Submitted:
31 August 2025
Posted:
01 September 2025
You are already at the latest version
Abstract
Dry eye disease is increasingly recognised as a condition driven by immune dysregulation at the ocular surface. Chronic inflammation, mediated by aberrant activation of both innate and adaptive immune pathways, underlies disease progression and symptom persistence. Neuroimmune interactions further amplify ocular surface inflammation, contributing to epithelial damage and impaired homeostatic regulation. This review synthesizes current literature on the immunopathogenesis of dry eye disease (DED), highlighting the complex interplay of molecular mechanisms of innate and adaptive immune activation, neuroimmune-mediated inflammation, and emerging molecular and cellular biomarkers. In addition, we examine existing and emerging therapeutic strategies that target these immune-molecular pathways, including precision immunomodulatory approaches, to inform future management of DED. By integrating mechanistic insights with clinical findings, this review aims to provide a comprehensive overview of the molecular mechanisms underlying the dysregulated immune response associated with DED.
Keywords:
dry eye disease
; ocular surface
; immunopathogenesis
; innate immunity
; adaptive immunity
; T cells
; tear biomarkers
1. Introduction
Dry eye disease (DED) is a prevalent and multifactorial disorder impacting the ocular surface (OS), and is characterized by tear film instability, hyperosmolarity, and inflammation, affecting approximately 10-20% of people over the age of 40 globally [1,2]. Once primarily viewed as a disorder of tear deficiency or excessive evaporation, this perspective has evolved to recognize the significant role of the loss of immune homeostasis and dysregulation of the innate and adaptive immune system in driving disease progression and symptom persistence [2,3].
Over the past decade, advances in immunology, molecular profiling, and animal models have reshaped our understanding of DED pathogenesis. Accumulating evidence has elucidated complex immunoregulatory dysfunction at the ocular surface, including aberrant activation of epithelial stress pathways, innate immune triggers, T cell polarization, and impaired regulatory networks [2,4,5,6]. Better understanding of the molecular mechanisms underlying DED has facilitated identification of potential precision immunomodulatory techniques to mitigate disease progression.
This review synthesizes the key literature on the biological processes underlying immune dysregulation in DED, drawing from both human studies and preclinical models. We highlight critical dysregulation of innate and adaptive immune pathways, the contribution of neuroimmune signaling, and novel therapeutic strategies that leverage this immunological insight to inform future treatment approaches.
2. Methodology
In this narrative review a comprehensive search strategy was developed to identify relevant publications focusing on the role of the immune system in the pathogenesis of DED. Literature searches were performed using electronic databases, including PubMed, Google Scholar, Web of Science, and Scopus, covering the period from January 2000 to July 2025. The following combinations of Medical Subject Headings (MeSH) terms and keywords were used: “dry eye disease,” “dry eye syndrome,” “ocular surface immunology,” “innate immunity,” “adaptive immunity,” “T cells,” “dendritic cells,” “cytokines,” “chemokines,” “tear biomarkers,” and “immunopathogenesis.”
3. OS Immune Homeostasis
The OS in healthy eyes is actively kept in an immune homeostasis by various regulatory mechanisms that when disrupted, can lead to inflammation seen in DED [3,7]. The tear film, produced by the lacrimal glands, meibomian glands and OS epithelia, is an immunologically active barrier that preserves corneal clarity through lubrication, suppresses local inflammation, and promotes wound healing [8,9] (Figure 1). It contains a diverse array of growth factors, antimicrobial peptides and antibodies, including immunoglobulin A (IgA) and G (IgG), which help to defend the OS from microorganisms and maintain homeostasis [9,10].
Conjunctival goblet cells (GCs) contribute to the mucinous layer of the tear film and aid in immunoregulatory homeostasis by secreting mucins for epithelial protection. GCs also release tolerogenic mediators such as tumor growth factor (TGF) beta and retinoic acid, which modulate antigen presenting cell (APC) activity and prevent pathological immune activation [10]. In addition, conjunctival associated lymphoid tissue (CALT), first described in 1994, contributes to ocular mucosal tolerance [3,7,11]. This dense follicular aggregate of T cells, particularly cluster differentiated T lymphocyte 4+ (CD4+), CD25+, Forkhead box protein P3+ (FoxP3+) and T regulatory cells (Tregs), can mediate antigen surveillance and initiate effector immune responses when appropriately stimulated [3,10,12]. Tregs can also reside in regional lymph nodes draining the ocular structures and are critical in immune modulation at the OS [13]. Treg release of immunosuppressive cytokines IL-10, TGF beta and IL-35 suppresses dendritic cell (DC) maturation and function. Furthermore, Treg expression of CD25 creates competition for IL-2 and thereby causing cytokine deprivation-mediated apoptosis [13].
The cornea has a unique immune-privileged status maintained through several mechanisms. Its avascular and alymphatic structure prevents the infiltration of circulating leukocytes and migration of APCs to regional lymphoid tissues, thereby dampening the adaptive immune response [3,5]. Furthermore, the cornea lacks mature resident APCs which express low levels of major histocompatibility complex class II (MHC II) and lacks co-stimulatory molecules such as CD80, CD86 and CD40, further contributing to corneal immunosenecense [3,14].
The corneal epithelium serves as a physical barrier (with glycocalyx and tight junctions) against microorganisms and environmental insults. It actively participates in immunoregulation by expressing immunomodulatory molecules such as programmed death ligand 1 (PD-L1), which promote apoptosis of active effector T cells [3,12]. Corneal and conjunctival epithelial cells express functional toll-like (TLR) and NOD-like receptors (NLR), enabling pathogen detection and recognition of danger associated molecular patterns (DAMPs) to initiate appropriate immune responses [12].
The cornea is a highly innervated surface and relies on neuropeptides derived from nerves such as substance P to help with immunoregulation by regulating virus and bacteria induced inflammation and maintaining corneal epithelial homeostasis [3,15]. However, oversecretion of neuropeptides seen in DED can lead to pathological amplification of the immune response and propagation of the ‘vicious cycle’ of inflammation [3].
4. Definition and Classification of DED
The classification of DED has evolved to aid diagnostic precision and guide targeted therapy by identifying the suspected etiology of the disease [16]. Originally divided into tear-deficient and evaporative types, this was refined in 2007 to distinguish between aqueous-deficient dry eye (ADDE) and evaporative dry eye (EDE), with ADDE often further subclassified into Sjögren’s syndrome (SS) and non-Sjögren’s etiologies [5,17,18]. ADDE is characterized by insufficient or reduced tear volume whilst EDE is due to over-evaporation of the tear film despite normal tear production. However, it is recognized that many patients exhibit overlapping features of both subtypes, with co-existing disturbance in tear quantity and quality, underscoring the complex and multifactorial nature of DED [2,16].
5. Pathophysiology of DED
Tear hyperosmolarity is a key feature of DED and underlies both reduced aqueous production and increased evaporation. It may be considered as the triggering factor leading to a cascade of OS damage due to inflammation and immune dysregulation (Figure 2) [2,4]. This hyperosmolar state activates an epithelial stress response, causing the release of proinflammatory cytokines and matrix metalloproteinases (MMP) which promote recruitment and activation of both innate and adaptive immune cells to the OS [2,19]. Type 1 helper (Th1) and type 17 helper (Th17) lymphocytes are the predominant T cell subtypes in DED and cause IFN-γ and IL-17–mediated epithelial damage [5,20]. This immune-driven disruption of the homeostasis of the OS further destabilizes the tear film, increases evaporation, and perpetuates the ‘vicious cycle’ that characterizes chronic DED [16,20]. Other extrinsic (e.g contact lens wear, LASIK surgery, use of systemic anti-cholinergics) and intrinsic factors (e.g., age, female sex, autoimmune conditions such as SS) can contribute to the cycle and further the chronicity of the disease [20].
6. Innate Immune System in DED
The innate immune system serves as the first line of defense at the ocular surface, mounting a rapid, nonspecific response to environmental insults [21]. In DED, the innate system’s dysregulation can be considered a critical initiating event, triggering proinflammatory signaling cascades, promoting immune cell recruitment, and priming the adaptive immune response [6,22,23,24].
Stressed corneal epithelial cells release a rapid and high concentration of pro-inflammatory cytokines which contribute to local inflammation [6,25]. Direct hyperosmolar stress to these cells rapidly activates mitogen-activated protein kinases (MAPKs), causing sustained activation of its three signaling pathways: c-Jun N-termnal kinase 1/2 (JNK1/2), extracellular signal-regulated kinase 1/2 (ERK1/2), and p38 [25,26]. These pathways transduce extracellular signals to intracellular responses which upregulate inflammation, apoptosis and immune response through expression of cytokines and MMPs [27,28].
MMP-9, a key proteolytic enzyme upregulated by proinflammatory cytokines IL-1β and TNF-α, contributes to OS damage by degrading epithelial basement membrane components and disrupting tight junction proteins such as Zonula Occludens-1 (ZO-1) and Occludin, thereby weakening corneal barrier integrity in DED [25,27,29]. Moreover, increased nuclear transcription factors like Nuclear Factor kappa-light-chain-enhancer of activated B cells (NF-κB ) and Nuclear Factor of Activated T cells 5 (NFAT5), drive the early upregulation of cytokines such as TNF-α, IL-1β, IL-6, and IL-20, creating a microenvironment conducive to immune cell activation and recruitment [25,30]. IL-20 specifically has been shown in both human and murine models of DED to promote macrophage recruitment and further inflammatory signaling at the OS [30].
Further T cell recruitment to the OS is promoted by C-C motif chemokine ligand 5 (CCL5), C-X-C motif chemokine ligand 9 (CXCL9), CXCL10 chemokines as well as Th17 inducing cytokines such as IL-6, TGF-β, IL-23 and IL-17A [6,13]. Recent work by Zhang et al. has identified the role of basal epithelial cells in driving OS inflammation as ‘non-professional APCs’ through upregulation of MHC Class II in DED and release of chemokines such as CCL2, thereby activating further adaptive immune responses [31].
Toll-like receptor 4 (TLR4) is expressed on various corneal and conjunctival epithelial cells and requires co-receptors CD14 and myeloid differentiation factor 2 (MD2) to detect bacterial lipopolysaccharide (LPS) [21]. Oversignaling is regulated through low TLR4 expression on apical cells and the absence of MD2 which prevents unnecessary immune responses to commensal bacteria [21]. Redfern et al. demonstrated that desiccating stress dysregulates TLR expression in human corneal epithelial cells, characterized by an increased TLR messenger ribonucleic Acid (mRNA) but decreased protein levels, suggesting impaired innate inflammatory signaling [32]. Moreover, they found consistent downregulation of TLR9 at both mRNA and protein levels, which may reduce immune regulation at the ocular surface [32]. The same group also showed that DAMPs such as high mobility group box 1 (HMGB1) and heat shock protein 60 (HSP-60) were elevated in the tear films of DED patients [33]. This further promoted the activation of the innate immune responses via their action on TLRs (especially TLR 4), further amplifying cytokine and MMP-9 production and perpetuating the vicious cycle of inflammation and subsequent OS damage [33].
