
Submitted:
11 August 2025
Posted:
13 August 2025
You are already at the latest version
Abstract
The widespread use of advanced imaging techniques has led to a rising incidence of ad-renal incidentalomas (AIs), asymptomatic adrenal masses discovered during imaging for non-adrenal-related conditions. AIs represent a diagnostic and therapeutic challenge due to their varied etiology, secretory potential, and potential for malignancy. Objective: This review aims to provide a comprehensive overview of the current knowledge on adrenal incidentalomas, focusing on their pathogenesis, diagnostic work-up, imaging features, hormonal evaluation, and evidence-based management, with a special emphasis on au-tonomous cortisol secretion (ACS). A thorough narrative review of the literature from the past two decades was conducted, synthesizing data from key international guidelines (ESE/ENSAT), observational studies, meta-analyses, and case series regarding the evalua-tion and treatment of AI. AI represents an increasingly relevant clinical condition requir-ing a multidisciplinary, personalized approach. Prompt endocrine and radiological eval-uation is essential to identify hormonally active or potentially malignant tumors. The complexity of the natural history of AI and the evolving understanding of ACS underline the need for tailored follow-up and management strategies.
Keywords:
adrenal incidentaloma
; autonomous cortisol secretion
; adrenal adenoma
; endocrine im-aging
; adrenalectomy
; subclinical Cushing’s syndrome
1. Introduction
The use of diagnostic imaging has extended dramatically during the last four decades due mainly to technological development in imaging techniques.
An adrenal incidentaloma (AI) is defined as a clinically silent or asymptomatic adrenal mass equal or greater than 1cm in diameter detected during imaging performed for reasons unrelated to suspected adrenal disease [1]. The concept of an AI does not include screening imaging in patients with genetic syndromes that can develop adrenal tumors. Additionally, adrenal masses found on imaging tests performed during tumor evaluation for extra-adrenal malignancies do not strictly fit the notion of an AI [2]. This endocrine entity requires a multidisciplinary approach for effective and personalized management.
2. Materials and Methods
Study design and search strategy: We conducted a narrative literature review on adrenal incidentaloma (AI) covering epidemiology, pathogenesis, imaging, biochemical work-up, and management. Literature searches were performed in PubMed/MEDLINE, Embase, Scopus, Web of Science, and the Cochrane Library up to 01 August 2025. Search terms combined controlled vocabulary and free-text words for “adrenal incidentaloma” and related functional entities (autonomous cortisol secretion, pheochromocytoma, primary aldosteronism, adrenocortical carcinoma) with imaging and diagnostic terms. Reference lists of included papers and guideline documents were screened manually for additional sources.
Eligibility criteria and study selection: We included clinical studies, guidelines, systematic reviews, and case reports relevant to AI in humans. Exclusions: animal/in vitro studies without clinical applicability, conference abstracts without full text, and non-relevant reports. Two reviewers screened titles/abstracts, assessed full texts, and resolved disagreements by consensus.
Data extraction and synthesis: Key information extracted included study type, population, diagnostic definitions, imaging/biochemical criteria, management approach, and outcomes. Data were synthesized qualitatively and compared with current guidelines; no meta-analysis was performed. The dataset and search strategies are available upon reasonable request.
The search identified 1,242 records; 312 duplicates were removed. 930 titles/abstracts were screened, of which 172 underwent full-text review. 118 records met inclusion criteria and were included in the synthesis. A PRISMA-style diagram was added.
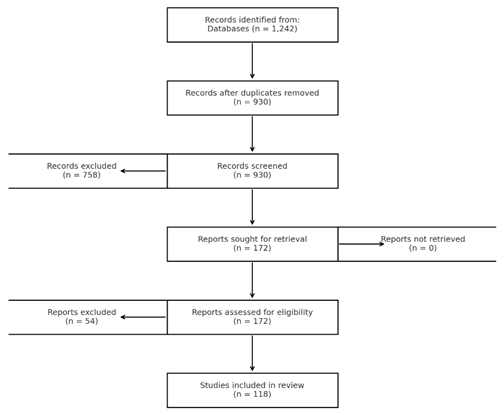
Ethics and AI disclosure: This review used only previously published material; no ethics approval or patient consent was required. All content was verified and approved by the authors. No generative artificial intelligence tools were used in the preparation of this manuscript.
3. Results
3.1. Prevalence
Adrenal masses are among the most prevalent adrenal tumors and their incidence increased by ten times in the last 20 years. When computed tomography (CT) scans or magnetic resonance imaging (MRI) are performed for other purposes, in about 75% of cases AI may be discovered serendipitously [2]. In a radiological study of 61,054 abdominal CT scans, 0.4 percent of all CT scans had diagnosed an AI [3]. Furthermore, the frequency of AI on CT scans was 4.4%, according to a later investigation using higher resolution scanners [4]. In older patients, AI prevalence is higher, about 7 to 10% [1,5]. Bilateral AIs may occur in 10% to 15% of cases, even though the majority of AIs are unilateral. The peak incidence is in the fifth and sixth decade and it is worth mentioning that AI is very rare under the age of thirty and unfrequent in childhood [6].
AI can be regarded as one of the public health challenges due to increasingly diagnosed cases in current medical practice [1,7].
The management of AI is still a clinical challenge because it is crucial to determine its secretory pattern (hormonal activity) and risk of malignancy, in orther to establish the proper treatment and the follow up period of time.
AI are mostly nonfunctioning adrenocortical adenomas, although they can potentially represent some specific endocrine pathology requiring surgical treatment (pheochromocytoma, adrenocortical carcinoma, hormone-producing adenoma or metastasis) [2]. In addition, authors reported the development of an adrenal metastasis on a pre-existing apparently benign AI, this being one of the cases in which the natural history of AI was one of malignancy [8].
When a clinician is faced with an AI, two important questions have to be addressed: if the tumour has malignant aspects and if the tumor has secretory hormonal activity [2].
3.2. Pathogenesis
Multiple mechanisms have been described in the pathogenesis of AI, but the molecular mechanisms that promote AI development are not well understood. Firstly, one of the pathophysiological mechanisms described in the literature is focal adrenal hyperplasia in response to ischemic lesions, as a natural phenomenon of aging. This mechanism could be a rational explanation for the increasing prevalence of AI among the elderly [9,10].
Secondly, increased insulin levels can induce AI development through mitogenic action on the adrenal cortex. This mechanism is cited in cases of AI in overweight people with insulin resistance [10].
Thirdly, changes in the sensitivity of the hypothalamic-pituitary-adrenal axis, which may lead to subtle but chronic stimulation of the adrenals by constantly elevated levels of ACTH, particularly in response to chronic stress, can lead to the development of nodular adrenal hyperplasia [11].
Furthermore, somatic or germline genetic mutations have been described as determining factors of AI. Particular genetic disorders predispose to adrenocortical tumors such as McCune-Albright, Beckwith-Wiedemann, Li-Fraumeni, and multiple endocrine neoplasia type 1 and type 4 (MEN-l and MEN-4) syndromes [11,12,13].
