Submitted:
29 July 2025
Posted:
30 July 2025
You are already at the latest version
Abstract
(1) Background: A number of viruses are oncogenic. These include the human papilloma virus (HPV), Epstein-Barr virus (EBV), Kaposi sarcoma human herpes virus 2/human herpes virus 8 (KSHHV/HHV8), hepatitis B virus, (HBV), hepatitis C virus (HCV), Merkel cell polyoma virus (McPyV) and the human T-cell leukemia virus type 1 (HTLV- 1). These viruses cause malignancies ranging from carcinomas, sarcomas, lymphomas to leukemias. This review aims to study the effects and efficacy of vaccines against these viruses and the cancers they cause in their prevention and treatment. (2) Methods: The literature in the past 30 years is searched employing Scopus and Google Scholar using the keywords "oncogenic viruses, HPV, EBV, KSHHV, HHV8, Polyoma virus, HTLV-1, COVID- 19, carcinoma, sarcoma, lymphoma, leukemia, anti-virus vaccines". (3) Results: Prophylactic vaccines against the HPV and HBV are highly effective in preventing and reducing the incidence of uterine cervical and hepatocellular carcinomas. Prophylactic vaccines against other oncogenic viruses have been less successful, though useful in some experimental animals. Therapeutic vaccines are still mostly under evaluation and development. (4) Conclusions: Identification of oncogenic viruses has rendered anti-viral vaccines conspicuous tools for preventing and treating cancers they cause. Much endeavor for development of such vaccines have been met with limited success, apart from the very effective anti-HPV and anti-HBV vaccines in universal vaccination programs. With the development of new vaccine technologies, it is hopeful that effective vaccines against other oncogenic viruses would be developed in future.
Keywords:
oncogenic viruses
; vaccines
; prevention
; therapy
1. Introduction
Oncogenic viruses contribute to cancer development by acting as oncogenes or epigenetically through oncogene activation, tumor suppressor gene inactivation, dysfunctional transcription, interfering cell signaling pathways and triggering oncogenic-enabling events in the microenvironment like inflammation [1,2,3,4]. Oncogenic viruses are among the rare known causes of cancer [1,5] and are often regarded as "low hanging fruits" amenable to cancer prophylaxis, diagnosis, prognostication and therapy [1,6,7]. Furthermore, most humans are infected with some of these viruses, such as the ubiquitous EBV [1,2]. Prophylaxis of the viral infections, especially latent infections, is an effective and economical means to cancer prevention and management [1,2,5,6,7]. Due to the prevalence of these viruses and their significant role in human oncogenesis, development and application of vaccines to prevent the infections and treat the virally caused cancers has attracted much interest and achieved variable success [6,7,8,9]. This review is geared to reviewing viral oncogenesis and the application of vaccinology in the prevention and treatment of the virally caused human cancers.
2. Vaccine Technologies and Platforms
The goal of vaccination is to induce immunological response to insulting agents [10,11]. Under the scope of this review, only oncogenic viruses and the established related cancers are discussed. The vaccination strategies include prophylaxis, thus averting both lytic and latent infections, and therapeutic for established cancers. The gist of vaccination is to introduce relevant insulting protein/antigen targets, including viral or cancer cell derived proteins, into humans to elicit immune humoral and cellular responses. An effective vaccine technology and platform is successful in presenting these antigens/proteins to immune cells to elicit effective robust immune cascades, production of specific neutralizing antibodies and cellular immunity. The immune cells involved include antigen presenting cells (APC), dendritic cells (DC), macrophages, B cells, T cells and natural killer (NK) cells [10,11].
2.1. Anti-Viral Vaccines
- i.
- Protein Platform
- 1.
- Whole Inactivated (killed) vaccines
This involves inactivation/killing of whole viruses by chemical or physical means or both [10,12]. The commonly use chemicals are formaldehyde, glutaraldehyde, ascorbic acid, hydrogen peroxide, beta-proiolactone and ethylenimine dervatives. Physical means commonly employed are heat, ultraviolet light and gamma irradiation [11,12]. These vaccines are safe, particularly in immunocompromised subjects, as the pathogenic viruses have been killed. They are, however, less immunogenic especially in eliciting cellular immunity. Adjuvants are usually required for enhancing vaccine. Nevertheless, immune responses to more viral protein targets compared to subunit vaccines may be produced. Vaccine production is, however, time consuming, more expensive and less amenable for rapid response to fight emerging outbreak pathogens [10,12].
- 2.
- Live Attenuated Vaccines
Viruses are attenuated through serial passaging under suboptimal conditions or temperatures and using genetic engineering methods that increase replication fidelity or codon de-optimization. These methods select mutations that disable their viral pathogenic capacities [10,12,13,14,15]. Live attenuated vaccines have enhanced immunogenicity and elicit potent cellular and innate responses. Most do not require adjuvants and a single vaccine dose can drive life long immunity. The disadvantages are their residual minor pathogenic potential especially in immunocompromised subjects [10,12,16,17], and the labor intensive and costly production [10,12,18].
- 3.
- Virus-like Particles (VLP) Vaccines
VLP are macromolecular assemblies of at least two viral structural components, meant to imitate the morphology and protein structure of the virus. VLP are bio-manufactured after transfection of bacterial, yeast, insect, plant or mammalian cells with one or more genetic constructs of the virus resulting in self-assembly into incomplete viral protein particles [19]. Fine tuning of VLP immunogenicity is possible through surface chemical modification and adding immunogenic peptides and adjuvants [20]. VLP vaccines are effectively targeted by B cells, DC and APC. Interaction within and increased avidity for innate immune cells increases VLP vaccine immunogenicity [21]. Manufacturing costs are however higher.
- 4.
- Synthetic Peptide Vaccines (SPV)
SPV contain immunodominant viral peptides which requires extensive in vitro screening and modeling to discover. SVP are synthesized using fragment condensation or solid-phase synthesis. These small peptides are mixed with or conjugated to carefully chosen adjuvants to avoid peptide denaturation and to enhance uptake by APC. SPV are safe and cause focused epitope-specific immune responses, though epitope restriction reduces response breadth and may result in escape variants of the virus [10,12].
- ii.
- Nucleic Acid Platforms (NAP)
NAP use selected nucleic acid sequences to generate viral proteins and antigens that are not disease-causing with production of potent and host immune responses [10,12]. Modular designs in NAP enable their application in outbreaks with rapid and large scale manufacturing at lower costs. NAP are useful against emerging and new pathogens, pandemic threats and escape viral variants [10,12].
- Bacterial and Viral Vectored Vaccines
These vaccines utilize non-pathogenic or attenuated pathogenic bacteria and viruses as carriers of nucleic acids sequences of the target pathogenic virus. Bacterial vectored vaccines are suitable for mucosal administration. Though there is risk of infection by the bacterial vector in the immunocompromised, the risk is much mitigated by identification and deletion of bacterial virulence genes with genetic engineering. Viral vectors are engineered to be replication incompetent while retaining infectivity and expression of the encoded target antigen. Replication incompetent vectors do not cause productive infection and are safe and easy to manufacture. Examples of bacterial vectors are non-pathogenic Lactobacillus sp and attenuated Yersinia pestis. Some viral vector examples are vesicular stomatitis virus, vaccinia, adenovirus, poxvirus and Newcastle disease virus [10,12,22,23,24].
- 2.
- Synthetic DNA Vaccines (SDNAV)
SDNAV are large, polyanionic and nuclease sensitive and require special methods of delivery to recipients, including the more commonly used electroporation and nanoparticle systems [10,25]. SDNAV is delivered into muscle tissue leading to transfection of recipient cells with DNA translocation to nuclei, mRNA transcription and target protein translation. SDNAV stimulates both humoral and cellular immunity causing robust immunogenicity and sustained potent responses. The theoretic risk viral DNA integration recipient cell genomic DNA has been shown to be speculative [26].
- 3.
- mRNA based Vaccines (mRNAV)
The drawbacks are RNA short half-life and reduced protein translation aggravated by the recipient inflammatory response, which are circumvented by loading the mRNA into liposomes [10,12,27]. Recent technological advances such as incorporation of modified nucleosides into in vitro transcribed mRNA, sequence engineering, codon optimization and utilization of safe lipid nanoparticle delivery systems further enhances the stability of mRNAV [28.29]. There are 3 main categories of mRNAV: conventional, self-amplifying (SA) and circular ( c) RNA. Extended protein translation is possible in SAmRNA due to viral derived molecular machines that enable intracellular mRNA sequence amplification [10,30]; and in cRNAV through addition of interval ribosaomal entry sites [10,31]. mRNAV elicit potent cellular and humoral immunity which is partly due to their delivery systems such as nanoparticles [27,32]. There is no risk of genomic DNA integration. They are easy to manufacture but require ultralow temperatures to store and transport.
2.2. Anti-Cancer Vaccines (ACV)
ACV are therapeutic vaccines developed against established cancers, including virus induced cancers. As for anti-viral vaccines, the aim of ACV is to elicit potent immunity, mostly cellular immunity, against cancer cell antigens that are exogenously or endogenously introduced. ACV may also augment host immunity or induce changes in the tumor microenvironment (TME). The former requires high quality cancer antigens for presentation to DC, helper and cytotoxic T cells and other cellular constituents of the TME. Another concern of ACV is possible development of intrinsic tumor resistance to the vaccine due to cancer cell genomic instability with alteration in expression and processing of tumor antigens [11,33,34]. In view of this, antigen agnostic in-situ vaccines (ISV) that modify and augment immune robustness in the TME and provide directly tumor-associated antigens by inducing in-situ cancer cell death are developed [11,35]. ISV, however, may also be compromised by tumor resistance through mechanisms of interfered T cell functions, development of inhibitory cytokines/regulatory cells and remodeled extracellular matrix [11,36,37].
ACV Technologies and Platforms
- Tumor Antigens
The quality of tumor antigens is at the center stage of ACV for robust effectiveness and efficacy. There are 2 commonly used categories of antigens.
