
Submitted:
18 August 2025
Posted:
20 August 2025
You are already at the latest version
Abstract
Biochemical characterization of proteins is fundamental to understanding their function. Typically, research in protein structure/function requires reasonable quantities of the protein of interest. Because of low abundance in their natural environment, heterogenous state of post-translational modifications or difficulty in obtaining the tissue containing the protein of interest, recombinant protein production is usually employed. One of the major difficulties impeding advances in biotechnological research is protein insolubility, undermining further downstream research and applications. Escherichia coli strains are popular hosts for protein production, but are often unfit for the expression of eukaryotic sequences due to the absence of proper post-translational modifications, some of which are crucial for protein folding and activity. Here, we showcase the challenges researchers may be confronted with when trying to produce proteins recombinantly, by using OsAPSE, an enzyme from rice, as an example of a difficult-to-produce protein. Several production hosts were explored and best results were obtained when OsAPSE was produced in E. coli combined with a solubility tag or when a higher eukaryotic system was used. This study highlights common pitfalls in protein research and provides strategies to overcome them, making it a case study for researchers facing similar challenges.
Keywords:
recombinant proteins
; protein solubility
; E. coli
; P. pastoris
; A. thaliana
; OsAPSE
1. Introduction
The term ‘protein’ was first coined in 1838 by Mulder and Berzelius, and referred to the ‘primitive organic oxides that are the basis for albumin and fibrin’. It is derived from the Greek ‘proteios’ meaning ‘the first’, referring to proteins as principal components of plant origin that herbivorous animals use as a primary source for nutrition [1,2]. Currently, proteins are defined as ‘unbranched linear polypeptide chains, comprising of 20 different types of amino acids, which are transcribed from genomic open reading frames’ [3]. Proteins are the cell’s workhorses and execute a wide range of physiological functions, including: structural support (f.i. collagen and keratin), transport of molecules (f.i. hemoglobin, membrane channels), (bio)chemical activity (i.e. enzymes), defense (f.i. immunoglobulins), cellular communication (f.i. receptors, several hormones) and storage (f.i. ferritin). Proteins are of crucial importance to every living organism, but are also subject to a wide range of research disciplines and industries [4].
Proteins in research and industrial applications are usually extracted from natural sources (i.e. plant organs, animal tissues) or produced recombinantly using living cells [5]. Protein production is generally considered as ‘successful’ if reasonable quantities of soluble and active protein of interest (POI) are obtained in a cost-effective and timely manner. Therefore, recombinant protein production is often opted when native expression levels are low or when higher protein abundances are required. Popular cell-based platforms for protein production include Escherichia coli, Bacillus spp., Pichia pastoris (syn. Komagataella phaffii), Saccharomyces spp. and Aspergillus spp. [6]. Protein production in living cells benefits from multiple advantages related to production costs, scalability, yield and downstream processing, depending on the considered system [7] (Table 1). There are, however, several important constraints to cell-based production strategies, limiting or even impeding successful protein production.
In general, production of prokaryotic proteins is confronted with least difficulties or challenges compared to production of eukaryotic proteins. Prokaryotic proteins are usually devoid of extensive post-translational modifications (PTMs). The opposite is true for eukaryotic proteins, as these often require N/O-glycosylation and/or disulfide bridges for oxidative protein folding, activity and solubility [12]. Interestingly, the largest share of recombinant proteins, either in industry or in research, are produced using a prokaryotic system [13,14], regardless of the protein’s origin (i.e. prokaryotic or eukaryotic). Indeed, the large majority (>80%) of all proteins present in the Protein Database (PDB) have been produced recombinantly in E. coli [15], while prokaryotic sequences only make up 36% of the sequences present in PDB.
The intrinsic ability of a protein to dissolve, usually in aqueous conditions, depends on the specific distribution of hydrophilic and hydrophobic residues, as well as the charge distribution, on the protein surface [16]. A protein polypeptide already starts to fold shortly after protein synthesis, as the polypeptide emerges from the ribosome, but complete folding can only occur once translation is finished [17]. Nascent polypeptides are exposed as random coils without defined structure and are in a partially folded and aggregation-prone state. Aggregation may occur when hydrophobic groups are exposed and mutually interact. Proteins have the intrinsic ability to fold into their native structure, by navigating through a funnel-shaped folding energy landscape. Local secondary structure elements (i.e. α-helices and β-sheets) are formed first and are stabilized by hydrogen bonding, which guides further folding. Along the folding process, proteins follow different folding pathways, assume various intermediate conformations, which are either energetically (un)favorable along the way to their desired native structure with lowest energy state and fully developed tertiary (and quaternary) structures [17]. Proteins can also assume partially folded states such as the ‘molten globule state’, in which secondary structures are largely formed but with less defined tertiary structures [18] or other stable non-native conformations [17]. In these cases, chaperones may help proteins to assume native folding and prevent aggregation by overcoming kinetic and thermodynamic barriers [19]. The folding route of a protein may be negatively inclined towards the misfolded state due to the absence of adequate PTMs, such as disulfide bridges, which directly impact protein structure. The host’s inability to create disulfide bridges is a strong predictor for protein misfolding. Misfolded proteins are kinetically and thermodynamically ‘trapped’ within the energy landscape and may aggregate and form so-called ‘inclusion bodies’ (IBs). IBs are typically observed in prokaryotic production systems and comprise of insoluble cytosolic aggregated and misfolded proteins, often due to the absence of PTMs or too high biosynthesis rates [20].
Protein insolubility and subsequent formation of IBs are considered one of the most important challenges in protein research. In most cases, IBs are undesired since soluble proteins are required for biochemical research or industrial applications [14,20]. Although the accumulation of a POI in IBs may seem like a failed experiment, this is not always the case, and in fact, IBs may even present researchers with unique opportunities. IBs contain insoluble but very pure forms of the POI and are easy to isolate through centrifugation. Furthermore, proteins in IBs are not necessarily (enzymatically) inactive, but may contain large quantities of active POI, as shown for insolubly produced β-galactosidase [21]. Unfolding and refolding strategies on misfolded proteins are widely performed [22]. However, solubilized and refolded proteins are no guarantee for protein activity, as the native structure is not always recovered [23]. Therefore, it is evident why prokaryotic proteins are produced most optimally in prokaryotic hosts and why eukaryotic proteins should be produced most successfully in eukaryotic hosts. Hence, it is strongly recommended to make use of the appropriate production system, taking the basic characteristics of every production host as well as the inherent characteristics of the POI into account. Prokaryotic production systems like E. coli lack the ability to perform PTMs. Eukaryotic production systems usually provide PTMs which affect protein solubility, structure and activity [12]. One should also consider PTM differences between eukaryotes. For instance, the glycosylation properties of animal proteins are very different from the glycosylation present in yeasts, insect or plant cells [24,25]. Although a general rule if to produce prokaryotic proteins in prokaryotes and eukaryotic proteins in eukaryotes, there are many exceptions, thereby emphasizing the unpredictability and uniqueness of every recombinant protein production.
Because of their ease of transformation, bacteria are usually the first platform of choice, even when the protein of interest is of eukaryotic origin. A plethora of examples demonstrate the successful recombinant production of eukaryotic proteins in prokaryotic hosts [26,27]. For instance, the α-D-Galactopyranosidase (AGAL) from Japanese rice was successfully produced in E. coli BL21 and used for crystallization and structure determination (PDB: 1UAS), even though this enzyme possesses 2 disulfide bridges [28]. When bacterial systems fail to produce a eukaryotic POI, yeasts are usually the next in line to experiment with. An important drawback for protein production in yeasts is the phenomenon of hyper-glycosylation, which could hamper downstream applications. To circumvent this problem, a mutant yeast strain which is devoid of the hyper-glycosylation can be used. Furthermore, yeast strains with plant-like and/or human-like N-glycosylation are available [29,30]. Other important eukaryotic protein platforms include mammalian cells, insect cells and plant cells (Table 1).
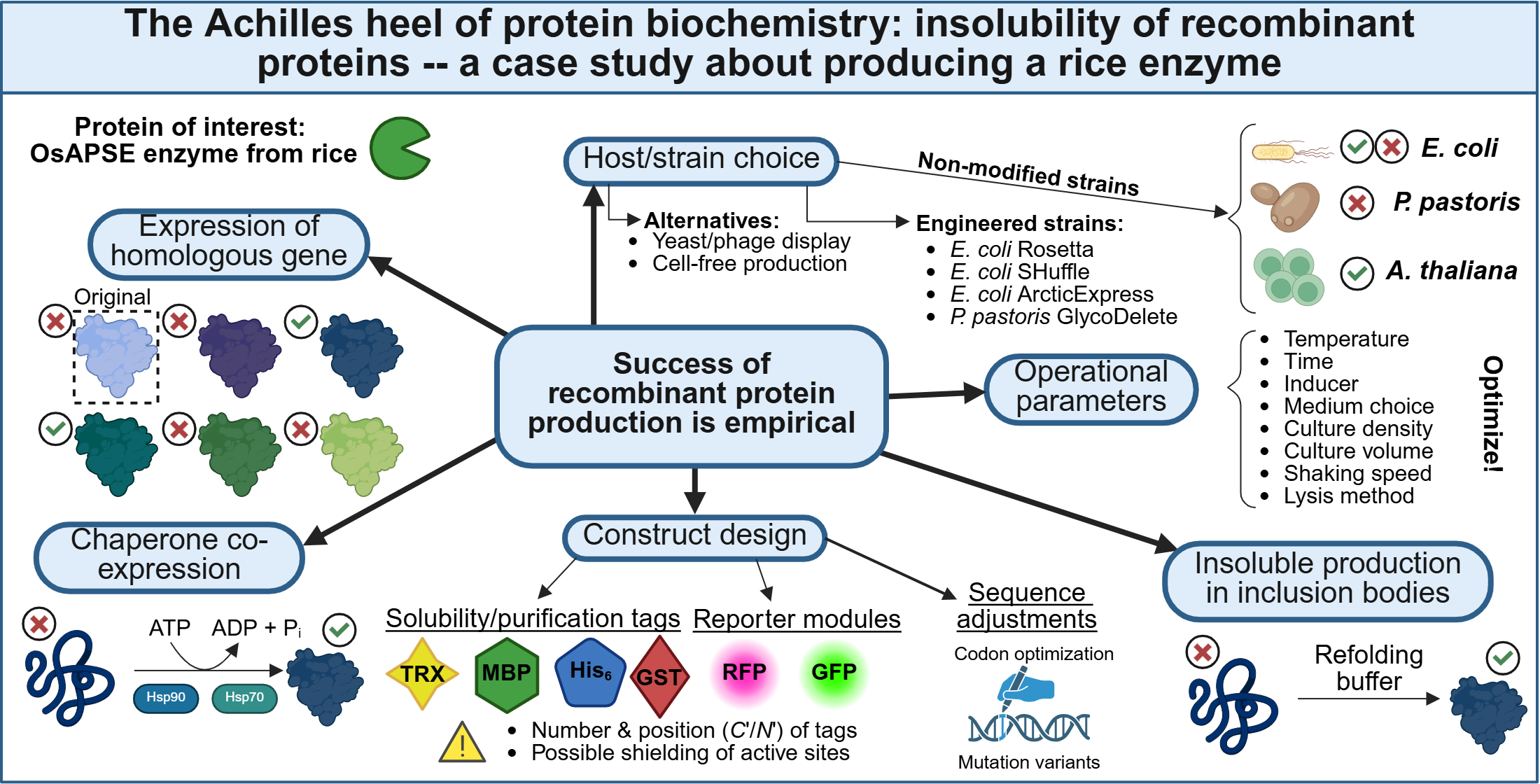
In contrast to several other protein characteristics, protein solubility is practically impossible to predict. Hence, empirical testing remains essential [31,32]. Therefore, when confronted with protein insolubility, several options can be considered to continue forward [14,33,34].
- First, it is recommended to optimize several operational production parameters such as the incubation temperature, shaking speed for aeration of the cell cultures, incubation time, concentration of inducer molecule for transcript expression, medium composition (f.i. presence of solubility enhancing additives, considering auto-induction medium), culture volume and cell lysis method [14,35]. In practice, it is advised to reduce the temperature during the induction because this reduces the protein biosynthesis rate and increases the chance of obtaining a soluble POI;
- Second, reconsideration of the expression construct may be advised. Researchers should take codon bias into account and optimize/harmonize the coding sequence. Several online tools make adjustments to the amino acid sequence, including deep learning and artificial intelligence, are available [36,37,38]. In addition, if non-optimized sequences are used in a prokaryotic system such as E. coli, the host strain Rosetta® could be considered. This strain is engineered with additional transfer-RNAs for enhancing translation of eukaryotic proteins with ‘rare codons’ [39]. Next to codon bias, the addition of solubility tags may be considered. Widely used solubility tags include the maltose-binding protein (MBP; 40 kDa), glutathione S-transferase (GST; 26 kDa) and thioredoxin (TRX; 12 kDa) [40]. Successful protein production is, however, not guaranteed when employing solubility tags. Several parameters exert an effect on the solubility of the new fusion protein [41,42], for instance the positioning (C or N terminal) of the solubility tag, the size of the tag, the number of tags … It should be taken into account that fusion with a large solubility tag may affect protein activity by sterically shielding active states. Finally, selection of a proper solubility tag and positioning towards the protein domain of interest often needs to be established and/or optimized empirically;
- Another option for prokaryotic protein production is to use modified host strains. Several modified hosts are available that may accommodate the researchers’ individual needs and are often equipped with additional chaperones. These chaperones are able to recognize unproperly folded proteins and prevent them from aggregation. Typical chaperons include the heat-shock proteins and have been engineered in strains to circumvent issues with protein aggregation [43], and may assist in proper protein folding [44]. The E. coli ArcticExpress® strain coproduces the Cpn10 and Cpn60 chaperonins from Oleispira antarctica, allowing protein production at lowered temperatures (4-10°C), potentially accommodating a lower protein biosynthesis rate and therefore limiting the risk of protein aggregation and IB formation [45]. Another example is the E. coli SHuffle® strain, which is equipped with the disulfide bond isomerase chaperone, allowing formation of disulfide bridges in the cytosol [46], hereby increasing solubility of proteins that require disulfide bridges [47];
- Next to usage of engineered host strains, it could be considered to co-express molecular chaperones that are situated upstream of downstream from the native gene of interest. There is sufficient evidence that these chaperones, mostly heat-shock proteins, are co-expressed under native conditions to ensure proper POI folding [48];
- A frequently utilized approach is to produce the POI in IBs and perform subsequent protein unfolding and refolding [22]. Protein refolding is controversial since the refolding step does not always restore the native folding, might trap the protein in a non-native state and could render it inactive. Protein refolding protocols require extensive optimization and are highly empirical [49,50,51]. Nevertheless, the performance of refolding strategies has been demonstrated may times before [20];
- Changing the expression host may be considered, since the success of recombinant protein production is for a large part determined by the host used. A study producing 29 human proteins in E. coli and P. pastoris demonstrated that all of the POI were soluble when using P. pastoris, compared to only 31% when using E. coli [52]. Eventually, cell-free production systems (CFPS) or phage/yeast display may be opted when traditional cell-based strategies are not successful [53]. CFPS systems make use of cell lysates and contain all the necessary component for protein synthesis. Both prokaryotic CFPS (f.i. cell lysates of E. coli, archaeans) and eukaryotic CFPS (f.i. tobacco Bright Yellow-2 lysates, rabbit reticulocyte lysates) systems exist, but similar to conventional recombinant protein production, the CFPS should be chosen carefully, taking into account the same considerations as mentioned above. Not unimportantly, CFPS may be confronted with reduced yields [54,55]. Page/yeast display has the advantage that the POI is produced by the host and presented at the cell surface, thereby removing the need for tedious or laborious optimization of protein production and purification. However, yeast/phage display may be confronted with similar issues a with traditional recombinant protein production, as the same constraints regarding non-native expression remain valid;
- A final option is considering to produce a homolog of the POI, as it was shown before that the success of recombinant protein production may vary between homologues [56].
