
Submitted:
11 August 2025
Posted:
13 August 2025
You are already at the latest version
Abstract
Carcinogenesis, while traditionally attributed to the accumulation of driver mutations in genes regulating cell proliferation and apoptosis, may also be explored as a consequence of fundamental metabolic reprogramming, an idea catalyzed by the Warburg effect, where cancer cells exhibit a paradoxical preference for glycolysis over the far more efficient oxi-dative phosphorylation, thereby implying that metabolic dysregulation may be a primary instigator of neoplastic transformation. Proposing an alternative hypothesis, informed by both metabolic dysfunction and evolutionary biology, it becomes apparent that a precipi-tous loss of cellular energy may stimulate an atavistic response, an evolutionarily con-served remnant of ancestral survival mechanisms inherited from unicellular organisms, wherein rapid proliferation and migration were triggered to enhance survival in fluctuat-ing environments, responses that in modern times lead to pathological angiogenesis and unchecked cell growth, thereby bridging the gap between genetic and metabolic models of cancer.
Keywords:
carcinogenesis
; mutations
; metabolic reprogramming
; Warburg effect
; oxidative phos-phorylation
; atavistic response
; cell migration
; angiogenesis
1. Two Competing Models for Carcinogenesis
Genomic analyses of diverse cancers have identified recurrent "driver mutations" in genes regulating cell proliferation, seemingly supporting the view that such mutations constitute initiating events in oncogenesis [1,2,3,4,5]. This interpretation assumes a linear progression from genetic alteration to malignant transformation. However, Otto Warburg, the noted German biochemist and Nobel Laureate, through biochemical investigations observed that many cancers preferentially utilize the pentose phosphate pathway over the tricarboxylic acid (TCA) cycle for energy production [6,7,8,9,10,11,12,13,14,15,16], a metabolic shift now exploited in positron emission tomography (PET) imaging [17], in which metastatic cancer is identified radiographically [17]. Warburg hypothesized that carcinogenesis originates primarily from metabolic dysfunction rather than mutations in proliferative pathways [9]. This long-standing dichotomy persists. Yet we propose a unifying framework that reconciles these perspectives, integrating contemporary findings to inform novel directions in cancer research and therapy while acknowledging the multifaceted nature of tumorigenesis [3]. We do not suggest that all cancers arise in this manner, just that a significant number do [3,18]. Carcinogenesis is a complex, many faceted processes [3]; we do not suggest otherwise.
Some genetic alterations are inferred to be initiating events in oncogenesis, leading to the accumulation of subsequent mutations [1,2,3,4,5]. Their identification supposedly relies on comparative genomic analyses across tumor samples to determine which mutations recur most frequently, with the most prevalent alteration classified as the driver mutation [1,2,3,4,5]. However, if an initial metabolic defect arises from multiple possible mutations, each inducing a similar metabolic disruption, the assumption that malignant transformation occurs as a linear progression from genetic alteration may be flawed [3]. Here, we hypothesize that this scenario underlies the observed discrepancies between genetic and metabolic models of carcinogenesis.
2. An Alternate Hypothesis for Carcinogenesis
It may, at first glance, seem paradoxical that cells facing an abrupt loss of available energy would respond by proliferating and migrating, both energy-intensive processes, yet this is precisely what we propose (Figure 1). First, as a response to sudden energy depletion, unicellular precursors to modern multicellular organisms react by rapid cell proliferation and migration, strategies designed to generate progeny better equipped to thrive in the altered environment or to seek out more hospitable surroundings [19].
In multicellular organisms, this ancient reflex, while potentially detrimental, was nonetheless conserved if it served a critical function, most notably wound healing [20]. However, its expression came under stringent regulatory control and underwent further evolution, including the induction of vascular endothelial growth factor (VEGF) [20]. This response is generated or augmented by adjacent cells undergoing this transformation, resulting in the copious production of lactate or its metabolites, pyruvate, and purines [21].
In contemporary carcinogenesis, the process commences with a mutation in one or more genes governing the TCA cycle enzymes or the mitochondrial electron transport chain [22]. The failure of one of these enzymes often precipitates the failure of all. As a consequence of this enzymatic collapse, oxidative phosphorylation and metabolism falter, leading to a precipitous decline in cellular energy production. The affected cell can then derive a mere two energy units (ATP molecules) per glucose molecule, a stark contrast to the thirty-six or thirty-eight ATPs typically generated. This metabolic shift is accompanied by a marked increase in low-energy adenosine monophosphate (AMP) [23].
The "abrupt loss of energy" response is thereby reactivated. This process is accelerated by the increased AMP levels activating AMP-activated protein kinase (AMPK) [23], triggering VEGF production and cellular proliferation through a mechanism that may be somewhat "sloppy," leading to the emergence of numerous mutant cells [20]. These cells simultaneously undergo migration, a phenomenon recognized in tumor cells as invasion [24]. Sooner or later, these cells manifest nuclear atypia, a defining characteristic of malignant cells and their precursors. These cancer cells subsequently exhibit mutations in cell proliferation and control genes, beginning with mutations later identified as "driver mutations" [25]. However, mutations in TCA cycle genes or mitochondrial electron transport chain genes are less readily recognized as driver mutations, primarily because any of a multitude of these genes may be mutated.
These mutations lead to the accumulation of oncometabolites resulting in epigenetic alterations and a pseudohypoxic state that promotes tumorigenesis [26]. Beyond glucose metabolism, cancer cells often exhibit alterations in amino acid metabolism, which are crucial for supporting rapid proliferation and survival. Amino acids such as glutamine, arginine, tryptophan, asparagine, and aspartate play significant roles in tumor growth and modulating the tumor microenvironment [27]. Interactions between amino acid metabolism and signaling molecules like VEGF and hypoxia-inducible factor 1-alpha (HIF-1α) further influence tumor progression and angiogenesis. Additionally, tumor-associated macrophages (TAMs) contribute to the immunosuppressive microenvironment through metabolic reprogramming, including altered amino acid metabolism, which supports tumor growth and metastasis.
Therefore, the collapse of the TCA cycle not only forces cells to rely on glycolysis but also drives compensatory metabolic rewiring, notably involving amino acid pathways such as glutaminolysis and asparagine metabolism. These pathways can replenish TCA intermediates (anaplerosis) and sustain redox balance, allowing cells to partially mitigate energy deficits and maintain biosynthesis, thus linking TCA failure directly to the upregulation of amino acid metabolism.
3. Manifestations of the Atavistic Reflex in Modern Multicellular Organisms
While we cannot fully reconstruct the environmental conditions of our unicellular ancestors, we can identify remnants of their adaptive responses that persist in modern multicellular organisms [28]. We propose that certain physiological manifestations reflect an evolutionarily conserved "abrupt loss of energy" reflex. Some of these responses may confer benefits, while others contribute to pathological processes. One such example is the rapid mitochondrial dysfunction leading to an abrupt decline in ATP production. This shift forces the cell to transition from oxidative phosphorylation to glycolysis, resulting in lactate accumulation. Lactate is subsequently converted into pyruvate and alanine, a non-essential amino acid. Fluctuations in these metabolites may trigger or amplify the "abrupt loss of energy" reflex through distinct regulatory mechanisms, potentially influencing carcinogenic processes.
Important players in this mechanism are HIFs, which are transcription factors that regulate cellular responses to decreased oxygen availability. It has been recognized for many years that hypoxia is a well-documented feature of various neoplasms, traditionally attributed to inadequate vascularization [29]. However, HIFs also respond to pseudohypoxia; oxygen-independent activation triggered by factors such as thiamine deficiency or mutations affecting TCA cycle enzymes or mitochondrial oxygen transport genes [30]. Both hypoxia and pseudohypoxia rapidly impair mitochondrial ATP production, potentially initiating this metabolic response [31].
