Submitted:
15 August 2025
Posted:
18 August 2025
You are already at the latest version
Abstract
Diabetes mellitus casts a long shadow over the nervous system, driving a spectrum of complications that extend far beyond glycemic control. Diabetic peripheral neuropathy and diabetes-associated cognitive decline—long viewed as separate clinical entities—are now recognized as intertwined expressions of a multi-organ, multi-pathway disease process. At the molecular core, hyperglycemia, oxidative stress, mitochondrial injury, immune activation, and vascular dysfunction con-verge with insulin resistance to erode neuronal and glial integrity.
These events are amplified by gut dysbiosis, which reshapes immune tone, dis-rupts neurovascular homeostasis, and fuels neurodegeneration via the gut–brain–nerve axis. Recent advances reveal that neuroinflammation, microvascular rare-faction, metabolic inflexibility, and lipid-mediated toxicity act not in isolation but as a self-reinforcing network linking peripheral and central injury. Yet, progress toward disease-modifying therapy remains slow, hindered by knowledge gaps in longitudinal human phenotyping, incomplete animal models, underdeveloped biomarkers, and neglected dimensions such as epigenetic memory, circadian disruption, and sex-specific biology.
This Review integrates emerging mechanistic insights—from endothelial dys-function and mitochondrial permeability transition to short-chain fatty acid sig-naling and ceramide neurotoxicity—with an urgent call for precision medicine approaches. By aligning metabolic, vascular, immune, and microbial interventions within longitudinal, biomarker-driven frameworks, the field stands poised to shift from symptom management to strategies that can slow, halt, or even reverse the neurological toll of diabetes.
Keywords:
diabetic neuropathy
; cognitive decline
; neuroinflammation
; gut–brain axis
; oxidative stress
; insulin resistance
; microvascular dysfunction
1. Introduction
Diabetes mellitus (DM), particularly type 2 diabetes (T2DM), is not solely a disorder of chronic hyperglycemia, it is a systemic condition that progressively compromises neural integrity and function. Beyond the well-characterized vascular, renal, and ocular complications, diabetes exerts substantial yet often underrecognized effects on the nervous system—both peripheral and central—leading to diminished quality of life, increased healthcare utilization, and adverse long-term outcomes [1]. Diabetic peripheral neuropathy (DPN) is the most prevalent neurological complication, affecting up to 50% of individuals with long-standing diabetes. Clinically, DPN encompasses small- and large-fiber neuropathies, painful neuropathy, and autonomic dysfunction, which frequently progress insidiously until neurological deficits become irreversible [2]. In parallel, diabetes is increasingly linked to central nervous system (CNS) complications, including mild cognitive impairment, accelerated brain aging, Alzheimer’s disease (AD)-like neurodegeneration, and mood disorders such as depression and anxiety. This constellation of findings has led to the broader concept of diabetes-associated neurodegeneration, with some investigators using the term “type 3 diabetes” to emphasize its neuropathological parallels with AD [3,4].
At the mechanistic level, hyperglycemia initiates multiple maladaptive biochemical pathways: activation of the polyol pathway, accumulation of advanced glycation end-products (AGEs), overactivation of protein kinase C (PKC), and increased flux through the hexosamine biosynthetic pathway. These processes converge to induce oxidative stress, mitochondrial dysfunction, endothelial injury, and impaired axonal transport [5]. Insulin resistance within neural tissue further exacerbates pathology by disrupting IRS–PI3K–Akt signaling, impairing synaptic plasticity, reducing neuronal survival, and imposing metabolic inflexibility—thereby rendering neurons more susceptible to injury and accelerating neurodegenerative changes [6,7].
Neuroinflammation serves as a critical amplifier of this injury. Chronic metabolic stress and immune dysregulation lead to activation of glial cells, infiltration of peripheral immune cells, and increased expression of proinflammatory cytokines such as TNF-α, IL-6, and IL-1β, which collectively exacerbate neural damage. The gut–brain axis emerges as another key contributor: diabetes-induced dysbiosis reduces beneficial taxa and microbial metabolites (e.g., short-chain fatty acids, indole derivatives), increases intestinal permeability, and promotes metabolic endotoxemia—thereby fueling systemic and neuroinflammation. Simultaneously, blood–brain barrier (BBB) integrity is compromised, particularly in hippocampal and cortical regions, allowing peripheral inflammatory mediators to access CNS compartments [8,9,10,11].
Current diagnostic modalities—such as nerve conduction studies, quantitative sensory testing, and standard neuroimaging—often detect damage only after it is well established [12]. Emerging biomarkers and tools, including neurofilament light chain (NfL) and glial fibrillary acidic protein (GFAP) as blood-based indicators of neuroaxonal and astroglial injury [13,14,15,16,17], corneal confocal microscopy for early small-fiber loss [18,19], and novel mitochondrial stress markers such as GDF15, FGF21, and circulating mitochondrial DNA [20,21], hold promise for earlier detection and risk stratification.
Therapeutic strategies remain largely palliative, focused on glycemic control and symptomatic management of neuropathic pain. However, a shift toward disease-modifying interventions is underway. Promising avenues include antioxidants and neurotrophic agents; incretin-based therapies such as GLP-1 receptor agonists with anti-inflammatory and neuroprotective effects; AMPK-activating compounds to enhance neuronal bioenergetics; and microbiota-targeted therapies to restore gut–brain axis homeostasis. When integrated with lifestyle-based interventions—structured exercise programs, cognitive training, and dietary modulation—these approaches may extend beyond symptom control to altering disease trajectory [1,14,22,23].
This review therefore begins with the clinical spectrum of diabetic neuropathies—including DPN, autonomic neuropathy, and CNS complications—before examining the underlying cellular and molecular mechanisms (oxidative stress, mitochondrial dysfunction, neuroinflammation, insulin resistance). It further explores the gut–brain axis as an integrator of systemic and neural pathology, and concludes with emerging diagnostic and therapeutic strategies. By integrating insights from endocrinology, neurology, immunology, and microbiome science, we aim to inform precision approaches that can transition diabetes-related neurodegeneration management from late-stage intervention to early, disease-modifying prevention [1,2].
2. Classification of Diabetes-Induced Neurological Disorders
Diabetes-induced neurological disorders encompass a diverse array of syndromes that can be broadly categorized based on the anatomical regions they affect: the peripheral nervous system (PNS) and the central nervous system (CNS). These disorders often overlap in pathogenesis but differ in their clinical manifestations, progression, and impact on functional outcomes. This section delineates the primary neurological complications of diabetes, including diabetic peripheral neuropathy (DPN), autonomic neuropathy, and central nervous system disorders, with a focus on their clinical classification and diagnostic features [24,25].
2.1. Diabetic Peripheral Neuropathy (DPN)
Diabetic peripheral neuropathy (DPN) is the most common neurological complication of diabetes, affecting ~50% of individuals with long-standing disease, particularly in the presence of poor glycemic control and cardiometabolic risk factors. Classically, it manifests as a length-dependent, symmetrical sensorimotor neuropathy—progressing in a “stocking–glove” distribution and predisposing to foot ulceration, infection, falls, and amputation [26].
The predominant form, distal symmetric polyneuropathy (DSPN), involves sensory, motor, and autonomic fibers, with sensory loss preceding motor deficits. Reduced ankle reflexes, impaired proprioception, and intrinsic foot muscle weakness are common, and electrophysiology typically reveals a mixed axonal–demyelinating pattern [26]. Small-fiber neuropathy—often an early stage—selectively affects Aδ and C fibers, producing burning pain, allodynia, hyperalgesia, and autonomic symptoms despite normal nerve conduction studies. Diagnosis relies on quantitative sensory testing, corneal confocal microscopy, and skin biopsy for intraepidermal nerve fiber density [27]. Large-fiber neuropathy impairs vibration and joint position sense, with gait instability and characteristic conduction slowing [26]. Less common focal and multifocal neuropathies include cranial mononeuropathies (oculomotor with pupil sparing), thoracolumbar radiculopathies, and entrapment syndromes. Diabetic amyotrophy presents with asymmetric proximal pain, weakness, and weight loss, typically improving over months to years [28]. Painful DPN (PDN), driven by both peripheral injury and central sensitization, manifests as burning, stabbing, or electric shock–like pain, often nocturnal and refractory to standard analgesics [26].
Diagnosis integrates symptom/sign assessment with nerve conduction studies, guided by validated tools such as the Michigan Neuropathy Screening Instrument and Toronto Clinical Neuropathy Score. For small-fiber disease, skin biopsy, corneal confocal microscopy, and laser-evoked potentials are recommended [27]. The ADA advises screening at T2DM diagnosis, 5 years after T1DM onset, and annually thereafter. Subtype identification informs prognosis and management [26].
2.2. Diabetic Autonomic Neuropathy (DAN)
Diabetic autonomic neuropathy (DAN) is a serious and underrecognized complication of both type 1 and type 2 diabetes, arising from chronic metabolic injury to autonomic nerve fibers. It impairs cardiovascular, gastrointestinal, genitourinary, and sudomotor regulation, and is associated with increased morbidity and mortality [29]. Risk increases with longer diabetes duration, poor glycemic control, and coexisting microvascular complications, and DAN may precede distal symmetric polyneuropathy in onset and pathophysiology.
Cardiovascular autonomic neuropathy (CAN) is the most prognostically severe manifestation, linked to sudden cardiac death, silent myocardial ischemia, and arrhythmias [30]. Clinical features include resting tachycardia, reduced heart rate variability (HRV), exercise intolerance, and orthostatic hypotension. Diagnosis relies on HRV analysis during deep breathing, the Valsalva maneuver, postural changes, and occasionally baroreflex sensitivity testing—yet early disease is often asymptomatic, necessitating proactive screening.
Gastrointestinal involvement reflects enteric and vagal neuropathy. Gastroparesis delays gastric emptying, causing nausea, vomiting, bloating, and early satiety; colonic dysmotility results in constipation, diarrhea, or fecal incontinence. Gastric scintigraphy remains the diagnostic gold standard, with wireless motility capsules and 13C-octanoic acid breath tests emerging as alternatives [31,32,33,34].
Genitourinary dysfunction includes erectile dysfunction, retrograde ejaculation, and reduced libido in men, and decreased lubrication, sexual dysfunction, and urinary incontinence in women [35]. Diabetic cystopathy presents with diminished bladder sensation, impaired detrusor contractility, and increased post-void residual volume, predisposing to retention and infection. Urodynamic testing and residual urine measurement aid diagnosis [36,37,38].
Sudomotor failure arises from degeneration of sympathetic cholinergic fibers, producing anhidrosis, hyperhidrosis, or patchy sweating, most pronounced in the feet [39]. Resultant xerosis increases the risk of ulcers, and impaired thermoregulation compounds systemic vulnerability. Quantitative sudomotor axon reflex testing (QSART), thermoregulatory sweat testing, and electrochemical skin conductance can detect early small-fiber autonomic damage [40,41,42].
Diagnosis incorporates standardized symptom questionnaires (e.g., COMPASS-31), Ewing’s battery, HRV testing, and autonomic reflex assessment [29]. Given its prognostic significance, routine autonomic evaluation should be integrated into diabetes care to enable early recognition, targeted intervention, and prevention of progression.
2.3. Central Nervous System (CNS) Complications of Diabetes
T2DM exerts a substantial yet often under-recognized impact on the central nervous system (CNS), spanning mild cognitive impairment, major depressive disorder, and increased Alzheimer’s disease (AD) risk. These complications often develop insidiously, remain asymptomatic for years, and ultimately impair quality of life, increase morbidity, and raise mortality [43,44,45].
Cognitive impairment is the most frequent CNS manifestation. Longitudinal studies, including ACCORD-MIND, Rotterdam, and Framingham Offspring, consistently report deficits in executive function, attention, processing speed, and memory in individuals with diabetes compared with non-diabetic peers [45]. Decline accelerates with longer disease duration, poor glycemic control, and vascular comorbidities. Mechanistically, brain insulin resistance disrupts neuronal survival signaling, synaptic plasticity, and energy metabolism; microvascular disease drives hypoperfusion, white-matter injury, and impaired neurogenesis; and microglial and astrocytic activation fuels chronic neuroinflammation [43,44,45,46,47]. The association between T2DM and AD is particularly strong, with epidemiological data indicating a ~50–100% higher risk of AD in T2DM[48]. This has prompted the term “type 3 diabetes” to describe insulin-resistant states within the brain. AD neuropathology includes reduced insulin and insulin-receptor signaling, impaired amyloid-β clearance due to insulin-degrading enzyme (IDE) competition, and GSK3β-mediated tau hyperphosphorylation—all amplified by oxidative stress and mitochondrial dysfunction [45,48,49,50].
Depression is another major comorbidity, with meta-analyses and prospective studies showing a two- to three-fold increased risk in diabetes. The relationship is bidirectional: depression worsens glycemic control and self-management, while diabetes promotes depression via HPA-axis dysregulation, elevated IL-6 and TNF-α, and reduced monoaminergic tone. Structural neuroimaging reveals overlapping reductions in hippocampal and prefrontal volumes in both conditions [51,52].
Neuroimaging across MRI, PET, and diffusion tensor modalities shows consistent diabetes-related changes: hippocampal and cortical atrophy correlating with memory deficits; white-matter hyperintensities and tract injury indicative of small-vessel ischemia; and reduced cerebral glucose metabolism and hypoperfusion, especially in temporoparietal and frontal regions. These abnormalities intensify with poorer glycemic control, greater glycemic variability, and longer disease duration [43,50,53]. Despite the prevalence and impact of CNS involvement, routine diabetes care rarely incorporates cognitive or mood screening, and patients may underreport symptoms. Brief, validated tools—Montreal Cognitive Assessment (MoCA) for cognition [54], Mini-Mental State Examination (MMSE) for global status [55], and Patient Health Questionnaire-9 (PHQ-9) for depression, enable rapid detection [56].
Overall, the CNS effects of diabetes reflect an interplay of metabolic, vascular, inflammatory, and neurodegenerative mechanisms. The clustering of cognitive decline, depression, and elevated AD risk highlights a systemic neural vulnerability likely to grow more clinically relevant as life expectancy increases in diabetes. Proactive, multidisciplinary care—linking neurology, endocrinology, and mental health—together with routine cognitive and mood screening, offers the best opportunity for early intervention [44,51].
3. Pathophysiological Mechanisms
3.1. Hyperglycemia and Glucotoxicity
Chronic hyperglycemia is the fundamental driver of diabetes-related complications and plays a pivotal role in the onset and progression of diabetic neuropathy and central nervous system (CNS) dysfunction. Sustained elevations in blood glucose initiate a cascade of maladaptive biochemical processes, collectively referred to as “glucotoxicity.” These processes are particularly detrimental to neurons and glial cells, which are highly metabolically active and vulnerable to oxidative stress [57,58,59]. Among the most studied pathways involved in hyperglycemia-induced neural damage are the polyol pathway, advanced glycation end-products (AGE)–receptor for AGE (RAGE) axis, the hexosamine biosynthetic pathway, and protein kinase C (PKC) activation. Each contributes uniquely to cellular dysfunction, inflammation, oxidative stress, and ultimately neurodegeneration [60,61].
3.1.1. Polyol Pathway Activation
The polyol pathway is a central contributor to hyperglycemia-induced injury in insulin-independent tissues such as peripheral nerves, retina, renal glomeruli, and the lens. Under euglycemic conditions, it accounts for minimal glucose metabolism; in chronic hyperglycemia, it becomes a major metabolic route, strongly implicated in diabetic peripheral neuropathy (DPN) and other microvascular complications [62,63,64].
Glucose flux through this pathway is catalyzed by two key enzymes. Aldose reductase reduces glucose to sorbitol using NADPH, followed by sorbitol dehydrogenase oxidation of sorbitol to fructose with NAD⁺ reduction to NADH [62,64]. While physiologically innocuous at low flux, hyperglycemia drives excessive sorbitol accumulation—particularly in Schwann cells and neurons, where membrane permeability to sorbitol is limited. This induces osmotic stress, leading to cell swelling, membrane instability, cytoskeletal disruption, and ultimately neurodegeneration [62,65,66].
Polyol pathway activation also perturbs cellular redox homeostasis. Aldose reductase activity depletes NADPH, impairing glutathione regeneration, while sorbitol dehydrogenase increases the NADH/NAD⁺ ratio, shifting redox balance and worsening mitochondrial dysfunction [64,65]. Furthermore, excessive glucose flux depletes myo-inositol, reducing phosphoinositide synthesis and impairing Na⁺/K⁺-ATPase activity—disturbances that alter membrane potentials, slow nerve conduction, and increase excitability risk. Classical studies demonstrated that aldose-reductase inhibition or myo-inositol supplementation restored Na⁺/K⁺-ATPase activity and conduction velocity [67,68,69,70]. In experimental models, heightened aldose-reductase activity correlates with early axonal swelling, segmental demyelination, and fiber loss, particularly in long peripheral nerves with high metabolic demand [66]. Aldose-reductase inhibitors such as epalrestat and ranirestat have shown partial clinical efficacy, especially when initiated early in the disease [71,72,73].
Collectively, these data position the polyol pathway as a pivotal mediator of glucose-induced neurotoxicity in diabetes. Its overactivation integrates osmotic imbalance, cofactor depletion, redox shifts, and ion-transport dysfunction into a unified pathogenic cascade, making it an enduring target for therapeutic intervention to prevent or slow the progression of DPN [62,64,74].
3.1.2. AGE–RAGE Signaling
Advanced glycation end-products (AGEs) are chemically stable adducts formed via the Maillard reaction—non-enzymatic glycation and oxidation of proteins, lipids, and nucleic acids—accelerated by chronic hyperglycemia and oxidative stress. Long-lived structural proteins, such as collagen and laminin in neural basement membranes and myelin-associated proteins, are preferential substrates, making neural and vascular tissues major depots of AGE accumulation [75,76,77].
AGEs profoundly alter tissue mechanics through covalent cross-linking, which stiffens extracellular matrices, thickens basal laminae, narrows capillary lumens, and reduces elasticity. In the peripheral nervous system (PNS), these changes disrupt the endoneurial microenvironment required for axonal conduction and regeneration, while glycation of myelin components compromises Schwann cell support and myelin stability, accelerating demyelination and axonal loss [76,77]. AGE-modified laminin and fibronectin impair neurite outgrowth in vitro, mechanistically linking matrix glycation to axonal failure [76] . The receptor for AGEs (RAGE) amplifies this injury. Expressed on neurons, Schwann cells, glia, and endothelium, RAGE is upregulated in diabetic peripheral nerves and dorsal root ganglia. AGE–RAGE engagement activates NF-κB, inducing TNF-α, IL-1β, and IL-6, and triggers MAPK signaling (ERK1/2, JNK, p38), establishing a self-reinforcing inflammatory loop [78,79,80]. Parallel activation of NADPH oxidase and mitochondrial ROS production promotes lipid peroxidation, DNA damage, and mitochondrial dysfunction [79,81,82].
