Submitted:
18 August 2025
Posted:
20 August 2025
You are already at the latest version
Abstract
Epstein-Barr virus (EBV) is implicated in the pathogenesis of different B-cell lymphomas and lymphoproliferative disorders, including diffuse large B-cell lymphoma (DLBCL) arising in immunodeficiency settings. Despite its clinical significance, the mechanisms of EBV-mediated lymphomagenesis across different disease subtypes remains poorly understood. Global DNA methylation profiling can provide insight into tumor heterogeneity and disease mechanisms. To further characterize the underlying biology of EBV(+) DLBCL, we performed a global methylome analysis of a cohort of EBV(+)/(-) DLBCL. Illumina MethylationEPIC array data were generated from a curated set of DLBCL tissue samples (n=43) from a rural patient population with defined EBV status and immunodeficiency background. Differential methylation analyses were conducted using linear mixed models to identify significant methylation changes associated with EBV status. Principle component analysis (PCA) and probe-level comparisons revealed a distinct, globally hypermethylated DNA methylome in EBV(+) DLBCL compared to EBV(-) cases, and an overall hypomethylated profile in all DLBCL relative to control tissues. We identified a total of 117,334 differentially methylated probes mapping to 1,557 cancer-associated genes in EBV(+) versus EBV(-) DLBCL, and 330,872 probes mapping to 4,230 cancer-associated genes in all DLBCL versus controls. Pathway enrichment analysis highlighted distinct biological processes in EBV(+) DLBCL, including P53 feedback loops (hypermethylated genes) and MAPK signaling (hypomethylated genes). These findings demonstrated that EBV(+) DLBCL is epigenetically distinct from EBV(-) disease, with alterations that may contribute to clinical heterogeneity and potentially serve as biomarkers for disease classification and therapeutic targeting.
Keywords:
Epstein-Barr virus
; EBV
; diffuse large B-cell lymphoma
; DLBCL
; methylation
; methylome
; epigenetics
1. Introduction
Epstein-Barr virus (EBV)-driven diffuse large B-cell lymphoma (DLBCL) represents a clinically aggressive tumor subtype that is often refractory to standard R-CHOP chemotherapy (Rituximab, Cyclophosphamide, Doxorubicin Hydrochloride, Vincristine Sulfate, Prednisone) [1,2,3]. Currently, no targeted treatment options exist for EBV(+) DLBCL. While EBV is generally accepted to play a role in the pathogenesis of DLBCL, the specific disease mechanisms and the significance of the viral contribution in vivo have not been clearly defined. Furthermore, no consensus standard exists for diagnosing DLBCL as EBV-positive using the conventional in situ hybridization tissue stain for EBV-encoded small RNA (EBER-ISH) [4,5,6,7,8,9]. Until EBV diagnostics and pathogenic mechanisms are elucidated, the development of targeted treatments for EBV(+) DLBCL will remain a challenge.
The role of EBV in modifying the epigenetic landscape of DLBCL offers promising avenues for understanding viral pathogenesis, identifying diagnostic biomarkers, and developing targeted therapeutics. In vitro studies using EBV-infected cell lines have shown that the virus directly modifies both the host cellular epigenome and its own genome through DNA methylation [10,11]. DNA methylation allows for dynamic changes in gene expression via hypermethylation (silencing) or hypomethylation (activation) of functional promoter regions. These alterations serve as epigenetic “switches” that can respond to external environmental cues, such as those present in the immunosuppressed tumor microenvironment. In vitro evidence has demonstrated the capacity of EBV to modulate the host epigenome by upregulating oncogene expression and downregulating tumor suppressor genes, thereby promoting B-cell malignant transformation [10,11,12]. Our study takes this investigation to in vivo clinical tissue samples of EBV(+)/(-) DLBCL to better define the tumor cell methylome and clarify the epigenetic role of EBV.
We characterized the DLBCL methylome in EBV-mediated disease using the high-throughput DNA methylation microarray, Illumina MethylationEPIC v2.0, which interrogates over 935,000 CpG sites across the human genome. This approach has been successfully employed across numerous in vivo cancer studies, including The Cancer Genome Atlas (TCGA), which profiled thousands of samples across diverse tumor types [13,14,15,16]. The Illumina EPIC methylation array is particularly well-suited for formalin-fixed tissue, allowing for a comprehensive genome-wide methylation profiling. Through this technology, the TCGA project has systematically characterized epigenomic alterations, yielding insight into oncogene activation, epigenetic regulation of gene expression, and tumor microenvironment. These insights have informed advances in diagnostics and targeted immunotherapies [14].
We applied the Illumina EPIC methylation array to a clinical cohort of EBV(+)/(-) DLBCL to identify distinct tumor methylomes associated with EBV-driven oncogenesis. This pilot study demonstrates that EBV(+) DLBCL harbors a unique signature compared to EBV(-) disease, underscoring the need for future investigation in larger cohorts.
2. Materials and Methods
2.1. Clinical Study Population and Tissue Sample Collection
A total of 31 clinical tissue cases of DLBCL from our institution, including 9 EBV(+) and 22 EBV(-), from 2000-2023 were selected for the study. In addition, 12 control tissue cases were selected over this period for comparison, including: 2 EBV(+) polymorphic lymphoproliferative disorders, 2 EBV(+) plasmacytic hyperplasia post-transplant lymphoproliferative disorders, 4 EBV(+) infectious mononucleosis tonsil/lymph nodes, and 4 EBV(-) reactive follicular hyperplasia lymph nodes. The cases originated from a wide range of immunosuppressive states including post solid organ/stem cell transplantation, systemic chemoradiation therapy-related (iatrogenic), autoimmune disease treated with systemic immunosuppressive pharmacotherapy (e.g. methotrexate, long-term steroid use, etc.), and primary immunodeficiency. The contribution of immunosenescence, as defined by age >65 years old, was likely in most tumor cases.
Representative hematoxylin and eosin-stained (H&E) slides for each case were reviewed by the study hematopathologist (Volaric) to confirm the diagnosis. Standardized automatic ISH staining platform (Leica Biosystems) for EBER-ISH was used in the evaluation. All cases were categorized as EBV(+) using EBER-ISH (ISH5687-A). EBV-positivity was defined in DLBCL or control cases as exhibiting at least 50% cellular staining in either tumor cells or lymphocytes for EBER-ISH.5 EBV-negative cases showed no cellular staining. Only cases in which diagnostic material was obtainable (formalin-fixed paraffin-embedded (FFPE) tissue blocks or unstained sections) were included. The study was approved by the UVM Institutional Review Board.
2.2. DNA Methylation Analysis
2.2.1. Global Methylation Profiling Using Illumina EPIC Methylation Array
FFPE tumor and control tissue sections were macro-dissected and sent to CD Genomics (NY, USA) for Illumina Infinium MethylationEPIC BeadChip array (935K). Samples were subjected to DNA extraction using protocols optimized for archival FFPE material. DNA quantity and quality were assessed prior to array hybridization. The array includes >935,000 CpG probes that span gene promoters, enhancers, gene bodies, and intergenic regions, and includes known cancer-associated loci and open chromatin regions. After hybridization and scanning, raw data were delivered in the form of IDAT files containing methylated and unmethylated signal intensities.
2.2.2. Bioinformatic Analysis
Illumina EPIC methylation analysis was performed using R (version 4.4.1). Raw IDAT files were processed using the SeSAMe package (v1.22.2) [17]. Initial quality filtering was conducted using the SeSAMe pOOBAH (p-value with out-of-band [OOB] array hybridization) method, which performs dye bias correction and calculates detection p-values. The background subtraction was done using the QCDPB criteria: Q, qualityMask Masking probes of poor design; C, inferInfiniumIChannel Infer channel for Infinium-I probes (needed for dye bias correction step); D, dyeBiasNL actual dye bias correction (non-linear); P, pOOBAH Detection p-value masking using oob; B noob background subtraction using oob. Only samples with median intensity >10.5 and mean detection p-values <0.05 were retained (n=43), resulting in 889,341 high-confidence probes for downstream analysis. Normalized beta values (ranging from 0 to 1) were used for downstream analysis. Principal component analysis (PCA) was performed using `prcomp()` to assess global methylome clustering. Differential methylation analysis was performed using linear models implemented in the ‘DML’ (differential methylation locus) function of the SeSAMe package, comparing EBV(+) vs. EBV(–) DLBCL and DLBCL vs. control tissues. Probe-level significance was adjusted using p-values of 0.05. Significant probes were annotated to genes using sesameAnno_attachManifest for EPICv2. Significant probes were aggregated at the gene-level to identify differentially methylated regions (DMRs) based on a log2 fold change cutoff and multiple testing thresholds. For enrichment analysis of gene sets, we used the testEnrichment function of the SeSAMe package, which performs a Fisher's exact test to determine if the number of significant probes associated with a gene is greater than expected by chance. We further used oncoEnrichR (v1.5.2), to map differentially methylated genes to curated cancer pathways. Immune cell composition inference employed IDOL Optimized CpGs from the Illumina Human Methylation Dataset package FlowSorted.Blood.EPIC (v2.8.0). This estimated the relative abundance of immune cell populations across samples. Differences in immune composition between EBV(+) and EBV(–) DLBCL were assessed using the Wilcoxon rank-sum test (p<0.05). Commercially available antibodies were used for immunohistochemical analysis of CD20 (Ventana Ultra, Thermo Fisher L26), CD3 (Leica, Biocare LN10), and CD56 (Ventana Ultra, Roche MRQ-42) on a representative case of EBV(+) and EBV(-) DLBCL.
3. Results
3.1. Clinicopathologic Characteristics of Study Cohort
To elucidate the DNA methylation profiles of EBV-mediated DLBCL, we established a clinical cohort of DLBCL (n=43) from our institution, consisting of EBV(+) DLBCL (n=9), EBV(-) DLBCL (n=22), and control cases [EBV(+) n=8; EBV(-) n=4]. EBV status for each case was determined by EBER-ISH chromogenic staining, whereby EBV(+) cases exhibited ≥50% positive nuclear staining in lesional cells, and EBV(-) cases showed no cellular staining. Representative EBV(+) and EBV(-) DLBCL cases, demonstrating EBER-ISH staining patterns, are shown in Figure 1.