- (a)
- Neoantigens
Neoantigens are mutated tumor antigens produced through non-synonymous somatic and frameshift mutations and in coding regions, human endogenous retroviruses, post-translational protein phosphorylation, citrullination and glycosylation [11,38,39,40,41]. This is usually applicable to tumors with high tumor mutation burden (TMB) which presumably provide large numbers of tumor-rejection neoantigens for vaccine targeting [11,42].
- (b) Shared antigens
These ACV utilize several shared tumor-associated antigens, usually applicable to tumors with low TMB [11,43,44].
- 2.
- ACV Platforms
These include direct antigen administration using neoantigens, RNA and synthetic long peptides. They could be administered intramuscularly, intravenously, by antigen delivery through loaded DC or non-cellular particles, nanoparticles, liopoplexes, amphophilic systems, liposomes or polyethy-eneimine silica microrods [11,47,48,49].
- 3.
- In-situ Vaccines (ISV)
ISV are antigen agnostic and not targeted at any individual tumor antigen. They aim to enhance endogenous anti-tumor responses by affecting the TME or distal sites by abscopal effect [11,35]. The TME cellular targets include APC, DC and T cells resulting in proliferation, activation and maturation of the cells leading to a robust anti-cancer cell immune response. Oncolytic viruses (such as herpes simplex virus, parvovirus, adenovirus) may be employed which are effective due to activation of immune cells and induction of tumor cell death thus releasing tumor-associated antigens. Further, oncolytic viruses enriched for the toll-like receptor agonist CpG (cytosine-phosphate-guanine) motifs strongly activate APC functions [11,35,50,51]. Oncolytic viruses administered to the TME in combination with agents that suppress the induction of immunosuppressive cells (regulatory T cells) also contribute to enhanced anti-tumor immunity. The agents used in combination with oncolytic viruses may include cyclophosphamide, temozolomide and immune checkpoint blocking antibodies [35,52,53,54,55].
- 4.
- Combined Vaccination
This strategy may be employed in advanced cancers as part of a multiphasic approach (surgery, chemotherapy, radiotherapy and vaccination) [11].
2.3. Adjuvants
Adjuvants are substances that enhance vaccine immunogenicity. While inactivated vaccines contain adjuvants introduced during vaccine production, most modern vaccines contain small components of the target proteins/antigens and may require additional adjuvants. Adjuvants range from aluminum salts, emulsions, liposomes, nanoparticles to VLP. Adjuvants target innate immune cells and activate pattern recognition signaling pathways [56,57,58]. mRNA vaccines further possess intrinsic adjuvant activity due to the nucleoside-unmodified RNA (in particular in double stranded RNA) which triggers innate immune signaling [52]. The delivery systems (such as lipid nanoparticles) used in modern vaccines are also contributory adjuvants [56,57,58].
3. The Oncogenic Virus Orchestra, Tumors and Vaccines
Viruses may cause cancer by acting as oncogenes, activating host oncogenes, creating host genomic instability, inhibiting host tumor suppressor genes and proteins, interfering cellular signaling pathways, increasing cell proliferation, resisting cell apoptosis, affecting DNA repair, contributing to tumor evasion through genetic or epigenetic mechanisms [1,2,3,4,59,60]. Oncogenic viruses is estimated to be responsible for up to 15% of cancers worldwide and about 20% in developing countries [1,5,59,60,61]. The best known 7 oncogenic viruses are described in the following sections. They may act as solos, duets or trios in the virus orchestra, with the coinfections contributing to higher cancer incidence or more advanced disease. The human immunodeficiency virus (HIV) is per se not oncogenic but plays an important role in cancer development when playing in the orchestra [1,2,59,60,61]. The SARS-Cov-2 virus, postulated to be possibly oncogenic, is also discussed below.
3.1. Human Papilloma Virus (HPV)
3.1.1. Virology and Oncogenesis
The HPV is a non-enveloped DNA virus with a double stranded genome containing about 8000 base pairs. The genome is enclosed in an icosahedral capsid made up of the structural proteins L1 and L2, which are encoded by the late region of the HPV genome. Non-structural proteins E1, E2, E3, E4, E5, E6 and E7 are encoded by the early region and E6 and E7 are most essential for oncogenesis. The HPV belongs to the family Papillomaviridae in which more than 400 HPV genotypes have been identified and categorized into the five genera: Alpha, Beta, Gamma, Mu and Nu. The Alpha HPVs contribute most to oncogenesis. HPV genotypes are classified according to their oncogenic potential into high risk (hrHPV) or low risk (lrHPV). The hrHPV are 16,18,31,33,35,39,45,51,52,56,58,59,66 and 68. HPV16 and 18 are responsible for more than 70% of global cervical cancer cases [62,63].
The protoype of HPV oncogenesis is uterine cervical cancer. Cervical cancer is the fourth leading cancer and cancer deaths in women in 2020 [62,63]. HPV infection is acquired through the basement of the affected epithelium and infects the basal cells where it remains in episomal form. It replicates in the upper epithelial layers, exploiting host tissue renewal mechanism. Most HPV infections resolve spontaneously. Persistent infection, however, results in disease and may lead to cervical intraepithelial neoplasia (CIN) or invasive cancer especially by HPV16 and 18. HPV DNA integration into the host genome causes genomic instability, gene alteration, loss of tumor suppressor gene function, chromatin reorganization, chromosomal rearrangement and epigenetic dysregulation. HPV integration usually occurs at hotspots (site-specific susceptibility) in the host genome. Examples are FH1T, KLF5, LINC00392 and MACROD2. MACROD2 dysfunction can cause dysregulation of PAR1 which is involved in cell differentiation, proliferation and tumor transformation. HPV integration is also dependent on host and other viral cofactors. These may be cervicovaginal microenvironment and microbiota, smoking, estrogen level, mental stress and coinfection with HIV.
HPV integration disrupts the E2 open-reading frame, causing loss of E1/E2 interaction for initiating viral replication, dysfunctional E2 regulation of viral early promoter and overexpression of oncoproteins E6 and E7. The integration percentage increases with disease progression from CIN to cervical cancer. Overexpression of E6 and E7 further promotes HPV integration in the journey to cancer development [60,61,62]. For the latter event, genomic alterations involving the PI3K/MAPK and TGF-beta signaling pathways and mutations in ERBB3(HER3), CASP8, HLA-A, SHKBP1 and TGFBR2 have been reported [63,64]. Despite HPV integration being the most important oncogenic event in cervical cancer, there are other oncogenic mechanisms other than HPV DNA integration. The episomal E2, E4 and E5 oncogenic pathway [62,65], AP1/Wnt/beta-catenin pathway [62,66], PI3K/AKT/mTOR [62,66] and E2 methylation [62,67,68] are non-HPV integration mechanisms that have been reported in cervical cancer development.
3.1.2. HPV-Associated Cancers
About 91% of HPV-associated cancers in women are cervical cancers which is the most common HPV-associated cancer subsequent to persistent infection in 5-15% affected women [62,63]. Cofactors are often involved including co-infection with HIV, Epstein-Barr virus (EBV), sexually transmitted diseases (Herpes Simplex, Chlamydia, Gonococcus), smoking, immunosuppression, young age at first pregnancy, hormone contraception and multiparity [60,61,62,63]. The most frequently responsible hrHPV are HPV16 and 18. Though approximately 90% of cervical cancers occur in low and middle income countries [61.63], the introduction of prophylactic HPV vaccination may potentially reshape its epidemiologic landscape.
HPV-associated cervical cancers are squamous cell carcinomas (SCC), classified histologically as non-keratinizing, keratinizing, basaloid, condylomatous, papillary or rarely lymphoepithelioma-like [69] (Figure 1). The prognosis of HPV-associated cervical cancer is better than the HPV-independent counterpart [69,70]. HPV-associated cancers also affects other anatomical sites. These include SCC of anus, 15-48% of vulval carcinoma, 78% of vaginal carcinoma, 53% of penile carcinoma, 13-60% of oropharyngeal carcinoma and <5% of laryngeal carcinoma [1,60,61], mostly caused by HPV16 [61]. Similar to cervical cancers, HPV-associated cancers of the head and neck region fare better than their HPV-independent counterparts [71].
3.1.3. Vaccines
These include prophylactic vaccines for disease prevention and therapeutic vaccines to treat established cancers.
- Prophylactic Vaccines (PV)
PV against HPV were first licensed in 2006. Currently, there are 6 licensed recombinant DNA PV, manufactured based on purified L1 structural protein. The proteins produced form HPV type specific empty shells or VLP which are potent immunogens bearing no pathogenic or infectious potential. They are fortified with adjuvants and all are against HPV16 and 18. The vaccines are bivalent (HPV16 and 18), quadrivalent (HPV6,11,16,18), nonavalent (HPV6,11,16,18,31,33,45,52,58) [61,72,73]. To date, 125 countries (64%) have introduced HPV vaccine in their universal immunization program for girls, and 47 countries (24%) for boys [61]. PV are aimed to be administered before sexual exposure and are indicated for females aged 9 or older. PV against HPV has been found to substantially reduce the incidence of CIN3 and cervical cancer, risk of invasive cervical cancer, and marked decrease in incidence of these diseases in socioeconomically deprived populations [7,61,72,73]. They are effective in HIV-positive subjects [61].
- 2.
- Therapeutic Vaccines (TV)
TV are based on mechanisms discussed under Section 2.2. Oncolytic viruses have been shown in preclinical settings to be an effective immunotherapy for HPV-associated cancers [74]. They could stimulate anti-tumor immunity and eradicate cancer cells [62,74]. No licensed TV agaist HPV, however, is available to date.
3.1.4. The Viral Orchestra
While HPV is the primary causative oncovirus and can cause cancer as a soloist on its own, coinfections with HIV and EBV which aggravate the incidence and severity of the cancer caused may act as duets or trios.