Diabetic vasculopathy intersects with these pathways. AGE–RAGE signaling reduces nitric oxide bioavailability, upregulates endothelial adhesion molecules (ICAM-1, VCAM-1), and increases permeability, compromising both the blood–nerve barrier (BNB) and blood–brain barrier (BBB) [83,84]. In brain microvascular endothelium and pericytes, AGEs downregulate tight junction proteins (claudin-5, occludin) via VEGF/MMP-2 and TGF-β autocrine signaling, while inducing basement membrane hypertrophy—changes that mirror BBB leakage in diabetic models [85,86]. At the BNB, similar processes promote endoneurial ischemia and hypoxia [85]. In the CNS, endothelial RAGE also facilitates amyloid-β transport across the BBB, linking diabetes to Alzheimer’s disease pathology [87].
Preclinical studies confirm causality: RAGE deletion or antagonism attenuates neural inflammation, preserves myelinated fiber density, improves conduction velocity, and enhances axonal regeneration in diabetic models [85,88]. At the organelle level, blocking AGE–RAGE signaling reduces ROS and protects mitochondrial function [81]. Clinically, circulating and tissue AGE levels correlate with neuropathy severity, while skin autofluorescence, a non-invasive marker of AGE burden, associates with large- and small-fiber deficits in type 2 diabetes. Higher AGE load correlates with slower conduction velocity, and longitudinal data link elevated AGE levels to faster cognitive decline, bridging glycation to both peripheral and central neurodegeneration [89,90,91].
Collectively, AGE–RAGE signaling represents a convergence point for hyperglycemia, oxidative stress, and inflammation in diabetic neurodegeneration. By remodeling extracellular matrices, driving glial and neuronal stress responses, and compromising neurovascular barriers, this pathway fuels both PNS injury and CNS pathology. Therapies targeting AGE formation, RAGE activation, or cross-link cleavage offer rational, mechanism-based strategies to modify disease trajectory [79,92].
3.1.3. Hexosamine Biosynthetic Pathway (HBP)
The hexosamine biosynthetic pathway (HBP) is a minor branch of glycolysis that exerts outsized regulatory influence. Under physiological conditions, only ~2–5% of intracellular glucose enters this pathway to generate UDP-N-acetylglucosamine (UDP-GlcNAc)—the donor substrate for O-GlcNAcylation, a dynamic post-translational modification of nuclear and cytosolic proteins [93,94]. In diabetes, excess glucose flux elevates UDP-GlcNAc levels, driving widespread hyper-O-GlcNAcylation and altering transcriptional, signaling, and metabolic programs [95].
O-GlcNAcylation modifies transcription factors such as Sp1 and NF-κB, enhancing DNA binding and promoting pro-inflammatory gene expression [96,97]. In neurons and glia, O-GlcNAcylation of insulin signaling intermediates—including IRS-1 and Akt—impairs phosphorylation dynamics, thereby attenuating downstream signaling [95]. Within the hippocampus, a key locus for memory processing, O-GlcNAcylation regulates vesicle trafficking, dendritic spine structure, and AMPA receptor function, with consequences for synaptic plasticity and cognitive performance [98,99]. Beyond transcriptional and synaptic effects, hyper-O-GlcNAcylation impacts mitochondrial function. Modification of electron transport chain subunits reduces ATP production and disrupts mitochondrial biogenesis, fission–fusion dynamics, and mitophagy, collectively impairing energy supply to long axons and compromising neuronal resilience [95,100]. Importantly, experimental modulation of O-GlcNAc cycling can reverse these deficits. Genetic or pharmacological manipulation of O-GlcNAc transferase (OGT) and O-GlcNAcase (OGA) alters hippocampal long-term potentiation (LTP) and memory in rodents [98,99]. In diabetic mice, preventing hyper-O-GlcNAcylation of the circadian regulator Bmal1 restores dendritic architecture and rescues cognitive function [101].
Thus, in the diabetic nervous system, the HBP functions as a nutrient sensor turned pathogenic effector, linking nutrient excess to impaired insulin signaling, mitochondrial dysfunction, disrupted neuroplasticity, and chronic inflammation. Targeting dysregulated O-GlcNAcylation represents a promising therapeutic avenue to restore neuronal function and cognitive health [93,95].
3.1.4. Protein Kinase C (PKC) Activation
Protein kinase C (PKC) is a family of serine/threonine kinases—conventional (α, βI, βII, γ) and novel (δ, ε, η, θ)—that regulate vascular tone, permeability, extracellular matrix turnover, and gene transcription. In diabetes, chronic hyperglycemia increases diacylglycerol (DAG) levels, persistently activating PKC isoforms and coupling metabolic stress to vascular and neural injury [102,103,104].
Within the vasa nervorum, PKC activation suppresses endothelial nitric oxide synthase (eNOS), reducing nitric oxide bioavailability and impairing vasodilation, while upregulating endothelin-1, a potent vasoconstrictor. The resulting ischemia diminishes endoneurial perfusion and oxygen delivery [105,106,107]. In parallel, PKC-driven induction of VEGF and other permeability mediators disrupts blood–nerve and blood–brain barrier integrity, facilitating plasma protein leakage, immune cell infiltration, neural edema, and glial activation [108,109,110,111,112,113]. PKC signaling also orchestrates pro-inflammatory transcriptional programs, activating NF-κB and AP-1 to upregulate TNF-α, IL-6, chemokines, and endothelial adhesion molecules [114,115,116,117]. This inflammatory milieu compounds axonal stress. Moreover, PKC impairs neurotrophic signaling—including NGF and BDNF pathways—and disrupts retrograde axonal transport, compromising neuronal survival and Schwann cell function [118,119,120,121]. Among PKC isoforms, PKCβ is most extensively implicated in diabetic microvascular complications. Pharmacological inhibition improves endothelial and neurovascular function in experimental models and early clinical studies. However, large randomized trials in diabetic neuropathy—such as those with ruboxistaurin—demonstrated safety and some subgroup benefit but failed to achieve consistent primary endpoint success [122,123,124,125]. PKC does not act in isolation. It intersects with the polyol pathway, AGE–RAGE axis, and hexosamine biosynthetic pathway, synergistically amplifying inflammation, oxidative stress, and mitochondrial dysfunction. Together, these interactions drive progressive injury to neurons, Schwann cells, endothelial cells, and glia, transitioning the neurovascular unit from metabolic imbalance to structural breakdown [102,126].
In summary, PKC hyperactivation is a central mediator of vascular dysfunction, inflammation, and neurovascular injury in diabetic neuropathy—and likely in diabetes-associated cognitive decline. While single-target PKC inhibition has yielded mixed results, multi-targeted therapeutic strategies addressing PKC alongside other pathogenic pathways remain a compelling avenue for disease modification [102,103,104].
3.2. Mitochondrial Dysfunction and Oxidative Stress
Mitochondria serve as the primary energy-generating organelles in neurons and glial cells, supplying ATP through oxidative phosphorylation and regulating calcium homeostasis, redox balance, and apoptosis [127,128]. Neurons are especially dependent on proper mitochondrial function due to their high metabolic demand and long axons, which require energy-intensive maintenance and repair [128]. In diabetes, chronic hyperglycemia and lipid overload lead to mitochondrial perturbations that initiate and sustain oxidative stress, bioenergetic failure, and ultimately, neurodegeneration [129].
3.2.1. Reactive Oxygen Species (ROS) Generation in Neurons and Glia
Hyperglycemia-driven mitochondrial dysfunction is a central initiating event in diabetic neurodegeneration. Excess glucose flux increases electron transport chain (ETC) activity, hyperpolarizing the mitochondrial membrane and promoting electron leakage from complexes I and III, generating superoxide anion (O₂⁻·). Manganese superoxide dismutase (MnSOD) rapidly converts superoxide to hydrogen peroxide (H₂O₂), which, in the presence of iron, undergoes Fenton chemistry to yield hydroxyl radicals. This reactive oxygen species (ROS) cascade damages lipids, proteins, and DNA—marked by 8-oxo-deoxyguanosine (8-oxo-dG)—and activates NF-κB and MAPK signaling, as well as NLRP3 inflammasome assembly in microglia and astrocytes, triggering release of IL-1β, IL-6, and TNF-α [130,131,132,133,134]. In axons, ROS destabilizes microtubules and impairs motor proteins, stalling organelle transport; in Schwann cells, oxidative injury disrupts myelin maintenance and neurotrophic support. Endogenous antioxidant defenses—glutathione (GSH), SOD, and catalase—are overwhelmed, perpetuating oxidative injury.
Chronic oxidative stress also impairs mitochondrial biogenesis, which PGC-1α orchestrates in cooperation with NRF1/NRF2 to induce TFAM and mitochondrial DNA transcription [135,136]. Pro-inflammatory cytokines and reduced AMPK activity suppress this program, decreasing mitochondrial content and ATP supply at energy-demanding sites such as synaptic terminals and distal axons [137,138]. Mitochondrial dynamics shift toward fission (DRP1) at the expense of fusion (MFN1/2, OPA1), producing fragmented, bioenergetically compromised organelles with diminished oxidative phosphorylation (OXPHOS) capacity, impaired calcium buffering, and heightened ROS output [138,139,140,141,142]. Mitophagy, the PINK1/Parkin-dependent clearance of damaged mitochondria, is likewise impaired in diabetes. Reduced PINK1/Parkin signaling, lysosomal dysfunction secondary to AGE and lipid toxicity, and defective autophagic flux allow dysfunctional mitochondria to persist, amplifying ROS production and precipitating apoptosis or necroptosis. Restoration of the FoxO3a–PINK1–Parkin axis in experimental painful diabetic neuropathy (PDN) improves mitophagy, reduces allodynia, and attenuates inflammasome activation; conversely, mitophagy failure exacerbates neuroinflammation [143,144,145]. Mitochondrial ROS is not only a direct effector of injury but also a master upstream trigger for multiple hyperglycemia-induced pathways, including the hexosamine biosynthetic pathway, PKC activation, and the AGE–RAGE axis, as well as inflammatory circuits that impair axonal transport and Schwann cell homeostasis [130,131].
Therapeutic targeting of mitochondrial dysfunction shows translational promise. AMPK activators (e.g., metformin) and interventions that upregulate PGC-1α can restore mitochondrial biogenesis and attenuate ROS production. Modulation of mitochondrial dynamics—such as DRP1 inhibition with mdivi-1—rescues fragmentation and improves neurovascular outcomes in diabetic models, though potential adverse effects from over-suppressed fission (including hippocampal dysfunction) necessitate caution. Redox-active agents such as melatonin and SIRT3 activators suppress excessive fission and restore FoxO3a–PINK1–Parkin-mediated mitophagy, alleviating neuropathic hypersensitivity in preclinical models [139,142,143,146].
3.2.2. Antioxidant Depletion and Redox Imbalance
Neurons and glia maintain high metabolic rates and rely on a robust antioxidant defense network to balance reactive oxygen species (ROS)—inevitable byproducts of mitochondrial respiration and cellular metabolism [147]. Key enzymatic systems include glutathione (GSH) and its partner glutathione peroxidase (GPx), superoxide dismutases (SODs), catalase, peroxiredoxins, and the thioredoxin (Trx) system, which sequentially detoxify superoxide (O₂⁻·) to hydrogen peroxide (H₂O₂) and then to water, thereby preserving DNA, protein, and lipid integrity [148,149].
In diabetes, this protective network is compromised at multiple levels. Hyperglycemia diverts glucose into the polyol pathway, where aldose reductase consumes NADPH, and concurrently activates NADPH oxidases (NOX2/NOX4), which also deplete NADPH while producing superoxide. NADPH scarcity limits regeneration of reduced GSH and Trx, increases oxidized glutathione (GSSG), and shifts the intracellular redox state toward pro-oxidative stress [62,65,150,151,152,153,154].
The master transcriptional regulator Nrf2, which normally translocates to the nucleus under stress to induce antioxidant genes such as SOD2, GPx, HO-1, and NQO1, is suppressed by chronic hyperglycemia and inflammation. This blunted activation limits inducible antioxidant capacity precisely when cytoprotective reinforcement is required [155,156,157]. Additionally, antioxidant enzymes themselves are susceptible to non-enzymatic glycation and dicarbonyl (e.g., methylglyoxal) modification, impairing their catalytic function and, in some cases, antigenicity—changes documented in tissues vulnerable to diabetic complications, including retinal microvasculature [158,159,160,161]. Within mitochondria, oxidative stress damages mtDNA, disrupts electron transport chain assembly, and promotes opening of the mitochondrial permeability transition pore (mPTP). The resulting loss of membrane potential (Δψm), swelling, and cytochrome c release initiate caspase-dependent apoptosis in neurons and glia, affecting both the peripheral and central nervous systems [162,163,164,165].
This progressive depletion of antioxidant reserves marks a transition from reversible metabolic dysfunction to entrenched neurodegeneration. Experimental interventions that restore redox balance—such as α-lipoic acid (ALA), coenzyme Q10 (CoQ10), N-acetylcysteine (NAC), or mitochondria-targeted antioxidants (e.g., mitoTEMPO)—reduce ROS, preserve mitochondrial integrity, and improve nerve conduction in preclinical models [166,167,168,169,170,171]. Clinical trials report symptomatic and conduction improvements, though effects are generally modest, variable, and short-lived, often limited by study design.
3.3. Neuroinflammation and Immune Dysregulation
Neuroinflammation is a key amplifier of neural injury in diabetes, linking metabolic stress to progressive degeneration in both the central (CNS) and peripheral nervous system (PNS). Chronic hyperglycemia and oxidative stress activate microglia, the brain’s resident immune sentinels, via pattern-recognition receptors including TLR4 and the receptor for advanced glycation end-products (RAGE). Activated microglia adopt amoeboid morphology, express CD11b, Iba1, and MHC-II, and initiate NF-κB and MAPK signaling, producing pro-inflammatory cytokines TNF-α, IL-1β, and IL-6. These mediators impair synaptic plasticity, suppress neurogenesis, and weaken the blood–brain barrier (BBB), facilitating peripheral immune cell entry [173,174].
Astrocytes, which normally regulate ion balance, clear glutamate, and support the BBB, undergo reactive gliosis marked by elevated GFAP. In diabetes, impaired glucose handling and reduced GLT-1 expression in hippocampal astrocytes compromise glutamate clearance, increasing excitotoxic risk and potentially contributing to cognitive decline. Insulin treatment can partially restore astrocytic function in experimental models [175,176,177].
In the PNS, disruption of the blood–nerve barrier (BNB) parallels CNS barrier breakdown. Diabetes reduces expression of the tight-junction protein claudin-1 in the perineurium, depletes vessel-associated macrophages, and increases permeability to small solutes—changes that sustain neuropathic pain [111,174]. Injured axons, Schwann cells, and endoneurial endothelial cells release monocyte chemoattractant protein-1 (MCP-1/CCL2), recruiting monocytes that differentiate into M1-polarized macrophages. These macrophages produce nitric oxide, ROS, TNF-α, and IL-1β, release matrix metalloproteinases, degrade myelin, and inhibit Schwann cell-mediated repair [111,178,179].
T-cell infiltration further shapes the inflammatory milieu. In human and experimental diabetic neuropathy, CD4⁺ T cells show activation markers CD69 and CD25, with a Th1 bias marked by interferon-γ production. Regulatory T cells (Tregs) are numerically and functionally reduced in diabetes, tilting immune balance toward sustained inflammation [180,181].
Three cytokines dominate the effector landscape. TNF-α sensitizes nociceptors and promotes neuronal apoptosis via TNFR1; inhibition of TNF signaling improves conduction velocity and reduces pain in diabetic neuropathy models [182,183]. IL-1β, generated via inflammasomes such as NLRP3, disrupts barrier integrity and induces pyroptosis, exacerbating neuroinflammation and axonal injury [184,185]. IL-6, although pleiotropic, can impair neuronal and glial insulin signaling via SOCS3 induction, thereby disrupting energy metabolism and neurotrophic support [186,187]. Sustained activation of microglia, astrocytes, macrophages, and T cells creates a self-reinforcing inflammatory loop that inhibits neurogenesis, remyelination, and axonal regeneration—mirroring the systemic low-grade inflammation of diabetes.
Therapeutic approaches targeting this axis are under active investigation. Anti-cytokine strategies (e.g., TNF-α blockade) show structural and functional benefit in models of diabetic neuropathy [183]. Minocycline, a microglial inhibitor, reduces spinal microglial activation and alleviates pain behaviors in streptozotocin-diabetic rats [188]. Restoration of Treg function using low-dose IL-2 has been proposed to rebalance immunity without promoting effector T-cell activation [189].
In summary, while hyperglycemia and oxidative stress initiate neural injury, it is chronic, dysregulated immune activation—driven by CNS glia, PNS macrophages, T cells, and the cytokines TNF-α, IL-1β, and IL-6—that sustains and amplifies neurodegeneration in diabetes. Strategies that restore barrier integrity, resolve glial overactivation, and re-establish immune regulation offer promising routes toward disease modification [111,173,174,190].
3.4. Vascular and Endothelial Dysfunction
Neural integrity depends on its microvascular networks—the vasa nervorum in the periphery and the brain’s capillary beds—delivering oxygen and glucose while clearing metabolic waste. In diabetes, these microvessels undergo structural and functional injury that precedes and accelerates both diabetic peripheral neuropathy (DPN) and diabetes-associated cognitive decline [191].
Peripheral microangiopathy is well documented in human sural nerve biopsies and experimental models, characterized by basement membrane thickening, luminal narrowing, endothelial hyperplasia, and perivascular support cell abnormalities [192,193,194]. These changes restrict blood flow and cause endoneurial hypoxia—confirmed by direct oxygen-tension measurements in diabetic nerves and by hypoxia markers such as HIF-1α induction [195,196,197,198]. Hypoxic axons exhibit impaired mitochondrial respiration and reduced ATP supply, compromising long, metabolically demanding fibers before overt axonal loss occurs.
Endothelial nitric oxide (NO) bioavailability is diminished in diabetes due to decreased eNOS expression and activation, accumulation of asymmetric dimethylarginine (ADMA), and eNOS uncoupling, which shifts the enzyme toward superoxide generation [199,200]. In peripheral nerve microvessels, this results in blunted arteriolar vasodilation and reduced perfusion; in the hippocampus, reduced NO impairs neurovascular coupling and contributes to early cognitive deficits [201,202,203].
In the CNS, blood–brain barrier (BBB) integrity is compromised. Diabetes downregulates tight-junction proteins (occludin, claudin-5, ZO-1) and increases barrier permeability, initially detected in hippocampal and cortical microvessels in diabetic rodents and confirmed in humans using dynamic contrast–enhanced MRI [9,204,205]. BBB breakdown allows entry of circulating cytokines, immune cells, and metabolites, propagating central inflammation and correlating with cognitive impairment [206].
Systemic endothelial activation further fuels neurovascular injury. High glucose increases endothelial expression of ICAM-1, VCAM-1, and selectins, promoting leukocyte adhesion, transendothelial migration, and impaired vascular repair—observed both in vitro and in people with type 2 diabetes [207,208]
Angiogenic imbalance is another hallmark. VEGF is often reduced in neuropathic target tissues, and exogenous VEGF delivery in experimental diabetes restores vasa nervorum density and improves nerve function [209,210]. Conversely, destabilizing signals such as angiopoietin-2 and thrombospondins are elevated in diabetic microangiopathy, favoring capillary regression and rarefaction [211].
