Submitted:
16 August 2025
Posted:
19 August 2025
You are already at the latest version
Abstract
Cellular senescence, a state of stable cell cycle arrest accompanied by a complex senescence-associated secretory phenotype (SASP), is a fundamental biological process implicated as a key driver of lung aging and lung age-related diseases (LARDs). This review provides a comprehensive overview of the rapidly evolving field of senotyping based on cellular heterogeneity in lung development and senotherapeutics in the field of lung biology and disease e.g. stages of COPD and IPF, which aim to mitigate the detrimental effects of senescent cell (SnC) accumulation. It also delves into the molecular mechanisms driving senescence and SASP production, highlighting pathways such as p53/p21, p16INK4a/RB, mTOR, and p38 MAPK as therapeutic targets. The involvement of various novel SASP proteins, such as GDP15, cytokines/chemokines, growth factors, and DNA damage response proteins. It also outlines two main therapeutic approaches: senolytics, which selectively trigger apoptosis in senescent cells (SnCs), and senomorphics (also known as senostatics), which mitigate the detrimental effects of the SASP without necessarily removing the senescent cells. It discusses various classes of senolytic and senomorphic agents and their deliveries, including natural products (e.g., quercetin, fisetin, resveratrol), repurposed drugs (e.g., dasatinib, navitoclax, metformin, rapamycin), and innovative approaches like HSP90 inhibitors, senolytic CAR-T cells, Antibody drug conjugate and galactose-modified prodrugs. Preclinical evidence and emerging data from early-phase human clinical trials, particularly with the combinatorial approach of Dasatinib & Quercetin (D+Q) and fisetin, demonstrate the therapeutic promise of these interventions in improving tissue function, alleviating LARDs, and potentially extending healthspan. The review also highlighted significant challenges, including SnC heterogeneity, immunosenescence, drug delivery, target specificity, long-term safety, and the need for robust biomarkers. Future perspectives, such as advanced delivery systems, and combination therapies, are considered critical for translating the potential of senotherapeutics into effective clinical applications for age-related pulmonary diseases/conditions.
Keywords:
cellular senescence
; senolytics
; senomorphics
; aging
; age related diseases
; senotherapeutics
; cellular heterogeneity
1. Introduction
Cellular senescence is defined as a stable and often irreversible arrest of the cell cycle, induced by a variety of stressors such as telomere attrition (replicative senescence), DNA damage, oncogenic signaling, and oxidative stress [1,2,3]. It is different from quiescence state, which is a reversible growth arrest and from terminal differentiation, which involves the acquisition of specialized cellular functions [1,2]. Senescent cells exhibit several hallmark features, including the upregulation of cyclin-dependent kinase inhibitors such as p16Ink4a and p21Cip1/Waf1, resistance to apoptosis, altered nuclear morphology, and increased activity of senescence-associated β-galactosidase (SA-β-gal) [1,2,3,4,5,6]. A central feature of many senescent cells is the development of a complex secretory profile known as the senescence-associated secretory phenotype (SASP) [7,8]. The SASP includes a broad range of secreted factors, such as pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines, growth factors, and matrix metalloproteinases (MMPs) [2,3,7,9]. Its composition and functional consequences are highly context-dependent, determined by the senescence-inducing stimulus, cell type heterogeneity, and local tissue microenvironment [9,10].
Cellular senescence is a heterogeneous process encompassing multiple distinct subtypes, each initiated by specific stimuli and governed by unique molecular mechanisms. Replicative senescence (RS) represents a classical form of permanent cell cycle arrest that arises after repeated cellular divisions. This process is primarily driven by progressive telomere shortening, which, upon reaching a critical length, is perceived as persistent DNA damage. The subsequent activation of the DNA damage response (DDR) engages key tumor suppressor pathways, particularly the p53/ p21Cip1/Waf1and p16Ink4a /retinoblastoma (Rb) axes, resulting in stable growth arrest and adoption of the senescent phenotype [11,12]. This mechanism is particularly relevant in the aging lung, where the accumulation of senescent alveolar epithelial cells (AECs) disrupts alveolar integrity and impairs tissue repair [13].
In contrast, stress-induced premature senescence (SIPS) arises independently of telomere attrition. It is driven by exogenous stressors such as cigarette smoke, reactive oxygen species (ROS), and persistent inflammation that are characteristic of chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) and idiopathic pulmonary fibrosis (IPF) [14,15,16]. Oncogene-induced senescence (OIS) constitutes another critical subtype, driven by hyperactivation of oncogenes, such as RAS or BRAF. This leads to replication stress and a robust DDR, which initially serves as a tumor-suppressive barrier [17]. However, in chronic contexts, the associated SASP may paradoxically facilitate tumor progression, particularly in the early stages of lung adenocarcinoma [7,18].
Therapy-induced senescence (TIS) has gained increasing recognition in the lungs of cancer survivors, where exposure to chemotherapeutic agents or thoracic radiation initiates senescence pathways that contribute to fibrosis and impaired regenerative capacity [19,20,21]. A non-canonical form, mitochondrial dysfunction-associated senescence (MiDAS), emerges from mitochondrial stress and altered metabolic homeostasis rather than direct DNA damage. This variant is characterized by disrupted NAD+/NADH ratios, activation of AMPK, and engagement of p53 signaling, often in the absence of elevated ROS, and exhibits a distinct SASP signature. MiDAS has been implicated in age-associated lung pathologies linked to mitochondrial decline [22].
Importantly, senescence also plays physiological roles; for instance, developmental senescence by transiently activating p21, transient oncogene-induced senescence during embryogenesis contributes to organ patterning via p21Cip1/Waf1-dependent but p53-independent mechanisms, without leading to pathological outcomes [23]. Additionally, paracrine or bystander senescence arises when SASP components propagate senescent signaling to neighboring non-senescent cells, amplifying local tissue dysfunction and senescence burden [24,25]. Further complicating the landscape is the concept of pseudosenescence, which is a reversible state in which cells exhibit certain senescence markers without undergoing complete cell cycle arrest posing challenges for biomarker interpretation and therapeutic precision [26,27].
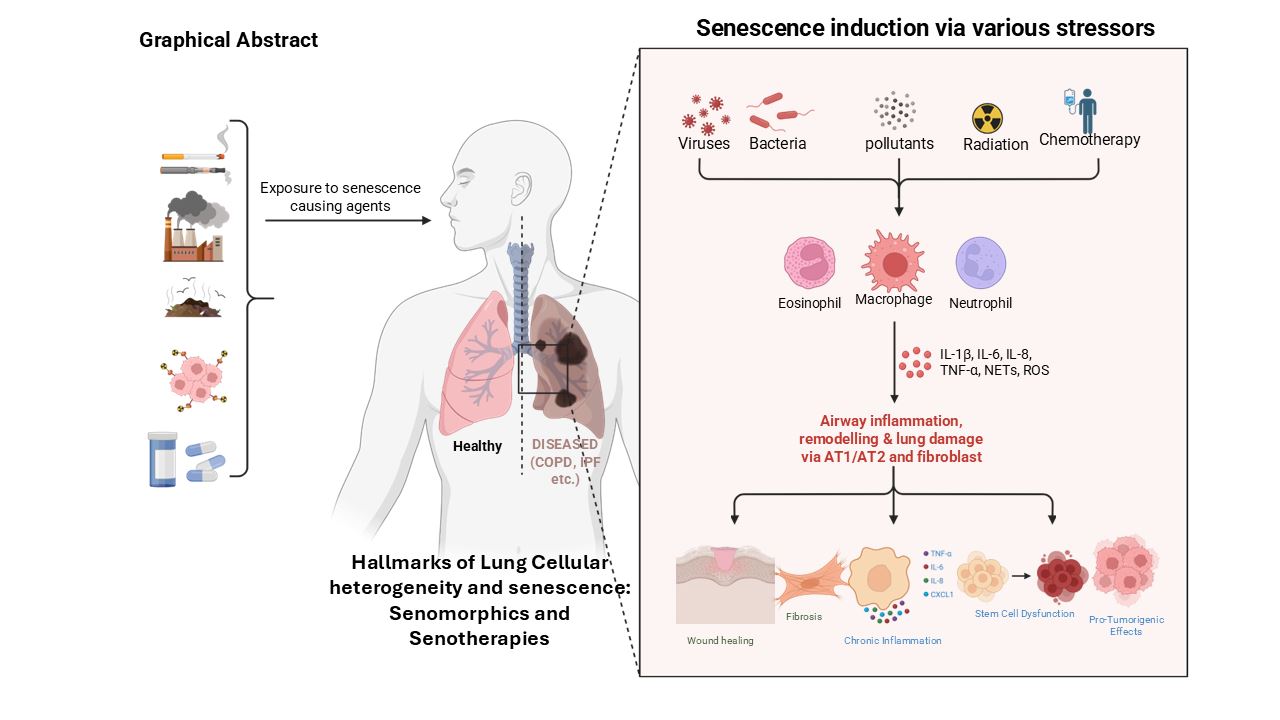
Cellular senescence plays a paradoxical role in tissue homeostasis, acting as both a protective and pathological mechanism. Under physiological conditions, senescence serves to suppress the proliferation of damaged, stressed, or oncogene-activated cells, thereby preserving genomic stability and preventing malignant transformation [28]. The lungs, continuously exposed to environmental insults such as cigarette smoke, particulate matter, ozone, pathogens, and occupational irritants, are particularly vulnerable to cellular damage and premature aging. These exposures elicit oxidative stress, DNA damage, mitochondrial dysfunction, and chronic inflammation all well-established inducers of cellular senescence [29,30,31,32]. In normal lung development and repair, transient senescence is functionally beneficial. Senescent cells contribute to wound healing and regeneration by secreting growth factors and remodeling components of the extracellular matrix via the SASP [23,33,34,35]. However, persistent accumulation of senescent cells particularly in post-mitotic tissues like the lung leads to homeostatic disruption. Chronically active SASP secretion by senescent alveolar epithelial cells, endothelial cells, and fibroblasts promotes sustained inflammation, matrix stiffening, and immune dysregulation, thereby impairing regenerative capacity and advancing diseases such as IPF and COPD [36,37].
Notably, senescent type II alveolar epithelial cells (AEC2s) lose their progenitor functionality, further compromising alveolar integrity and repair [13,38]. The inherently slow turnover of distal alveolar regions exacerbates this vulnerability, as insufficient regenerative input allows the accumulation of damaged or senescent cells over time [39,40,41,42]. Chronic environmental exposure also induces stress-induced premature senescence (SIPS) in both epithelial and stromal lung cells [31,43], leading to persistent SASP signaling that fuels inflammation, hinders tissue repair, and promotes fibrotic remodeling, particularly in COPD and IPF [44,45]. In COPD, cigarette smoke triggers senescence in airway epithelial cells and fibroblasts, resulting in a pro-inflammatory phenotype characterized by the secretion of cytokines, such as IL-6, IL-8, and MMPs, which degrade the extracellular matrix and sustain chronic inflammation [44,46]. Senescent endothelial cells also contribute to vascular remodeling and emphysematous changes in COPD lungs [44]. In IPF, senescence of alveolar type II (AT2) cells is driven by telomere dysfunction, inherited mutations (e.g., in TERT or RTEL1), and environmental stressors. These senescent AT2 cells lose regenerative potential and produce SASP components that activate immune cells and fibroblasts, thereby fostering fibrogenesis and inhibiting epithelial repair [13,38,47]. Senescence also intersects with tumorigenesis. Oncogene-induced senescence (OIS) acts as an early tumor-suppressive mechanism; however, chronic SASP signaling can create a permissive tumor microenvironment that facilitates immune evasion and promotes malignant progression [7,48]. More recently, senescence has been implicated in acute lung injury (ALI) and COVID-19-associated lung pathology. In these contexts, viral infection and inflammation provoke transient senescence in epithelial cells, which impairs resolution and contributes to fibrosis and long-term pulmonary dysfunction [49,50]. Collectively, emerging evidence underscores that the chronic accumulation of senescent cells in the lung disrupts tissue homeostasis, amplifies inflammatory signaling, and impairs regenerative capacity positioning senescence not merely as a consequence of disease, but as a central driver of pulmonary pathology [36,38]. This paradigm shift has catalyzed growing interest in senotherapeutics, including senolytics agents that selectively eliminate senescent cells and senomorphics, which suppress the deleterious effects of the SASP and other mediators [36,51].
Recent findings suggest that cellular senescence in lung disease extends well beyond epithelial cell cycle arrest. Senescence affects a broad spectrum of cell types including fibroblasts, endothelial cells, immune cells, and alveolar type I and II (AT1 and AT2) epithelial cells (Figure 1), each contributing uniquely to the progression of inflammation, matrix remodeling, and failure of tissue repair [38,52]. However, substantial heterogeneity exists within senescent cell populations, leading to variable SASP profiles and regenerative capacity. The spatial and functional diversity of these senescent subsets across lung compartments remains incompletely characterized [53,54,55]. Furthermore, metabolic reprogramming and mitochondrial dysfunction particularly in AT2 cells have been implicated in reinforcing fibrogenic signaling and destabilizing epithelial integrity, though these pathways require deeper mechanistic insight [22,56,57,58]. Challenging the traditional view of senescence as irreversible, recent studies indicate that senescent phenotypes may be reversible in certain lung cell types, particularly under the influence of pharmacological agents or microenvironmental cues, opening new therapeutic possibilities [59]. Underexplored signaling networks such as the YAP/TAZ-driven mechano-transduction axis, activated by extracellular matrix (ECM) stiffening, have been shown to reinforce fibroblast senescence and activation in fibrotic lung tissue, and merit further investigation [60]. Additionally, persistent senescence may be sustained by impaired immune surveillance, particularly due to aging-associated dysfunction in senescent cell clearance mechanisms [61]. The potential role of the lung microbiome in modulating cellular senescence and SASP expression remains largely speculative but could significantly influence disease trajectory [62].
2. Molecular Mechanisms of Cellular Senescence.
This is briefly discussed here based on the discovery of cellular senescence and its phenotypes. The induction and maintenance of cellular senescence are orchestrated by a complex network of molecular pathways activated in response to diverse intrinsic and extrinsic stressors, including telomere attrition, DNA damage, and oncogenic signaling [1,2,3]. These stress signals ultimately converge on tumor suppressor networks that enforce a durable cell cycle arrest [1,2]. Among these, the DNA damage response (DDR) represents a central and historically pivotal mechanism, particularly in the theme of replicative senescence, where progressive telomere shortening activates the DDR and triggers downstream effectors [12,63]. This response primarily engages two key tumor suppressor pathways: the p53/ p21Cip1/Waf1 axis and the p16Ink4a/retinoblastoma (Rb) pathway. Together, these signaling cascades reinforce and stabilize the senescent phenotype by halting cell cycle progression and preventing proliferation of damaged cells [12,63]. In addition to canonical DDR-mediated senescence, alternative mechanisms such as mitochondrial dysfunction, epigenetic regulation have also been implicated. Specifically, metabolic stress, histone modifications, and imbalances in NAD+/NADH ratios can induce a senescence program characterized by altered energy metabolism and a distinct SASP profile [22,64,65].
3. The Dichotomous Role of Senescence in Health and Disease
Cellular senescence exemplifies the principle of antagonistic pleiotropy, exerting both beneficial and deleterious effects that are highly dependent on physiological context and temporal dynamics based on cellular heterogeneity in lung development and diseases [7,10,66].
- Beneficial Roles
Transient cellular senescence plays an important role in normal physiological processes. During embryonic development, programmed senescence contributes to tissue patterning and morphogenesis [1,67]. In the context of acute tissue injury, senescent cells support wound healing and limit fibrotic remodeling by secreting reparative factors such as PDGF-AA and matrix metalloproteinases (MMPs), which promote tissue repair and recruit immune cells for the clearance of damaged or apoptotic cells [2,7,10,68]. Arguably, the most well-established beneficial function of senescence is tumor suppression. This occurs through both cell-autonomous mechanisms, via stable cell cycle arrest, and non-cell-autonomous mechanisms, through SASP-mediated immune surveillance that facilitates the clearance of potentially malignant cells [1,2,7,69].
- Detrimental Roles
In contrast, the chronic accumulation of senescent cells particularly with aging or in the context of unresolved tissue stress has deleterious effects [1,10,66]. This accumulation is exacerbated by immunosenescence, the age-associated decline in immune surveillance capacity, which impairs the effective clearance of senescent cells [1,4]. The resulting persistent SASP induces a chronic, low-grade inflammatory state known as "inflammaging," a hallmark of aging and numerous age-related disorders [1,10]. This unresolved pro-inflammatory milieu disrupts tissue architecture, promotes paracrine senescence in neighboring cells, depletes tissue-resident stem cell populations, and fosters a microenvironment conducive to tumorigenesis. Consequently, senescence contributes to the pathophysiology of a broad spectrum of diseases, including cancer, type 2 diabetes, cardiovascular disease, idiopathic pulmonary fibrosis, liver fibrosis, osteoarthritis, neurodegenerative disorders such as Alzheimer’s disease, and clinical frailty [1,2,7,10,66,67,70,71,72,73,74]. Understanding the dynamic balance between the beneficial and harmful effects of senescence across tissue types and temporal contexts is critical for designing effective, context-specific therapeutic interventions [2].
4. Roles of Senescence in Lung Diseases
- Role of cellular senescence in lung diseases based on cellular heterogeneity
Cellular senescence is recognized as a key contributor to the onset and progression of several chronic lung diseases, including COPD, IPF, and asthma [38,75,76,77]. The SASP, senescent cells accumulate in response to stressors, such as oxidative damage, telomere shortening, and environmental exposures like cigarette smoke [16, 28, 31, 44, 78]. The milieu of the SASP is highly heterogeneous based on heterogeneous nature of senotypes, and is determined by both the originating cell type and the surrounding disease microenvironment.
In fibroblasts, the senescence-associated phenotype includes the secretion of IL-6, IL-8, and matrix metalloproteinases (MMPs), which promote inflammation, extracellular matrix (ECM) remodeling, and even tumor progression [7, 79, 80]. In alveolar type II (AT2) epithelial cells, particularly in IPF senescence, results in a pro-fibrotic SASP enriched in TGF-β and PAI-1, contributing to fibrosis and impaired regenerative responses [81]. In COPD, senescent airway epithelial cells secrete IL-6, IL-8, CCL2, and MMP-12, exacerbating chronic inflammation and alveolar destruction [76, 82-84]. Notably, in post-COVID-19 lung fibrosis, epithelial SASP profiles including IL-1β, CCL2, and TGF-β resemble those observed in IPF [85].
Across multiple lung diseases, senescence of distinct cell types contributes to pathology through impaired tissue regeneration, persistent in[16,28,31,44,78flammation, and structural remodeling [36,38,84,86]. In IPF, senescent AT2 cells display DNA damage and reduced proliferative capacity, impairing alveolar repair and promoting fibrotic remodeling [38]. In COPD, bronchial epithelial cells, fibroblasts, and endothelial cells undergo senescence, driving inflammation, alveolar wall thinning, and progressive loss of lung function [87]. In asthma, especially in elderly individuals, senescent airway epithelial and smooth muscle cells accumulate, leading to abnormal tissue remodeling and airway dysfunction [88]. Following severe COVID-19, persistent senescence in AT2 cells, transitional epithelial cells, and endothelial cells has been documented, contributing to fibrosis and recapitulating IPF-like features [77]. Similarly, in radiation-induced lung fibrosis, senescent epithelial, fibroblast, and vascular endothelial cells exhibit stable growth arrest and fail to support effective tissue repair [36,89].
These disease-specific patterns underscore the role of cell type-dependent senescence in lung pathology and emphasize the therapeutic potential of targeting senescence at the cellular level. In pulmonary hypertension and post-COVID complications, senescence of endothelial and epithelial cells further contributes to vascular remodeling and fibrotic progression [90,91,92,93]. Far from being a passive byproduct of aging, cellular senescence actively drives disease processes. As such, it represents a compelling target for novel interventions including senolytics and senomorphics which aim to reduce the burden of senescent cells or modulate the SASP to restore pulmonary homeostasis [94,95].
- Age-dependent biomarkers in lung diseases
- Disease-dependent biomarkers
Although direct evidence on age-dependent changes in lung cellular senescence remains limited, a substantial body of literature supports the role of senescence in various pulmonary pathologies, including IPF, COPD, and lung cancer. In the following sections, we provide an overview of key senescence markers identified in each of these disease contexts, with a particular focus on their expression within distinct lung cell populations.
4.1. Senescence in COPD
COPD is a progressive lung disorder predominantly associated with underlying inflammation, alveolar destruction, aging and characterized by airflow limitation, and extensive structural remodeling of the airways and lung parenchyma. Clinically, COPD encompasses phenotypes such as chronic bronchitis and emphysema, with small airway disease being a central pathological feature. Accumulating evidence demonstrates increased cellular senescence in key lung cell populations including type II alveolar epithelial cells, endothelial cells, and fibroblasts in emphysematous lungs [31,96]. A meta-analysis involving 6,378 individuals further supported this association, revealing a significant correlation between shortened leukocyte telomere length and increased COPD risk [97] (Figure 2).
Senescence propagation may occur through extracellular vehicles (EVs) carrying senescence-promoting microRNAs. In a recent study, EVs from COPD patients were shown to carry miR-34a, which induced senescence in healthy small airway epithelial cells (SAECs) by downregulating SIRT1 and upregulating p21Cip1/Waf1, suggesting a mechanism for both local disease exacerbation and systemic comorbidities [98]. Recombinant human CC16 (rhCC16) has demonstrated senescence-reducing effects in lung epithelial cells and COPD mouse models by downregulating senescence markers such as β-galactosidase, p16Ink4a, p21Cip1/Waf1, and reactive oxygen species (ROS). These effects are mediated via activation of the AMPK/SIRT1-PGC-1α signaling pathway, thereby restoring mitochondrial function and offering promise as a senescence-modulating therapy [99].
Given that cigarette smoke is the primary etiologic factor in COPD, several in vitro studies have investigated its role in epithelial senescence. These studies report upregulation of senescence-associated markers including p21Cip1/Waf1, p53, SA-β-Gal, CXCL5, CXCL8, VEGF, and the DNA damage marker γH2A.X [84,100]. However, the role of p16Ink4a remains debated. While murine models show increased p16Ink4a expression in COPD, prior work by Sundar et al. (2018) showed that genetic ablation of p16Ink4a alone did not protect against smoke-induced senescence. Subsequent studies, however, suggest that p16Ink4a contributes to the regulation of smoke-mediated senescence responses [76,101]. Additionally, Woldhuis et al. (2020) demonstrated elevated p16Ink4a expression in COPD-derived fibroblasts, which inversely correlated with decorin expression, linking accelerated senescence with extracellular matrix (ECM) dysregulation.
A clinical trial supplementing COPD patient with nicotinamide riboside (NR) a precursor of NAD⁺ reported reduced airway inflammation (IL-8), increased systemic NAD⁺ levels, and improvements in genomic integrity and epigenetic aging, highlighting its therapeutic potential in targeting cellular senescence [102]. Bioinformatics studies using machine learning and weighted gene co-expression network analysis (WGCNA) have identified four key genes EP300, MTOR, NFE2L1, and TXN as molecular links between COPD and aging. A diagnostic model based on these genes, developed using artificial neural networks, showed high predictive accuracy for COPD [103]. A six-month physical activity intervention in COPD patients reduced the proportion of senescent T-lymphocytes, improved their proliferative capacity, and reduced their ability to induce fibroblast senescence, thereby improving immune function [104].