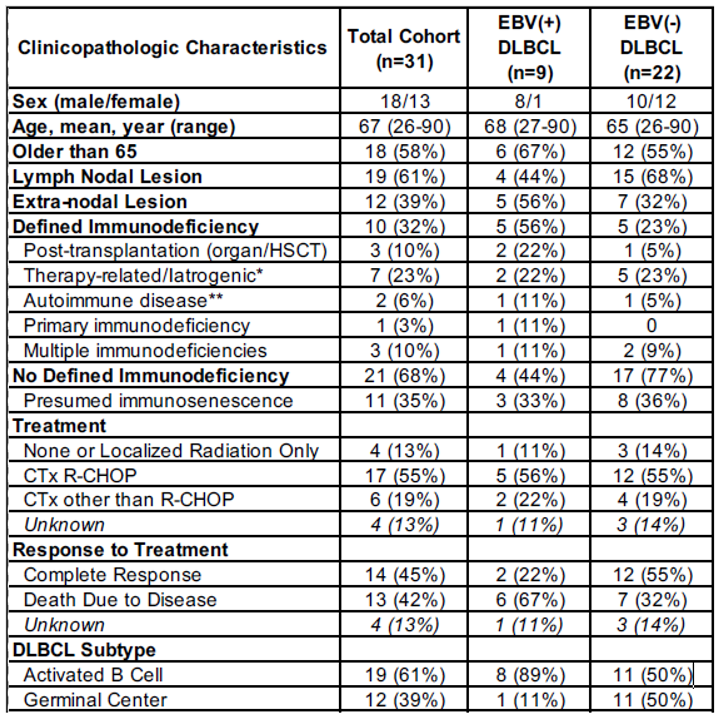
The clinicopathologic characteristics of the EBV(+)/(-) DLBCL cohort (n=31) are summarized in Table 1. Most patients (58%) were aged >65 years old, and roughly one-third had a defined immunodeficiency, with post-transplantation (22%) and therapy-related/iatrogenic (22%) immunodeficiency states particularly enriched in EBV(+) DLBCL cases. The majority of cases (55%) were treated with standard R-CHOP chemotherapy. Less than half of cases responded to therapy, with disease-related mortality occurring in 67% of EBV(+) DLBCL cases compared to 32% of EBV(-) DLBCL cases.
3.2. Global DNA Methylation Analysis of Clinical Cohort
Global DNA methylation analysis was performed on the EBV(+)/(-) DLBCL cohort (n=31) and controls (n=12) using the Ilumina EPIC methylation array, which interrogates approximately 950,000 CpG loci across the human genome. Quality control was carried out using the SeSAMe package to ensure data reliability and minimize technical artifacts.17 Based on signal intensity and detection p-value assessments, samples and probes failing established thresholds were removed (Table S1). Probes were filtered out if they had detection p-values >0.05, or were poorly designed (Figure S1). Following quality control and filtering, 889,341 probes were retained for analysis.
3.3. Methylome Patterns: All DLBCL versus Control Cases
All EBV(+)/(-) DLBCL (n=31) were directly compared to control cases (n=12), which included: 2 EBV(+) polymorphic lymphoproliferative disorders, 2 EBV(+) plasmacytic hyperplasia post-transplant lymphoproliferative disorders, 4 EBV(+) infectious mononucleosis tonsil/lymph nodes, and 4 EBV(-) reactive follicular hyperplasia lymph nodes. PCA revealed a clear separation between DLBCL and controls (Figure 2A). A comparison of the Beta values representing methylation levels of all probes, overall showed a trend of hypomethylation in DLBCL compared to control cases (Figure 2B), suggesting global methylation associated with DLBCL pathogenesis.
Differential methylation analysis, performed using the statistical probe filtering and normalization pipeline of the SeSAMe package [17], identified 330,872 differentially methylated CpG sites (p <0.05) between DLBCL and controls (Table S1; Figure S2A). Gene-level aggregation of probes identified 4,110 hypermethylated genes and 120 hypomethylated genes in DLBCL (FDR <0.05 and log2 fold change of 2; Table S2A), consistent with prior reports of global hypomethylation in cancer (Figure 2C) [18,19,20,21]. Many of these genes, including tumor suppressors such as CDKN2A, HLA-A, and FAT4, have established roles in hematolymphoid malignancies (Figure 2D). Pathway enrichment analysis of hypermethylated genes revealed significant involvement of cancer-related pathways, notably Wnt/β-catenin and Cadherin signaling, which have complex roles in lymphomagenesis (Figure 2E) [22,23,24,25,26,27]. No significant enrichment pertaining to cancer-associated pathways was discovered in the hypo-methylated gene set. The tumor suppressor gene CDKN2A, known to contribute to aggressive disease behavior when inactivated [28,29,30,31,32,33], was significantly hypermethylated in DLBCL compared to controls (Figure 2F).
3.4. Methylome Patterns: EBV(+) DLBCL versus EBV(-) DLBCL
We next sought to identify methylation differences between EBV(+) and EBV(-) DLBCL. PCA analysis revealed clear separation between EBV(+) (n=9) and EBV(-) cases (n=22) (Figure 3A). Differential methylation analysis using SeSAMe identified 117,334 differentially methylated CpG sites (p<0.05) between EBV(+) DLBCL and EBV(-) DLBCL (Table S1; Figure S2B). Comparison of beta values across all probes indicated an overall hypermethylation pattern in EBV(+) DLBCL relative to EBV(-) DLBCL (Figure 3B). Gene-level methylation analysis identified 2,114 differentially methylated genes between EBV(+) DLBCL and EBV(-) DLBCL (FDR <0.05 and log2 fold change of 2; Table S2B; Figure 3C). The distribution was skewed towards hypomethylation in EBV(+) cases (630 hypermethylated vs. 1,484 hypomethylated genes). Of these, 1,557 genes had cancer-related functions, with a similar skew towards hypomethylation (367 hypermethylated and 1,190 hypomethylated).
Among the hypermethylated genes were both lymphoid oncogenes and tumor suppressor genes, including 15 oncogenes, 4 tumor suppressor genes, and 6 with dual oncogenic and tumor suppressor roles (Figure 4A). Notably, hypermethylated tumor suppressor genes in EBV(+) DLBCL included CBFA2T3, CSNK1E, and HDAC10, all of which are associated with hematolymphoid malignancies [34,35,36,37,38]. Pathway enrichment analysis of the hypermethylated genes revealed involvement in tumor suppressor pathways such as P53 feedback loops and TGF-beta signaling [2,39,40,41,42], oncogenic pathways including P38 MAPK and Ras pathways [43], and immune signaling pathways like IFN-gamma signaling.[44,45]. The HDAC10 gene showed significant hypermethylation in EBV(+) DLBCL (Figure 4C). While HDAC10 gene can function as either an oncogene or tumor suppressor depending on cellular context, it has been reported to exert tumor-suppressive effects in B-cell lymphomagenesis [38,46,47,48].
We also characterized hypomethylated genes in EBV(+) DLBCL. The top 25 hypomethylated genes included 6 oncogenes, 8 tumor suppressor genes, and 11 genes with dual functions (Figure 4D). These included oncogenes FLT3 and HDAC6, as well as dual-function genes NPM1 and BTK, all implicated in lymphomagenesis [49,50,51,52,53,54,55]. Pathway analysis showed that hypomethylated genes were enriched in oncogenic cascades such as Insulin/IGF MAPK cascade, DNA replication, and RAS pathway (Figure 4E) [43,56,57]. The BTK gene, a well-established oncogene in B-cell lymphomagenesis, exhibited pronounced hypomethylation in EBV(+) DLBCL compared to EBV(-) DLBCL (Figure 4F) [58].
3.5. Immune Cell Composition in EBV(+) DLBCL
The immune cell composition of DLBCL plays an important role in the clinicopathologic progression of the disease. To assess differences between EBV(+) and EBV(-) DLBCL, we performed immune cell inference through deconvolution of the methylation data. This analysis revealed a distinct immune cell composition profile for EBV(+) DLBCL, with an enrichment of certain immune cell types and relative depletion of others. Specifically, EBV(+) DLBCL demonstrated a significant predominance of CD8(+) T-cells (p=0.01), with a relative predominance of monocytes (p=0.11), neutrophils (p=0.08), and NK cells (p=0.11). In contrast, B-cells (p=0.07) and CD4(+) T cells (p=0.63) were relatively depleted compared to EBV(-) DLBCL (Figure 5A). These findings were supported by immunohistochemical staining for B-cells (CD20), T cells (CD3), and NK cells (CD56) in representative cases of EBV(+) and EBV(-) DLBCL cases (Figure 5B and 5C, respectively). EBV(+) tumors showed an inflammatory background enriched in CD8(+) T cells and NK cells, while EBV(–) cases exhibited higher B-cell content.
4. Discussion
EBV(+) DLBCL is more clinically aggressive, resulting in worse overall survival and treatment response compared to EBV(-) disease [1,2,3]. However, there is no targeted therapy or prognostic biomarker for EBV(+) DLBCL. Few studies have investigated the epigenomic heterogeneity of EBV-mediated DLBCL, with most studies focused heavily on overall DLBCL pathogenesis and non-viral disease mechanisms [59,60,61,62,63]. The epigenetic landscape of B-cell lymphomas is increasingly being recognized as crucial in disease pathogenesis and therapeutic targeting [64,65,66,67]. EBV epigenetically modifies the host cellular genome through selective DNA methylation, potentially promoting lymphomagenesis [10,11]. To better understand the influence of EBV on the DNA methylome of DLBCL, this study provides a systematic epigenetic characterization of EBV(+) DLBCL in relation to EBV(-) DLBCL and controls. Our results indicate that EBV(+) DLBCL exhibits a distinct DNA methylome from EBV(-) DLBCL, possibly influencing the observed clinical heterogeneity in EBV(+) tumors while also revealing therapeutic and prognostic biomarkers of disease.
A total of 2,114 differentially methylated genes, corresponding to 1,557 cancer-associated genes, were identified in EBV(+) DLBCL compared to EBV(-) DLBCL, defining a biologically distinct DNA methylome. This trend was also observed in the DLBCL-versus-controls analysis, where DLBCL cases exhibited global hypomethylation across all probe sites relative to controls. Global DNA hypomethylation is a well-documented feature of tumors compared to normal tissue and is considered a driver of tumorigenesis by enabling transcriptional activation of oncogene promoters, promoting genomic instability, and increasing tumor heterogeneity [19]. In DLBCL specifically, hypomethylation relative to normal B-cells has been reported and can serve as both a diagnostic and prognostic biomarker, capable of distinguishing molecular subtypes [21]. The study by Chambwe et al. demonstrated that increased DNA methylation variability in DLBCL correlates with worse clinical outcomes and overall survival. This variability reflects a loss of characteristic bimodal methylation distribution seen in normal tissue, a phenomenon observed across multiple tumor types [21,68] Such epigenetic variability is closely linked to clinical outcome and tumor heterogeneity as part of tumorigenesis [68]. Our findings confirm the presence of epigenetic variability in all DLBCL cases relative to control tissue, as well as within EBV-defined subgroups, supporting the utility of DNA methylation patterns as a global biomarker of tumor heterogeneity.