In summary, vascular injury precedes neural degeneration. Vasa-nervorum ischemia and BBB disruption set the stage for metabolic and inflammatory damage; NO loss impairs perfusion and neurovascular coupling; endothelial activation and skewed angiogenic signaling perpetuate a microenvironment hostile to axons and glia. These vascular pathologies are therefore not mere accomplices but early, actionable targets for interventions aimed at preventing or reversing DPN and diabetes-related cognitive decline [191].
3.5. Insulin Resistance and Metabolic Inflexibility in the Nervous System
Insulin signaling is essential for neural metabolism, synaptic function, and structural integrity. Neurons and glia express insulin receptors, and ligand binding activates the canonical IRS–PI3K–Akt cascade, promoting glucose uptake, inhibiting GSK3β, regulating mTOR activity, and supporting both synaptic plasticity and mitochondrial function [212,213]. In diabetes, chronic hyperinsulinemia and hyperglycemia induce brain and peripheral nerve insulin resistance, impairing metabolic flexibility—the ability to switch between fuel substrates in response to demand [212,214].
In the healthy CNS, insulin facilitates translocation of glucose transporters, activates pro-survival pathways, and enhances plasticity. Astrocytes adjust glycolysis and lactate shuttling to match neuronal needs, while oligodendrocytes, under the insulin/IGF axis, maintain myelin and lipid metabolism [215,216,217,218]. In diabetes, inflammatory cytokines TNF-α and IL-6 activate JNK and IKK, inducing serine phosphorylation of IRS-1, which blocks PI3K recruitment and diminishes Akt activity. Reduced Akt allows GSK3β hyperactivation, a tau kinase implicated in Alzheimer’s disease–like pathology and neuronal loss [219,220,221,222]. Glucose transport is further compromised. Neuronal GLUT3—normally insulin-independent but responsive to stress—and astrocytic/BBB GLUT1 both decline in experimental diabetes, limiting glucose entry into neural tissue and producing cellular energy deficit despite systemic hyperglycemia [223,224]. Energy scarcity at axon terminals impairs vesicle cycling, ion pump activity, and axonal transport, slowing conduction and weakening synaptic transmission [225,226].
Dyslipidemia compounds these deficits. Insulin-resistant adipose tissue releases excess fatty acids that enter the CNS and PNS. Under mitochondrial stress, incomplete β-oxidation yields diacylglycerols (DAGs) and ceramides, bioactive lipids that activate PKC isoforms or PP2A, further suppressing Akt signaling, increasing ROS generation, promoting mitochondrial outer membrane permeabilization, and triggering caspase-dependent apoptosis [227,228,229]. Sphingolipid dysregulation, particularly ceramide accumulation, associates with neuropathic pain, myelin instability, and cognitive decline, linking insulin resistance to oxidative stress and inflammatory activation [230]. Therapeutic approaches to restore insulin sensitivity and metabolic flexibility show emerging potential. Intranasal insulin and GLP-1 receptor agonists have demonstrated preliminary benefits for cognition and neural metabolism, although results are mixed and mechanisms remain multifactorial [231,232,233,234,235]. Strategies to preserve GLUT1/GLUT3 expression at the BBB and in astrocytes, and to limit DAG/ceramide synthesis or block PP2A/PKC-mediated Akt inhibition, are rational targets [223].
In summary, insulin resistance and metabolic inflexibility represent a central node linking synaptic dysfunction, axonal transport failure, and lipid-mediated injury in the diabetic nervous system. Preserving insulin signaling, maintaining glucose transporter function, and mitigating sphingolipid-driven Akt suppression may collectively protect neuronal and glial viability.
3.6. The Gut–Brain Axis in Diabetes-Induced Neuropathy
The gut–brain–nerve axis is increasingly recognized as a modulator of diabetic neurodegeneration, with intestinal dysbiosis serving as an upstream driver of metabolic and neural injury. In diabetes, SCFA-producing taxa such as Faecalibacterium and Roseburia decline, while pro-inflammatory species including Ruminococcus gnavus and Bacteroides vulgatus expand [8,236,237,238]. Reduced butyrate availability compromises epithelial tight junctions, increases intestinal permeability, and promotes metabolic endotoxemia, wherein lipopolysaccharide (LPS) activates TLR4 signaling to induce inflammatory cytokine cascades that propagate through neural and glial compartments [239].
SCFAs have direct neuroimmune significance. Microglia require SCFA signals for maturation and homeostatic regulation; depletion shifts them toward pro-inflammatory phenotypes, increasing synaptic remodeling and inflammatory signaling. Replenishing SCFAs or stimulating their receptors (FFAR2/3) restores microglial homeostasis, while SCFA-driven enteroendocrine signaling enhances GLP-1 release, indirectly supporting neural metabolism [240,241]. Microbial tryptophan metabolites, particularly indole-3-propionic acid (IPA), act via the aryl hydrocarbon receptor (AhR) to suppress NF-κB–mediated inflammatory programs in astrocytes. Human studies link higher IPA levels to reduced type 2 diabetes risk, and in vitro work demonstrates that IPA reduces LPS-induced cytokine production in human astrocytes, implicating indole restoration as a potential anti-inflammatory strategy within the CNS [242,243,244].
Bile acids, reshaped by microbial metabolism, also contribute to neuroimmune modulation. Through TGR5 and FXR signaling in neurons and glia, they can suppress neuroinflammation and, in preclinical models, alleviate neuropathic pain behaviors—suggesting that altered bile acid composition in diabetes may have direct neurological consequences [245,246]. The vagus nerve provides a direct neural conduit linking gut and brain. In diabetes, reduced vagal tone—reflected in diminished heart rate variability—weakens the cholinergic anti-inflammatory reflex, allowing gut-derived immune activation to propagate unchecked. Restoration of vagal tone therefore holds potential as both a cardiovascular and neuroimmune intervention [237,247].
Causality is supported by interventional studies. In rodent models, fecal microbiota transplantation (FMT) from lean donors or direct butyrate supplementation improves gut barrier integrity, lowers systemic cytokines, and normalizes mechanical and thermal nociceptive thresholds. Early clinical evidence suggests that FMT may ameliorate painful DPN, while in metabolic diabetes models it rebalances microbial composition, metabolite profiles, and systemic inflammation [248,249].
In summary, diabetes-associated dysbiosis reduces SCFAs, compromises epithelial integrity, and promotes LPS–TLR4-mediated inflammation. SCFA depletion disrupts microglial regulation and GLP-1–mediated metabolic support; loss of indole signals impairs astrocytic immune control; altered bile acid signaling skews neuroimmune balance; and diminished vagal tone removes an essential anti-inflammatory checkpoint. These gut-derived mechanisms are modifiable—microbiota restoration, targeted metabolite supplementation, and neural–microbial signaling enhancement represent promising strategies to prevent or mitigate diabetic neuropathy and cognitive decline, complementing but distinct from mitochondrial, vascular, inflammatory, or insulin-resistance–focused interventions.
- Linking Mechanisms to Clinical Phenotypes in Diabetic Neurodegeneration
| Pathogenic Mechanism | Mechanism | Clinical Phenotypes | Key References |
| Polyol Pathway Overdrive | AR-mediated glucose-to-sorbitol conversion consuming NADPH, causing osmotic and redox stress (myo-inositol depletion; Na⁺/K⁺-ATPase impairment) | Length-dependent DPN; early small-fiber symptoms; slowed conduction | [64,65] |
| AGE–RAGE Axis | Tissue glycation, ECM stiffening, RAGE-mediated NF-κB/MAPK activation and oxidative stress | DPN (axonal/demyelination); painful DPN; cognitive decline | [250] |
| PKC Activation (via DAG) | Impaired eNOS/NO, elevated endothelin-1, VEGF-induced permeability | Ischemic DPN; microvascular dysfunction; autonomic neuropathy | [104] |
| Oxidative Stress & Mitochondrial Dysfunction | ETC superoxide generation; imbalance of mitochondrial fission/fusion; impaired biogenesis | Axonal transport impairment; PDN; cognitive slowing | [133] |
| Compromised Antioxidant Defenses | Nrf2 suppression, enzyme glycation (SOD, catalase, GPx), NADPH depletion | Accelerated DPN progression; CNS vulnerability | [155,251,252] |
| Neuroinflammation (Microglia/Astrocytes, Cytokines) | TLR4/RAGE induction; astrocytic GLT-1 downregulation; release of TNF-α, IL-1β, IL-6 | Painful DPN; cognitive impairment; barrier dysfunction | [173] |
| Neurovascular Dysfunction (vasa nervorum & BBB) | Microangiopathy; reduced eNOS/mitigated NO; BBB tight-junction breakdown | DPN ischemia; cognitive decline | [191,204] |
| Insulin Resistance & Metabolic Inflexibility | IRS-1 Ser-phosphorylation; GLUT1/GLUT3 downregulation; lipotoxic DAG/ceramide accumulation | Cognitive decline; synaptic fatigue; DPN progression | [212] |
| Gut–Brain–Nerve Axis (Dysbiosis) | Loss of SCFA-producing species; LPS-TLR4 endotoxemia; impaired SCFA/indole/bile signaling; vagal dysfunction | PDN; cognitive symptoms; systemic inflammation | [236,242] |
| Small-Fiber Predominant Pathology | Early Aδ/C-fiber involvement; autonomic sudomotor fiber loss | Painful DPN; autonomic features | [18] |
| Large-Fiber Predominant Pathology | Axonal degeneration ± demyelination | Numbness, gait instability | [26] |
| CNS Cognitive/Affective Manifestations | BBB leak; insulin resistance; neuroinflammation | MCI; depression/anxiety | [54] |
| Autonomic Neuropathy (CAN & GI Dysmotility) | Autonomic fiber loss; impaired vagal signaling; enteric dysfunction | Orthostatic hypotension; gastroparesis | [253] |
4. Knowledge Gaps and Future Directions
Diabetes-induced neurological disorders arise from a convergence of mechanistic insults that act in concert across the peripheral and central nervous systems. Hyperglycemia and oxidative stress initiate a cascade that activates microglia via AGE–RAGE and TLR4 signaling, triggering NF-κB and MAPK pathways and releasing pro-inflammatory cytokines (TNF-α, IL-1β, IL-6) that impair synaptic plasticity, weaken the blood–brain barrier, and promote neurodegeneration [173,174]. Astrocytic gliosis further disrupts glutamate clearance and barrier integrity, while in the periphery, blood–nerve barrier leakage and macrophage recruitment amplify demyelination and axonal injury [111,181].
Vascular pathology compounds this injury. Microangiopathy of the vasa nervorum, reduced nitric oxide bioavailability, and capillary rarefaction generate chronic endoneurial hypoxia, mirrored in the brain by impaired neurovascular coupling and BBB breakdown [191]. Concurrent insulin resistance within neurons and glia disrupts PI3K–Akt signaling, reduces GLUT1/GLUT3-mediated glucose transport, and drives lipid-mediated toxicity through diacylglycerol and ceramide accumulation [212,254].
The gut–brain–nerve axis adds another layer: dysbiosis diminishes the production of short-chain fatty acids, such as butyrate, weakening microglial maturation and anti-inflammatory tone, while reduced microbial indole metabolites destabilize astrocytic homeostasis [236,242]. Altered bile acid signaling through TGR5/FXR and diminished vagal tone further skew neuroimmune balance [237,245]. These interconnected systems form a multiorgan network of degeneration, where vascular, metabolic, immune, and microbial pathways amplify one another over time.
Yet critical knowledge gaps persist. Few longitudinal human studies track individuals from pre-symptomatic metabolic dysfunction to overt neuropathy or cognitive decline with integrated multimodal measures—combining neuroimaging, electrophysiology, omics profiling, and microbiome analysis [255,256]. Commonly used rodent models incompletely recapitulate the chronic, low-grade inflammation and gradual sensory-cognitive decline of human disease, underscoring the need for standardized phenotyping and humanized microbiota platforms [257,258,259].
Biomarker development is another frontier. Circulating neurofilament light (NfL) and GFAP show potential for early detection and monitoring, while corneal nerve fiber metrics offer a window into small-fiber pathology and regeneration [260,261]. These should be validated across ancestries and combined with cytokine, endothelial, and metabolic signatures for patient stratification. Mechanistic gaps also include the underexplored roles of epigenetic programming (“metabolic memory”) and circadian disruption in neural vulnerability [262,263], as well as sex-specific differences in immune tone, pain processing, and mitochondrial function—factors too often ignored in study design [264].
Toward Precision-Based, Multi-Targeted Therapy
Addressing these gaps will require interdisciplinary convergence—linking endocrinology, neurology, immunology, microbiology, and systems biology. Precision medicine can integrate advanced biomarkers, neuroimaging, and microbiome profiling with metabolic, vascular, and neuroimmune interventions. Lifestyle-based strategies—structured exercise, dietary modulation, cognitive training—should be systematically combined with pharmacologic approaches, including GLP-1 receptor agonists, intranasal insulin, anti-inflammatory agents, and microbiota-modulating therapies.
The field must also invest in longitudinal, stratified cohorts that sample brain, nerve, blood, and gut over time; in preclinical models that reflect human chronicity and complexity; and in mechanistic mapping of inter-organ networks linking gut, liver, vasculature, and nervous tissue. Only by aligning these strands—metabolism and immunity, vasculature and mitochondria, gut and brain—can the goal shift from symptomatic control toward slowing, halting, or reversing the neurodegenerative trajectory in people living with diabetes.
Acknowledgments
We utilized AI-based tools, to assist with grammar and language refinement in this manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Feldman, E.L. , et al., Diabetic neuropathy. Nat Rev Dis Primers, 2019. 5(1): p. 42.
- Pop-Busui, R.; Boulton, A.J.; Feldman, E.L.; Bril, V.; Freeman, R.; Malik, R.A.; Sosenko, J.M.; Ziegler, D. Diabetic Neuropathy: A Position Statement by the American Diabetes Association. Diabetes Care 2016, 40, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A. , et al., Diabetes and Cognitive Impairment. Curr Diab Rep, 2016. 16(9): p. 87.
- Kciuk, M.; Kruczkowska, W.; Gałęziewska, J.; Wanke, K.; Kałuzińska-Kołat, Ż.; Aleksandrowicz, M.; Kontek, R. Alzheimer’s Disease as Type 3 Diabetes: Understanding the Link and Implications. Int. J. Mol. Sci. 2024, 25, 11955. [Google Scholar] [CrossRef] [PubMed]
- Giacco, F. and M. Brownlee, Oxidative stress and diabetic complications. Circ Res, 2010. 107(9): p. 1058-70.
- Talbot, K. , et al., Demonstrated brain insulin resistance in Alzheimer's disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest, 2012. 122(4): p. 1316-38.
- Zhao, F.; Siu, J.J.; Huang, W.; Askwith, C.; Cao, L. Insulin Modulates Excitatory Synaptic Transmission and Synaptic Plasticity in the Mouse Hippocampus. Neuroscience 2019, 411, 237–254. [Google Scholar] [CrossRef]
- Cani, P.D. , et al., Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes, 2007. 56(7): p. 1761-72.
- Starr, J.M.; Wardlaw, J.; Ferguson, K.; MacLullich, A.; Deary, I.J.; Marshall, I. Increased blood-brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J. Neurol. Neurosurg. Psychiatry 2003, 74, 70–76. [Google Scholar] [CrossRef]
- Qiao, J.; Lawson, C.M.; Rentrup, K.F.G.; Kulkarni, P.; Ferris, C.F. Evaluating blood–brain barrier permeability in a rat model of type 2 diabetes. J. Transl. Med. 2020, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Shimu, S.J. and S. Islam, Gender Differences in Drug Addiction: Neurobiological, Social, and Psychological Perspectives in Women–A Systematic Review. Journal of Primeasia, 2025. 6(1): p. 1-13.
- Burgess, J.; Frank, B.; Marshall, A.; Khalil, R.S.; Ponirakis, G.; Petropoulos, I.N.; Cuthbertson, D.J.; Malik, R.A.; Alam, U. Early Detection of Diabetic Peripheral Neuropathy: A Focus on Small Nerve Fibres. Diagnostics 2021, 11, 165. [Google Scholar] [CrossRef]
- Määttä, L.L.; Andersen, S.T.; Parkner, T.; Hviid, C.V.; Bjerg, L.; Kural, M.A.; Charles, M.; Søndergaard, E.; Kuhle, J.; Tankisi, H.; et al. Longitudinal Change in Serum Neurofilament Light Chain in Type 2 Diabetes and Early Diabetic Polyneuropathy: ADDITION-Denmark. Diabetes Care 2024, 47, 986–994. [Google Scholar] [CrossRef]
- Fridman, V.; Sillau, S.; Ritchie, A.; Bockhorst, J.; Coughlan, C.; Araya, P.; Espinosa, J.M.; Smith, K.; Lange, E.M.; Lange, L.A.; et al. Plasma neurofilament light chain concentrations are elevated in youth-onset type 2 diabetes and associated with neuropathy. J. Peripher. Nerv. Syst. 2023, 28, 460–470. [Google Scholar] [CrossRef]
- Mielke, M.M.; Evans, J.K.; Neiberg, R.H.; Molina-Henry, D.P.; Marcovina, S.M.; Johnson, K.C.; Carmichael, O.T.; Rapp, S.R.; Sachs, B.C.; Ding, J.; et al. Alzheimer Disease Blood Biomarkers and Cognition Among Individuals With Diabetes and Overweight or Obesity. JAMA Netw. Open 2025, 8, e2458149. [Google Scholar] [CrossRef]
- Maalmi, H.; Strom, A.; Petrera, A.; Hauck, S.M.; Strassburger, K.; Kuss, O.; Zaharia, O.-P.; Bönhof, G.J.; Rathmann, W.; Trenkamp, S.; et al. Serum neurofilament light chain: a novel biomarker for early diabetic sensorimotor polyneuropathy. Diabetologia 2022, 66, 579–589. [Google Scholar] [CrossRef]
- Karger, A.B.; Nasrallah, I.M.; Braffett, B.H.; Luchsinger, J.A.; Ryan, C.M.; Bebu, I.; Arends, V.; Habes, M.; Gubitosi-Klug, R.A.; Chaytor, N.; et al. Plasma Biomarkers of Brain Injury and Their Association With Brain MRI and Cognition in Type 1 Diabetes. Diabetes Care 2024, 47, 1530–1538. [Google Scholar] [CrossRef]
- Perkins, B.A.; Lovblom, L.E.; Lewis, E.J.; Bril, V.; Ferdousi, M.; Orszag, A.; Edwards, K.; Pritchard, N.; Russell, A.; Dehghani, C.; et al. Corneal Confocal Microscopy Predicts the Development of Diabetic Neuropathy: A Longitudinal Diagnostic Multinational Consortium Study. Diabetes Care 2021, 44, 2107–2114. [Google Scholar] [CrossRef]
- Gad, H.; Petropoulos, I.N.; Khan, A.; Ponirakis, G.; MacDonald, R.; Alam, U.; A Malik, R. Corneal confocal microscopy for the diagnosis of diabetic peripheral neuropathy: A systematic review and meta-analysis. J. Diabetes Investig. 2021, 13, 134–147. [Google Scholar] [CrossRef]
- Billeson, K.; Baldimtsi, E.; Wahlberg, J.; Whiss, P.A. Growth Differentiation Factor 15 and Matrix Metalloproteinase 3 in Plasma as Biomarkers for Neuropathy and Nephropathy in Type 1 Diabetes. Int. J. Mol. Sci. 2024, 25, 7328. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, I.S. , Pluripotent Stem Cells for Cell Therapy. Methods Mol Biol, 2021. 2269: p. 25-33.