Senescence also appears to bridge COPD and cancer pathogenesis. A 2018 study showed that exposure of human bronchial epithelial cells (HBECs) to serum from COPD patients led to increased markers of senescence SA-β-Gal, γH2A.X, p21Cip1/Waf1, and ROS. The conditioned medium from these cells contained elevated CXCL5, CXCL8/IL-8, and VEGF, which promoted adhesion, proliferation, and migration of hypersecretory cells. These findings suggest that COPD-associated senescence may create a pro-tumorigenic microenvironment, independent of smoking status [84].
A pilot study revealed that induced sputum (IS) cells in COPD patients exhibit higher biological aging than peripheral blood leukocytes, as measured by DNA methylation age (DNAmAge) and age acceleration (AgeAcc). While telomere length did not show strong correlation, lung function (FEV₁%) and use of inhaled corticosteroids were associated with reduced biological aging in leukocytes suggesting a clinical relevance for site-specific aging markers [105]. While the concept of targeting senescence in COPD is compelling, it remains an emerging field requiring further validation. COPD is a highly heterogeneous disease, with variability in clinical manifestations depending on the location of injury, type of insult, patient age, sex, and comorbid conditions. Although senescence is a critical component of accelerated lung aging, it is not the sole driver of COPD pathogenesis. Importantly, senescence also serves physiological roles in tissue repair and immune regulation. Therefore, indiscriminate elimination of senescent cells may lead to adverse effects. A more nuanced approach focused on identifying and targeting pathogenic senescent cell subtypes that contribute to irreversible tissue damage, immune dysfunction, and stem cell depletion is essential for developing effective senescence-directed therapies in COPD.
Senescence Across Different COPD Stages and Severities
The role and extent of cellular senescence in COPD appear to vary based on disease severity and clinical phenotype. While systemic factors capable of inducing senescence are present across all stages of COPD, accumulating evidence suggests that the burden of senescent cells in the lung correlates with disease progression and severity (Figure 2).
Studies analyzing patient-derived cells and tissues indicate that the accumulation of senescent cells is more pronounced in severe forms of COPD, particularly in early-onset cases. In a study by Woldhuis et al., fibroblasts isolated from patients with severe, early-onset COPD (SEO-COPD) and those with mild-to-moderate COPD were examined for classical hallmarks of senescence. COPD-derived fibroblasts showed elevated levels of senescence markers, γ-H2A.X-positive nuclei (indicative of DNA damage), and oxidative stress when compared to healthy controls. Importantly, these effects were most significant in the SEO-COPD subgroup. For example, senescence-associated β-galactosidase (SA-β-Gal) positivity was significantly increased only in SEO-COPD fibroblasts, and elevated expression of p21Cip1/Waf1 in lung tissue was similarly confined to this group [52]. These findings align with genetic studies indicating that telomerase mutations are a risk factor for early-onset emphysema [82], supporting the hypothesis that intrinsic susceptibility to senescence may underlie more aggressive disease forms.
In contrast, studies exploring systemic senescence-inducing factors present a more nuanced picture. Kuźnar-Kamińska et al. investigated whether serum from COPD patients across GOLD stages 1-4 and risk groups A-D could induce a senescent phenotype in human bronchial epithelial cells (HBECs). Their findings revealed that COPD patient serum significantly increased the expression of senescence markers (SA-β-Gal, γ-H2A.X, p21Cip1/Waf1) in HBECs when compared to serum from healthy controls. However, no significant differences in senescence marker expression were observed between different GOLD stages or risk groups. Moreover, the levels of SASP components secreted by the exposed HBECs, including VEGF, CXCL8, and CXCL5 also showed no variation with disease severity [84]. These findings suggest a two-tiered model of senescence in COPD. Systemic senescence-promoting factors may be present early in disease and consistent across stages, constituting an underlying pro-senescent milieu. Localized accumulation of senescent cells within lung tissues, particularly in patients with severe or early-onset COPD may drive progressive tissue remodeling, inflammation, and functional decline. This model highlights the importance of distinguishing between senescence-inducing stimuli and the actual tissue burden of senescent cells. It further underscores the need for stratified therapeutic approaches that consider COPD phenotype, disease stage, and cell-type specific senescence profiles when designing senescence-targeted interventions.
4.2. Senescence in Idiopathic Pulmonary Fibrosis (IPF)
Senescent lung fibroblasts are increasingly recognized as contributors to the pathogenesis of IPF, a progressive and irreversible interstitial lung disease marked by excessive extracellular matrix (ECM) deposition and alveolar remodeling [106]. While telomere shortening and replicative stress have long been implicated in promoting fibroblast senescence in IPF, emerging evidence highlights the additional roles of oxidative stress, mitochondrial dysfunction, and chronic inflammation in driving this phenotype [107]. Multiple studies have investigated biomarkers of fibroblast senescence in IPF. Sanders and colleagues found that elevated plasma levels of GDF15, IL-6, CRP, and TNFRII are associated with an increased risk of interstitial lung abnormalities (ILA), a precursor state to pulmonary fibrosis [108,109]. Primary lung fibroblasts derived from IPF patients exhibit increased expression of canonical senescence markers including SA-β-gal, p53, p16Ink4a, and p21Cip1/Waf1, alongside a robust SASP [110,111].
Recent studies have implicated STAT3 activation in driving early senescence and IPF progression, suggesting that modulation of STAT3 signaling could mitigate fibroblast dysfunction [106]. Senescent fibroblasts in IPF also exhibit increased expression of anti-apoptotic Bcl-2 family proteins (Bcl-W, Bcl-2, Bcl-XL) and decreased expression of pro-apoptotic factors (Bak, Bax), conferring resistance to cell death [112,113]. Additionally, age-associated upregulation of plasminogen activator inhibitor-1 (PAI-1) has been observed in murine lung fibroblasts. PAI-1 promotes fibrotic remodeling via a vitronectin-independent pathway mediated through its interaction with SorLA (sortilin-related receptor 1), which is upregulated in both mouse models and human IPF tissue [114,115]. Targeting the PAI-1–SorLA axis may thus represent a novel therapeutic approach in IPF.
At the tissue level, senescent IPF fibroblasts display expanded SASP profiles, including pro-inflammatory cytokines (IL-1β, IL-6, TNF-α), profibrotic growth factors (TGF-β, CTGF, PDGF), chemokines (MCP-1, CXCL1), and matrix-degrading enzymes (MMP-2, MMP-9, MMP-12), which collectively foster a self-reinforcing fibrotic niche [110,116]. Notably, CTGF has been shown to induce fibroblast senescence via ROS accumulation and p53-mediated induction of p16Ink4a [117]. Proteomic analyses further support a senescence-associated chromatin remodeling phenotype, including the accumulation of NF-κB subunit p65 on chromatin in senescent fibroblasts [118].
Advances in single-cell transcriptomics have revealed substantial heterogeneity within the fibroblast compartment in IPF lungs. Studies by Tsukui et al. (2020) and Habermann et al. (2020) identified diverse fibroblast subtypes, including homeostatic fibroblasts, pathogenic myofibroblasts, and peribronchial fibroblasts [119,120]. However, it remains unclear which of these subpopulations are most prone to senescence and responsible for sustaining a chronic SASP. Integrating high-resolution single-cell RNA sequencing with spatial transcriptomics and senescence marker profiling may enable precise mapping of senescent fibroblast niches and clarify their interactions with neighboring senescent epithelial cells [119,121,122].
Additionally, applying RNA velocity and lineage tracing techniques may help determine whether activated fibroblast subtypes transition into a senescent state and whether this state is reversible under therapeutic pressure. These tools could illuminate the plasticity of fibroblast senescence in IPF and inform the development of subpopulation-specific senolytic therapies and targeted delivery systems. Taken together, senescent fibroblasts play a central role in shaping the profibrotic microenvironment of the IPF lung. Understanding the heterogeneity, plasticity, and molecular underpinnings of fibroblast senescence remains essential for designing precision interventions aimed at halting or reversing fibrotic progression (Figure 1).
4.3. Lung Cancer
In the context of lung cancer particularly non-small cell lung cancer (NSCLC) and its predominant adenocarcinoma subtype cellular senescence exhibits a distinctly paradoxical or “dual” role. In early carcinogenesis, senescence functions as a potent tumor-suppressive mechanism, interfering the proliferation of cells with oncogenic alterations through stable cell cycle arrest and immune-mediated clearance. However, in advanced or established tumors, senescent cells especially those that persist can adopt a pro-tumorigenic phenotype via SASP. This chronic SASP output promotes inflammation, tumor progression, immune evasion, metastatic spread, and resistance to therapy [123].
Oncogene-induced senescence (OIS) represents a critical tumor-suppressive barrier that acts to prevent malignant transformation at the earliest stages of carcinogenesis. In lung adenocarcinoma, activation of oncogenes such as KRAS or BRAF elicits a potent senescence response characterized by stable cell cycle arrest, effectively halting the proliferation of transformed cells [17]. This protective mechanism, often referred to as the “friend” role of senescence, helps to confine lesions to a pre-malignant or early malignant state. However, for tumor progression to occur, cancer cells must bypass this senescence barrier, frequently through the acquisition of loss-of-function mutations in tumor suppressors, such as p53 or p16Ink4a [124].
Paradoxically, once senescent cells persist particularly in the setting of therapy-induced senescence (TIS) they often adopt a “foe” role in cancer progression. Senescent cells develop a robust SASP, which is a complex cocktail of pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines, growth factors, and proteases that remodel the tumor microenvironment (TME) [7]. This remodeling can promote epithelial-mesenchymal transition (EMT), angiogenesis, and invasion, thereby enhancing tumor aggressiveness [125,126].
Recent studies have demonstrated that senescent lung fibroblasts such as those found in IPF, a known risk factor for lung cancer, secrete exosomes enriched in matrix metalloproteinase-1 (MMP-1). These exosomes are readily internalized by NSCLC cells, where they activate the PI3K–AKT–mTOR pathway, stimulating cellular proliferation and clonogenicity [127]. The persistence of senescent cells following chemotherapy is therefore increasingly linked to adverse clinical outcomes, including tumor relapse and metastasis [128].
SASP also exerts profound effects on the immune landscape of the TME. While initially capable of recruiting immune cells for senescent cell clearance, a chronic SASP may shift the microenvironment toward immunosuppression. This phenomenon has been exemplified in KRAS-driven lung cancer models, where senescent macrophages have been identified as a major pro-tumorigenic population [129]. These macrophages, which are also found in aged lungs and pre-malignant human lung lesions, exhibit a unique SASP that promotes tumor progression. Notably, senolytic clearance of these cells using agents such as ABT-737 was shown to reduce tumor burden, enhance anti-tumor immunity, and extend survival in murine models [129].
These findings show the context-dependent duality of senescence in lung cancer. While senescence can initially restrain malignant transformation, its persistence especially in the TME facilitates tumor evolution and immune evasion. Critically, these effects are mediated by specific, targetable senescent cell populations, reinforcing the therapeutic potential of senolytics and SASP modulators in lung cancer treatment.
4.4. Senescence in Acute Lung Injury and LARDS (Including COVID-19)
The role of cellular senescence in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS) has emerged as a critical area of investigation, particularly in the wake of the COVID-19 pandemic. In contrast to chronic lung diseases where senescence is well established as a pathogenic driver the function of senescence in acute injury is more context-dependent and paradoxical. It often exhibits characteristics of a “double-edged sword”, exerting both protective and deleterious effects depending on the timing, duration, and cellular context of its activation [91,130,131].
In response to acute insults, such as viral infection or severe inflammation, the lung initiates a transient senescence response that serves as an early protective mechanism [132]. This form of acute senescence acts to arrest the proliferation of damaged or infected cells, thereby limiting the spread of pathogens and preserving tissue integrity. Unlike apoptosis, this temporary growth arrest allows for cellular survival and immune-mediated clearance, preventing further tissue disruption [130].
A clear illustration of this process is observed in the context of severe COVID-19, where SARS-CoV-2 infection has been shown to induce a pronounced senescent phenotype in alveolar type II (AT2) epithelial cells. These infected senescent cells exhibit elevated expression of key markers such as p16Ink4a and the DNA damage marker γ-H2AX, indicating the activation of a canonical senescence program [93]. In the setting of ALI and LARDS, the SASP plays a dual role in immunomodulation. Initially, the secretion of pro-inflammatory cytokines and chemokines facilitates a beneficial immune response, aiding in the recruitment of immune cells to eliminate pathogens, clear apoptotic or damaged cells, and remove senescent cells once their protective role is fulfilled [130]. This acute inflammatory signaling is prominently observed in severe COVID-19, where senescent alveolar type II (AT2) epithelial cells exhibit markedly elevated expression of SASP cytokines such as IL-1β and IL-6. These factors contribute directly to the hyperinflammatory state that characterizes severe disease [130].
However, when senescent cells are not efficiently cleared, this initially protective SASP can become pathogenic. Persistent SASP signaling can promote chronic inflammation, impair tissue repair, and exacerbate lung damage. This failure of clearance is particularly pronounced in the context of immunosenescence, a state of age-associated immune dysfunction which limits the immune system’s capacity to resolve inflammation and remove senescent cells [130]. Thus, the context, duration, and immune competence of the host are critical determinants of whether the SASP functions as a beneficial or deleterious mediator in ALI and LARDS.
When the acute senescence program fails to resolve appropriately, it transitions from a protective mechanism into a pathogenic driver of chronic lung damage. This unresolved senescent state contributes to delayed resolution, fibrotic remodeling, and long-term impairment of pulmonary function [132]. The persistent secretion of SASP factors from an accumulating burden of senescent cells perpetuates a chronic inflammatory milieu, which affects with normal tissue regeneration and promotes the deposition of extracellular matrix, ultimately leading to fibrosis [130]. Ihe accumulation of senescent alveolar epithelial cells significantly compromises the host’s capacity to recover, diminishing epithelial integrity and regenerative potential in acute lung injury [133].
This chronic, unresolved senescent state characterized by persistent SASP signaling, immune dysfunction, and a loss of reparative capacity is increasingly recognized as a central mechanism underlying the post-acute sequelae observed in survivors of ARDS and severe COVID-19 [93,132]. These findings emphasize the need for therapeutic strategies aimed at modulating senescence resolution, restoring immune surveillance, and preserving epithelial regeneration in the aftermath of acute lung injury.
4.5. Cystic Fibrosis (CF)
Cellular senescence is a critical pathological feature in cystic fibrosis (CF) a genetic disease marked by chronic neutrophilic inflammation, mucus accumulation, and progressive lung tissue damage [134,135]. While CF was traditionally considered a pediatric disorder, advancements in treatment have significantly extended patient lifespan, uncovering hallmarks of premature lung aging in this population [134]. The CF airway epithelium exhibits a distinct senescence-associated signature, with elevated expression of canonical senescence markers including p16Ink4a, γH2A.X, and phospho-Chk2, when compared to non-CF controls [136]. Notably, this senescent phenotype is spatially and cell-type restricted. Recent single-cell RNA sequencing revealed that basal and secretory epithelial cells within CF airways show the highest expression of senescence markers [137]. Intriguingly, even in the absence of inflammatory stimuli, CFTR-deficient bronchial epithelial cells exhibit a senescent-like phenotype characterized by elevated p21Cip1/Waf1 levels and reduced proliferative capacity, suggesting that CFTR dysfunction itself acts as a cell-intrinsic senescence Among the extrinsic inducers of senescence in CF, neutrophil elastase (NE) is particularly important. NE, abundantly released into the CF airway due to sustained neutrophil infiltration, has been shown to induce cellular senescence in airway epithelial cells via p16Ink4a mediated CDK4 inhibition [136,138,139]. This establishes a self-reinforcing loop, as senescent cells develop a senescence-associated secretory phenotype that exacerbates inflammation by recruiting additional immune cells, thus amplifying tissue damage and further promoting senescence [139,140]. Additionally, neutrophils isolated from bronchoalveolar lavage fluid of CF patients have been shown to express p21Cip1/Waf1, which may prolong their survival and contribute to persistent inflammation [135].
Emerging studies have begun to elucidate novel pathways that sustain senescence in CF beyond CFTR loss. The FGFR-MAPK-p38 axis has been implicated in promoting senescence in CF bronchial epithelium. Pharmacologic inhibition of FGFR4 significantly reduced senescence marker expression and improved mucociliary clearance in preclinical models, indicating this pathway's therapeutic potential [137]. These findings suggest that although CFTR dysfunction initiates disease, secondary senescence may be maintained by CFTR-independent mechanisms, possibly explaining persistent inflammation in CF patients treated with highly effective modulator therapies [141]. Altogether, the accumulation of senescent cells in CF lungs not only accelerates structural damage but also contributes to chronic inflammation and impaired repair. As such, targeting cellular senescence represents a promising therapeutic avenue to mitigate disease progression, especially in the aging CF population [140].
4.6. Pulmonary Hypertension
Pulmonary hypertension (PH) is a progressive and proliferative vascular disorder in which cellular senescence is increasingly recognized as a central pathological mechanism [142,143]. The accumulation of senescent cells, particularly pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs), contributes to the vascular remodeling that characterizes PH pathogenesis [144].
In both human idiopathic PH (iPAH) and animal models, elevated expression of senescence markers such as p16Ink4a, p21Cip1/Waf1, and γH2AX has been consistently reported in vascular tissues [142,145]. Notably, hypoxia-induced senescence in PASMCs leads to paracrine secretion of IL-6, promoting the proliferation of neighboring PASMCs via the mTOR/S6K1 signaling axis [146].
In addition to paracrine signaling, juxtacrine interactions between senescent endothelial cells (ECs) and PASMCs have been shown to drive disease progression. Specifically, senescent ECs upregulate Notch ligands (e.g., JAG1, DLL4), which activate Notch signaling in adjacent PASMCs. This mechanism was confirmed in EC-specific progeroid mouse models, where pharmacological inhibition of Notch signaling attenuated disease severity [147]. Transcriptomic analyses have further identified YWHAZ as a central hub gene in hypoxic PAECs and PH models; its silencing promotes autophagy, suggesting a potential strategy to facilitate senescent cell clearance [148].
Senescence also appears to demarcate the irreversibility of PH progression. In a congenital heart disease-associated PH (PAH-CHD) rat model, the transition from reversible to irreversible pathology was marked by a shift toward a senescent vascular phenotype, with increased expression of p16Ink4a, p21Cip1/Waf1, MMP2, and IL-6. Remarkably, senolytic therapy with ABT263 reversed advanced PH in this model, supporting a causal role for senescent cell accumulation in disease progression [149]. This pro-fibrotic and pro-remodeling function of senescence is echoed in findings that frataxin deficiency, a known inducer of mitochondrial dysfunction, promotes endothelial senescence and PH, which is also amenable to senolytic intervention [143].
Paradoxically, recent data suggest that senescence may play a protective or homeostatic role in certain contexts. In contrast to the above findings, genetic (p16Ink4a-ATTAC model) and pharmacological (ABT263, FOXO4-DRI) clearance of senescent cells in various rodent PH models resulted in worsened hemodynamics and enhanced vascular remodeling [145]. These effects were attributed to the loss of senescent pulmonary endothelial cells (P-ECs), which represent a significant portion of the senescent population even in healthy lungs and may serve essential homeostatic and anti-proliferative functions [144,145].
- Key proteins implicated in Lung cellular senescence
An array of proteins is critically involved in the complex process of cellular senescence in the lungs. These proteins participate in various pathways, including cell cycle regulation, DNA damage response, and the secretion of senescence-associated secretory phenotype (SASP) factors, all of which contribute to lung aging and the pathogenesis of lung age-related diseases (Table 1).
IL-10 (Interleukin 10): Interleukin-10 is an anti-inflammatory cytokine. Studies in mice with IL-10 gene knockouts have revealed that a deficiency in this protein exacerbates cellular senescence and accelerates lung fibrosis. These mice exhibit heightened inflammatory responses, and the introduction of recombinant IL-10 can mitigate these effects by reducing the expression of senescence markers p16 and p21. In vitro experiments have further demonstrated that the absence of IL-10 promotes the secretion of SASP factors from lung fibroblasts [150]. In the context of COPD, some studies have noted that mice with a deficiency in the IL-10 gene show an increase in senescent cells. Another report showed that IL-10 levels are reduced in the airways of patients with COPD, suggesting its deficiency may contribute to the chronic inflammation characteristic of the disease [151,152].
TP53 (p53): The tumor suppressor protein p53 is a central figure in cellular senescence, primarily by governing cell cycle arrest and apoptosis in response to cellular stress. In conditions such as IPF and COPD, increased levels of p53 are observed in lung tissues. This upregulation of p53 is associated with the induction of apoptosis in alveolar epithelial cells, which can impair the regenerative capacity of the lung. It’s role in cell cycle arrest is mediated through the transcriptional activation of CDKN1A (p21). In human lung fibroblasts, TP53 is also implicated in cell cycle arrest in the context of acute respiratory distress syndrome (ARDS) [36,100,153,154,155,156,157].
H2AX (H2A.X variant histone): H2A.X is a well-established marker of DNA double-strand breaks and is closely linked to cellular senescence. In aging and COPD, an increase in γ-H2AX in the lungs indicates an accumulation of DNA damage that can trigger cellular senescence [34,74,158]. Studies have shown that γ-H2AX accumulates at telomeres and in cells undergoing senescence. In IPF, a disease characterized by accelerated lung aging, increased levels of γ-H2AX are found in lung cells [158].
CDKN2A (p16INK4a): This gene encodes the p16INK4a protein, a key inhibitor of cyclin-dependent kinases that plays a crucial role in inducing and maintaining cell cycle arrest in senescent cells. Knockout or silencing of CDKN2A in mouse models has been shown to affect cellular senescence in the lungs. The expression of p16INK4a is considered one of the most specific markers of senescent cells and is frequently used to identify them in aging tissues [76,101,156,157,159,160,161].
GDF15 (Growth Differentiation Factor 15): GDF15 is a member of the transforming growth factor-beta (TGF-β) superfamily and is recognized as a component of the SASP. Its expression increases under cellular stress and in chronic lung diseases like COPD and IPF. In these diseases, elevated GDF15 levels serve as a biomarker and are associated with disease severity and progression [109,162,163,164,165].
CDKN1A (p21): p21 is a potent cyclin-dependent kinase inhibitor that is a downstream target of p53. Following cellular stress and DNA damage, p21 is induced and enforces cell cycle arrest, a hallmark of senescence. In lung fibrosis and ARDS, an increase in p21 expression is observed. While it can limit acute lung injury by preventing apoptosis, its sustained expression can also promote fibrosis by suppressing alveolar regeneration [31,34,36,100,153,156,157,166,167].
TNFRSF1B (TNF receptor superfamily member 1B): TNFRSF1B shows increased expression in the lungs in hyperoxia-induced senescence [34]. This suggests a role for this receptor in inflammatory signaling pathways contributing to the SASP.
Bcl2 L1 (BCL2 like 1): BCL2L1, an anti-apoptotic protein, shows decreased expression in fibroblasts in the context of IPF [113].
CXCL8 (IL-8): CXCL8 is a component of the SASP whose expression is increased in senescent lung cells, particularly in IPF and COPD [151,168,169]. Produced by various cell types, CXCL8 is a chemoattractant for neutrophils, contributing to chronic inflammation and tissue damage in lung diseases.
IL1A (Interleukin 1 alpha): IL1A is a pro-inflammatory cytokine that shows increased expression in senescent epithelial cells in the lungs, particularly in IPF [36,156,157,170]. Damaged epithelial cells release IL-1A, triggering an inflammatory response in neighboring fibroblasts, promoting fibrosis.
MMP12 (Matrix Metallopeptidase 12): MMP12 is a protease increasingly expressed in the lungs of IPF patients [36]. It contributes to tissue remodeling and fibrosis and is secreted by senescent alveolar epithelial cells. Its expression also increases after cigarette smoke exposure [157].