- The duplet HPV and HIV
The incidence of HPV-associated anal, cervical and oropharyngeal cancer is 80, 22 and 6 times higher in HIV-infected compared to HIV-uninfected subjects [75,76,77,78]. The incidence of oral, head and neck, liver, lung, testicular and kidneys cancers is also increased in HIV-infected individuals even when the cancers are HIV-negative [79]. While HIV may increase the risk of cancer development by attenuating local and systemic immunity, it has been observed that HIV may induce epithelial-mesenchymal transition (EMT) which is a process of losing the epithelial characteristics: apical polarity, cell tight junctions, epithelial immunophenotype (such as E-cadherin) and cell adhesion. This is followed by expression of mesenchymal (such as vimentin) and stemness phenotype (such as CD133 and CD44), dedifferentiation, cancer development and invasiveness [78]. EMT is regulated by transforming growth factor [80] and the MAPK [81] signaling pathways. Inhibition of these pathways may prevent HIV-associated acceleration of malignancy. It has been demonstrated that prolonged exposure of HPV-16 immortalized and HPV-uninfected epithelial cells to cell free HIV-1 virions or the HIV-1 viral proteins gp120 and tat (trans-activator of transcription) results in promotion of EMT and cancer invasiveness [78]. Though HIV plays a role in HPV-associated cancers, the incidence these cancers has not been reduced by anti-retroviral therapy (ART) in HIV-infected populations [77]. This may be due to limited penetration of ART drugs into solid tissues [82]. Effective anti-HIV vaccines, however, have regrettably been unavailable [83].
- 2.
- The duplet HPV and EBV
Co-infection with EBV has been documented in HPV-associated CIN and cervical cancers [84,85,86,87,88]. While EBV has been considered a co-factor [86], it is also etiologically related to HPV-associated cervical cancers [87]. Co-infection with EBV increases HPV16/18 integration into the host genome [85,86,87], thus promoting cancer development [60,61,62]. EBV oncoproteins (such as Epstein-Barr nuclear antigen 1, EBNA1) promote EMT, angiogenesis and activation of the signaling pathways NF-KB (nuclear factor kappa-light-chain-enhancer of activated B cells), MAPK (mitogen-activated protein kinase), JAK/STAT (Janus kinase/signal transducers and activators of transcription). Other EBV latency products may also play a role. LMP1 (latency membrane protein 1) suppresses host immunity and BARF1 (BAMHI-A rightward frame 1) promotes cell proliferation [60,87,88].
- 3.
- The trios HPV, HIV and EBV
3.2. Epstein-Barr Virus (EBV)
3.2.1. Virology and Oncogenesis
EBV is a ubiquitous gamma herpes enveloped double stranded DNA virus
Also known as human herpes virus 4 (HHV4). It infects over 90% of the world human population and spreads by oral secretions. Most infections occur during early childhood and are asymptomatic. Development of infectious mononucleosis is rare though more frequent in well developed countries [2,8,91]. After cell entry, EBV DNA enters the host cell nucleus and forms a stable episome for long term survival and latent infection [2,91]. The latent infection is implicated in cancer development and it is estimated that EBV-related malignancies are responsible for 0.2-0.3 million new cancer cases and 0.1 -0.2 million cancer deaths in 2020 [91,92]. EBV is tropic for B lymphocytes and epithelial cells. Viral entry is through interaction with CD21 on B lymphocytes and integrin on epithelial cells [2,93]. EBV also infects NK and T cells, probably mediated by CD21 acquired from infected B cells by synaptic transfer [2].
There are 3 patterns of latency with differential expression of 6 nuclear proteins (EBNA1, 2, 3A, 3B, 3C and leader protein EBNA-LP), 3 latent membrane proteins (LMP1, 2A and 2B), BART mRNA and EBER 1 and 2 (EBV small non-coding non-adenylated RNA). Restricted EBV products expression is present in latency 1 (EBNA1, EBERs and BARTs) and latency 2 (LMP1, LMP2, EBNA1, EBERs and BARTs). Latency 1 is observed in Burkitt lymphoma. Latency 2 expression is present in nasopharyngeal carcinoma (NPC), gastric cancer (GC), Hodgkin lymphoma (HL) and NK/T-cell lymphoma (NK/TL). Latency 3 expresses the full repertoire of EBV products and is observed in lymphomas that develop in transplant recipients and immunocompromised subjects [2,60,94,95,96].
Among the latency proteins, LMP1 is the major transforming protein mimicking constitutive CD40 receptor signaling causing cell proliferation. Other EBV proteins interact with signaling pathways, cause c-myc activation and dysregulation, induce oxidative stress, suppress MHC-II gene expression, enhance PD-L1 expression, recruit SWI/SNF complex and synergize with EZH2. This leads to cell proliferation, increased cell survival, decreased apoptosis, immune evasion, TME changes, cell transformation and cancer development [2,3,91,92,94,95,96,97]. The 6 major signaling pathways involved in EBV oncogenesis are NF-KB, PI3K (phosphoinositide-3-kinase), JAK/STAT, MAPK, TGF-beta (transforming growth factor-beta) and Wnt/beta-catenin pathways [2,3,4,60,96,98].
Apart from latency products, EBV lytic products also contribute to oncogenesis [60,94,96,99,100,101]. There is decrease in oncogenesis in cell cultures and experimental animals where the genes BZL1, BRLF1 and BALF5 encoding early lytic proteins are knocked out. The early lytic proteins BZLF1, BRLF1 and BLLF3 increase expression of cytokines IL6, IL8, IL10, IL13 and IL1B, thus leading to immune modulation and evasion. BHRF1 and BALF1 are bcl2 homologs (vbcl2) and overexpression inhibits apoptosis. Early lytic proteins may also contribute to oncogenesis and invasiveness by causing genomic instability and angiogenesis. The lytic phase microRNAs (BART and BHRF1-2) contribute by inhibiting tumor suppressors (PTEN and PRDM1) and ILR-1 receptor [60,94,96,99,100,101].
3.2.2. EBV-Associated Cancers
- Nasopharyngeal Carcinoma (NPC)
NPC is a rare cancer worldwide but shows high prevalence in southern China, south-east Asia, among inuit populations in Alaska and Canada and some populations in northern Africa (Algeria, Tunisia) [102,103]. It affects more commonly the NP lateral wall (especially the fossa of Rosenmuller) and may present with symptoms of an NP mass (blood-stained post nasal drip, epistaxis, nasal obstruction), aural symptoms, diploplia, facial numbness, headache or regional lymph node enlargement. There are 3 histological types: non-keratinizing SCC (NKSCC), keratinizing SCC (KSCC) and basaloid SCC. The NKSCC may show a rich intratumoral lymphoplasmacytic infiltrate, qualifying the designation “lymphoepithelioma-like carcinoma (LELC)”. NKSCC is characterized by invasive syncytia of large epithelioid or spindle tumor cells with indistinct cell membranes forming whorls, fascicles or reticular pattern [102] (Figure 2). EBV is practically always associated with NKSCC with demonstrable ERBER. Positive EBV serology and elevated plasma EBV DNA are demonstrated in most NKSCC and are useful as an independent prognostic factor, marker of therapeutic response, tumor surveillance, relapse and metastasis prediction. [102,104]. Etiologically, the ethnicity prediction of NPC indicates genetic susceptibility with strong linkage of risk to MHC Class I variants (HLA-A, HLA-B, HLA-C) in Chinese patients [105], which may be related to dysfunctional immunosurveillance of EBV. Apart from the oncogenic mechanisms discussed before, interaction with host factors may also be important. Recognized host genetic factors are single nucleotide polymorphism in MDS-EV11, TNFRSF19, CDKN2A, CDKN2B, TERTICLPTM1L and inactivated tumor suppressors CDKN2A and TGFBR2 [101,106,107].
- 2.
- Gastric Cancer (GC)
EBV-associated GC accounts for <10% of GC worldwide depending on the region and detection methodology [108,109,110,111,112]. This is designated gastric adenocarcinoma with lymphoid stroma in the WHO Classification [108], and is also referred to as LELC and medullary carcinoma [108,112]. It forms sheets, trabeculae, ill-defined tubules or syncytia of large tumors with prominent lymphoid stroma. Other EBV-associated GC histologic subtypes have been reported which differ in genetic alterations and PD-L1 expression [111].
EBV-positive GC is one of the molecular GC subtypes proposed by the Cancer Genome Atlas (TCGA) Research Network. It displays PIK3CA, ARID1A mutations, genome-wide hypermethylation and amplification of the CD274 (PD-L1) gene [108,109,110,111,112].
There is predilection for the proximal stomach or gastric stump and the male sex. The prognosis of EBV-associated GC is better than the non-EBV-associated counterparts. Treatment is surgical resection in early stage and advanced stage GC may require DNA methylation inhibitors, proteasome inhibitors, histone deacetylase inhibitors, EBNA1 inhibitors and EBV-specific cytotoxic T cell infusion [108,113].
- 3.
- Lymphoepithelial carcinoma of the lung (LEC)
LEC is classified as a poorly differentiated SCC in the WHO Classification [114]. The carcinoma accounts for <1% of non-small cell lung cancer (NSCLC) and predominantly affects younger (median age 51 years) female Asian non-smokers [114,115] EBV association is >90% in Asian patients but much less in Europeans. EBV serology titer is correlated with burden and stage [116]. LEC does not harbor the genetic alterations common to conventional NSCLC. Histologically, there is resemblance to NKSCC of the NP, often with pushing borders (Figure 2). It is therefore important to exclude secondary from NP NKSCC. Survival is better compared to other NSCLC. Treatment is surgical resection, with neoadjuvant or adjuvant chemotherapy, chemoradiotherapy or immunotherapy in advanced disease [116].
- 4.