While the genes in EBV(+) DLBCL were skewed towards hypomethylation, the overall methylome across all probe sites exhibited a hypermethylated state in EBV(+) compared to EBV(-) tumors. Furthermore, although the global methylome in DLBCL showed a hypomethylation bias toward controls, the subset of genes of oncologic significance was predominantly hypermethylated, with no significant hypomethylated genes detected in this group. These findings are consistent with several studies demonstrating EBV’s role in promoting global hypermethylation of the host genome during carcinogenesis [10,69]. Both in vitro and clinical studies have shown that EBV actively modifies its own genome and the host cellular genome to establish viral latency, evade immune surveillance, and persist in the host B-cell [10,11,64,69]. EBV-mediated lymphoblastoid transformation is thought to result from accumulated pathogenic mutations and epigenetic modifications that drive oncogenesis. Hypermethylation of promoter regions in key tumor suppressor genes, coupled with hypomethylation of oncogenes, has been cited as a critical epigenetic mechanism in lymphomagenesis [11,64]. In DLBCL, such differential methylation patterns can define molecular subtypes and, in some cases, dictate prognosis [21,63,65,70]. Our findings demonstrate that EBV-mediated DLBCL harbors both hyper- and hypomethylated states within its tumor methylome, suggesting potential drivers of aggressive lymphomagenesis compared to EBV(-) disease.
We have characterized distinct methylomes defining DLBCL from control tissue and EBV(+) from EBV(-) tumors. The differential methylation of oncogenic genes in DLBCL, overall, skewed towards a hypermethylated state. However, we have also identified a significant set of hypomethylated genes in EBV(+) DLBCL as well. These patterns support distinct tumor heterogeneity and differences in prognosis in viral-mediated DLBCL. Hypermethylated genes of DLBCL included both oncogenic and tumor suppressor genes, with several exhibiting both functions. Overall, however, there were more tumor suppressor genes that were hypermethylated in DLBCL relative to controls. For EBV(+) DLBCL, we identified a similar mix of oncogenes, tumor suppressor genes, and genes with both functions that were hypermethylated. The hypomethylated genes also exhibited a mix of oncologic function with a predominance of genes with both oncogenic and tumor suppressor functions. All genes identified across the compared groups correlated to significant oncologic function in hematolymphoid malignancy and other human cancers. These defining genetic patterns correlated with unique gene pathways. DLBCL showed enrichment for hypomethylated genes involved in Wnt/β-catenin and Cadherin signal transduction pathways, which are known to play complex roles in lymphomagenesis [22,23,24,25,26,27]. Upregulation of Wnt/β-catenin signaling can cause more aggressive clinical behavior in DLBCL by promoting the endothelial to mesenchymal transition [25,26]. The tumor suppressor gene, CDKN2A, was significantly hypermethylated in DLBCL relative to controls, suggesting downregulation and loss of function. The functional loss of CDKN2A is linked to dysregulated P53 expression leading to more aggressive lymphomagenesis and worse clinical outcome in DLBCL despite standard chemotherapy [28,29,30,31,32,33].
Both hypermethylated and hypomethylated oncologic genes were detected in EBV(+) DLBCL relative to EBV(-) tumors, reflecting the tumor heterogeneity of viral-mediated disease. Hypermethylated oncologic genes correlated to tumor suppressive genetic pathways including P53 feedback loops and TGF-beta signaling. Loss of normal P53 function is associated with DLBCL occurrence and worse overall prognosis even in the setting of standard R-CHOP chemotherapy [28,39]. Transforming growth factor beta (TGF-beta) signaling is highly conserved and important for organismal development, but is shown to be exploited by various human cancer types to promote tumorigenesis, immune system evasion, and microenvironment modification [71]. In its normal state, TGF-beta functions as a tumor suppressor in B-cells, promoting cryostasis and modulating cellular differentiation and apoptotic pathways [42,71]. However, in vitro studies show that EBV-infected B-cell lymphoma cell lines dysregulate TGF-beta receptor signaling making the cells impervious to the anti-tumor effects of endogenous TGF-beta [40,72]. Further, these studies link the dysregulation of TGF-beta signaling with constitutive activation of the P38 MAPK oncogenic pathway, which we also found to be enriched in the hypermethylated gene set of EBV(+) DLBCL [40]. Interestingly, the IFN-gamma pathway was also enriched, suggesting functional suppression of its immunogenic anti-viral effects in EBV-mediated lymphomagenesis [45].
Finally, we highlighted the HDAC10 gene as significantly hypermethylated and potentially suppressed in EBV(+) DLBCL relative to EBV(-) disease. The HDAC10 gene encodes histone deacetylase 10, a class II family, which has crucial roles in epigenetic regulation. Depending on cellular context, HDAC10 can induce both oncogenic and tumor suppressive effects across an array of human cancer types. In B-cell lymphoma, it can exert tumor suppressive effects in concert with the dysregulation of the TGF-beta pathway [38,73]. The hypermethylation of this gene in EBV(+) DLBCL implies suppression of the HDAC10 pathway and loss of inherent tumor suppressive function.
Several pathways were identified significantly enriched in the hypomethylated gene set in EBV(+) DLBCL compared to EBV(-) DLBCL. These pathways included prominent oncogenic signaling like the MAPK cascade, DNA replication, and RAS pathway, which are highly represented across human cancer types [43,56,57]. In addition, the known oncogenes FLT3, HDAC6, NPM1, and BTK were significantly hypomethylated in EBV(+) DLBCL, suggesting activation [49,50,51,52,53,54,55] In particular, Bruton’s tyrosine kinase (BTK) is highlighted as significantly hypomethylated in EBV(+) DLBCL. BTK is a well-characterized and important non-receptor tyrosine kinase that functions in B-cell signaling pathway. Abnormal upregulation of BTK can turn on its oncogenic function in promoting B-cell proliferation, activation, survival, and differentiation [54,58]. BTK inhibitors are powerful targeted therapies used in an array of B-cell malignancies, including DLBCL [53,55]. BTK activates a signaling cascade which involves the RAS pathway and downstream transcription initiation of MYC oncogene [54].
The immune cell microenvironment of DLBCL plays a critical role in tumor aggression and response to therapy. Inference of immune cell composition in EBV(+) and EBV(-) DLBCL was made using the global DNA methylation data. EBV(+) DLBCL exhibited a significant predominance of cytotoxic CD8(+) T-cells relative to EBV(-) DLBCL (p=0.01), as well as a relative predominance in neutrophils, monocytes, and NK cells comprising the background inflammatory microenvironment. In contrast, EBV(-) DLBCL showed a relative predominance in B-cells and CD4(+) T-cells. These findings were confirmed using immunohistochemical stains for B-cells (CD20), T-cells (CD3), and NK cells (CD56). This unique tumor microenvironment composition of EBV(+) DLBCL with a relative increase in inflammatory cells, CD8(+) T-cells, and reduced B-cells is confirmed by multiple studies [74,75,76]. The “immune-rich” environment of EBV(+) DLBCL is observed across immunodeficiency states and is associated with the activated B-cell (ABC) phenotype. The cellular composition includes immunologically exhausted cytotoxic T-cells and NK cells and the upregulation of immune checkpoint proteins (PD-L1) and associated monocytes/macrophages [74,75]. These constellation of factors all lead to increased tumor heterogeneity, an immunosuppressive profile, and worse clinical prognosis in EBV(+) DLBCL across various states of immunodeficiency [74,76].
While this study represents a novel characterization of EBV-mediated DLBCL using global methylation analysis, there are several limitations that should be noted. First, while this is a single-institutional pilot study, the cohort size of EBV(+) DLBCL is small (n=9), limiting comparability of the findings with EBV(-) tumors. Second, the nature of DLBCL as a systemic disease with limited clinical sampling of diagnostic tissue specimens makes obtainment of control tissue from the same patient very difficult in this retrospective approach. In turn, we obtained EBV(+)/(-) normal lymph node/tonsillar tissue as well as comparison cases of other EBV(+) B-cell lymphoproliferative disorders. We acknowledge that the control tissue from different patients limits comparability of the DLBCL findings. Lastly, the global EPIC methylation array employed by this study, although comprehensive, has its limitations in understanding more granular nuances of epigenetic change between compared datasets. Our study is a pilot initiative with the intent to first characterize the overall methylome of EBV-mediated DLBCL. Future, more extensive studies are necessary on a larger clinical cohort utilizing an integrative analysis of epigenetic and whole transcriptomic approaches. This will help elucidate specific genetic pathways and epigenetic alterations that may lead to diagnostic and prognostic biomarker identification in EBV(+) DLBCL.
5. Conclusions
Using a global DNA methylation array, we found that the methylome of EBV(+) DLBCL across immunodeficiency states is epigenetically distinct from EBV(-) disease, and that DLBCL as a whole, is epigenetically distinct from control tissue cases. Both EBV(+) DLBCL and all DLBCL cases exhibited distinct patterns of hyper- and hypomethylation, which corresponded to specific gene pathways with complex roles in lymphomagenesis. EBV(+) DLBCL demonstrated greater hypermethylation across all probe sets compared to EBV(-) tumors, with enrichment of hypermethylated genes involved in tumor-suppressive pathways such as P53 feedback loops and TGF-beta signaling. The immune cell microenvironment of EBV(+) DLBCL, inferred from methylation data, displayed an immune-rich yet immunosuppressive profile, characterized by increased cytotoxic T cells and NK cells relative to the neoplastic population. These findings support the conclusion that EBV(+) DLBCL, across various immunodeficiency settings, harbors an epigenetically distinct methylome that may contribute to the observed tumor heterogeneity and clinical aggressiveness of this disease. Larger studies are warranted to more comprehensively characterize the methylation landscape of EBV-mediated DLBCL, with the goal to identify targetable diagnostic and prognostic biomarkers.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Funding
TGIR: P20 GM125498, UVM Pathology Research Support Award
References
- Ahn, J.S.; Yang, D.H.; Duk Choi, Y.; Jung, S.H.; Yhim, H.Y.; Kwak, J.Y.; Sung Park, H.; Shin, M.G.; Kim, Y.K.; Kim, H.J.; Lee, J.J. Clinical Outcome of Elderly Patients with Epstein–Barr Virus Positive Diffuse Large B-Cell Lymphoma Treated with a Combination of Rituximab and CHOP Chemotherapy. American Journal of Hematology 2013, 88, 774–779. [Google Scholar] [CrossRef]
- Ok, C.Y.; Li, L.; Xu-Monette, Z.Y.; Visco, C.; Tzankov, A.; Manyam, G.C.; Montes-Moreno, S.; Dybaer, K.; Chiu, A.; Orazi, A.; Zu, Y.; Bhagat, G.; Chen, J.; Richards, K.L.; Hsi, E.D.; Choi, W.W. L.; Van Krieken, J.H.; Huh, J.; Ai, W.; Ponzoni, M.; Ferreri, A.J. M.; Farnen, J.P.; Møller, M.B.; Bueso-Ramos, C.E.; Miranda, R.N.; Winter, J.N.; Piris, M.A.; Medeiros, L.J.; Young, K.H. Prevalence and Clinical Implications of Epstein-Barr Virus Infection in de Novo Diffuse Large B-Cell Lymphoma in Western Countries. Clinical Cancer Research 2014, 20, 2338–2349. [Google Scholar] [CrossRef]
- Lu, T.X.; Liang, J.H.; Miao, Y.; Fan, L.; Wang, L.; Qu, X.Y.; Cao, L.; Gong, Q.X.; Wang, Z.; Zhang, Z.H.; Xu, W.; Li, J.Y. Epstein-Barr Virus Positive Diffuse Large B-Cell Lymphoma Predict Poor Outcome, Regardless of the Age. Scientific reports 2015, 5. [Google Scholar] [CrossRef]
- Editorial Board., W.C. of. Haematolymphoid Tumours [Internet; Beta Version Ahead of Print]; International Agency for Research on Cancer: Lyon (France), 2022.
