
Submitted:
18 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Phytoplasmas ('Candidatus Phytoplasma') are intracellular pleomorphic plant pathogens belonging to the class Mollicutes. They colonize both plant hosts and insect vectors in their life cycle. Apple proliferation (AP) is one of the most important phytoplasmoses present in Europe causing significant economic losses in apple production. The causal agent, ‘Ca. P. mali', was identified in apple and Cacopsylla picta samples using both real-time PCR and nested PCR based on the amplification of 16S rDNA. The objective of this study was to gain deeper insights into the epidemiology of apple proliferation in Croatia. Variability of genetic markers other than 16S rRNA was used for characterization of strains. Four molecular markers differing in level of conservation, aceF, pnp, imp and secY, were selected in line with previously typed fruit tree phytoplasmas. New genotypes were discerned for each genetic marker and 20 different sequence types were revealed in the Croatian isolates of ‘Ca. P. mali’. On the basis of this comprehensive analysis, the founder sequence type ST1 (A13-P10-S12-I21) can be proposed. This is the first extensive research and multigene typing performed on Croatian ‘Ca. P. mali’ isolates. Obtained results reveal considerable genetic diversity of epidemiological relevance limited to only two locations in north-western Croatia. Additionally, novel primers were constructed to amplify fragments larger than the entire coding region for all four genes in order to further expand phytoplasma multi locus sequence typing scheme.
Keywords:
apple proliferation
; Cacopsylla picta
; aceF
; pnp
; imp
; secY
; genotype
; sequence type
1. Introduction
Phytoplasmas ('Candidatus Phytoplasma') are wall-less prokaryotes from the class Mollicutes that induce diseases in more than a thousand plant species worldwide [1]. They are limited to the phloem conducting elements in host plants, while in insect vectors they can be found in various organs and tissues, including the hemolymph and reproductive organs [1,2]. Their phylogeny is mainly based on the common bacterial phylogenetic marker 16S rRNA gene [3]. Phytoplasma genomes are highly reduced and are among the smallest bacterial genomes described to date, regardless of their adaptation to both plant and insect hosts. Inability to cultivate phytoplasmas in axenic culture still hinders their detailed characterization as well as the acquisition of high-quality genomic DNA. Despite many difficulties, more than 50, either draft or complete, genomes have been sequenced in the last 20 years [4,5], with the first one being OY-M strain of the subgroup 16SrI-B [6].
Fruit tree phytoplasmas occurring in Europe, ‘Ca. P. mali’, ‘Ca. P. pyri’ and ‘Ca. P. prunorum’, belong to the same phylogenetic cluster [7]. Their chromosomes are linear, which is an unusual feature for phytoplasmas and bacteria in general [8]. Multi-locus sequence typing (MLST) was proposed as a method applicable for characterization of many pathogenic bacteria [9]. This approach was used to differentiate phytoplasma strains and enhance understanding of molecular epidemiology of European fruit tree phytoplasmas belonging to the ribosomal group 16SrX [10].
Characteristic symptoms of apple proliferation (AP) include the formation of "witches' brooms", enlarged stipules and reduced fruit size with elongated pedicels [11]. The described symptoms were observed in apple orchards in north-western Croatia more than 40 years ago and pleomorphic mycoplasma-like cells were detected in phloem tissues [12]. Although surveys of both ‘Ca. P. mali’ and ‘Ca. P. pyri’ presence in Croatian orchards started in 2003, ‘Ca. P. mali’, the causal agent of AP, was not confirmed in apple samples until 2011 [13,14]. Two psyllid species, Cacopsylla picta (Förster) [15] and C. melanoneura (Förster) [16] are identified as vectors of ‘Ca. P. mali’. Population dynamics of psyllids was monitored from 2005 to 2007 and both C. melanoneura and C. picta were found to be present and widespread [17]. With the availability of ‘Ca. P. mali’ genome [8], this study was designed to optimize the MLST scheme, enabling more accurate and robust molecular characterization of this phytoplasma. In parallel, it aimed to improve the understanding of apple proliferation epidemiology in Croatian apple orchards.
2. Materials and Methods
Plant and Insect Samples
Samples infected with ‘Ca. P. mali’ from a previous study, one C. picta and 60 apple samples, were used for MLST [14]. In 2016, additional 176 C. picta, 34 C. melanoneura and 12 apple samples were collected mainly in western continental part of Croatia where the AP disease pressure was the highest, with a particular focus on psyllid monitoring. Samples were tested for ‘Ca. P. mali’ using both real-time PCR [18] and nested PCR/RFLP methods [19,20,21] as previously described [14]. A total of 423 samples were tested for phytoplasma presence, of which 74 positive samples were used for MLST (Table A.1.).
MLST Primers and PCR Conditions
Primers designed to amplify the entire aceF gene were used for both direct PCR and sequencing, as previously described [14]. New primers were designed based on the available AP phytoplasma genome [8] to amplify fragments longer than complete coding regions of imp, secY and pnp genes. Due to the amplicon size, additional primer pairs were needed for both nested PCR and sequencing for pnp and secY genes (Table 1). In a nested PCR reaction, 1 µl of direct PCR mix was used as a template. Both direct and nested PCR conditions for aceF, pnp and secY amplification were the same: initial denaturation at 94°C for 4 min, followed by 35 cycles of denaturation at 94°C for 1 min, annealing at 52°C for 2 min, extension at 68°C for 3 min and a final extension step of 7 min at 68°C. To amplify the imp fragment in a direct PCR, the annealing and extension temperatures were 51°C and 66°C, respectively, with the same duration and number of cycles. Amplicons were custom sequenced (Macrogen Europe, The Netherlands) on both strands for aceF and imp. For pnp and secY additional internal primers were used for sequencing to increase coverage due to the amplicon size. After the initial sequencing results were analyzed, a third sequencing primer pair was designed to increase the coverage of the most variable region of secY gene (Table 1).