The McCune-Albright syndrome is caused by a somatic activating mutation in the GNAS gene, defined by early puberty, cafe au lait patches, polyostotic fibrous dysplasia, and hyperfunction of numerous endocrine glands (adrenals, thyroid, pituitary). Five percent of patients can develop cortisol secretion, which is linked to macronodular adrenocortical hyperplasia [9,11].
Newborn gigantism, omphalocele, macroglossia, visceromegaly, hemihypertrophy, neonatal hypoglycemia, and a number of cancers, including nephroblastoma, adrenocortical carcinoma, neuroblastoma, and hepatoblastoma, are all symptoms of the hereditary condition known as Beckwith-Wiedemann syndrome [11,14].
Li-Fraumeni syndrome, which is caused by hereditary mutations in the p53 gene, is characterized by an elevated risk for breast carcinoma, soft-tissue sarcoma, brain tumor, osteosarcoma, leukemia, and adrenocortical carcinoma. Adrenocortical carcinoma is the primary malignancy in less than 5% of the cases, occuring especially in young people [11,13].
MEN1 is an autosomal dominant disease, caused by a genetic mutation of the menin gene located on chromosome 11, leading to the development of endocrine tumors with the classic name known by the "3 P's" tumours – parathyroid tumours, pituitary tumours, pancreatic neuroendocrine tumours [15]. In addition, in the last decade, the clinical spectrum of this syndrome has been extended and completed with the appearance of adrenal tumours, pheocromocythomas, carcinoid tumours, thymic tumours but also non-endocrine tumours such as angiofibromas, lipomas, meningiomas, collagenomas, ependymomas [16,17,18]. Furthermore, in MEN1 syndrome, in about 40% of cases, non-functional AI development has been cited [18].
In 2008 the MEN-4 syndrome was described and discovered through studies on MEN syndrome in rats (called MENX in rats). In this pathology, patients can develop parathyroid (81%) and anterior pituitary (42%) tumours due to mutations in the CDKN1B gene encoding p27, a cyclin-dependent kinase inhibitor that regulates the transition of cells from G1 to S phase, thus preventing cell cycle progression. Patients may also develop gastric and bronchial NETs, gastrinomas, lymphomas, breast neoplasms, testicular, renal and adrenal tumours [19,20].
In addition, adrenal tumors that produce cortisol have been linked to inactivating PRKAR1A mutations. Inactivating mutations result in constitutive activation of the cAMP–protein kinase A pathway. Somatic PRKAR1A mutations have been reported in some sporadic adrenocortical cancers, despite the fact that mutations in this gene were initially identified in Carney complex [6].
Numerous studies in the literature have attempted to find molecules involved in AI pathogenesis. Some authors have tried to find a link between inappropriate Jak2 signaling in a group of AIs, knowing the involvement of JAK2 in neoplasia, but the results were negative [21]. Furthermore, it is well known that bisphenol-A (BPA) is an endocrine-disrupting chemical and authors stated that increased BPA exposure as a consequence of industrialization may play a role in AI development [22].
3.3. Diagnostic Features
Along with ruling out neoplasia, determining the endocrine state of individuals with AI is a critical component of clinical management. Personalized care is supported by a hormonal evaluation, clinical examination correlated with possible excess hormone secretion, focusing on suggestive signs and symptoms and related comorbidities [2,6].
According to a study regarding AI, adenomas account for 41% of total AI, metastases for 19%, adrenal cortical carcinoma for 10%, myelolipomas for 9%, and pheochromocytomas for 8%, with other more rare entities such as adrenal cysts, ganglioneuromas, hematomas, and infectious or infiltrative lesions. With a prevalence ranging from 5% to 30%, cortisol excess is the most prevalent endocrine abnormality in people with AI [10,23]. In adults, an AI can represent many endocrine disorders, which are synthesized in Table 1 (adapted from [5]).
In addition, one case of AI was reported which on pathological examination proved to be Kaposi's sarcoma [24]. Similarly, adrenal oncocytoma may be a rare etiology of AI [25]. The endocrine assessment should exclude pheochromocythomas, cortisol producing adenomas and sexosteroid producing tumours.
Cortisol excess is the most frequent endocrine exces among patients with AI. Consecutively, it is recommended that all patients with AI have a screening suppression test with dexamethasone 1 mg overnight to rule out cortisol excess. Serum cortisol levels post dexamethasone 1 mg overnight ≤50nmol/L (≤1.8µg/dL) are a criterion for exclusion of autonomic cortisol secretion (ACS).
The term "subclinical Cushing's syndrome" was used to refere to people who have biochemical indicators of cortisol excess but lacked the so-called "specific" clinical symptoms of the condition, such as central obesity, moon face, myopathy and skin fragility.
Therefore, for patients without obvious clinical signs of Cushing's syndrome but with a serum cortisol level post 1mg dexamethasone overnight > 138nmol/L (> 5µg/dL), the term "autonomic cortisol secretion" (ACS) was proposed in 2016 by the European Society of Endocrinology, replacing the previous term. It is necessary to do additional biochemical tests to confirm the degree of cortisol excess. Urinary free cortisol levels that are higher than the assay's normal range, serum cortisol levels that are higher than 1.8 ug/dl (50 nmol/liter) after taking 1 mg of dexamethasone overnight or after 2 mg/day dexamethasone for 48-hours, and late-night salivary cortisol levels that are higher than 145 ng/dl are all diagnostic indicators of overt Cushing's syndrome, accompanied with classical features of hypercortisolism [26].
The Endocrine Society recommends the use of 2 of 3 highly sensitive screening tests for diagnosing overt Cushing syndrome: 24-hours urine free cortisol (UFC) excretion, late-night salivary cortisol levels and plasma cortisol following an overnight 1 mg dexamethasone suppression test or low-dose dexamethasone test [26]. As it is already known, cases in which an ACTH-independent Cushing's syndrome is attested by an adrenal secreting tumor show suppressed ACTH, suppressed DHEAS, increased 24-hour urinary free cortisol, increased late night cortisol and lack of suppression on dexamethasone 1 mg overnight or dexamethasone 2 mg/2-day tests. Confounding variables in the evaluation of cotisol excess in patients with AI include patient comorbidities causing physiological hypercortisolism and various assays artefacts (radioimmunoassay, enzyme-linked immunosorbent assay, automated chemiluminescence, high-performance liquid chromatography or mass spectrometry), thus making the assessment of excess cortisol very challenging for the clinician [6].
In case of ACS it is important to do the screening for diabetes, hypertension, asymptomatic vertebral fractures, obesity and dyslipidemia [27,28]. In addition, subclinical atherosclerosis, thromboembolic events, infectious diseases and sarcopenia were cited in patients with ACS [28] .
In addition, patients with serum cortisol level after 1mg dexamethasone overnight between 1.9-5 µg/dl are considered to have (possible) "autonomic cortisol secretion" (PACS). Furthermore, all individuals with PACS/ACS should be evaluated for hypertension and type 2 diabetes mellitus [29].