The aim of this article is to highlight the difficulties associated with recombinant protein production. We will use the production of OsAPSE, a bifunctional enzyme from Japanese rice (Oryza sativa subsp. Japonica) [57] as a case study to demonstrate the challenges of protein expression in active and soluble form. This article also illustrates the difficulty of biotechnological research, how scientist can be confronted with setbacks but also how to turn negative and unexpected results into a learning ground for peers.
2. Results and Discussion
2.1. Characteristics of OsAPSE
OsAPSE is a multi-domain enzyme from Japanese rice (Figure 1A), containing 649 amino acid residues, with a calculated molecular weight of 73.1 kDa and an iso-electrical point at pH = 6.1 [57]. The protein was named after AtAPSE, its homolog in Arabidopsis thaliana [58]. The coding sequence of OsAPSE consists of an N-terminal signal peptide (residues 1-32), a glycoside hydrolase (GH) family 27 domain (residues 42-387), a ricin-B-like lectin domain (residues 498-563) and a C-terminal GH-all-beta domain (residues 563-649) (Figure 1B). In silico predictions (Figure 1C) revealed that the OsAPSE sequence possibly contains 3 sites for N-glycosylation, 5 sites for O-glycosylation and 3 disulfide bridges. In short, the N-terminal signal peptide is of importance for protein secretion under native conditions. The GH27 domain assumes a TIM-barrel fold and confers dual AGAL and β-L-Arabinopyranosidase (ARAP) activity towards substrates with α-D-Galp and β-L-Arap side chains from plant cell wall polysaccharides [57]. The ricin-B-like domain is a key determinant for the phylogeny of OsAPSE in its homologues within the GH27 family. This lectin-like domain is truncated and shows many similarities towards ricin-B lectins as well as proteins with a carbohydrate-binding module of family 13, which typically have a pseudo-symmetric threefold β-trefoil fold [59,60,61]. The C-terminal GH-all-beta domain is often observed in carbohydrate-active enzymes and confers structural stability and assists in protein folding [62,63].
2.2. Expression in E. coli Leads to Mostly Insoluble Proteins
Several experiments making use of diverse expression constructs and experimental conditions have been executed in attempt to obtain soluble OsAPSE (Table 2). Although, experiments to optimize the expression conditions (i.e. incubation temperature and duration, concentration of inducer) have also been carried out, these are not reported in this study to limit redundancy. We will focus on the results from experiments 1, 14, 17, 19, 21 and 26 (Table 2). In general, almost every experiment with E. coli as a host resulted in the expression of recombinant OsAPSE or its subdomains, being present in the insoluble cytosolic fraction as part of IBs. Figure 2 illustrates the outcomes of OsAPSE expression in different E. coli strains. Most constructs yielded insoluble protein, except for the MBP-tagged GH27 domain (experiment 26).
2.2.1. Effect of Codon Optimization
The factors leading to an insoluble protein are not completely understood and considered a ‘black box’, although there is a lot of empirical evidence that certain experimental parameters can improve protein solubility. Very often, proteins are produced recombinantly, using a host organism that differs from the sequence origin. It is widely accepted to produce a synthetic DNA sequence for which codon bias between the native organism and the expression host have been taken into account [64]. Codon optimization encompasses substituting native codons according to the codon preference/bias of the production host. Native codon usage is measured by the Relative Synonymous Codon Usage (RSCU) value, which is a percentage, indicating the relative usage of a codon for a particular amino acid. RSCU values are used to calculate the Codon Adaptation Index (CAI) for a particular POI in a chosen host. The CAI is normalized against RSCU values of highly expressed proteins, yielding a value between 0-1, in which 1 means that the POI is produced with the same easy as highly expressed proteins, and 0 means that the POI will not be produced at all [65]. Several reports indicate that taking codon bias into account through codon optimization will significantly enhance protein solubility, which was observed for the production of, for instance, KRAS4B, a human protein associated with MAPK signaling in cancers [64]. The CAI of OsAPSE for expression in E. coli equals 0.54, indicating that OsAPSE would be expressed with an efficiency of 54% compared to highly expressed proteins. Indeed, upon analysis of rare codons, it turned out that the native OsAPSE sequence from rice contained multiple (n = 89; 13.7%) rare codons, which could be difficult to express in a prokaryotic system. After codon optimization, the codon quality was improved considerably, reaching a CAI up to 0.89 (Supplementary File S1). The codon optimized sequence was used throughout all experiments in E. coli, except in experiments 17-22 where harmonization, alternative codon optimization algorithms and PROSS were used. However, codon optimization could not improve the solubility of OsAPSE (Figure 2A).
2.2.2. Utilization of E. coli Strains Capable of Synthesizing Disulfide Bridges
The possibility to create disulfide bridges by using the E. coli SHuffle® strain did not shift the solubility state of the GH27 domain of OsAPSE (Figure 2B). It was reported before that the use of this strain may lead to variable results in terms of expression level and protein solubility, emphasizing the need for optimizing for each recombinant protein [46].
2.2.3. Exploration of Protein Refolding
Because the solubility of OsAPSE remained a bottleneck, protein unfolding and refolding was attempted using a series of refolding buffers. Recombinant OsAPSE was unfolded and obtained a protein concentration of 2.8 mg/mL (Supplementary File S2.1). Best results were obtained for alkaline buffers (i.e. 50 mM bis-Tris pH 9 or 50 mM piperazine pH 10) in combination with 15 mM beta-mercaptoethanol, salts (i.e. 1-20 mM KCl, 20-250 mM NaCl, 5 mM CaCl2, 5 mM MgCl2), 2 mM glutathione and glutathione disulfide in a 10:1 ratio, 10% (v/v) glycerol, 400 mM L-arginine, 0.01% (w/v) PEG-1000 and 0.2% (v/v) CHAPS (Supplementary File S2.2), although A405 was just above the set threshold of 0.05. Similar to experiment 1, unfolding and refolding of the POI was executed at GenScript (experiment 14) (Figure 2C).
2.2.4. Effect of Codon Harmonization and Mutational Variants
In contrast to codon optimization, codon harmonization encompasses the mimicking of the relative codon frequency of the native gene of interest in the chosen production host, thereby ensuring ‘translational pauses’ that would occur naturally in the native background. Unfortunately, codon harmonization did not yield a soluble POI. Both codon harmonization and optimization yielded insoluble proteins (Table 2), indicating that the solubility issue was more deeply rooted than merely codon usage. Therefore, another strategy, utilizing the PROSS tool to create mutation variants of the codon-optimized OsAPSE sequence was considered. The underlying principle of PROSS it the introduction of multiple wisely considered mutations, based on atomistic modeling and phylogenetic sequence information, possibly resulting in proteins with a more energetically favorable folding state resembling the native state [17,18,38]. The PROSS algorithm has been proven useful in the past for recombinant production of human acetylcholinesterase, histone deacetylase and DNA methyltransferase, and delivered good results with considerably increased expression level and improved protein stability [36].
We obtained 9 PROSS variants of OsAPSE that differed between 1.3 and 7.2% compared to the original sequence, but were structurally very similar (Table 3). Most common mutations included H92P, N103V, L114P, F117W, G155A, I309P, E341N, S344L/P and S642C. Remarkably, several native residues were substituted by proline residues, which are known to drastically disrupt protein structure by introducing kinks in α-helices [66]. However, the mutations were not applied in regions with secondary structure elements. The introduction of proline residues did not distort protein structure meaningfully compared to the native OsAPSE (Table 3), but could impede catalytic sites, although these sites were shielded from mutations during the PROSS pipeline. However, the introduction of mutations that could be beneficial for the energy state of the protein folding did not contribute to a solubility shift. It should be noted that PROSS works based on 3D structures of the POI together with the structures of homologs. However, at present, no resolved structures for OsAPSE or its natural homologs exist, thereby likely reducing the efficacy of PROSS in our case. We used the AlphaFold model of OsAPSE, although this structure contains a highly uncertain region, i.e. the ricin-B-like domain, as indicated in Figure 1.

Table 2.
Overview of the PROSS variants of OsAPSE and their solubility when expressed. .
| WT | Variant 1 | Variant 2 | Variant 3 | Variant 4 | Variant 5 | Variant 6 | Variant 7 | Variant 8 | Variant 9 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Sequence identity compared to OsAPSE | 100 | 98.7 | 97.8 | 97.7 | 96.5 | 95.7 | 95.5 | 94.6 | 93.5 | 92.8 |
| Number of mutated amino acid residues | 0 | 8 | 14 | 15 | 23 | 28 | 29 | 35 | 42 | 47 |
| RMSD (Å) compared to OsAPSE | 0 | 0.0684 | 0.0825 | 0.0881 | 0.0908 | 0.1017 | 0.1030 | 0.0963 | 0.1081 | 0.1113 |
| POI produced recombinantly? | Yes | Yes | No | Yes | No | Yes | No | Yes | No | No |
| Soluble POI? | No | No | No | No | No | No | No | No | No | No |
2.2.5. Usage of Solubility Tags
Experiment 26 was the only one that yielded a soluble recombinant protein for the GH27 domain of OsAPSE, in combination with an N-terminal MBP tag and TEV protease cleavage site, and a C-terminal 3xFLAG and His6 tag (Figure 2D). The combination with the MBP solubility tag resulted in highly abundant protein production in the soluble phase. Interestingly, we noticed an important effect of the cell lysis technique. When lysing E. coli cells by using a lysis buffer, the GH27 domain ended up to be insoluble, while soluble POI was obtained when using sonication in combination with the TGH1 buffer (Figure 2D). MBP is considered to be one of the most effective fusion tags enhancing protein solubility, although the exact mechanism how MBP increases the solubility of its fusion partner is not completely understood [67,68]. It is hypothesized that MBP possesses chaperone-like properties and stabilized the folding process of its fusion partner, thereby reducing the likelihood of misfolding and protein aggregation. In addition, MBP by itself is naturally secreted to the periplasm through the general secretory pathway [69,70]. There are also several other solubility enhancing tags available, such as GST and TRX [71]. Our results indicate that the presence of MBP in itself is not a determinant for OsAPSE solubility, since drastic changes in protein solubility were observed only when the lysis method was changed. We assume that multiple factors are at play that work together and synergistically, including construct design (i.e. position and number of solubility tags), experimental conditions (i.e. production temperature, culture density, inducer concentration) and lysis method [72].
2.2.6. Enzymatic Activity of Soluble GH27_OsAPSE and Refolded OsAPSE
The refolded OsAPSE proteins from experiment 14 and the soluble GH27 domain obtained in experiment 26 were submitted to discontinuous enzymatic assays screening for AGAL activity (Table 2). Activity assays with the non-purified MBP-tagged GH27 domain showed no measurable AGAL activity (Supplementary File S3.1). In fact, the absorbance values decreased over time. In contrast, low AGAL activity was observed when the refolded OsAPSE proteins from experiment 14 were submitted to an enzymatic assay (Supplementary File S3.2). The increase in absorbance over time was dependent on the substrate concentration, as expected for enzymes obeying to Michaelis-Menten kinetics (Figure 3A). An increase of 53 µOD/s, 84.6 µOD/s, 111.6 µOD/s and 181 µOD/s were observed for the substrate concentrations of 1 mM, 5 mM, 10 mM and 20 mM pNP-α-D-Galp, respectively. These observations allowed estimation of the KM and Vmax value for the protein, based on the linearization methods of Hanes-Woolf and Eadie-Hofstee (Figure 3B).
Both linearization methods yielded the same maximum velocity and Michaelis constant, Vmax = 10.3 nM/s and KM = 44 mM, respectively. The reported Vmax value is mainly depending on the enzyme concentration used (i.e. 50 µg/mL ~ 65.5 nM), while the KM is an intrinsic parameter reflecting the affinity of the enzyme for the substrate. The calculated value for KM is considerably higher than that observed for the GH27 domain of OsAPSE produced by a CFPS system, i.e. KM = 0.67-4.68 mM and also the calculated Vmax is higher compared to the CFPS experiment (i.e. Vmax = 3.9-6.3 nM/s) but within the same order of magnitude in both cases [57]. The KM value of other plant GH27 AGALs for the synthetic pNP-α-D-Galp substrate is varying between 0.67-105 mM, with most reported KM values being situated below 3 mM [73,74,75,76,77,78]. The calculated turnover number was kcat = 0.15 s-1 for the used experimental setup. It is very likely that the high values for KM are due to suboptimal protein folding, which is often observed in protein refolding experiments. Indeed, it was reported before that achieving the native protein structure upon refolding is often difficult and that the POI can get trapped in a stable but non-native state, with aberrant protein folding, and therefore also activity, which could explain the increased KM [17,79,80]. Despite the divergent KM value, we were still able to demonstrate and validate the AGAL activity of OsAPSE, as was also observed in the CFPS experiment.
