
Submitted:
23 August 2025
Posted:
25 August 2025
You are already at the latest version
Abstract
Glioblastoma (GB) is an extremely aggressive tumour for which effective therapy is still in its infancy. Although several candidate therapeutics have been identified in functional preclinical assays, clinical trials have not supported their effectiveness in GB patients. One reason for their poor clinical efficacy is that available preclinical models poorly mimic GB characteristics in patients. In this review article, we provide a comprehensive overview of GB preclinical models, which are classified according to their origin (animal or human), type and modelling strategy (2D or 3D cell culture, in vivo grafting or in silico modelling). Moreover, the article compares developing cut-ting-edge technologies, including GB derived organoids, bioprinting, mi-crofluidic devices, and their multimodal integration in tumor-on-chip sys-tems, which aim to replicate the tumour microenvironment with high pre-cision. In silico and in vivo approaches are also reviewed, including zebrafish transplantation models. The respective costs, benefits and purposes of each model system and/or modelling strategy are discussed in detail and com-pared. We highlight that the most appropriate, or combination of, GB pre-clinical models must be selected (or even customized) based on the specific aims and constraints of each study. Finally, to improve the reliability and translational relevance of GB research, we propose a practical roadmap that addresses critical challenges in preclinical assay development, ranging from short-term adjustments to long-term strategic planning.
Keywords:
glioblastoma
; preclinical models
; clinical translation
; strengths and weaknesses analysis
; organoids
; microfluidics
; tumor on chip
; bioprinting
; animal models
; in silico modeling
1. Introduction
Glioblastoma (GB) is the most frequent and aggressive primary malignant tumor of the central nervous system in adults, classified as grade IV by the World Health Organization. Despite standard treatment, which includes surgical resection, followed by radiotherapy and concomitant and adjuvant temozolomide (TMZ) chemotherapy, median overall survival remains approximately with a prognosis of 12–15 months and just 3–5% of survival over 5 years [1,2,3]. The treatment of GB poses significant therapeutic challenges due to several factors: the tumor’s highly invasive nature, rapid development of resistance to radio- and chemotherapy, early recurrence, marked heterogeneity and the presence of the blood-brain barrier (BBB), which most pharmacological compounds are unable to penetrate [3,4]. GB cells are capable of invading healthy brain tissue by degrading the extracellular matrix (ECM), allowing them to access both the brain parenchyma and perivascular spaces. Their highly migratory nature enables them to evade complete removal during surgery, with the result that tumor recurrence is frequently observed in close proximity to the original site [5,6]. The invasive behaviour is closely linked to the organization and directionality of cell movement. This process involves the remodelling of the ECM and the cytoskeleton, interactions through cell-cell and cell-ECM adhesion, and the expression of epithelial-mesenchymal transition (EMT) features [7,8]. Recurrence is also related to the tumor’s intrinsic and acquired chemoresistance mechanisms, including the expression of DNA repair enzymes (e.g., O-6-methylguanine-DNA methyltransferase, MGMT), altered drug efflux systems, metabolic reprogramming, and the survival of therapy-resistant glioma stem-like cells (GSCs), which play a key role in tumor growth and in therapeutic resistance, exhibiting low responsiveness to both chemotherapies and radiotherapies, which further decreases following repeated chemoradiation treatments [4,9,10,11]. Another major challenge for GB treatment is its extreme cellular and molecular heterogeneity, that includes genetic mutations, different epigenetic profiles and complex interactions with the tumor microenvironment. This heterogeneity makes it extremely difficult not only to treat effectively multiple patients but also to target all tumor cells of the same patient at a given time within different regions or over time as the tumor evolves [3,12,13,14]. Therapeutic development is further hindered by the presence of the BBB, which is a highly specialized and selective interface that preserves the central nervous system (CNS) homeostasis by shielding the brain from detrimental substances circulating in the bloodstream, including pathogens like viruses. Its complex architecture consists of several layers, primarily composed of non-fenestrated endothelial cells of the brain microvasculature, tightly connected by junctional complexes. The BBB's protective function arises from a combination of physical, transport-related, and metabolic mechanisms within the endothelial cells, which are finely regulated through interactions with various vascular, immune, and neural cell populations [15,16] . While this barrier is crucial for maintaining brain health and function, in pathological conditions like GB, it poses a major obstacle to the effective delivery of therapeutic agents, thereby reducing treatment efficacy [16,17,18,19]. Even though the integrity of the BBB is significantly impaired in GB, this is not beneficial for therapeutic drug delivery. Instead, the leakage of the BBB, also named the blood-tumor barrier (BTB), which has unique features respect to BBB, allows infiltration of tumor cells, inflammatory mediators, and pro-tumor signals, contributing towards the GB highly invasive nature [20,21,22]. Due to all these features, clinical management of GB patient is nowadays extremely challenging. Figure 1 summarises the main characteristics of GB and the principal therapeutic treatment currently used, that however, are still inefficient in avoiding tumor recurrence.
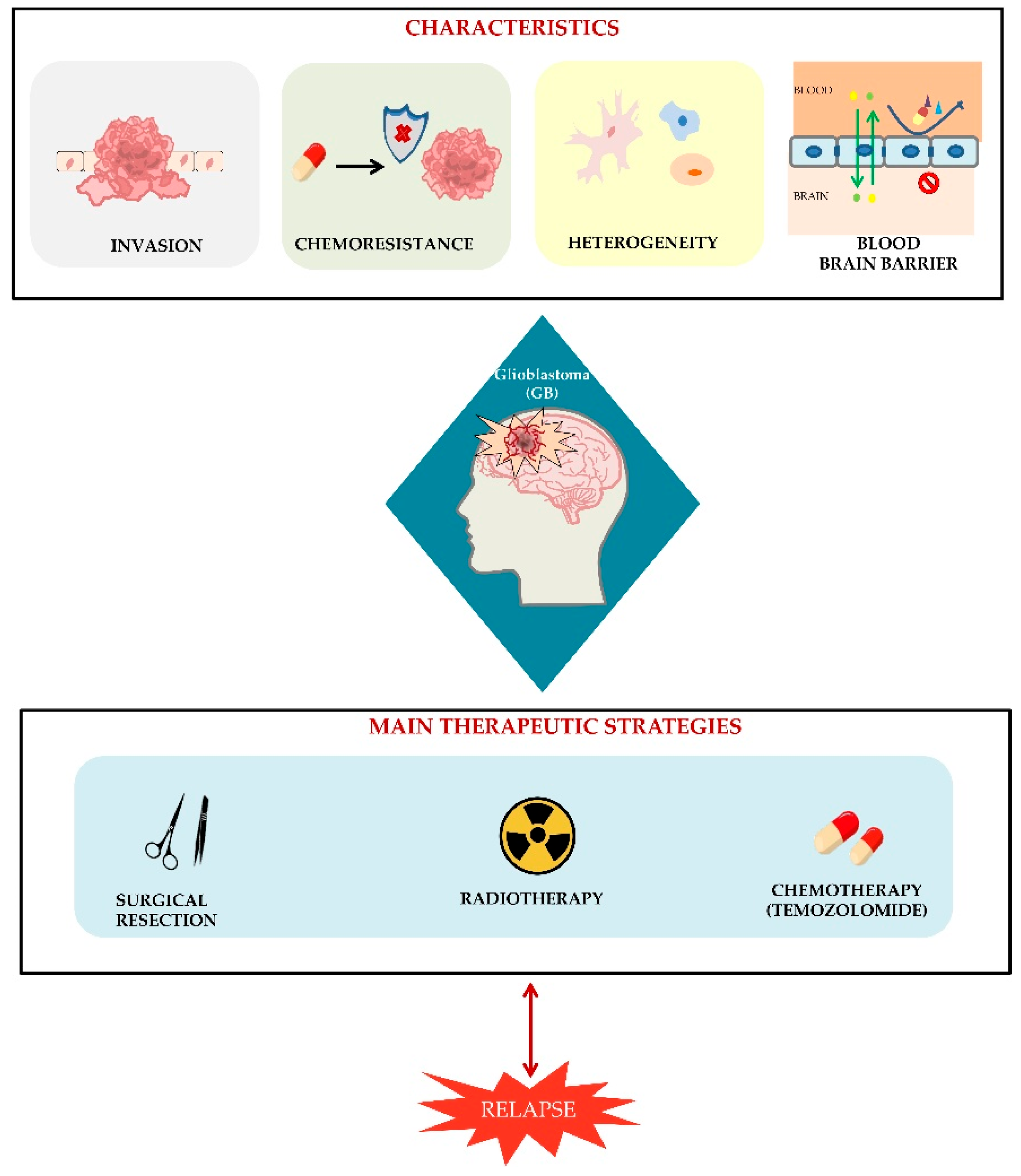
Figure 1.
Schematic representation of the main hallmarks and current therapeutic strategies for glioblastoma (GB). GB is characterized by aggressive and invasive growth, high resistance to chemotherapeutic agents (chemoresistance), cellular heterogeneity and the presence of a highly selective blood–brain barrier (BBB) that limits drug delivery to the tumor site. Standard therapies include surgical resection, radiotherapy and temozolomide-based chemotherapy. Despite this multimodal approach, tumor relapse remains common.
Figure 1.
Schematic representation of the main hallmarks and current therapeutic strategies for glioblastoma (GB). GB is characterized by aggressive and invasive growth, high resistance to chemotherapeutic agents (chemoresistance), cellular heterogeneity and the presence of a highly selective blood–brain barrier (BBB) that limits drug delivery to the tumor site. Standard therapies include surgical resection, radiotherapy and temozolomide-based chemotherapy. Despite this multimodal approach, tumor relapse remains common.
Emerging therapeutic strategies involve immunotherapies, nanoparticles and targeted therapy [3,23]. Immunotherapy proposes to use check point inhibitors (CPIs), vaccines or chimeric antigen receptor T (CAR-T) to promote immune response against cancer cells [21,24,25]. Nanoparticles include extracellular vesicles naturally produced by cells or biomimetic nanostructures, combining the advantages of both artificial and natural nanocarriers. Both seem promising candidates for drug-delivery to counteract GB progression and invasion [4,26,27]. Targeted therapy is designed to selectively inhibit molecular pathways dysregulated in GB cells without impair normal cells, as for example EGFR, PDGFR, VEGF or PI3K/AKT/mTOR that arecritical for tumor progression [28,29].
However, despite considerable efforts to elucidate GB tumorigenesis pathways and develop novel treatments, many therapies that show promise in preclinical studies do not translate into effective patient outcomes. Differences observed between preclinical and clinical outcome underscore the importance of developing a robust translational platform capable of faithfully mimicking the biological, molecular, and structural aspects of GB (including its heterogeneity, invasiveness, and interactions within the brain microenvironment) An international panel of clinicians and researchers convened by the Cancer Research UK identified already in 2019, the development of more accurate and predictive preclinical models as one of the main challenges to face for curing patients with brain tumors [30]. This is the context in which our review is located, providing a comprehensive and updated overview of the current models used in GB research. The review aims to highlight advantages, limitations and applicability of the different models in the context of translational oncology. Our final goal is promoting an aware choice of the current GB model for biological and therapeutic assays as well as suggesting future directions to develop a multimodal integrated approach to GB preclinical research.
2. Glioblastoma Preclinical Models: An Overview
Preclinical models play a key role in elucidating GB pathology and testing the efficacy, dosage and safety features of therapeutic factors prior to entering clinical phase trials. To achieve an ideal experimental setup, GB preclinical models should satisfy the following specific standards: (i) their genetic characteristics and intratumoral heterogeneity should resemble those of patients with GB; they should effectively replicate the GB microenvironment and its relationship with the human brain; and they should be reproducible and consistent over time [31]. Although the first GB models were unable to meet all these requirements, they enabled scientists to begin understanding and analysing the characteristics of GB, and to start testing potential therapeutic treatments. Today, increasingly sophisticated and complex GB models are being developed that closer and closer resemble human GB tumors.
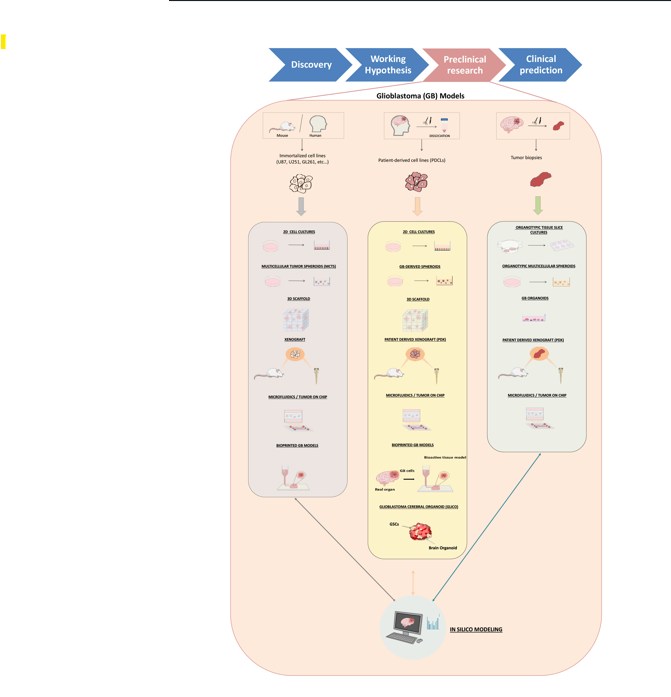
In the following paragraphs, GB models are first divided according to whether they originate from animals or humans, and then according to the different modelling strategies in the absence or presence of the tumour microenvironment (TME) cells. The specific strengths, weaknesses, purposes and examples of applications have been discussed. Figure 2 offers a panoramic of how the different GB model systems are represented in the literature, by showing the number of articles that mention each specific one in the title or abstract, broken down by one (August 2020–July 2025) or two (August 2015–July 2020 and August 2020–July 2025) time periods.
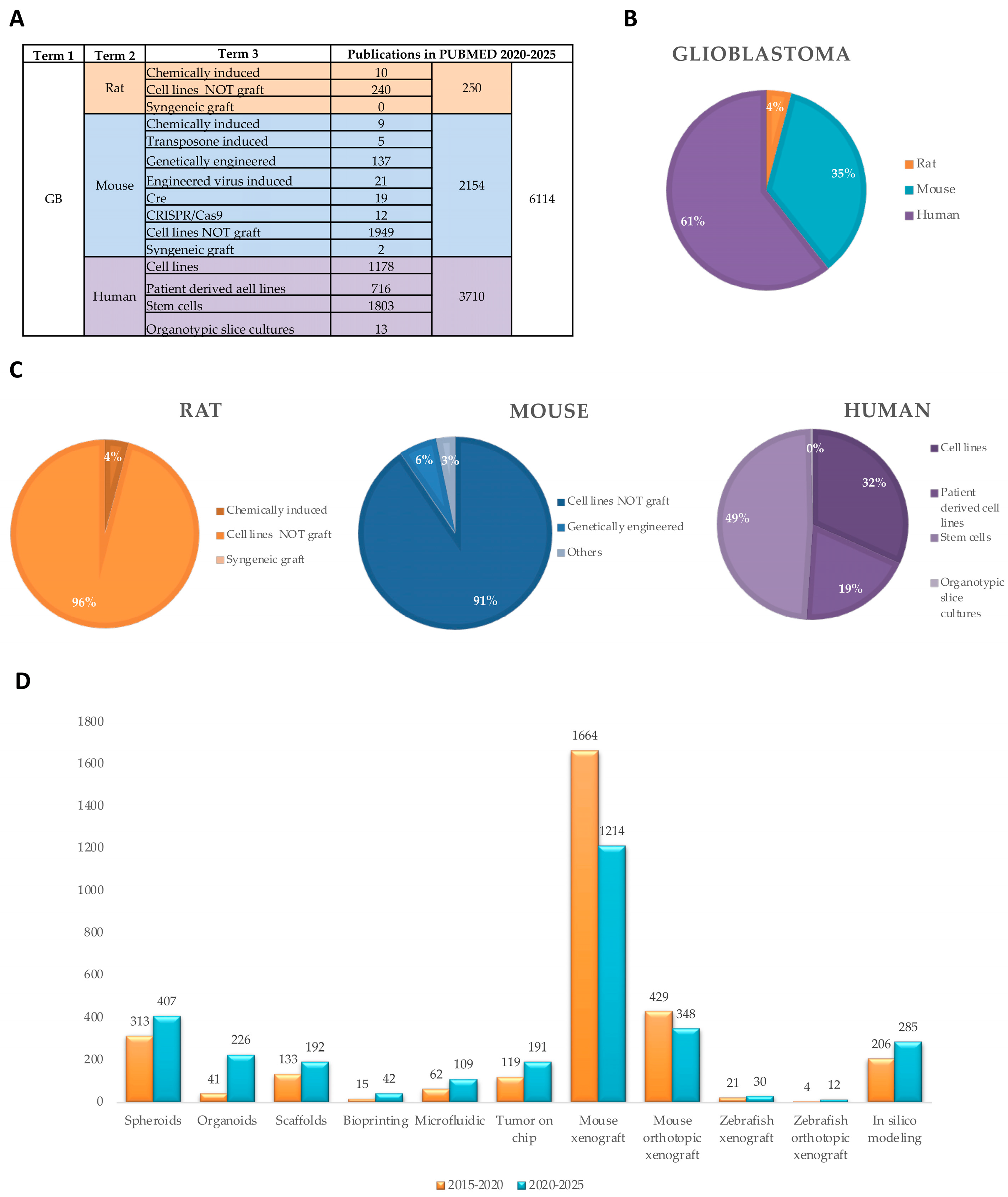
Figure 2.
Mentioning in the literature of the different GB models. (A) Table summarizing the number of articles published in the last five years (August 2020-July 2025) by searching PUBMED with the indicated terms. (B) Percentage of publications on Glioblastoma divided for origin (rat, mouse or human). (C) Percentage of publications on Glioblastoma rat (orange), mouse (blue) or human (purple) models, divided for model type (i.e. cell lines, chemically induced animals or syngenic grafts). (D) Number of publications mentioning the different types of GB modeling strategies (3D, in vivo and in silico) in the indicated time periods.
