
Submitted:
25 August 2025
Posted:
27 August 2025
You are already at the latest version
Abstract
Introduction: Langerhans cell histiocytosis (LCH) is a rare clonal myeloid disorder characterized by tissue infiltration with pathological antigen-presenting cells morphologically and phenotypically similar to Langerhans cells. Activating mutations in the MAPK/ERK signaling pathway play a key role in the pathogenesis of LCH. The aim of this study was to characterize the morphological and molecular features of the neoplastic process. Materials and Methods: This study presents a clinical case of a 34-year-old patient with a rare mixed form of LCH combined with Erdheim-Chester disease (ECD), involving multiple organs including kidneys, skin, bones, ENT organs, eyes, and the central nervous system. The patient also had diabetes insipidus, chronic external otitis, and presumptive hereditary forms of anemia and hemochromatosis. A comprehensive histopathological evaluation was performed, including histochemical, immunohistochemical, and immunofluorescent analyses, alongside molecular genetic testing using whole exome sequencing (WES) of blood and skin samples. The patient demonstrated a partial clinical and laboratory response to targeted therapy with trametinib and desmopressin. Results: Comparison of WES data from blood and skin revealed 71,953 shared variants consistent with likely germline changes, as well as 3,081 unique somatic variants in skin and 2,633 unique variants in blood. Low-level mosaic BRAF V600E mutation (VAF = 1%) was detected in the skin, alongside pathogenic germline variants in the STEAP3 (c.523-2A>T, NM_182915.3) and SLC40A1 (c.332T>A, p.Met111Lys, NM_014585.6) genes. Immunofluorescence analysis of the trephine biopsy revealed diverse cellular populations, including sparse tryptase-positive mast cells, which showed minimal contact with each other or other labeled cells. The populations of Vimentin+CD11b–CD34+ and Vimentin+CD11b+CD34– cells (excluding endothelial cells) accounted for median values of 0.68% and 7.78%, respectively, of the total Vimentin+ cells. Regarding CD68 and S100 expression, CD68+ cells predominated (median = 55.61%), S100+ cells constituted a median of 43.02%, whereas CD68+S100+ cells were the rarest (median = 2.174%), reflecting the transition of Langerhans cells to LCH cells, encompassing up to 4.81% of the cellular population. This hybrid microenvironment paralleled the patient’s systemic inflammatory activity (elevated acute-phase reactants), multi-organ involvement, and renal interstitial fibrosis/tubular atrophy with reduced kidney function, without serological or histological evidence of vasculitis. Conclusion: This case illustrates the histological heterogeneity and hybrid pathology of mixed histiocytosis, where canonical dendritic LCH cells coexist with macrophage-rich ECD-like stroma. An integrated approach involving a multidisciplinary clinical team, thorough molecular genetic testing, and the application of targeted therapy is essential for improving prognosis and quality of life in young patients.
Keywords:
histiocytosis
; BRAF
; Langerhans cells
; anemia
; hemochromatosis
Introduction
Langerhans cell histiocytosis (LCH) belongs to the group of histiocytic neoplasms and is characterized by clonal proliferation of antigen-presenting cells exhibiting the Langerhans cell (LC) phenotype. Lesions typically consist of pathological LC with Birbeck granules, T-lymphocytes, eosinophils, and macrophages. Pathological LC express CD1a++ and CD207+++ similarly to normal epidermal LC but are less differentiated [1]. Immunohistochemical (IHC) analysis of lesional tissue reveals expression of CD1a (membranous), S100 (nuclear and cytoplasmic), CD207/langerin (granular cytoplasmic) and CD68 (Golgi dot-like). The Ki-67 proliferation index usually ranges below 10% [2]. Absence of expression for CD1c, B- and T-cell markers, CD23, and CD30 is expected.
Macrophages and dendritic cells (including LC) may originate from CD34+ hematopoietic stem cells in bone marrow (including transplanted marrow) or circulating monocytes. Initial histiocytosis classification relied on Langerhans (CD1a, CD207) vs. macrophage (CD68, CD163) markers, but hybrid forms have since been described (e.g., combined LCH, Erdheim-Chester disease (ECD), and Rosai-Dorfman disease) often sharing a single mutation, suggesting common origin [3]. Additionally, single LCH lesions can contain heterogeneous cell populations [4]. It is hypothesized that LC derive from oligopotent BM progenitors, recruited to inflamed tissues, where they differentiate into pathological cells [5]. A recent study suggests DC3/monocytes as LCH precursors, based on iPSC models with BRAF V600E mutation that differentiated into CD14low/CD1c+ precursors, then into CD1a+/CD207+ LCH-like cells [6].
LCH incidence in children under 15 ranges from 2.6 to 8.9 per million per year (median diagnosis age: 3 years [7]; M:F ratio = 1.2:1 [1]), while adult incidence is estimated at 1–2 cases per million annually [8]. Pediatric LCH is often systemic with fever, hepatosplenomegaly, cytopenia, and sometimes deafness or chronic otorrhea. Adults show more pulmonary involvement (dyspnea, cough), tooth loss, and diabetes insipidus in both groups [3]. Adolescents (14–17 years) present with adult-like features: more frequent pituitary and pulmonary involvement, less cutaneous and hematologic manifestations [9].
Although hereditary etiology is unconfirmed, a genome-wide association study of familial LCH identified variant rs12438941 in SMAD6 as significantly associated with disease risk. SMAD6 inhibits BMP/TGF-β pathways involved in LC differentiation; thus, polymorphisms may dysregulate myeloid differentiation [10].
Modern molecular research has shown that activating mutations in the MAPK/ERK pathway are central to LCH pathogenesis. The most frequent and clinically relevant mutation is BRAF V600E (exon 15), which constitutively activates the BRAF kinase domain independently of RAS signaling [11,12]. This mutation is found in ~50–65% of LCH cases and is classified as oncogenic (Level 2; FDA Level 3), responsive to selective BRAF inhibitors such as vemurafenib and dabrafenib [13]. Notably, BRAF V600E is detectable in trephine biopsies and myeloid precursors from blood but absent in peripheral mononuclear cells, supporting its somatic nature [1]. BRAF V600E in early myeloid cells (CD11c+, CD14+, CD34+) correlates with multisystem involvement (liver, spleen, bone marrow) and higher relapse risk, while its restriction to differentiated cells (CD207+ only) is associated with localized disease and better prognosis [14]. MEK inhibitors such as cobimetinib and trametinib are effective in LCH with MAPK-pathway mutations (BRAF, MAP2K1, NRAS) and recommended as monotherapy by NCCN. MAP2K1 mutations (10%-20%) activate ERK1/2 and are second most frequent, while NRAS mutations are rarer [15,16,17]. In a cohort of 99 Russian LCH patients, 44% were BRAF-positive [18]. Genes analyzed included BRAF, ARAF, MAP2K1, MAP3K1, NRAS, PIK3CA, KIT, ERBB3, KRAS, PIK3CD, and HRAS.
Unlike early phenotype-based classification, the current system (Goyal et al., 2022) is based on clinical-radiological features and organ involvement, enhancing clinical utility. Four LCH subtypes are defined: unifocal (single-organ), unisystem pulmonary (lung-only, often smoking-related), unisystem multifocal (>1 site in a single organ), and multisystem (>2 organs/systems).
LCH diagnosis is established via clinical presentation, histopathological evaluation, computed tomography (CT)/magnetic resonance imaging (MRI) (for bone, lung, central nervous system (CNS) assessment), and molecular-genetic testing (Table 1) [20].
Treatment depends on disease extent, organ involvement, and genetic profile [20]. Localized (single-system) forms are managed surgically or with radiotherapy. Systemic forms are treated with targeted BRAF and MEK inhibitors, in combination with vinblastine and prednisone. In cases involving risk organs (liver, spleen, bone marrow), cladribine or cytarabine is used. Evidence suggests that MAPK inhibitors effectively reduce mature CD14+ cell populations but do not affect myeloid progenitors [6]. Hormone replacement therapy is prescribed in cases of pituitary insufficiency; pulmonary rehabilitation is indicated for lung fibrosis. The 5-year survival rate for localized forms exceeds 90%. In multisystem forms with risk organ involvement, prognosis depends on therapy response and relapse frequency.
Case Report
Patient N., a 34-year-old male, had been under observation by specialists of various disciplines since 2019 due to a progressive array of symptoms.
Anamnesis vitae: Full-term birth from an uncomplicated pregnancy. Normal growth and developmental milestones. Holds a university degree; currently unemployed. From 1986 to 1998, resided in an area classified as a zone of preferential socio-economic status due to radioactive contamination by cesium-137 (1–5 Ci/m2) following the Chernobyl disaster. Smoked up to 2 cigarettes per day for 10 years; quit in December 2024. Denies history of tuberculosis, sexually transmitted infections, or viral hepatitis. Vaccinated per national immunization schedule. Denies prior trauma, surgeries, or excessive sun exposure. Medical history includes varicella and upper respiratory infections. No known hereditary or allergic conditions.
Anamnesis morbi: The patient considers himself ill since 2019, when he first experienced headaches and dizziness. He consulted a local general practitioner and was diagnosed with arterial hypertension (maximum blood pressure 160/120 mmHg). Prescribed perindopril 5 mg/day. Simultaneously, he noted severe thirst but did not undergo further evaluation.
By 2020, the patient developed ear pain and significant discharge of white epidermoid material from the external auditory canals. Chronic bilateral otitis media was diagnosed. Topical antibacterial treatment yielded partial symptom relief, reducing pain but not otorrhea. Otolaryngological cleaning of the ear canals was performed every 3–4 months.
In spring 2022, the patient began experiencing persistent, intense headaches (mainly in the right parietal region) with a shooting character. Treatment with B vitamins, nootropics, and nonsteroidal anti-inflammatory drugs (NSAIDs) produced only temporary relief. Around the same time, he reported pain in the right shoulder and elbow joints. Rheumatoid factor was within normal limits. A rheumatologist prescribed chondroprotectors, muscle relaxants, and a 3-month course of therapy with mild improvement.
By summer 2022, the patient noticed ulcerations on the oral mucosa and erosions in the groin area. He used topical antibacterial and antifungal agents with minimal effect.
In December 2022, an endocrinologist diagnosed central diabetes insipidus. No pharmacological treatment was initiated; further investigation was recommended.
In January 2023, the patient consulted a dermatologist due to a scalp rash with dense crust formation (Figure 1). Dermatological evaluation: The patient reported itchy rashes on the scalp, groin folds, and lower legs. Status localis: On the scalp, the pathological changes included small papules and pustules on an erythematous background with numerous confluent massive serous crusts. Hair was of normal structure without pathological changes. Bilateral groin folds exhibited maceration and linear erosions. Skin of both lower legs appeared dry, tense, with scaling and excoriations. Differential diagnosis included seborrheic dermatitis, psoriasis, and dermatomycosis. Topical treatments with antibacterial, antifungal, corticosteroid, and combination therapies (ketoconazole, betamethasone dipropionate with salicylic acid, sertaconazole, dioxomethyltetrahydropyrimidine with chloramphenicol, clotrimazole + beclomethasone dipropionate + gentamicin sulfate, methylprednisolone aceponate) showed no effect. Minor improvement in the groin area was noted following treatment with 1% aqueous methylene blue solution.
In February 2023, the patient experienced a recurrence of rashes and erosions in the inguinal area and on the oral mucosa. Intramuscular injections of calcium gluconate were prescribed with minimal clinical effect.
In spring 2023, there was a recurrence of arthralgia in the elbow and shoulder joints, as well as headaches.
In April 2024, the patient reported episodes of white flashes of light in both eyes. Ophthalmologic examination confirmed bilateral optic disc edema and peripheral retinal hemorrhages.
Between May and June 2024, laboratory tests revealed anemia with hemoglobin level of 112 g/L (reference: 130–150), increased serum creatinine to 136.5 µmol/L (estimated glomerular filtration rate [eGFR] by CKD-EPI: 58 mL/min/1.73 m2), elevated C-reactive protein (CRP) to 148.21 mg/L (0–5), ferritin to 450 µg/L (20–300), and decreased serum iron to 7.1 µmol/L (11–28) (see Table 2). A single outpatient intravenous infusion of 500 mg ferric carboxymaltose was administered. Thyroid-stimulating hormone was 0.72 µIU/mL (0.35–4.96). Ultrasound examination of the kidneys revealed normal size and parenchymal thickness with parapelvic cysts. Ambulatory blood pressure monitoring (ABPM) showed average values of 140/90 mmHg. Brain MRI revealed a few small supratentorial foci in the frontal and parietal lobes, likely of vascular or residual origin; early signs of an “empty sella” (reduction in pituitary height with herniation of the suprasellar cistern into the sella turcica); a pineal cyst up to 0.5 × 0.6 cm; no cerebrospinal fluid flow disturbances.