2.3. Expression in P. pastoris Yields Inactive Proteins of Interest
Although several attempts were undertaken to produce OsAPSE in the eukaryotic host P. pastoris (Table 2), most experiments did not yield production of the POI. However, the GH27 domain of OsAPSE, in combination with an N-terminal MBP tag, TEV protease site and mCherry RFP reporter tag, combined with a C-terminal His6 tag, was successfully produced (experiment 32). A total of 22 putatively transformed Pichia colonies were cultivated and protein production was induced. Afterwards, red fluorescence intensity was measured for every culture (Figure 4A) and was obvious in cultures C10, C12 and C18.
The presence of pink-colored proteins was even more obvious after centrifugation (Figure 4B) and is indicative for the production of the mCherry module, which was present in the MBP-TEV-mCherry-GH27_OsAPSE-His6 fusion protein (Figure 4C). Unfortunately, the produced fusion protein did not show measurable AGAL activity (Supplementary File S3.3). In contrast, an AGAL activity of 0.692 mOD/min was measured in the wild type negative control, which is attributed to background AGAL activity [81]. The inactivity of the GH27 domain, may be explained by steric hinderance caused by either the MBP tag or the mCherry modules, which have a molecular weight of 40 kDa and 26.7 kDa, respectively. Attempts to remove the mCherry tag using TEV protease cleavage were not successful, indicating that the MBP and/or mCherry modules are possibly also shielding the TEV cleavage site. Misfolding of the fusion protein is not likely, since the protein of interest was produced in a eukaryotic background, promoting oxidative disulfide bridge formation. P. pastoris typically produces proteins with hyper-mannose N-glycans [24], which are considerably different compared to the native N-glycans of plant proteins. It was already demonstrated that the large hyper-mannose glycans can significantly affect the enzyme activity through steric hinderance of the catalytic site [82,83]. Protein production and purification experiments with the mutant GlycoDelete strain, which creates truncated and plant-like N-glycans [30], were not successful (Table 2).
2.4. Expression in A. thaliana PSB-D Cell Cultures Results in Low Yields
A. thaliana PSB-D cell cultures were transformed with the pK7WG2D vector, co-producing the ER-localized EGFP, separately from the full-length OsAPSE protein or its individual subdomains (Table 2). Only the production of full-length OsAPSE was successful (experiment 34). Green fluorescent signals were observed in the ER and in vesicular structures (Figure 5A), which is indicative for transcription and translation of ER-localized EGFP and OsAPSE. Since OsAPSE was produced with its native signal peptide, it was hypothesized that OsAPSE would be secreted to the extracellular medium. Therefore, the medium from the plant cell culture was collected, and extracellular proteins in the medium were precipitated. A small but distinct protein band around 70 kDa was observed for the medium at 60% (390 g/L) and 80% (561 g/L) (NH4)2SO4 saturation (Figure 5B). Subsequently, AGAL activity was measured during a discontinuous enzymatic assay (Supplementary File S3.4), making use of the crude, non-purified, precipitates. The highest AGAL activity was observed in the precipitate at 80% saturation, reaching an enzymatic activity of 0.80 U/L (Figure 5C), although we could not repeat the measurements due to discontinuation of the cell cultures. The transformed A. thaliana PSB-D cultures were discontinued for different reasons. The used MSMO medium contains high levels of sucrose and is therefore prone to bacterial and fungal contamination. More important, the expression of a transgene in plant cell cultures is typically limited in time as the expression of transgenes if often (epi)genetically altered over successive generations and passages [84], until the point that the transgenic cells are outcompeted by native non-transformed cells.
3. Conclusions
This study exemplifies the difficulties researchers may face in recombinant protein production, particularly when working with eukaryotic proteins in non-native host systems. Different hosts for recombinant protein production are available and have specific advantages and limitations, requiring careful consideration when designing recombinant protein experiments [15]. The most important rule of thumb remains to ‘know your protein’ and select the production host based on the requirements of the POI with respect to structural characteristics and PTMs [27]. While E. coli is the most commonly used production host in research and industry [85], its limitations – particularly protein insolubility and improper folding – are major bottlenecks. Misfolded proteins often aggregate into IBs due to a lack of appropriate PTMs or too high protein biosynthesis rates. This study highlights the limitations of standard solubility-enhancing strategies, such as codon optimization, chaperone co-expression, or lowering expression temperatures. While solubility tags such as MBP may improve solubility, they introduce potential steric hinderance that complicates or impedes downstream applications.
Eukaryotic proteins are typically equipped with extensive PTMs which are important for protein structure and function, but also ensure solubility. Hence, the use of a eukaryotic system is recommended in the case the POI is of eukaryotic origin. However, eukaryotic protein production platforms are associated with their own difficulties and challenges in terms of ease of cultivation, yield, scalability, complexity, production costs and need for downstream processing. For example, P. pastoris often exhibits hyper-mannosylation, which can sterically hinder active sites, while plant cell systems typically suffer from low expression yields and slow growth cycles, complicating its scalability. Further fine-tuning in terms of construct design, promotor choice, high/low copy expression plasmid, is recommended for every production host.
In summary, our findings reinforce the highly empirical nature of recombinant protein production, where even well-established optimization strategies may fail depending on the POI. Despite testing multiple host systems and production conditions, OsAPSE expression remained challenging, illustrating the trial-and-error nature of protein biochemistry. We explored a multitude of experimental conditions across several production hosts, but these were mostly unsuccessful because the POI was usually produced in high abundance but insolubly. Usage of solubility tags and/or refolding approaches provided the best results, yet active enzyme recovery was still not straightforward. Despite the diverse and mixed results, we can confirm that it is possible to produce OsAPSE recombinantly in the soluble and active state, albeit with difficulties and challenges, under very specific conditions.
Our results emphasize the importance of negative data in protein research, as these can guide future optimization efforts and methodological refinements. Our findings provide practical insight for researchers facing similar challenges, underscoring the need for systematic screening, flexible experimental design and a realistic approach to optimizing recombinant protein production.
4. Materials and Methods
4.1. Construct Design and Host Transformation
The coding sequence of OsAPSE (UniProt: Q5QLK3) was codon-optimized for expression in different hosts (Table 2) and synthesized using the GeneArt Gene Synthesis service (Thermo Fisher Scientific, Waltham (MA), USA). Prior removal of the native signal peptide, addition of restriction sites and stop codons were performed when necessary. The Codon Harmonizer of the University of Graz, Austria was employed (http://biocatalysis.uni-graz.at/sites/codonharmonizer.html). For codon optimization, 4 algorithms were used from different websites: Integrated DNA Technologies (https://eu.idtdna.com/CodonOpt), EUROFINS (https://eurofinsgenomics.eu/), VectorBuilder (https://en.vectorbuilder.com/tool/codon-optimization.html) and NovoPro (https://www.novoprolabs.com/tools/codon-optimization). Codon bias was studied by using the Biologics International Corporation Rare Codon Analyzer tool [86] and the Rare Codon Caltor (https://people.mbi.ucla.edu/sumchan/caltor.html). The Protein Repair One-Stop Shop (PROSS) tool (https://pross.weizmann.ac.il/step/pross-terms/) was used to generate mutational variants of OsAPSE. The effect of the mutations on the resulting protein structure was assessed in PyMol v2.5.4 using calculation of root-mean square deviation (RMSD) values [87,88].
Expression in E. coli and P. pastoris was attempted using inducible expression plasmids containing the T7 promotor and the alcohol oxidase 1 promotor, respectively [89,90]. For expression in A. thaliana cell suspension cultures, plasmids enabling constitutive expression under the influence of the 35S promotor were used [91,92] (Table 2).
As indicated in Table 2, several expression constructs contained solubility tags at different positions, aiming to enhance the solubility of the POI, including the GST, MBP and TRX tags. Furthermore, several expression constructs contain additional tags that aid in protein detection, such as the enhanced green fluorescent protein (EGFP) and the mCherry red fluorescent protein (RFP) tags. The MBP and His6 purification tags were often used, but the 3xFLAG and 3xHA tags were also considered. Different cloning methods were utilized throughout this study: restriction and ligation using type I restriction enzymes (New England Biolabs, Ipswich (MA), USA), Gibson assembly combined with golden/green gate cloning [93,94], VersaTile cloning [95] and Gateway cloning [96]. Colony PCR and DNA sequencing of the inserted fragments (Biosearch/LGC Genomics GmbH, Berlin, Germany) were executed with vector-specific primers (Supplementary File S4). E. coli cells were transformed by heat-shock transformation [97]. P. pastoris cells were transformed through electroporation [98]. Cell suspension cultures of A. thaliana PSB-D were transformed through Agrobacterium-mediated transformation [99].
4.2. Protein Production and Extraction
4.2.1. Escherichia Coli
Overnight cultures (5 mL) with sterile selective lysogeny broth (LB) (Duchefa Biochemie, The Netherlands), were inoculated with a single colony and incubated at 37°C for 16-18 hours while shaking at 220 rpm. The overnight cultures were sub-cultured in sterile LB (upscaling in 1/20 ratio of overnight culture to sterile LB) and incubated at 37°C. the optical density at 600 nm (OD600) was measured regularly. When OD600 = 0.6-0.8, the culture was divided in several flasks and protein production was induced by adding isopropyl β-D-1-thiogalactopyranoside (Chem-Lab, Zedelgem, Belgium) to a final concentration of 0.5-1 mM. In the case of E. coli BL21-AI, arabinose (Thermo Fisher Scientific) was added to a final concentration of 0.2% (w/v). The cultures were incubated at different temperatures (37°C, 28°C, 21°C, 16°C or 4°C), depending on the used strain, for 20-24 hours while shaking at 220 rpm. When the pET-22b(+) plasmid was used, POIs were produced with an N-terminal pelB signal sequence, supposedly guiding the produced proteins to the periplasm and medium. Medium protein fractions were isolated by means of precipitation with 100% trichloroacetic acid (TCA) (Carl-Roth), rinsing with ice cold acetone and restitution of the pellet in 50 µL non-buffered Tris. Proteins present in the periplasm were collect by administering an osmotic shock by incubating the cell pellet in a TSE buffer containing 50 mM Tris-HCl pH 7.5, 30% (w/v) sucrose (Sigma-Aldrich) and 10 mM EDTA (Carl-Roth), while stirring gently for 30 minutes. The cells are collected through centrifugation (30 minutes, 15000 g, 4°C) and resuspended in ice-cold 5 mM MgSO4 (Chem-Lab) and incubated in an ice bath while shaking gently. During the shaking step, the proteins are released from the periplasmic fraction. The released periplasmic proteins are then collected by TCA precipitation, acetone rinsing and restitution in non-buffered Tris.
Throughout this study, multiple methods for protein extraction were utilized. Mostly, cell lysis was achieved through mechanical rupture using a sonicator (amplitude 30%, total sonication time of 15 minutes, on ice), or using glass beads (ø 0.2-0.5 mm) (Carl Roth, Karlsruhe, Germany), combined with chemical lysis using an extraction buffer: either the NEB-Express® E. coli Lysis Reagent (New England Biolabs) or the TGH1 (i.e. Triton-Glycerol-HEPES) aqueous extraction buffer containing 1% (w/v) Triton X-100 (Sigma-Aldrich, Saint-Louis (MO), USA), 10% glycerol (Chem-Lab) and 50 mM HEPES (Santa Cruz Biotechnology, Dallas (TX), USA), 300 mM NaCl (Chem-Lab), supplemented with 2 tablets of EDTA-free cOmpleteTM Protease Inhibitor (Hoffmann-La Roche, Basel, Switzerland) per 100 mL of extraction buffer and 0.1 mM phenylmethyl-sulfonyl fluoride (PMSF) (Thermo Fisher Scientific). Extractions were executed at room temperature for 20-30 minutes. Afterwards, the soluble and insoluble fractions were separated by centrifugation for 30 minutes at 4000g (4°C). The soluble cytoplasmic protein fraction is then present in the supernatant and the insoluble cytoplasmic protein fraction resides in the pellet.
4.2.2. Pichia Pastoris
Experiments using P. pastoris strains were limited to small-scale screenings in deep-well plates (24 wells, 2 mL per well) and did not proceed to the upscaling stage. Transformed P. pastoris cells bearing methanol-inducible expression plasmids were resuspended in selective BMGY medium containing 1 g/L yeast extract (Merck, Darmstadt, Germany), 2 g/L peptone (Merck), 1.34% (w/v) yeast nitrogen bae without amino acids but with (NH4)2SO4 (Chem-Lab), 100 mM KH2PO4/K2HPO4 (Merck) buffer at pH 6 and 10% (w/v) glycerol. The Pichia cultures were incubated at 28-30°C for 3 days in darkness, sealed with Millipore tape, while shaking at 240 rpm for adequate aeration. Afterwards, the cells were washed with BMGY and resuspended in BMMY medium, which contains 10% (v/v) sterile methanol (Chem-Lab), instead of glycerol. The following 2 days, the Pichia cultures were periodically spiked with methanol to a final concentration of 1% (v/v). For the constructs co-expressing an mCherry RFP reporter, protein production was screened by investigating red fluorescence intensity. This was done by analyzing 50 µL of Pichia culture in 96-well plates using a TECAN Infinite 200 PRO (TECAN, Männedorf, Switzerland) plate reader. RFP was exited at 560 ± 20 nm and emitted light was detected at 590 ± 10 nm. Pichia cultures showing high red fluorescence intensities were considered for further analysis and harvested by means of centrifugation. Extracellular proteins, secreted to the medium (total volume of 2 mL), were collected through acid precipitation using 100% TCA and washing with 100% ice-cold acetone (Chem-Lab). The protein pellet was collected by centrifugation (10 minutes, 10000 g, 4°C), after which it was dried to the air and dissolved in 50 µL of 1 M non-buffered Tris (MP Biomedicals, Irvine (CA), USA). For the intracellular proteins, the cultures (2 mL volume) were harvested through centrifugation (10 minutes, 10000 g, room temperature) and dissolved in TGH1 extraction buffer combined with glass beads (ø 0.2-0.5 mm), vigorous vortexing and cooling on ice, to achieve cell lysis. The lysates were centrifuged (30 minutes, 10000 g, 4°C) and the intracellular proteins were present in the supernatant. The pellet contained mainly cell debris and insoluble proteins.
4.2.3. Arabidopsis Thaliana PSB-D Cell Cultures
Continuous cultures of A. thaliana PSB-D cells were maintained and sub-cultured weekly in fresh selective Murashige and Skoog medium with Minimal Organics (MSMO) [99]. Simultaneously, 2 mL of the cell suspension was added to fresh selective MSMO plates and incubated at 21°C in darkness, as a back-up for failing or contaminated cultures. We made use of the pK7WG2D expression vector, which contains an endoplasmic reticulum (ER)-localized version of EGFP as reporter protein [91]. For each passage of the cell cultures, the presence of green fluorescence was assessed through confocal microscopy [100] using a Nikon A1R confocal laser scanning microscope (Nikon Instruments, Tokyo, Japan) and a CFI Plan Apo VC 60x WI DIC (NA1.2) objective. GFP was excited using the 488 nm argon laser line and detected using an 515-530 nm emission filter. Images were processed using the ImageJ software [101]. All original microscopy raw data is included in Supplementary File S5 and the processed images in Supplementary File S6.