Sequence Analysis
Raw nucleotide sequences were assembled and edited using both the Sequencher® 4.7 software (Gene Codes Corporation, Ann Arbor, MI USA, http://www.genecodes.com/) and Geneious 10.1.3 ([22], http://www.geneious.com), and aligned with ClustalX 2.0 [23]. Consensus sequences of all four genes for each sample were concatenated using the Geneious 10.1.3 [22] and analyzed using the PhyloViz 2.0 [24]. Phylogenetic analyses were performed with MEGA 11 [25] using maximum parsimony method with bootstrap test (500 replicates) to support the inferred clades [26]. As amplicons and sequenced fragments exceeded the coding region for all four genes, both ExPasy [27] (http://web.expasy.org/translate/) and MEGA 11 [25] were used to identify open reading frames for each consensus sequence. Obtained sequences were trimmed in silico in two ways: first, to the length of reference sequences for each genotype of ‘Ca. P. mali’ [10] to enable comparison and continuation of the genotype labeling system, and second, to the length of the coding region to verify the accuracy of the consensus sequence. For each sample, a combination of four genotypes was used to form a sequence type (ST), with the assigned ST number in the order of their prevalence.
3. Results
In a five-year period (2011-2014 and 2016) a total of 204 apple and 219 psyllid samples were analyzed using both nested PCR/RFLP and real-time PCR methods. Along with previously published 61 positive samples [14], two apple and 11 C. picta samples were found to be infected with the phytoplasma in additional monitoring in 2016 (Table A1.). None of the tested C. melanoneura harbored the phytoplasma. Out of 74 positive samples that were included in MLST, all four markers were successfully typed for 64 (Table A2.).
aceF, pnp, imp and secY Genotyping
For the aceF fragment of approximately 790 bp in length, five of the eight previously described genotypes [10,28] were identified in Croatian samples, along with a new one labeled A27 (Table A2. and Figure B1.). The predominant genotype, A13, was present in 67% of the analyzed samples. The unrooted phylogenetic tree obtained through the analysis of the full-length (1260 bp) aceF shows strong support for A27 genotype divergence (Figure B5.).
Analysis of a 512 bp pnp gene fragment revealed seven distinct genotypes (Figure B2.). Of the five previously described genotypes [10], only P9 and P10 are present in our samples, with the P10 being the prevalent one in almost 70% of the samples (Table A2.). Two new genotypes were identified, P17 and P18, with P18 found in only one apple sample (Figure B2. and Table A2.). Unrooted phylogenetic tree based on the analysis of the complete pnp gene expectedly showed greater diversity because of the more than four times longer sequence (Figure B6.).
Out of ten already described imp genotypes [10,28], five were found in Croatia together with six new ones (I36 – I41). It is expectedly the most variable of the four typed genes (Figure B3. and Table A2.). The prevalent genotype, I21, was found in almost 50% of the samples (Table A2.). BLAST search of the new genotype sequences in GenBank nucleotide database revealed imp sequences identical to the new genotypes (Table A4.) that had not been labeled according to this MLST system [29].
Analysis of nucleotide sequences of the secY gene fragment resulted in a phylogenetic tree separating into 12 genotypes (Figure B4.). In addition to seven known [10,28], five new genotypes (S17 – S21) were identified in the samples from this study (Table A2.). The genotype S12 was dominant and found in over 67% of the samples. Within the variable region of the secY gene, compared to the reference sequence of the strain AT ‘Ca. P. mali’ with a length of 1245 bp, there was either an insertion of three nucleotides (S17, sample 392) or deletions of multiple nucleotides (genotypes S10 and S12). As a result, the coding region length varies from 1227 bp (S11, S12, S19, and S18) to 1248 bp (S17). The secY gene fragment covers the most variable region (positions 397–438/1248 bp) and only a small number of significant nucleotide changes are found outside this section. Therefore, the unrooted phylogenetic tree based on the 1227 - 1248 bp secY gene sequence largely supports the tree obtained from the phylogenetic analysis of the shorter fragment (Figure B7.).
Erroneously, labels I31 and S13 that had already been used for ‘Ca. P. pyri’ imp and secY genotypes from Azerbaijan [10] were used for the new genotypes in a more recent study [28]. Therefore, we propose correcting the genotype labels to avoid ambiguity, starting with the first available I35 and S16, respectively. These proposed labels are used throughout figures and tables in this study (Table A2. and A4., Figures B3. and B4.).
Sequence Types (STs) and Analysis of Concatenated Sequences
The obtained profile of genotypes combination for each sample was grouped to a sequence type (ST) and labeled in order of their frequency (Table A2.). There are 20 different sequence types, with ST1 (A13–P10–I23–S12) represented in 25% of the Croatian samples (Figure 1.). The ST4 (A15-P17-I21-S10) contains the new genotype P17 and is present in more than 10% of the samples. The ST2 profile (A13–P10–I21–S12) is the most common in C. picta, present in 50% of the insect samples (Figure 1., Table A2.). Ten out of 20 STs are represented in a single sample, mainly due to imp and secY variability, although ST11 and ST14 harbor known genotypes that are not frequent in Croatia (Figure 1., Table A2.).
Concatenating all four gene sequences allowed simultaneous and comprehensive analysis of the ‘Ca. P. mali’ variability targeted in MLST. Analysis of concatenated sequences resulted in a finer resolution because all nucleotide differences are considered. In addition to interconnectedness shown in the phylogenetic network, the number of strains of the same profile is also considered and graphically represented by the node size (Figure 2.). The frequency of each profile is used to select the founder ST for each cluster. Therefore, ST1 is considered as an overall founder sequence type, and five clusters are suggested by analyzing this dataset (Figure 2.). Unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp and secY sequences (Figure 3.) largely supports the results represented in the network (Figure 2.), with the presence of four main clusters.
4. Discussion
Following the results of systematic survey of apple orchards, efforts were made to characterize Croatian 'Ca. P. mali' strains in more detail in order to get a better insight in the epidemiology of this important plant pathogen. However, since the trees exhibiting symptoms reminiscent of apple proliferation were preferentially sampled, the percentage of infected apple trees in this study does not necessarily represent the overall apple infection rate in the country. The frequency of naturally infected psyllids, C. picta and C. melanoneura, as well as their ability to transmit the disease, differs significantly in countries and regions where the disease is present and well-studied [16,30]. In Croatia, C. melanoneura populations are more numerous than C. picta, and yet C. picta adults are present in apple orchards for a longer period before migration to overwintering coniferous hosts. This possibly increases their potential for phytoplasma acquisition [17]. Transovarial transmission of the phytoplasma to offspring has been proven for C. picta [31], thus bypassing the process of acquisition from the infected plant and multiplication in the vector. With more than 200 psyllid samples tested, we can positively conclude that the major AP phytoplasma vector in Croatia is C. picta. This vector infection pattern is consistent with research results from Germany, Switzerland, and France [32].