- Shrikanth, C.; Nandini, C. AMPK in microvascular complications of diabetes and the beneficial effects of AMPK activators from plants. Phytomedicine 2020, 73, 152808. [Google Scholar] [CrossRef] [PubMed]
- Zilliox, L.A.; Russell, J.W. Physical activity and dietary interventions in diabetic neuropathy: a systematic review. Clin. Auton. Res. 2019, 29, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Kajiyama, S.; Kawamoto, M.; Shiraishi, S.; Gaus, S.; Matsunaga, A.; Suyama, H.; Yuge, O. Spinal orexin-1 receptors mediate anti-hyperalgesic effects of intrathecally-administered orexins in diabetic neuropathic pain model rats. Brain Res. 2005, 1044, 76–86. [Google Scholar] [CrossRef]
- Berti-Mattera, L.; Larkin, B.; Hourmouzis, Z.; Kern, T.; Siegel, R. NF-κB subunits are differentially distributed in cells of lumbar dorsal root ganglia in naïve and diabetic rats. Neurosci. Lett. 2011, 490, 41–45. [Google Scholar] [CrossRef]
- Dyck, P.J.; Albers, J.W.; Andersen, H.; Arezzo, J.C.; Biessels, G.; Bril, V.; Feldman, E.L.; Litchy, W.J.; O'BRien, P.C.; Russell, J.W.; et al. Diabetic polyneuropathies: update on research definition, diagnostic criteria and estimation of severity. Diabetes/Metabolism Res. Rev. 2011, 27, 620–628. [Google Scholar] [CrossRef]
- Gu, H. , et al., Controlled chattering--a new 'cutting-edge' technology for nanofabrication. Nanotechnology, 2010. 21(35): p. 355302.
- Yaribeygi, H.; Ashrafizadeh, M.; Henney, N.C.; Sathyapalan, T.; Jamialahmadi, T.; Sahebkar, A. Neuromodulatory effects of anti-diabetes medications: A mechanistic review. Pharmacol. Res. 2020, 152, 104611. [Google Scholar] [CrossRef]
- Robinson, C.S.; Sharp, P. Tighter Accuracy Standards within Point-of-Care Blood Glucose Monitoring: How Six Commonly Used Systems Compare. J. Diabetes Sci. Technol. 2012, 6, 547–554. [Google Scholar] [CrossRef]
- Kouri, A.; Boulet, L.-P.; Kaplan, A.; Gupta, S. An evidence-based, point-of-care tool to guide completion of asthma action plans in practice. Eur. Respir. J. 2017, 49, 1602238. [Google Scholar] [CrossRef] [PubMed]
- Pozzi, M.; Rivolta, M.; Gelosa, M.; Capra, M.; Poggioli, G.; Bernasconi, A.; Lomuscio, G. Upper gastrointestinal involvement in diabetes mellitus: Study of esophagogastric function. Acta Diabetol. 1988, 25, 333–341. [Google Scholar] [CrossRef]
- Bharucha, A.E., Y. C. Kudva, and D.O. Prichard, Diabetic Gastroparesis. Endocr Rev, 2019. 40(5): p. 1318-1352.
- Usai-Satta, P.; Bellini, M.; Morelli, O.; Geri, F.; Lai, M.; Bassotti, G. Gastroparesis: New insights into an old disease. World J. Gastroenterol. 2020, 26, 2333–2348. [Google Scholar] [CrossRef] [PubMed]
- von Gerichten, J. , et al., The [(13) C]octanoic acid breath test for gastric emptying quantification: A focus on nutrition and modeling. Lipids, 2022. 57(4-5): p. 205-219.
- Agochukwu-Mmonu, N.; Pop-Busui, R.; Wessells, H.; Sarma, A.V. Autonomic neuropathy and urologic complications in diabetes. Auton. Neurosci. 2020, 229, 102736–102736. [Google Scholar] [CrossRef]
- Shaw, L.C.; Barnes, M.P.; Rodgers, H. Response to Letter by Munin et al Regarding Article, “Botulinum Toxin for the Upper Limb After Stroke (BoTULS) Trial: Effect on Impairment, Activity Limitation, and Pain”. Stroke 2011, 42. [Google Scholar] [CrossRef]
- Martonosi, Á.R.; Pázmány, P.; Kiss, S.; Dembrovszky, F.; Oštarijaš, E.; Szabó, L. Urodynamics in Early Diagnosis of Diabetic Bladder Dysfunction in Women: A Systematic Review and Meta-Analysis. Med Sci. Monit. 2022, 28, e937166–1. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Z.; Tang, Z.; He, C.; Tang, W. Diabetic cystopathy: A review 综述:糖尿病性膀胱病. J. Diabetes 2015, 7, 442–447. [Google Scholar] [CrossRef]
- Akbar, M.; Wandy, A.; Soraya, G.V.; Goysal, Y.; Lotisna, M.; Basri, M.I. Sudomotor dysfunction in diabetic peripheral neuropathy (DPN) and its testing modalities: A literature review. Heliyon 2023, 9, e18184. [Google Scholar] [CrossRef]
- Kaur, H.; Bal, A.; Sandhir, R. Curcumin supplementation improves mitochondrial and behavioral deficits in experimental model of chronic epilepsy. Pharmacol. Biochem. Behav. 2014, 125, 55–64. [Google Scholar] [CrossRef]
- Shimada, H.; Kihara, M.; Kosaka, S.; Ikeda, H.; Kawabata, K.; Tsutada, T.; Miki, T. Comparison of SSR and QSART in early diabetic neuropathy—the value of length-dependent pattern in QSART. Auton. Neurosci. 2001, 92, 72–75. [Google Scholar] [CrossRef] [PubMed]
- Krieger, S.-M.; Reimann, M.; Haase, R.; Henkel, E.; Hanefeld, M.; Ziemssen, T. Sudomotor Testing of Diabetes Polyneuropathy. Front. Neurol. 2018, 9, 803. [Google Scholar] [CrossRef]
- Tan, Z.S. , et al., Association of metabolic dysregulation with volumetric brain magnetic resonance imaging and cognitive markers of subclinical brain aging in middle-aged adults: the Framingham Offspring Study. Diabetes Care, 2011. 34(8): p. 1766-70.
- Launer, L.J.; E Miller, M.; Williamson, J.D.; Lazar, R.M.; Gerstein, H.C.; Murray, A.M.; Sullivan, M.; Horowitz, K.R.; Ding, J.; Marcovina, S.; et al. Effects of intensive glucose lowering on brain structure and function in people with type 2 diabetes (ACCORD MIND): a randomised open-label substudy. Lancet Neurol. 2011, 10, 969–977. [Google Scholar] [CrossRef] [PubMed]
- Ott, A. , et al., Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology, 1999. 53(9): p. 1937-42.
- Bahniwal, M.; Little, J.P.; Klegeris, A. High Glucose Enhances Neurotoxicity and Inflammatory Cytokine Secretion by Stimulated Human Astrocytes. Curr. Alzheimer Res. 2017, 14, 731–741. [Google Scholar] [CrossRef]
- Meng, F.; Fu, J.; Zhang, L.; Guo, M.; Zhuang, P.; Yin, Q.; Zhang, Y. Function and therapeutic value of astrocytes in diabetic cognitive impairment. Neurochem. Int. 2023, 169, 105591. [Google Scholar] [CrossRef]
- Steen, E. , et al., Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer's disease--is this type 3 diabetes? J Alzheimers Dis, 2005. 7(1): p. 63-80.
- Farris, W. , et al., Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci U S A, 2003. 100(7): p. 4162-7.
- Willette, A.A.; Bendlin, B.B.; Starks, E.J.; Birdsill, A.C.; Johnson, S.C.; Christian, B.T.; Okonkwo, O.C.; La Rue, A.; Hermann, B.P.; Koscik, R.L.; et al. Association of Insulin Resistance With Cerebral Glucose Uptake in Late Middle–Aged Adults at Risk for Alzheimer Disease. JAMA Neurol. 2015, 72, 1013–1020. [Google Scholar] [CrossRef]
- Anderson, R.J. , et al., The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care, 2001. 24(6): p. 1069-78.
- Mezuk, B.; Eaton, W.W.; Golden, S.H. Depression and Type 2 Diabetes Over the Lifespan: A Meta-Analysis. Diabetes Care 2009, 32, e57–e57. [Google Scholar] [CrossRef]
- Sanjeev, S.; Murthy, M.K.; Devi, M.S.; Khushboo, M.; Renthlei, Z.; Ibrahim, K.S.; Kumar, N.S.; Roy, V.K.; Gurusubramanian, G. Isolation, characterization, and therapeutic activity of bergenin from marlberry (Ardisia colorata Roxb.) leaf on diabetic testicular complications in Wistar albino rats. Environ. Sci. Pollut. Res. 2019, 26, 7082–7101. [Google Scholar] [CrossRef]
- Nasreddine, Z.S.; Phillips, N.A.; Bédirian, V.; Charbonneau, S.; Whitehead, V.; Collin, I.; Cummings, J.L.; Chertkow, H. The Montreal Cognitive Assessment, MoCA: A Brief Screening Tool For Mild Cognitive Impairment. J. Am. Geriatr. Soc. 2005, 53, 695–699. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F., S. E. Folstein, and P.R. McHugh, "Mini-mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res, 1975. 12(3): p. 189-98.
- Kroenke, K., R. L. Spitzer, and J.B. Williams, The PHQ-9: validity of a brief depression severity measure. J Gen Intern Med, 2001. 16(9): p. 606-13.
- Kawahito, S.; Kitahata, H.; Oshita, S. Problems associated with glucose toxicity: Role of hyperglycemia-induced oxidative stress. World J. Gastroenterol. 2009, 15, 4137–4142. [Google Scholar] [CrossRef]
- Calcutt, N.A. Diabetic neuropathy and neuropathic pain: a (con)fusion of pathogenic mechanisms? PAIN® 2020, 161, S65–S86. [Google Scholar] [CrossRef]
- Rojas, D.R.; Tegeder, I.; Kuner, R.; Agarwal, N. Hypoxia-inducible factor 1α protects peripheral sensory neurons from diabetic peripheral neuropathy by suppressing accumulation of reactive oxygen species. J. Mol. Med. 2018, 96, 1395–1405. [Google Scholar] [CrossRef]
- Oshitari, T. Advanced Glycation End-Products and Diabetic Neuropathy of the Retina. Int. J. Mol. Sci. 2023, 24, 2927. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Wu, J.; Jing, S.; Yan, L.-J. Hyperglycemic Stress and Carbon Stress in Diabetic Glucotoxicity. Aging Dis. 2016, 7, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Oates, P.J. Polyol pathway and diabetic peripheral neuropathy. Int. Rev. Neurobiol. 2002, 50, 325–392. [Google Scholar] [CrossRef]
- Malamas, M.S.; Hohman, T.C.; Millen, J. Novel Spirosuccinimide Aldose Reductase Inhibitors Derived from Isoquinoline-1,3-diones: 2-[(4-Bromo-2-fluorophenyl)methyl]-6-fluorospiro[isoquinoline-4(11H),3'-pyrrolidine]-1,2',3,5'(2H)-tetrone and Congeners. 1. J. Med. Chem. 1994, 37, 2043–2058. [Google Scholar] [CrossRef]
- Singh, M.; Kapoor, A.; Bhatnagar, A. Physiological and Pathological Roles of Aldose Reductase. Metabolites 2021, 11, 655. [Google Scholar] [CrossRef] [PubMed]
- Niimi, N.; Yako, H.; Takaku, S.; Chung, S.K.; Sango, K. Aldose Reductase and the Polyol Pathway in Schwann Cells: Old and New Problems. Int. J. Mol. Sci. 2021, 22, 1031. [Google Scholar] [CrossRef]
- A Greene, D.; Chakrabarti, S.; A Lattimer, S.; A Sima, A. Role of sorbitol accumulation and myo-inositol depletion in paranodal swelling of large myelinated nerve fibers in the insulin-deficient spontaneously diabetic bio-breeding rat. Reversal by insulin replacement, an aldose reductase inhibitor, and myo-inositol. J. Clin. Investig. 1987, 79, 1479–1485. [Google Scholar] [CrossRef]
- Greene, D.A. and A.M. Mackway, Decreased myo-inositol content and Na+-K+-ATPase activity in superior cervical ganglion of STZ-diabetic rat and prevention by aldose reductase inhibition. Diabetes, 1986. 35(10): p. 1106-8.
- Lattimer, S.A.; Sima, A.A.; Greene, D.A. In vitro correction of impaired Na+-K+-ATPase in diabetic nerve by protein kinase C agonists. Am. J. Physiol. Metab. 1989, 256, E264–E269. [Google Scholar] [CrossRef]
- Gillon, K.R.W.; Hawthorne, J.N.; Tomlinson, D.R. Myo-inositol and sorbitol metabolism in relation to peripheral nerve function in experimental diabetes in the rat: The effect of aldose reductase inhibition. Diabetologia 1983, 25, 365–371. [Google Scholar] [CrossRef]
- Sima, A.A. , et al., Supplemental myo-inositol prevents L-fucose-induced diabetic neuropathy. Diabetes, 1997. 46(2): p. 301-6.
- Goto, Y.; Hotta, N.; Shigeta, Y.; Sakamoto, N.; Kikkawa, R. Effects of an aldose reductase inhibitor, epalrestat, on diabetic neuropathy. Clinical benefit and indication for the drug assessed from the results of a placebo-controlled double-blind study. Biomed. Pharmacother. 1995, 49, 269–277. [Google Scholar] [CrossRef]
- Kawai, T. , et al., Effects of epalrestat, an aldose reductase inhibitor, on diabetic peripheral neuropathy in patients with type 2 diabetes, in relation to suppression of N(ɛ)-carboxymethyl lysine. J Diabetes Complications, 2010. 24(6): p. 424-32.
- Sekiguchi, K.; Kohara, N.; Baba, M.; Komori, T.; Naito, Y.; Imai, T.; Satoh, J.; Yamaguchi, Y.; Hamatani, T. ; the Ranirestat Group Aldose reductase inhibitor ranirestat significantly improves nerve conduction velocity in diabetic polyneuropathy: A randomized double-blind placebo-controlled study in Japan. J. Diabetes Investig. 2018, 10, 466–474. [Google Scholar] [CrossRef]
- Gabbay, K.H. , Aldose reductase inhibition in the treatment of diabetic neuropathy: where are we in 2004? Curr Diab Rep, 2004. 4(6): p. 405-8.
- Twarda-Clapa, A.; Olczak, A.; Białkowska, A.M.; Koziołkiewicz, M. Advanced Glycation End-Products (AGEs): Formation, Chemistry, Classification, Receptors, and Diseases Related to AGEs. Cells 2022, 11, 1312. [Google Scholar] [CrossRef] [PubMed]
- Prommer, E.E. Methadone Isomers and Their Role in Pain Management. Am. J. Hosp. Palliat. Med. 2009, 26, 149–150. [Google Scholar] [CrossRef] [PubMed]
- The CRASH trial protocol (Corticosteroid randomisation after significant head injury) [ISRCTN74459797]. BMC Emerg Med, 2001. 1(1): p. 1.
- Toth, C.; Rong, L.L.; Yang, C.; Martinez, J.; Song, F.; Ramji, N.; Brussee, V.; Liu, W.; Durand, J.; Nguyen, M.D.; et al. Receptor for Advanced Glycation End Products (RAGEs) and Experimental Diabetic Neuropathy. Diabetes 2008, 57, 1002–1017. [Google Scholar] [CrossRef]
- Win, M.T.T.; Yamamoto, Y.; Munesue, S.; Saito, H.; Han, D.; Motoyoshi, S.; Kamal, T.; Ohara, T.; Watanabe, T.; Yamamoto, H. Regulation of RAGE for Attenuating Progression of Diabetic Vascular Complications. Exp. Diabetes Res. 2011, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Plieth, C. Peroxide-Induced Liberation of Iron from Heme Switches Catalysis during Luminol Reaction and Causes Loss of Light and Heterodyning of Luminescence Kinetics. ACS Omega 2019, 4, 3268–3279. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, M.T.; Thorburn, D.R.; Penfold, S.A.; Laskowski, A.; Harcourt, B.E.; Sourris, K.C.; Tan, A.L.; Fukami, K.; Thallas-Bonke, V.; Nawroth, P.P.; et al. RAGE-Induced Cytosolic ROS Promote Mitochondrial Superoxide Generation in Diabetes. J. Am. Soc. Nephrol. 2009, 20, 742–752. [Google Scholar] [CrossRef]
- Talabi, O.O.; Dorfi, A.E.; O’nEil, G.D.; Esposito, D.V. Membraneless electrolyzers for the simultaneous production of acid and base. Chem. Commun. 2017, 53, 8006–8009. [Google Scholar] [CrossRef]
- DeMarco, S.J.; Strehler, E.E. Plasma Membrane Ca2+-ATPase Isoforms 2b and 4b Interact Promiscuously and Selectively with Members of the Membrane-associated Guanylate Kinase Family of PDZ (PSD95/Dlg/ZO-1) Domain-containing Proteins. J. Biol. Chem. 2001, 276, 21594–21600. [Google Scholar] [CrossRef]
- Ernst, E. , Frankincense: systematic review. Bmj, 2008. 337: p. a2813.
- Shimizu, F.; Sano, Y.; Haruki, H.; Kanda, T. Advanced glycation end-products induce basement membrane hypertrophy in endoneurial microvessels and disrupt the blood–nerve barrier by stimulating the release of TGF-β and vascular endothelial growth factor (VEGF) by pericytes. Diabetologia 2011, 54, 1517–1526. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, F.; Sano, Y.; Tominaga, O.; Maeda, T.; Abe, M.-A.; Kanda, T. Advanced glycation end-products disrupt the blood–brain barrier by stimulating the release of transforming growth factor–β by pericytes and vascular endothelial growth factor and matrix metalloproteinase–2 by endothelial cells in vitro. Neurobiol. Aging 2013, 34, 1902–1912. [Google Scholar] [CrossRef]
- Wenk, M.R.; Lucast, L.; Di Paolo, G.; Romanelli, A.J.; Suchy, S.F.; Nussbaum, R.L.; Cline, G.W.; I Shulman, G.; McMurray, W.; De Camilli, P. Phosphoinositide profiling in complex lipid mixtures using electrospray ionization mass spectrometry. Nat. Biotechnol. 2003, 21, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Juranek, J.K.; Geddis, M.S.; Song, F.; Zhang, J.; Garcia, J.; Rosario, R.; Yan, S.F.; Brannagan, T.H.; Schmidt, A.M. RAGE Deficiency Improves Postinjury Sciatic Nerve Regeneration in Type 1 Diabetic Mice. Diabetes 2013, 62, 931–943. [Google Scholar] [CrossRef]
- Vasconcelos, M.; Crego, A.; Rodrigues, R.; Almeida-Antunes, N.; López-Caneda, E. Effects of the COVID-19 Mitigation Measures on Alcohol Consumption and Binge Drinking in College Students: A Longitudinal Survey. Int. J. Environ. Res. Public Heal. 2021, 18, 9822. [Google Scholar] [CrossRef]
- Charles, M. , et al., Low peripheral nerve conduction velocities and amplitudes are strongly related to diabetic microvascular complications in type 1 diabetes: the EURODIAB Prospective Complications Study. Diabetes Care, 2010. 33(12): p. 2648-53.