Figure 2.
Mentioning in the literature of the different GB models. (A) Table summarizing the number of articles published in the last five years (August 2020-July 2025) by searching PUBMED with the indicated terms. (B) Percentage of publications on Glioblastoma divided for origin (rat, mouse or human). (C) Percentage of publications on Glioblastoma rat (orange), mouse (blue) or human (purple) models, divided for model type (i.e. cell lines, chemically induced animals or syngenic grafts). (D) Number of publications mentioning the different types of GB modeling strategies (3D, in vivo and in silico) in the indicated time periods.
3. Glioblastoma Origin
3.1. Animal
Here we provide an overview of the main GB models derived from animal source, showcasing their origin, key features. strengths, weaknesses and purpose, as also summarized in Table 1. The development along time of the GB models of animal origin is reported in Figure 3.

Table 1.
Main Glioblastoma Models of animal origin.
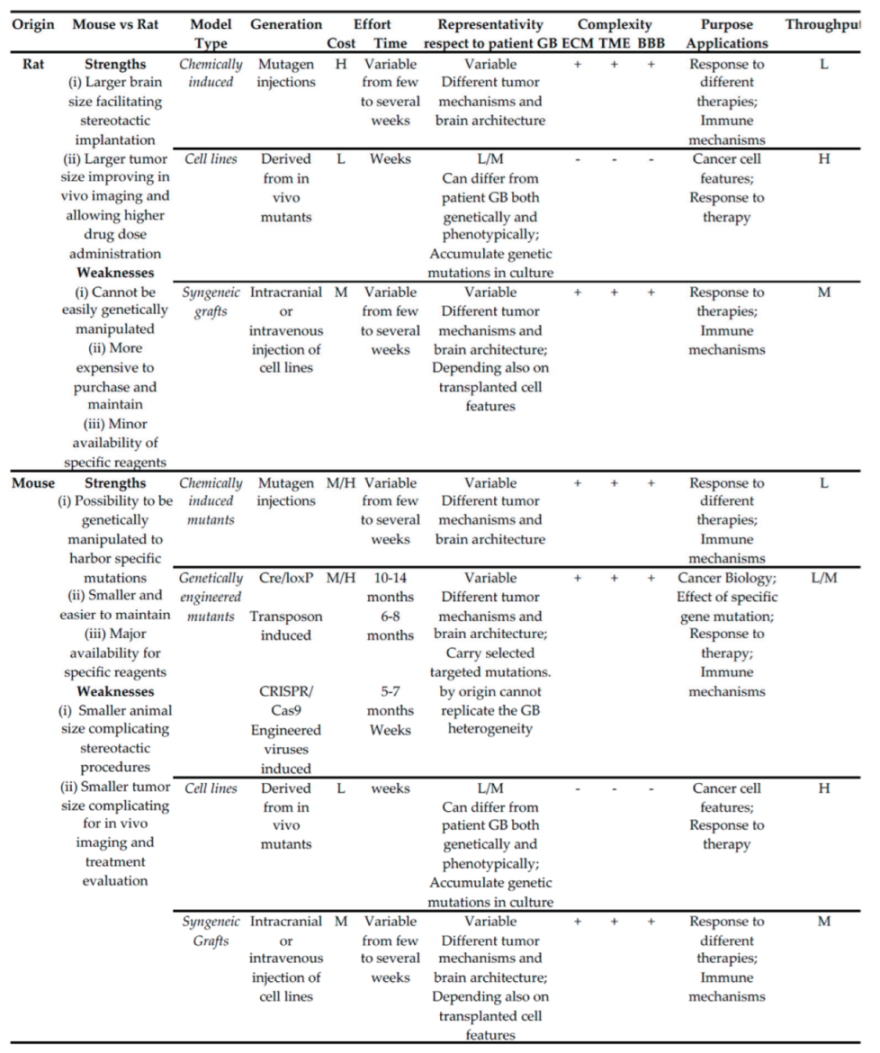 |
Abbreviations: BBB, blood-brain barrier; ECM, extracellular matrix; H, high; L, low; M, medium; TME, tumor microenvironment.
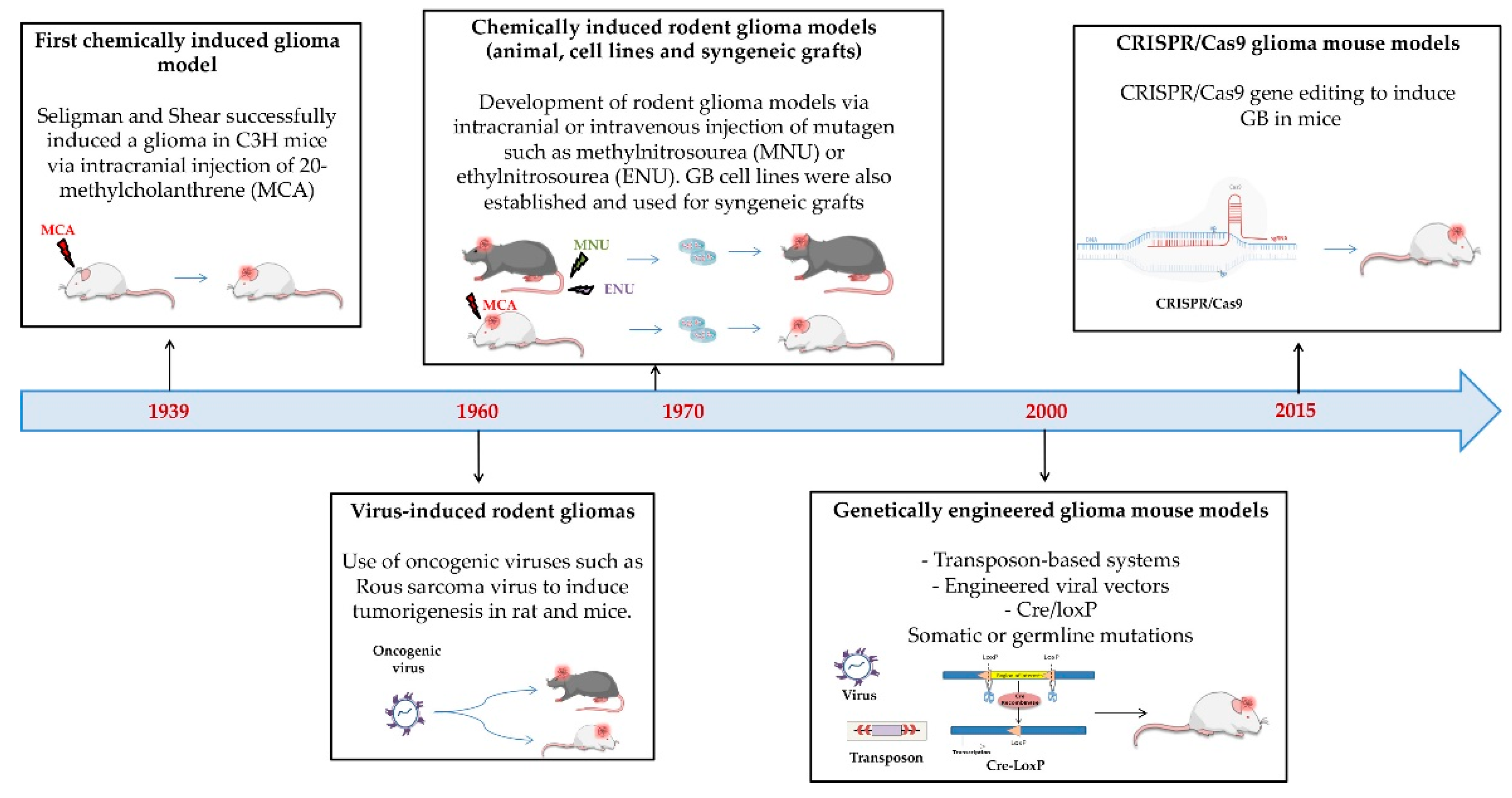
Figure 3.
Timeline emphasizing the principal milestones in the development of GB models derived from animal sources.
Figure 3.
Timeline emphasizing the principal milestones in the development of GB models derived from animal sources.
3.1.1. Origin and Characteristics
- Chemically and Genetically Induced In Vivo Models
For several human pathologies, including cancer, research has attempted to establish animal models that could resemble the pathological features of human diseases. According to a general consensus, valid GB animal models should possess the following requisites: (1) origin from glial cells; (3) predictable and reproducible tumor growth rate; (4) glioma-like growth characteristics within the brain such as BBB leakage, an invasive behavior and neovascularization, (5) sufficient host survival time for in vivo analysis and therapeutic studies; (6) possibility to propagate and clone them in vitro as continuous cell lines and in vivo by serial transplantation [32]. Different GB models have been first developed in rodents by directly inducing tumor formation through mutagen injections intracranially or intravenously. In 1939 the first glioma tumor was successfully induced by Seligman and Shear, after implanting the mutagen 20-methylcholanthrene intracranially into C3H mice [33]. Since the 1960s viruses, such as Rous sarcoma virus, were also used to induce tumorigenesis in both rats and mice [34,35], even though with several limitations, such as the incomplete penetrance of the induced tumor, safety concerns and extremely higher costs [36]. Since the late 1960s, several rat GB models (i.e. C6, 9L, T9, RG2, F98, CNS-1, BT4C and RT-2) have been produced by specific mutagen injection and used to study GB biology as well as for evaluating the efficacy of various therapeutic strategies [32,37]. Among the most studied, C6 glioma rat models were produced by repetitively administering methylnitrosourea (MNU) over a period of approximately 8 months to adult outbred Wistar rats, whereas 9L gliosarcoma tumor model was obtained by injecting MNU for 26 weeks into inbred Fisher rats [38,39]. RG2 and F98 gliomas were both chemically induced by a single ethylnitrosourea (ENU) injection to pregnant Fischer rats, the progeny of which developed brain tumors that subsequently were propagated in vitro and cloned [40]. Until 2000s, rat tumor models were wider used than mouse ones, that were mainly limited to the GL261 mouse models, obtained by intracerebral injection of methylcholanthrene into C57Bl/6 mice, followed by serial intracranial and subcutaneous transplantation of tumor fragments [41,42]. C6 and 9L rat glioma models had a circumscribed growth pattern, whereas both rat F98 and GL261 developed infiltrative tumors [37]. With the incoming possibility of genetically manipulate mouse embryonic stem cells, transgenic cancer mouse models definitely gained the upper hand. Genetically engineered mouse (GEM) models mice can been produced by different methods, including Cre/loxP traditional or inducible systems, transposon usage, CRISPR/Cas9 systems and engineered viruses (both retrovirus and lentivirus) [36,37,43]. The turnaround time is extremely variable from 10-14 months for Cre/loxP system to few months for the CRISPR/Cas9 strategy and few weeks in the case of transgene delivery through engineered viral vectors. The genetic alteration for most systems can be in the germline or somatic, in the case of direct virus injection it is only somatic [36,43].
Cell Lines
In most cases, cells were derived from the GB animal models obtained, propagated in vitro, and cloned to obtain stable cell lines. Cell lines were named after the glioma tumor model from which they were derived, and used in both in vitro testing and in vivo transplants. The most used rat cell lines were the C6, 9L and F98, each one carrying specific gene mutations, some of them in common with the ones found in human GB [32,37]. C6 cell line, for example, shows higher expression of Ras oncogene with consequent Ras pathway activation [44], whereas 9L cells carry mutation on p53 tumor suppressors gene and higher EGFR expression [45,46], and F98, overexpressing Ras, PDGFb, cyclin D1, cyclin D2, and EGFR and downregulating BRCA1 is the most similar to human GB [46,47]. Among the most used mouse cell lines there were GL261, sharing many genetic mutations with human GB, including Ras point mutations and activation along with loss of oncosoppressor genes such as p53 [48]. Even though chemical-induced animal GB models have long been outdated, several mouse and rat cell lines established from those tumors have been used for in vitro assays and allograft models [36] as well as for the set-up of GB 3D culture models .
Syngeneic Grafting Models
Grafting of tumor cells inside an animal model is able to ensure a steady and constant supply of nutrient, growth factors and oxygen, ideal to study tumor progression. Syngeneic grafts, allografts and xenografts can be distinguished, depending if the donor and the host have the same genetic background (syngeneic), have different genetic background but are of the same species (allografts) or belong to different species (xenografts). Moreover, transplants can be heterotopic, if occur in a different location respect to the site of origin, mainly subcutaneously, or, orthotopic, when engrafted cells are implanted in the same location, which for GB is the brain [49]. Syngeneic orthotopic GB grafts have been first used in rat of the same strains, to study GB progression and response to therapy, avoiding immune rejection [32,36]. Among the most widely studied, there was the 9L rat gliosarcoma model, characterised by a high immunogenicity and a circumscribed tumor growth pattern, with sharp margins and poor invasion into the contiguous normal brain and F98, showing exactly the opposite characteristics [37]. For this reason, F98 is definitely more similar to human GBM, characterised by high invasiveness. For C6 instead, having been generated in outbred Wister rats, there is no syngeneic host in which it can be propagated, but allografts in Wister rats have been performed. C6 cells are highly immunogenic and poorly invasive [50]. Mouse cell lines used in allograft mouse model include CT-2A and 4C8, but especially GL261, resembling closer human GB features, and characterised by high immunogenicity [36].
3.1.2. Purposes, Strengths and Weaknesses
The use of animal models has the strong advantage to allow the study of the interaction between the developing tumor and the TME as well as the behaviour of the BBB or BTB. These aspects are particularly relevant respect to both GB pathogenesis and therapeutic development. Rat and mouse brain tumor models have been the most widely used in experimental neuro-oncology and led to the development of several approaches for the treatment of human GB. Rats were wider used than mice until 2000s, despite higher costs of purchase and maintenance. In fact, they had undoubted advantages, such as the larger size of their brain (1200mg vs 400 mg) facilitating intracranial stereotactic procedures, and consequently larger tumor size, longer time until death, which allowed better in vivo imaging and prolonged testing of therapeutic agents [32]. However, with the onset of genetic manipulation, which is much easier in mouse than in rat, transgenic mouse models prevailed. Independently from the used strategy, transgenic mice were based on the introduction of a specific mutation in the target gene(s). Therefore, differently from the tumor models generated by a general and untargeted mutagenesis, transgenics have a precise known genetic signature. This feature has the advantage of allowing the study of the effect of specific targeted genetic mutations, but at the same time, cannot reflect the typical GB heterogeneity. Syngeneic transplant models in both mouse and rats have been used to study the immune mechanism for radiation therapy, immune checkpoint therapy, vascular endothelial growth factor (VEGF) therapy and vaccine therapy [36]
Rodent tumor models have been mainly use in neuro-onocology to evaluate the efficacy of a plethora of therapeutic treatments, including chemotherapy, radiation therapy, antiangiogenic therapy, photodynamic therapy, oncolytic viral therapy, gene therapy and treatment with proteosome inhibitors or toxins [36,37,47,51,52,53,54,55,56,57]. However, the strong limitation of both chemically induced, genetically engineered and syngeneic grafting rodent models, despite a similar genome, relies on the genetic and phenotypic difference between rodent and human tumorigenesis process. Tumor growth and progression might use different paths and, therefore, targeting molecules and therapeutics having a positive antitumoral outcome in animal models, might fail to function in patients [58]. Moreover, even though rodent and human brain architecture has several similarities, human brain and neocortex is significantly more complex, and human glia possesses different features respect to mice [59,60,61]. For these reasons, deriving and studying human GB cells from patients to compare the different results obtained is fundamental. Ethical concerns have also contributed to put aside these animal models in favour of cutting-edge technologies able to reproduce tumor interactions with its microenvironment.
3.2. Human
Here, we describe the main GB models originating from human sources, including their origin, main attributes, benefits, constraints, and applications. Table 2 provides a summary of the more relevant info, whereas Figure 4 summarizes the key milestones in the development of GB models from human sources.
Table 2.
Main Glioblastoma Models of human origin.
 |
Abbreviations: BBB, blood-brain barrier; bFGF, basic fibroblast growth factor; ECM, extracellular matrix; EGF epidermal growth factor; H, high; iPS, induced pluripotent stem cells; L, low; M, medium; TME, tumor microenvironment.
