native L-ASNase with Conjugates
Comparison of non-liposomal preparations revealed a notable alteration in the activity of the enzyme as a result of the conjugation. EcA-spm exhibited a marked increase in activity (380 U/mg) compared to the native EcA (330 U/mg). This suggests that the attachment of spermine may positively influence the enzyme’s conformational state or its accessibility to the substrate, potentially due to its charged nature or specific interaction with the enzyme’s active site or surrounding microenvironment. In contrast, the conjugate (EcA-PEI-g-PEG) showed a similar activity (327 U/mg) relative to the native enzyme (330 U/mg). This is due to the bulky PEI-PEG polymer chains, which might partially shield the active site or impede substrate binding. However, the overall change in activity for the PEI-PEG conjugate is minimal, indicating that the conjugation process did not severely compromise the enzyme’s catalytic function. Importantly, kinetic analysis showed that the Michaelis constant (KM) values for all preparations remained practically unchanged, indicating that substrate affinity was not significantly affected by the conjugation. This confirms that the modifications predominantly influence enzyme stability and activity without compromising substrate binding.
Activities comparison of Liposomal Formulations with Non-Liposomal Preparations
Encapsulation of the conjugates within liposomes also influenced their enzymatic activity. Generally, liposomal formulations, especially those with smaller vesicle sizes, showed a trend towards slightly enhancing the activity observed in their non-liposomal counterparts, although with some variations.
For the L-ASNase-spermine conjugate, liposomal encapsulation (both 100 nm and 400 nm) resulted in activities (370 U/mg) that were comparable to the non-liposomal conjugate (380 U/mg). The 400 nm liposomes in particular showed the highest activity among all preparations, suggesting that the specific liposomal environment at this size might be conducive to enzyme function, perhaps by stabilizing the enzyme or facilitating substrate diffusion.
In the case of the L-ASNase-PEI-g-PEG conjugate, the activity within the 100 nm liposomes (306 U/mg) was slightly reduced compared to its non-liposomal form (327 U/mg). This could be due to factors like partial enzyme denaturation during the encapsulation process or limitations in substrate diffusion through the liposomal membrane. However, encapsulation within the 400 nm liposomes led to recovery of activity (350 U/mg), bringing it closer to the native enzyme’s performance and even exceeding the activity of the non-liposomal PEI-PEG conjugate. This suggests that the larger liposomal structure may offer a more favorable microenvironment for this particular conjugate, mitigating the potential steric hindrance from the PEI-PEG chains.
It is important to note that the observed variations in activity upon conjugation and encapsulation are common phenomena in enzyme delivery systems. The aim of these modifications is often not only to increase intrinsic activity but to improve pharmacokinetic properties such as stability, circulation time, and targeting.
Stability of Liposomal L-ASNase Formulations during Storage
The stability of L-asparaginase (L-ASNase) formulations is a critical parameter influencing their therapeutic viability and shelf-life. To evaluate this, we assessed the residual enzymatic activity of different formulations after one month of storage at +4 °C in PBS (Table 1). Our findings indicate that liposomal encapsulation significantly enhances the stability of L-ASNase compared to non-liposomal counterparts.
For the non-liposomal enzyme formulations, the residual activity after one month decreased to 63%, 83%, and 75% of the initial activity for EcA, EcA-spm, and EcA-PEI-g-PEG, respectively. Encapsulation within PC/Cl 80/20 liposomes provided a protective effect, as observed by higher retention of enzymatic activity. Specifically, 100 nm liposomes preserved 84%, 93%, and 91% of initial activity for EcA, EcA-spm, and EcA-PEI-g-PEG, respectively, superseding their non-liposomal analogs. This enhanced stability likely stems from the liposomal bilayer shielding the enzyme from environmental factors such as proteolytic degradation or denaturation.
Formulations encapsulated in larger, 400 nm PC/Cl 80/20 liposomes also demonstrated improved stability, though with a slightly lower retention for EcA (77%) and EcA-PEI-g-PEG (81%) compared to their smaller counterparts. The retention of activity for EcA-spm remained relatively high (90%) in 400 nm liposomes, indicating that polymer conjugation combined with encapsulation can synergistically contribute to enzyme preservation.
Overall, these data suggest that liposomal encapsulation, especially within 100 nm vesicles, substantially stabilizes L-ASNase formulations during storage at +4 °C, reducing enzymatic loss over time. Moreover, polymer modifications such as spermine (spm) conjugation or PEI-g-PEG grafting further enhance stability, potentially by improving enzyme-lipid interactions or providing steric protection.
3.2.3. Circular Dichroism Spectra and Secondary Structure Analysis of EcA Formulations
Changes in enzymatic activity were observed upon encapsulation of the enzymes into liposomes. To elucidate the possible molecular basis of these effects, we investigated the structural characteristics of the enzymes and their formulations. Circular dichroism (CD) spectroscopy in the far-UV region is a sensitive and widely used method for probing protein secondary structure and detecting conformational changes resulting from chemical modifications or incorporation into delivery systems such as liposomes. In this study, far-UV CD spectroscopy was employed to assess the structural integrity and conformational alterations of EcA upon conjugation with polymers and subsequent liposomal encapsulation. This allowed evaluation of the impact of formulation strategies on the enzyme’s secondary structure.
Figure 4 shows the far-UV CD spectra of native EcA, its conjugates, and their liposomal formulations (phosphatidylcholine/cardiolipin, 80/20). The spectra exhibit characteristic features typical of protein secondary structure. Specifically, native EcA displays two negative minima near 209–210 nm and 222 nm, along with a positive maximum around 193–195 nm, consistent with substantial α-helical content.
Spermine conjugation has minimal impact on the secondary structure of EcA, and encapsulation of EcA-spm conjugates into liposomes induces only negligible structural changes. Notably, the EcA-PEI-g-PEG conjugate exhibited an increased intensity at the 222 nm band, suggesting a slight enhancement or stabilization of α-helical segments upon conjugation.
In contrast, the spectrum of native EcA incorporated into liposomes shows pronounced deviations from that of the free enzyme. It exhibits a relatively stronger negative band near 209 nm and a reduction in the 222 nm signal, indicative of a decrease in α-helical content coupled with an increase in random coil structures. These changes suggest conformational modifications induced by liposomal encapsulation.
Secondary structure content was quantitatively assessed via deconvolution of the CD spectra using the CDNN software package, with results summarized in Figure 4b. Native EcA and the EcA-spm conjugate both exhibit substantial α-helical content (~42–43%), minor β-structures content and a significant proportion of random coil. The EcA-PEI-g-PEG conjugate reveals an increase in α-helix (48%) alongside a slight decrease in random coil.
In contrast, the liposomal formulations show a reduced α-helix proportion, particularly for EcA-spm and EcA-PEI-g-PEG in liposomes (27% and 25%, respectively). This decrease is accompanied by an augmentation of the antiparallel beta-sheet content by 13–16%, as well as a moderate increase in the random coil fraction. This is due to the formation of protein clusters on the surface of the bilayer, which appears as aggregated protein structures. These findings suggest that incorporation into liposomes induces specific secondary structural changes likely arising from protein–lipid interactions, which enhance the enzyme’s stability and activity.
3.3. In Vitro L-ASNase Release Kinetics
A critical attribute of an effective drug delivery system is its ability to provide sustained release, thereby maintaining therapeutic drug concentrations over an extended period. Our in vitro release studies (Figure 5, Table 2) demonstrate that liposomal encapsulation profoundly alters the release profile of L-ASNase.
Kinetic curves of cumulative L-ASNase release clearly show a shift from rapid release for free EcA to a significantly prolonged, sustained release for the liposomal formulations, with the EcA-spermine conjugate showing the greatest retention.
The free enzyme EcA exhibited rapid diffusion through the dialysis membrane, with a half-release time (T50%) of only 24 ± 2 minutes and 80% of the enzyme released within 52 ± 5 minutes. This rapid release mimics the fast clearance expected in vivo.
In stark contrast, encapsulation of EcA in 100 nm liposomes dramatically retarded its release. The T50% nearly doubled to 45 ± 5 minutes, and the time to 80% release (T80%) was extended almost five-fold to 240 ± 20 minutes. This sustained release is attributed to the physical barrier imposed by the lipid bilayer, which the large enzyme molecule must traverse to be released.
The most significant prolongation of release was achieved for conjugates in liposomes. Liposomal conjugates displayed the slowest release kinetics, with a T50% of 56 – 70 minutes and a T80% of 330 – 420 min. This enhanced retention can be explained by a dual mechanism. In addition to the physical barrier of the liposome, there are strong electrostatic interactions between the positively charged polyamine-conjugated enzyme and the negatively charged cardiolipin headgroups in the inner leaflet of the liposome bilayer. This ionic tethering further hinders the enzyme’s diffusion out of the vesicle, leading to a more controlled and prolonged release profile, which is highly desirable for improving therapeutic efficacy and reducing dosing frequency.
3.5. Cytotoxicity Evaluation of Liposomal Formulations of EcA and Its Conjugates
The ultimate goal of these formulations is to enhance the killing of cancer cells. The cytotoxicity assay (Figure 6, Table 3) confirmed that all L-ASNase formulations induced a dose-dependent reduction in the viability of Raji lymphoma cells. However, the delivery vehicle and modifications had a profound impact on efficacy. Across all tested preparations, cell viability decreased as enzyme concentration increased, confirming dose-dependent their cytotoxic efficacy.