In August 2024, the patient developed lower leg and foot edema, papular eruptions on the chest and face, and isolated small papules on the lower legs. Fungal cultures from the scalp and inguinal folds were negative. Abdominal ultrasound showed signs of hepatic steatosis, gallbladder angulation, and diffuse pancreatic heterogeneity. Esophagogastroduodenoscopy revealed erosive gastritis and superficial bulbitis, not associated with Helicobacter pylori. Otolaryngological examination: bloody discharge in the nasal passages, hyperkeratotic masses in the external auditory canals. Cultures from the external auditory canals were negative for bacterial and fungal pathogens. Diagnosis: chronic bilateral external otitis, eczematous form; fibrotic atresia of the external auditory canals; subatrophic rhinitis. Chest radiography showed no pathology. Findings included elbow joint osteoarthritis, thoracic spine scoliosis, bilateral coxarthrosis (stage II, Kellgren–Lawrence), osteoarthritis of both sacroiliac joints, and early degenerative-dystrophic changes in the pubic symphysis.
In September 2024, nephrology consultation was performed. Given the involvement of the optic nerves, ENT organs, kidneys, and markedly elevated systemic inflammation markers, systemic vasculitis was suspected (with involvement of kidneys, eyes, and ENT structures). Laboratory testing: Rheumatoid factor, ANCA (c-ANCA, p-ANCA), antinuclear factor, aquaporin-4 antibodies, and complement component C3 were within reference ranges. C4 complement component was elevated at 0.44 g/L (0.1–0.4). Prostate-specific antigen and carcinoembryonic antigen levels were within reference ranges.
In October 2024, new papular eruptions appeared on the inner surfaces of the lower limbs.
In November 2024, X-rays of long bones revealed symmetrical mottled intramedullary osteosclerosis with relatively preserved epiphyses, cortical thickening – most prominent in the distal femoral diaphysis – with undulating cortical contours. These findings are characteristic of ECD. Bone scintigraphy showed pathological radiotracer uptake in the facial bones and long tubular bones (humerus, radius, ulna, femur, tibia, fibula bilaterally), as well as in both shoulder, elbow, wrist, knee, ankle, and tarsal joints – findings most consistent with ECD (Figure 2).
Repeat ophthalmologic evaluation revealed optic disc edema and peripheral retinal dystrophy in both eyes, interpreted as further manifestations of ECD disease.
Abdominal and retroperitoneal MRI with contrast revealed bilateral hydronephrosis, left adrenal hyperplasia, and focal bone marrow lesions in the scanned regions.
Renal ultrasound showed bilateral dilation of the pelvicalyceal systems: calyces up to 14 mm, left renal pelvis up to 28 mm, right up to 24 mm. Renal sinus cysts (11 mm and 12 mm) in the left kidney, diffuse changes in renal echotexture, and bilateral calicopyeloectasia were noted.
On November 19, 2024, a renal biopsy was performed (Figure 3A):
Light microscopy: The sample included a small cortical fragment and perinephric fat. Of the 5 glomeruli, none were sclerotic; one appeared ischemic. Glomeruli were of normal size, without mesangial or endocapillary hypercellularity. Basement membranes were thin and single-contoured. Interstitial fibrosis and tubular atrophy were present but not quantifiable due to limited cortical representation (approx. 20–30% of sample area). Multifocal mild mononuclear interstitial infiltrates without tubulitis. Small arteries appeared unremarkable; no large-caliber arteries were present. Mild arteriolar hyalinosis. Moderate multifocal mononuclear infiltration in perinephric fat. Conclusion: Interstitial fibrosis and tubular atrophy (grade not determined).
Immunofluorescence (IF): IgG, IgM, IgA, C3c, C1q, kappa, lambda, fibrinogen – all negative.
IHC: CD138, IgG, IgG4 – negative.
On December 24, 2024, a scalp skin biopsy was performed (Figure 3B):
Light microscopy: Sections of skin with “crusts”, epidermis showed parakeratosis, focal acanthosis, and spongiosis. The dermis (papillary and reticular layers) contained an infiltrate of medium-sized cells with grooved/folded nuclei and moderately pale cytoplasm, among eosinophilic granulocytes and lymphoplasmacytic infiltration. The infiltrating cells expressed CD1a (membranous pattern), Langerin (fine granular cytoplasmic pattern), S100 (nuclear and cytoplasmic staining), CD68 (diffuse and perinuclear dot-like positivity). Ki-67 proliferation index: 6%. Negative for CD21. Rare cells expressed ERK-1 (very weak nuclear/cytoplasmic staining). Conclusion: Histopathological features consistent with LCH.
In December 2024, the patient began treatment with trametinib 0.1 mg once daily and desmopressin 0.1 mg twice daily, as prescribed by the attending physician.
In February 2025, the patient consulted a dermatologist with complaints of pink-colored rashes on the cheeks and nasolabial folds, chest, and scalp; periodic edema of the feet and lower legs; erythematous-papular eruptions and scaling on the lower legs. He associated the facial and chest rashes with trametinib intake.
Status localis: erythematous papulopustular eruptions with crusting on the scalp; isolated erythematous-papular lesions on the face, lower legs, and sternum. In the inguinal folds, erythema and maceration were observed (Figure 4). Visible mucous membranes appeared pink and unremarkable.
Electrocardiogram (February 2025): sinus rhythm, regular, heart rate 69 bpm. Normal electrical axis. QT interval 0.41 s, QTc 0.443 s.
Echocardiography (February 2025): left ventricular hypertrophy – interventricular septal thickness 1.15 cm (ref: 0.6–0.9 cm), posterior wall thickness 1.03 cm (ref: 0.6–0.9 cm), LV mass index 136 g/m2. Grade II diastolic dysfunction, moderate left ventricular dilatation. Left ventricular ejection fraction 50%. No regional wall motion abnormalities. Mild mitral regurgitation (Grade I), mild aortic regurgitation (Grade I), mild right ventricular dilatation, tricuspid regurgitation (Grade I–II), pulmonary regurgitation (Grade II).
Status praesens: On physical examination, the general condition was satisfactory. Height 170 cm, weight 87 kg, BMI 28.7 kg/m2. Skin was of normal color and moisture. Nasal breathing was free. Respiratory rate 16/min. Percussion: clear pulmonary sound. Auscultation: vesicular breath sounds, no rales. Heart sounds were muffled, rhythm regular, no murmurs. HR 70 bpm. BP 160/120 mmHg. SpO2 97%. Peripheral pulses were palpable and of satisfactory quality. Tongue moist, not coated. Abdomen soft, painless, no signs of peritoneal irritation. Liver at the costal margin. Spleen not palpable. Renal percussion tenderness negative bilaterally.
Clinical diagnosis, based on history, investigations, and examination:
Primary disease: Mixed histiocytosis: LCH + ECD (associated with BRAF V600E mutation), with involvement of: kidneys (perinephric fibrosis, calicopyeloectasia with impaired nitrogen excretion; histologically: interstitial fibrosis, tubular atrophy), skin and mucous membranes (erythematous and ulcerative lesions), CNS (diabetes insipidus, optic disc edema), ocular system (peripheral retinal dystrophy OU), long bones (osteosclerosis and osteolysis in distal segments of humeral and femoral bones), joints (shoulders, elbows, wrists, knees, ankles, tarsal joints), bone marrow, non-specific manifestations (acute-phase inflammatory response, asthenia).
Comorbidities: chronic hypochromic microcytic anemia associated with STEAP3 (chr2:119247677A>T); type 4 hereditary hemochromatosis (SLC40A1, chr2:189572901A>T; heterozygous, resulting in amino acid substitution p.Met111Lys, NM_014585.6); chronic bilateral eczematous otitis externa, fibrotic atresia of external auditory canals; moderate myopia, myopic astigmatism.
The patient is under multidisciplinary follow-up and continues treatment with trametinib 1 mg orally once daily and desmopressin 0.1 mg twice daily. For skin lesions, topical therapy is used as needed: metronidazole gel and betamethasone cream. Over a 6-month period, partial regression of scalp and inguinal rashes, as well as improvement in acute-phase markers and a downward trend in serum creatinine, have been observed.
A visual timeline of key diagnostic, therapeutic, and monitoring events is shown in Figure 5.
Materials and methods
Genomic DNA Extraction and Quality Control
The skin sample was minced using sterile scalpels. The minced tissue was then transferred to a lysis mixture containing 180 µL of ATL buffer and 20 µL of Proteinase K. The sample was incubated overnight at 56°C in a thermoshaker at 1500 rpm. Following incubation, 200 µL of AL buffer and 200 µL of 96% ethanol were added to the lysate, and the mixture was loaded onto a DNA-binding spin column. DNA purification was carried out using AW1 and AW2 wash buffers according to the manufacturer’s protocol (Qiagen, Hilden, Germany). After the final wash and drying step, DNA was eluted in 50 µL of AE buffer. The quality of the extracted DNA was assessed by measuring the concentration using the Qubit dsDNA HS Assay Kit (Biodynami, Huntsville, AL, USA), which yielded a concentration of 33,5 ng/µL. DNA integrity was evaluated by electrophoresis on a 1.5% agarose gel (Qiagen, Hilden, Germany).
For the blood sample, genomic DNA was extracted using standard protocols and subjected to downstream library preparation.
Library Preparation, Exome Сapture and Sequencing
Ultrasonic fragmentation was performed with Covaris S220 (Covaris, Inc., Woburn, MA, USA). For the skin sample, library preparation was conducted using VAHTS Universal DNA Library Prep Kit for MGI (Vazyme, China). Blood DNA libraries were prepared using the MGIEasy Universal DNA Library Prep Set (MGI Tech, Shenzhen, China). Enrichment of exonic sequences was achieved using Agilent All Exon v8 (Agilent Technologies, Santa Clara, CA, USA) [22]. Sequencing of protein-coding regions was performed by paired-end sequencing (PE100) on the G-400 (MGI Tech, Shenzhen, China) platform, achieving 100× mean coverage.
Bioinformatic Processing and Variant Analysis
Raw sequence data quality was evaluated using FastQC v0.11.9 [23]. Sequencing quality reports identified imbalanced bases at read starts, which were trimmed using BBDuk (BBMap v38.96) [24]. Processed reads were then aligned to the GRCh38.p14 reference genome assembly employing bwa-mem2 v2.2.1 [25], with subsequent file format conversion and coordinate sorting implemented through SAMtools v1.9 [26]. Duplicates were marked using Picard v2.22.4 [27]. Variant calling was executed through a dual approach incorporating bcftools mpileup v1.9 [28] and DeepVariant v1.5.0 [29], followed by variant decomposition into biallelic representations using vt decompose v0.5772 [30] and normalization via vt normalize v0.5772. Filtering criteria included coverage depth ≥3 and FILTER=PASS for DeepVariant. The resultant VCF files were merged using bcftools-1.9 and annotated via ANNOVAR [29]. The lower threshold for variant allele frequency (VAF) was set at 5%. Clinical interpretation followed ACMG guidelines [32], incorporating minimum coverage thresholds (14×) and population frequency cutoffs (gnomAD v4.1.0 < 1%), with additional consideration of Russian-specific allele frequencies obtained from the RUSeq browser and the Database of Population Frequencies [33,34,35]. Somatic variant analysis was performed using MuTect2 with default parameters [36].
Validation of BRAF V600E Mutation
Validation was performed to assess the presence of BRAF V600E mutation in NGS BRAF V600E positive sample using EntroGen’s BRAF-RT64 kit (EntroGen, Woodland Hills, CA, USA). PCR-based assay uses allele-specific probes was performed to identify the presence of the BRAF V600E, V600K, V600D, V600R, V600M and V600G mutations.
The testing procedure involves three steps: 1) Isolation of DNA from blood sample. 2) Amplification of regions of the BRAF gene using allele-specific probes. 3) Detection of amplification product on a Real-Time PCR instrument CFX 96 (BioRad, Saint Petersburg, Russia).