- Thymus Epithelial Tumors (TET)
TET encompass thymomas and thymic carcinomas. Despite isolated reports of EBV-association in thymomas, the involvement of EBV in thymomas remains controversial [117,118,119]. EBV etiological involvement in thymomas is not recognized in the WHO Classification. The WHO and others recognize LEC to be EBV-associated [117,118,120]. About half is etiologically related to EBV. EBV is almost always positive children and young adults only only uncommon in those >30 years of age [120]. It is a poorly differentiated SCC with a rich lymphoplascytic infiltrate, similar to NP NKSCC and lung LEC. Lymphocyte poor cases are recognized and diagnosis is supported by EBER-positivity. Thymic LEC accounts for 1-6% of thymic carcinomas, and affects more males with median age of 41 years (range 4-76 years) [120,121]. Most advanced cases present with superior vena caval syndrome. There is no association with myasthenia gravis. LEC spreads locally to neighboring organs. Lung, liver and bone are the more frequent metastatic sites [119]. Prognosis is poor which is independent of EBV status [120].
- 5.
- Burkitt Lymphoma (BL)
The role of EBV in BL has recently been elucidated. The defining molecular event in BL is the juxtaposition of the myc oncogene to the immunoglobulin gene heavy chain(IGH) locus t(8.14)(q24;q32) and less commonly to the IG lambda (IGL) or kappa (IGK) locus t(8,22)(q24;q11) or t(2,8)(p12;q24). This translocation activates the myc gene leading to lymphogenesis [122]. EBV infection in BL contributes to suppression of myc induced apoptosis through the latency proteins EBNA1, EBNA2, ENA3A, EBNA3C, LMP1, LMP2A, the latency microRNAs BARTs, EBERs and the lytic proteins BHRF1, BALF1 [94,99].
EBV is associated with almost 100% of BL in endemic areas (sub-Saharan Africa, Malawi, Uganda, Cameroon), but shows much lower association (20%) in other areas (sporadic BL) or immunodeficiency-associated BL [123]. BL may be typed as EBV-positive and EBV-negative, with differing biology and pathogenesis. EBV-negative BL features higher frequency of mutations in TCF3 and its negative regulator ID3, resulting in tonic B-cell receptor (BCR) signaling. In EBV-positive BL, intense antigenic pressure induced by EBV causing TME interactions on BCR leads to BCR chronic stimulation, B cell clonal expansion and neoplasia [124,125]. Other cofactors in BL oncogenesis are co-infection with malaria or HIV [96,127]. EBV-negative BL, however, may be the result of a “hit-and-run” initial EBV infection that plays an initiating role to be followed by loss of the EBV genome on acquisition of more stable genetic and epigenetic status in the lymphoma cells [128].
BL is characterized histologically by invasive sheets of immunophenotypically B lymphoma cells with squared off cytoplasmic borders, round nuclei, fine clumped chromatin, brisk mitotic activity, often with interspersed tangible body histiocytes to impart the characteristic “starry sky” appearance (Figure 3) [122,129]. Employing contemporary immunochemotherapy, prognosis is good with overall survival of 90% in children and 80% in adults [122,129,130].
- 6.
- Hodgkin Lymphoma (HL)
HL is classified into classic HL (CHL) and nodular lymphocyte predominant HL (NLPHL). The neoplastic cells in CHL are the Hodgkin and Reed-Sternberg (HRS) cells while those in NLPHL are germinal center derived B cells known as LP (lymphocyte predominant) cells [131]. CHL is usually EBV-associated while NLPHL is not. The neoplastic HRS cells are crippled germinal center (GC) cells plagued with high loads of somatic mutations in the rearranged IGH gene. These deleterious mutations usually cause apoptosis. The HRS cells, however, are salvaged by survival signals including those from EBV infection (vbcl2) [101,132]. HRS cells have mostly lost their B-cell phenotype due to the remarkable genetic and metabolic aberrancies. The mixed cellularity and lymphocyte depleted CHL subtypes are most frequently EBV-positive, while the nodular sclerosing type is mostly EBV-negative [131]. The histopathology of CHL varies with the subtypes but all show HRS cells in variable amounts with a TME of reactive lymphocytes, eosinophils, histiocytes, neutrophils, plasma cells and fibrosis in varying proportions according to the subtype [131]. CD30 and CD15 immunopositivity is frequent in the HRS cells (Figure 3). A great majority of CHL can be cured with modern polychemotherapy and radiotherapy [131].
- 7.
- Extranodal NK/T –cell lymphoma (ENNKTL)
Most (60-90%) ENNKTL are derived from NK cells and the remaining 10-40% from T cells [2,132]. Eighty percent are nasal and 20% non-nasal. Nasal NNKTL presents with midfacial disfiguring destructive lesions of the upper aerodigestive tract and affects males in the 4th to 5th decades. It is histologicallycharacterized by diffuse sheets of atypical lymphoid cells, usually with a broad range of sizes and irregular nuclei. Epithelial invasion, angiocentricity, agioinvasion and extensive geographical necrosis are typical features. The NK-cell type is negative for surface CD3, T-cell receptor (TCR), positive for CD56 with no TCR rearrangement. The T-cell type is surface CD3+, TCR+ with rearranged TCR. There is frequent cytotoxic molecules expression (Figure 4). A strong etiologic relationship with EBV (latency 2) is present [129,133]. A multitude of alterations in genes encoding proteins of the JAK/STAT and NF-KB pathways and epigenetic changes including dysregulated microRNA, tumor suppressor genes, myc overexpression and effects of EBV latency products (LMP1 contributes to immune evasion) are operative [2,129,133]. Treatment involves concurrent or sequential chemoradiotherapy usingL-asparaginase containing non-anthracycline regimes. Immune check point inhibitors and stem cell transplantation may be required [2,129,134]. Plasma EBV DMA level is an indicator of chemosensitivity and prognosis [2,129,135].
- 8.
- EBV+ Nodal T- and NK-cell Lymphoma (NTNKL)
EBV lays a major etiologic role in EBV+ NTNKL [2,135]. Most display latency 2, though LMP1 is often undetectable. Over 80% is of T-cell lineage, mostly CD8+ with alpha-beta TCR phenotype, though a gamma-delta TCR phenotype with inferior survival may be present. Expression of cytotoxic molecules is frequent. The involved lymph nodes are infiltrated by medium-sized to large blastic tumor cells. Angiocenctricity and necrosis are not typical. NTNKL is genomically unstable with alterations in genes related to the JAK/STAT and NF-KB pathways. The degree of genomic instability is lower than ENNKTL. Prognosis is grave [2,136].
- 9.
- Systemic EBV+ T-cell Lymphoma of Childhood (SEBVTCLC)
There is strong EBV etiologic association related to an undefined genetic defect in the host immune response to EBV [137]. Transformation and progression from systemic chronic active EBV disease (CAEBV) may occur [2]. The involved tissues show infiltration by atypical lymphoid cells of a broad cytologic spectrum. Most cases are CD8+, while those arising from systemic CAEBV are CD4+ [2]. Distinction from fulminant systemic CAEBV and hemophagocytic lymphohistiocytosis is often difficult [2]. SEBVTCLC is highly aggressive with grave prognosis.
- 10.
- Aggressive NK-cell Leukemia (ANKL)
ANKL are rare and almost always EBV-associated. About 20 cases of EBV-negative ANKL have been reported [2,138]. It is a systemic disease causing hepatosplenomegaly with involvement of the peripheral blood and bone marrow. The atypical leukemia cells may show granular or agranular cytoplasm with a broad spectrum of atypical nuclei. They are CD56+, cytotoxic molecules+ and surface CD3-. Genetically, there are alterations in genes involved in the JAK/STAT and RAS/MAPK pathways. Prognosis is grave with no established treatment protocol [2].
- 11.
- EBV+ Inflammatory Folicular Dendritic Cell Sarcoma (IFDCS)
EBV+ IFDCS is rare with only about 80 reported cases and is distinct from EBV-negative FDCS. It affects predominantly the spleen and liver, though GIT and upper aerodigestive tract cases have been reported [139]. Asians are mostly affected, with female predominance. The neoplastic cells are spindle to oval occurring singly or form loose fascicles and whorls. In IFDCS, there is a prominent lymphoplasmacytic infiltrate, due to which the tumor has been interpreted as an inflammatory psudotumor [140]. A recent study identified 3 subtypes of IFDCS: classic, lymphoma-like and hemangioma-like, thus often causing diagnostic confusion [141]. Surgery is the mainstay treatment and most patients have favorable outcomes [139,140,141].
- 12.
- Other EBV-associated lymphoproliferative diseases (LPD)
These include EBV-positive mucocutaneous ulcer (MCU), lymphomatoid granulomatosis (LyG), EBV-positive diffuse large B-cell lymphoma (DLBL) and plasmablastic lymphoma (PBL). These conditions are and pathogenetically related to immunodeficiency and dysfunction (IDD). In EBV+ DLBL, which affects mostly the elderly, IDD may be due to immunosenescence which may also account for IDD in LyG. In EBV+ MCU, IDD may be inborn or acquired. IDD in PBL is acquired (as in HIV infection or immunosuppressive therapy) or related to immunosenescence. IDD causes abnormal host response to EBV. EBV+ MCU affects the mucosal and cutaneous tissue only, while LyG affects the lungs predominantly. Both MCU and LyG show invasion by polymorphous atypical lymphoid cells with frequent angioinvasion and necrosis (in high grade LyG). EBV+ DLBL may be nodal or extranodal with polymorphic or monomorphic lymphoid infiltrates dominated blastic cells exhibiting the activated B-cell immunophenotype (IRF4/MUM1+). EBV+ MCU may regress spontaneously after correction of IDD. The prognosis of LyG and EBV+ DLBL is variable. PBL is prognostically poor [142,143,144,145].
3.2.3. EBV Vaccines
Considering the multitude of cancers etiologically and pathogenetically related to EBV infection, it is insightful to prevent EBV infection to avoid latency and subsequent oncogenesis [8]. Throughout the past decades, much effort had been made to develop prophylactic EBV vaccines, albeit with limited success. The following is a summary of the current state of EBV vaccine development.