The half-maximal inhibitory concentration (IC₅₀) values, summarized in Table 3, reveal significant differences in potency contingent upon formulation type and liposome size. For the non-liposomal preparations, EcA exhibited an IC₅₀ of 35 ± 5 U/mL, whereas its conjugates EcA-SPM and EcA-Chit-PEI demonstrated markedly enhanced cytotoxicity with IC₅₀ values reduced to 8 ± 1 U/mL and 7 ± 1 U/mL, respectively. This enhancement likely reflects increased cellular uptake or improved enzyme stability imparted by surface conjugation.
Incorporation into liposomes further modulated the cytotoxic effects of L-asparaginase formulations. Notably, EcA encapsulated in 100 nm phosphatidylcholine/cardiolipin (PC/CL, 80/20) liposomes exhibited a twofold decrease in IC₅₀ to 20 ± 5 U/mL compared to the free enzyme, demonstrating the advantage of liposomal delivery in enhancing cellular efficacy. Importantly, control liposomal formulations without enzyme show no cytostatic activity against the cells, confirming that the observed effects derive from the active payload.
Strikingly, the 100 nm liposomal formulations of EcA-spm and EcA-Chit-PEI conjugates displayed even greater cytotoxicity, with IC₅₀ values of 4.5 ± 0.8 U/mL and 4.7 ± 0.5 U/mL, respectively, underscoring the synergistic benefit of polymer conjugation combined with liposomal encapsulation.
For 400 nm EcA liposomes IC₅₀ is 9 ± 2 U/mL—an improvement over free EcA is observed but it is still less active than the 100 nm formulation—suggesting that smaller liposomes may facilitate more efficient cellular interaction and uptake.
Conversely, 400 nm liposomal conjugated formulations exhibited slightly higher IC₅₀ values compared to their 100 nm counterparts (EcA-spm: 11 ± 1 U/mL; EcA-Chit-PEI: 13 ± 2 U/mL), despite showing higher enzymatic activity in vitro (Table 1). This discrepancy may result from altered biodistribution or increased steric hindrance in larger liposomes, potentially impeding cellular internalization and thus reducing cytotoxic efficacy.
Smaller liposomes (100 nm) likely benefit from enhanced cellular internalization due to favorable size for endocytosis, while larger liposomes (400 nm) may offer prolonged circulation or altered biodistribution but with less efficient immediate uptake. Overall, these results underscore the promise of conjugation and liposomal encapsulation strategies in improving L-asparaginase therapeutic efficacy against lymphoma cells. Optimization of liposome size and surface functionality is critical for maximizing cytotoxic potential while minimizing off-target effects.
3.5. Cellular L-ASNase and Liposome Uptake and Colocalization by CLSM
To understand the mechanism behind enhanced cytotoxicity, we used CLSM to visualize the interaction of the formulations with Raji cells. The fluorescence spectra in Figure 7 confirm that the chosen dyes (Eosin and BODIPY) have distinct emission profiles, and their combination in the liposomal formulation does not lead to significant signal quenching or unwanted FRET (Förster Resonance Energy Transfer), validating their use as independent reporters for the enzyme and the liposome.
Specifically, liposomes containing EcA-spm exhibited more pronounced quenching of eosin fluorescence compared to those with EcA-Chit-PEI. This enhanced quenching likely reflects closer spatial proximity or stronger interactions between the eosin-labeled enzyme molecules and the liposomal bilayer in the EcA-spm formulation. Such effects may result from differences in enzyme orientation or distribution on versus within the liposomal membrane, stabilized by electrostatic interactions with cardiolipin. It is plausible that the PEI-PEG polymer conjugates predominantly localize on the external surface of the liposomes, whereas EcA-spm is incorporated more internally, within the aqueous core or at the inner leaflet of the bilayer. This spatial arrangement could explain the stronger fluorescence quenching observed for EcA-spm.
Conversely, the EcA-Chit-PEI formulation in liposomes displayed a distinct bathochromic (red) shift in the eosin emission maximum compared to EcA-spm liposomes, indicating an altered local polarity or microenvironment around the eosin probe. This red shift suggests modifications in the electronic environment, possibly arising from differences in polymer coating or the nature of conjugate-liposome interactions. Specifically, eosin experience within EcA-Chit-PEI liposomes corresponds to a more hydrophobic or sterically constrained environment relative to eosin-labeled EcA in aqueous solution. In contrast, eosin fluorescence in EcA-spm liposomes remains largely unchanged relative to free EcA, supporting the hypothesis that the PEI-PEG conjugated enzyme is mainly surface-associated—likely adsorbed onto the hydrophobic lipid layer—while EcA-spm is embedded more internally in a predominantly aqueous environment.
The CLSM images (Figure 8) provide a vivid illustration of the drug delivery process. Cells treated with free EcA-eosin showed only weak, diffuse red fluorescence, indicating poor cellular association and/or uptake. In contrast, all liposomal formulations led to a dramatic increase in cell-associated fluorescence. The signal appeared as bright, distinct puncta within the cells, a classic pattern for uptake via endocytosis, where the liposomes are internalized into endosomes and lysosomes.
Crucially, the merged images for the liposomal formulations show extensive colocalization (yellow signal) of the enzyme (red) and the liposome (green). This is direct evidence that the enzyme remains encapsulated during cellular uptake and is delivered into the cell as part of the liposomal package. The most visually striking result was for the EcA-spermine-eosin in liposomes-BODIPY sample. These cells displayed the most intense green and red fluorescence, which merged to create a brilliant yellow signal, indicating a massive uptake of the intact drug-loaded liposomes.
The data presented in Figure 9 demonstrate a clear enhancement in the binding efficiency of L-asparaginase (L-ASNase) to Raji cancer cells when the enzyme is encapsulated within liposomes compared to its free form. Free EcA-eosin showed a moderate fluorescence signal-to-background ratio of 1.67, indicating baseline cellular association. Incorporation of EcA-eosin into BODIPY-labeled liposomes significantly increased this ratio to 3.17, suggesting improved enzyme delivery and retention on or within the cancer cells. Notably, the EcA-spm-eosin liposomal formulation exhibited the highest signal ratio of 3.57, highlighting that spermine conjugation further enhances enzyme interaction or uptake by the cells. This formulation appears to penetrate more efficiently inside the cells, supporting enhanced internalization.
In contrast, the EcA-Chit-PEI-eosin liposomes displayed a more moderate increase to 2.19, still surpassing free enzyme but lower than the spermine conjugate. Microscopy observations revealed that EcA-Chit-PEI tends to remain associated with the cell surface, with visible particles approximately 1 μm in size representing aggregates or intact liposomal formulations bound extracellularly. Interestingly, the BODIPY fluorescence, reflecting liposome presence, was highest for plain liposomes (4.08) and somewhat reduced for both conjugated formulations (3.14 for EcA-spm and 2.93 for EcA-Chit-PEI), which may be attributed to altered liposome-cell interactions or changes in lipid probe exposure due to conjugation. Taken together, these findings suggest that liposomal encapsulation substantially improves L-ASNase targeting Raji cells, with spermine conjugation providing an additional advantage through enhanced cellular internalization, while Chit-PEI conjugates primarily remain surface-bound.
3.6. Correlation Between Binding to Raji Cells and Cytotoxicity of L-ASNase Formulations
Figure 10 illustrates the relationship between the extent of binding of L-asparaginase (L-ASNase) formulations to Raji cells, expressed as the fluorescence signal-to-background ratio (binding index), and their cytotoxic potency, represented by IC₅₀ values against the same cell line. The data reveal a clear inverse correlation between binding efficiency and IC₅₀, indicating that stronger association with Raji cells corresponds to enhanced cytotoxic activity.
Our data clearly indicate a correlation between binding efficiency and cytostatic activity (IC₅₀): formulations with higher binding indices exhibit enhanced cytotoxic effects. For instance, free EcA shows a modest binding index of approximately 1.67 and a correspondingly high IC₅₀ value of 35 U/mL, suggesting relatively weak cell association and limited cytotoxicity. Incorporation of EcA into liposomes substantially increased binding to 3.17, with IC₅₀ improving nearly two-fold to 20 U/mL, demonstrating the beneficial effect of liposomal encapsulation on cell targeting and therapeutic activity.
Notably, liposomal formulation of EcA conjugates with polyamines falls out of the correlation: EcA-spm without liposomes demonstrated a high binding index of 4.51 but a somewhat higher IC₅₀ of 8 U/mL compared to its liposomal counterpart. This suggests that while conjugation increases cellular association, liposomal encapsulation additionally enhances delivery, through improved cellular uptake and due to enzyme stabilization within the Raji cells. EcA-spm in liposomes achieved the intermediate binding ratio among еру tested formulations (3.57) but id show the lowest IC₅₀ (4.5 U/mL), indicating significantly enhanced cytotoxic potential upon encapsulation in the liposomes. Similarly, EcA-Chit-PEI in liposomes exhibited an intermediate binding index (2.19) but higher cytotoxicity (IC₅₀ of 4.7 U/mL) compared to expected, which is probably due to differences in cellular uptake mechanisms.
Additional data from related studies on polyamine-modified L-ASNase (our prior work [22]) further support these conclusions. For example, free EcA mixed with spermidine caproate (spd-caproate) showed a binding index of 1.89 and IC₅₀ of 25 U/mL. In contrast, EcA conjugated to spermine increased binding affinity to 4.06 with a lowered IC₅₀ of 15 U/mL, confirming the advantageous role of polyamine-based targeting moieties. Interestingly, EcA-spm conjugate in the presence of concurrent inhibitor of polyamine transport system PTS spd-caproate exhibited lower binding (2.38) and reduced cytotoxicity (IC₅₀ of 40 U/mL) due to blocking of PTS for EcA-spm conjugate [22].