SERPINE1 (Plasminogen Activator Inhibitor-1, PAI-1)
SERPINE1 is found at elevated levels in the lungs in contexts of cellular senescence, including IPF [36,155,158,171]. Increased SERPINE1 expression in alveolar type II cells can induce senescence via the p53-p21-Rb pathway [155].
TGFβ1 (Transforming Growth Factor Beta 1): TGFβ1 can induce senescence and is key to fibrosis. In mouse models, TGF-β1 signaling mediates senescence-associated pulmonary fibrosis [154]. In alveolar type-2 epithelial cells, TGFβ signaling is linked to a DNA damage response and senescence [156]. In IPF, the fibrogenic secretome of senescent cells highlights TGFB1’s role in disease progression [36].
TNF (Tumor Necrosis Factor): TNF is implicated in the SASP. In human and mouse models of IPF, increased senescence biomarkers are observed, and the secretome of senescent fibroblasts includes TNF, which is fibrogenic [36].
IL-6 (Interleukin 6): IL-6 is a prominent SASP factor. In COPD, senescent cells secrete elevated IL-6, contributing to chronic inflammation [151,169]. In IPF and cigarette smoke exposure models, IL-6 is also increased and is part of the fibrogenic secretome [36,157,170].
IL-1beta (Interleukin 1 beta): IL-1beta is a pro-inflammatory cytokine and SASP mediator. In COPD, increased IL-1beta is found in the lungs and is associated with inflammatory processes [151].
MMP-8 (Matrix Metallopeptidase 8): MMP-8 is a protease involved in extracellular matrix degradation. In COPD, senescent cells secrete MMP8, contributing to tissue remodeling [172].
VEGFA (Vascular Endothelial Growth Factor A): VEGFA promotes angiogenesis and is part of the SASP in COPD. Increased VEGFA secretion by senescent cells contributes to pathological vascular remodeling [169].
5.1. Senomorphics
While the elimination of SnCs via senolytics has garnered significant attention, an alternative and potentially complementary therapeutic strategy involves modulating the characteristics of SnCs without inducing cell death (Table 2). This approach utilizes agents termed senomorphics or senostatics.16,17 The core principle behind senomorphism is the suppression or alteration of the detrimental aspects of the SASP, a complex secretome produced by many SnCs that significantly contributes to chronic inflammation, tissue dysfunction, and the progression of aging and age-related diseases [1,66,173].
The rationale for developing senomorphics stems from several considerations. Firstly, the SASP itself is recognized as a major driver of the deleterious effects associated with SnC accumulation [1,173]. Targeting the SASP directly offers a way to mitigate these effects. Secondly, SnCs are not uniformly detrimental; they play essential roles in physiological processes such as tumor suppression, embryonic development, and wound healing [10,173]. Senolytic therapies, by eliminating SnCs wholesale, risk ablating these beneficial functions. Senomorphics, by preserving the SnCs while neutralizing their harmful secretions, might offer a more nuanced intervention. Thirdly, the heterogeneity of SnCs and their SASP profiles across different tissues, cell types, and inducing stimuli [9,173] presents a challenge for broadly effective senolytics. Senomorphic agents targeting common SASP regulatory pathways might provide a more universally applicable approach. By altering the SnC phenotype rather than inducing cell death, senomorphics aim to achieve a state of "senostasis," reducing the pro-aging impact of senescent cells [174] (Figure 3).
5.2. Mechanisms of Senomorphic Action: Targeting SASP Regulation
Senomorphic compounds exert their effects primarily by interfering with the signaling pathways and molecular machinery responsible for producing and secreting SASP components. The SASP is remarkably complex, comprising hundreds of distinct factors including pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines (e.g., MCP-1/CCL2), growth factors, proteases (e.g., matrix metalloproteinases-MMPs), bioactive lipids, extracellular vesicles, and other molecules [9,175]. The production of this diverse secretome is tightly regulated at multiple levels, transcriptional, post-transcriptional, and translational, providing various points for senomorphic intervention.
Several key intracellular signaling pathways are implicated in SASP regulation and serve as primary targets for senomorphic drugs [173]:
NF-κB Pathway: The nuclear factor-κB (NF-κB) pathway is a master regulator of inflammation and immunity and is critically involved in the transcription of numerous SASP factors, particularly pro-inflammatory cytokines like IL-6 and IL-8 [118,173,175]. Persistent DNA damage response (DDR) signaling, often present in SnCs, can lead to chronic NF-κB activation. Senomorphics targeting NF-κB signaling, either by inhibiting upstream kinases like IKK or preventing NF-κB nuclear translocation, can effectively dampen a significant portion of the inflammatory SASP [173].
mTOR Pathway: The mammalian target of rapamycin (mTOR) pathway, particularly mTOR complex 1 (mTORC1) regulates cell growth, proliferation, and metabolism. It also plays a significant role in SASP regulation, primarily at the translational level. mTORC1 promotes the translation of specific mRNAs encoding SASP components, including the upstream SASP regulator IL-1α [173,175,176]. Inhibitors of mTOR, such as rapamycin, are potent senomorphics that suppress SASP production [173,174].
p38 MAPK Pathway: The stress-activated p38 mitogen-activated protein kinase (MAPK) pathway is triggered by various cellular stresses, including those that induce senescence. p38 MAPK contributes to both senescence entry and SASP regulation, partly through stabilizing mRNAs of SASP factors and potentially converging with NF-κB activation [63]. Inhibitors of p38 MAPK have demonstrated senomorphic activity by reducing SASP secretion [173].
JAK/STAT Pathway: The Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway is implicated in regulating specific SASP components, particularly those involved in immunosuppression [175]. JAK inhibitors, such as ruxolitinib, have shown promise as senomorphics by suppressing SASP factors and alleviating frailty in preclinical models [177].
ATM Pathway: Ataxia telangiectasia mutated (ATM), a key kinase in the DDR pathway, is persistently activated in many SnCs. Beyond its role in cell cycle arrest, ATM contributes to SASP regulation, potentially through modulating NF-κB activity [178]. ATM inhibitors have shown senomorphic potential by reducing SASP expression in vitro and in vivo [173].
By targeting these central regulatory nodes, senomorphics can potentially normalize the microenvironment perturbed by SnCs, reduce chronic inflammation, and restore aspects of tissue homeostasis.
5.3. Major Classes and Examples of Senomorphic Agents
Research into senomorphics has identified promising candidates from diverse sources, including natural products, repurposed drugs originally developed for other indications, and novel synthetic molecules specifically designed to target senescence pathways (Table 3).
- Natural Compounds and Derivatives
Rapamycin (Sirolimus): Isolated from Streptomyces hygroscopicus found on Easter Island, rapamycin is the archetypal senomorphic [174]. Its mechanism involves inhibiting mTORC1 signaling [176]. Extensive preclinical studies have demonstrated its ability to suppress SASP components, reduce cellular senescence markers, ameliorate age-related dysfunction in various models, and extend lifespan in species ranging from yeast to mice [173,174]. Despite its promise, clinical use for broad anti-aging purposes is hampered by potential side effects including metabolic dysregulation and immunosuppression, possibly linked to off-target inhibition of mTORC2 [173]. Development of rapalogs with improved specificity and pharmacokinetic profiles is ongoing [173].
Resveratrol: This polyphenol, found in grapes and other plants, activates SIRT1, an NAD+-dependent deacetylase involved in regulating metabolism, stress resistance, and aging [173]. Resveratrol exhibits complex, dose-dependent effects. At lower concentrations, it often acts as a senomorphic, suppressing SASP factors (e.g., by inhibiting NF-κB and activating Nrf2 pathways) and preventing senescence induction [173]. At higher concentrations, it can act as a pro-oxidant and induce senescence or apoptosis [173]. Its efficacy in extending lifespan in mice appears context-dependent (e.g., beneficial on high-fat diets but not standard diets), and its poor bioavailability remains a challenge [173]. Sirtuin-activating compounds (STACs) with potentially improved properties are under development [173].
Curcumin: The primary bioactive compound in turmeric, curcumin exhibits broad biological activities. It has demonstrated senomorphic potential by down-regulating Nrf2 and NF-κB pathways involved in SASP production [173]. However, like resveratrol, curcumin suffers from poor bioavailability, limiting its systemic efficacy. Its analogue EF-24, while having improved bioavailability, is primarily characterized as a senolytic targeting BCL-2 family proteins [173].
Other Flavonoids (Apigenin, Kaempferol, Quercetin): Apigenin and kaempferol have shown senomorphic activity, potentially via inhibiting IRAK1/IκBα/NF-κB signaling [173]. Quercetin, while predominantly known as a senolytic, also exhibits senostatic properties in certain contexts, such as suppressing proinflammatory responses when delivered via functionalized nanoparticles [173,179]. This highlights the potential dual roles of some compounds.
Niacinamide (Vitamin B3) and Hyaluronic Acid: A recent study demonstrated that a topical formulation combining 6% niacinamide and hyaluronic acid fragments exerted senomorphic effects in human skin biopsies taken after two months of clinical application [180]. This was evidenced by significant downregulation of SASP-related genes, including the DAMPs S100A8 and S100A9, MMP12, and the chemokine CXCL9. These molecular changes correlated positively with observed clinical improvements in skin radiance, smoothness, and homogeneity, providing a direct link between senomorphic action and cosmetic benefit in skin aging [180].
5.4. Repurposed Drugs
Metformin: This biguanide drug, a cornerstone of type 2 diabetes treatment, is perhaps the most extensively studied repurposed drug for geroprotection [173,174]. Its senomorphic effects are thought to be mediated through multiple pathways, including AMPK activation, inhibition of NF-κB signaling, and modulation of STAT3, DICER1, and Nrf2/GPx7 activity [181]. It reduces SASP factors in various cell types and improves health span in model organisms [173,174]. The ongoing TAME trial is specifically designed to test its efficacy in targeting fundamental aging processes in humans [173].
Statins: Primarily used for lowering cholesterol, certain statins (atorvastatin, pravastatin, pitavastatin, simvastatin) have shown potential to inhibit oxidative stress-induced endothelial senescence and suppress SASP factors [173]. Proposed mechanisms include Akt activation leading to eNOS/SIRT1 upregulation, and inhibition of protein prenylation affecting Rho GTPase activity, which can modulate actin dynamics and potentially SASP secretion [173]. However, potential side effects like muscle problems and increased diabetes risk need consideration [173].
Aspirin: This common non-steroidal anti-inflammatory drug has shown complex, context-dependent effects on senescence. While some studies suggest it can inhibit senescence and SASP in endothelial cells, others indicate it can induce senescence in colorectal cancer cells [173]. Its senomorphic potential requires further investigation to clarify dose- and cell-type-specific effects.
JAK Inhibitors (Ruxolitinib): Given the role of JAK/STAT signaling in mediating responses to SASP cytokines and potentially regulating immunosuppressive SASP components, JAK inhibitors like ruxolitinib have been tested as senomorphics. Ruxolitinib suppressed SASP production and alleviated frailty in aged mice [173], supporting this pathway as a viable senomorphic target.
ATM Inhibitors (KU-55933, KU-60019): Since persistent ATM activity contributes to senescence and SASP, its inhibition has been explored. ATM inhibitors reduced NF-κB activation and SASP expression, alleviating senescence phenotypes in vitro and in Ercc1−/Δ progeroid mice [173]. However, concerns about potentially increasing cancer risk due to inhibiting a key DNA repair kinase necessitate careful evaluation [173].
p38 MAPK Inhibitors (SB203580, UR13756, BIRB796): Targeting the p38 MAPK pathway, which is activated by senescence-inducing stress and contributes to SASP regulation, is another senomorphic strategy. Inhibitors of p38 MAPK or its downstream effector MK2 effectively suppressed SASP secretion in various SnC models [173].
- Novel Synthetic Compounds
NF-κB Inhibitors (SR12343): Directly targeting the central SASP regulator NF-κB is a rational approach. The small molecule SR12343, developed as a specific IKK/NF-κB inhibitor, successfully reduced senescence and SASP in vitro and improved tissue pathologies and healthspan in aged and progeroid mouse models, demonstrating the potential of synthetic senomorphics [173].
Research is actively pursuing other synthetic molecules targeting SASP regulatory pathways or specific SASP components, aiming for greater potency, selectivity, and improved drug-like properties compared to existing agents.
5.5. Therapeutic Potential and Limitations of Senomorphism
Senomorphics offer a distinct therapeutic paradigm for tackling aging and age-related diseases. By focusing on neutralizing the harmful environment created by SnCs rather than eliminating the cells themselves, they may provide a safer long-term strategy, particularly considering the potential beneficial roles of SnCs and the risks associated with broad senolysis [173,174]. This approach could be particularly valuable for chronic conditions where sustained suppression of inflammation and tissue degradation is desired. Evidence from preclinical models using agents like rapamycin, metformin, and specific pathway inhibitors (NF-κB, JAK, ATM) suggests that senomorphic interventions can indeed improve healthspan and alleviate symptoms associated with aging and specific diseases [173]. The recent demonstration of clinical benefit from a topical senomorphic formulation in skin aging further supports this potential [180].
Despite this promise, the development and application of senomorphics face significant hurdles [173]. A primary challenge lies in the complexity and context-dependency of the SASP [9]. Indiscriminate suppression of all SASP factors may be detrimental, as some components might have protective or necessary physiological functions (e.g., immune recruitment, tissue repair signals). Achieving selective modulation of only the pathogenic SASP components requires a much deeper understanding of SASP composition and regulation in different physiological and pathological contexts. Long-term safety is a major concern, especially for agents targeting fundamental pathways like mTOR, NF-κB, or ATM, which have vital roles beyond SASP regulation. Chronic inhibition could lead to unforeseen adverse effects, including impaired immunity, metabolic disturbances, or potentially increased cancer risk [173]. Developing robust biomarkers to specifically monitor senomorphic activity (i.e., effective SASP suppression) in vivo and in clinical trials is crucial for assessing efficacy and guiding dosing regimens, yet such biomarkers are currently lacking. Furthermore, optimizing drug delivery to ensure adequate and sustained levels of senomorphic agents in target tissues while minimizing systemic exposure remains a significant pharmacokinetic challenge [173]. Finally, the heterogeneity of senescence itself means that the effectiveness of a given senomorphic might vary considerably depending on the specific type of SnCs present and the underlying cause of their senescence. Overcoming these limitations will require continued basic research into SASP biology, careful preclinical safety and efficacy testing, innovative drug design and delivery strategies, and thoughtfully designed clinical trials.
5.6. Senolytics
Senotherapeutics encompass two main strategies for targeting cellular senescence: senolysis and senomorphism. Senolytics are compounds designed to selectively induce apoptosis in SnCs, thereby reducing the burden of these cells in tissues [173,174]. This approach contrasts with senomorphics (or senostatics), which aim to suppress the deleterious SASP without necessarily eliminating the cells themselves [173,179]. The rationale behind senolysis is that the removal of SnCs can alleviate their detrimental contributions to aging and age-related diseases, potentially improving tissue function and extending healthspan [173,174] (Figure 4).
5.7. Mechanisms of Senolytic Action
The selective action of senolytics relies on exploiting the vulnerabilities inherent in the senescent state. To survive despite accumulating cellular damage and producing a potentially self-toxic SASP, SnCs upregulate a network of pro-survival pathways, collectively termed senescent cell anti-apoptotic pathways (SCAPs) [173,182,183]. These pathways confer resistance to apoptosis. Senolytics function by transiently disabling these specific SCAPs, rendering SnCs susceptible to their pro-apoptotic microenvironment or internal damage signals [173,174].
Bioinformatic analyses of senescent versus non-senescent cells identified several key SCAPs, including pathways involving Ephrins, PI3K/AKT, p53/p21Cip1/Waf1/Serpins, HIF-1α, and notably, the BCL-2 family of anti-apoptotic proteins [173,174,182]. The dependency on specific SCAPs can vary between different types of SnCs, highlighting the heterogeneity of senescence and suggesting why certain senolytics exhibit cell-type specificity [173,182]. For instance, senescent human umbilical vein endothelial cells (HUVECs) rely on BCL-xL, while senescent human preadipocytes do not, making the former susceptible to BCL-xL inhibitors while the latter are resistant [182].
- Major Classes and Examples of Senolytics
Several classes of compounds have been identified with senolytic activity, ranging from repurposed drugs to natural products and novel modalities.
- Early Senolytics (Dasatinib and Quercetin)
The first senolytics reported, dasatinib (D) and quercetin (Q), were identified through a hypothesis-driven, bioinformatics-based approach targeting SCAPs [183]. Dasatinib is a tyrosine kinase inhibitor used clinically for leukemia, while quercetin is a natural flavonoid known to interact with PI3K and Bcl-2 family members [173,174,184]. While each compound has senolytic activity against specific cell types (e.g., dasatinib against senescent preadipocytes, quercetin against senescent HUVECs), their combination (D+Q) exhibits broader efficacy, targeting a wider range of SnCs and often showing synergistic effects [173,174]. Preclinical studies demonstrated that D+Q administration can alleviate numerous age-related dysfunctions in mice, including improving cardiovascular function, reducing osteoporosis, mitigating frailty, and improving physical function even when administered late in life [174]. Furthermore, D+Q treatment has shown benefits in models of atherosclerosis, pulmonary fibrosis, hepatic steatosis, neurodegeneration, and metabolic dysfunction associated with diabetes or obesity [72,173,174]. D+Q treatment also protected against cisplatin-induced ovarian injury by removing SnCs [73].
- Natural Products
Fisetin: A flavonoid structurally related to quercetin, found in fruits and vegetables, has shown potent senolytic activity [173,185]. It targets multiple pathways including PI3K/AKT and Bcl-2 family members [185]. Fisetin selectively induces apoptosis in senescent HUVECs and certain fibroblasts, but not primary human preadipocytes [173,185]. In vivo studies reported that fisetin extends healthspan and lifespan in mice [186].
Gingerenone A: A component isolated from ginger extract, was recently identified as a novel senolytics [187]. It selectively eliminated senescent human fibroblasts (WI-38) without significant effect on proliferating cells. Its mechanism involves inducing apoptosis via caspase-3 activation and reducing the anti-apoptotic protein Bcl-xL, acting independently of p53. Gingerenone A also exhibited senomorphic properties by suppressing aspects of the SASP, including IL-6 secretion [187].
Other natural products like piperlongumine and curcumin analogues (EF-24) have also been reported as senolytics [173,174,188].
- Bcl-2
- l-2 Family Inhibitors
Given the role of anti-apoptotic Bcl-2 family proteins (Bcl-2, Bcl-xL, Bcl-w) in SnC survival, inhibitors of these proteins have been investigated as senolytics.
Navitoclax (ABT-263): An orally bioavailable inhibitor of Bcl-2, Bcl-xL, and Bcl-w, demonstrated senolytic activity in multiple cell types (senescent HUVECs, IMR90 fibroblasts, MEFs) but notably not in primary human preadipocytes [182]. In vivo, Navitoclax treatment reduced SnC burden and rejuvenated aged hematopoietic stem cells in mice [189]. However, its clinical translation for senolysis is hampered by significant dose-limiting toxicities, particularly thrombocytopenia and neutropenia, likely due to its inhibition of Bcl-xL in platelets and Bcl-2 in neutrophils [173,182].
Selective Bcl-xL Inhibitors: Agents like A1331852 and A1155463, which primarily target Bcl-xL, also exhibit senolytic activity in HUVECs and IMR90 cells but not preadipocytes [173,174,185]. By sparing Bcl-2, they are hypothesized to have reduced hematological toxicity compared to navitoclax, although this requires further investigation [173].
- HSP90
- P90 Inhibitors:
Heat shock protein 90 (HSP90) is a chaperone crucial for stabilizing numerous client proteins involved in cell survival and proliferation, including AKT [190]. Several HSP90 inhibitors, such as 17-DMAG (alvespimycin), geldanamycin, and 17-AAG (tanespimycin), originally developed as anti-cancer agents, were identified as a novel class of senolytics [174,191]. These compounds induce apoptosis in various types of senescent cells (e.g., MEFs, human fibroblasts, HUVECs). Mechanistically, they disrupt the HSP90-AKT interaction, leading to the destabilization and degradation of active AKT [190,191]. In vivo, intermittent treatment with 17-DMAG reduced tissue senescence markers and extended healthspan in the Ercc1−/Δ progeroid mouse model [191].
- Novel Therapeutic Modalities
Beyond small molecules, innovative approaches are being developed.
Senolytic CAR-T Cells: Chimeric antigen receptor (CAR) T cell technology has been adapted for senolysis. By redirecting T cells to target proteins upregulated on the surface of SnCs, such as urokinase plasminogen activator receptor (uPAR), senolytic CAR T cells can effectively eliminate SnCs [192]. In preclinical models of physiological aging and diet-induced obesity, a single administration of anti-uPAR CAR T cells led to long-lasting clearance of uPAR-positive senescent cells, improved metabolic function (including glucose tolerance and insulin sensitivity), and enhanced physical capacity. Notably, these CAR T cells persisted for extended periods and demonstrated prophylactic effects, preventing metabolic decline when administered early in life [193]. This approach highlights the potential for durable effects from cell-based senolytic therapies.
- Other Classes:
Additional classes with senolytic activity include cardiac glycosides (e.g., ouabain, digoxin) which inhibit Na+/K+-ATPase [193], and compounds targeting the p53 pathway, such as FOXO4-DRI peptides which disrupt the FOXO4-p53 interaction [194], and inhibitors of MDM2 or USP7 which stabilize p53 [173]. Aspirin has also been reported to have senolytic properties in certain contexts [174].
Beyond the knoqn classes of senolytics such as tyrosine kinase inhibitors (Dasatinib), flavonoids (Quercetin, Fisetin), and Bcl-2 family inhibitors (Navitoclax), several novel strategies have recently emerged to improve selectivity and efficacy while minimizing off-target effects. For instance, β-galactosidase–activated prodrugs such as Galacto-Navitoclax exploit the elevated SA-β-Gal activity in senescent cells to release cytotoxic payloads selectively, demonstrating enhanced precision in preclinical models [195]. Similarly, PROTAC-based senolytics like ARV825 have been designed to degrade anti-apoptotic or epigenetic survival proteins specifically in senescent cell populations, offering a promising next-generation approach [192].
Additionally, MDM2 inhibitors such as Nutlin-3a can reactivate p53 pathways to induce apoptosis in senescent or stressed cells [189]. Selective targeting of other anti-apoptotic proteins is also gaining traction; for example, Mcl-1 inhibitors (e.g., S63845) have shown potential for removing resistant senescent cells, though primarily explored in oncology thus far [196]. Finally, the development of nanoparticle-encapsulated senolytics, including lung-targeted formulations of Quercetin or Dasatinib, represents an innovative strategy to localize senolytic activity, potentially reducing systemic toxicity and improving therapeutic index for lung diseases [197].
Together, these emerging modalities expand the senotherapeutic arsenal and underscore the need for disease-specific delivery systems and rigorous preclinical validation, particularly in fibrotic lung diseases such as IPF.
- Delivery Mechanisms for Senotherapeutics
Effective and targeted delivery remains a challenge for many senotherapeutics, particularly natural products with poor bioavailability or agents with potential off-target toxicities. Nanotechnology offers promising solutions.
Nanoparticles: Various nanoparticle formulations are being explored to improve senolytic delivery, stability, and targeting [179,184]. For example, quercetin surface-functionalized magnetic iron oxide nanoparticles (MNPQ) exhibited both senolytic and senostatic activity in cultured human fibroblasts undergoing oxidative stress-induced senescence, mediated partly through AMPK activation [179]. Nanoparticles can be engineered with specific ligands or properties to enhance accumulation in target tissues or uptake by SnCs [174,184].