- Prophylactic Vaccine
EBV infects primarily B cells and epithelial cells. The envelope proteins gH/gL , gB (The core fusion machinery) and gp350 bind to CD21 on B cell surface followed by cell entry. Entry of EBV into epithelial cells involves binding of EBV BMRF2 to epithelial cell integrins, followed by gH/gL and gB binding. To prevent EBV infection, these envelope proteins pose obvious targets for vaccine development [8,88,93,146]. Early efforts have focused on gp350, which is the most abundant envelope glycoprotein of the virus. The anti-gp350 vaccine, however, did not protect against EBV infection, probably reflecting that gp350 is not strictly required for EBV cell entry [8,93,146,147]. Subsequent vaccines developed against the core fusion machinery proteins gH/gL and gB induce markedly higher EBV neutralizing antibodies compared to gp350 in humanized mice [146]. gH/gL and gB packed in nanoparticles or presented as VLP, possibly through inducing high titers of neutralizing antibodies, may disrupt native conformational epitopes of the EBV envelope proteins. The antibodies induced by these vaccine platforms are quantitatively though not necessarily qualitatively robust [146]. Recombinant vaccines express native epitopes and may also elicit high antibody responses both quantitatively and qualitatively and are potentially ideal for EBV prophylactic vaccination [146,148]. The use of multiple cell surface receptors by EBV to establish an infection adds to the difficulty of developing an effective vaccine [96]. Despite success in humanized mice [146], further trials in human subjects are warranted for safe efficacious use. A phase 1 clinical trial comprising mRNA coding gH, gL, g42 and g220 glycoproteins has been initiated and effected increased antibody titers and decreased EBV copy numbers in the enrolled subjects. It is important also to evaluate the durability of the elicited humoral and cellular immune responses [149,150]. As EBV is a ubiquitous virus infecting over 90% of the world population, there is also the logistic difficulty of identifying target age groups and populations to be vaccinated [2,8]. There is to date no approved effective prophylactic vaccine for EBV infection.
- 2.
- Therapeutic Vaccine
Phase 1 trials have been conducted for a peptide vaccine and an mRNA vaccine for NPC [150]. No therapeutic vaccine has been developed for EBV-driven cancers.
3.2.4. The Viral Orchestra
- The duplet EBV and HPV
The interplay of this duplet in uterine cervical intraepithelial neoplasia and SCC has been discussed under 3.1.4.
- 2.
- The duplet EBV and Kaposi sarcoma-associated herpes virus (KSHV)
This will be discussed in the following sections.
- 3.
- The duplet EBV and HIV
HIV coinfection dampens the host immune response against EBV, thus aggravating the cancer severity and incidence. EBV+ BL and HL occurs more frequently in HIV co-infected subjects. The inflammatory microenvironment and the HIV antigens promote growth of EBV-associated lymphoma cells. This repertoire induces increased mutation rate and myc translocation (in BL) [88.96].
3.3. Kaposi Sarcoma-Associated Herpes Virus (KSHV)
3.3.1. Virology and Oncogenesis
KSHV, also known as human herpes virus 8 (HHV8) was identified as herpes-like DNA sequences in HIV-associated Kaposi sarcoma [151]. It is a gamma 2 herpesvirus, which unlike its widely prevalent counterpart EBV, occurs with a much lower worldwide incidence of 2-5%. It is prevalent in endemic areas of sub-Saharan Africa and the Mediterranean [152]. KSHV is transmitted by saliva primarily in childhood, or through sex (especially men with men sex). Co-infection with HIV and EBV may occur. Control of HIV with antiretrovirals reduces the prevalence of HIV-associated KS [153]. Host cell infection is by interaction of heparin sulfate receptor with viral K8.1 envelope glycoprotein [152]. KSHV produces both lytic and latent infections, both important for persistent infection and oncogenesis [154]. KSHV is etiologically associated with KS, primary effusion lymphoma (PEL), multicentric Castleman disease (MCCD), KSHV-positive DLBL and KSHV-positive germinotropic lymphoproliferative disorder [96,152,154,155,156,157,158,159,160,161,162,163,164]. The latent KSHV gene oncogenic products are latency associated nuclear antigen (LANA), viral (v) cyclin, vFLIP, Kaposin A, B &C, microRNA and ORFK1. LANA dysregulates cell growth, survival and contributes to viral persistence and immune evasion. There are oncogenic lytic products related to inflammation (vGPCR, vIL6, K15), angiogenesis (vIL6, vGPCR, vCCL1, vCCL2), cell growth (vIL6, vGPCR, K1) and apoptosis inhibition (vCCL1, vCCL2. vbcl2, vIRF1, K1) [152,154]. The virus also causes expression of immunoregulatory genes that hinder host innate and adaptive antiviral responses [166,167,168]. KSHHV affects endothelial cells or their progenitors, conferring them characteristics of lymphatic endothelium [165]. The world-wide incidence of KSHV exceeds that of KS and factors other than the virus may be at play in oncogenesis. Host chronic inflammation and immune dysfunction play an important part in oncogenesis and tumor progression [60,157,166,167,168].
3.3.2. KSHV-Associated Cancers
The non-neoplastic KSHV-associated MCCD and KSHV-positive positive germinotropic lymphoproliferative disease (LPD) [156] will not be discussed below, though these may progress to B-cell lymphoma.
- Kaposi Sarcoma (KS)
KS is a vascular neoplasm of intermediate malignant potential and behavior, varying from indolent to locally aggressive. It is named after Moritz Kaposi who first described the disease as “idiopathic multiple pigmented sarcoma of the skin” in 1872 [169]. KS affects the skin, lymph nodes or other viscera (lungs, GIT). Involved sites vary with the epidemiologic type of KS: classical (skin), endemic (lymph nodes in children, skin in adults), transplant-related and HIV epidemic (skin, mucosa, viscera) [153].
Lymph nodes may be primarily involved or secondarily involved when draining a nearby KS. The early lesion occurs in the nodal capsule, with extension along fibrous trabeculae of the lymph node. The early lesion is subtle and difficult to recognize. In the tumoral stage, there are interlacing fascicles of deceptively bland-looking mitotically inactive spindle cells forming a sieve-like pattern with erythrocyte filled vascular spaces. The tumor cells express endothelial cell immunophenotype. The diagnostic marker is LANA detectable by immunohistochemistry [152,154] (Figure 5). LANA staining is preferred over polymerase chain reaction for viral DNA as the associated inflammatory cells may also harbor the virus [166]. Similar histological features are found in other involved organs.
There is at present no curative treatment for KS [158]. Optimal control of HIV with anti-retrovirus therapy (ART) alleviates severity. In iatrogenic KS, reduction of immunosuppressive therapy is helpful. Surgery may be indicated for cosmetic reasons. Targeting at endothelial cells may be the future therapeutic direction [157,158,167].
- 2.
- Primary Effusion Lymphoma (PEL)
PEL is a large B-cell lymphoma presenting as lymphomatous effusions and rarely as body cavity related tumor masses. Most are HIV-positive with frequent EBV co-infection [96,159,160,161,170,171]. There is often co-occurrence of other KSHV-associated diseases (KS and MCCD) in 50% of cases [160,161]. PEL can occur in the elderly without HIV infection, and may be related to immunosenescence [162,163,164]. The tumor cells are blastic or anaplastic among apoptotic debris [161,163]. Demonstration of LANA is essential for diagnosis. PEL is aggressive with poor prognosis. EBV-associated PEL may show better outcome [164].
- 3.
- KSHV-positive Diffuse Large B-cell Lymphoma (DLBL)
KSHV+ DLBL are rare and occurs in a backdrop of severe immunodeficiency involving lymph nodes and/or spleen with possible extranodal and peripheral blood dissemination. It affects mostly HIV+ men aged 30-40 years. There is infiltration by sheets of blastic B cells positive for LANA. It is extremely aggressive with poor prognosis [172].
3.3.3. KSHV Vaccines
KSHV vaccines target HSHV structural glycoproteins that are important for host cell attachment, fusion and entry. These are K8.1, gB, gH, gL, gM and gN [173,174]. K8.1. gB and gH/gL have been used in VLP based vaccines and resulted in neutralizing antibody responses in mice and rabbits that prevented in vitro infection of epithelial, endothelial, fibroblastic and B cells [173,175,176]. Other KSHV antigen targets include non-structural proteins unique to the virus. LANA is a maintenance protein expressed in all KSHV infected cells and is an obvious target for vaccine development. However, LANA is an intracellular antigen and is unlikely to be targeted by humoral immunity. It is therefore a good target candidate for mRNA vaccines for eliciting cellular immunity (CI) when the antigen is modified to eliminate its CI-escape motif [177]. There are several other viral surface expressed non-structural proteins that may be targeted. They are K1 protein, v!L6, G protein-coupled receptor and viral chemokines [173]. Short of a dominant viral antigen target, the myriad structural and non-structural viral proteins suggest that vaccines developed against multiple viral antigens is the direction to follow [173]. There is hitherto no approved effective vaccine against KSHV [173].
3.3.4. The Viral Orchestra
- The duplet KSHV and HIV
This is of common occurrence in KS, HSHK+ PEL and DLBL, as discussed above.
- 2.
- The duplet HSHV and EBV
This may be observed in PEL, where the outcome may be better [164]. It may also be possible that the viruses are supportive of each other [170,178,179].
- 3.
- The trios KSHV, HIV and EBV
This may occur in KSHV+ DLBL, where EBV occurs in 80% of cases. Co-infection with EBV in HIV+ KS of the conjunctiva has also been reported [180].
