Submitted:
31 August 2025
Posted:
01 September 2025
Read the latest preprint version here
Abstract
One-carbon metabolism (also known as the methylation pathway) is a fundamental biochemical process involving the transfer of methyl groups from one molecule to another, to carry out many cellular functions, including DNA, RNA, and protein methylation and phosphatidylcholine synthesis. Many chronic diseases are linked to its dysfunction. The pathway centers on S-adenosylmethionine (SAMe), the universal methyl donor. The methyl group is sourced from methyl-tetrahydrofolate, and methionine, glycine, serine, and formaldehyde are carriers of one-carbon units. An overlooked aspect is the crucial role that the gut microbiome plays in assuring that the one-carbon units are virtually free of deuterium. Deuterium is a natural heavy isotope of hydrogen, and it damages the ATPase pumps in the mitochondria. Microbes produce extremely deuterium depleted (deupleted) hydrogen gas which they use as a reducing agent to convert carbon dioxide into organic molecules, such as acetate, butyrate and formate. We show here through an in-depth review of methylation and demethylation processes how they are used to deliver deupleted protons to the mitochondria, to protect them from damage. Toxic exposures to organophosphate insecticides and deficiencies in nutrients such as methionine, choline, betaine, and serine can impair methylation pathways, causing disease through co-causality of mitochondrial dysfunction.
Keywords:
gut dysbiosis
; methanogens
; acetogens
; archaea
; butyrate
; methionine
; choline
; hydrogen gas
; deuterium
; mitochondria
1. Introduction
Myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS) is a debilitating chronic disease characterized by extreme fatigue, post-exertional malaise, impaired memory, pain, and gastrointestinal and sleep disturbances [1]. Its incidence may be increasing since the SARS-CoV-2 pandemic, because the condition known as long COVID has significant overlapping symptoms [2]. Mitochondrial dysfunction associated with oxidative stress is a major factor in both conditions [3,4].
A comprehensive multi-omics analysis involving 106 cases of ME/CFS and 91 healthy controls revealed major disturbances in the gut microbiome of patients compared to controls. Most notable was a reduction in the prevalence of the common butyrate-producing microbes Faecalibacterium prausnitzii and Eubacterium rectale, along with a deficiency in butyrate. The abundance of F. prausnitzii was inversely associated with fatigue severity. The acetate-producing bacterium Coprobacter secundus was also reduced in prevalence, along with reduced acetate production [5].
Ruminococcus gnavus, also known as Mediterraneibacter gnavus, a mucin-degrading bacterium [5], was elevated in the ME/CFS patients [6]. R. gnavus is strongly associated with Crohn’s disease, and it produces an inflammatory polysaccharide called glucorhamnan, formed from the sugars rhamnose and glucose [7]. Another species that was elevated in association with ME/CFS was Clostridium bolteae, also known as Enterocloster bolteae, whose overgrowth has also been linked to autism [8], as well as IgG-4 related disease and systemic sclerosis [9], and fatty liver disease [10,11]. Like R. gnavus, it also produces an inflammatory immunogenic polysaccharide [8]. Interestingly, both E. bolteae and R. gnavus are alcohol producers, and increased levels of ethanol in the feces along with elevated levels of these species are associated with nonalcoholic steatohepatitis [11].
All the microbially produced nutrients mentioned above - lactate, ethanol, acetate, and butyrate - are low deuterium nutrients, and, for this reason, all are likely vital for the health of the mitochondria in the host [12]. Deuterium is a heavy isotope of protium, and it is a natural element found in seawater at 155 parts per million (ppm). Metabolic processes inherently favor a significantly deuterium depleted (deupleted) environment within the mitochondria, because deuterium damages the ATPase pumps, causing inefficiencies in ATP production and increasing the release of reactive oxygen species (ROS) [13]. The microbes play an essential role in the supply of deupleted nutrients to the host, and they accomplish this feat primarily through the synthesis of hydrogen gas that has lost 80% of the deuterium normally present in water [14]. This hydrogen gas is then used as a reducing agent to produce deupleted nutrients, especially acetate and butyrate, but also, as we shall see, formaldehyde, formate, glycine, serine, methionine, choline, and methyl-tetrahydrofolate.
While not derived directly from hydrogen gas, lactate does possess a deupleted proton. Lactate carries a “reducing equivalent” via the reversible reaction catalyzed by lactate dehydrogenase, which converts nicotinamide adenine dinucleotide (NADH) to NAD+ to produce lactate, and reconverts NAD+ back to NADH in the mitochondria when lactate is taken up from the medium [15]. Lactate dehydrogenase is a member of a large class of proteins called flavoproteins, which work with a cofactor flavin adenine dinucleotide (FAD) to facilitate the transfer of hydride ions, and these enzymes typically have a high deuterium kinetic isotope effect (KIE) due to their use of proton tunneling [16]. Deuterons are not nearly as efficient in this physical phenomenon as are protons due to their larger size. Sutcliffe et al. wrote in 2006: “It is now widely accepted that enzyme-catalyzed CH bond breakage occurs by quantum mechanical tunnelling” [16]. In the mitochondrial matrix, deupleted protons carried by NADH are released during the enzymatic action of NADH dehydrogenase (Complex II), another flavoprotein, which converts NADH back to NAD+ [17]. This enzyme is possibly the most important direct supplier of deupleted protons to the mitochondrial ATPase pumps.
Ethanol plays a similar role in providing NADH to the mitochondria, through the activity of the mitochondrial enzyme acetaldehyde dehydrogenase. However, acetaldehyde, an intermediary in ethanol metabolism, is highly reactive, so ethanol is not an ideal nutrient for this reason [18,19].
F. prausnitzii is widely recognized as an important health-promoting butyrate-producing bacterium in the human colon, although efforts to produce F. prausnitzii probiotics are challenging due to its sensitivity to oxygen and dependence on other microbes for critical micronutrients [20,21]. Inflammatory conditions cause luminal oxygen levels to rise, and this is problematic for F. prausnitzii, a strict anaerobe [21,22]. Furthermore, F. prausnitzii are auxotrophic for folate, meaning that they depend upon either the diet or other microbes for its supply, and folate is essential for them to thrive [23]. Folate plays a central role in methylation pathways, the main topic of this paper, because, as methyl-tetrahydrofolate (CH3-THF), it supplies methyl groups to S-adenosyl methionine (SAMe), the universal methyl donor. The methyl groups will be highly unlikely to carry deuterium if they are derived from microbial metabolism.
In a study involving 60 patients with chronic fatigue, half of the patients were found to have serum levels of folate that were deficient (below 3.0 micrograms/liter) [24]. As early as 1968 it was already recognized that folate deficiency is a common feature in Crohn’s disease [25], and this deficiency was later confirmed in a paper published in 2010 [26]. The human gut microbiome has a very efficient capacity to convert dietary folates into CH3-THF, but the microbes are unable to reduce supplemental folic acid to folate and subsequently to CH3-THF. Folic acid is delivered unmodified to the liver, which then reduces it to folate and converts it to CH3-THF. Excessive doses of folic acid stress the liver, causing pseudo-MTHFR (CH2-THF reductase) deficiency and liver injury in mice [27]. High doses of folic acid appear unmodified in the circulation and are likely not useful for supporting methylation pathways [28]. Gut dysbiosis is now recognized to be linked to a long list of debilitating chronic diseases, including type 2 diabetes, obesity, non-alcoholic fatty liver disease, hypertension, arthritis, autism, depression, anxiety, and sleep disorder [29]. A common thread may be decreased acetate and butyrate production, concurrent with insufficient methylation capacity and increased production of lactate, ethanol, and inflammatory polysaccharides.
2. Hydrogen Gas Recycling by Gut Microbes: A Critical Role for Formaldehyde
The production of hydrogen and methane gasses by gut microbes is an essential step in the recycling of organic matter from carbon dioxide (CO2), through multiple organic intermediates, and back to CO2. Gut microbes can use hydrogen gas to reduce CO2 to both methane and acetate, and they can then oxidize methane in a series of steps yielding methanol, formaldehyde, and formate, and finally returning to CO2 in a complete cycle. In the process, the hydrogen atoms originally in the hydrogen gas are retained in the biosynthesized small organic molecules, and they become valuable to the host because they are extremely deupleted. This is because of the under-appreciated fact that the hydrogen gas has much less deuterium than is typical in the natural world, probably in part because deuterons preferentially stay in the liquid phase. The microbes that produce abundant H2 become enriched in deuterium internally as a consequence, but microbes are much more resilient to high deuterium load than mammals are [30]. In fact, it is possible to cultivate deuterium-tolerant strains of Escherichia coli via an adaptation protocol, and these microbes have proven to be useful for synthesizing highly deuterated biological molecules for experimental use. Via such methods, O Paliy et al. succeeded in creating three highly deuterium-tolerant strains of E. coli [31]. E. coli possesses four membrane-associated [Ni-Fe]-hydrogenases that can produce H2 gas from glucose, formate, glycerol, and other small organic molecules, via anaerobic fermentation [32].
Strict anaerobes likely play an essential role in providing deupleted nutrients to the host. Interestingly, research comparing the deuterium content of lipids in sediments sourced from deep marine water vs shallow water revealed that certain lipids produced at depth by strict anaerobes had significantly less deuterium content, with δD values up to -348‰ (parts per thousand), which is about 100 ppm as compared to 155 ppm. This same study found significantly enriched deuterium in hopanoids and branched chain fatty acids, components of bacterial membranes produced by bacteria at the surface [33].
2.1. Production of Methane Gas by Archaea and the Recycling Between Methane and CO2
Evolution has created the three domains of life, which are the Archaea, the Bacteria, and the Eukarya. Archaea are a remarkable group of microorganisms with unique capabilities that play an essential role in supporting the other domains. Methanogens are anaerobic archaea that can synthesize methane from carbon dioxide and hydrogen gas using a unique set of enzymes not present in the other two domains. Methane production (methanogenesis) is the only biochemical pathway that methanogens use to generate ATP [34]. The unique enzyme complex, methyl-coenzyme M reductase, catalyzes the final step that reduces methyl-coenzyme M to methane [35]. These remarkable microbes can produce methane not only from CO2 but also from formate, methanol, methylamines, and acetate by a pathway involving a unique set of coenzymes [36].
Table 1 shows the reaction sequence that begins with four molecules of deupleted hydrogen gas and, through a series of steps, converts CO2 to a sequence of small organic molecules, finally returning to CO2 production in a complete cycle. Formaldehyde dehydrogenase depends on the cofactor NAD+, which it converts to NADH as another product of the reaction [37]. Methanol dehydrogenases also use NAD+ as a cofactor in some cases, but other cofactors are also found, such as pyrroloquinoline quinone (PQQ) [38]. Formate dehydrogenase can also convert NAD+ to NADH, but there is an alternative pathway that uses a hydrogenase to produce hydrogen gas. The formate hydrogenylase complex (FHL), expressed by E. coli, catalyzes the decomposition of formate into CO2 and H2 gas, restoring one of the four hydrogen molecules that were consumed in this cycle. This is the primary path by which hydrogen gas is originally produced by enteric bacterial fermentation processes that often begin with glucose [39].
The overall result of the cycle, as shown in Table 1, is the production of 2 molecules of deupleted NADH from NAD+, two molecules of deupleted water, and 2 deupleted protons. All these products, accumulating in the gut lumen, become valuable in helping to maintain low deuterium in the host mitochondria.
Not all of the formaldehyde is converted to CO2 -- some of it is siphoned off to convert tetrahydrofolate (THF) to methylene-tetrahdrofolate (CH2-THF). The two protons in the methylene group will be extremely deupleted due to the fact that they are sourced from formaldehyde, which traces back to H2 gas. Both methylotrophs and methanotrophs possess the necessary enzymes to conjugate formaldehyde with THF to produce CH2-THF. This is an important point that we will return to later in this paper, because it implies that the methyl groups carried by methionine will be extremely deupleted. Formaldehyde can also react spontaneously with THF, although the enzymatic pathway is favored in the microbes [40].
2.2. Synthesis of Deupleted Hydrogen Gas Through Fermentation
The hydrogen gas that archaea use to synthesize methane from CO2 is produced by many species of anaerobic bacteria in the colon through fermentation of dietary fiber and sugars. The primary hydrogen-producing bacteria are members of the Bacteroides and Firmicutes phyla [41,42]. A highly significant finding is that hydrogen gas produced by microbes from small organic molecules is likely severely depleted in deuterium, compared to the typical ratio of deuterium to hydrogen in nature [12]. It has been found experimentally that microbially produced hydrogen gas had a deuterium fraction that was 80% lower than the reference amount normally found in seawater (155 parts per million) [14]. While this fractionation is partially due to the fact that deuterium tends to stay in the liquid phase, hydrogenases, the class of enzymes that perform this feat, are members of the flavoprotein class of enzymes that exploit proton tunneling to favor hydrogen over deuterium in their reaction product [43]. Hydrogenases can have a very high deuterium kinetic isotope effect (KIE), as high as 43 in an acidic environment [44].
2.3. Other Pathways Besides Methane Production for H2 Consumption
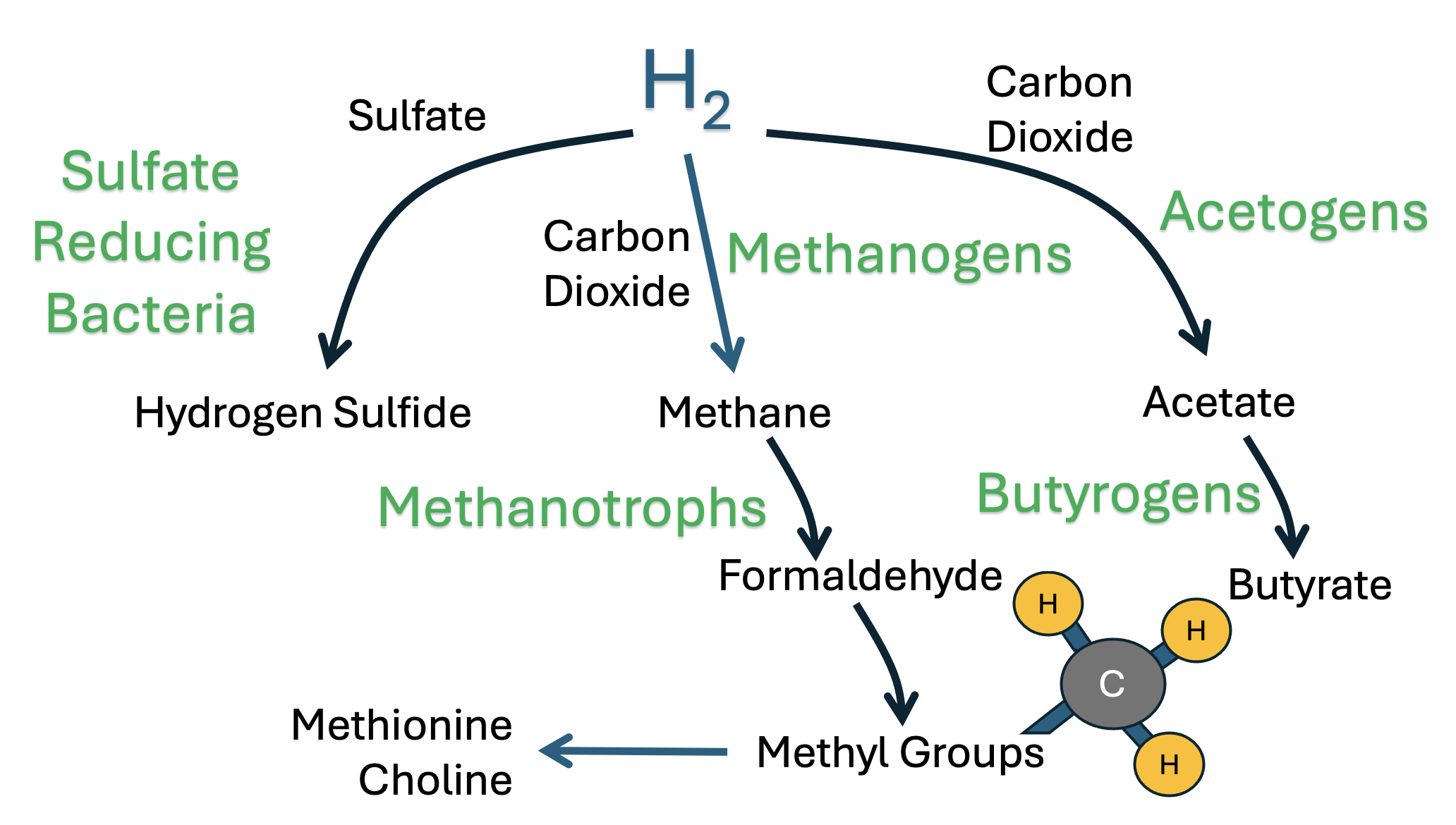
Besides methane production, H2 can also be used to reduce CO2 to acetate and to reduce sulfate to hydrogen sulfide gas, H2S. Both the acetate and the H2S are therefore likely to be sources of deupleted protons. Acetogenic bacteria are a specialized group of strictly anaerobic species that can convert two molecules of CO2 to acetate using H2 as a reducing agent in the Wood-Ljungdahl pathway. They are primarily members of the Firmicutes phylum [45].