In summary, these results consistently show that enhancing the binding affinity of L-ASNase formulations to Raji cells through liposomal encapsulation and targeted polymer conjugations correlate strongly with improved cytotoxicity. This underscores the importance of formulation strategies that maximize cellular interactions to optimize therapeutic outcomes in cancer treatment.
4. Conclusions
In this study, we have successfully engineered a novel dual-strategy delivery platform for L-asparaginase, designed to overcome the well-documented clinical limitations of the native enzyme. By combining the covalent modification of E. coli L-asparaginase (EcA) with cationic polymers and subsequent encapsulation within phosphatidylcholine/cardiolipin liposomes, we have developed a system with significantly superior formulation characteristics and therapeutic efficacy.
Our approach resulted in a dramatic improvement in both enzyme entrapment efficiency, reaching up to 97%, and a significantly higher final enzyme-to-lipid payload ratio compared to conventional methods. This structural optimization translated directly into enhanced functional performance. The liposomal formulations demonstrated a sustained-release profile, prolonging enzyme availability, and more importantly, exhibited a nearly eight-fold increase in cytotoxic potency against Raji lymphoma cells, with the IC₅₀ value dropping from 35 U/mL for free EcA to as low as 4.5 U/mL for the 100 nm EcA-spermine liposomal conjugate.
Crucially, our mechanistic investigations using confocal microscopy provided direct visual evidence that this profound enhancement in cytotoxicity is unequivocally linked to an increase in the cellular internalization of the enzyme. The colocalization of the enzyme and the lipid carrier confirmed that EcA is delivered into cancer cells as an intact liposomal package. This finding underscores a critical insight: the therapeutic success of L-asparaginase is more dependent on efficient intracellular delivery and subsequent internal asparagine depletion than on its extracellular enzymatic activity.
Taken together, the synergistic benefits of cationic polymer conjugation with polycations and encapsulation in anionic liposomal represent a significant advancement in L-ASNase delivery. This platform not only enhances enzyme loading and stability but also fundamentally improves the mechanism of action by ensuring the enzyme reaches its intracellular target. These findings position our polyamine-conjugated liposomal formulations as a highly promising next-generation therapeutic strategy, holding the potential to improve clinical outcomes in the treatment of acute lymphoblastic leukemia and other sensitive malignancies.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, E.V.K. and I.D.Z.; methodology, I.D.Z., A.A.E., D.A.B. and E.V.K.; formal analysis, I.D.Z., A.A.E.; investigation, I.D.Z., A.A.E., D.A.B, AVL, AVB and E.V.K.; data curation, I.D.Z.; writing—original draft preparation, I.D.Z.; writing—review and editing, E.V.K.; project supervision, E.V.K.; funding acquisition, E.V.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation, grant number 24-25-00104.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the main text and Supplementary Materials.
Acknowledgments
This work was performed using the equipment FTIR microscope MICRAN-3 (Simex, Novosibirsk, Russia), FTIR spectrometer Bruker Tensor 27 (Bruker, Ettlingen, Germany), Jasco J-815 CD Spectrometer (JASCO, Tokyo, Japan), AFM microscope NTEGRA II (NT-MDT Spectrum Instruments, Moscow, Russia), and confocal laser scanning microscope FluoView FV1000 (Olympus, Tokyo, Japan) of the program of Moscow State University development. This work was performed with the support of the DIA-M company, which provided key equipment, including, a lyophilizer and extruder for the liposome formation, critical for the quantitative and reproducible aspects of this research.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ALL | Acute lymphoblastic leukemia |
| CD | Circular dichroism |
| CL | Cardiolipin |
| FTIR | Fourier-transform infrared |
| L-ASNase | L-asparaginase |
| PC | Phosphatidylcholine |
| PEG | Polyethylene glycol |
| spm | spermine |
References
- Wriston, J.C.; Yelln, T.O. L-Asparaginase : A Review. Adv. Enzymol. Relat. Areas Mol. Biol. 1973, 39, 185–248. [Google Scholar]
- Schmidt, M.P.; Ivanov, A.V.; Coriu, D.; Miron, I.C. L-Asparaginase Toxicity in the Treatment of Children and Adolescents with Acute Lymphoblastic Leukemia. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef]
- Zlotnikov, I.D.; Kudryashova, E. V A Novel Approach for the Activity Assessment of L-Asparaginase Formulations When Dealing with Complex Biological Samples. Int. J. Mol. Sci. 2025, 26, 5227. [Google Scholar] [CrossRef]
- Piek, C.J.; Putteman, G.R.; Teske, E. Evaluation of the Results of a L-Asparaginase-Based Continuous Chemotherapy Protocol versus a Short Doxorubicin-Based Induction Chemotherapy Protocol in Dogs with Malignant Lymphoma. Vet. Q. 1999, 21, 44–49. [Google Scholar] [CrossRef]
- Fernandez, C.A.; Cai, X.; Elozory, A.; Liu, C.; Panetta, J.C.; Jeha, S. High-Throughput Asparaginase Activity Assay in Serum of Children with Leukemia. Int. J. Clin. Exp. Med. 2013, 6, 478–487. [Google Scholar]
- Simas, R.G.; Krebs Kleingesinds, E.; Pessoa Junior, A.; Long, P.F. An Improved Method for Simple and Accurate Colorimetric Determination of L-Asparaginase Enzyme Activity Using Nessler’s Reagent. J. Chem. Technol. Biotechnol. 2021, 96, 1326–1332. [Google Scholar] [CrossRef]
- Borsakova, D. V.; Sinauridze, E.I. L-Asparaginase: New Approaches to Improve Pharmacological Characteristics. Pediatr. Hematol. Immunopathol. 2018, 17, 82–99. [Google Scholar] [CrossRef]
- de Moura, W.A.F.; Schultz, L.; Breyer, C.A.; de Oliveira, A.L.P.; Tairum, C.A.; Fernandes, G.C.; Toyama, M.H.; Pessoa-Jr, A.; Monteiro, G.; de Oliveira, M.A. Functional and Structural Evaluation of the Antileukaemic Enzyme L-Asparaginase II Expressed at Low Temperature by Different Escherichia Coli Strains. Biotechnol. Lett. 2020, 42, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Juluri, K.R.; Siu, C.; Cassaday, R.D. Asparaginase in the Treatment of Acute Lymphoblastic Leukemia in Adults: Current Evidence and Place in Therapy. Blood Lymphat. Cancer Targets Ther. 2022, 12, 55–79. [Google Scholar] [CrossRef]
- Malakhova, M.A.; Pokrovskaya, M. V.; Alexandrova, S.S.; Sokolov, N.N.; Kudryashova, E. V. Regulation of Catalytic Activity of Recombinant L-Asparaginase from Rhodospirillum Rubrum by Conjugation with a PEG-Chitosan Copolymer. Moscow Univ. Chem. Bull. 2018, 73, 185–191. [Google Scholar] [CrossRef]
- Holcenberg, J.S.; Ericsson, L.; Roberts, J. Amino Acid Sequence of the Diazooxonorleucine Binding Site of Acinetobacter and Pseudomonas 7A Glutaminase--Asparaginase Enzymes. Biochemistry 1978, 17, 411–417. [Google Scholar] [CrossRef]