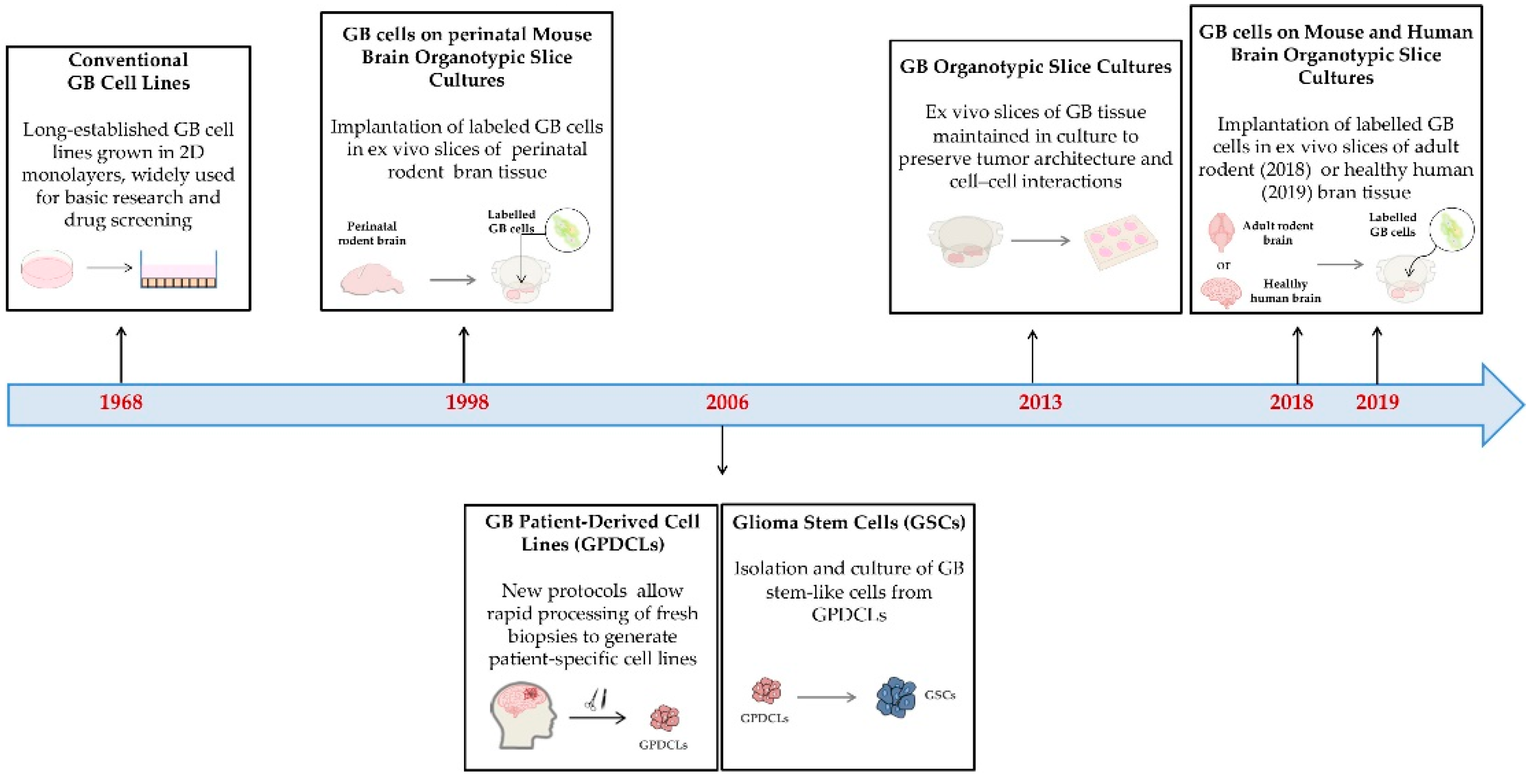
Figure 4.
Timeline showcasing the key milestones in the development of GB models derived from human sources.
Figure 4.
Timeline showcasing the key milestones in the development of GB models derived from human sources.
3.2.1. Origin and Characteristics
Conventional Cell Lines
In 1968, glioma human cell lines were first isolated to obtain immortalized and stabilized long term cultures [62]. During the decades, several cell lines (i.e. U87-MG, U251-MG, T98G, LN229, GBM) have been established from patient GB and immortalized to make them proliferate indefinitely. Despite several common features, the different cell lines show high genotypic and phenotypic heterogeneity, largely due to the heterogeneity of the tumors of origin likely as well as of the different cell types inside the same tumor [63]. Human tumor cell lines have been traditionally used and are still in use to understand the molecular basis of human GB and to test candidate drugs affecting specific GB features [64,65,66,67,68]. Among GB cell lines, the most widely studied are the U87-MG, isolated from malignant gliomas from a male patient, and characterized by an epithelial morphology. U87-MG have been studied in a plethora of in vitro functional assays, to test cell viability, proliferation, migration and chemoresistance of GB cells. Moreover, they have been successfully transplanted subcutaneously and intracranially in mouse models, where they show fast growth, with a median survival around 22 days [69]. U251-MG is a pleomorphic/astrocytoid cell line also used for both in vitro assays and mouse xenografts, where they grow and form tumor very fast with a median survival around 28,5 days [69]. The different cell lines differ for gene expression profile, and therefore functional characteristics including proliferation and migration rate, invasion, and colony formation capacities. Therefore, several studies have compared different cell lines, highlighting similarities and discrepancies [63,70].
Patient-Derived Cell Lines
Cell lines derived from patient GB have been first established in 2006 by Lee and coworkers [71]. GB patient-derived cell lines (GPDCLs) differ from traditional cell lines because they derive from fresh biopsies processed with 2-3 hours using specific protocols that allow to preserve in vivo biology of the tumor of origin. Therefore, GPDCLs have better chance to recapitulate GB genetic profile, gene expression pattern and, then, its histological features [72,73,74]. Interesting, since 2015 at least two biobanks have been developed collecting annotated and validated GPDCLs, classified according the transcriptional GB subtypes, mesenchymal, proneural, neural and classical [75,76].These collections are open resources which undoubtedly constitute a precious opportunity for both basic and translational GB research. Culture conditions used for deriving GPDCLs preserve also GSC identity and features [69].
- Glioma
- oma stem cells
GSCs, due to activated signaling pathways (i.e. Wnt/-catenin, Notch, PI3K, and JAK/STAT pathway), are the principal responsible for resistance to radiation and conventional chemotherapeutics, as well of the invasive GB behavior, being often localized to the leading edge of the tumor, and, last but not least, of tumor relapse. These properties render GSCs particularly relevant for GB therapy. Moreover, GSCs are capable to differentiate into non-tumorigenic cells, such as endothelial cells, that are part of the tumor microenvironment [77]. Therefore, recent studies specifically aimed to enrich GSCs in immortalized or patient-derived cell lines, by both 2D as well as 3D culture, [78,79,80,81]. Other approaches for obtaining GSCs are based on the reprogramming of human-induced pluripotent stem cells (hiPSC) to neural progenitor cells that can acquire the characteristic of GSCs, including the ability to form GB tumor when orthotopically transplanted into mouse brains [82,83].
- Patient-derived organotypic slice cultures
The organotypic slice cultures originate from GB tissues acquired during neurosurgical procedures, cut into thick slices (hundreds of m of diameter), and maintained on membranes in 6-well plates for up to four weeks maintaining their cellular structure. This approach was set-up in 2013 by Metz and coworkers, and was first used to test the therapeutic effect of TMZ, and X-rays or carbon-ion radiation therapy, monitoring cell proliferation, cell death, and DNA double-strand breaks [84]. The study revealed the suitability of the slices as an experimental model to dissect mechanisms of therapy resistance, as well as an assay platform for novel personalized treatments. An alternative approach to studying GB cell invasion is to derive organotypic slices from healthy mouse or rat brain tissue and study the migration behaviour of previously labelled GB tumour cells (single cells or tumor spheroids) that have been implanted. This approach was first used in 1998 on slices from perinatal rat brain [85], and then optimized from adult mouse brain which better recapitulates features of adult GB tumor microenvironment [86]. In 2019, slices were obtained from human healthy tissues surrounding the excised GB. Implantation of patient-derived GB cells on the healthy brain tissues of the same patients may represent a powerful model to develop novel personalized treatments [87,88].
3.2.2. Purpose, Strengths and Weaknesses
Human immortalized cell lines constitute a very simple and well-characterized system that can be easily cultured and studied by using the most widely diffused proliferation, migration and invasion assays. Cell line cultures are commercially available, not particular expensive, and require only basic cell biology expertise. Moreover, they do not pose any ethical concern related to the use of animal models or human biopsies. Cell lines are also suitable for the use in high-throughput screening, for an initial evaluation of huge amount of compounds with possible antitumoral activities. Therefore, in vitro assays with GB human cell lines are still valuable complementary options, providing a simple and easy-to-get basis for further ex-vivo and in-vivo research. However, cell lines are not able to resemble the heterogeneity and complexity of GB tumor and their intricate relationships with TME. Most cell lines were established decades ago and may have lost key features respect to the tumors they were derived from. Moreover, traditional 2D models have very limited ability to simulate the in vivo tumor conditions. To have more reliable results, it is advisable to utilize at least multiple cell lines with different genetic backgrounds. The origin of the cell line, the number of passages in culture and the absence of Mycoplasma are also key parameter to check to improve reliability and reproducibility of the results. Finally, 3D culture models might also improve the reliability of the cell culture system [69,89] .
Primary cells and tissues, which derive directly from patient, possess invaluable features that render them extremely precious for preclinical studies. Maintaining biological relevance, GDPCs and GB organotypic slice cultures closely reflect the tumor of origin, and the complex architecture of in vivo conditions. Moreover, thanks to their donor-specific variability, primary cells and tissues allow researchers to explore the impact of genotypic and phenotypic diversity on tumor cell behavior, and therapy response, opening the way to the development of personalised treatments [90].
GPDCLs are an important resource for the development of clinically relevant ex vivo models, since they are both genotypically and phenotypically closer to the original tumors they were derived from. However, GPDCLs are difficult to establish and maintain in tissue culture, requiring each time specific experimental procedures closely depending on patient and tumor [84] . Culturing methods, 2D or 3D also influence genetic stability, with 3D culture more efficient in preventing genetic changes compared to the tumor of origin [91]. Moreover, these cell lines require a very long time to form in vivo tumors in xenograft animal models, varying from 2 to 11 months [69,92]. Therefore, difficulties of obtainment, time constraints and inconsistent culture, limit their use. GSCs, on the contrary, are a very interesting system, which brings together the main GB features related to a poor prognosis (chemoresistance, invasiveness, relapse), and, therefore, is a suitable model for therapeutic investigation. However, culture conditions might strongly affect GSC homogeneity and propagation and, even more importantly, may cause the loss of specific mutations, including the EGFR ones, which are present in almost 50% of the GB biopsies, weakening consequent analysis [77].
The organotypic slice cultures include several advantages, such as the presence of the organotypic matrix and components of the tumor microenvironment (ii) open access allowing treatment and direct observation over extended periods of time; and (iii) collection of supernatants for analysis. On the other side, slice cultures are cost and time consuming, and due to their intrinsic nature and derivation, individually unique and less suitable for well-controlled reference experimental systems [69,92]. Both patient derived cell lines and organotypic slices were used to study GB cell proliferation, migration, invasion, and susceptibility to treatment, and are suitable to test novel personalized therapeutic strategies. In particular, implantation of labelled patient-derived GB cells on brain organotypic slice culture derived from the healthy tissues of the same patient can offer the unparalleled opportunity to study the interaction among tumor cells and the brain microenvironment, comprising both glia, vascular and immune cells, and to monitor personalized response to different therapies.
4. Cell Culture Modeling Strategies
4.1. 2D Models
4.1.1. Origin and Characteristics
The 2D monolayer cell culture is the conventional culture system of cells growing in adhesion in culture medium containing serum and all the nutrients necessary for cells to grow and proliferate.
4.1.2. Purpose, Strengths and Limitations
2D cell culture the easier and cost-effective way of culturing, for whom standardized protocols and commercially available reagents and tests are available, and characterized by high homogeneity of cell population, in which all cells has the same access to oxygen and nutrients [90]. 2D models have been used in a plethora of assay, including GB cell proliferation, toxicity and migration assays and have greatly contributed to drug discovery and understanding of GB. However, they have two great biases, that 3D culture systems allow to bypass. First of all, 2D culture models are subject to both genotypic and phenotypic drift, after being subjected to repeated passaging in culture for a considerable amount. This is true for commercial cell lines, but also for patient-derived ones. Second, they fail to represent the complexity of the cell to cell interactions which characterise the tumor and its microenvironment. These limitations have strongly restricted the usage of 2D culturing systems during time, especially of immortalized cell lines, to preliminary and/or high-throughput assays. In the meantime, more suitable and complex models have been developed and their usage in preclinical assays is increasing [43,49,77].
4.2. 3D Models
4.2.1. Origin and Characteristics
Recently, various types of GB 3D models have been developed to culture established cell lines, primary tumor cells as well as tumor biopsies. All 3D culture models are characterized by complex interactions among tumor cells, which aggregate, then resembling the tumor growth inside the organism. However, they differ for the presence or not of ECM components or external scaffold which influence and guide tumor cell aggregation and growth. Here we report the different 3D model systems actually available, in order of increasing complexity, and concomitant similarity to in vivo tumorigenesis. The key milestones in the development of GB modelling strategies are shown in Figure 5, whereas specific highlights, pitfalls and benefits are resumed in Table 3.
Figure 5.
Timeline showcasing the key milestones in the development of GB modeling strategies.
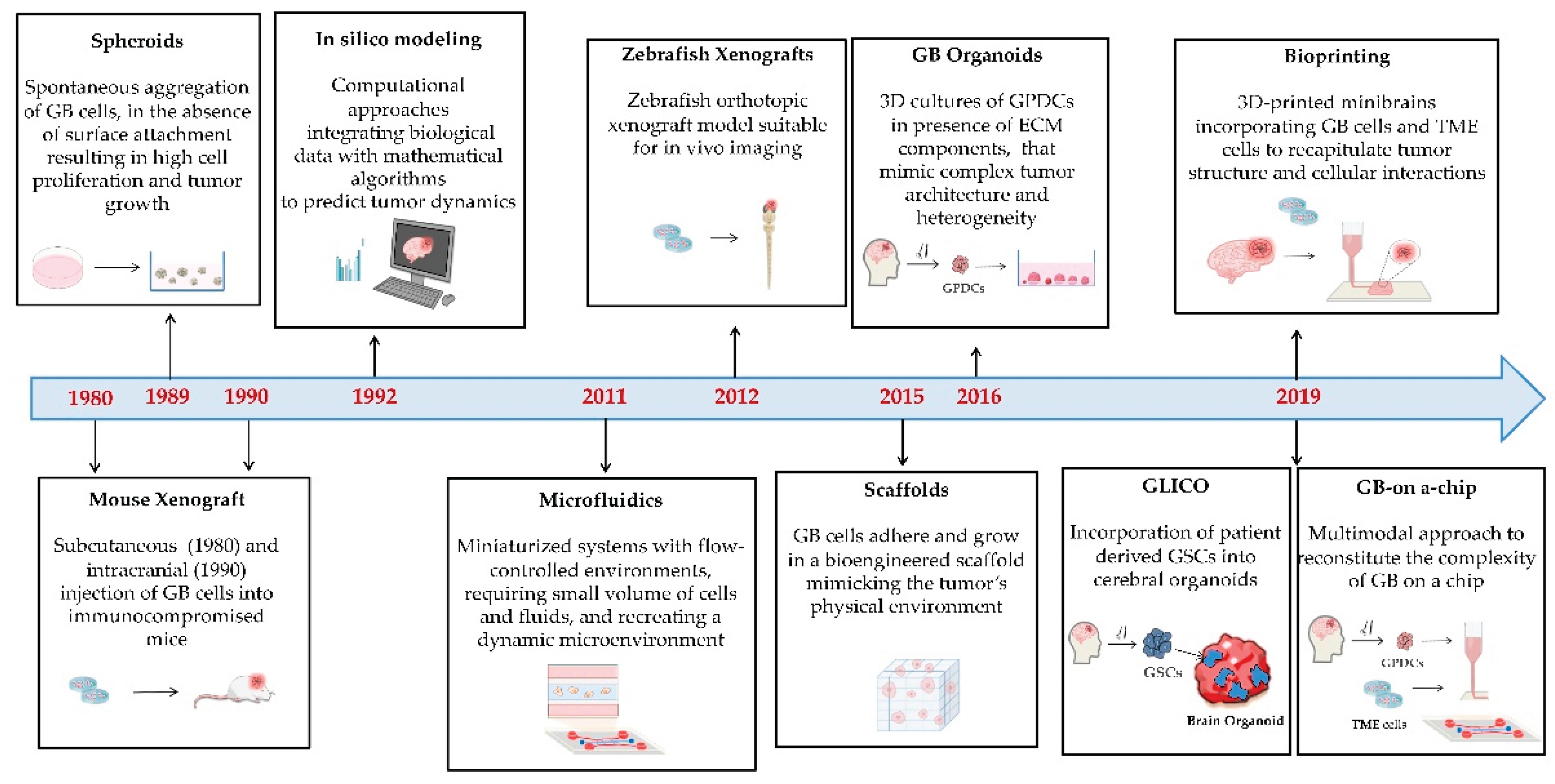
Table 3.