Exosomes: These naturally occurring extracellular vesicles can act as delivery vehicles. Engineering exosomes to carry senolytic drugs represents another strategy for targeted therapy, although primarily explored in cancer contexts thus far [198].
Galactose-Modified Prodrugs: This approach leverages the increased senescence-associated β-galactosidase (SA-β-gal) activity found in the lysosomes of many SnCs. Cytotoxic drugs are conjugated to a galactose moiety, rendering them inactive. Upon uptake by SnCs, the elevated SA-β-gal activity cleaves the galactose, releasing the active drug selectively within the senescent cell [173,174]. Examples include Nav-Gal (a navitoclax prodrug) and SSK1 (a gemcitabine prodrug), which demonstrated enhanced specificity and reduced toxicity compared to the parent compounds in preclinical models [199].
Antibody-drug conjugates (ADCs): Building on recent findings by Amor et al. (2021), ADCs that target senescent cell surface markers, such as β₂-microglobulin, offer a promising strategy for treating lung diseases. The study showed that selective clearance of senescent cells using a B2M-directed ADC effectively reduced SASP-mediated inflammation and tissue remodeling. This precision-targeted approach holds therapeutic potential for managing chronic lung conditions, such as IPF and COPD, while minimizing off-target effects. Such strategies may ultimately help restore lung homeostasis and counter age-related pulmonary decline [200].
- Therapeutic Applications and Strategies
The broad involvement of SnCs in numerous pathologies suggests that senolytics could have wide-ranging therapeutic applications, including for osteoarthritis, neurodegenerative diseases (e.g., Alzheimer's disease), metabolic disorders (e.g., type 2 diabetes, obesity-related dysfunction), cardiovascular diseases, idiopathic pulmonary fibrosis, and frailty [66,72,173,174,193].
Given the heterogeneity of SnCs and their SCAPs, combination strategies are being considered. Combining senolytics with different mechanisms or targets (e.g., D+Q) may broaden the spectrum of SnCs eliminated and enhance overall efficacy [173,174]. Additionally, combining senolytics with senomorphics could offer a dual approach, reducing SnC burden while also suppressing the SASP from remaining or newly formed SnCs [173].
A key strategic advantage proposed for senolytics is the potential for intermittent dosing. Since SnCs accumulate relatively slowly, periodic administration ("hit-and-run" approach) might be sufficient to maintain a low SnC burden and achieve therapeutic benefits, while minimizing the potential for side effects associated with continuous drug exposure [72,174]. The long-lasting effects observed with senolytic CAR T cells further support the feasibility of intermittent or even single-dose curative therapies [193].
Despite significant progress, the development of senolytics faces challenges. The heterogeneity of SnCs necessitates careful selection of senolytic agents based on the specific cell types involved in a particular disease [173,174]. Off-target toxicity remains a concern, as exemplified by navitoclax, underscoring the need for highly selective agents or targeted delivery systems [182]. Furthermore, long-term safety data, particularly concerning potential effects on tissue repair, immune function, and tumorigenesis, are still needed [66,174]. Ongoing research focusing on identifying more specific senolytic targets, developing novel delivery platforms like PROTACs and advanced nanocarriers, refining immunotherapy approaches, and conducting well-designed clinical trials will be crucial for realizing the full therapeutic potential of senolytics in combating age-related diseases [173,174,193].
- Current Status of Clinical Trials
The field of senotherapeutics has witnessed rapid translation from preclinical discoveries to initial human clinical trials, representing a significant step towards targeting fundamental aging processes for the treatment of age-related diseases [173,175,201]. While still in early stages, clinical investigations are exploring the potential of senolytic agents, which selectively eliminate SnCs, and senomorphic agents, which modulate the SASP, across a spectrum of conditions [173,174].
Several senolytic agents, primarily repurposed drugs or natural compounds, are currently under investigation in human clinical trials [173,201]. The combination of the tyrosine kinase inhibitor dasatinib (D) and the flavonoid quercetin (Q) was among the first senolytic strategies to enter clinical testing [173,201]. Early phase trials have yielded encouraging, albeit preliminary, results. The first-in-human, open-label pilot study investigated intermittent D+Q administration in patients with idiopathic pulmonary fibrosis (IPF), a fatal lung condition associated with SnC accumulation [173,201]. This study reported improvements in physical function, such as gait speed and chair-rise times, although direct evidence of SnC clearance in the lung was not assessed [94,201]. Another phase 1 trial evaluated D+Q in patients with diabetic kidney disease (DKD). Findings indicated that a short course of D+Q reduced the burden of senescent cells, measured by markers like p16Ink4a, p21Cip1/Waf1, and SA-β-gal activity, in subcutaneous adipose tissue biopsies collected 11 days after treatment cessation [201,202]. This study also observed a reduction in circulating SASP factors [201]. Ongoing trials are further evaluating D+Q in conditions including Alzheimer's disease, chronic kidney disease, frailty in hematopoietic stem cell transplant survivors, and skeletal health [173,201].
Fisetin, another naturally occurring flavonoid related to quercetin, has also entered clinical investigation based on promising preclinical data demonstrating its senotherapeutic potential and ability to extend healthspan and lifespan in mice [186]. Current trials are assessing fisetin's efficacy in conditions including frailty, chronic kidney disease, osteoarthritis, and COVID-19 [201]. Additionally, trials involving adult survivors of childhood cancer are planned [201]. Results from these fisetin trials are largely pending but anticipated with significant interest.
Other senolytic agents, such as BCL-2 family inhibitors (for example, navitoclax (ABT-263)), have also been identified and show promise in preclinical settings [173,175,182], although their clinical development specifically for senescence-related indications is less advanced compared to D+Q and fisetin, partly due to concerns about side effects like thrombocytopenia [203]. The development pipeline includes diverse agents targeting various SCAPs [173,174].
- Senotherapeutics Challenges and Opportunities
Despite the promising advances, the clinical development of senotherapeutics faces significant challenges [173,174]. One major hurdle is the delivery and targeting of senotherapeutics. Achieving selective elimination of senescence cells (SnCs) while minimizing damage to healthy, non-senescent cells is critical for safety and efficacy [66,175]. The heterogeneity of SnCs across different tissues, developmental stages, and disease contexts further complicates targeted delivery [175]. Innovations in drug delivery systems offer potential solutions. Nanotechnology, including the use of nanoparticles and exosomes, is being explored to enhance the targeted delivery of senotherapeutics [179,184,198]. For instance, quercetin functionalized onto Fe3O4 nanoparticles has shown senolytic activity in vitro [179], and engineered exosomes have been investigated as natural drug delivery vehicles for cancer therapy, a concept potentially adaptable to senotherapeutics [198]. Furthermore, galactose-modified prodrugs, designed to be activated specifically by the high β-galactosidase activity characteristic of many SnCs, represent another promising strategy to improve target specificity [204].
Safety considerations are paramount, particularly as senotherapeutics may eventually be considered for chronic or preventive use in aging populations [66,201]. Potential off-target effects remain a concern, as senolytics often target pathways crucial for the survival of other cell types, exemplified by the thrombocytopenia associated with certain BCL-2 inhibitors like navitoclax [173,203]. Modulation of the immune system, either directly by the drug or indirectly via alteration of the SASP or removal of SnCs that interact with immune cells, requires careful evaluation [66]. Long-term safety data are currently lacking, as clinical trials have involved relatively short-term interventions [201]. Robust biomarkers are needed not only to demonstrate target engagement (SnC clearance) but also to monitor potential adverse effects during clinical development [9,175].
- Biomarker gaps: Need for in vivo markers of senescence in lungs
Despite increasing interest in targeting senescence for the treatment of chronic lung diseases, a major translational barrier lies in the absence of robust, specific, and non-invasive in vivo biomarkers capable of reliably detecting senescent cells within lung tissue. Current identification strategies predominantly rely on ex vivo or histological markers such as p16Ink4a, p21Cip1/Waf1, senescence-associated β-galactosidase (SA-β-gal) activity, and DNA damage indicators like γH2AX [1]. However, these markers are hindered by limited specificity, restricted tissue accessibility, and an inability to differentiate between transient (e.g., pseudosenescent) and truly senescent states, especially in complex tissues such as lung.
Efforts to develop in vivo imaging tools, including PET tracers that detect SA-β-gal activity (e.g., Galacto-rhodamine-based probes), are underway but currently lack validation in lung-specific disease models [205]. Meanwhile, transcriptomic-based senescence classifiers, such as SenMayo and SeneScore, have shown promise in characterizing senescent cell populations using bulk and single-cell RNA-sequencing data across tissues [9,206,207,208,209]. Nevertheless, these molecular signatures remain largely experimental and not yet adaptable for clinical use, particularly in the lung.
Emerging fluid-based biomarkers in bronchoalveolar lavage fluid (BALF), sputum, and serum such as SASP components and circulating mitochondrial DNA (mtDNA) released by senescent cells represent a promising non-invasive approach. However, these candidates remain insufficiently validated for lung-specific applications, and their capacity to distinguish between deleterious versus reparative senescence is unclear.
Altogether, these limitations underscore an urgent need for the development of lung-specific, senescence-selective biomarkers that can: discriminate senescent from non-senescent cells in vivo, & differentiate pathologic from physiologic senescence, and serve as pharmacodynamic markers for therapeutic response to senolytic or senomorphic agents.
6. Underexplored Aspects
Building upon these foundational insights, several emerging areas have garnered increasing attention, underscoring the need for further investigation to comprehensively delineate the multifaceted role of cellular senescence in lung disease pathogenesis and progression. Key among these are: (i) the dynamic crosstalk between senescent cells and the immune system, including mechanisms of immune evasion and immune-mediated clearance; (ii) cell type specific senescence trajectories across distinct pulmonary compartments; (iii) the reversibility and phenotypic plasticity of senescent states under varying physiological and pathological conditions; and (iv) the contribution of biomechanical stressors such as altered matrix stiffness and disrupted tissue architecture to senescence induction and maintenance. In parallel, recent advances in high-dimensional omics platforms (e.g., single-cell transcriptomics, spatial proteomics) and therapeutic innovations (e.g., senolytics, senomorphics, and nanoparticle-mediated drug delivery) are paving the way for precision-targeted interventions and the identification of lung-specific senescence biomarkers. Collectively, these developments hold promise for reshaping our understanding of senescence as both a pathogenic driver and a therapeutic target in chronic lung diseases.
6.1. Senescence and Immune Cell Crosstalk in the Lung
Senescent epithelial and stromal cells in the lung actively modulate the immune microenvironment by secreting SASP factors that influence both innate and adaptive immune cell function.
Modulation of Alveolar Macrophages
Senescent epithelial and stromal cells profoundly reshape immune dynamics in the lung through the secretion of a complex SASP, comprising pro-inflammatory cytokines (e.g., IL-6, IL-8), chemokines (e.g., CCL2, CXCL1), growth factors, matrix-remodeling enzymes, and extracellular vesicles. A principal target of this signaling is the alveolar macrophage, which is both recruited and reprogrammed by SASP factors into a dysfunctional or M1-like pro-inflammatory state. These macrophages exhibit impaired phagocytic capacity and increased production of inflammatory mediators, thereby establishing a self-perpetuating loop of chronic inflammation and defective resolution. This phenomenon has been demonstrated in aging lungs and KRAS-driven lung cancer models [129,210,211].
Senescent stromal cells, particularly fibroblasts and mesenchymal stromal cells, contribute to this reprogramming by secreting key SASP components such as IL-6, TNF-α, GM-CSF, and CCL2, which not only enhance macrophage recruitment but also skew their phenotype toward a pro-inflammatory state. In the context of fibrosis and lung aging, these macrophages adopt an M1-like profile that sustains inflammation and impairs tissue repair. This maladaptive feedback loop has been well documented in models of idiopathic pulmonary fibrosis and senescence-associated lung injury [212,213,214,215].
Modulation of T Cells
Senescent stromal cells significantly impact adaptive immunity, particularly by modulating T cell responses. While early SASP-associated chemokines can facilitate T cell infiltration, sustained exposure to SASP factors leads to T cell dysfunction. Senescent stromal cells upregulate immune checkpoint ligands such as PD-L1 and secrete immunosuppressive cytokines including TGF-β and IL-10, which collectively suppress CD8⁺ T cell proliferation, cytotoxic activity, and promote the expansion of regulatory T cells. This immunosuppressive signaling contributes to T cell exhaustion and blunted anti-tumor responses in both chronic lung disease and malignancy settings [216,217,218,219].
In the tumor microenvironment, senescent cancer-associated fibroblasts (CAFs) have been shown to suppress T cell function through both paracrine SASP signaling and direct immune checkpoint engagement. This facilitates immune evasion and supports tumor progression [216,217,220]. Such chronic immunosuppression also impairs tissue regeneration and promotes persistent pathological remodeling in non-malignant fibrotic lung disease.
Modulation of Neutrophils
Senescent epithelial and stromal cells are potent recruiters of neutrophils through the secretion of SASP-associated chemokines such as IL-8 (CXCL8), CXCL1, and CXCL5 [221,222]. Upon recruitment, neutrophils become hyperactivated, releasing neutrophil elastase, matrix metalloproteinases (MMPs), and reactive oxygen species (ROS), which contribute to extracellular matrix degradation and tissue injury. This cascade has been well-characterized in both acute lung injury and chronic inflammatory models, where senescent cells amplify neutrophilic infiltration and drive progressive damage [139,223].
Recent studies further underscore this mechanism; for example, senescent fibroblasts were shown to enhance neutrophil recruitment and activation, perpetuating a feed-forward loop of inflammation and tissue destruction [224].
The Role of Immunosenescence in Worsening Chronic Lung Disease Outcomes
Immunosenescence plays a pivotal role in the progression and exacerbation of chronic lung diseases, such as COPD, IPF, and post-viral lung injury [225]. This process is characterized by impaired immune surveillance, a persistent low-grade inflammatory state termed “inflammaging,” and compromised tissue repair mechanisms.
In the innate immune compartment, aging leads to functional deficits in alveolar macrophages, neutrophils, and dendritic cells. Aged alveolar macrophages exhibit impaired phagocytosis and efferocytosis, resulting in defective clearance of pathogens and apoptotic cells. These cells also adopt a pro-inflammatory phenotype, producing elevated levels of TNF-α, IL-6, and IL-1β, thereby perpetuating tissue injury and fibrosis [226]. Similarly, aged neutrophils demonstrate delayed apoptosis, enhanced degranulation, and excessive reactive oxygen species (ROS) production, all of which contribute to alveolar damage and extracellular matrix degradation [227,228,229].
The adaptive immune system is equally affected by aging. CD4⁺ and CD8⁺ T cells show reduced clonal diversity, increased expression of exhaustion markers such as PD-1 and KLRG1, and diminished effector cytokine secretion, leading to impaired antiviral responses and delayed resolution of inflammation [230,231]. Age-related B cell dysfunction results in decreased antibody diversity and reduced responsiveness to infections and vaccinations. Notably, immunosenescence exacerbates the SASP generated by both immune and stromal cells. This sustained inflammatory milieu fosters epithelial mesenchymal transition (EMT), fibroblast activation, and extracellular matrix deposition key processes driving chronic inflammation and fibrosis in the lung [13,36,53,226,232].
SASP as a Driver of Immune Dysfunction in Aging Lungs
In the aging lung, persistent activation of the SASP plays a pivotal role in disrupting immune homeostasis. SASP chemokines actively recruit innate immune cells such as neutrophils and monocytes; however, the chronic inflammatory environment they establish often skews these cells toward dysfunctional or even senescent states. For instance, alveolar macrophages in aged lungs exhibit impaired phagocytic function and elevated production of inflammatory cytokines, exacerbating tissue damage rather than facilitating resolution [226].
In parallel, SASP components negatively impact adaptive immunity by impairing T cell activation, survival, and effector function. This occurs through the upregulation of immune checkpoint molecules (e.g., PD-L1) and the enrichment of immunosuppressive cytokines, ultimately promoting T cell exhaustion and blunted antiviral responses in older individuals [216,233].
Furthermore, SASP factors compromise epithelial barrier integrity and disrupt tissue repair mechanisms, facilitating increased neutrophilic infiltration and perpetuating local inflammation. This sustained, low-grade inflammatory state termed "inflammaging" has been implicated in the pathogenesis and progression of several age-related pulmonary diseases, including COPD, IPF, and post-infectious fibrotic remodeling [229,234,235]. Collectively, these effects position the SASP as a central driver of immune dysregulation in the aging lung, transforming a homeostatic and regenerative immune landscape into one marked by chronic inflammation, impaired resolution, and progressive tissue dysfunction.
6.2. Senescence Heterogeneity Across Lung Cell Types
Senescence in the lung represents a highly heterogeneous phenomenon, with distinct molecular signatures, functional consequences, and SASP profiles emerging across diverse cell types. Single-cell transcriptomic analyses have demonstrated that lung epithelial cells, endothelial cells, fibroblasts, and immune cells each adopt unique senescence programs, shaped by their specialized functions and stress responses within the tissue microenvironment [55,236,237]. The advent of high-throughput single-cell and single-nucleus RNA sequencing has enabled unprecedented resolution in characterizing these senescent states and identifying novel cell-type-specific markers [54].
For instance, using high-content imaging, Neri et al. examined senescence marker expression in primary human endothelial cells and fibroblasts and identified that G2-arrested senescent cells exhibited higher expression of canonical markers, greater IL-6 secretion, and increased sensitivity to the senolytic ABT263, compared to G1-arrested counterparts [238]. Functionally, senescent alveolar epithelial cells impair barrier integrity and reduce surfactant production, whereas senescent endothelial cells contribute to vascular permeability and leukocyte extravasation through pro-inflammatory SASP activity [57,239,240,241]. Senescent fibroblasts frequently adopt a pro-fibrotic phenotype characterized by secretion of TGF-β, IL-6, and extracellular matrix proteins that promote fibrogenesis [242].
Beyond intercellular differences, intra-lineage heterogeneity has also been documented. Single-cell analyses utilizing tools such as SenePy have revealed that even within the same cell type, distinct senescent “states” exist, each defined by variable transcriptional outputs and responses to stress stimuli [55,243]. These differences are influenced by the nature of the senescence-inducing insult whether DNA damage, oxidative stress, or oncogenic activation each eliciting a context-specific senescence program. Proteomic and transcriptomic profiling confirm that the SASP composition is not only cell-type specific but also inducer-dependent, with senescent fibroblasts producing a cytokine and matrix-modifying profile distinct from that of senescent epithelial or endothelial cells [6].
To better navigate this complexity, transcriptional marker panels such as SenMayo have been developed to detect senescent cells across multiple lung cell populations by targeting conserved features of the senescence program. Nonetheless, expression of individual markers, such as CDKN2A or p21Cip1/Waf1, can still vary substantially based on both cell lineage and senescence trigger [244]. Collectively, these findings emphasize that lung cellular senescence is a non-uniform, context-dependent process that requires precise, cell-type aware identification and interpretation for both mechanistic studies and the development of targeted senotherapeutics.
Fibroblasts, endothelial cells, pericytes, and club cells may undergo functionally distinct senescence.
Senescence manifests in a functionally distinct and cell-type specific manner across various structural and stromal compartments of the lung, contributing to the overall heterogeneity of the senescent landscape. Among these, fibroblasts are well-characterized for acquiring a pro-fibrotic and pro-inflammatory phenotype upon senescence. This phenotype is typified by elevated secretion of TGF-β, IL-6, and matrix metalloproteinases (MMPs), which not only reinforce fibrotic matrix deposition but also influence immune cell recruitment and extracellular matrix (ECM) remodeling processes prominently observed in idiopathic pulmonary fibrosis [245].
In contrast, senescent endothelial cells are primarily implicated in the disruption of vascular homeostasis. They promote vascular permeability, rarefaction, and leukocyte adhesion through a distinct SASP profile enriched in chemokines such as CXCL1 and adhesion molecules like ICAM-1, collectively contributing to endothelial dysfunction and inflammation [246].
Pericytes, although less extensively studied, have been shown to undergo senescence in response to oxidative and mechanical stress. This impairs their ability to maintain capillary integrity, destabilizes endothelial-pericyte interactions, and promotes vascular leakage, thereby exacerbating pulmonary hypertension and microvascular injury [247].
Club cells, the non-ciliated epithelial progenitors residing in the bronchioles, represent another important but underappreciated senescent population. Following environmental injury or viral infection, club cells may enter a senescent state by loss of proliferative capacity and lack of regenerative potential. These senescent club cells secrete cytokines such as IL-33 and amphiregulin, which alter epithelial–mesenchymal signaling and contribute to chronic airway remodeling [248]. These cell-type-specific senescence responses underscore the complexity and functional diversity of senescent cell populations in the lung, each shaping disease pathogenesis through distinct mechanisms and SASP profiles.
Mapping senescent cell subtypes with single-cell RNA-seq or spatial transcriptomics
Recent advances in single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have profoundly enhanced our understanding of senescence heterogeneity in the lung, enabling high-resolution mapping of senescent cell subtypes within their native tissue microenvironments. A landmark single-cell atlas of the aging lung revealed that senescence-associated transcriptional changes are strikingly cell-type specific, with alveolar macrophages and monocytes displaying the most pronounced alterations, while epithelial and endothelial cells followed distinct senescence trajectories [249, 250]. In aged, influenza-infected mouse lungs, integrated scRNA-seq and spatial transcriptomics approaches identified spatial clustering of senescent cells particularly within fibrotic and peribronchial regions and uncovered cell-type-specific SASP signatures, including CXCL1 secretion by senescent endothelial cells [251].
The SenePy platform, applied to both human and murine lung datasets, identified type II alveolar epithelial cells and fibroblasts as key senescent populations in aged lungs, each expressing distinct senescence-associated genes such as CDKN1A and GDF15 [55]. SenMayo, a robust transcriptomic senescence signature platform, derived from human bulk RNA-seq data, has demonstrated cross-tissue applicability, facilitating senescent cell identification in the lung and other aging organs [244]. To overcome challenges in detecting senescence across diverse biological settings, the human universal senescence index (hUSI) was developed. This integrative framework enables robust identification of senescent states across aging, COVID-19, and cancer by leveraging large-scale transcriptomic datasets and highlights prognostic senescence-linked pathways [252].
Further, scDOT, a multimodal integrative tool, links cellular transcriptomes to spatial coordinates in lung tissues. When applie[249,250d to human IPF samples, it revealed distinct senescent fibroblast niches and their interactions with neighboring immune and epithelial cells [253]. In parallel, SenCID, a machine learning based classification framework, was trained on 602 samples from 52 senescence transcriptome datasets spanning 30 cell types. SenCID categorizes senescent cells into six major senescence identities (SIDs), enabling reconstruction of dynamic senescence trajectories across contexts such as physiological aging, chronic lung diseases, and SARS-CoV-2 infection [243].
6.3. Reversibility of Senescence and Plasticity
While cellular senescence has long been regarded as a terminal, irreversible growth arrest, emerging evidence challenges this notion by revealing that certain senescent states can, under specific conditions, be at least partially reversible. This plasticity appears to be influenced by factors including cell type, the nature and severity of the inducing stressor, the duration of senescence, and the metabolic and epigenetic landscape of the cell.
Experimental studies have shown that transient senescence, particularly in response to sublethal DNA damage or oxidative stress, may be reversible. For example, oxidative stress-induced senescence in lung epithelial cells and hepatic stellate cells can be reversed upon removal of the insult or through modulation of the p53/p21Cip1/Waf1 pathway [254,255]. Similarly, mesenchymal stem cells exposed to reversible senescence stimuli demonstrate restored proliferative capacity when treated with epigenetic modulators [256].