While excessive H2S is toxic to human cells, H2S is also a signaling gas, and it can be utilized by mitochondria to facilitate ATP production. In fact, human cells endogenously synthesize H2S from cysteine under stress conditions, and it is released into the mitochondrial intermembrane space, where it is metabolized to sulfate and thiosulfate [46]. H2S oxidation in the mitochondria could be a mechanism to decrease the deuterium burden in the mitochondria through the supply of two deupleted protons sourced from H2S.
Figure 1 illustrates the three primary pathways where bacteria consume H2 to produce useful organic products. Sulfate-reducing bacteria convert sulfate to hydrogen sulfide gas, methanogens convert carbon dioxide to methane, and acetogens convert carbon dioxide to acetate. We argue here that all three of these products are useful sources of deupleted protons. Acetate can be converted to acetyl-Coenzyme A (acetyl-CoA) and metabolized in the citric acid cycle in the mitochondria to yield CO2 and water. Acetate is also the precursor to butyrate, a short-chain fatty acid that is the primary energy source of the colonocytes [47]. Methane, through the intermediary formaldehyde, becomes a source of deupleted methyl groups that are ultimately transferred from CH3-THF to homocysteine to form methionine, the universal methyl donor, via the cobalamin-dependent enzyme, methionine synthase [48]. The step that adds the third proton to convert CH2-THF to CH3-THF is catalyzed by the enzyme MTHFR, a flavoprotein that can select protons over deuterons through proton tunneling. Thus, all the protons in the methyl group attached to methionine’s sulfur atom can be expected to be deupleted. H2S, the product of the third pathway, can provide two deupleted protons to the intermembrane space of the mitochondria when it is oxidized to sulfate and thiosulfate.
2.4. An Important Role for Methylotrophs
The human archaeome (archaeal microbes that colonize the human gut) is an underestimated contributor to the gut microbiome [49]. Thus far, we have shown that archaea can use hydrogen gas as a reducing agent to produce severely deupleted methane gas, which is then utilized by gut bacteria to produce formate, NADH, and deupleted water, and to supply methylene units to the methylation pathways. The methanogenic order of archaea called Methanomassiliicoccales are exclusively obligate anaerobes. These unique organisms produce methane by hydrogen-dependent reduction of methanol or methylamines [50]. Members of this clade inhabit the human gut, where they can metabolize tri-, di-, and monomethylamine to produce methane gas. The enzyme trimethylamine (TMA) methyltransferase (MttB) transfers a methyl group from TMA to a corrinoid protein, from which it is then transferred by coenzyme M methyltransferase to coenzyme M. Methyl Coenzyme M reductase then produces methane, the final product [51,52,53].
While Methanobrevibacter smithii is the dominant archaeal methanogen in the human gut, Methanomassiliicoccales are also found at significant levels, and it is intriguing that their levels rise with age [54]. It has been suggested that they would likely be beneficial as a probiotic because of their ability to clear TMA from the gut [34]. This would lead to a reduction in the production of trimethylamine oxide (TMAO) by the liver. Elevated serum levels of TMAO are linked to cardiovascular disease [34] and to fatty liver disease [55]. The pathway from choline to trimethylamine to methane gas may ultimately be a significant contributor to the supply of deupleted NADH and deupleted methyl groups in the methylation pathways.
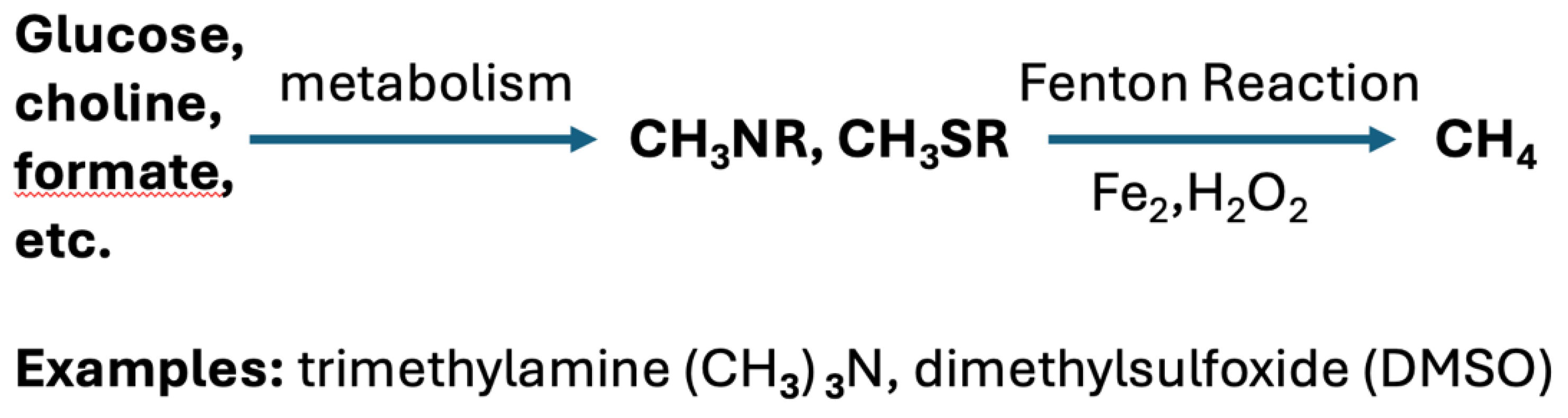
While it is traditionally said that methane is produced mainly by enzymatic decomposition of organic matter by anaerobic archaea, it has been shown recently that many organisms, including bacteria, cyanobacteria, plants, fungi, algae, and even human cells, can produce methane nonenzymatically in the presence of reactive oxygen. Methane production by Bacillus subtilis and E. coli is triggered by free iron and reactive oxygen species (ROS) under conditions of oxidative stress. A seminal paper by Ernst et al. demonstrated that methyl groups bound to nitrogen and sulfur are substrates for this spontaneous reaction, where they are converted to reactive methyl radicals as an intermediary [56]. This process is shown schematically in Figure 2. An editorial on the Ernst et al. paper made bold predictions as follows: “An in-depth understanding of the various factors that control cellular methane formation and consumption and a thorough understanding of its dual function and bioactivity may bring significant benefits to the domain of human health and disease.” [57].
It is tempting to speculate that oxidative stress induced by deuterium overload in the mitochondria leads to the productive release of methane gas derived from methyl groups that can then be recycled into useful sources of deupleted protons through the action of resident gut methanotrophs. Oxidative stress in the gut linked to inflammatory bowel disease would likely impair the activity of strictly anaerobic species [21,22], and it is therefore fortuitous that methane can be sourced from methyl groups under such conditions to compensate for the reduction in anaerobic methane production.
Figure 2.
Especially under conditions of oxidative stress, several gut microbial strains, and even human cells, can convert methyl groups attached to nitrogen or sulfur to methane gas. Possible substrates include methionine, trimethylamine (TMA) and dimethylsulfoxide (DMSO).
Figure 2.
Especially under conditions of oxidative stress, several gut microbial strains, and even human cells, can convert methyl groups attached to nitrogen or sulfur to methane gas. Possible substrates include methionine, trimethylamine (TMA) and dimethylsulfoxide (DMSO).
3. Choline Plays a Central Role in Methylation Pathways
In this section, we trace the metabolic pathways associated with choline, a nutrient that has been shown to have many benefits to human health. Choline carries three methyl groups attached to its nitrogen atom, which are sourced from SAMe. As phosphatidylcholine, it populates plasma membranes. As acetylcholine, it acts as an excitatory neurotransmitter in neurons. Gut microbes can break it down into interesting metabolites that likely serve to further deplete deuterium in derived nutrients. But the liver produces a metabolite called trimethylamine oxide (TMAO) whose elevation in the serum has been linked to many chronic diseases.
3.1. Choline Synthesis
Choline, also known as trimethylethanolamine, is a remarkable nutrient known to be essential for brain health. Phosphatidylcholine (PC) is the most abundant phospholipid in the outer leaflet of mammalian plasma membranes, and it serves as a storage form of choline. PC localizes to the outer layer of the lipid bilayer. PC hydrolysis by phospholipase D (PLD) releases free choline, which can then be taken up by neurons and converted to acetylcholine, a powerful neurotransmitter.
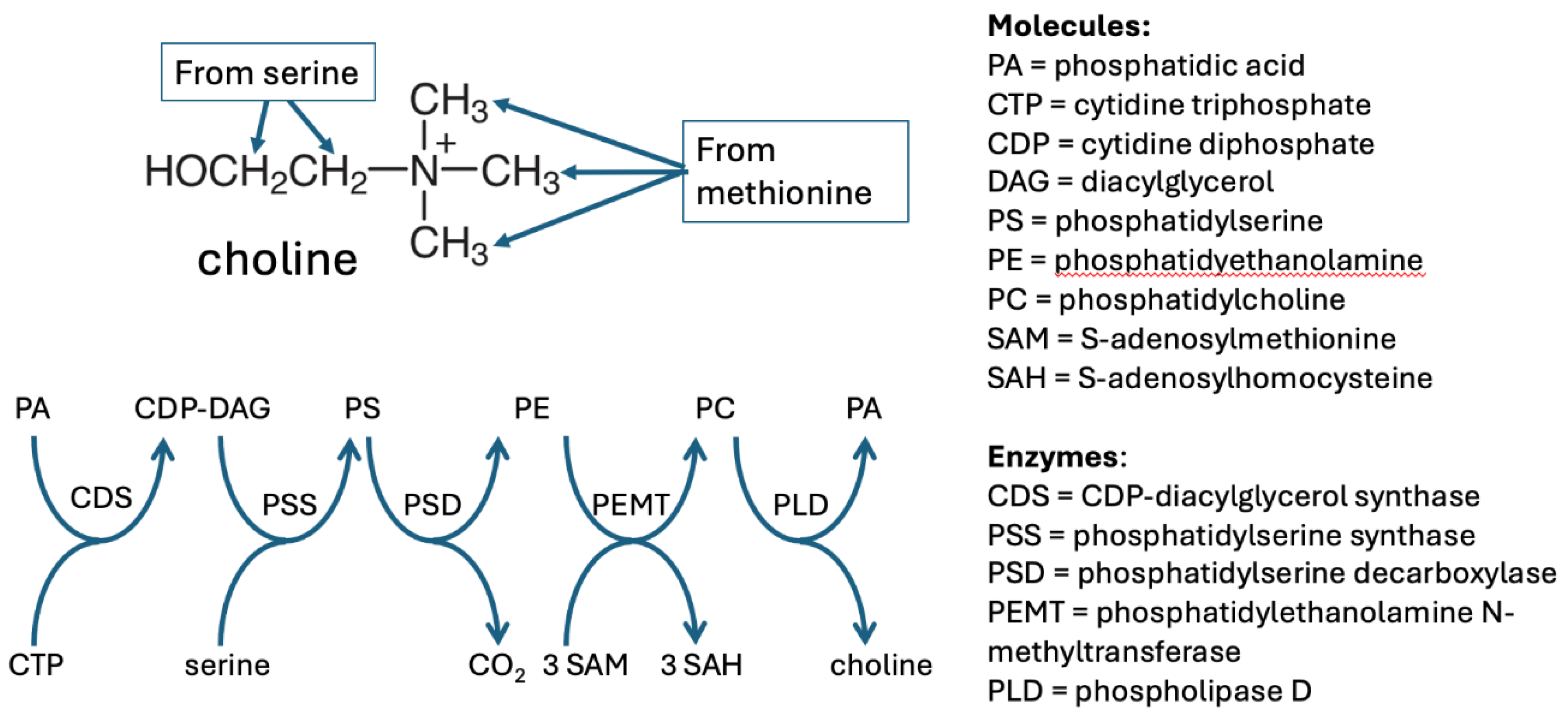
De novo choline synthesis as PC takes place primarily in the liver, in a series of steps that involve three phospholipids -- phosphatidyl serine (PS), phosphatidylethanolamine (PE) and phosphatidylcholine (PC), via the Kennedy pathway [58]. Phosphatidylserine decarboxylase (PSD) converts PS to PE, and then PE becomes PC via the transfer of three methyl groups from SAMe to PE, catalyzed by phosphatidylethanolamine N-methyl transferase (PEMT). Since the methyl groups in SAMe are derived from CH3-THF, they too will be deupleted. These pathways are illustrated in Figure 3.
PC can also be synthesized directly from dietary choline, which normally accounts for about 70% of the PC molecules in the body. Choline is an essential nutrient because the liver does not normally synthesize enough to serve the body’s needs. Humans with insufficient dietary choline develop fatty liver disease [59]. However, PEMT plays an essential supportive role as well, since Pemt-/- mice develop non-alcoholic fatty liver disease and cholestasis when fed a high fat diet [60].
Plants synthesize choline via a somewhat different pathway, which begins with phosphoethanolamine instead of PE. But the enzyme that catalyzes the reaction, phosphoethanolamine N-methyltransferase (PEAMT) also transfers methyl groups from SAMe, just as in animal-based synthesis [61].
Figure 3.
The series of reactions that lead to choline synthesis, beginning with PA. Three different membrane phospholipids (PE, PS, and PC) act as intermediaries. PE and PS localize mainly to the inner membrane of the lipid bilayer in the plasma membrane. Once three methyl groups are attached to ethanolamine via enzymatic action of PEMT, the product PC translocates to the outer membrane. PLD is the enzyme that detaches choline from PA in the outer membrane to release it into the circulation.
Figure 3.
The series of reactions that lead to choline synthesis, beginning with PA. Three different membrane phospholipids (PE, PS, and PC) act as intermediaries. PE and PS localize mainly to the inner membrane of the lipid bilayer in the plasma membrane. Once three methyl groups are attached to ethanolamine via enzymatic action of PEMT, the product PC translocates to the outer membrane. PLD is the enzyme that detaches choline from PA in the outer membrane to release it into the circulation.