- Ottobre, M.; Colitto, L.; Marcone, M.I.; Krystal, E.; Moran, L.; Wittmund, L.E.; Gimenez, V.; Makiya, M. A Pediatric Laboratory Experience: Measurement of L-Asparaginase Activity in Patients with Acute Lymphoblastic Leukemia. a Multicentric Study. Blood 2024, 144, 2826–2826. [Google Scholar] [CrossRef]
- Lubkowski, J.; Wlodawer, A. Structural and Biochemical Properties of L-Asparaginase. FEBS J. 2021, 288, 4183–4209. [Google Scholar] [CrossRef] [PubMed]
- Ehrman, M.; Cedar, H.; Schwartz, J.H. L-Asparaginase II of Escherichia Coli. J. Biol. Chem. 1971, 246, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Mashburn, L.T.; Wriston, J.C. Tumor Inhibitory Effect of L-Asparaginase from Escherichia Coli. Arch. Biochem. Biophys. 1964, 105, 450–453. [Google Scholar] [CrossRef]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Pegaspargase (Oncaspar) for the First-Line Treatment of Children with Acute Lymphoblastic Leukemia (ALL). Onkologist 2007, 12, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.J.; Beier, R.; Da Palma, J.; Lanvers, C.; Ahlke, E.; Von Schütz, V.; Gunkel, M.; Horn, A.; Schrappe, M.; Henze, G.; et al. PEG-Asparaginase (Oncaspar) 2500 u/M2 BSA in Reinduction and Relapse Treatment in the ALL/NHL-BFM Protocols. Cancer Chemother. Pharmacol. 2002, 49, 149–154. [Google Scholar] [CrossRef]
- Würthwein, G.; Lanvers-Kaminsky, C.; Hempel, G.; Gastine, S.; Möricke, A.; Schrappe, M.; Karlsson, M.O.; Boos, J. Population Pharmacokinetics to Model the Time-Varying Clearance of the PEGylated Asparaginase Oncaspar® in Children with Acute Lymphoblastic Leukemia. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 955–963. [Google Scholar] [CrossRef]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Pegaspargase (Oncaspar®) for the First-Line Treatment of Children with Acute Lymphoblastic Leukemia (ALL). Oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef]
- Oncaspar, P.; Ettinger, A.R. Pharmacology. 1995, 46–48. [Google Scholar]
- Borghorst, S.; Hempel, G.; Poppenborg, S.; Franke, D.; König, T.; Baumgart, J. Comparative Pharmacokinetic/Pharmacodynamic Characterisation of a New Pegylated Recombinant E. Coli L-Asparaginase Preparation (MC0609) in Beagle Dog. Cancer Chemother. Pharmacol. 2014, 74, 367–378. [Google Scholar] [CrossRef]
- Zlotnikov, I.D.; Ezhov, A.A.; Kudryashova, E. V. Receptor-Mediated Internalization of L-Asparaginase into Tumor Cells Is Suppressed by Polyamines. Int. J. Mol. Sci. 2025, 26, 6749. [Google Scholar] [CrossRef]
- Vorselen, D.; Piontek, M.C.; Roos, W.H.; Wuite, G.J.L. Mechanical Characterization of Liposomes and Extracellular Vesicles, a Protocol. Front. Mol. Biosci. 2020, 7, 1–14. [Google Scholar] [CrossRef]
- Lukyanov, A.N.; Elbayoumi, T.A.; Chakilam, A.R.; Torchilin, V.P. Tumor-Targeted Liposomes: Doxorubicin-Loaded Long-Circulating Liposomes Modified with Anti-Cancer Antibody. J. Control. Release 2004, 100, 135–144. [Google Scholar] [CrossRef]
- de Souza Guimarães, M.; Cachumba, J.J.M.; Bueno, C.Z.; Torres-Obreque, K.M.; Lara, G.V.R.; Monteiro, G.; Barbosa, L.R.S.; Pessoa, A.; Rangel-Yagui, C. de O. Peg-Grafted Liposomes for L-Asparaginase Encapsulation. Pharmaceutics 2022, 14. [Google Scholar] [CrossRef] [PubMed]
- Harijan, M.; Singh, M. Zwitterionic Polymers in Drug Delivery: A Review. J. Mol. Recognit. 2022, 35, 1–13. [Google Scholar] [CrossRef]
- Barenholz, Y. Doxil® - The First FDA-Approved Nano-Drug: Lessons Learned. J. Control. Release 2012, 160, 117–134. [Google Scholar] [CrossRef]
- Arranja, A.G.; Pathak, V.; Lammers, T.; Shi, Y. Tumor-Targeted Nanomedicines for Cancer Theranostics. Pharmacol. Res. 2017, 115, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Jain, N.K.; Mishra, V.; Mehra, N.K. Targeted Drug Delivery to Macrophages. Expert Opin. Drug Deliv. 2013, 10, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Cruz, M.E.M.; Gaspar, M.M.; Lopes, F.; Jorge, J.S.; Perez-Soler, R. Liposomal L-Asparaginase: In Vitro Evaluation. Int. J. Pharm. 1993, 96, 67–77. [Google Scholar] [CrossRef]
- Jorge, J.C.S.; Perez-Soler, R.; Morais, J.G.; Cruz, M.E.M. Liposomal Palmitoyl-L-Asparaginase: Characterization and Biological Activity. Cancer Chemother. Pharmacol. 1994, 34, 230–234. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Perez-Soler, R.; Cruz, M.E.M. Biological Characterization of L-Asparginase Liposomal Formulations. Cancer Chemother. Pharmacol. 1996, 38, 373–377. [Google Scholar] [CrossRef]
- De, A.; Venkatesh, D.N. Design and Evaluation of Liposomal Delivery System for L-Asparaginese. J. Appl. Pharm. Sci. 2012, 2, 112–117. [Google Scholar] [CrossRef]
- Dobryakova, N. V.; Zhdanov, D.D.; Sokolov, N.N.; Aleksandrova, S.S.; Pokrovskaya, M. V.; Kudryashova, E. V. Improvement of Biocatalytic Properties and Cytotoxic Activity of L-Asparaginase from Rhodospirillum Rubrum by Conjugation with Chitosan-Based Cationic Polyelectrolytes. Pharmaceuticals 2022, 15, 406. [Google Scholar] [CrossRef] [PubMed]
- Sukhoverkov, K. V.; Sokolov, N.N.; Abakumova, O.Y.; Podobed, O. V.; Kudryashova, E. V. The Formation of Conjugates with PEG–Chitosan Improves the Biocatalytic Efficiency and Antitumor Activity of L-Asparaginase from Erwinia Carotovora. Moscow Univ. Chem. Bull. 2016, 71, 122–126. [Google Scholar] [CrossRef]
- Zlotnikov, I.D.; Kudryashova, E. V Targeted Polymeric Micelles System, Designed to Carry a Combined Cargo of L-Asparaginase and Doxorubicin, Shows Vast Improvement in Cytotoxic Efficacy. Polymers (Basel). 2024, 16, 2132. [Google Scholar] [CrossRef] [PubMed]
- Dobryakova, N. V.; Zhdanov, D.D.; Sokolov, N.N.; Aleksandrova, S.S.; Pokrovskaya, M. V.; Kudryashova, E. V. Rhodospirillum Rubrum L-Asparaginase Conjugates with Polyamines of Improved Biocatalytic Properties as a New Promising Drug for the Treatment of Leukemia. Appl. Sci. 2023, 13, 3373. [Google Scholar] [CrossRef]
- Liang, W.; W. Lam, J.K. Endosomal Escape Pathways for Non-Viral Nucleic Acid Delivery Systems. Mol. Regul. Endocytosis 2012. [Google Scholar] [CrossRef]
- Zlotnikov, I.D.; Shishparyonok, A.N.; Pokrovskaya, M. V; Alexandrova, S.S.; Zhdanov, D.D.; Kudryashova, E. V Structural Features Underlying the Mismatch Between Catalytic and Cytostatic Properties in L-Asparaginase from Rhodospirillum Rubrum. Catalysts 2025, 15, 476. [Google Scholar] [CrossRef]
- Kudryashova, E. V.; Sukhoverkov, K. V. “Reagent-Free” l-Asparaginase Activity Assay Based on CD Spectroscopy and Conductometry. Anal. Bioanal. Chem. 2016, 408, 1183–1189. [Google Scholar] [CrossRef]
Figure 1.
A schematic representation of the process of preparing L-ASNase conjugates and their incorporation into liposomal formulations.
Figure 1.
A schematic representation of the process of preparing L-ASNase conjugates and their incorporation into liposomal formulations.
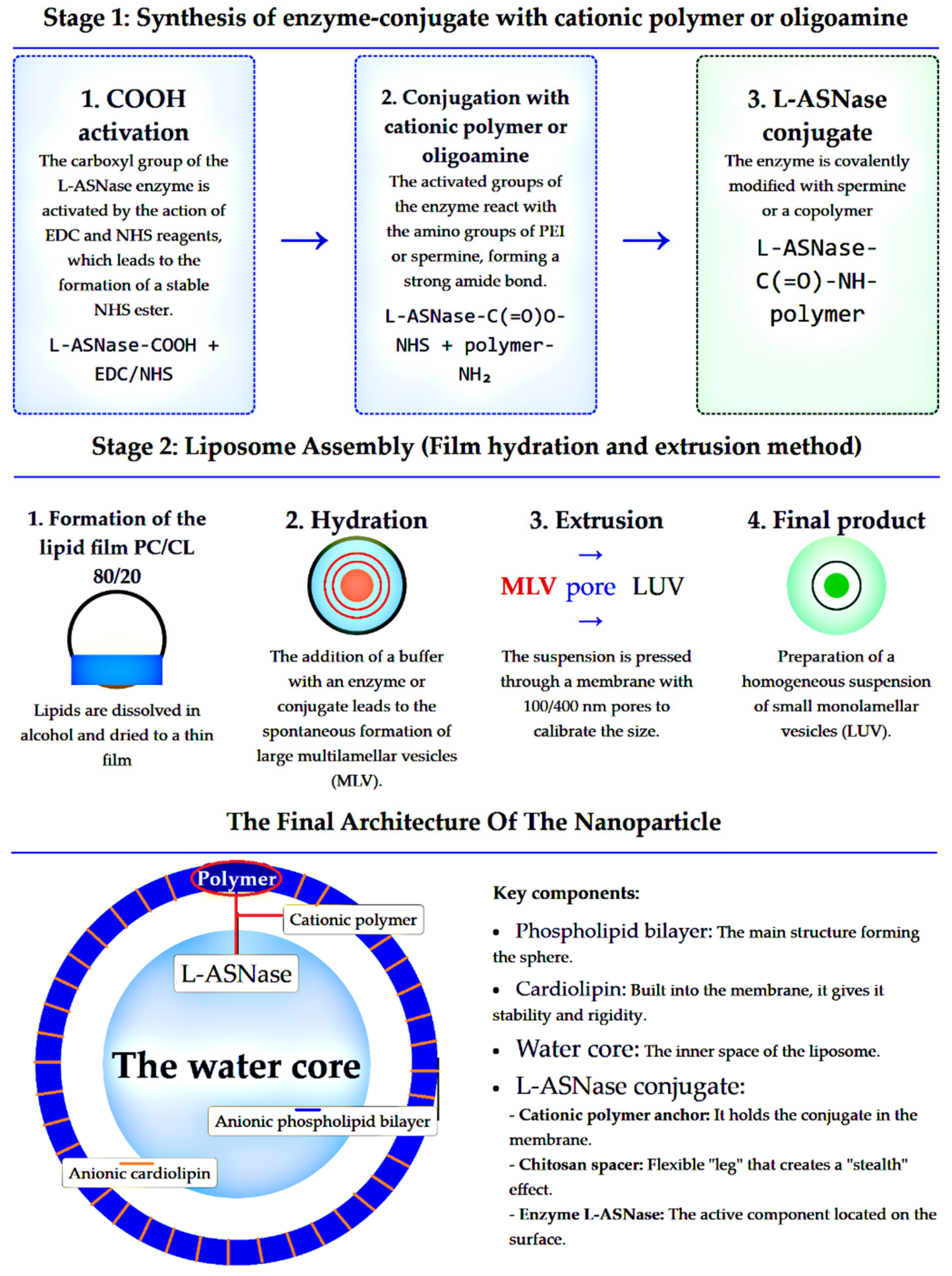
Figure 2.