In addition, AMP-activated protein kinase (AMPK) functions as a cellular energy sensor, and it is activated under conditions of low ATP levels [32]. In specific contexts, AMPK activation can facilitate cancer cell survival and proliferation under metabolic stress, highlighting its dual role in carcinogenesis [32]. Indeed, AMPK activation promotes ATP production by increasing catabolism while decreasing anabolism [32]. This response serves to balance energy production and consumption.
Moreover, glycolysis produces abundant lactate and its metabolites, pyruvate and alanine [11,33]. These metabolites engage in a complex network of cellular signaling, including the reinforcement of the abrupt loss of energy reflex, amplifying the ongoing metabolic response [34]. As loss of muscle pyruvate dehydrogenase (PDH) demonstrates, this rapid increase in glycolysis and subsequent accumulation of pyruvate and alanine contributes to severe lactic acidosis [34]. This, then, becomes a self-perpetuating cycle, reminiscent of a runaway train, underscoring the significance of these metabolites in modulating cellular responses to energy stress.
4. Manifestations of the Atavistic Reflex Beyond Cancer
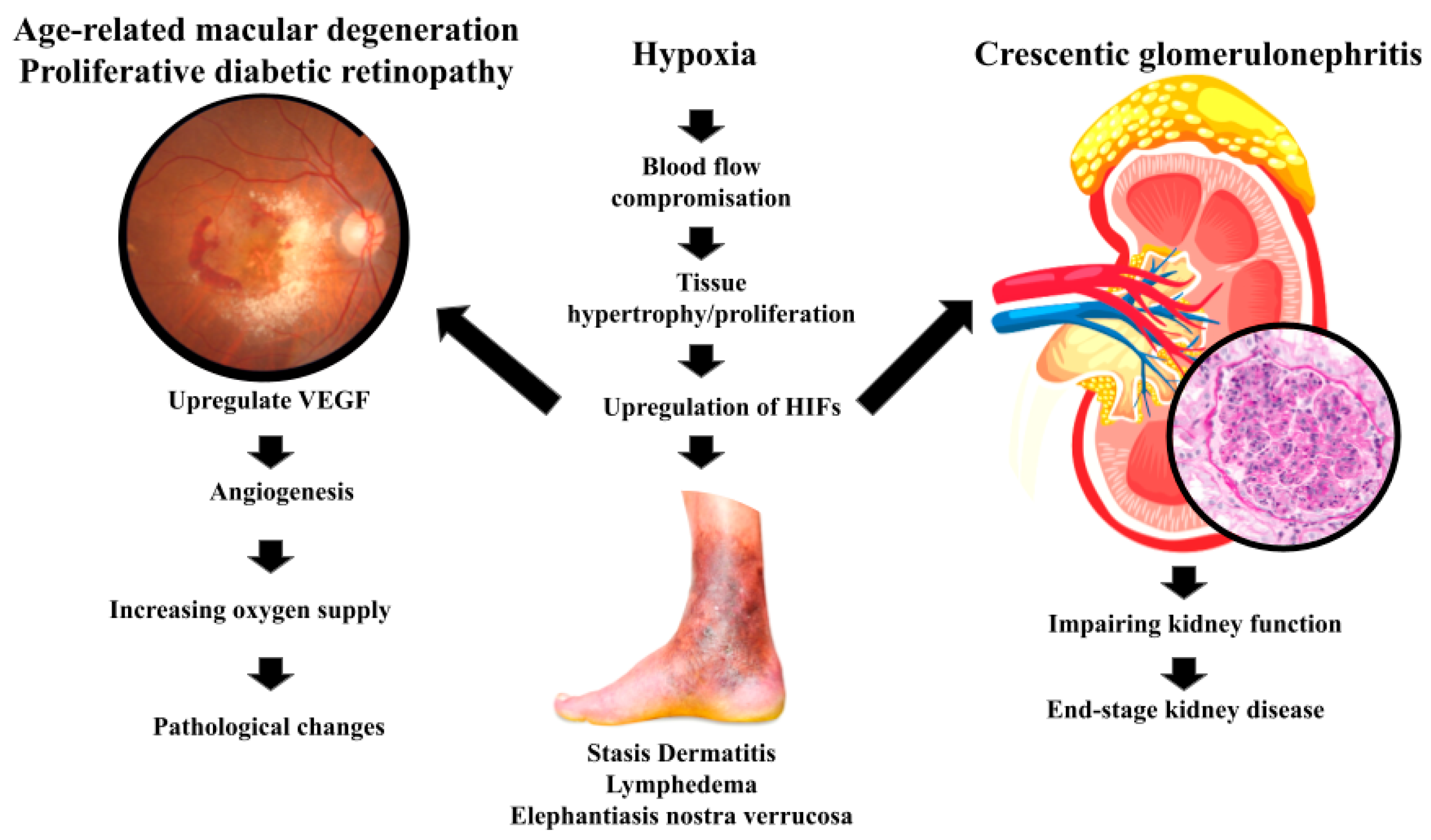
Carcinogenesis, understood as the misfiring of an ancient survival mechanism, offers a unifying explanation for the metabolic and genetic chaos that defines cancer [35]. Yet, this perspective, which frames tumorigenesis as an evolutionary relic of ancestral survival mechanisms, may also extend beyond the confines of cancer to encompass a spectrum of pathological conditions [35]. Indeed, the atavistic loss-of-energy reflex, originally an adaptive response to hypoxia and metabolic stress, appears to drive pathological proliferation in diverse diseases [35]. As will be explored below (Figure 2), this conserved mechanism enables cells to survive energy deprivation by initiating proliferation and migration; however, in the complex environment of modern multicellular organisms, this reflex can manifest in maladaptive ways, contributing to disease pathogenesis rather than promoting survival [35]. It may be that certain physiological manifestations reflect an evolutionarily conserved "abrupt loss of energy" reflex, with the balance between benefit and harm often determined by the specific tissue context and the degree of regulatory control [35].
Numerous studies have noted that oxygen tension in healing wounds is significantly reduced [35]. In this context, the proliferation and migration of fibroblasts to the wound site, a key step in tissue repair, may be driven by the activation of this atavistic reflex [36]. Fibroblasts are essential for the formation of granulation tissue [36]. They migrate into the wound area and deposit matrix de novo, with migration and proliferation rate-limiting steps to repair wounds [37]. It may be that this ancient survival response, vital for the survival of multicellular organisms, is necessarily preserved, even if it carries the risk of dysregulation [38]. As such, therapies that restore mitochondrial function, inhibit glycolysis, or modulate metabolic pathways could prevent the initiation or progression of cancer [39,40,41].
In lymphedema and elephantiasis nostra verrucosa, areas with compromised lymphatic drainage and poor blood flow, and therefore reduced oxygenation, exhibit tissue hypertrophy [35]. As a result of this oxygen deficit, the skin undergoes massive hyperplasia, paradoxically increasing the tissue's oxygen demand in an area already struggling with hypoxia [42]. The chronic, progressive accumulation of protein-rich fluid within the interstitium and the fibro-adipose tissue exceeds the capacity of the lymphatic system to transport the fluid [43]. This creates a vicious cycle, wherein the initial insult of reduced blood flow triggers a proliferative response that ultimately exacerbates the underlying oxygen deficiency [42].
Stasis dermatitis presents a seemingly contradictory picture. While blood flow in affected areas is often increased, the permeability of blood vessels is markedly reduced, leading to decreased oxygen tension in the tissues [44]. This reduced oxygenation triggers hyperplasia of the affected skin, further compromising oxygen delivery, and, in some cases, leading to the more severe manifestation of elephantiasis nostra verrucosa [35]. If left untreated, the condition can lead to more serious conditions including venous ulcerations [44]. As such, specific oxygen dosing as a function of tissue hypoxia is key to a successful outcome [45].
