Submitted:
31 July 2025
Posted:
31 July 2025
You are already at the latest version
Abstract
AMOG/β2, the β2 isoform of the sodium pump (Na⁺,K⁺-ATPase), functions as an adhesion
molecule on glial cells, mediating critical neuron–astrocyte interactions during central
nervous system (CNS) development. Despite its established role in glial adhesion, the
neuronal receptor that partners with AMOG/β2 remains unknown. This review examines
the structural and functional properties of AMOG/β2 including its capacity to form trans
dimers, both homophilic and potentially heterophilic—drawing comparisons with the β1
subunit, a well-characterized adhesion molecule. By integrating computational modeling,
in vitro data, and structural predictions, we explore how factors such as N-glycosylation
and cis-membrane interactions influence β2-mediated adhesion. We further consider candidate neuronal partners, including TSPAN31 and RTN4, and speculate on their potential
roles in mediating heterophilic AMOG/β2 interactions. Finally, we discuss the broader implications of AMOG/β2 in neuron-glia communication, synaptic organization, neurodevelopment, and CNS disorders such as glioblastoma. Identifying the binding partner of
AMOG/β2 holds promise not only for understanding the molecular basis of CNS adhesion
but also for uncovering novel mechanisms of neuroglial regulation in health and disease.
Keywords:
AMOG/β2
; Na/K‐ATPase
; CAMs
; neuro‐glia interaction
; glioblastoma
1. Introduction
The central nervous system (CNS), comprising the brain and spinal cord, is crucial for processing information and regulating bodily functions. Interactions among neurons, glial cells (astrocytes, oligodendrocytes, and microglia), and endothelial cells of the blood-brain barrier are essential for CNS structure and function. Neurons communicate through synapses, transmitting electrical signals critical for brain activity, learning, and memory [1]. Glial cells, particularly astrocytes, regulate neurotransmitter levels, maintain the blood-brain barrier, and support neuronal metabolism [2].
During neurogenesis, glial cells provide scaffolding for neuronal migration and growth cones, offering guidance cues and potentially aiding neuronal proliferation [3]. Oligodendrocytes are responsible for myelination, enhancing nerve conduction speeds [4], while microglia act as immune cells, protecting the CNS from pathogens and clearing debris [5]. Cell-cell interactions are vital for synaptic plasticity, which underlies learning and memory [6,7]. These interactions are also critical for maintaining ionic balance, nutrient supply, and waste removal, essential for CNS homeostasis [8,9]. This complex network of interconnected cells relies on the expression of selective molecules known as cell adhesion molecules (CAMs), which mediate various forms of contact between neural cell surfaces. Notable families of CAMs include immunoglobulin superfamily cell adhesion molecules (IgCAMs), cadherins, integrins, and C-type lectin-like domain proteins (CTLDs).
Adhesion molecules are key in cell junctions throughout various tissues, including interneural synapses and neuron-glia junctions in the CNS. They help maintain tissue integrity through extracellular interactions and modulate intracellular signaling pathways important for cellular homeostasis. Altered expression of these molecules can disrupt signals that regulate cell growth, contributing to tumor formation. Understanding CAMs expands our knowledge of cellular inter-actions in neural tissues, revealing their roles in synapse formation and neuron-glia communication. The adhesion molecule on glia (AMOG) was first identified in 1987 [10] and later found to be an isoform of the β-subunit of Na+/ K⁺-ATPase (NKA), essential for maintaining sodium and potassium gradients across cell membranes [11,12,13]. The renaming to AMOG/β2 reflects its dual role in adhesion and ionic transport processes. The β-subunit serves as an accessory protein that aids in protein localization within the plasma membrane and K⁺ binding [14]. The two isoforms of the β-subunit, β1 and β2, act as adhesion molecules in epithelial tissues and the nervous system, respectively. In epithelial cells, β1 maintains intercellular junctions through homophilic β1-β1 adhesions, sup-porting cell polarity and transepithelial transport [15,16,17,18,19,20,21]. In the nervous system, β2 is primarily expressed in astrocytes, cerebellar granule cells, and photoreceptors, allowing NKA to function as both an ionic pump and a recognition molecule that mediates interactions between neurons and glial cells during brain development, promoting neurite outgrowth and cell migration [22].
Despite initial studies suggesting a heterophilic interaction between AMOG/β2 and a neural adhesion molecule (receptor), the specific pathways by which AMOG/β2 contributes to cell adhesion or the receptor molecule identity remain to be fully elucidated. This review examines the role of AMOG/β2 in the nervous system and poses the question: Does AMOG/β2 interact with an unidentified receptor on neurons?
2. Background
2.1. Adhesion Molecules in CNS
Adhesion molecules in the CNS are crucial for synapse formation, neuron-glia communication, and brain development. They facilitate cell-cell interactions, guiding neuronal migration and axon targeting. During synaptogenesis, adhesion molecules stabilize synaptic connections and regulate plasticity, essential for learning and memory. Disruptions in these molecules can lead to neurodevelopmental disorders, highlighting their importance in maintaining brain integrity and function [23,24]. Like epithelial junctions, cell-cell interactions in the CNS are mediated by various transmembrane proteins. These adhesion molecules can be found in interneural synapses and neuron-glia junctions, serving as important regulators of axon guidance and synapse formation [25]. We highlight key CAMs involved in synapse formation and astrocyte-synapse interactions, focusing on their homophilic and heterophilic characteristics.
2.1.1. Classic Cadherins
Classic cadherins, such as N-cadherin, are calcium-dependent homophilic adhesion molecules critical for synaptic stability. They form symmetrical adhesion structures in synaptic junctions (puncta adherentia junctions) across most regions of the nervous system. Notably, classic cadherins exhibit binding specificity and region-specific distribution [26]. In the brain, various subtypes of classic cadherins are expressed by functionally connected nuclei and laminas [27,28].
2.1.2. Proto-Cadherins
This extensive family of cadherins is essential for establishing synaptic specificity. The clustered protocadherin (Pcdh) genes encode diverse cell-surface assemblies. Their combinatorial expression patterns generate the numerous address codes necessary for neuronal identity, allowing neurons to discriminate self from non-self, thereby contributing to the organization of neural circuits [29].
2.1.3. Nectins
Nectins are a family of Ca²⁺-independent immunoglobulin (Ig)-like cell-cell adhesion molecules comprising four members (Nec1-Nec4). They form homo- or hetero-trans-dimers, with heterotypic binding resulting in stronger adhesion than homotypic binding. Nectins are integral to cell adhesion within synapses, particularly in puncta adherentia junctions (PAJs), and help cluster other adhesion molecules and signaling proteins to promote effective communication between neurons [30].
2.1.4. Nectin-Like Molecules (Necls)
Necl-2 acts as a homophilic adhesion molecule and exhibits heterophilic adhesion activity with Necl-1 and nectin-3. Like nectins, Necls facilitate interactions between neurons and supportive cells like astrocytes. Necl-2 localizes at synapses and is crucial for presynaptic differentiation and stabilization, which are essential for synapse formation and maintenance [31].
2.1.5. NCAM (Neural Cell Adhesion Molecule)
The neural cell adhesion molecule (NCAM) contains five Ig-like domains and two fibronectin type III repeats, engaging in both homophilic and heterophilic interactions with various ligands at synapses, including fibroblast growth factor receptor (FGFR), L1, TAG-1/axonin-1, and heparan sulfate proteoglycans. NCAM is expressed widely in the developing and adult brain, playing critical roles in migration, axon pathfinding, and synaptic plasticity. It influences neuron-neuron and neuron-glia interactions, impacting early synaptogenesis and subsequent maturation [32,33].
2.1.6. Integrins
Integrins are cell surface receptors that interact with the extracellular matrix (ECM) and transduce signals from the ECM to the cell. Comprising α- and β-subunits, integrins at the contacts between neurons and astrocytes promote synaptogenesis. During CNS development, ECM receptors and their ligands serve as guidance molecules, informing neurons where and when to extend axons and dendrites, thus establishing synapses. Once stable synapses are formed, ECM receptors transition to regulate the maintenance of these connections and influence synaptic plasticity, with their activity being strongly affected by ECM composition [34].
2.1.7. NgCAM (Neuron-Glia Cell Adhesion Molecule)
NgCAM facilitates adhesion between neurons and glial cells, contributing to nervous system development and the structure and signaling pathways of synapses.
2.1.8. Contactins
Contactins (CNTNs) are a subfamily of the Ig superfamily of neural cell-adhesion molecules (Ig-CAMs), consisting of six members (CNTN1-6) [35].
2.1.9. TAG-1 (Transient Axonal Glycoprotein-1)
In the embryonic nervous system, Contactin-2/TAG-1 plays important roles in axonal elongation, axonal guidance, and cellular migration. In the postnatal nervous system, it also plays an essential role in the formation of myelinated nerve fibers [36].
2.1.10. SYG-1 and SYG-2
2.1.11. Sidekicks
Related to the immunoglobulin superfamily, sidekicks function as homophilic adhesion molecules in vitro and are highly concentrated at synapses in vivo. They play a role in specifying and stabilizing synaptic connections, which is crucial for neuronal communication [39].
2.1.12. Neuroligins and Neurexins
Neuroligins are located on the postsynaptic side, interacting with presynaptic neurexins. β-Neurexin binds neuroligins trans-synaptically, inducing the formation of glutamatergic and GABAergic presynaptic specializations in vitro. This interaction influences synaptic strength and plasticity, which are essential for learning and memory [40,41].
2.2. The Na⁺,K⁺-ATPase in Neuron-Astrocyte Interactions
The molecule of interest, AMOG, is the β2 isoform of NKA. This transmembrane heterodimer consists of an α and a β subunit, potentially modulated by a third FXYD family subunit. The catalytic α subunit facilitates the exchange of intracellular Na⁺ for extracellular K⁺, while the glycosylated β subunit regulates the assembly and insertion of the α/β heterodimer into the plasma membrane. Characterized in mammalian cells are four α, three β, and seven FXYD isoforms, demonstrating tissue-dependent distributions [42,43,44,45,46,47]. In the nervous system, the expression of NKA isoforms is complex, with neurons primarily producing the α3 polypeptide [48,49,50,51], and glial cells expressing α2 [52,53,54]. β isoforms also exhibit tissue-dependent distributions [54], with β2 found in skeletal muscle [55], pineal gland [56], Astrocytes [57] and granular cells [46] while β3 is present in testis, retina, liver, and lung [58,59]. Expression patterns are influenced by developmental and hormonal regulation and can be altered in disease contexts [60,61,62,63,64].