The increased release and production of pro-inflammatory cytokines and chemokines in DED causes abnormal immune cell infiltration and functioning [22,25]. Neutrophils contribute to inflammation not only through phagocytosis and reactive oxygen species (ROS) production but also by releasing neutrophil extracellular traps (NETs) in response to tear hyperosmolarity [22,34]. These NETs can form in the absence of infection, perpetuating sterile inflammation and promoting chronic OS damage through sustained cytokine expression and type I IFN signaling [34,35]. Kwon et al. identified the presence of anti-citrullinated protein autoantibodies (ACPA) in the OS wash of 40% of patients with DED, likely generated through peptidylarginine deiminase 4 (PAD4)-mediated citrullination of proteins during neutrophil NETosis[36]. These ACPAs were found to subsequently induce OS inflammation in murine models [36].
Macrophages are versatile innate immune cells involved in pathogen clearance, tissue repair, and immune regulation. Differing activation states exist including Classically activated (M1) pro-inflammatory macrophages and alternatively activated (M2) anti-inflammatory macrophages, depending on their environment [22]. In DED, hyperosmolar stress skews macrophage polarization toward a pro-inflammatory M1 state while suppressing anti-inflammatory M2 activity, promoting Th1 and Th17 infiltration and amplifying OS immune dysregulation [37].
A recent study by Alam et al. using single-cell RNA sequencing in murine models of DED showed a three-fold increase in resident macrophages [38]. Moreover, the phenotypic shift of macrophages towards pain sensitization via gene expression of CXCL1 and loss of homeostatic gene expression was observed [38]. Increased production of neurosensitizing factors such as CXCL1 can activate transient receptor potential vanilloid 1 (TRPV-1), A disintegrin, and metalloproteinase 17 (ADAM17), contributing to ocular pain and epithelial barrier disruption associated with DED [38]. Complementing these findings, recent single-cell transcriptomic and epigenomic profiling has revealed distinct conjunctival macrophage subsets in DED mouse models, including a regulatory retinoid X receptor alpha (RXRα), a nuclear receptor modulating immune gene expression that suppresses inflammation via IL-10 signaling, the depletion of which exacerbates goblet cell loss and Th17-mediated pathology [39].
Natural killer (NK) cells play a pivotal role in ocular surface immunity by secreting large amounts of IFN-γ, which activates surrounding macrophages and T cells [22]. In addition to their immunomodulatory function, NK cells exhibit cytotoxic activity via granzyme and perforin release, contributing to tissue damage [22]. The role of NK cells in the early stages of DED pathogenesis has been suggested, as their activation promotes IFN-γ–mediated inflammation and drives APC maturation, ultimately priming adaptive immune responses [40]. In murine models of DED, conjunctival NK and natural killer T cells (NKT) produce IL-6 and IL-23, activating DCs and enhancing Th17 responses [41]. Notably, NK cell depletion reduces cytokine levels and preserves corneal integrity, underscoring a critical NK–DC–Th17 axis in early DED.
7. Adaptive Immune System in DED
Once primed, the adaptive immune system plays a central role in the chronicity of DED. Autoreactive T cells drive sustained inflammation of the OS, in particular, CD4⁺ T helper subsets Th1 and Th17 cells [25]. These mediate further cytokine-driven epithelial damage and perpetuate immune dysregulation and are unimpeded by impaired T regulatory cells [25].
The activation, migration and interaction of APCs with naive T cells is central to the initiation of the adaptive immune response in DED and has been well established [25,42]. In murine models, the migration of APCs towards draining lymph nodes is facilitated by the upregulated expression of C-C chemokine receptor-7 (CCR7) which guides their exit from the OS to lymph nodes by responding to specific ligands CCL19 and CCL21, found in high density in corneal lymphatics and draining lymph nodes [43,44]. Within the lymph node microenvironment, APCs prime naive T cells by MHC–antigen interaction and costimulatory engagement (e.g., CD80/CD86 with CD28) towards CD4+ Th1 and Th17 effectors [45,46].
7.1. Th1 Cells
The effect of Th1 cells in DED was initially noted to be more pronounced in the acute phase, as evidenced by the presence of its related cytokine IFN-γ in murine models of early DED and its reduced prominence later in disease progression [47]. DED-primed Th1 cells upregulate chemokine receptors CCR5 and CXCR3, facilitating their targeted migration from the lymph nodes back to the inflamed OS via corresponding chemokine gradients, as demonstrated in murine models [48].
IFN-γ was initially thought to amplify its own production by upregulating IL-12 receptor expression, promoting further Th1 differentiation and inducing chemokines (CXCL9, CXCL10, CXCL11), which recruit and retain Th1 cells in inflamed tissues [49]. IFN-γ itself exerts its effects in DED by inducing GC loss and reducing mucin production, thus worsening tear film instability and perpetuating the vicious cycle of inflammation [50]. Moreover, loss of conjunctival GC likely disrupts local immune tolerance by impairing APC tolerance, leading to enhanced IL-12 production and pathogenic Th1/Th17 polarization as evidenced in murine models [51]. However, a review by Chen et al. scrutinizing the varied roles of Th-17 cells in DED concluded that the exact source of IFN-γ in acute phase of DED may be associated with NK cells, and its continued secretion throughout the course of the disease may be related to IFN-γ+IL-17+ “double-positive” Th17/1 cells [25].
7.2. Th17 Cells
The role of Th-17 cells in DED is significant and central to the pathogenesis of DED. Differentiation of Th-17 cells is largely dependent on the microenvironment and is initiated by the signal transducer and activator of transcription 3 (STAT3) signaling pathway after exposure to IL-6 and TGF-beta secreted by APCs [52,53]. This is further regulated and promoted by the transcription factor retinoic acid–related orphan receptor gamma t (RORγt) which was found to be significantly upregulated in the OS tissues after exposure to desiccating stress in murine models [54]. Th17 cells then migrate back to the OS, specifically the conjunctiva, via chemokine receptors CCR6 and CCL20, where they undergo further differentiation following exposure to IL-1 and IL-23 [55].
A key cytokine produced by Th17 is IL-17, found in greater concentrations in tears of both non-SS and SS-DED patients, which stimulates MMP production and causes damage to the corneal epithelium [49,56]. However, there are several other key Th-17 subsets, including IL-10 producing Th17 cells, Th17/Th1 cells which co-produce IL-17 and IFN gamma, Th-17 producing Granulocyte-macrophage colony-stimulating factor (Th17GM-CSF), and more recently interleukin 17 receptor E (IL-17RE) and CCR10 producing Th17 cells in murine models [57,58,59,60].
Whilst IL-10 producing Th-17 cells may potentially play a regulatory role in DED (as seen in murine models), other subsets are more detrimental [57]. Co-producing IL-17 and IFNγ Th-17 cells are significantly pathogenic and drive epithelial apoptosis, lymphangiogenesis, APC maturation and potentially IFNγ production seen in in chronic DED [25,57,58].
Granulocyte-macrophage colony-stimulating factor (GM-CSF) exerts its role in DED through stimulation of monocytic cells to produce pro-inflammatory cytokines such as IL-1β, IL-6, and IL-23. IL-6 and IL-23 further perpetuate Th17 differentiation [59]. In a murine model of DED, the IL-17RE^highCCR10⁺ Th17 subset exhibited enhanced JNK and p38 MAPK signaling. This is likely mediated through IL-17C/IL-17RE interaction, which reinforces and perpetuates their Th17 phenotype by sustaining IL-17A expression in vitro [60]. The presence of multiple subsets of Th-17 cells and their more hybrid phenotypes such as Th17/Th1 and CCR10 expression (typically found on T helper 22 cells) may suggest plasticity of Th17 cells which can transdifferentiate depending on the microenvironment as seen in murine models [60].
Alam et al. identified γδ T cells as a significant source of IL-17 in RXRα-deficient Pinkie mouse models, with elevated IL-17A and IL-17F expression that exacerbated DED [61]. They also demonstrated that 9-cis retinoic acid, the natural ligand for RXRα, suppresses IL-17 production from both γδ T cells and monocytes in vitro, suggesting that RXRα may act as a negative regulator of IL-17-driven inflammation [61] .
7.3. Memory T Cells
Chronic DED is predominantly driven by a persistent Th17 response, particularly by effector memory Th17 cells [62]. Chen et al. used murine models to demonstrate that OS inflammation persists despite the absence of desiccating stress and is largely driven by continued IL-17 secretion from this population of cells [62]. Th17 memory cells also continue to promote further migration of effector T cells from lymph nodes (LN) to the OS via pathological lymphangiogenesis [62]. The same group also identified in pre-clinical models that IL-7 and IL-15 are critical for the maintenance of Th17 memory cells and promote continued survival via STAT5 and phosphoinositide 3-kinase–Akt (PI3K-Akt) pathways [63]. Moreover, IL-23 was found to promote the transition of Th17 effector cells into memory cells whilst IL-2 had an inhibiting effect on this pathway as demonstrated in murine models [64].
Ageing has also been identified as a risk factor for increased severity of DED as seen in mice models which demonstrated higher amounts of memory Th-17 cells compared with their younger counterparts [65]. Furthermore, an increase in Th-17 cells correlated with increased disease severity upon re-exposure to desiccating stress in the aged mice [65].
7.4. Tregs
The impairment of Tregs cells in suppressing pathogenic T effector cell priming and activity is a crucial aspect of immune dysfunction central to DED. CD25+CD4+Foxp3+ Tregs are peripheral Tregs with important mechanisms of action including: 1) granzyme-B and perforin mediated cytolysis, 2) the release of immunosuppressive cytokines including IL-10, TGF beta and IL-35, 3) the inhibition of DC maturation and function via the transendocytosis of co-stimulatory molecules (CD80/86) and 4) suppression via metabolic competition [13,66,67]. In DED murine models, their reduction led to increased severity of SS-like DED [68]. Conversely, the transfer of in vitro expanded Tregs into two different strains of DED murine models, BALB/c and C57BL/6, showed suppression of pathogenic CD4+ effector T cell mediated inflammation [69].
Interestingly, Chauchan et al. reported that Treg levels in DED murine models remained unchanged but impaired ability to suppress T effector cells, particularly Th17 cells, potentially due to Th17 cells secretion of IL-17A [70]. Moreover, the high IL-6 microenvironment in DED has an inhibitory effect on Treg differentiation [71].