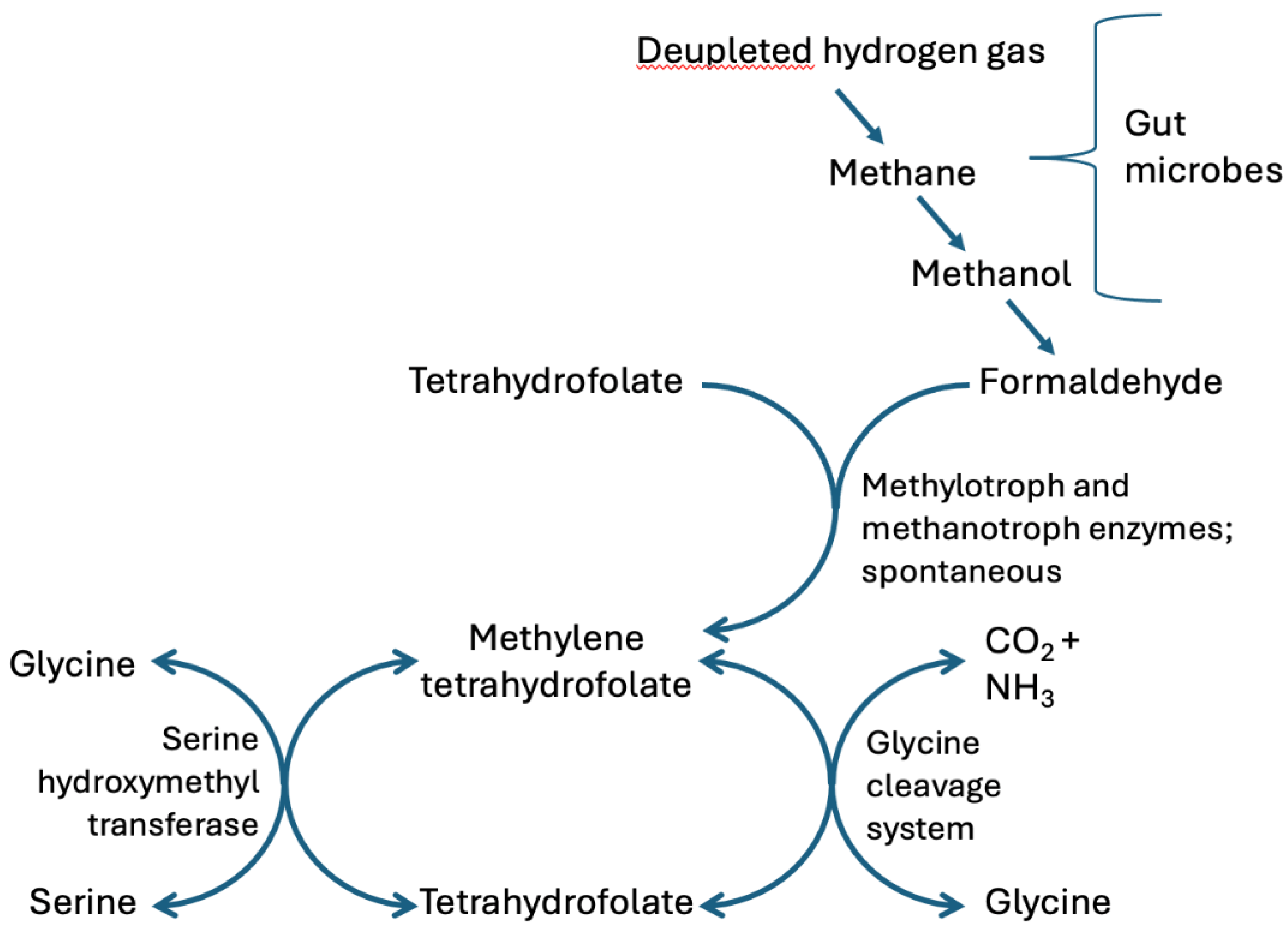
All the protons bound to the carbon atoms in choline can be predicted to be deupleted, given their origins. This may be the primary reason why choline is so important in metabolism. Both the α carbon and the β carbon in serine can be traced back to formaldehyde because they are sourced from CH2-THF via the glycine cleavage system and serine hydroxymethyltransferase (SHMT) (see Figure 4). Both reactions take place primarily in the mitochondrial inner membrane.
serine + THF ←→ glycine + CH2-THFSHMT
glycine + THF + NAD+ ←→ CH2-THF + CO2 + NH3 +NADH + H+glycine cleavage
The glycine cleavage system is widely expressed in animals, plants, and bacteria. The reaction is reversible, so it can be used both to synthesize and to degrade glycine [62]. Whether glycine is obtained from the diet or synthesized by gut microbes or human cells, the protons bound to its α carbon atom will be deupleted, because of their origin in formaldehyde. Glycine sourced from a synthetic supplement is an exception. The glycine cleavage system is the most prominent pathway in serine and glycine catabolism in humans and other vertebrates [62].
Figure 4.
Glycine and serine are both mobile carriers of deupleted methylene groups transferred from methylene tetrahydrofolate. Both the α carbon and the β carbon of serine can be expected to be deupleted because they trace back to formaldehyde produced by gut microbes from methane gas, where the protons were sourced from extremely deupleted hydrogen gas. Formaldehyde feeds into the system either through a spontaneous reaction or through enzymatic catalysis by enzymes expressed by methylotrophs and methanotrophs.
Figure 4.
Glycine and serine are both mobile carriers of deupleted methylene groups transferred from methylene tetrahydrofolate. Both the α carbon and the β carbon of serine can be expected to be deupleted because they trace back to formaldehyde produced by gut microbes from methane gas, where the protons were sourced from extremely deupleted hydrogen gas. Formaldehyde feeds into the system either through a spontaneous reaction or through enzymatic catalysis by enzymes expressed by methylotrophs and methanotrophs.
3.2. Choline Metabolism
Dietary choline can be metabolized via several different pathways, many of which involve gut microbial enzymes. Up to 60% of the methyl groups in a typical human diet are derived from choline [63]. Choline is first converted to betaine (trimethylglycine) via the intermediary glycine betaine aldehyde (GlyBA). Choline oxidase, a microbial enzyme, converts choline to GlyBA, which is then converted to betaine by betaine-aldehyde dehydrogenase (BADH), primarily expressed in the liver. Betaine then donates its three methyl groups to three molecules of homocysteine, regenerating three methionine molecules.
Another pathway for choline metabolism begins with the microbial enzyme choline trimethylamine-lyase (CutC), which anaerobic bacteria use to convert choline into trimethylamine (TMA) and acetaldehyde [64]. TMA can be further metabolized by gut microbes to produce either three molecules of methane (via the enzyme methyl-coenzyme M reductase (MCR) expressed in archaea), or three molecules of formaldehyde, via the microbial enzyme trimethylamine dehydrogenase (TMADH), found in methylotrophs. Both the methane and the formaldehyde potentially become excellent sources of deupleted protons to fuel the mitochondria in the colonocytes. Any TMA that does not get further metabolized by gut microbes makes its way to the liver, where it is oxidized to trimethylamine oxide (TMAO). Elevated levels of TMAO in the blood are associated with both fatty liver disease and increased risk to cardiovascular disease [4]. Archaea protect from the synthesis of TMAO in the liver by metabolizing TMA to produce methane gas [65].
Most intriguing is the fact that TMA dehydrogenase is a flavoprotein with a high deuterium KIE due to vibrationally assisted hydrogen tunneling [66]. A study on its activity on deuterated dimethylamine showed that the KIE for Kcat/Km had a strong dependency on pH and rose to as high as 8 at pH 9 or above [67]. What this could mean is that it selectively removes molecules of TMA that are free of deuterium, leaving behind a pool that is enriched in deuterium, which can then get oxidized in the liver to produce deuterium-enriched TMAO.
As shown in Figure 5, the metabolism of choline ultimately to glycine, via the gut microbes and the liver, results in the production of one molecule of NADH, one molecule of H2O2, and three molecules of methionine. All of these are useful sources of deupleted protons, assuming that the protons in choline are deupleted. Laboratory-synthesized choline is problematic because it is not expected to have deupleted protons.
3.3. Dietary Versus Synthetic Supplemental Choline
The study of dietary and supplemental choline and its potential therapeutic impact came to prominence in the 1970s. A 1977 study was the first to show that choline supplements given to patients with tardive dyskinesia brought about a significant reduction in choreic movements in 9 of 20 patients over the course of 2 weeks of supplementation. The benefit was presumed to be due to an increase in brain acetylcholine in response to the supplemental choline [68]. Since that time, hundreds of studies have sought to clarify a wide range of potential health benefits of dietary or supplemental choline. The most consistently identified benefit has been with respect to improved memory and/or attention in patients with Alzheimer’s or other forms of cognitive decline, which is again attributed to its ability to increase brain acetylcholine production [69].
The growing recognition of choline’s role in maintaining health led to the National Institute of Health including it among the nutrients for which it established a Dietary Reference Intake (DRI) in 1998 [70]. Sales data for choline over time are not available, but sales of cognition-enhancing nutrients called “nootropics,” of which choline is one, are projected to have a compounded annual growth rate globally of nearly 15% between 2023 and 2030 [71].
It is within this context that we feel it is important to bring to wider awareness the processes utilized for the manufacture of choline sold as a dietary supplement, and the potential that deuterium introduced incidentally through these purely synthetic manufacturing processes could impact the efficacy of these products, for the reasons explained in detail in this paper.
One method of producing choline at scale involves the application of solvents such as ethanol, hexane, or acetone, to natural choline sources such as arctic krill, soybeans, sunflower seeds, or egg yolks [72]. We would expect the choline derived from these natural sources to be relatively deupleted due to the KIEs of the enzymes involved in its biosynthesis as we have described throughout this paper. Consequently, TMA, a microbial metabolite of choline described previously, would also be deupleted in its three methyl groups. As mentioned, if the anaerobic methylotrophs are thriving in the gut, those methyl groups go on to contribute to methane gas or formaldehyde production, both of which act as substrates for the methylation cycle and SAMe production.
A purely synthetic method for choline production was patented as long ago as the 1950s, wherein trimethylamine was reacted with ethylene oxide to produce choline that was considered pure enough for human consumption [73]. Since that time, several other techniques have been developed for the production of various forms of choline salts sold as dietary supplements, such as choline chloride, choline bitartrate, and choline citrate. These salts are formed predominantly by reacting trimethylamine (which is itself synthetically produced) with the relevant acid to generate the salt, leaving behind trimethylamine oxide (TMAO) as a contaminant that must be removed in order to purify the choline salt before consumption.
It is interesting to note that supplementation with the synthetically produced choline bitartrate, but not dietary choline or naturally derived phosphatidylcholine, resulted in a significant increase in serum TMAO levels [74]. These authors wrote in the Conclusion of their abstract: “Despite high choline content in egg yolks, healthy participants consuming four eggs daily showed no significant increase in TMAO or platelet reactivity. However, choline bitartrate supplements providing comparable total choline raised both TMAO and platelet reactivity, demonstrating that the form and source of dietary choline differentially contributes to systemic TMAO levels and platelet responsiveness.” The authors of that study hypothesized that phosphatidylcholine might be less available to the gut microbes for metabolism into TMA, compared to a choline salt. In a study aptly titled, “Gut microbe-generated trimethylamine N-oxide from dietary choline is prothrombotic in subjects”, both vegetarians and omnivores were given a supplement containing 500 mg choline bitartrate twice a day over a period of two months. Both groups showed a ten-fold increase in TMAO levels already after one month of supplementation, and this correlated with increased platelet aggregation associated with thrombotic risk [75].
We suggest that the cause of these differences in choline’s differential relationship with TMAO production may have to do with the relative excess of deuterium that would be introduced into the synthetically produced choline compared to the deuterium depletion inherent within a naturally derived choline source. The methyl groups associated with the TMA microbially generated from a high deuterium supplement would be less favored enzymatically for donation as methyl groups. Consequently, more would be transported from the gut to the liver to be oxidized into TMAO, resulting in the risks associated with that metabolite. Furthermore, the TMA that does not get metabolized by the archaea (and is therefore converted to TMAO by the liver) becomes relatively enriched in deuterium, because the high KIE of the enzymes involved results in the selective removal of the deupleted molecules of TMA. It may be that TMAO is exploited by the body as a signal of deuterium overload in the methylation pathways, and metabolic policies are altered to reflect this issue.
3.4. The Pathogenic Mechanisms of TMAO that Induce Tissue Damage and Disease
Choline is considered an essential nutrient for human health. However, the metabolites of choline produced by the gut microbes and the liver enzymes, TMA and subsequently TMAO in the liver, respectively, are responsible for the development of human diseases such as those involving liver, kidney, and cardiovascular damage [76]. Elevated levels of TMAO in the blood stream result in systemic inflammation, endoplasmic reticulum (ER) stress, mitochondrial stress, and disruption of normal physiological functions [77]. TMAO causes its harmful effects on cells by modulating the autophagy mechanisms. Autophagy for normal cells is a housekeeping mechanism to degrade an excess of damaged organelles and through recycling to provide new cellular constituents for regeneration and self-sustainability. However, when dysregulated, autophagy can lead to cell death, tissue damage, and disease [78]. Nevertheless, autophagy is also a life-rescuing mechanism for cancer cells [79]. Therefore, a fine balance of autophagic mechanisms is required to maintain homeostasis.
TMAO’s harmful effects caused by modulation of autophagy are bidirectional, even for the same organ. TMAO’s inhibition of autophagy produces damage to the kidneys through the activation of NLRP3 inflammasome and mitochondrial dysfunction [80]. Overall, these two pathogenic processes are interrelated [81]. On the other hand, again regarding kidney disease, when it promotes autophagy, TMAO also promotes cell death and thus kidney tissue damage through the aggravation of overwhelming deposition of calcium oxalate crystals that induce the production of ROS and inflammation [82].
Regarding cardiometabolic disease, TMAO, by inhibiting autophagy, activates the p13K/AKT/mTOR pathway and interferes with the activity of p62 protein, provoking the expression of other interrelated genes that promote atherosclerosis [83]. Indeed, other studies on human aortic cells have shown that TMAO, apart from its action as an inflammatory pathway mediator (by phosphorylating numerous kinases in the cytosol) is a mitochondrial gene modulator, and a promoter of intracellular adhesion molecule (ICAM) expression that is tightly linked to atherosclerosis [84]. TMAO induces alterations in healthy metabolic processes and inflammation through the generation of ROS [85]. These mechanisms involve the ER and mitochondrial stress, and the disturbance of innate immunity.
On the other hand, as in the kidney tissue cases with TMAO pathogenic activity, TMAO, by increasing the autophagic degradation of an essential calcium transporter, the sarcoplasmic reticulum Ca2+-ATPase (SERCA2a), affects calcium homeostasis. TMAO induced cardiac hypertrophy in the animals tested in the B Lei et al. study by increasing the intracellular calcium concentrations [86]. Moreover, the reversal of autophagic (lysosomal) induction by TMAO restored the SERCA2a-mediated calcium influx and helped to alleviate cardiac hypertrophy. However, the pathogenic outcome was not only attributed to TMAO’s promotion of autophagic degradative capacity, but also to the alteration of essential cellular regulators that protect from cardiac myopathy on a genetic level. The response to TMAO in this study was found not only to downregulate the expression of the SERCA-2a gene but also to upregulate the expression of the ANP and MYH7 genes. The myosin heavy chain 7 (MYH7) gene expression has been found to be responsible for severe cardiac and other skeletal myopathy complications [87], whereas the atrial natriuretic peptide (ANP) gene encodes for a hormone-like peptide, ANP, which is often released in conditions of heart failure in humans [88]. Moreover, the downregulation of p62 expression by TMAO in the same study indicates the loss of p62 cardioprotection. p62 has been found to protect from hypoxia and ischemic disease in other animal studies [89].
In summary, TMAO as a toxic metabolite is pathogenic for cardiac and kidney tissues. The pathology induced by TMAO in these organs is interrelated [90]. TMAO functions to induce pathology not only by influencing the fine balance between autophagy and cell death but also as a gene expression modulator that promotes heart myopathy that leads to heart failure. Finally, TMAO’s role to promote atherosclerosis is a major contributor to vascular damage and heart disease. Recent studies attribute to TMAO an important pathogenic role in promoting cancer and metastasis [77]. The autophagic and gene expression modulations by TMAO that have been identified to produce the already mentioned diseases can prove to be important factors that also promote and lead to cancer. The mitochondrial dysfunction and accelerated aging induced by TMAO [91], like that found in cardiac mitochondria [92] can be the common link between the autophagy interference and the pathology promotion of the diseases caused by metabolic reprogramming [93].
4. Metabolism of Methyl Groups Supports Mitochondrial Health
We have seen that SAMe donates methyl groups to several different molecules, e.g., to produce phosphatidylcholine, and also to methylate proteins, DNA and RNA. We have also seen that phosphatidylcholine ultimately delivers its methyl groups back to the gut microbes, which can then recycle the methyl groups to restore CH3THF, while further scrubbing deuterium from the molecules. The process of demethylation is not simply the removal of a methyl group to be attached to some other molecule. Instead, it results in the full metabolism of the methyl group, which directly or indirectly provides deupleted protons to the mitochondria.
In this section, we will critically examine the various ways in which methyl groups are removed from organic molecules. We will consider the removal of methyl groups from histones and from DNA, and the metabolism of the methyl group in methionine that results in its conversion back to formaldehyde. Finally, we will consider whether the studies in rats that found that dietary methionine restriction supports longevity were flawed in their design.
4.1. Histone Methylation and Demethylation
Histone methylation is an epigenetic modification that can regulate gene expression. Histones can be methylated through a transfer of a methyl group from SAMe to either their arginine or their lysine residues. Lysine can pick up as many as three methyl groups, and arginine can hold up to two. All these methyl groups are bound to nitrogen atoms in the residues [94].