AMOG/β2 functions as an adhesion molecule on glia and is expressed in mature brain astrocytes, focusing on interactions between astrocytes and other cell types. These bidirectional interactions play a crucial role in nervous system functioning. Astrocytes provide structural support to neurons and actively participate in synaptic modulation, neuronal development, and metabolism. They are involved in synapse formation and long-term plasticity related to learning [65,66,67,68], communicating with neural networks through gliotransmitters like gamma-aminobutyric acid (GABA) and adenosine triphosphate (ATP) to modulate neuronal activity and synaptic transmission [69,70]. Astrocytes also play a crucial role in regulating endothelial cells forming the blood-brain barrier (BBB), which protects the brain and controls substance passage between the brain and blood. Astrocytes regulate BBB permeability, nutrient supply, and waste removal [8,9,71]. NKA is essential in neuron-astrocyte interactions and astrocyte-vascular endothelium interactions. It maintains electrochemical gradients and participates in adhesion processes mediated by its β2 subunit [72]. Ongoing research aims to clarify how AMOG/β2 on astrocytes influences neuronal function and contributes to brain homeostasis, providing insights into the pathophysiology of neurological disorders and guiding therapeutic strategies to restore normal astrocytic function.
2.3. AMOG as a Heterophilic Adhesion Molecule
In 1987, Melitta Schachner’s group identified AMOG (Adhesion Molecule On Glia), a novel adhesion molecule distinct from known neural cell adhesion molecules [10]. This ~50 kD glycosylated integral membrane protein, primarily expressed by astrocytes, facilitates cerebellar granule cell migration by ensuring contact with Bergmann glial fibers. After migration, AMOG is prominently expressed in the internal granular layer, suggesting it may halt granule cell movement there [10,57,69]
Monoclonal AMOG antibodies showed specificity for glial surfaces during granule cell migration, indicating that AMOG does not participate in homophilic binding with neurons [10]. This was supported by findings that AMOG antibodies disrupted astrocyte-neuron adhesion in vitro but not astrocyte-astrocyte adhesion. Co-purification with AMOG suggested a neuronal receptor binding to AMOG is likely among these co-purified proteins. Subsequent studies demonstrated AMOG-containing liposomes could adhere to cultured granule cells, leading to the cloning of the AMOG gene and its identification as a β-subunit isoform of NKA, renamed AMOG/β2 [12,57].
The cloning of AMOG/β2 [11,73] allowed further investigation of its function in transfected cells. Muller-Husmann et al. [74] used AMOG-transfected L-cells as substrates for cerebellar neuron neurite outgrowth, observing increased neurite length—a response inhibited by antibodies against AMOG/β2. The soluble recombinant extracellular domain of AMOG/β2 partially blocked neurite outgrowth on AMOG/β2-expressing L-cells, while L-cells transfected with the mouse β1 subunit did not affect neurite extension. These findings indicate that AMOG/β2 interacts with an unidentified neuronal receptor to enhance neurite growth, likely through signal transduction pathways.
To elucidate AMOG’s physiological role, β2 knockout mice were generated. At postnatal day 15, these mice exhibited motor coordination abnormalities, tremors, and paralysis, leading to death by days 17-18 due to dysfunction in critical brain structures [75]. Morphological analyses revealed enlarged ventricles and swollen astrocytic end feet, yet several brain regions appeared normal. This raises questions about AMOG/β2’s potential role in brain development. Interestingly, AMOG/β2 antibodies interfere with granule cell migration in vitro [10], suggesting other molecules might compensate for the lack of AMOG/β2 in AMOG(−/−) mutants.
To differentiate pump activity from adhesion molecule function, Weber et al. [76] generated β2/β1 knock-in mutant mice, where β1 expression replaced β2 expression. Unlike β2-deficient animals, these knock-in mutants had a normal lifespan and did not exhibit swollen end feet. Photoreceptor cell degeneration was reduced compared to β2 null mutants, suggesting the β1 subunit can partially substitute for β2 function. However, the role of AMOG/β2 as an adhesion molecule remains to be fully elucidated.
Exploring the long-term consequences of AMOG/β2 deficiency, Isenmann et al. [77] grafted parts of the embryonic telencephalic anlage from deficient mice into wild-type mice, analyzing up to 500 days post-transplantation. The grafts developed normally, forming solid neural tissue indistinguishable from controls. No signs of degeneration were observed.
By the late 1990s, the understanding of AMOG/β2 remained incomplete. It had been identified as a glial adhesion molecule, prominently expressed on Bergmann glial cells during cerebellar granule cell migration and in astrocytes of the mature cerebellum. AMOG/β2 was also detected in cerebellar granular cells and was recognized as a heterophilic adhesion molecule interacting with an as-yet-unidentified neuronal receptor that played a critical role in brain development. Studies involving β2 knockout mice showed that these animals died shortly after birth, exhibiting severe neurological deficits, while β2/β1 knock-in mice survived. This survival in knock-in mice highlighted that the observed deficits were linked to altered Na⁺ and K⁺ pumping, which directly affected neuronal activity. Despite these findings, the identity of the specific neuronal receptor for AMOG/β2 remains unknown, leaving a key aspect of its function unresolved.
3. Current Understanding and Knowledge Gaps
3.1. β1-Subunit as a Homophilic Adhesion Molecule in Epithelia
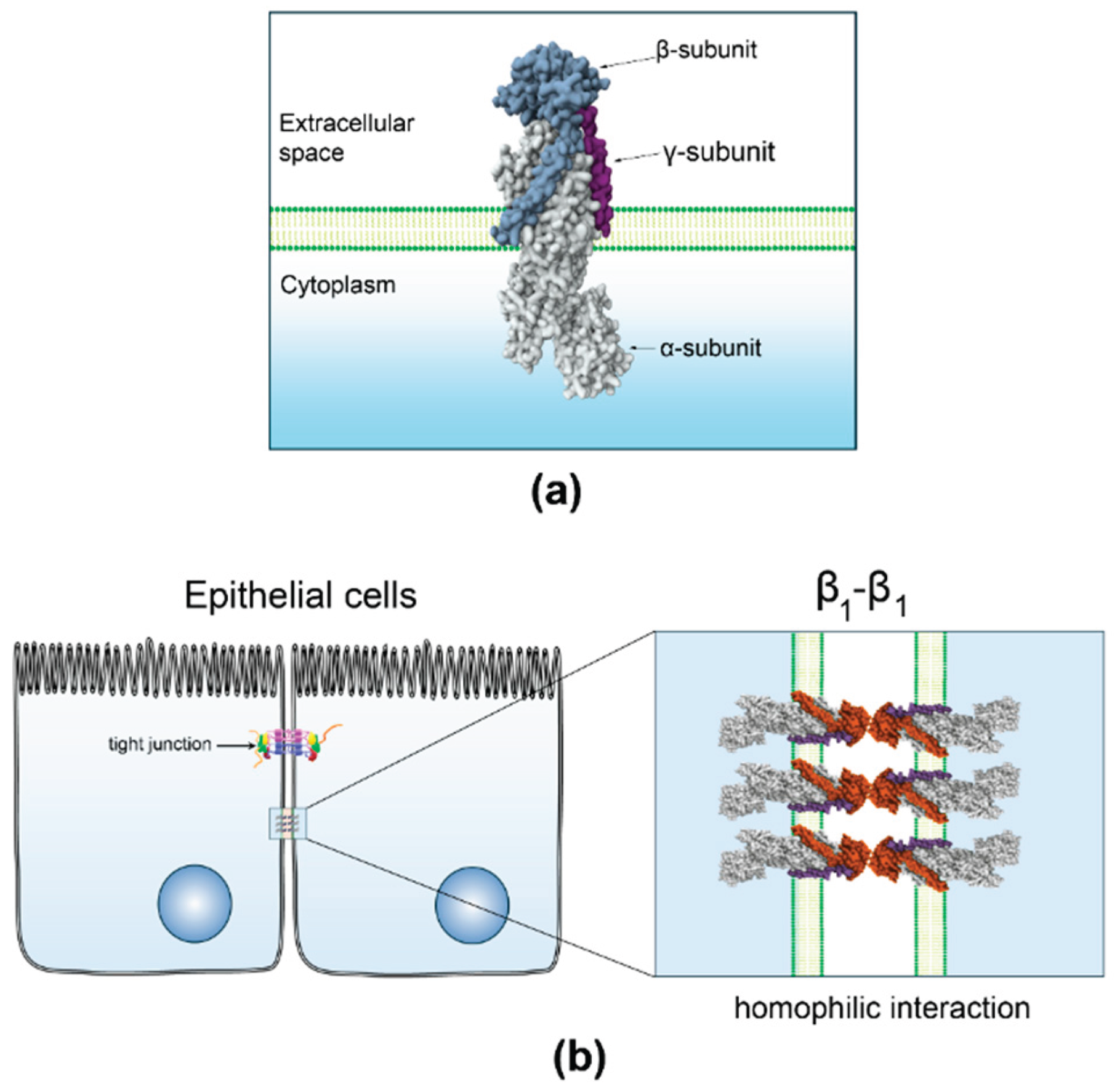
Numerous studies have shown that the β1 subunit functions as a homotypic cell-adhesion molecule. Its interaction with neighboring cells is essential for the lateral polarization of the pump and the apico-basal polarization of epithelial cells (Figure 1). Epithelial cells deficient in the β1 subunit undergo epithelial-mesenchymal transition (EMT), but transfection with the β1 subunit restores their adhesive properties and enhances polarization [15].
At the start of the 21st century, research on epithelial cells highlighted the role of the β1 isoform of NKA, linking β1 subunit expression to cell-cell adhesion and cell polarity [18,44], further studies established that the β1 subunit acts as a homotypic cell-adhesion molecule, with its interaction critical for both the lateral polarization of the pump [16], and cell-cell adhesion [17,19,21]. Molecular research identified a specific 10 amino-acid sequence (198-207) crucial for trans-dimerization of β1 subunits and cell-cell adhesion. This sequence explains the lack of stable interaction between β1 subunits from different species, such as rats and dogs [20]. Using in-silico methods, residues at the interface of β1-subunits from dog epithelium were identified, confirming their role in protein-protein interactions through site mutagenesis [78].
3.2. Gaps and Unresolved Questions
Despite progress in understanding AMOG/β2, several gaps and questions remain about its roles and interactions in the nervous system. If AMOG/β2 is a heterophilic adhesion molecule, identifying its receptor molecule in neurons remains a significant gap. Early studies by Schachner’s group attempted to determine if known adhesion molecules like L1, NCAM, or NAG were the receptors, but none were identified [10,11]. Although many new CAMs have been identified in the CNS, none appear to interact with AMOG/β2.