Literature data states that patients with ACS/PACS very rarely develop overt Cushing's syndrome, in about <0.1% cases [30] , which is associated with severe morbidity and increased mortality rates [31,32].
Adrenal incidentalomas rarely have overt hormone excess, however up to 30% of patients may have ACS [6,33]. Thus, all individuals with AI need to be evaluated with 1 mg dexamethasone overnight suppression test since autonomous cortisol release is linked to higher cardiovascular morbidity and metabolic problems [10,33].
Early diagnosis of pheochromocytoma is imperative because if left undiagnosed and untreated, cardiovascular morbidity and mortality rates are high [34].
Therefore, excluding pheochromocytoma by measurement of plasma-free metanephrines or 24 hours urinary fractionated metanephrines are the ESE/ENSAT recommendations for all patients with AI. Although in lipid-rich cortical adenomas, documented on CT scans, some authors postulated that it may not be necessary [31]. When considering whether to undertake biochemical testing for screening pheochromocytoma on a patient with an unenhanced CT attenuation of 10 HU or less, additional CT scan markers to consider include tumor heterogeneity, tumor necrosis and geriatric patients [2,6]. It is noteworthy that there is a risk of false positive results if concomitant medications like sympathomimetic pharmaceuticals or interfering substances are used, for exemple acetaminophen use, antipsychotics, glucocorticoids, tricyclic antidepressants, etc[35] . Additionally, a number of foods should be avoided for 3 to 5 days before the urine collection, including bananas, cheeses, coffee, yogurt, cured meats, soy sauce, red wine, fava beans, and chocolate [29]. When measuring plasmatic free metanephrines, it is preferably using blood samples drawn while the patient is in supine position, after a period of physical rest with concomitant dosing of urinary creatinine [36]. Additionally, the plasmatic dosage of 3-methoxytyramine is advised in suspected dopamine-secreting tumors [37,38,39].
If the initial biochemical evaluation indicates an increase 3-fold or greater than the upper limits of metanephrines, the next clinical step should be imaging studies for tumor localization [40]. To detect metastatic disease, computed tomography is the ideal initial imaging technique, followed by magnetic resonance imaging, and 123I-metaiodobenzylguanidine (MIBG) scintigraphy is advised [41].
A case of AI was reported in an asymptomatic young man in whom further investigation proved that the adrenal tumour was in fact a phaeochromocytoma [42]. Thus, regardless of the non-apparent clinical picture, it is necessary to strictly adhere to the hormonal evaluation protocol.
In addition, it is relatively rare to find primary hyperaldosteronism in an AI's context. As evidence, a recent prospective study was performed to determine the prevalence and clinical characteristics of primary aldosteronism in patients with AI. Of the total 269 participants, only 9 had primary aldosteronism, indicating a prevalence of 3.35 percent in the study group [43]. In patients with concomitant hypertension or unexplained hypokalemia, the ESE/ENSAT guideline recommend the use of the aldosterone/renin ratio collected under standard conditions and continue with confirmatory test to exclude primary aldosteronism [2,44]. Initial evaluation using plasma aldosterone levels, plasma renin activity, or direct renin concentration and use of the aldosterone/renin ratios have a sensitivity of 68 to 94 percent and a negative predictive value that is close to 100 percent [44]. Before testing, serum potassium levels should be returned to normal, and interference-causing medications should be stopped: angiotensin converting enzyme inhibitors, angiotensin receptor blockers, and beta blockers need to be tapered off for 2–4 weeks while all diuretics need to be stopped for 4–6 weeks [29].
Elevated sex steroids or steroid precursors in patients with suggestive clinical or imaging aspects are extremely suspicious for adrenocortical carcinoma and diagnostic evaluation is necessary [2]. Androgen-secreting tumors in women can exhibit signs of virilization such excessive facial hair development, acneic skin, a deeper voice, clitoromegaly, male pattern baldness and primary amenorrhea in adolescent [45]. Women who have estrogen-secreting tumors may experience painful breasts and irregular uterine bleeding. Furthermore, reduced libido, testicular regression, and gynecomastia are three major clinical signs of feminization in males who have estrogen-secreting tumors [29].
In order to avoid an inappropriate adrenalectomy, the diagnosis of congenital adrenal hyperplasia (CAH) must be taken into account in a patient who presents with an AI [46] and especially in bilateral AI, the clinician should measure 17 -hydroxy progesterone level.
Adrenal insufficiency can occur in patients with bilateral adrenal masses, also valid for patients with large bilateral adrenal metastases, so all patients should be tested according to ESE/ENSAT guideline [2,27].
The radiological assessment begins with unenhanced CT, which is used as the initial test, with an attenuation value of percent 10 Hounsfield units (HU) used to distinguish between adenomas and non-adenomas. When CT is unclear, positron emission tomography (PET) or PET/CT should be investigated, although fine needle aspiration biopsy should only be done in specific circumstances where metastasis is suspected (after biochemical exclusion of pheochromocytoma). A benign adrenal adenoma or other benign tumors are consistent with a HU of less than 10 (myelolipomas, lipomas) because these type of benign tumours are rich in lipids [29].
Only 0.5% of pheochromocytomas have an unenhanced CT attenuation of 10 HU, with mean HU scores for pheochromocytomas ranging from 30 to 35 [6,34,47]. Consequently, some authors came to the conclusion that it is fair to postpone biochemical testing for pheochromocytoma in cases of AI with an unenhanced attenuation value less than 10 HU [47].
Furthermore, positron emission tomography with 18F-2-deoxy-d-glucose (mostly combined with CT; FDGPET/CT) is mainly used for the detection of malignant disease, whereas CT and MRI are techniques mainly aiming to identify benign lesions [2,48]. Furthermore, it's essential to measure the washout time during an enhanced CT scan when a suspected case is present. A wash-out > 60% in the arterial phase or a relative washout > 40% in the delayed pictures (15 min for the venous phase) suggest a benign form with a sensitivity > 82% and specificity > 92% [29].
A tertiary care medical facility conducted a 15-year retrospective analysis in which 244 patients with AI were discovered. The results showed that mass tumour size >4 cm and HU less than 10 are important radiological predictors for functional AI, together with hypertension as a key clinical feature [49].
In addition, according to the European guidelines, in case of a CT scan with benign appearance (HU≤10), with homogeneous mass lesser than 4cm, no further imaging is required because it is most likely a benign lipid-rich adrenal adenoma.In contrast, for an indeterminate non-contrast, non-functional adrenal mass on CT, treatment options should be considered by a multidisciplinary team and adrenal imaging should be repeated in 6-12 months if after the initial assessment the option is against surgical therapy[2].