Marginally increased Treg count was found in the conjunctiva of patients with DED associated with SS without a correlated increase in signs and symptoms of DED, further supporting Treg role in DED may be a functional rather than quantitative issue [72]. This was further evidenced in stromal interaction molecule 1/2 (STIM1/2) Foxp3 mice, where targeted deletion of calcium-sensing proteins STIM1/2 in Foxp3+ Tregs resulted in a fulminant SS-like phenotype characterized by lacrimal gland inflammation, lymphocytic infiltration, and IFN-γ–dominated transcriptional signatures. [73]. Transcriptomic analyses revealed downregulation of core memory Treg genes in both mouse and human SS, suggesting that functional Treg impairment, not mere reduction, is a conserved and critical step in disease development [73].
Ageing is a significant driver of Treg dysregulation and plasticity in DED [13,74]. In aged murine models, Tregs were found to maintain Foxp3 expression without loss of suppressive capacity and aberrantly produced IL-17A and IFN-γ [75]. Adoptive transfer experiments demonstrate that these Tregs may contribute to tissue inflammation, suggesting a phenotypic conversion toward a pro-inflammatory effector state [75].
8. Neuroimmune Mediated Inflammation in DED
Neuroimmune dysregulation significantly contributes to the pathogenesis of DED by aberrantly modulating both innate and adaptive immune responses. As proposed in a review by Huang et al., this may be due to a bidirectional feedback loop in which proinflammatory cytokines released by immune cells damage peripheral nerves, triggering excessive neuropeptide release [76]. These neuropeptides, in turn, further activate immune pathways, amplifying OS inflammation and perpetuating chronic disease [76]. Yu et al. have underscored the role of the neuropeptide substance P (SP) from sensory nerve endings in enhancing MHC-II expression on DCs, thereby promoting subsequent T cell priming [42]. Moreover, the SP–neurokinin 1 receptor (NK1R) axis directly acts on pathogenic Th17GM-CSF cells, significantly increasing GM-CSF production and exacerbating dry eye disease severity in murine models [77]. This highlights a novel neuroimmune pathway through which neuropeptides enhance effector T cell activity and OS inflammation [77].
In addition, SP/NK1R signaling has been implicated in pathological lymphangiogenesis in DED, where it upregulates Vascular Endothelial Growth Factor (VEGF)-C, VEGF-D, and VEGFR-3 expression, promoting lymphatic vessel growth and facilitating APC trafficking to draining lymph nodes [78]. Moreover, whilst Tregs expressing NK1R are found in increased quantities in DED, their abnormal expression of critical immunomodulatory markers CTLA-4, PD-1, TGF-β, and IL-10 demonstrate an impaired suppressive capacity against effector T cells, suggesting a compromised regulatory phenotype [79,80]. SP may also be implicated in the promotion and maintenance of memory Th17 cells [81]. In vitro studies have shown increased conversion of effector T cells to memory Th17 cells when cultured with SP [81]. Further, when cultured with Th17 memory cells, SP continued to preserve the cells [81].
Calcitonin gene-related peptide (CGRP) plays a potentially more complex and unclear role in DED, exhibiting both immunosuppressive and proinflammatory roles depending on the microenvironment [76,82]. Its immunosuppressive activity includes the inhibition of APCs by Langerhans cells and attenuation of contact hypersensitivity through the suppression of mast cell-derived tumor necrosis factor [83]. However, clinical data on CGRP and SP remain conflicting. Some studies report reduced tear concentrations of CGRP and SP, particularly in severe or chronic DED, where corneal nerve loss may impair neuropeptide production and secretion, thereby diminishing their homeostatic and immunomodulatory functions [83,84,85]. Conversely, other studies demonstrate increased neuropeptide levels, particularly in DED subtypes with prominent neuroinflammation or post-surgical neuropathic pain, suggesting upregulation in response to inflammatory stimuli or nerve sensitization [79,86]. These discrepancies possibly reflect underlying disease heterogeneity, variation in nerve integrity, and differences in immune activation across DED phenotypes.
In murine models, tear hyperosmolarity alone disrupted neuroimmune homeostasis via TRPV1, NF-κB activation in conjunctival epithelium, leading to DC maturation, memory CD4⁺ T cell priming, corneal nerve loss, and impaired mucosal tolerance [87]. Adoptive transfer of these T cells induced DED in naive mice, establishing hyperosmolarity as a direct pathogenic trigger [87]. Complementing these findings, studies in guinea pig models of aqueous tear deficiency demonstrate that chronic dryness also sensitizes TRPV1-expressing corneal nociceptors, enhancing blink reflexes and neuronal calcium responses to capsaicin [88]. This neuroplasticity likely contributes to the ocular hyperalgesia and discomfort characteristic of DED [88].
9. Tear Biomarkers of DED
The pathophysiology of dry eye disease involves a complex interplay of immune, epithelial, and neuronal factors at the ocular surface. Numerous biomarkers have been identified that reflect distinct aspects of disease activity, including inflammatory mediators, chemokines, cytokines, matrix metalloproteinases, and regulatory molecules (Table 1). They play roles in promoting or modulating ocular surface inflammation, epithelial barrier disruption, and immune cell recruitment.
10. Therapeutic Implications
While the initiating events in DED remain unclear, sustained immune dysregulation, marked by aberrant activation of innate and adaptive immune pathways, plays a central role in perpetuating OS inflammation. This understanding has driven a shift from symptomatic treatments to targeted immunomodulation, aimed at disrupting specific molecular mediators of inflammation and restoring immune homeostasis.
Several currently approved agents exert immunomodulatory effects, albeit with variable efficacy [89]. Cyclosporine A, a topical calcineurin inhibitor, suppresses IL-2–mediated T cell activation, reduces epithelial apoptosis, and has been shown to restore GC density in DED [90]. Lifitegrast, a lymphocyte function-associated antigen-1 (LFA-1) antagonist, blocks T cell adhesion and migration through competitive inhibition of the LFA-1/ intercellular adhesion molecule 1 (ICAM-1) interaction on the OS [90]. Topical corticosteroids, though effective in rapidly reducing inflammatory mediators such as MMP-9, IL-1β, and TNF-α via suppression of NF-κB and activator protein 1 (AP-1), carry well-documented risks with prolonged use, including ocular hypertension, cataract formation, and susceptibility to infection [90].
Therapeutic advances have shifted focus toward precision immunomodulation targeting the upstream molecular and cellular drivers of innate immune dysregulation in DED. Modulating macrophage polarization is a potential therapeutic target, with murine studies demonstrating that shifting macrophages from a pro-inflammatory M1 phenotype to an anti-inflammatory M2 phenotype can contribute to reduction in pro-inflammatory cells and increase in anti-inflammatory factors [37]. In a benzalkonium chloride (BAC)-induced murine model of DED, treatment with M2 macrophage–derived extracellular vesicles (M2-EVs) improved tear production, preserved corneal integrity, and downregulated inflammatory cytokines [91]. Zhou et al. highlighted the α7 nicotinic acetylcholine receptor (α7nAChR) as a regulator of macrophage-driven inflammation; activation of this receptor reduced OS inflammation via neuroimmune crosstalk in DED murine models [92].
Endogenous counter-regulatory molecules such as pigment epithelium-derived factor (PEDF) have also demonstrated immunosuppressive effects [93]. In both murine and human cells, elevated PEDF expression in the corneal epithelium and tear film suppressed key proinflammatory cytokines including IL-1β, IL-6, TNF-α, and IL-17A, and reduced Th17 cell density through inhibition of the p38 MAPK and JNK pathways [93]. A ROS-responsive microneedle patch (CE-MN), loaded with cyclosporin A and the antioxidant epigallocatechin gallate (EGCG), enabled sustained periocular delivery to the lacrimal gland. In an SS-DED model, CE-MN suppressed macrophage activation and oxidative stress, while attenuating downstream Th1 and Th17-mediated inflammation more effectively than conventional eye drops [94].
Several potential strategies that target the adaptive immune response have emerged. A number of murine studies have shown that neutralizing IL-17A or blocking its upstream regulators improves OS quality. Local CCL20 neutralization reduces Th17 cell infiltration and inflammation in vitro and in vivo, while RXRα agonism with 9-cis retinoic acid, attenuates Th17-driven inflammation and preserves goblet cells in the Pinkie mouse model [55,61]. Topical inhibition of phosphodiesterase type-4 (PDE4) with Cilomilast significantly suppressed IL-17 and IL-23 expression in conjunctival tissue and draining lymph nodes, reduced CD11b⁺ APC infiltration, and downregulated IL-1β, IL-6, and TNF-α [95]. This improved clinical outcomes, with therapeutic efficacy comparable or superior to both dexamethasone and cyclosporine in DED murine models [95]. However, when Secukinumab (a human monoclonal antibody that neutralizes IL-17A) was utilized systemically in patients with DED, it showed no significant amelioration of DED symptoms [96]. A phase II clinical trial focusing on topical administration of an IL-17A antagonist has yet to release its results [97].
In chronic DED, targeting Il-7 and IL-15 with topical anti-IL-7 and anti-IL15 antibody treatments in vivo murine models showed amelioration of disease severity, however, this could not target memory Th17 cells in draining lymph nodes [63]. Moreover, a murine study employing CCL22-releasing microspheres demonstrated increased recruitment of endogenous Tregs to the lacrimal gland, leading to improved epithelial integrity, reduced IFN-γ–mediated inflammation, and a restored Treg:effector T cell ratio [98].
Targeting neuroimmune signaling in DED is another therapeutic avenue that has been explored in the last decade. Topical blockade of NK1R using antagonists such as spantide and CP-99,994 significantly suppressed Th17 responses, reduced corneal lymphangiogenesis, and improved clinical outcomes [42,78]. Pyroptosis, a form of proinflammatory programmed cell death, and its associated polo-like kinase 1–cell division cycle 25C–cyclin-dependent kinase 1 (Plk1–Cdc25c–Cdk1) axis, can also be targeted using CP-99,994 [99]. This has led to reduced IL-6, IL-1β, and TNF-α levels, and further ameliorating OS inflammation in murine DED model [99].
More recently, mesenchymal stem cell (MSC) therapy has emerged as a promising immunomodulatory strategy for the treatment of DED. MSCs derived from bone marrow, adipose tissue, and umbilical cord modulate ocular inflammation through both cell-mediated and paracrine mechanisms [100]. In a murine T cell–driven model of DED, local infusion of human or mouse MSCs suppressed CD4⁺ T cell proliferation and IFN-γ production, reducing OS inflammation and restoring GC density and tear secretion [101]. These effects occurred independently of Treg induction or indoleamine 2,3-dioxygenase (IDO)-mediated tryptophan metabolism, suggesting alternative regulatory pathways, including transient recruitment of other immunosuppressive cells or species-specific mechanisms such as inducible nitric oxide synthase in murine MSCs [101]. Similarly, in a murine SS-DED model, bone marrow–derived mouse MSCs improved lacrimal gland secretory function and increased aquaporin 5 expression, while reducing lymphocytic foci size without significant changes in Foxp3⁺ Tregs or stromal cell-derived factor 1 (SDF-1) /CXCR4 signaling [102].