Histones are demethylated via the enzymatic action of histone demethylases. But, in this reverse step, the methyl group is not simply attached to another carrier molecule but rather is released as formaldehyde. While formaldehyde is known to be a cancer risk due to its high reactivity, it is usually very quickly cleared through metabolism [95]. As has been described, in the gut, formaldehyde can donate its methylene unit to tetrahydrofolate (THF) to produce CH2-THF, or it can be processed by anerobic bacteria to produce further deupleted hydrogen gas, which can then be used to synthesize SCFAs.
Besides the microbial pathways, human cells can exploit formaldehyde to reduce NAD+ to NADH. Alcohol dehydrogenase 5 metabolizes formaldehyde to formate, while converting NAD+ to NADH. Formate is a precursor for de novo synthesis of purine nucleotides, in a folate-independent pathway [96]. Formate can also be further metabolized to produce CO2 and H2O, through the enzyme formate dehydrogenase, producing yet another molecule of NADH. Assuming that SAMe carried a deupleted methyl group, both of these molecules of NADH can provide deupleted protons to the mitochondria.
All these reactions are beneficial as they support lower deuterium exposure in the mitochondria.
4.2. DNA Methylation and Demethylation
SAMe is also responsible for methylating the cytosine base in DNA, forming 5-methylcytosine (5mC), in both eukaryotes and bacteria, and this is a key epigenetic modification that alters gene expression, occurring mainly at CpG dinucleotides [97,98].
Just as for histones, the process of removing methyl groups from DNA does not involve a simple methyl transfer reaction. The process begins with a family of ten-eleven translocation (TET) enzymes, which remove the protons from the methyl groups, one by one, finally yielding 5-carboxylcytosine (5caC). Thymine DNA glycosylase then swaps out 5caC for a fresh intact cytosine base, initiating the process of base excision repair [99]. TET enzymes are key players in the control of cellular differentiation and transformation in mammalian cells [100]. It has only recently been demonstrated that 5caC can also be directly decarboxylated in place, without requiring the machinery that has to rely on DNA breaks and repairs. Direct decarboxylation is a rapid process that occurs in diverse mammalian cells [101].
An interesting aspect of the reaction catalyzed by TET enzymes is that other reaction products are succinate and CO2, which are derived from α-ketoglutarate, a substrate [102]. The deupleted proton that is extracted is presumably released into the medium, helping to lower the deuterium content there. Each methyl group that is ultimately removed yields three succinate molecules and three free protons.
Mitochondrial DNA can also be methylated, and TET2 is expressed in mitochondria. A study demonstrated that TET2 levels were upregulated in mitochondria following acute brain ischemia, and the levels of hydroxymethylcytosine increased in parallel, peaking two days after the injury. There was a corresponding increase in the levels of ATP in the mitochondria, suggesting that TET activity facilitated ATP production [103]. The synthesis of succinate to support Complex II, as well as the release of deupleted protons into the medium, should both be beneficial to mitochondrial health. Defects in TET2 are linked to cancer. Mutations in TET2 are present in 7% to 28% of adult acute myeloid leukemia (AML) patients [104].
TET proteins are members of a class of proteins called dioxygenases that are known to have a high deuterium KIE for the removal of a proton from a methyl group. TET was shown experimentally to have a deuterium KIE of 9 for 5-methyl-cytosine, and 29 for 5-hydroxymethyl-cytosine [102]. This not only assures that the proton that is removed is deupleted but also predicts that 5-methyl-cytosines containing deuterium in the methyl group would tend to remain unmetabolized.
Myelodysplastic syndrome is a hematopoietic disorder that frequently progresses to AML. Patients with TET2 mutations exhibit lower survival rates and a greater potential to progress to AML. TET2 is one of the most frequently mutated genes in myeloid malignancies. It has been proposed that pharmacological enhancement of TET2 activity might be a good strategy for blocking malignant transformation of leukemia cancer cells [105].
4.3. Methionine Metabolism to Methanethiol and Beyond
Several commensal bacteria in the human gut are capable of producing methanethiol as a breakdown product of methionine [106,107]. Methanethiol, also known as methylmercaptan, is a one-carbon organic sulfur compound with the formula CH3SH. It has a distinctive unpleasant odor resembling rotten cabbage, and it is a major contributor to the smell of feces. Human cells express an enzyme called selenium-binding protein 1 (SELENBP1), which metabolizes methanethiol into H2O2, H2S, and formaldehyde [108]. This collaborative activity between the gut microbes and the human host is thus another way to produce formaldehyde, which can then be further metabolized, as we’ve discussed. The reaction product H2O2 may also be significant. We have argued in previous research that H2O2 may be an important supplier of deupleted protons, as it is a gas that freely diffuses past the mitochondrial outer and inner membranes, and it is converted into two molecules of deupleted water by glutathione peroxidase, highly expressed in the mitochondria [109]. Being a gas, it is likely further stripped of any deuterium during its synthesis.
SELENBP1, a selenoprotein that uses copper as a cofactor, is highly expressed in the colon, liver, and lungs. SELENBP1 is a tumor suppressor involved in the regulation of cell proliferation, senescence, migration and apoptosis [110]. Many tumor cells express low levels of SELENBP1, and this reduced expression is often associated with tumor progression, poor prognosis, and resistance to anti-cancer therapy in many types of cancer [110,111]. It is conceivable that SELENBP1 protects from cancer through its ability to release small molecules that support the delivery of low-deuterium protons to the mitochondria.
Germ-free mice and germ-free pigs were observed to have elevated levels of methionine in their colon compared to controls [112]. This may reflect the fact that methionine is no longer being converted to methanethiol, due to the absence of gut microbes.
4.4. Dietary Methionine Restriction and Longevity
Senescence and aging are complex processes directly related to homeostasis during the human lifespan [113]. It has been experimentally proven for nematodes that increased growth rate through metabolic reprogramming comes at the cost of reduced life expectancy (longevity) [114]. Therefore, it would not be surprising for molecules involved in metabolic (aging) stress responses and that affect growth rate to also affect life expectancy [115].
In a paper published in 1993, Orentreich et al. reported on an experiment where rats fed a low-methionine diet experienced a 30% increase in life span [116]. This is surprising, since methionine is an essential amino acid. Variations on this experiment have been repeated many times over the years, and the results consistently show that methionine deficiency, while it slows growth rate, supports longevity in rats [116,117,118,119]. It appears that the suppression of growth hormone production by dietary methionine restriction is the primary mechanism by which it extends lifespan, since mice that overexpress growth hormone experience a 50% reduction in lifespan [120].
Eighty percent methionine restriction in rats and mice increases longevity and strongly improves mitochondrial function, but it also slows growth rate. Caro et al., in a paper published in 2009, were interested in assessing whether a 40% reduction in dietary methionine would be sufficient to improve the lifespan without causing slow growth. Indeed, their experiment demonstrated that this was possible. They quantified results specifically showing that the restricted rats had decreased ROS production and leak at complex I in the mitochondria and reduced oxidative stress in the kidneys and brain. In the abstract, these authors boldly concluded that “methionine is the only dietary factor responsible for the decrease in mitochondrial ROS production and oxidative stress, and likely for part of the longevity extension effect, occurring in DR [dietary restriction]” [117].
What is especially significant to us is that the authors specified in detail in the Methods section that both the methionine-restricted and the control rats were supplied with precise amounts of synthetic amino acids in lieu of natural proteins. They wrote: “The diets contained a mixture of amino acids instead of protein, and their precise composition is shown in Table 1” [117]. The restricted group received only 60% of the synthetic methionine compared to the control group.
While human cells cannot synthesize methionine from cysteine, gut microbes can, and they source the methyl group from CH3-THF [121]. Nearly all bacterial species possess biosynthetic pathways for methionine synthesis [122]. The dietary deficiency in methionine would be expected to induce methionine synthesis from cysteine by the gut microbes to compensate for the deficiency. Assuming that the methyl group in CH3-THF is derived from formaldehyde, then the microbial biosynthesis of methionine will yield a methionine molecule carrying deupleted protons in its methyl group. This could explain why the rats with reduced dietary methionine had improved function of mitochondrial oxidative phosphorylation, due to the reduced deuterium burden in the mitochondria.
In a mouse study published in 2023, it was found that restricted dietary methionine could reduce the levels of TMAO resulting from a high-choline diet. A choline-rich diet combined with methionine restriction led to an alteration in the gut microbes resulting in increased production of butyrate and reduced TMA production by the bacteria [123]. This can be explained by the hypothesis that choline provided the precursors for methionine synthesis from homocysteine via the intermediary glycine beta aldehyde, to correct for methionine deficiency. This left behind less choline to get converted to TMA, while also allowing more hydrogen gas to be used as a reducing agent by acetogenic bacteria, to boost butyrate production.
5. Does Acetylcholine Provide Acetate to Synaptic Mitochondria?
Acetylcholine is a neurotransmitter that plays an important role in the synapse by signaling to acetylcholine receptors to effect various responses in the receptor neuron or muscle cell. Nicotinic acetylcholine receptors (nAChRs) are ligand-gated ion channels that transmit the signal from nerve to muscle that elicits muscle contraction. Acetylcholine is released from the axon of a motor neuron into the synaptic cleft, and acetylcholine then diffuses across the synaptic cleft and binds to the nAChRs on the muscle cell membrane. nAChRs are also expressed throughout the peripheral and central nervous system, where they are involved in fast synaptic transmission [124]. Muscarinic acetylcholine receptors are G-protein-coupled receptors with complex physiological responses, present in synapses in both central and peripheral nerves [125,126].
The enzyme acetylcholinesterase very quickly breaks down acetylcholine into acetate and choline, terminating the signaling response. Choline is then reabsorbed by the presynaptic neuron through a high-affinity choline transporter and reused for further acetylcholine synthesis. However, acetate is released into the medium, and it is plausible that it may be a significant source of fuel for the synapse-resident mitochondria. Mitochondria are enriched at synapses due to the high energy requirements of synaptic activity and plasticity. Being isolated from the cytoplasm, they would likely be challenged to meet their energy needs. Acetate transport into mitochondria does not require a carnitine shuttle [127]. Acetate is small enough to readily permeate the mitochondrial inner membrane. Inside the mitochondrial matrix, acetate is converted to acetyl-CoA by acetyl-CoA synthetase (ACSS1). Interestingly, cells in many organs, but especially the heart, skeletal muscles, and brown adipose tissue, can deacetylate acetylated proteins and convert the acetate into acetyl-CoA in their mitochondria [128]. Acetate serves as a nutrient source under nutrient-deprived conditions. In fact, mitochondria in tumor cells often overexpress ACSS1, which allows them to reduce their dependence on glucose and glutamine [129].
Synaptic mitochondrial dysfunction is an early manifestation of the disease process in a mouse model of Alzheimer’s disease, due to their increased vulnerability to stressors [130]. It is interesting that synaptosomes do not readily take up acetate, compared to astrocytes, and this may be a strategy to assure that acetate remains in the synapse for its use by the resident mitochondria [131]. Cholinergic neurons have a greater need for acetyl-CoA due to their supply of acetylcholine to the synapse. They are therefore more susceptible to death due to deficiencies in acetyl-CoA and/or choline [132]. Cholinergic neurons in the basal forebrain are severely lost in Alzheimer’s disease [133].
Several organophosphate insecticides induce their toxic effects by suppressing acetyl- cholinesterase, causing hyperstimulation of both muscarinic and nicotinic receptors. This overstimulation can result in a cholinergic crisis, characterized by symptoms that include muscle weakness, seizures, respiratory failure, and even death [134]. A popular organophosphate insecticide, pirimiphos-methyl, has been shown to disrupt spermatogenesis and reduce fertility in male rats [135]. It is conceivable that an aspect of their toxicity is that they deprive the synaptic mitochondria of an important fuel supply.
6. Benefits of a High Fiber Diet
A high fiber diet leads to increased fermentation by gut microbes, which can then promote the synthesis of deupleted SCFAs by the gut microbes. Of these, butyrate has garnered the most attention for its beneficial role not only in gut health but also throughout the organism. It is well established that butyrate is a histone deacetylase inhibitor, but it is also a ligand for G-protein coupled receptors and, of course, a valuable energy source for the mitochondria [136]. It is conceivable that much of the benefit of butyrate stems from its value as a supplier of low-deuterium protons to the mitochondria. There are several distinct gut-brain communication channels by which SCFAs could modulate brain function, including immune, endocrine, vagal, and humoral pathways [137].
A promising prebiotic containing wheat bran extract that is enriched in arabinoxylan oligosaccharides (AXOS) has been shown to have striking positive effects on the gut microbiome in an obese population. Arabinoxylan is a plant hemicellulose found in the cell walls of cereal grains. These oligosaccharides are not digested by human enzymes, but they are easily fermented by beneficial gut bacteria, notably Bifidobacteria. A multiomics study showed that this prebiotic supplement led to an increase in not only Bifidobacteria but also the beneficial strain Prevotella in the gut of overweight subjects with previous signs of metabolic syndrome. The authors observed an increase in the production of butyrate associated with the supplement, along with a boost in “a wide diversity of butyrate producers.” Notably, there was an increase in plasma levels of phosphatidyl-choline, which they hypothesized was due to a greater availability of dietary choline following a reduction in the availability of microbial enzymes that convert choline to TMA. This implies a reduced synthesis of TMAO in the liver, and therefore protection from cardiovascular disease [138].
7. Butyrate Is a Universal Protective/Essential Nutrient for the Human Organism
The short chain fatty acids, predominantly acetate, propionate, and butyrate, are produced by gut microbiota of animals and humans mainly through fermentation of dietary fiber. Human and animal cells do not possess the necessary enzymatic machinery for butyrate biosynthesis. The overall health effects of butyrate and how these are exerted are described in a study by K Hodkinson et al. [139]. Butyrate is readily produced in the gut, in multiple ways, by the gut colonizers [140]. A question arising, however, is whether butyrate produced by the gut microbiome remains localized to the GI tract, or if it is distributed throughout the organism. Undeniably, butyrate produced in mammals (including humans) is mainly the result of microbial fermentation in the large intestine [140]. Recent studies have shown that butyrate, apart from its anticancer effects, by controlling cellular proliferation of colonocytes, is readily transported by the blood stream to reach not only the hepatic and kidney tissues but far more distantly, reaching the human brain tissues and the skin [141].
In the human brain, butyrate can potentially reach adequate amounts from the gut to explain its beneficial health effects [142,143]. Adults that show low colonisation of butyrate microbial fermenters, and therefore low production of butyrate in the gut, have increased risk to develop inflammatory bowel disease (IBD) [144]. Moreover, the loss of butyrate production by gut fermenters has been linked to the development of various systemic autoimmune disorders, amongst them are rheumatoid arthritis, lupus erythematosus, and allergic asthma [145,146].
It is probably clinical cases involving infants that best indicate how important butyrate synthesis by the intestinal colonizers is, not only for GI tract health, but also for more distant organs. Butyrate produced in the large intestine is mainly metabolized by colonocytes, while the remainder is transported via the liver portal system through monocarboxylate transporters and metabolized by hepatocytes. Butyrate may extend its regulatory functions well beyond the GI tract in distant organs like the liver, the pancreas, the brain, the adipose tissue, and the immune system [143]. Moreover, there is evidence to suggest that butyrate is metabolized in organs distant from the GI tract; however more studies are needed to clarify this [147]. From infancy until the age of 2, butyrate production in the gut is a significant factor in conferring a reduced risk of developing atopic diseases in the respiratory tract and the skin [148,149]. Butyrate has found therapeutic value in treating depression, as well as neurodegenerative diseases and cognitive behavior [150]. Particularly for skin diseases, including psoriasis, the presence of gut-produced butyrate is essential for their prevention [151]. Moreover, the potential health effects of butyrate on human skin healing and re-epithelization are remarkable. A study of E Bachar-Wikstrom et al. indicates that butyrate exerts its healing activity on the skin by lowering the endoplasmic stress markers and thus producing re-epithelization of skin tissue [152].
Furthermore, the health-inducing activity of butyrate against brain tissue disorders that involve neural inflammation (e.g., cognitive impairment) and evolve from mitochondrial respiratory dysfunction, may be due to butyrate’s direct deacetylation activity that affects the normalisation of gene transcription [136].