- Ohashi, A.; Kato, S.; Okamoto, A.; Inaguma, Y.; Satou, A.; Tsuzuki, T.; Emi, N.; Okamoto, M.; Nakamura, S. Reappraisal of Epstein–Barr Virus (EBV) in Diffuse Large B-Cell Lymphoma (DLBCL): Comparative Analysis between EBV-Positive and EBV-Negative DLBCL with EBV-Positive Bystander Cells. Histopathology 2017, 71, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmann-Laeisz, C.; Szczepanowski, M.; Borchert, A.; Brüggemann, M.; Klapper, W. Epstein-Barr Virus-Negative Diffuse Large B-Cell Lymphoma Hosts Intra- and Peritumoral B-Cells with Activated Epstein-Barr Virus. Virchows Arch 2015, 466, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Hofscheier, A.; Ponciano, A.; Bonzheim, I.; Adam, P.; Lome-Maldonado, C.; Vela, T.; Cortes, E.; Ortiz-Hidalgo, C.; Fend, F.; Quintanilla-Martinez, L. Geographic Variation in the Prevalence of Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma of the Elderly: A Comparative Analysis of a Mexican and a German Population. Mod Pathol 2011, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; Schiavoni, G.; Segreto, G.; Onyango, N.; Nyagol, J.A.; Amato, T.; Bellan, C.; Anagnostopoulos, I.; Falini, B.; Leoncini, L.; Tiacci, E.; Lazzi, S. Frequent Traces of EBV Infection in Hodgkin and Non-Hodgkin Lymphomas Classified as EBV-Negative by Routine Methods: Expanding the Landscape of EBV-Related Lymphomas. Modern Pathology 2020, 33, 2407–2421. [Google Scholar] [CrossRef]
- Volaric, A.K.; Fedoriw, Y. Epstein-Barr Virus-Associated B-Cell Lymphoproliferative Disorders and Lymphomas: Diagnostic Overlaps and Defining Features. Human Pathology 2024, 105697. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, R.; Xie, Z. The Roles of DNA Methylation on the Promotor of the Epstein–Barr Virus (EBV) Gene and the Genome in Patients with EBV-Associated Diseases. Applied Microbiology and Biotechnology 2022, 106, 4413–4426. [Google Scholar] [CrossRef]
- Saha, A.; Jha, H.C.; Upadhyay, S.K.; Robertson, E.S. Epigenetic Silencing of Tumor Suppressor Genes during in Vitro Epstein-Barr Virus Infection. Proceedings of the National Academy of Sciences of the United States of America 2015, 112, E5199–E5207. [Google Scholar] [CrossRef]
- Ghosh Roy, S.; Robertson, E.S.; Saha, A. Epigenetic Impact on EBV Associated B-Cell Lymphomagenesis. Biomolecules 2016, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R. M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer Analysis Project. Nat Genet 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Ding, W.; Kaur, D.; Horvath, S.; Zhou, W. Comparative Epigenome Analysis Using Infinium DNA Methylation BeadChips. Brief Bioinform 2023, 24, bbac617. [Google Scholar] [CrossRef] [PubMed]
- Fan, S.; Tang, J.; Li, N.; Zhao, Y.; Ai, R.; Zhang, K.; Wang, M.; Du, W.; Wang, W. Integrative Analysis with Expanded DNA Methylation Data Reveals Common Key Regulators and Pathways in Cancers. npj Genomic Med 2019, 4, 1–11. [Google Scholar] [CrossRef]
- Berglund, A.; Yamoah, K.; Eschrich, S.A.; Falahat, R.; Mulé, J.J.; Kim, S.; Matta, J.; Dutil, J.; Ruiz-Deya, G.; Ortiz Sanchez, C.; Wang, L.; Park, H.; Banerjee, H.N.; Lotan, T.; Barry, K.H.; Putney, R.M.; Kim, S.J.; Gwede, C.; Kresovich, J.K.; Kim, Y.; Lin, H.; Dhillon, J.; Chakrabarti, R.; Park, J.Y. Epigenome-wide Association Study of Prostate Cancer in African American Men Identified Differentially Methylated Genes. Cancer Medicine 2024, 13, e70044. [Google Scholar] [CrossRef]
- Zhou, W.; Triche, T.J., Jr; Laird, P.W.; Shen, H. SeSAMe: Reducing Artifactual Detection of DNA Methylation by Infinium BeadChips in Genomic Deletions. Nucleic Acids Research 2018, 46, e123. [Google Scholar] [CrossRef]
- Nishiyama, A.; Nakanishi, M. Navigating the DNA Methylation Landscape of Cancer. Trends in Genetics 2021, 37, 1012–1027. [Google Scholar] [CrossRef]
- Besselink, N.; Keijer, J.; Vermeulen, C.; Boymans, S.; de Ridder, J.; van Hoeck, A.; Cuppen, E.; Kuijk, E. The Genome-Wide Mutational Consequences of DNA Hypomethylation. Sci Rep 2023, 13, 6874. [Google Scholar] [CrossRef]
- Ehrlich, M. DNA Hypomethylation in Cancer Cells. Epigenomics 2009, 1, 239–259. [Google Scholar] [CrossRef]
- Chambwe, N.; Kormaksson, M.; Geng, H.; De, S.; Michor, F.; Johnson, N.A.; Morin, R.D.; Scott, D.W.; Godley, L.A.; Gascoyne, R.D.; Melnick, A.; Campagne, F.; Shaknovich, R. Variability in DNA Methylation Defines Novel Epigenetic Subgroups of DLBCL Associated with Different Clinical Outcomes. Blood 2014, 123, 1699–1708. [Google Scholar] [CrossRef]
- Drillenburg, P.; Pals, S.T. Cell Adhesion Receptors in Lymphoma Dissemination. Blood 2000, 95, 1900–1910. [Google Scholar] [CrossRef]
- Ashton-Key, M.; Cowley, G.P.; Smith, M.E. Cadherins in Reactive Lymph Nodes and Lymphomas: High Expression in Anaplastic Large Cell Lymphomas. Histopathology 1996, 28, 55–59. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front Oncol 2019, 9, 989. [Google Scholar] [CrossRef]
- Yu, S.; Han, R.; Gan, R. The Wnt/β-Catenin Signalling Pathway in Haematological Neoplasms. Biomarker Research 2022, 10, 74. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Fang, J.; Ye, F.; Zhang, S.; Huang, H.; Hou, J.; Wang, T. Diffuse Large B-Cell Lymphoma Promotes Endothelial-to-Mesenchymal Transition via WNT10A/Beta-Catenin/Snail Signaling. Front Oncol 2022, 12, 871788. [Google Scholar] [CrossRef] [PubMed]
- Mansoor, A.; Kamran, H.; Akhter, A.; Seno, R.; Torlakovic, E.E.; Roshan, T.M.; Shabani-Rad, M.-T.; Elyamany, G.; Minoo, P.; Stewart, D. Identification of Potential Therapeutic Targets for Plasmablastic Lymphoma Through Gene Expression Analysis: Insights into RAS and Wnt Signaling Pathways. Modern Pathology 2023, 36, 100198. [Google Scholar] [CrossRef] [PubMed]
- Jardin, F.; Jais, J.-P.; Molina, T.-J.; Parmentier, F.; Picquenot, J.-M.; Ruminy, P.; Tilly, H.; Bastard, C.; Salles, G.-A.; Feugier, P.; Thieblemont, C.; Gisselbrecht, C.; de Reynies, A.; Coiffier, B.; Haioun, C.; Leroy, K. Diffuse Large B-Cell Lymphomas with CDKN2A Deletion Have a Distinct Gene Expression Signature and a Poor Prognosis under R-CHOP Treatment: A GELA Study. Blood 2010, 116, 1092–1104. [Google Scholar] [CrossRef]
- Cobbers, J.M. J. L.; Wolter, M.; Reifenberger, J.; Ring, G.U.; Jessen, F.; An, H.; Niederacher, D.; Schmidt, E.E.; Ichimura, K.; Floeth, F.; Kirsch, L.; Borchard, F.; Louis, D.N.; Collins, V.P.; Reifenberger, G. Frequent In Activation of CDKN2A and Rare Mutation of TP53 in PCNSL. Brain Pathol 2006, 8, 263–276. [Google Scholar] [CrossRef]
- Maura, F.; Dodero, A.; Carniti, C.; Bolli, N.; Magni, M.; Monti, V.; Cabras, A.; Leongamornlert, D.; Abascal, F.; Diamond, B.; Rodriguez-Martin, B.; Zamora, J.; Butler, A.; Martincorena, I.; Tubio, J.M. C.; Campbell, P.J.; Chiappella, A.; Pruneri, G.; Corradini, P. CDKN2A Deletion Is a Frequent Event Associated with Poor Outcome in Patients with Peripheral T-Cell Lymphoma Not Otherwise Specified (PTCL-NOS). Haematologica 2021, 106, 2918–2926. [Google Scholar] [CrossRef]
- Laharanne, E.; Chevret, E.; Idrissi, Y.; Gentil, C.; Longy, M.; Ferrer, J.; Dubus, P.; Jouary, T.; Vergier, B.; Beylot-Barry, M.; Merlio, J.-P. CDKN2A–CDKN2B Deletion Defines an Aggressive Subset of Cutaneous T-Cell Lymphoma. Modern Pathology 2010, 23, 547–558. [Google Scholar] [CrossRef]
- Alhejaily, A.; Day, A.G.; Feilotter, H.E.; Baetz, T.; LeBrun, D.P. Inactivation of the CDKN2A Tumor-Suppressor Gene by Deletion or Methylation Is Common at Diagnosis in Follicular Lymphoma and Associated with Poor Clinical Outcome. Clinical Cancer Research 2014, 20, 1676–1686. [Google Scholar] [CrossRef]