Primers that were designed in this study (Table 1.) efficiently amplified targeted fragments of all four genes for most samples. New genotypes were recorded for all of the four genes used in MLST. For aceF, the dominant genotype was A13 (Figure B1. And Table A2.) which corresponds to findings in isolates from France, Italy, and Germany [10]. For the 'Ca. P. prunorum', two aceF genotypes, A6 and A8 [10], are associated with previously described hypovirulent phytoplasma strains [33]. However, since the biological properties of the Croatian 'Ca. P. mali' strains are not known, it is currently not possible to draw this type of conclusion from our results. Phylogenetic analysis of the pnp gene fragment revealed two new genotypes - P17 and P18 (Figure B2. and Table A2.). Genotype P17 was fairly frequent and is present in eight samples, while P18 was recorded in only one apple sample. The prevalent genotype P10 was present in almost 70% of the samples. From the results of genotyping European isolates, the dominant genotype was P11 [10], which is not present in Croatia. When the entire coding region was analyzed, samples that grouped within genotypes P9 and P10 separated into distinct branches, which is not surprising given that the complete coding region is four times longer than the fragment usually used in genotyping [10]. The imp gene was expectedly the most variable of the four typed genes (Figure B3., Table A2.). This gene encodes the immunodominant membrane protein that is located on the cell surface and is thus exposed to positive selective pressure. Due to its role in interactions with both the apple and the insect vector, it is relevant in the molecular pathology of the disease [34,35]. Six novel genotypes (I36 – I41) were identified in Croatia (Figure B3. and Table A2.). This variability is common and consistent with studies of immunodominant membrane proteins from other phytoplasma species [35,36,37,38]. As many as 87 out of approximately 507 nucleotide positions in the imp gene sequence are variable, with the most significant difference being deletions within the sequence, causing the length to vary from 498 to 507 bp. The dominant genotype is I21, the same as at the European level [10,28]. The three genotypes identified in the largest number of samples (I21, I22, and I23) group in the same cluster (Figure B3.). Interestingly, all new genotypes were found exclusively in apple samples from two locations less than 5 kilometers apart (Table A2.), while in C. picta only I21 and I23 genotypes were present (Table A2.). Changes in the imp gene in apple samples probably bring advantages, such as evading the host immune response [34]. In C. picta phytoplasma must pass through various tissues and multiply to a sufficient concentration for successful transmission from salivary glands to the host [35,39,40]. This suggests that these two genotypes might be optimal for this translocation. The secY gene, which encodes the central subunit of the membrane transport system, has long been used as an additional phylogenetic marker for better differentiation of closely related phytoplasmas within the same 16S rRNA group [41]. Phylogenetic analyses of the obtained sequences show higher than expected variability (Figure B4. and Table A2.). Such variability of the secY gene is not straightforward and was not expected for this housekeeping gene, especially since MLST of the European fruit tree phytoplasma isolates had shown that this gene is less variable than aceF and pnp [10]. The previously described genotype S12, which was dominant in 'Ca. P. mali' isolates from five European countries [10], is present in by far the largest number of Croatian samples too (almost 70%) (Figure 1., Table A2.). In this study, five new genotypes (S17 – S21) were described. The same as with the imp gene, all new genotypes are present exclusively in apple samples from two locations in north-western part of Croatia (Table A2.). Introducing and naming new genotypes does not come without difficulties and we encountered inadvertently introduced erroneous labels for newly recorded genotypes for both imp and secY [28]. To ensure labeling accuracy moving forward, we propose reassigning the labels to the first available ones, namely I35 and S16, respectively.
Results of MLST revealed new genotypes and great diversity in two close-by locations in the north-western part of Croatia, Donji Mihaljevec and Sveta Marija (Table A2.). Apple has been traditionally grown in this region for a long time, and there are records indicating that 'Ca. P. mali' has been present here at least since the 1980s when symptoms were first observed [12]. This long-lasting coevolution of the host and pathogen is one possible explanation for this diversification. Interestingly, this diversity is not represented in C. picta samples that harbor only one new genotype, P17 (Table A2.). Results of genotyping individual genes were combined to form 20 different sequence types (Figure 1., Table A2.). ST1 (A13–P10–I23–S12) is the most frequent one and is represented in 25% of the Croatian samples (Figure 1., Table A2.). The profile ST2 (A13–P10–I21–S12) is the most common in C. picta, present in more than 50% of the insect samples (Figure 1., Table A2.). Two sequence types differ only in imp genotype (I23 vs. I21). Since ST2 is dominant in psyllid samples, it can be hypothesized that I21 is significant in vector colonization and transmission. This is consistent with findings in Slovenia, where only the novel genotype I35 was found in C. melanoneura sample [28]. Ten out of 20 STs are represented in a single sample, mainly due to imp and secY variability (Figure 1., Table A2.).
Analysis of concatenated sequences suggested that the most frequent ST1 can be considered as the founder sequence type (Figure 1, and ). Unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp and secY sequences (Figure 3) largely supports the results represented in the network (Figure 2), with four main clusters. With only 18 previously fully genotyped isolates from Austria, Switzerland, Germany, France, Italy, and Romania [10] and 64 as a part of this study, the dataset was still insufficient to create clear conclusions on the geographic distribution, origin, and virulence of individual strains. Nevertheless, the results of this study contribute to understanding the prevalence of individual genotypes in relation to the plant or insect host.
To improve and expand genotyping for phytoplasmas, it would be essential to agree on a MLST system for genes that have proven to be very informative, like hflB [42], SAP11 [43] or ribosomal protein (rp) genes [44]. The genome of 'Ca. P. mali' has been fully sequenced and annotated [8], enabling the search for additional, epidemiologically relevant and evolutionary interesting genes. Furthermore, the creation of a database with representative genotype sequences according to uniform criteria and protocols would allow for the structured comparison of an increasing number of isolates from across Europe. This would advance future applied research, such as the selection of apple cultivars resistant or tolerant to apple proliferation disease. The use of specific primers that allow amplification of the entire coding region for all four genes provided deeper insights into molecular characterization and may serve as a foundation for further in silico research of protein structure. Genotyping results are useful for understanding epidemiology of phytoplasmas and this study is a step toward creating a standardized MLST system for 'Ca. P. mali'.