MSC-derived extracellular vesicles (MSC-EVs) demonstrate comparable efficacy: umbilical cord MSC-EVs downregulated the IRAK1/TAB2/NF-κB pathway via miRNAs including miR-125b and let-7b, suppressing proinflammatory cytokines in murine DED models [103]. Adipose-derived MSC exosomes (mADSC-Exos) attenuated hyperosmotic stress–induced inflammation by reducing IL-1β, IL-6, and NOD-, LRR- and Pyrin domain-containing protein 3 (NLRP3) inflammasome activation in murine models, while promoting tear film stability and epithelial repair [104]. Additionally, human umbilical cord MSC–derived exosomal microRNA-146a (miR-146a) suppressed apoptosis and inflammation in hyperosmotic-stressed human corneal epithelial cells and a murine DED model by upregulating sequestosome 1 (SQSTM1), revealing a novel miRNA-mediated protective axis [105].
11. Conclusions
Our understanding of the molecular and cellular mechanisms underlying dry eye disease has evolved significantly, revealing how dysregulated innate and adaptive immune pathways contribute to the perpetuation of ocular surface inflammation and neuroimmune dysfunction.
12. Future Directions
Advances in molecular and translational research have identified promising targeted immunomodulatory strategies such as Th17 inhibition, macrophage modulation, neuropeptide receptor blockade, and stem cell therapies. Clinical translation remains limited by disease heterogeneity, lack of robust biomarkers, and potentially incomplete understanding of human ocular immunopathology. Future research may focus on precise immunophenotyping to enable personalized treatment, development of reliable biomarkers for diagnosis and monitoring, and well-designed clinical trials exploring novel topical and cell-free immunotherapies to restore immune homeostasis and improve patient outcomes.
Author Contributions
Conceptualization: M.W., H.R.., C.R., P.S.; Methodology: S.S., M.N.T., L.F.; Writing—original draft preparation: S.S., M.N.T. Writing—review and editing: M.W., L.F., C.R., H.R. and P.S.; Illustrations: M.N.T. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Conflicts of Interest
No conflicts of interest were declared
Abbreviations
| Abbreviation | Full Term |
| ACPA | Anti-citrullinated protein autoantibodies |
| ADAM17 | A Disintegrin And Metalloproteinase 17 |
| ADDE | Aqueous-deficient dry eye |
| AP-1 | Activator Protein 1 |
| α7nAChR | α7 Nicotinic Acetylcholine Receptor |
| BAC | Benzalkonium Chloride |
| BALB/c | BALB/c mouse strain |
| CCL5 | C-C motif chemokine ligand 5 |
| CCR7 | C-C chemokine receptor 7 |
| CGRP | Calcitonin Gene-Related Peptide |
| CE-MN | Reactive Oxygen Species (ROS)-responsive Microneedle Patch |
| CP-99,994 | N1KR antagonist |
| CXCL9 | C-X-C motif chemokine ligand 9 |
| DC | Dendritic Cell |
| DAMPs | Danger Associated Molecular Patterns |
| EDE | Evaporative Dry Eye |
| EGCG | Epigallocatechin Gallate |
| FoxP3 | Forkhead Box Protein P3 |
| HMGB1 | High Mobility Group Box 1 |
| HSP-60 | Heat Shock Protein 60 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IDO | Indoleamine 2,3-dioxygenase |
| IL | Interleukin |
| IL-17C | Interleukin 17C |
| IL-17RE | Interleukin 17 Receptor E |
| JNK1/2 | c-Jun N-terminal kinase 1/2 |
| LFA-1 | Lymphocyte Function-Associated Antigen-1 |
| LN | Lymph Nodes |
| MAPKs | Mitogen-Activated Protein Kinases |
| M1 | Classically activated (pro-inflammatory) macrophage |
| M2 | Alternatively activated (anti-inflammatory) macrophage |
| M2-EVs | M2 Macrophage–Derived Extracellular Vesicles |
| MD2 | Myeloid Differentiation Factor 2 |
| MHC | Major Histocompatibility Complex |
| miR-146a | MicroRNA-146a |
| MSC | Mesenchymal Stem Cell |
| MSC-EVs | MSC-Derived Extracellular Vesicles |
| mADSC-Exos | Adipose-Derived Mesenchymal Stem Cell Exosomes |
| mRNA | Messenger Ribonucleic Acid |
| NF-κB | Nuclear Factor kappa-light-chain-enhancer of activated B cells |
| NFAT5 | Nuclear Factor of Activated T Cells 5 |
| NK | Natural Killer Cells |
| NKT | Natural Killer T Cells |
| NETs | Neutrophil Extracellular Traps |
| NLRP3 | NOD-, LRR- and Pyrin Domain-Containing Protein 3 |
| PAD4 | Peptidylarginine Deiminase 4 |
| PEDF | Pigment Epithelium-Derived Factor |
| PI3K-Akt | Phosphoinositide 3-Kinase–Akt |
| Plk1–Cdc25c–Cdk1 | Polo-Like Kinase 1 – Cell Division Cycle 25C – Cyclin-Dependent Kinase 1 |
| PDE4 | Phosphodiesterase Type-4 |
| PD-L1 | Programmed Death Ligand 1 |
| RASγt | Retinoic Acid–Related Orphan Receptor Gamma t |
| RXRα | Retinoid X Receptor Alpha |
| ROS | Reactive Oxygen Species |
| RORγt | Retinoic Acid–Related Orphan Receptor Gamma t |
| SDF-1 | Stromal Cell-Derived Factor 1 |
| SP | Substance P |
| STIM1/2 | Stromal Interaction Molecule 1/2 |
| SQSTM1 | Sequestosome 1 |
| STAT3 | Signal Transducer and Activator of Transcription 3 |
| Th17GM-CSF | Th-17 Producing Granulocyte-Macrophage Colony-Stimulating Factor |
| TGF | Tumor Growth Factor |
| Tregs | T Regulatory Cells |
| TRPV1 | Transient Receptor Potential Vanilloid 1 |
| ZO-1 | Zonula Occludens-1 |
References
- Britten-Jones, A.C.; Wang, M.T.M.; Samuels, I.; Jennings, C.; Stapleton, F.; Craig, J.P. Epidemiology and Risk Factors of Dry Eye Disease: Considerations for Clinical Management. Medicina (B Aires) 2024, 60, 1458. [Google Scholar] [CrossRef]
- Bron, A.J.; de Paiva, C.S.; Chauhan, S.K.; Bonini, S.; Gabison, E.E.; Jain, S.; Knop, E.; Markoulli, M.; Ogawa, Y.; Perez, V.; et al. TFOS DEWS II Pathophysiology Report. Ocular Surface 2017, 15, 438–510. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, S.; Alemi, H.; Dohlman, T.; Dana, R. Immune Regulation of the Ocular Surface. Exp Eye Res 2022, 218, 109007. [Google Scholar] [CrossRef]
- Stapleton, F.; Argüeso, P.; Argüeso, A.; Asbell, P.; Azar, D.; Bosworth, C.; Chen, W.; Ciolino, J.; Craig, J.P.; Gallar, J.; et al. TFOS DEWS III Digest Report. Am J Ophthalmol 2025, 0. [Google Scholar] [CrossRef]
- Barabino, S.; Chen, Y.; Chauhan, S.; Dana, R. Ocular Surface Immunity: Homeostatic Mechanisms and Their Disruption in Dry Eye Disease. Prog Retin Eye Res 2012, 31, 271. [Google Scholar] [CrossRef]
- Periman, L.M.; Perez, V.L.; Saban, D.R.; Lin, M.C.; Neri, P. The Immunological Basis of Dry Eye Disease and Current Topical Treatment Options. Journal of Ocular Pharmacology and Therapeutics 2020, 36, 137. [Google Scholar] [CrossRef]
- Galletti, J.G.; de Paiva, C.S. Age-related Changes in Ocular Mucosal Tolerance: Lessons Learned from Gut and Respiratory Tract Immunity. Immunology 2021, 164, 43. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.E.; Schaumburg, C.S.; Pflugfelder, S.C. Dry Eye as a Mucosal Autoimmune Disease. Int Rev Immunol 2013, 32, 19. [Google Scholar] [CrossRef]
- Zhou, L.; Beuerman, R.W. Tear Analysis in Ocular Surface Diseases. Prog Retin Eye Res 2012, 31, 527–550. [Google Scholar] [CrossRef] [PubMed]
- de Paiva, C.S.; St. Leger, A.J.; Caspi, R.R. Mucosal Immunology of the Ocular Surface. Mucosal Immunol 2022, 15, 1143. [Google Scholar] [CrossRef] [PubMed]
- Dua, H.S.; Donoso, L.A.; Laibson, P.R. Conjunctival Instillation of Retinal Antigens Induces Tolerance Does It Invoke Mucosal Tolerance Mediated via Conjunctiva Associated Lymphoid Tissues (CALT)? Ocul Immunol Inflamm 1994, 2, 29–36. [Google Scholar] [CrossRef]
- Galletti, J.G.; Guzmán, M.; Giordano, M.N. Mucosal Immune Tolerance at the Ocular Surface in Health and Disease. Immunology 2017, 150, 397–407. [Google Scholar] [CrossRef]
- Foulsham, W.; Marmalidou, A.; Amouzegar, A.; Coco, G.; Chen, Y.; Dana, R. The Function of Regulatory T Cells at the Ocular Surface: Review. Ocul Surf 2017, 15, 652. [Google Scholar] [CrossRef]
- Forrester, J. V.; McMenamin, P.G. Evolution of the Ocular Immune System. Eye 2024, 39, 468. [Google Scholar] [CrossRef] [PubMed]
- Irmina, J.M.; Bartosz, M.; Dorota, W.P.; Adrian, S. The Role of Substance P in Corneal Homeostasis. Biomolecules 2025, 15, 729. [Google Scholar] [CrossRef] [PubMed]
- Craig, J.P.; Nichols, K.K.; Akpek, E.K.; Caffery, B.; Dua, H.S.; Joo, C.K.; Liu, Z.; Nelson, J.D.; Nichols, J.J.; Tsubota, K.; et al. TFOS DEWS II Definition and Classification Report. Ocular Surface 2017, 15, 276–283. [Google Scholar] [CrossRef]
- Lemp, M.A.; Baudouin, C.; Baum, J.; Dogru, M.; Foulks, G.N.; Kinoshita, S.; Laibson, P.; McCulley, J.; Murube, J.; Pflugfelder, S.C.; et al. The Definition and Classification of Dry Eye Disease: Report of the Definition and Classification Subcommittee of the International Dry Eye WorkShop (2007). Ocular Surface 2007, 5, 75–92. [Google Scholar] [CrossRef]
- Report of the National Eye Institute/Industry Workshop on Clinical Trials in Dry Eyes - PubMed Available online:. Available online: https://pubmed.ncbi.nlm.nih.gov/8565190/ (accessed on 8 June 2025).