- Yaffe, K. , et al., Advanced glycation end product level, diabetes, and accelerated cognitive aging. Neurology, 2011. 77(14): p. 1351-6.
- Takahashi, S.; Kurimura, Y.; Hashimoto, J.; Uehara, T.; Hiyama, Y.; Tsukamoto, T.; Iwasawa, A.; Nishimura, M.; Sunaoshi, K.; Takeda, K.; et al. Antimicrobial susceptibility and penicillin-binding protein 1 and 2 mutations in Neisseria gonorrhoeae isolated from male urethritis in Sapporo, Japan. J. Infect. Chemother. 2013, 19, 50–56. [Google Scholar] [CrossRef]
- Akella, N.M.; Ciraku, L.; Reginato, M.J. Fueling the fire: emerging role of the hexosamine biosynthetic pathway in cancer. BMC Biol. 2019, 17, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, X.; He, L.; Zhu, S.; Lai, S.; Zhang, X.; Huang, Z.; Yu, B.; Cui, C.; Wang, Q. Empagliflozin improves renal ischemia–reperfusion injury by reducing inflammation and enhancing mitochondrial fusion through AMPK–OPA1 pathway promotion. Cell. Mol. Biol. Lett. 2023, 28, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Hart, G.W. ProteinO-GlcNAcylation in diabetes and diabetic complications. Expert Rev. Proteom. 2013, 10, 365–380. [Google Scholar] [CrossRef]
- Donovan, K.; Alekseev, O.; Qi, X.; Cho, W.; Azizkhan-Clifford, J. O-GlcNAc Modification of Transcription Factor Sp1 Mediates Hyperglycemia-Induced VEGF-A Upregulation in Retinal Cells. Investig. Opthalmology Vis. Sci. 2014, 55, 7862–7873. [Google Scholar] [CrossRef]
- Yang, W.H.; Park, S.Y.; Nam, H.W.; Kim, D.H.; Kang, J.G.; Kang, E.S.; Kim, Y.S.; Lee, H.C.; Kim, K.S.; Cho, J.W. NFκB activation is associated with its O -GlcNAcylation state under hyperglycemic conditions. Proc. Natl. Acad. Sci. 2008, 105, 17345–17350. [Google Scholar] [CrossRef] [PubMed]
- Tallent, M.K.; Varghis, N.; Skorobogatko, Y.; Hernandez-Cuebas, L.; Whelan, K.; Vocadlo, D.J.; Vosseller, K. In Vivo Modulation of O-GlcNAc Levels Regulates Hippocampal Synaptic Plasticity through Interplay with Phosphorylation. J. Biol. Chem. 2009, 284, 174–181. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.W.; Wang, K.; Nelson, A.R.; Bredemann, T.M.; Fraser, K.B.; Clinton, S.M.; Puckett, R.; Marchase, R.B.; Chatham, J.C.; McMahon, L.L. O-GlcNAcylation of AMPA Receptor GluA2 Is Associated with a Novel Form of Long-Term Depression at Hippocampal Synapses. J. Neurosci. 2013, 34, 10–21. [Google Scholar] [CrossRef]
- Jóźwiak, P.; Oracz, J.; Dziedzic, A.; Szelenberger, R.; Żyżelewicz, D.; Bijak, M.; Krześlak, A. Increased O-GlcNAcylation by Upregulation of Mitochondrial O-GlcNAc Transferase (mOGT) Inhibits the Activity of Respiratory Chain Complexes and Controls Cellular Bioenergetics. Cancers 2024, 16, 1048. [Google Scholar] [CrossRef] [PubMed]
- Hui, Y.; Zhong, Y.; Kuang, L.; Xu, J.; Hao, Y.; Cao, J.; Zheng, T. O-GlcNAcylation of circadian clock protein Bmal1 impairs cognitive function in diabetic mice. EMBO J. 2024, 43, 5667–5689. [Google Scholar] [CrossRef]
- Geraldes, P.; King, G.L. Activation of Protein Kinase C Isoforms and Its Impact on Diabetic Complications. Circ. Res. 2010, 106, 1319–1331. [Google Scholar] [CrossRef]
- Farb, M.G.; Ganley-Leal, L.; Mott, M.; Liang, Y.; Ercan, B.; Widlansky, M.E.; Bigornia, S.J.; Fiscale, A.J.; Apovian, C.M.; Carmine, B.; et al. Arteriolar function in visceral adipose tissue is impaired in human obesity. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 467–473. [Google Scholar] [CrossRef]
- Dasevcimen, N.; King, G. The role of protein kinase C activation and the vascular complications of diabetes. Pharmacol. Res. 2007, 55, 498–510. [Google Scholar] [CrossRef]
- Tabit, C.E.; Shenouda, S.M.; Holbrook, M.; Fetterman, J.L.; Kiani, S.; Frame, A.A.; Kluge, M.A.; Held, A.; Dohadwala, M.M.; Gokce, N.; et al. Protein Kinase C-β Contributes to Impaired Endothelial Insulin Signaling in Humans With Diabetes Mellitus. Circulation 2013, 127, 86–95. [Google Scholar] [CrossRef]
- Naruse, K.; Rask-Madsen, C.; Takahara, N.; Ha, S.-W.; Suzuma, K.; Way, K.J.; Jacobs, J.R.; Clermont, A.C.; Ueki, K.; Ohshiro, Y.; et al. Activation of Vascular Protein Kinase C-β Inhibits Akt-Dependent Endothelial Nitric Oxide Synthase Function in Obesity-Associated Insulin Resistance. Diabetes 2006, 55, 691–698. [Google Scholar] [CrossRef]
- Park, J.Y.; Takahara, N.; Gabriele, A.; Chou, E.; Naruse, K.; Suzuma, K.; Yamauchi, T.; Ha, S.W.; Meier, M.; Rhodes, C.J.; et al. Induction of endothelin-1 expression by glucose: an effect of protein kinase C activation. Diabetes 2000, 49, 1239–1248. [Google Scholar] [CrossRef]
- Harhaj, N.S.; Felinski, E.A.; Wolpert, E.B.; Sundstrom, J.M.; Gardner, T.W.; Antonetti, D.A. VEGF Activation of Protein Kinase C Stimulates Occludin Phosphorylation and Contributes to Endothelial Permeability. Investig. Opthalmology Vis. Sci. 2006, 47, 5106–5115. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.B.; Milne, L.; Nelson, N.; Eddie, S.; Brown, P.; Atanassova, N.; O'BRyan, M.K.; O'DOnnell, L.; Rhodes, D.; Wells, S.; et al. KATNAL1 Regulation of Sertoli Cell Microtubule Dynamics Is Essential for Spermiogenesis and Male Fertility. PLOS Genet. 2012, 8, e1002697. [Google Scholar] [CrossRef] [PubMed]
- Fleegal, M.A.; Hom, S.; Borg, L.K.; Davis, T.P. Activation of PKC modulates blood-brain barrier endothelial cell permeability changes induced by hypoxia and posthypoxic reoxygenation. Am. J. Physiol. Circ. Physiol. 2005, 289, H2012–H2019. [Google Scholar] [CrossRef]
- Richner, M.; Ferreira, N.; Dudele, A.; Jensen, T.S.; Vaegter, C.B.; Gonçalves, N.P. Functional and Structural Changes of the Blood-Nerve-Barrier in Diabetic Neuropathy. Front. Neurosci. 2019, 12, 1038. [Google Scholar] [CrossRef]
- Takeshita, Y.; Sato, R.; Kanda, T. Blood–Nerve Barrier (BNB) Pathology in Diabetic Peripheral Neuropathy and In Vitro Human BNB Model. Int. J. Mol. Sci. 2020, 22, 62. [Google Scholar] [CrossRef]
- Shimu, S.J.; Patil, S.M.; Dadzie, E.; Tesfaye, T.; Alag, P.; Więckiewicz, G. Exploring Health Informatics in the Battle against Drug Addiction: Digital Solutions for the Rising Concern. J. Pers. Med. 2024, 14, 556. [Google Scholar] [CrossRef] [PubMed]
- Shanmugam, N. , et al., High glucose-induced expression of proinflammatory cytokine and chemokine genes in monocytic cells. Diabetes, 2003. 52(5): p. 1256-64.
- Chen, S. , et al., High glucose-induced, endothelin-dependent fibronectin synthesis is mediated via NF-kappa B and AP-1. Am J Physiol Cell Physiol, 2003. 284(2): p. C263-72.
- Venkatesan, B.; Valente, A.J.; Das, N.A.; Carpenter, A.J.; Yoshida, T.; Delafontaine, J.-L.; Siebenlist, U.; Chandrasekar, B. CIKS (Act1 or TRAF3IP2) mediates high glucose-induced endothelial dysfunction. Cell. Signal. 2013, 25, 359–371. [Google Scholar] [CrossRef]
- Pieper, G.M. and H. Riaz ul, Activation of nuclear factor-kappaB in cultured endothelial cells by increased glucose concentration: prevention by calphostin C. J Cardiovasc Pharmacol, 1997. 30(4): p. 528-32.
- Ozsarac, N.; Weible, M.; Reynolds, A.J.; Hendry, I.A. Activation of protein kinase C inhibits retrograde transport of neurotrophins in mice. J. Neurosci. Res. 2003, 72, 203–210. [Google Scholar] [CrossRef]
- Ding, W.-L.; Zhu, H.; Yu, W.-J.; Le, Y.; Wang, W.-J.; Li, F.; Gui, T.; Wang, Y.; Shi, W.-D.; Fan, X.-Q. High glucose levels increase the expression of neurotrophic factors associated with p-p42/p44 MAPK in Schwann cells in vitro. Mol. Med. Rep. 2012, 6, 179–184. [Google Scholar] [CrossRef]
- Naruse, K. Schwann Cells as Crucial Players in Diabetic Neuropathy. Adv. Exp. Med. Biol. 2019, 345–356. [Google Scholar] [CrossRef]
- Zuo, L.; Qu, M.; Zhang, M.; Cheng, P.; Guo, M.; Selvarajah, D.; Tesfaye, S.; Wu, J. High glucose mediates diabetic peripheral neuropathy by inducing Schwann cells apoptosis through the Dgkh/PKC-α signaling pathway. Acta Diabetol. 2025, 1–11. [Google Scholar] [CrossRef]
- Mehta, N.N.; Sheetz, M.; Price, K.; Comiskey, L.; Amrutia, S.; Iqbal, N.; Mohler, E.R.; Reilly, M.P. Selective PKC Beta Inhibition with Ruboxistaurin and Endothelial Function in Type-2 Diabetes Mellitus. Cardiovasc. Drugs Ther. 2008, 23, 17–24. [Google Scholar] [CrossRef]
- Beckman, J.A.; Goldfine, A.B.; Gordon, M.B.; Garrett, L.A.; Creager, M.A. Inhibition of Protein Kinase Cβ Prevents Impaired Endothelium-Dependent Vasodilation Caused by Hyperglycemia in Humans. Circ. Res. 2002, 90, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Casellini, C.M. , et al., A 6-month, randomized, double-masked, placebo-controlled study evaluating the effects of the protein kinase C-beta inhibitor ruboxistaurin on skin microvascular blood flow and other measures of diabetic peripheral neuropathy. Diabetes Care, 2007. 30(4): p. 896-902.
- Vinik, A.I. , et al., Treatment of symptomatic diabetic peripheral neuropathy with the protein kinase C beta-inhibitor ruboxistaurin mesylate during a 1-year, randomized, placebo-controlled, double-blind clinical trial. Clin Ther, 2005. 27(8): p. 1164-80.
- Baird, A.A. and J.A. Fugelsang, The emergence of consequential thought: evidence from neuroscience. Philos Trans R Soc Lond B Biol Sci, 2004. 359(1451): p. 1797-804.
- Clemente-Suárez, V.J.; Redondo-Flórez, L.; Beltrán-Velasco, A.I.; Ramos-Campo, D.J.; Belinchón-Demiguel, P.; Martinez-Guardado, I.; Dalamitros, A.A.; Yáñez-Sepúlveda, R.; Martín-Rodríguez, A.; Tornero-Aguilera, J.F. Mitochondria and Brain Disease: A Comprehensive Review of Pathological Mechanisms and Therapeutic Opportunities. Biomedicines 2023, 11, 2488. [Google Scholar] [CrossRef]
- Dash, U.C.; Bhol, N.K.; Swain, S.K.; Samal, R.R.; Nayak, P.K.; Raina, V.; Panda, S.K.; Kerry, R.G.; Duttaroy, A.K.; Jena, A.B. Oxidative stress and inflammation in the pathogenesis of neurological disorders: Mechanisms and implications. Acta Pharm. Sin. B 2024, 15, 15–34. [Google Scholar] [CrossRef]
- Fernyhough, P.; Chowdhury, S.K.R.; E Schmidt, R. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef]
- Nishikawa, T.; Edelstein, D.; Du, X.L.; Yamagishi, S.-I.; Matsumura, T.; Kaneda, Y.; Yorek, M.A.; Beebe, D.J.; Oates, P.J.; Hammes, H.-P.; et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000, 404, 787–790. [Google Scholar] [CrossRef] [PubMed]
- Du, X.-L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef]
- Qiu, Y.-Y.; Tang, L.-Q. Roles of the NLRP3 inflammasome in the pathogenesis of diabetic nephropathy. Pharmacol. Res. 2016, 114, 251–264. [Google Scholar] [CrossRef] [PubMed]
- Sifuentes-Franco, S. , et al., The Role of Oxidative Stress, Mitochondrial Function, and Autophagy in Diabetic Polyneuropathy. J Diabetes Res, 2017. 2017: p. 1673081.
- Eckersley, L. Role of the Schwann cell in diabetic neuropathy. Int. Rev. Neurobiol. 2002, 50, 293–321. [Google Scholar] [CrossRef]
- Chen, S. , et al., New insights into the role of mitochondrial dynamics in oxidative stress-induced diseases. Biomed Pharmacother, 2024. 178: p. 117084.
- Rius-Pérez, S. , et al., PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid Med Cell Longev, 2020. 2020: p. 1452696.
- Vargas-Mendoza, N.; Angeles-Valencia, M.; Morales-González, Á.; Madrigal-Santillán, E.O.; Morales-Martínez, M.; Madrigal-Bujaidar, E.; Álvarez-González, I.; Gutiérrez-Salinas, J.; Esquivel-Chirino, C.; Chamorro-Cevallos, G.; et al. Oxidative Stress, Mitochondrial Function and Adaptation to Exercise: New Perspectives in Nutrition. Life 2021, 11, 1269. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, H.; Lin, S.; Lv, Y.; Lin, Y.; Liu, Y.; Peng, R.; Jin, H. New therapeutic directions in type II diabetes and its complications: mitochondrial dynamics. Front. Endocrinol. 2023, 14, 1230168. [Google Scholar] [CrossRef]
- Chen, L.; Qin, Y.; Liu, B.; Gao, M.; Li, A.; Li, X.; Gong, G. PGC-1α-Mediated Mitochondrial Quality Control: Molecular Mechanisms and Implications for Heart Failure. Front. Cell Dev. Biol. 2022, 10, 871357. [Google Scholar] [CrossRef]
- Guo, L.; Chen, X.; Yu, B.; Shen, L.; Zhang, X. Delayed Intracerebral Hemorrhage Secondary to Ventriculoperitoneal Shunt: A Retrospective Study. World Neurosurg. 2017, 107, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Gureev, A.P.; Shaforostova, E.A.; Popov, V.N. Regulation of Mitochondrial Biogenesis as a Way for Active Longevity: Interaction Between the Nrf2 and PGC-1α Signaling Pathways. Front. Genet. 2019, 10, 435. [Google Scholar] [CrossRef]
- Zhang, M.-Y.; Zhu, L.; Bao, X.; Xie, T.-H.; Cai, J.; Zou, J.; Wang, W.; Gu, S.; Li, Y.; Li, H.-Y.; et al. Inhibition of Drp1 ameliorates diabetic retinopathy by regulating mitochondrial homeostasis. Exp. Eye Res. 2022, 220, 109095. [Google Scholar] [CrossRef]
- Yang, J.; Yu, Z.; Jiang, Y.; Zhang, Z.; Tian, Y.; Cai, J.; Wei, M.; Lyu, Y.; Yang, D.; Shen, S.; et al. SIRT3 alleviates painful diabetic neuropathy by mediating the FoxO3a-PINK1-Parkin signaling pathway to activate mitophagy. CNS Neurosci. Ther. 2024, 30, e14703. [Google Scholar] [CrossRef]
- Charmpilas, N.; Fang, E.F.; Palikaras, K. Mitophagy and Neuroinflammation: A Compelling Interplay. Curr. Neuropharmacol. 2023, 21, 1477–1481. [Google Scholar] [CrossRef]
- Figley, M.D.; Thomas, A.; Gitler, A.D. Evaluating noncoding nucleotide repeat expansions in amyotrophic lateral sclerosis. Neurobiol. Aging 2014, 35, 936.e1–936.e4. [Google Scholar] [CrossRef] [PubMed]
- Hussain, A.R.; Siraj, A.K.; Ahmed, M.; Bu, R.; Pratheeshkumar, P.; Alrashed, A.M.; Qadri, Z.; Ajarim, D.; Al-Dayel, F.; Beg, S.; et al. XIAP over-expression is an independent poor prognostic marker in Middle Eastern breast cancer and can be targeted to induce efficient apoptosis. BMC Cancer 2017, 17, 640–640. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M. , et al., An Overview of Oxidative Stress, Neuroinflammation, and Neurodegenerative Diseases. Int J Mol Sci, 2022. 23(11).
- Averill-Bates, D.A. , The antioxidant glutathione. Vitam Horm, 2023. 121: p. 109-141.