Cells from a one-week old culture (or two-week old calli from MS plates) were harvested and cryogenically crushed to a fine cell powder using liquid nitrogen, a mortar and pestle. Approximately 0.5-1.0 g of cell powder was combined with 0.5-1.0 mL extraction buffer (f.i. TGH1) and incubated for 20-30 minutes at room temperature, after which the cell debris was separated from the protein solution through centrifugation (30 minutes, 4000 g, 4°C). The soluble intracellular protein fraction was collected in the supernatant, whereas the pellet contains the insoluble protein fraction. Thereafter, the supernatant was submitted to (NH4)2SO4 (Chem-Lab) precipitation at 80% saturation (561 g/L) to precipitate the secreted protein fraction for 3-4 days at 4°C. NaN3 (Sigma-Aldrich) to a final concentration of 0.1-0.5% (w/v) was added to prevent microbial growth in the protein solution. Protein precipitates were harvested through centrifugation (30 minutes, 4000 g, 4°C) and dissolved in 1-2 mL of 50 mM Tris-HCl pH 7.5.
4.3. Protein Analysis
4.3.1. Protein Concentration
Protein concentrations were determined using either the colorimetric Bradford assay [102] at 595 nm or UV spectrophotometry at 280 nm. Bovine serum albumin (BSA) was used as a reference protein in standard curves between 0-1 mg/mL.
4.3.2. SDS-PAGE and Western Blot
Discontinuous acrylamide gels with 0.01% SDS (MP Biomedicals) were prepared, containing 4% acrylamide in the stacking gel (pH 6.8) and 15% in the separating gel (pH 8.8). Protein samples were incubated at 98°C for 10 minutes in 4X sample buffer containing 1 M Tris-HCl pH 6.8, 8% (w/v) SDS, 40% (w/v) glycerol, 0.4% (w/v) bromophenol blue and 1.125 M beta-mercaptoethanol (Carl Roth), prior to electrophoretic analysis in a continuous electric field (180-200 V) for 1 hour. Running buffer containing 25 mM Tris, 200 mM glycine (Merck) and 0.1% (w/v) SDS was used. Gels were stained with acidic 0.1% (w/v) Coomassie Brilliant Blue R250 (Merck) and destained with destaining solution containing 2.5 M technical ethanol (Chem-Lab) and 1.3 M glacial acetic acid (Chem-Lab).
After SDS-PAGE, proteins were blotted on methanol-activated PVDF membranes (GE Healthcare, Chicago (IL), USA) by semi-dry electroblotting (Bio-Rad, Hercules (CA), USA) in Towbin buffer containing 25 mM Tris, 20% (v/v) methanol and 192 mM glycine. Membranes were incubated in 5% (w/v) milk powder solution (AppliChem GmbH, Darmstadt, Germany). His6-tagged proteins were detected using consecutively 1/5000 THETM His-tag monoclonal antibody (GenScript, Piscataway (NJ), USA), 1/1000 polyclonal rabbit anti-mouse antibody conjugated with horseradish peroxidase (Agilent/DAKO, Santa Clara (CA), USA), 1/300 peroxidase anti-peroxidase antibody (Sigma-Aldrich). All antibodies were incubated for 1 hour, except the peroxidase anti-peroxidase, which was incubated for 45 minutes. For detection, 100 mM Tris-HCl pH 7.6 containing 1 mM 3,3’-diaminobenzidine (Thermo Fisher Scientific) containing 320 µM H2O2 (Acros Organics, Geel, Belgium) was used. Trissaline containing 10 mM Tris, 150 mM NaCl and 0.1% (v/v) Triton X-100 was used as a diluent for all antibodies and for membrane washes (3x5 minutes) in between antibody incubations.
All original SDS-PAGE gels and blots are included in Supplementary File S6.
4.4. Downstream Analyses
4.4.1. Protein Refolding
Protein refolding was executed after recombinant protein production of certain expression constructs in E. coli (Table 2). Cells from a 2 L bacterial culture were harvested and resuspended in 50 mL sonication buffer (50 mM Tris, 100 mM NaCl, 1 mM PMSF, pH 8). The suspension was placed in ice water for temperature control and sonicated at 30% amplitude in pulses of 5 seconds, alternated with 5 seconds of pause (QSonica LLC, Newton (CT), USA), for a total sonication time of 15 minutes. After sonication, the IBs were isolated by means of centrifugation (30 minutes, 15000 rpm, 4°C). The IBs were thoroughly washed and vortexed, twice, with washing buffer 1 (i.e. extraction buffer supplemented with 0.1% (v/v) Triton X-100). Finally, the IBs were washed with washing buffer 2 (i.e. extraction buffer without PMSF) and collected through centrifugation (30 minutes, 15000 rpm, 4°C). The IBs were then solubilized by using a solubilization buffer containing 20 mM NaH2PO4, 500 mM NaCl, 5 mM beta-mercaptoethanol, 6 M guanidine hydrochloride (MP Biomedicals), 5 mM imidazole (Merck) at pH 7.5. Thereafter, the solubilized IBs were purified by nickel affinity chromatography, using column buffer 1 containing 20 mM Na2HPO4, 500 mM NaCl, 5 mM beta-mercaptoethanol, 8 M urea (Merck), 20 mM imidazole at pH 7.5, for column equilibration and washing. Column buffer 2 (i.e. column buffer 1 supplemented with 300 mM imidazole), was used for elution of the unfolded proteins. Finding the optimal refolding conditions need to be determined empirically, as there is no universal refolding buffer. Usually, a combination of a buffer system (f.i. PBS, Tris, HEPES, MES, citrate buffer, piperazine buffer, ...) at a pH between 4-10, supplemented with combinations of additives (f.i. salts, metal ion chelators, polyols, reducing agents, detergents, polymers, chaotropic agents, amino acids and monosaccharides) is considered [22,49,50,51,103,104]. Semi high-throughput screening methods have been developed in the past and have been proven useful for reference proteins including lysozyme, carbonic anhydrase B and glutamate receptor R2 [49,50,51]. Refolding conditions leading to soluble or insoluble proteins were evaluated by measuring the turbidity of the mixtures spectrophotometrically in the range of 300-400 nm. Low turbidity values (A < 0.05) indicate that the refolding buffer is a good solvent for the POI and does not cause insolubility and aggregation. We used different combinations of buffers and additives, as used in [50,51], similar to [104], using 10 µL of purified, unfolded proteins, combined with 190 µL of refolding buffer, and measured the turbidity at 405 ± 10 nm after overnight incubation at room temperature. The success of the refolding is usually confirmed through dynamic light scattering or circular dichroism, but can also be confirmed through other downstream analyses, such as enzymatic activity analysis (§4.4.2), since activity is determined by the extent to which the native protein structure has been restored [105].
4.4.2. Enzymatic Activity Assays
The success of protein refolding after protein production in E. coli, or when soluble proteins were obtained in other production hosts (Table 2), was assessed by performing enzymatic activity tests. Activity tests made use of the synthetic substrate p-4-nitrophenol-α-D-Galactopyranoside (pNP-α-D-Galp) (Thermo Fisher Scientific) at a final concentration of 25 mM to screen for AGAL activity. The enzyme solution at a final working concentration of 50 µg/mL (i.e. 66 nM) was used in 50 mM Tris-HCl pH 8 buffer. Discontinuous enzymatic assays were set up. Samples of 100 µL were taken every 5 minutes and inactivated in 100 µL of 0.2 M Na2CO3 (pH 11). Afterwards, absorbance measurements at 405 ± 10 nm were executed. Kinetic parameters were calculated by using Eadie-Hofstee [106] and Hanes-Woolf [107] linearization methods.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. S1: analysis of codon bias in E. coli; S2: protein refolding screening data; S3: enzymatic assay data; S4: used oligonucleotides; S5: raw microscopy data; S6: original SDS-PAGE gels, blots and microscopy images.
Author Contributions
Conceptualization: T.D.C.; methodology: T.D.C. and E.J.M.V.D.; formal analysis: T.D.C.; investigation: T.D.C.; resources: H.V.; data curation: T.D.C.; writing – original draft preparation: T.D.C.; writing – review and editing: H.V. and E.J.M.V.D.; visualization: T.D.C.; supervision: H.V. and E.J.M.V.D.; project administration: E.J.M.V.D.; funding acquisition: E.J.M.V.D.
Funding
This work was supported by funding from Fonds voor Wetenschappelijk Onderzoek Vlaanderen, grant number G008619N.
Data Availability Statement
The original contributions presented in this study are included in the article and supplementary materials. Further inquiries can be directed to the corresponding author.
Acknowledgments
The authors wish to acknowledge all students that were helping in exploring different strategies to produce OsAPSE and/or its subdomains as part of their bachelor/master training: Gythe Vandebriel, Chloé Vanden Herrewegen, Brahim Vandormael, Anne-Sophie Chys, Jarno Van de Geuchte and Zoé Suffys.
Conflicts of Interest
The authors declare no conflicts of interest
Abbreviations
The following abbreviations are used in this manuscript:
| AGAL | α-D-Galactopyranosidase |
| BSA | Bovine Serum Albumin |
| CAI | Codon Adaptation Index |
| CFPS | Cell-Free Production System |
| EGFP | Enhanced Green Fluorescent Protein |
| ER | Endoplasmic Reticulum |
| GST | Glutathione S-Transferase |
| IB | Inclusion Body |
| LB | Lysogeny Broth |
| MBP | Maltose-Binding Protein |
| MSMO | Murashige and Skoog medium with Minimal Organics |
| OD600 | Optical Density at 600 nm |
| PMSF | Phenyl Methyl Sulfonyl Fluoride |
| pNP-α-D-Galp | p-4-nitrophenol-α-D-Galactopyranoside |
| POI | Protein Of Interest |
| PROSS | Protein Repair One-Stop Shop |
| PSB-D | Plant Systems Biology – Dark |
| PTM | Post-Translational Modification |
| RSCU | Relative Synonymous Codon Usage |
| RFP | Red Fluorescent Protein |
| RMSD | Root-Mean Square Deviation |
| TCA | Trichloroacetic acid |
| TEV | Tobacco Etch Virus |
| TGH | Tris-Glycerol-HEPES |
| TRX | Thioredoxin |
References
- Mulder, G. Sur La Composition de Quelques Substances Animales. Bulletin des Sciences Physiques et Naturelles en Néerlande 1838, 129–151. [Google Scholar]
- Hartley, H. Origin of the Word “Protein”. Nature 1951, 168, 244. [Google Scholar] [CrossRef] [PubMed]
- Parisi, G.; Palopoli, N.; Tosatto, S.C.E.; Fornasari, M.S.; Tompa, P. “Protein” No Longer Means What It Used To. Current Research in Structural Biology 2021, 3, 146–152. [Google Scholar] [CrossRef] [PubMed]
- Corsetti, G.; Pasini, E.; Scarabelli, T.M.; Romano, C.; Singh, A.; Scarabelli, C.C.; Dioguardi, F.S. Importance of Energy, Dietary Protein Sources, and Amino Acid Composition in the Regulation of Metabolism: An Indissoluble Dynamic Combination for Life. Nutrients 2024, 16, 2417. [Google Scholar] [CrossRef]
- Puetz, J.; Wurm, F.M. Recombinant Proteins for Industrial versus Pharmaceutical Purposes: A Review of Process and Pricing. 2019. [Google Scholar]
- Sewalt, V.; Shanahan, D.; Gregg, L.; La Marta, J.; Carrillo, R. The Generally Recognized as Safe (GRAS) Process for Industrial Microbial Enzymes. Industrial Biotechnology 2016, 12, 295–302. [Google Scholar] [CrossRef]
- Francis, D.M.; Page, R. Strategies to Optimize Protein Expression in E. Coli. CP Protein Science 2010, 61. [Google Scholar] [CrossRef]
- Balen, B.; Krsnik-Rasol, M. N-Glycosylation of Recombinant Therapeutic Glycoproteins in Plant Systems. 2007. [Google Scholar]
- Karbalaei, M.; Rezaee, S.A.; Farsiani, H. Pichia Pastoris : A Highly Successful Expression System for Optimal Synthesis of Heterologous Proteins. Journal Cellular Physiology 2020, 235, 5867–5881. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Liu, H.; Lv, B.; Li, C. Regulating Strategies for Producing Carbohydrate Active Enzymes by Filamentous Fungal Cell Factories. Front. Bioeng. Biotechnol. 2020, 8, 691. [Google Scholar] [CrossRef]
- Schütz, A.; Bernhard, F.; Berrow, N.; Buyel, J.F.; Ferreira-da-Silva, F.; Haustraete, J.; van den Heuvel, J.; Hoffmann, J.-E.; de Marco, A.; Peleg, Y.; et al. A Concise Guide to Choosing Suitable Gene Expression Systems for Recombinant Protein Production. STAR Protocols 2023, 4. [Google Scholar] [CrossRef]
- Lee, J.M.; Hammarén, H.M.; Savitski, M.M.; Baek, S.H. Control of Protein Stability by Post-Translational Modifications. Nat Commun 2023, 14, 201. [Google Scholar] [CrossRef]
- Overton, T.W. Recombinant Protein Production in Bacterial Hosts. Drug Discovery Today 2014, 19, 590–601. [Google Scholar] [CrossRef] [PubMed]