Differentiating between benign and malignant adrenocortical tumors is the main problem for clinicians. In 5 to 8 percent of individuals with AI, malignancy is identified; the risk is higher in young patients, in those with a history of extra-adrenal malignancy, for patients with large adrenal tumours, with an indefinite radiological aspect and for patients with bilateral adrenal tumors [33,50,51]. Additionally, a clinical picture suggestive of malignancy, such as rapid weight loss despite appetite, should be a cause for concern [29]. Moreover, authors stated that there is a link between tumor size and the likelihood of developing adrenocortical cancer: AIs under 4 cm have a 2% risk, those between 4.1 and 6 cm have a 6% risk, and those over 6 cm have a 25% risk [52]. Thus, a large diameter (>6 cm), discontinuous border, inhomogeneity, high unenhanced CT attenuation values (>20 HU), "washout" of contrast after 15 min of less than 40%, and calcifications on CT scan can indicate the presence of adrenal cancer [5,53]. Additionally, in the study of 4085 patients with adrenal tumors of which 13% were found to have adrenocortical carcinomas, in multivariate analysis, the following factors were found to be statistically significant predictors of malignancy: older age at diagnosis, male gender, incidental mode of discovery, larger tumour size and an attenuation ≥10 Hounsfield units on CT imaging [54].
A diagnostic CT guided fine-needle aspiration (FNA) biopsy may be necessary in a patient with a newly diagnosed adrenal mass who has a known primary malignancy elsewhere and imaging phenotypes compatible with metastatic illness, but only after ruling out pheochromocytoma with biochemical tests. If the patient is already known to have widespread metastatic illness, an adrenal biopsy is not necessary [55].
3.4. Management and Follow up of AI
According on the type of the tumour, accompanying comorbidities, and patient desire, several approaches are taken into considerations for the management of AI.
Adrenal gland surgery is complex. According to literature data, only referral institutions and surgeons with experience in open and laparoscopic adrenal surgery and who perform more than 15 benign and malignant adrenalectomies annually should perform surgery in patients with adrenal pathology, for good clinical management and a favorable outcome and prognosis [56,57].
Surgical approach is the gold standard therapy for the following adrenal tumour types: hyperfunctional adrenal tumours, adrenal tumours suspected of malignancy and indeterminate adrenal masses. Laparoscopic adrenalectomy is recommended in patients with unilateral adrenal masses with benign imaging appearance or in adrenal masses with radiological features suspicious of malignancy and diameter ≤6cm, but without imaging evidence of local invasion. Open adrenalectomy is usually performed for unilateral adrenal masses with radiological aspects suspicious of malignancy and with imaging evidence of local invasion [2,48].
Additionaly, for patients with ACS who also have comorbidities that are potentially related to cortisol excess, surgical treatment should be considered in an individualized approach [6]. Annual clinical evaluations for up to 5 years, are advised as well as ongoing, customized risk assessments for possible surgical intervention for patients with ACS [58].
Current guidelines recommendations postulated that is desirable to avoid doing surgery on patients who have a unilateral non-functional AI that is asymptomatic and clearly benign on imaging tests [2]. Furthermore, publications claim that patients with bilateral benign AIs have a higher frequency of subclinical hypercortisolemia than patients with unilateral tumors. It is currently unclear if surgery is necessary for all individuals with bilateral adrenal tumors with ACS and, if so, which tumor needs to be taken out first [59].
Regarding preoperative management, in case of PACS/ACS patients, perioperative glucocorticoid treatment at major surgical stress doses are recommended by guidelines [2].
If the initial radiological and hormonal evaluations decide on IA monitoring, this is done periodically every 6-12 months or customised according to the tumour type. If during tumour imaging monitoring, an increase in size (by more than 20% along with at least a 5 mm increase in maximum diameter) is detected or signs and/or symptoms of hormonal excess appear, surgery may be recommended. Additional imaging after other 6–12 months should be carried out if the lesion grows below this threshold [58]. Similarly, congruent with ESE/ENSAT guideline recomandations if a patient's initial hormonal work-up is normal, it should not be repeated until there are new clinical markers of endocrine activity or comorbidities worsens [2].
In the particular case of bilateral AI, the recomandations of endocrine work-up are similar to ones of unilateral AI and the evaluation of comorbidities that could be associated to ACS follows the same pattern [27,29].
Furthermore, due to an increased risk of adrenocortical neoplasia in case of diagnosing of an AI, particular age groups such as young adults (below the age of forty), children and pregnant women are indicated to have an urgent assessment of an AI, with the predilect use of MRI rather than CT scan [48].
The natural evolution of an AI was studied in a meta-analysis ant the results concluded that neither non-functional AI, neither adenomas causing mild autonomous cortisol excess do not exhibit clinically significant changes in size or in hormonal function over follow-up. In addition, the same authors stated that the monitorised patients had a low probability (0.1%) of developing a clinically obvious hormone excess (Cushing's syndrome) [30].
3.5. Clinical Implications of AI
The medical literature abounds with numerous studies conducted to evaluate the hypothesis that non-functioning AI increase the risk of cardiovascular events and have significant metabolic consequences compared to the absence of adrenal tumors. In that direction, results from a study published in 2019 indicate that vascular changes precede the development of cardiovascular disease and may increase morbidity and mortality in patients with AI. In addition, the same authors conclude that apparently hormonally inactive adrenal tumors may indeed produce small amounts of glucocorticoids that have metabolic and cardiovascular implications [10].
Concurrent, clinical consequences of both PACS/ACS involves effects especially on the cardiovascular system, metabolism and bone architecture [32].
In ACS the pathogenesis of cardiometabolic events appears to be the consequence of hemodynamic changes, vascular inflammatory pathways initiated and maintained by hypercortisolic status, pancreatic β-cell dysfunction, insulin resistance and visceral obesity [28,32].
There is evidence that cortisol excess in these specific endocrine disorders might be associated with comorbidities such as glucose intolerance or diabetes, hypertension, osteoporosis, obesity and dyslipidemia [2].
Furthermore, hypertension, coronary heart disease, stroke, increased left ventricular hypertrophy, subclinical atherosclerosis and arterial stiffness, and fatal or nonfatal myocardial infarction are the cardiovascular morbidities that are cited in association with cortisol excess [58,60].
Some authors postulated that in comparing with patients with non-functional AI, those with ACS had a prevalence of cardiovascular events that was more than three times higher. Similarly, it was discovered that ACS was linked to greater rates of diabetes and hypertension [61].
Increased cardiovascular morbidity (43 percent vs 8.8 percent, P .005) and mortality (22.6 percent vs 2.5 percent, P .02) in ACS patients compared to those with nonfunctioning AI have been described, with cardiovascular events and pulmonary infections as the two leading causes of death [6,61].
It is widely known that diabetes mellitus and glucose intolerance are frequent symptoms of overt Cushing's syndrome. Furthermore, improved glycemic control (and, in some cases, diabetes reversal) following surgery has been reported to occur in 10% to 69 percent of patients with ACS who have impaired glucose tolerance or diabetes [62,63].