Summing up, all the above studies indicate that butyrate’s production by the gut colonizers has a highly beneficial effect on human health. Some of these benefits come indirectly through sensitization and transportation of nerve signals from the GI tract. Other benefits stem from its direct transport to and action on distant tissues, far away from the GI tract. This establishes the production of butyrate in the gut as a far more essential factor for human health than was once thought. One of its basic properties seems to be the maintenance of mitochondrial respiratory chain health, as demonstrated in a study on cell lines taken from autistic boys, in order to combat neurodegeneration [153].
Sarcopenia is the loss of skeletal muscle mass during aging, which can lead to injury due to falls. A study on aged female mice demonstrated remarkable abilities of butyrate to protect against muscle atrophy in hindlimb muscles. It also increased markers of mitochondrial biogenesis in skeletal muscle, and it reduced markers of oxidative stress and apoptosis [154]. Butyric acid is severely reduced in the serum of patients with acetylcholine signaling deficiency due to myasthenia gravis. Butyrate’s presence induces autophagy of the T regulatory cells in these patients by inhibiting the activity of mammalian target of rapamycin (mTOR) [155]. In turn, the autophagy induction activates their differentiation, and this enables the T cells to combat the myasthenia disease.
These studies indicate that butyrate’s direct activity must be exerted in far distant organs from the gut. Butyrate’s localized effect such as increased histone acetylation must involve many human tissues, although its transport from the intestinal lumen is minimal. Butyrate activity in alleviating neurological disorders (Alzheimer’s disease (AD), Parkinson’s disease (PD), autism spectrum disorder (ASD), and Huntington’s disease (HD)), however, is a far more distant phenomenon [156]. Furthermore, as far as neural tissue is concerned, butyrate has proved to influence the production of acetylcholine by enhancing the activity of choline acetyltransferase, its synthesizing enzyme [157].
8. Creatinine Is a Waste Product, but Is It Really?
Creatine is a nutrient found only in animal-based foods. It has become quite popular as a supplement, especially for people seeking athletic fitness. Phosphocreatine acts as an energy buffer to support rapid synthesis of ATP during extreme physical activity. Creatine spontaneously converts to its metabolite creatinine, which forms a ring structure that may offer a unique opportunity to trap and secure deuterium. It is interesting that creatinine is not further metabolized by human cells but rather is simply excreted through the urine.
8.1. Creatine Synthesis Is a Major Pathway by Which One-Carbon Units Are Lost
Creatine is an endogenously produced molecule that is also available from the diet which plays an important role in powering muscle fibers. It can only be sourced from animal- based foods, which can be an issue for vegetarians. Many athletes and fitness enthusiasts take high doses of creatine supplements to increase muscle strength and endurance [158]. Creatine is synthesized from glycine, methionine, and arginine, in a two-step process. An amidino group is first transferred from arginine to glycine to produce guanidinoacetate, catalyzed by L-arginine:glycine amidinotransferase (AGAT). Guanidinoacetate then picks up a methyl group from methionine catalyzed by the enzyme guanidinoacetate N-methyltransferase (GAMT). Creatine is released into the circulation by the liver and is readily taken up by muscle cells, the main consumers of creatine [159]. Both the methylene unit derived from glycine and the methyl group supplied by methionine will be deupleted, as we have previously argued in this paper. Creatine synthesis accounts for the loss of about 40% of all methyl groups derived from SAMe, and it also consumes 20-30% of arginine’s amidino groups. Vegetarians obtain no creatine from the diet, putting greater strain on methylation pathways [160].
Creatine phosphate is the main high-energy phosphate-storage molecule in mammals, and it has recently been found that muscle cells synthesize a substantial amount of it themselves [161]. Creatine is converted to creatine phosphate in the mitochondria of muscle cells, via the activity of creatine kinase, while also converting ATP to ADP. When ATP levels drop, a high energy phosphate is transferred from creatine phosphate to ADP to reform ATP in a reverse reaction that replenishes ATP, producing a waste product called creatinine. Creatinine can also form spontaneously from creatine, and it involves the loss of a water molecule and the formation of a ring structure called an imidazolidinone ring. Creatinine then gets excreted into the circulation and eventually is passed out through the kidneys. It is not further metabolized, so the one-carbon units originally coming from both glycine and methionine are lost. In the next section, we will hypothetically describe how, via its imidazolidinone ring, creatinine may play a useful role in trapping deuterium for excretion through the kidneys.
8.2. Can the Imidazolidinone Ring in Creatinine Sequester Deuterium?
Normally, nitrogen and oxygen atoms in organic molecules can freely exchange their bound protons with deuterons from the medium, but they are unable to secure a deuteron for very long because it quickly exchanges back. Carbon, on the other hand, rarely gives up its protons in exchange for deuterons. However, there are exceptions, and these may be significant for a potential to trap and sequester deuterium [162].
In previous published work, we have pointed out the unusual fact that certain carbon- and nitrogen-containing ring structures have a unique property that a carbon atom in the ring can exchange its bound protons with deuterons from the water-based medium. Rings with this property include the imidazole ring in histidine [5] and the pyrrolidine ring in proline [162,163]. The breakdown product of creatine, namely creatinine, is formed nonenzymatically along with the release of a water molecule. It reconfigures into a carbon- and nitrogen-containing ring called an imidazolidinone ring, which bears a strong family resemblance to an imidazole ring, since both are five-membered rings containing two nitrogen atoms. Interestingly, imidazolidinone derivatives have shown promise as anti-cancer agents [164].
The five-membered ring structure in imidazolidinone contains two nitrogen atoms and a carbon atom derived from glycine in a one-carbon unit bound to two protons. A study published in 2020 showed that the protons bound to this α carbon atom, under basic conditions, could freely exchange with deuterons when immersed in heavy water [165]. These authors wrote: “We presented a new method of synthesis of deuterated creatinine analogues, using organic base and D2O. The study allowed us to obtain the Cre [creatinine] analogue, containing two deuterons, which were introduced after 60 min incubation in a 1% solution of TEA [trimethylamine] in D2O at room temperature. The introduced deuterons did not undergo back-exchange under acidic and neutral conditions” [165]. If creatinine is then able to trap deuterium, this would justify it being a waste product, since it would no longer be suitable as a deupleted nutrient in the mitochondria.
A central aspect of the beneficial effects of creatine supplementation relates to its ability to improve mitochondrial health. Creatine supplements have been shown to be beneficial in treating a long list of diseases that are associated with mitochondrial dysfunction, including both acute traumatic mitochondrial dysfunction and chronic atraumatic mitochondrial dysfunction. These conditions include traumatic brain injury, fatty liver disease, diabetes, obesity, cardiovascular disease, neurodegenerative disorders, chronic fatigue syndrome, and long COVID [166]. It could be that the sequestering of deuterium in creatinine in the mitochondria plays a role in improving mitochondrial health, systemically.
Curiously, brown adipocytes in mice express a “futile creatine cycle” in their mitochondria that involves robust phosphocreatine phosphatase activity by tissue nonspecific alkaline phosphatase (TNAP). This thermogenic reaction wastes the energy in the phosphate anion by converting it to heat. Creatine kinase then restores creatine phosphate in the futile cycle. Inhibition of futile creatine recycling leads to obesity in mice [167]. We can therefore postulate that this recycling process may facilitate the acquisition of deuterium by creatinine inside the mitochondria, as creatine kinase has been shown to accelerate the phosphocreatine proton-deuterium exchange rate [168].
9. Conclusions
Gut dysbiosis is widely recognized as a contributing factor to a long list of chronic diseases and conditions. Mitochondrial dysfunction is also a common thread among many diseases. In this paper, we present evidence that these two pathologies are intimately intertwined, primarily because the gut microbiome is responsible for fueling the host with a multitude of deupleted nutrients. Furthermore, microbial ability to carry out this service is severely compromised when the microbes are disturbed by nutritional deficiencies and toxic exposures. The mitochondrial ATPase pumps are very sensitive to deuterium, which interferes with their production of ATP and induces the release of reactive oxygen species.
We have shown that the metabolite butyrate, derived from dietary fiber and produced by obligate anaerobes, is an essential nutrient for the colonocytes, and that butyrate deficiency has been found in association with many diseases. The microbes produce butyrate and other short chain fatty acids (SCFAs) through a process that involves recycling severely deupleted hydrogen gas: producing it from simple organic molecules such as glucose and formate and using it to reduce carbon dioxide to acetate and butyrate and to generate the one-carbon units that feed the methylation pathways. Inflammation in the gut interferes with the production of SCFAs.
Methylation pathways play many roles both in metabolism and in altering gene expression. We have described the methylation pathways in great detail, but through a lens that reflects on the way in which they can work to minimize the exposure of mitochondria to deuterium. Choline, methionine, and formaldehyde are central players in methylation pathways, and an intricate collaboration between the microbes and the host assures a healthy outcome, as long as nutritional support is sufficient and toxic exposures are minimized.
A novelty of our hypothesis is that it may be able to explain why dietary choline and dietary methionine are much more beneficial to health than synthetic versions of these molecules, simply because the biosynthesized dietary forms are deupleted and the synthetic forms are not. A diet that is rich in prebiotics (e.g., fiber), probiotics (e.g., fermented foods), and adequate natural sources of methionine and choline is likely to maintain healthy mitochondria and to promote longevity.
Disclosure of Interest
The authors have no conflicts of interest to declare.
Funding
Stephanie Seneff received funding for this research by Quanta Computer, Inc. in Taoyuan, Taiwan under contract number 6950759. The other authors received no funding for this work.
References
- Fukuda K, Straus SE, Hickie I, Sharpe MC, Dobbins JG, Komaroff A. The chronic fatigue syndrome: a comprehensive approach to its definition and study. International Chronic Fatigue Syndrome Study Group. Ann Intern Med. 1994 Dec 15;121(12):953-9. [CrossRef]
- Wood E, Hall KH, Tate W. Role of mitochondria, oxidative stress and the response to antioxidants in myalgic encephalomyelitis/chronic fatigue syndrome: A possible approach to SARS-CoV-2 ’long-haulers’? Chronic Dis Transl Med. 2021 Mar;7(1):14-26. [CrossRef]
- Syed AM, Karius AK, Ma J, Wang P-Y, Hwang PM. Mitochondrial dysfunction in myalgic encephalomyelitis/chronic fatigue syndrome. Physiology 2025; 40: 319-328. [CrossRef]
- Molnar T, Lehoczki A, Fekete M, Varnai R, Zavori L, Erdo-Bonyar S, Simon D, Berki T, Csecsei P, Ezer E. Mitochondrial dysfunction in long COVID: mechanisms, consequences, and potential therapeutic approaches. Geroscience. 2024 Oct;46(5):5267-5286. [CrossRef]
- Crost EH, Tailford LE, Monestier M, Swarbreck D, Henrissat B, Crossman LC, and Juge N. (). The mucin-degradation strategy of Ruminococcus gnavus: the importance of intramolecular trans-sialidases. Gut Microbes 7, 302312. 10.1080/19490976.2016.1186334.
- Guo C, Che X, Briese T, Ranjan A, Allicock O, Yates RA, Cheng A, March D, Hornig M, Komaroff AL, Levine S, Bateman L, Vernon SD, Klimas NG, Montoya JG, Peterson DL, Lipkin WI, Williams BL. Deficient butyrate-producing capacity in the gut microbiome is associated with bacterial network disturbances and fatigue symptoms in ME/CFS. Cell Host Microbe. 2023 Feb 8;31(2):288-304.e8. [CrossRef]
- Henke MT, Kenny DJ, Cassilly CD, Vlamakis H, Xavier RJ, Clardy J. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc Natl Acad Sci U S A. 2019 Jun 25;116(26):12672-12677. [CrossRef]
- Li L, Hu J, Qin C, Cai J, Zou X, Tian G, Seeberger PH, Yin J. Immunological evaluation of the autism-related bacterium Enterocloster bolteae capsular polysaccharide driven by chemical synthesis. Chinese Chemical Letters 2025; 36(9): 110797. [CrossRef]
- Plichta DR, Somani J, Pichaud M, Wallace ZS, Fernandes AD, Perugino CA, Lhdesmki H, Stone JH, Vlamakis H, Chung DC, Khanna D, Pillai S, Xavier RJ. Congruent microbiome signatures in fibrosis-prone autoimmune diseases: IgG4-related disease and systemic sclerosis. Genome Med. 2021 Feb 28;13(1):35. [CrossRef]
- Ruuskanen MO, berg F, Mnnist V, Havulinna AS, Mric G, Liu Y, Loomba R, Vzquez-Baeza Y, Tripathi A, Valsta LM, Inouye M, Jousilahti P, Salomaa V, Jain M, Knight R, Lahti L, Niiranen TJ. Links between gut microbiome composition and fatty liver disease in a large population sample. Gut Microbes. 2021 Jan-Dec;13(1):1-22. [CrossRef]
- Mbaye B, Magdy Wasfy R, Borentain P, Tidjani Alou M, Mottola G, Bossi V, Caputo A, Gerolami R, Million M. Increased fecal ethanol and enriched ethanol-producing gut bacteria Limosilactobacillus fermentum, Enterocloster bolteae, Mediterraneibacter gnavus and Streptococcus mutans in nonalcoholic steatohepatitis. Front Cell Infect Microbiol. 2023 Nov 16;13:1279354. [CrossRef]
- Seneff S, Kyriakopoulos A. Cancer, deuterium, and gut microbes: A novel perspective. Endocrine and Metabolic Science 2025; 17: 100215. [CrossRef]
- Olgun, A., 2007. Biological effects of deuteronation: ATP synthase as an example. Theor. Biol. Med. Model. 4, 9. [CrossRef]
- Krichevsky MI, Friedman I,Newell MF, Sisler FD. Deuterium fractionation during molecular hydrogen formation in marine pseudomonad. J Biol Chem 1961; 236: 2520-2525.
- Kane DA. Lactate oxidation at the mitochondria: a lactate-malate-aspartate shuttle at work. Front Neurosci. 2014 Nov 25;8:366. [CrossRef]
- Sutcliffe MJ, Masgrau L, Roujeinikova A, Johannissen LO, Hothi P, Basran J, Ranaghan KE, Mulholland AJ, Leys D, Scrutton NS. Hydrogen tunnelling in enzyme-catalysed H-transfer reactions: flavoprotein and quinoprotein systems. Philos Trans R Soc Lond B Biol Sci. 2006 Aug 29;361(1472):1375-86. [CrossRef]
- Yagi T, Matsuno-Yagi A. The proton-translocating NADH-quinone oxidoreductase in the respiratory chain: the secret unlocked. Biochemistry. 2003 Mar 4;42(8):2266-74. [CrossRef]
- Waddell J, McKenna MC, Kristian T. Brain ethanol metabolism and mitochondria. Curr Top Biochem Res. 2022;23:1-13.