Main Glioblastoma modeling strategies.
| Modeling strategy | Generation | Strengths | Weaknesses | Purpose/Applications | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| CP/D | CM/I | DA | RT | TME | BBB | T | |||||
| 2D | Monolayer Culture | Culture of adherent cells in medium with serum and nutrients | Easy; Cost effective; High availability of standardized protocols and commercial reagents | Subject to genetic drift; Not reproducing the spatial complexity and intricate cell relationships of in vivo GB | + | + | + | + | H | ||
| 3D | Spheroids | Spontaneous aggregation in suspension | |||||||||
| MCTS | Mainly GB cell lines | Easy to culture and genetically manipulate | Low histological similarity to in vivo GB | + | + | + | + | M/H | |||
| Gliomaspheres | GB primary cells | Representing genetic and phenotypic in vivo GB heterogeneity; Suitable for personalized medicine |
Low architecture complexity respect to in vivo GB | + | + | + | + | L/M | |||
| OMS | Tumor and not tumor primary cells | More similar to in vivo GB; reproducing TME; Suitable for personalized medicine | Low architecture complexity respect to in vivo GB | + | + | + | + | + | L/M | ||
| GB Organoids | Free self-assemblement of GB primary cells | High complexity; Representing heterogeneity of in vivo GB; Suitable for personalized medicine; Versatile and customizable |
Low control and reproducibility | + | + | + | + | M | |||
| GLICO | Incorporation of primary GSCs into brain organoids | High correlation with in vivo GB; Reproducing interaction with TME; Suitable for personalized medicine | High cost, Low control | + | + | + | + | L | |||
| Scaffold | Embedding and growth of primary GB cells on defined scaffolds | Representing heterogeneity of in vivo GB; Reproducible; Suitable for personalized medicine | Scaffold biocompatibility | + | + | + | + | M | |||
| Bioprinting | Combination of GB cells (and eventually TME cells) in a bioreactor with a bioink | Highly controlled; Reproducible; Depending on the types of bioprinted cells can highly represent GB complexity and TME | High cost; High specialization; Need of suitable bioinks to mimic TME complexity | + | + | + | + | +/- | +/- | M | |
| Microfluidic | Very small volumes of cells and fluids are combined for perfusion culturing | Low amounts of cells and materials; Reproducible; Dynamic environments closer resembling in vivo GB; Depending on the types of cells can highly represent GB complexity and TME | High cost and specialization; Need of performant materials to build the devices; Challenging downstream sample analysis |
+ | + | + | + | +/- | M/H | ||
| GB-on-a-chip | Integration on different technologies in a chip | Controlled; Reproducible; Highly resembling in vivo GB; Suitable for personalized medicine | High cost and specialization; Need of performant materials to build the devices; Challenging downstream sample analysis |
+ | + | + | + | + | +/- | M | |
| In vivo | Mouse xenograft | Injection of GB cells (cell lines, GPDCS or GB tumor pieces) | L/M | ||||||||
| - Heterotopic | Intravenous | Simple; High efficiency | High costs of housing for immunodeficient mice; Different tumor environment | + | + | + | L/M | ||||
| - Orthotopic | Intracranial | Representative of the in vivo physiological TME | High costs of housing for immunodeficient mice, Complex; High mortality | + | + | + | + | + | L/M | ||
| Zebrafish xenograft | Injection of GB cells (cell lines, GPDCS or GB tumor pieces) | M/H | |||||||||
| - Heterotopic | Intra yolk sac | Simple, High efficiency, Low cost of housing,;Transparency; In vivo imaging | Different tumor environment | + | + | + | M/H | ||||
| - Orthotopic | Intracranial or intrablastula (injected cells incorporate into the brain) | Low cost of housing, Representative of the in vivo physiological TME; In vivo imaging; Transparency; | Complex | + | + | + | + | + | + | M | |
| In silico | Discrete Models | simulation at single cell level | Can accurately describe the behaviour of single cells in simpler contexts | Need of experimental data to create, validate and optimize the model | + | + | + | ||||
| Continuum Models | simulation at tissue level | Can accurately describe patient GB | Need of experimental data to create, validate and optimize the model | + | + | + | + | + | |||
Abbreviations: BBB, blood-brain barrier; CM, cell migration; CP/D cell proliferation and death; DA, drug assays; GLICO, glioblastoma cerebral organoids; GB, glioblastoma; GBO, glioblastoma organoids; GPDCs, glioblastoma patient derived cells, GSCs, glioblastoma stem-like cells; H, high; L, low; M, medium; MCTS multicellular tumor spheroids, OMS, organotypic multicellular spheroids; PDMS, polydimethylsiloxane; RT resistance to therapy; T, throughput TME, tumor microenvironment.
Spheroids
Tumor spheroids form by spontaneous aggregation of tumor cells, in the absence of any surface attachment, and even ECM components, thus resulting in high cell proliferation and tumor growth [43,77]. Following intercellular contact, tumor cells produce adhesion molecules, such as cadherins and integrins, on the cell surface, which promote cell survival and allow growing of spheroids as a compact structure [93,94]. Tumor spheroids were first generated in the early 1970s by the study of Sutherland and coworkers [95], and since then, a plethora of spheroid methods have been developed. Three different spheroid models can be identified, based on the cellular sources and methodology used. Multicellular tumor spheroids (MCTS) were the first GB spheroid model to be obtained, in 1989 [96]. They are typically established from cancer cell lines in conventional media supplemented with FBS, and, differently from conventional 2D models, are grown as spheres in a suspension culture or other conditions that promote cell–cell adhesion [96].
MCTS are clonal, easy to culture and to genetically manipulate and, even though show little histological resemblance to the primary cancer, are still able to mimic its metabolic, proliferative and chemoresistance properties [79,97], and for these reasons, they are used in high-throughput drug screening [98]. On the other side, GB-derived spheroids or gliomaspheres have been generated by mechanical or enzymatic dissociation of tumor specimens into a single cell suspension followed by growth in absence of serum and in presence of supplements such as B27 and N2 nutrient mix, epidermal growth factor (EGF) and/or basic fibroblast growth factor (bFGF) to positively select GSCs [78,80]. Finally, organotypic multicellular spheroids (OMS), are established after extremely gentle mechanical or enzymatic dissociation of cancer tissues, in a way to save also surrounding non tumor cells, such as stromal, vascular and immune components of the tumor microenvironment. As a result, OMSs, differently form the other two types, are similar to ex vivo explants, generally retaining many histological features and the cellular heterogeneity of the primary GB and faithfully reproduce TME [78,80].
Tumor-like Organoids or Tumoroids
Organoids are complex structures aiming to recapitulate the whole organ generated starting from stem cells (embryonic or induced pluripotent stem cells) let to develop and differentiate in 3D culture systems [89,99]. In particular, in 2013, neural organoids or minibrains were generated for human induced pluripotent stem cell [100]. Tumoroids are a particular type of organoids that are derived from patient cancer tissues. GB cells self-assemble and interact with each other to form 3D-tissue like structures, following specific protocols, usually involving Matrigel or other components of the ECM [43,77]. The presence of ECM components results in a higher level of structural complexity of the organoids respect to spheroids, and the acquisition of an architecture closer to the tumor of origin [94]. The first GB organoids (GBO) were generated in 2016, by adapting the methodology previously used for mini-brains [101]. Successfully implanted into mouse brains, these organoids produced more sophisticated and representative models than basic GPDCLs. A biobank of patient-derived GB organoids, with each one showing parental tumor heterogeneity, was established as a precious resource for personalized GB therapy [102]. However, derivation of organoids from patient tumors is challenging in term of both cost and time. An alternative strategy has been developed based on the induction of GB into brain organoids through genetic engineering by CRISPR/Cas9 system [103]. Recently, technological progress has also enabled the development of more precise, customizable and physiologically relevant GBOs.
Cerebral Organoid Glioma (GLICO)
In 2019 a brilliant system was set-up by Linkous and coworkers, based on the co-culture of GSCs derived from patients into human cerebral organoids obtained by human embryonic stem cells or human induced pluripotent stem cells [104]. Patient-derived GSCs, labelled with GFP, were successfully incorporated after one week into the miniature brains and formed infiltrative tumors with a pattern strongly resembling the one commonly found in surgical GB specimens. The GLICO model, in addition, reflects the same genetic mutations and activated signaling pathway of the tumor of origin [104]. GLICO model was found to have the highest correlation with primary patient tumor respect to other three GSC-derived models, that are gliomaspheres, GBOs and patient-derived xenografts (PDXs) [105]. This model, therefore, represents a valuable opportunity to study GB progression and infiltration into the brain tissues, as well as to test potential therapeutics [106].
Scaffold-Based Models
Differently from previous ones, scaffold-based models take advantage of biomimetic ECM-like materials, which can support and structure 3D cell culture, miming the interaction of cancer cells with each other and the surrounding ECM [43,90]. Therefore, GB cells do not freely assemble as in GBOs, but adhere and then grow and maintain following a scaffold structure in which are embedded. In 2015 Heffernan and coauthors developed a bioengineerd 3D scaffold, a hydrogel obtained by cross-linking of jaluronic acid and gelatin, to study GB proliferation and invasion [107]. Since then, a great effort has been produced to develop more and more suitable cell supporting materials able to closely resemble physiological ECM, in terms of pore size and porosity. Moreover, scaffold with post-manufacturing tunability, reproducing the ECM continuous remodeling abilities have been also proposed [77]. Supports can be divided, based on the type of polymer, into natural- (i.e. bio-polyamide, bio-polyethylene, protein- or polysaccharide-based) or synthetic-derived (i.e. polyamide, polystyrene, polyethylene, polycaprolactone, polyanhydrides), and, depending on the rigidity of the material, into solid and hydrogels [77,90]. Synthetic scaffolds, however, can have less biocompatibility than natural and might require surface modification for cells to attach and growth on them. A very interesting type of scaffold has been recently developed, based on decellularized tissues, in which cells were removed, leaving only natural ECM components [90]. Coculture with non tumor cells is also possible and can mimic interaction within the TME [90].
4.2.2. Purpose, Strenghts and Limitations
2D cell cultures are not able to mimic the in vivo spatial complexity of the in vivo environments, whereas 3D system models have better chances. Tumor cell adhesion to form 3D aggregates resembles in vivo tumor growth, with the same variability in the access to oxygen, nutrient, metabolite, and growth factors, and often the acquisition of different polarity and phenotype from the various tumor cells inside the same tumor aggregate. GB spheroids are the simplest and most popular 3D cell cultures. They show a multilayered structure with an inner layer of hypoxic and necrotic cells, an outer layer of actively proliferating cells, and a middle layer of quiescent cells. This structure resembles the in vivo tumor structure and recapitulated tumor properties, including response to therapy. For these reasons, GB spheroids have been largely utilized in drug screening assays [94].
Organoids are characterized by higher cell density, more physiological intercellular interactions and a more complex spatial organization than spheroids. Moreover, due to its formation process in absence of a support, organoid culture offer major versatility and customizability in terms of culture protocols and experimental design, but on the other side, show high heterogeneity of cell composition and structure and lower control and reproducibility than scaffold-based models [4,90]. GLICO inherit the same disadvantages of organoids, with cost and time to culture being some of the major challenges to face. However, GLICO provides a precious method of studying GB biology in a human brain microenvironment, in particular GB growth and infiltration into brain tissues and response to therapy [106]. Finally, respect to GBO, the use of a scaffold on which tumor cells adhere and grow allow the development of more controlled and reproducible tumor models. Moreover, bioactive signals or candidate drugs can be incorporated into scaffolds to allow a concentration gradient and controlled release to guide tumor formation or to study drug response, respectively [90]. GLICO and OMS are the ones that, by origin, contain non tumoral cells of the TME. In the other models, however, tumor cells can be cocultured with stromal, immune or endothelial cells, to get closer to in vivo physiological GB. In the following paragraph, modelling strategies more suitable to mimic and study TME interactions are described.
5. Modeling of the Interactions with Glioblastoma Microenvironment
5.1. In Vitro or Ex Vivo
5.1.1. Origin and Characteristics
Bioprinting
As scaffold-based models, bioprinted models also use biomaterials in which cells are dispersed. The generation of bioprinted GB models is based on the classical steps of the 3D printing procedure. First, a programmed outline of the desired structure is required, followed by image segmentation for the successive printing of the different layers that form the planned 3D tumor structure [108,109]. There are different types of bioprinters, including inkjet printers, which are supplied with a heater; extrusion-based printers, which are supplied with a piston; and laser-assisted printers. All are able to combine the bioprinting material, different types of cells, associated biomolecules and the chosen biomaterial or bioink in the bioreactor to form a complex 3D tumor-TME structure [108]. As bioinks, are commonly used derivatives form hyaluronic acid, gelatin methacrylate (GelMA), collagen with or without alginate, which allow to build an hydrogel scaffold in which the cells grow and interact [110]. The exploitation of novel biomaterials and cutting-edge technologies for tissue engineering allowed the formation of complex 3D structures, resembling the complex in vivo tumor organization, in which tumor cells establish intricate relationships with each other, the non-tumor cells of the microenvironment and the extracellular matrix. These extremely advanced structure are able to model more complex processes involved in tumorigenesis, as for example the reciprocal interaction and conditioning of tumor cells and the surrounding niche, including endothelial and immune cells, and the related phenomena of angiogenesis, neovascularization, BBB leakage, and immune escape [43,109,110]. Moreover, the progress in the field of biomaterial and bioengineering assures a high control over the cellular and biomaterial layers, resulting in a high level of reproducibility of the bio-printed models, therefore suitable for preclinical drug sensitivity assays [111,112]. The design of the 3D bioprinted model can be based on clinical images to perfectly mimic in vivo tumors from real patients or can be planned to model specific simplified conditions and cell interactions, for instance between GB tumor cells and macrophages or glioma associated stromal cells [111,112]. In 2019 Heinrich and collaborators engineered 3D-bioprinted miniature brains encapsulating the RAW264.7 mouse macrophage cell line and harbouring a large cavity were first obtained and then the cavity was filled with GL261 mouse GB cell line embedded in the hydrogel bioink. The minibrains unravelled the complex relations between GB cells and macrophages, showing that GB cells were able to recruit and condition macrophages, polarizing them in GB-associated macrophages or GAMs, which in turns induce GB cell invasiveness. Interestingly, drugs able to inhibit the interaction between GB cells and GAMs were able to reduce tumor growth and increase chemosensitivity [111]. Moreover, by utilizing 4D bioprinting to incorporate glioblastoma patient-derived cells (GPDCs) and ECM biomaterials, Chadwick and colleagues have created dynamic GBOs that can model interactions within the TME over time [113]. This method offers a novel way to investigate the evolution of GB heterogeneity and therapy resistance as they respond to environmental changes, and, ultimately, provides a strong and scalable foundation for drug screening at high throughput [31].
Microfluidic
Microfluidic models are based on submillimetre-scale devices equipped with etched channels, in which extremely small volumes of cells and fluids can be combined for perfusion culturing. These miniature scales come with numerous advantages, such as requiring minimal amounts reagents, high sensitivity and resolution, and laminar flow [69,114]. Microfluidic technology had a great impact for biological applications, especially in the cancer field, allowing the controlled placement of cells and precise delivery of factors. Devices were widely constructed through a photolithographic process with polydimethylsiloxane (PDMS), characterized by high biocompatibility, flexibility, optical transparency, imaging resolution and low cost [31,69,114]. The first GB microfluidic-based model was developed by Huang and coworkers in 2011 and was composed by three communicating compartments, a seeding chamber, a receiving chamber and bridging microchannels with the aim to evaluate the migratory ability of previously isolated GSCs [115]. After that, microfluidic devices have been evolved to resemble closer and closer the in vivo tumor. 2D or 3D biomimetic hydrogel (i.e. collagen, jaluronic acid, chondroitin sulphate proteoglycans) have been also integrated in the device to mimic in vivo ECM and TME [116,117,118,119]. Moreover, concentration gradient generator microchannels and a precision syringe pump were used to allow circulation of media of strictly controlled composition (nutrients, growth factors, substances to test) into the device, thus miming vasculature function, and generating a dynamic and more realistic GB microenvironment [116,117]. Another development direction aims in coculturing different cell types resembling the complexity of the TME. Cui and colleagues combined mouse GB cell lines, macrophages and endothelial cells into a microfluidic angiogenesis model with controllable and biomimetic immunosuppressive conditions to study immune-vascular and cell-matrix interaction and the mechanism leading to the failure of current anti-angiogenic therapy of GB [120]. Microfluidic technology was first set-up for commercial GB cell lines and then improved for culturing GPDCs [121,122]. In 2018, researchers were able to expand GPDC cell cultures and grow them as spheroids, with high cell viability, high volumetric yield and maintaining GSC features [121]. Moreover, different combinations and concentrations of chemotherapy drugs could be screened to identify the optimal combination tailored to individual GB patient [122]. The results obtained were consistent with the inherent TMZ GB resistance in patients. More recently microfluidic technology was used to maintain entire GB patient-derived tissues in culture. Olubajo and coworkers (2020) developed a device composed of two layers of glass bonded together to contain a tissue chamber and a network of microchannels for permanent tissue perfusion. The device was used on 128 GB biopsies from 33 patients and allowed tumor tissue maintenance for an average of 3 days with high cell viability and histological similarity to fresh biopsies [123]. Microfluidic devices filled with GPDCs in culture open the way to personalized medicine approaches. Nowadays, commercial microfluidic devices are also available and have been successfully utilized to evaluate the antitumoral effects of new candidate drugs [124].
GB-on-a-chip
GB-on-a chip-models aim to recreate in vitro the complexity of the in vivo tumor, by coculturing in a sophisticated 3D environment supported by analogues of ECM, both GPDCs, and non tumor cells (stromal, macrophages, endothelial cells) in a dynamic relation. In 2019 Yi and colleagues bioprinted a GB tumour co-culturing GPDCs, endothelial cells and decellularized ECM from brain tissue in a compartmentalized cancer-stroma concentric-ring structure sustaining a radial oxygen gradient. The bioprinted model recapitulated the structural, biochemical and biophysical properties together with the patient-specific tumor resistances to treatment [125]. On the other side, thanks to microfluidic devices, GPDCs can be cultured in a dynamic 3D environment together with stromal, immune and endothelial cells, mimicking both the TME cellular interactions, the in vivo oxygen and nutrient gradients as well as drug diffusion and permeability. Truong and coauthors combined GPDCs in Matrigel and HUVECs in fibrin gel in a GB-on-a-chip to mimic the GB vascular niche and study the effect of endothelial cells on GSC proliferation, migration and molecular features. The model was validated as physiologically relevant thorough comparative analysis with in vivo orthotopic mouse PDX model [126]. Finally, microfluidics, tissue engineering, biomaterial research, bioprinting, and more recently biosensors for better downstream analysis, have been converging to develop more and more sophisticated GB-on-a-chip models. In 2020, Ozturk and coworkers created a 3D microfluidic platform comprising of bioprinted patient derived GB spheroids integrated within two perfused vascular channels to analyse the long-term effects of TMZ treatment. Noteworthy this study highlighted the need for multi-model treatment strategies to understand and bypass GB resistance and recurrence [127]. All these findings show the suitability of the tumor-on-a-chip models for the development of personalized therapeutic strategies.
5.2. In Vivo Graft Models
5.2.1. Origin and Characteristics
Nowadays, the GB animal models used are basically generated by xenografts of the different human GB sources discussed above, including established cell lines (genetically manipulated or not), patient-derived cell lines or biopsies (fresh or pre-cultured [49]. Most xenografts are orthotopic, in which the brain tumor microenvironment with its own cellular and extracellular components, organ architectures and anatomical barriers, is the natural one for grafted GB cells, mimicking the GB progression inside the patient [49].