Research suggests the interaction between AMOG/β2 and its elusive neuronal receptor may activate pathways promoting neurite outgrowth and cell migration. Some studies have linked specific signaling pathways to AMOG/β2 [79,80,81]. However, these generally assume activation through an unidentified receptor without identifying it. Litan and coworkers [81] demonstrated that cerebellar granule cells express α1, α2, β1, and β2 isoforms during postnatal differentiation. They proposed a model where AMOG/β2 activates the mTOR/p70S6 kinase pathway, associated with cell size regulation, though it remains unclear whether the neighboring cell is another granule cell or an astrocyte expressing AMOG/β2 [81]. Antonicek et al. [10] suggested that AMOG does not facilitate astrocyte adhesion, ruling out homophilic interactions. Yet, U87-MG glioma cells transfected with AMOG/β2 form aggregates, indicating AMOG/β2 can engage in homophilic interactions [79]. This raises the question: if AMOG can engage in homophilic interactions, why not in astrocytes?
Figure 1.
β1 subunit functions as a homophilic cell-adhesion molecule in epithelia. (a) 3D structure of NKA, created from PB ID 7WYT [82]. b) Homophilic and species-specific interaction of β1-subunits of NKA localized at the lateral membrane of neighboring epithelial cells.
Figure 1.
β1 subunit functions as a homophilic cell-adhesion molecule in epithelia. (a) 3D structure of NKA, created from PB ID 7WYT [82]. b) Homophilic and species-specific interaction of β1-subunits of NKA localized at the lateral membrane of neighboring epithelial cells.
Zlokovic et al. [83] identified β1 and β2 isoforms in rat cerebral microvessels. Boer et al. [84] described AMOG/β2 expression in human cerebral cortex development, showing high perivascular abundance and glial end feet staining, suggesting a role in vascular integrity. This prompts the question of whether AMOG/β2 facilitates adhesion at the blood-brain barrier between astrocytes and endothelial cells via homophilic interactions.
Observations from AMOG/β2 knockout mice reveal they do not survive, displaying motor incoordination by postnatal day 15 and dying by days 17-18. Interestingly, their cerebella appear unaffected, with cortical layer thickness like wild-type mice. This suggests AMOG/β2 may not be essential for early cell migration. Instead, its role may be in maintaining the osmotic balance for neuronal activity [75]. This raises a crucial question: how can we differentiate between these two functions of AMOG/β2?
4. Recent Findings
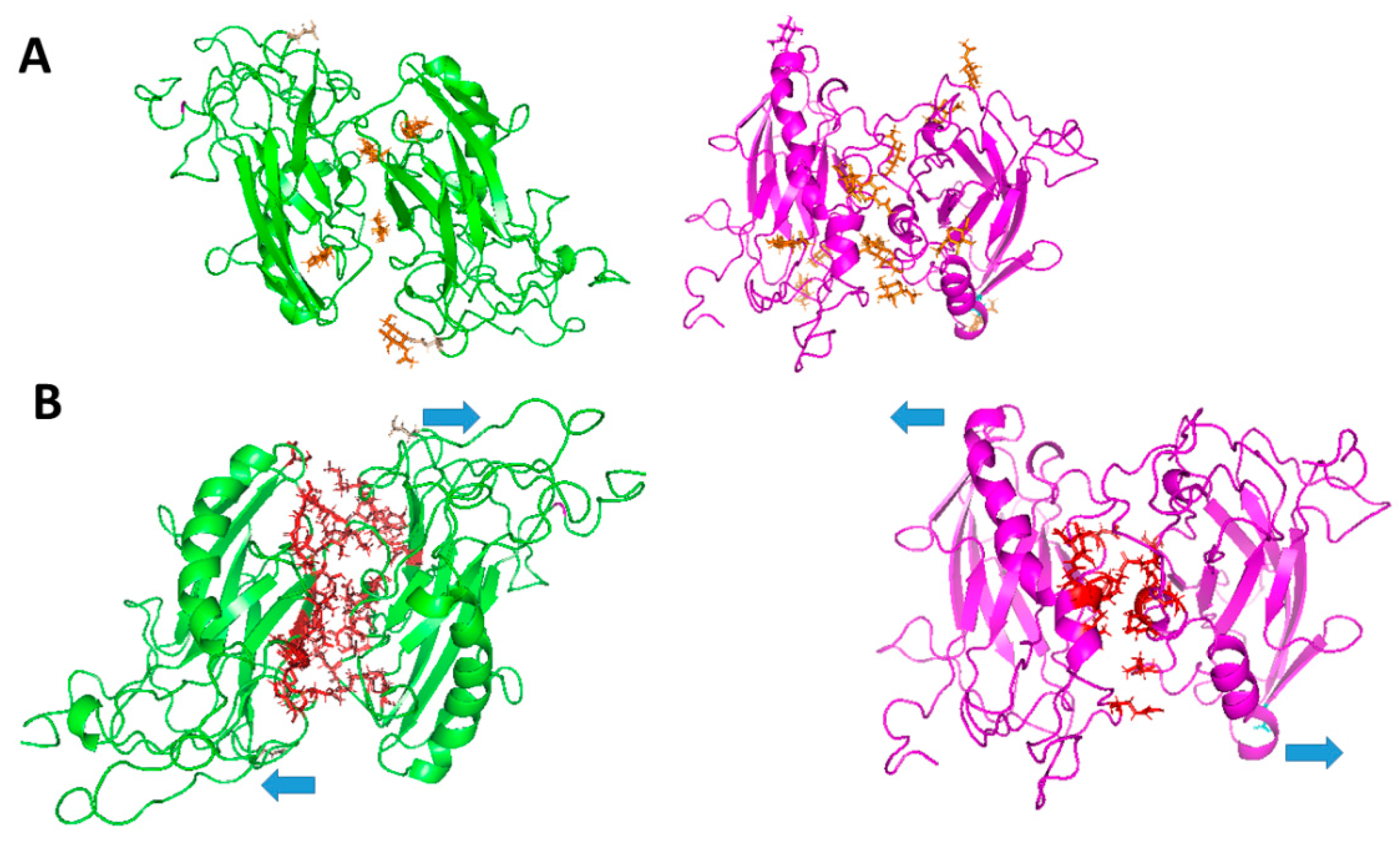
Although the neuronal receptor for AMOG/β2 remains unidentified, studies suggest that AMOG/β2 may also function as a homophilic cell adhesion molecule. Recent research hypothesizes that AMOG/β2, like the β1 subunit in epithelial cells, could mediate homophilic interactions. Given the distinct expression patterns of NKA subunits [85,86], both homophilic and heterophilic interactions at the CNS are plausible. Thus, the β2 subunit may engage in heterophilic interactions with neurons at tripartite synapses (Figure 2A), in homophilic interactions with cerebellar granule cells during migration (Figure 2B), and in homophilic interactions with endothelial cells at the blood-brain barrier (Figure 2C) [30]. Roldan et al. demonstrated that YFP-tagged AMOG/β2 in CHO and MDCK cells facilitates aggregation through β2-β2 interactions [87] (Figure 3). Dilution experiments confirmed that aggregation was proportional to β2 expression, though heterotypic interactions were also considered. Pull-down assays revealed no interaction in vitro; however, co-culture experiments detected β2-β2 interactions, emphasizing the importance of cellular context [87]. Cerebral organoids may be an effective model to study such interactions in vivo. β1 subunit homophilic interactions are well-documented [21,45] with species-specific residues critical for adhesion [20,78]. Roldan et al. [87] and Ramirez et al. [88] identified glycosylated extracellular domains in AMOG/β2 forming likely homodimers, though β2-β2 interfaces were smaller and weaker compared to β1-β1. Molecular modeling and molecular dynamics (MD) simulations revealed key residues in β1-β1 dimers (e.g., Gly225 and Leu266) and distributed hot spots in β2-β2 dimers. Binding free energies were calculated as -22671.13 kcal/mol for β1-β1 and -19707.5 kcal/mol for β2-β2 dimers. Glycosylation plays a vital role in stabilizing β1-β1 and β2-β2 interactions. While β1 has three conserved glycosylation sites, β2 contains four additional sites, totaling seven (Figure 4). These glycosylation sites are essential for adhesion, with distinct contributions in β1 and β2 dimers. For instance, Asn265 in β1-β1 mediates intramolecular interactions, while Asn153 in β2-β2 facilitates both intra- and intermolecular contacts [87,88].
5. Looking for the Partner
The hypothesis that AMOG/β2 requires a specific neuronal partner or receptor is supported by evidence suggesting its adhesive function may depend on a heterophilic interaction with a neuronal surface protein. While some studies have indicated potential homophilic β2–β2 interactions, especially in heterologous systems like CHO and MDCK cells [87], such binding has not been observed between astrocytes in vivo [10], or in biochemical assays with purified proteins. This discrepancy suggests that AMOG/β2 may need a specific neuronal binding partner, which is absent in astrocytes but present at the neuron–astrocyte interface during CNS development or maintenance.
5.1. Experimental Strategies
To identify the elusive AMOG/β2 receptor involved in neuron–astrocyte adhesion, several complementary experimental strategies can be pursued:
Co-Immunoprecipitation and Mass Spectrometry: This technique allows for the isolation of protein complexes associated with AMOG/β2 from astrocyte-neuron co-cultures or brain membrane fractions. By immunoprecipitating AMOG/β2 under native conditions and analyzing co-precipitated proteins, potential binding partners may be identified. However, this method can miss transient or low-affinity interactions, especially those involving membrane proteins. Careful optimization of detergents is required to preserve native protein structures [89].
Proximity Labeling Techniques (BioID or APEX): These methods involve fusing AMOG/β2 with an enzyme that biotinylates neighboring proteins in live cells. When expressed in astrocytes or neuron–astrocyte co-cultures, these tags label proteins near AMOG/β2, which can then be purified using streptavidin beads and identified by mass spectrometry. This approach is particularly useful for capturing transient interactions in their native membrane environment, providing insights into the proteins that interact with AMOG/β2 in vivo [90,91].
Functional Screening with Adhesion Assays: This strategy involves expressing AMOG/β2 in non-adhesive cells, such as CHO or HEK cells, and co-culturing them with a membrane protein cDNA library derived from neurons. Cells that adhere selectively can be isolated, and the corresponding cDNA identified, providing direct functional evidence of interaction. This method can reveal novel interaction partners that mediate AMOG/β2-dependent adhesion.