It is likely that patients with AI have diminished bone mass due to PACS/ACS as osteoporosis is a well-known side effect of endogenous and exogenous glucocorticoid excess. In terms of bone fracture, a meta-analysis revealed that individuals with ACS had a 63.6 percent prevalence of vertebral fracture [64]. Contrary, other authors concluded that the slight glucocorticoid excess associated with AI does not increase the risk of osteoporosis [65]. Additional factors including decreased androgens and sarcopenia appear to increase the risk of fractures but further studies are needed to elucidate the exact correlations [28].
There have been reports of metabolic improvement following adrenalectomy, including weight loss, reduced blood pressure, improved glucose tolerance and decreased cholesterol levels [66]. Moreover, adrenalectomy improved cardiovascular prognosis and death in ACS patients, according to a study [61].
A personalized strategy must take into account the level of hypercortisolism within the ACS definition, the severity of the clinical characteristics described previously, and the risks of surgery, especially in an elderly patient [6].
Furthermore, in the clinical picture of ACS, thromboembolic events and infectious diseases were also described, through direct and indirect effects of cortisol. Moreover, a considerable clinical impact appears to be played by altered circadian cortisol rhythm and co-secretion of aldosterone and steroid precursors cited in ACS [28].
3.6. Particular Diagnostic Markers in AI
Adrenal tumors have the ability to secrete hormones and induce clinical syndromes. Although most AI are clinically quiet, some could be functional in terms of cosecretion of different biochemical substances. The non-specific marker of neuroendocrine tumors is chromogranin A (CgA). Chromaffin cells in the adrenal medulla produce CgA. Studies on the plasma levels of CgA in adrenocortical adenomas have postulated inconsistent results. Because it is not expressed at the immuno-histochemical level and has not been related to noticeably increased plasma levels associated with adrenocortical tumors, CgA does not appear to be involved in cortical carcinogenesis, according to multiple studies. Other researchers, however, did not discover any connection between adrenocortical adenomas and high serum CgA concentrations, even when the tumors were stained with the protein using immunohistochemistry. On contrary, some studies suggested that cortisol-secreting adenomas had higher blood CgA values. Some authors postulated that high CgA levels are not a reliable predictor of AI's risk for malignancy [38,67,68,69,70].
In addition, in the study of steroid metabolome analysis, the patients with primary aldosteronism, have proven to have a glucocorticoid excess, using mass spectrometry to examine the 24-hour urinary output of steroid metabolites. The cosecretion was demonstrated biochemically by the rise in various glucocorticoid precursor levels in urine samples and clinically by the onset of adrenal insufficiency following surgery [71]. Particularly, the cosecretion of adrenal androgens and glucocorticoid metabolites was cited as developing into a sensitive diagnostic tool for adrenocortical carcinoma [6].
4. Discussion
The evaluation and management of adrenal incidentalomas (AIs) remain a dynamic field, shaped by evolving diagnostic tools, improved imaging resolution, and a deeper understanding of subclinical hormone excess. Our synthesis of the literature reinforces the pivotal role of a combined biochemical and radiological assessment in determining both functional status and malignancy risk. This aligns with established guidelines [2,4,5], which recommend a systematic approach to exclude hormonally active tumors and characterize lesion morphology.
One of the most significant findings across studies is the prevalence and clinical impact of autonomous cortisol secretion (ACS), even in the absence of overt Cushing’s syndrome. Multiple reports [10,11,12,13] confirm that mild cortisol excess is associated with increased cardiometabolic morbidity, including hypertension, impaired glucose tolerance, and osteoporosis. While adrenalectomy can reverse some of these effects, the decision to operate in ACS remains controversial, given the variable risk-benefit profile and the absence of universal surgical criteria. This underscores the need for individualized patient management, integrating hormonal activity, comorbidities, and patient preferences.
Radiological evaluation—particularly the use of unenhanced CT attenuation values, washout characteristics, and size thresholds—remains a cornerstone in differentiating benign from malignant lesions [5,8,9]. However, false positives and negatives occur, particularly in lipid-poor adenomas and atypical metastases, highlighting the potential utility of advanced imaging modalities such as chemical-shift MRI and PET tracers targeting steroidogenesis. Future prospective studies are needed to refine risk stratification tools, potentially incorporating radiomics and artificial intelligence–driven image analysis.
Our review also highlights the unique challenges posed by bilateral AIs, which may reflect bilateral adenomas, hyperplasia, or metastatic disease. The biochemical complexity of these cases, particularly in the setting of mild cortisol excess or concomitant aldosteronism, requires a multidisciplinary approach and often long-term follow-up.
In the broader context, the incidental detection of adrenal lesions is expected to increase further with the rising use of cross-sectional imaging for unrelated conditions. This trend magnifies the clinical importance of developing clear, evidence-based algorithms to avoid both under-treatment of clinically significant lesions and over-investigation of benign findings. Future research should focus on multicenter, longitudinal studies to determine the long-term cardiovascular and metabolic outcomes of various management strategies, especially in ACS.
Overall, while the diagnostic framework for AIs is well-established, important uncertainties persist regarding surgical thresholds, follow-up intervals, and optimal care pathways for hormonally inactive lesions. Advances in molecular profiling, imaging biomarkers, and integrated risk scoring systems may help resolve these debates and enable more personalized management.
5. Conclusions
Over the past few years, there has been an increase in the literature on adrenal incidentaloma. Formulating exact diagnostic and therapy procedures is unfortunately challenging given the paucity of controlled studies. Every patient with AI should be evaluated for possible hormonal excess, along with clinical examination focusing on suggestive signs and symptoms. In most cases, AI are non-functioning adrenocortical adenomas, with a higher prevalence in elderly patients. Autonomic cortisol secretion is the most common cause of hormonal activity in AI, causing numerous clinical implications.
Pheochromocytoma and cortisol hypersecretion should be excluded in all patients diagnosed with AI, whereas primary aldosteronism should be evaluated in hypertensive and/or hypokalemic patients.
For unilateral adrenal tumors with clinically substantial hormone excess and adrenocortical carcinoma, adrenalectomy is the gold standard of therapy.
Incidental adrenalomas are extremely rare in the paediatric population and should be evaluated urgently because of the high malignant risk.
A multidisciplinary team approach combining doctors with experience in endocrinology, imaging, surgery, and pathology is necessary to optimise the therapy of patients with adrenal incidentalomas due to the complexity of the condition. A personalised strategy based on image analysis, endocrine workup, and clinical assessment is necessary for adequate clinical management.
Author Contributions
Conceptualization, A.M. and D-M.T.; methodology, A.M. and D-M.T.; software, D.L.P.; validation, S.C.P., A.M.A., and V.N.; formal analysis, D.L.P. and S.C.P.; investigation, A.M., D-M.T., and A.M.A.; resources, V.N. and S.C.P.; data curation, A.M.A. and V.N.; writing—original draft preparation, A.M., D-M.T., and D.L.P.; writing—review and editing, S.C.P., A.M.A., and V.N.; visualization, D.L.P.; supervision, A.M. and D-M.T.; project administration, A.M. and D-M.T. All authors contributed equally to this work and have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This article is a review and does not report or generate any new research data. Therefore, no new datasets were created or analyzed in this study. All data referenced in this review are available from the original sources cited throughout the article.