- Krishnamurthy, H.K.; Pereira, M.; Rajavelu, I.; Jayaraman, V.; Krishna, K.; Wang, T.; Bei, K.; Rajasekaran, J.J. Oxidative stress: fundamentals and advances in quantification techniques. Front. Chem. 2024, 12, 1470458. [Google Scholar] [CrossRef]
- Gupta, J.K. The Role of Aldose Reductase in Polyol Pathway: An Emerging Pharmacological Target in Diabetic Complications and Associated Morbidities. Curr. Pharm. Biotechnol. 2024, 25, 1073–1081. [Google Scholar] [CrossRef]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.J.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: implications in type 2 diabetic nephropathy. Am. J. Physiol. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef]
- Tu, D.; Velagapudi, R.; Gao, Y.; Hong, J.-S.; Zhou, H.; Gao, H.-M. Activation of neuronal NADPH oxidase NOX2 promotes inflammatory neurodegeneration. Free. Radic. Biol. Med. 2023, 200, 47–58. [Google Scholar] [CrossRef]
- Lin, Q.; Li, K.; Chen, Y.; Xie, J.; Wu, C.; Cui, C.; Deng, B. Oxidative Stress in Diabetic Peripheral Neuropathy: Pathway and Mechanism-Based Treatment. Mol. Neurobiol. 2023, 60, 4574–4594. [Google Scholar] [CrossRef]
- Mendez, M.M.; Folgado, J.; Tormo, C.; Artero, A.; Ascaso, M.; Martinez-Hervás, S.; Chaves, F.J.; Ascaso, J.F.; Real, J.T. Altered glutathione system is associated with the presence of distal symmetric peripheral polyneuropathy in type 2 diabetic subjects. J. Diabetes its Complicat. 2015, 29, 923–927. [Google Scholar] [CrossRef]
- Neagu, M. , et al., Diabetic neuropathy: A NRF2 disease? J Diabetes, 2024. 16(9): p. e13524.
- Kumar, A.; Mittal, R. Nrf2: a potential therapeutic target for diabetic neuropathy. Inflammopharmacology 2017, 25, 393–402. [Google Scholar] [CrossRef]
- van der Oost, J. , et al., The ferredoxin-dependent conversion of glyceraldehyde-3-phosphate in the hyperthermophilic archaeon Pyrococcus furiosus represents a novel site of glycolytic regulation. J Biol Chem, 1998. 273(43): p. 28149-54.
- Yan, H.; Harding, J.J. Glycation-induced inactivation and loss of antigenicity of catalase and superoxide dismutase. Biochem. J. 1997, 328, 599–605. [Google Scholar] [CrossRef] [PubMed]
- Polykretis, P.; Luchinat, E.; Boscaro, F.; Banci, L. Methylglyoxal interaction with superoxide dismutase 1. Redox Biol. 2020, 30, 101421. [Google Scholar] [CrossRef] [PubMed]
- Najjar, F.M.; Taghavi, F.; Ghadari, R.; Sheibani, N.; Moosavi-Movahedi, A.A. Destructive effect of non-enzymatic glycation on catalase and remediation via curcumin. Arch. Biochem. Biophys. 2017, 630, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Begum, M.; Choubey, M.; Tirumalasetty, M.B.; Arbee, S.; Sadik, S.; Mohib, M.M.; Srivastava, S.; Minhaz, N.; Alam, R.; Mohiuddin, M.S. Exploring the Molecular Link Between Diabetes and Erectile Dysfunction Through Single-Cell Transcriptome Analysis. Genes 2024, 15, 1596. [Google Scholar] [CrossRef]
- Cai, L. , et al., Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes, 2002. 51(6): p. 1938-48.
- Kowalczyk, P.; Sulejczak, D.; Kleczkowska, P.; Bukowska-Ośko, I.; Kucia, M.; Popiel, M.; Wietrak, E.; Kramkowski, K.; Wrzosek, K.; Kaczyńska, K. Mitochondrial Oxidative Stress—A Causative Factor and Therapeutic Target in Many Diseases. Int. J. Mol. Sci. 2021, 22, 13384. [Google Scholar] [CrossRef]
- Norat, P.; Soldozy, S.; Sokolowski, J.D.; Gorick, C.M.; Kumar, J.S.; Chae, Y.; Yağmurlu, K.; Prada, F.; Walker, M.; Levitt, M.R.; et al. Mitochondrial dysfunction in neurological disorders: Exploring mitochondrial transplantation. npj Regen. Med. 2020, 5, 1–9. [Google Scholar] [CrossRef]
- Xu, X.; Pang, Y.; Fan, X. Mitochondria in oxidative stress, inflammation and aging: from mechanisms to therapeutic advances. Signal Transduct. Target. Ther. 2025, 10, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Ametov, A.S. , et al., The sensory symptoms of diabetic polyneuropathy are improved with alpha-lipoic acid: the SYDNEY trial. Diabetes Care, 2003. 26(3): p. 770-6.
- Ziegler, D. , et al., Oral treatment with alpha-lipoic acid improves symptomatic diabetic polyneuropathy: the SYDNEY 2 trial. Diabetes Care, 2006. 29(11): p. 2365-70.
- Han, T. , et al., A systematic review and meta-analysis of α-lipoic acid in the treatment of diabetic peripheral neuropathy. Eur J Endocrinol, 2012. 167(4): p. 465-71.
- Heidari, N.; Sajedi, F.; Mohammadi, Y.; Mirjalili, M.; Mehrpooya, M. Ameliorative Effects Of N-Acetylcysteine As Adjunct Therapy On Symptoms Of Painful Diabetic Neuropathy. J. Pain Res. 2019, ume 12, 3147–3159. [Google Scholar] [CrossRef]
- Fakhrabadi, M.A.; Ghotrom, A.Z.; Mozaffari-Khosravi, H.; Nodoushan, H.H.; Nadjarzadeh, A. Effect of Coenzyme Q10 on Oxidative Stress, Glycemic Control and Inflammation in Diabetic Neuropathy: A Double Blind Randomized Clinical Trial. Int. J. Vitam. Nutr. Res. 2014, 84, 252–260. [Google Scholar] [CrossRef]
- Celen, M.C.; Tuncer, S.; Akkoca, A.; Dalkilic, N. Enhancing diabetic rat peripheral nerve conduction velocity parameters through the potent effects of MitoTEMPO. Heliyon 2024, 10, e41045. [Google Scholar] [CrossRef] [PubMed]
- Schemmel, K.E.; Padiyara, R.S.; D'SOuza, J.J. Aldose reductase inhibitors in the treatment of diabetic peripheral neuropathy: a review. J. Diabetes its Complicat. 2010, 24, 354–360. [Google Scholar] [CrossRef] [PubMed]
- Vargas-Soria, M.; García-Alloza, M.; Corraliza-Gómez, M. Effects of diabetes on microglial physiology: a systematic review of in vitro, preclinical and clinical studies. J. Neuroinflammation 2023, 20, 1–30. [Google Scholar] [CrossRef]
- Ben-Kraiem, A.; Sauer, R.-S.; Norwig, C.; Popp, M.; Bettenhausen, A.-L.; Atalla, M.S.; Brack, A.; Blum, R.; Doppler, K.; Rittner, H.L. Selective blood-nerve barrier leakiness with claudin-1 and vessel-associated macrophage loss in diabetic polyneuropathy. J. Mol. Med. 2021, 99, 1237–1250. [Google Scholar] [CrossRef]
- Coleman, E.; Judd, R.; Hoe, L.; Dennis, J.; Posner, P. Effects of diabetes mellitus on astrocyte GFAP and glutamate transporters in the CNS. Glia 2004, 48, 166–178. [Google Scholar] [CrossRef]
- Coleman, E.S.; Dennis, J.C.; Braden, T.D.; Judd, R.L.; Posner, P. Insulin treatment prevents diabetes-induced alterations in astrocyte glutamate uptake and GFAP content in rats at 4 and 8 weeks of diabetes duration. Brain Res. 2010, 1306, 131–141. [Google Scholar] [CrossRef]
- Jiao, X.; Wan, J.; Wu, W.; Ma, L.; Chen, C.; Dong, W.; Liu, Y.; Jin, C.; Sun, A.; Zhou, Y.; et al. GLT-1 downregulation in hippocampal astrocytes induced by type 2 diabetes contributes to postoperative cognitive dysfunction in adult mice. CNS Neurosci. Ther. 2024, 30, e70024. [Google Scholar] [CrossRef] [PubMed]
- Biswas, J.; Pijewski, R.S.; Makol, R.; Miramontes, T.G.; Thompson, B.L.; Kresic, L.C.; Burghard, A.L.; Oliver, D.L.; Martinelli, D.C.; Shore, S.E. C1ql1 is expressed in adult outer hair cells of the cochlea in a tonotopic gradient. PLOS ONE 2021, 16, e0251412. [Google Scholar] [CrossRef] [PubMed]
- Raka, M.A. , et al., Inhibitory role of resveratrol in the development of profibrogenesis and fibrosis mechanisms. Immunology, Endocrine & Metabolic Agents in Medicinal Chemistry (Formerly Current Medicinal Chemistry-Immunology, Endocrine and Metabolic Agents), 2018. 18(1): p. 80-104.
- Younger, D.S. , et al., Diabetic peripheral neuropathy: a clinicopathologic and immunohistochemical analysis of sural nerve biopsies. Muscle Nerve, 1996. 19(6): p. 722-7.
- O'Brien, J.A.; McGuire, H.M.; Shinko, D.; Groth, B.F.d.S.; Russo, M.A.; Bailey, D.; Santarelli, D.M.; Wynne, K.; Austin, P.J. T lymphocyte and monocyte subsets are dysregulated in type 1 diabetes patients with peripheral neuropathic pain. Brain, Behav. Immun. - Heal. 2021, 15, 100283. [Google Scholar] [CrossRef] [PubMed]
- Dogrul, A.; Gul, H.; Yesilyurt, O.; Ulas, U.H.; Yildiz, O. Systemic and spinal administration of etanercept, a tumor necrosis factor alpha inhibitor, blocks tactile allodynia in diabetic mice. Acta Diabetol. 2010, 48, 135–142. [Google Scholar] [CrossRef]
- Shi, X.; Chen, Y.; Nadeem, L.; Xu, G. Beneficial effect of TNF-α inhibition on diabetic peripheral neuropathy. J. Neuroinflammation 2013, 10, 836–69. [Google Scholar] [CrossRef]
- Su, W.-J.; Li, J.-M.; Zhang, T.; Cao, Z.-Y.; Hu, T.; Zhong, S.-Y.; Xu, Z.-Y.; Gong, H.; Jiang, C.-L. Microglial NLRP3 inflammasome activation mediates diabetes-induced depression-like behavior via triggering neuroinflammation. Prog. Neuro-Psychopharmacology Biol. Psychiatry 2023, 126, 110796. [Google Scholar] [CrossRef] [PubMed]
- Al Mamun, A.; Shao, C.; Geng, P.; Wang, S.; Xiao, J. Pyroptosis in Diabetic Peripheral Neuropathy and its Therapeutic Regulation. J. Inflamm. Res. 2024, ume 17, 3839–3864. [Google Scholar] [CrossRef]
- Rehman, K.; Akash, M.S.H.; Liaqat, A.; Kamal, S.; Qadir, M.I.; Rasul, A. Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit. Rev. Eukaryot. Gene Expr. 2017, 27, 229–236. [Google Scholar] [CrossRef]
- Callanan, A.; Davis, N.; McGloughlin, T.; Walsh, M. The effects of stent interaction on porcine urinary bladder matrix employed as stent-graft materials. J. Biomech. 2014, 47, 1885–1893. [Google Scholar] [CrossRef]
- Sun, J.-S.; Yang, Y.-J.; Zhang, Y.-Z.; Huang, W.; Li, Z.-S. Minocycline attenuates pain by inhibiting spinal microglia activation in diabetic rats. Mol. Med. Rep. 2015, 12, 2677–2682. [Google Scholar] [CrossRef]
- Dong, S.; Hiam-Galvez, K.J.; Mowery, C.T.; Herold, K.C.; Gitelman, S.E.; Esensten, J.H.; Liu, W.; Lares, A.P.; Leinbach, A.S.; Lee, M.; et al. The effect of low-dose IL-2 and Treg adoptive cell therapy in patients with type 1 diabetes. J. Clin. Investig. 2021, 6. [Google Scholar] [CrossRef] [PubMed]
- Austin, P.J.; Moalem-Taylor, G. The neuro-immune balance in neuropathic pain: Involvement of inflammatory immune cells, immune-like glial cells and cytokines. J. Neuroimmunol. 2010, 229, 26–50. [Google Scholar] [CrossRef]
- Cameron, N.E.; Eaton, S.E.M.; Cotter, M.A.; Tesfaye, S. Vascular factors and metabolic interactions in the pathogenesis of diabetic neuropathy. Diabetologia 2001, 44, 1973–1988. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Dyck, P.J. Abnormalities of endoneurial microvessels and sural nerve pathology in diabetic neuropathy. Neurology 1987, 37, 20–20. [Google Scholar] [CrossRef] [PubMed]
- Powell, H.C.; Rosoff, J.; Myers, R.R. Microangiopathy in human diabetic neuropathy. Acta Neuropathol. 1985, 68, 295–305. [Google Scholar] [CrossRef]
- Sima, A.A. , et al., Endoneurial microvessels in human diabetic neuropathy. Endothelial cell dysjunction and lack of treatment effect by aldose reductase inhibitor. Diabetes, 1991. 40(9): p. 1090-9.
- Newrick, P.G. , et al., Sural nerve oxygen tension in diabetes. Br Med J (Clin Res Ed), 1986. 293(6554): p. 1053-4.
- Tuck, R.R.; Schmelzer, J.D.; Low, P.A. ENDONEURIAL BLOOD FLOW AND OXYGEN TENSION IN THE SCIATIC NERVES OF RATS WITH EXPERIMENTAL DIABETIC NEUROPATHY. Brain 1984, 107, 935–950. [Google Scholar] [CrossRef]
- Kan, H.-W.; Hsieh, J.-H.; Chien, H.-F.; Lin, Y.-H.; Yeh, T.-Y.; Chao, C.-C.; Hsieh, S.-T. CD40-mediated HIF-1α expression underlying microangiopathy in diabetic nerve pathology. Dis. Model. Mech. 2018, 11. [Google Scholar] [CrossRef]
- Mohib, M.M.; Rabe, S.; Nolze, A.; Rooney, M.; Ain, Q.; Zipprich, A.; Gekle, M.; Schreier, B. Eplerenone, a mineralocorticoid receptor inhibitor, reduces cirrhosis associated changes of hepatocyte glucose and lipid metabolism. Cell Commun. Signal. 2024, 22, 1–18. [Google Scholar] [CrossRef]
- Toutouzas, K.; Riga, M.; Stefanadi, E.; Stefanadis, C. Asymmetric Dimethylarginine (ADMA) and other Endogenous Nitric Oxide Synthase (NOS) Inhibitors as an Important Cause of Vascular Insulin Resistance. Horm. Metab. Res. 2008, 40, 655–659. [Google Scholar] [CrossRef]
- Thum, T.; Fraccarollo, D.; Schultheiss, M.; Froese, S.; Galuppo, P.; Widder, J.D.; Tsikas, D.; Ertl, G.; Bauersachs, J. Endothelial Nitric Oxide Synthase Uncoupling Impairs Endothelial Progenitor Cell Mobilization and Function in Diabetes. Diabetes 2007, 56, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Kihara, M. and P.A. Low, Impaired vasoreactivity to nitric oxide in experimental diabetic neuropathy. Exp Neurol, 1995. 132(2): p. 180-5.
- Gonçalves, J.S.; Seiça, R.M.; Laranjinha, J.; Lourenço, C.F. Impairment of neurovascular coupling in the hippocampus due to decreased nitric oxide bioavailability supports early cognitive dysfunction in type 2 diabetic rats. Free. Radic. Biol. Med. 2022, 193, 669–675. [Google Scholar] [CrossRef]
- Ellemann, K.; Thorsteinsson, B.; Fugleberg, S.; Feldt-Rasmussen, B.; Andersen, O.O.; Grönbæk, P.; Binder, C. KINETICS OF INSULIN DISAPPEARANCE FROM PLASMA IN CORTISONE-TREATED NORMAL SUBJECTS. Clin. Endocrinol. 1987, 26, 623–628. [Google Scholar] [CrossRef]
- Shahaf, G. A Possible Common Neurophysiologic Basis for MDD, Bipolar Disorder, and Schizophrenia: Lessons from Electrophysiology. Front. Psychiatry 2016, 7, 94. [Google Scholar] [CrossRef]
- Xu, Z.; Zeng, W.; Sun, J.; Chen, W.; Zhang, R.; Yang, Z.; Yao, Z.; Wang, L.; Song, L.; Chen, Y.; et al. The quantification of blood-brain barrier disruption using dynamic contrast-enhanced magnetic resonance imaging in aging rhesus monkeys with spontaneous type 2 diabetes mellitus. NeuroImage 2017, 158, 480–487. [Google Scholar] [CrossRef]
- Batzias, K.; Antonopoulos, A.S.; Oikonomou, E.; Siasos, G.; Bletsa, E.; Stampouloglou, P.K.; Mistakidi, C.-V.; Noutsou, M.; Katsiki, N.; Karopoulos, P.; et al. Effects of Newer Antidiabetic Drugs on Endothelial Function and Arterial Stiffness: A Systematic Review and Meta-Analysis. J. Diabetes Res. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Piconi, L. , et al., Intermittent high glucose enhances ICAM-1, VCAM-1, E-selectin and interleukin-6 expression in human umbilical endothelial cells in culture: the role of poly(ADP-ribose) polymerase. J Thromb Haemost, 2004. 2(8): p. 1453-9.
- Siddiqui, K.; George, T.P.; Mujammami, M.; Isnani, A.; Alfadda, A.A. The association of cell adhesion molecules and selectins (VCAM-1, ICAM-1, E-selectin, L-selectin, and P-selectin) with microvascular complications in patients with type 2 diabetes: A follow-up study. Front. Endocrinol. 2023, 14, 1072288. [Google Scholar] [CrossRef]
- Quattrini, C.; Jeziorska, M.; Boulton, A.J.; Malik, R.A. Reduced Vascular Endothelial Growth Factor Expression and Intra-Epidermal Nerve Fiber Loss in Human Diabetic Neuropathy. Diabetes Care 2008, 31, 140–145. [Google Scholar] [CrossRef]
- Schratzberger, P.; Walter, D.H.; Rittig, K.; Bahlmann, F.H.; Pola, R.; Curry, C.; Silver, M.; Krainin, J.G.; Weinberg, D.H.; Ropper, A.H.; et al. Reversal of experimental diabetic neuropathy by VEGF gene transfer. J. Clin. Investig. 2001, 107, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y., A. Yabluchanskiy, and M.L. Lindsey, Thrombospondin-1: the good, the bad, and the complicated. Circ Res, 2013. 113(12): p. 1272-4.