- Gaudio, F.; Dicataldo, M.; Di Giovanni, F.; Cazzato, G.; d’Amati, A.; Perrone, T.; Masciopinto, P.; Laddaga, F.E.; Musto, P.; Maiorano, E.; Ingravallo, G. Prognostic Role of CDKN2A Deletion and P53 Expression and Association With MIPIb in Mantle Cell Lymphoma. Clinical Lymphoma Myeloma and Leukemia 2023, 23, 599–605. [Google Scholar] [CrossRef]
- Salaverria, I.; Akasaka, T.; Gesk, S.; Szczepanowski, M.; Burkhardt, B.; Harder, L.; Damm-Welk, C.; Oschlies, I.; Klapper, W.; Dyer, M.J. S.; Siebert, R. The CBFA2T3/ACSF3 Locus Is Recurrently Involved in IGH Chromosomal Translocation t(14;16)(Q32;Q24) in Pediatric B-Cell Lymphoma with Germinal Center Phenotype. Genes Chromosomes Cancer 2012, 51, 338–343. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Cheney, K.M.; Neilsen, P.M.; Schulz, R.B.; Callen, D.F. CBFA2T3–ZNF651, like CBFA2T3–ZNF652, Functions as a Transcriptional Corepressor Complex. FEBS Letters 2010, 584, 859–864. [Google Scholar] [CrossRef] [PubMed]
- Micci, F.; Thorsen, J.; Panagopoulos, I.; Nyquist, K.B.; Zeller, B.; Tierens, A.; Heim, S. High-Throughput Sequencing Identifies an NFIA/CBFA2T3 Fusion Gene in Acute Erythroid Leukemia with t(1;16)(P31;Q24). Leukemia 2013, 27, 980–982. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Wang, M.; Li, S.; Niu, J.; Xue, J.; Li, J.; Li, X. Identification of Hub Genes and Key Pathways Associated with Peripheral T-Cell Lymphoma. CURR MED SCI 2020, 40, 885–899. [Google Scholar] [CrossRef]
- Sahakian, E.; Shah, B.D.; Powers, J.; Deng, S.; Merino, O.; Gill, A.S.; Rock-Klotz, J.; Woan, K.V.; Vazquez, L.; Wang, H.; Chen-Kiang, S.; Tao, J.; Villagra, A.; Pinilla-Ibarz, J.; Sotomayor, E.M. The Opposing Role of Histone Deacetylase 10 (HDAC10) and HDAC11 in Proliferation/Survival of Mantle Cell Lymphoma (MCL) and Chronic Lymphocytic Leukemia (CLL). Blood 2011, 118, 1363. [Google Scholar] [CrossRef]
- Wen, W.; Zhang, W.-L.; Tan, R.; Zhong, T.-T.; Zhang, M.-R.; Fang, X.-S. Progress in Deciphering the Role of P53 in Diffuse Large B-Cell Lymphoma: Mechanisms and Therapeutic Targets. Am J Cancer Res 2024, 14, 3280–3293. [Google Scholar] [CrossRef]
- Bakkebø, M.; Huse, K.; Hilden, V.I.; Smeland, E.B.; Oksvold, M.P. TGF-β-Induced Growth Inhibition in B-Cell Lymphoma Correlates with Smad1/5 Signalling and Constitutively Active P38 MAPK. BMC Immunology 2010, 11, 57. [Google Scholar] [CrossRef]
- Chen, G.; Ghosh, P.; Osawa, H.; Sasaki, C.Y.; Rezanka, L.; Yang, J.; O’Farrell, T.J.; Longo, D.L. Resistance to TGF-Β1 Correlates with Aberrant Expression of TGF-β Receptor II in Human B-Cell Lymphoma Cell Lines. Blood 2007, 109, 5301–5307. [Google Scholar] [CrossRef]
- Lebman, D.A.; Edmiston, J.S. The Role of TGF-β in Growth, Differentiation, and Maturation of B Lymphocytes. Microbes and Infection 1999, 1, 1297–1304. [Google Scholar] [CrossRef] [PubMed]
- Bahar, M.E.; Kim, H.J.; Kim, D.R. Targeting the RAS/RAF/MAPK Pathway for Cancer Therapy: From Mechanism to Clinical Studies. Sig Transduct Target Ther 2023, 8, 455. [Google Scholar] [CrossRef] [PubMed]
- Ruvolo, V.; Navarro, L.; Sample, C.E.; David, M.; Sung, S.; Swaminathan, S. The Epstein-Barr Virus SM Protein Induces STAT1 and Interferon-Stimulated Gene Expression. J Virol 2003, 77, 3690–3701. [Google Scholar] [CrossRef] [PubMed]
- Morrison, T.E.; Mauser, A.; Wong, A.; Ting, J.P.; Kenney, S.C. Inhibition of IFN-Gamma Signaling by an Epstein-Barr Virus Immediate-Early Protein. Immunity 2001, 15, 787–799. [Google Scholar] [CrossRef]
- Powers, J.; Lienlaf, M.; Perez-Villarroel, P.; Deng, S.; Knox, T.; Villagra, A.; Sahakian, E. Expression and Function of Histone Deacetylase 10 (HDAC10) in B Cell Malignancies. Methods Mol Biol 2016, 1436, 129–145. [Google Scholar] [CrossRef]
- Wu, C.; Song, Q.; Gao, S.; Wu, S. Targeting HDACs for Diffuse Large B-Cell Lymphoma Therapy. Sci Rep 2024, 14, 289. [Google Scholar] [CrossRef]
- Mieland, A.O.; Petrosino, G.; Dejung, M.; Chen, J.-X.; Fulzele, A.; Mahmoudi, F.; Tu, J.-W.; Mustafa, A.-H. M.; Zeyn, Y.; Hieber, C.; Bros, M.; Schnöder, T.M.; Heidel, F.H.; Najafi, S.; Oehme, I.; Hofmann, I.; Schutkowski, M.; Hilscher, S.; Kosan, C.; Butter, F.; Bhatia, S.; Sippl, W.; Krämer, O.H. The Protein Deacetylase HDAC10 Controls DNA Replication in Malignant Lymphoid Cells. Leukemia 2025, 39, 1756–1768. [Google Scholar] [CrossRef]
- Medina, K.L. Flt3 Signaling in B Lymphocyte Development and Humoral Immunity. Int J Mol Sci 2022, 23, 7289. [Google Scholar] [CrossRef]
- Tobón, G.J.; Renaudineau, Y.; Hillion, S.; Cornec, D.; Devauchelle-Pensec, V.; Youinou, P.; Pers, J.-O. The Fms-like Tyrosine Kinase 3 Ligand, a Mediator of B Cell Survival, Is Also a Marker of Lymphoma in Primary Sjögren’s Syndrome. Arthritis Rheum 2010, 62, 3447–3456. [Google Scholar] [CrossRef]
- Yang, J.; Li, D.; Zhou, J. Histone Deacetylase 6 as a Therapeutic Target in B Cell-Associated Hematological Malignancies. Front Pharmacol 2020, 11, 971. [Google Scholar] [CrossRef]
- Chen, Y.; Hu, J. Nucleophosmin1 (NPM1) Abnormality in Hematologic Malignancies, and Therapeutic Targeting of Mutant NPM1 in Acute Myeloid Leukemia. Ther Adv Hematol 2020, 11, 2040620719899818. [Google Scholar] [CrossRef]
- Wang, H.; Guo, H.; Yang, J.; Liu, Y.; Liu, X.; Zhang, Q.; Zhou, K. Bruton Tyrosine Kinase Inhibitors in B-Cell Lymphoma: Beyond the Antitumour Effect. Experimental Hematology & Oncology 2022, 11, 60. [Google Scholar] [CrossRef]
- Mehra, S.; Nicholls, M.; Taylor, J. The Evolving Role of Bruton’s Tyrosine Kinase Inhibitors in B Cell Lymphomas. Int J Mol Sci 2024, 25, 7516. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Zhu, Y.; Cheng, Y.; Hou, J.; Jin, F.; Li, M.; Jia, W.; Cheng, Z.; Xing, H.; Liu, M.; Han, T. BTK Kinase Activity Is Dispensable for the Survival of Diffuse Large B-Cell Lymphoma. J Biol Chem 2022, 298, 102555. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Jeffrey, M.L.; Li, Y.; Li, J.; Young, K.H. Genetic Alterations and Their Clinical Implications in DLBCL. Nature Reviews. Clinical Oncology 2019, 16, 634–652. [Google Scholar] [CrossRef]
- Loo, S.K.; Ab. Hamid, S.S.; Musa, M.; Wong, K.K. DNMT1 Is Associated with Cell Cycle and DNA Replication Gene Sets in Diffuse Large B-Cell Lymphoma. Pathology - Research and Practice 2018, 214, 134–143. [Google Scholar] [CrossRef]
- Pal Singh, S.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s Tyrosine Kinase in B Cells and Malignancies. Molecular Cancer 2018, 17, 57. [Google Scholar] [CrossRef]
- Tang, C.-L.; Li, X.-Z.; Zhou, T.; Deng, C.-M.; Jiang, C.-T.; Zhang, Y.-M.; Liao, Y.; Wang, T.-M.; He, Y.-Q.; Xue, W.-Q.; Jia, W.-H.; Zheng, X.-H. EBV DNA Methylation Profiles and Its Application in Distinguishing Nasopharyngeal Carcinoma and Nasal NK/T-Cell Lymphoma. Clinical Epigenetics 2024, 16, 11. [Google Scholar] [CrossRef]
- Healy, J.A.; Dave, S.S. The Role of EBV in the Pathogenesis of Diffuse Large B Cell Lymphoma. Current topics in microbiology and immunology 2015, 390, 315–337. [Google Scholar] [CrossRef]
- Liu, F.; Tian, S.; Liu, Q.; Deng, Y.; He, Q.; Shi, Q.; Chen, G.; Xu, X.; Yuan, J.; Nakamura, S.; Karube, K.; Wang, Z. Comparison of Genomic Alterations in Epstein–Barr Virus-Positive and Epstein–Barr Virus-Negative Diffuse Large B-Cell Lymphoma. Cancer Medicine 2024, 13, e6995. [Google Scholar] [CrossRef]