5. Conclusions
New genotypes are described in Croatian apple and psyllid samples for all four genes used in MLST: one for aceF, two for pnp, six for imp and five for secY genes. All new imp and secY genotypes are found exclusively in apple samples. Twenty different sequence types (ST) are described, with 50% represented with only one sample mainly due to imp and secY variability. Dominant ST1 was found in 25% of the samples, yet in C. picta samples ST2 was present in more than half of the samples. Specific primers enabled amplification of full coding region for all four genes, improving molecular characterization and supporting future in silico protein studies. The genotyping results provide valuable insights into the epidemiology of ‘Ca. P. mali’, with the dominance of ST1 highlighting its potential role in disease persistence and spread. This study contributes to the development of an expanded and improved MLST system for more precise characterization of phytoplasma strains.
Supplementary Materials
The following supporting information can be downloaded at: Preprints.org, Appendix A: Tables A1.–A4.; Appendix B: Figures B1.–B7.
Author Contributions
Conceptualization, M.Š.M, I.K. and D.Š.; methodology M.Š.M; formal analysis, I.K. and M.Š.M.; investigation I.K., M.Š.M. and J.P.; writing—original draft preparation, I.K.; writing—review and editing, I.K., D.Š., M.Š.M. and J.P.; visualization, I.K. and M.Š.M.; supervision, M.Š.M. and D.Š.; funding acquisition, I.K., M.Š.M and D.Š. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Croatian Ministry of Agriculture to I.K. and the University of Zagreb to M.Š.M. and D.Š.
Data Availability Statement
The consensus sequences presented in this study are openly available in NCBI GenBank at (https://www.ncbi.nlm.nih.gov/nucleotide/) with accession numbers listed in table A3.
Acknowledgments
Authors wish to thank Drs. Dario Ivić for collecting apple samples and Željko Budinšćak for psyllid collection and identification. We appreciate the assistance of colleagues from the Croatian Phytosanitary Inspection and Agricultural Extension Service in sampling and liaising with farmers. We are especially grateful to Dr. Xavier Foissac for his valuable advice and continued support throughout the study.
The authors wish to honor the memory of our esteemed colleague and friend Jean-Luc Danet whose dedication to phytoplasma research and collaborative spirit continue to inspire our work.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| AP | Apple proliferation |
| MLST | Multi-locus sequence typing |
| ST | Sequence type |
Appendix A. Tables
Table A1. Overall number of tested, positive and typed samples from previous (Križanac et al. 2017) and this study.
Table A2. Genotyping results for all four loci (aceF, pnp, imp and secY), sequence types (ST) and their respective labels. Assigned new genotype labels from this study and sequence types represented with a single sample are in bold and shaded. Regions in Croatia are labeled CW (continental west), CE (continental east) and AD (Adriatic).
Table A3. GenBank accession numbers of representative sequences from this study. Previously published sequences (Križanac et al. 2017) are shaded and new genotypes are in bold.
Table A4. Sequences from GenBank used in this study. Reference sequences for ‘Ca. P. mali’ aceF, pnp, imp and secY genotypes are in bold and shaded (Danet et al. 2011). Sequences blasted from GenBank blast search (https://blast.ncbi.nlm.nih.gov/Blast.cgi) identical to genotypes assigned in this study are underlined (Dermastia et al. 2018; Fránová et al. 2019; Kube et al. 2008; Seemüller et al. 2010).
Appendix B. Figures
Figure B1. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of partial aceF gene sequences representative for each genotype (Tables A.3. and A.4.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Genotypes present in Croatia are marked with an asterisk* and the new genotype, A27, is marked with a red triangle.
Figure B2. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of partial pnp gene sequences representative for each genotype (Tables A.3. and A.4.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Genotypes present in Croatia are marked with an asterisk* and new genotypes, P17 and P18, are marked with red triangles.
Figure B3. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of partial imp gene sequences representative for each genotype (Tables A.3. and A.4.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Genotypes present in Croatia are marked with an asterisk* and new genotypes, I36 – I41, are marked with red triangles.
Figure B4. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of partial secY gene sequences representative for each genotype (Tables A.3. and A.4.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Genotypes present in Croatia are marked with an asterisk* and new genotypes, S17 – S21, are marked with red triangles.
Figure B5. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of complete aceF gene sequences (Tables A2. and A3.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Samples belonging to a novel A27 genotype are marked with red triangles.
Figure B6. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of complete pnp gene sequences (Table A2. and A3.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Samples belonging to novel P17 and P18 genotypes are marked with red triangles.
Figure B7. Unrooted phylogenetic tree inferred using the maximum parsimony analysis of complete secY gene sequences (Tables A2. and A3.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Samples belonging to novel S17 – S21 genotypes are marked with red triangles.