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191–a009191. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- E Kelley, D.; Mandarino, L.J. Fuel selection in human skeletal muscle in insulin resistance: a reexamination. Diabetes 2000, 49, 677–683. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Zeger, M.; Popken, G.; Zhang, J.; Xuan, S.; Lu, Q.R.; Schwab, M.H.; Nave, K.; Rowitch, D.; D'ERcole, A.J.; Ye, P. Insulin-like growth factor type 1 receptor signaling in the cells of oligodendrocyte lineage is required for normal in vivo oligodendrocyte development and myelination. Glia 2006, 55, 400–411. [Google Scholar] [CrossRef]
- Begum, M.; Choubey, M.; Tirumalasetty, M.B.; Arbee, S.; Mohib, M.M.; Wahiduzzaman; Mamun, M. A.; Uddin, M.B.; Mohiuddin, M.S. Adiponectin: A Promising Target for the Treatment of Diabetes and Its Complications. Life 2023, 13, 2213. [Google Scholar] [CrossRef]
- Mohib, M.; Rabby, S.F.; Paran, T.Z.; Hasan, M.; Ahmed, I.; Hasan, N.; Sagor, A.T.; Mohiuddin, S.; Hsu, T.-C. Protective role of green tea on diabetic nephropathy—A review. Cogent Biol. 2016, 2. [Google Scholar] [CrossRef]
- Kim, H.-S.; Brill, S.J. Rfc4 Interacts with Rpa1 and Is Required for Both DNA Replication and DNA Damage Checkpoints in Saccharomyces cerevisiae. Mol. Cell. Biol. 2001, 21, 3725–3737. [Google Scholar] [CrossRef]
- Gao, Z.; Hwang, D.; Bataille, F.; Lefevre, M.; York, D.; Quon, M.J.; Ye, J. Serine Phosphorylation of Insulin Receptor Substrate 1 by Inhibitor κB Kinase Complex. J. Biol. Chem. 2002, 277, 48115–48121. [Google Scholar] [CrossRef]
- Zhang, Y. , et al., Diabetes mellitus and Alzheimer's disease: GSK-3β as a potential link. Behav Brain Res, 2018. 339: p. 57-65.
- Talbot, K. Brain Insulin Resistance in Alzheimer's Disease and its Potential Treatment with GLP-1 Analogs. Neurodegener. Dis. Manag. 2014, 4, 31–40. [Google Scholar] [CrossRef]
- Hou, W.-K.; Xian, Y.-X.; Zhang, L.; Lai, H.; Hou, X.-G.; Xu, Y.-X.; Yu, T.; Xu, F.-Y.; Song, J.; Fu, C.-L.; et al. Influence of blood glucose on the expression of glucose transporter proteins 1 and 3 in the brain of diabetic rats. Chin. Med J. 2007, 120, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.K. , et al., Predicting the efficiency of prime editing guide RNAs in human cells. Nat Biotechnol, 2021. 39(2): p. 198-206.
- Lete, M.G. , et al., Histones and DNA compete for binding polyphosphoinositides in bilayers. Biophys J, 2014. 106(5): p. 1092-100.
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-Driven Local ATP Synthesis Is Required for Synaptic Function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef]
- Lyu, K.; Zhang, Y.; Zhang, D.; Kahn, M.; ter Horst, K.W.; Rodrigues, M.R.; Gaspar, R.C.; Hirabara, S.M.; Luukkonen, P.K.; Lee, S.; et al. A Membrane-Bound Diacylglycerol Species Induces PKCϵ-Mediated Hepatic Insulin Resistance. Cell Metab. 2020, 32, 654–664.e5. [Google Scholar] [CrossRef] [PubMed]
- A Stoica, B.; A Movsesyan, V.; Iv, P.M.L.; I Faden, A. Ceramide-induced neuronal apoptosis is associated with dephosphorylation of Akt, BAD, FKHR, GSK-3β, and induction of the mitochondrial-dependent intrinsic caspase pathway. Mol. Cell. Neurosci. 2003, 22, 365–382. [Google Scholar] [CrossRef] [PubMed]
- AT Sagor, M. , et al., Angiotensin-II, a potent peptide, participates in the development of hepatic dysfunctions. Immunology, Endocrine & Metabolic Agents-Medicinal ChemistryCurrent Medicinal Chemistry-Immunology, Endocrine & Metabolic Agents), 2016. 16(3): p. 161-177.
- El-Aarag, B.; Shalaan, E.S.; Ahmed, A.A.; Sayed, I.E.T.E.; Ibrahim, W.M. Cryptolepine Analog Exhibits Antitumor Activity against Ehrlich Ascites Carcinoma Cells in Mice via Targeting Cell Growth, Oxidative Stress, and PTEN/Akt/mTOR Signaling Pathway. Anti-Cancer Agents Med. Chem. 2024, 24, 436–442. [Google Scholar] [CrossRef]
- Kellar, D.; Lockhart, S.N.; Aisen, P.; Raman, R.; Rissman, R.A.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer's Disease. J. Prev. Alzheimer's Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Craft, S. , et al., Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol, 2012. 69(1): p. 29-38.
- Kopp, K.O.; Glotfelty, E.J.; Li, Y.; Greig, N.H. Glucagon-like peptide-1 (GLP-1) receptor agonists and neuroinflammation: Implications for neurodegenerative disease treatment. Pharmacol. Res. 2022, 186, 106550–106550. [Google Scholar] [CrossRef]
- Hölscher, C. Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin. Investig. Drugs 2020, 29, 333–348. [Google Scholar] [CrossRef]
- Sultana, A.; Shimu, S.J.; Faika, M.J.; Islam, T.; Ferdous, N.-E.; Nessa, A. Bangladeshi parents’ knowledge and awareness about cervical cancer and willingness to vaccinate female family members against human papilloma virus: a cross sectional study. Int. J. Community Med. Public Heal. 2023, 10, 3446–3453. [Google Scholar] [CrossRef]
- Coopmans, C.; Zhou, T.L.; Henry, R.M.; Heijman, J.; Schaper, N.C.; Koster, A.; Schram, M.T.; van der Kallen, C.J.; Wesselius, A.; Engelsman, R.J.D.; et al. Both Prediabetes and Type 2 Diabetes Are Associated With Lower Heart Rate Variability: The Maastricht Study. Diabetes Care 2020, 43, 1126–1133. [Google Scholar] [CrossRef]
- Bonaz, B.; Bazin, T.; Pellissier, S. The Vagus Nerve at the Interface of the Microbiota-Gut-Brain Axis. Front. Neurosci. 2018, 12, 49. [Google Scholar] [CrossRef]
- Hong, J.; Fu, T.; Liu, W.; Du, Y.; Bu, J.; Wei, G.; Yu, M.; Lin, Y.; Min, C.; Lin, D. An Update on the Role and Potential Molecules in Relation to Ruminococcus gnavus in Inflammatory Bowel Disease, Obesity and Diabetes Mellitus. Diabetes, Metab. Syndr. Obesity: Targets Ther. 1235; 17. [Google Scholar] [CrossRef]
- Leite, A.Z.; Rodrigues, N.d.C.; Gonzaga, M.I.; Paiolo, J.C.C.; de Souza, C.A.; Stefanutto, N.A.V.; Omori, W.P.; Pinheiro, D.G.; Brisotti, J.L.; Junior, E.M.; et al. Detection of Increased Plasma Interleukin-6 Levels and Prevalence of Prevotella copri and Bacteroides vulgatus in the Feces of Type 2 Diabetes Patients. Front. Immunol. 2017, 8, 1107. [Google Scholar] [CrossRef]
- Erny, D. , et al., Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci, 2015. 18(7): p. 965-77.
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-Like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371. [Google Scholar] [CrossRef] [PubMed]
- Rothhammer, V.; Mascanfroni, I.D.; Bunse, L.; Takenaka, M.C.; Kenison, J.E.; Mayo, L.; Chao, C.-C.; Patel, B.; Yan, R.; Blain, M.; et al. Type I interferons and microbial metabolites of tryptophan modulate astrocyte activity and central nervous system inflammation via the aryl hydrocarbon receptor. Nat. Med. 2016, 22, 586–597. [Google Scholar] [CrossRef]
- de Mello, V.D. , et al., Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci Rep, 2017. 7: p. 46337.
- Garcez, M.L.; Tan, V.X.; Heng, B.; Guillemin, G.J. Sodium Butyrate and Indole-3-propionic Acid Prevent the Increase of Cytokines and Kynurenine Levels in LPS-induced Human Primary Astrocytes. Int. J. Tryptophan Res. 2020, 13. [Google Scholar] [CrossRef]
- Keitel, V.; Görg, B.; Bidmon, H.J.; Zemtsova, I.; Spomer, L.; Zilles, K.; Häussinger, D. The bile acid receptor TGR5 (Gpbar-1) acts as a neurosteroid receptor in brain. Glia 2010, 58, 1794–1805. [Google Scholar] [CrossRef]
- Wu, Y.; Qiu, Y.; Su, M.; Wang, L.; Gong, Q.; Wei, X. Activation of the bile acid receptors TGR5 and FXR in the spinal dorsal horn alleviates neuropathic pain. CNS Neurosci. Ther. 2023, 29, 1981–1998. [Google Scholar] [CrossRef] [PubMed]
- Benichou, T.; Pereira, B.; Mermillod, M.; Tauveron, I.; Pfabigan, D.; Maqdasy, S.; Dutheil, F.; A Malik, R. Heart rate variability in type 2 diabetes mellitus: A systematic review and meta–analysis. PLOS ONE 2018, 13, e0195166. [Google Scholar] [CrossRef] [PubMed]
- Bonomo, R.R.; Cook, T.M.; Gavini, C.K.; White, C.R.; Jones, J.R.; Bovo, E.; Zima, A.V.; Brown, I.A.; Dugas, L.R.; Zakharian, E.; et al. Fecal transplantation and butyrate improve neuropathic pain, modify immune cell profile, and gene expression in the PNS of obese mice. Proc. Natl. Acad. Sci. 2020, 117, 26482–26493. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Guo, L.; Feng, S.; Wang, C.; Cui, Z.; Wang, S.; Lu, Q.; Chang, H.; Hang, B.; Snijders, A.M.; et al. Fecal microbiota transplantation ameliorates type 2 diabetes via metabolic remodeling of the gut microbiota in db/db mice. BMJ Open Diabetes Res. Care 2023, 11, e003282. [Google Scholar] [CrossRef]
- Sugimoto, K.; Yasujima, M.; Yagihashi, S. Role of Advanced Glycation End Products in Diabetic Neuropathy. Curr. Pharm. Des. 2008, 14, 953–961. [Google Scholar] [CrossRef]
- Chi, H.; Sun, Y.; Lin, P.; Zhou, J.; Zhang, J.; Yang, Y.; Qiao, Y.; Liu, D.; Chiefari, E. Glucose Fluctuation Inhibits Nrf2 Signaling Pathway in Hippocampal Tissues and Exacerbates Cognitive Impairment in Streptozotocin-Induced Diabetic Rats. J. Diabetes Res. 2024, 2024, 5584761. [Google Scholar] [CrossRef]
- Leng, J.; Li, X.; Tian, H.; Liu, C.; Guo, Y.; Zhang, S.; Chu, Y.; Li, J.; Wang, Y.; Zhang, L. Neuroprotective effect of diosgenin in a mouse model of diabetic peripheral neuropathy involves the Nrf2/HO-1 pathway. BMC Complement. Med. Ther. 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Ewing, D.J.; Martyn, C.N.; Young, R.J.; Clarke, B.F. The Value of Cardiovascular Autonomic Function Tests: 10 Years Experience in Diabetes. Diabetes Care 1985, 8, 491–498. [Google Scholar] [CrossRef]
- Mohib, M.M.; Rabe, S.; Nolze, A.; Rooney, M.; Ain, Q.; Zipprich, A.; Gekle, M.; Schreier, B. Eplerenone, a mineralocorticoid receptor inhibitor, reduces cirrhosis associated changes of hepatocyte glucose and lipid metabolism. Cell Commun. Signal. 2024, 22, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Pop-Busui, R. , Cardiac autonomic neuropathy in diabetes: a clinical perspective. Diabetes Care, 2010. 33(2): p. 434-41.
- Braffett, B.H.; Gubitosi-Klug, R.A.; Albers, J.W.; Feldman, E.L.; Martin, C.L.; White, N.H.; Orchard, T.J.; Lopes-Virella, M.; Lachin, J.M.; Pop-Busui, R.; et al. Risk Factors for Diabetic Peripheral Neuropathy and Cardiovascular Autonomic Neuropathy in the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (DCCT/EDIC) Study. Diabetes 2020, 69, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- O'Brien, P.D., S. A. Sakowski, and E.L. Feldman, Mouse models of diabetic neuropathy. Ilar j, 2014. 54(3): p. 259-72.
- Doty, M.; Yun, S.; Wang, Y.; Hu, M.; Cassidy, M.; Hall, B.; Kulkarni, A.B. Integrative multiomic analyses of dorsal root ganglia in diabetic neuropathic pain using proteomics, phospho-proteomics, and metabolomics. Sci. Rep. 2022, 12, 1–15. [Google Scholar] [CrossRef]
- Mohib, M. , et al. Hypoxia induces activation of Mineralocorticoid receptor. in ACTA PHYSIOLOGICA. 2022. WILEY 111 RIVER ST, HOBOKEN 07030-5774, NJ USA.
- Latif-Hernandez, A.; Yang, T.; Butler, R.R.; Losada, P.M.; Minhas, P.S.; White, H.; Tran, K.C.; Liu, H.; Simmons, D.A.; Langness, V.; et al. A TrkB and TrkC partial agonist restores deficits in synaptic function and promotes activity-dependent synaptic and microglial transcriptomic changes in a late-stage Alzheimer's mouse model. Alzheimer's Dement. 2024, 20, 4434–4460. [Google Scholar] [CrossRef]
- Pritchard, N.; Edwards, K.; Russell, A.W.; Perkins, B.A.; Malik, R.A.; Efron, N. Corneal Confocal Microscopy Predicts 4-Year Incident Peripheral Neuropathy in Type 1 Diabetes. Diabetes Care 2015, 38, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Reddy, M.A.; Zhang, E.; Natarajan, R. Epigenetic mechanisms in diabetic complications and metabolic memory. Diabetologia 2014, 58, 443–455. [Google Scholar] [CrossRef]
- Stenvers, D.J.; Scheer, F.A.J.L.; Schrauwen, P.; la Fleur, S.E.; Kalsbeek, A. Circadian clocks and insulin resistance. Nat. Rev. Endocrinol. 2018, 15, 75–89. [Google Scholar] [CrossRef]
- Elliott, J.; Sloan, G.; Stevens, L.; Selvarajah, D.; Cruccu, G.; Gandhi, R.A.; Kempler, P.; Fuller, J.H.; Chaturvedi, N.; Tesfaye, S.; et al. Female sex is a risk factor for painful diabetic peripheral neuropathy: the EURODIAB prospective diabetes complications study. Diabetologia 2023, 67, 190–198. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Chronic hyperglycemia-driven neuroinflammation and neurodegeneration in the CNS and PNS. Chronic hyperglycemia induces advanced glycation end-products (AGEs), disrupting the blood–brain barrier (BBB) and enabling peripheral cytokine infiltration. In the CNS, oxidative stress and AGEs activate microglia (↑MHC-II, CD11b, Iba1) via NF-κB and MAPK signaling, triggering TNF-α, IL-1β, IL-6, and MCP-1 release. These cytokines stimulate reactive astrocytes to produce IL-6, MCP-1, and S100B, causing glutamate accumulation, excitotoxicity, and neuronal apoptosis. In the PNS, activated macrophages release TNF-α, IL-1β, and MCP-1, recruiting CD4⁺ T lymphocytes, which impair Schwann cells, induce demyelination, and promote axonal degeneration. Together, these processes mediate diabetes-associated neuroinflammation, demyelination, and neuronal loss.
Figure 1.
Chronic hyperglycemia-driven neuroinflammation and neurodegeneration in the CNS and PNS. Chronic hyperglycemia induces advanced glycation end-products (AGEs), disrupting the blood–brain barrier (BBB) and enabling peripheral cytokine infiltration. In the CNS, oxidative stress and AGEs activate microglia (↑MHC-II, CD11b, Iba1) via NF-κB and MAPK signaling, triggering TNF-α, IL-1β, IL-6, and MCP-1 release. These cytokines stimulate reactive astrocytes to produce IL-6, MCP-1, and S100B, causing glutamate accumulation, excitotoxicity, and neuronal apoptosis. In the PNS, activated macrophages release TNF-α, IL-1β, and MCP-1, recruiting CD4⁺ T lymphocytes, which impair Schwann cells, induce demyelination, and promote axonal degeneration. Together, these processes mediate diabetes-associated neuroinflammation, demyelination, and neuronal loss.
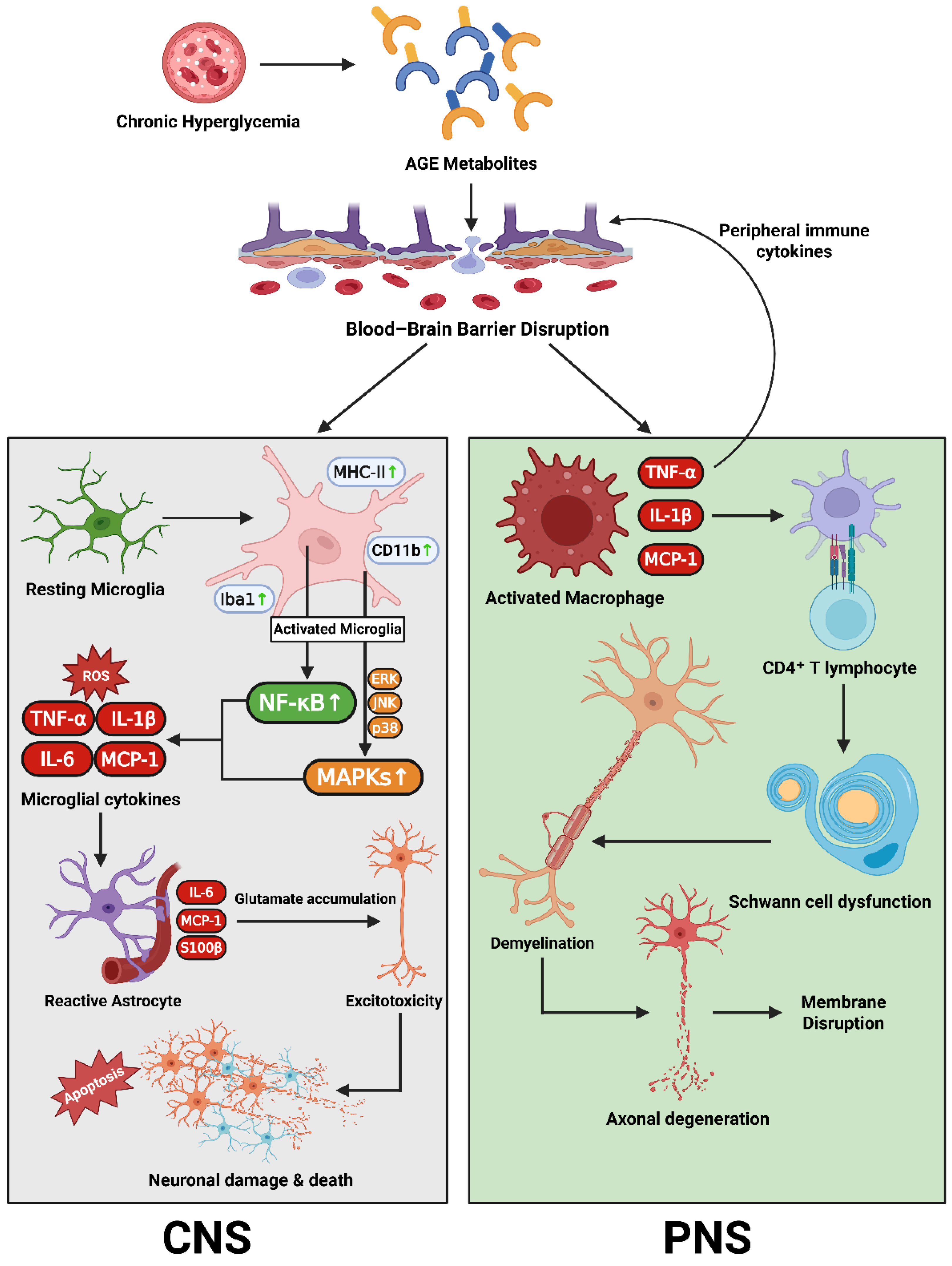
Figure 2.