FTIR spectra of EcA L-ASNase, PC/CL (Phosphatidylcholine / cardiolipin, 80/20 w/w) liposomes and EcA liposomal formulation.
Figure 2.
FTIR spectra of EcA L-ASNase, PC/CL (Phosphatidylcholine / cardiolipin, 80/20 w/w) liposomes and EcA liposomal formulation.
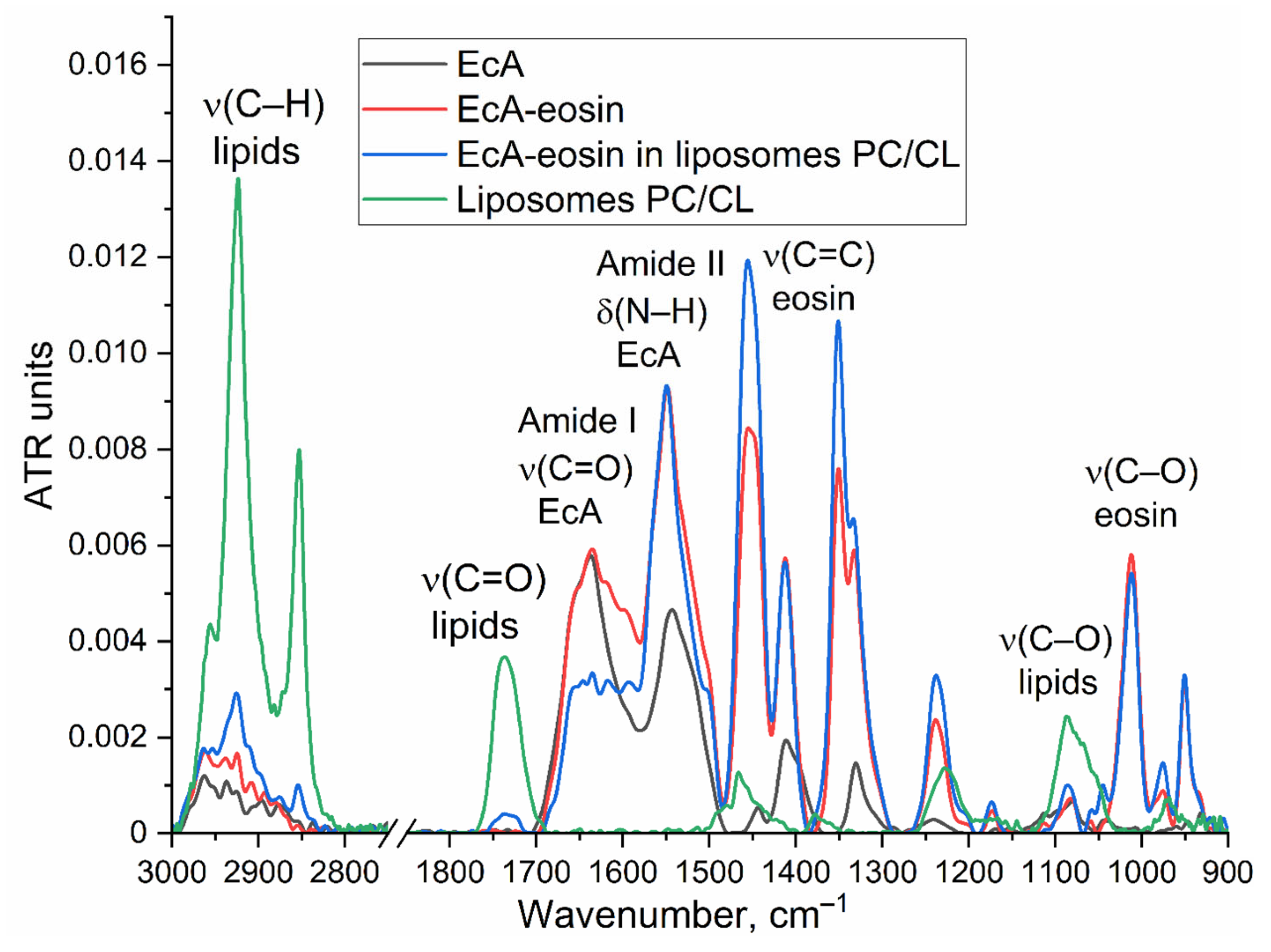
Figure 3.
Representative kinetic curves of L-asparagine hydrolysis by EcA determined by CD spectroscopy. The CD signal is shown in black, and the optical density signal at 210 nm is shown in red. Conditions: C(Asn) = 15 mM, PBS (0.01M, pH 7.4), T = 37 °C, λ=210 nm.
Figure 3.
Representative kinetic curves of L-asparagine hydrolysis by EcA determined by CD spectroscopy. The CD signal is shown in black, and the optical density signal at 210 nm is shown in red. Conditions: C(Asn) = 15 mM, PBS (0.01M, pH 7.4), T = 37 °C, λ=210 nm.
Figure 4.
(a) Far-UV circular dichroism spectra of L-asparaginase (EcA), its conjugates, and liposomal formulations (phosphatidylcholine/cardiolipin 80/20) recorded in 5 mM sodium phosphate buffer. T = 37 °C. (b) Quantitative secondary structure composition of the EcA formulations obtained by deconvolution of the CD spectra using the CDNN program.
Figure 4.
(a) Far-UV circular dichroism spectra of L-asparaginase (EcA), its conjugates, and liposomal formulations (phosphatidylcholine/cardiolipin 80/20) recorded in 5 mM sodium phosphate buffer. T = 37 °C. (b) Quantitative secondary structure composition of the EcA formulations obtained by deconvolution of the CD spectra using the CDNN program.
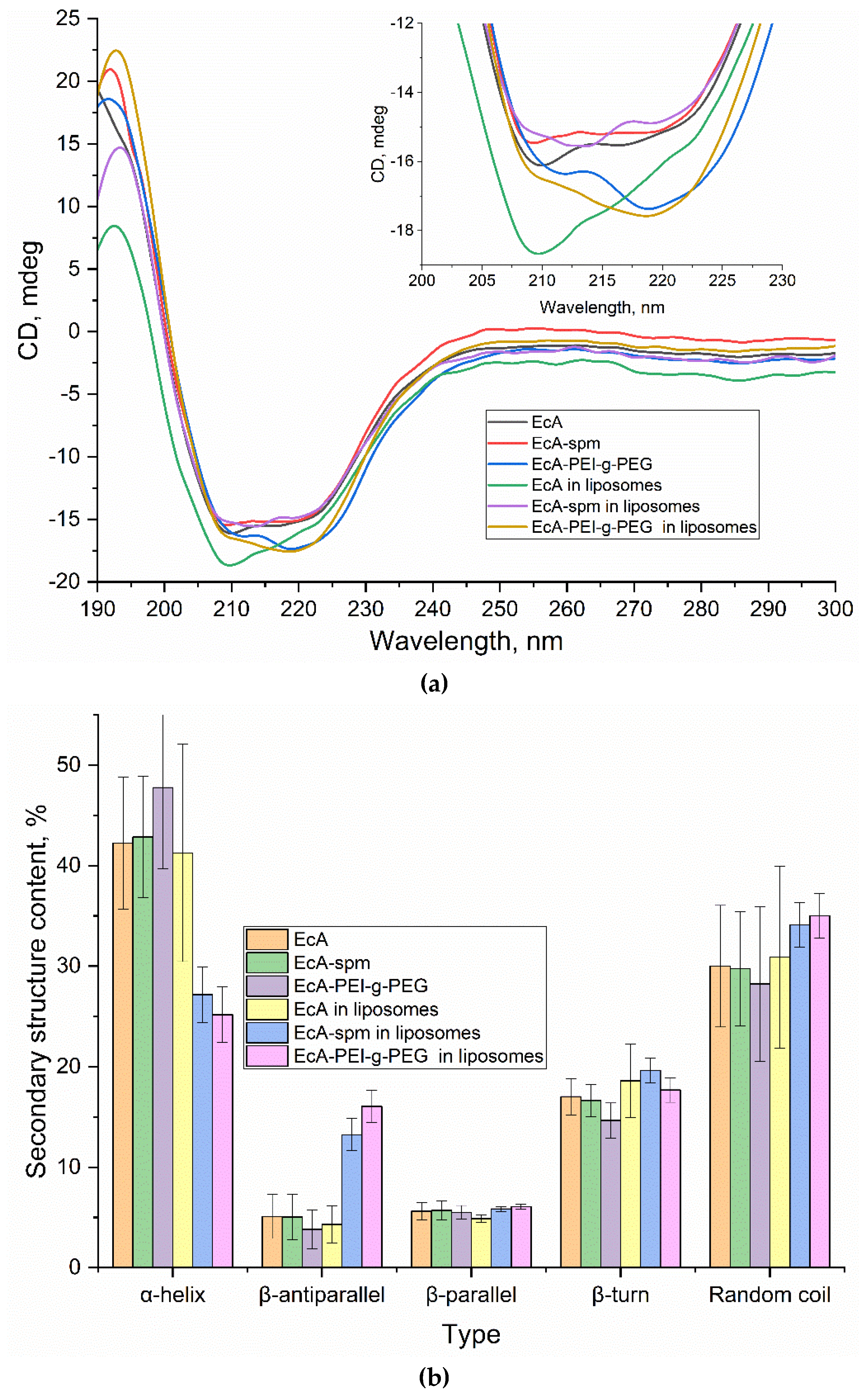
Figure 5.