Mouse Xenografts
Experimental GB mouse models can be rapidly generated thorough GB cell injection at both embryonic or post-natal stages Converging
The origin of human cells implanted and the site of implantation are key conditions which determine the features of the developing tumors in terms of genetic and phenotypic and similarity to human GB. Experimental mouse heterograft is simpler to perform, via a subcutaneous injection, and to follow during time (tumor mass can be directly measured being the tumor easily accessible through the skin). Ortho transplantation, instead, requires a stereotactical intracranial injection, therefore, is more invasive for the animal, with a higher risk of complicacies. Engrafted GB cells can be established cell lines or patient-derived xenografts also named PDXs. Cell lines due to long time in culture can be very dissimilar from the original GB tumor, and then lacking some GB features, such as the infiltrative potential. The PDX mouse model involves transplanting patient-derived tumor cells, either directly or after in vitro culture, even though genetic modification may occur in the latter case, causing the tumour cells to diverge from the original explanted tumor [72,128]. The past few decades have been dominated by heterotopic graft models in which GB cell lines were injected subcutaneously into mice. Primary xenograft GB lines were established and maintained thorough direct implantation of GB biopsies heterotopically into the flank of nude mice and subsequent serial passage of these tumors in the flank of mice [129]. However, the most representative GB graft model is the patient-derived orthotopic graft (PDOX), in which GB cells are injected into the brain, providing the natural stromal support and tumor environment. An adopted strategy was to maintain GB PDXs by serial subcutaneous passaging in nude mice and then use them to generate a panel of GBM PDOXs.
A library of PDOX models using surgical GB samples has been also established, recapitulating biological and histopathological features of human glioblastoma in situ [130]. The use of tumor fragments, without passage in culture, has the great advantage of preserving the genetic and phenotypic features of the tumor of origin, but shows also important technical limitations, such as an extremely variable success rate, especially using small tumor fragments, and a long tumor latency [128]. Possible compromise might be obtained by intermediate steps, such as the derivation of gliomasphers in vitro, which, when injected in mouse brain, form tumors with a high efficiency and speed, even though these passages can lower the similarity with the tumor of origin.
Immunocompromised mouse strains, such as athymic nude mice, severe combined immunodeficient (SCID) mice, non-obese diabetic (NOD) or NOD-SCID mice are usually used, to avoid immune rejection of grafted cells [36]. The use of immunodeficient mice, however, enables the study of the interactions between grafted human GB cells and the immune system as well as evaluation of candidate immunotherapies; for this reason, different approaches have been developed to use also immunocompetent mice. Hoffmann and coauthors took advantage of the immune-privileged developmental time window, to successfully inject human GB cells into the telencephalic ventricle of wild-type embryos at 12.5 days of development [131]. As an alternative approach, GB humanized mice have been obtained by engrafting into immunodeficient mice human stromal/hematopoietic stem cells which then differentiate into various immune cell types [132]. Finally, an orthotopic xenotransplant model of human GB cells was also obtained into inbred strain immunocompetent mice [133,134].
Zebrafish Xenografts
Xenografted human GB cells or PDXs can be also injected into zebrafish models. Using zebrafish as model systems has several advantages, including animal small size, high fecundity, ex utero development, and cost-efficient husbandary. Moreover, as in mouse, adaptive immune system does not form until embryonic day 21 makes it an ideal animal model for xenotransplantation [135]. Microinjection is performed at the embryonic stage when larvae are transparent and easily accessible. At this stage, as with mouse embryos, immune surveillance has not yet developed. Microtumors form in three, four days, respect to the weeks necessary in rodents. Moreover, the use of fluorescently labelled cells renders also in vivo imaging easy to perform [136,137]. First attempts used transplantation of adherent GB cell lines into the yolk sac, demonstrating formation of tumors, angiogenesis and allowing to study response to treatments [135,138,139]. Further studies xenotrasplanted conventional GB cell lines orthotopically into zebrafish larva brain to analyze tumor invasion, angiogenesis and the impact of Wnt signaling during GB cell differentiation [140,141]. This approach was technically challenging due to the high lethality of transplanted embryos and has been also standardized to have more reliable and reproducible results and follow tumor growth and invasion with real time imaging [142]. Recently, novel simpler methods have been developed to overcome intracranial transplantation of GB cells into single zebrafish embryos, based on cell grafting at blastula stage. This methods allow for easy, fast, and automatable orthotopic injection, opening the way to high-throughput drug screenings [143].
In addition to wild-type zebrafish, transgenic strains were also used to simplify and fast the in vivo analysis. Among them, Casper mutants, which remain transparent into adulthood due to the inhibition of melanophore formation. and Tg(fli1:EGFP), in which enhanced green fluorescent protein (EGFP) marks the endogenous vasculature. This strain is commonly utilized for research on vasculogenesis, angiogenesis, and the development of metastases [137]. U87 cells were often xenografted in GB angiogenesis studies, whereas U251 cells in GB proliferation studies, and PDXs were employed for preclinical studies with clinical relevance. Zebrafish can be an excellent model for large-scale drug discovery, and its predictive ability has been harnessed in multiple high-throughput screening studies.
5.2.2. Purpose, Strengths and Limitations
Both bioprinting and microfluidic technologies allow to assemble multiple cell types to study interactions among tumor cells and the other cells of the TME. The presence in the 3D model of components of the tumor microenvironment, such as ECM and non-tumoral cells (stromal, endothelial and immune), that strongly impact on tumor growth, controlling cell morphology, and function is undoubtedly a high added value. The possibility to mimic not only the complex interactions among tumor cells with each other but also with stromal cells and ECM components and the relative signaling gradients render the models more interesting for studying tumor cell behavior and drug response than simple spheroids [81]. This added value, however, has also a high cost, in term of time, human and economic resources. Moreover, 3D bioprinting has currently various limitations, including the need to improve printing resolution and to develop suitable printing biomaterials with both the mechanical properties necessary for printing and the physiological properties for mimicking in vivo tumor [112].
Microfluidic technology, respect to bioprinting, give also the opportunity to create multiplex physical and chemical gradients to better mimic the dynamic environment which resembles closer the physiological situation in which tumor cells continually receive oxygen, nutrients and factors. Moreover, the device transparency allows real-time visualisation and high-resolution in vivo imaging. The advantages of microfluidics are several, included minimal amount of cell samples and biomaterials required, high sensitivity, and rapid analysis [60]. However, microfluidics possesses some limitations linked to the material of the device, that often is PDMS, which can absorb hydrophobic components, altering their gradient inside the device and then affecting drug response and reliability of the results. Moreover, microfluidic devices cannot restructure the shear stress observed in GB in vivo, and to ensure the equilibrium of cytokines in the growing cells, sub-channels linking the microwells to the main channel should be included [110]. On the other side, one major benefit of bioprinting respect to microfluidics is the possibility of adopting a one-step production process, thus enabling customisable architectural designs, with less labour and advanced precision. Finally, GB-on-chips models have the potential to improve knowledge of human cancer biology, fast drug discovery and explore pharmacokinetics of GB therapeutics, as well as offering promising applications in personalised medicine. Multimodal systems, incorporating microfluidics, 3D bioprinting and biosensors, have demonstrated promise in evaluating various GB therapeutic response in a physiologically representative environment [31,144]
Even though cell-based models are fundamental for the study of GB pathogenesis, at present, they have limited utility in the context of drug development. The efficacy of candidate GB therapeutics has to be assessed in physiologically relevant systems, such as laboratory animals, also due to the presence of the BBB, which does influence drug permeation to the brain [128,145].
Mouse orthotopic xenograft models have been widely used to study drug performance in an in vivo context, even though the use of immunodeficient mice to suppress host immune rejection enables the study of physiologically- human immune responses as well as of the impact of immunotherapies. Zebrafish xenograft models are e a valid alternative to mouse due to several strengths such as lower cost of husbandry, transparency in the early stages, which facilitates injection and tumor visualization; fast tumor formation, high number, rapid development and small size of the offspring, suitable for implementation of high-throughput screening [69]. Zebrafish graft models is used to study a plethora of tumorigenesis process, including cell proliferation, migration, angiogenesis, tumor invasion and BBB permeability [136,146,147]. However, animal xenograft models have also important limitations, besides obvious ethical concerns. Despite similarities in the overall cellular architecture, humans have a more complex and evolved neocortex, stark differences in gene expression patterns [59], especially of non-neuronal cells such as microglia, which is thought to play a key role in GB progression [60,61] . Ultimately, brain tumours are not cell autonomous, and they can be influenced by the host brain environment [148], thus decreasing reliability and predictive ability of animal models.
5.3. In Silico Modeling
5.3.1. Origin and Characteristics
In vitro, ex vivo and animal tumor models demand significant effort and time, and they yield a substantial amount of data that needs to be systematically analyzed and understood to be meaningful in a clinical context. Noteworthy, mathematical and computational models can be employed to help in depicting the intricate progression of tumors, as they can handle the multiscale aspects of the biological processes, as well as incorporate data from various imaging technologies, clinical assessments and biological experiments [49,149,150,151]. Remarkably, predictions from models can also lead to the design of targeted experiments to confirm incoming hypotheses [49]. Based on the scale at which the tumor is represented, mathematical oncology models can be divided, into discrete or stochastic models and continuum or analytical ones. The discrete models simulate the behaviour of individual cancer cells, capturing their interactions with the microenvironment and the randomness of cellular events. In 1992, Dutching and coworkers published one of the first discrete developed computer models describing 3D cell growth inside tumor spheroids [152]. This approach is more appropriate for detailing in vitro studies and tumors of small size [49,153]. On the other side, the continuum models are used to depict tumors at tissue level, approximating tumor cells and tumor microenvironment as continuous variables, and describing the collective, averaged behavior of tumor cells. These models can accurate describe patient tumors and their response to therapy, having potentially a high clinical relevance [49,153]. Among the first continuum models, there were the ones developed in the first 2000s by Swanson and coauthors to describe spatio-temporal growth, diffusion and invasion of GB cells into human brain [154,155].
5.3.2. Purpose, Strengths and Limitations
The use of computational simulations allows to model tumor biology to understand tumor disease progression, as well as molecular and functional dynamics. In silico modelling can be also used to predict treatment response, therapeutic outcomes, and eventually guide clinical decisions. This is particularly relevant in the case of GB, due to its complex pathophysiology, heterogeneity and extremely poor prognosis [49,153]. Several mathematical models have been developed to describe GB progression and its main features such as tumor heterogeneity, invasiveness, irregular angiogenesis, and also treatment response [151,156,157,158,159,160,161]. Since they are defined as such, in silico models lack experimental errors, are affordable, precise, non-invasive, and usually save time by providing a fast method to systematically analyze the influence of various cellular components in diverse environmental conditions. Though aimed at patients, however, the majority of in silico models tend to be either theoretical or generic, as they require data that are impossible to obtain at the appropriate spatial and temporal scales for proper patient-specific parametrization. Mathematical models are typically parameterized by incorporating pre-existing data from the literature. Although recent efforts are guiding research toward this goal, the challenge persists in identifying a plausible mechanistic model and properly parametrizing it to quantitatively account for a large dataset not involved in calibration and to forecast clinical or experimental results. Regardless of the mathematical method used, however, biological data are crucial for parameterizing and validating all computational models to ensure they accurately reflect biological processes [49,109,153].
In the second half of 2000s studies combining mathematical modeling and experimental results emerged, describing GB cell radially symmetric migration and invasiveness, based on data collected from U87 2D in vitro culture dispersion and U87 tumor spheroid 3D model, respectively [162,163]. During time, more and more sophisticated approaches integrating wet data and in silico modeling have been developed to study response to predict treatment response [164,165,166] or evolutionary dynamics guiding tumor growth [167].
6. Future Perspectives and Conclusions
Nowadays, it is clear that different GB models have their own peculiarities, ethical concerns, strengths and weaknesses. No single model can be considered the ideal choice for every study purpose. Improving the reliability and predictability of GB models is essential for achieving successful clinical outcomes. To overcome this challenge, we propose defining a roadmap that addresses the most relevant issues in preclinical assays. We believe that the issues to be solved are illustrated below, starting with the easiest and working up to the long-term ones.
- 1.
- Choice of the most suitable GB model(s)
The number of available GB models and related protocols is increasing, as are the different modelling options. It is crucial to choose the best model or set of models to guarantee high reliability and reproducibility of results, as well as increasing the chances of therapeutic success in clinical studies. Various factors must be considered, including the model's performance, complexity, stability and reproducibility. Other relevant practical criteria include the management of time, human resources and financial investment, the presence of the necessary expertise and/or collaborations, and the number of therapeutic agents to be tested, as well as the amount of drug needed for the specific model — a factor which can sometimes be extremely important. Finally, depending on the tumour feature being investigated and the purpose of the study, one model may be more relevant and suitable than others. The weight of each criterion can differ from case to case; therefore, it is too simplistic to attempt to identify the best model for GB studies. The model(s) to use, as well as the utilisation pipeline, must be carefully and specifically evaluated to avoid wasting resources, using inappropriate animal models and achieving poor outcomes. Using specific multi-criteria decision-making (MCDM) matrices, which have been employed in various contexts within the life sciences in both basic and applied research, as an efficient, customisable, easy-to-use tool [168,169,170,171] may also be helpful.
- 2.
- Identification of guidelines and standardization of procedures for model usage
Optimising, standardising and sharing procedures is fundamental to ensuring the reliability and reproducibility of preclinical models and relative results. Currently, raising awareness among the European scientific community and various stakeholders regarding standards, data reliability and reproducibility is encouraging collaborative efforts to identify and share good practices and standard operating procedures[172]. Several international scientific societies and networks have initiated discussions to establish their own standards and encourage adherence to these within their respective communities [173,174,175]. This is particularly relevant and challenging in the case of GB and, more generally, tumor preclinical models for two reasons. Firstly, this research is becoming increasingly transdisciplinary, based on the integration of novel expertise and complex technologies from various disciplines for which standardized protocols must be defined [31,144]. Secondly, the outcome of preclinical research is the identification of novel diagnostic or therapeutic solutions to be translated into clinical practice. The reliability of the outcomes, the standardization of the procedures and the accuracy of clinical predictions are all fundamental to fostering effective and efficient clinical translation, as well as compliance with related regulatory systems. Therefore, we strongly advocate a collective effort within the GB community to identify and promote initiatives in this direction.
- 3.
- Open platforms and integrations of the different models.
A conscious and well-designed strategy that integrates different approaches can help to discover and analyze the most effective anti-GB therapies, as well as generating more comprehensive and reliable preclinical data. Implementing open platforms and resources that focus on optimizing, validating and sharing increasingly sophisticated technologies, such as GB-on-chip, is fundamental to enhancing the use of these cutting-edge models and fostering preclinical GB research, drug discovery and validation. In this context, biobanks collecting GPDC or GBO from patients [75,76,102] are undoubtedly a valuable resource. Furthermore, comparative analyses of outcomes collected using different GB models to investigate specific GB features and the therapeutic effects of different drugs, as in the work of Truong and coauthors [126], are also extremely valuable contributions.
- 4.
- Development of novel multimodal integrated approach
The integration of various technologies and strategies can greatly enhance our understanding of GB etiology and transform drug development by minimizing animal testing and improving clinical translation. Microfluidics, bioprinting and bioengineering are converging to create more sophisticated yet simple and reproducible GB-on-a-chip systems that address GB pathophysiology and therapeutic treatment. To this end, creating transdisciplinary networks and projects that address the issue from different theoretical and methodological perspectives is fundamental, as is establishing dedicated funding programmes.
Continued development and improvement of suitable GB models, alongside the availability and utilization of open modelling platforms, will undoubtedly increase our basic knowledge of GB pathophysiology and the success rate of translational research. However, strict quality control of the input data and variables is essential to ensure the reliability and reproducibility of the outcome. This is particularly important when using complex, heterogeneous model systems, either ex vivo or in vivo, as any significant difference in the input variables may be amplified. The production, isolation, collection, quantification and storage of candidate drugs for preclinical research must also be carefully standardized, validated and monitored through a quality assurance process. Last but not least, sample batches must also pass quality control before they can be used for preclinical assays.
Funding
This work was funded by the European Union—NextGenerationEU—under the CEVITA project within the framework of the AMICO 2 program of CNR—UVR supported by the PoC—PNRR measure of the Ministry of Enterprise and Made in Italy.
Data Availability Statement
No data were created.
Acknowledgments
The authors acknowledge Mariarosaria Aletta for bibliographic support.”.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the writing of the manuscript.
References
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant Astrocytic Glioma: Genetics, Biology, and Paths to Treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef]
- Stupp, R.; Warren Mason, P.; Bent, M.J. van den B.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus Concomitant and Adjuvant Temozolomide for Glioblastoma in Elderly Patients. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Singh, S.; Dey, D.; Barik, D.; Mohapatra, I.; Kim, S.; Sharma, M.; Prasad, S.; Wang, P.; Singh, A.; Singh, G. Glioblastoma at the Crossroads: Current Understanding and Future Therapeutic Horizons; Springer US, 2025; Vol. 10; ISBN 4139202502299.