Acknowledgments
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AI | Adrenal Incidentaloma |
| ACS | autonomous cortisol secretion |
| CT | computed tomography |
| MRI | Magnetic resonance imaging |
| UFC | 24-hours urine free cortisol |
| PET | positron emission tomography |
| FNA | fine-needle aspiration |
| HU | Hounsfield units |
References
- Terzolo, M.; Stigliano, A.; Chiodini, I.; Loli, P.; Furlani, L.; Arnaldi, G.; et al. AME position statement on adrenal incidentaloma. Eur J Endocrinol 2011, 164, 851–70. [Google Scholar] [CrossRef]
- Martin Fassnacht Wiebke Arlt Irina Bancos, H.D.; John Newell-Price Anju Sahdev Antoine Tabarin, M.T.; Dekkers Stylianos Tsagarakis, O.M. European Society of Endocrinology Clinical Practice Guideline in collaboration with the European Network for the Study of Adrenal 2016.
- Herrera, M.F.; Grant, C.S.; van Heerden, J.A.; Sheedy, P.F.I.D. Incidentally discovered adrenal tumors: an institutional perspective. Surgery (United States) 1991, 110, 101. [Google Scholar]
- Bovio, S.; Cataldi, A.; Reimondo, G.; Sperone, P.; Novello, S.; Berruti, A.; Borasio, P.; Fava, C.; Dogliotti, L.; Scagliotti, G.V.; Angeli, A.T.M. Prevalence of adrenal incidentaloma in a contemporary computerized tomography series. J Endocrinol Invest 2006, 29, 298. [Google Scholar] [CrossRef] [PubMed]
- Lynnette, K. Nieman. Approach to the Patient with an Adrenal Incidentaloma. J CLIN ENDOCRINOL METAB 2010, 4106–13. [Google Scholar]
- Mark Sherlock Andrew Scarsbrook Afroze Abbas Sheila Fraser Padiporn Limumpornpetch Rosemary Dineen, P. S. Adrenal Incidentaloma. Endocr Rev 2020, 775–820. [CrossRef]
- Becker, J.; Woloszyn, J.; Bold, R.; Campbell, M.J. The Adrenal Incidentaloma: An Opportunity to Improve Patient Care. J Gen Intern Med 2018, 33, 256–7. [Google Scholar] [CrossRef] [PubMed]
- Müller, A.; Ingargiola, E.; Solitro, F.; Bollito, E.; Puglisi, S.; Terzolo, M.; et al. May an adrenal incidentaloma change its nature? J Endocrinol Invest 2020, 43, 1301–7. [Google Scholar] [CrossRef]
- Giorgio Arnaldi, M.B. Adrenal incidentaloma. Best Pract Res Clin Endocrinol Metab 2012, 405–419. [Google Scholar] [CrossRef]
- Szychlińska, M.; Baranowska-Jurkun, A.; Matuszewski, W.; Wołos-Kłosowicz, K.; Bandurska-Stankiewicz, E. Markers of subclinical cardiovascular disease in patients with adrenal incidentaloma. Medicina (Lithuania) 2020, 56, 69. [Google Scholar] [CrossRef] [PubMed]
- Gicquel, C.; Bertherat, J.; Le Bouc, Y.; Bertagna, X. Pathogenesis of adrenocortical incidentalomas and genetic syndromes associated with adrenocortical neoplasms. Endocrinol Metab Clin North Am 2000, 29, 1–13. [Google Scholar] [CrossRef]
- Espiard, S.; Bertherat, J. The Genetics of Adrenocortical Tumors. Endocrinol Metab Clin North Am 2015, 44, 311–34. [Google Scholar] [CrossRef]
- Faillot, S.; Assie, G. The genomics of adrenocortical tumors. Eur J Endocrinol 2016, 174, R249–65. [Google Scholar] [CrossRef]
- Assié, G.; Jouinot, A.; Bertherat, J. The “omics” of adrenocortical tumours for personalized medicine. Nat Rev Endocrinol 2014, 10, 215–28. [Google Scholar] [CrossRef]
- Powell, A.C.; Libutti, S.K. Multiple endocrine neoplasia type 1: Clinical manifestations and management. Cancer Treat Res 2010, 153, 287–302. [Google Scholar] [CrossRef]
- Agarwal, S.K. Multiple endocrine neoplasia type 1. Front Horm Res 2013, 41, 1–15. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Stratakis, C.A. Multiple endocrine neoplasia type 1 (MEN1): An update and the significance of early genetic and clinical diagnosis. Front Endocrinol (Lausanne) 2019, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Thakker, R.V.; Newey, P.J.; Walls, G.V.; Bilezikian, J.; Dralle, H.; Ebeling, P.R.; et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). Journal of Clinical Endocrinology and Metabolism 2012, 97, 2990–3011. [Google Scholar] [CrossRef] [PubMed]
- Rami Alrezk, Fady Hannah-Shmouni CA, Stratakis. MEN4 and CDKN1B mutations: The latest of the MEN syndromes. Endocr Relat Cancer 2017, T195–T208. [CrossRef]
- El-Maouche, D.; Welch, J.; Agarwal, S.K.; Weinstein, L.S.; Simonds, W.F.M.S. A patient with MEN1 typical features and MEN2-like features. Int J Endocr Oncol 2016, 89–95. [Google Scholar] [CrossRef]
- Ekinci, F.; Soyaltin, U.E.; Kutbay, Y.B.; Yaşar, H.Y.; Demirci Yıldırım, T.; Akar, H. Jak2 v617f mutation scanning in patients with adrenal incidentaloma. Acta Endocrinol (Copenh) 2017, 13, 150–3. [Google Scholar] [CrossRef] [PubMed]
- Eker, F.; Gungunes, A.; Durmaz, S.; Kisa, U.; Celik, Z.R. Nonfunctional adrenal incidentalomas may be related to bisphenol-A. Endocrine 2021, 71, 459–66. [Google Scholar] [CrossRef]
- Mansmann, G.; Lau, J.; Balk, E.; Rothberg, M.; Miyachi, Y.; Bornstein, S.R. The clinically inapparent adrenal mass: Update in diagnosis and management. Endocr Rev 2004, 25, 309–40. [Google Scholar] [CrossRef]
- de Risi-Pugliese, T.; Genc, S.; Bertherat, J.; Larousserie, F.; Bollet, M.; Bassi, C.; etal. Classic Kaposi Sarcoma: An exceptional cause of adrenal incidentaloma. J Endocr Soc 2017, 1, 737–41. [Google Scholar] [CrossRef]
- Kedia, R.R.; Muinov, L.; Lele, S.M.; Shivaswamy, V. Adrenal oncoctyoma of uncertain malignant potential: A rare etiology of adrenal incidentaloma. Clin Case Rep 2016, 4, 303–4. [Google Scholar] [CrossRef]
- Nieman, L.K.; Biller, B.M.K.; Findling, J.W.; Newell-Price, J.; Savage, M.O.; Stewart, P.M.; et al. The diagnosis of Cushing’s syndrome: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 2008, 93, 1526–40. [Google Scholar] [CrossRef]