Kinetic curves of L-ASNase half-release through 300 nm membrane. PBS (0.01M, pH 7.4), T = 37 °C. L-ASNase concentration were measured fluorimetrically: λex = 290 nm, λemi = 450 nm.
Figure 5.
Kinetic curves of L-ASNase half-release through 300 nm membrane. PBS (0.01M, pH 7.4), T = 37 °C. L-ASNase concentration were measured fluorimetrically: λex = 290 nm, λemi = 450 nm.
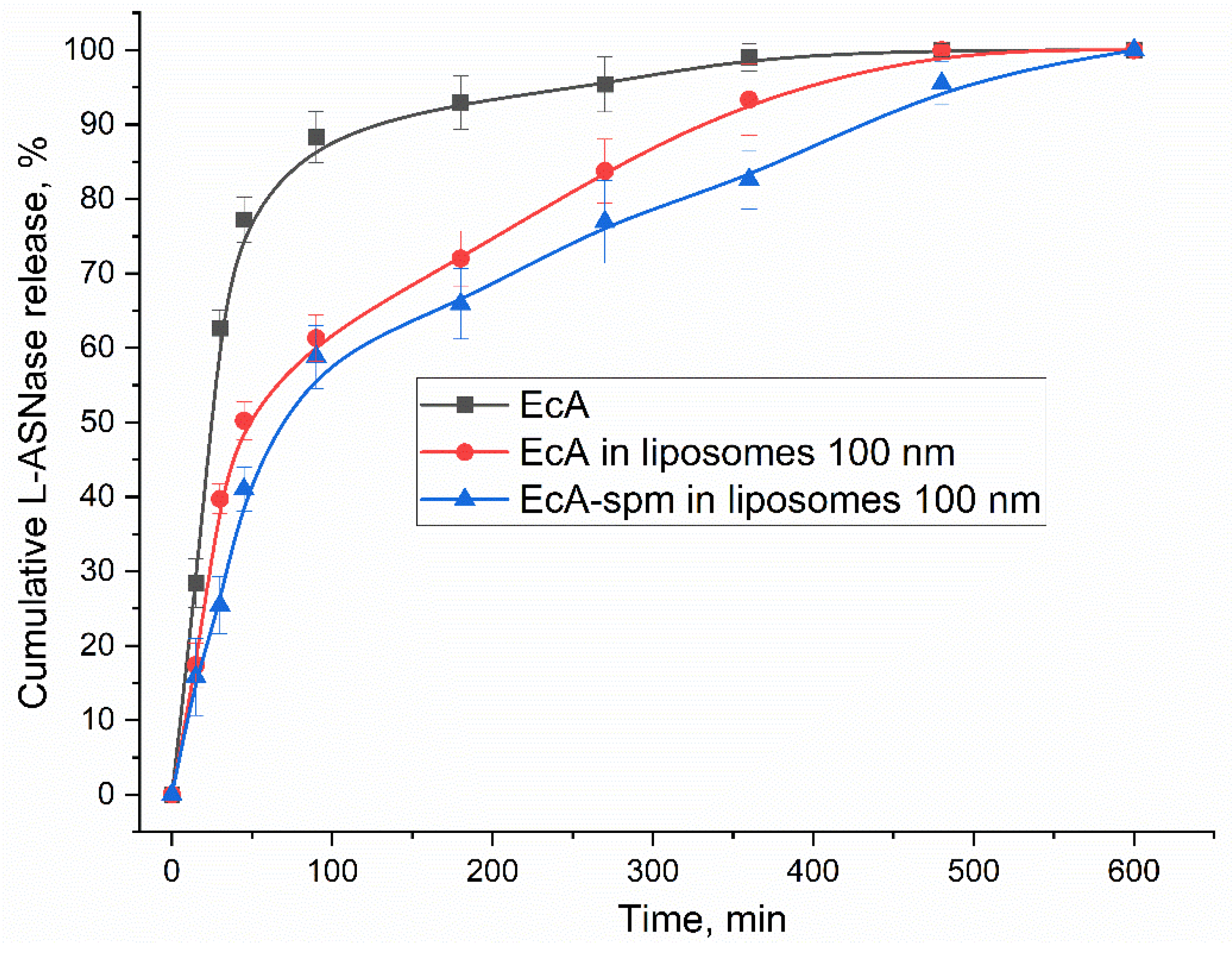
Figure 6.
Dose-dependent viability of Raji cells. RPMI-1640, 5% CO2, T = 37 °C.
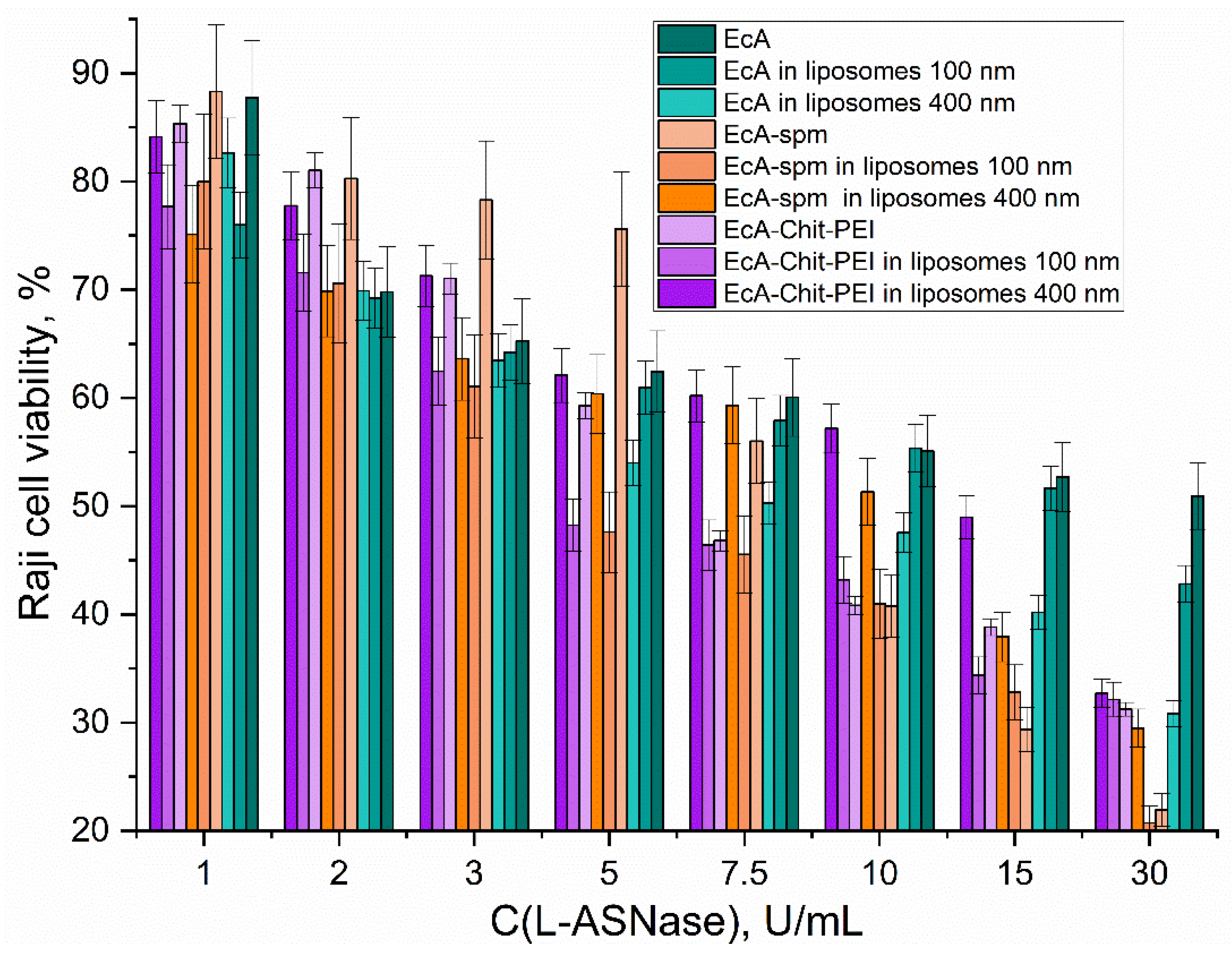
Figure 7.
Fluorescence emission spectra of liposome labeled components. PBS (0.01M, pH 7.4), T = 37 °C. λex = 488 nm.
Figure 7.
Fluorescence emission spectra of liposome labeled components. PBS (0.01M, pH 7.4), T = 37 °C. λex = 488 nm.