- Baudouin, C.; Messmer, E.M.; Aragona, P.; Geerling, G.; Akova, Y.A.; Benítez-Del-Castillo, J.; Boboridis, K.G.; Merayo-Lloves, J.; Rolando, M.; Labetoulle, M. Revisiting the Vicious Circle of Dry Eye Disease: A Focus on the Pathophysiology of Meibomian Gland Dysfunction. British Journal of Ophthalmology 2016, 100, 300–306. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; de Paiva, C.S. The Pathophysiology of Dry Eye Disease: What We Know and Future Directions for Research. Ophthalmology 2017, 124, S4. [Google Scholar] [CrossRef]
- Bolaños-Jiménez, R.; Navas, A.; López-Lizárraga, E.P.; Ribot, F.M. de; Peña, A.; Graue-Hernández, E.O.; Garfias, Y. Ocular Surface as Barrier of Innate Immunity. Open Ophthalmol J 2015, 9, 49. [Google Scholar] [CrossRef]
- Reyes, J.L.; Vannan, D.T.; Eksteen, B.; Avelar, I.J.; Rodríguez, T.; González, M.I.; Mendoza, A.V. Innate and Adaptive Cell Populations Driving Inflammation in Dry Eye Disease. Mediators Inflamm 2018, 2018, 2532314. [Google Scholar] [CrossRef] [PubMed]
- Luo, L.; Li, D.Q.; Doshi, A.; Farley, W.; Corrales, R.M.; Pflugfelder, S.C. Experimental Dry Eye Stimulates Production of Inflammatory Cytokines and MMP-9 and Activates MAPK Signaling Pathways on the Ocular Surface. Invest Ophthalmol Vis Sci 2004, 45, 4293–4301. [Google Scholar] [CrossRef]
- Li, D.Q.; Luo, L.; Chen, Z.; Kim, H.S.; Song, X.J.; Pflugfelder, S.C. JNK and ERK MAP Kinases Mediate Induction of IL-1β, TNF-α and IL-8 Following Hyperosmolar Stress in Human Limbal Epithelial Cells. Exp Eye Res 2006, 82, 588–596. [Google Scholar] [CrossRef]
- Chen, Y.; Dana, R. Autoimmunity in Dry Eye Disease – an Updated Review of Evidence on Effector and Memory Th17 Cells in Disease Pathogenicity. Autoimmun Rev 2021, 20, 102933. [Google Scholar] [CrossRef]
- Moustardas, P.; Aberdam, D.; Lagali, N. MAPK Pathways in Ocular Pathophysiology: Potential Therapeutic Drugs and Challenges. Cells 2023, 12, 617. [Google Scholar] [CrossRef]
- Li, S.; Lu, Z.; Huang, Y.; Wang, Y.; Jin, Q.; Shentu, X.; Ye, J.; Ji, J.; Yao, K.; Han, H. Anti-Oxidative and Anti-Inflammatory Micelles: Break the Dry Eye Vicious Cycle. Advanced Science 2022, 9, 2200435. [Google Scholar] [CrossRef] [PubMed]
- De Paiva, C.S.; Corrales, R.M.; Villarreal, A.L.; Farley, W.J.; Li, D.Q.; Stern, M.E.; Pflugfelder, S.C. Corticosteroid and Doxycycline Suppress MMP-9 and Inflammatory Cytokine Expression, MAPK Activation in the Corneal Epithelium in Experimental Dry Eye. Exp Eye Res 2006, 83, 526–535. [Google Scholar] [CrossRef]
- Li, D.Q.; Lokeshwar, B.L.; Solomon, A.; Monroy, D.; Ji, Z.; Pflugfelder, S.C. Regulation of MMP-9 Production by Human Corneal Epithelial Cells. Exp Eye Res 2001, 73, 449–459. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.H.; Chen, W.Y.; Huang, Y.H.; Hsu, S.M.; Tsao, Y.P.; Hsu, Y.H.; Chang, M.S. Interleukin-20 Is Involved in Dry Eye Disease and Is a Potential Therapeutic Target. J Biomed Sci 2022, 29. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, L.; Chen, B. Characterization of T Cells in the Progression of Dry Eye Disease Using Single-Cell RNA Sequencing in Mice. Eur J Med Res 2025, 30, 338. [Google Scholar] [CrossRef]
- Redfern, R.L.; Barabino, S.; Baxter, J.; Lema, C.; McDermott, A.M. Dry Eye Modulates the Expression of Toll-Like Receptors on the Ocular Surface. Exp Eye Res 2015, 134, 80. [Google Scholar] [CrossRef] [PubMed]
- Alven, A.; Lema, C.; Redfern, R.L. Impact of Low Humidity on Damage Associated Molecular Patterns at the Ocular Surface during Dry Eye Disease. Optom Vis Sci 2021, 98, 1231. [Google Scholar] [CrossRef] [PubMed]
- Tibrewal, S.; Ivanir, Y.; Sarkar, J.; Nayeb-Hashemi, N.; Bouchard, C.S.; Kim, E.; Jain, S. Hyperosmolar Stress Induces Neutrophil Extracellular Trap Formation: Implications for Dry Eye Disease. Invest Ophthalmol Vis Sci 2014, 55, 7961. [Google Scholar] [CrossRef]
- Sonawane, S.; Khanolkar, V.; Namavari, A.; Chaudhary, S.; Gandhi, S.; Tibrewal, S.; Jassim, S.H.; Shaheen, B.; Hallak, J.; Horner, J.H.; et al. Ocular Surface Extracellular DNA and Nuclease Activity Imbalance: A New Paradigm for Inflammation in Dry Eye Disease. Invest Ophthalmol Vis Sci 2012, 53, 8253–8263. [Google Scholar] [CrossRef]
- Kwon, J.; Surenkhuu, B.; Raju, I.; Atassi, N.; Mun, J.; Chen, Y.F.; Sarwar, M.A.; Rosenblatt, M.; Pradeep, A.; An, S.; et al. Pathological Consequences of Anti-Citrullinated Protein Antibodies in Tear Fluid and Therapeutic Potential of Pooled Human Immune Globulin-Eye Drops in Dry Eye Disease. Ocular Surface 2020, 18, 80–97. [Google Scholar] [CrossRef]
- Yingming, W.; Jing, G.; Tianhong, W.; Zhenyu, W. M2 Macrophages Mitigate Ocular Surface Inflammation and Promote Recovery in a Mouse Model of Dry Eye. Exp Eye Res 2025, 110439. [Google Scholar] [CrossRef]
- Alam, J.; Yaman, E.; Silva, G.C.V.; Chen, R.; de Paiva, C.S.; Stepp, M.A.; Pflugfelder, S.C. Single Cell Analysis of Short-Term Dry Eye Induced Changes in Cornea Immune Cell Populations. Front Med (Lausanne) 2024, 11, 1362336. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Yaman, E.; de Paiva, C.S.; Li, D.Q.; Villalba Silva, G.C.; Zuo, Z.; Pflugfelder, S.C. Changes in Conjunctival Mononuclear Phagocytes and Suppressive Activity of Regulatory Macrophages in Desiccation Induced Dry Eye. Ocular Surface 2024, 34, 348–362. [Google Scholar] [CrossRef]
- Chen, Y.; Chauhan, S.K.; Saban, D.R.; Sadrai, Z.; Okanobo, A.; Dana, R. Interferon-γ-Secreting NK Cells Promote Induction of Dry Eye Disease. J Leukoc Biol 2011, 89, 965. [Google Scholar] [CrossRef]
- Zhang, X.; Volpe, E.A.; Gandhi, N.B.; Schaumburg, C.S.; Siemasko, K.F.; Pangelinan, S.B.; Kelly, S.D.; Hayday, A.C.; Li, D.Q.; Stern, M.E.; et al. NK Cells Promote Th-17 Mediated Corneal Barrier Disruption in Dry Eye. PLoS One 2012, 7, e36822. [Google Scholar] [CrossRef]
- Yu, M.; Lee, S.M.; Lee, H.; Amouzegar, A.; Nakao, T.; Chen, Y.; Dana, R. Neurokinin-1 Receptor Antagonism Ameliorates Dry Eye Disease by Inhibiting Antigen-Presenting Cell Maturation and T Helper 17 Cell Activation. American Journal of Pathology 2020, 190, 125–133. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Li, W.; Cheng, H.; Zhong, L.; Deng, J.; Ling, S. The Important Role of the Chemokine Axis CCR7-CCL19 and CCR7-CCL21 in the Pathophysiology of the Immuno-Inflammatory Response in Dry Eye Disease. Ocul Immunol Inflamm 2021, 29, 266–277. [Google Scholar] [CrossRef]
- Kodati, S.; Chauhan, S.K.; Chen, Y.; Dohlman, T.H.; Karimian, P.; Saban, D.; Dana, R. CCR7 Is Critical for the Induction and Maintenance of Th17 Immunity in Dry Eye Disease. Invest Ophthalmol Vis Sci 2014, 55, 5871–5877. [Google Scholar] [CrossRef]
- Hamrah, P.; Huq, S.O.; Liu, Y.; Zhang, Q.; Dana, M.R. Corneal Immunity Is Mediated by Heterogeneous Population of Antigen-Presenting Cells. J Leukoc Biol 2003, 74, 172–178. [Google Scholar] [CrossRef]
- Lee, H.S.; Amouzegar, A.; Dana, R. Kinetics of Corneal Antigen Presenting Cells in Experimental Dry Eye Disease. BMJ Open Ophthalmol 2017, 1, e000078. [Google Scholar] [CrossRef]
- Chen, Y.; Chauhan, S.K.; Soo Lee, H.; Saban, D.R.; Dana, R. Chronic Dry Eye Disease Is Principally Mediated by Effector Memory Th17 Cells. Mucosal Immunology 2014 7:1 2013, 7, 38–45. [Google Scholar] [CrossRef]