EntroGen’s BRAF Codon 600 mutation analysis kits (BRAFX-RT64) are available for research (RUO) and diagnostic (CE-IVD) purposes. It is highly sensitive and specific to detects as little as 1% mutation load. According to the manufacturer's instructions, the difference between the Ct values of the wild and mutant allele less than 11 confirms the presence of a mutation in the sample being tested.
Histopathological Examination
Biopsies were obtained from the scalp skin with erythematous-desquamative rash (0.7 × 0.2 × 0.2 cm), RBM (1.1 × 0.3 × 0.3 cm), and kidney (1.0 × 0.3 × 0.2 cm). Standard histological processing was performed, with paraffin embedding of all samples. Kidney biopsy sections were additionally stained using PAS and Masson’s trichrome methods. IHC was performed on the VENTANA BenchMark ULTRA platform (Roche Diagnostics, Rotkreuz, Switzerland) using the following primary antibodies: CD138, IgG, and IgG4 (for kidney samples); CD1a, CD21, CD68, Ki-67, Langerin, S-100, and ERK-1 (for skin biopsy). All antibodies were used at standard dilutions recommended by the manufacturers. For kidney biopsy samples, direct IF analysis was carried out using antibodies against IgG, IgA, IgM, C3c, C1q, kappa, lambda, and fibrinogen. For RBM samples, IF staining targeted CD4, CD8, CD34, CD68, tryptase, S-100, vimentin, and CD11b [37]. The following secondary antibodies were used: Goat Anti-Mouse IgG H&L conjugated with Alexa Fluor® 488 (ab150113), 555 (ab150114), and 647 (ab150115), all from Abcam (Cambridge, MA, USA). Additionally, OPAL650 fluorophore (Akoya Biosciences, Marlborough, MA, USA) was used for multiplex detection. Nuclei were counterstained with Fluoroshield Mounting Medium containing DAPI (ab104139, Abcam). Brightfield microscopy was performed using an Olympus CX43 microscope equipped with an Olympus SC50 camera (Olympus Co., Tokyo, Japan). Multiplex IF imaging was performed using the Mantra 2 Quantitative Pathology Imaging System (Akoya Biosciences) based on an Olympus BX43 microscope, equipped with a scientific-grade multispectral 12-bit monochrome CCD camera and a liquid crystal tunable filter. Quantification and analysis of bone marrow cell populations were performed using QuPath v0.5.1 software.
Results
Genetic Analysis Results
Comparison of paired blood–skin biopsy samples revealed 71 953 shared variants, with 3,081 unique to the skin (Table S1) and 2 633 germline variants unique to the blood (Figure 6A). Annotation of somatic skin variants showed VAF ranging from 0.032 to 1.000 (median = 0.318, IQR = 0.278). The distribution was right-skewed (skewness = 1.170), indicating a predominance of low-VAF variants (Figure 6B). In the blood sample, VAF ranged from 0.061 to 1.000 (median = 0.539, IQR = 0.549), with a near-symmetric distribution (skewness = 0.242). A similar pattern was observed for all skin cell mutations: VAF range 0.032–1.000 (median = 0.527, IQR = 0.535), with a slightly positively skewed distribution (skewness = 0.298). All three variant types were predominantly associated with non-exonic substitutions, especially heterozygous and homozygous ones (>80%) (Figure 6C). Nonframeshift deletions predominated among heterozygous variants (52.31%), while nonsynonymous (63.94%), synonymous (60.71%), and variants of unknown significance (80%) were more common among mosaic variants. The greatest number of mutations were detected in MUC3A, HLA-DRB5, PRSS2, TCAF2, MUC2, HLA-DRB1, FCGBP, GOLGA6L2, PRSS3; however, these are regions known to be problematic in next-generation sequencing (NGS) and are not considered pathogenetically relevant. Therefore, only genes from Osipov D.S. et al. [18] were included in further analysis. All variants with VAF >5% were benign (Figure 6D). Additionally, a PIK3CD variant at chr1:9721087G>C was found only in the skin (VAF = 25%) and is likely a sequencing artifact.
Skin biopsy specimens were analyzed for known histiocytosis-related mutations [18,38]. A BRAF V600E variant (VAF 1%; 3/251 reads) was detected in the skin but not in blood and was subsequently validated (Figure 7). Additionally, a pathogenic mosaic variant NRAS(NM_002524.5):c.181C>G (p.Gln61Glu) was present at low VAF in both blood (1/105 reads) and skin (3/199 reads); this missense variant has previously been reported in a single Russian histiocytosis patient [18].
Whole exome sequencing performed on May 5, 2025, identified a novel intronic variant in STEAP3 (chr2:119247677A>T) in heterozygous state, disrupting a splice site (c.523-2A>T, NM_182915.3). Such variants are known causes of hypochromic microcytic anemia type II (OMIM: 615234). This variant is absent in gnomAD and the FMBA Russian variant database. All aggregated in silico prediction tools (BayesDel noAF, BayesDel addAF) classify it as pathogenic.
A previously undescribed heterozygous missense variant was also detected in SLC40A1 (chr2:189572901A>T), resulting in a p.Met111Lys amino acid change (c.332T>A; NM_014585.6). Such variants are a known cause of type IV hereditary hemochromatosis (OMIM: 606069). Its allele frequency is 0.000002478 in gnomAD (4 heterozygotes) and 0.00000828 in the FMBA database (2 heterozygotes). All in silico predictors classify this variant as pathogenic (BayesDel noAF, BayesDel addAF, MetaRNN, REVEL, MetaLR, MetaSVM).
A heterozygous variant of uncertain significance in KCNH2(NM_000238.4):c.273G>T (p.Glu91Asp) associated with epilepsy [39] was also identified, though the patient exhibited no neurological symptoms. All other clinically annotated variants were benign carrier states.
Pathomorphological Analysis of the Trephine Biopsy
IF showed CD34 expression in endothelial cells and fibroblast-like cells (Figure 8A). Vimentin+CD11b–CD34+ and Vimentin+CD11b+CD34– cell populations comprised 3 [1.25–20] (Me = 0.6814% of Vimentin-positive cells) and 15 [0–56.25] (Me = 7.778%) cells per high-power field (HPF), respectively (excluding endothelial cells).
Analysis of bone marrow IF images stained for CD8, CD68, and tryptase (Figure 8B) revealed a density of Tr+CD68– cells of 0.02596 [0.01536–0.1915] per 1000 µm2 and 10.5 [8.25–19.75] per HPF (356 371.896 µm2), corresponding to Me = 0.75% of all cells. CD8+ cell counts were 89.5 [29.25–131.3] (median 5.945%), CD68+ cells 41 [23.75–57.25] (2.953%), and Tr+CD68+ cells 19 [15.25–20.0] (1.506%). Contacts (< 1 µm) were observed between 2 mast cells in two HPFs and 3 mast cells in another HPF. The number of Tr+CD68– cells contacting CD8+ cells was 0.5 [0–3], and that of Tr+CD68+ cells – 2.5 [2–4].
Analysis of CD68 and S100 markers demonstrated predominance of CD68+ granulocytic cells, amounting to 117.5 [33.5–250] per HPF (Me = 55.61% of all immunopositive cells), followed by S100+ cells – 110 [57–137.5] (Me = 43.02%) (Figure 8C). Their median area was 38.75 µm2 [24.50–60.00], with a minimum of 8.75 µm2 and a maximum of 240.3 µm2 (skewness = 1.717). CD68+S100+ cells were less frequent, 4 [1.5–6.25] per HPF (Me = 2.174%). Notably, two types of CD68+ staining were observed: perinuclear dot-like, typical for LCH cells, and diffuse staining in smaller-volume cells, most likely monocytes or macrophages.
Discussion
Both ECD and LCH share the somatic activating mutation BRAF V600E, found in approximately 50% of cases, suggesting overlap in pathogenetic pathways and explaining their coexistence in mixed histiocytosis [40].
According to the 2022 LCH classification [19], the patient fits the multisystem subtype, with involvement of >2 organs: bones, skin/mucosa, kidneys, endocrine system, CNS, eyes, and bone marrow.
CNS involvement manifested as diabetes insipidus via pituitary insufficiency and MRI-confirmed pituitary atrophy. Zhou et al. described a patient with isolated pituitary LCH presenting with polydipsia, polyuria, and MRI pituitary stalk thickening [41]. In our case, the pituitary insufficiency may be tied to the primary diagnosis.
No hepatosplenomegaly or cholestatic liver failure characteristic of LCH [19] was found, though hepatic steatosis was noted. Ear involvement is uncommon in LCH, but our patient had chronic bilateral eczematous otitis externa, fibrotic atresia of the external auditory canals, and subatrophic rhinitis. Savina et al. reported a similar chronic otitis case attributed to LCH [42]. Given the long-standing resistance to conventional therapy and response to trametinib, a connection to LCH is plausible.
According to Pires et al., the age at diagnosis of mixed histiocytosis varies widely, ranging from 41 to 74 years [43]. In our case, the diagnosis was established at the age of 34, which is likely due to the early manifestation of the disease (the first symptoms appeared at age 29), as well as a pronounced clinical presentation predominantly involving the skin and bones.
Pulmonary involvement in LCH is relatively common (observed in more than 50% of cases with unifocal LCH). The at-risk group includes males aged 20–40 years. The only proven triggering factor is tobacco smoke – more than 90% of patients with pulmonary LCH are smokers [44]. In our case, although the patient fully met the above criteria, only centrilobular emphysema in the upper lung zones – characteristic of smokers – was detected.
A similar case of mixed histiocytosis was described by Tsai JW et al. in a 34-year-old female patient who presented with ulcerative skin lesions on both thighs, osteosclerosis of the distal tibiae, and involvement of the soft tissues of the chest wall [45]. Our patient exhibited almost identical symptoms; however, in our case, the skin lesions were more prominent on the scalp, whereas in the case described by Tsai JW et al., ulcerative lesions were mainly observed on the lower extremities.
Treatment of patients with mixed histiocytosis is particularly challenging. In pulmonary LCH, smoking cessation is strongly recommended to slow disease progression. Reported treatment regimens include glucocorticoids, interferon-α, systemic chemotherapy, radiation therapy, and in certain cases, cladribine (an immunosuppressive agent) and vemurafenib (a protein kinase inhibitor) [43]. Patient N. is currently receiving therapy with trametinib (an allosteric inhibitor of mitogen-activated protein kinases 1 and 2) and desmopressin (a hormone analogue related to hypothalamic-pituitary regulation, including gonadotropins and their antagonists).
Histological and IF data in our patient provided a deeper insight into the disease biology. Light microscopy revealed an infiltrate of medium-sized cells with grooved nuclei and pale cytoplasm, displaying CD1a, Langerin, S100, and perinuclear dot-like CD68 expression – classic hallmarks of LCH. However, the additional presence of diffuse CD68+ macrophages and weak ERK-1 nuclear staining highlights a pathological spectrum extending beyond conventional LCH morphology. The Ki-67 index of 6% indicated low proliferative activity, consistent with a smoldering inflammatory process rather than an aggressive neoplasm.
IF analysis further revealed a structured yet functionally imbalanced microenvironment. The low prevalence of CD34+ cells argues against a blast-driven process, while the relative enrichment of CD11b+ elements is more consistent with chronic inflammatory infiltration than with neoplastic proliferation. Mast cells (~2.26% of all cells) were sparse and rarely engaged in close contacts with other immune populations, suggesting a limited regulatory role. CD8+ T lymphocytes were present but modest in proportion (~6% of all cells), while the predominance of CD68+ macrophages created a pro-inflammatory milieu that was nonetheless insufficient to eliminate pathological clones, yet capable of sustaining tissue injury. Of particular note, the coexistence of perinuclear-dot CD68+ histiocytes with diffusely CD68+ macrophages reflects both canonical LCH cells and activated myeloid infiltrates, bridging the histological features of LCH and ECD within one tissue context. Thus, the histology and IF together illustrate a “hybrid” lesion in which classical dendritic LCH cells are embedded in an abnormal macrophage-rich stroma, providing a morphological correlate of the mixed histiocytosis phenotype.