- Liguori, G.L. Challenges and Promise for Glioblastoma Treatment through Extracellular Vesicle Inquiry †. Cells 2024, 13, 336. [Google Scholar] [CrossRef]
- Hou, L.C.; Veeravagu, A.; Hsu, A.R.; Tse, V.C.K. Recurrent Glioblastoma Multiforme: A Review of Natural History and Management Options. Neurosurg. Focus 2006, 20. [Google Scholar] [CrossRef]
- Sherriff, J.; Tamangani, J.; Senthil, L.; Cruickshank, G.; Spooner, D.; Jones, B.; Brookes, C.; Sanghera, P. Patterns of Relapse in Glioblastoma Multiforme Following Concomitant Chemoradiotherapy with Temozolomide. Br. J. Radiol. 2013, 86. [Google Scholar] [CrossRef] [PubMed]
- Liguori, G.L.; Kralj-Iglič, V. Pathological and Therapeutic Significance of Tumor-Derived Extracellular Vesicles in Cancer Cell Migration and Metastasis. Cancers (Basel). 2023, 15, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Nieto, M.A.; Huang, R.Y.Y.J.; Jackson, R.A.A.; Thiery, J.P.P. Emt: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Hegi, M.E.; Diserens, A.-C.; Gorlia, T.; Hamou, M.-F.; de Tribolet, N.; Weller, M.; Kros, J.M.; Hainfellner, J.A.; Mason, W.; Mariani, L.; et al. MGMT Gene Silencing and Benefit from Temozolomide in Glioblastoma. N. Engl. J. Med. 2005, 352, 997–1003. [Google Scholar] [CrossRef]
- Vlashi, E.; Lagadec, C.; Vergnes, L.; Matsutani, T.; Masui, K.; Poulou, M.; Popescu, R.; Della Donna, L.; Evers, P.; Dekmezian, C.; et al. Metabolic State of Glioma Stem Cells and Nontumorigenic Cells. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 16062–16067. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer Stem Cells in Glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B. V; Curry, W.T.; Martuza, R.L.; et al. Single-Cell RNA-Seq Highlights Intratumoral Heterogeneity in Primary Glioblastoma. Science (80-. ). 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Reardon, D.A.; Wen, P.Y. Glioma in 2014: Unravelling Tumour Heterogeneity-Implications for Therapy. Nat. Rev. Clin. Oncol. 2015, 12, 69–70. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The Blood–Brain Barrier. Cold Spring Harb. Perspect. Biol. 2015, 7. [Google Scholar] [CrossRef]
- Luo, H.; Shusta, E. V. Blood–Brain Barrier Modulation to Improve Glioma Drug Delivery. Pharmaceutics 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Ramos-Zaldívar, H.M.; Polakovicova, I.; Salas-Huenuleo, E.; Corvalán, A.H.; Kogan, M.J.; Yefi, C.P.; Andia, M.E. Extracellular Vesicles through the Blood–Brain Barrier: A Review. Fluids Barriers CNS 2022, 19, 1–15. [Google Scholar] [CrossRef]
- Wang, J.; Liu, Y.; Liu, F.; Gan, S.; Roy, S.; Hasan, I.; Zhang, B.; Guo, B. Emerging Extracellular Vesicle-Based Carriers for Glioblastoma Diagnosis and Therapy. Nanoscale 2023, 15, 10904–10938. [Google Scholar] [CrossRef]
- Agarwal, S.; Sane, R.; Oberoi, R.; Ohlfest, J.R.; Elmquist, W.F. Delivery of Molecularly Targeted Therapy to Malignant Glioma, a Disease of the Whole Brain. Expert Rev. Mol. Med. 2011, 13, 1–27. [Google Scholar] [CrossRef]
- Yalamarty, S. S. K. , Filipczak, N., Li, X., Subhan, M. A., Parveen, F., Ataide, J. A.,... & Torchilin, V.P. Mechanisms of Resistance and Current Treatment Options For. Cancers (Basel). 2023, 15, 2116. [Google Scholar]
- Yu, M.W.; Quail, D.F. Immunotherapy for Glioblastoma: Current Progress and Challenge. Front. Immunol. 2021, 12, 1–12. [Google Scholar] [CrossRef]
- Mathew-Schmitt, S.; Peindl, M.; Neundorf, P.; Dandekar, G.; Metzger, M.; Nickl, V.; Appelt-Menzel, A. Blood-Tumor Barrier in Focus - Investigation of Glioblastoma-Induced Effects on the Blood-Brain Barrier. J. Neurooncol. 2024, 170, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Angom, R.S.; Nakka, N.M.R.; Bhattacharya, S. Advances in Glioblastoma Therapy: An Update on Current Approaches. Brain Sci. 2023, 13. [Google Scholar] [CrossRef] [PubMed]
- Medikonda, R.; Dunn, G.; Rahman, M.; Fecci, P.; Lim, M. A Review of Glioblastoma Immunotherapy. J. Neurooncol. 2021, 151, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Sferruzza, G.; Consoli, S.; Dono, F.; Evangelista, G.; Giugno, A.; Pronello, E.; Rollo, E.; Romozzi, M.; Rossi, L.; Pensato, U. A Systematic Review of Immunotherapy in High-Grade Glioma: Learning from the Past to Shape Future Perspectives. Neurol. Sci. 2024, 45, 2561–2578. [Google Scholar] [CrossRef]
- Mantile, F.; Franco, P.; Stoppelli, M.P.; Liguori, G.L. Biological Role and Clinical Relevance of Extracellular Vesicles as Key Mediators of Cell Communication in Cancer. In Biological Membrane Vesicles: Scientific, Biotechnological and Clinical Considerations. Advances in Biomembranes and Lipid Self-Assembly; Elsiever, 2020; Vol. 33.
- Wu, J.Y.; Li, Y.J.; Hu, X. Bin; Huang, S.; Luo, S.; Tang, T.; Xiang, D.X. Exosomes and Biomimetic Nanovesicles-Mediated Anti-Glioblastoma Therapy: A Head-to-Head Comparison. J. Control. Release 2021, 336, 510–521. [Google Scholar] [CrossRef]
- Jain, K.K. A Critical Overview of Targeted Therapies for Glioblastoma. Front. Oncol. 2018, 8, 1–19. [Google Scholar] [CrossRef]
- Rusak, A.; Wiatrak, B.; Krawczyńska, K.; Górnicki, T.; Zagórski, K.; Zadka, Ł.; Fortuna, W. Starting Points for the Development of New Targeted Therapies for Glioblastoma Multiforme. Transl. Oncol. 2025, 51. [Google Scholar] [CrossRef]
- Aldape, K.; Brindle, K.M.; Chesler, L.; Chopra, R.; Gajjar, A.; Gilbert, M.R.; Gottardo, N.; Gutmann, D.H.; Hargrave, D.; Holland, E.C.; et al. Challenges to Curing Primary Brain Tumours. Nat. Rev. Clin. Oncol. 2019, 16, 509–520. [Google Scholar] [CrossRef]
- Thomas, G.; Rahman, R. Evolution of Preclinical Models for Glioblastoma Modelling and Drug Screening. Curr. Oncol. Rep. 2025, 27, 601–624. [Google Scholar] [CrossRef]
- Barth, R.F.; Kaur, B. Rat Brain Tumor Models in Experimental Neuro-Oncology: The C6, 9L, T9, RG2, F98, BT4C, RT-2 and CNS-1 Gliomas. J. Neurooncol. 2009, 94, 299–312. [Google Scholar] [CrossRef]
- Seligman, A.M.; Shear, M.J.; Alexander, L. Studies in Carcinogenesis: VIII. Experimental Production of Brain Tumors in Mice with Methylcholanthrene. Am. J. Cancer 1939, 37, 364–395. [Google Scholar]
- Wilfong, R.F.; Bigner, D.D.; Self, D.J.; Wechsler, W. Brain Tumor Types Induced by the Schmidt-Ruppin Strain of Rous Sarcoma Virus in Inbred Fischer Rats. Acta Neuropathol. 1973, 25, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hoshino, H.; Kiuchi, Y.; Nakano, G.; Nagamachi, Y. Potential Usefulness of a Cultured Glioma Cell Line Induced by Rous Sarcoma Virus in B10.A Mouse as an Immunotherapy Model. Jpn. J. Exp. Med. 1989, 59, 173–180. [Google Scholar] [PubMed]
- Jin, F.; Jin-Lee, H.J.; Johnson, A.J. Mouse Models of Experimental Glioblastoma; 2021; ISBN 9780645001747.
- Sahu, U.; Barth, R.F.; Otani, Y.; McCormack, R.; Kaur, B. Rat and Mouse Brain Tumor Models for Experimental Neuro-Oncology Research. J. Neuropathol. Exp. Neurol. 2022, 81, 1–18. [Google Scholar] [CrossRef]
- Schmidek, H.H.; Nielsen, S.L.; Schiller, A.L.; Messer, J. Morphological Studies of Rat Brain Tumors Induced by N-Nitrosomethylurea. J. Neurosurg. 1971, 34, 335–340. [Google Scholar] [CrossRef]
- Benda, P.; Lightbody, J.; Sato, G.; Levine, L.; Sweet, W. Differentiated Rat Glial Cell Strain in Tissue Culture. Science (80-. ). 1968, 161, 370–371. [Google Scholar] [CrossRef]
- Ko, L.; Koestner, A.; Wechsler, W. Morphological Characterization of Nitrosourea-Induced Glioma Cell Lines and Clones. Acta Neuropathol. 1980, 51, 23–31. [Google Scholar] [CrossRef]
- Ausman, J.I.; Shapiro, W.R.; Rall, D.P. Studies on the Chemotherapy of Experimental Brain Tumors: Development of an Experimental Model. Cancer Res. 1970, 30, 2394–2400. [Google Scholar]
- Fomchenko, E.I.; Holland, E.C. Mouse Models of Brain Tumors and Their Applications in Preclinical Trials. Clin. Cancer Res. 2006, 12, 5288–5297. [Google Scholar] [CrossRef]
- Pasupuleti, V.; Vora, L.; Prasad, R.; Nandakumar, D.N.; Khatri, D.K. Glioblastoma Preclinical Models: Strengths and Weaknesses. Biochim. Biophys. Acta - Rev. Cancer 2024, 1879, 189059. [Google Scholar] [CrossRef]
- Schlegel, J.; Piontek, G.; Kersting, M.; Schuermann, M.; Kappler, R.; Scherthan, H.; Weghorst, C.; Buzard, G.; Mennel, H. The P16/Cdkn2a/Ink4a Gene Is Frequently Deleted in Nitrosourea-Induced Rat Glial Tumors. Pathobiology 1999, 67, 202–206. [Google Scholar] [CrossRef]
- Asai, A.; Miyagi, Y.; Sugiyama, A.; Gamanuma, M.; Hong, S.H.; Takamoto, S.; Nomura, K.; Matsutani, M.; Takakura, K.; Kuchino, Y. Negative Effects of Wild-Type P53 and s-Myc on Cellular Growth and Tumorigenicity of Glioma Cells. Implication of the Tumor Suppressor Genes for Gene Therapy. J. Neurooncol. 1994, 19, 259–268. [Google Scholar] [CrossRef]
- Sibenaller, Z.A.; Etame, A.B.; Ali, M.M.; Barua, M.; Braun, T.A.; Casavant, T.L.; Ryken, T.C. Genetic Characterization of Commonly Used Glioma Cell Lines in the Rat Animal Model System. Neurosurg. Focus 2005, 19, E1. [Google Scholar] [CrossRef] [PubMed]
- Mannino, S.; Molinari, A.; Sabatino, G.; Ciafrè, S.A.; Colone, M.; Maira, G.; Anile, C.; Arancia, G.; Mangiola, A. Intratumoral vs Systemic Administration of Meta-Tetrahydroxyphenylchlorin for Photodynamic Therapy of Malignant Gliomas: Assessment of Uptake and Spatial Distribution in C6 Rat Glioma Model.File:///Users/Giovanna/Downloads/16734735.Nbib. Int. J. Immunopathol. Pharmacol. 2008, 21, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Szatmári, T.; Lumniczky, K.; Désaknai, S.; Trajcevski, S.; Hídvégi, E.J.; Hamada, H.; Sáfrány, G. Detailed Characterization of the Mouse Glioma 261 Tumor Model for Experimental Glioblastoma Therapy. Cancer Sci. 2006, 97, 546–553. [Google Scholar] [CrossRef] [PubMed]
- Oraiopoulou, M.E.; Tzamali, E.; Papamatheakis, J.; Sakkalis, V. Phenocopying Glioblastoma: A Review. IEEE Rev. Biomed. Eng. 2023, 16, 456–471. [Google Scholar] [CrossRef]
- Giakoumettis, D.; Kritis, A.; Foroglou, N. C6 Cell Line: The Gold Standard in Glioma Research. Hippokratia 2018, 22, 105–112. [Google Scholar]
- Doblas, S.; Saunders, D.; Kshirsagar, P.; Pye, Q.; Oblander, J.; Gordon, B.; Kosanke, S.; Floyd, R.A.; Towner, R.A. Phenyl-Tert-Butylnitrone Induces Tumor Regression and Decreases Angiogenesis in a C6 Rat Glioma Model. Free Radic. Biol. Med. 2008, 44, 63–72. [Google Scholar] [CrossRef]
- Solly, F.; Fish, R.; Simard, B.; Bolle, N.; Kruithof, E.; Polack, B.; Pernod, G. Tissue-Type Plasminogen Activator Has Antiangiogenic Properties without Effect on Tumor Growth in a Rat C6 Glioma Model. Cancer Gene Ther. 2008, 15, 685–692. [Google Scholar] [CrossRef]
- Ahmed, A.E.; Jacob, S.; Nagy, A.A.; Abdel-Naim, A.B. Dibromoacetonitrile-Induced Protein Oxidation and Inhibition of Proteasomal Activity in Rat Glioma Cells. Toxicol. Lett. 2008, 179, 29–33. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, X.; Zhang, J.; Zhang, J.; Zou, H.; Liu, Y.; Dong, X.; Sun, X. Intravenous Administration of Arsenic Trioxide Encapsulated in Liposomes Inhibits the Growth of C6 Gliomas in Rat Brains. J. Chemother. 2008, 20, 253–262. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, J.; Ionescu, A.; Pouratian, N.; Hamilton, D.K.; Schlesinger, D.; Oskouian, R.J.J.; Sansur, C. Use of Trans Sodium Crocetinate for Sensitizing Glioblastoma Multiforme to Radiation: Laboratory Investigation. J. Neurosurg. 2008, 108, 972–978. [Google Scholar] [CrossRef] [PubMed]
- Towner, R.A.; Gillespie, D.L.; Schwager, A.; Saunders, D.G.; Smith, N.; Njoku, C.E.; Krysiak, R.S. 3rd; Larabee, C.; Iqbal, H.; Floyd, R.A.; et al. Regression of Glioma Tumor Growth in F98 and U87 Rat Glioma Models by the Nitrone OKN-007. Neuro. Oncol. 2013, 15, 330–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Barth, R.F.; Wu, G.; Ciesielski, M.J.; Fenstermaker, R.A.; Moffat, B.A.; Ross, B.D.; Wikstrand, C.J. Development of a Syngeneic Rat Brain Tumor Model Expressing EGFRvIII and Its Use for Molecular Targeting Studies with Monoclonal Antibody L8A4. Clin. cancer Res. an Off. J. Am. Assoc. Cancer Res. 2005, 11, 341–350. [Google Scholar] [CrossRef]
- Merlo, A. Genes and Pathways Driving Glioblastomas in Humans and Murine Disease Models. Neurosurg. Rev. 2003, 26, 145–158. [Google Scholar] [CrossRef]
- Zhang, Q.; Zeng, Y.; Zhang, T.; Yang, T. Comparison Between Human and Rodent Neurons for Persistent Activity Performance: A Biologically Plausible Computational Investigation. Front. Syst. Neurosci. 2021, 15, 1–10. [Google Scholar] [CrossRef]
- Fattorelli, N.; Martinez-Muriana, A.; Wolfs, L.; Geric, I.; De Strooper, B.; Mancuso, R. Stem-Cell-Derived Human Microglia Transplanted into Mouse Brain to Study Human Disease. Nat. Protoc. 2021, 16, 1013–1033. [Google Scholar] [CrossRef]
- Geribaldi-Doldán, N.; Fernández-Ponce, C.; Quiroz, R.N.; Sánchez-Gomar, I.; Escorcia, L.G.; Velásquez, E.P.; Quiroz, E.N. The Role of Microglia in Glioblastoma. Front. Oncol. 2021, 10, 1–8. [Google Scholar] [CrossRef]
- PONTÉN, J.A.N.; MACINTYRE, E.H. LONG TERM CULTURE OF NORMAL AND NEOPLASTIC HUMAN GLIA. Acta Pathol. Microbiol. Scand. 1968, 74, 465–486. [Google Scholar] [CrossRef]
- Bigner, D.D.; Bigner, S.H.; Pontén, J.; Westermark, B.; Mahaley, M.S.; Ruoslahti, E.; Herschman, H.; Eng, L.F.; Wikstrand, C.J. Heterogeneity of Genotypic and Phenotypic Characteristics of Fifteen Permanent Cell Lines Derived from Human Gliomas. J. Neuropathol. Exp. Neurol. 1981, 40, 201–229. [Google Scholar] [CrossRef]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Mantile, F.; Kisovec, M.; Adamo, G.; Romancino, D.P.; Hočevar, M.; Božič, D.; Bedina Zavec, A.; Podobnik, M.; Stoppelli, M.P.; Kisslinger, A.; et al. A Novel Localization in Human Large Extracellular Vesicles for the EGF-CFC Founder Member CRIPTO and Its Biological and Therapeutic Implications. Cancers (Basel). 2022, 14. [Google Scholar] [CrossRef]
- Camerino, I.; Franco, P.; Bajetto, A.; Thellung, S.; Florio, T.; Stoppelli, M.P.; Colucci-D’Amato, L. Ruta Graveolens, but Not Rutin, Inhibits Survival, Migration, Invasion, and Vasculogenic Mimicry of Glioblastoma Cells. Int. J. Mol. Sci. 2024, 25. [Google Scholar] [CrossRef]
- Casciati, A.; Tanori, M.; Gianlorenzi, I.; Rampazzo, E.; Persano, L.; Viola, G.; Cani, A.; Bresolin, S.; Marino, C.; Mancuso, M.; et al. Effects of Ultra-Short Pulsed Electric Field Exposure on Glioblastoma Cells. Int. J. Mol. Sci. 2022, 23. [Google Scholar] [CrossRef] [PubMed]
- Mirabelli, P.; Coppola, L.; Salvatore, M. Cancer Cell Lines Are Useful Model Systems for Medical Research. Cancers (Basel). 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Griffiths, S.; Veljanoski, D.; Vaughn-Beaucaire, P.; Speirs, V.; Brüning-Richardson, A. Preclinical Models of Glioblastoma: Limitations of Current Models and the Promise of New Developments. Expert Rev. Mol. Med. 2021, 23. [Google Scholar] [CrossRef] [PubMed]