At the molecular level, senescence reversal is associated with restoration of mitochondrial function, metabolic reprogramming, and epigenetic remodeling. Specifically, replenishment of NAD⁺ levels or improvement in mitochondrial health has been shown to suppress SASP expression and restore proliferative competence in certain senescent cell types [257]. These findings underscore that not all senescent states represent a permanent cell fate, and that therapeutic interventions aimed at modulating or reversing early or context-dependent senescence may hold promise in treating fibrosis, cancer, and age-associated dysfunction.
Pseudosenescence: Senescence Marker Expression Without Stable Growth Arrest
The concept of pseudosenescence refers to a transient, senescence-like cellular state in which cells express canonical markers such as p16Ink4a, p21Cip1/Waf1, and SA-β-gal activity without undergoing irreversible growth arrest. Unlike bona fide senescent cells, pseudosenescent cells retain the capacity to re-enter the cell cycle, particularly upon withdrawal of the inducing stimulus or in response to specific microenvironmental signals.
This phenomenon has been observed in epithelial progenitor cells, hematopoietic stem cells, and immune cells, particularly during tissue regeneration or repair processes [26]. For instance, during wound healing, transient expression of p21Cip1/Waf1 and selected SASP components facilitates a regenerative inflammatory response, which resolves as progenitor cells resume proliferation [258].
Pseudosenescence is also implicated in therapy-induced senescence (TIS). Tumor cells exposed to sublethal doses of radiation or chemotherapy may transiently acquire a senescent-like phenotype but subsequently escape arrest, often exhibiting increased malignancy and therapy resistance [259]. This reversibility complicates therapeutic targeting, as pseudosenescent cells may evade senolytic strategies designed to eliminate truly senescent populations. Importantly, pseudosenescence underscores the limitations of relying solely on marker-based definitions of senescence. Functional criteria such as stable growth arrest, irreversible chromatin remodeling (e.g., SAHF formation), and resistance to mitogenic signals are essential to distinguish true senescence from temporary or adaptive phenotypes.
6.4. Role of Mechanical Stress and ECM Stiffness
Changes in the biomechanical properties of the extracellular matrix (ECM) particularly increased stiffness in fibrotic lungs and loss of structural integrity in emphysematous tissue play a critical role in both initiating and reinforcing cellular senescence. In pulmonary fibrosis, excessive deposition of collagen and cross-linked ECM proteins leads to increased tissue stiffness, which exerts abnormal mechanical stress on resident fibroblasts, epithelial cells, and endothelial cells. This mechanical cue is transduced via integrins, focal adhesion kinase (FAK), and the YAP/TAZ signaling axis, culminating in senescence-associated growth arrest and the upregulation of SASP components [260]. Fibroblasts cultured on stiff substrates or isolated from fibrotic lung tissue exhibit a robust senescent phenotype, marked by increased expression of p16Ink4a, IL-6, TGF-β, and matrix metalloproteinases, thereby further promoting ECM remodeling and fibrosis [261].
In contrast, emphysematous lungs are characterized by progressive ECM degradation particularly the loss of elastin and collagen resulting in weakened alveolar architecture and aberrant cell-ECM interactions. This destabilized mechanical environment induces abnormal cellular stretching, mitochondrial dysfunction, and oxidative stress, collectively driving epithelial cell senescence and impairing regenerative capacity [262]. Importantly, senescent cells within these mechanically altered tissues demonstrate enhanced resistance to apoptosis and maintain persistent SASP activity, establishing a self-perpetuating loop of matrix remodeling, inflammation, and senescence [36].
These findings emphasize that ECM mechanics are not mere consequences of chronic lung pathology, but active determinants of senescence induction and maintenance. Targeting mechanotransduction pathways or ECM remodeling processes offers a promising avenue for senescence-modulating therapies in fibrotic and degenerative lung diseases.
6.5. Senescence-Associated Metabolic Reprogramming
Cellular senescence in the lung is intimately associated with profound metabolic reprogramming that encompasses alterations in glycolysis, lipid metabolism, and mitochondrial bioenergetics. These metabolic shifts not only accompany the senescent phenotype but actively contribute to its initiation, maintenance, and pathological effects.
A prominent metabolic hallmark of senescent cells is a shift toward enhanced glycolysis often described as a “Warburg-like” effect even in the absence of proliferation. In senescent lung fibroblasts and epithelial cells, glycolytic reprogramming supports increased NADPH production and fuels the biosynthetic and pro-inflammatory output of the SASP [263]. In parallel, senescent cells exhibit dysregulated lipid metabolism, characterized by the accumulation of intracellular lipid droplets and upregulation of lipogenic enzymes such as fatty acid synthase. These changes are particularly evident in alveolar type II epithelial cells and fibroblasts from aged or fibrotic lungs, where altered lipid handling contributes to lipotoxic stress, mitochondrial dysfunction, and fibrotic activation [264,265,266].
At the mitochondrial level, senescent lung cells display altered oxidative phosphorylation (OXPHOS), elevated ROS generation, and disrupted mitochondrial dynamics, including increased fission and reduced mitophagy. These mitochondrial defects exacerbate the DNA damage response and reinforce senescence-associated growth arrest [267]. Furthermore, mitochondrial dysfunction amplifies SASP output through NF-κB activation and inflammasome signaling, linking bioenergetic collapse to persistent inflammation [22].
Importantly, interventions that target mitochondrial regulators such as PGC-1α or enhance mitophagy have been shown to attenuate senescence phenotypes and restore epithelial homeostasis in preclinical lung models [268]. These findings suggest that the metabolic rewiring of senescent cells is not merely epiphenomenal but represents a core driver of lung aging and disease progression.
Potential target for therapy: metabolic senolytics.
Recent studies have elucidated distinct metabolic dependencies in senescent cells, paving the way for the development of metabolic senolytics agents that selectively eliminate senescent cells by exploiting their altered bioenergetic state. Senescent lung cells exhibit a unique metabolic phenotype characterized by enhanced glycolysis, impaired mitochondrial oxidative phosphorylation (OXPHOS), elevated reactive oxygen species (ROS) production, and reliance on specific survival pathways [32,267,269,270]. These adaptations render senescent cells particularly vulnerable to therapeutic strategies that disrupt redox homeostasis, NAD⁺ metabolism, or mitochondrial function.
One of the most extensively studied metabolic senolytics is navitoclax (ABT-263), a BCL-2/BCL-xL inhibitor that induces apoptosis preferentially in senescent cells by targeting their heightened mitochondrial priming [189]. Similarly, the glycolytic inhibitor 2-deoxy-D-glucose (2-DG) has demonstrated selective cytotoxicity against senescent cells with elevated glycolytic flux [271]. In preclinical lung models, pharmacologic modulation of mitochondrial health using agents such as metformin or mitophagy enhancers like urolithin A has successfully reduced senescent cell burden and mitigated fibrotic remodeling [272,273].
Additionally, peptide-based approaches targeting senescence-specific survival pathways have shown promise. For instance, the FOXO4-DRI peptide disrupts the interaction between p53 and FOXO4 a critical axis for senescent cell survival leading to selective apoptosis of senescent cells, including in lung tissues [194]. These findings underscore the therapeutic potential of metabolic senolytics in treating chronic lung diseases, such as COPD, IPF, and post-COVID-19 pulmonary fibrosis, conditions where persistent senescent cells perpetuate inflammation and tissue remodeling. Future strategies that combine metabolic senolytics with anti-fibrotic or pro-regenerative therapies may yield synergistic effects, restoring lung homeostasis and improving clinical outcomes.
7. Future Directions and Unmet Needs
The integration of artificial intelligence (AI)-based histological analysis and machine learning (ML) algorithms is emerging as a transformative strategy for the detection and spatial characterization of senescent cells within complex tissues such as the lung. Conventional senescence markers including p16Ink4a, p21Cip1/Waf1, and SA-β-gal suffer from limited specificity and are challenging to interpret reliably in heterogeneous tissue environments. To address these limitations, AI-driven image analysis platforms, particularly deep learning-based convolutional neural networks (CNNs), have been utilized to detect senescence-associated morphological features such as cellular enlargement, nuclear irregularities, and lipofuscin accumulation directly from histological and immunofluorescence slides [274,275].
For instance, AI-assisted digital pathology has been employed to distinguish senescent fibroblasts in fibrotic lung tissues by integrating shape and texture-based descriptors with immunostaining for senescence markers [276]. Parallel advances in ML applications to single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have enabled the identification of senescent subpopulations based on transcriptional signatures, including upregulation of CDKN2A, GDF15, and key SASP components across diverse lung cell types [275].
Integrated platforms such as SenNet are being developed to combine histological features, spatial transcriptomic profiles, and imaging-derived markers, thereby enhancing the predictive power and resolution of in vivo senescence detection [53]. These approaches not only improve diagnostic capabilities but also offer tools for real-time monitoring of senotherapeutic efficacy in both preclinical models and clinical settings. Collectively, AI- and ML-based technologies represent a critical advancement in overcoming current biomarker limitations and hold significant promise for accelerating the translation of senescence research into precision pulmonary medicine.
Personalized medicine: identifying senescent cell burden in individual patients
The concept of personalized medicine in the context of lung senescence seeks to tailor therapeutic strategies according to an individual's senescent cell burden, spatial distribution, and cell type-specific senescence profiles. Senescent cells accumulate heterogeneously across lung compartments including alveolar epithelium, fibroblasts, endothelium, and immune infiltrates with this variation shaped by intrinsic factors such as aging and extrinsic stressors like environmental pollutants, viral infections, and fibrotic remodeling [277]. However, the clinical implementation of senescence profiling remains limited by the absence of validated in vivo biomarkers.
Transcriptomic tools such as SenMayo and SenePy developed from bulk and single-cell RNA sequencing datasets have shown promise in estimating senescent cell signatures from biopsy-derived samples [55,244]. Additional modalities, including machine learning-based histological classifiers, circulating SASP factors, and senescence-associated extracellular vesicles, are under active investigation as non-invasive surrogates of senescence burden [278].
The integration of these approaches with clinical and molecular disease staging holds potential for stratifying patients into senescence-enriched or senescence-low subgroups. For example, in IPF, profiling patients with elevated senescent fibroblast signatures could guide the selective use of senotherapeutic agents alongside existing antifibrotic therapies [279]. Such personalized approaches may optimize treatment efficacy, minimize off-target toxicity, and enable longitudinal monitoring of therapeutic response.
Role of environmental pollutants, microbiome, and circadian rhythms in lung senescence
Senescence in the lung is intricately influenced by environmental exposures, microbiome dynamics, and circadian rhythm regulation, each acting as a modifiable contributor to senescence induction and progression in chronic respiratory diseases.
Environmental pollutants such as cigarette smoke, particulate matter (PM2.5), ozone, and diesel exhaust are well-established inducers of senescence, triggering oxidative stress, DNA damage, and mitochondrial dysfunction in epithelial cells, fibroblasts, and endothelial cells [280,281,282,283]. For instance, cigarette smoke induces premature senescence through the activation of p16Ink4a and p21Cip1/Waf1, alongside a proinflammatory SASP that perpetuates tissue injury and inflammation in COPD [46].
The lung microbiome is another key regulator. Dysbiosis marked by reduced microbial diversity and increased abundance of pro-inflammatory mediators can amplify alveolar macrophage activation and epithelial dysfunction, fueling chronic inflammation and promoting senescence via low-grade immune activation [284]. Microbial metabolites, such as short-chain fatty acids (SCFAs) and lipopolysaccharides (LPS) may further influence SASP output in a context-dependent manner [285].
Circadian rhythm disruption, common in aging, shift work, or chronic stress, also contributes to lung senescence. Core clock genes (e.g., BMAL1, CLOCK, Nrf2) regulate redox homeostasis, mitochondrial health, and DNA repair. Their dysregulation has been linked to impaired alveolar regeneration and increased susceptibility to fibrotic remodeling via enhanced senescence and inflammatory signaling [286,287,288]. Thus, this highlights the importance of incorporating exposome and chrono-biological context into senescence-related research and therapeutic design.
Clinical Translation of Senescence-Targeted Therapies: Challenges and Progresses
Although senescence presents a promising therapeutic target in chronic lung diseases, its clinical translation is hindered by several unresolved challenges. A primary obstacle is the lack of reliable, non-invasive biomarkers for senescence in human lungs, limiting patient stratification, therapeutic monitoring, and outcome assessment [1]. Moreover, the cellular heterogeneity of senescence across lung compartments necessitates cell type- and context-specific interventions, as no universal senolytic agent is effective across all senescent cell types [9].
Current senolytic agents such as navitoclax (ABT-263) exhibit significant off-target toxicity, particularly hematologic effects, posing risks for elderly patients and those with comorbidities [289]. These safety concerns have constrained the advancement of senolytics in clinical settings.
Nevertheless, promising advances are underway. Early-phase clinical trials of dasatinib+quercetin (D+Q) have shown improved physical performance and reduced circulating SASP components in IPF patients [290]. Other compounds under development include UBX1325 (a BCL-xL inhibitor) and FOXO4-DRI peptides, which target senescent cells in fibrotic and post-COVID lung injury models. Concurrently, AI-driven histopathological scoring systems and integrative multi-omics approaches are being tested to stratify patients and predict treatment responses [291,292].
Moving forward, success in this field will rely on: Development of lung-specific senescence biomarkers; Optimization of inhalable senolytics for localized delivery; Advanced patient stratification based on molecular senescence subtypes; Long-term safety profiling and regulatory oversight to mitigate off-target risks. In sum, while hurdles persist, the clinical translation of senotherapeutics in lung disease is entering a promising phase-I, driven by molecular precision, computational insight, and a growing appreciation for the complex biology of senescence.
8. Conclusions
Cellular senescence exerts a paradoxical influence on lung health, functioning as both a protective mechanism and a contributor to lung development and disease pathogenesis. Transient or acute senescence plays a beneficial role by limiting tissue injury, facilitating wound repair, and suppressing tumorigenesis. In contrast, the chronic persistence of senescent cells is increasingly implicated in driving sustained inflammation, fibrotic remodeling, and impaired tissue regeneration across various pulmonary disorders. This dual nature highlights the necessity of delineating the contextual, temporal, and cell type specific dynamics of the senescence response.
Despite substantial progress in the field, several pivotal knowledge gaps remain. These include the absence of reliable in vivo biomarkers to track senescent cell populations, limited understanding of the reversibility of senescence and their cellular heterogeneity, and an incomplete characterization of senescent cell subsets within distinct lung compartments. Advances in high-resolution technologies such as single-cell RNA sequencing, spatial transcriptomics, metabolomic profiling, and artificial intelligence enhanced histopathological analysis are beginning to unravel the complexity and plasticity of senescence phenotypes in the lung microenvironment.
Collectively, these insights pave the way for the development of therapeutic strategies aimed at modulating senescence. Approaches such as targeted senolytics, suppression of the senescence-associated secretory phenotype, senotyping, metabolic reprogramming, and enhancement of immune-mediated senescent cell clearance hold considerable promise for restoring lung homeostasis. Such interventions may offer clinical benefit in conditions including COPD, IPF, and post-viral fibrotic remodeling. Realizing this potential within the framework of precision medicine will require the integration of molecular profiling, rational drug design, and individualized therapeutic approaches in lung diseases associated with aging.
Author Contributions
S.A.O. and M.I.F. contributed to writing of the main manuscript, hence equal authorship. G.K. contributed to COPD-associated senescence, S.B.S. assisted with IPF-related senescence, and K.U.I.G. supported the work on virus-induced senescence. All three provided valuable scientific insights and critical feedback during the proofreading and revision stages. I.R. secured funding, guided the overall study design and layout, and performed the final review of the manuscript.
Funding
This study was supported by the National Institutes of Health (NIH) 1R01ES029177, 1R01HL158316, 1R01HL167655, 1RO1HL147715, and TriState SenNet U54 AG075931, The funding agencies had no role in design of the study, data collection, analysis, and interpretation of data and in writing the manuscript.
Acknowledgments
We would like to extend our heartfelt gratitude to all the lab members for their continuous support and valuable contributions during the preparation of this review.
Conflicts of Interest
The authors have declared no competing interest.
Abbreviations
| Abbreviation | Full Form |
| DDR | DNA Damage Response |
| SASP | Senescence-Associated Secretory Phenotype |
| COPD | Chronic Obstructive Pulmonary Disease |
| IPF | Idiopathic Pulmonary Fibrosis |
| OIS | Oncogene-Induced Senescence |
| TIS | Therapy-Induced Senescence |
| MiDAS | Mitochondrial Dysfunction-Associated Senescence |
| AMPK | AMP-activated Protein Kinase |
| AT2 | Alveolar Type II Cells |
| ECM | Extracellular Matrix |
| ROS | Reactive Oxygen Species |
| PDGF-AA | Platelet-Derived Growth Factor-AA |
| MMPs | Matrix Metalloproteinases |
| p53 | Tumor Suppressor Protein 53 |
| p21 | Cyclin-Dependent Kinase Inhibitor 1 |
| p16 | Cyclin-Dependent Kinase Inhibitor 4A |
| Rb | Retinoblastoma |
| ALI | Acute Lung Injury |
| ARDS | Acute Respiratory Distress Syndrome |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome Coronavirus 2 |
| EVs | Extracellular Vesicles |
| rhCC16 | Recombinant Human Clara Cell Protein 16 |
| SIRT1 | Sirtuin 1 |
| PAI-1 | Plasminogen Activator Inhibitor-1 |
| TGF-β | Transforming Growth Factor Beta |
| CTGF | Connective Tissue Growth Factor |
| PDGF | Platelet-Derived Growth Factor |
| MMP | Matrix Metalloproteinase |
| PAECs | Pulmonary Artery Endothelial Cells |
| PASMCs | Pulmonary Artery Smooth Muscle Cells |
| iPAH | Idiopathic Pulmonary Arterial Hypertension |
| JAG1 | Jagged-1 |
| DLL4 | Delta-Like 4 |
| YWHAZ | Tyrosine 3-Monooxygenase/Tryptophan 5-Monooxygenase Activation Protein Zeta |
| ABT-737 | Senolytic Therapy Drug |
| FOXO4-DRI | Forkhead Box O4-Drug Resistance Inhibitor |
| SA-β-Gal | Senescence-Associated β-Galactosidase |
| γH2A.X | Gamma-Histone H2A Variant X |
| DNAmAge | DNA Methylation Age |
| AgeAcc | Age Acceleration |
| WGCNA | Weighted Gene Co-Expression Network Analysis |
| IL-10 | Interleukin 10 |
| TP53 | Tumor Protein 53 |
| H2AX | H2A.X Variant Histone |
| CDKN2A | Cyclin-Dependent Kinase Inhibitor 2A |
| GDF15 | Growth Differentiation Factor 15 |
| CDKN1A | Cyclin-Dependent Kinase Inhibitor 1A |
| TNFRSF1B | Tumor Necrosis Factor Receptor Superfamily Member 1B |
| Bcl2 L1 | BCL2 Like 1 |
| CXCL8 | C-X-C Motif Chemokine Ligand 8 |
| IL1A | Interleukin 1 Alpha |
| MMP12 | Matrix Metallopeptidase 12 |
| SERPINE1 | Serine Protease Inhibitor, Clade E, Member 1 (Plasminogen Activator Inhibitor-1) |
| TGFβ1 | Transforming Growth Factor Beta 1 |
| TNF | Tumor Necrosis Factor |
| IL-6 | Interleukin 6 |
| IL-1beta | Interleukin 1 Beta |
| MMP-8 | Matrix Metallopeptidase 8 |
| VEGFA | Vascular Endothelial Growth Factor A |
| SnCs | Senescent Cells |
| NF-κB | Nuclear Factor Kappa B |
| mTOR | Mammalian Target of Rapamycin |
| p38 MAPK | p38 Mitogen-Activated Protein Kinase |
| JAK/STAT | Janus Kinase/Signal Transducer and Activator of Transcription |
| ATM | Ataxia Telangiectasia Mutated |
| STACs | Sirtuin-Activating Compounds |
| Nrf2 | Nuclear Factor Erythroid 2-Related Factor 2 |
| DAMPs | Damage-Associated Molecular Patterns |
| TAME | Targeting Aging with Metformin |
| eNOS | Endothelial Nitric Oxide Synthase |
| Ruxolitinib | JAK Inhibitor |
| Rapalogs | Rapamycin Analogs |
| SR12343 | NF-κB Inhibitor |
References
- Gorgoulis, V. , et al. , Cellular Senescence: Defining a Path Forward. Cell, 2019, 179, 813–827. [Google Scholar]
- Herranz, N. and J. Gil, Mechanisms and functions of cellular senescence. J Clin Invest, 2018, 128, 1238–1246. [Google Scholar] [PubMed]
- Wei, W. and S. Ji, Cellular senescence: Molecular mechanisms and pathogenicity. J Cell Physiol, 2018, 233, 9121–9135. [Google Scholar] [PubMed]
- Calabrò, A. , et al., Senotherapeutics to Counteract Senescent Cells Are Prominent Topics in the Context of Anti-Ageing Strategies. Int J Mol Sci, 2024, 25(3).
- Dimri, G.P. , et al. , A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A, 1995, 92, 9363–7. [Google Scholar] [PubMed]
- Bitencourt, T.C. , et al. , Subcellular structure, heterogeneity, and plasticity of senescent cells. Aging Cell, 2024, 23, e14154. [Google Scholar]
- Coppé, J.P. , et al., The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol, 2010, 5: 99-118.
- Wang, B. , et al. , The senescence-associated secretory phenotype and its physiological and pathological implications. Nat Rev Mol Cell Biol, 2024, 25, 958–978. [Google Scholar]
- Basisty, N. , et al. , A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol, 2020, 18, e3000599. [Google Scholar]
- Borghesan, M. , et al. , A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol, 2020, 30, 777–791. [Google Scholar]
- d'Adda di Fagagna, F. , et al. , A DNA damage checkpoint response in telomere-initiated senescence. Nature, 2003, 426, 194–8. [Google Scholar]
- Hayflick, L. and P.S. Moorhead, The serial cultivation of human diploid cell strains. Exp Cell Res, 1961, 25: 585-621.
- Parimon, T. , et al. , Senescence of alveolar epithelial progenitor cells: a critical driver of lung fibrosis. Am J Physiol Cell Physiol, 2023, 325, C483–c495. [Google Scholar]
- Okuda, R. , et al. , Cellular senescence and senescence-associated secretory phenotype: comparison of idiopathic pulmonary fibrosis, connective tissue disease-associated interstitial lung disease, and chronic obstructive pulmonary disease. J Thorac Dis, 2019, 11, 857–864. [Google Scholar] [PubMed]
- Sundar, I.K. , et al. , Genetic Ablation of p16(INK4a) Does Not Protect against Cellular Senescence in Mouse Models of Chronic Obstructive Pulmonary Disease/Emphysema. Am J Respir Cell Mol Biol, 2018, 59, 189–199. [Google Scholar]
- Rashid, K. , et al. , Lung cellular senescence is independent of aging in a mouse model of COPD/emphysema. Sci Rep, 2018, 8, 9023. [Google Scholar]
- Serrano, M. , et al. , Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell, 1997, 88, 593–602. [Google Scholar]
- Pribluda, A. , et al. , A senescence-inflammatory switch from cancer-inhibitory to cancer-promoting mechanism. Cancer Cell, 2013, 24, 242–56. [Google Scholar]
- Ewald, J.A. , et al. , Therapy-induced senescence in cancer. J Natl Cancer Inst, 2010, 102, 1536–46. [Google Scholar] [CrossRef]
- Schmitt, C.A. , et al. , A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell, 2002, 109, 335–46. [Google Scholar]
- Bajtai, E. , et al. , Therapy-induced senescence is a transient drug resistance mechanism in breast cancer. Mol Cancer, 2025, 24, 128. [Google Scholar]
- Wiley, C.D. , et al. , Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab, 2016, 23, 303–14. [Google Scholar] [PubMed]
- Muñoz-Espín, D. , et al. , Programmed cell senescence during mammalian embryonic development. Cell, 2013, 155, 1104–18. [Google Scholar] [PubMed]
- Acosta, J.C. , et al. , A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol, 2013, 15, 978–90. [Google Scholar] [PubMed]
- Rattanavirotkul, N., K. Kirschner, and T. Chandra, Induction and transmission of oncogene-induced senescence. Cell Mol Life Sci, 2021, 78, 843–852. [Google Scholar]
- Mastri, M. , et al. , A Transient Pseudosenescent Secretome Promotes Tumor Growth after Antiangiogenic Therapy Withdrawal. Cell Rep, 2018, 25, 3706–3720. [Google Scholar]
- Reimann, M., S. Lee, and C.A. Schmitt, Cellular senescence: Neither irreversible nor reversible. J Exp Med, 2024, 221(4).