- Bhatwa, A.; Wang, W.; Hassan, Y.I.; Abraham, N.; Li, X.-Z.; Zhou, T. Challenges Associated With the Formation of Recombinant Protein Inclusion Bodies in Escherichia Coli and Strategies to Address Them for Industrial Applications. Front. Bioeng. Biotechnol. 2021, 9, 630551. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Miralles, N.; Saccardo, P.; Corchero, J.L.; Garcia-Fruitós, E. Recombinant Protein Production and Purification of Insoluble Proteins. In Insoluble Proteins; Garcia Fruitós, E., Arís Giralt, A., Eds.; Methods in Molecular Biology; Springer US: New York, NY, 2022; Volume 2406, pp. 1–31. ISBN 978-1-07-161858-5. [Google Scholar]
- Kramer, R.M.; Shende, V.R.; Motl, N.; Pace, C.N.; Scholtz, J.M. Toward a Molecular Understanding of Protein Solubility: Increased Negative Surface Charge Correlates with Increased Solubility. Biophysical Journal 2012, 102, 1907–1915. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative Strategies to Treat Protein Misfolding in Inborn Errors of Metabolism: Pharmacological Chaperones and Proteostasis Regulators. J Inherit Metab Dis 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Onuchic, J.N.; Luthey-Schulten, Z.; Wolynes, P.G. Theory of Protein Folding: The Energy Landscape Perspective. Annu. Rev. Phys. Chem 1997, 48, 545–600. [Google Scholar] [CrossRef]
- Finkelstein, A.V.; Bogatyreva, N.S.; Ivankov, D.N.; Garbuzynskiy, S.O. Protein Folding Problem: Enigma, Paradox, Solution. Biophys Rev 2022, 14, 1255–1272. [Google Scholar] [CrossRef]
- García-Fruitós, E.; González-Montalbán, N.; Morell, M.; Vera, A.; Ferraz, R.M.; Arís, A.; Ventura, S.; Villaverde, A. Aggregation as Bacterial Inclusion Bodies Does Not Imply Inactivation of Enzymes and Fluorescent Proteins. Microb Cell Fact 2005, 4, 27. [Google Scholar] [CrossRef]
- Flores, S.S.; Nolan, V.; Perillo, M.A.; Sánchez, J.M. Superactive β-Galactosidase Inclusion Bodies. Colloids and Surfaces B: Biointerfaces 2019, 173, 769–775. [Google Scholar] [CrossRef]
- Singh, A.; Upadhyay, V.; Upadhyay, A.K.; Singh, S.M.; Panda, A.K. Protein Recovery from Inclusion Bodies of Escherichia Coli Using Mild Solubilization Process. Microb Cell Fact 2015, 14, 41. [Google Scholar] [CrossRef]
- Vallejo, L.F.; Rinas, U. Strategies for the Recovery of Active Proteins through Refolding of Bacterial Inclusion Body Proteins. Microb Cell Fact 2004, 3, 11. [Google Scholar] [CrossRef]
- Chung, C.-Y.; Majewska, N.I.; Wang, Q.; Paul, J.T.; Betenbaugh, M.J. SnapShot: N-Glycosylation Processing Pathways across Kingdoms. Cell 2017, 171, 258–258.e1. [Google Scholar] [CrossRef] [PubMed]
- Joshi, H.J.; Narimatsu, Y.; Schjoldager, K.T.; Tytgat, H.L.P.; Aebi, M.; Clausen, H.; Halim, A. SnapShot: O-Glycosylation Pathways across Kingdoms. Cell 2018, 172, 632–632.e2. [Google Scholar] [CrossRef]
- Sørensen, H.P.; Mortensen, K.K. Soluble Expression of Recombinant Proteins in the Cytoplasm of Escherichia Coli. Microb Cell Fact 2005, 4, 1. [Google Scholar] [CrossRef]
- Martínez-Alarcón, D.; Blanco-Labra, A.; García-Gasca, T. Expression of Lectins in Heterologous Systems. IJMS 2018, 19, 616. [Google Scholar] [CrossRef]
- Fujimoto, Z.; Kaneko, S.; Momma, M.; Kobayashi, H.; Mizuno, H. Crystal Structure of Rice α-Galactosidase Complexed with D-Galactose. Journal of Biological Chemistry 2003, 278, 20313–20318. [Google Scholar] [CrossRef]
- Meuris, L.; Santens, F.; Elson, G.; Festjens, N.; Boone, M.; Dos Santos, A.; Devos, S.; Rousseau, F.; Plets, E.; Houthuys, E.; et al. GlycoDelete Engineering of Mammalian Cells Simplifies N-Glycosylation of Recombinant Proteins. Nat Biotechnol 2014, 32, 485–489. [Google Scholar] [CrossRef]
- Piron, R.; Santens, F.; De Paepe, A.; Depicker, A.; Callewaert, N. Using GlycoDelete to Produce Proteins Lacking Plant-Specific N-Glycan Modification in Seeds. Nat Biotechnol 2015, 33, 1135–1137. [Google Scholar] [CrossRef]
- Habibi, N.; Mohd Hashim, S.Z.; Norouzi, A.; Samian, M.R. A Review of Machine Learning Methods to Predict the Solubility of Overexpressed Recombinant Proteins in Escherichia Coli. BMC Bioinformatics 2014, 15, 134. [Google Scholar] [CrossRef] [PubMed]
- Bhandari, B.K.; Gardner, P.P.; Lim, C.S. Solubility-Weighted Index: Fast and Accurate Prediction of Protein Solubility. Bioinformatics 2020, 36, 4691–4698. [Google Scholar] [CrossRef]
- Gutiérrez-González, M.; Farías, C.; Tello, S.; Pérez-Etcheverry, D.; Romero, A.; Zúñiga, R.; Ribeiro, C.H.; Lorenzo-Ferreiro, C.; Molina, M.C. Optimization of Culture Conditions for the Expression of Three Different Insoluble Proteins in Escherichia Coli. Sci Rep 2019, 9, 16850. [Google Scholar] [CrossRef] [PubMed]
- Mital, S.; Christie, G.; Dikicioglu, D. Recombinant Expression of Insoluble Enzymes in Escherichia Coli: A Systematic Review of Experimental Design and Its Manufacturing Implications. Microb Cell Fact 2021, 20, 208. [Google Scholar] [CrossRef]
- Atroshenko, D.L.; Sergeev, E.P.; Golovina, D.I.; Pometun, A.A. Additivities for Soluble Recombinant Protein Expression in Cytoplasm of Escherichia Coli. Fermentation 2024, 10, 120. [Google Scholar] [CrossRef]
- Goldenzweig, A.; Goldsmith, M.; Hill, S.E.; Gertman, O.; Laurino, P.; Ashani, Y.; Dym, O.; Unger, T.; Albeck, S.; Prilusky, J.; et al. Automated Structure- and Sequence-Based Design of Proteins for High Bacterial Expression and Stability. Molecular Cell 2016, 63, 337–346. [Google Scholar] [CrossRef]
- Mignon, C.; Mariano, N.; Stadthagen, G.; Lugari, A.; Lagoutte, P.; Donnat, S.; Chenavas, S.; Perot, C.; Sodoyer, R.; Werle, B. Codon Harmonization – Going beyond the Speed Limit for Protein Expression. FEBS Letters 2018, 592, 1554–1564. [Google Scholar] [CrossRef]
- Listov, D.; Goverde, C.A.; Correia, B.E.; Fleishman, S.J. Opportunities and Challenges in Design and Optimization of Protein Function. Nat Rev Mol Cell Biol 2024, 25, 639–653. [Google Scholar] [CrossRef] [PubMed]
- Novy, R.; Drott, D.; Yaeger, K.; Mierendorf, R. inNovations; 2001; pp. 1–3. [Google Scholar]
- Bell, M.R.; Engleka, M.J.; Malik, A.; Strickler, J.E. To Fuse or Not to Fuse: What Is Your Purpose? Protein Science 2013, 22, 1466–1477. [Google Scholar] [CrossRef]
- Sommer, B.; Friehs, K.; Flaschel, E.; Reck, M.; Stahl, F.; Scheper, T. Extracellular Production and Affinity Purification of Recombinant Proteins with Escherichia Coli Using the Versatility of the Maltose Binding Protein. Journal of Biotechnology 2009, 140, 194–202. [Google Scholar] [CrossRef]
- Raran-Kurussi, S.; Keefe, K.; Waugh, D.S. Positional Effects of Fusion Partners on the Yield and Solubility of MBP Fusion Proteins. Protein Expression and Purification 2015, 110, 159–164. [Google Scholar] [CrossRef]
- Piette, F.; Struvay, C.; Feller, G. The Protein Folding Challenge in Psychrophiles: Facts and Current Issues. Environmental Microbiology 2011, 13, 1924–1933. [Google Scholar] [CrossRef] [PubMed]
- Saibil, H. Chaperone Machines for Protein Folding, Unfolding and Disaggregation. Nat Rev Mol Cell Biol 2013, 14, 630–642. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Chernikova, T.N.; Timmis, K.N.; Golyshin, P.N. Expression of a Temperature-Sensitive Esterase in a Novel Chaperone-Based Escherichia Coli Strain. Appl Environ Microbiol 2004, 70, 4499–4504. [Google Scholar] [CrossRef]
- Lobstein, J.; Emrich, C.A.; Jeans, C.; Faulkner, M.; Riggs, P.; Berkmen, M. SHuffle, a Novel Escherichia Coli Protein Expression Strain Capable of Correctly Folding Disulfide Bonded Proteins in Its Cytoplasm. Microb Cell Fact 2012, 11, 753. [Google Scholar] [CrossRef]
- Kauzmann, W.; Douglas, R.G. The Effect of Disulfide Bonding on the Solubility of Unfolded Serum Albumin in Salt Solutions. Archives of Biochemistry and Biophysics 1956, 65, 106–119. [Google Scholar] [CrossRef]
- An, L.; Gao, H.; Zhong, Y.; Liu, Y.; Cao, Y.; Yi, J.; Huang, X.; Wen, C.; Tong, R.; Pan, Z.; et al. Molecular Chaperones HSP40, HSP70, STIP1, and HSP90 Are Involved in Stabilization of Cx43. Cytotechnology 2023, 75, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.-Q.; Gouaux, E. Overexpression of a Glutamate Receptor (GluR2) Ligand Binding Domain in Escherichia Coli : Application of a Novel Protein Folding Screen. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 13431–13436. [Google Scholar] [CrossRef]
- Armstrong, N.; Lencastre, A.D.; Gouaux, E. A New Protein Folding Screen: Application to the Ligand Binding Domains of a Glutamate and Kainate Receptor and to Lysozyme and Carbonic Anhydrase. Protein Science 1999, 8, 1475–1483. [Google Scholar] [CrossRef] [PubMed]
- Vincentelli, R.; Canaan, S.; Campanacci, V.; Valencia, C.; Maurin, D.; Frassinetti, F.; Scappucini-Calvo, L.; Bourne, Y.; Cambillau, C.; Bignon, C. High-throughput Automated Refolding Screening of Inclusion Bodies. Protein Science 2004, 13, 2782–2792. [Google Scholar] [CrossRef] [PubMed]
- Lueking, A.; Holz, C.; Gotthold, C.; Lehrach, H.; Cahill, D. A System for Dual Protein Expression in Pichia Pastoris and Escherichia Coli. Protein Expression and Purification 2000, 20, 372–378. [Google Scholar] [CrossRef]
- Tsai, S.-L.; DaSilva, N.A.; Chen, W. Functional Display of Complex Cellulosomes on the Yeast Surface via Adaptive Assembly. ACS Synth. Biol. 2013, 2, 14–21. [Google Scholar] [CrossRef]
- Harbers, M. Wheat Germ Systems for Cell-free Protein Expression. FEBS Letters 2014, 588, 2762–2773. [Google Scholar] [CrossRef]
- Zemella, A.; Thoring, L.; Hoffmeister, C.; Kubick, S. Cell-Free Protein Synthesis: Pros and Cons of Prokaryotic and Eukaryotic Systems. ChemBioChem 2015, 16, 2420–2431. [Google Scholar] [CrossRef] [PubMed]
- Morel, N.; Massoulié, J. Comparative Expression of Homologous Proteins. Journal of Biological Chemistry 2000, 275, 7304–7312. [Google Scholar] [CrossRef]
- De Coninck, T.; Verbeke, I.; Rougé, P.; Desmet, T.; Van Damme, E.J.M. OsAPSE Modulates Non-Covalent Interactions between Arabinogalactan Protein O-Glycans and Pectin in Rice Cell Walls. Frontiers in Plant Science 2025, 16. [Google Scholar] [CrossRef]
- Imaizumi, C.; Tomatsu, H.; Kitazawa, K.; Yoshimi, Y.; Shibano, S.; Kikuchi, K.; Yamaguchi, M.; Kaneko, S.; Tsumuraya, Y.; Kotake, T. Heterologous Expression and Characterization of an Arabidopsis β-L-Arabinopyranosidase and α-D-Galactosidases Acting on β-L-Arabinopyranosyl Residues. Journal of Experimental Botany 2017, 68, 4651–4661. [Google Scholar] [CrossRef]
- Fujimoto, Z. Structure and Function of Carbohydrate-Binding Module Families 13 and 42 of Glycoside Hydrolases, Comprising a β-Trefoil Fold. Bioscience, Biotechnology, and Biochemistry 2013, 77, 1363–1371. [Google Scholar] [CrossRef]
- Taylor, M.E.; Drickamer, K. Convergent and Divergent Mechanisms of Sugar Recognition across Kingdoms. Current Opinion in Structural Biology 2014, 28, 14–22. [Google Scholar] [CrossRef] [PubMed]
- De Coninck, T.; Gippert, G.P.; Henrissat, B.; Desmet, T.; Van Damme, E.J.M. Investigating Diversity and Similarity between CBM13 Modules and Ricin-B Lectin Domains Using Sequence Similarity Networks. BMC Genomics 2024, 25, 643. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, M.; Karnas, S.; Lepock, J.R.; Cabelli, D.E.; Tainer, J.A.; Getzoff, E.D.; Hallewell, R.A. Function of the Greek Key Connection Analysed Using Circular Permutants of Superoxide Dismutase. The EMBO Journal 1997, 16, 2171–2178. [Google Scholar] [CrossRef]
- Kemplen, K.R.; De Sancho, D.; Clarke, J. The Response of Greek Key Proteins to Changes in Connectivity Depends on the Nature of Their Secondary Structure. Journal of Molecular Biology 2015, 427, 2159–2165. [Google Scholar] [CrossRef]
- Ranaghan, M.J.; Li, J.J.; Laprise, D.M.; Garvie, C.W. Assessing Optimal: Inequalities in Codon Optimization Algorithms. BMC Biology 2021, 19. [Google Scholar] [CrossRef]
- Sharp, P.M.; Li, W.-H. The Codon Adaptation Index-a Measure of Directional Synonymous Codon Usage Bias, and Its Potential Applications. Nucleic Acids Research 1987, 15, 1281–1295. [Google Scholar] [CrossRef]
- Morgan, A.A.; Rubenstein, E. Proline: The Distribution, Frequency, Positioning, and Common Functional Roles of Proline and Polyproline Sequences in the Human Proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef]