3.4. Human Immunodeficiency Virus (HIV)
3.4.1. Virology and Role in Oncogenesis
The HIV is an RNA retrovirus belonging to the subgroup lentivirus [181,182]. It came to light after reports of KS and pneumocystis pneumonia among men who had sex with men (MSM) in 1981 [183]. HIV targets active CD4+ cells and macrophages. Most of the initial viral replication occurs in lymph nodes [181]. HIV possesses an envelope containing 2 glycoproteins gp41 (transmembrane) and gp120 (surface) [181,182]. The viral capsid is constituted by p24, which encloses 2 viral RNA strands of the viral genome. Gp16, a matrix protein, is anchored to the internal surface of the envelope. p9 is a nucleocapsid protein not covalently attached to the viral RNA. There are 6 regulatory proteins: tat, reverse transcriptase (RT), Nef, Vif, Vpu and Vpr, which are essential for viral replication. HIV enters lymphocytes and monocytes through viral protein gp120 cognate recognition of the CD4 molecule and chemokine receptor (CXCR4/CCR). This is followed by fusion between the viral envelope and the host cell membrane, ensuing in release of viral capsid into the cell cytoplasm [181,182]. Though HIV does not replicate in B cells, it produces severe B-cell dysfunction [182].
HIV has not been identified to cause a specific cancer. The role of HIV in oncogenesis is mostly through immune dysfunction, persistent immune activation and chronic inflammation caused by the infection. Co-infection with HIV with other oncogenic viruses positively contributes to cancer development [184,185,186,187]. There is however evidence that some HIV proteins may directly ne involved in oncogenesis. The 5 HIV proteins that may be oncogenic are tat, Nef, gp120, p17 and RT. They contribute to oxidative stress, enhanced EMT, decreased tumor suppressor gene expression, glycolysis stimulation, cell proliferation, migration, survival, angiogenesis and immunomodulation [185,186].
3.4.2. HIV Vaccines
Identification of the various viral proteins and their roles in host cell infection has spawned much research in HIV vaccine development. More than 20 candidate vaccines developed against the proteins gp120, gp160, multimeric gp120 and V3 peptide have been tried in human subjects [182]. However, there are hitherto no approved effective HIV vaccines.
3.4.3. The Viral Orchestra
Co-infection with HIV with the oncogenic viruses HPV, EBV and KSHV results in increased incidence and severity of the cancers caused, as discussed above. The role of HIV co-infection in oncogenesis induced by the hepatitis viruses B and C will be discussed below.
3.5. Hepatitis Viruses
3.5.1. Virology and Oncogenesis
- Hepatitis B (HBV)
HBV is a partial double strand DNA virus belonging to the Hepadnaviridae family. There 8 genotypes A to H. It consists of one complete coding minus(-) strand and one incomplete non-coding plus(+) strand. There is encapsulation by a lipoprotein membrane constituted by 3 hepatitis B surface antigens (HBsAg): large, middle and small forms. The nucleocapsid is composed of the hepatitis B core protein (HBc), viral polymerase (pol) and the viral genome DNA. Entry into host cell entails low specificity binding between HBsAg and heparin sulfate proteoglycans on the hepatocytic surface, followed by high affinity interaction between the HBV envelope and sodium taurocholate co-transporting polypeptide receptor on hepatocytes, endocytosis of the virion and release of relaxed circular DNA (rcDNA) into the hepatocytic cytoplasm. rcDNA is converted into covalently closed circular DNA (cccDNA) which encodes for RNA then translated to produce structural and non-structural viral proteins. Persistence and stability of cccDNA is key to HBV chronic infection. cccNA also transcribes to pregenomic RNA (pgRNA) which is reversely transcribed to double stranded linear DNA (dlsDNA). The latter is responsible for integration of the viral genome into host DNA, which is important for hepatocytic damage and carcinogenesis. While viral integration was believed to be random, there is recent evidence that host cancer associated gene segments may be favored sites for integration. These are fragile chromosomal sites containing HBV preferred CpG islands. These sites are TERT, KMT2B, CCNE1, PAK3, CCND1 and FGF19. TERT appears to be most favored for viral integration, leading to overexpression of telomerase, conservation of telomere length and thus inhibition of cell senescence and tumor cell growth promotion [188,189,190]. Viral integration also causes chromosomal instability, modulation of host oncogenes and signaling pathways, expression of truncated HBV mutant proteins (such as hepatitis B virus X protein – HBX), inhibition of apoptosis, induction of stemness and host immune dysfunction [188,189,190].
HBV is aclass I carcinogen and is amajor cause of hepatocellular carcinoma (HCC) [191]. HCC ranks fifth in global cancer incidence and is third in leading causes of cancer deaths [188,192].
- 2.
- Hepatitis C (HCV)
HCV is appositive-stranded RNA virus belonging to the Flaviviridae family. It consists of the structural proteins E1 and E2, and non-structural proteins P7, NS2, NS3, NS4A, NS4B, NS5A and NS5B. There is extraordinary genetic diversity with 7 genotypes and more than 80 subtypes [193]. It is a major cause of HCC, and may also cause cancers of the oral cavity, oropharynx, intrahepatic bile ducts, pancreas, kidney and B-cell lymphoma [194]. Unlike HBV, HCV cannot integrate into host cell DNA. It causes cancer through direct interaction of viral proteins with the host immune system, tumor suppressor genes and oncogenes [194,195,196]. This results in persistent cellular proliferation, promotion of stemness, inhibition of apoptosis, angiogenesis and an inflammatory milieu. This involves promoting tyrosine kinase receptor signaling, Hedgehog signaling, NF-KB and STAT3 signaling, p53 and pRB tumor suppressor protein cytoplasmic retention and EMT promotion [194].
3.5.2. Hepatitis Virus – Associated Cancers
HBV and BCV are major causes of HCC. Other cancers that can be caused y HCV will not be covered in this section. HCC accounts for 75-85% of primary liver cancers, being the 6th commonest cancer and the 4th leading cause of cancer deaths worldwide. It is more common in Africa, China and southeast Asia [197]. HCC develops in a backdrop of chronic liver disease which are often also caused by the hepatitis viruses. Other HCC predisposing chronic liver diseases and conditions are chronic alcoholism, exogenous fungal toxins, steatohepatitis and inherited metabolic diseases [198,199]. HCC can be multifocal and spreads by lymphatic and hematogenous routes [199]. Histologically, HCC is characterized by hepatocytic differentiation with variable degrees of cytologic atypia, increased arterialization and sinusoidal capillarization, and varying trabecular, solid, pseudoglandular, macrotrabecular and mixed patterns (Figure 5). Some HCC subtypes are distinct clinicopathologic and molecular entities: fibrolamellar, schirrhous, clear cell, steatohepatitic, massive macrotrabecular, chromophobe, neutrophil-rich and lymphocyte-rich. Histologic grading is by degree of likeness to normal hepatocytes. However, a rigorously defined, reproducible and easy to use grading system is yet to be defined [199,200]. Prognosis of HCC is poor, especially in advanced disease [199].
3.5.3. Hepatitis Virus Vaccines
- HBV
Several Food and Drug Administration (FDA) approved anti-HBV vaccines are available. These are prophylactic neutralizing recombinant vaccines developed against the HBsAg, specifically against the major hydrophilic region (MHR) containing all 3 forms of HBsAg. They provide protection against all HBV genotypes (A to H). However, alterations of residues within the MHR can result in replication of the mutated virus which would evade protection by the vaccines [201,202]. The FDA approved single vaccines including Engerix-B, Recombivax HB, HEPLISAV-B, and PreHevbrio. There are 3 approved combination vaccines: Pediatrix, Twinrix, Vaxelis where vaccines against other infections are combined. HBV vaccines are used in universal vaccination programs at birth in many parts of the world. This resulted in profound reduction of HBV infections, HBV related chronic disease and HCC [6,191,203,204,205,206,207].
- 2.
- HCV
Despite knowledge and understanding of the structural envelope, non-structural and the core proteins of the virus, no approved effective anti-HCV vaccines are available to date. This may be related to the great virus genetic diversity and subtype variability, poor understanding of the host immune response to HCV and difficulty in recruiting study cohorts due social, psychological and marginalization issues [193,208]. Vaccines using recombinant virus vector or VLP platforms against structural and non-structural proteins have been studied without success [193,208]. A minicircle-based vaccine against chimeric HBV-HCV VLP has been reported to induce potent T-cell and antibody response against HCV in BALB/c mice [209].
3.5.4. The Viral Orchestra
- The Duplet HBV and HIV or HCV and HIV
Apart from HIV-induced immunodeficiency causing decreased clearance of HBV or HCV from the liver, HIV may contribute to oncogenesis through increased liver damage, inflammation and fibrosis. HIV infection results in elevated levels of circulating lipopolysaccharides (LPS) in the portal and systemic circulation. LPS activates hepatic stellate cells (HSC) and Kupffer cells (KFC), thus inducing oxidative damage, proinflammatory cytokines release and fibrosis. HIV can directly infect HSC and KFC leading to similar pro-oncogenic results. In HIV/HCV co-infected individuals, HCC occurs at a yonger age with risk of HCC development being increased by 11% each year compared to HIV-negative individuals [183,185,210,211].
- 2.
- The Duplet HBV and HCV
Co-infections of HBV and HCV increase the risk of HCC compared to the mono-infections. The co-infections cause greater progression to advanced chronic liver disease and HCC. This may be related to long chronic disease duration, high levels of liver fibrosis and carbohydrate intolerance [212,213]. HCV dominance is more common than HBV dominance in the co-infection, which is relevant to treatment protocols [214].
- 3.
- The trios
Co-infectons of HBV, HCV and HIV carry higher risks of HCC development compared to HBV or HCV mono-infections. The triple infection, however, carries a lower rate of HCC compared to co-infections with HCV and HIV [215].