References
- Marcone, C. Molecular Biology and Pathogenicity of Phytoplasmas. Ann. Appl. Biol 2014, 165, 199–221. [Google Scholar] [CrossRef]
- Lee, I.-M.; Davis, R.E.; Gundersen-Rindal, D.E. Phytoplasma: Phytopathogenic Mollicutes. Annu. Rev. Microbiol. 2000, 54, 221–255. [Google Scholar] [CrossRef]
- IRPCM ‘Candidatus Phytoplasma’, a Taxon for the Wall-Less, Non-Helical Prokaryotes That Colonize Plant Phloem and Insects. Int. J. Syst. Evol. Microbiol. 2004, 54, 1243–1255. [CrossRef]
- Wei, W.; Zhao, Y. Phytoplasma Taxonomy: Nomenclature, Classification, and Identification. Biology (Basel) 2022, 11, 1119. [Google Scholar] [CrossRef]
- Kirdat, K.; Tiwarekar, B.; Sathe, S.; Yadav, A. From Sequences to Species: Charting the Phytoplasma Classification and Taxonomy in the Era of Taxogenomics. Front. Microbiol. 2023, 14. [Google Scholar] [CrossRef]
- Oshima, K.; Kakizawa, S.; Nishigawa, H.; Jung, H.-Y.; Wei, W.; Suzuki, S.; Arashida, R.; Nakata, D.; Miyata, S.; Ugaki, M.; Namba, S. Reductive Evolution Suggested from the Complete Genome Sequence of a Plant-Pathogenic Phytoplasma. Nat. Genet. 2004, 36, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Seemüller, E.; Schneider, B. ‘Candidatus Phytoplasma mali’, ‘Candidatus Phytoplasma pyri’ and ‘Candidatus Phytoplasma prunorum’, the Causal Agents of Apple Proliferation, Pear Decline and European Stone Fruit Yellows, respectively. Int. J. Syst. Evol. Microbiol. 2004, 54, 1217–1226. [Google Scholar] [CrossRef]
- Kube, M.; Schneider, B.; Kuhl, H.; Dandekar, T.; Heitmann, K.; Migdoll, A.M.; Reinhardt, R.; Seemüller, E. The Linear Chromosome of the Plant-Pathogenic Mycoplasma “Candidatus Phytoplasma mali. ” BMC Genomics 2008, 9, 306. [Google Scholar] [CrossRef] [PubMed]
- Urwin, R.; Maiden, M.C.J. Multi-Locus Sequence Typing: A Tool for Global Epidemiology. Trends Microbiol. 2003, 11, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Danet, J.L.; Balakishiyeva, G.; Cimerman, A.; Sauvion, N.; Marie-Jeanne, V.; Labonne, G.; Laviňa, A.; Batlle, A.; Križanac, I.; Škorić, D.; et al. Multilocus Sequence Analysis Reveals the Genetic Diversity of European Fruit Tree Phytoplasmas and Supports the Existence of Inter-Species Recombination. Microbiology (NY) 2011, 157, 438–450. [Google Scholar] [CrossRef]
- Janik, K.; Barthel, D.; Oppedisano, T.; Anfora, G.; Schuler, H. Apple Proliferation—A Joint Review. Janik, K., Barthel, D., Oppedisano, T., Anfora, G., Eds.; Laimburg Research Centre: Stadio - Laimburg, Italy, 2020; ISBN 9788878430549. [Google Scholar]
- Šarić, A.; Cvjetković, B. Mycoplasma-like Organism Associated with Apple Proliferation and Pear Decline – like Disease of Pears. Agric. Conspec. Sci. 1985, 68, 61–65. [Google Scholar]
- Križanac, I.; Mikec, I.; Budinščak, Z.; Šeruga Musić, M.; Krajačić, M.; Škorić, D. Pomaceous Fruit Tree Phytoplasmas and Their Potential Vectors in Croatia. Acta Hortic. 2008, 477–482. [Google Scholar] [CrossRef]
- Križanac, I.; Plavec, J.; Budinšćak, Ž.; Ivić, D.; Škorić, D.; Šeruga Musić, M. Apple Proliferation Disease in Croatian Orchards: A Molecular Characterization of “Candidatus Phytoplasma mali. ” J. Plant Pathol. 2017, 99, 95–101. [Google Scholar]
- Frisinghelli, C.; Delaiti, L.; Grando, M.S.; Forti, D.; Vindimian, M.E. Cacopsylla Costalis (Flor 1861), as a Vector of Apple Proliferation in Trentino. J. Phytopath. 2000, 148, 425–431. [Google Scholar] [CrossRef]
- Tedeschi, R.; Bosco, D.; Alma, A. Population Dynamics of Cacopsylla melanoneura; (Homoptera: Psyllidae), a Vector of Apple Proliferation Phytoplasma in Northwestern Italy. J. Econ. Entomol. 2002, 95, 544–551. [Google Scholar] [CrossRef]
- Budinšćak, Ž. Lisne Buhe – Vektori Fitoplazme Proliferacije Jabuke u Hrvatskoj. GBZ 2021, 21, 587–603. [Google Scholar]
- Baric, S.; Dalla-Via, J. A New Approach to Apple Proliferation Detection: A Highly Sensitive Real-Time PCR Assay. J. Microbiol. Methods 2004, 57, 135–145. [Google Scholar] [CrossRef]
- Deng, S.; Hiruki, C. Amplification of 16S RRNA Genes from Culturable and Nonculturable Mollicutes. J. Microbiol. Methods 1991, 14, 53–61. [Google Scholar] [CrossRef]
- Schneider, B.; Seemueller, E.; Smart, C.D.; Kirkpatrick, B.C. Phylogenetic Classification of Plant Pathogenic Mycoplasma-like Organisms or Phytoplasmas. In Molecular and Diagnostic Procedures in Mycoplasmology; Razin, S. and T.J.G., Ed.; Elsevier, 1995; Vol. Vol. 1, pp. 369–380.