- Demircan, T.; Yavuz, M.; Kaya, E.; Akgül, S.; Altuntaş, E. Cellular and Molecular Comparison of Glioblastoma Multiform Cell Lines. Cureus 2021, 13, 0–6. [Google Scholar] [CrossRef]
- Lee, J.; Kotliarova, S.; Kotliarov, Y.; Li, A.; Su, Q.; Donin, N.M.; Pastorino, S.; Purow, B.W.; Christopher, N.; Zhang, W.; et al. Tumor Stem Cells Derived from Glioblastomas Cultured in BFGF and EGF More Closely Mirror the Phenotype and Genotype of Primary Tumors than Do Serum-Cultured Cell Lines. Cancer Cell 2006, 9, 391–403. [Google Scholar] [CrossRef]
- Da Hora, C.C.; Schweiger, M.W.; Wurdinger, T.; Tannous, B.A. Patient-Derived Glioma Models: From Patients to Dish to Animals. Cells 2019, 8. [Google Scholar] [CrossRef]
- Seidel, S.; Garvalov, B.K.; Acker, T. Isolation and Culture of Primary Glioblastoma Cells from Human Tumor Specimens. Methods Mol. Biol. 2015, 1235, 263–275. [Google Scholar] [CrossRef]
- Frenster, J.D.; Placantonakis, D.G. Establishing Primary Human Glioblastoma Tumorsphere Cultures from Operative Specimens. Methods Mol. Biol. 2018, 1741, 63–69. [Google Scholar] [CrossRef]
- Xie, Y.; Bergström, T.; Jiang, Y.; Johansson, P.; Marinescu, V.D.; Lindberg, N.; Segerman, A.; Wicher, G.; Niklasson, M.; Baskaran, S.; et al. The Human Glioblastoma Cell Culture Resource: Validated Cell Models Representing All Molecular Subtypes. EBioMedicine 2015, 2, 1351–1363. [Google Scholar] [CrossRef]
- Stringer, B.W.; Day, B.W.; D’Souza, R.C.J.; Jamieson, P.R.; Ensbey, K.S.; Bruce, Z.C.; Lim, Y.C.; Goasdoué, K.; Offenhäuser, C.; Akgül, S.; et al. A Reference Collection of Patient-Derived Cell Line and Xenograft Models of Proneural, Classical and Mesenchymal Glioblastoma. Sci. Rep. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Paolillo, M.; Comincini, S.; Schinelli, S. In Vitro Glioblastoma Models: A Journey into the Third Dimension. Cancers (Basel). 2021, 13, 1–25. [Google Scholar] [CrossRef]
- Hong, X.; Chedid, K.; Kalkanis, S.N. Glioblastoma Cell Line-Derived Spheres in Serum-Containing Medium versus Serum-Free Medium: A Comparison of Cancer Stem Cell Properties. Int. J. Oncol. 2012, 41, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, T.; Ohata, H.; Sato, A.; Yamawaki, K.; Enomoto, T.; Okamoto, K. Tumor-Derived Spheroids: Relevance to Cancer Stem Cells and Clinical Applications. Cancer Sci. 2017, 108, 283–289. [Google Scholar] [CrossRef] [PubMed]
- Galli, R.; Binda, E.; Orfanelli, U.; Cipelletti, B.; Gritti, A.; De Vitis, S.; Fiocco, R.; Foroni, C.; Dimeco, F.; Vescovi, A. Isolation and Characterization of Tumorigenic, Stem-like Neural Precursors from Human Glioblastoma. Cancer Res. 2004, 64, 8130. [Google Scholar] [CrossRef] [PubMed]
- Conti, L.; Crisafulli, L.; Caldera, V.; Tortoreto, M.; Brilli, E.; Conforti, P.; Zunino, F.; Magrassi, L.; Schiffer, D.; Cattaneo, E. REST Controls Self-Renewal and Tumorigenic Competence of Human Glioblastoma Cells. PLoS One 2012, 7, 1–13. [Google Scholar] [CrossRef]
- Sancho-Martinez, I.; Nivet, E.; Xia, Y.; Hishida, T.; Aguirre, A.; Ocampo, A.; Ma, L.; Morey, R.; Krause, M.N.; Zembrzycki, A.; et al. Establishment of Human IPSC-Based Models for the Study and Targeting of Glioma Initiating Cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Bagó, J.R.; Alfonso-Pecchio, A.; Okolie, O.; Dumitru, R.; Rinkenbaugh, A.; Baldwin, A.S.; Miller, C.R.; Magness, S.T.; Hingtgen, S.D. Therapeutically Engineered Induced Neural Stem Cells Are Tumour-Homing and Inhibit Progression of Glioblastoma. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef]
- Merz, F.; Gaunitz, F.; Dehghani, F.; Renner, C.; Meixensberger, J.; Gutenberg, A.; Giese, A.; Schopow, K.; Hellwig, C.; Schäfer, M.; et al. Organotypic Slice Cultures of Human Glioblastoma Reveal Different Susceptibilities to Treatments. Neuro. Oncol. 2013, 15, 670–681. [Google Scholar] [CrossRef]
- Ohnishi, T.; Matsumura, H.; Izumoto, S.; Hiraga, S.; Hayakawa, T. A Novel Model of Glioma Cell Invasion Using Organotypic Brain Slice Culture. Cancer Res. 1998, 58, 2935–2940. [Google Scholar]
- Eisemann, T.; Costa, B.; Strelau, J.; Mittelbronn, M.; Angel, P.; Peterziel, H. An Advanced Glioma Cell Invasion Assay Based on Organotypic Brain Slice Cultures. BMC Cancer 2018, 18, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Henrik Heiland, D.; Ravi, V.M.; Behringer, S.P.; Frenking, J.H.; Wurm, J.; Joseph, K.; Garrelfs, N.W.C.; Strähle, J.; Heynckes, S.; Grauvogel, J.; et al. Tumor-Associated Reactive Astrocytes Aid the Evolution of Immunosuppressive Environment in Glioblastoma. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Joseph, K.; Wurm, J.; Behringer, S.; Garrelfs, N.; d’Errico, P.; Naseri, Y.; Franco, P.; Meyer-Luehmann, M.; Sankowski, R.; et al. Human Organotypic Brain Slice Culture: A Novel Framework for Environmental Research in Neuro-Oncology. Life Sci. Alliance 2019, 2, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Boccellato, C.; Rehm, M. Glioblastoma, from Disease Understanding towards Optimal Cell-Based in Vitro Models. Cell. Oncol. 2022, 45, 527–541. [Google Scholar] [CrossRef]
- Abuwatfa, W.H.; Pitt, W.G.; Husseini, G.A. Scaffold-Based 3D Cell Culture Models in Cancer Research. J. Biomed. Sci. 2024, 31. [Google Scholar] [CrossRef]
- De Witt Hamer, P.C.; Van Tilborg, A.A.G.; Eijk, P.P.; Sminia, P.; Troost, D.; Van Noorden, C.J.F.; Ylstra, B.; Leenstra, S. The Genomic Profile of Human Malignant Glioma Is Altered Early in Primary Cell Culture and Preserved in Spheroids. Oncogene 2008, 27, 2091–2096. [Google Scholar] [CrossRef]
- Boccellato, C.; Rehm, M. Glioblastoma, from Disease Understanding towards Optimal Cell-Based in Vitro Models. Cell. Oncol. 2022, 45, 527–541. [Google Scholar] [CrossRef]
- Bates, R.C.; Edwards, N.S.; Yates, J.D. Spheroids and Cell Survival. Crit. Rev. Oncol. Hematol. 2000, 36, 61–74. [Google Scholar] [CrossRef]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers (Basel). 2021, 13, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas2. JNCI J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Slika, H.; Karimov, Z.; Alimonti, P.; Abou-Mrad, T.; De Fazio, E.; Alomari, S.; Tyler, B. Preclinical Models and Technologies in Glioblastoma Research: Evolution, Current State, and Future Avenues. Int. J. Mol. Sci. 2023, 24. [Google Scholar] [CrossRef] [PubMed]
- Durand, R.E.; Olive, P.L. Resistance of Tumor Cells to Chemo- and Radiotherapy Modulated by the Three-Dimensional Architecture of Solid Tumors and Spheroids. Methods Cell Biol. 2001, 64, 211–233. [Google Scholar] [CrossRef]
- Friedrich, J.; Seidel, C.; Ebner, R.; Kunz-Schughart, L.A. Spheroid-Based Drug Screen: Considerations and Practical Approach. Nat. Protoc. 2009, 4, 309–324. [Google Scholar] [CrossRef]
- Andreatta, F.; Beccaceci, G.; Fortuna, N.; Celotti, M.; Felice, D. De; Lorenzoni, M.; Foletto, V.; Genovesi, S.; Rubert, J.; Alaimo, A. The Organoid Era Permits the Development of New Applications to Study Glioblastoma. Cancers (Basel). 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Madeline A., Lancaster; Juergen, A. Knoblich1 Generation of Cerebral Organoids from Human Pluripotent Stem Cells. Nat Protoc 2014, 9, 2329–2340. [Google Scholar]
- Hubert, C.G.; Rivera, M.; Spangler, L.C.; Wu, Q.; Mack, S.C.; Prager, B.C.; Couce, M.; McLendon, R.E.; Sloan, A.E.; Rich, J.N. A Three-Dimensional Organoid Culture System Derived from Human Glioblastomas Recapitulates the Hypoxic Gradients and Cancer Stem Cell Heterogeneity of Tumors Found in Vivo. Cancer Res. 2016, 76, 2465–2477. [Google Scholar] [CrossRef]
- Jacob, F.; Salinas, R.D.; Zhang, D.Y.; Nguyen, P.T.T.; Schnoll, J.G.; Wong, S.Z.H.; Thokala, R.; Sheikh, S.; Saxena, D.; Prokop, S.; et al. A Patient-Derived Glioblastoma Organoid Model and Biobank Recapitulates Inter- and Intra-Tumoral Heterogeneity. Cell 2020, 180, 188–204.e22. [Google Scholar] [CrossRef]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211.e5. [Google Scholar] [CrossRef] [PubMed]
- Pine, A.R.; Cirigliano, S.M.; Nicholson, J.G.; Hu, Y.; Linkous, A.; Miyaguchi, K.; Edwards, L.; Singhania, R.; Schwartz, T.H.; Ramakrishna, R.; et al. Tumor Microenvironment Is Critical for the Maintenance of Cellular States Found in Primary Glioblastomas. Cancer Discov. 2020, 10, 964–979. [Google Scholar] [CrossRef] [PubMed]
- Weth, F.R.; Peng, L.; Paterson, E.; Tan, S.T.; Gray, C. Utility of the Cerebral Organoid Glioma ‘GLICO’ Model for Screening Applications. Cells 2023, 12. [Google Scholar] [CrossRef] [PubMed]
- Heffernan, J.M.; Overstreet, D.J.; Le, L.D.; Vernon, B.L.; Sirianni, R.W. Bioengineered Scaffolds for 3D Analysis of Glioblastoma Proliferation and Invasion. Ann. Biomed. Eng. 2015, 43, 1965–1977. [Google Scholar] [CrossRef]
- Shafiee, A.; Atala, A. Printing Technologies for Medical Applications. Trends Mol. Med. 2016, 22, 254–265. [Google Scholar] [CrossRef]
- Stanković, T.; Ranđelović, T.; Dragoj, M.; Stojković Burić, S.; Fernández, L.; Ochoa, I.; Pérez-García, V.M.; Pešić, M. In Vitro Biomimetic Models for Glioblastoma-a Promising Tool for Drug Response Studies. Drug Resist. Updat. 2021, 55. [Google Scholar] [CrossRef]
- Orcheston-Findlay, L.; Bax, S.; Utama, R.; Engel, M.; Govender, D.; O’neill, G. Advanced Spheroid, Tumouroid and 3d Bioprinted in-Vitro Models of Adult and Paediatric Glioblastoma. Int. J. Mol. Sci. 2021, 22, 1–19. [Google Scholar] [CrossRef]
- Heinrich, M.A.; Bansal, R.; Lammers, T.; Zhang, Y.S.; Michel Schiffelers, R.; Prakash, J. 3D-Bioprinted Mini-Brain: A Glioblastoma Model to Study Cellular Interactions and Therapeutics. Adv. Mater. 2019, 31, 1–9. [Google Scholar] [CrossRef]
- Hermida, M.A.; Kumar, J.D.; Schwarz, D.; Laverty, K.G.; Di Bartolo, A.; Ardron, M.; Bogomolnijs, M.; Clavreul, A.; Brennan, P.M.; Wiegand, U.K.; et al. Three Dimensional in Vitro Models of Cancer: Bioprinting Multilineage Glioblastoma Models. Adv. Biol. Regul. 2020, 75, 100658. [Google Scholar] [CrossRef]
- Chadwick, M.; Yang, C.; Liu, L.; Gamboa, C.M.; Jara, K.; Lee, H.; Sabaawy, H.E. Rapid Processing and Drug Evaluation in Glioblastoma Patient-Derived Organoid Models with 4D Bioprinted Arrays. iScience 2020, 23, 101365. [Google Scholar] [CrossRef]
- Huang, Y.; Agrawal, B.; Sun, D.; Kuo, J.S.; Williams, J.C. Microfluidics-Based Devices: New Tools for Studying Cancer and Cancer Stem Cell Migration. Biomicrofluidics 2011, 5, 1–17. [Google Scholar] [CrossRef]
- Huang, Y.; Agrawal, B.; Clark, P.A.; Williams, J.C.; Kuo, J.S. Evaluation of Cancer Stem Cell Migration Using Compartmentalizing Microfluidic Devices and Live Cell Imaging. J. Vis. Exp. 2011, 1–7. [Google Scholar] [CrossRef]
- Pedron, S.; Polishetty, H.; Pritchard, A.M.; Mahadik, B.P.; Sarkaria, J.N.; Harley, B.A.C. Spatially Graded Hydrogels for Preclinical Testing of Glioblastoma Anticancer Therapeutics. MRS Commun. 2017, 7, 442–449. [Google Scholar] [CrossRef]
- Ma, J.; Li, N.; Wang, Y.; Wang, L.; Wei, W.; Shen, L.; Sun, Y.; Jiao, Y.; Chen, W.; Liu, J. Engineered 3D Tumour Model for Study of Glioblastoma Aggressiveness and Drug Evaluation on a Detachably Assembled Microfluidic Device. Biomed. Microdevices 2018, 20, 80. [Google Scholar] [CrossRef]
- Logun, M.T.; Bisel, N.S.; Tanasse, E.A.; Zhao, W.; Gunasekera, B.; Mao, L.; Karumbaiah, L. Glioma Cell Invasion Is Significantly Enhanced in Composite Hydrogel Matrices Composed of Chondroitin 4- and 4,6-Sulfated Glycosaminoglycans. J. Mater. Chem. B 2016, 4, 6052–6064. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.H.; Lee, K.H.; Lee, J.; Choi, H.; Lee, D.; Park, Y.; Lee, S.-H. Integration of Microfluidic Chip with Biomimetic Hydrogel for 3D Controlling and Monitoring of Cell Alignment and Migration. J. Biomed. Mater. Res. A 2014, 102, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Morales, R.-T.T.; Qian, W.; Wang, H.; Gagner, J.-P.; Dolgalev, I.; Placantonakis, D.; Zagzag, D.; Cimmino, L.; Snuderl, M.; et al. Hacking Macrophage-Associated Immunosuppression for Regulating Glioblastoma Angiogenesis. Biomaterials 2018, 161, 164–178. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Lin, H.; Rauch, J.; Deleyrolle, L.P.; Reynolds, B.A.; Viljoen, H.J.; Zhang, C.; Gu, L.; Van Wyk, E.; Lei, Y. Scalable Culturing of Primary Human Glioblastoma Tumor-Initiating Cells with a Cell-Friendly Culture System. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Akay, M.; Hite, J.; Avci, N.G.; Fan, Y.; Akay, Y.; Lu, G.; Zhu, J.J. Drug Screening of Human GBM Spheroids in Brain Cancer Chip. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef]
- Olubajo, F.; Achawal, S.; Greenman, J. Development of a Microfluidic Culture Paradigm for Ex Vivo Maintenance of Human Glioblastoma Tissue: A New Glioblastoma Model? Transl. Oncol. 2020, 13, 1–10. [Google Scholar] [CrossRef]
- Park, J.H.; Kim, M.J.; Kim, W.J.; Kwon, K.-D.; Ha, K.-T.; Choi, B.T.; Lee, S.-Y.; Shin, H.K. Isolinderalactone Suppresses Human Glioblastoma Growth and Angiogenic Activity in 3D Microfluidic Chip and in Vivo Mouse Models. Cancer Lett. 2020, 478, 71–81. [Google Scholar] [CrossRef]
- Yi, H.-G.; Jeong, Y.H.; Kim, Y.; Choi, Y.-J.; Moon, H.E.; Park, S.H.; Kang, K.S.; Bae, M.; Jang, J.; Youn, H.; et al. A Bioprinted Human-Glioblastoma-on-a-Chip for the Identification of Patient-Specific Responses to Chemoradiotherapy. Nat. Biomed. Eng. 2019, 3, 509–519. [Google Scholar] [CrossRef]
- Truong, D.; Fiorelli, R.; Barrientos, E.S.; Melendez, E.L.; Sanai, N.; Mehta, S.; Nikkhah, M. A Three-Dimensional (3D) Organotypic Microfluidic Model for Glioma Stem Cells - Vascular Interactions. Biomaterials 2019, 198, 63–77. [Google Scholar] [CrossRef]
- Ozturk, M.S.; Lee, V.K.; Zou, H.; Friedel, R.H.; Intes, X.; Dai, G. High-Resolution Tomographic Analysis of in Vitro 3D Glioblastoma Tumor Model under Long-Term Drug Treatment. Sci. Adv. 2020, 6, eaay7513. [Google Scholar] [CrossRef]
- Patrizii, M.; Bartucci, M.; Pine, S.R.; Sabaawy, H.E. Utility of Glioblastoma Patient-Derived Orthotopic Xenografts in Drug Discovery and Personalized Therapy. Front. Oncol. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Carlson, B.L.; Pokorny, J.L.; Schroeder, M.A.; Sarkaria, J.N. Establishment, Maintenance and in Vitro and in Vivo Applications of Primary Human Glioblastoma Multiforme (GBM) Xenograft Models for Translational Biology Studies and Drug Discovery.; 2011; Vol. Chapter 14; ISBN 0471141755.