- Campisi, J. and F. d'Adda di Fagagna, Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol, 2007, 8, 729–40. [Google Scholar] [PubMed]
- Chang-Chien, J. , et al. , Particulate matter causes telomere shortening and increase in cellular senescence markers in human lung epithelial cells. Ecotoxicol Environ Saf, 2021, 222, 112484. [Google Scholar]
- Eckhardt, C.M. and H. Wu, Environmental Exposures and Lung Aging: Molecular Mechanisms and Implications for Improving Respiratory Health. Curr Environ Health Rep, 2021, 8, 281–293. [Google Scholar]
- Tsuji, T., K. Aoshiba, and A. Nagai, Cigarette smoke induces senescence in alveolar epithelial cells. Am J Respir Cell Mol Biol, 2004, 31, 643–9. [Google Scholar]
- Miwa, S. , et al., Mitochondrial dysfunction in cell senescence and aging. J Clin Invest, 2022, 132(13).
- O'Reilly, S., E. Markiewicz, and O. C. Idowu, Aging, senescence, and cutaneous wound healing-a complex relationshiFront Immunol, 2024, 15, 1429716. [Google Scholar]
- Yao, H. , et al. , Timing and cell specificity of senescence drives postnatal lung development and injury. Nat Commun, 2023, 14, 273. [Google Scholar]
- Dennery, P.A. and H. Yao, Emerging role of cellular senescence in normal lung development and perinatal lung injury. Chin Med J Pulm Crit Care Med, 2024, 2, 10–16. [Google Scholar] [PubMed]
- Schafer, M.J. , et al. , Cellular senescence mediates fibrotic pulmonary disease. Nat Commun, 2017, 8, 14532. [Google Scholar]
- Woldhuis, R.R. , et al. , COPD-derived fibroblasts secrete higher levels of senescence-associated secretory phenotype proteins. Thorax, 2021, 76, 508–511. [Google Scholar]
- Yao, C. , et al. , Senescence of Alveolar Type 2 Cells Drives Progressive Pulmonary Fibrosis. Am J Respir Crit Care Med, 2021, 203, 707–717. [Google Scholar]
- Barkauskas, C.E. , et al. , Type 2 alveolar cells are stem cells in adult lung. J Clin Invest, 2013, 123, 3025–36. [Google Scholar]
- Ruysseveldt, E., K. Martens, and B. Steelant, Airway Basal Cells, Protectors of Epithelial Walls in Health and Respiratory Diseases. Front Allergy, 2021, 2, 787128. [Google Scholar] [PubMed]
- Desai, T.J., D. G. Brownfield, and M.A. Krasnow, Alveolar progenitor and stem cells in lung development, renewal and cancer. Nature, 2014, 507, 190–4. [Google Scholar]
- Han, S., G. R.S. Budinger, and C.J. Gottardi, Alveolar epithelial regeneration in the aging lung. J Clin Invest, 2023, 133(20).
- Wisman, M. , et al. , Lower levels of senescence in human lung mesenchymal stromal cells compared with lung fibroblasts: implications for tissue regeneration in COPD. Am J Physiol Lung Cell Mol Physiol, 2025, 328, L858–l865. [Google Scholar]
- Tsuji, T., K. Aoshiba, and A. Nagai, Alveolar cell senescence in patients with pulmonary emphysema. Am J Respir Crit Care Med, 2006, 174, 886–93. [Google Scholar]
- Yanai, H. , et al. , Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY), 2015, 7, 664–72. [Google Scholar] [PubMed]
- Nyunoya, T. , et al. , Cigarette smoke induces cellular senescence. Am J Respir Cell Mol Biol, 2006, 35, 681–8. [Google Scholar]
- Alder, J.K. , et al. , Short telomeres are a risk factor for idiopathic pulmonary fibrosis. Proc Natl Acad Sci U S A, 2008, 105, 13051–6. [Google Scholar]
- Baek, K.H. , et al. , Thrombospondin-1 mediates oncogenic Ras-induced senescence in premalignant lung tumors. J Clin Invest, 2013, 123, 4375–89. [Google Scholar]
- Tripathi, U. , et al. , SARS-CoV-2 causes senescence in human cells and exacerbates the senescence-associated secretory phenotype through TLR-3. Aging (Albany NY), 2021, 13, 21838–21854. [Google Scholar]
- Hendrickson, C.M. and M. A. Matthay, Viral pathogens and acute lung injury: investigations inspired by the SARS epidemic and the 2009 H1N1 influenza pandemic. Semin Respir Crit Care Med, 2013, 34, 475–86. [Google Scholar]
- Kellogg, D.L. , et al. , Cellular Senescence in Idiopathic Pulmonary Fibrosis. Curr Mol Biol Rep, 2021, 7, 31–40. [Google Scholar]
- Woldhuis, R.R. , et al. , Link between increased cellular senescence and extracellular matrix changes in COPD. Am J Physiol Lung Cell Mol Physiol, 2020, 319, L48–l60. [Google Scholar]
- Gurkar, A.U. , et al. , Spatial mapping of cellular senescence: emerging challenges and opportunities. Nat Aging, 2023, 3, 776–790. [Google Scholar] [PubMed]
- Cohn, R.L. , et al. , The heterogeneity of cellular senescence: insights at the single-cell level. Trends Cell Biol, 2023, 33, 9–17. [Google Scholar] [PubMed]
- Sanborn, M.A. , et al. , Unveiling the cell-type-specific landscape of cellular senescence through single-cell transcriptomics using SenePy. Nat Commun, 2025, 16, 1884. [Google Scholar] [PubMed]
- Bueno, M. , et al. , Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol, 2020, 33, 101509. [Google Scholar]
- Liu, S. , et al. , Mitochondrial dysfunction and alveolar type II epithelial cell senescence: The destroyer and rescuer of idiopathic pulmonary fibrosis. Front Cell Dev Biol, 2025, 13, 1535601. [Google Scholar]
- Cuevas-Mora, K. , et al. , Hermansky-Pudlak syndrome-2 alters mitochondrial homeostasis in the alveolar epithelium of the lung. Respir Res, 2021, 22, 49. [Google Scholar]
- Su, W. , et al. , YAP1 inhibits the senescence of alveolar epithelial cells by targeting Prdx3 to alleviate pulmonary fibrosis. Exp Mol Med, 2024, 56, 1643–1654. [Google Scholar]
- Yu, Y. , et al. , YAP/TAZ activation mediates PQ-induced lung fibrosis by sustaining senescent pulmonary epithelial cells. Respir Res, 2024, 25, 212. [Google Scholar] [PubMed]
- Ovadya, Y. , et al. , Impaired immune surveillance accelerates accumulation of senescent cells and aging. Nat Commun, 2018, 9, 5435. [Google Scholar]
- Parikh, P. , et al. , Cellular senescence in the lung across the age spectrum. Am J Physiol Lung Cell Mol Physiol, 2019, 316, L826–l842. [Google Scholar] [CrossRef]
- Freund, A., C. K. Patil, and J. Campisi, p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. Embo j, 2011, 30, 1536–48. [Google Scholar]
- Ajoolabady, A. , et al. , Hallmarks and mechanisms of cellular senescence in aging and disease. Cell Death Discovery, 2025, 11, 364. [Google Scholar]
- Di Micco, R. , et al. , Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nature Reviews Molecular Cell Biology, 2021, 22, 75–95. [Google Scholar]
- Zhang, L. , et al., Cellular senescence: a key therapeutic target in aging and diseases. J Clin Invest, 2022, 132(15).
- Paez-Ribes, M. , et al. , Targeting senescent cells in translational medicine. EMBO Mol Med, 2019, 11, e10234. [Google Scholar]
- Demaria, M. , et al. , An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell, 2014, 31, 722–33. [Google Scholar]
- Krizhanovsky, V. , et al. , Senescence of activated stellate cells limits liver fibrosis. Cell, 2008, 134, 657–67. [Google Scholar]
- Ferrucci, L. and M. Zampino, A mitochondrial root to accelerated ageing and frailty. Nat Rev Endocrinol, 2020, 16, 133–134. [Google Scholar]
- Cho, H.J. , et al. , Nintedanib induces senolytic effect via STAT3 inhibition. Cell Death Dis, 2022, 13, 760. [Google Scholar] [PubMed]
- Palmer, A.K. , et al. , Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell, 2019, 18, e12950. [Google Scholar]
- Du, D. , et al. , Senotherapy Protects against Cisplatin-Induced Ovarian Injury by Removing Senescent Cells and Alleviating DNA Damage. Oxid Med Cell Longev, 2022, 2022, 9144644. [Google Scholar] [PubMed]
- Lee, S. , et al. , Molecular programs of fibrotic change in aging human lung. Nat Commun, 2021, 12, 6309. [Google Scholar]
- Wu, J. , et al. , Central role of cellular senescence in TSLP-induced airway remodeling in asthma. PLoS One, 2013, 8, e77795. [Google Scholar]
- Cottage, C.T. , et al. , Targeting p16-induced senescence prevents cigarette smoke-induced emphysema by promoting IGF1/Akt1 signaling in mice. Commun Biol, 2019, 2, 307. [Google Scholar]
- Sinha, S. , et al. , COVID-19 lung disease shares driver AT2 cytopathic features with Idiopathic pulmonary fibrosis. EBioMedicine, 2022, 82, 104185. [Google Scholar]
- Alder, J.K. , et al. , Telomere dysfunction causes alveolar stem cell failure. Proc Natl Acad Sci U S A, 2015, 112, 5099–104. [Google Scholar]
- Orjalo, A.V. , et al. , Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A, 2009, 106, 17031–6. [Google Scholar]
- Ortiz-Montero, P., A. Londoño-Vallejo, and J. Vernot, Senescence-associated IL-6 and IL-8 cytokines induce a self- and cross-reinforced senescence/inflammatory milieu strengthening tumorigenic capabilities in the MCF-7 breast cancer cell line. Cell Commun Signal, 2017, 15, 17. [Google Scholar]
- Rana, T. , et al. , PAI-1 Regulation of TGF-β1-induced Alveolar Type II Cell Senescence, SASP Secretion, and SASP-mediated Activation of Alveolar Macrophages. Am J Respir Cell Mol Biol, 2020, 62, 319–330. [Google Scholar]
- Rivas, M. , et al., Senescence: Pathogenic Driver in Chronic Obstructive Pulmonary Disease. Medicina (Kaunas), 2022, 58(6).
- Zhou, F. , et al. , Epithelial cell senescence impairs repair process and exacerbates inflammation after airway injury. Respir Res, 2011, 12, 78. [Google Scholar] [PubMed]
- Kuźnar-Kamińska, B. , et al. , Serum from patients with chronic obstructive pulmonary disease induces senescence-related phenotype in bronchial epithelial cells. Sci Rep, 2018, 8, 12940. [Google Scholar]
- Wendisch, D. , et al. , SARS-CoV-2 infection triggers profibrotic macrophage responses and lung fibrosis. Cell, 2021, 184, 6243–6261. [Google Scholar]
- Tsuji, T., K. Aoshiba, and A. Nagai, Alveolar cell senescence exacerbates pulmonary inflammation in patients with chronic obstructive pulmonary disease. Respiration, 2010, 80, 59–70. [Google Scholar] [PubMed]
- Bateman, G. , et al., Airway Epithelium Senescence as a Driving Mechanism in COPD Pathogenesis. Biomedicines, 2023, 11(7).
- Aghali, A. , et al. , Cellular senescence is increased in airway smooth muscle cells of elderly persons with asthma. Am J Physiol Lung Cell Mol Physiol, 2022, 323, L558–l568. [Google Scholar] [PubMed]
- Hernandez-Gonzalez, F. , et al., Cellular Senescence in Lung Fibrosis. Int J Mol Sci, 2021, 22(13).
- Kyi, P. , et al. , Endothelial senescence mediates hypoxia-induced vascular remodeling by modulating PDGFB expression. Front Med (Lausanne), 2022, 9, 908639. [Google Scholar]
- Schmitt, C.A. , et al. , COVID-19 and cellular senescence. Nat Rev Immunol, 2023, 23, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M. , et al., Senolytic drugs target alveolar epithelial cell function and attenuate experimental lung fibrosis ex vivo. Eur Respir J, 2017, 50(2).
- Evangelou, K. , et al., Pulmonary infection by SARS-CoV-2 induces senescence accompanied by an inflammatory phenotype in severe COVID-19: possible implications for viral mutagenesis. Eur Respir J, 2022, 60(2).
- Justice, J.N. , et al. , Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine, 2019, 40, 554–563. [Google Scholar]
- Childs, B.G. , et al. , Senescent cells: an emerging target for diseases of ageing. Nat Rev Drug Discov, 2017, 16, 718–735. [Google Scholar]
- MacNee, W. , Is Chronic Obstructive Pulmonary Disease an Accelerated Aging Disease? Ann Am Thorac Soc, 2016, 13 Suppl 5: S429-s437.
- Wang, T. , et al. , The association between leukocyte telomere length and chronic obstructive pulmonary disease is partially mediated by inflammation: a meta-analysis and population-based mediation study. BMC Pulm Med, 2022, 22, 320. [Google Scholar]
- Devulder, J.V., et al., COPD Airway Epithelial Cell-derived Extracellular Vesicles Spread Cellular Senescence via MicroRNA-34a. Am J Respir Cell Mol Biol, 2025.
- Ren, Y.J. , et al. , rhCC16 Suppresses Cellular Senescence and Ameliorates COPD-Like Symptoms by Activating the AMPK/Sirt1-PGC-1-α-TFAM Pathway to Promote Mitochondrial Function. J Cell Mol Med, 2025, 29, e70566. [Google Scholar] [PubMed]
- Xiaofei, Y. , et al. , Erythromycin attenuates oxidative stress-induced cellular senescence via the PI3K-mTOR signaling pathway in chronic obstructive pulmonary disease. Front Pharmacol, 2022, 13, 1043474. [Google Scholar]
- Kaur, G., T. Muthumalage, and I. Rahman, Clearance of senescent cells reverts the cigarette smoke-induced lung senescence and airspace enlargement in p16-3MR mice. Aging Cell, 2023, 22, e13850. [Google Scholar]
- Norheim, K.L. , et al. , Effect of nicotinamide riboside on airway inflammation in COPD: a randomized, placebo-controlled trial. Nat Aging, 2024, 4, 1772–1781. [Google Scholar]
- Luo, X. , et al. , Multi-modal transcriptomic analysis reveals metabolic dysregulation and immune responses in chronic obstructive pulmonary disease. Sci Rep, 2024, 14, 22699. [Google Scholar]
- Alfaro, E. , et al. , Effect of physical activity in lymphocytes senescence burden in patients with COPD. Am J Physiol Lung Cell Mol Physiol, 2024, 327, L464–l472. [Google Scholar] [PubMed]
- Campisi, M. , et al. , DNA Methylation-Based Age Prediction and Telomere Length Reveal an Accelerated Aging in Induced Sputum Cells Compared to Blood Leukocytes: A Pilot Study in COPD Patients. Front Med (Lausanne), 2021, 8, 690312. [Google Scholar]
- He, Y. , et al. , Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res, 2019, 209, 14–21. [Google Scholar]
- Borie, R., B. Crestani, and H. Bichat, Prevalence of telomere shortening in familial and sporadic pulmonary fibrosis is increased in men. Am J Respir Crit Care Med, 2009, 179, 1073. [Google Scholar]
- Oldham, J.M. , Interstitial Lung Abnormalities and Aging Biomarkers: A Mediation. Am J Respir Crit Care Med, 2021, 203, 1058–1060. [Google Scholar] [CrossRef] [PubMed]
- Sanders, J.L. , et al. , The Association of Aging Biomarkers, Interstitial Lung Abnormalities, and Mortality. Am J Respir Crit Care Med, 2021, 203, 1149–1157. [Google Scholar]
- Álvarez, D. , et al. , IPF lung fibroblasts have a senescent phenotype. Am J Physiol Lung Cell Mol Physiol, 2017, 313, L1164–l1173. [Google Scholar] [CrossRef] [PubMed]
- Ramos, C. , et al. , Fibroblasts from idiopathic pulmonary fibrosis and normal lungs differ in growth rate, apoptosis, and tissue inhibitor of metalloproteinases expression. Am J Respir Cell Mol Biol, 2001, 24, 591–8. [Google Scholar]
- Yosef, R., et al., Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat Commun, 2016, 7: 11190.
- Sanders, Y.Y. , et al. , Histone deacetylase inhibition promotes fibroblast apoptosis and ameliorates pulmonary fibrosis in mice. Eur Respir J, 2014, 43, 1448–58. [Google Scholar]
- Sisson, T.H. , et al., PAI-1 interaction with sortilin-related receptor 1 is required for lung fibrosis. JCI Insight, 2025, 10(11).
- Marudamuthu, A.S. , et al. , Plasminogen activator inhibitor-1 suppresses profibrotic responses in fibroblasts from fibrotic lungs. J Biol Chem, 2015, 290, 9428–41. [Google Scholar] [PubMed]
- Wiley, C.D. , et al., Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight, 2019, 4(24).
- Jun, J.I. and L.F. Lau, CCN2 induces cellular senescence in fibroblasts. J Cell Commun Signal, 2017, 11, 15–23. [Google Scholar]
- Chien, Y. , et al. , Control of the senescence-associated secretory phenotype by NF-κB promotes senescence and enhances chemosensitivity. Genes Dev, 2011, 25, 2125–36. [Google Scholar] [PubMed]
- Tsukui, T. , et al. , Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun, 2020, 11, 1920. [Google Scholar]
- Habermann, A.C. , et al. , Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv, 2020, 6, eaba1972. [Google Scholar]
- Reyfman, P.A. , et al. , Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med, 2019, 199, 1517–1536. [Google Scholar]
- Muñoz-Espín, D. and M. Serrano, Cellular senescence: from physiology to pathology. Nat Rev Mol Cell Biol, 2014, 15, 482–96. [Google Scholar]
- Prasanna, P.G. , et al. , Therapy-Induced Senescence: Opportunities to Improve Anticancer Therapy. J Natl Cancer Inst, 2021, 113, 1285–1298. [Google Scholar]
- Saleh, T. , et al., Therapy-Induced Senescence: An "Old" Friend Becomes the Enemy. Cancers (Basel), 2020, 12(4).
- Coppé, J.P. , et al. , Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol, 2008, 6, 2853–68. [Google Scholar]
- Wang, L., L. Lankhorst, and R. Bernards, Exploiting senescence for the treatment of cancer. Nat Rev Cancer, 2022, 22, 340–355. [Google Scholar]
- Lei, Y. , et al. , Senescent lung fibroblasts in idiopathic pulmonary fibrosis facilitate non-small cell lung cancer progression by secreting exosomal MMP1. Oncogene, 2025, 44, 769–781. [Google Scholar] [PubMed]
- Demaria, M. , et al. , Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov, 2017, 7, 165–176. [Google Scholar] [PubMed]
- Haston, S. , et al. , Clearance of senescent macrophages ameliorates tumorigenesis in KRAS-driven lung cancer. Cancer Cell, 2023, 41, 1242–1260. [Google Scholar]
- Huidobro, C. , et al. , Cellular and molecular features of senescence in acute lung injury. Mech Ageing Dev, 2021, 193, 111410. [Google Scholar]
- Wissler Gerdes, E.O. , et al. , Role of senescence in the chronic health consequences of COVID-19. Transl Res, 2022, 241, 96–108. [Google Scholar]
- Lee, S. , et al. , Virus-induced senescence is a driver and therapeutic target in COVID-19. Nature, 2021, 599, 283–289. [Google Scholar] [PubMed]
- Torres Acosta, M.A. and B.D. Singer, Pathogenesis of COVID-19-induced ARDS: implications for an ageing population. Eur Respir J, 2020, 56(3).
- Künzi, L., et al., Cystic Fibrosis Lung Disease in the Aging Population. Front Pharmacol, 2021, 12: 601438.
- Bezzerri, V. , et al. , Is cellular senescence involved in cystic fibrosis? Respir Res, 2019, 20, 32. [Google Scholar] [PubMed]
- Fischer, B.M. , et al. , Increased expression of senescence markers in cystic fibrosis airways. Am J Physiol Lung Cell Mol Physiol, 2013, 304, L394–400. [Google Scholar] [CrossRef]
- Easter, M. , et al., FGF receptors mediate cellular senescence in the cystic fibrosis airway epithelium. JCI Insight, 2024, 9(15).
- Laucirica, D.R., L. W. Garratt, and A. Kicic, Progress in Model Systems of Cystic Fibrosis Mucosal Inflammation to Understand Aberrant Neutrophil Activity. Front Immunol, 2020, 11, 595. [Google Scholar]
- Voynow, J.A. and M. Shinbashi, Neutrophil Elastase and Chronic Lung Disease. Biomolecules, 2021, 11(8).
- Barnes, P.J., J. Baker, and L.E. Donnelly, Cellular Senescence as a Mechanism and Target in Chronic Lung Diseases. Am J Respir Crit Care Med, 2019, 200, 556–564. [Google Scholar]
- Wang, S. , et al. , Inflammatory Activity of Epithelial Stem Cell Variants from Cystic Fibrosis Lungs Is Not Resolved by CFTR Modulators. Am J Respir Crit Care Med, 2023, 208, 930–943. [Google Scholar]
- Roger, I. , et al., Senescence Alterations in Pulmonary Hypertension. Cells, 2021, 10(12).
- Culley, M.K. and S. Y. Chan, Endothelial Senescence: A New Age in Pulmonary Hypertension. Circ Res, 2022, 130, 928–941. [Google Scholar]
- Safaie Qamsari, E. and D. J. Stewart, Cellular senescence in the pathogenesis of pulmonary arterial hypertension: the good, the bad and the uncertain. Front Immunol, 2024, 15, 1403669. [Google Scholar]
- Born, E. , et al. , Eliminating Senescent Cells Can Promote Pulmonary Hypertension Development and Progression. Circulation, 2023, 147, 650–666. [Google Scholar]
- Wang, A.P. , et al. , Pulmonary Artery Smooth Muscle Cell Senescence Promotes the Proliferation of PASMCs by Paracrine IL-6 in Hypoxia-Induced Pulmonary Hypertension. Front Physiol, 2021, 12, 656139. [Google Scholar]
- Ramadhiani, R. , et al. , Endothelial cell senescence exacerbates pulmonary hypertension by inducing juxtacrine Notch signaling in smooth muscle cells. iScience, 2023, 26, 106662. [Google Scholar] [PubMed]
- Meng, Z.Y. , et al. , Identification and experimental verification of senescence-related gene signatures and molecular subtypes in idiopathic pulmonary arterial hypertension. Sci Rep, 2024, 14, 22157. [Google Scholar] [PubMed]
- van der Feen, D.E. , et al., Cellular senescence impairs the reversibility of pulmonary arterial hypertension. Sci Transl Med, 2020, 12(554).