- Yan T, Zhao Y, Jiang Z, Chen J. Acetaldehyde induces cytotoxicity via triggering mitochondrial dysfunction and overactive mitophagy. Mol Neurobiol. 2022 Jun;59(6):3933-3946. [CrossRef]
- Ferreira-Halder CV, Faria AVS, Andrade SS. Action and function of Faecalibacterium prausnitzii in health and disease. Best Pract Res Clin Gastroenterol. 2017 Dec;31(6):643-648. [CrossRef]
- Khan MT, van Dijl JM, Harmsen HJM. Antioxidants keep the potentially probiotic but highly oxygen-sensitive human gut bacterium Faecalibacterium prausnitzii alive at ambient air. PLoS ONE 2014; 9(5): e96097. [CrossRef]
- Zeng MY, Inohara N, Nuez G. Mechanisms of inflammation-driven bacterial dysbiosis in the gut. Mucosal Immunol. 2017 Jan;10(1):18-26. [CrossRef]
- Soto-Martin EC, Warnke I, Farquharson FM, Christodoulou M, Horgan G, Derrien M, Faurie JM, Flint HJ, Duncan SH, Louis P. Vitamin biosynthesis by human gut butyrate-producing bacteria and cross-feeding in synthetic microbial communities. mBio. 2020 Jul 14;11(4):e00886-20. [CrossRef]
- Jacobson W, Saich T, Borysiewicz LK, Behan WM, Behan PO, Wreghitt TG. Serum folate and chronic fatigue syndrome. Neurology. 1993 Dec;43(12):2645-7. [CrossRef]
- Hoffbrand AV, Stewart JS, Booth CC, Mollin DL. Folate deficiency in Crohn’s disease: incidence, pathogenesis, and treatment. Br Med J. 1968 Apr 13;2(5597):71-5. [CrossRef]
- Yakut M, Ustün Y, Kabaçam G, Soykan I. Serum vitamin B12 and folate status in patients with inflammatory bowel diseases. Eur J Intern Med. 2010 Aug;21(4):320-3. [CrossRef]
- Christensen KE, Mikael LG, Leung KY, Lévesque N, Deng L, Wu Q, Malysheva OV, Best A, Caudill MA, Greene ND, Rozen R. High folic acid consumption leads to pseudo-MTHFR deficiency, altered lipid metabolism, and liver injury in mice. Am J Clin Nutr. 2015 Mar;101(3):646-58. [CrossRef]
- Patanwala I, King MJ, Barrett DA, Rose J, Jackson R, Hudson M, Philo M, Dainty JR, Wright AJ, Finglas PM, Jones DE. Folic acid handling by the human gut: implications for food fortification and supplementation. Am J Clin Nutr. 2014 Aug;100(2):593-9. [CrossRef]
- Madhogaria B, Bhowmik P, Kundu A. Correlation between human gut microbiome and diseases. Infect Med (Beijing). 2022 Aug 24;1(3):180-191. [CrossRef]
- Xie X, Zubarev RA. Effects of low-level deuterium enrichment on bacterial growth. PLoS One. 2014 Jul 17;9(7):e102071. [CrossRef]
- Paliy O, Bloor D, Brockwell D, Gilbert P, Barber J. Improved methods of cultivation and production of deuterated proteins from E. coli strains grown on fully deuterated minimal medium. J Appl Microbiol. 2003;94(4):580-6. [CrossRef]
- Trchounian K, Trchounian A. Escherichia coli hydrogen gas production from glycerol: Effects of external formate. Renewable Energy, 2015; 83: 345-351.
- Li C, Sessions AL, Kinnaman FS, Valentine DL. Hydrogen-isotopic variability in lipids from Santa Barbara Basin sediments. Geochim Cosmochim Acta 2009; 73: 4803-4823. [CrossRef]
- Gaci N, Borrel G, Tottey W, O’Toole PW, Brugre JF. Archaea and the human gut: new beginning of an old story. World J Gastroenterol. 2014 Nov 21;20(43):16062-78. [CrossRef]
- Thauer RK. Methyl (alkyl)-Coenzyme M reductases: Nickel F-430-containing enzymes involved in anaerobic methane formation and in anaerobic pxidation of methane or of short chain alkanes. Biochemistry. 2019 Dec 31;58(52):5198-5220. [CrossRef]
- Thauer RK, Kaster AK, Seedorf H, Buckel W, Hedderich R. Methanogenic archaea: ecologically relevant differences in energy conservation. Nat Rev Microbiol. 2008 Aug;6(8):579-91. [CrossRef]
- Liao Y, Chen S, Wang D, Zhang W, Wang S, Ding J, Wang Y, Cai L, Ran X, Wang X, Zhu H. Structure of formaldehyde dehydrogenase from Pseudomonas aeruginosa: the binary complex with the cofactor NAD+. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013 Sep;69(Pt 9):967-72. [CrossRef]
- Keltjens JT, Pol A, Reimann J, Op den Camp HJ. PQQ-dependent methanol dehydrogenases: rare-earth elements make a difference. Appl Microbiol Biotechnol. 2014;98(14):6163-83. [CrossRef]
- McDowall JS, Murphy BJ, Haumann M, Palmer T, Armstrong FA, Sargent F. Bacterial formate hydrogenlyase complex. Proc Natl Acad Sci U S A. 2014 Sep 23;111(38):E3948-56. [CrossRef]
- Chen NH, Djoko KY, Veyrier FJ, McEwan AG. Formaldehyde stress responses in bacterial pathogens. Front Microbiol. 2016 Mar 3;7:257. [CrossRef]
- Ichikawa Y, Yamamoto H, Hirano SI, Sato B, Takefuji Y, Satoh F. The overlooked benefits of hydrogen-producing bacteria. Med Gas Res. 2023 Jul-Sep;13(3):108-111. [CrossRef]
- Smith NW, Shorten PR, Altermann EH, Roy NC, McNabb WC. Hydrogen cross- feeders of the human gastrointestinal tract. Gut Microbes. 2019;10(3):270-288. [CrossRef]
- Hay S, Pudney CR, Scrutton NS. Structural and mechanistic aspects of flavoproteins: probes of hydrogen tunnelling. FEBS J. 2009 Aug;276(15):3930-41. [CrossRef]
- Greene BL, Wu CH, McTernan PM, Adams MW, Dyer RB. Proton-coupled electron transfer dynamics in the catalytic mechanism of a [NiFe]-hydrogenase. J. Am. Chem. Soc. 2015; 137(13): 45584566. [CrossRef]
- Frolov EN, Elcheninov AG, Gololobova AV, Toshchakov SV, Novikov AA, Lebedinsky AV, Kublanov IV. Obligate autotrophy at the thermodynamic limit of life in a new acetogenic bacterium. Front Microbiol. 2023 May 12;14:1185739. [CrossRef]
- Fu M, Zhang W, Wu L, Yang G, Li H, Wang R. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci U S A. 2012 Feb 21;109(8):2943-8. [CrossRef]
- Gasaly N, Hermoso MA, Gotteland M. Butyrate and the fine-tuning of colonic homeostasis: Implication for inflammatory bowel diseases. Int J Mol Sci. 2021 Mar 17;22(6):3061. [CrossRef]
- Froese DS, Fowler B, Baumgartner MR. Vitamin B12, folate, and the methionine remethylation cycle-biochemistry, pathways, and regulation. J Inherit Metab Dis. 2019 Jul;42(4):673-685. [CrossRef]
- Borrel G, Brugre JF, Gribaldo S, Schmitz RA, Moissl-Eichinger C. The host-associated archaeome. Nat Rev Microbiol. 2020 Nov;18(11):622-636. [CrossRef]
- Lang K, Schuldes J, Klingl A, Poehlein A, Daniel R, Brunea A. New mode of energy metabolism in the seventh order of methanogens as revealed by comparative genome analysis of Candidatus methanoplasma termitum. Appl Environ Microbiol. 2015 Feb;81(4):1338-52. [CrossRef]
- Bueno de Mesquita CP, Wu D, Tringe SG. Methyl-based methanogenesis: an eco- logical and genomic review. Microbiol Mol Biol Rev. 2023 Mar 21;87(1):e0002422. [CrossRef]
- de la Cuesta-Zuluaga J, Spector TD, Youngblut ND, Ley RE. Genomic insights into adaptations of trimethylamine-utilizing methanogens to diverse habitats, including the human gut. mSystems. 2021 Feb 9;6(1):e00939-20. [CrossRef]
- Chen YR, Chen LD, Zheng LJ. Exploring the trimethylamine-degrading genes in the human gut microbiome. AMB Express. 2025 Jun 12;15(1):91. [CrossRef]
- Mohammadzadeh R, Mahnert A, Shinde T, Kumpitsch C, Weinberger V, Schmidt H, Moissl-Eichinger C. Age-related dynamics of predominant methanogenic archaea in the human gut microbiome. BMC Microbiol. 2025 Apr 4;25(1):193. [CrossRef]
- Chen YM, Liu Y, Zhou RF, Chen XL, Wang C, Tan XY, Wang LJ, Zheng RD, Zhang HW, Ling WH, Zhu HL. Associations of gut-flora-dependent metabolite trimethylamine- N-oxide, betaine and choline with non-alcoholic fatty liver disease in adults. Sci Rep. 2016 Jan 8;6:19076. [CrossRef]
- Ernst L, Steinfeld B, Barayeu U, Klintzsch T, Kurth M, Grimm D, Dick TP, Rebelein JG, Bischofs IB, Keppler F. Methane formation driven by reactive oxygen species across all living organisms. Nature. 2022 Mar;603(7901):482-487. [CrossRef]
- Keppler F, Ernst L, Polag D, Zhang J, Boros M. ROS-driven cellular methane formation: Potential implications for health sciences. Clin Transl Med. 2022 Jul;12(7):e905. [CrossRef]
- Gibellini F, Smith TK. The Kennedy pathway – De novo synthesis of phosphatidylethanolamine and phosphatidylcholine. IUBMB Life. 2010 Jun;62(6):414-28. [CrossRef]
- Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012 Mar;28(2):159-65. [CrossRef]
- Wan S, Kuipers F, Havinga R, Ando H, Vance DE, Jacobs RL, van der Veen JN. Impaired hepatic phosphatidylcholine synthesis leads to cholestasis in mice challenged with a high-fat diet. Hepatol Commun. 2019 Jan 2;3(2):262-276. [CrossRef]
- McNeil SD, Nuccio ML, Ziemak MJ, Hanson AD. Enhanced synthesis of choline and glycine betaine in transgenic tobacco plants that overexpress phosphoethanolamine N-methyltransferase. Proc Natl Acad Sci U S A. 2001 Aug 14;98(17):10001-5. [CrossRef]
- Kikuchi G, Motokawa Y, Yoshida T, Hiraga K. Glycine cleavage system: reaction mechanism, physiological significance, and hyperglycinemia. Proc Jpn Acad Ser B Phys Biol Sci. 2008;84(7):246-63. [CrossRef]
- Niculescu MD, Zeisel SH. Diet, methyl donors and DNA methylation: interactions between dietary folate, methionine and choline. J Nutr. 2002 Aug;132(8 Suppl):2333S-2335S. [CrossRef]
- Craciun S, Balskus EP. Microbial conversion of choline to trimethylamine requires a glycyl radical enzyme. Proc Natl Acad Sci U S A. 2012 Dec 26;109(52):21307-12. [CrossRef]
- Ramezani A, Nolin TD, Barrows IR, Serrano MG, Buck GA, Regunathan-Shenk R, West RE 3rd, Latham PS, Amdur R, Raj DS. Gut colonization with methanogenic archaea lowers plasma trimethylamine N-oxide concentrations in apolipoprotein e-/- mice. Sci Rep. 2018 Oct 3;8(1):14752. [CrossRef]
- Basran J, Sutcliffe MJ, Scrutton NS. Deuterium isotope effects during carbon-hydrogen bond cleavage by trimethylamine dehydrogenase. Implications for mechanism and vibrationally assisted hydrogen tunneling in wild-type and mutant enzymes. J Biol Chem. 2001 Jul 6;276(27):24581-7. [CrossRef]
- Wanninayake US, Subedi B, Fitzpatrick PF. pH and deuterium isotope effects on the reaction of trimethylamine dehydrogenase with dimethylamine. Arch Biochem Biophys. 2019 Nov 15;676:108136. [CrossRef]
- Growdon JH, Hirsch MJ, Wurtman RJ, Wiener W. Oral choline administration to patients with tardive dyskinesia. N Engl J Med. 1977 Sep 8;297(10):524-7. [CrossRef]
- Kansakar U, Trimarco V, Mone P, Varzideh F, Lombardi A, Santulli G. Choline supplements: An update. Front Endocrinol (Lausanne). 2023 Mar 7;14:1148166. [CrossRef]
- National Academies of Sciences, Engineering, and Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. Washington, DC: The National Academies Press. 1998. [CrossRef]
- Grand View Research, Inc. Nootropics Market Size, Share and Trends Report, 2030. 2025. [last accessed August 15,2025]. https://www.grandviewresearch.com/industry-analysis/nootropics-market.
- Zhao J, Wei S, Liu F, Liu D. Separation and characterization of acetone-soluble phosphatidylcholine from Antarctic krill (Euphausia superba) oil. Eur Food Res Technol 2014; 238: 1023–1028. [CrossRef]
- Blackett EG, Soliday AJ. Preparation of choline base and choline salts. US Patent #US2774759A. 1955. [last accessed August 15, 2025]. https://patents.google.com/patent/US2774759A/en.