In decubitus ulcers, where sustained pressure deprives the skin of blood flow, tissue might die over time. Examination of these lesions reveals that the surrounding skin often exhibits marked hypertrophy, driven by increased cell proliferation [46]. This hypertrophic response, while perhaps initially having the effect of protecting the underlying tissue, increases the oxygen demand in an area already suffering from severe ischemia, ultimately contributing to the formation of deep, necrotic ulcers [46]. As a result, current research has suggested, decreasing pressure and keeping skin clean can reduce these ulcers [46].
Actinic keratoses (AKs) are characterized by keratinocytes mutated by chronic sunlight exposure [47]. These lesions carry the potential to evolve into squamous cell carcinoma, a distinctly malignant entity [47]. Stressed in modern dermatopathology textbooks, AK keratinocytes, though not themselves malignant, exhibit a tendency for affected epidermal cells to migrate into the underlying dermis [48,49]. It may be that this migratory behavior, reminiscent of the invasive properties of cancer cells, represents another manifestation of the atavistic loss-of-energy reflex, reactivated by the cellular stress induced by UV radiation [47].
Although Age-related macular degeneration (AMD) is not a neoplastic condition, it exemplifies how hypoxia-induced VEGF expression, often driven by underlying mitochondrial dysfunction and energy deprivation, can result in pathological angiogenesis. This parallel reinforces our central hypothesis, that the atavistic ‘loss-of-energy’ reflex, triggered by metabolic disruption and hypoxia, may drive cellular behaviors such as proliferation and angiogenesis in various pathologies, including but not limited to cancer. As a leading cause of vision loss in the elderly, AMD particularly highlights this complex interplay [50]. The neovascular (wet) form of AMD involves abnormal blood vessel growth in the retina, driven, in part, by hypoxia-induced expression of VEGF [51]. As noted in the literature, VEGF promotes angiogenesis in the initial stage of choroidal neovascularization that further leads to increased vascular permeability [51]. Evidence suggests that mitochondrial dysfunction, specifically mitochondrial DNA damage and dysfunction in retinal pigment epithelial cells, plays a key role in AMD [52]. This mitochondrial impairment leads to energy deficits and oxidative stress, further contributing to disease progression [52]. Hypoxia in retinal cells leads to the stabilization of HIFs, which, in turn, upregulate VEGF expression [51]. VEGF promotes angiogenesis, increasing oxygen supply but also leading to pathological changes, a balance between benefit and harm which is central to our hypothesis [51]. Indeed, anti-VEGF therapies have proven effective in treating wet AMD, underscoring the role of hypoxia-induced metabolic changes in driving the disease [51].
Proliferative diabetic retinopathy (PDR), a complication of advanced diabetes mellitus, provides another compelling example [53]. In DM-associated PDR, restricted blood flow through small vessels supplying oxygen to the retina leads to hypoxia [54]. This hypoxia, in turn, drives the proliferation of non-light reactive cells, ultimately leading to blindness as these cells obscure the photoreceptors [55]. It may be that hypoxia-driven pathways are involved, paralleling mechanisms observed in cancer and AMD, reflecting the conserved nature of the cellular response to energy deprivation [35].
Finaly, in certain severe forms of glomerulonephritis, most notably crescentic glomerulonephritis, a striking proliferation of parietal epithelial cells occurs along Bowman's capsule [56]. This proliferation, which occupies Bowman's space, gives rise to cellular crescents that compress the glomerular tuft, ultimately impairing kidney function [56], and often leads to end-stage kidney disease [57].
In crescentic glomerulonephritis, inflammation leads to occlusion of capillaries and disruption of normal blood flow. This results in a state of hypoxia and energy deprivation in glomerular cells, particularly the parietal epithelial cells lining Bowman's capsule. The response to this deprivation includes the upregulation of HIFs. It may well be that the hypoxic environment and energy scarcity within the glomerulus activate an ancient, atavistic cellular reaction, prompting proliferation. In this scenario, the parietal epithelial cells of Bowman's capsule respond to energy deprivation by proliferating and migrating into the urinary space, forming crescents. In effect, this process mirrors the proposed mechanism wherein cells, facing a sudden energetic crisis, revert to a more primitive survival strategy, prioritizing proliferation and migration, even at the expense of normal function.
Support for this perspective comes from a variety of sources. The increased expression of HIF-1α and VEGF in glomeruli affected by crescentic glomerulonephritis suggests the involvement of hypoxia-driven pathways in these pathological changes. Moreover, studies indicating mitochondrial damage in glomerular cells during severe glomerulonephritis point to decreased ATP production and increased reactive oxygen species (ROS) generation, thereby underscoring the role of metabolic dysfunction in initiating cellular responses. Perhaps most compelling is the evidence that experimental treatments designed to reduce hypoxia or inhibit HIFs and VEGF have shown promise in ameliorating glomerular injury and proliferation in animal models [58,59]. Further investigation of such interventions may prove invaluable.
5. Metabolic Reprogramming in Cancer
It has become increasingly clear that cancer cells, in their relentless pursuit of unchecked proliferation, exhibit a remarkable plasticity in their metabolic strategies. This metabolic dysregulation is hallmarked by a markedly increased glucose uptake, coupled with what some might term a rather profligate production of lactate, even when oxygen is readily available [60]. This preference for anerobic glycolysis, a phenomenon often referred to as the Warburg effect, is not merely an inefficient means of energy production; rather, it is a carefully orchestrated metabolic reprogramming that serves the biosynthetic and energy requirements of rapidly dividing cancer cells [61].
Emerging evidence suggests that mitochondrial dysfunction may play a more pivotal role in cancer development than previously appreciated, potentially acting as an instigator of genomic instability. Recent studies have illuminated that mitochondrial dysfunction can precede and, perhaps more alarmingly, actively promote genetic mutations [62]. As it was demonstrated, defects in mitochondrial respiration can unleash a torrent of ROS, those molecular wrecking balls that inflict damage upon DNA and incite genomic instability [62]. This cascade of events raises the unsettling possibility that mitochondrial dysfunction is not merely a consequence of cancer, but rather an initiating event in carcinogenesis, a spark that ignites the inferno of uncontrolled cellular growth.
Metabolic alterations can trigger a cascade of epigenetic modifications that profoundly affect the fate of a cell [63]. Fluctuations in the levels of key metabolic cofactors, such as NAD+, acetyl-CoA, and α-ketoglutarate, can directly influence the activity of epigenetic enzymes, including sirtuins, histone acetyltransferases, and DNA demethylases [64]. These epigenetic changes can either unleash the oncogenic potential of proto-oncogenes or, conversely, silence the tumor suppressor genes, thereby contributing to the initiation of cancer [63]. As others have noted, the interplay between glucose metabolism and histone acetylation illustrates this point [64].
In cancer, HIFs are often found to be upregulated, not solely as a response to hypoxia within tumors, but also as a consequence of mitochondrial dysfunction [65]. This aberrant activation of HIFs triggers an increased expression of VEGF and other factors that promote angiogenesis, glucose metabolism, and cell survival, all of which contribute to the insidious growth and metastatic spread of tumors [65]. These upregulated factors create an environment ripe for tumor progression.
Perhaps one of the most intriguing aspects of cancer metabolism is the phenomenon of pseudohypoxia, a state in which cells behave as if they are starved of oxygen, even when oxygen is readily available [66].
Mutations in key enzymes of the TCA cycle, such as succinate dehydrogenase (SDH) and fumarate hydratase (FH), can lead to the accumulation of succinate and fumarate, respectively [66]. These oncometabolites, as they are sometimes called, inhibit prolyl hydroxylases, the very enzymes that regulate HIF degradation, leading to HIF stabilization even under normoxic conditions [66].