Genome-Wide CRISPR Knockout Screens: By employing CRISPR technology in neurons, researchers can identify genes required for adhesion to AMOG/β2-expressing astrocytes. Loss of adhesion in co-culture systems would suggest that the disrupted gene encodes a critical component of the receptor complex. This unbiased approach can uncover novel interactors that might not be predicted based on sequence homology or prior knowledge [92]
In Situ Chemical Crosslinking: Applying membrane-permeable crosslinkers to live neuron–astrocyte cultures can stabilize protein complexes in their native context. Following crosslinking, AMOG/β2 complexes can be immunoprecipitated and analyzed via proteomics. This method provides a snapshot of direct physical interactions that occur at the cell surface in vivo [93].
FRET or Bimolecular Fluorescence Complementation (BiFC): If specific candidate receptors are available, these techniques can be used to assess interactions in live cells. They rely on fluorescent signal restoration when two halves of a split fluorophore are brought together by the interaction of two fused proteins, enabling spatial and temporal visualization of the interaction [94].
To confirm the physiological relevance of any identified candidates, cerebral organoids can be employed. These three-dimensional structures, derived from human pluripotent stem cells, mimic aspects of the human brain’s architecture and function. They provide a valuable model for studying brain development, disease mechanisms, and drug responses. Cerebral organoids exhibit layered structures similar to those found in the developing brain, including regions resembling the cortex, hippocampus, and other areas [95,96]. They contain neurons, astrocytes, and other glial cells, allowing for the study of intercellular interactions. Knockouts of the putative receptor can be used to determine whether loss of the candidate protein disrupts astrocyte-neuron adhesion in vivo, mimicking the phenotype observed in AMOG/β2-deficient animals. This comprehensive approach, ranging from biochemical isolation and proximity labeling to functional genetic screens and in vivo validation, forms a robust framework for identifying the receptor or complex responsible for AMOG/β2-mediated adhesion in the central nervous system.
5.2. Candidates for AMOG/β2 Receptor
Two intriguing candidates for the AMOG/β2 receptor are Tetraspanin 31 (TSPAN31) and Reticulon 4 (RTN4/NogoA). Both have emerged from protein–protein interaction databases [97,98].
- TSPAN31: As a member of the Tetraspanin family, TSPAN31 is notable for its role in organizing membrane microdomains and mediating lateral interactions between cell surface proteins. Tetraspanins act as molecular scaffolds, clustering adhesion molecules, integrins, and signaling receptors into functional complexes [99,100,101]. TSPAN31 has been implicated in cell adhesion, migration, and membrane signaling functions aligning closely with AMOG/β2 activities. TSPAN31 might associate in cis with a neuronal adhesion receptor, creating a complex that interacts in trans with AMOG/β2 on astrocytes. Alternatively, TSPAN31 might directly stabilize or present the neuronal partner required for AMOG/β2 recognition. Its potential involvement raises intriguing questions about how these microdomain organizations contribute to AMOG/β2 function.
- RTN4 (Nogo-A): Known for inhibiting neurite outgrowth, RTN4 has a complex topology and is present not only in the endoplasmic reticulum but also on the plasma membrane of axons and dendrites [102]. Its interactions with membrane proteins suggest it could serve as a scaffold or modulator for a receptor complex capable of interacting with AMOG/β2. If RTN4 is enriched in specific neuronal compartments, such as dendritic spines or axon terminals, its spatial distribution could explain the specificity and context-dependence of AMOG/β2-mediated adhesion during synaptogenesis or glial ensheathment. RTN4’s role in membrane dynamics and its interaction network make it a compelling candidate for further investigation.
Together, TSPAN31 and RTN4 represent distinct but not mutually exclusive modes of interaction with AMOG/β2: one through microdomain organization and lateral associations (TSPAN31), and the other via scaffolding or presenting transmembrane receptors (RTN4). Both could act directly or indirectly to facilitate the molecular handshake between astrocytes and neurons. Elucidating whether either—or both—of these proteins interact functionally with AMOG/β2 will require targeted biochemical, cellular, and in vivo studies. Nonetheless, their candidacy reinforces the emerging view that AMOG/β2 operates not as a lone adhesion molecule but as part of a larger multiprotein complex that coordinates glia–neuron communication at the cell surface.
6. Functional Implications in the CNS
The implications of AMOG/β2-mediated adhesion in the CNS extend beyond its role in NKA. Positioned in astrocytes, AMOG/β2 influences neuron–glia communication, contributing to processes such as synaptic organization, neurogenesis, and potentially pathological conditions like glioblastoma.
A key role of AMOG/β2 is facilitating communication at the tripartite synapse. Astrocytes are crucial for synapse formation, maintenance, and plasticity, interacting closely with neuronal components [65,66,67,68]. AMOG/β2 may enhance the alignment of astrocytic processes with synaptic sites, optimizing neurotransmitter uptake, ion homeostasis, and gliotransmitter release. Its involvement in metabolic coupling [103] suggests a role in supporting synaptic interfaces.
During neurogenesis, AMOG/β2-mediated adhesion could aid neuronal migration and synaptic connection formation, anchoring developing neurons to astrocytic scaffolds. Disruption in this mechanism might lead to aberrant synapse formation, linking AMOG/β2 to neurodevelopmental disorders. In glioblastoma, changes in adhesion molecules often affect tumor invasiveness and glial cell transformation. AMOG/β2 dysregulation could influence tumor progression, possibly facilitating cell detachment and invasion or altering tumor-microenvironment interactions [104,105,106,107]. Beyond glioblastoma, AMOG/β2 could impact other neuropathological conditions [105]. Impaired function may disrupt astrocytic support in neurodegenerative diseases, affecting synaptic stability and metabolic coupling. Altered expression or interactions might also influence neuroinflammation, where astrocytes play a key role.
AMOG/β2 acts as a regulator of cell–cell communication and structural organization in the CNS. Future research to identify its neuronal receptor(s) and signaling pathways is essential for understanding its influence on neuronal function and CNS health. These mechanisms could reveal therapeutic targets for neurological disorders and gliomas involving adhesion dynamics.
7. Future Directions
7.1. Research Avenues and Implications
Identifying the binding partner(s) of AMOG/β2 opens several promising research directions. In neurodevelopment, understanding how AMOG/β2 mediates neuron-glia interactions could illuminate mechanisms guiding neuronal migration, circuit formation, and synaptic stabilization. Insights into these processes may clarify how changes in adhesion dynamics contribute to developmental disorders such as autism spectrum disorders or cortical malformations [108]. Understanding these pathways could lead to interventions that stabilize synaptic connections and support proper neural circuit formation.
In neurodegeneration, where synaptic loss and glial reactivity are significant pathological features, altered AMOG/β2 expression or function might influence disease progression. Investigating its role in maintaining neuron-glia contacts could shed light on early events in diseases like Alzheimer’s or ALS, where supportive glial functions become compromised [109]. Understanding these interactions may offer new strategies to preserve synaptic integrity and delay neurodegenerative processes.
In cancer, particularly glioblastoma, the progressive loss of AMOG/β2 is associated with increased tumor invasiveness [80,105]. Uncovering its adhesion partners may help define the molecular switch from organized glial tissue to a disaggregated, migratory tumor phenotype. This could lead to identifying biomarkers for tumor progression or new molecular checkpoints vulnerable to therapeutic intervention, potentially improving prognosis and treatment options.
7.2. Therapeutic Targeting Potential
Studying AMOG/β2’s adhesion mechanisms could inspire novel therapeutic strategies. For instance, developing mimetics or stabilizers of AMOG/β2-mediated adhesion might restore normal glia-neuron or glia-endothelial interactions in degenerative diseases. By enhancing these interactions, it may be possible to improve neuronal support and function. Conversely, in glioblastoma, artificially reactivating AMOG/β2 expression or mimicking its adhesive function might reduce invasiveness or sensitize tumor cells to treatment, potentially inhibiting tumor growth and spread. This approach could lead to therapies that specifically target the adhesive properties of tumor cells, reducing their ability to migrate and invade surrounding tissues.
Targeting the β2 subunit and its receptor pair could also represent a new class of adhesion-modulating therapies, especially relevant in CNS conditions where adhesion loss precedes or drives pathology. As AMOG/β2 bridges ion transport and adhesion signaling, it offers dual-entry points for therapeutic modulation—both electrochemical and structural. This dual functionality presents unique opportunities for designing interventions that can modulate both cellular adhesion and ion homeostasis, addressing multiple facets of CNS disorders simultaneously, electrochemical and structural.
8. Summary
The β2 subunit of the Na⁺/K⁺-ATPase (NKA), also known as AMOG/β2, integrates ion transport and cell adhesion functions. Initially characterized for its role in promoting neuron–glia adhesion during development, AMOG/β2 is now recognized as a key player in central nervous system (CNS) physiology and pathology. Despite its established adhesive role, the identity of its binding partner(s) in the CNS remains unknown—a central question addressed in this review.
Elucidating the molecular partners of AMOG/β2 is crucial to understanding how adhesive interactions shape neurodevelopmental processes. Such insights may also clarify how disruptions in these interactions contribute to CNS disorders, including neurodegeneration and glioblastoma. Notably, AMOG/β2 expression is reduced in high-grade gliomas, supporting its role as a malignancy suppressor and a marker of glial differentiation.
Adhesion molecules like β2 are essential for organizing CNS architecture, orchestrating cell–cell interactions, modulating signaling pathways, and adapting to developmental and pathological cues. Investigating AMOG/β2 may therefore advance our understanding of neurobiology and reveal novel therapeutic targets.
Identifying AMOG/β2’s binding partners and elucidating its mechanisms of action may ultimately reshape our understanding of structural connectivity and signal integration in the CNS, with implications for brain development, plasticity, and disease.
9. Concluding Remarks
Understanding AMOG/β2’s role in the CNS opens new research avenues in neurodevelopment, neurodegeneration, and glioma biology. Identifying its binding partners will clarify its function in neuron-glia interactions and its disruption in disease. As a dual-function target, AMOG/β2 offers potential therapeutic strategies to modulate adhesion in developmental disorders, neurodegenerative diseases, and brain tumors.
Author Contributions
All authors have contributed equally to this review. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank the technical. Support of Rosalia Aguirre and the secretarial service of Marisela Garzon Gonzalez.