The molecular and cellular mechanisms underlying the pathogenesis of LCH are associated with impaired differentiation and function of antigen-presenting cells. Specifically, elevated levels of CD34+ cells have been identified in the blood of LCH patients; these cells are capable of differentiating into LC-like dendritic cells (CD1a+CD83+Lag+) a subset of which express CD2 and exhibit reduced viability in vitro. Although these cells elicit an allogeneic T-cell response, they often fail to initiate a primary antigen-specific response, indicating functional defects and suggesting that LCH may originate at the level of hematopoietic progenitors [46].
CD34 is a transmembrane sialomucin molecule predominantly expressed on hematopoietic stem/progenitor cells, endothelial cells, and embryonic fibroblasts. It is also present at later stages of hematopoietic development, including pro-T/B cells, monocyte/mast cell progenitors, megakaryocyte colony-forming units (CFU-Mk), and erythroid precursors (BFU-E). Furthermore, CD34 expression is retained in mature NK cells [47]. The precise functions of CD34 remain unclear; it is hypothesized to regulate interactions with the bone marrow microenvironment and control cell proliferation, although data are conflicting and depend on the experimental model used. At present CD34 is primarily viewed as a marker of functional cellular subsets, without a well-established functional role in haematopoietic stem cells [48].
CD68 is a glycoprotein highly expressed in macrophages and other monocyte-lineage cells. It resides primarily in lysosomes and participates in intracellular functions, including phagocytosis. CD68 expression is minimal in lymphocytes, dendritic cells, non-mesenchymal cells (fibroblasts, endothelial cells) and tumor cells [49]. CD11b is the α-subunit of the β2-integrin heterodimer that binds ICAM-1, iC3b, and fibrinogen. It plays a crucial role in phagocytosis, adhesion, and the regulation of inflammatory responses. Deficiency of β2-integrin leads to impaired phagocytic function and compromised immune defense. In contrast, CD11b knockout in mice impairs complement-mediated phagocytosis and vasculitis but does not significantly affect neutrophil migration [50]. CD11b is also a key regulator of antitumor immunity, modulating macrophage polarization and shaping the immune microenvironment. Loss of CD11b enhances the immunosuppressive phenotype of tumor-associated macrophages (TAMs), promotes accumulation of FoxP3+ regulatory T cells, reduces CD8+ T-cell infiltration, and accelerates tumor growth. Pharmacological activation of CD11b by the agonist LA1 repolarizes TAMs toward a pro-inflammatory phenotype, enhances T-cell responses, and suppresses tumor progression. CD11b also contributes to vasculogenesis by regulating vessel maturation and nutrient supply to the tumor [51]. Neither CD68 nor CD11b are characteristic markers of resting LCs; their expression indicates pathological activation in LCH [52]. CD11b expression has been identified in LCH tissue samples from skin, mucosa, bone, and lymph nodes [53]. The active phase of LCH is associated with an increase in CD11bhigh and CD11bint/low populations, including CD207+CD1a+ cells, detectable in peripheral blood [54]. In the present case, the detection of LCH/ECD with systemic inflammation, vasculitis-like features, and multiorgan involvement is consistent with pathological activation of CD11b+ myeloid cells. This supports the concept that CD11b-driven immune dysregulation may underlie both the chronic inflammatory response and the extensive tissue damage observed in the patient.
Bone marrow involvement occurs in 2–33% of LCH cases, more commonly in infants with multisystem disease. It is associated with anemia, presence of CD1a+ histiocytes in bone marrow trephines, and poor prognosis [55].
The patient’s anemia is likely related to the identified STEAP3 splice-site variant c.523-2A>T. A nonsense mutation in the same gene (c.300C>A, p.Cys100Ter) was described in a Pakistani family with three siblings affected by severe anemia and iron overload [56]. First described in 2011, the disorder also manifests as hypogonadism and elevated serum ferritin levels before transfusions begin. The pathogenesis involves disruption of STEAP3, an endosomal ferrireductase that converts Fe3+ to Fe2+ in the transferrin-dependent iron uptake pathway in erythroid cells. Impaired ferrireductase activity reduces iron availability for incorporation into protoporphyrin (via ferrochelatase), leading to microcytic hypochromic anemia and compensatory iron accumulation in the reticuloendothelial system (in siderosomes and mitochondria). The disease is presumed to manifest in compound heterozygosity with one null allele and one weakly expressed allele (allele-specific regulation).
A study of 3,205 healthy Chinese donors demonstrated that STEAP3 mutations affecting the NAD(P)H-binding motif (p.Arg59Cys) or heme-binding sites (p.His316Asn) significantly impaired ferrireductase function. However, heterozygous carriers, including those with the p.Cys261Ter nonsense mutation, showed no clinical signs of anemia or iron metabolism abnormalities. The mutant mRNA was not fully degraded, and the truncated protein formed aberrant aggregates in endosomes and on the plasma membrane [57]. Our patient also carried the heterozygous STEAP3(NM_182915.3):c.-24G>A variant, which is common in gnomAD (including in homozygous individuals; n = 126,453), suggesting uncertain pathogenicity. Thus, the roles of c.523-2A>T and c.-24G>A in this patient’s clinical presentation require further evaluation.
Knockout of ANXA10 has been shown to increase TFRC (transferrin receptor) levels by inhibiting its autophagic degradation, leading to intracellular iron (Fe2+) accumulation via the TFRC–STEAP3–SLC11A2 axis. Concurrently, expression of SLC40A1 (ferroportin) is downregulated, reducing iron export and triggering ferroptosis [58]. In addition to iron overload, lipid ROS accumulation is a critical factor in ferroptotic cell death [59,60].
Ferroportin disease (type IV hereditary hemochromatosis) is the second most common genetic iron overload disorder after HFE-hemochromatosis. It is caused by heterozygous mutations in SLC40A1, encoding ferroportin, the only known cellular Fe2+ exporter in humans, primarily localized to the basolateral membrane of duodenal enterocytes (less active in the colon) and macrophage membranes. Ferroportin expression and function are regulated by intracellular iron via an iron-responsive element (IRE) in the 5'UTR of its mRNA, which binds iron-responsive proteins (IRPs) [61].
The disease manifests in two major phenotypes. The classical (macrophage) phenotype results from loss-of-function mutations (e.g., Δ162, Δ160–162, G323V, G490D) that impair cellular localization and membrane trafficking, reducing iron export and leading to macrophage iron accumulation. It is associated with high serum ferritin, normal or low transferrin saturation, and may include anemia. The course is typically indolent, complicating early diagnosis. The nonclassical (hepatocellular) phenotype involves gain-of-function mutations resistant to hepcidin inhibition (e.g., N144H, N144T, N144D). Ferroportin remains membrane-bound and exports iron constitutively, resulting in elevated transferrin saturation and hepatic iron deposition, similar to classical hemochromatosis. Ferroportin functions as a multimer; mutant proteins exert a dominant-negative effect on wild-type ferroportin, disrupting iron export regulation [62].
SLC40A1 interacts with JAK2, which in turn interacts with BRAF (co-expression and experimentally determined interaction per STRING [63]). JAK2 is also directly linked to BRAF via RAF1, NRAS, KRAS, and HRAS. Although no direct experimental interaction between SLC40A1 and STEAP3 has been documented, both appear alongside SMAD6 (previously implicated in LCH [10]) in the “Negative regulation of programmed cell death subnetwork” pathway.
Conclusion
The presented case illustrates a rare overlapping phenotype of LCH and ECD with early-onset, multisystem involvement, and severe anemia. The key pathogenetic driver is the somatic BRAF V600E mutation, supporting clonal proliferation of histiocytes, with additional contribution from rare NRAS alteration. Germline variants in STEAP3 and SLC40A1 create a background of iron dysregulation and chronic inflammation, which likely modulates the course and severity of the disease. Morphological and immunophenotypic data confirm the coexistence of true Langerhans-type cells and a dominant macrophage/stromal component, reflecting the mixed pathogenesis. The immune microenvironment is characterized by prevailing innate activation and limited cytotoxic T-cell involvement, favoring persistence of the histiocytic clone. This complex architecture explains the partial response to MAPK-targeted therapy and highlights the need for strategies addressing both clonal and reactive compartments. Timely integration of clinical, pathological, and molecular findings is crucial for accurate diagnosis. Multidisciplinary discussion remains essential for personalized treatment planning and improvement of patient outcomes.
Ethics
This study was conducted in compliance with local legislation and institutional requirements. Written informed consent was obtained from the patient both for participation in the study and for the publication of potentially identifiable images or data.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Table S1. Somatic variants detected in the skin biopsy.
Author Contributions
Buianova A.A., Gaydina T.A., Reznik E.V. – clinical assessment; Buianova A.A. – analysis of exome sequencing and pathomorphological data; Kuznetsova A.A., Belova V.A. – sequencing; Repinskaia Zh.A. – bioinformatic analysis; Ignatyuk M.A., Volodkin A.V. – histological section processing; Shatalov P.A., Shinkarina A.P. – skin sample validation of BRAF mutation; Buianova A.A., Gaydina T.A., Reznik E.V., Iarovoi M.D. – manuscript preparation; Atiakshin D.A., Korostin D.O. – supervision.
Funding
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (Federal scientific and technical program for the development of genetic technologies for 2019–2030, agreement № 075-15-2025-598).
Data Availability Statement
Corresponding author upon request.
References
- Gulati, N.; Allen, C.E. Langerhans cell histiocytosis: Version 2021. Hematol Oncol. 2021, 39 Suppl 1, 15–23. [Google Scholar] [CrossRef]
- Yoon, S.O. Pathologic characteristics of histiocytic and dendritic cell neoplasms. Blood Res. 2024, 59, 18. [Google Scholar] [CrossRef]
- Emile, J.F.; Cohen-Aubart, F.; Collin, M.; et al. Histiocytosis. Lancet. 2021, 398, 157–170. [Google Scholar] [CrossRef]
- Halbritter, F.; Farlik, M.; Schwentner, R.; et al. Epigenomics and Single-Cell Sequencing Define a Developmental Hierarchy in Langerhans Cell Histiocytosis. Cancer Discov. 2019, 9, 1406–1421. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; van Halteren, A.G.S.; Lei, X.; et al. Bone marrow-derived myeloid progenitors as driver mutation carriers in high- and low-risk Langerhans cell histiocytosis. Blood. 2020, 136, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Abagnale, G.; Schwentner, R.; Ben Soussia-Weiss, P.; et al. BRAFV600E induces key features of LCH in iPSCs with cell type-specific phenotypes and drug responses. Blood. 2025, 145, 850–865. [Google Scholar] [CrossRef]
- Rodriguez-Galindo, C.; Allen, C.E. Langerhans cell histiocytosis. Blood. 2020, 135, 1319–1331. [Google Scholar] [CrossRef]
- Allen, C.E.; Merad, M.; McClain, K.L. Langerhans-Cell Histiocytosis. N Engl J Med. 2018, 379, 856–868. [Google Scholar] [CrossRef]
- Cai, H.C.; Chen, J.; Liu, T.; et al. Langerhans cell histiocytosis in adolescent patients: a single-centre retrospective study. Orphanet J Rare Dis. 2022, 17, 268. [Google Scholar] [CrossRef]
- Peckham-Gregory, E.C.; Chakraborty, R.; Scheurer, M.E.; et al. A genome-wide association study of LCH identifies a variant in SMAD6 associated with susceptibility. Blood. 2017, 130, 2229–2232. [Google Scholar] [CrossRef]
- Ozer, E.; Sevinc, A.; Ince, D.; Yuzuguldu, R.; Olgun, N. BRAF V600E Mutation: A Significant Biomarker for Prediction of Disease Relapse in Pediatric Langerhans Cell Histiocytosis. Pediatr Dev Pathol. 2019, 22, 449–455. [Google Scholar] [CrossRef]
- Feng, S.; Han, L.; Yue, M.; et al. Frequency detection of BRAF V600E mutation in a cohort of pediatric langerhans cell histiocytosis patients by next-generation sequencing. Orphanet J Rare Dis. 2021, 16, 272. [Google Scholar] [CrossRef]
- Whitlock, J.A.; Geoerger, B.; Dunkel, I.J.; et al. Dabrafenib, alone or in combination with trametinib, in BRAF V600-mutated pediatric Langerhans cell histiocytosis. Blood Adv. 2023, 7, 3806–3815. [Google Scholar] [CrossRef]
- Berres, M.L.; Lim, K.P.; Peters, T.; et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med, 2014; 211, 669–683correction in J Exp Med. 2015 Feb 9;212, 281. https://doi.org/10.1084/jem.2013097701202015c. [Google Scholar] [CrossRef]
- Chakraborty, R.; Hampton, O.A.; Shen, X.; et al. Mutually exclusive recurrent somatic mutations in MAP2K1 and BRAF support a central role for ERK activation in LCH pathogenesis. Blood. 2014, 124, 3007–3015. [Google Scholar] [CrossRef] [PubMed]
- Alayed, K.; Medeiros, L.J.; Patel, K.P.; et al. BRAF and MAP2K1 mutations in Langerhans cell histiocytosis: a study of 50 cases. Hum Pathol. 2016, 52, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Novosad, O.; Skrypets, T.; Pastushenko, Y.; et al. MAPK/ERK signal pathway alterations in patients with Langerhans Cell Histiocytosis. Změny v signální dráze MAPK/ERK u pacientů s histiocytózou Langerhansových buněk. Klin Onkol. 2018, 31, 130–136. [Google Scholar] [CrossRef]
- Osipova, D.S.; Raykina, E.V.; Kozlova, Y.A.; et al. Clinicogenomic associations in patients with Langerhans cell histiocytosis: a cohort study. Pediatric Hematology/Oncology and Immunopathology. 2023, 22, 102–107. [Google Scholar] [CrossRef]
- Goyal, G.; Tazi, A.; Go, R.S.; Rech, K.L.; Picarsic, J.L.; Vassallo, R.; Young, J.R.; Cox, C.W.; Van Laar, J.; Hermiston, M.L.; et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults. Blood. 2022 Apr 28;139, 2601-2621. [CrossRef] [PubMed] [PubMed Central]
- McKinney, R.A.; Wang, G. Langerhans Cell Histiocytosis and Other Histiocytic Lesions. Head Neck Pathol. 2025, 19, 26. [Google Scholar] [CrossRef]
- Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-Caballero L, Jordan MB, Abdel-Wahab O, Allen CE, et al; Histiocyte Society. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016, 127, 2672–2681. [CrossRef] [PubMed] [PubMed Central]
- Belova, V.; Pavlova, A.; Afasizhev, R.; et al. System analysis of the sequencing quality of human whole exome samples on BGI NGS platform. Sci. Rep. 2022, 12, 609. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 20 April 2025).