The resulting aberrant activation of HIFs then promotes the very oncogenic pathways that fuel the relentless growth and spread of cancer [66]. Therefore, several genes are upregulated, including glycolysis (e.g., LDHA, GLUT1, PDK1), angiogenesis (VEGF), and survival pathways [67,68,69]. This contributes to therapy resistance and tumor progression.
Some therapeutic approaches discussed in the literature include, direct HIF inhibition (e.g. PX-478 inhibits HIF-1α deubiquitination. In addition, PT2385 and Belzutifan selectively antagonize HIF-2α, disrupting dimerization and transcriptional activity) [70]. Another strategy includes targeting metabolic enzymes (e.g. Dichloroacetate (DCA) inhibits PDK1, reversing glycolysis [71], IDH mutant inhibitors (e.g., Enasidenib, Ivosidenib) decrease 2-hydroxyglutarate levels and restore PHD function [72], LDH and monocarboxylate transporter (MCT) inhibitors impair lactate flux and reduce immune suppression [73].
Although primarily developed for hypoxic tumors, these strategies also mitigate pseudohypoxia by improving oxygen sensing and metabolism [68]. Given the metabolic plasticity of cancer cells, monotherapy may lead to compensatory adaptations. Therefore, combination therapies targeting HIFs, metabolic pathways, and the tumor microenvironment may enhance efficacy [74]. Additionally, the integration of these strategies with immunotherapy (e.g., immune checkpoint inhibitors) may overcome immune evasion in pseudohypoxic tumors [75].
Mutations in mitochondrial DNA (mtDNA) are commonplace in the chaotic landscape of cancer [76]. These mutations can wreak havoc on oxidative phosphorylation complexes, leading to a dysfunction of the respiratory chain [76]. Somatic mtDNA mutations can affect the functions of tRNAVal (T1659C), tRNAAla (G5650A), ND1 (G3842A), ND4 (11032delA, A11708G), ND5 (12418insA), COI (T6787C), COII (G7976A), and COIII (A9263G, G9267A) [76]. In the face of mitochondrial dysfunction, cancer cells are compelled to rely on glycolysis for their survival [77]. This reliance on glycolysis, known as the Warburg effect, provides the metabolic intermediates necessary for biosynthesis, the essential building blocks that fuel the rapid cell proliferation and ensure survival under the harsh conditions that prevail within the tumor microenvironment [77]. Mitochondrial dysfunction often begets a surge in the production of ROS [78]. These ROS can inflict damage upon DNA, proteins, and lipids, thereby contributing to genomic instability [78].
6. Additional Supporting Evidence: Stem Cells, Autophagy, and AMPK
Ito and Suda [79] have shown that hypoxic conditions and metabolic stress can maintain the quiescence of hematopoietic stem cells, while shifts in metabolism can propel them toward proliferation and differentiation. The dysregulation of these finely tuned processes can lead to the uncontrolled proliferation of cells, as seen in leukemogenesis.
Autophagy has emerged as a key player in cancer metabolism [80]. Under conditions of metabolic stress, autophagy can provide a lifeline to cells, supplying them with essential nutrients by recycling cellular components [80]. However, this very same process, if dysregulated, can also contribute to the initiation and progression of cancer, highlighting the multifaceted and often paradoxical role of autophagy in the disease process [80]. An important addition to the autophagic process would be its ability to provide a link to the tumor microenvironment [81].
Adenosine monophosphate-activated protein kinase (AMPK) is activated when ATP levels plummet, triggering a cascade of events aimed at restoring energy balance [82]. Activation of AMPK can inhibit cell proliferation by modulating metabolic pathways, essentially putting the brakes on uncontrolled growth [82]. However, it is important to consider that AMPK activation, in certain contexts, can also promote cancer cell survival under metabolic stress, suggesting that its role in carcinogenesis is far more complex than a simple good-versus-evil dichotomy [82].
7. Animal Models and Human Trials Targeting Metabolic Pathways
Animal models have provided compelling evidence linking metabolic dysfunction to cancer [83]. For instance, it has been demonstrated that mice with mutations in the mitochondrial polymerase gamma (POLG) gene, the very enzyme responsible for replicating mtDNA, accumulate mtDNA mutations and exhibit increased tumor formation [78]. Although the majority of evidence supports a role of mtDNA mutations in tumorigenesis and malignant progression, there is not a clear connection between mtDNA mutations and tumorigenesis [83]. These findings underscore the notion that tinkering with the delicate metabolic balance within cells can have profound consequences for their propensity to develop into tumors.
The insights gleaned from preclinical studies have spurred a wave of clinical trials exploring therapies that target cancer metabolism directly [84]. Inhibitors of isocitrate dehydrogenase (IDH) mutations have shown promise in treating certain leukemias and gliomas, offering a glimpse of the therapeutic potential of targeting cancer's metabolic underbelly [84]. Metformin, a drug known for its ability to activate AMPK, is also under investigation for its anticancer properties, owing to its multifaceted effects on cellular metabolism [82]. However, the success of these interventions is often dependent on the patient population and specific type of cancer [84].
8. Implications for Cancer Research and Therapy
Framing carcinogenesis as a process initiated by metabolic dysfunction unlocks new avenues for both the prevention and treatment of this insidious disease. As such, this reframing presents several potential therapeutic targets. Therapies that restore mitochondrial function, inhibit glycolysis, or modulate key metabolic pathways could disrupt the atavistic reflex, preventing the initiation or progression of cancer [85]. As seen with dichloroacetate (DCA) and other such molecules, such agents can significantly alter cellular homeostasis [85].
Targeting the epigenetic changes induced by metabolic alterations may offer a means of reversing aberrant gene expression patterns, silencing oncogenes, and reactivating tumor suppressor genes [86]. Reducing ROS levels, may minimize DNA damage and genomic instability, thereby slowing the accumulation of mutations that drive cancer progression [87]. Integrating metabolic inhibitors with traditional chemotherapy or targeted therapies could enhance treatment efficacy by simultaneously attacking cancer cells from multiple angles.
However, although the atavistic reflex may represent a conserved response to metabolic stress, its manifestation likely varies with the tissue type, genetic background, and degree of mitochondrial dysfunction. This may explain why only certain metabolic drugs (e.g., IDH inhibitors, metformin) show efficacy, highlighting the need for individualized metabolic profiling before therapy selection.
The atavistic reflex hypothesis may also inform biomarker development. Levels of HIF-1α, VEGF, lactate, or AMPK activation could serve as proxies for energy stress and metabolic reprogramming. In clinical settings, elevated expression of these markers could predict sensitivity to therapies targeting glycolysis, angiogenesis, or mitochondrial function, providing a basis for stratified treatment approaches.
9. Obesity, Sugary Food and Drink, and Cancer
Recent evidence has increasingly suggested that elevated serum glucose levels, as seen in obesity and the overconsumption of sugary foods or beverages, may function as a key metabolic component predisposing individual to the development of cancer [88]. Indeed, this association has been observed in at least twelve human internal cancers, comprising a staggering 40 percent of all internal human cancers, results that have been further corroborated in animal models [89].
While the precise etiopathogenesis remains to be fully elucidated, we can envision several potential mechanisms at play. Perhaps the most straightforward explanation is that elevated glucose levels provide a rich and readily available fuel source for those glucose-hungry cancer cells, accelerating their proliferation and fueling their relentless expansion [89]. It is also possible that the loss of a Krebs cycle enzyme or a gene controlling mitochondrial electron transport could lead to a more abrupt decrease in energy and trigger that aforementioned atavistic reflex [89].