- Sun, P.; Tropea, J.E.; Waugh, D.S. Enhancing the Solubility of Recombinant Proteins in Escherichia Coli by Using Hexahistidine-Tagged Maltose-Binding Protein as a Fusion Partner. In Heterologous Gene Expression in E.coli; Evans, T.C., Xu, M.-Q., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2011; Volume 705, pp. 259–274. ISBN 978-1-61737-966-6. [Google Scholar]
- Reuten, R.; Nikodemus, D.; Oliveira, M.B.; Patel, T.R.; Brachvogel, B.; Breloy, I.; Stetefeld, J.; Koch, M. Maltose-Binding Protein (MBP), a Secretion-Enhancing Tag for Mammalian Protein Expression Systems. PLoS ONE 2016, 11, e0152386. [Google Scholar] [CrossRef]
- Kellermann, O.; Szmelcman, S. Active Transport of Maltose in Escherichia Coli K12: Involvement of a “Periplasmic” Maltose Binding Protein. European Journal of Biochemistry 1974, 47, 139–149. [Google Scholar] [CrossRef]
- Lénon, M.; Ke, N.; Ren, G.; Meuser, M.E.; Loll, P.J.; Riggs, P.; Berkmen, M. A Useful Epitope Tag Derived from Maltose Binding Protein. Protein Science 2021, 30, 1235–1246. [Google Scholar] [CrossRef]
- Costa, S.; Almeida, A.; Castro, A.; Domingues, L. Fusion Tags for Protein Solubility, Purification and Immunogenicity in Escherichia Coli: The Novel Fh8 System. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef]
- Gao, K.; Rao, J.; Chen, B. Plant Protein Solubility: A Challenge or Insurmountable Obstacle. Advances in Colloid and Interface Science 2024, 324, 103074. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, B.; Ramachandra, G.; Virupaksha, T.K. Alpha-Galactosidase of Germinating Seeds of Cassia Sericea Sw. J Food Biochemistry 1990, 14, 45–59. [Google Scholar] [CrossRef]
- Gao, Z.; Schaffer, A.A. A Novel Alkaline α-Galactosidase from Melon Fruit with a Substrate Preference for Raffinose. Plant Physiology 1999, 119, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Chien, S.-F.; Chen, S.-H.; Chien, M.-Y. Cloning, Expression, and Characterization of Rice α-Galactosidase. Plant Mol Biol Rep 2008, 26, 213–224. [Google Scholar] [CrossRef]
- Peters, S.; Egert, A.; Stieger, B.; Keller, F. Functional Identification of Arabidopsis ATSIP2 (At3g57520) as an Alkaline -Galactosidase with a Substrate Specificity for Raffinose and an Apparent Sink-Specific Expression Pattern. Plant and Cell Physiology 2010, 51, 1815–1819. [Google Scholar] [CrossRef]
- Sakharayapatna Ranganatha, K.; Venugopal, A.; Chinthapalli, D.K.; Subramanyam, R.; Nadimpalli, S.K. Purification, Biochemical and Biophysical Characterization of an Acidic α-Galactosidase from the Seeds of Annona Squamosa (Custard Apple). International Journal of Biological Macromolecules 2021, 175, 558–571. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, Y.; Dai, H.; Miao, M. Characteristics and Expression Patterns of Six α-Galactosidases in Cucumber (Cucumis Sativus L.). PLoS ONE 2021, 16, e0244714. [Google Scholar] [CrossRef]
- Khrapunov, S.; Cheng, H.; Hegde, S.; Blanchard, J.; Brenowitz, M. Solution Structure and Refolding of the Mycobacterium Tuberculosis Pentapeptide Repeat Protein MfpA. Journal of Biological Chemistry 2008, 283, 36290–36299. [Google Scholar] [CrossRef]
- Michaux, C.; Pomroy, N.C.; Privé, G.G. Refolding SDS-Denatured Proteins by the Addition of Amphipathic Cosolvents. Journal of Molecular Biology 2008, 375, 1477–1488. [Google Scholar] [CrossRef] [PubMed]
- Dulermo, R.; Legras, J.-L.; Brunel, F.; Devillers, H.; Sarilar, V.; Neuvéglise, C.; Nguyen, H.-V. Truncation of Gal4p Explains the Inactivation of the GAL/MEL Regulon in Both Saccharomyces Bayanus and Some Saccharomyces Cerevisiae Wine Strains. FEMS Yeast Research 2016, 16, fow070. [Google Scholar] [CrossRef] [PubMed]
- Tang, H.; Wang, S.; Wang, J.; Song, M.; Xu, M.; Zhang, M.; Shen, Y.; Hou, J.; Bao, X. N-Hypermannose Glycosylation Disruption Enhances Recombinant Protein Production by Regulating Secretory Pathway and Cell Wall Integrity in Saccharomyces Cerevisiae. Sci Rep 2016, 6, 25654. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Li, Q.; Tan, H.; Jiang, H.; Li, K.; Zhang, L.; Shi, Q.; Yin, H. Unique N-Glycosylation of a Recombinant Exo-Inulinase from Kluyveromyces Cicerisporus and Its Effect on Enzymatic Activity and Thermostability. J Biol Eng 2019, 13, 81. [Google Scholar] [CrossRef]
- Tanurdzic, M.; Vaughn, M.W.; Jiang, H.; Lee, T.-J.; Slotkin, R.K.; Sosinski, B.; Thompson, W.F.; Doerge, R.W.; Martienssen, R.A. Epigenomic Consequences of Immortalized Plant Cell Suspension Culture. PLoS Biol 2008, 6, e302. [Google Scholar] [CrossRef]
- Tungekar, A.A.; Castillo-Corujo, A.; Ruddock, L.W. So You Want to Express Your Protein in Escherichia Coli ? Essays in Biochemistry 2021, 65, 247–260. [Google Scholar] [CrossRef]
- Agarwal, S.; Agarwal, S.; Biancucci, M.; Satchell, K.J.F. Induced Autoprocessing of the Cytopathic Makes Caterpillars Floppy-like Effector Domain of the Vibrio Vulnificus MARTX Toxin: A Novel Cysteine Peptidase in Toxin Autoprocessing. Cellular Microbiology 2015, 17, 1494–1509. [Google Scholar] [CrossRef]
- Shindyalov, I.N.; Bourne, P.E. Protein Structure Alignment by Incremental Combinatorial Extension (CE) of the Optimal Path. Protein Engineering Design and Selection 1998, 11, 739–747. [Google Scholar] [CrossRef]
- Kufareva, I.; Abagyan, R. Methods of Protein Structure Comparison. In Homology Modeling; Methods in Molecular Biology; Orry, A.J.W., Abagyan, R., Eds.; Humana Press: Totowa, NJ, 2011; Volume 857, pp. 231–257. ISBN 978-1-61779-587-9. [Google Scholar]
- Ahmad, M.; Hirz, M.; Pichler, H.; Schwab, H. Protein Expression in Pichia Pastoris: Recent Achievements and Perspectives for Heterologous Protein Production. Appl Microbiol Biotechnol 2014, 98, 5301–5317. [Google Scholar] [CrossRef] [PubMed]
- Rosano, G.L.; Ceccarelli, E.A. Recombinant Protein Expression in Escherichia Coli: Advances and Challenges. Frontiers in Microbiology 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Inzé, D.; Depicker, A. GATEWAYTM Vectors for Agrobacterium-Mediated Plant Transformation. Trends in Plant Science 2002, 7, 193–195. [Google Scholar] [CrossRef]
- Gerasimova, S.V.; Smirnova, O.G.; Kochetov, A.V.; Shumnyi, V.K. Production of Recombinant Proteins in Plant Cells. Russ J Plant Physiol 2016, 63, 26–37. [Google Scholar] [CrossRef]
- Lampropoulos, A.; Sutikovic, Z.; Wenzl, C.; Maegele, I.; Lohmann, J.U.; Forner, J. GreenGate - A Novel, Versatile, and Efficient Cloning System for Plant Transgenesis. PLoS ONE 2013, 8, e83043. [Google Scholar] [CrossRef]
- Chen, Y.; Vermeersch, M.; Van Leene, J.; De Jaeger, G.; Li, Y.; Vanhaeren, H. A Dynamic Ubiquitination Balance of Cell Proliferation and Endoreduplication Regulators Determines Plant Organ Size. Sci. Adv. 2024, 10, eadj2570. [Google Scholar] [CrossRef] [PubMed]
- Gerstmans, H.; Grimon, D.; Gutiérrez, D.; Lood, C.; Rodríguez, A.; Van Noort, V.; Lammertyn, J.; Lavigne, R.; Briers, Y. A VersaTile-Driven Platform for Rapid Hit-to-Lead Development of Engineered Lysins. Sci. Adv. 2020, 6, eaaz1136. [Google Scholar] [CrossRef]
- Hartley, J.L. DNA Cloning Using In Vitro Site-Specific Recombination. Genome Research 2000, 10, 1788–1795. [Google Scholar] [CrossRef]
- De Zaeytijd, J.; Rougé, P.; Smagghe, G.; Van Damme, E.J.M. Structure and Activity of a Cytosolic Ribosome-Inactivating Protein from Rice. Toxins 2019, 11, 325. [Google Scholar] [CrossRef]
- Al Atalah, B.; Fouquaert, E.; Vanderschaeghe, D.; Proost, P.; Balzarini, J.; Smith, D.F.; Rougé, P.; Lasanajak, Y.; Callewaert, N.; Van Damme, E.J.M. Expression Analysis of the Nucleocytoplasmic Lectin ‘Orysata’ from Rice in Pichia Pastoris. The FEBS Journal 2011, 278, 2064–2079. [Google Scholar] [CrossRef]
- Van Leene, J.; Eeckhout, D.; Persiau, G.; Van De Slijke, E.; Geerinck, J.; Van Isterdael, G.; Witters, E.; De Jaeger, G. Isolation of Transcription Factor Complexes from Arabidopsis Cell Suspension Cultures by Tandem Affinity Purification. In Plant Transcription Factors; Yuan, L., Perry, S.E., Eds.; Methods in Molecular Biology; Humana Press: Totowa, NJ, 2011; Volume 754, pp. 195–218. ISBN 978-1-61779-153-6. [Google Scholar]
- Dubiel, M.; De Coninck, T.; Osterne, V.J.S.; Verbeke, I.; Van Damme, D.; Smagghe, G.; Van Damme, E.J.M. The ArathEULS3 Lectin Ends up in Stress Granules and Can Follow an Unconventional Route for Secretion. IJMS 2020, 21, 1659. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Analytical Biochemistry 1976, 72, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Beygmoradi, A.; Homaei, A.; Hemmati, R.; Fernandes, P. Recombinant Protein Expression: Challenges in Production and Folding Related Matters. International Journal of Biological Macromolecules 2023, 233, 123407. [Google Scholar] [CrossRef]
- Dechavanne, V.; Barrillat, N.; Borlat, F.; Hermant, A.; Magnenat, L.; Paquet, M.; Antonsson, B.; Chevalet, L. A High-Throughput Protein Refolding Screen in 96-Well Format Combined with Design of Experiments to Optimize the Refolding Conditions. Protein Expression and Purification 2011, 75, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Illergård, K.; Ardell, D.H.; Elofsson, A. Structure Is Three to Ten Times More Conserved than Sequence—A Study of Structural Response in Protein Cores. Proteins 2009, 77, 499–508. [Google Scholar] [CrossRef]
- Hofstee, B.H.J. Non-Inverted versus Inverted Plots in Enzyme Kinetics. Nature 1959, 184, 1296–1298. [Google Scholar] [CrossRef]
- Hanes, C.S. Studies on Plant Amylases: The Effect of Starch Concentration upon the Velocity of Hydrolysis by the Amylase of Germinated Barley. Biochemical Journal 1932, 26, 1406–1421. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(continued on next page). OsAPSE is a multi-domain protein composed of an N-terminal signal peptide (purple), a GH27 domain (red), a ricin-B-like domain (blue) and a C-terminal GH-all-beta domain (green). The model of OsAPSE was obtained through AlphaFold and was constructed with high confidence (predicted Local Distance Difference Test value = 83.36). A: 3D representation of OsAPSE shown as ribbon diagram and protein surface. The sites for N/O-glycosylation as well as the catalytic sites are indicated. B: Domain modularity representation of OsAPSE. The confidence of structure prediction is indicated on a sliding scale between orange (low confidence) to blue (high confidence). The domain boundaries and InterPro annotations are shown. C: Primary and secondary structure of OsAPSE, showing strands, helices and coils. The confidence of the secondary structure prediction is shown on a sliding scale between light blue (low confidence) to dark blue (high confidence). Putative N/O-glycosylation sites as well as cysteines possibly involved in disulfide bridge formation are shown with a red N, green O, or blue S. The cysteine residues from the same disulfide bridges are enumerated with the same number (S1, S2, S3). The catalytic residues D226 and D282 are indicated with asterisks.
Figure 1.
(continued on next page). OsAPSE is a multi-domain protein composed of an N-terminal signal peptide (purple), a GH27 domain (red), a ricin-B-like domain (blue) and a C-terminal GH-all-beta domain (green). The model of OsAPSE was obtained through AlphaFold and was constructed with high confidence (predicted Local Distance Difference Test value = 83.36). A: 3D representation of OsAPSE shown as ribbon diagram and protein surface. The sites for N/O-glycosylation as well as the catalytic sites are indicated. B: Domain modularity representation of OsAPSE. The confidence of structure prediction is indicated on a sliding scale between orange (low confidence) to blue (high confidence). The domain boundaries and InterPro annotations are shown. C: Primary and secondary structure of OsAPSE, showing strands, helices and coils. The confidence of the secondary structure prediction is shown on a sliding scale between light blue (low confidence) to dark blue (high confidence). Putative N/O-glycosylation sites as well as cysteines possibly involved in disulfide bridge formation are shown with a red N, green O, or blue S. The cysteine residues from the same disulfide bridges are enumerated with the same number (S1, S2, S3). The catalytic residues D226 and D282 are indicated with asterisks.