3.6. The Merkel Cell Polyoma Virus (McPyV)
3.6.1. Virology and Oncogenesis
The McPyV is the only oncogenic virus among th e 14 human polyomaviruses. It was discovered in 2008 [216,217,218,219] and is a small non-enveloped DNA virus. There is a circular double-stranded genome of approximately 5400 base pairs (bp)[217,218]. It contains an early region which encodes the oncoproteins large T (LT) , small T (sT) and the 57kT protein. The role of the 57kT protein is still nebulous. The late region of the viral genome encodes the capsid structural proteins VP1, VP2, VP3 and microRNAs [217,218,220]. McPyV enters the host cell through attachment of VP1 to sulfated glycosaminoglycans (particularly heparin sulfate), followed by attachment to sialylated glycan co-receptor for gene transduction. Viral integration is important for oncogenesis. This results in mutations leading to truncation of LT [217,218,219]. The truncated LT is capable of binding to and inactivates the Rb protein [217,219,220]. The sT induces hyperphosphorylation of the cellular translation factor 4E-BP1 in an mTOR-dependent manner, causing cell cycle progression and transformation [217,219,220].
3.6.2. The McPyV-Associated Cancers
The major McPyV-associated cancer is Merkel Cell Carcinoma (MCC), though other malignancies including skin SCC, basal cell carcinoma, melanoma and cutaneous B- and T-cell lymphomas have also be related to McPyV [218]. Though McPyV is named after Merkel cells, the cell origin of McPyV-associated MCC is debatable [220]. Other possible histogenetic origins including epidermal progenitor cells, pre/pro B cells and dermal fibroblasts have been proposed [218,220]. MCC is arare highly aggressive primary cancer of the skin affecting the dermis and subcutis in large tumors. The epidermis may be the only involved site and pure epidermal MCC have been described in McPyV-negative cases [220,221,222]. There are invasive sheets of small rpund cells which are positive for CK20 and neuroendocrine markers [218,222]. The risk factors include old age, sun exposure, white skinned, male sex, immunosuppression and history of skin malignancies [218,221,222]. About 80% of MCC is McPyV-positive with the remaining McPyV-negative cases related to uv exposure or chronic arsenicism. McPyV-negative MCC exhibits more frequent mutations and poorer outcome [218,219,220,221]. There is higher association of McPyV-negativity in MCC combined with other types of skin cancers [222].
3.6.3. McPyV Vaccines
Despite knowledge of the McPyV capsid structural proteins V1, V2 and V3 and the oncoproteins Lt and sT, no effective approved anti-McPyV vaccine has become available [223]. A prophylactic vaccine against the LT antigen has been found to be useful in C57BL/6 mice [224]. Therapeutic vaccines targeted to enhance host immunity and augment ant-tumor/ant-virus effects in the TME have been the focus of research in McPyV-positive MCC. The viral targets used are mostly LT and more recently the capsid protein V1. Increased CD4/CD8 cellular responses have been demonstrated using various platforms (recombinant protein, mRNA, DNA and oncolytic viruses0 [223,224,225,226,227]. In addition, potent antibodies against the V1 protein was demonstrated in one study [223]. However, most of the work was done on experimental animals (BALB/c mice) [223,224,225,226,227], though some phase 1 or 2 preclinical and clinical trials have been conducted on human subjects [224].
3.6.4. The Viral Orchestra
Co-infection with HIV interplays with McPyV in oncogenesis. The incidence of MCC in HIV infected individuals is 13 times higher than the general population [221,228,229]. MCC is also more frequent in immunocompromised subjects (organ transplant or B-cell malignancy patients) and this may also be operative in HIV infected individuals [221,228,229].
3.7. The Human T-Cell Leukemia Virus Type-1 (HTLV-1)/Human Tlymphotropic Virus
3.7.1. Virology and Oncogenesis
HTLV-1 is a retrovirus belonging to the family retroviridiae and genus Deltaretrovirus [230,231,232,233,234,235]. It is spherical, measures 100-120 nm with the proteins gp21 and gp46 making up the envelope. The capsid is constituted by the proteins p15, p19 and p24, which encloses the viral genome. The latter is made up of 2 identical single-stranded RNA with protease (p10), RT (p55), integrase (p32) and RNAase. The viral genome encodes the structural proteins (envelope and capsid) and the regulatory proteins Tax and HBZ (HTLV-1 basic leucine zipper factor). The regulatory proteins contribute to control of cell proliferation and immune inflammatory responses [230,231,232,233,234,235]. The viral genome is integrated into the host genome as the provirus, facilitated by integrase. The integration occurs more frequently near some “hotspots” [234]. HTLV-1 infects mostly CD4+ T cells, though CD8+ lymphocytes, dendritic cells and macrophages may also be infected [230,234]. The infected cells do not undergo lysis and the proviral genome remain latently integrated, achieving host immune evasion [230,231,232,233,234]. HTLV-1 has 6 reported subtypes A to F with most infections caused by subtype A. There are 3 other HTLV viruses: HTLV-2, HTLV-3 and HTLV-4, but only HTLV-1 is convincing related to human diseases [230—233].
Oncogenesis is related to 2 factors: duration of infection and a high proviral load (PVL). PVL is the percentage of peripheral blood mononuclear cells infected by HTLV-1. The infected cell clones pass through many mitotic events, especially associated with a long infection duration, become plagued with replication errors and mutations. Some of these replicative errors are deleterious and lead to oncogenesis [234]. Recurrent somatic mutations detected in adult T-cell leukemia/lymphoma involve genes encoding proteins of the T-cell signaling pathway NF-KB: PRKBB, PLCGL1, VAV and CARD11. Mutations in transcription factor encoding genes IRF4, GATA3, IKZF2 and chemoreceptor genes CCR4, CCR7 and GPR183 are also implicated [231,234]. A modulated host immune response is also contributory [234].
The HTLV-1 regulatory protein Tax and NZB contribute by causing genomic instability and conferring survival or proliferative advantage to the infected clones. Tax expression is however frequently lost in 50% of Adult T-cell leukemia/lymphoma (ATL) clones and may thus be contributory only in the early stages of oncogenesis. HBZ, on the other hand, is retained in all ATL clones and may be a good target for vaccine development [234].
3.7.2. HTLV-1-Associated Cancer
HTLV-1 is prevalent in Japan, Africa, the Caribbean islands, Central and South America [233]. The global distribution of HTLV-1 infection is estimated to be 5 to 10 million persons [233,235]. Transmission is predominantly from mother to child through breastfeeding, sexual activity and blood contact [233]. There are 4 subtypes of ATL: smouldering, chronic, lymphomatous and acute. In the most aggressive acute and lymphomatous subtypes, there is lymphadenopathy, hepatosplenomegaly, skin, bone, lung and multifocal visceral infiltration with hypercalcemia. In the chronic and smouldering subtypes, there are non-specific symptoms and no tumor mass formation. There may be prominent skin involvement. Strongyloidosis is common in all subtypes [233]. The peripheral blood and involved organs show infiltration by markedly pleomorphic medium to large lymphoid cells with marked nuclear abnormalities causing the so-called “flower cells” or hallmark cells with horseshoe or kidney-shaped nuclei. Immunophenotypically, the cells are CD3+, CD4+, CD25+, CCR4+ and GATA3+ [236]. No effective treatment is available. Inhibition of cell pathway proteins AKT, BET, phosphatidylinositol 3-kinase, NF-KB have been used with success in mice [234].
3.7.3. HTLV-1 Vaccine
Despite tremendous efforts to develop an effective vaccine, through employing multiple platforms and targeting structural and non-structural proteins of the virus, no effective vaccines have become available [235]. Most studies have been performed on experimental animals. One study on ATL patients using a therapeutic vaccine containing autologous dendritic cells along with Tax peptide and HTLV-1-specific cytotoxic T lymphocytes showed clinical improvement in 2 patients with one eventually achieving complete recovery has been reported [237]. The presence of target populations (pregnant women) in endemic regions may represent research focus in future development of prophylactic vaccines in future [233,234,235].
3.8. SARS-Cov-2 Virus
3.8.1. Virology and Possible Oncogenetic Mechanisms
The SARS-Cov-2 virus is a coronavirus belonging to the genus Betacoronvirus. It was responsible for the pandemic COVID 19 (coronavirus infectious disease 2019) and caused infections of hundreds of millions and death toll of millions of people [238,239]. It is a single-stranded RNA (ssRNA) enveloped zoonotic virus and among the 7 coronaviruses hat attack humans. The ssRNA encode structural proteins including the membrane glycoprotein, envelope protein, nucleocapsid and the spike (S) proteins. The S protein is responsible for host cell invasion through interaction with the angiotensin-converting enzyme (ACE) on the host cell surface [238,239,240,241]. SARS-Cov-2 infection typically causes multi-organ disease ensuing in respiratory and GI symptoms, loss of taste and smell [238,239]. COVID 19 may resolve clinically or may result in death due to the severe host immune response and upregulated cytokines and severe inflammation in various organs, especially in the respiratory system [238,239,240]. In convalescence, however, a variety of long lasting symptoms may persist as the so-called “long COVID 19”[242]. It is theoretically possible that cancer development may occur, as latent viral infection predisposes to oncogenesis [1,2]. A recent report suggests that latent SARS-Cov-2 infection may occur in the testes [242,243]. There is, however, so far no unequivocal evidence of latency in SARS-Cov-2 infection. Multiple mechanisms of oncogenesis have been postulated for a possible role of SARS-Cov-2. These include activation of oncogenes, inhibition of tumor suppressor activity (nsp3 and nsp15 degrades p53 and pRb), cell cycle and signaling pathways modulation, extracellular vesicles, host genomic instability, epigenetic changes, inflammatory cascade, reactive oxygen species and EMT [240,241,242,244,245]. There have been reports of developing hepatic EBV-negative DLBL [246] and progression of KS [247] after SARS-Cov-2 infection. However, there are also reports of oncolytic properties of SARS-Cov-2 virus in various malignancies: NK cell lymphoma, HL and mycosis fungoides [244,245,248,249,250]. These contradictory reports on possible oncogenesis of SARS-Cov-2 renders further studies on the issue mandatory.