10. Implications of These Findings for Disorders Other than Cancer
The metabolic responses proposed herein may also be relevant to understanding, and therapeutic intervention, in other human or animal diseases. For instance, phocomelia, which is a rare congenital anomaly characterized by the absence of intermediate segments of the extremity [90], may respond to site-directed pyruvate injections, perhaps even delivered in utero [91]. Similarly, one can envision a future in which we are able to regenerate organs damaged by disease or injury, thereby obviating the need for the often-difficult and traumatic process of organ transplantation [90].
11. Addressing Counterarguments
It is certainly true that driver mutations in cell proliferation genes are frequently observed in cancers, a point often raised by those skeptical of the primacy of metabolic dysfunction in tumorigenesis. However, these mutations may well be secondary events, downstream consequences of the initial metabolic dysregulation, rather than the primary instigators of cancer [92]. The sheer diversity of mutations in metabolic genes, and the relatively small role of each mutation, makes them less apparent in genomic studies that focus on recurrent mutations [92].
Skeptics might also point to the well-established metabolic flexibility exhibited by cancer cells, their remarkable ability to adapt their energy production strategies based on the ever-changing environmental conditions within the tumor microenvironment [93]. Yet, this metabolic shape-shifting, aligns perfectly with our hypothesis. The initial metabolic dysfunction forces cells to adopt alternative pathways, to scramble for energy and resources, thereby promoting their survival and, ultimately, their proliferation. In other words, the plasticity itself is a consequence of the initial metabolic insult. It should also be noted that hypotheses that rely on the proliferation of cellular control genes do not account for the prevalence of the Warburg effect [86].
Conclusion
It seems increasingly likely that carcinogenesis often begins with subtle but significant metabolic alterations, leading to energy scarcity, and triggering an evolutionarily conserved atavistic reflex that drives proliferation and migration. Observations in a variety of medical conditions support the idea that hypoxia induces cell proliferation. Integrating these findings, we propose that metabolic dysregulation frequently precedes the genetic mutations in oncogenes and tumor suppressor genes that are hallmarks of cancer.
This is not to say that genetics is not important, but rather, that genetics may often be the cart, rather than the horse. Further research into mitochondrial function, metabolic pathways, hypoxia responses, and their intricate roles in pathological cell proliferation is urgently warranted. Targeting these pathways may offer novel and, hopefully, more effective strategies for cancer prevention and therapy. If our hypothesis holds true, then cancer is not simply a matter of bad genes, but rather, a metabolic misstep, a price we pay for the very evolutionary processes that have allowed us to flourish and evolve.
Author Contributions
All authors contributed to concept, design and writing of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
None.
Conflicts of Interest
None.
References
- Sinkala, M.; et al. Mutational Landscape of Cancer-Driver Genes across Human Cancers. Sci Rep, 2023, 13, 12742. [Google Scholar] [CrossRef]
- Peters, L.; Venkatachalam, A.; Ben-Neriah, Y.; et al. Tissue-Predisposition to Cancer Driver Mutations. Cells. [CrossRef]
- Ostroverkhova, D.; Przytycka, T. M.; Panchenko, A. R.; et al. Cancer Driver Mutations: Predictions and Reality. Trends Mol Med 2023, 29, 554–566. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H.; Gonzalez-Perez, A.; Lopez-Bigas, N.; et al. A Compendium of Mutational Cancer Driver Genes. Nat Rev Cancer, 2020, 20, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M. H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M. C.; Kim, J.; Reardon, B.; Kwok-Shing Ng, P.; Jeong, K. J.; Cao, S.; Wang, Z.; Gao, J.; Gao, Q.; Wang, F.; Liu, E. M.; Mularoni, L.; Rubio-Perez, C.; Nagarajan, N.; Cortés-Ciriano, I.; Zhou, D. C.; Liang, W. W.; Hess, J. M.; Yellapantula, V. D.; Tamborero, D.; Gonzalez-Perez, A.; Suphavilai, C.; Ko, J. Y.; Khurana, E.; Park, P. J.; Van Allen, E. M.; Liang, H.; Lawrence, M. S.; Godzik, A.; Lopez-Bigas, N.; Stuart, J.; Wheeler, D.; Getz, G.; Chen, K.; Lazar, A. J.; Mills, G. B.; Karchin, R.; Ding, L.; Group, M. W.; Network, C. G. A. R.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell, 2018, 174, 1034–1035. [Google Scholar] [CrossRef] [PubMed]
- Gu, F.; Wu, Q.; et al. Quantitation of Dynamic Total-Body PET Imaging: Recent Developments and Future Perspectives. Eur J Nucl Med Mol Imaging, 2023, 50, 3538–3557. [Google Scholar] [CrossRef] [PubMed]
- Lambert, W. C.; Truong, T. M.; Gagna, C. E.; Lambert, M. W.; Lea, M.; et al. Otto Warburg. Skinmed, 2021, 19, 412–413. [Google Scholar]
- BURK, D.; SCHADE, A. L.; et al. On Respiratory Impairment in Cancer Cells. Science, 1956, 124, 270–272. [Google Scholar] [CrossRef]
- Koppenol, W. H.; Bounds, P. L.; Dang, C. V.; et al. Otto Warburg’s Contributions to Current Concepts of Cancer Metabolism. Nat Rev Cancer 2011, 11, 325–337. [Google Scholar] [CrossRef]
- Burns, J. S.; Manda, G.; et al. Metabolic Pathways of the Warburg Effect in Health and Disease: Perspectives of Choice, Chain or Chance. Int J Mol Sci. [CrossRef]
- Liberti, M. V.; Locasale, J. W.; et al. Correction to: “The Warburg Effect: How Does It Benefit Cancer Cells?”: [Trends in Biochemical Sciences, 41 (2016) 211]. Trends Biochem Sci, 2016, 41, 287. [Google Scholar] [CrossRef]