- Malinina, A. , et al. , IL10 deficiency promotes alveolar enlargement and lymphoid dysmorphogenesis in the aged murine lung. Aging Cell, 2020, 19, e13130. [Google Scholar] [PubMed]
- Gessner, C. , et al. , Exhaled breath condensate cytokine patterns in chronic obstructive pulmonary disease. Respir Med, 2005, 99, 1229–40. [Google Scholar]
- Lim, S. , et al., Balance of matrix metalloprotease-9 and tissue inhibitor of metalloprotease-1 from alveolar macrophages in cigarette smokers. Regulation by interleukin-10. Am J Respir Crit Care Med, 2000, 162(4 Pt 1): 1355-60.
- Blázquez-Prieto, J. , et al. , Activation of p21 limits acute lung injury and induces early senescence after acid aspiration and mechanical ventilation. Transl Res, 2021, 233, 104–116. [Google Scholar]
- Kobayashi, Y. , et al. , Persistence of a regeneration-associated, transitional alveolar epithelial cell state in pulmonary fibrosis. Nat Cell Biol, 2020, 22, 934–946. [Google Scholar]
- Jiang, C. , et al. , Serpine 1 induces alveolar type II cell senescence through activating p53-p21-Rb pathway in fibrotic lung disease. Aging Cell, 2017, 16, 1114–1124. [Google Scholar]
- Chen, H. , et al. , TGF-β1/IL-11/MEK/ERK signaling mediates senescence-associated pulmonary fibrosis in a stress-induced premature senescence model of Bmi-1 deficiency. Exp Mol Med, 2020, 52, 130–151. [Google Scholar]
- Kaur, G., I. K. Sundar, and I. Rahman, p16-3MR: A Novel Model to Study Cellular Senescence in Cigarette Smoke-Induced Lung Injuries. Int J Mol Sci, 2021, 22(9).
- Yamada, Z. , et al. , Senescence of alveolar epithelial cells impacts initiation and chronic phases of murine fibrosing interstitial lung disease. Front Immunol, 2022, 13, 935114. [Google Scholar]
- Reyes, N.S. , et al. , Sentinel p16(INK4a+) cells in the basement membrane form a reparative niche in the lung. Science, 2022, 378, 192–201. [Google Scholar]
- Zhou, Y. , et al. , Expression of p16 and p53 in non-small-cell lung cancer: clinicopathological correlation and potential prognostic impact. Biomark Med, 2019, 13, 761–771. [Google Scholar]
- Keow, J. , et al. , Digital quantification of p16-positive foci in fibrotic interstitial lung disease is associated with a phenotype of idiopathic pulmonary fibrosis with reduced survival. Respir Res, 2022, 23, 147. [Google Scholar]
- Verhamme, F.M. , et al. , Elevated GDF-15 contributes to pulmonary inflammation upon cigarette smoke exposure. Mucosal Immunol, 2017, 10, 1400–1411. [Google Scholar] [PubMed]
- Jiang, G., C. T. Liu, and W.D. Zhang, IL-17A and GDF15 are able to induce epithelial-mesenchymal transition of lung epithelial cells in response to cigarette smoke. Exp Ther Med, 2018, 16, 12–20. [Google Scholar]
- Radwanska, A. , et al., Increased expression and accumulation of GDF15 in IPF extracellular matrix contribute to fibrosis. JCI Insight, 2022, 7(16).
- Zhang, Y. , et al. , GDF15 is an epithelial-derived biomarker of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol, 2019, 317, L510–l521. [Google Scholar] [CrossRef] [PubMed]
- Lv, X. , et al. , The cell cycle inhibitor P21 promotes the development of pulmonary fibrosis by suppressing lung alveolar regeneration. Acta Pharm Sin B, 2022, 12, 735–746. [Google Scholar]
- Ma, J.H. , et al., K63 Ubiquitination of P21 Can Facilitate Pellino-1 in the Context of Chronic Obstructive Pulmonary Disease and Lung Cellular Senescence. Cells, 2022, 11(19).
- Russo, R.C. , et al. , Role of the chemokine receptor CXCR2 in bleomycin-induced pulmonary inflammation and fibrosis. Am J Respir Cell Mol Biol, 2009, 40, 410–21. [Google Scholar]
- Kristan, S.S. , et al. , Airway angiogenesis in stable and exacerbated chronic obstructive pulmonary disease. Scand J Immunol, 2012, 75, 109–14. [Google Scholar]
- Shaghaghi, H. , et al. , A model of the aged lung epithelium in idiopathic pulmonary fibrosis. Aging (Albany NY), 2021, 13, 16922–16937. [Google Scholar]
- Khan, S.S. , et al. , A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv, 2017, 3, eaao1617. [Google Scholar]
- Betsuyaku, T. , et al. , Neutrophil granule proteins in bronchoalveolar lavage fluid from subjects with subclinical emphysema. Am J Respir Crit Care Med, 1999, 159, 1985–91. [Google Scholar]
- Zhang, L. , et al. , Targeting cellular senescence with senotherapeutics: senolytics and senomorphics. Febs j, 2023, 290, 1362–1383. [Google Scholar] [PubMed]
- Robbins, P.D. , et al. , Senolytic Drugs: Reducing Senescent Cell Viability to Extend Health Span. Annu Rev Pharmacol Toxicol, 2021, 61, 779–803. [Google Scholar] [PubMed]
- Di Micco, R. , et al. , Cellular senescence in ageing: from mechanisms to therapeutic opportunities. Nat Rev Mol Cell Biol, 2021, 22, 75–95. [Google Scholar]
- Laberge, R.M. , et al. , Author Correction: MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat Cell Biol, 2021, 23, 564–565. [Google Scholar] [PubMed]
- Xu, M. , et al. , JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A, 2015, 112, E6301–10. [Google Scholar]
- Van Nguyen, T. , et al. , DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: a mouse model. J Exp Med, 2007, 204, 1453–61. [Google Scholar]
- Lewinska, A. , et al. , AMPK-mediated senolytic and senostatic activity of quercetin surface functionalized Fe(3)O(4) nanoparticles during oxidant-induced senescence in human fibroblasts. Redox Biol, 2020, 28, 101337. [Google Scholar]
- Bogdanowicz, P. , et al. , Senomorphic activity of a combination of niacinamide and hyaluronic acid: correlation with clinical improvement of skin aging. Sci Rep, 2024, 14, 16321. [Google Scholar]
- Moiseeva, O. , et al. , Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell, 2013, 12, 489–98. [Google Scholar]
- Zhu, Y. , et al. , Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell, 2016, 15, 428–35. [Google Scholar]
- Zhu, Y. , et al. , The Achilles' heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell, 2015, 14, 644–58. [Google Scholar] [PubMed]
- Wang, W. , et al. , The biological activities, chemical stability, metabolism and delivery systems of quercetin: A review. Trends in Food Science & Technology, 2016, 56, 21–38. [Google Scholar]
- Zhu, Y. , et al. , New agents that target senescent cells: the flavone, fisetin, and the BCL-X(L) inhibitors, A1331852 and A1155463, Aging (Albany NY), 2017, 9, 955–963. [Google Scholar]
- Yousefzadeh, M.J. , et al. , Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine, 2018, 36, 18–28. [Google Scholar] [PubMed]
- Moaddel, R. , et al. , Identification of gingerenone A as a novel senolytic compound. PLoS One, 2022, 17, e0266135. [Google Scholar]
- Rawat, L. , et al. , Piperlongumine induces ROS mediated cell death and synergizes paclitaxel in human intestinal cancer cells. Biomed Pharmacother, 2020, 128, 110243. [Google Scholar]
- Chang, J. , et al. , Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med, 2016, 22, 78–83. [Google Scholar]
- Fuhrmann-Stroissnigg, H., L. J. Niedernhofer, and P.D. Robbins, Hsp90 inhibitors as senolytic drugs to extend healthy aging. Cell Cycle, 2018, 17, 1048–1055. [Google Scholar]
- Fuhrmann-Stroissnigg, H. , et al. , Identification of HSP90 inhibitors as a novel class of senolytics. Nat Commun, 2017, 8, 422. [Google Scholar]
- Amor, C. , et al. , Senolytic CAR T cells reverse senescence-associated pathologies. Nature, 2020, 583, 127–132. [Google Scholar]
- Amor, C. , et al. , Prophylactic and long-lasting efficacy of senolytic CAR T cells against age-related metabolic dysfunction. Nat Aging, 2024, 4, 336–349. [Google Scholar]
- Baar, M.P. , et al. , Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell, 2017, 169, 132–147. [Google Scholar]
- González-Gualda, E. , et al. , A guide to assessing cellular senescence in vitro and in vivo. Febs j, 2021, 288, 56–80. [Google Scholar] [PubMed]
- Kotschy, A. , et al. , The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature, 2016, 538, 477–482. [Google Scholar]
- Partridge, L., M. Fuentealba, and B. K. Kennedy, The quest to slow ageing through drug discovery. Nat Rev Drug Discov, 2020, 19, 513–532. [Google Scholar]
- Gomari, H., M. Forouzandeh Moghadam, and M. Soleimani, Targeted cancer therapy using engineered exosome as a natural drug delivery vehicle. Onco Targets Ther, 2018, 11, 5753–5762. [Google Scholar]
- Cai, Y. , et al. , Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice. Cell Res, 2020, 30, 574–589. [Google Scholar]
- Poblocka, M. , et al. , Targeted clearance of senescent cells using an antibody-drug conjugate against a specific membrane marker. Sci Rep, 2021, 11, 20358. [Google Scholar] [PubMed]
- Wissler Gerdes, E.O., et al., Strategies for late phase preclinical and early clinical trials of senolytics. Mech Ageing Dev, 2021, 200: 111591.
- Hickson, L.J. , et al. , Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine, 2019, 47, 446–456. [Google Scholar] [PubMed]
- de Vos, S. , et al. , Safety and efficacy of navitoclax, a BCL-2 and BCL-X(L) inhibitor, in patients with relapsed or refractory lymphoid malignancies: results from a phase 2a study. Leuk Lymphoma, 2021, 62, 810–818. [Google Scholar]
- Cai, Y. , et al. , The landscape of aging. Sci China Life Sci, 2022, 65, 2354–2454. [Google Scholar] [CrossRef]
- Muñoz-Espín, D. , et al., A versatile drug delivery system targeting senescent cells. EMBO Mol Med, 2018, 10(9).
- Schafer, M.J. , et al., The senescence-associated secretome as an indicator of age and medical risk. JCI Insight, 2020, 5(12).
- Zhou, L. , et al. , Senescence as a dictator of patient outcomes and therapeutic efficacies in human gastric cancer. Cell Death Discov, 2022, 8, 13. [Google Scholar]
- Bian, R., et al., CDKN1A as a target of senescence in heart failure: insights from a multiomics study. Front Pharmacol, 2024, 15: 1446300.
- Suryadevara, V. , et al. , SenNet recommendations for detecting senescent cells in different tissues. Nat Rev Mol Cell Biol, 2024, 25, 1001–1023. [Google Scholar]
- Prieto, L.I. , et al. , Senescent alveolar macrophages promote early-stage lung tumorigenesis. Cancer Cell, 2023, 41, 1261–1275,e6. [Google Scholar]
- Smith, R. , et al. , A new model and precious tool to study molecular mechanisms of macrophage aging. Aging (Albany NY), 2024, 16, 12697–12725. [Google Scholar]
- Maus, M. , et al. , Iron accumulation drives fibrosis, senescence and the senescence-associated secretory phenotype. Nat Metab, 2023, 5, 2111–2130. [Google Scholar] [PubMed]
- Su, L. , et al. , Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis, 2021, 12, 527. [Google Scholar]
- Zhou, B.W. , et al. , The role of macrophage polarization and cellular crosstalk in the pulmonary fibrotic microenvironment: a review. Cell Commun Signal, 2024, 22, 172. [Google Scholar]
- Campbell, R.A. , et al. , The Role of Ageing and Parenchymal Senescence on Macrophage Function and Fibrosis. Front Immunol, 2021, 12, 700790. [Google Scholar]
- Salminen, A. , Inhibitory immune checkpoints suppress the surveillance of senescent cells promoting their accumulation with aging and in age-related diseases. Biogerontology, 2024, 25, 749–773. [Google Scholar] [CrossRef]
- Majewska, J. , et al. , p16-dependent increase of PD-L1 stability regulates immunosurveillance of senescent cells. Nat Cell Biol, 2024, 26, 1336–1345. [Google Scholar] [PubMed]
- Marin, I., M. Serrano, and F. Pietrocola, Recent insights into the crosstalk between senescent cells and CD8 T lymphocytes. NPJ Aging, 2023, 9, 8. [Google Scholar]
- Melo-Narváez, M.C. , et al., Lung regeneration: implications of the diseased niche and ageing. Eur Respir Rev, 2020, 29(157).
- Ruhland, M.K. , et al. , Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat Commun, 2016, 7, 11762. [Google Scholar] [PubMed]
- Li, X. , et al. , Inflammation and aging: signaling pathways and intervention therapies. Signal Transduct Target Ther, 2023, 8, 239. [Google Scholar]
- Favaretto, G. , et al. , Neutrophil-activating secretome characterizes palbociclib-induced senescence of breast cancer cells. Cancer Immunol Immunother, 2024, 73, 113. [Google Scholar] [PubMed]
- Rolas, L. , et al. , Senescent endothelial cells promote pathogenic neutrophil trafficking in inflamed tissues. EMBO Rep, 2024, 25, 3842–3869. [Google Scholar]
- Cai, C. , et al. , The role of fibroblast-neutrophil crosstalk in the pathogenesis of inflammatory diseases: a multi-tissue perspective. Front Immunol, 2025, 16, 1588667. [Google Scholar]
- Fulop, T. , et al. , Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol, 2017, 8, 1960. [Google Scholar]
- Wynn, T.A. and K. M. Vannella, Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity, 2016, 44, 450–462. [Google Scholar]
- Simmons, S.R. , et al., Older but Not Wiser: the Age-Driven Changes in Neutrophil Responses during Pulmonary Infections. Infect Immun, 2021, 89(4).
- Yang, S.C. , et al. , Understanding the role of neutrophils in acute respiratory distress syndrome. Biomed J, 2021, 44, 439–446. [Google Scholar] [PubMed]
- Wang, Y. , et al. , The aging lung: microenvironment, mechanisms, and diseases. Front Immunol, 2024, 15, 1383503. [Google Scholar]
- Soto-Heredero, G. , et al. , KLRG1 identifies regulatory T cells with mitochondrial alterations that accumulate with aging. Nat Aging, 2025, 5, 799–815. [Google Scholar] [PubMed]
- Liu, Z. , et al. , Immunosenescence: molecular mechanisms and diseases. Signal Transduct Target Ther, 2023, 8, 200. [Google Scholar] [PubMed]
- Han, S., Q. Lu, and X. Liu, Advances in cellular senescence in idiopathic pulmonary fibrosis (Review). Exp Ther Med, 2023, 25, 145. [Google Scholar]
- Wang, Y. , et al. , Immunosenescence, aging and successful aging. Front Immunol, 2022, 13, 942796. [Google Scholar]
- Devulder, J.V. , Unveiling mechanisms of lung aging in COPD: A promising target for therapeutics development. Chin Med J Pulm Crit Care Med, 2024, 2, 133–141. [Google Scholar] [CrossRef]
- Xie, C. , et al., Role of cellular senescence in inflammatory lung diseases. Cytokine Growth Factor Rev, 2023, 70: 26-40.
- Wan, R. , et al., Cellular Senescence: A Troy Horse in Pulmonary Fibrosis. Int J Mol Sci, 2023, 24(22).
- Sun, Y. , An updated landscape of cellular senescence heterogeneity: Mechanisms, technologies and senotherapies. Translational Medicine of Aging, 2023, 7, 46–51. [Google Scholar] [CrossRef]
- Neri, F., et al., Senescent cell heterogeneity and responses to senolytic treatment are related to cell cycle status during cell growth arrest. bioRxiv, 2024.
- D'Agnillo, F. , et al. , Lung epithelial and endothelial damage, loss of tissue repair, inhibition of fibrinolysis, and cellular senescence in fatal COVID-19. Sci Transl Med, 2021, 13, eabj7790. [Google Scholar]
- Zhou, S. , et al. , Alveolar type 2 epithelial cell senescence and radiation-induced pulmonary fibrosis. Front Cell Dev Biol, 2022, 10, 999600. [Google Scholar]
- Najari Beidokhti, M. , et al. , Lung endothelial cell senescence impairs barrier function and promotes neutrophil adhesion and migration. Geroscience, 2025, 47, 2655–2671. [Google Scholar]
- Mebratu, Y.A. , et al. , The aged extracellular matrix and the profibrotic role of senescence-associated secretory phenotype. Am J Physiol Cell Physiol, 2023, 325, C565–c579. [Google Scholar]
- Tao, W., Z. Yu, and J. J. Han, Single-cell senescence identification reveals senescence heterogeneity, trajectory, and modulators. Cell Metab, 2024, 36, 1126–1143. [Google Scholar]
- Saul, D. , et al. , A new gene set identifies senescent cells and predicts senescence-associated pathways across tissues. Nat Commun, 2022, 13, 4827. [Google Scholar] [PubMed]
- Melo-Narváez, M.C. , et al., Stimuli-Specific Senescence of Primary Human Lung Fibroblasts Modulates Alveolar Stem Cell Function. Cells, 2024, 13(13).
- Liao, Y.L. , et al. , Senescent endothelial cells: a potential target for diabetic retinopathy. Angiogenesis, 2024, 27, 663–679. [Google Scholar] [PubMed]
- Morton, L. , et al. , Pericytes and Extracellular Vesicle Interactions in Neurovascular Adaptation to Chronic Arterial Hypertension. J Am Heart Assoc, 2025, 14, e038457. [Google Scholar] [PubMed]
- Blackburn, J.B. , et al. , An update in club cell biology and its potential relevance to chronic obstructive pulmonary disease. Am J Physiol Lung Cell Mol Physiol, 2023, 324, L652–l665. [Google Scholar]
- Jia, M. , et al. , Transcriptional changes of the aging lung. Aging Cell, 2023, 22, e13969. [Google Scholar]
- Jia, H. , et al. , A single-cell atlas of lung homeostasis reveals dynamic changes during development and aging. Commun Biol, 2024, 7, 427. [Google Scholar]
- Kasmani, M.Y. , et al. , A spatial sequencing atlas of age-induced changes in the lung during influenza infection. Nat Commun, 2023, 14, 6597. [Google Scholar] [PubMed]
- Wang, J. , et al. , A transcriptome-based human universal senescence index (hUSI) robustly predicts cellular senescence under various conditions. Nat Aging, 2025, 5, 1159–1175. [Google Scholar] [PubMed]
- Nguyen, N.D. , et al. , scDOT: optimal transport for mapping senescent cells in spatial transcriptomics. Genome Biol, 2024, 25, 288. [Google Scholar]
- Goyal, A. , et al., Targeting p53-p21 signaling to enhance mesenchymal stem cell regenerative potential. Regen Ther, 2025, 29: 352-363.
- Martínez-Zamudio, R.I. , et al. , AP-1 imprints a reversible transcriptional programme of senescent cells. Nat Cell Biol, 2020, 22, 842–855. [Google Scholar]
- Ashraf, H.M., B. Fernandez, and S. L. Spencer, The intensities of canonical senescence biomarkers integrate the duration of cell-cycle withdrawal. Nat Commun, 2023, 14, 4527. [Google Scholar]
- Nacarelli, T. , et al. , NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol, 2019, 21, 397–407. [Google Scholar]
- Ritschka, B. , et al. , The senotherapeutic drug ABT-737 disrupts aberrant p21 expression to restore liver regeneration in adult mice. Genes Dev, 2020, 34, 489–494. [Google Scholar]
- Saleh, T. , et al., Tumor cell escape from therapy-induced senescence. Biochem Pharmacol, 2019, 162: 202-212.
- Sladitschek-Martens, H.L. , et al. , YAP/TAZ activity in stromal cells prevents ageing by controlling cGAS-STING. Nature, 2022, 607, 790–798. [Google Scholar]
- Blokland, K.E.C. , et al., Senescence of IPF Lung Fibroblasts Disrupt Alveolar Epithelial Cell Proliferation and Promote Migration in Wound Healing. Pharmaceutics, 2020, 12(4).
- Suki, B., J. H. T. Bates, and E. Bartolák-Suki, Remodeling of the Aged and Emphysematous Lungs: Roles of Microenvironmental Cues. Compr Physiol, 2022, 12, 3559–3574. [Google Scholar]
- Zhang, Y. , et al. , Metabolic reprogramming in cancer and senescence. MedComm (2020), 2025, 6, e70055. [Google Scholar]
- Lian, H. , et al., Fatty acid synthase inhibition alleviates lung fibrosis via β-catenin signal in fibroblasts. Life Sci Alliance, 2025, 8(2).
- Schneider, J.L. , et al. , The aging lung: Physiology, disease, and immunity. Cell, 2021, 184, 1990–2019. [Google Scholar]
- Liu, B. , et al. , Lipid and glucose metabolism in senescence. Front Nutr, 2023, 10, 1157352. [Google Scholar]
- Correia-Melo, C. , et al. , Mitochondria are required for pro-ageing features of the senescent phenotype. Embo j, 2016, 35, 724–42. [Google Scholar]
- Kang, Y. , et al. , Telomere Dysfunction Disturbs Macrophage Mitochondrial Metabolism and the NLRP3 Inflammasome through the PGC-1α/TNFAIP3 Axis. Cell Rep, 2018, 22, 3493–3506. [Google Scholar] [PubMed]
- Sabbatinelli, J., et al., Where Metabolism Meets Senescence: Focus on Endothelial Cells. Front Physiol, 2019, 10: 1523.
- Wiley, C.D. and J. Campisi, The metabolic roots of senescence: mechanisms and opportunities for intervention. Nat Metab, 2021, 3, 1290–1301. [Google Scholar] [PubMed]
- Singh, R. , et al. , 2-Deoxy-D-Glucose: A Novel Pharmacological Agent for Killing Hypoxic Tumor Cells, Oxygen Dependence-Lowering in Covid-19, and Other Pharmacological Activities. Adv Pharmacol Pharm Sci, 2023, 2023, 9993386. [Google Scholar] [PubMed]
- Zong, Y. , et al. , Mitochondrial dysfunction: mechanisms and advances in therapy. Signal Transduct Target Ther, 2024, 9, 124. [Google Scholar]
- Jiménez-Loygorri, J.I. , et al. , Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat Commun, 2024, 15, 830. [Google Scholar]
- Duran, I. , et al. , Detection of senescence using machine learning algorithms based on nuclear features. Nat Commun, 2024, 15, 1041. [Google Scholar] [PubMed]
- Abdelmohsen, K. , et al. , Identification of senescent cell subpopulations by CITE-seq analysis. Aging Cell, 2024, 23, e14297. [Google Scholar]
- Viswanathan, V.S. , et al. , The state of the art for artificial intelligence in lung digital pathology. J Pathol, 2022, 257, 413–429. [Google Scholar]
- Kokosi, M.A., G. A. Margaritopoulos, and A.U. Wells, Personalised medicine in interstitial lung diseases: Number 6 in the Series "Personalised medicine in respiratory diseases" Edited by Renaud Louis and Nicolas Roche. Eur Respir Rev, 2018, 27(148).
- Misawa, T. , et al., Identification of Novel Senescent Markers in Small Extracellular Vesicles. Int J Mol Sci, 2023, 24(3).