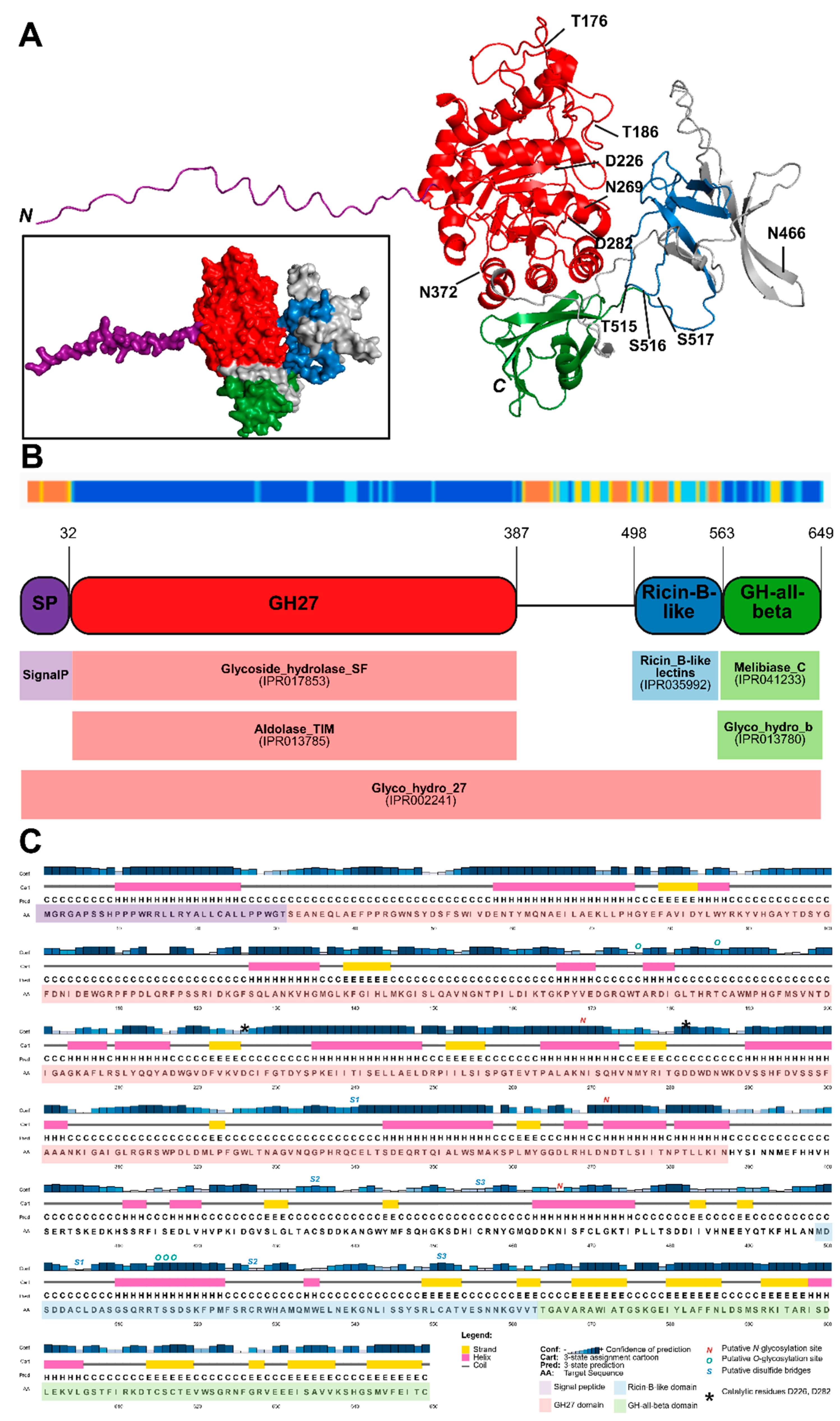
Figure 2.
Expression experiments in different E. coli strains usually resulted in insoluble protein of interest. Results from experiment 1 (A), experiment 7 (B), experiment 14 (C) and experiment 26 (D) are shown. For experiment 26, protein samples were run across multiple gels. The contents of every lane is highlighted below or next to the SDS-PAGE gel or immunoblot. The protein of interest is indicated with an arrow.
Figure 2.
Expression experiments in different E. coli strains usually resulted in insoluble protein of interest. Results from experiment 1 (A), experiment 7 (B), experiment 14 (C) and experiment 26 (D) are shown. For experiment 26, protein samples were run across multiple gels. The contents of every lane is highlighted below or next to the SDS-PAGE gel or immunoblot. The protein of interest is indicated with an arrow.
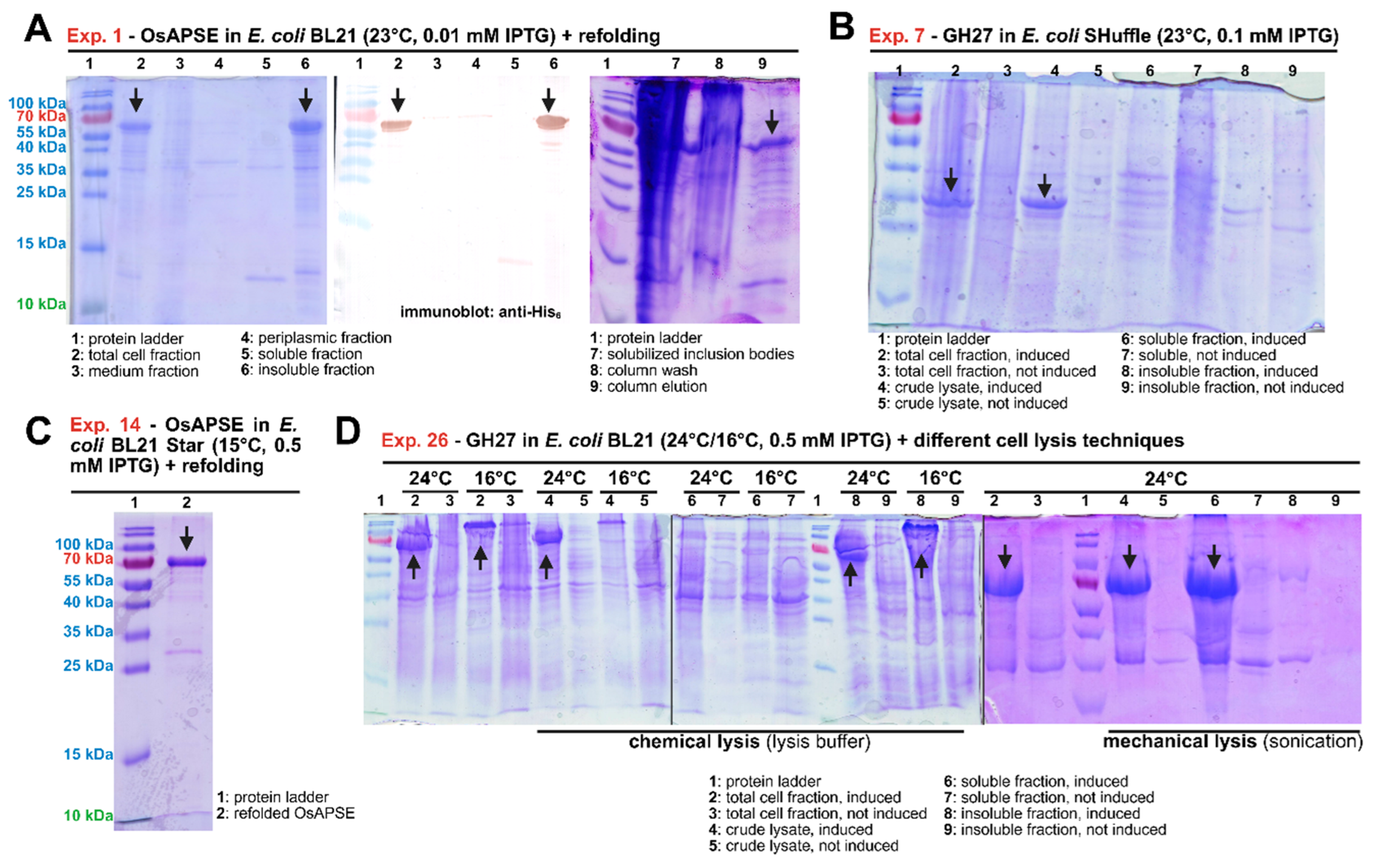
Figure 3.
Enzymatic assays of refolded OsAPSE (experiment 14). The reaction rate expressed as increase of absorbance over time (A) is used in Hanes-Woolf (blue) and Eadie-Hofstee (orange) linearization methods (B) to calculate the enzymatic paramters KM and Vmax. The legend displays the substrate concentration (A) or the linearization method (B).
Figure 3.
Enzymatic assays of refolded OsAPSE (experiment 14). The reaction rate expressed as increase of absorbance over time (A) is used in Hanes-Woolf (blue) and Eadie-Hofstee (orange) linearization methods (B) to calculate the enzymatic paramters KM and Vmax. The legend displays the substrate concentration (A) or the linearization method (B).
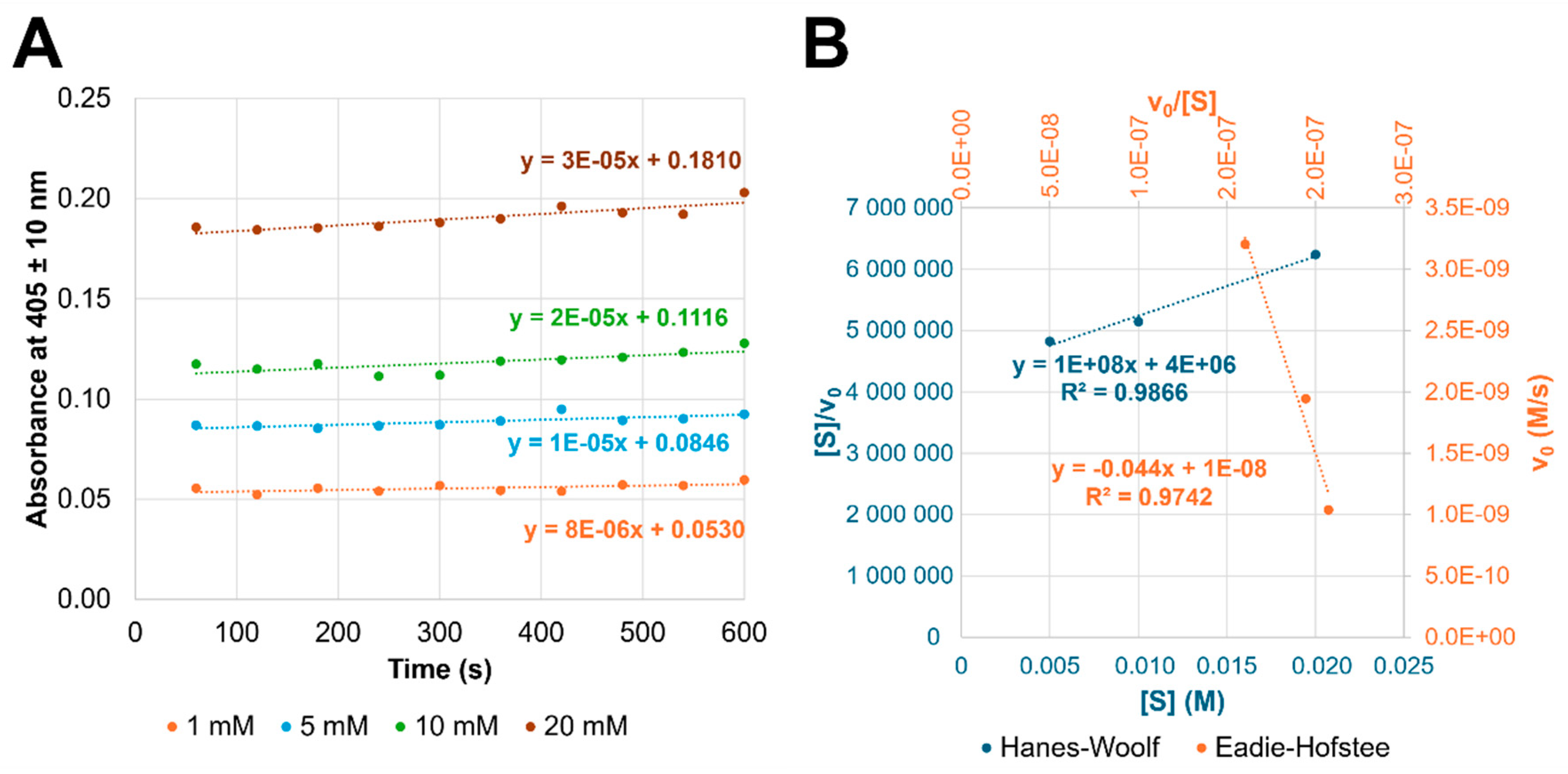
Figure 4.
Expression of GH27_OsAPSE in Pichia pastoris X-33 cells. Pichia cultures containing the MBP-TEV-mCherry-GH27_OsAPSE-His6 fusion protein were analyzed for the presence of red fluorescence (A), which was obvious in centrifuged samples (B). The fusion proteins were detected with antibodies against the MBP tag (C). The positive control (p.c.) (C23) comprised a Pichia culture producing an antibody (without MBP/mCherry tag and is therefore not visible on the blot). The negative control (n.c.) (C24) included non-transformed P. pastoris X-33 cells (which also do not possess MBP-tagged proteins and are thus not visible on the blot). The error bars represent standard deviations from n = 3 technical replicates.
Figure 4.
Expression of GH27_OsAPSE in Pichia pastoris X-33 cells. Pichia cultures containing the MBP-TEV-mCherry-GH27_OsAPSE-His6 fusion protein were analyzed for the presence of red fluorescence (A), which was obvious in centrifuged samples (B). The fusion proteins were detected with antibodies against the MBP tag (C). The positive control (p.c.) (C23) comprised a Pichia culture producing an antibody (without MBP/mCherry tag and is therefore not visible on the blot). The negative control (n.c.) (C24) included non-transformed P. pastoris X-33 cells (which also do not possess MBP-tagged proteins and are thus not visible on the blot). The error bars represent standard deviations from n = 3 technical replicates.