- Lee, I.-M.; Gundersen, D.E.; Hammond, R.W.; Davis, R.E. Use of Mycoplasmalike Organism (MLO) Group-Specific Oligonucleotide Primers for Nested-PCR Assays to Detect Mixed-MLO Infections in a Single Host Plant. Phytopathology 1994, 84, 559–566. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An Integrated and Extendable Desktop Software Platform for the Organization and Analysis of Sequence Data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef]
- Thompson, J. The CLUSTAL_X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Analysis Tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Francisco, A.P.; Vaz, C.; Monteiro, P.T.; Melo-Cristino, J.; Ramirez, M.; Carriço, J.A. PHYLOViZ: Phylogenetic Inference and Data Visualization for Sequence Based Typing Methods. BMC Bioinformatics 2012, 13, 87. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. Confidence Limits on Phylogenies: An Approach Using the Bootstrap. Evolution (NY) 1985, 39, 783. [Google Scholar] [CrossRef]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as Designed by Its Users. Nucleic Acids Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef]
- Dermastia, M.; Dolanc, D.; Mlinar, P.; Mehle, N. Molecular Diversity of ‘Candidatus Phytoplasma mali’ and ‘Ca. P. prunorum’ in Orchards in Slovenia. Eur. J. Plant. Pathol. 2018, 152, 791–800. [Google Scholar] [CrossRef]
- Seemüller, E.; Kiss, E.; Sule, S.; Schneider, B. Multiple Infection of Apple Trees by Distinct Strains of ‘Candidatus Phytoplasma mali’ and Its Pathological Relevance. Phytopathology 2010, 100, 863–870. [Google Scholar] [CrossRef]
- Mayer, C.J.; Jarausch, B.; Jarausch, W.; Jelkmann, W.; Vilcinskas, A.; Gross, J. Cacopsylla melanoneura has no Relevance as Vector of Apple Proliferation in Germany. Phytopathology 2009, 99, 729–738. [Google Scholar] [CrossRef]
- Mittelberger, C.; Obkircher, L.; Oettl, S.; Oppedisano, T.; Pedrazzoli, F.; Panassiti, B.; Kerschbamer, C.; Anfora, G.; Janik, K. The Insect Vector Cacopsylla picta Vertically Transmits the Bacterium ‘Candidatus Phytoplasma mali’ to Its Progeny. Plant Pathol. 2017, 66, 1015–1021. [Google Scholar] [CrossRef]
- Jarausch, B.; Schwind, N.; Fuchs, A.; Jarausch, W. Characteristics of the Spread of Apple Proliferation by Its Vector Cacopsylla picta. Phytopathology 2011, 101, 1471–1480. [Google Scholar] [CrossRef]
- Kison, H.; Seemüller, E. Differences in Strain Virulence of the European Stone Fruit Yellows Phytoplasma and Susceptibility of Stone Fruit Trees on Various Rootstocks to This Pathogen. J. Phytopathol. 2001, 149, 533–541. [Google Scholar] [CrossRef]
- Konnerth, A.; Krczal, G.; Boonrod, K. Immunodominant Membrane Proteins of Phytoplasmas. Microbiology (NY) 2016, 162, 1267–1273. [Google Scholar] [CrossRef]
- Kakizawa, S.; Oshima, K.; Namba, S. Diversity and Functional Importance of Phytoplasma Membrane Proteins. Trends Microbiol. 2006, 14, 254–256. [Google Scholar] [CrossRef] [PubMed]
- Barbara, D.J.; Morton, A.; Clark, M.F.; Davies, D.L. Immunodominant Membrane Proteins from Two Phytoplasmas in the Aster Yellows Clade (Chlorante Aster Yellows and Clover Phyllody) Are Highly Divergent in the Major Hydrophilic Region. Microbiology (NY) 2002, 148, 157–167. [Google Scholar] [CrossRef]
- Bohunická, M.; Valentová, L.; Suchá, J.; Nečas, T.; Eichmeier, A.; Kiss, T.; Cmejla, R. Identification of 17 ‘Candidatus Phytoplasma pyri’ Genotypes Based on the Diversity of the Imp Gene Sequence. Plant Pathol. 2018, 67, 971–977. [Google Scholar] [CrossRef]
- Alessio, F.I.; Bongiorno, V.A.; Marcone, C.; Conci, L.R.; Fernandez, F.D. Genetic Diversity in Phytoplasmas from X-Disease Group Based in Analysis of IdpA and Imp Genes. Microorganisms 2025, 13, 1170. [Google Scholar] [CrossRef]
- Weintraub, P.G.; Beanland, L. Insect Vectors of Phytoplasmas. Annu. Rev. Entomol. 2006, 51, 91–111. [Google Scholar] [CrossRef]
- Rashidi, M.; Galetto, L.; Bosco, D.; Bulgarelli, A.; Vallino, M.; Veratti, F.; Marzachì, C. Role of the Major Antigenic Membrane Protein in Phytoplasma Transmission by Two Insect Vector Species. BMC Microbiol. 2015, 15, 193. [Google Scholar] [CrossRef]
- Lee, I.-M.; Bottner-Parker, K.D.; Zhao, Y.; Davis, R.E.; Harrison, N.A. Phylogenetic Analysis and Delineation of Phytoplasmas Based on SecY Gene Sequences. Int. J. Syst. Evol. Microbiol. 2010, 60, 2887–2897. [Google Scholar] [CrossRef]
- Seemüller, E.; Sule, S.; Kube, M.; Jelkmann, W.; Schneider, B. The AAA+ ATPases and HflB/FtsH Proteases of ‘Candidatus Phytoplasma mali’: Phylogenetic Diversity, Membrane Topology, and Relationship to Strain Virulence. MPMI 2013, 26, 367–376. [Google Scholar] [CrossRef]
- Bai, X.; Correa, V.R.; Toruño, T.Y.; Ammar, E.-D.; Kamoun, S.; Hogenhout, S.A. AY-WB Phytoplasma Secretes a Protein That Targets Plant Cell Nuclei. Mol. Plant-Microbe Interact. 2009, 22, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Lee, I.-M.; Bottner, K.D.; Zhao, Y.; Botti, S.; Bertaccini, A.; Harrison, N.A.; Carraro, L.; Marcone, C.; Khan, A.J.; et al. Ribosomal Protein Gene-Based Phylogeny for Finer Differentiation and Classification of Phytoplasmas. Int. J. Syst. Evol. Microbiol. 2007, 57, 2037–2051. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Prevalence of ‘Ca. P. mali’ sequence types (ST) in Croatian samples. MLST analysis based on the aceF, pnp, imp and secY genotyping revealed 20 different sequence types. ST1 (A13–P10–I23–S12) is prevalent and ST10 – ST20 are represented in a single sample.
Figure 1.
Prevalence of ‘Ca. P. mali’ sequence types (ST) in Croatian samples. MLST analysis based on the aceF, pnp, imp and secY genotyping revealed 20 different sequence types. ST1 (A13–P10–I23–S12) is prevalent and ST10 – ST20 are represented in a single sample.