- Waters, D.W. , et al. , Fibroblast senescence in the pathology of idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol, 2018, 315, L162–l172. [Google Scholar]
- Wu, Q. , et al. , Cigarette Smoke Induces Human Airway Epithelial Senescence via Growth Differentiation Factor 15 Production. Am J Respir Cell Mol Biol, 2016, 55, 429–38. [Google Scholar]
- Xu, S. , et al. , Assessment of cellular senescence potential of PM2.5 using 3D human lung fibroblast spheroids in vitro model. Toxicol Res (Camb), 2024, 13, tfae037. [Google Scholar]
- Chew, S. , et al. , Impairment of mitochondrial function by particulate matter: Implications for the brain. Neurochem Int, 2020, 135, 104694. [Google Scholar]
- Martic, I. , Jansen-Dürr, and M. Cavinato, Effects of Air Pollution on Cellular Senescence and Skin Aging. Cells, 2022, 11(14).
- Yang, D. , et al. , The impact of lung microbiota dysbiosis on inflammation. Immunology, 2020, 159, 156–166. [Google Scholar] [PubMed]
- Jang, D.H. , et al. , The connection between aging, cellular senescence and gut microbiome alterations: A comprehensive review. Aging Cell, 2024, 23, e14315. [Google Scholar]
- Zhang, W. , et al. , Emerging Insight Into the Role of Circadian Clock Gene BMAL1 in Cellular Senescence. Front Endocrinol (Lausanne), 2022, 13, 915139. [Google Scholar] [PubMed]
- Chhunchha, B., E. Kubo, and D.Singh, Clock Protein Bmal1 and Nrf2 Cooperatively Control Aging or Oxidative Response and Redox Homeostasis by Regulating Rhythmic Expression of Prdx6. Cells, 2020, 9(8).
- Wang, Q. , et al. , Circadian clock molecule REV-ERBα regulates lung fibrotic progression through collagen stabilization. Nat Commun, 2023, 14, 1295. [Google Scholar]
- Fu, T.E. and Z. Zhou, Senescent cells as a target for anti-aging interventions: From senolytics to immune therapies. J Transl Int Med, 2025, 13, 33–47. [Google Scholar]
- Nambiar, A. , et al. , Senolytics dasatinib and quercetin in idiopathic pulmonary fibrosis: results of a phase I, single-blind, single-center, randomized, placebo-controlled pilot trial on feasibility and tolerability. EBioMedicine, 2023, 90, 104481. [Google Scholar]
- Mansfield, L. , et al. , Emerging insights in senescence: pathways from preclinical models to therapeutic innovations. NPJ Aging, 2024, 10, 53. [Google Scholar] [PubMed]
- Bendani, H. , et al. , Revolutionizing breast cancer immunotherapy by integrating AI and nanotechnology approaches: review of current applications and future directions. Bioelectron Med, 2025, 11, 13. [Google Scholar]
Figure 1.
Schematic representation of alveolar alterations during normal, reversible, and irreversible lung damage in COPD & IPF progression. Panels (a) and (b) depict healthy alveolar architecture and a moderately stressed state, respectively. Under mild injury, immune cell infiltration may support tissue repair, allowing for reversibility. In contrast, panels (c) and (d) illustrate irreversible damage observed in advanced COPD and IPF respectively. COPD progression is characterized by alveolar epithelial cell loss and emphysematous changes driven by SASP-associated senescence and endothelial dysfunction. In IPF, fibroblast accumulation and excessive collagen deposition dominate the pathology. Addressing both epithelial damage and fibrotic remodeling remains a significant therapeutic challenge in chronic lung diseases.
Figure 1.
Schematic representation of alveolar alterations during normal, reversible, and irreversible lung damage in COPD & IPF progression. Panels (a) and (b) depict healthy alveolar architecture and a moderately stressed state, respectively. Under mild injury, immune cell infiltration may support tissue repair, allowing for reversibility. In contrast, panels (c) and (d) illustrate irreversible damage observed in advanced COPD and IPF respectively. COPD progression is characterized by alveolar epithelial cell loss and emphysematous changes driven by SASP-associated senescence and endothelial dysfunction. In IPF, fibroblast accumulation and excessive collagen deposition dominate the pathology. Addressing both epithelial damage and fibrotic remodeling remains a significant therapeutic challenge in chronic lung diseases.
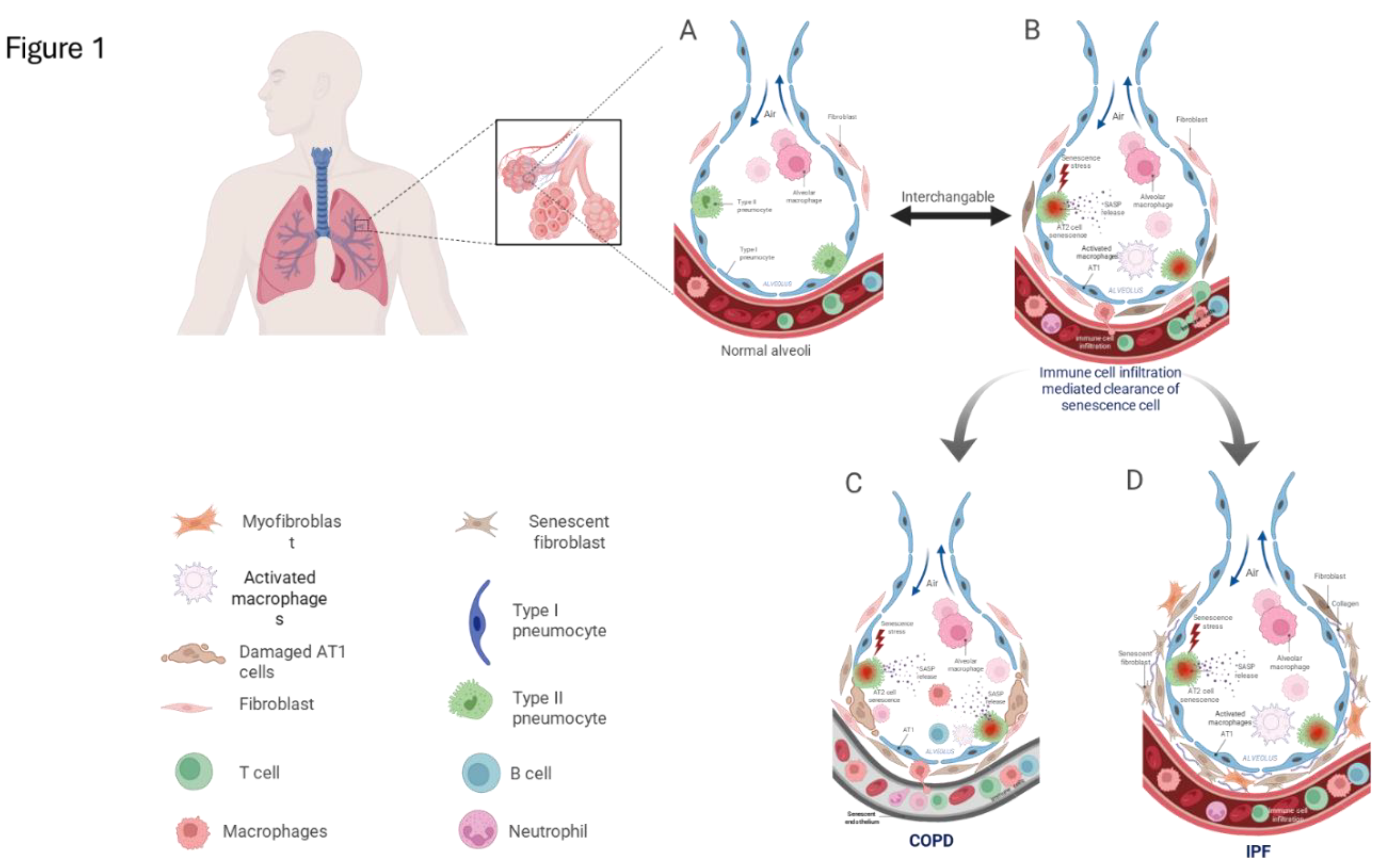
Figure 2.
Stage-wise representation of COPD progression and associated senescence markers. The figure illustrates the progression of COPD across GOLD stages 1 to 4 (x-axis), with corresponding decline in FEV₁ (% predicted) depicted along the y-axis. The upper trajectory highlights the cumulative tissue damage over time, while the lower segmented arrow represents the GOLD classification of COPD severity, showing an inverse relationship with FEV₁ levels. As disease severity increases with age-related decline in lung function, there is a concomitant rise in the expression of senescence-associated markers, indicating a potential link between disease progression and cellular senescence.
Figure 2.
Stage-wise representation of COPD progression and associated senescence markers. The figure illustrates the progression of COPD across GOLD stages 1 to 4 (x-axis), with corresponding decline in FEV₁ (% predicted) depicted along the y-axis. The upper trajectory highlights the cumulative tissue damage over time, while the lower segmented arrow represents the GOLD classification of COPD severity, showing an inverse relationship with FEV₁ levels. As disease severity increases with age-related decline in lung function, there is a concomitant rise in the expression of senescence-associated markers, indicating a potential link between disease progression and cellular senescence.
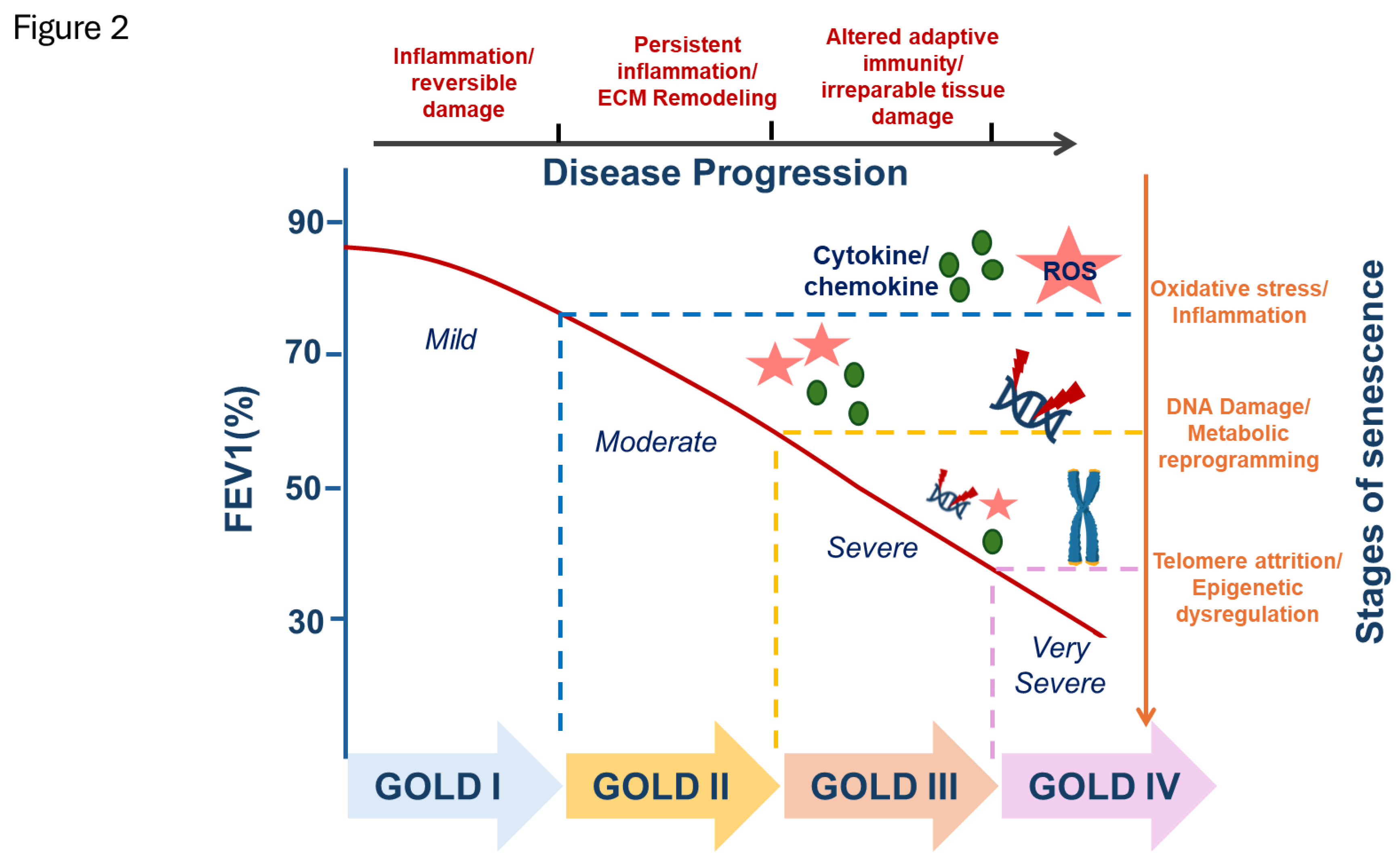
Figure 3.
Mechanisms of action of senomorphic drugs targeting inflammatory and stress-related pathways in senescent cells. This schematic illustrates key intracellular pathways modulated by senomorphic agents to suppress the senescence-associated secretory phenotype (SASP). Stress signals activate the p38 MAPK–MK2 axis in the cytoplasm, which stabilizes SASP mRNAs and promotes inflammatory cytokine expression. Compounds such as SB203580, UR13756, and BIRB796 inhibit p38 MAPK to prevent downstream activation of MK2 and SASP production at the post-transcriptional level. Other senomorphic agents (e.g., rapamycin, metformin, resveratrol, curcumin) act by modulating mTOR, AMPK, NF-κB, STAT3, or Nrf2 pathways, affecting SASP at transcriptional or regulatory levels. Drug targets are shown relative to their subcellular localization, and inhibitory actions are indicated by blunt arrows (⊣).
Figure 3.
Mechanisms of action of senomorphic drugs targeting inflammatory and stress-related pathways in senescent cells. This schematic illustrates key intracellular pathways modulated by senomorphic agents to suppress the senescence-associated secretory phenotype (SASP). Stress signals activate the p38 MAPK–MK2 axis in the cytoplasm, which stabilizes SASP mRNAs and promotes inflammatory cytokine expression. Compounds such as SB203580, UR13756, and BIRB796 inhibit p38 MAPK to prevent downstream activation of MK2 and SASP production at the post-transcriptional level. Other senomorphic agents (e.g., rapamycin, metformin, resveratrol, curcumin) act by modulating mTOR, AMPK, NF-κB, STAT3, or Nrf2 pathways, affecting SASP at transcriptional or regulatory levels. Drug targets are shown relative to their subcellular localization, and inhibitory actions are indicated by blunt arrows (⊣).
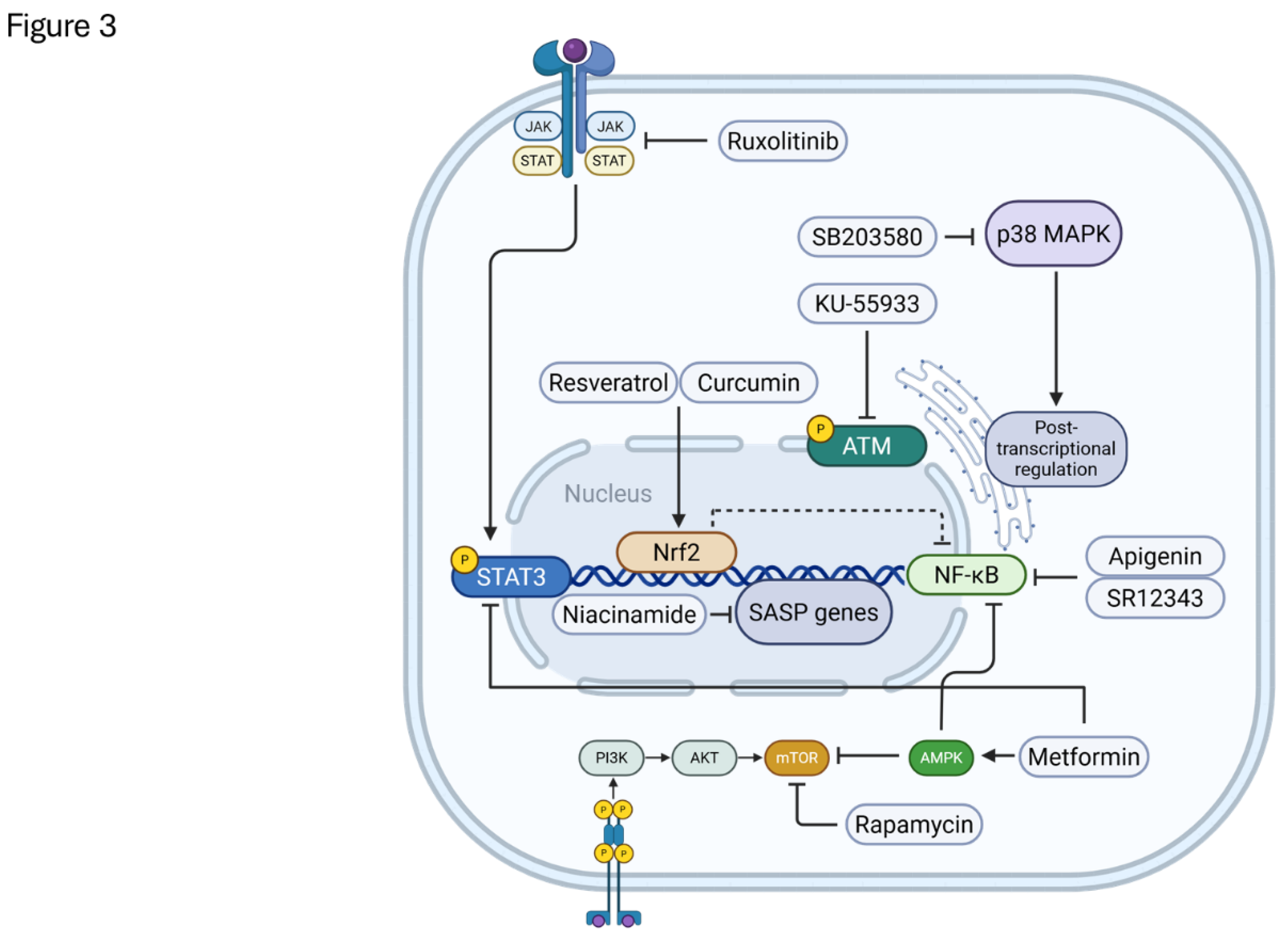
Figure 4.
Mechanisms of action of senolytic drugs targeting pro-survival pathways in senescent cells. This schematic illustrates the primary intracellular mechanisms targeted by senolytic agents to selectively induce apoptosis in senescent cells. Senescent cells rely on distinct senescent cell anti-apoptotic pathways (SCAPs) for survival, including the Bcl-2 family, PI3K/AKT, tyrosine kinases, HSP90, and FOXO4-p53 interactions. Compounds such as navitoclax, fisetin, quercetin, and dasatinib disrupt these SCAPs, promoting mitochondrial-mediated cell death. Other agents like ouabain, digoxin, and CAR T cells targeting uPAR eliminate senescent cells through plasma membrane or immune recognition mechanisms. Drug actions are depicted relative to their subcellular targets, with inhibitory effects shown using blunt arrows (⊣).
Figure 4.
Mechanisms of action of senolytic drugs targeting pro-survival pathways in senescent cells. This schematic illustrates the primary intracellular mechanisms targeted by senolytic agents to selectively induce apoptosis in senescent cells. Senescent cells rely on distinct senescent cell anti-apoptotic pathways (SCAPs) for survival, including the Bcl-2 family, PI3K/AKT, tyrosine kinases, HSP90, and FOXO4-p53 interactions. Compounds such as navitoclax, fisetin, quercetin, and dasatinib disrupt these SCAPs, promoting mitochondrial-mediated cell death. Other agents like ouabain, digoxin, and CAR T cells targeting uPAR eliminate senescent cells through plasma membrane or immune recognition mechanisms. Drug actions are depicted relative to their subcellular targets, with inhibitory effects shown using blunt arrows (⊣).
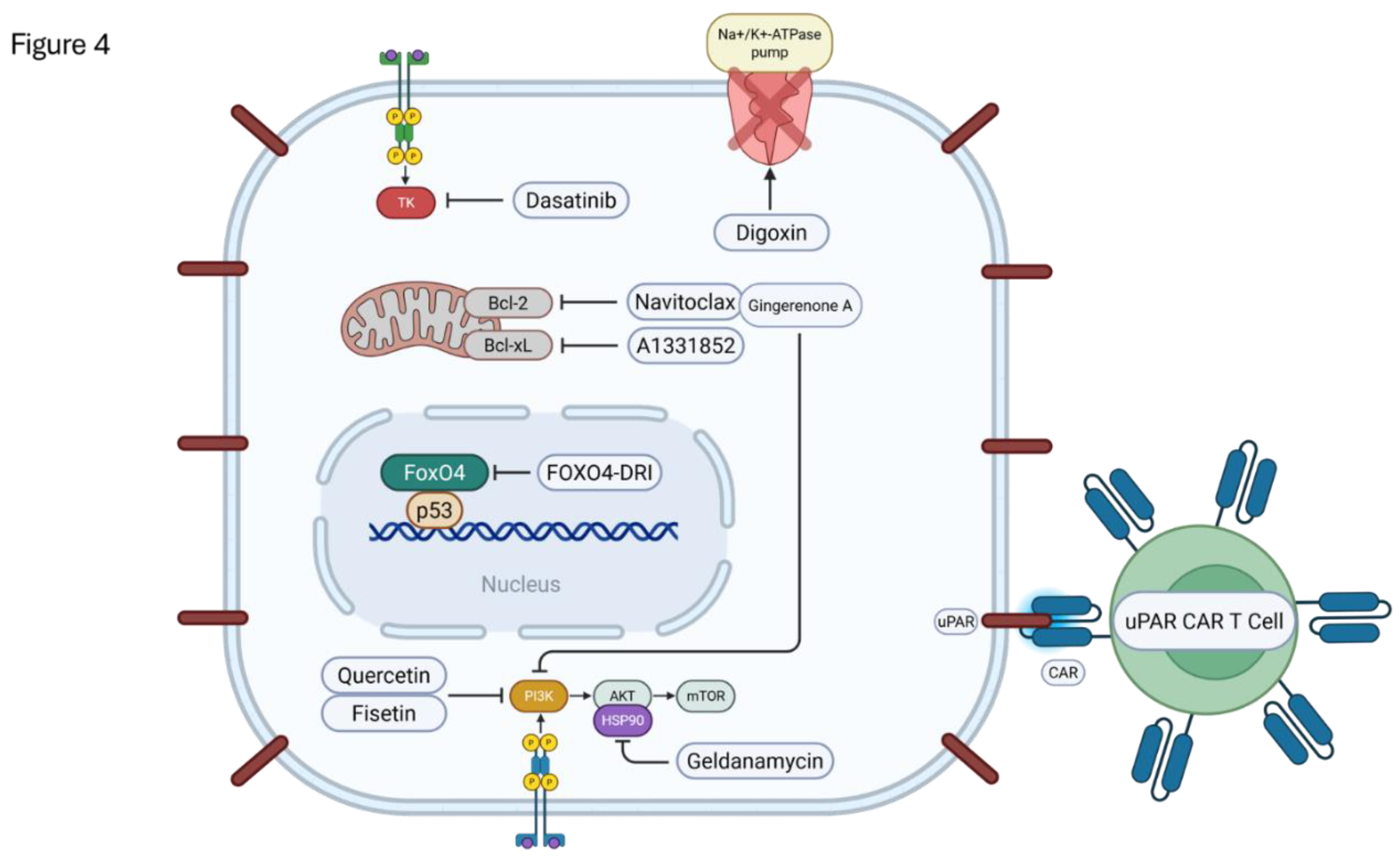
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