- Annan, J. El; Chauhan, S.K.; Ecoiffier, T.; Zhang, Q.; Saban, D.R.; Dana, R. Characterization of Effector T Cells in Dry Eye Disease. Invest Ophthalmol Vis Sci 2009, 50, 3802. [Google Scholar] [CrossRef]
- Pflugfelder, S.C.; Corrales, R.M.; de Paiva, C.S. T Helper Cytokines in Dry Eye Disease. Exp Eye Res 2013, 117, 10.1016. [Google Scholar] [CrossRef]
- Jackson, D.C.; Zeng, W.; Wong, C.Y.; Mifsud, E.J.; Williamson, N.A.; Ang, C.S.; Vingrys, A.J.; Downie, L.E. Tear Interferon-Gamma as a Biomarker for Evaporative Dry Eye Disease. Invest Ophthalmol Vis Sci 2016, 57, 4824–4830. [Google Scholar] [CrossRef] [PubMed]
- Ko, B.Y.; Xiao, Y.; Barbosa, F.L.; de Paiva, C.S.; Pflugfelder, S.C. Goblet Cell Loss Abrogates Ocular Surface Immune Tolerance. JCI Insight 2018, 3, e98222. [Google Scholar] [CrossRef] [PubMed]
- Mills, K.H.G. Induction, Function and Regulation of IL-17-Producing T Cells. Eur J Immunol 2008, 38, 2636–2649. [Google Scholar] [CrossRef] [PubMed]
- Chu, L.; Wang, C.; Zhou, H. Inflammation Mechanism and Anti-Inflammatory Therapy of Dry Eye. Front Med (Lausanne) 2024, 11, 1307682. [Google Scholar] [CrossRef]
- Zheng, X.; de Paiva, C.S.; Li, D.Q.; Farley, W.J.; Pflugfelder, S.C. Desiccating Stress Promotion of Th17 Differentiation by Ocular Surface Tissues through a Dendritic Cell-Mediated Pathway. Invest Ophthalmol Vis Sci 2010, 51, 3083. [Google Scholar] [CrossRef]
- Dohlman, T.H.; Chauhan, S.K.; Kodati, S.; Hua, J.; Chen, Y.; Omoto, M.; Sadrai, Z.; Dana, R. The CCR6/CCL20 Axis Mediates Th17 Cell Migration to the Ocular Surface in Dry Eye Disease. Invest Ophthalmol Vis Sci 2013, 54, 4081. [Google Scholar] [CrossRef]
- Tan, X.; Sun, S.; Liu, Y.; Zhu, T.; Wang, K.; Ren, T.; Wu, Z.; Xu, H.; Zhu, L. Analysis of Th17-Associated Cytokines in Tears of Patients with Dry Eye Syndrome. Eye (Basingstoke) 2014, 28, 608–613. [Google Scholar] [CrossRef]
- Hong, Q.; Chen, Y.; Inomata, T.; Liu, R.; Dana, R. Potential Role of IL-10-Producing Th17 Cells in Pathogenesis of Dry Eye Disease. Invest Ophthalmol Vis Sci 2017, 58, 1022–1022. [Google Scholar]
- Chen, Y.; Chauhan, S.K.; Shao, C.; Omoto, M.; Inomata, T.; Dana, R. Interferon-γ-Expressing Th17 Cells Are Required for Development of Severe Ocular Surface Autoimmunity. J Immunol 2017, 199, 1163. [Google Scholar] [CrossRef] [PubMed]
- Dohlman, T.H.; Ding, J.; Dana, R.; Chauhan, S.K. T Cell–Derived Granulocyte-Macrophage Colony-Stimulating Factor Contributes to Dry Eye Disease Pathogenesis by Promoting CD11b+ Myeloid Cell Maturation and Migration. Invest Ophthalmol Vis Sci 2017, 58, 1330. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gong, J.; Yang, Q.; Wang, L.; Jian, Y.; Wang, P. Interleukin-17 Receptor E and C-C Motif Chemokine Receptor 10 Identify Heterogeneous T Helper 17 Subsets in a Mouse Dry Eye Disease Model. American Journal of Pathology 2022, 192, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Yazdanpanah, G.; Ratnapriya, R.; Borcherding, N.; de Paiva, C.S.; Li, D.Q.; Guimaraes de Souza, R.; Yu, Z.; Pflugfelder, S.C. IL-17 Producing Lymphocytes Cause Dry Eye and Corneal Disease With Aging in RXRα Mutant Mouse. Front Med (Lausanne) 2022, 9, 849990. [Google Scholar] [CrossRef]
- Chen, Y.; Chauhan, S.K.; Soo Lee, H.; Saban, D.R.; Dana, R. Chronic Dry Eye Disease Is Principally Mediated by Effector Memory Th17 Cells. Mucosal Immunol 2013, 7, 38. [Google Scholar] [CrossRef]
- Chen, Y.; Chauhan, S.K.; Tan, X.; Dana, R. Interleukin-7 and -15 Maintain Pathogenic Memory Th17 Cells in Autoimmunity. J Autoimmun 2016, 77, 96. [Google Scholar] [CrossRef]
- Chen, Y.; Shao, C.; Fan, N.W.; Nakao, T.; Amouzegar, A.; Chauhan, S.K.; Dana, R. The Functions of IL-23 and IL-2 on Driving Autoimmune Effector T-Helper 17 Cells into the Memory Pool in Dry Eye Disease. Mucosal Immunol 2020, 14, 177. [Google Scholar] [CrossRef]
- Foulsham, W.; Mittal, S.K.; Taketani, Y.; Chen, Y.; Nakao, T.; Chauhan, S.K.; Dana, R. Aged Mice Exhibit Severe Exacerbations of Dry Eye Disease with an Amplified Memory Th17 Cell Response. Am J Pathol 2020, 190, 1474. [Google Scholar] [CrossRef] [PubMed]
- Shevach, E.M.; Thornton, A.M. TTregs, PTregs, and ITregs: Similarities and Differences. Immunol Rev 2014, 259, 88. [Google Scholar] [CrossRef]
- Sakaguchi, S.; Ono, M.; Setoguchi, R.; Yagi, H.; Hori, S.; Fehervari, Z.; Shimizu, J.; Takahashi, T.; Nomura, T. Foxp3+CD25+CD4+ Natural Regulatory T Cells in Dominant Self-Tolerance and Autoimmune Disease. Immunol Rev 2006, 212, 8–27. [Google Scholar] [CrossRef] [PubMed]
- Niederkorn, J.Y.; Stern, M.E.; Pflugfelder, S.C.; Cintia, ‡; De Paiva, S.; Corrales, R.M.; Gao, J.; Siemasko, K. Desiccating Stress Induces T Cell-Mediated Sjögren’s Syndrome-Like Lacrimal Keratoconjunctivitis. The Journal of Immunology 2006, 176, 3950–3957. [Google Scholar] [CrossRef] [PubMed]
- Siemasko, K.F.; Gao, J.; Calder, V.L.; Hanna, R.; Calonge, M.; Pflugfelder, S.C.; Niederkorn, J.Y.; Stern, M.E. In Vitro Expanded CD4+CD25+Foxp3+ Regulatory T Cells Maintain a Normal Phenotype and Suppress Immune-Mediated Ocular Surface Inflammation. Invest Ophthalmol Vis Sci 2008, 49, 5434–5440. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, S.K.; El Annan, J.; Ecoiffier, T.; Goyal, S.; Zhang, Q.; Saban, D.R.; Dana, R. Autoimmunity in Dry Eye Is Due to Resistance of Th17 to Treg Suppression. J Immunol 2009, 182, 1247. [Google Scholar] [CrossRef]
- Fan, N.W.; Dohlman, T.H.; Foulsham, W.; McSoley, M.; Singh, R.B.; Chen, Y.; Dana, R. The Role of Th17 Immunity in Chronic Ocular Surface Disorders. Ocul Surf 2020, 19, 157. [Google Scholar] [CrossRef]
- Kuklinski, E.J.; Yu, Y.; Ying, G.S.; Asbell, P.A. Association of Ocular Surface Immune Cells With Dry Eye Signs and Symptoms in the Dry Eye Assessment and Management (DREAM) Study. Invest Ophthalmol Vis Sci 2023, 64, 7. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Li, W.; McDermott, M.; Son, G.-Y.; Maiti, G.; Zhou, F.; Tao, A.Y.; Raphael, D.; Moreira, A.L.; Shen, B.; et al. IFN-γ-Producing Th1 Cells and Dysfunctional Regulatory T Cells Contribute to the Pathogenesis of Sjögren’s Disease. Sci Transl Med 2024, 16, eado4856. [Google Scholar] [CrossRef]
- de Souza, R.G.; de Paiva, C.S.; Alves, M.R. Age-Related Autoimmune Changes in Lacrimal Glands. Immune Netw 2019, 19, e3. [Google Scholar] [CrossRef]
- Coursey, T.G.; Bian, F.; Zaheer, M.; Pflugfelder, S.C.; Volpe, E.A.; De Paiva, C.S. Age-Related Spontaneous Lacrimal Keratoconjunctivitis Is Accompanied by Dysfunctional T Regulatory Cells. Mucosal Immunol 2016, 10, 743. [Google Scholar] [CrossRef] [PubMed]
- Huang, D.; Li, Z. Multidimensional Immunotherapy for Dry Eye Disease: Current Status and Future Directions. Frontiers in Ophthalmology 2024, 4, 1449283. [Google Scholar] [CrossRef]
- Rong, H.; Yang, H.; Liu, Q.; Zhang, H.; Wang, S. Substance P and Neurokinin 1 Receptor Boost the Pathogenicity of Granulocyte-Macrophage Colony-Stimulating Factor-Producing T Helper Cells in Dry Eye Disease. Scand J Immunol 2025, 101, e13434. [Google Scholar] [CrossRef]
- Lee, S.J.; Im, S.T.; Wu, J.; Cho, C.S.; Jo, D.H.; Chen, Y.; Dana, R.; Kim, J.H.; Lee, S.M. Corneal Lymphangiogenesis in Dry Eye Disease Is Regulated by Substance P/Neurokinin-1 Receptor System through Controlling Expression of Vascular Endothelial Growth Factor Receptor 3. Ocul Surf 2021, 22, 72–79. [Google Scholar] [CrossRef]