- Wilcox J, Skye SM, Graham B, Zabell A, Li XS, Li L, Shelkay S, Fu X, Neale S, O'Laughlin C, Peterson K, Hazen SL, Tang WHW. Dietary choline supplements, but not eggs, raise fasting TMAO levels in participants with normal renal function: A randomized clinical trial. Am J Med. 2021 Sep;134(9):1160-1169.e3. [CrossRef]
- Zhu W, Wang Z, Tang WHW, Hazen SL. Gut microbe-generated trimethylamine N-oxide from dietary choline is prothrombotic in subjects. Circulation. 2017 Apr 25;135(17):1671-1673. [CrossRef]
- Arias N, Arboleya S, Allison J, Kaliszewska A, Higarza SG, Gueimonde M, Arias JL. The relationship between choline bioavailability from diet, intestinal microbiota composition, and its modulation of human diseases. Nutrients. 2020 Aug 5;12(8):2340. [CrossRef]
- Zhou Y, Zhang Y, Jin S, Lv J, Li M, Feng N. The gut microbiota derived metabolite trimethylamine N-oxide: Its important role in cancer and other diseases. Biomed Pharmacother. 2024 Aug;177:117031. [CrossRef]
- Alharbi YM, Bima AI, Elsamanoudy AZ. An overview of the perspective of cellular autophagy: mechanism, regulation, and the role of autophagy dysregulation in the pathogenesis of diseases. J Microsc Ultrastruct. 2021 Jan 9;9(2):47-54. [CrossRef]
- Bhutia SK, Mukhopadhyay S, Sinha N, Das DN, Panda PK, Patra SK, Maiti TK, Mandal M, Dent P, Wang XY, Das SK, Sarkar D, Fisher PB. Autophagy: cancer's friend or foe? Adv Cancer Res. 2013;118:61-95. [CrossRef]
- Su JQ, Wu XQ, Wang Q, Xie BY, Xiao CY, Su HY, Tang JX, Yao CW. The microbial metabolite trimethylamine N-oxide and the kidney diseases. Front Cell Infect Microbiol. 2025 Mar 11;15:1488264. [CrossRef]
- Praharaj PP, Singh A, Patil S, Dhiman R, Bhutia SK. Mitochondrial dysfunction as a driver of NLRP3 inflammasome activation and its modulation through mitophagy for potential therapeutics. Int J Biochem Cell Biol. 2021 Jul;136:106013. [CrossRef]
- Dong F, Jiang S, Tang C, Wang X, Ren X, Wei Q, Tian J, Hu W, Guo J, Fu X, Liu L, Patzak A, Persson PB, Gao F, Lai EY, Zhao L. Trimethylamine N-oxide promotes hyperoxaluria-induced calcium oxalate deposition and kidney injury by activating autophagy. Free Radic Biol Med. 2022 Feb 1;179:288-300. [CrossRef]
- Atherosclerosis by regulating low-density lipoprotein-induced autophagy in vascular smooth muscle cells through PI3K/AKT/mTOR pathway. Int Heart J. 2023;64(3):462-469. [CrossRef]
- Davies MJ, Gordon JL, Gearing AJ, Pigott R, Woolf N, Katz D, Kyriakopoulos A. The expression of the adhesion molecules ICAM-1, VCAM-1, PECAM, and E-selectin in human atherosclerosis. J Pathol. 1993 Nov;171(3):223-9. [CrossRef]
- Saaoud F, Liu L, Xu K, Cueto R, Shao Y, Lu Y, Sun Y, Snyder NW, Wu S, Yang L, Zhou Y, Williams DL, Li C, Martinez L, Vazquez-Padron RI, Zhao H, Jiang X, Wang H, Yang X. Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. JCI Insight. 2023 Jan 10;8(1):e158183. [CrossRef]
- Lei D, Liu Y, Liu Y, Jiang Y, Lei Y, Zhao F, Li W, Ouyang Z, Chen L, Tang S, Ouyang D, Li X, Li Y. The gut microbiota metabolite trimethylamine N-oxide promotes cardiac hypertrophy by activating the autophagic degradation of SERCA2a. Commun Biol. 2025 Apr 10;8(1):596. [CrossRef]
- Fiorillo C, Astrea G, Savarese M, Cassandrini D, Brisca G, Trucco F, Pedemonte M, Trovato R, Ruggiero L, Vercelli L, D'Amico A, Tasca G, Pane M, Fanin M, Bello L, Broda P, Musumeci O, Rodolico C, Messina S, Vita GL, Sframeli M, Gibertini S, Morandi L, Mora M, Maggi L, Petrucci A, Massa R, Grandis M, Toscano A, Pegoraro E, Mercuri E, Bertini E, Mongini T, Santoro L, Nigro V, Minetti C, Santorelli FM, Bruno C; Italian Network on Congenital Myopathies. MYH7-related myopathies: clinical, histopathological and imaging findings in a cohort of Italian patients. Orphanet J Rare Dis. 2016 Jul 7;11(1):91. [CrossRef]
- Nakagawa Y, Nishikimi T, Kuwahara K. Atrial and brain natriuretic peptides: Hormones secreted from the heart. Peptides. 2019 Jan;111:18-25. [CrossRef]
- Ghosh R, Fatahian AN, Rouzbehani OMT, Hathaway MA, Mosleh T, Vinod V, Vowles S, Stephens SL, Chung SD, Cao ID, Jonnavithula A, Symons JD, Boudina S. Sequestosome 1 (p62) mitigates hypoxia-induced cardiac dysfunction by stabilizing hypoxia-inducible factor 1α and nuclear factor erythroid 2-related factor 2. Cardiovasc Res. 2024 Apr 30;120(5):531-547. [CrossRef]
- Zixin Y, Lulu C, Xiangchang Z, Qing F, Binjie Z, Chunyang L, Tai R, Dongsheng O. TMAO as a potential biomarker and therapeutic target for chronic kidney disease: A review. Front Pharmacol. 2022 Aug 12;13:929262. [CrossRef]
- Varzideh F, Farroni E, Kaunsakar U, Eiwaz M, Jankauskas SS, Santulli G. TMAO accelerates cellular aging by disrupting endoplasmic reticulum integrity and mitochondrial unfolded protein response. Cell Mol Life Sci. 2025 Jan 21;82(1):53. [CrossRef]
- Makrecka-Kuka M, Volska K, Antone U, Vilskersts R, Grinberga S, Bandere D, Liepinsh E, Dambrova M. Trimethylamine N-oxide impairs pyruvate and fatty acid oxidation in cardiac mitochondria. Toxicol Lett. 2017 Feb 5;267:32-38. [CrossRef]
- García-Miranda A, Montes-Alvarado JB, Sarmiento-Salinas,FL, Vallejo-Ruiz V, Castañeda-Saucedo E, Navarro-Tito N, Maycotte P. Regulation of mitochondrial metabolism by autophagy supports leptin-induced cell migration. Sci Rep 2024; 14: 1408. [CrossRef]
- Greer EL, Shi Y. Histone methylation: a dynamic mark in health, disease and inheritance. Nat Rev Genet. 2012 Apr 3;13(5):343-57. [CrossRef]
- Li T, Wei Y, Qu M, Mou L, Miao J, Xi M, Liu Y, He R. Formaldehyde and de/methylation in age-related cognitive impairment. Genes (Basel). 2021 Jun 13;12(6):913. [CrossRef]
- Bae S, Chon J, Field MS, Stover PJ. Alcohol dehydrogenase 5 is a source of formate for de novo purine biosynthesis in HepG2 cells. J Nutr. 2017 Apr;147(4):499-505. [CrossRef]
- Yin Y, Morgunova E, Jolma A, Kaasinen E, Sahu B, Khund-Sayeed S, Das PK, Kivioja T, Dave K, Zhong F, Nitta KR, Taipale M, Popov A, Ginno PA, Domcke S, Yan J, Schbeler D, Vinson C, Taipale J. Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science. 2017 May 5;356(6337):eaaj2239. [CrossRef]
- Jang HS, Shin WJ, Lee JE, Do JT. CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes (Basel). 2017 May 23;8(6):148. [CrossRef]
- Zhang L, Lu X, Lu J, Liang H, Dai Q, Xu GL, Luo C, Jiang H, He C. Thymine DNA glycosylase specifically recognizes 5-carboxylcytosine-modified DNA. Nat Chem Biol. 2012 Feb 12;8(4):328-30. [CrossRef]
- Scourzic L, Mouly E, Bernard OA. TET proteins and the control of cytosine demethylation in cancer. Genome Med. 2015 Jan 29;7(1):9. [CrossRef]
- Feng Y, Chen JJ, Xie NB, Ding JH, You XJ, Tao WB, Zhang X, Yi C, Zhou X, Yuan BF, Feng YQ. Direct decarboxylation of ten-eleven translocation-produced 5-carboxylcytosine in mammalian genomes forms a new mechanism for active DNA demethylation. Chem Sci. 2021 Jul 21;12(34):11322-11329. [CrossRef]
- Jonasson NSW, Daumann LJ. 5-Methylcytosine is oxidized to the natural metabolites of TET enzymes by a biomimetic iron(IV)-oxo complex. Chemistry. 2019 Sep 18;25(52):12091-12097. doi: 10.1002/chem.201902340. Erratum in: Chemistry. 2019 Nov 22;25(65):15004. [CrossRef]
- Ji F, Zhao C, Wang B, Tang Y, Miao Z, Wang Y. The role of 5-hydroxymethylcytosine in mitochondria after ischemic stroke. J Neurosci Res. 2018 Oct;96(10):1717-1726. [CrossRef]
- Gao Q, Shen K, Xiao M. TET2 mutation in acute myeloid leukemia: biology, clinical significance, and therapeutic insights. Clin Epigenetics. 2024 Nov 9;16(1):155. [CrossRef]
- Huang F, Sun J, Chen W, Zhang L, He X, Dong H, Wu Y, Wang H, Li Z, Ball B, Khaled S, Marcucci G, Li L. TET2 deficiency promotes MDS-associated leukemogenesis. Blood Cancer J. 2022 Oct 4;12(10):141. doi: 10.1038/s41408-022-00739-w. Erratum in: Blood Cancer J. 2023 Nov 28;13(1):174. [CrossRef]
- Oliphant K, Allen-Vercoe E. Macronutrient metabolism by the human gut microbiome: major fermentation by-products and their impact on host health. Microbiome. 2019 Jun 13;7(1):91. [CrossRef]
- Tangerman A. Measurement and biological significance of the volatile sulfur compounds hydrogen sulfide, methanethiol and dimethyl sulfide in various biological matrices. J Chromatogr B Analyt Technol Biomed Life Sci. 2009 Oct 15;877(28):3366-77. Epub 2009 May 21. [CrossRef] [PubMed]
- Philipp TM, Gernoth L, Will A, Schwarz M, Ohse VA, Kipp AP, Steinbrenner H, Klotz LO. Selenium-binding protein 1 (SELENBP1) is a copper-dependent thiol oxidase. Redox Biol. 2023 Sep;65:102807. [CrossRef]
- Seneff S, Kyriakopoulos AM. Deuterium trafficking, mitochondrial dysfunction, copper homeostasis, and neurodegenerative disease. Front Mol Biosci. 2025 Jul 22;12:1639327. [CrossRef]
- Schott M, de Jel MM, Engelmann JC, Renner P, Geissler EK, Bosserhoff AK, Kuphal S. Selenium-binding protein 1 is down-regulated in malignant melanoma. Oncotarget. 2018 Jan 2;9(12):10445-10456. [CrossRef]
- Zhang S, Li F, Younes M, Liu H, Chen C, Yao Q. Reduced selenium-binding protein 1 in breast cancer correlates with poor survival and resistance to the anti-proliferative effects of selenium. PLoS One. 2013 May 21;8(5):e63702. [CrossRef]
- Wu X, Han Z, Liu B, Yu D, Sun J, Ge L, Tang W, Liu S. Gut microbiota contributes to the methionine metabolism in host. Front Microbiol. 2022 Dec 22;13:1065668. [CrossRef]
- McHugh D, Gil J. Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol. 2018 Jan 2;217(1):65-77. [CrossRef]
- Lee WS, Monaghan P, Metcalfe NB. Experimental demonstration of the growth rate--lifespan trade-off. Proc Biol Sci. 2012 Dec 12;280(1752):20122370. [CrossRef]
- Haigis MC, Yankner BA. The aging stress response. Mol Cell. 2010 Oct 22;40(2):333-44. [CrossRef]
- Orentreich N, Matias JR, DeFelice A, Zimmerman JA. Low methionine ingestion by rats extends life span. J Nutr. 1993 Feb;123(2):269-74. [CrossRef]
- Caro P, Gomez J, Sanchez I, Naudi A, Ayala V, Lpez-Torres M, Pamplona R, Barja G. Forty percent methionine restriction decreases mitochondrial oxygen radical production and leak at complex I during forward electron flow and lowers oxidative damage to proteins and mitochondrial DNA in rat kidney and brain mitochondria. Rejuvenation Res. 2009 Dec;12(6):421-34. [CrossRef]
- Yang Y, Zhang Y, Xu Y, Luo T, Ge Y, Jiang Y, Shi Y, Sun J, Le G. Dietary methionine restriction improves the gut microbiota and reduces intestinal permeability and inflammation in high-fat-fed mice. Food Funct. 2019 Sep 1;10(9):5952-5968. [CrossRef]
- Nagarajan A, Lasher AT, Morrow CD, Sun LY. Long term methionine restriction: Influence on gut microbiome and metabolic characteristics. Aging Cell. 2024 Mar;23(3):e14051. [CrossRef]
- Brown-Borg, H. M., Rakoczy, S. G., Wonderlich, J. A., Rojanathammanee, L., Kopchick, J. J., Armstrong, V., Raasakka, D. Growth hormone signaling is necessary for lifespan extension by dietary methionine. Aging Cell 2014; 13: 10191027. [CrossRef]
- Kim YT, Kwon JG, O’Sullivan DJ, Lee JH. Regulatory mechanism of cysteine-dependent methionine biosynthesis in Bifidobacterium longum: insights into sulfur metabolism in gut microbiota. Gut Microbes. 2024 Jan-Dec;16(1):2419565. [CrossRef]
- Ferla MP, Patrick WM. Bacterial methionine biosynthesis. Microbiology (Reading). 2014 Aug;160(Pt 8):1571-1584. [CrossRef]
- Lu M, Yang Y, Xu Y, Wang X, Li B, Le G, Xie Y. Dietary methionine restriction alleviates choline-induced tri-methylamine-N-oxide (TMAO) elevation by manipulating gut microbiota in mice. Nutrients. 2023 Jan 1;15(1):206. [CrossRef]
- Hogg RC, Raggenbass M, Bertrand D. Nicotinic acetylcholine receptors: from structure to brain function. Rev Physiol Biochem Pharmacol. 2003;147:1-46. [CrossRef]
- Santiago LJ, Abrol R. Understanding G protein selectivity of muscarinic acetylcholine receptors using computational methods. Int J Mol Sci. 2019 Oct 24;20(21):5290. [CrossRef]
- Kruse AC, Kobilka BK, Gautam D, Sexton PM, Christopoulos A, Wess J. Muscarinic acetylcholine receptors: novel opportunities for drug development. Nat Rev Drug Discov. 2014 Jul;13(7):549-60. Epub 2014 Jun 6. [CrossRef] [PubMed] [PubMed Central]
- Arduini A, Zammit V. Acetate transport into mitochondria does not require a carnitine shuttle mechanism. Magn Reson Med. 2017 Jan;77(1):11. [CrossRef]
- Moffett JR, Puthillathu N, Vengilote R, Jaworski DM, Namboodiri AM. Acetate revisited: a key biomolecule at the nexus of metabolism, epigenetics and oncogenesis-part 1: acetyl-CoA, acetogenesis and acyl-CoA short-chain synthetases. Front Physiol. 2020 Nov 12;11:580167. [CrossRef]
- Hlavaty SI, Salcido KN, Pniewski KA, Mukha D, Ma W, Kannan T, Cassel J, Srikanth YVV, Liu Q, Kossenkov A, Salvino JM, Chen Q, Schug ZT. ACSS1-dependent acetate utilization rewires mitochondrial metabolism to support AML and melanoma tumor growth and metastasis. Cell Rep. 2024 Dec 24;43(12):114988. [CrossRef]
- Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS. Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A. 2010 Oct 26;107(43):18670-5. [CrossRef]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998 Jul 15;18(14):5225-33. [CrossRef] [PubMed] [PubMed Central]
- Szutowicz A, Bielarczyk H, Jankowska-Kulawy A, Paweczyk T, Ronowska A. Acetyl-CoA the key factor for survival or death of cholinergic neurons in course of neurodegenerative diseases. Neurochem Res. 2013 Aug;38(8):1523-42. [CrossRef]
- Ferreira-Vieira TH, Guimaraes IM, Silva FR, Ribeiro FM. Alzheimer's disease: Targeting the cholinergic system. Curr Neuropharmacol. 2016;14(1):101-15. [CrossRef]
- Aroniadou-Anderjaska V, Figueiredo TH, de Araujo Furtado M, Pidoplichko VI, Braga MFM. Mechanisms of organophosphate toxicity and the role of acetylcholinesterase inhibition. Toxics. 2023 Oct 18;11(10):866. [CrossRef]
- Ngoula F, Watcho P, Dongmo MC, Kenfack A, Kamtchouing P, Tchoumboué J. Effects of pirimiphos-methyl (an organophosphate insecticide) on the fertility of adult male rats. Afr Health Sci. 2007 Mar;7(1):3-9. [CrossRef]
- Bourassa MW, Alim I, Bultman SJ, Ratan RR. Butyrate, neuroepigenetics and the gut microbiome: Can a high fiber diet improve brain health? Neurosci Lett. 2016 Jun 20;625:56-63. [CrossRef]
- Dalile B, Van Oudenhove L, Vervliet B, Verbeke K. The role of short-chain fatty acids in microbiota-gut-brain communication. Nat Rev Gastroenterol Hepatol. 2019. Aug;16(8):461-478. [CrossRef]
- Benítez-Páez A, Kjølbæk L, Gómez Del Pulgar EM, Brahe LK, Astrup A, Matysik S, Schött HF, Krautbauer S, Liebisch G, Boberska J, Claus S, Rampelli S, Brigidi P, Larsen LH, Sanz Y. A Multi-omics approach to unraveling the microbiome-mediated effects of arabinoxylan oligosaccharides in overweight humans. mSystems. 2019 May 28;4(4):e00209-19. [CrossRef]
- Hodgkinson K, El Abbar F, Dobranowski P, Manoogian J, Butcher J, Figeys D, Mack D, Stintzi A. Butyrate's role in human health and the current progress towards its clinical application to treat gastrointestinal disease. Clin Nutr. 2023 Feb;42(2):61-75. [CrossRef]
- Guilloteau P, Martin L, Eeckhaut V, Ducatelle R, Zabielski R, Van Immerseel F. From the gut to the peripheral tissues: the multiple effects of butyrate. Nutr Res Rev. 2010 Dec;23(2):366-84. [CrossRef]
- Anshory M, Effendi RMRA, Kalim H, Dwiyana RF, Suwarsa O, Nijsten TEC, Nouwen JL, Thio HB. Butyrate properties in immune-related diseases: Friend or foe? Fermentation 2023; 9: 205. [CrossRef]
- Bachmann C, Colombo JP, Berüter J. Short chain fatty acids in plasma and brain: quantitative determination by gas chromatography. Clin Chim Acta. 1979 Mar 1;92(2):153-9. [CrossRef]
- Liu H, Wang J, He T, Becker S, Zhang G, Li D, Ma X. Butyrate: A double-edged sword for health? Adv Nutr. 2018 Jan 1;9(1):21-29. [CrossRef]
- Recharla N, Geesala R, Shi XZ. Gut microbial metabolite butyrate and its therapeutic role in inflammatory bowel disease: A literature review. Nutrients. 2023 May 11;15(10):2275. [CrossRef]
- Coccia C, Bonomi F, Lo Cricchio A, Russo E, Peretti S, Bandini G, Lepri G, Bartoli F, Moggi-Pignone A, Guiducci S, Del Galdo F, Furst DE, Matucci Cerinic M, Bellando-Randone S. The potential role of butyrate in the pathogenesis and treatment of autoimmune rheumatic diseases. Biomedicines. 2024 Aug 5;12(8):1760. [CrossRef]
- Yip W, Hughes MR, Li Y, Cait A, Hirst M, Mohn WW, McNagny KM. Butyrate shapes immune cell fate and function in allergic asthma. Front Immunol. 2021 Feb 15;12:628453. [CrossRef]
- Zhang L, Liu C, Jiang Q, Yin Y. Butyrate in energy metabolism: There is still more to learn. Trends Endocrinol Metab. 2021 Mar;32(3):159-169. [CrossRef]
- Milani C, Duranti S, Bottacini F, Casey E, Turroni F, Mahony J, Belzer C, Delgado Palacio S, Arboleya Montes S, Mancabelli L, Lugli GA, Rodriguez JM, Bode L, de Vos W, Gueimonde M, Margolles A, van Sinderen D, Ventura M. The first microbial colonizers of the human gut: Composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev. 2017 Nov 8;81(4):e00036-17. [CrossRef]
- Fujimura KE, Sitarik AR, Havstad S, Lin DL, Levan S, Fadrosh D, Panzer AR, LaMere B, Rackaityte E, Lukacs NW, Wegienka G, Boushey HA, Ownby DR, Zoratti EM, Levin AM, Johnson CC, Lynch SV. Neonatal gut microbiota associates with childhood multisensitized atopy and T cell differentiation. Nat Med. 2016 Oct;22(10):1187-1191. [CrossRef]
- Stilling RM, van de Wouw M, Clarke G, Stanton C, Dinan TG, Cryan JF. The neuropharmacology of butyrate: The bread and butter of the microbiota-gut-brain axis? Neurochem Int. 2016 Oct;99:110-132. [CrossRef]
- Coppola S, Avagliano C, Sacchi A, Laneri S, Calignano A, Voto L, Luzzetti A, Berni Canani R. Potential clinical applications of the postbiotic butyrate in human skin diseases. Molecules. 2022 Mar 12;27(6):1849. [CrossRef]
- Bachar-Wikstrom E, Manchanda M, Bansal R, Karlsson M, Kelly-Pettersson P, Sköldenberg O, Wikstrom JD. Endoplasmic reticulum stress in human chronic wound healing: Rescue by 4-phenylbutyrate. Int Wound J. 2021 Feb;18(1):49-61. [CrossRef]
- Rose S, Bennuri SC, Davis JE, Wynne R, Slattery JC, Tippett M, Delhey L, Melnyk S, Kahler SG, MacFabe DF, Frye RE. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl Psychiatry. 2018 Feb 2;8(1):42. [CrossRef]
- Walsh ME, Bhattacharya A, Sataranatarajan K, Qaisar R, Sloane L, Rahman MM, Kinter M, Van Remmen H. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell. 2015 Dec;14(6):957-70. [CrossRef]
- He L, Zhong Z, Wen S, Li P, Jiang Q, Liu F. Gut microbiota-derived butyrate restores impaired regulatory T cells in patients with AChR myasthenia gravis via mTOR-mediated autophagy. Cell Commun Signal. 2024 Apr 3;22(1):215. [CrossRef]
- Kalkan AE, BinMowyna MN, Raposo A, Ahmad MF, Ahmed F, Otayf AY, Carrascosa C, Saraiva A, Karav S. Beyond the gut: Unveiling butyrate's global health impact through gut health and dysbiosis-related conditions: a narrative review. Nutrients. 2025 Apr 9;17(8):1305. [CrossRef]
- Casper D, Davies P. Stimulation of choline acetyltransferase activity by retinoic acid and sodium butyrate in a cultured human neuroblastoma. Brain Res. 1989 Jan 23;478(1):74-84. [CrossRef]
- Wu SH, Chen KL, Hsu C, Chen HC, Chen JY, Yu SY, Shiu YJ. Creatine supplementation for muscle growth: A scoping review of randomized clinical trials from 2012 to 2021. Nutrients. 2022 Mar 16;14(6):1255. [CrossRef]
- Bonilla DA, Kreider RB, Stout JR, Forero DA, Kerksick CM, Roberts MD, Rawson ES. Metabolic basis of creatine in health and disease: A bioinformatics-assisted review. Nutrients. 2021 Apr 9;13(4):1238. [CrossRef]
- Brosnan JT, da Silva RP, Brosnan ME. The metabolic burden of creatine synthesis. Amino Acids. 2011 May;40(5):1325-31. Epub 2011 Mar 9. [CrossRef] [PubMed]
- Ostojic SM. Creatine synthesis in the skeletal muscle: The times they are a-changin. Am J Physiol Endocrinol Metab. 2021 Feb 1;320(2):E390E391. [CrossRef]
- Seneff S, Nigh G, Kyriakopoulos AM. Is deuterium sequestering by reactive carbon atoms an important mechanism to reduce deuterium content in biological water? FASEB Bioadv. 2025 May 14;7(6):e70019. [CrossRef]
- Sletten MR, Schoenheimer R. The metabolism of 1(-)-proline studied with the aid of deuterium and isotopicnitrogen. J Biol Chem 1944; 153: 113-13.