- Joint Genome Institute. Available online: https://jgi.doe.gov/data-and-tools/software-tools/bbtools/bb-tools-user-guide/bbduk-guide/ (accessed on 20 April 2025).
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; et al. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar]
- Picard Toolkit. version 2.22.4; Broad Institute: Cambridge, MA, USA, 2019; Available online: https://broadinstitute.github.io/picard/ (accessed on 20 April 2025).
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; et al. A universal SNP and small-indel variant caller using deep neural networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.; Abecasis, G.R.; Kang, H.M. Unified Representation of Genetic Variants. Bioinformatics 2015, 31, 2202–2204. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Barbitoff, Y.A.; Khmelkova, D.N.; Pomerantseva, E.A.; Slepchenkov, A.V.; Zubashenko, N.A.; Mironova, I.V.; et al. Expanding the Russian allele frequency reference via cross-laboratory data integration: insights from 7452 exome samples. Natl Sci Rev. 2024, 11, nwae326. [Google Scholar] [CrossRef]
- Database of Population Frequencies of Genetic Variants of the Population of the Russian Federation. [Internet]. FMBA of Russia.; 2024. Available online: https://gdbpop.nir.cspfmba.ru/ (accessed on 20 April 2025).
- Cibulskis, K.; Lawrence, M.S.; Carter, S.L.; Sivachenko, A.; Jaffe, D.; Sougnez, C.; et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol. 2013, 31, 213–219. [Google Scholar] [CrossRef]
- Buchwalow, I.; Samoilova, V.; Boecker, W.; Tiemann, M. Multiple immunolabeling with antibodies from the same host species in combination with tyramide signal amplification. Acta Histochem. 2018, 120, 405–411. [Google Scholar] [CrossRef]
- Jouenne, F.; Chevret, S.; Bugnet, E.; et al. Genetic landscape of adult Langerhans cell histiocytosis with lung involvement. Eur Respir J. 2020, 55, 1901190. [Google Scholar] [CrossRef]
- Li, X.; Liu, N.; Bai, R. Variant frequencies of KCNQ1, KCNH2, and SCN5A in a Chinese inherited arrhythmia cohort and other disease cohorts undergoing genetic testing. Ann Hum Genet. 2020, 84, 161–168. [Google Scholar] [CrossRef]
- Hervier B, Haroche J, Arnaud L, Charlotte F, Donadieu J, Néel A, Lifermann F, Villabona C, Graffin B, et al; French Histiocytoses Study Group. Association of both Langerhans cell histiocytosis and Erdheim-Chester disease linked to the BRAFV600E mutation. Blood. 2014, 124, 1119–1126. [CrossRef] [PubMed]
- Zhou, W.; Rao, J.; Li, C. Isolated Langerhans cell histiocytosis in the hypothalamic-pituitary region: a case report. BMC Endocr Disord. 2019, 19, 143. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Savina VYu Solovyeva, S.E.; Dolzhansky, O.V.; Metelin, A.V. Clinical and morphological analysis of Langerhans cell histiocytosis in a child of early age. Pirogov Russian Journal of Surgery. [CrossRef]
- Pires, Y.; Jokerst, C. E.; Panse, P. M.; Kipp, B. R.; Tazelaar, H. D. Combined Erdheim-Chester Disease and Langerhans Cell Histiocytosis in the Lung: A Report of 2 Patients With Overlap Syndrome. AJSP: Reviews and Reports 2020, 25, 33–39. [Google Scholar] [CrossRef]
- Potapenko, V.G.; Baykov, V.V.; Zinchenko, A.V.; Potikhonova, N.A. Langerhans cell histiocytosis in adults: literature review. Onkogematologiya = Oncohematology 2022, 17, 16–32. [Google Scholar] [CrossRef]
- Tsai, J.W.; Tsou, J.H.; Hung, L.Y.; Wu, H.B.; Chang, K.C. Combined Erdheim-Chester disease and Langerhans cell histiocytosis of skin are both monoclonal: a rare case with human androgen-receptor gene analysis. J Am Acad Dermatol. 2010, 63, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Misery, L.; Rougier, N.; Crestani, B.; et al. Presence of circulating abnormal CD34+ progenitors in adult Langerhans cell histiocytosis. Clin Exp Immunol. 1999, 117, 177–182. [Google Scholar] [CrossRef]
- Raghav, P. K. , Gangenahalli G. (). Hematopoietic Stem Cell Molecular Targets and Factors Essential for Hematopoiesis. J. Stem Cel Res Ther 2018, 8, 4412. [Google Scholar] [CrossRef]
- Rix, B.; Maduro, A.H.; Bridge, K.S.; Grey, W. Markers for human haematopoietic stem cells: The disconnect between an identification marker and its function. Front Physiol. 2022, 13, 1009160. [Google Scholar] [CrossRef]
- Chistiakov, D.A.; Killingsworth, M.C.; Myasoedova, V.A.; Orekhov, A.N.; Bobryshev, Y.V. CD68/macrosialin: not just a histochemical marker. Lab Invest. 2017, 97, 4–13. [Google Scholar] [CrossRef]
- Duan, M.; Steinfort, D.P.; Smallwood, D.; et al. CD11b immunophenotyping identifies inflammatory profiles in the mouse and human lungs. Mucosal Immunol. 2016, 9, 550–563. [Google Scholar] [CrossRef]
- Schmid, M.C.; Khan, S.Q.; Kaneda, M.M.; et al. Integrin CD11b activation drives anti-tumor innate immunity. Nat Commun. 2018, 9, 5379. [Google Scholar] [CrossRef]
- Emile, J.F.; Fraitag, S.; Leborgne, M.; de Prost, Y.; Brousse, N. Langerhans' cell histiocytosis cells are activated Langerhans' cells. J Pathol. 1994, 174, 71–76. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, J.H.; Tamminga, R.Y.; Kamps, W.A.; Timens, W. Expression of cellular adhesion molecules in Langerhans cell histiocytosis and normal Langerhans cells. Am J Pathol. 1995, 147, 1161–1171. [Google Scholar] [PubMed]
- Carrera Silva, E.A.; Nowak, W.; Tessone, L.; et al. CD207+CD1a+ cells circulate in pediatric patients with active Langerhans cell histiocytosis. Blood. 2017, 130, 1898–1902. [Google Scholar] [CrossRef]
- Kumar, M.; Updesh Singh Sachdeva, M.; Naseem, S.; et al. Bone marrow infiltration in Langerhan's cell histiocytosis - An unusual but important determinant for staging and treatment. Int J Hematol Oncol Stem Cell Res. 2015, 9, 193–197. [Google Scholar]
- Grandchamp, B.; Hetet, G.; Kannengiesser, C.; et al. A novel type of congenital hypochromic anemia associated with a nonsense mutation in the STEAP3/TSAP6 gene. Blood. 2011, 118, 6660–6666. [Google Scholar] [CrossRef]
- Liu, D.; Yi, S.; Zhang, X.; et al. Human STEAP3 mutations with no phenotypic red cell changes. Blood. 2016, 127, 1067–1071. [Google Scholar] [CrossRef]
- Wang, X.; Zhou, Y.; Ning, L.; Chen, J.; Chen, H.; Li, X. Knockdown of ANXA10 induces ferroptosis by inhibiting autophagy-mediated TFRC degradation in colorectal cancer Cell Death Dis. 2023, 14, 588. https://10.1038/s41419-023-06114-2. Correction in Cell Death Dis. 2023, 14, 683. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Y.; Wu, P.; Tian, Y.; Liu, B.; Wang, J.; Bihl, J.; Shi, H. Inhibition of Ferroptosis Alleviates Early Brain Injury After Subarachnoid Hemorrhage In Vitro and In Vivo via Reduction of Lipid Peroxidation. Cell Mol Neurobiol. 2021 Mar;41, 263-278. [CrossRef] [PubMed]
- Fang, Y.; Gao, S.; Wang, X.; Cao, Y.; Lu, J.; Chen, S.; Lenahan, C.; Zhang, J.H.; Shao, A.; Zhang, J. Programmed Cell Deaths and Potential Crosstalk With Blood-Brain Barrier Dysfunction After Hemorrhagic Stroke. Front Cell Neurosci. 2020, 14, 68. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Montalbetti, N.; Simonin, A.; Kovacs, G.; Hediger, M.A. Mammalian iron transporters: families SLC11 and SLC40. Mol Aspects Med. 2013, 34, 270–287. [Google Scholar] [CrossRef]
- Mayr, R.; Janecke, A.R.; Schranz, M.; et al. Ferroportin disease: a systematic meta-analysis of clinical and molecular findings. J Hepatol. 2010, 53, 941–949. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; et al. The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
Figure 1.
a, b – Macroscopic images of the patient’s scalp (2023): small papules and pustules on an erythematous background, numerous confluent massive serous crusts. c – Macroscopic image of the scalp after initiation of trametinib therapy.
Figure 1.
a, b – Macroscopic images of the patient’s scalp (2023): small papules and pustules on an erythematous background, numerous confluent massive serous crusts. c – Macroscopic image of the scalp after initiation of trametinib therapy.