- Joo, K.M.; Kim, J.; Jin, J.; Kim, M.; Seol, H.J.; Muradov, J.; Yang, H.; Choi, Y. La; Park, W.Y.; Kong, D.S.; et al. Patient-Specific Orthotopic Glioblastoma Xenograft Models Recapitulate the Histopathology and Biology of Human Glioblastomas In Situ. Cell Rep. 2013, 3, 260–273. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, N.; Fernández, V.; Pereira, R.C.; Rancati, S.; Pelizzoli, R.; De Pietri Tonelli, D. A Xenotransplant Model of Human Brain Tumors in Wild-Type Mice. iScience 2020, 23. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.; Labani-Motlagh, A.; Chen, A.; Bohorquez, J.A.; Qin, B.; Dodda, M.; Yang, F.; Ansari, D.; Patel, S.; Ji, H.; et al. Development of a Human Glioblastoma Model Using Humanized DRAG Mice for Immunotherapy. Antib. Ther. 2023, 6, 253–264. [Google Scholar] [CrossRef]
- Garcia, C.; Dubois, G.G.; Xavier, L.L.; Geraldo, H.H.; da Fonseca, C.C.C.; Correia, H.H.; Meirelles, F.; Ventura, G.; Romão, L.; Canedo, S.H.S.; et al. The Orthotopic Xenotransplant of Human Glioblastoma Successfully Recapitulates Glioblastoma-Microenvironment Interactions in a Non-Immunosuppressed Mouse Model. BMC Cancer 2014, 14, 1–11. [Google Scholar] [CrossRef]
- Valdor, R.; García-Bernal, D.; Bueno, C.; Ródenas, M.; Moraleda, J.M.; Macian, F.; Martínez, S. Glioblastoma Progression Is Assisted by Induction of Immunosuppressive Function of Pericytes through Interaction with Tumor Cells. Oncotarget 2017, 8, 68614–68626. [Google Scholar] [CrossRef]
- Vittori, M.; Motaln, H.; Turnšek, T.L. The Study of Glioma by Xenotransplantation in Zebrafish Early Life Stages. J. Histochem. Cytochem. 2015, 63, 749–761. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Patron, L.A.; Agudelo-Dueñãs, N.; Madrid-Wolff, J.; Venegas, J.A.; González, J.M.; Forero-Shelton, M.; Akle, V. Xenotransplantation of Human Glioblastoma in Zebrafish Larvae: In Vivo Imaging and Proliferation Assessment. Biol. Open 2019, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pliakopanou, A.; Antonopoulos, I.; Darzenta, N.; Serifi, I.; Simos, Y.V.; Katsenos, A.P.; Bellos, S.; Alexiou, G.A.; Kyritsis, A.P.; Leonardos, I.; et al. Glioblastoma Research on Zebrafish Xenograft Models: A Systematic Review. Clin. Transl. Oncol. 2024, 26, 311–325. [Google Scholar] [CrossRef]
- Geiger, G.A.; Fu, W.; Kao, G.D. Temozolomide-Mediated Radiosensitization of Human Glioma Cells in a Zebrafish Embryonic System. Cancer Res. 2008, 68, 3396–3404. [Google Scholar] [CrossRef]
- Lally, B.E.; Geiger, G.A.; Kridel, S.; Arcury-Quandt, A.E.; Robbins, M.E.; Kock, N.D.; Wheeler, K.; Peddi, P.; Georgakilas, A.; Kao, G.D.; et al. Identification and Biological Evaluation of a Novel and Potent Small Molecule Radiation Sensitizer via an Unbiased Screen of a Chemical Library. Cancer Res. 2007, 67, 8791–8799. [Google Scholar] [CrossRef]
- Lal, S.; La Du, J.; Tanguay, R.L.; Greenwood, J.A. Calpain 2 Is Required for the Invasion of Glioblastoma Cells in the Zebrafish Brain Microenvironment. J. Neurosci. Res. 2012, 90, 769–781. [Google Scholar] [CrossRef]
- Rampazzo, E.; Persano, L.; Pistollato, F.; Moro, E.; Frasson, C.; Porazzi, P.; Della Puppa, A.; Bresolin, S.; Battilana, G.; Indraccolo, S.; et al. Wnt Activation Promotes Neuronal Differentiation of Glioblastoma. Cell Death Dis. 2013, 4, 1–14. [Google Scholar] [CrossRef]
- Welker, A.M.; Jaros, B.D.; Puduvalli, V.K.; Imitola, J.; Kaur, B.; Beattie, C.E. Erratum: Standardized Orthotopic Xenografts in Zebrafish Reveal Glioma Cell-Line-Specific Characteristics and Tumor Cell Heterogeneity (Disease Models & Mechanisms (2016) 9 (199-210). DMM Dis. Model. Mech. 2016, 9, 1063–1065. [Google Scholar] [CrossRef] [PubMed]
- Pudelko, L.; Edwards, S.; Balan, M.; Nyqvist, D.; Al-Saadi, J.; Dittmer, J.; Almlöf, I.; Helleday, T.; Bräutigam, L. An Orthotopic Glioblastoma Animal Model Suitable for High-Throughput Screenings. Neuro. Oncol. 2018, 20, 1475–1484. [Google Scholar] [CrossRef]
- Maity, S.; Bhuyan, T.; Jewell, C.; Kawakita, S.; Sharma, S.; Nguyen, H.T.; Hassani Najafabadi, A.; Ermis, M.; Falcone, N.; Chen, J.; et al. Recent Developments in Glioblastoma-On-A-Chip for Advanced Drug Screening Applications. Small 2025, 21. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M. The Blood-Brain Barrier: Bottleneck in Brain Drug Development. NeuroRx 2005, 2, 3–14. [Google Scholar] [CrossRef]
- Morad, G.; Carman, C. V.; Hagedorn, E.J.; Perlin, J.R.; Zon, L.I.; Mustafaoglu, N.; Park, T.E.; Ingber, D.E.; Daisy, C.C.; Moses, M.A. Tumor-Derived Extracellular Vesicles Breach the Intact Blood-Brain Barrier via Transcytosis. ACS Nano 2019, 13, 13853–13865. [Google Scholar] [CrossRef]
- Zeng, A.; Ye, T.; Cao, D.; Huang, X.; Yang, Y.; Chen, X.; Xie, Y.; Yao, S.; Zhao, C. Identify a Blood-Brain Barrier Penetrating Drug-TNB Using Zebrafish Orthotopic Glioblastoma Xenograft Model. Sci. Rep. 2017, 7, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.M.; Will, P.; Kueckelhaus, J.; Sun, N.; Joseph, K.; Salié, H.; Vollmer, L.; Kuliesiute, U.; von Ehr, J.; Benotmane, J.K.; et al. Spatially Resolved Multi-Omics Deciphers Bidirectional Tumor-Host Interdependence in Glioblastoma. Cancer Cell 2022, 40, 639–655.e13. [Google Scholar] [CrossRef] [PubMed]
- Swanson, K.R.; Rockne, R.C.; Claridge, J.; Chaplain, M.A.; Alvord, E.C.; Anderson, A.R.A. Quantifying the Role of Angiogenesis in Malignant Progression of Gliomas: In Silico Modeling Integrates Imaging and Histology. Cancer Res. 2011, 71, 7366–7375. [Google Scholar] [CrossRef] [PubMed]
- Sakkalis, V.; Sfakianakis, S.; Tzamali, E.; Marias, K.; Stamatakos, G.; Misichroni, F.; Ouzounoglou, E.; Kolokotroni, E.; Dionysiou, D.; Johnson, D.; et al. Web-Based Workflow Planning Platform Supporting the Design and Execution of Complex Multiscale Cancer Models. IEEE J. Biomed. Heal. Informatics 2014, 18, 824–831. [Google Scholar] [CrossRef]
- Yankeelov, T.E.; Atuegwu, N.; Hormuth, D.; Weis, J.A.; Barnes, S.L.; Miga, M.I.; Rericha, E.C.; Quaranta, V. Clinically Relevant Modeling of Tumor Growth and Treatment Response. Sci. Transl. Med. 2013, 5, 187ps9–187ps9. [Google Scholar] [CrossRef]
- Düchting, W.; Ulmer, W.; Lehrig, R.; Ginsberg, T.; Dedeleit, E. Computer Simulation and Modelling of Tumor Spheroid Growth and Their Relevance for Optimization of Fractionated Radiotherapy. Strahlentherapie und Onkol. Organ der Dtsch. Rontgengesellschaft... [et al] 1992, 168, 354–360. [Google Scholar]
- Falco, J.; Agosti, A.; Vetrano, I.G.; Bizzi, A.; Restelli, F.; Broggi, M.; Schiariti, M.; Dimeco, F.; Ferroli, P.; Ciarletta, P.; et al. In Silico Mathematical Modelling for Glioblastoma: A Critical Review and a Patient-Specific Case. J. Clin. Med. 2021, 10. [Google Scholar] [CrossRef]
- Swanson, K.R.; Alvord, J.; Murray, J.D. A Quantitative Model for Differential Motility of Gliomas in Grey and White Matter. Cell Prolif. 2000, 33, 317–329. [Google Scholar] [CrossRef]
- Swanson, K.R.; Alvord, E.C.; Murray, J.D. Virtual Brain Tumours (Gliomas) Enhance the Reality of Medical Imaging and Highlight Inadequacies of Current Therapy. Br. J. Cancer 2002, 86, 14–18. [Google Scholar] [CrossRef]
- Jiao, Y.; Torquato, S. Emergent Behaviors from a Cellular Automaton Model for Invasive Tumor Growth in Heterogeneous Microenvironments. PLoS Comput. Biol. 2011, 7, 10–12. [Google Scholar] [CrossRef]
- Alfonso, J.C.L.; Talkenberger, K.; Seifert, M.; Klink, B.; Hawkins-Daarud, A.; Swanson, K.R.; Hatzikirou, H.; Deutsch, A. The Biology and Mathematical Modelling of Glioma Invasion: A Review. J. R. Soc. Interface 2017, 14. [Google Scholar] [CrossRef]
- Khalafallah, A.M.; Huq, S.; Jimenez, A.E.; Serra, R.; Bettegowda, C.; Mukherjee, D. “Zooming in” on Glioblastoma: Understanding Tumor Heterogeneity and Its Clinical Implications in the Era of Single-Cell Ribonucleic Acid Sequencing. Neurosurgery 2021, 88. [Google Scholar] [CrossRef]
- Frieboes, H.B.; Jin, F.; Chuang, Y.L.; Wise, S.M.; Lowengrub, J.S.; Cristini V, V. Three-Dimensional Multispecies Nonlinear Tumor Growth-II: Tumor Invasion and Angiogenesis. J. Theor. Biol. 2010, 264, 1254–1278. [Google Scholar] [CrossRef] [PubMed]
- Azuaje, F. Computational Models for Predicting Drug Responses in Cancer Research. Brief. Bioinform. 2017, 18, 820–829. [Google Scholar] [CrossRef] [PubMed]
- Dogra, N.; Singh, P.; Kumar, A. A Multistep In Silico Approach Identifies Potential Glioblastoma Drug Candidates via Inclusive Molecular Targeting of Glioblastoma Stem Cells. Mol. Neurobiol. 2024, 61, 9253–9271. [Google Scholar] [CrossRef]
- Zhang, B.; Nguyen, L.X.T.; Li, L.; Zhao, D.; Kumar, B.; Wu, H.; Lin, A.; Pellicano, F.; Hopcroft, L.; Su, Y.L.; et al. Bone Marrow Niche Trafficking of MiR-126 Controls the Self-Renewal of Leukemia Stem Cells in Chronic Myelogenous Leukemia. Nat. Med. 2018, 24, 450–462. [Google Scholar] [CrossRef]
- Kim, Y.; Lawler, S.; Nowicki, M.O.; Chiocca, E.A.; Friedman, A. A Mathematical Model for Pattern Formation of Glioma Cells Outside the Tumor Spheroid Core. J. Theor. Biol. 2009, 260, 359–371. [Google Scholar] [CrossRef]
- Stein, S.; Zhao, R.; Haeno, H.; Vivanco, I.; Michor, F. Mathematical Modeling Identifies Optimum Lapatinib Dosing Schedules for the Treatment of Glioblastoma Patients. PLoS Comput. Biol. 2018, 14, 1–24. [Google Scholar] [CrossRef]
- Rabé, M.; Dumont, S.; Álvarez-Arenas, A.; Janati, H.; Belmonte-Beitia, J.; Calvo, G.F.; Thibault-Carpentier, C.; Séry, Q.; Chauvin, C.; Joalland, N.; et al. Identification of a Transient State during the Acquisition of Temozolomide Resistance in Glioblastoma. Cell Death Dis. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Oraiopoulou, M.E.; Tzamali, E.; Tzedakis, G.; Liapis, E.; Zacharakis, G.; Vakis, A.; Papamatheakis, J.; Sakkalis, V. Integrating in Vitro Experiments with in Silico Approaches for Glioblastoma Invasion: The Role of Cell-to-Cell Adhesion Heterogeneity. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef]
- Pérez-García, V.M.; Calvo, G.F.; Bosque, J.J.; León-Triana, O.; Jiménez, J.; Pérez-Beteta, J.; Belmonte-Beitia, J.; Valiente, M.; Zhu, L.; García-Gómez, P.; et al. Universal Scaling Laws Rule Explosive Growth in Human Cancers. Nat. Phys. 2020, 16, 1232–1237. [Google Scholar] [CrossRef]
- Bongiovanni, A.; Colotti, G.; Liguori, G.L.; Di Carlo, M.; Digilio, F.A.; Lacerra, G.; Mascia, A.; Cirafici, A.M.; Barra, A.; Lanati, A.; et al. Applying Quality and Project Management Methodologies in Biomedical Research Laboratories: A Public Research Network’s Case Study. Accredit. Qual. Assur. 2015, 20, 203–213. [Google Scholar] [CrossRef]
- Digilio, F.A.; Lanati, A.; Bongiovanni, A.; Mascia, A.; Di Carlo, M.; Barra, A.; Cirafici, A.M.; Colotti, G.; Kisslinger, A.; Lacerra, G.; et al. Quality-Based Model for Life Sciences Research Guidelines. Accredit. Qual. Assur. 2016, 21. [Google Scholar] [CrossRef]
- Picciotto, S.; Barone, M.E.; Fierli, D.; Aranyos, A.; Adamo, G.; Božič, D.; Romancino, D.P.; Stanly, C.; Parkes, R.; Morsbach, S.; et al. Isolation of Extracellular Vesicles from Microalgae: Towards the Production of Sustainable and Natural Nanocarriers of Bioactive Compounds. Biomater. Sci. 2021. [Google Scholar] [CrossRef]
- Loria, F.; Picciotto, S.; Adamo, G.; Zendrini, A.; Raccosta, S.; Manno, M.; Bergese, P.; Liguori, G.L.; Bongiovanni, A.; Zarovni, N. A Decision-Making Tool to Navigate through Extracellular Vesicle Research and Product Development. JEV 2023, 2023.11.16.567368. [Google Scholar] [CrossRef]
- Hollmann, S.; Regierer, B.; D’Elia, D.; Kisslinger, A.; Liguori, G.L. Toward the Definition of Common Strategies for Improving Reproducibility, Standardization, Management, and Overall Impact of Academic Research. Adv. Biomembr. Lipid Self-Assembly 2022, 35, 2–24. [Google Scholar]
- Lacroix, R.; Judicone, C.; Mooberry, M.; Boucekine, M.; Key, N.S.; Dignat-George, F.; Ambrozic, A.; Bailly, N.; Buffat, C.; Buzas, E.; et al. Standardization of Pre-Analytical Variables in Plasma Microparticle Determination: Results of the International Society on Thrombosis and Haemostasis SSC Collaborative Workshop. J. Thromb. Haemost. 2013. [Google Scholar] [CrossRef]
- Welsh, J.A.; Goberdhan, D.C.I.; O’Driscol, L.; Buzas, E.I.; Blenkiron, C.; Bussolati, B.; Cai, H.; Vizio, D. Di; Driedonks, T.A.P.; Erdbrügger, U.; et al. Minimal Information for Studies of Extracellular Vesicles (MISEV2023): From Basic to Advanced Approaches. J. Extracell. Vesicles 2023, 12. [Google Scholar] [CrossRef]
- Bustin, S.A.; Beaulieu, J.F.; Huggett, J.; Jaggi, R.; Kibenge, F.S.B.; Olsvik, P.A.; Penning, L.C.; Toegel, S. MIQE Précis: Practical Implementation of Minimum Standard Guidelines for Fluorescence-Based Quantitative Real-Time PCR Experiments. BMC Mol. Biol. 2010, 11. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