- Dewangan S, Rawat V, Thakur AS. Imidazolidinones derivatives as pioneering anti-cancer agents: Unraveling insights through synthesis and structure-activity exploration. Chemistry Europe 2024; 9(23): e202401379. [CrossRef]
- Bąchor R, Konieczny A, Szewczuk Z. Preparation of isotopically labelled standards of creatinine via H/D exchange and their application in quantitative analysis by LC-MS. Molecules. 2020 Mar 26;25(7):1514. [CrossRef]
- Marshall RP, Droste JN, Giessing J, Kreider RB. Role of creatine supplementation in conditions involving mitochondrial dysfunction: A narrative review. Nutrients. 2022 Jan 26;14(3):529. [CrossRef]
- Sun Y, Rahbani JF, Jedrychowski MP, Riley CL, Vidoni S, Bogoslavski D, Hu B, Dumesic PA, Zeng X, Wang AB, Knudsen NH, Kim CR, Marasciullo A, Milln JL, Chouchani ET, Kazak L, Spiegelman BM. Mitochondrial TNAP controls thermogenesis by hydrolysis of phosphocreatine. Nature. 2021 May;593(7860):580-585. [CrossRef]
- Kupriyanov VV, Steinschneider AYa, Ruuge EK, Smirnov VN, Saks VA. 31P-NMR spectrum of phosphocreatine: Deuterium-induced splitting of the signal. Biochem Biophys Res Commun. 1983 Aug 12;114(3):1117-25. [CrossRef]
Figure 1.
Schematic of three primary pathways where bacteria utilize hydrogen gas as a reducing agent. All three pathways yield valuable metabolites that can help the host mitochondria maintain low levels of deuterium. The Gibbs free energy (ΔG) values show that hydrogen sulfide production is the most thermodynamically favorable among the three pathways.
Figure 1.
Schematic of three primary pathways where bacteria utilize hydrogen gas as a reducing agent. All three pathways yield valuable metabolites that can help the host mitochondria maintain low levels of deuterium. The Gibbs free energy (ΔG) values show that hydrogen sulfide production is the most thermodynamically favorable among the three pathways.
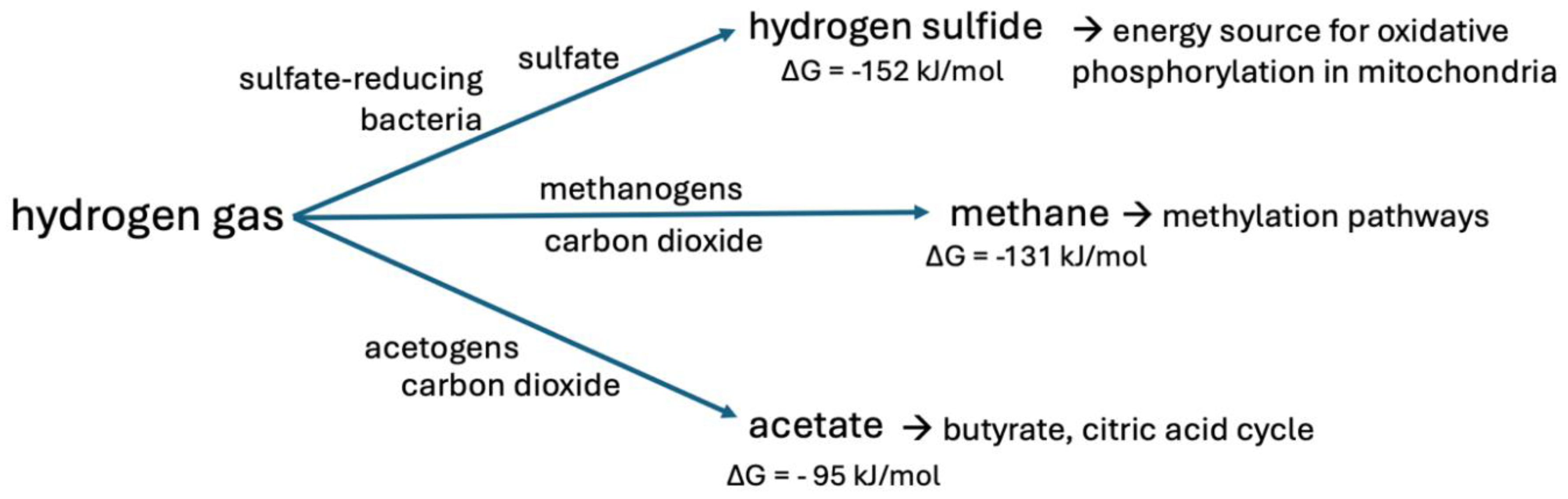
Figure 5.
Metabolic pathways in the gut, liver and brain that are involved in choline metabolism. Anaerobic microbes convert choline to TMA. When the archaeal methylogens that express MCR are deficient, the liver converts TMA to TMAO, a metabolite whose elevation in the circulation is linked to fatty liver disease and increased cardiovascular risk. Gut microbes convert choline to TMG, while releasing hydrogen peroxide and converting NAD+ to NADH. The three methyl groups in TMG are metabolized sequentially in the liver and kidneys, leaving glycine as the final reaction product. This pathway regenerates three methionine molecules from homocysteine. Choline is reversibly converted to acetylcholine in the brain, and acetylcholine is a powerful neurotransmitter that may be crucial in supplying acetate to the mitochondria in the synapse.
Figure 5.
Metabolic pathways in the gut, liver and brain that are involved in choline metabolism. Anaerobic microbes convert choline to TMA. When the archaeal methylogens that express MCR are deficient, the liver converts TMA to TMAO, a metabolite whose elevation in the circulation is linked to fatty liver disease and increased cardiovascular risk. Gut microbes convert choline to TMG, while releasing hydrogen peroxide and converting NAD+ to NADH. The three methyl groups in TMG are metabolized sequentially in the liver and kidneys, leaving glycine as the final reaction product. This pathway regenerates three methionine molecules from homocysteine. Choline is reversibly converted to acetylcholine in the brain, and acetylcholine is a powerful neurotransmitter that may be crucial in supplying acetate to the mitochondria in the synapse.
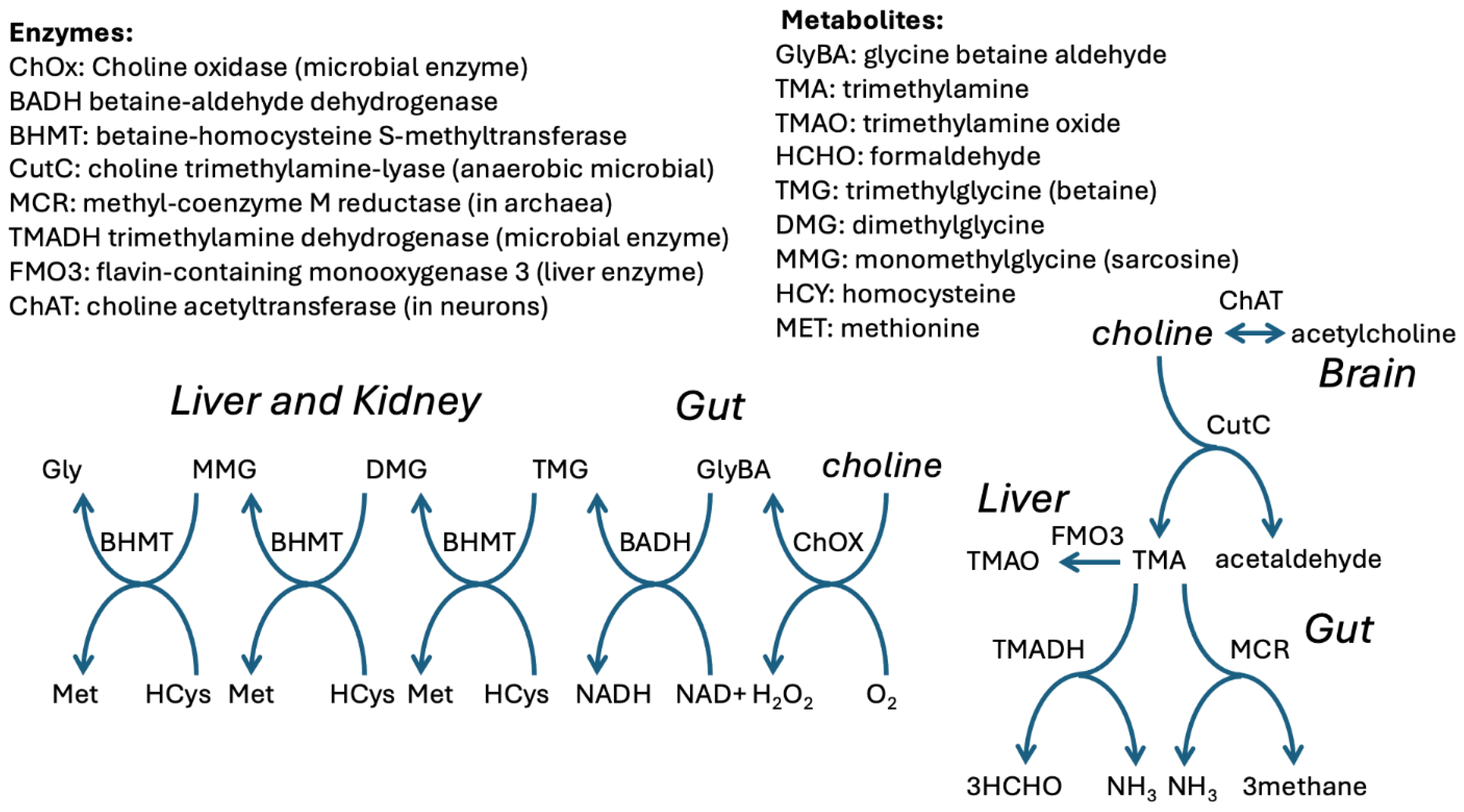

Table 1.
The reaction sequence that is common among gut microbes, which starts by converting carbon dioxide and extremely deupleted hydrogen gas into methane gas and water. A series of dehydrogenases extract protons from methane, ultimately converting it to formate, while reducing NAD+ to NADH, or converting oxygen to water while oxidizing NADH to NAD+. The last step in the cycle restores carbon dioxide, to come full circle. In the end, two molecules of NAD+ are reduced to NADH, two molecules of deupleted water are produced, and two deupleted protons are released into the medium. Some of the formaldehyde that is produced gets siphoned off to convert tetrahydrofolate to methylene-tetrahydrofolate, thus seeding methylation pathways with deupleted protons originally sourced from hydrogen gas.
Table 1.
The reaction sequence that is common among gut microbes, which starts by converting carbon dioxide and extremely deupleted hydrogen gas into methane gas and water. A series of dehydrogenases extract protons from methane, ultimately converting it to formate, while reducing NAD+ to NADH, or converting oxygen to water while oxidizing NADH to NAD+. The last step in the cycle restores carbon dioxide, to come full circle. In the end, two molecules of NAD+ are reduced to NADH, two molecules of deupleted water are produced, and two deupleted protons are released into the medium. Some of the formaldehyde that is produced gets siphoned off to convert tetrahydrofolate to methylene-tetrahydrofolate, thus seeding methylation pathways with deupleted protons originally sourced from hydrogen gas.
| Reaction | Enzymes involved |
|---|---|
| 4H2 + CO2 → CH4 + 2H2O | Methanogenesis by archaea |
| CH4 + O2 + NADH + H+ → CH3OH + NAD+ + H2O | Methane monooxygenase |
| CH3OH + NAD+ → HCHO + NADH + H+ | Methanol dehydrogenase |
| HCHO + NAD+ + H2O → HCOOH + NADH + H+ | Formaldehyde dehydrogenase |
| HCOOH + NAD+ → CO2 + NADH + H+ | Formate dehydrogenase |
| 4H2 + 2NAD+ + O2→ 2NADH + 2H+ + 2H2O | Overall Reaction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