Hyperglycemia-driven mitochondrial overload, oxidative stress, and redox failure. Chronic hyperglycemia increases glucose flux into mitochondria (left), over-reducing the electron transport chain (ETC). The heightened reducing pressure promotes electron leak to O₂ (bottom), generating superoxide (O₂•–), which is converted to H₂O₂ and •OH; diminished SOD2 activity limits detoxification, amplifying oxidative damage and provoking neuronal inflammation. In parallel, mitochondrial quality control is impaired (top): ↓ TFAM and ↓ PGC-1α/Nrf2 suppress biogenesis; ↑ DRP1-mediated fission with mitophagy yields a fragmented, shrunken mitochondrial pool with reduced capacity. Antioxidant defenses are concurrently depleted (right): ↓ Nrf2-dependent genes (HO-1, SOD, GPx) weaken redox buffering, while ↑ NF-κB and ↑ NOS favor peroxynitrite (ONOO–) formation, further escalating ROS and mitochondrial injury. Together, these processes form a self-reinforcing cycle of oxidative stress, mitochondrial dysfunction, and inflammation.
Figure 2.
Hyperglycemia-driven mitochondrial overload, oxidative stress, and redox failure. Chronic hyperglycemia increases glucose flux into mitochondria (left), over-reducing the electron transport chain (ETC). The heightened reducing pressure promotes electron leak to O₂ (bottom), generating superoxide (O₂•–), which is converted to H₂O₂ and •OH; diminished SOD2 activity limits detoxification, amplifying oxidative damage and provoking neuronal inflammation. In parallel, mitochondrial quality control is impaired (top): ↓ TFAM and ↓ PGC-1α/Nrf2 suppress biogenesis; ↑ DRP1-mediated fission with mitophagy yields a fragmented, shrunken mitochondrial pool with reduced capacity. Antioxidant defenses are concurrently depleted (right): ↓ Nrf2-dependent genes (HO-1, SOD, GPx) weaken redox buffering, while ↑ NF-κB and ↑ NOS favor peroxynitrite (ONOO–) formation, further escalating ROS and mitochondrial injury. Together, these processes form a self-reinforcing cycle of oxidative stress, mitochondrial dysfunction, and inflammation.
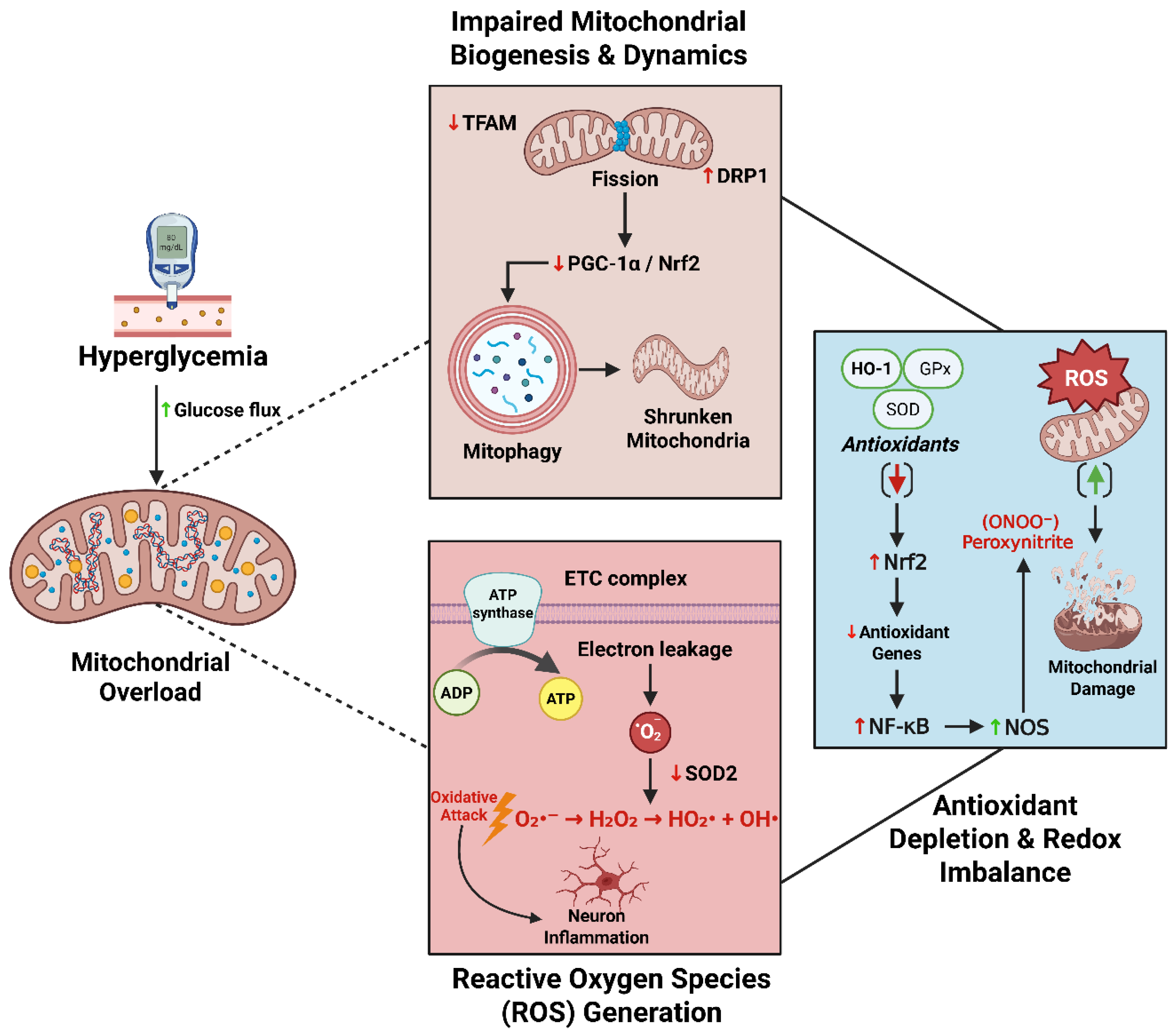
Figure 3.
Hyperglycemia-induced neuroinflammation, vascular injury, and insulin resistance in the nervous system. Chronic hyperglycemia activates microglia (↑CD11b, ↑Iba1, ↑MHC-II), triggering NF-κB/MAPK signaling, release of IFN-γ, CXCL10, IL-17, and upregulation of iNOS, COX-2, and ROS. Astrocyte activation disrupts glutamate balance and releases IL-6, MCP-1, and S100β, driving excitotoxic neuronal apoptosis. Concurrently, AGEs, ROS, and ceramides increase VCAM-1, ICAM-1, and MMP-9, leading to endothelial injury, cytokine release (IL-17A, IFN-γ, CXCL10), upregulation of Endothelin-1, and eNOS dysfunction. Reduced NO promotes vasoconstriction, ischemia, and blood–brain barrier disruption, enabling inflammatory infiltration, hypoxia, and further neuron damage. In parallel, impaired IRS-1–PI3K–Akt signaling results in lower NO production, increased lipid accumulation, and the production of pro-inflammatory cytokines (IFN-γ, IL-12, and leptin), as well as reduced mitochondrial oxidative phosphorylation, leading to decreased ATP and neuronal apoptosis. Together, these mechanisms create a reinforcing cycle of oxidative stress, vascular dysfunction, inflammation, and neurodegeneration in diabetes.
Figure 3.
Hyperglycemia-induced neuroinflammation, vascular injury, and insulin resistance in the nervous system. Chronic hyperglycemia activates microglia (↑CD11b, ↑Iba1, ↑MHC-II), triggering NF-κB/MAPK signaling, release of IFN-γ, CXCL10, IL-17, and upregulation of iNOS, COX-2, and ROS. Astrocyte activation disrupts glutamate balance and releases IL-6, MCP-1, and S100β, driving excitotoxic neuronal apoptosis. Concurrently, AGEs, ROS, and ceramides increase VCAM-1, ICAM-1, and MMP-9, leading to endothelial injury, cytokine release (IL-17A, IFN-γ, CXCL10), upregulation of Endothelin-1, and eNOS dysfunction. Reduced NO promotes vasoconstriction, ischemia, and blood–brain barrier disruption, enabling inflammatory infiltration, hypoxia, and further neuron damage. In parallel, impaired IRS-1–PI3K–Akt signaling results in lower NO production, increased lipid accumulation, and the production of pro-inflammatory cytokines (IFN-γ, IL-12, and leptin), as well as reduced mitochondrial oxidative phosphorylation, leading to decreased ATP and neuronal apoptosis. Together, these mechanisms create a reinforcing cycle of oxidative stress, vascular dysfunction, inflammation, and neurodegeneration in diabetes.
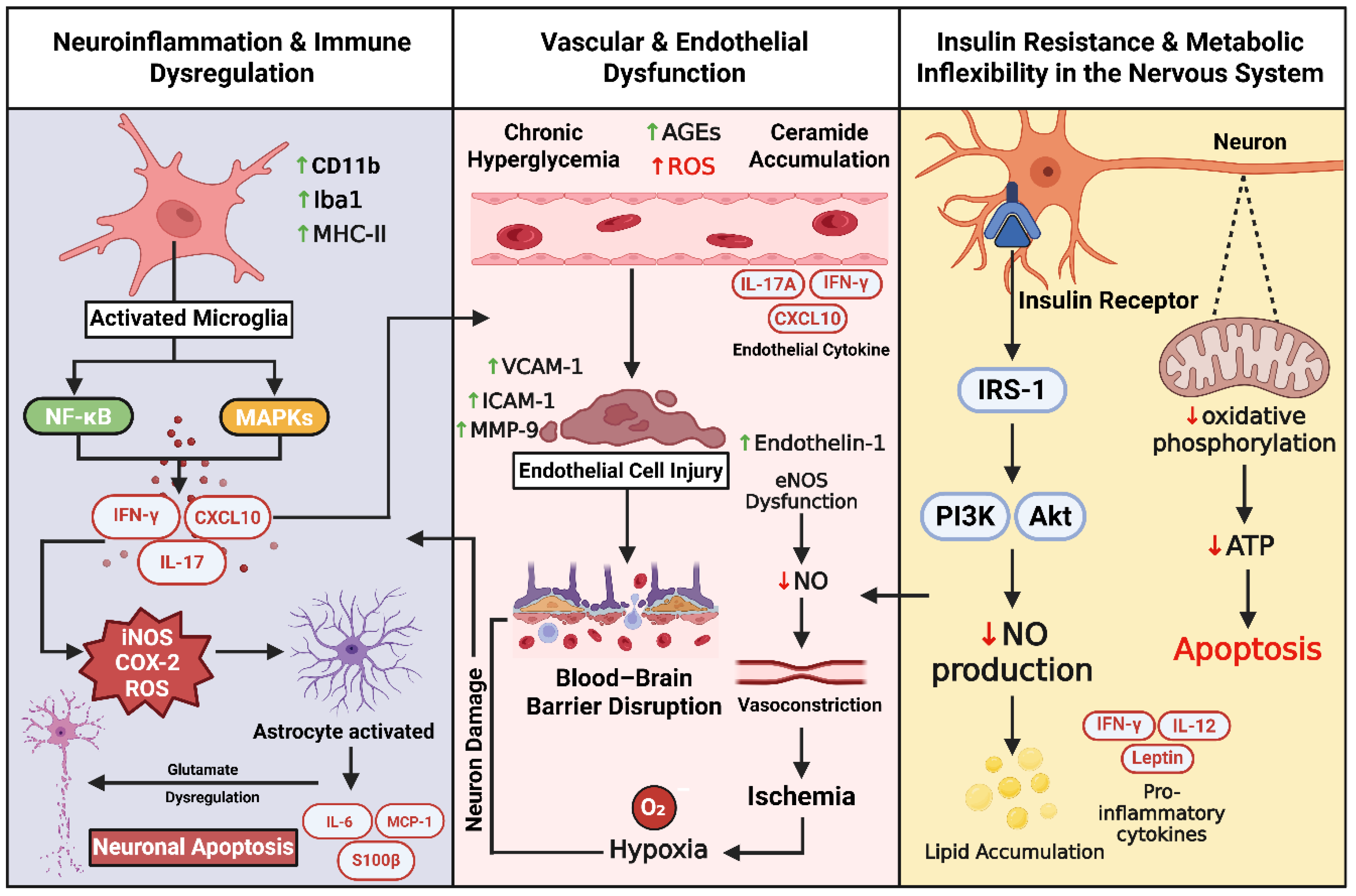
Figure 4.
Gut dysbiosis–induced systemic inflammation and neuroimmune injury via the gut–brain axis. Pro-inflammatory gut taxa (Desulfovibrio, Ruminococcus gnavus, Bacteroides vulgatus, Clostridium) reduce beneficial SCFAs (butyrate, propionate) and neuroprotective metabolites (indole-3-propionic acid). Barrier disruption (leaky gut) allows bacterial components (LPS, flagellin, peptidoglycan) to activate TLR4, driving systemic inflammation with ↑IL-17A, IFN-γ, CXCL10. These mediators disrupt the blood–brain barrier (↑MMP-9, VCAM-1, ICAM-1), trigger microglial activation, and promote neuroinflammation. Parallel immune activation damages the peripheral nervous system, causing Schwann cell and dorsal root ganglion injury. The vagus nerve provides a bidirectional route for gut-derived signals to influence central and peripheral neuroimmune responses.
Figure 4.
Gut dysbiosis–induced systemic inflammation and neuroimmune injury via the gut–brain axis. Pro-inflammatory gut taxa (Desulfovibrio, Ruminococcus gnavus, Bacteroides vulgatus, Clostridium) reduce beneficial SCFAs (butyrate, propionate) and neuroprotective metabolites (indole-3-propionic acid). Barrier disruption (leaky gut) allows bacterial components (LPS, flagellin, peptidoglycan) to activate TLR4, driving systemic inflammation with ↑IL-17A, IFN-γ, CXCL10. These mediators disrupt the blood–brain barrier (↑MMP-9, VCAM-1, ICAM-1), trigger microglial activation, and promote neuroinflammation. Parallel immune activation damages the peripheral nervous system, causing Schwann cell and dorsal root ganglion injury. The vagus nerve provides a bidirectional route for gut-derived signals to influence central and peripheral neuroimmune responses.
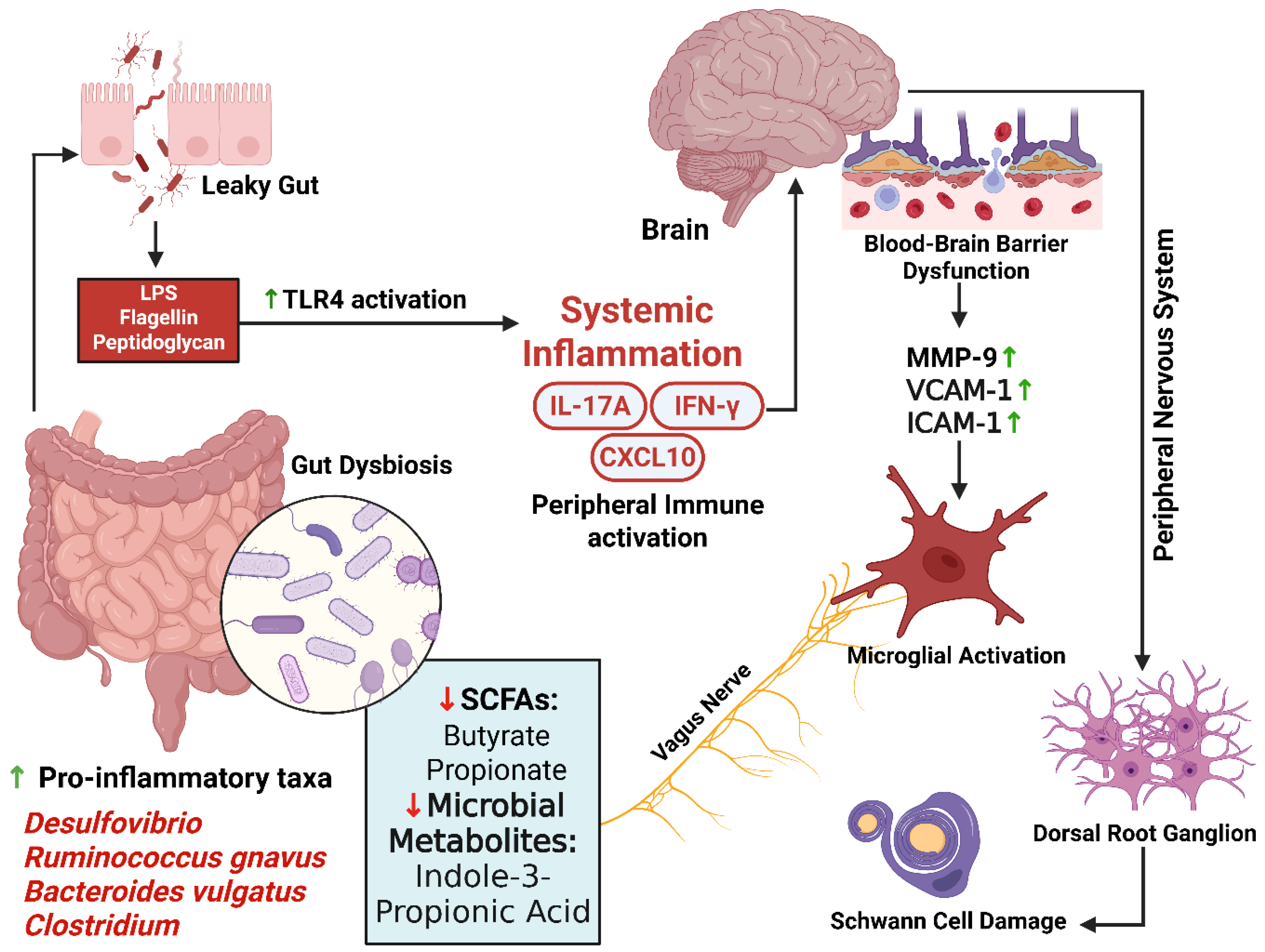
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