- Lee, J.M.; Kim, M.K.; Ko, S.H.; Koh, J.M.; Kim, B.Y.; Kim, S.W.; et al. Clinical guidelines for the management of adrenal incidentaloma. Endocrinology and Metabolism 2017, 32, 200–18. [Google Scholar] [CrossRef]
- Di Dalmazi, G. Adrenal Incidentaloma: Picking out the High-Risk Patients. Experimental and Clinical Endocrinology and Diabetes 2019, 127, 178–84. [Google Scholar] [CrossRef]
- Ceccato, F.; Barbot, M.; Scaroni, C.; Boscaro, M. Frequently asked questions and answers (if any) in patients with adrenal incidentaloma. J Endocrinol Invest 2021, 44, 2749–63. [Google Scholar] [CrossRef]
- Yasir, S. Elhassan, Fares Alahdab, Alessandro Prete, Danae A. Delivanis, Aakanksha Khanna, Larry Prokop, Mohammad H. Murad, Michael W. O’ReillyWiebke Arlt IB. Natural History of Adrenal Incidentalomas With and Without Mild Autonomous Cortisol Excess. Ann Intern Med 2019. [Google Scholar] [CrossRef]
- Cawood, T.J.; Hunt, P.J.; O’Shea, D.; Cole, D.; Soule, S. Recommended evaluation of adrenal incidentalomas is costly, has high false-positive rates and confers a risk of fatal cancer that is similar to the risk of the adrenal lesion becoming malignant; time for a rethink? Eur J Endocrinol 2009, 161, 513–27. [Google Scholar] [CrossRef]
- Debono, M.; Bradburn, M.; Bull, M.; Harrison, B.; Ross, R.J.; Newell-Price, J. Cortisol as a marker for increased mortality in patients with incidental adrenocortical adenomas. Journal of Clinical Endocrinology and Metabolism 2014, 99, 4462–70. [Google Scholar] [CrossRef]
- Irina Bancos, A.P. Approach to the Patient With Adrenal Incidentaloma. J Clin Endocrinol Metab 2021, 106, 3331–3353. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Eisenhofer, G. Update on Modern Management of Pheochromocytoma and Paraganglioma. Endocrinology and Metabolism 2017, 32, 152. [Google Scholar] [CrossRef]
- Mirică AIBet, a.l. Current approaches and recent developments in the clinical management of catecholamine-producing neuroendocrine tumors. Curr Health Sci J 2016, 42, 9–14. [Google Scholar]
- Farrugia, F.A.; charalampopoulos, A. Pheochromocytoma. Endocr Regul 2019, 53, 191–212. [Google Scholar] [CrossRef] [PubMed]
- Paun, D.L.; Mirica, A. Pheochromocytoma a focus on genetic. 2016.
- Păun, D.L.; Mirica, A. Pheochromocytomas and Paragangliomas: Genotype-Phenotype Correlations. IntechOpen, 2021.
- Rao, D.; Peitzsch, M.; Prejbisz, A.; Hanus, K.; Fassnacht, M.; Beuschlein, F.; et al. Plasma methoxytyramine: Clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol 2017, 177, 103–13. [Google Scholar] [CrossRef] [PubMed]
- Lenders, J.W.M.; Duh, Q.Y.; Eisenhofer, G.; Gimenez-Roqueplo, A.P.; Grebe, S.K.G.; Murad, M.H.; et al. Pheochromocytoma and paraganglioma: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 2014, 99, 1915–42. [Google Scholar] [CrossRef] [PubMed]
- Adapa, S.; Naramala, S.; Gayam, V.; Gavini, F.; Dhingra, H.; Hazard, F.K.G.; et al. Adrenal Incidentaloma: Challenges in Diagnosing Adrenal Myelolipoma. J Investig Med High Impact Case Rep 2019, 7, 0–3. [Google Scholar] [CrossRef]
- Vurallı, D.; Kandemir, N.; Clark, G.; Orhan, D.; Alikaşifoğlu, A.; Gönç, N.; et al. A pheochromocytoma case diagnosed as adrenal incidentaloma. Turkish Journal of Pediatrics 2017, 59, 200–6. [Google Scholar] [CrossRef]
- Stavropoulos, K.; Imprialos, K.P.; Katsiki, N.; Petidis, K.; Kamparoudis, A.; Petras, P.; et al. Primary aldosteronism in patients with adrenal incidentaloma: Is screening appropriate for everyone? J Clin Hypertens 2018, 20, 942–8. [Google Scholar] [CrossRef]
- Funder, J.W.; Carey, R.M.; Mantero, F.; Murad, M.H.; Reincke, M.; Shibata, H.; et al. The management of primary aldosteronism: Case detection, diagnosis, and treatment: An endocrine society clinical practice guideline. Journal of Clinical Endocrinology and Metabolism 2016, 101, 1889–916. [Google Scholar] [CrossRef]
- Rodríguez-Gutiérrez, R.; Bautista-Medina, M.A.; Teniente-Sanchez, A.E.; Zapata-Rivera, M.A.; Montes-Villarreal, J. Pure Androgen-Secreting Adrenal Adenoma Associated with Resistant Hypertension. Case Rep Endocrinol 2013, 2013, 1–4. [Google Scholar] [CrossRef]
- Buitenwerf, E.; Links, T.P.; Kema, I.P.; Haadsma, M.L.; Kerstens, M.N. Congenital adrenal hyperplasia as a cause of adrenal incidentaloma. Netherlands Journal of Medicine 2017, 75, 298–300. [Google Scholar]
- Azoury Saïd, Nagarajan Neeraja; Youn Allen, Mathur Aarti; Prescott Jason; Fishma Elliot ZM. Computed Tomography in the Management of Adrenal Tumors: Does Size Still Matter? J Comput Assist Tomogr 2017, 41, 628–32. [CrossRef] [PubMed]
- Pogorzelski, R.; Celejewski, K.; Toutounchi, S.; Krajewska, E.; Woloszko, T.; Szostek, M.; et al. Adrenal incidentaloma - diagnostic and treating problem - own experience. Open Medicine 2018, 13, 281–4. [Google Scholar] [CrossRef] [PubMed]
- Muangnoo, N.; Manosroi, W.; Leelathanapipat, N.; Meejun, T.; Chowchaiyaporn, P.; Teetipsatit, P. Predictive Factors of Functioning Adrenal Incidentaloma: A 15-Year Retrospective Study. Medicina (Lithuania) 2022, 58, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mirică Aet, a.l. A rare case of metastasized non-functional pancreatic neuroendocrine tumor with a good long-term survival. J Med Life 2016, 9, 369–72. [Google Scholar] [CrossRef]