The authors have reviewed and edited the output and take full responsibility for the content of this publication.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AMOG/β2 | Adhesion molecule on Glia |
| CNS | Central nervous system |
| TSPAN31 | Tetraspanin 31 |
| RTN4 | Reticulon 4 |
| CAMs | Cell adhesion molecules |
| IgCAMs | Inmunoglobulin superfamily cell adhesion molecules |
| CTLDs | C-type lectin-like domain proteins |
| Pcdh | Protocadherin |
| PAJs | Puncta adherentia junctions |
| Necls | Nectin-like molecules |
| NCAM | Neural Cell Adhesion molecule |
| FGFR | Fibroblast growth factor receptor |
| ECM | Extracellular matrix |
| NgCAM | Neuron-glia cell adhesion molecule |
| CNTNs | Contactins |
| TAG-1 | Transient Axonal Glycoprotein-1 |
| GABA | Gamma-aminobutyric acid |
| ATP | Adenosine triphosphate |
| BBB | Blood-brain barrier |
| EMT | Epithelial-mesenchymal transition |
| U87-MG | Uppsala 87 malignant glioma |
| YFP | Yellow fluorescence protein |
| CHO | Chinese hamster ovary cells |
| MDCK | Madin-Darby Canine Kidney |
| MD | Molecular dynamic |
| HEK | Human embryonic kidney |
| CRISPR | Clustered Regularly Interspaced short palindromic repeats |
| FRET | Förster resonance energy transfer |
| BiFC | Biomolecular Fluorescence Complementation |
References
- Kennedy, M.B. Synaptic Signaling in Learning and Memory. Cold Spring Harb. Perspect. Biol. 2016, 8, a016824. [CrossRef]
- Noriega-Prieto, J.A.; Araque, A. Sensing and Regulating Synaptic Activity by Astrocytes at Tripartite Synapse. Neurochem. Res. 2021, 46, 2580–2585. [CrossRef]
- Accogli, A.; Addour-Boudrahem, N.; Srour, M. Chapter 4 - Neurogenesis, Neuronal Migration, and Axon Guidance. In Handbook of Clinical Neurology; Gallagher, A., Bulteau, C., Cohen, D., Michaud, J.L., Eds.; Neurocognitive Development: Normative Development; Elsevier, 2020; Vol. 173, pp. 25–42.
- Affrald R, J.; Narayan, S. A Review: Oligodendrocytes in Neuronal Axonal Conduction and Methods for Enhancing Their Performance. Int. J. Neurosci. 0, 1–22. [CrossRef]
- Norris, G.T.; Kipnis, J. Immune Cells and CNS Physiology: Microglia and Beyond. J. Exp. Med. 2018, 216, 60–70. [CrossRef]
- Thalhammer, A.; Cingolani, L.A. Cell Adhesion and Homeostatic Synaptic Plasticity. Neuropharmacology 2014, 78, 23–30. [CrossRef]
- Ben Achour, S.; Pascual, O. Glia: The Many Ways to Modulate Synaptic Plasticity. Neurochem. Int. 2010, 57, 440–445. [CrossRef]
- Durkee, C.A.; Araque, A. Diversity and Specificity of Astrocyte–Neuron Communication. Neuroscience 2019, 396, 73–78. [CrossRef]
- Benarroch, E.E. Neuron-Astrocyte Interactions: Partnership for Normal Function and Disease in the Central Nervous System. Mayo Clin. Proc. 2005, 80, 1326–1338. [CrossRef]
- Antonicek, H.; Persohn, E.; Schachner, M. Biochemical and Functional Characterization of a Novel Neuron-Glia Adhesion Molecule That Is Involved in Neuronal Migration. J. Cell Biol. 1987, 104, 1587–1595. [CrossRef]
- Gloor, S.; Antonicek, H.; Sweadner, K.J.; Pagliusi, S.; Frank, R.; Moos, M.; Schachner, M. The Adhesion Molecule on Glia (AMOG) Is a Homologue of the Beta Subunit of the Na,K-ATPase. J. Cell Biol. 1990, 110, 165–174. [CrossRef]
- Martin-Vasallo, P.; Dackowski, W.; Emanuel, J.R.; Levenson, R. Identification of a Putative Isoform of the Na,K-ATPase β Subunit: Primary Structure and Tissue-Specific Expression. J. Biol. Chem. 1989, 264, 4613–4618. [CrossRef]
- Contreras, R.G.; Torres-Carrillo, A.; Flores-Maldonado, C.; Shoshani, L.; Ponce, A. Na+/K+-ATPase: More than an Electrogenic Pump. Int. J. Mol. Sci. 2024, 25, 6122. [CrossRef]
- Lutsenko, S.; Kaplan, J.H. An Essential Role for the Extracellular Domain of the Sodium-Potassium-ATPase .Beta.-Subunit in Cation Occlusion. Biochemistry 1993, 32, 6737–6743. [CrossRef]
- Rajasekaran, S.A.; Palmer, L.G.; Moon, S.Y.; Peralta Soler, A.; Apodaca, G.L.; Harper, J.F.; Zheng, Y.; Rajasekaran, A.K. Na,K-ATPase Activity Is Required for Formation of Tight Junctions, Desmosomes, and Induction of Polarity in Epithelial Cells. Mol. Biol. Cell 2001, 12, 3717–3732. [CrossRef]
- Shoshani, L.; Contreras, R.G.; Roldán, M.L.; Moreno, J.; Lázaro, A.; Balda, M.S.; Matter, K.; Cereijido, M. The Polarized Expression of Na+,K+-ATPase in Epithelia Depends on the Association between β-Subunits Located in Neighboring Cells. Mol. Biol. Cell 2005, 16, 1071–1081. [CrossRef]
- Padilla-Benavides, T.; Roldán, M.L.; Larre, I.; Flores-Benitez, D.; Villegas-Sepúlveda, N.; Contreras, R.G.; Cereijido, M.; Shoshani, L. The Polarized Distribution of Na+,K+-ATPase: Role of the Interaction between β Subunits. Mol. Biol. Cell 2010, 21, 2217–2225. [CrossRef]
- Vagin, O.; Dada, L.A.; Tokhtaeva, E.; Sachs, G. The Na-K-ATPase A1β1 Heterodimer as a Cell Adhesion Molecule in Epithelia. Am. J. Physiol.-Cell Physiol. 2012, 302, C1271–C1281. [CrossRef]
- Vagin, O.; Tokhtaeva, E.; Sachs, G. The Role of the Β1 Subunit of the Na,K-ATPase and Its Glycosylation in Cell-Cell Adhesion *. J. Biol. Chem. 2006, 281, 39573–39587. [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Sun, H.; Dada, L.A.; Sznajder, J.I.; Vagin, O. Identification of the Amino Acid Region Involved in the Intercellular Interaction between the Β1 Subunits of Na+/K+-ATPase. J. Cell Sci. 2012, 125, 1605–1616. [CrossRef]
- Tokhtaeva, E.; Sachs, G.; Souda, P.; Bassilian, S.; Whitelegge, J.P.; Shoshani, L.; Vagin, O. Epithelial Junctions Depend on Intercellular Trans-Interactions between the Na,K-ATPase Β1 Subunits*. J. Biol. Chem. 2011, 286, 25801–25812. [CrossRef]
- Antonicek, H.; Schachner, M. The Adhesion Molecule on Glia (AMOG) Incorporated into Lipid Vesicles Binds to Subpopulations of Neurons. J. Neurosci. 1988, 8, 2961–2966. [CrossRef]
- Togashi, H.; Sakisaka, T.; Takai, Y. Cell Adhesion Molecules in the Central Nervous System. Cell Adhes. Migr. 2009, 3, 29–35. [CrossRef]
- Saint-Martin, M.; Goda, Y. Astrocyte–Synapse Interactions and Cell Adhesion Molecules. FEBS J. 2023, 290, 3512–3526. [CrossRef]
- Shapiro, L.; Love, J.; Colman, D.R. Adhesion Molecules in the Nervous System: Structural Insights into Function and Diversity. Annu. Rev. Neurosci. 2007, 30, 451–474. [CrossRef]
- Uchida, N.; Honjo, Y.; Johnson, K.R.; Wheelock, M.J.; Takeichi, M. The Catenin/Cadherin Adhesion System Is Localized in Synaptic Junctions Bordering Transmitter Release Zones. J. Cell Biol. 1996, 135, 767–779. [CrossRef]
- Hirano, S.; Takeichi, M. Cadherins in Brain Morphogenesis and Wiring. Physiol. Rev. 2012, 92, 597–634. [CrossRef]
- de Agustín-Durán, D.; Mateos-White, I.; Fabra-Beser, J.; Gil-Sanz, C. Stick around: Cell–Cell Adhesion Molecules during Neocortical Development. Cells 2021, 10, 118. [CrossRef]
- Wu, Q.; Jia, Z. Wiring the Brain by Clustered Protocadherin Neural Codes. Neurosci. Bull. 2021, 37, 117–131. [CrossRef]
- Mizoguchi, A.; Nakanishi, H.; Kimura, K.; Matsubara, K.; Ozaki-Kuroda, K.; Katata, T.; Honda, T.; Kiyohara, Y.; Heo, K.; Higashi, M.; et al. Nectin : An Adhesion Molecule Involved in Formation of Synapses. J. Cell Biol. 2002, 156, 555–565. [CrossRef]
- Mizutani, K.; Miyata, M.; Shiotani, H.; Kameyama, T.; Takai, Y. Nectins and Nectin-like Molecules in Synapse Formation and Involvement in Neurological Diseases. Mol. Cell. Neurosci. 2021, 115, 103653. [CrossRef]
- Jozsef Zoltan Kiss; Dominique Müller Contribution of the Neural Cell Adhesion Molecule to Neuronal and Synaptic Plasticity. Rev. Neurosci. 2001, 12, 297–310. [CrossRef]
- Walsh, F.S.; Doherty, P. NEURAL CELL ADHESION MOLECULES OF THE IMMUNOGLOBULIN SUPERFAMILY: Role in Axon Growth and Guidance. Annu. Rev. Cell Dev. Biol. 1997, 13, 425–456. [CrossRef]
- Kerrisk, M.E.; Cingolani, L.A.; Koleske, A.J. Chapter 5 - ECM Receptors in Neuronal Structure, Synaptic Plasticity, and Behavior. In Progress in Brain Research; Dityatev, A., Wehrle-Haller, B., Pitkänen, A., Eds.; Brain Extracellular Matrix in Health and Disease; Elsevier, 2014; Vol. 214, pp. 101–131.