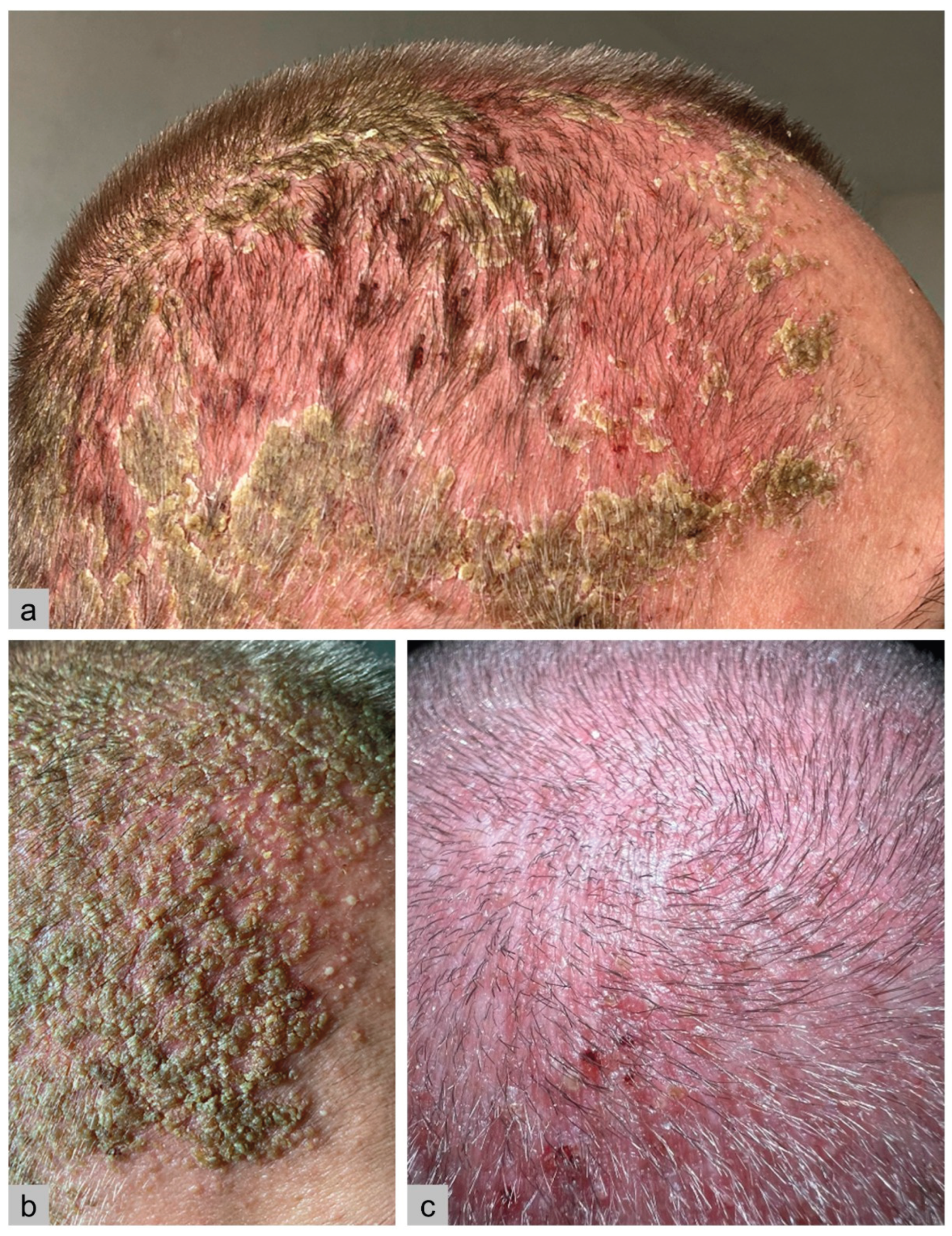
Figure 2.
Bone scintigraphy.
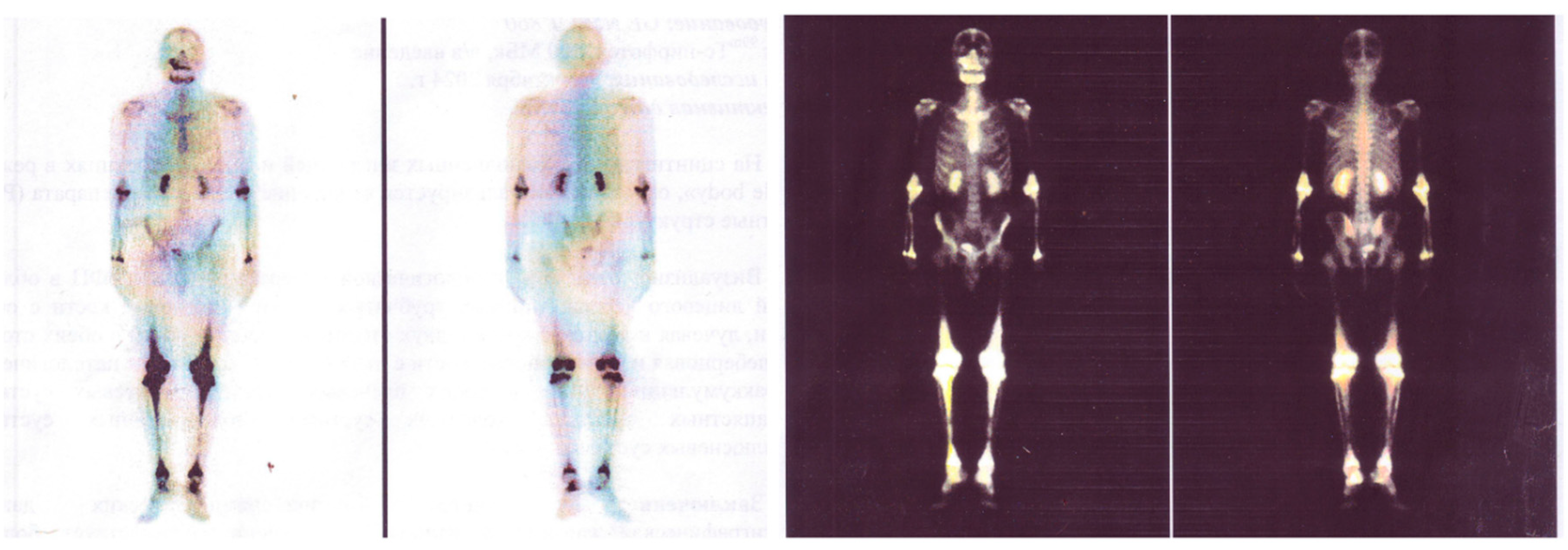
Figure 3.
Morphological and immunohistochemical characteristics of lesions in Langerhans cell histiocytosis. a – Kidney biopsy: multifocal mononuclear cell infiltration in the cortical interstitium and perinephric fat tissue without signs of glomerulopathy. b – Skin biopsy: dermal infiltrate composed of medium-sized cells with bean-shaped nuclei surrounded by eosinophils. Infiltrating cells show strong membranous expression of CD1a and weak nuclear-cytoplasmic Ki-67 expression (6%), confirming the diagnosis of Langerhans cell histiocytosis. CD68 demonstrates diffuse and perinuclear dot-like positivity. Abbreviations: H&E – hematoxylin and eosin; PAS – Periodic acid–Schiff. Scale bar: 100 μm.
Figure 3.
Morphological and immunohistochemical characteristics of lesions in Langerhans cell histiocytosis. a – Kidney biopsy: multifocal mononuclear cell infiltration in the cortical interstitium and perinephric fat tissue without signs of glomerulopathy. b – Skin biopsy: dermal infiltrate composed of medium-sized cells with bean-shaped nuclei surrounded by eosinophils. Infiltrating cells show strong membranous expression of CD1a and weak nuclear-cytoplasmic Ki-67 expression (6%), confirming the diagnosis of Langerhans cell histiocytosis. CD68 demonstrates diffuse and perinuclear dot-like positivity. Abbreviations: H&E – hematoxylin and eosin; PAS – Periodic acid–Schiff. Scale bar: 100 μm.
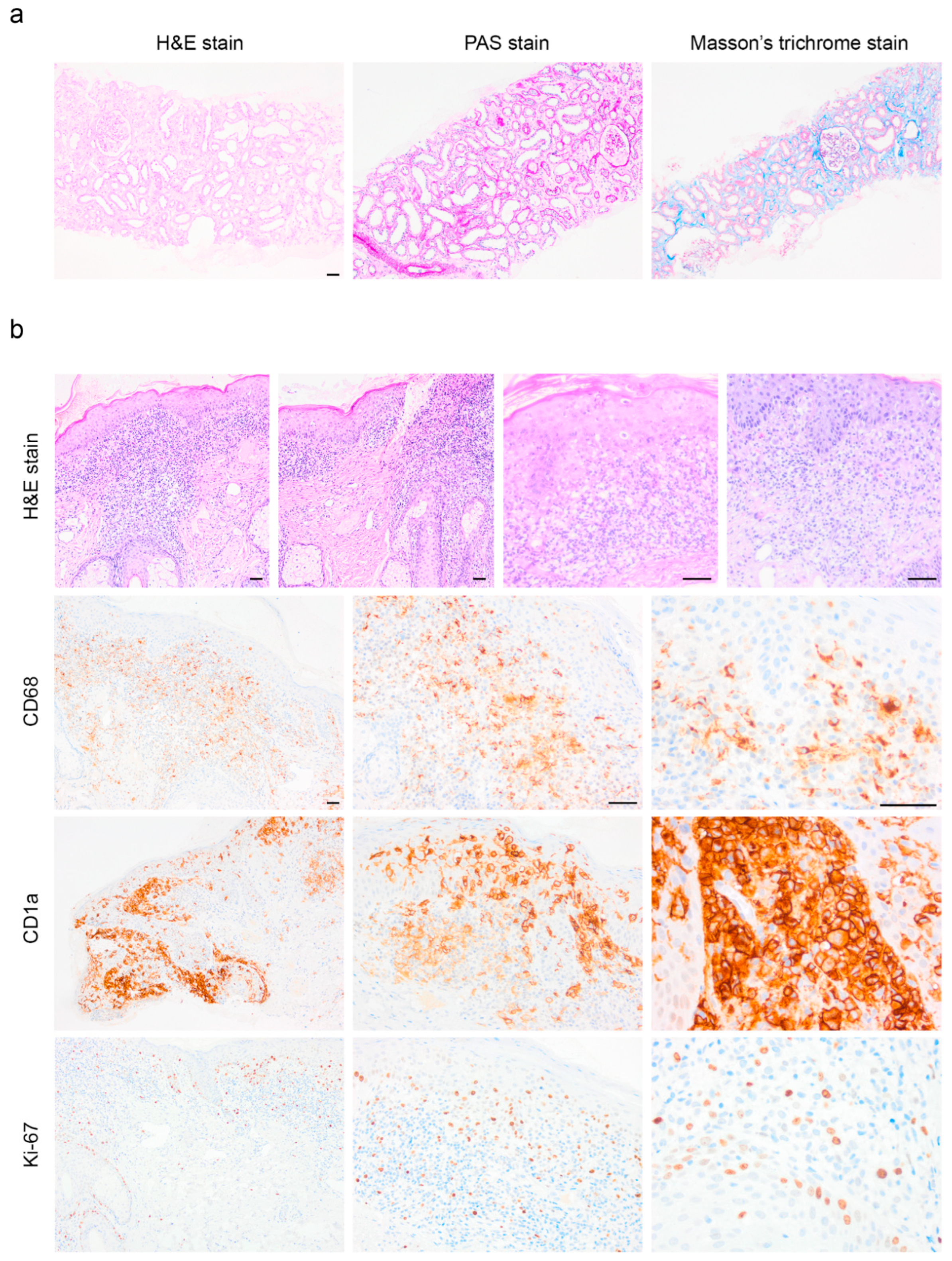
Figure 4.
a – Macroscopic image of the patient’s lower legs: multiple symmetrical papular eruptions and excoriations. The red circle marks the punch biopsy site. b – Macroscopic image of the right lower leg with the punch biopsy site marked. c – Dermatoscopic image of the rash on the right lower leg at the biopsy site. The image was taken using an optical adapter (Handyscope, Germany) compatible with a smartphone, 20× magnification.
Figure 4.
a – Macroscopic image of the patient’s lower legs: multiple symmetrical papular eruptions and excoriations. The red circle marks the punch biopsy site. b – Macroscopic image of the right lower leg with the punch biopsy site marked. c – Dermatoscopic image of the rash on the right lower leg at the biopsy site. The image was taken using an optical adapter (Handyscope, Germany) compatible with a smartphone, 20× magnification.