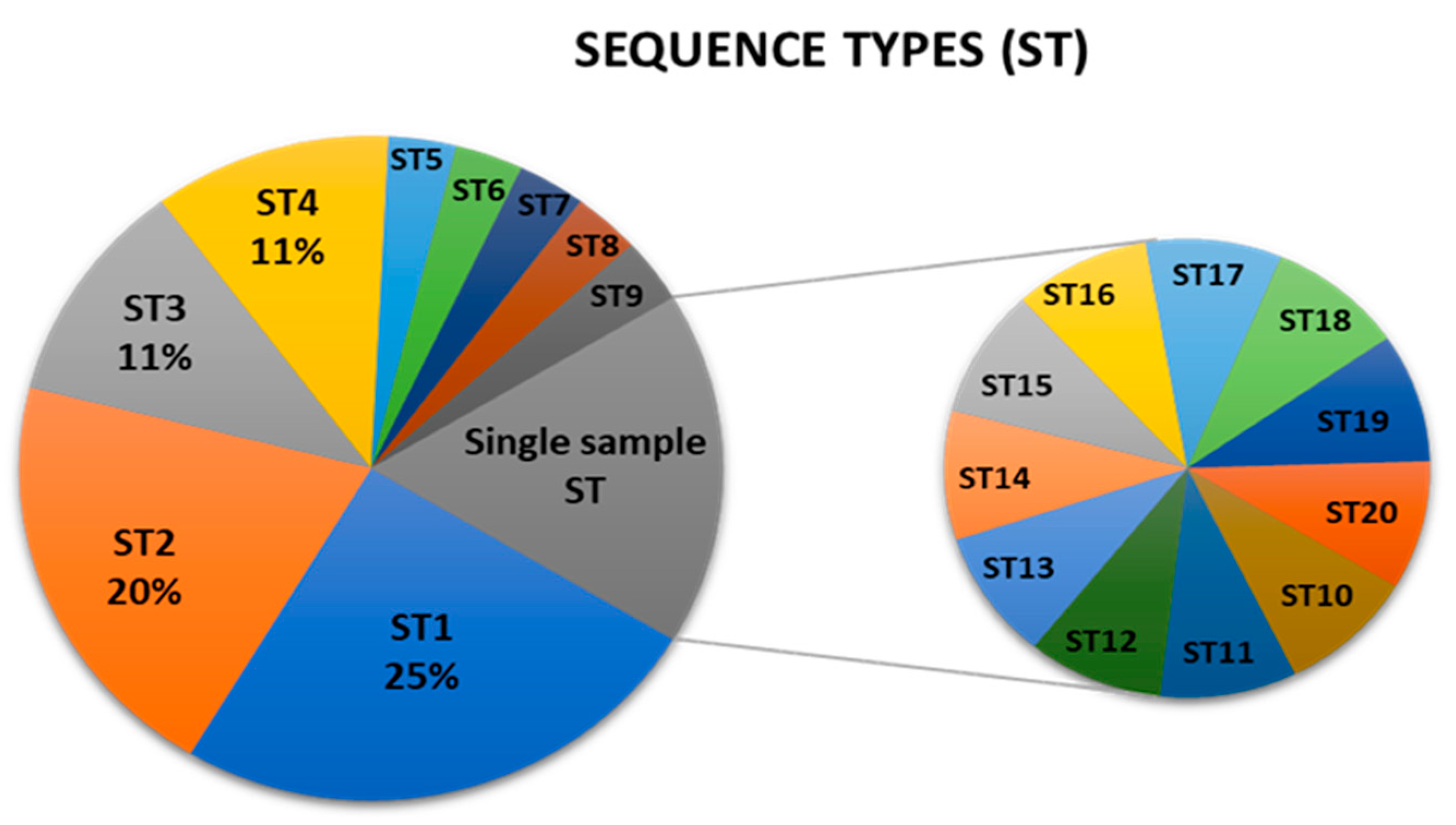
Figure 2.
Graphical representation of concatenated sequences (ST) dataset analysis using Phyloviz 2.0. [24]. Size of a node represents the number of samples within the ST. Founder STs for each cluster are in green. Along with the sequence type label, a representative sample for the ST is shown for each node.
Figure 2.
Graphical representation of concatenated sequences (ST) dataset analysis using Phyloviz 2.0. [24]. Size of a node represents the number of samples within the ST. Founder STs for each cluster are in green. Along with the sequence type label, a representative sample for the ST is shown for each node.
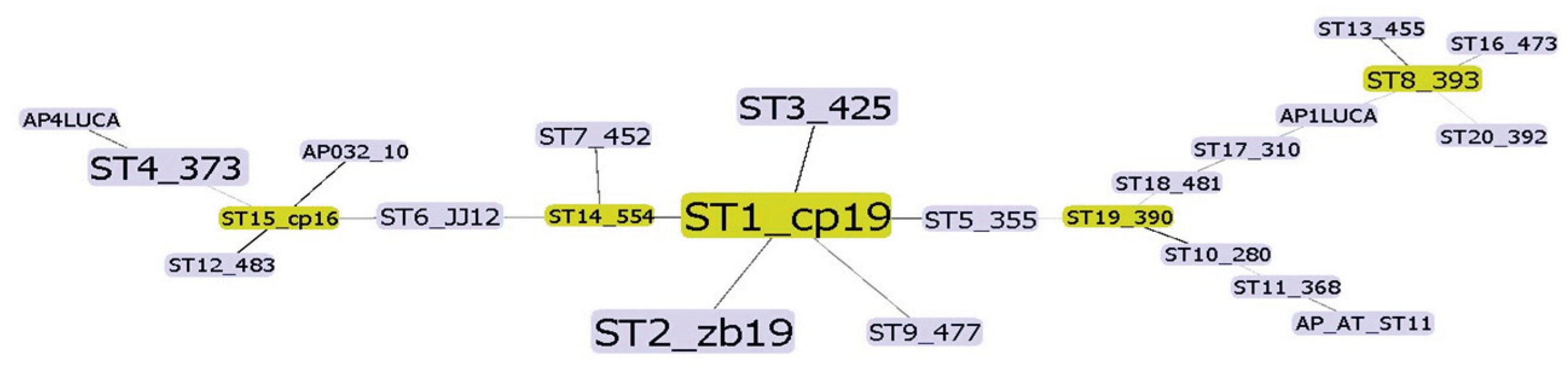
Figure 3.
Unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp and secY sequences (Table A2). Numbers in brackets represent the number of samples for each sequence type (ST). Samples from previous study with all four available sequences are included in the analysis (Table A3.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Prevalent sequence type in overall samples, ST1, is marked with a red dot. Prevalent sequence type in Cacopsylla picta samples, ST2, is marked with a blue dot.