- Zuko, A.; Kleijer, K.T.E.; Oguro-Ando, A.; Kas, M.J.H.; van Daalen, E.; van der Zwaag, B.; Burbach, J.P.H. Contactins in the Neurobiology of Autism. Eur. J. Pharmacol. 2013, 719, 63–74. [CrossRef]
- Masuda, T. Contactin-2/TAG-1, Active on the Front Line for Three Decades. Cell Adhes. Migr. 2017, 11, 524–531. [CrossRef]
- Shen, K.; Fetter, R.D.; Bargmann, C.I. Synaptic Specificity Is Generated by the Synaptic Guidepost Protein SYG-2 and Its Receptor, SYG-1. Cell 2004, 116, 869–881. [CrossRef]
- Shen, K.; Bargmann, C.I. The Immunoglobulin Superfamily Protein SYG-1 Determines the Location of Specific Synapses in C. Elegans. Cell 2003, 112, 619–630. [CrossRef]
- Goodman, K.M.; Yamagata, M.; Jin, X.; Mannepalli, S.; Katsamba, P.S.; Ahlsén, G.; Sergeeva, A.P.; Honig, B.; Sanes, J.R.; Shapiro, L. Molecular Basis of Sidekick-Mediated Cell-Cell Adhesion and Specificity. eLife 2016, 5, e19058. [CrossRef]
- Craig, A.M.; Kang, Y. Neurexin–Neuroligin Signaling in Synapse Development. Curr. Opin. Neurobiol. 2007, 17, 43–52. [CrossRef]
- Bang, M.L.; Owczarek, S. A Matter of Balance: Role of Neurexin and Neuroligin at the Synapse. Neurochem. Res. 2013, 38, 1174–1189. [CrossRef]
- Clausen, M.V.; Hilbers, F.; Poulsen, H. The Structure and Function of the Na,K-ATPase Isoforms in Health and Disease. Front. Physiol. 2017, 8. [CrossRef]
- Álvarez, J.A.L.; Murillo, T. del C.L.; Nestor, C.A.V.; Gutierrez, M.L.R.; Gómez, O.P.; Shoshani, L.; Álvarez, J.A.L.; Murillo, T. del C.L.; Nestor, C.A.V.; Gutierrez, M.L.R.; et al. Epithelial Na+,K+-ATPase — A Sticky Pump. In Cell Biology - New Insights; IntechOpen, 2016 ISBN 978-953-51-2242-5.
- Cereijido, M.; Contreras, R.G.; Shoshani, L.; Larre, I. The Na+-K+-ATPase as Self-Adhesion Molecule and Hormone Receptor. Am. J. Physiol.-Cell Physiol. 2012, 302, C473–C481. [CrossRef]
- Blanco, G. Na,K-ATPase Subunit Heterogeneity as a Mechanism for Tissue-Specific Ion Regulation. Semin. Nephrol. 2005, 25, 292–303. [CrossRef]
- Peng, L.; Martin-Vasallo, P.; Sweadner, K.J. Isoforms of Na,K-ATPase α and β Subunits in the Rat Cerebellum and in Granule Cell Cultures. J. Neurosci. 1997, 17, 3488–3502. [CrossRef]
- Levenson, R. Isoforms of the Na,K-ATPase: Family Members in Search of Function. In Reviews of Physiology, Biochemistry and Pharmacology, Volume 123: Volume: 123; Springer: Berlin, Heidelberg, 1994; pp. 1–45 ISBN 978-3-540-48217-8.
- Lingrel, J.B. Na,K-ATPase: Isoform Structure, Function, and Expression. J. Bioenerg. Biomembr. 1992, 24, 263–270. [CrossRef]
- Lingrel, J.B.; Orlowski, J.; Shull, M.M.; Price, E.M. Molecular Genetics of Na,K-ATPase. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Moldave, K., Eds.; Academic Press, 1990; Vol. 38, pp. 37–89.
- Sweadner, K.J. Overlapping and Diverse Distribution of Na–K ATPase Isozymes in Neurons and Glia. Can. J. Physiol. Pharmacol. 1992, 70, S255–S259. [CrossRef]
- Brines, M.L.; Robbins, R.J. Cell-Type Specific Expression of Na+,K+-ATPase Catalytic Subunits in Cultured Neurons and Glia: Evidence for Polarized Distribution in Neurons. Brain Res. 1993, 631, 1–11. [CrossRef]
- Cameron, R.; Klein, L.; Shyjan, A.W.; Rakic, P.; Levenson, R. Neurons and Astroglia Express Distinct Subsets of Na,K-ATPase α and β Subunits. Mol. Brain Res. 1994, 21, 333–343. [CrossRef]
- Fink, D.; Knapp, P.E.; Mata, M. Differential Expression of Na,K-ATPase Isoforms in Oligodendrocytes and Astrocytes. Dev. Neurosci. 1996, 18, 319–326. [CrossRef]
- Wetzel, R.K.; Arystarkhova, E.; Sweadner, K.J. Cellular and Subcellular Specification of Na,K-ATPase α and β Isoforms in the Postnatal Development of Mouse Retina. J. Neurosci. 1999, 19, 9878–9889. [CrossRef]
- Lavoie, L.; Levenson, R.; Martin-Vasallo, P.; Klip, A. The Molar Ratios of α and β Subunits of the Na+−K+-ATPase Differ in Distinct Subcellular Membranes from Rat Skeletal Muscle. Biochemistry 1997, 36, 7726–7732. [CrossRef]
- Shyjan, A.W.; Ceña, V.; Klein, D.C.; Levenson, R. Differential Expression and Enzymatic Properties of the Na+,K(+)-ATPase Alpha 3 Isoenzyme in Rat Pineal Glands. Proc. Natl. Acad. Sci. 1990, 87, 1178–1182. [CrossRef]
- Pagliusi, S.R.; Schachner, M.; Seeburg, P.H.; Shivers, B.D. The Adhesion Molecule on Glia (AMOG) Is Widely Expressed by Astrocytes in Developing and Adult Mouse Brain. Eur. J. Neurosci. 1990, 2, 471–480. [CrossRef]
- Malik, N.; Canfield, V.A.; Beckers, M.-C.; Gros, P.; Levenson, R. Identification of the Mammalian Na,K-ATPase Β3 Subunit *. J. Biol. Chem. 1996, 271, 22754–22758. [CrossRef]
- Arystarkhova, E.; Sweadner, K.J. Tissue-Specific Expression of the Na,K-ATPase Β3 Subunit: THE PRESENCE OF Β3 IN LUNG AND LIVER ADDRESSES THE PROBLEM OF THE MISSING SUBUNIT *. J. Biol. Chem. 1997, 272, 22405–22408. [CrossRef]
- Book, C.B.; Wilson, R.P.; Ng, Y.C. Cardiac Hypertrophy in the Ferret Increases Expression of the Na(+)-K(+)-ATPase Alpha 1- but Not Alpha 3-Isoform. Am. J. Physiol.-Heart Circ. Physiol. 1994, 266, H1221–H1227. [CrossRef]
- Charlemagne, D.; Swynghedauw, B. Myocardial Phenotypic Changes in Na+, K+ATPase in Left Ventricular Hypertrophy: Pharmacological Consequences. Eur. Heart J. 1995, 16, 20–23. [CrossRef]
- Charlemagne, D.; Orlowski, J.; Oliviero, P.; Rannou, F.; Sainte Beuve, C.; Swynghedauw, B.; Lane, L.K. Alteration of Na,K-ATPase Subunit mRNA and Protein Levels in Hypertrophied Rat Heart. J. Biol. Chem. 1994, 269, 1541–1547. [CrossRef]
- Ewart, H.S.; Klip, A. Hormonal Regulation of the Na(+)-K(+)-ATPase: Mechanisms Underlying Rapid and Sustained Changes in Pump Activity. Am. J. Physiol.-Cell Physiol. 1995, 269, C295–C311. [CrossRef]
- Zahler, R.; Gilmore-Hebert, M.; Sun, W.; Benz, E.J. Na, K-ATPase Isoform Gene Expression in Normal and Hypertrophied Dog Heart. Basic Res. Cardiol. 1996, 91, 256–266. [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G.; Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite Synapses: Glia, the Unacknowledged Partner. Trends Neurosci. 1999, 22, 208–215. [CrossRef]
- Chung, W.-S.; Allen, N.J.; Eroglu, C. Astrocytes Control Synapse Formation, Function, and Elimination. Cold Spring Harb. Perspect. Biol. 2015, 7, a020370. [CrossRef]
- Allen, N.J.; Eroglu, C. Cell Biology of Astrocyte-Synapse Interactions. Neuron 2017, 96, 697–708. [CrossRef]
- Baldwin, K.T.; Eroglu, C. Molecular Mechanisms of Astrocyte-Induced Synaptogenesis. Curr. Opin. Neurobiol. 2017, 45, 113–120. [CrossRef]
- Kilb, W.; Kirischuk, S. GABA Release from Astrocytes in Health and Disease. Int. J. Mol. Sci. 2022, 23, 15859. [CrossRef]
- Harada, K.; Kamiya, T.; Tsuboi, T. Gliotransmitter Release from Astrocytes: Functional, Developmental and Pathological Implications in the Brain. Front. Neurosci. 2016, 9. [CrossRef]
- Abbott, N.J.; Rönnbäck, L.; Hansson, E. Astrocyte–Endothelial Interactions at the Blood–Brain Barrier. Nat. Rev. Neurosci. 2006, 7, 41–53. [CrossRef]
- Pietrobon, D.; Conti, F. Astrocytic Na+, K+ ATPases in Physiology and Pathophysiology. Cell Calcium 2024, 118, 102851. [CrossRef]
- Pagliusi, S.; Antonicek, H.; Gloor, S.; Frank, R.; Moos, M.; Schachner, M. Identification of a cDNA Clone Specific for the Neural Cell Adhesion Molecule AMOG. J. Neurosci. Res. 1989, 22, 113–119. [CrossRef]
- Müller-Husmann, G.; Gloor, S.; Schachner, M. Functional Characterization of Beta Isoforms of Murine Na,K-ATPase. The Adhesion Molecule on Glia (AMOG/Beta 2), but Not Beta 1, Promotes Neurite Outgrowth. J. Biol. Chem. 1993, 268, 26260–26267. [CrossRef]
- Magyar, J.P.; Bartsch, U.; Wang, Z.Q.; Howells, N.; Aguzzi, A.; Wagner, E.F.; Schachner, M. Degeneration of Neural Cells in the Central Nervous System of Mice Deficient in the Gene for the Adhesion Molecule on Glia, the Beta 2 Subunit of Murine Na,K-ATPase. J. Cell Biol. 1994, 127, 835–845. [CrossRef]
- Weber, P.; Bartsch, U.; Schachner, M.; Montag, D. Na,K-ATPase Subunit Β1 Knock-in Prevents Lethality of Β2 Deficiency in Mice. J. Neurosci. 1998, 18, 9192–9203. [CrossRef]