- Singh, R.B.; Naderi, A.; Cho, W.; Ortiz, G.; Musayeva, A.; Dohlman, T.H.; Chen, Y.; Ferrari, G.; Dana, R. Modulating the Tachykinin: Role of Substance P and Neurokinin Receptor Expression in Ocular Surface Disorders. Ocul Surf 2022, 25, 142. [Google Scholar] [CrossRef]
- Taketani, Y.; Marmalidou, A.; Dohlman, T.H.; Singh, R.B.; Amouzegar, A.; Chauhan, S.K.; Chen, Y.; Dana, R. Restoration of Regulatory T-Cell Function in Dry Eye Disease by Antagonizing Substance P/Neurokinin-1 Receptor. Am J Pathol 2020, 190, 1859. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Naderi, A.; Kahale, F.; Ortiz, G.; Forouzanfar, K.; Chen, Y.; Dana, R. Substance P Regulates Memory Th17 Cell Generation and Maintenance in Chronic Dry Eye Disease. J Leukoc Biol 2024, 116, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Zhu, X.; Ye, H.; Yang, K.; Zhou, X.; Hong, J. Tear Neuropeptides Are Associated with Clinical Symptoms and Signs of Dry Eye Patients. Ann Med 2025, 57, 2451194. [Google Scholar] [CrossRef] [PubMed]
- Yoon, H.-J.; Jang, W.-H.; An, S.; Ji, Y.S.; Yoon, K.C. Tear Neuromediators in Subjects with and without Dry Eye According to Ocular Sensitivity. Chonnam Med J 2022, 58, 37. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, H.; Zhu, X.; Ye, H.; Yang, K.; Zhou, X.; Hong, J. Tear Neuropeptides Are Associated with Clinical Symptoms and Signs of Dry Eye Patients. Ann Med 2025, 57, 2451194. [Google Scholar] [CrossRef]
- Lambiase, A.; Micera, A.; Sacchetti, M.; Cortes, M.; Mantelli, F.; Bonini, S. Alterations of Tear Neuromediators in Dry Eye Disease. Archives of Ophthalmology 2011, 129, 981–986. [Google Scholar] [CrossRef]
- Blanco-Vázquez, M.; Vázquez, A.; Fernández, I.; Novo-Diez, A.; Martínez-Plaza, E.; García-Vázquez, C.; González-García, M.J.; Sobas, E.M.; Calonge, M.; Enríquez-de-Salamanca, A. Inflammation-Related Molecules in Tears of Patients with Chronic Ocular Pain and Dry Eye Disease. Exp Eye Res 2022, 219. [Google Scholar] [CrossRef] [PubMed]
- Guzmán, M.; Miglio, M.; Keitelman, I.; Shiromizu, C.M.; Sabbione, F.; Fuentes, F.; Trevani, A.S.; Giordano, M.N.; Galletti, J.G. Transient Tear Hyperosmolarity Disrupts the Neuroimmune Homeostasis of the Ocular Surface and Facilitates Dry Eye Onset. Immunology 2020, 161, 148–161. [Google Scholar] [CrossRef]
- Masuoka, T.; Yamashita, Y.; Nakano, K.; Takechi, K.; Niimura, T.; Tawa, M.; He, Q.; Ishizawa, K.; Ishibashi, T. Chronic Tear Deficiency Sensitizes Transient Receptor Potential Vanilloid 1-Mediated Responses in Corneal Sensory Nerves. Front Cell Neurosci 2020, 14, 598678. [Google Scholar] [CrossRef]
- Sheppard, J.; Shen Lee, B.; Periman, L.M. Dry Eye Disease: Identification and Therapeutic Strategies for Primary Care Clinicians and Clinical Specialists. Ann Med 2022, 55, 241. [Google Scholar] [CrossRef]
- Jones, L.; Downie, L.E.; Korb, D.; Benitez-del-Castillo, J.M.; Dana, R.; Deng, S.X.; Dong, P.N.; Geerling, G.; Hida, R.Y.; Liu, Y.; et al. TFOS DEWS II Management and Therapy Report. Ocul Surf 2017, 15, 575–628. [Google Scholar] [CrossRef]
- Yang, C.; Gao, Q.; Liu, J.; Wu, Y.; Hou, X.; Sun, L.; Zhang, X.; Lu, Y.; Yang, Y. M2 Macrophage-Derived Extracellular Vesicles Ameliorate Benzalkonium Chloride-Induced Dry Eye. Exp Eye Res 2024, 247. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, Y.; Zhang, Y.; Chu, B.; Yang, K.; Hong, J.; He, Y. Targeting A7 Nicotinic Acetylcholine Receptor for Modulating the Neuroinflammation of Dry Eye Disease Via Macrophages. Invest Ophthalmol Vis Sci 2025, 66, 13. [Google Scholar] [CrossRef]
- Ma, B.; Zhou, Y.; Liu, R.; Zhang, K.; Yang, T.; Hu, C.; Gao, Y.; Lan, Q.; Liu, Y.; Yang, X.; et al. Pigment Epithelium-Derived Factor (PEDF) Plays Anti-Inflammatory Roles in the Pathogenesis of Dry Eye Disease. Ocular Surface 2021, 20, 70–85. [Google Scholar] [CrossRef]
- Mu, J.; Ding, X.; Song, Y.; Mi, B.; Fang, X.; Chen, B.; Yao, B.; Sun, X.; Yuan, X.; Guo, S.; et al. ROS-Responsive Microneedle Patches Enable Peri-Lacrimal Gland Therapeutic Administration for Long-Acting Therapy of Sjögren’s Syndrome-Related Dry Eye. Advanced Science 2025, 12, 2409562. [Google Scholar] [CrossRef]
- Sadrai, Z.; Stevenson, W.; Okanobo, A.; Chen, Y.; Dohlman, T.H.; Hua, J.; Amparo, F.; Chauhan, S.K.; Dana, R. PDE4 Inhibition Suppresses IL-17–Associated Immunity in Dry Eye Disease. Invest Ophthalmol Vis Sci 2012, 53, 3584. [Google Scholar] [CrossRef]
- Grosskreutz, C.L.; Hockey, H.U.; Serra, D.; Dryja, T.P. Dry Eye Signs and Symptoms Persist during Systemic Neutralization of IL-1b by Canakinumab or IL-17A by Secukinumab. Cornea 2015, 34, 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Study Details | A Study to Evaluate the Safety, Tolerability, and Efficacy of A197 in Subjects With Dry Eye Disease | ClinicalTrials. Available online: https://clinicaltrials.gov/study/NCT05238597 (accessed on 2 July 2025).
- Ratay, M.L.; Glowacki, A.J.; Balmert, S.C.; Acharya, A.P.; Polat, J.; Andrews, L.P.; Fedorchak, M. V.; Schuman, J.S.; Vignali, D.A.A.; Little, S.R. Treg-Recruiting Microspheres Prevent Inflammation in a Murine Model of Dry Eye Disease. J Control Release 2017, 258, 208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Zou, Y.; Li, H.; Li, D.; Liu, Y.; Gong, B.; Yu, M. NK1R Antagonist, CP-99,994 Ameliorates Dry Eye Disease via Inhibiting the Plk1-Cdc25c-Cdk1 Axis. J Biochem Mol Toxicol 2025, 39. [Google Scholar] [CrossRef] [PubMed]
- Surico, P.L.; Barone, V.; Singh, R.B.; Coassin, M.; Blanco, T.; Dohlman, T.H.; Basu, S.; Chauhan, S.K.; Dana, R.; Zazzo, A. Di Potential Applications of Mesenchymal Stem Cells in Ocular Surface Immune-Mediated Disorders. Surv Ophthalmol 2025, 70, 467–479. [Google Scholar] [CrossRef]
- Joung Lee, M.; Young Ko, A.; Hwa Ko, J.; Ju Lee, H.; Kum Kim, M.; Ryang Wee, W.; In Khwarg, S.; Youn Oh, J. Mesenchymal Stem/Stromal Cells Protect the Ocular Surface by Suppressing Inflammation in an Experimental Dry Eye. 2014. [CrossRef]
- Aluri, H.S.; Samizadeh, M.; Edman, M.C.; Hawley, D.R.; Armaos, H.L.; Janga, S.R.; Meng, Z.; Sendra, V.G.; Hamrah, P.; Kublin, C.L.; et al. Delivery of Bone Marrow-Derived Mesenchymal Stem Cells Improves Tear Production in a Mouse Model of Sjögren’s Syndrome. Stem Cells Int 2017, 2017, 3134543. [Google Scholar] [CrossRef]
- Wang, L.; Wang, X.; Chen, Q.; Wei, Z.; Xu, X.; Han, D.; Zhang, Y.; Chen, Z.; Liang, Q. MicroRNAs of Extracellular Vesicles Derived from Mesenchymal Stromal Cells Alleviate Inflammation in Dry Eye Disease by Targeting the IRAK1/TAB2/NF-ΚB Pathway. Ocul Surf 2023, 28, 131–140. [Google Scholar] [CrossRef]
- Wang, G.; Li, H.; Long, H.; Gong, X.; Hu, S.; Gong, C. Exosomes Derived from Mouse Adipose-Derived Mesenchymal Stem Cells Alleviate Benzalkonium Chloride-Induced Mouse Dry Eye Model via Inhibiting NLRP3 Inflammasome. Ophthalmic Res 2022, 65, 40–51. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Gu, C.; Yang, Y.; He, T.; Zhang, Q. Exosomal MiR-146a Derived from Human Umbilical Cord Mesenchymal Stem Cells Alleviates Inflammation and Apoptosis in Dry Eye Disease by Targeting SQSTM1. Exp Eye Res 2025, 110490. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
The components of the tear film.
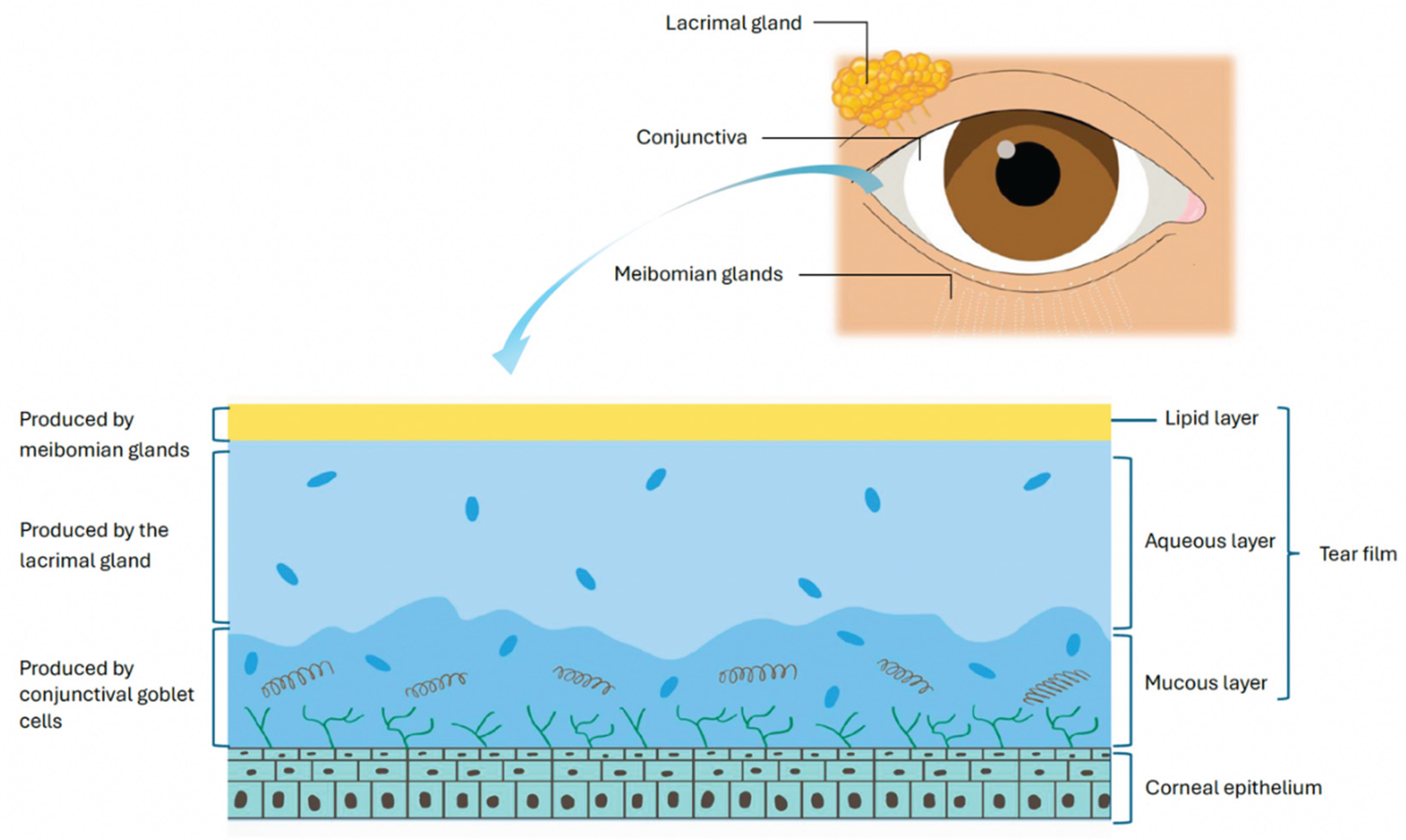
Figure 2.