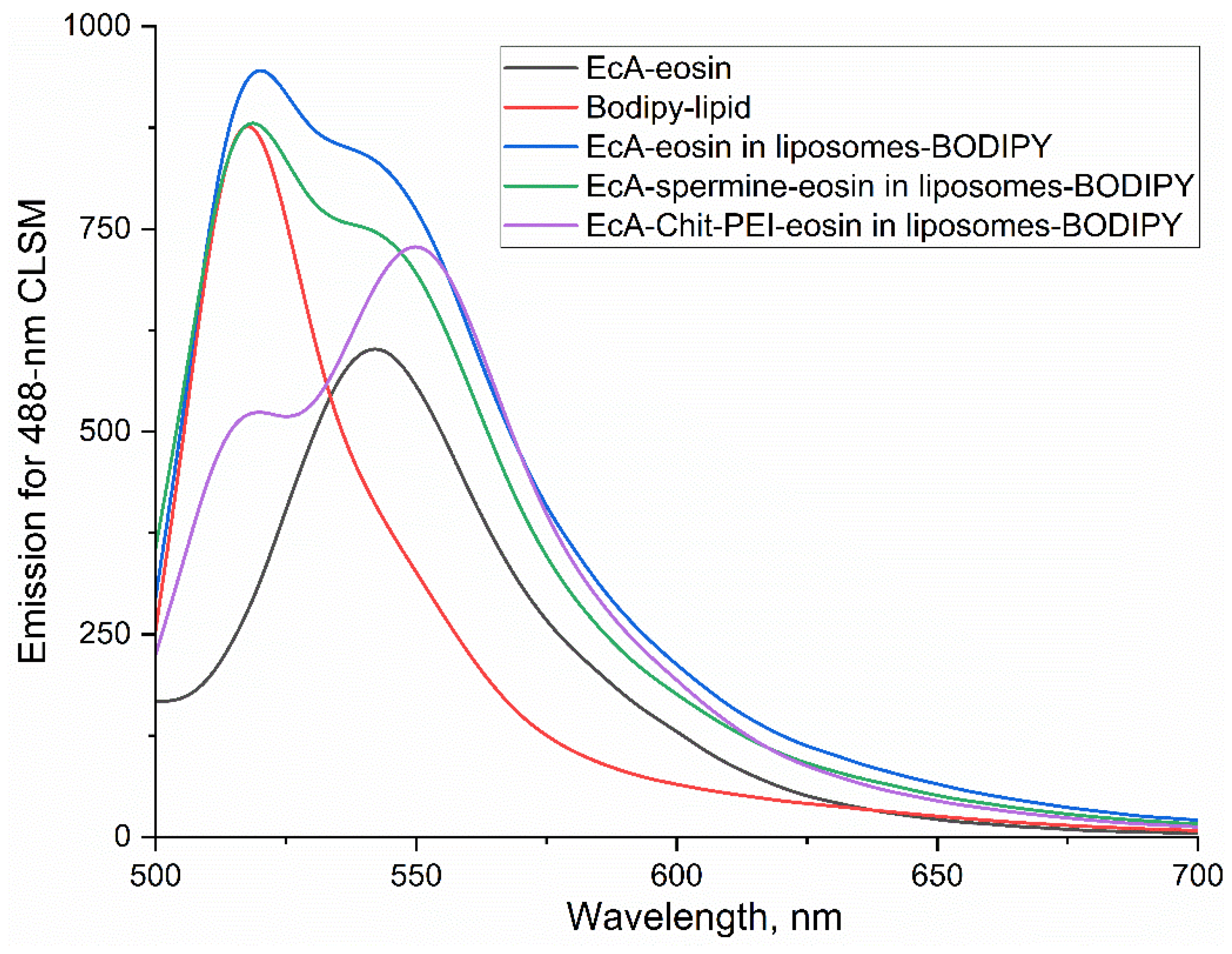
Figure 8.
CLSM images of Raji cells treated with labelled EcA liposomal formulations. Enzyme (red) and liposome (green) fluorescence signal are shown.
Figure 8.
CLSM images of Raji cells treated with labelled EcA liposomal formulations. Enzyme (red) and liposome (green) fluorescence signal are shown.
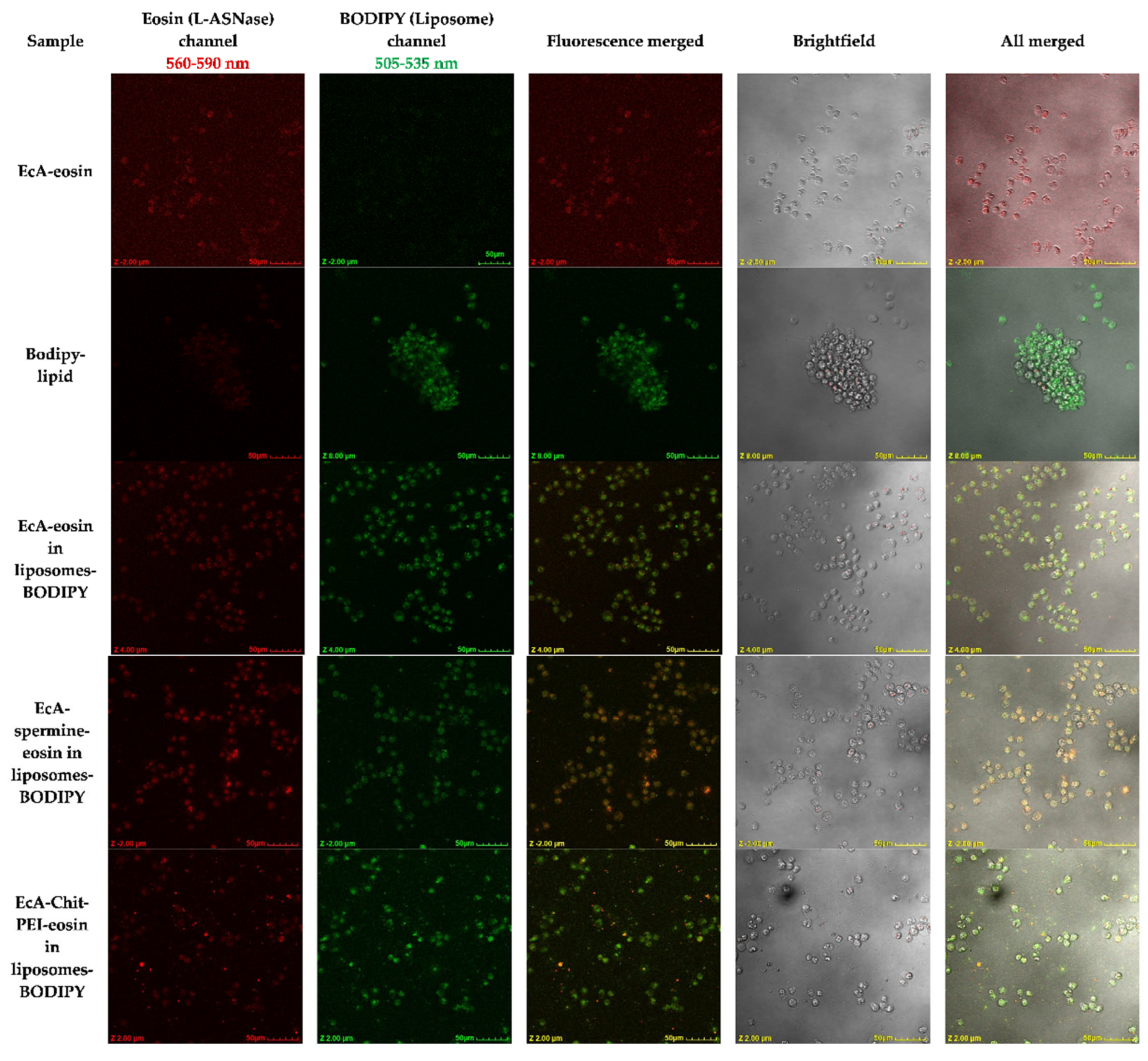
Figure 9.
Quantitative analysis of Mean Fluorescence Intensity (MFI) from CLSM images.
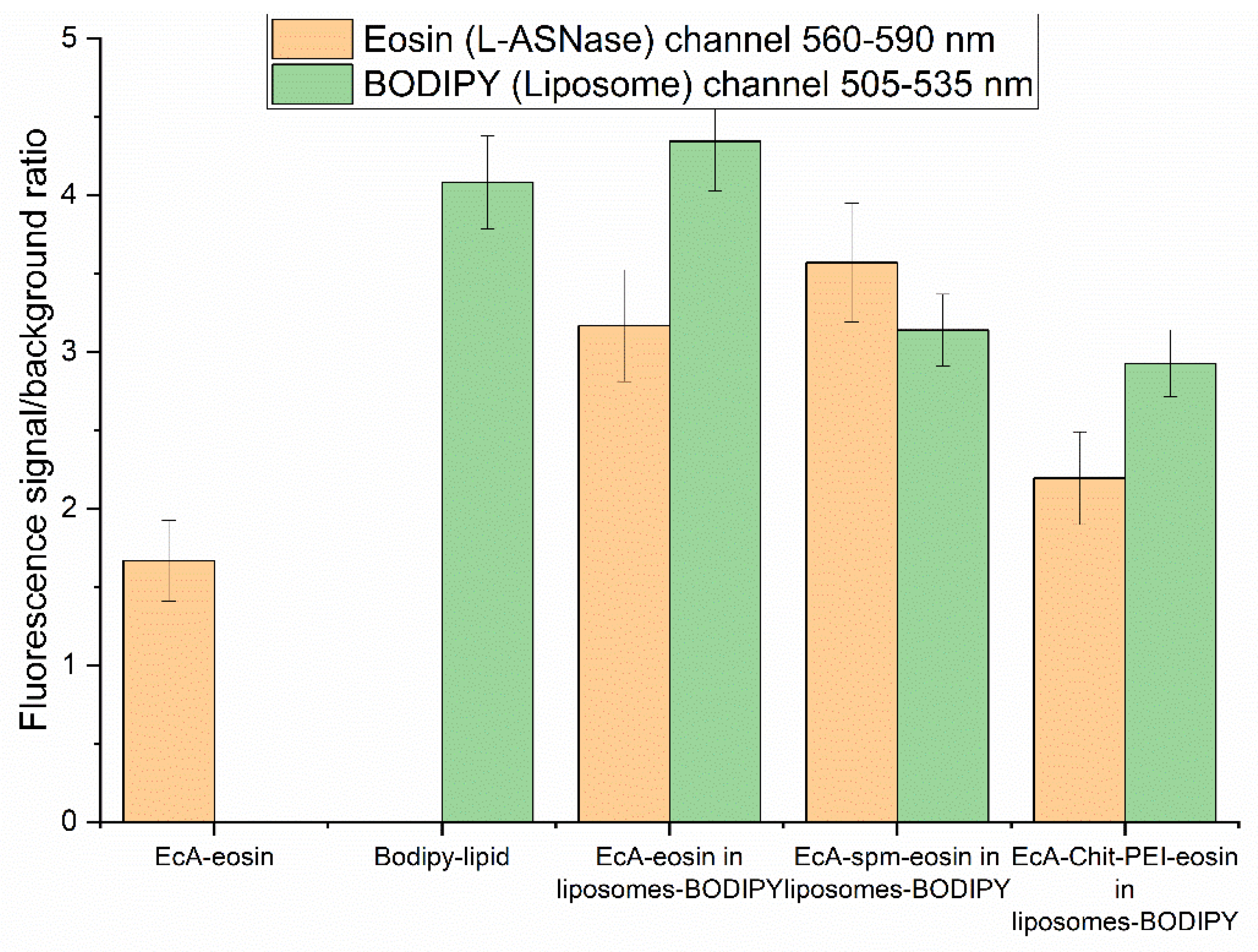
Figure 10.