- Isenmann, S.; Molthagen, M.; Brandner, S.; Bartsch, U.; Kühne, G.; Magyar, J.P.; Sure, U.; Schachner, M.; Aguzzi, A. The AMOG/Β2 Subunit of Na, K-ATPase Is Not Necessary for Long-Term Survival of Telencephalic Grafts. Glia 1995, 15, 377–388. [CrossRef]
- Páez, O.; Martínez-Archundia, M.; Villegas-Sepúlveda, N.; Roldan, M.L.; Correa-Basurto, J.; Shoshani, L. A Model for the Homotypic Interaction between Na+,K+-ATPase Β1 Subunits Reveals the Role of Extracellular Residues 221–229 in Its Ig-Like Domain. Int. J. Mol. Sci. 2019, 20, 4538. [CrossRef]
- Scheidenhelm, D.K.; Cresswell, J.; Haipek, C.A.; Fleming, T.P.; Mercer, R.W.; Gutmann, D.H. Akt-Dependent Cell Size Regulation by the Adhesion Molecule on Glia Occurs Independently of Phosphatidylinositol 3-Kinase and Rheb Signaling. Mol. Cell. Biol. 2005, 25, 3151–3162. [CrossRef]
- Barreto, N.; Caballero, M.; Bonfanti, A.P.; de Mato, F.C.P.; Munhoz, J.; da Rocha--e--Silva, T.A.A.; Sutti, R.; Vitorino-Araujo, J.L.; Verinaud, L.; Rapôso, C. Spider Venom Components Decrease Glioblastoma Cell Migration and Invasion through RhoA-ROCK and Na+/K+-ATPase Β2: Potential Molecular Entities to Treat Invasive Brain Cancer. Cancer Cell Int. 2020, 20, 576. [CrossRef]
- Litan, A.; Li, Z.; Tokhtaeva, E.; Kelly, P.; Vagin, O.; Langhans, S.A. A Functional Interaction Between Na,K-ATPase Β2-Subunit/AMOG and NF2/Merlin Regulates Growth Factor Signaling in Cerebellar Granule Cells. Mol. Neurobiol. 2019, 56, 7557–7571. [CrossRef]
- Kanai, R.; Ogawa, H.; Vilsen, B.; Cornelius, F.; Toyoshima, C. Crystal Structure of a Na+-Bound Na+,K+-ATPase Preceding the E1P State. Nature 2013, 502, 201–206. [CrossRef]
- Zlokovic, B.V.; Mackic, J.B.; Wang, L.; McComb, J.G.; McDonough, A. Differential Expression of Na,K-ATPase Alpha and Beta Subunit Isoforms at the Blood-Brain Barrier and the Choroid Plexus. J. Biol. Chem. 1993, 268, 8019–8025. [CrossRef]
- Boer, K.; Spliet, W.G.M.; van Rijen, P.C.; Jansen, F.E.; Aronica, E. Expression Patterns of AMOG in Developing Human Cortex and Malformations of Cortical Development. Epilepsy Res. 2010, 91, 84–93. [CrossRef]
- McGrail, K.M.; Phillips, J.M.; Sweadner, K.J. Immunofluorescent Localization of Three Na,K-ATPase Isozymes in the Rat Central Nervous System: Both Neurons and Glia Can Express More than One Na,K-ATPase. J. Neurosci. 1991, 11, 381–391. [CrossRef]
- Watts, A.G.; Sanchez-Watts, G.; Emanuel, J.R.; Levenson, R. Cell-Specific Expression of mRNAs Encoding Na+,K(+)-ATPase Alpha- and Beta-Subunit Isoforms within the Rat Central Nervous System. Proc. Natl. Acad. Sci. 1991, 88, 7425–7429. [CrossRef]
- Roldán, M.L.; Ramírez-Salinas, G.L.; Martinez-Archundia, M.; Cuellar-Perez, F.; Vilchis-Nestor, C.A.; Cancino-Diaz, J.C.; Shoshani, L. The Β2-Subunit (AMOG) of Human Na+, K+-ATPase Is a Homophilic Adhesion Molecule. Int. J. Mol. Sci. 2022, 23, 7753. [CrossRef]
- Ramírez-Salinas, G.; Shoshani, L.; Rosas-Trigueros, J.L.; Huerta, C.S.; Martínez-Archundia, M. In Silico Studies Provide New Structural Insights into Trans-Dimerization of Β1 and Β2 Subunits of the Na+, K+-ATPase. PLOS ONE 2025, 20, e0321064. [CrossRef]
- Goulding, S.P.; Szumlinski, K.K.; Contet, C.; MacCoss, M.J.; Wu, C.C. A Mass Spectrometry-Based Proteomic Analysis of Homer2-Interacting Proteins in the Mouse Brain. J. Proteomics 2017, 166, 127–137. [CrossRef]
- Roux, K.J.; Kim, D.I.; Burke, B.; May, D.G. BioID: A Screen for Protein-Protein Interactions. Curr. Protoc. Protein Sci. 2018, 91, 19.23.1-19.23.15. [CrossRef]
- Mannix, K.M.; Starble, R.M.; Kaufman, R.S.; Cooley, L. Proximity Labeling Reveals Novel Interactomes in Live Drosophila Tissue. Development 2019, 146, dev176644. [CrossRef]
- Tsiami, F.; Lago, C.; Pozza, N.; Piccioni, F.; Zhao, X.; Lülsberg, F.; Root, D.E.; Tiberi, L.; Kool, M.; Schittenhelm, J.; et al. Genome-Wide CRISPR-Cas9 Knockout Screens Identify DNMT1 as a Druggable Dependency in Sonic Hedgehog Medulloblastoma. Acta Neuropathol. Commun. 2024, 12, 125. [CrossRef]
- Charrin, S.; Naour, F.L.; Oualid, M.; Billard, M.; Faure, G.; Hanash, S.M.; Boucheix, C.; Rubinstein, E. The Major CD9 and CD81 Molecular Partner: IDENTIFICATION AND CHARACTERIZATION OF THE COMPLEXES *. J. Biol. Chem. 2001, 276, 14329–14337. [CrossRef]
- Hegazy, M.; Cohen-Barak, E.; Koetsier, J.L.; Najor, N.A.; Arvanitis, C.; Sprecher, E.; Green, K.J.; Godsel, L.M. Proximity Ligation Assay for Detecting Protein-Protein Interactions and Protein Modifications in Cells and Tissues in Situ. Curr. Protoc. Cell Biol. 2020, 89, e115. [CrossRef]
- Mateos-Martínez, P.; Coronel, R.; Sachse, M.; González-Sastre, R.; Maeso, L.; Rodriguez, M.J.; Terrón, M.C.; López-Alonso, V.; Liste, I. Human Cerebral Organoids: Cellular Composition and Subcellular Morphological Features. Front. Cell. Neurosci. 2024, 18. [CrossRef]
- Rakotomamonjy, J.; Rylaarsdam, L.; Fares-Taie, L.; McDermott, S.; Davies, D.; Yang, G.; Fagbemi, F.; Epstein, M.; Fairbanks-Santana, M.; Rozet, J.-M.; et al. PCDH12 Loss Results in Premature Neuronal Differentiation and Impeded Migration in a Cortical Organoid Model. Cell Rep. 2023, 42. [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the Human Interactome Defines Protein Communities and Disease Networks. Nature 2017, 545, 505–509. [CrossRef]
- Huttlin, E.L.; Bruckner, R.J.; Navarrete-Perea, J.; Cannon, J.R.; Baltier, K.; Gebreab, F.; Gygi, M.P.; Thornock, A.; Zarraga, G.; Tam, S.; et al. Dual Proteome-Scale Networks Reveal Cell-Specific Remodeling of the Human Interactome. Cell 2021, 184, 3022-3040.e28. [CrossRef]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a Glance. J. Cell Sci. 2014, 127, 3641–3648. [CrossRef]
- Ovalle, S.; Gutiérrez-López, M.D.; Olmo, N.; Turnay, J.; Lizarbe, M.A.; Majano, P.; Molina-Jiménez, F.; López-Cabrera, M.; Yáñez-Mó, M.; Sánchez-Madrid, F.; et al. The Tetraspanin CD9 Inhibits the Proliferation and Tumorigenicity of Human Colon Carcinoma Cells. Int. J. Cancer 2007, 121, 2140–2152. [CrossRef]
- Wang, J.; Zhou, Y.; Li, D.; Sun, X.; Deng, Y.; Zhao, Q. TSPAN31 Is a Critical Regulator on Transduction of Survival and Apoptotic Signals in Hepatocellular Carcinoma Cells. FEBS Lett. 2017, 591, 2905–2918. [CrossRef]
- Chen, M.S.; Huber, A.B.; van der Haar, M.E.; Frank, M.; Schnell, L.; Spillmann, A.A.; Christ, F.; Schwab, M.E. Nogo-A Is a Myelin-Associated Neurite Outgrowth Inhibitor and an Antigen for Monoclonal Antibody IN-1. Nature 2000, 403, 434–439. [CrossRef]
- Kleene, R.; Loers, G.; Langer, J.; Frobert, Y.; Buck, F.; Schachner, M. Prion Protein Regulates Glutamate-Dependent Lactate Transport of Astrocytes. J. Neurosci. 2007, 27, 12331–12340. [CrossRef]
- Rotoli, D.; Cejas, M.-M.; Maeso, M.-C.; Pérez-Rodríguez, N.-D.; Morales, M.; Ávila, J.; Mobasheri, A.; Martín-Vasallo, P. The Na, K-ATPase β-Subunit Isoforms Expression in Glioblastoma Multiforme: Moonlighting Roles. Int. J. Mol. Sci. 2017, 18, 2369. [CrossRef]
- Sun, M.Z.; Kim, J.M.; Oh, M.C.; Safaee, M.; Kaur, G.; Clark, A.J.; Bloch, O.; Ivan, M.E.; Kaur, R.; Oh, T.; et al. Na+/K+-ATPase Β2-Subunit (AMOG) Expression Abrogates Invasion of Glioblastoma-Derived Brain Tumor-Initiating Cells. Neuro-Oncol. 2013, 15, 1518–1531. [CrossRef]
- Lefranc, F.; Mijatovic, T.; Kiss, R. The Sodium Pump Could Constitute a New Target to Combat Glioblastomas. Bull. Cancer (Paris) 2008, 95, 271–281. [CrossRef]
- Senner, V.; Schmidtpeter, S.; Braune, S.; Püttmann, S.; Thanos, S.; Bartsch, U.; Schachner, M.; Paulus, W. AMOG/Β2 and Glioma Invasion: Does Loss of AMOG Make Tumour Cells Run Amok? Neuropathol. Appl. Neurobiol. 2003, 29, 370–377. [CrossRef]
- Kim, Y.S.; Choi, J.; Yoon, B.-E. Neuron-Glia Interactions in Neurodevelopmental Disorders. Cells 2020, 9, 2176. [CrossRef]
- Phatnani, H.; Maniatis, T. Astrocytes in Neurodegenerative Disease. Cold Spring Harb. Perspect. Biol. 2015, 7, a020628. [CrossRef]
Figure 2.