- Dinnes, J.; Bancos, I.; Di Ruffano, L.F.; Chortis, V.; Davenport, C.; Bayliss, S.; et al. Management of endocrine disease: Imaging for the diagnosis of malignancy in incidentally discovered adrenal masses: A systematic review and meta-analysis. Eur J Endocrinol 2016, 175, R51–64. [Google Scholar] [CrossRef] [PubMed]
- Sherlock, M.; Scarsbrook, A.; Abbas, A.; Fraser, S.; Limumpornpetch, P.; Dineen, R.; et al. Adrenal incidentaloma. vol. 41. 2020. [CrossRef]
- Ye, Y.L.; Yuan, X.X.; Chen, M.K.; Dai, Y.P.; Qin, Z.K.; Zheng, F.F. Management of adrenal incidentaloma: The role of adrenalectomy may be underestimated. BMC Surg 2016, 16, 1–6. [Google Scholar] [CrossRef]
- Iñiguez-Ariza, N.M.; Kohlenberg, J.D.; Delivanis, D.A.; Hartman, R.P.; Dean, D.S.; Thomas, M.A.; et al. Clinical, Biochemical, and Radiological Characteristics of a Single-Center Retrospective Cohort of 705 Large Adrenal Tumors. Mayo Clin Proc Innov Qual Outcomes 2018, 2, 30–9. [Google Scholar] [CrossRef]
- Plouin, P.F.; Amar, L.; Dekkers, O.M.; Fassnach, M.; Gimenez-Roqueplo, A.P.; Lenders, J.W.M.; et al. European Society of Endocrinology Clinical Practice Guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol 2016, 174, G1–10. [Google Scholar] [CrossRef]
- Gaujoux, S.; Mihai, R.; Carnaille, B.; Dousset, B.; Fiori, C.; Porpiglia, F.; et al. European Society of Endocrine Surgeons (ESES) and European Network for the Study of Adrenal Tumours (ENSAT) recommendations for the surgical management of adrenocortical carcinoma. British Journal of Surgery 2017, 104, 358–76. [Google Scholar] [CrossRef]
- Mirica, R.M.; Paun, S. Surgical Approach in Pheochromocytoma. IntechOpen, 2021. [CrossRef]
- Morelli, V.; Reimondo, G.; Giordano, R.; Della Casa, S.; Policola, C.; Palmieri, S.; et al. Long-term follow-up in adrenal incidentalomas: An Italian multicenter study. Journal of Clinical Endocrinology and Metabolism 2014, 99, 827–34. [Google Scholar] [CrossRef] [PubMed]
- L. P, J. C, M. R, P. G, M. O, A. K-Z. Bilateral adrenal incidentaloma with subclinical hypercortisolemia: Indications for surgery. Pol Arch Med Wewn 2014, 124, 387–94. [CrossRef]
- Yener, S.; Ertilav, S.; Secil, M.; Akinci, B.; Demir, T.; Kebapcilar, L.; et al. Increased risk of unfavorable metabolic outcome during short-term follow-up in subjects with nonfunctioning adrenal adenomas. Medical Principles and Practice 2012, 21, 429–34. [Google Scholar] [CrossRef]
- Park, J.; De Luca, A.; Dutton, H.; Malcolm, J.C.; Doyle, M.A. Cardiovascular outcomes in autonomous cortisol secretion and nonfunctioning adrenal adenoma: A systematic review. J Endocr Soc 2019, 3, 996–1008. [Google Scholar] [CrossRef]
- Di Dalmazi, G. Update on the risks of benign adrenocortical incidentalomas. Curr Opin Endocrinol Diabetes Obes 2017, 24, 193–9. [Google Scholar] [CrossRef] [PubMed]
- Giordano, R.; Guaraldi, F.; Berardelli, R.; Karamouzis, I.; D’Angelo, V.; Marinazzo, E.; et al. Glucose metabolism in patients with subclinical Cushing’s syndrome. Endocrine 2012, 41, 415–23. [Google Scholar] [CrossRef]
- Chiodini, I.; Vainicher, C.E.; Morelli, V.; Palmieri, S.; Cairoli, E.; Salcuni, A.S.; et al. Endogenous subclinical hypercortisolism and bone: A clinical review. Eur J Endocrinol 2016, 175, R265–82. [Google Scholar] [CrossRef] [PubMed]
- Osella, G.; Reimondo, G.; Peretti, P.; Alì, A.; Paccotti, P.; Angeli, A.; et al. The patients with incidentally discovered adrenal adenoma (incidentaloma) are not at increased risk of osteoporosis. Journal of Clinical Endocrinology and Metabolism 2001, 86, 604–7. [Google Scholar] [CrossRef]
- Chiodini, I.; Morelli, V.; Salcuni, A.S.; Eller-Vainicher, C.; Torlontano, M.; Coletti, F.; et al. Beneficial metabolic effects of prompt surgical treatment in patients with an adrenal incidentaloma causing biochemical hypercortisolism. Journal of Clinical Endocrinology and Metabolism 2010, 95, 2736–45. [Google Scholar] [CrossRef]
- Mirica, A.; Badarau, I.A.; Stefanescu, A.M.; Mirica, R.; Paun, S.; Andrada, D.; et al. The Role of Chromogranin A in Adrenal Tumors. REVCHIM(Bucharest) 2018, 69, 34–6. [Google Scholar] [CrossRef]
- Mirica A BI et al. Clinical use of plasma chromogranin A in neuroendocrine tumors. Current Health Science Journal 2015, 41, 69–76.
- Glinicki Piotr Wojciech Jeske, L.B.; KasperlikZałuska, A.; Elżbieta Rosłonowska, M.G.; Zgliczyński, W. Chromogranin A (CgA) in adrenal tumours. Endokrynol Pol 2013, 64, 358–362. [Google Scholar] [CrossRef] [PubMed]
- Bernini, G.; Moretti, A.; Fontana, V.; Orlandini, C.; Miccoli, P.; Berti, P.; et al. Plasma chromogranin A in incidental non-functioning, benign, solid adrenocortical tumors. Eur J Endocrinol 2004, 151, 215–22. [Google Scholar] [CrossRef] [PubMed]
- Arlt, W.; Lang, K.; Sitch, A.J.; Dietz, A.S.; Rhayem, Y.; Bancos, I.; et al. Steroid metabolome analysis reveals prevalent glucocorticoid excess in primary aldosteronism. JCI Insight 2017, 2, 1–14. [Google Scholar] [CrossRef]

Table 1.
Functional and non-functional adrenal masses.
| Adrenal masses associated with hormonal activity | Adrenal masses whitout hormonal secretion |
|---|---|
| Adrenal Adenoma –cortisol/aldosterone secretion | Lymphoma |
| Pheochromocytoma | Metastases |
| Primary bilateral macronodular adrenal hyperplasia | Myelolipoma |
| Nodular variant of Cushing’s disease | Neuroblastoma |
| Congenital adrenal hyperplasia | Hemangioma |
| Adrenal carcinoma | Cyst |
| Adrenal masses associated with hormonal activity | Hemorrhage |
| Granuloma | |
| Amyloidosis | |
| Ganglioneuroma | |
| Infiltrative disease |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