- Vaitheesvaran, B.; Xu, J.; Yee, J.; Q-Y, L.; Go, V. L.; Xiao, G. G.; Lee, W. N.; et al. The Warburg Effect: A Balance of Flux Analysis. Metabolomics, 2015, 11, 787–796. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E.; et al. THE METABOLISM OF TUMORS IN THE BODY. J Gen Physiol, 1927, 8, 519–530. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O.; et al. THE CHEMICAL CONSTITUTION OF RESPIRATION FERMENT. Science, 1928, 68, 437–443. [Google Scholar] [CrossRef]
- WARBURG, O.; et al. On Respiratory Impairment in Cancer Cells. Science, 1956, 124, 269–270. [Google Scholar] [CrossRef] [PubMed]
- WEINHOUSE, S.; et al. On Respiratory Impairment in Cancer Cells. Science, 1956, 124, 267–269. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Lee, D.; Shim, H.; et al. Metabolic Positron Emission Tomography Imaging in Cancer Detection and Therapy Response. Semin Oncol 2011, 38, 55–69. [Google Scholar] [CrossRef]
- Lambert, W. C.; Truong, T. M.; Gagna, C. E.; Lambert, M. W.; Lea, M.; et al. Otto Warburg versus Molecular Biologists: Who Is Correct About Human Carcinogenesis, and Why Does It Matter to Dermatologists? Skinmed 2021, 19, 412–413. [Google Scholar]
- Nishikawa, T.; Gulbahce, N.; Motter, A. E.; et al. Spontaneous Reaction Silencing in Metabolic Optimization. PLoS Comput Biol 2008, 4. [Google Scholar] [CrossRef]
- Bao, P.; Kodra, A.; Tomic-Canic, M.; Golinko, M. S.; Ehrlich, H. P.; Brem, H.; et al. The Role of Vascular Endothelial Growth Factor in Wound Healing. J Surg Res 2009, 153, 347–358. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Zhang, B.; Lin, X.; Fu, X.; An, Y.; Zou, Y.; Wang, J.-X.; Wang, Z.; Yu, T.; et al. Lactate Metabolism in Human Health and Disease. Sig Transduct Target Ther 2022, 7. [Google Scholar] [CrossRef]
- Przybyla-Zawislak, B.; Gadde, D. M.; Ducharme, K.; McCammon, M. T.; et al. Genetic and Biochemical Interactions Involving Tricarboxylic Acid Cycle (TCA) Function Using a Collection of Mutants Defective in All TCA Cycle Genes. Genetics 1999, 152, 153–166. [Google Scholar] [CrossRef]
- Marín-Aguilar, F.; Pavillard, L. E.; Giampieri, F.; Bullón, P.; Cordero, M. D.; et al. Adenosine Monophosphate (AMP)-Activated Protein Kinase: A New Target for Nutraceutical Compounds. Int J Mol Sci 2017, 18. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.-S.; Jiang, J.; Chen, B.-J.; Wang, K.; Tang, Y.-L.; Liang, X.-H.; et al. Plasticity of Cancer Cell Invasion: Patterns and Mechanisms. Transl Oncol 2021, 14. [Google Scholar] [CrossRef] [PubMed]
- Novikov, N. M.; Zolotaryova, S. Y.; Gautreau, A. M.; Denisov, E. V.; et al. Mutational Drivers of Cancer Cell Migration and Invasion. Br J Cancer 2021, 124, 102–114. [Google Scholar] [CrossRef] [PubMed]
- Cardaci S; Ciriolo MR; et al. TCA Cycle Defects and Cancer: When Metabolism Tunes Redox State. Int J Cell Biol. 2012, 161837. [CrossRef]
- Chen, J.; Cui, L.; Lu, S.; Xu, S.; et al. Amino Acid Metabolism in Tumor Biology and Therapy. Cell Death Dis 2024, 15. [Google Scholar] [CrossRef]
- Gray, L. R.; Tompkins, S. C.; Taylor, E. B.; et al. Regulation of Pyruvate Metabolism and Human Disease. Cell Mol Life Sci 2014, 71, 2577–2604. [Google Scholar] [CrossRef]
- Peng, G.; Liu, Y.; et al. Hypoxia-Inducible Factors in Cancer Stem Cells and Inflammation. Trends Pharmacol Sci 2015, 36, 374–383. [Google Scholar] [CrossRef]
- Zera, K.; Zastre, J.; et al. Thiamine Deficiency Activates Hypoxia Inducible Factor-1α to Facilitate pro-Apoptotic Responses in Mouse Primary Astrocytes. PLoS One 2017, 12. [Google Scholar] [CrossRef]
- Zera, K.; Zastre, J.; et al. Stabilization of the Hypoxia-Inducible Transcription Factor-1 Alpha (HIF-1α) in Thiamine Deficiency Is Mediated by Pyruvate Accumulation. Toxicol Appl Pharmacol 2018, 355, 180–188. [Google Scholar] [CrossRef]
- Mihaylova, M. M.; Shaw, R. J.; et al. The AMPK Signalling Pathway Coordinates Cell Growth, Autophagy and Metabolism. Nat Cell Biol 2011, 13, 1016–1023. [Google Scholar] [CrossRef]
- Liberti, M. V.; Locasale, J. W.; et al. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem Sci, 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Gopal, K.; Abdualkader, A. M.; Li, X.; Greenwell, A. A.; Karwi, Q. G.; Altamimi, T. R.; Saed, C.; Uddin, G. M.; Darwesh, A. M.; Jamieson, K. L.; Kim, R.; Eaton, F.; Seubert, J. M.; Lopaschuk, G. D.; Ussher, J. R.; Al Batran, R.; et al. Loss of Muscle PDH Induces Lactic Acidosis and Adaptive Anaplerotic Compensation via Pyruvate-Alanine Cycling and Glutaminolysis. J Biol Chem 2023, 299. [Google Scholar] [CrossRef] [PubMed]
- Yip, W. L.; et al. Influence of Oxygen on Wound Healing. Int Wound J 2015, 12, 620–624. [Google Scholar] [CrossRef] [PubMed]
- Cen, R.; Wang, L.; He, Y.; Yue, C.; Tan, Y.; Li, L.; Lei, X.; et al. Dermal Fibroblast Migration and Proliferation Upon Wounding or Lipopolysaccharide Exposure Is Mediated by Stathmin. Front Pharmacol 2021, 12, 781282. [Google Scholar] [CrossRef] [PubMed]
- Knoedler, S.; Broichhausen, S.; Guo, R.; Dai, R.; Knoedler, L.; Kauke-Navarro, M.; Diatta, F.; Pomahac, B.; Machens, H.-G.; Jiang, D.; Rinkevich, Y.; et al. Fibroblasts - the Cellular Choreographers of Wound Healing. Front Immunol 2023, 14, 1233800. [Google Scholar] [CrossRef]
- Atlante, A.; Valenti, D.; et al. Mitochondria Have Made a Long Evolutionary Path from Ancient Bacteria Immigrants within Eukaryotic Cells to Essential Cellular Hosts and Key Players in Human Health and Disease. CIMB 2023, 45, 4451–4479. [Google Scholar] [CrossRef]