The vicious cycle of DED.
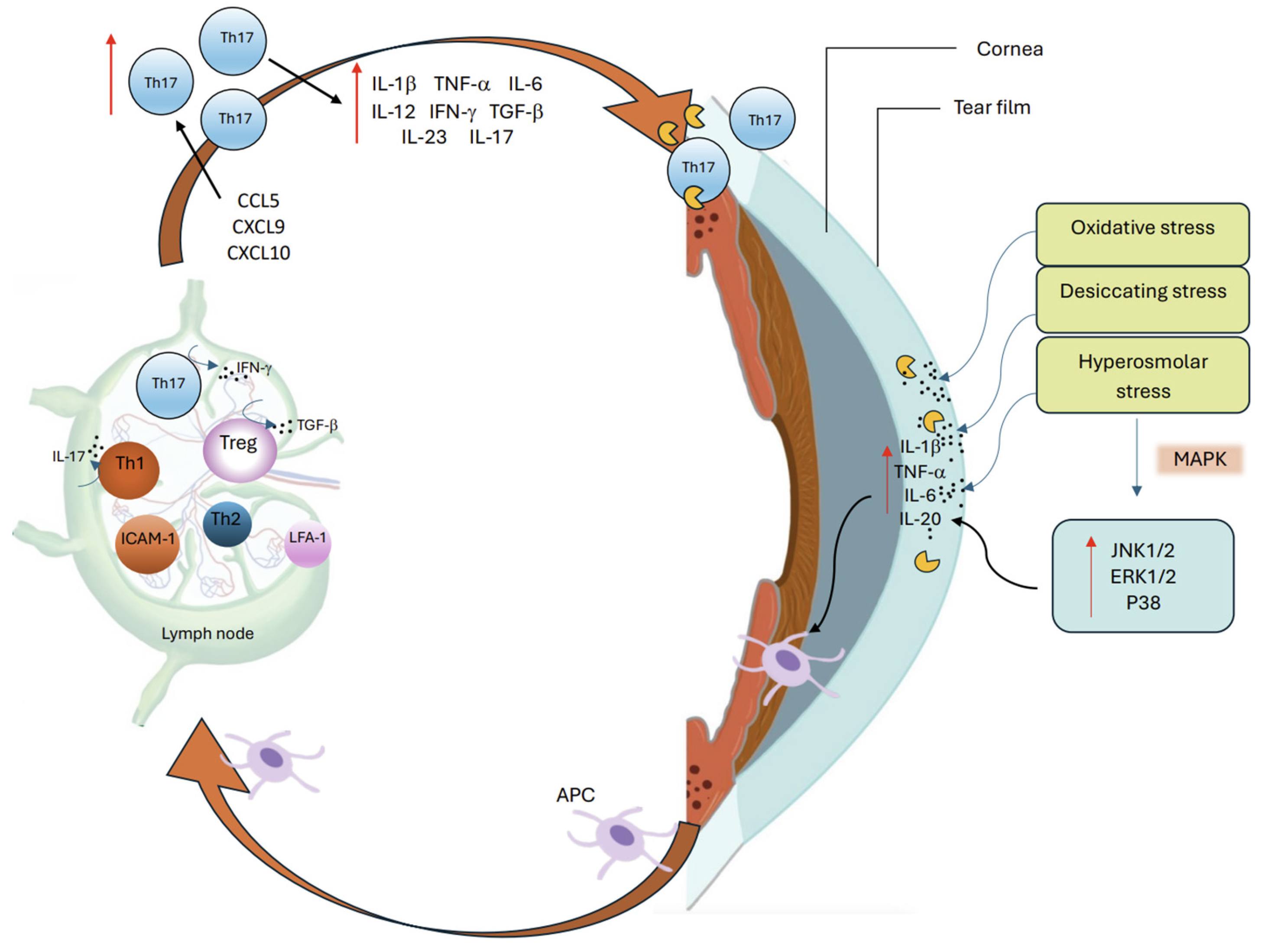

Table 1.
Up and downregulated biomarkers in DED and their role in disease activity.
| Biomarker | Response in DED | Role | Ref. |
|---|---|---|---|
| ACPA | ↑ | Generated during neutrophil NETosis, induces OS inflammation in murine models | [36] |
| ADAM17 | ↑ | Contributes to ocular pain and epithelial barrier disruption | [38] |
| CCL2 | ↑ | Drives basal epithelial cells in acting as ‘non-professional APCs’ in further activating immune response | [31] |
| CCL20 | ↑ | Aids migration of Th17 cells back to OS, specifically the conjunctiva | [55] |
| CCL5 | ↑ | Promotes T cell recruitment | [6,13] |
| CCR5 | ↑ | Facilitates Th1 targeted migration from lymph nodes back to inflamed OS | [48] |
| CCR6 | ↑ | Aids migration of Th17 cells back to OS, specifically the conjunctiva | [55] |
| CGRP | ↑ and ↓ | Exhibits both an immunosuppressive and proinflammatory role depending on the microenvironment; Inhibits APCs through suppression of mast cell-derived TNF; Upregulated in response to inflammatory stimulation or nerve sensitization. | [76,82,83,86] |
| CTLA-4 | ↑ | Impairs suppressive capacity against effector T cells | [79,80] |
| CXCL1 | ↑ | Activates TRPV1 and ADAM17 which contributes to ocular pain and epithelial barrier disruption | [38] |
| CXCL10 | ↑ | Recruits Th1 cells to the OS through CXCR3 signaling, amplifying local inflammation | [6,13] |
| CXCL9 | ↑ | Activates T cells and sustains chronic inflammatory responses via CXCR3 | [6,13] |
| CXCR3 | ↑ | Facilitates migration of DED-primed Th1 cells from lymph nodes back to inflamed OS | [48] |
| GM-CSF | ↑ | Stimulates monocytic cells to produce pro-inflammatory cytokines such as IL-1β, IL-6, and IL-23 with IL-6 and IL-23 further perpetuating Th17 differentiation | [59] |
| HMGB1 | ↑ | Activates TLR pathways and induces proinflammatory cytokine and MMP-9 release | [33] |
| HSP-60 | ↑ | Activates TLR pathways, leading to cytokine release and immune cell recruitment | [33] |
|
IFN-γ |
↑ in early DED ↓ in later disease progression |
Induces epithelial damage and disrupts homeostasis of OS NK activation promotes IFN- γ-mediated inflammation and drives APC maturation which primes adaptive immune response; Induces GC loss and reduces mucin production |
[40,47,50] |
| IL-1 | ↑ | Allows Th17 cells to undergo further differentiation at the conjunctiva. | [55] |
| IL-10 | ↓ | Exacerbates goblet cell loss and Th17 mediated pathology contribute to impaired suppressive capacity against effector T cells. | [39,79,80] |
| IL-12 | ↑ | Leads to further Th1 polarization | [51] |
| IL-15 | ↑ | Maintains Th17 memory cells and promotes continued survival | [63] |
| IL-17 | ↑ | Disrupts corneal epithelium barrier integrity, stimulates MMP production and promotes inflammation and apoptosis, | [49,56] |
| IL-17C | ↑ | Enhances JNK and p38 MAPK signaling though IL-17C/IL17RE interaction therefore reinforces and perpetuates Th17 phenotype | [60] |
| IL-1β | ↑ | Promotes epithelial damage, upregulates proinflammatory mediators, and enhances immune cell activation | [25,27,29] |
| IL-2 | - | Inhibits differentiation of Th17 effector cells into memory cells | [64] |
| IL-20 | ↑ | Promotes macrophage recruitment and increases inflammatory signaling in OS | [30] |
| IL-23 | ↑ | Allows Th17 cells to undergo further differentiation at the conjunctiva and promotes transition into memory cells | [6,13,41,55,64] |
| IL-6 | ↑ | Activates DCs and enhances Th17 responses; Initiates Th-17 cell differentiation via STAT3 signaling pathways; Exhibits inhibitory effect on Treg differentiation. | [6,13,41,52,53,71] |
| IL-7 | ↑ | Helps maintain Th17 memory cells and promotes continued survival | [63] |
| IL17A | ↑ | Promotes neutrophil recruitment, epithelial barrier disruption, and proinflammatory cytokine production at OS | [61,70] |
| IL17F | ↑ | Stimulates epithelial cells and immune cells to release inflammatory mediators and chemokines | [61] |
| MMP-9 | ↑ | Degrades epithelial basement membrane components and disrupts tight junction proteins | [25,27,29] |
| NF-κB | ↑ | Drives early upregulation of proinflammatory cytokines, promoting immune cell activation | [25,30] |
| NFAT5 | ↑ | Promotes early cytokine upregulation and immune cell activation | [25,30] |
| PD-1 | ↑ | Impairs suppressive capacity against effector T cells | [79,80] |
| RORγt | ↑ | Regulates and promotes Th-17 cell differentiation | [54] |
| RXRa | ↓ | Exacerbates goblet cell loss and Th17 mediated pathology. | [39] |
| TGF-β | ↑ | Induces Th17 cells and contributes to impaired suppressive capacity against effector T cells. | [6,13,79,80] |
| TLR4 | ITLR mRNA ↑ TLR protein levels ↓ |
Recognizes DAMPs (like HMGB1) and microbial products, activating NF-κB and driving cytokine/chemokine release | [21] |
| TLR9 | TLR9 mRNA ↓ TLR9 protein ↓ |
Impairs local immune regulatory function at OS | [32] |
| TNF-β | ↑ | Initiates Th-17 cell differentiation via STAT3 signaling pathways. | [52,53] |
| TNF-α | ↑ | upregulates proinflammatory cytokines, disrupting epithelial barrier integrity, and amplifying immune cell infiltration | [25,27,29] |
| TRPV1 | ↑ | Contributes to ocular pain and epithelial barrier disruption | [38] |
| VEGF, VEGF-D, VEGFR-3 | ↑ | Promotes lymphatic vessel growth and facilitates APC trafficking to draining lymph nodes. | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