Homophilic and heterophilic interactions of AMOG/β2 are plausible at the CNS. (a) AMOG/β2 may engage in heterophilic interactions with neurons at tripartite synapses (b) AMOG/β2 may engage in homophilic interactions with cerebellar granule cells during migration; (c) AMOG/β2 may engage in homophilic interactions with endothelial cells at the blood-brain barrier.
Figure 2.
Homophilic and heterophilic interactions of AMOG/β2 are plausible at the CNS. (a) AMOG/β2 may engage in heterophilic interactions with neurons at tripartite synapses (b) AMOG/β2 may engage in homophilic interactions with cerebellar granule cells during migration; (c) AMOG/β2 may engage in homophilic interactions with endothelial cells at the blood-brain barrier.
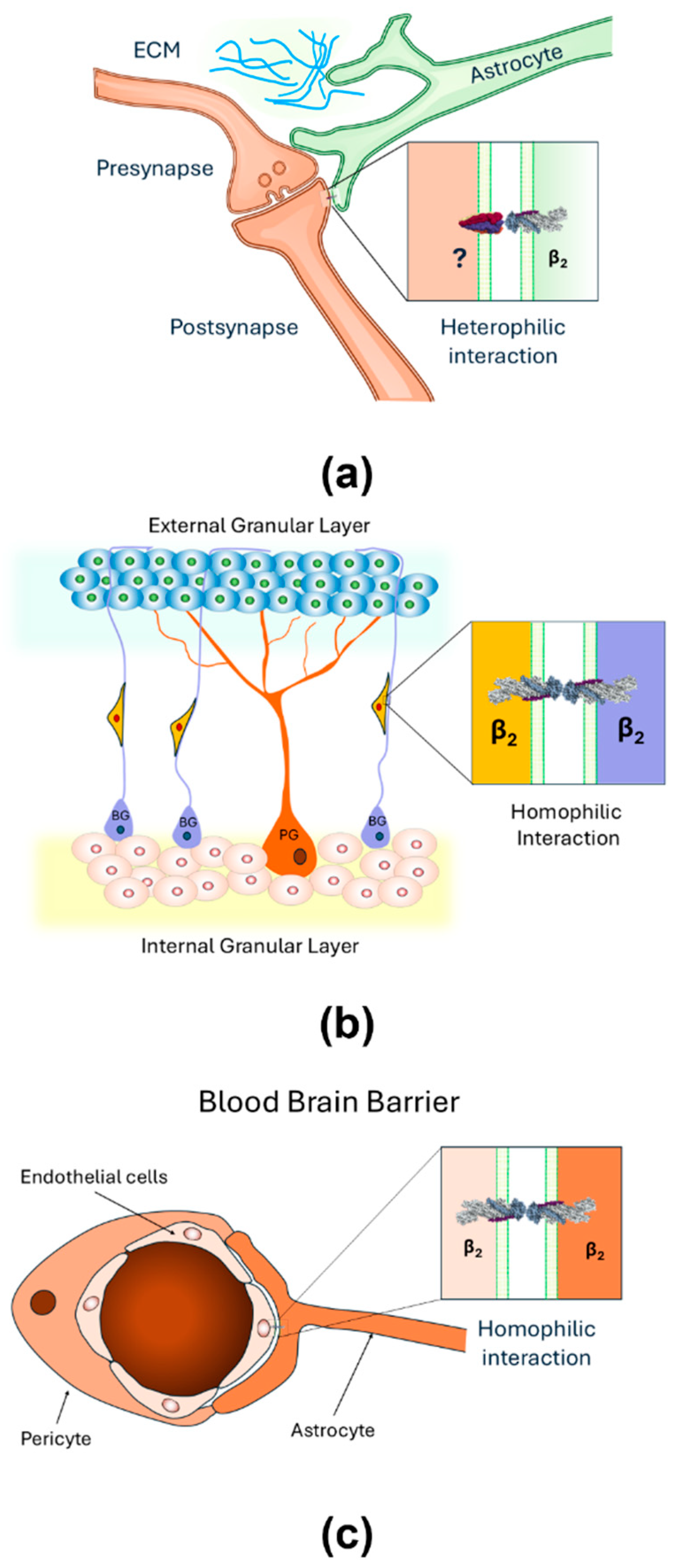
Figure 3.
AMOG/β2 transfected in CHO fibroblasts facilitates aggregation through β2-β2 interactions. (a) A simple illustration of the aggregation assay of transfected or untransfected CHO cells. (b) Images taken by light microscopy of dispersed cells of untransfected CHO fibroblast (left) and big aggregates of AMOG/β2 transfected CHO cells (right).
Figure 3.
AMOG/β2 transfected in CHO fibroblasts facilitates aggregation through β2-β2 interactions. (a) A simple illustration of the aggregation assay of transfected or untransfected CHO cells. (b) Images taken by light microscopy of dispersed cells of untransfected CHO fibroblast (left) and big aggregates of AMOG/β2 transfected CHO cells (right).
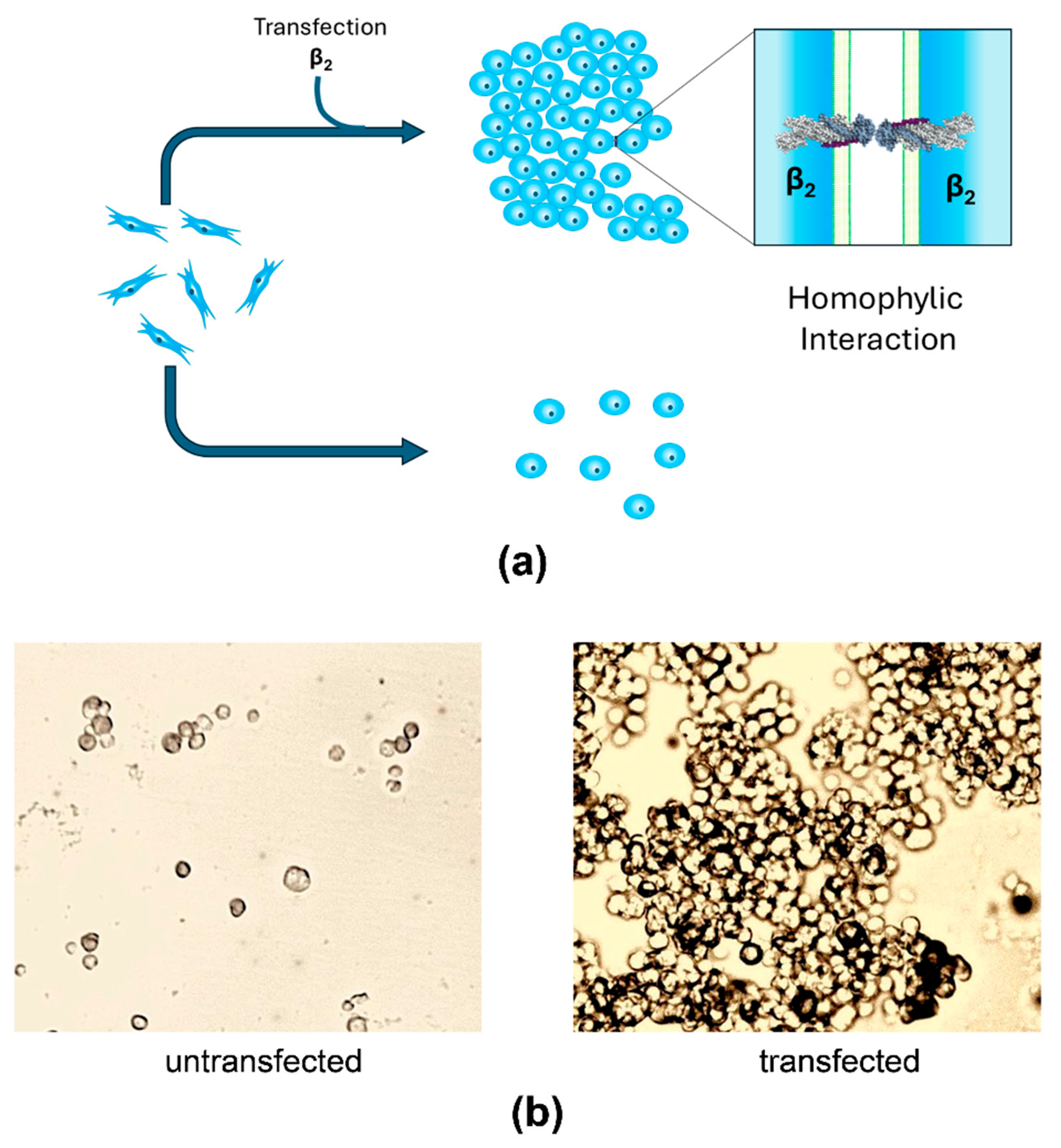
Figure 4.
3D models of trans-dimers of β1 and β2 subunits of human Na+/K+-ATPase. (A) N-Glycosylation sites of β1 and β2 dimers. N-Glycosylations are depicted in orange. (B) Protein-protein interfaces of β1 and β2 trans-dimers. The protein interface is highlighted in red in both models. The 3D models of β1 and β2 are shown in green and magenta, respectively.
Figure 4.
3D models of trans-dimers of β1 and β2 subunits of human Na+/K+-ATPase. (A) N-Glycosylation sites of β1 and β2 dimers. N-Glycosylations are depicted in orange. (B) Protein-protein interfaces of β1 and β2 trans-dimers. The protein interface is highlighted in red in both models. The 3D models of β1 and β2 are shown in green and magenta, respectively.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