- Stine, Z. E.; Schug, Z. T.; Salvino, J. M.; Dang, C. V.; et al. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat Rev Drug Discov 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Pelicano, H.; Martin, D. S.; Xu, R.-H.; Huang, P.; et al. Glycolysis Inhibition for Anticancer Treatment. Oncogene 2006, 25, 4633–4646. [Google Scholar] [CrossRef]
- Icard, P.; Loi, M.; Wu, Z.; Ginguay, A.; Lincet, H.; Robin, E.; Coquerel, A.; Berzan, D.; Fournel, L.; Alifano, M.; et al. Metabolic Strategies for Inhibiting Cancer Development. Advances in Nutrition 2021, 12, 1461–1480. [Google Scholar] [CrossRef]
- Liaw, F.-Y.; Huang, C.-F.; Wu, Y.-C.; Wu, B.-Y.; et al. Elephantiasis Nostras Verrucosa: Swelling with Verrucose Appearance of Lower Limbs. Can Fam Physician 2012, 58, e551–553. [Google Scholar]
- Sleigh, B. C.; Manna, B.; et al. Lymphedema. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- Yosipovitch, G.; Nedorost, S. T.; Silverberg, J. I.; Friedman, A. J.; Canosa, J. M.; Cha, A.; et al. Stasis Dermatitis: An Overview of Its Clinical Presentation, Pathogenesis, and Management. Am J Clin Dermatol 2023, 24, 275–286. [Google Scholar] [CrossRef]
- Castilla, D. M.; Liu, Z.-J.; Velazquez, O. C.; et al. Oxygen: Implications for Wound Healing. Adv Wound Care (New Rochelle) 2012, 1, 225–230. [Google Scholar] [CrossRef]
- Litin, S. C. , Nanda, S., Mayo Clinic, Eds.; et al. Mayo Clinic Family Health Book, Fifth edition. Mayo Clinic: Rochester, MN, 2018. [Google Scholar]
- Lee, Y. B.; Kim, J.-I.; et al. Genetic Studies of Actinic Keratosis Development: Where Are We Now? Ann Dermatol 2023, 35, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Patterson, J. W.; Weedon, D.; et al. Weedon’s Skin Pathology, Fifth edition.; Elsevier: Amsterdam, 2021. [Google Scholar]
- Calonje, E. , Brenn, T., Lazar, A. J., MacKee, P. H., Billings, S. D., Eds.; et al. McKee’s Pathology of the Skin: With Clinical Correlations, Fifth edition. Elsevier: Philadelphia, PA, 2020. [Google Scholar]
- Ferrington, D. A.; Fisher, C. R.; Kowluru, R. A.; et al. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol Med 2020, 26, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Shahidatul-Adha, M.; Zunaina, E.; Aini-Amalina, M. N.; et al. Evaluation of Vascular Endothelial Growth Factor (VEGF) Level in the Tears and Serum of Age-Related Macular Degeneration Patients. Sci Rep 2022, 12. [Google Scholar] [CrossRef] [PubMed]
- Ferrington, D. A.; Fisher, C. R.; Kowluru, R. A.; et al. Mitochondrial Defects Drive Degenerative Retinal Diseases. Trends Mol Med 2020, 26, 105–118. [Google Scholar] [CrossRef]
- Yumnamcha, T.; Guerra, M.; Singh, L. P.; Ibrahim, A. S.; et al. Metabolic Dysregulation and Neurovascular Dysfunction in Diabetic Retinopathy. Antioxidants (Basel) 2020, 9. [Google Scholar] [CrossRef]
- Majidova, S. R.; et al. Evaluation of Hypoxia and Microcirculation Factors in the Progression of Diabetic Retinopathy. Invest Ophthalmol Vis Sci 2024, 65. [Google Scholar] [CrossRef]
- Forrester, J. V.; Shafiee, A.; Schröder, S.; Knott, R.; McIntosh, L.; et al. The Role of Growth Factors in Proliferative Diabetic Retinopathy. Eye 1993, 7, 276–287. [Google Scholar] [CrossRef]
- Delbet, J.-D.; Anquetil, V.; Saitoski, K.; Toso, A.; Baumert, T.; Toovey, S.; Manenti, L.; Iacone, R.; Ulinski, T.; Lenoir, O.; Teixeira, G.; Tharaux, P.-L.; et al. #2837 Novel Therapeutic for Crescentic Glomerulonephritis through Targeting CLDN1 in Parietal Epithelial Cells. Nephrology Dialysis Transplantation 2024, 39 (Supplement_1), gfae069-0018–2837. [Google Scholar] [CrossRef]
- Chen, A.; Lee, K.; D’Agati, V. D.; Wei, C.; Fu, J.; Guan, T.-J.; He, J. C.; Schlondorff, D.; Agudo, J.; et al. Bowman’s Capsule Provides a Protective Niche for Podocytes from Cytotoxic CD8+ T Cells. J Clin Invest 2018, 128, 3413–3424. [Google Scholar] [CrossRef]
- Masuda, Y.; Shimizu, A.; Mori, T.; Ishiwata, T.; Kitamura, H.; Ohashi, R.; Ishizaki, M.; Asano, G.; Sugisaki, Y.; Yamanaka, N.; et al. Vascular Endothelial Growth Factor Enhances Glomerular Capillary Repair and Accelerates Resolution of Experimentally Induced Glomerulonephritis. Am J Pathol 2001, 159, 599–608. [Google Scholar] [CrossRef]
- Mayer, G.; et al. Capillary Rarefaction, Hypoxia, VEGF and Angiogenesis in Chronic Renal Disease. Nephrol Dial Transplant 2011, 26, 1132–1137. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Xiao, Z.; Chen, T.; Liang, S. H.; Guo, H.; et al. Glucose Metabolism on Tumor Plasticity, Diagnosis, and Treatment. Front Oncol 2020, 10, 317. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M. V.; Locasale, J. W.; et al. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem Sci 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Ma, J.; Lu, W.; et al. The Significance of Mitochondrial Dysfunction in Cancer. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Chen, Y.; Oyang, L.; Lin, J.; Xia, L.; Peng, M.; Wu, N.; Tang, Y.; Li, J.; Liao, Q.; Zhou, Y.; et al. Metabolic Reprogramming and Epigenetic Modifications in Cancer: From the Impacts and Mechanisms to the Treatment Potential. Exp Mol Med 2023, 55, 1357–1370. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, C.; Wang, X.; Sun, Y.; Zhang, J.; Chen, J.; Shi, Y.; et al. An Epigenetic Role of Mitochondria in Cancer. Cells 2022, 11. [Google Scholar] [CrossRef]
- Wicks, E. E.; Semenza, G. L.; et al. Hypoxia-Inducible Factors: Cancer Progression and Clinical Translation. J Clin Invest 2022, 132. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, B.; Li, F.; Zhang, Y.; Gao, X.; Wang, Y.; Zhou, H.; et al. The Connection between Tricarboxylic Acid Cycle Enzyme Mutations and Pseudohypoxic Signaling in Pheochromocytoma and Paraganglioma. Front Endocrinol (Lausanne) 2023, 14, 1274239. [Google Scholar] [CrossRef]