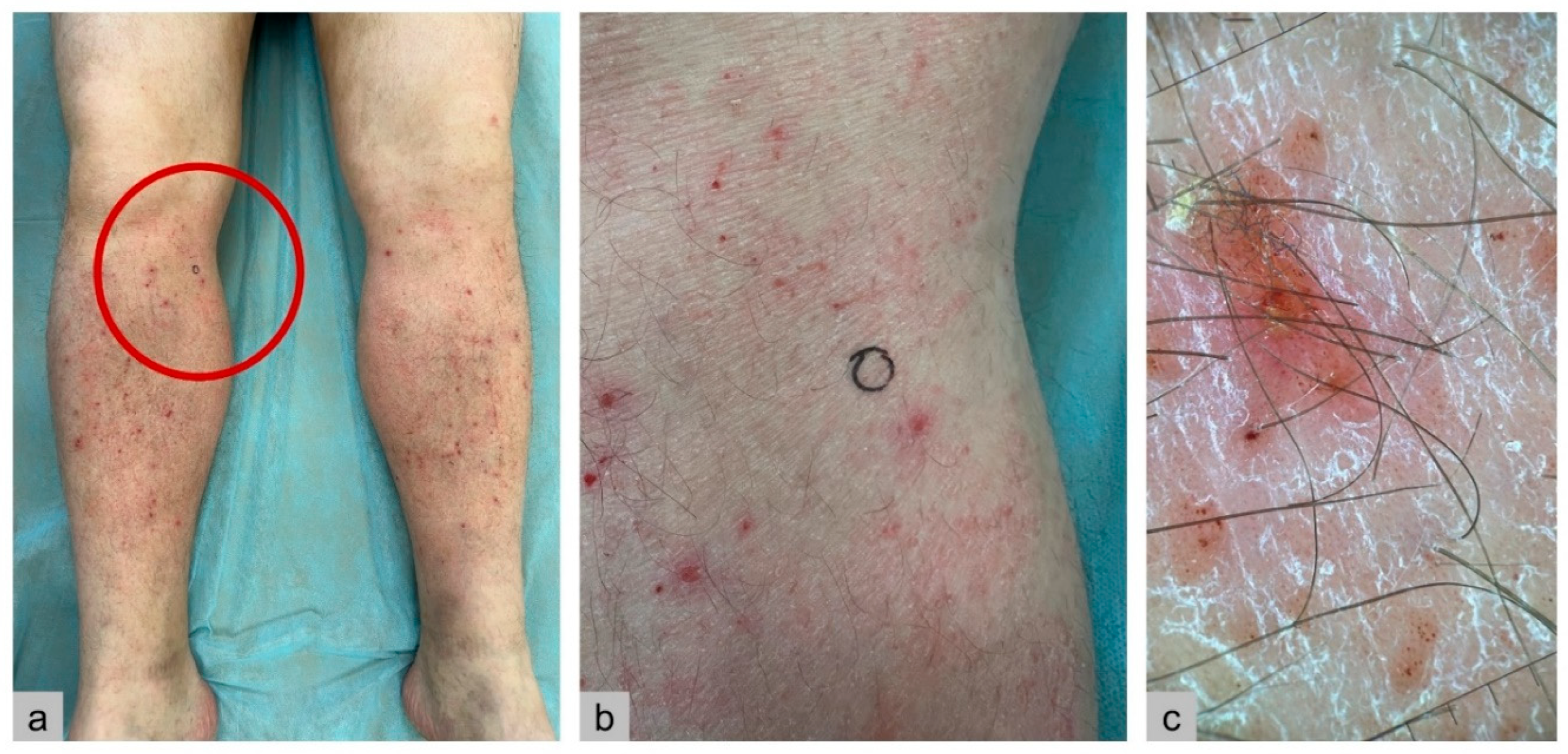
Figure 5.
Timeline of patient N. findings and management (2019-2024).
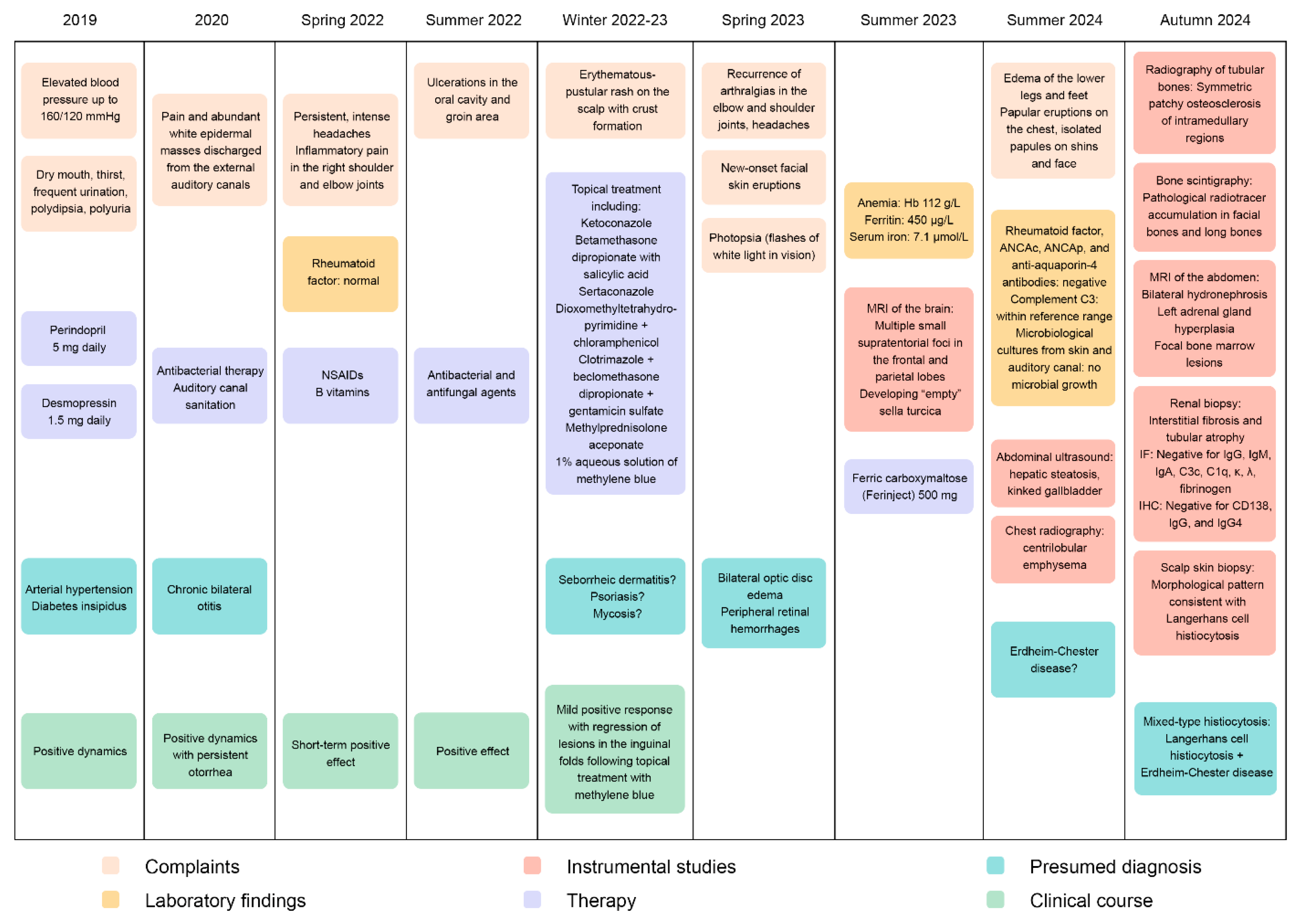
Figure 6.
Comparative analysis of somatic (skin only) and germline (blood) variants in a patient with histiocytosis (VAF > 5%). Only reference transcripts were considered. a – Venn diagram showing variant overlap between tissues. b – Distribution of VAF values. c – Variant localization and type distribution. d – Mutation frequency across candidate histiocytosis-associated genes. Abbreviation: VAF – variant allele frequency.
Figure 6.
Comparative analysis of somatic (skin only) and germline (blood) variants in a patient with histiocytosis (VAF > 5%). Only reference transcripts were considered. a – Venn diagram showing variant overlap between tissues. b – Distribution of VAF values. c – Variant localization and type distribution. d – Mutation frequency across candidate histiocytosis-associated genes. Abbreviation: VAF – variant allele frequency.
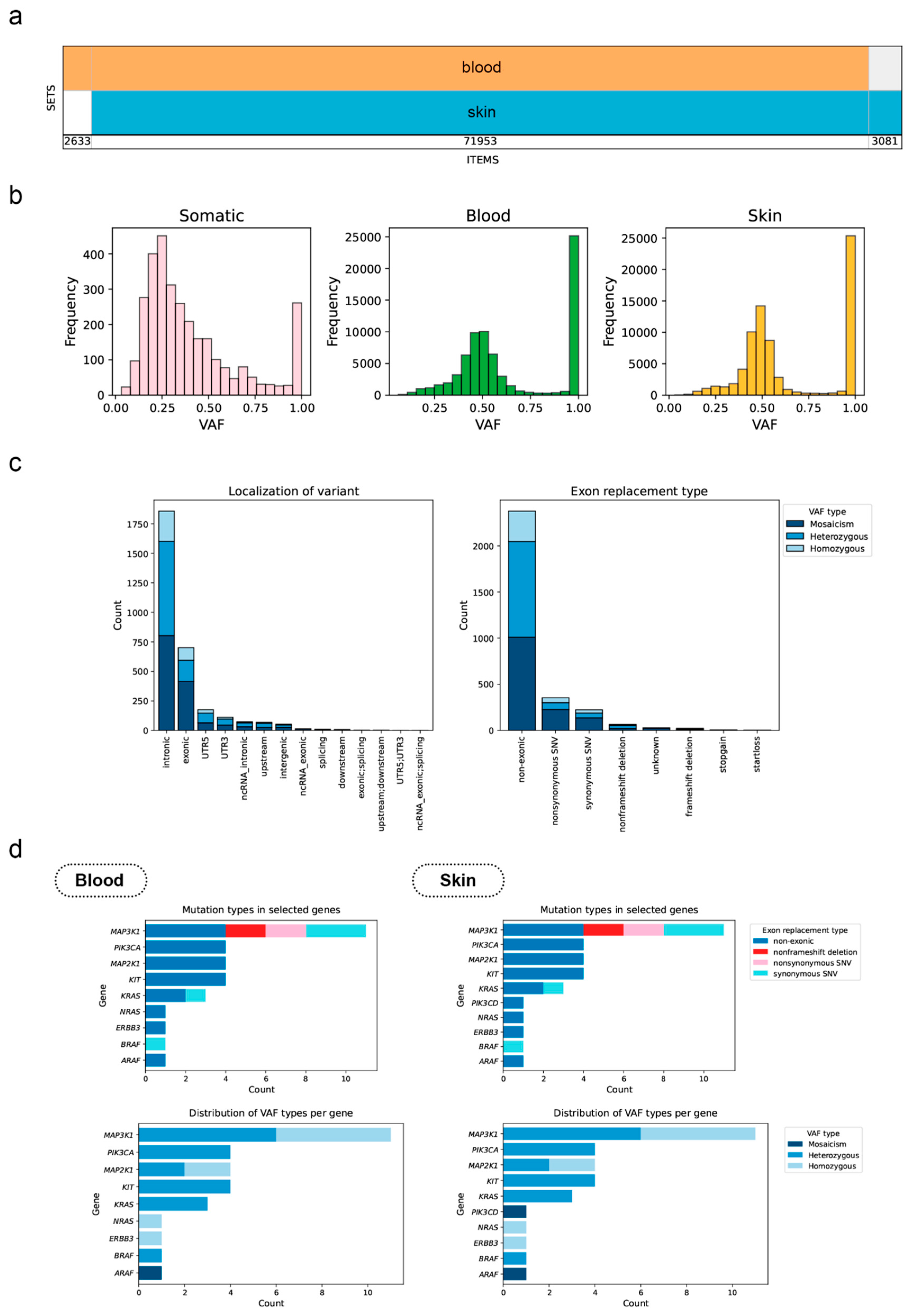
Figure 7.
Amplification curve of BRAF V600E mutation detection. Ct (Internal control) = 27. Ct (V600E mutation) = 34.3. Positive for a mutation if: Ct (V600E mutation)- Ct (Internal control) = 7.3 < 11.
Figure 7.
Amplification curve of BRAF V600E mutation detection. Ct (Internal control) = 27. Ct (V600E mutation) = 34.3. Positive for a mutation if: Ct (V600E mutation)- Ct (Internal control) = 7.3 < 11.