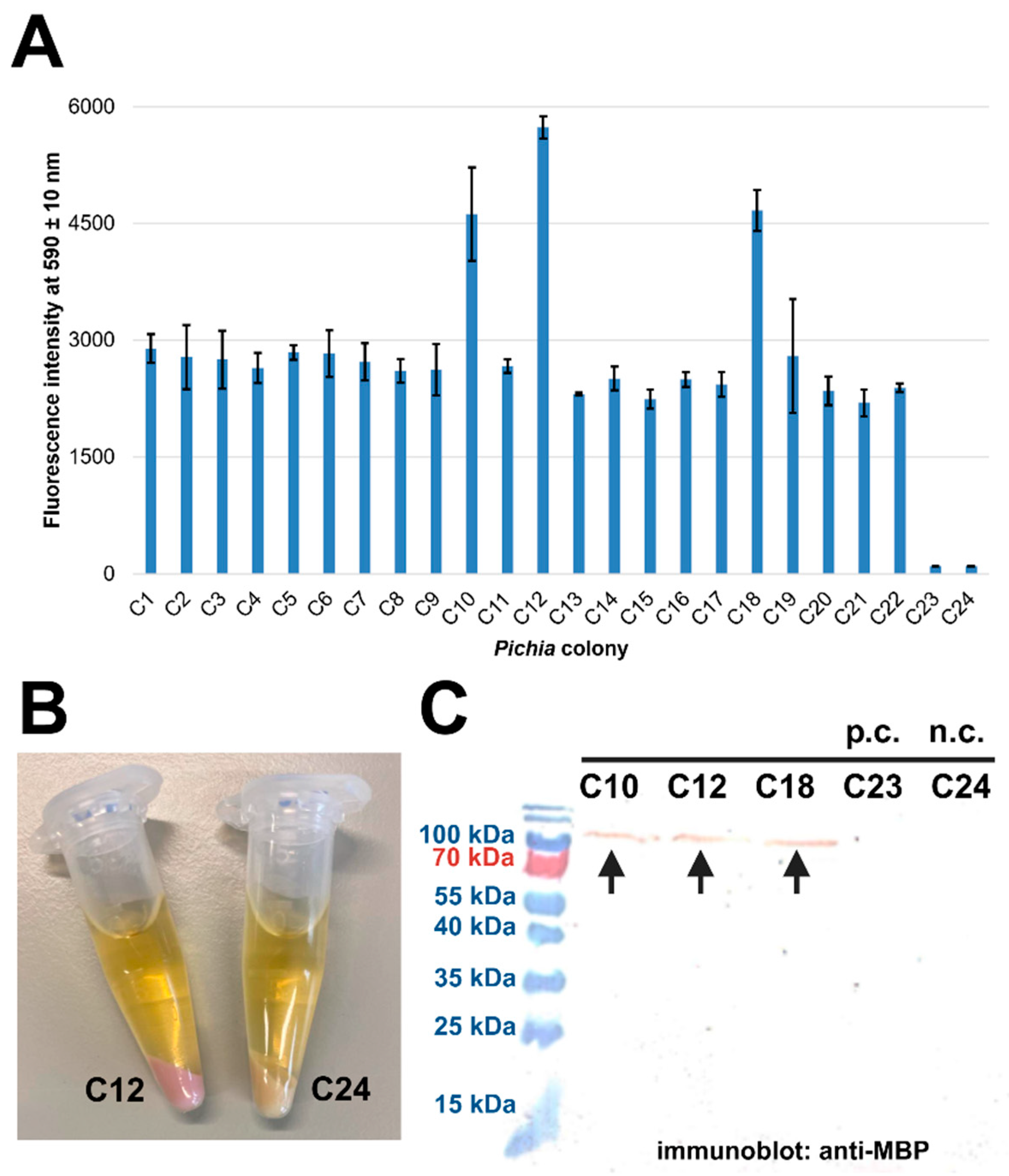
Figure 5.
Production of OsAPSE in Arabidopsis thaliana PSB-D cell cultures. A: Confocal fluorescence microscopy image of 600x magnified A. thaliana PSB-D cells co-expressing ER-localized EGFP with OsAPSE. A. thaliana PSB-D cells typically grow in clumps. The outlined box shows vesicular structures. The scale bar indicates a size of 50 µm. B: Analysis of produced proteins in A. thaliana PSB-D cells by means of a Coomassie-stained SDS-PAGE gel. C: Discontinuous enzymatic assays on different protein extracts from A. thaliana PSB-D cells transformed with pK7FWG2D::OsAPSE compared to non-transformed A. thaliana PSB-D cells. The observations in panel C could not be repeated due to discontinuation of the A. thaliana PSB-D cultures. .
Figure 5.
Production of OsAPSE in Arabidopsis thaliana PSB-D cell cultures. A: Confocal fluorescence microscopy image of 600x magnified A. thaliana PSB-D cells co-expressing ER-localized EGFP with OsAPSE. A. thaliana PSB-D cells typically grow in clumps. The outlined box shows vesicular structures. The scale bar indicates a size of 50 µm. B: Analysis of produced proteins in A. thaliana PSB-D cells by means of a Coomassie-stained SDS-PAGE gel. C: Discontinuous enzymatic assays on different protein extracts from A. thaliana PSB-D cells transformed with pK7FWG2D::OsAPSE compared to non-transformed A. thaliana PSB-D cells. The observations in panel C could not be repeated due to discontinuation of the A. thaliana PSB-D cultures. .
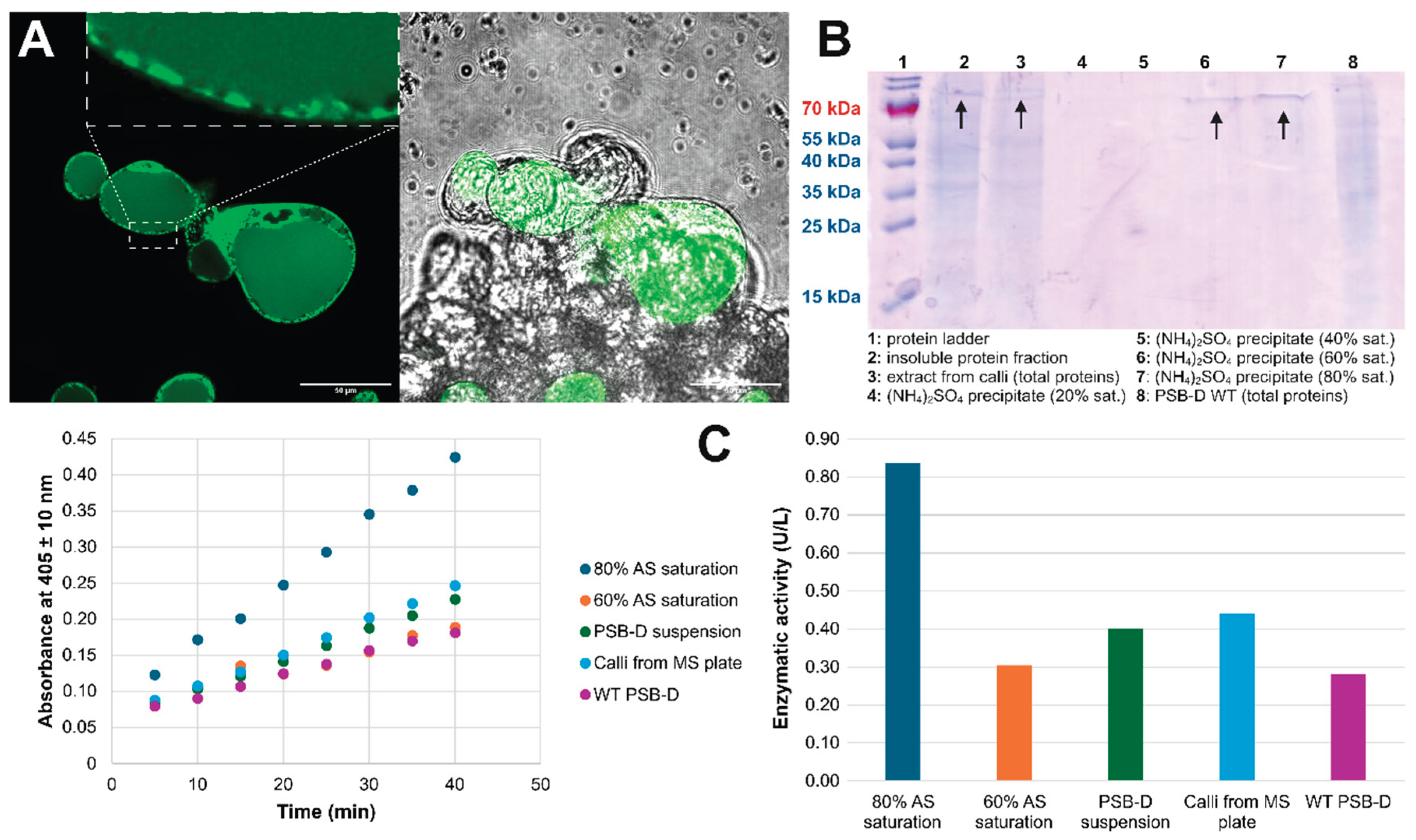
| Criterion | Bacterial cells | Yeast/fungal cells | Plant cells | Insect cells | Mammalian cells |
|---|---|---|---|---|---|
| Common example | Escherichia coli | Pichia pastoris | Arabidopsis PSB-D | Spodoptera frugiperda | Chinese Hamster Ovaries |
| Ease of genetic manipulation | Easy | Moderate | Difficult | Moderate | Difficult |
| Time for protein production | 1-2 days | 3-5 days | weeks | 7-10 days | Weeks to months |
| Cell doubling time | Very fast (20 minutes) |
Moderate (1-2 hours) |
Slow (20-48 hours) |
Slow (18-24 hours) |
Slow (16-24 hours) |
| Cultivation costs | Very low | Low to moderate | Moderate to high | High | Very high |
| Complexity of growth media | Simple | Moderate | Complex | Very complex | Very complex |
| Expression (mg protein per L of medium) | 100-5000 | 100-5000 | 10-100 | 10-1000 | 10-1000 |
| Scalability | High | High | Moderate | Moderate | High |
| Disulfide bridges | No | Yes | Yes | Yes | Yes |
| Glycosylation type | None | High-mannose N-glycans | Complex (plant-specific) N-glycans | Partial (insect-specific) N-glycans | Fully human-like N-glycans |
| Protein secretion | Periplasmic is possible but requires secretion signals | Possible | Possible | Possible | Possible |
| Protein stability and degradation risk | High – risk of inclusion bodies | Moderate – secreted proteins are more stable | Risk of proteolysis | Moderate | High – minimal proteolysis |
| Suitability for complex eukaryotic proteins | Usually not | Yes | Yes | Yes | Yes |
| Regulatory approval and industrial use | Widely used for research, not for therapeutics | Approved for some enzymes and vaccines | Limited biopharma use | Used in some vaccines | Industry standards, FDA/EMA approved |
Table 2.
Overview of the executed recombinant protein production experiments in this study.
| EXP | Host organism |
Strain or type | Expression plasmid | CDS a | CDS adjustments |
Tags | Cloning method | Lysis method | Result | Refolding |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | E. coli | BL21 | pET-22b(+) | OsAPSE | Codon Opt. | N-pelB+C-His6 | Res. & Lig | LyB | Ins. | Yes |
| 2 | BL21-AI | Ins. | No | |||||||
| 3 | pLysS | Ins. | No | |||||||
| 4 | Rosetta | Ins. | No | |||||||
| 5 | ArcticExpress | Ins. | No | |||||||
| 6 | Shuffle | N.P. | No | |||||||
| 7 | GH27 | Ins. | No | |||||||
| 8 | E. coli | BL21 | pET-28a(+) | OsAPSE | Codon Opt. | C-His6 | Res. & Lig | LyB | Ins. | No |
| 9 | ArcticExpress | Ins. | No | |||||||
| 10 | E. coli | BL21 | pVTD13 | OsAPSE | Codon Opt. | N-GST+C-His6 | VersaTile cloning | LyB | N.P. | No |
| 11 | GH27 | N.P. | No | |||||||
| 12 | ricin-B | Ins. | No | |||||||
| 13 | GH-all-beta | Ins. | No | |||||||
| 14 | E. coli | BL21 Star | pET-32a(+) b | OsAPSE | Codon Opt. | N-TRX+C-His6 | Res. & Lig | Son. | Ins. | Yes |
| 15 | ArcticExpress | Ins. | No | |||||||
| 16 | Rosetta | Ins. | No | |||||||
| 17 | E. coli | BL21 | pET-21a(+) | OsAPSE | Harm. c | C-His6 | Res. & Lig | LyB | Ins. | No |
| 18 | Rosetta | N.P. | No | |||||||
| 19 | BL21 | Codon Opt. d | Ins. | No | ||||||
| 20 | Shuffle | N.P. | No | |||||||
| 21 | BL21 | PROSS e | Ins. | No | ||||||
| 22 | Shuffle | N.P. | No | |||||||
| 23 | E. coli | BL21 | pDEST | OsAPSE | Codon Opt. | N-MBP+TEV site+C-FLAG3 or C-HA3 or C-His6 | GG cloning |
LyB | Ins. | No |
| 24 | ricin-B | Ins. | No | |||||||
| 25 | GH-β | Ins. | No | |||||||
| 26 | GH27 | LyB + Son. | Soluble | No | ||||||
| 27 | N/C-MBP2+C-His6 | LyB | Ins. | No | ||||||
| 28 | N-GST+C-His6 | Ins. | No | |||||||
| 29 | P. pastoris | X-33 | pPICZαA | OsAPSE | Codon Opt. | N-α-factor+C-His6 | Res. & Lig | Beads + LyB | N.P. | No |
| 30 | GlycoDelete | N.P. | No | |||||||
| 31 | KM71H | N.P. | No | |||||||
| 32 | P. pastoris | X-33 | Modified pPICZαA | GH27 | Codon Opt. | N-MBP+C-RFP | GG cloning |
Beads + LyB | Soluble | No |
| 33 | N/C-MBP2+C-His6 | N.P. | No | |||||||
| 34 | A. thaliana | PSB-D cell culture | pK7WG2D | OsAPSE | Codon Opt. | Reporter EGFP+C-His6 | GW cloning |
Cryo + LyB | Soluble | No |
| 35 | GH27 | N.P. | No | |||||||
| 36 | ricin-B | N.P. | No | |||||||
| 37 | GH-β | N.P. | No | |||||||
| 38 | BY-2 CFPS | ALiCE | pALiCE02 | GH27 | Codon Opt. | C-His6 | Res. & Lig | None | Soluble f | No |
Abbreviations: CDS (coding sequence), CFPS (cell-free production system), Cryo (cryogenic crushing), EXP (experiment), GG (Golden/Green Gate), GW (Gateway), Harm. (harmonization), Ins. (insoluble), LyB (lysis buffer), N.P. (not produced), Opt. (optimization), Res. & Lig. (restriction and ligation), Son. (sonication), TEV (Tobacco Etch Virus). Remarks: The terminal positions of tags are indicated with N’ and C’. Notes: a OsAPSE refers to the full-length coding sequence, while GH27, ricin-B and GH-β refer to the subdomains of OsAPSE; b outsourced to GenScript; c 2 harmonization variants were used; d 4 optimization algorithms available on IDT, EUROFINS, VectorBuilder and NovoPro were utilized; e Protein Repair One-Stop Shop; f not part of this study but recently published [57].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