- Nong, S.; Han, X.; Xiang, Y.; Qian, Y.; Wei, Y.; Zhang, T.; Tian, K.; Shen, K.; Yang, J.; Ma, X.; et al. Metabolic Reprogramming in Cancer: Mechanisms and Therapeutics. MedComm 2023, 4. [Google Scholar] [CrossRef]
- Paredes, F.; Williams, H. C.; San Martin, A.; et al. Metabolic Adaptation in Hypoxia and Cancer. Cancer Letters 2021, 502, 133–142. [Google Scholar] [CrossRef]
- Kluckova, K.; Tennant, D. A.; et al. Metabolic Implications of Hypoxia and Pseudohypoxia in Pheochromocytoma and Paraganglioma. Cell Tissue Res 2018, 372, 367–378. [Google Scholar] [CrossRef]
- Koh, M. Y.; Spivak-Kroizman, T.; Venturini, S.; Welsh, S.; Williams, R. R.; Kirkpatrick, D. L.; Powis, G.; et al. Molecular Mechanisms for the Activity of PX-478, an Antitumor Inhibitor of the Hypoxia-Inducible Factor-1α. Molecular Cancer Therapeutics 2008, 7, 90–100. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, W. Y.; McGee, S. L.; Connor, T.; Mottram, B.; Wilkinson, A.; Whitehead, J. P.; Vuckovic, S.; Catley, L.; et al. Dichloroacetate Inhibits Aerobic Glycolysis in Multiple Myeloma Cells and Increases Sensitivity to Bortezomib. Br J Cancer 2013, 108, 1624–1633. [Google Scholar] [CrossRef] [PubMed]
- Tian, W.; Zhang, W.; Wang, Y.; Jin, R.; Wang, Y.; Guo, H.; Tang, Y.; Yao, X.; et al. Recent Advances of IDH1 Mutant Inhibitor in Cancer Therapy. Front. Pharmacol. 2022, 13, 982424. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J. R.; Cleveland, J. L.; et al. Targeting Lactate Metabolism for Cancer Therapeutics. J. Clin. Invest. 2013, 123, 3685–3692. [Google Scholar] [CrossRef]
- Mao, Y.; Xia, Z.; Xia, W.; Jiang, P.; et al. Metabolic Reprogramming, Sensing, and Cancer Therapy. Cell Reports 2024, 43. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D.; et al. Therapeutic Targeting of Hypoxia and Hypoxia-Inducible Factors in Cancer. Pharmacology & Therapeutics 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Smith, A. L. M.; Whitehall, J. C.; Greaves, L. C.; et al. Mitochondrial DNA Mutations in Ageing and Cancer. Mol Oncol 2022, 16, 3276–3294. [Google Scholar] [CrossRef]
- Liberti, M. V.; Locasale, J. W.; et al. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem Sci 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Luo, Y.; Ma, J.; Lu, W.; et al. The Significance of Mitochondrial Dysfunction in Cancer. Int J Mol Sci 2020, 21. [Google Scholar] [CrossRef]
- Ito, K.; Suda, T.; et al. Metabolic Requirements for the Maintenance of Self-Renewing Stem Cells. Nat Rev Mol Cell Biol 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, J.; Levine, B.; Debnath, J.; et al. Autophagy and Cancer Metabolism. Methods Enzymol 2014, 542, 25–57. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M. V.; Locasale, J. W.; et al. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem Sci 2016, 41, 211–218. [Google Scholar] [CrossRef]
- Li, W.; Saud, S. M.; Young, M. R.; Chen, G.; Hua, B.; et al. Targeting AMPK for Cancer Prevention and Treatment. Oncotarget 2015, 6, 7365–7378. [Google Scholar] [CrossRef]
- Zhang, B.-T.; Xu, J.-Y.; Wang, W.; Zeng, Y.; Jiang, J.; et al. Obesity and Cancer: Mouse Models Used in Studies. Front Oncol 2023, 13, 1125178. [Google Scholar] [CrossRef]
- Stine, Z. E.; Schug, Z. T.; Salvino, J. M.; Dang, C. V.; et al. Targeting Cancer Metabolism in the Era of Precision Oncology. Nat Rev Drug Discov 2022, 21, 141–162. [Google Scholar] [CrossRef]
- Bhat, T. A.; Kumar, S.; Chaudhary, A. K.; Yadav, N.; Chandra, D.; et al. Restoration of Mitochondria Function as a Target for Cancer Therapy. Drug Discov Today 2015, 20, 635–643. [Google Scholar] [CrossRef]
- Xu, X.; Peng, Q.; Jiang, X.; Tan, S.; Yang, Y.; Yang, W.; Han, Y.; Chen, Y.; Oyang, L.; Lin, J.; Xia, L.; Peng, M.; Wu, N.; Tang, Y.; Li, J.; Liao, Q.; Zhou, Y.; et al. Metabolic Reprogramming and Epigenetic Modifications in Cancer: From the Impacts and Mechanisms to the Treatment Potential. Exp Mol Med 2023, 55, 1357–1370. [Google Scholar] [CrossRef]
- Srinivas, U. S.; Tan, B. W. Q.; Vellayappan, B. A.; Jeyasekharan, A. D.; et al. ROS and the DNA Damage Response in Cancer. Redox Biol 2019, 25, 101084. [Google Scholar] [CrossRef]
- Crawley, D. J.; Holmberg, L.; Melvin, J. C.; Loda, M.; Chowdhury, S.; Rudman, S. M.; Van Hemelrijck, M.; et al. Serum Glucose and Risk of Cancer: A Meta-Analysis. BMC Cancer 2014, 14, 985. [Google Scholar] [CrossRef]
- Duan, W.; Shen, X.; Lei, J.; Xu, Q.; Yu, Y.; Li, R.; Wu, E.; Ma, Q.; et al. Hyperglycemia, a Neglected Factor during Cancer Progression. Biomed Res Int 2014, 2014, 461917. [Google Scholar] [CrossRef]
- Davis, D. D.; Kane, S. M.; et al. Phocomelia. In StatPearls; StatPearls Publishing: Treasure Island (FL), 2025. [Google Scholar]
- Ferriero, R.; Manco, G.; Lamantea, E.; Nusco, E.; Ferrante, M. I.; Sordino, P.; Stacpoole, P. W.; Lee, B.; Zeviani, M.; Brunetti-Pierri, N. Phenylbutyrate Therapy for Pyruvate Dehydrogenase Complex Deficiency and Lactic Acidosis. Sci Transl Med 2013, 5. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Tseng, L.-M.; Lee, H.-C.; et al. Role of Mitochondrial Dysfunction in Cancer Progression. Exp Biol Med (Maywood) 2016, 241, 1281–1295. [Google Scholar] [CrossRef]
- Fendt, S.-M.; Frezza, C.; Erez, A.; et al. Targeting Metabolic Plasticity and Flexibility Dynamics for Cancer Therapy. Cancer Discov 2020, 10, 1797–1807. [Google Scholar] [CrossRef]
Figure 1.
summarizes our alternate hypothesis.
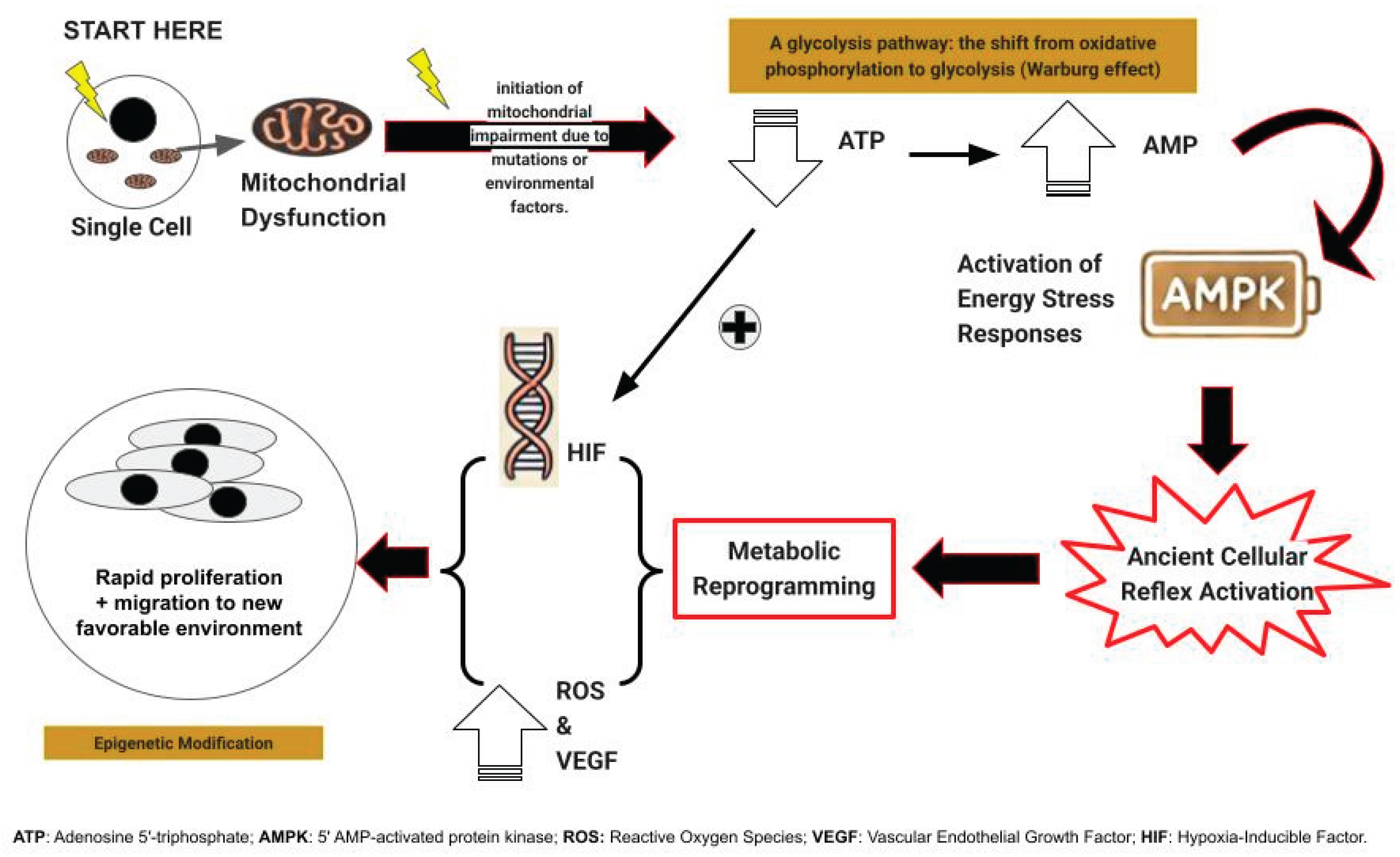
Figure 2.
summarizes the manifestations of the atavistic reflex beyond cancer.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