- Chapman, J.R.; Bouska, A.C.; Zhang, W.; Alderuccio, J.P.; Lossos, I.S.; Rimsza, L.M.; Maguire, A.; Yi, S.; Chan, W.C.; Vega, F.; Song, J.Y. EBV-Positive HIV-Associated Diffuse Large B Cell Lymphomas Are Characterized by JAK/STAT (STAT3) Pathway Mutations and Unique Clinicopathologic Features. British journal of haematology 2021, 194, 870–878. [Google Scholar] [CrossRef]
- Frontzek, F.; Staiger, A.M.; Wullenkord, R.; Grau, M.; Zapukhlyak, M.; Kurz, K.S.; Horn, H.; Erdmann, T.; Fend, F.; Richter, J.; Klapper, W.; Lenz, P.; Hailfinger, S.; Tasidou, A.; Trautmann, M.; Hartmann, W.; Rosenwald, A.; Quintanilla-Martinez, L.; Ott, G.; Anagnostopoulos, I.; Lenz, G. Molecular Profiling of EBV Associated Diffuse Large B-Cell Lymphoma. Leukemia 2023, 37, 670–679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Melnick, A. The Epigenetic Basis of Diffuse Large B-Cell Lymphoma. Semin Hematol 2015, 52, 86–96. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, M.L.; Reyes-Garau, D.; Armengol, M.; Fernández-Serrano, M.; Roué, G. Recent Advances in the Targeting of Epigenetic Regulators in B-Cell Non-Hodgkin Lymphoma. Front. Genet. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- Esteller, M. Epigenetics in Cancer. New England Journal of Medicine 2008, 358, 1148–1159. [Google Scholar] [CrossRef]
- Hassler, M.R.; Schiefer, A.-I.; Egger, G. Combating the Epigenome: Epigenetic Drugs against Non-Hodgkin’s Lymphoma. Epigenomics 2013, 5, 397–415. [Google Scholar] [CrossRef]
- Hansen, K.D.; Timp, W.; Bravo, H.C.; Sabunciyan, S.; Langmead, B.; McDonald, O.G.; Wen, B.; Wu, H.; Liu, Y.; Diep, D.; Briem, E.; Zhang, K.; Irizarry, R.A.; Feinberg, A.P. Increased Methylation Variation in Epigenetic Domains across Cancer Types. Nat Genet 2011, 43, 768–775. [Google Scholar] [CrossRef]
- Fiches, G.N.; Zhou, D.; Kong, W.; Biswas, A.; Ahmed, E.H.; Baiocchi, R.A.; Zhu, J.; Santoso, N. Profiling of Immune Related Genes Silenced in EBV-Positive Gastric Carcinoma Identified Novel Restriction Factors of Human Gammaherpesviruses. PLoS Pathog 2020, 16, e1008778. [Google Scholar] [CrossRef]
- Shaknovich, R.; Geng, H.; Johnson, N.A.; Tsikitas, L.; Cerchietti, L.; Greally, J.M.; Gascoyne, R.D.; Elemento, O.; Melnick, A. DNA Methylation Signatures Define Molecular Subtypes of Diffuse Large B-Cell Lymphoma. Blood 2010, 116, e81–e89. [Google Scholar] [CrossRef]
- Massagué, J. TGFβ in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef]
- Inman, G.J.; Allday, M.J. Resistance to TGF-Β1 Correlates with a Reduction of TGF-β Type II Receptor Expression in Burkitt’s Lymphoma and Epstein–Barr Virus-Transformed B Lymphoblastoid Cell Lines. Journal of General Virology 2000, 81, 1567–1578. [Google Scholar] [CrossRef]
- Cheng, F.; Zheng, B.; Wang, J.; Zhao, G.; Yao, Z.; Niu, Z.; He, W. Histone Deacetylase 10, a Potential Epigenetic Target for Therapy. Biosci Rep 2021, 41, BSR20210462. [Google Scholar] [CrossRef]
- Cerchietti, L. Genetic Mechanisms Underlying Tumor Microenvironment Composition and Function in Diffuse Large B-Cell Lymphoma. Blood 2024, 143, 1101–1111. [Google Scholar] [CrossRef]
- Cioroianu, A.I.; Stinga, P.I.; Sticlaru, L.; Cioplea, M.D.; Nichita, L.; Popp, C.; Staniceanu, F. Tumor Microenvironment in Diffuse Large B-Cell Lymphoma: Role and Prognosis. Analytical Cellular Pathology 2019, 2019, 8586354. [Google Scholar] [CrossRef]
- He, M.; Liu, M.; Yuan, J.; Lv, J.; Li, W.; Yan, Q.; Tang, Y.; Wang, L.; Guo, L.; Liu, F. Spatial Transcriptomics Reveals Tumor Microenvironment Heterogeneity in EBV Positive Diffuse Large B Cell Lymphoma. Sci Rep 2025, 15, 15878. [Google Scholar] [CrossRef]
Figure 1.
Representative Histological Images of EBV(+) and EBV(-) DLBCL. (A-B) EBV(+) DLBCL showing prominent large B-cells with a mixed inflammatory background of small lymphocytes, granulocytes, and apoptotic debris (A, H&E, 40X). EBER in situ hybridization highlighting >80% nuclear staining in tumor B-cells (B, EBER, 20X). (C-D) EBV(-) DLBCL composed predominantly of sheets of large B-cells with mitotic figures and apoptotic debris (C, H&E, 40X). EBER stain is negative for nuclear cellular staining in tumor cells (D, EBER, 20X).
Figure 1.
Representative Histological Images of EBV(+) and EBV(-) DLBCL. (A-B) EBV(+) DLBCL showing prominent large B-cells with a mixed inflammatory background of small lymphocytes, granulocytes, and apoptotic debris (A, H&E, 40X). EBER in situ hybridization highlighting >80% nuclear staining in tumor B-cells (B, EBER, 20X). (C-D) EBV(-) DLBCL composed predominantly of sheets of large B-cells with mitotic figures and apoptotic debris (C, H&E, 40X). EBER stain is negative for nuclear cellular staining in tumor cells (D, EBER, 20X).
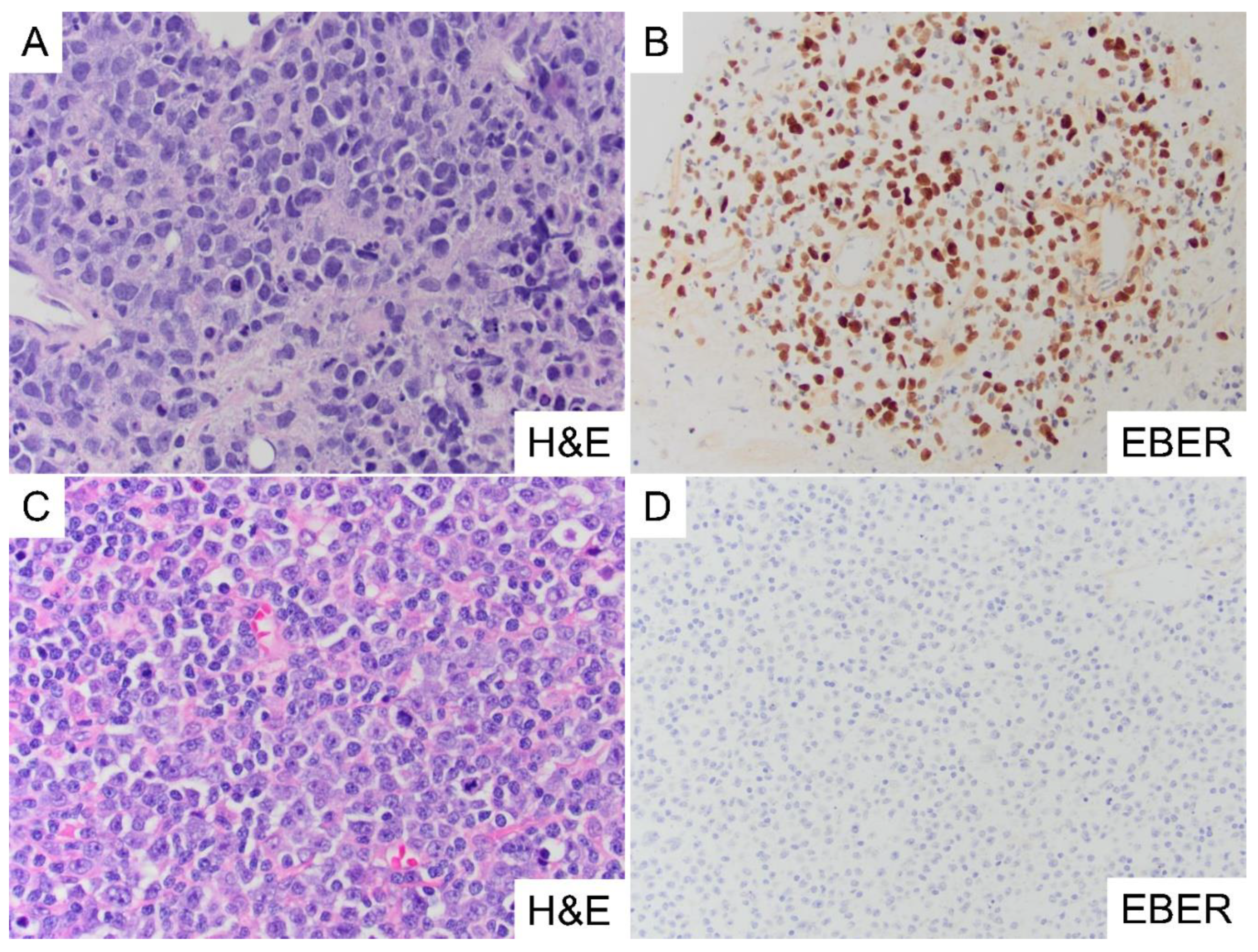
Figure 2.
Differential Methylation in All DLBCL Compared to Control Cases. A) Principal component analysis (PCA) of all EBV(+)/(-) DLBCL (n=30, blue) versus all EBV(+)/(-) control cases (n=12, orange) across 330,872 CpG sites, showing distinct separation of tumor from controls. B) Beta density scatter plot showing average differences in methylation between DLBCL (x-axis) and control cases (y-axis) across all probe sites. C) Volcano plot comparing methylation between DLBCL and controls with colors based on p-values (p=0.05) and log-fold change highlighting significantly differentially methylated genes. D) Heatmap of genes hypermethylated in DLBCL compared to controls, annotated for associations with human cancers using oncoEnrichR analysis. Hypermethylated genes highly represented in hematolymphoid malignancies (red box) include tumor suppressor genes (cyan) CDKN2A, HLA-A, and FAT4. Hypomethylated genes were uninformative. E) Pathway enrichment analysis showing cancer-related pathways, including Wnt/β catenin and Cadherin signaling, that are hypermethylated in DLBCL compared to controls. F) Probe-level methylation map of tumor suppressor gene CDKN2A, showing specific CpG sites hypermethylated in DLBCL relative to controls. The heatmap y-axis shows a colored bar delineating control (orange) from DLBCL (blue) cases. OG= oncogene (black), TS= tumor suppressor (cyan), Both= both TS/OG functions (magenta).
Figure 2.
Differential Methylation in All DLBCL Compared to Control Cases. A) Principal component analysis (PCA) of all EBV(+)/(-) DLBCL (n=30, blue) versus all EBV(+)/(-) control cases (n=12, orange) across 330,872 CpG sites, showing distinct separation of tumor from controls. B) Beta density scatter plot showing average differences in methylation between DLBCL (x-axis) and control cases (y-axis) across all probe sites. C) Volcano plot comparing methylation between DLBCL and controls with colors based on p-values (p=0.05) and log-fold change highlighting significantly differentially methylated genes. D) Heatmap of genes hypermethylated in DLBCL compared to controls, annotated for associations with human cancers using oncoEnrichR analysis. Hypermethylated genes highly represented in hematolymphoid malignancies (red box) include tumor suppressor genes (cyan) CDKN2A, HLA-A, and FAT4. Hypomethylated genes were uninformative. E) Pathway enrichment analysis showing cancer-related pathways, including Wnt/β catenin and Cadherin signaling, that are hypermethylated in DLBCL compared to controls. F) Probe-level methylation map of tumor suppressor gene CDKN2A, showing specific CpG sites hypermethylated in DLBCL relative to controls. The heatmap y-axis shows a colored bar delineating control (orange) from DLBCL (blue) cases. OG= oncogene (black), TS= tumor suppressor (cyan), Both= both TS/OG functions (magenta).