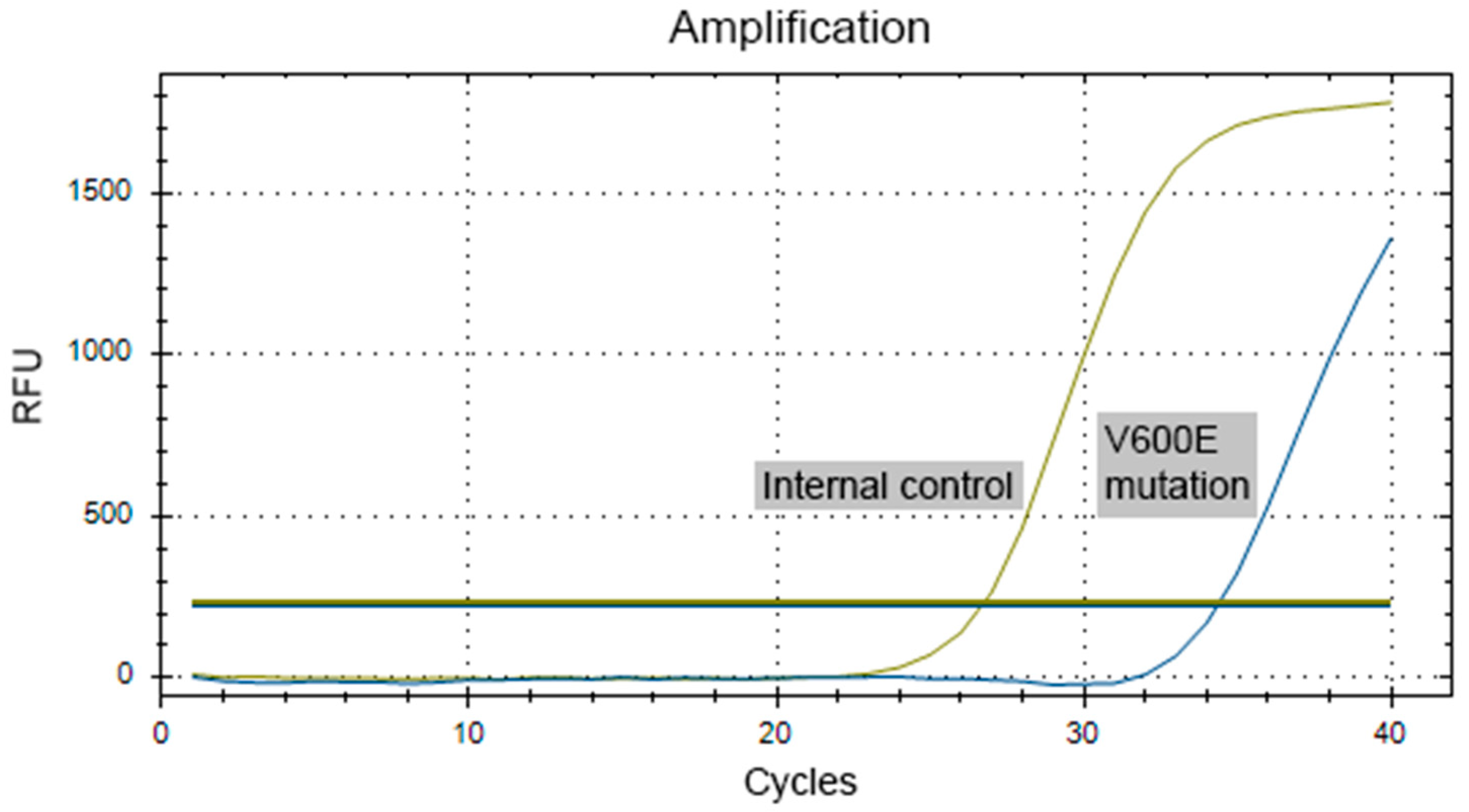
Figure 8.
Immunofluorescence analysis of the bone marrow trephine biopsy. a – CD11b (yellow), CD34 (green), Vimentin (red). The population of Vimentin+CD11b+CD34– cells was 11.4 times greater than that of Vimentin+CD11b–CD34+ cells (as a percentage of all Vimentin+ cells). b – CD8 (yellow), CD68 (red), tryptase (green). Scant Tr+ mast cells are shown, which hardly interact with other immunopositive cells. c – CD68 (green), S100 (yellow). CD68+ cells predominated (~56% of immunopositive cells), while CD68+S100+ cells were less frequent (~2%). Two types of CD68 staining were observed: perinuclear, characteristic of Langerhans cells, and diffuse, likely representing cells of the mononuclear phagocyte lineage. Scale bar: 100 μm.
Figure 8.
Immunofluorescence analysis of the bone marrow trephine biopsy. a – CD11b (yellow), CD34 (green), Vimentin (red). The population of Vimentin+CD11b+CD34– cells was 11.4 times greater than that of Vimentin+CD11b–CD34+ cells (as a percentage of all Vimentin+ cells). b – CD8 (yellow), CD68 (red), tryptase (green). Scant Tr+ mast cells are shown, which hardly interact with other immunopositive cells. c – CD68 (green), S100 (yellow). CD68+ cells predominated (~56% of immunopositive cells), while CD68+S100+ cells were less frequent (~2%). Two types of CD68 staining were observed: perinuclear, characteristic of Langerhans cells, and diffuse, likely representing cells of the mononuclear phagocyte lineage. Scale bar: 100 μm.
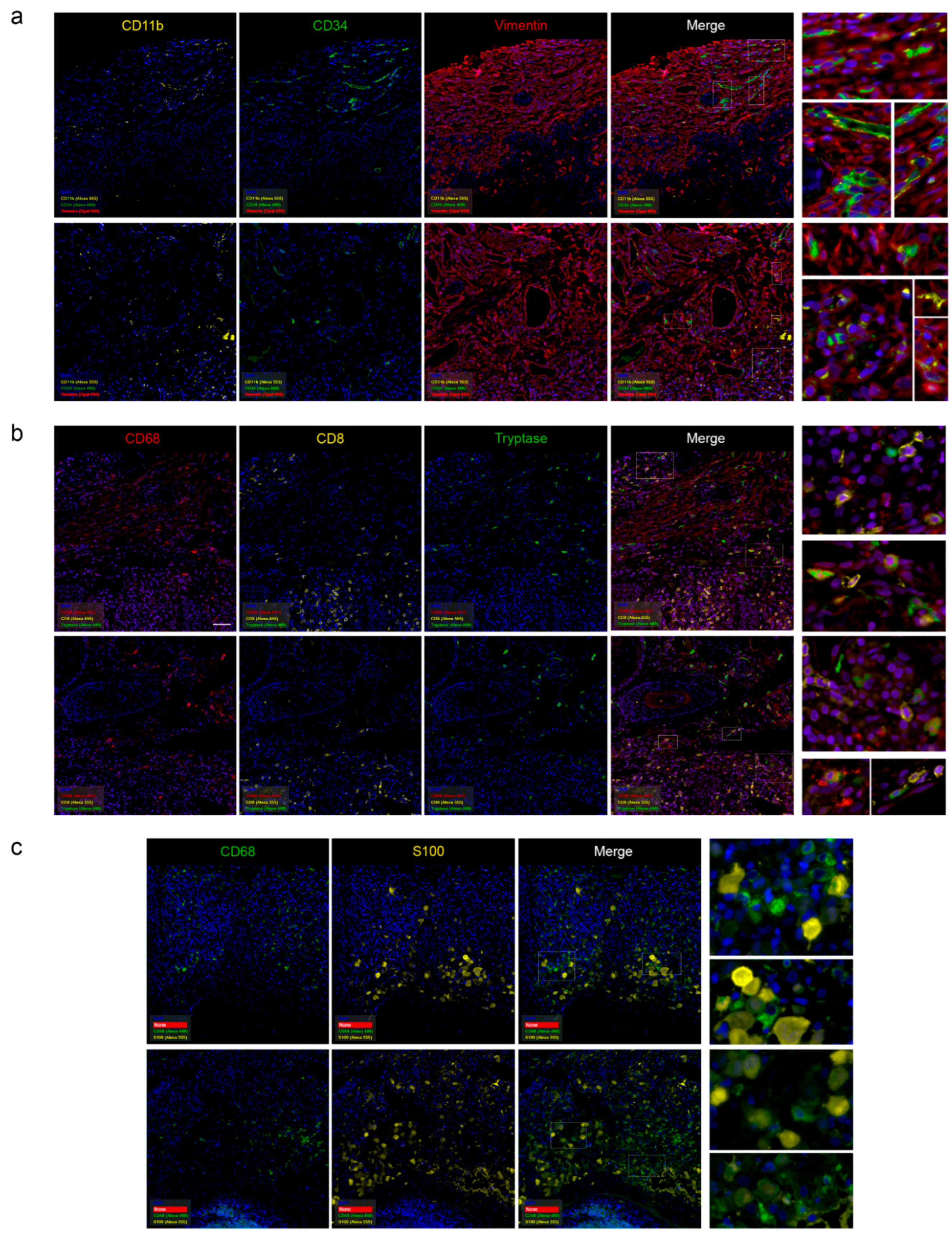

Table 1.
Clinical Manifestations of LCH [21].
Table 1.
Clinical Manifestations of LCH [21].
| Affected Organ/System | Clinical and Radiologic Features |
|---|---|
| Bones | Lytic bone lesions observed in 30–50% of cases on positron emission tomography-computed tomography (PET-CT). |
| Skin | Erythematous, papular eruptions; less commonly, involvement of oral, genital, or perianal mucosa. |
| Endocrine system | Pituitary insufficiency. |
| Nervous system | Focal or extensive lesions of the pituitary stalk, hypothalamus, or pineal gland. Less commonly, infiltration of the brainstem or cerebellum with development of ataxia and dysarthria. |
| Lungs | Cystic or nodular formations presenting with obstructive, restrictive, or mixed changes, pneumothorax. |
| Liver, spleen | Early-stage: parenchymal infiltration (hepatomegaly, tumor-like nodules, mild cholestasis). Late-stage: sclerosing cholangitis-like damage (severe cholestasis) rapidly progressing to terminal liver failure and death. |
| Bone marrow | Involvement with changes in complete blood count. |
| Lymph nodes | Lymphadenopathy. |
| Gastrointestinal tract | Diarrhea, abdominal discomfort, colonic polyps. |
| Cardiovascular system | Arterial stenosis, more common in mixed forms. |
Table 2.
Dynamics of laboratory parameters from May 2024 to January 2025.
| Parameter, units | 05/ 2024 |
06/ 2024 |
07/ 2024 |
08/ 2024 |
09/ 2024 |
10/ 2024 |
12/ 2024 |
01/ 2025 |
Reference Range |
|---|---|---|---|---|---|---|---|---|---|
| Hemoglobin, g/L | 112 | 108 | 114 | 123 | 141 | 126 | 131 | 130-150 | |
| MCV, fL | 83 | 89.1 | 85.9 | 80-99 | |||||
| MCH, pg | 28.3 | 28.9 | 27-31 | ||||||
| Leukocytes, × 109/L | 7.53 | 5.9 | 7.97 | 6.69 | 4-9 | ||||
| Platelets, × 109/L | 350 | 327 | 331 | 224 | 160-370 | ||||
| ESR, mm/h | 40 | 40 | 19 | 0-15 | |||||
| Ferritin, µg/L | 450 | 303.5 | 385 | 20-306 | |||||
| Serum Iron, µmol/L | 7.1 | 8.6 | 8.1 - 28.3 | ||||||
| Creatinine, µmol/L | 136.5 | 123.9 | 142.6 | 148.6 | 200 | 184.3 | 155.5 | 62-115 | |
| eGFR (CKD-EPI), mL/min/1.73 m2 | 58 | 65 | 55 | 52 | 36 | 41 | 49 | ||
| Urea, mmol/L | 5.46 | 7.43 | 8.3 | 9.12 | 6.5 | 2.8-7.2 | |||
| C-reactive protein, mg/L | 58.91 | 148.21 | 77.72 | 90.2 | 51.1 | 49.2 | 69.1 | 19.06 | 0-5 |
| Total bilirubin, µmol/L | 6.6 | 6.2 | 11.2 | 7.6 | 2-21 | ||||
| Direct bilirubin, µmol/L | 1.9 | 0-5 | |||||||
| Alkaline phosphatase, U/L | 97.4 | 106 | 30-120 | ||||||
| Urine specific gravity | 1007 | 1004 | 1012 | 1008-1025 | |||||
| Urine pH | 5.0 | 7.0 | 7.0 | 5.0-7.0 | |||||
| Urine sediment | No | No | No | ||||||
| Urine protein | 0 | 0 | 0 | 0-0.15 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