Figure 3.
Unrooted phylogenetic tree inferred using the maximum parsimony analysis of concatenated aceF, pnp, imp and secY sequences (Table A2). Numbers in brackets represent the number of samples for each sequence type (ST). Samples from previous study with all four available sequences are included in the analysis (Table A3.). The percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (500 replicates) are shown above the branches. Prevalent sequence type in overall samples, ST1, is marked with a red dot. Prevalent sequence type in Cacopsylla picta samples, ST2, is marked with a blue dot.
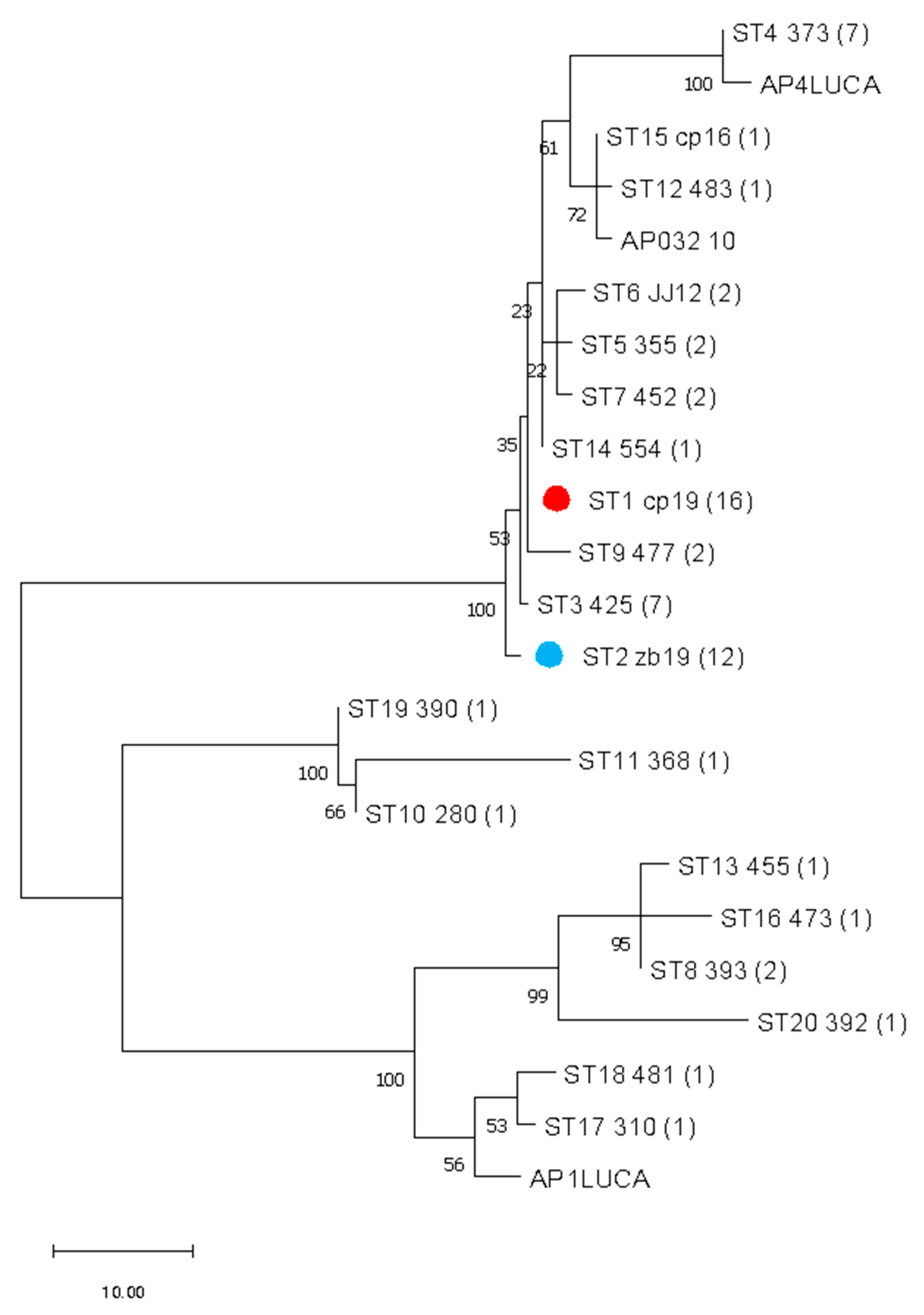

Table 1.
Specific primers amplifying entire coding regions of 'Ca. P. mali' aceF*, pnp, imp and secY (this study) genes used in MLST. Both amplicon size and coding region lengths are variable.
Table 1.
Specific primers amplifying entire coding regions of 'Ca. P. mali' aceF*, pnp, imp and secY (this study) genes used in MLST. Both amplicon size and coding region lengths are variable.
| Gene | Method | Primer name | Primer sequence (5’-3’) | Amplicon size (bp) | coding region (bp) |
|---|---|---|---|---|---|
| aceF* | PCR/ sequencing |
acoB_F | CTGCTCCATCTAGAGTTAC | 1600 | 1260 |
| lpd_R0m | GCTAGCTTTTATAGCAGCT | ||||
| pnp | PCR | pnpF | GCTCAGTTGGTAGAGCAT | ||
| pnpR | AGACACAAACACTACATACAT | ||||
| nPCR/ sequencing |
pnpF1 | GGTAGAGCATCTGACTGTT | 2400 | 2187 | |
| pnpR1 | CCCTCGATCGCCTTCTAT | ||||
| imp | PCR/ sequencing |
impF | CGTAGAACCAAATGATAAAG | 1000 | 507 |
| impR | GACATAGACATCGTTACGA | ||||
| secY | PCR | secYF1 | CAGAGAATTCCTAAACGTG | ||
| secYR | GAATACCGTGAACAACTAC | ||||
| nPCR/ sequencing |
secYF2 | GTTAATCTAGGTGCTTTAGA | 1800 | 1227-1248 | |
| secYR2 | GTGAACAACTACTTCATTAAC | ||||
| sequencing | secYF3 | AGTTGGTGGCAATATTGA | |||
| secYR3 | CTACTTCATTAACAGAAGTACA |
*Previously published primers [14].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