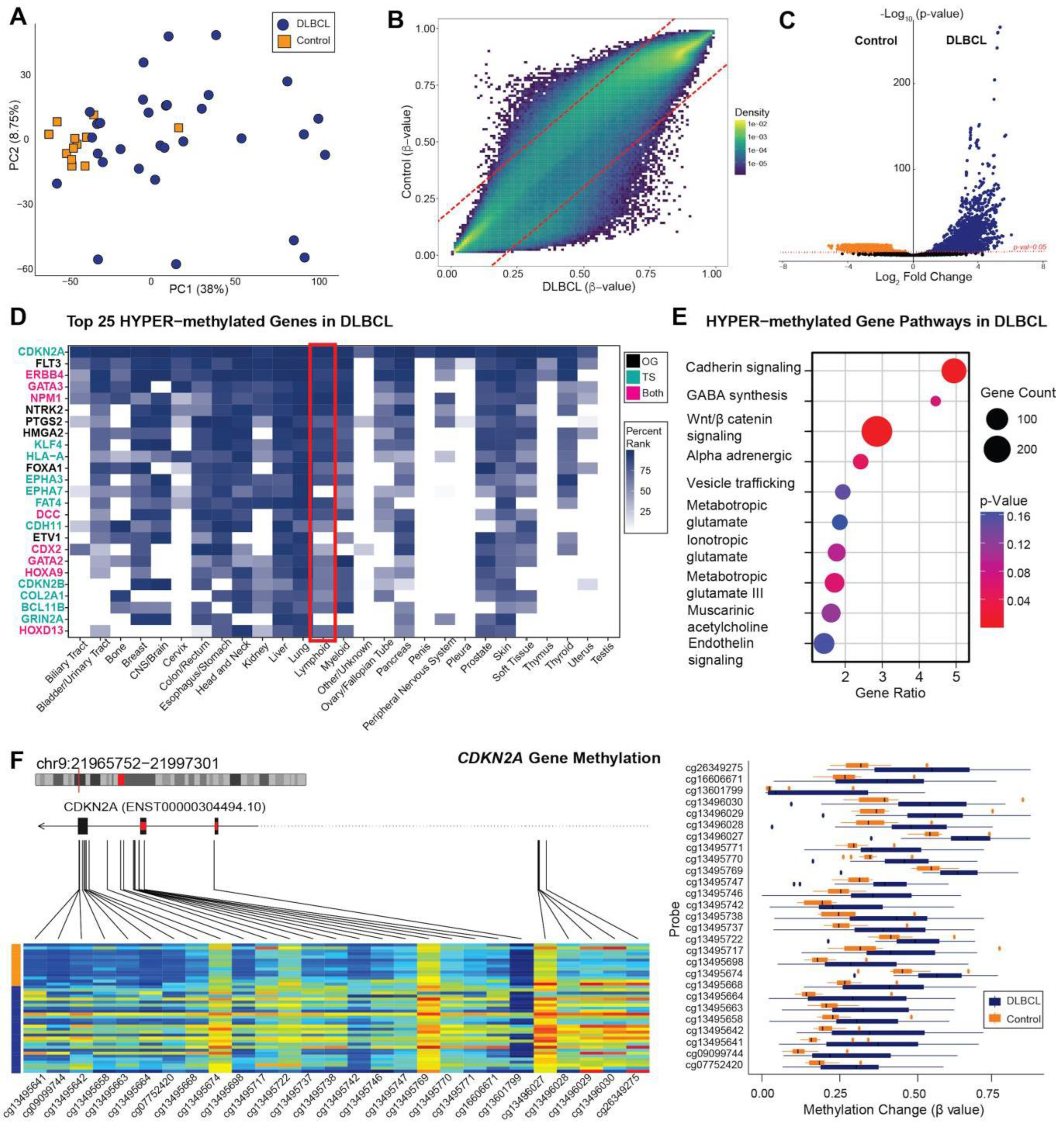
Figure 3.
Differential Methylation Patterns in EBV(+) DLBCL versus EBV(-) DLBCL. A) PCA of EBV(+) DLBCL (n=9, purple) versus EBV(-) DLBCL (n=22, yellow) across 420,159 CpG sites, showing distinct clustering of EBV(+) from EBV(-) tumors. B) Beta density scatter plot showing average methylation differences between EBV(+) DLBCL (x-axis) and EBV(-) DLBCL (y-axis) across all probe sites, with a distinct population of probes hypermethylated in EBV(+) DLBCL. C) Volcano plot comparing significantly differentially methylated genes between EBV(+) DLBCL (purple) and EBV(-) DLBCL (yellow). Colors are based on p-values (p=0.05) and log2 fold change thresholds. .
Figure 3.
Differential Methylation Patterns in EBV(+) DLBCL versus EBV(-) DLBCL. A) PCA of EBV(+) DLBCL (n=9, purple) versus EBV(-) DLBCL (n=22, yellow) across 420,159 CpG sites, showing distinct clustering of EBV(+) from EBV(-) tumors. B) Beta density scatter plot showing average methylation differences between EBV(+) DLBCL (x-axis) and EBV(-) DLBCL (y-axis) across all probe sites, with a distinct population of probes hypermethylated in EBV(+) DLBCL. C) Volcano plot comparing significantly differentially methylated genes between EBV(+) DLBCL (purple) and EBV(-) DLBCL (yellow). Colors are based on p-values (p=0.05) and log2 fold change thresholds. .
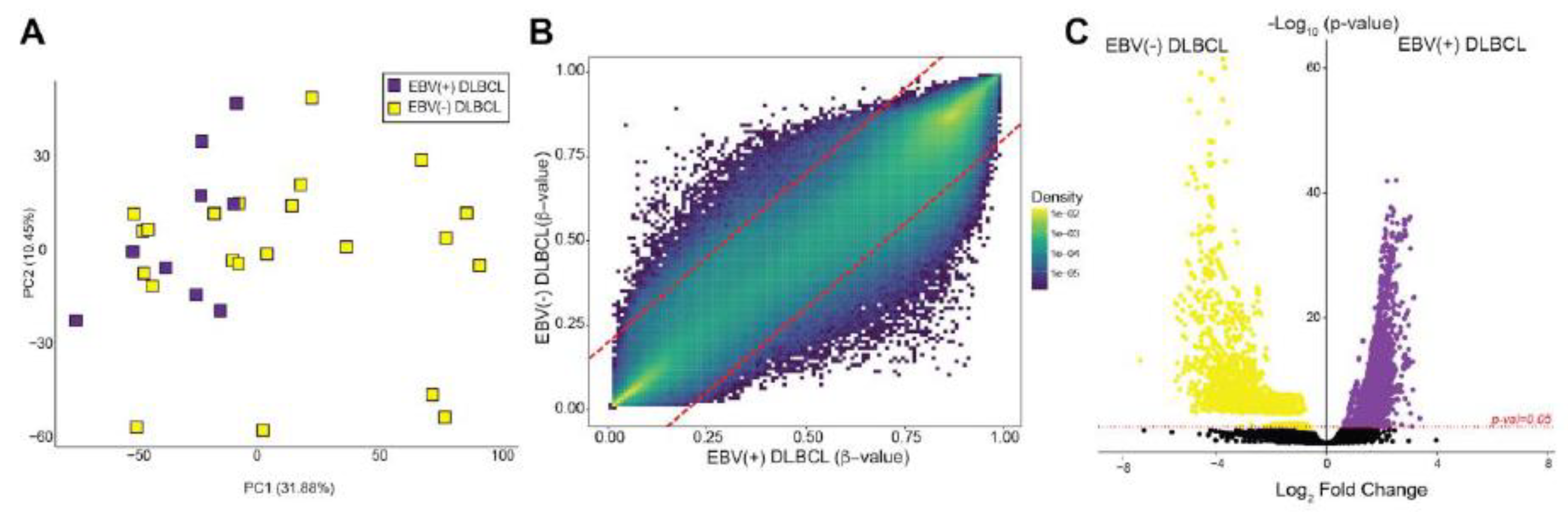
Figure 4.
Methylation States of Top Genes and Gene Pathways in EBV(+) DLBCL versus EBV(-) DLBCL. A) Heatmap of genes hypermethylated in EBV(+) DLBCL relative to EBV(-) DLBLCL, annotated for associations with human cancers using oncoEnrichR analysis. The red box highlights genes significantly represented in hematolymphoid malignancies, including genes dual-function genes (magenta) such as HDAC10 and only tumor suppressor function (cyan) such as CBFA2T3 and CSNK1E. B) Pathway enrichment analysis of hypermethylated genes in EBV(+) DLBCL. C) Probe-level methylation map of HDAC10 gene, a gene with both tumor suppressor and oncogenic roles, showing significant hypermethylation in EBV(+) DLBCL. D) Heatmap of genes hypomethylated in EBV(+) DLBCL compared to EBV(-) DLBCL. The red box highlights genes significantly associated with hematolymphoid malignancies, including dual-function genes (BTK and NPM1) and oncogenes (FLT3 and HDAC6). E) Pathway enrichment analysis of hypomethylated in EBV(+) DLBCL. F) Probe-level methylation map of BTK, hypomethylated in EBV(+) DLBCL relative to EBV(-) DLBCL. BTK exhibits both tumor suppressive and oncogenic functions and is strongly associated with B-cell lymphomagenesis.OG= oncogene, TS= tumor suppressor, Both= both TS and OG functions.
Figure 4.
Methylation States of Top Genes and Gene Pathways in EBV(+) DLBCL versus EBV(-) DLBCL. A) Heatmap of genes hypermethylated in EBV(+) DLBCL relative to EBV(-) DLBLCL, annotated for associations with human cancers using oncoEnrichR analysis. The red box highlights genes significantly represented in hematolymphoid malignancies, including genes dual-function genes (magenta) such as HDAC10 and only tumor suppressor function (cyan) such as CBFA2T3 and CSNK1E. B) Pathway enrichment analysis of hypermethylated genes in EBV(+) DLBCL. C) Probe-level methylation map of HDAC10 gene, a gene with both tumor suppressor and oncogenic roles, showing significant hypermethylation in EBV(+) DLBCL. D) Heatmap of genes hypomethylated in EBV(+) DLBCL compared to EBV(-) DLBCL. The red box highlights genes significantly associated with hematolymphoid malignancies, including dual-function genes (BTK and NPM1) and oncogenes (FLT3 and HDAC6). E) Pathway enrichment analysis of hypomethylated in EBV(+) DLBCL. F) Probe-level methylation map of BTK, hypomethylated in EBV(+) DLBCL relative to EBV(-) DLBCL. BTK exhibits both tumor suppressive and oncogenic functions and is strongly associated with B-cell lymphomagenesis.OG= oncogene, TS= tumor suppressor, Both= both TS and OG functions.