Correlation between the binding affinity of L-ASNase formulations to Raji cells, expressed as fluorescence signal-to-background ratio (Raji cells binding index), and their cytotoxicity, represented by IC₅₀ values (U/mL). Additionally, the data from our last article is provided [22].
Figure 10.
Correlation between the binding affinity of L-ASNase formulations to Raji cells, expressed as fluorescence signal-to-background ratio (Raji cells binding index), and their cytotoxicity, represented by IC₅₀ values (U/mL). Additionally, the data from our last article is provided [22].
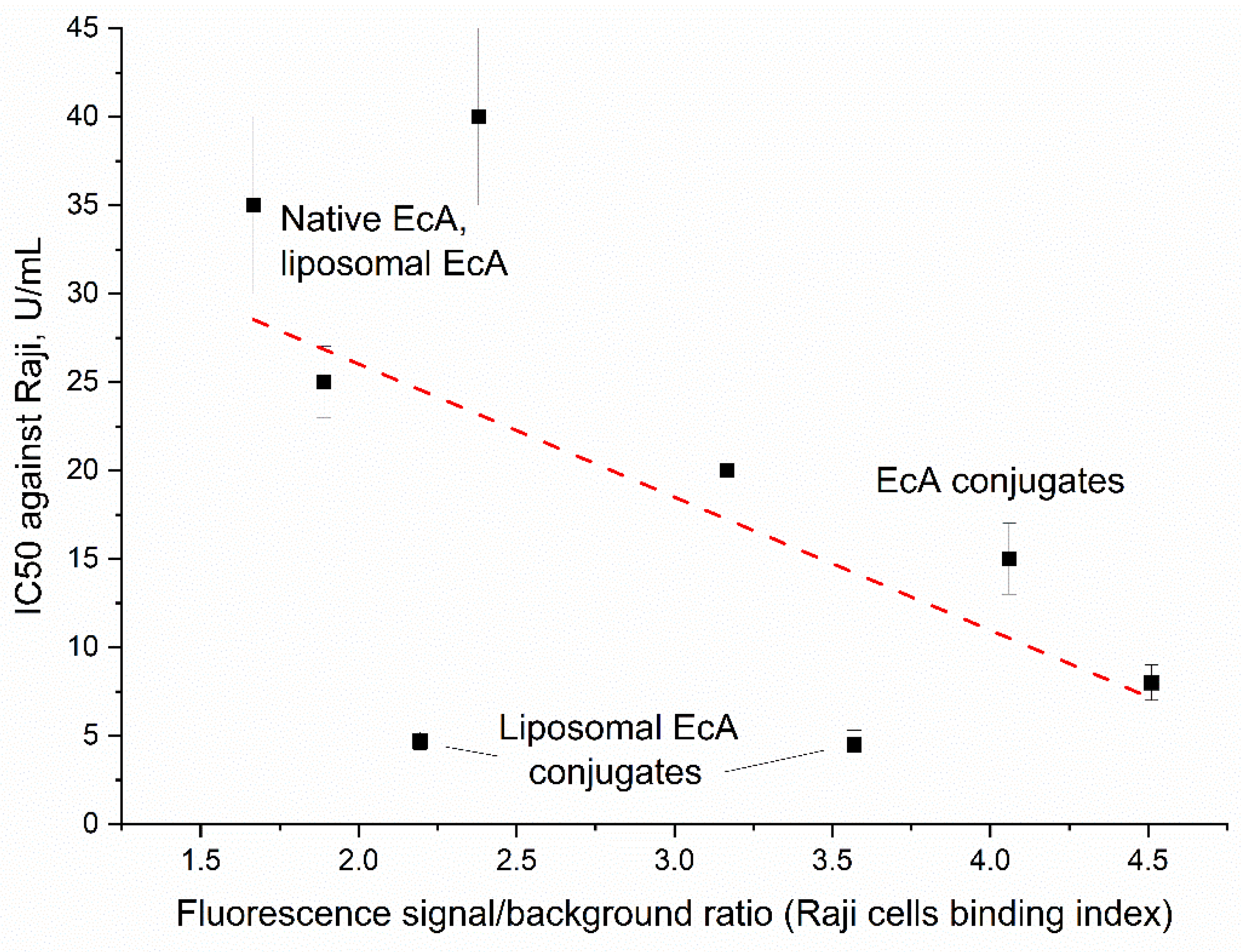

Table 1.
Physico-chemical properties of L-ASNase liposomes. L-ASNase activity (U/mg) of preparations determined by CD spectroscopy. Conditions: C(Asn) = 15 mM, PBS (0.01M, pH 7.4), λ=210 nm.
Table 1.
Physico-chemical properties of L-ASNase liposomes. L-ASNase activity (U/mg) of preparations determined by CD spectroscopy. Conditions: C(Asn) = 15 mM, PBS (0.01M, pH 7.4), λ=210 nm.
| L-ASNase entrapment efficiency, % | EcA | EcA-spm | EcA-PEI-g-PEG |
| In PC/Cl 80/20 100 nm liposomes | 63±2 | 89±1 | 82±2 |
| In PC/Cl 80/20 400 nm liposomes | 70±5 | 97±1 | 94±1 |
| Final L-ASNase / lipid ratio, µg/µmol | EcA | EcA-spm | EcA-PEI-g-PEG |
| In PC/Cl 80/20 100 nm liposomes | 85±3 | 120±2 | 111±3 |
| In PC/Cl 80/20 400 nm liposomes | 95±7 | 131±2 | 127±2 |
| L-ASNase activity, U/mg | EcA | EcA-spm | EcA-PEI-g-PEG |
| Non liposomal | 330±20 | 380±14 | 327±15 |
| In PC/Cl 80/20 100 nm liposomes | 355±18 | 365±13 | 306±22 |
| In PC/Cl 80/20 400 nm liposomes | 364±23 | 370±25 | 350±18 |
| L-ASNase residual activity after a month of storage at +4 °C in PBS, U/mg | EcA | EcA-spm | EcA-PEI-g-PEG |
| Non liposomal | 208 (63% from initial) | 315 (83% from initial) | 245 (75% from initial) |
| In PC/Cl 80/20 100 nm liposomes | 298 (84% from initial) | 339 (93% from initial) | 278 (91% from initial) |
| In PC/Cl 80/20 400 nm liposomes | 280 (77% from initial) | 333 (90% from initial) | 284 (81% from initial) |
Table 2.
Time of L-ASNase half-release and time of 80%-cumulative L-ASNase release. The conditions are similar to those presented in Figure 5. 100-nm liposomes were studied.
Table 2.
Time of L-ASNase half-release and time of 80%-cumulative L-ASNase release. The conditions are similar to those presented in Figure 5. 100-nm liposomes were studied.
| Parameter | EcA | EcA in liposomes | EcA-spm in liposomes | EcA-PEI-g-PEG in liposomes |
| T50%, min | 24±2 | 45±5 | 56±7 | 70±5 |
| T80%, min | 52±5 | 240±20 | 330±35 | 420±50 |
Table 3.
The half-maximal inhibitory concentration (IC₅₀) values of EcA and its formulations when acting on Raji cells (U/mL). RPMI-1640, 5% CO2, T = 37 °C.
Table 3.
The half-maximal inhibitory concentration (IC₅₀) values of EcA and its formulations when acting on Raji cells (U/mL). RPMI-1640, 5% CO2, T = 37 °C.
| IC50, U/mL | EcA | EcA-spm | EcA-Chit-PEI |
| Non liposomal | 35±5 | 8±1 | 7±1 |
| In PC/Cl 80/20 100 nm liposomes | 20±5 | 4.5±0.8 | 4.7±0.5 |
| In PC/Cl 80/20 400 nm liposomes | 9±2 | 11±1 | 13±2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