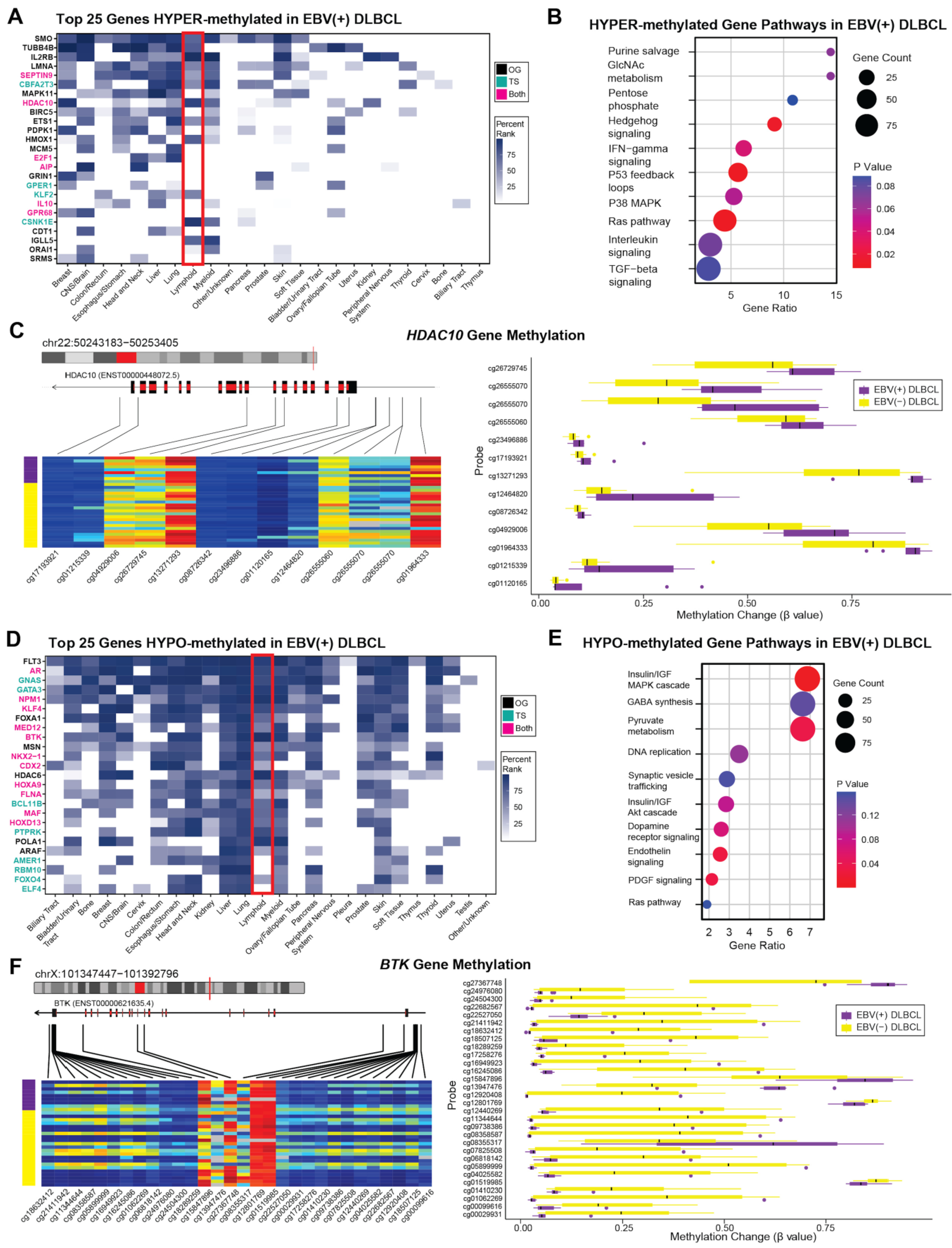
Figure 5.
Immune Cell Composition of EBV(+) versus EBV(-) DLBCL. A) Immune cell type inference revealed enrichment of CD8(+) T-cells (p=0.01), monocytes (p=0.11), neutrophils (p=0.08), and NK cells (p=0.11) in EBV(+) DLBCL. In contrast, EBV(-) DLBCL showed an enrichment of B-cells (p=0.07) and CD4(+) T-cells (p=0.63) relative to EBV(+) tumors. Statistical comparisons were performed using the Wilcoxon test. B) Immunohistochemistry of EBV(+) DLBCL showing an inflammatory microenvironment enriched in CD3(+) T cells and CD56(+) NK cells, with reduced CD20(+) B-cells. C) Immunohistochemistry of EBV(-) DLBCL showing enrichment of CD20(+) B-cells, with relatively fewer CD3(+) T cells and absence of CD56(+) NK cells.
Figure 5.
Immune Cell Composition of EBV(+) versus EBV(-) DLBCL. A) Immune cell type inference revealed enrichment of CD8(+) T-cells (p=0.01), monocytes (p=0.11), neutrophils (p=0.08), and NK cells (p=0.11) in EBV(+) DLBCL. In contrast, EBV(-) DLBCL showed an enrichment of B-cells (p=0.07) and CD4(+) T-cells (p=0.63) relative to EBV(+) tumors. Statistical comparisons were performed using the Wilcoxon test. B) Immunohistochemistry of EBV(+) DLBCL showing an inflammatory microenvironment enriched in CD3(+) T cells and CD56(+) NK cells, with reduced CD20(+) B-cells. C) Immunohistochemistry of EBV(-) DLBCL showing enrichment of CD20(+) B-cells, with relatively fewer CD3(+) T cells and absence of CD56(+) NK cells.
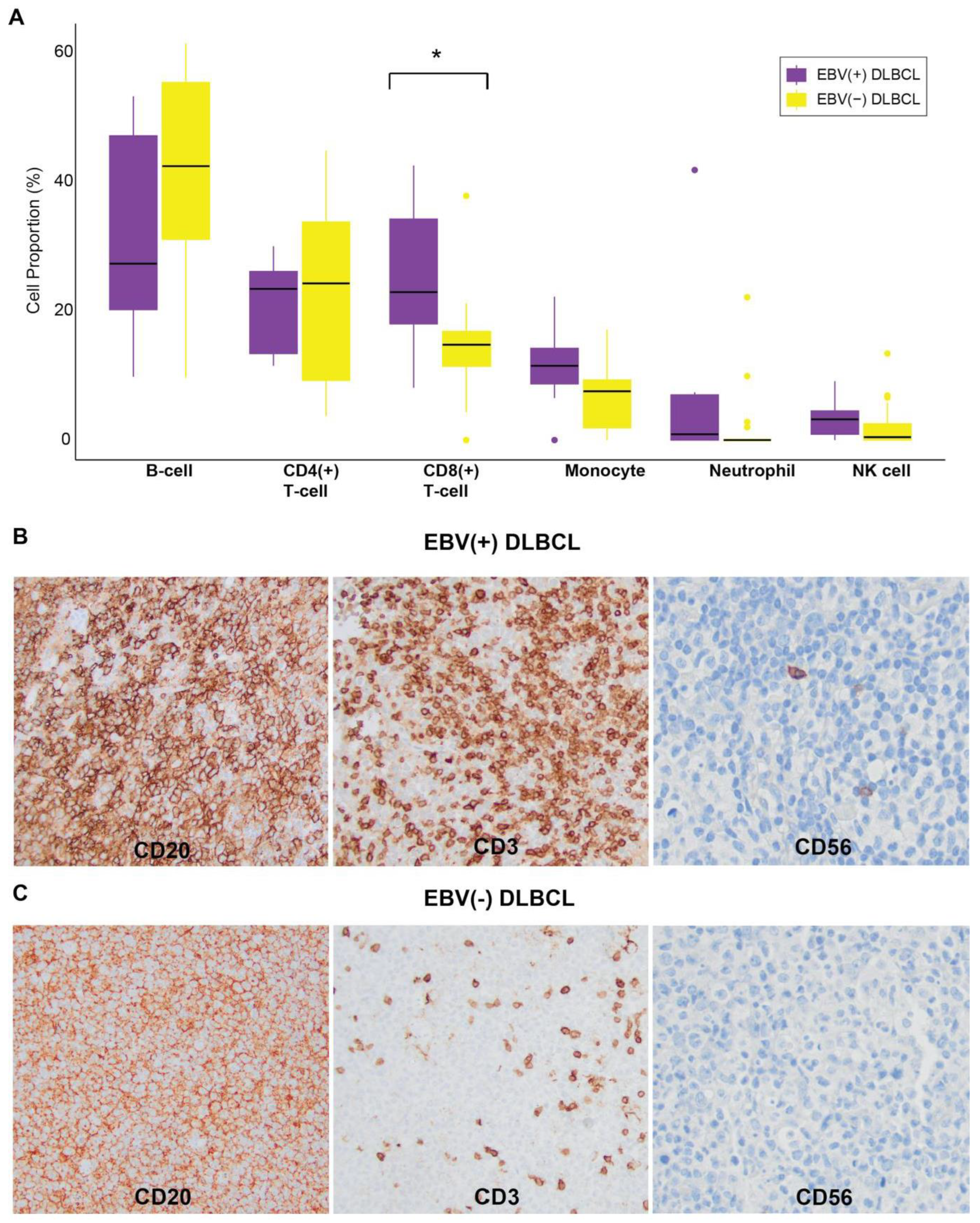

Table 1.
Clinicopathologic and Immunophenotypic Characteristics of EBV(+) and EBV(-)DLBCL (n=31). Not included are EBV(+)/(-) control cases (n=12).*Therapy-related/Iatrogenic immunodeficiency includes post cytotoxic chemotherapy/radiation therapy for prior malignancy either concurrent or within prior 15 years to DLBCL. **Autoimmune disease immunodeficiency includes anti-TNF/methotrexate and systemic corticosteroid therapy either concurrent or within prior 15 years to DLBCL.
Table 1.
Clinicopathologic and Immunophenotypic Characteristics of EBV(+) and EBV(-)DLBCL (n=31). Not included are EBV(+)/(-) control cases (n=12).*Therapy-related/Iatrogenic immunodeficiency includes post cytotoxic chemotherapy/radiation therapy for prior malignancy either concurrent or within prior 15 years to DLBCL. **Autoimmune disease immunodeficiency includes anti-TNF/methotrexate and systemic corticosteroid therapy either concurrent or within prior 15 years to DLBCL.
 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