3.8.2. Vaccines
The surprise and rapid onset of the COV-19 pandemic triggered accelerated development of anti-SARS-Cov-2 vaccines [12]. Various types were developed and mRNA vaccines have proved to be superior in effectiveness and efficacy. Five years have elapsed since the initial outbreak in China and many countries have adopted various vaccination strategies and programs against the virus [18,251,252].
4. Conclusions
Oncogenic viruses are responsible for 12% of human cancers. Seven viruses including, HPV, EBV, KSHV, HBV, HCV, McPyV and HTLV-1 are well recognized in causing cancers ranging from, carcinoma, sarcoma to lymphoma/leukemia. As viruses are among the rare known causes of cancer, it is intuitively possible to prevent the cancers caused by developing and using prophylactic vaccines. Recognition of the appropriate target virus antigens/proteins, employment of efficacious vaccine platforms and effective use of adjuvants are important attributes of successful vaccines. The development and use of prophylactic anti-HPV and anti-HBV vaccines in universal vaccination programs for target at risk populations have contributed profoundly to preventing the infections (HPV and HBV) and incidence of the cancers caused (cervical and hepatocellular carcinomas). Development of prophylactic vaccines against the other oncogenic viruses have been much less successful. This may be related to less successful recognition of useful viral target antigens/proteins that are instrumental in causing disease, or the lack of clear at risk target populations such as due to ubiquity of the infections (as in EBV). Therapeutic vaccines, on the other hand, have been useful in treating various cancers in animals, and may constitute an applicable tool in cancer treatment. HIV though probably not oncogenic per se, plays important collaborative roles in oncogenesis in various cancers caused by viruses. Prevention of HIV infection is important in alleviating virus-related cancer incidence and severity in HIV prevalent areas. Regrettably, there are still no effective anti-HIV vaccines. Co-infection by multiple oncogenic viruses is not unusual and contributes to their collaboration and reinforcement in oncogenesis. The role of the SARS-Cov-2 virus in oncogenesis is theoretically plausible. It remains to be seen in long term follow up studies on patients with “long COVID-19 disease” whether an oncogenic role of the SARS-Cov-2 virus could be documented.
Author Contributions
The single author was responsible for conception, literature research, curation and interpretation, preparation, writing and finalization of this manuscript. Author have read and agreed to the published version of the manuscript.
Funding
No external funding was obtained for this work.
Acknowledgments
The author thanks Dr. JL Qi, Pathology, First Affiliated Hospital, Guangzhou Medical University, Guangzhou, China, for supplying images of ENNKTL in Figure 4.
Conflicts of Interest
The author declares that there is no conflict of interest in this work.
Appendix A

Table A1.
Oncogenic Viruses, Cancers and Vaccines.
| Viruses | Oncogenicity | Cancers Caused | Cancer promoting products | Vaccines |
|---|---|---|---|---|
| HPV | Yes | ● Uterine cervical SCC ● Vaginal, vulval, penile, anal, oropharyngeal SCC ● Urinary bladder |
E2, E4, E5,E6, E7 | ● Gardasil (bi-,quadri-, nona-valent) ● Cervarix (bivalent) |
| EBV | Yes | ● Burkitt lymphoma ● Hodgkin lymphoma ● ENNKTL ● NPC ● GC ● LEC, lung and thymus ● IFDCS ● ANKL ● EBV+DLBL ● EBV+ nodal T and NK lymphoma ● SEBVTCLC |
● Latency products: LMP1, LMP2, EBNA1, EBNA2, EBER, microRNA ● Lytic products: BZL1, BHRF1, BALF1, vbcl2, BZLF1, BRLF1, BLLF3, microRNA (BART, BHRF1-2) |
No available vaccine |
| KSHV | Yes | ● KS ● PEL |
LANA, Kaposin, v-cyclin, vFLIP, vGPCR, K1, K15, vIL-6, microRNA | No available vaccine |
| HBV | Yes | Hepatocellular carcinoma (HCC) | HBx | ● Energix-B ● Recombivax HB ● HEPLISAV-B# ● PREHEVBRIO ● Twinrix# ● Pediarix# ● Vaxelis# |
| HCV | Yes | HCC | Core, NS3, NS4, NS5 | No available vaccine |
| McPyV | Yes | Merkel cell carcinoma | Small T, truncated large T | No available vaccine |
| HTLV-1 | Yes | Adult T-cell leukemia/lymphoma | Tax, HBZ | No available vaccine |
# Combination vaccines.
References
- Gaglia, M.M.; Munger, K. More than just oncogenes: mechanisms of tumorigenesis by human viruses. Curr. Opin. Virol. 2018, 32, 48–59. [Google Scholar] [CrossRef] [PubMed]
- Ng, C.S. From the midfacial destructive drama to the unfolding EBV story: a short history of EBV-positive NK-cell and T-cell lymphoproliferative diseases. Pathology 2024, 56, 773–785. [Google Scholar] [CrossRef] [PubMed]
- Mahdavi, P.; Javaherdehi, A.P.; Khanjanpoor, P.; Aminian, H.; Zakeri, M.; Zafarani, A.; Razizadeh, M.H. The role of c-Myc in Epstein-Barr virus-associated cancers: Mechanistic insights and therapeutic implications. Microb. Pathog. 2024, 197, 107025. [Google Scholar] [CrossRef]
- Allday, M.J. How does Epstein–Barr virus (EBV) complement the activation of Myc in the pathogenesis of Burkitt's lymphoma? Semin. Cancer Biol. 2009, 19, 366–376. [Google Scholar] [CrossRef]
- Moore, P.S.; Chang, Y. Why do viruses cause cancer? Highlights of the first century of human tumour virology. Nat. Rev. Cancer 2010, 10, 878–889. [Google Scholar] [CrossRef]
- Park, N.H.; Chung, Y.-H.; Lee, H.-S. Impacts of Vaccination on Hepatitis B Viral Infections in Korea over a 25-Year Period. Intervirology 2010, 53, 20–28. [Google Scholar] [CrossRef]
- Schiller, J.T.; Lowy, D.R. Understanding and learning from the success of prophylactic human papillomavirus vaccines. Nat. Rev. Microbiol. 2012, 10, 681–692. [Google Scholar] [CrossRef]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-Barr Virus: An Important Vaccine Target for Cancer Prevention. Sci. Transl. Med. 2011, 3, 107fs7–107fs7. [Google Scholar] [CrossRef]
- Casper, C.; Corey, L.; Cohen, J.I.; Damania, B.; Gershon, A.A.; Kaslow, D.C.; Krug, L.T.; Martin, J.; Mbulaiteye, S.M.; Mocarski, E.S.; et al. KSHV (HHV8) vaccine: promises and potential pitfalls for a new anti-cancer vaccine. npj Vaccines 2022, 7, 1–10. [Google Scholar] [CrossRef]
- Ghattas, M.; Dwivedi, G.; Lavertu, M.; Alameh, M.-G. Vaccine Technologies and Platforms for Infectious Diseases: Current Progress, Challenges, and Opportunities. Vaccines 2021, 9, 1490. [Google Scholar] [CrossRef]
- Saxena, M.; van der Burg, S.H.; Melief, C.J.M.; Bhardwaj, N. Therapeutic cancer vaccines. Nat. Rev. Cancer 2021, 21, 360–378. [Google Scholar] [CrossRef] [PubMed]
- Bayani, F.; Hashkavaei, N.S.; Arjmand, S.; Rezaei, S.; Uskoković, V.; Alijanianzadeh, M.; Uversky, V.N.; Siadat, S.O.R.; Mozaffari-Jovin, S.; Sefidbakht, Y. An overview of the vaccine platforms to combat COVID-19 with a focus on the subunit vaccines. Prog. Biophys. Mol. Biol. 2023, 178, 32–49. [Google Scholar] [CrossRef] [PubMed]
- Folorunso, OS; Sebolai, OM. Overview of the Development, Impacts and Challenges of Live-attenuated Oral Rotavirus Vaccines. Vaccines 2020, 8, 341.
- Vignuzzi, M.; Wendt, E.; Andino, R. Engineering attenuated virus vaccines by controlling replication fidelity. Nat. Med. 2008, 14, 154–161. [Google Scholar] [CrossRef]
- Groenke, N.; Trimpert, J.; Merz, S.; Conradie, A.M.; Wyler, E.; Zhang, H.; Hazapis, O.-G.; Rausch, S.; Landthaler, M.; Osterrieder, N.; et al. Mechanism of Virus Attenuation by Codon Pair Deoptimization. Cell Rep. 2020, 31, 107586. [Google Scholar] [CrossRef]
- Mak, TW; Saunders, ME. Vaccines and Clinical Immunization. In: The Immune Response. Mak, TW; Saunders, ME (Eds). Academic Press, Burlington, MA, USA. 2006, 695-749.
- Fehlner-Gardiner, C.; Nadin-Davis, S.; Armstrong, J.; Muldoon, F.; Bachmann, P.; Wandeler, A. ERA VACCINE-DERIVED CASES OF RABIES IN WILDLIFE AND DOMESTIC ANIMALS IN ONTARIO, CANADA, 1989–2004. J. Wildl. Dis. 2008, 44, 71–85. [Google Scholar] [CrossRef]
- Frederiksen, L.S.F.; Zhang, Y.; Foged, C.; Thakur, A. The Long Road Toward COVID-19 Herd Immunity: Vaccine Platform Technologies and Mass Immunization Strategies. Front. Immunol. 2020, 11, 1817. [Google Scholar] [CrossRef]
- Syomin, B.V.; Ilyin, Y.V. Virus-Like Particles as an Instrument of Vaccine Production. Mol. Biol. 2019, 53, 323–334. [Google Scholar] [CrossRef]
- Cimica, V.; Galarza, J.M. Adjuvant formulations for virus-like particle (VLP) based vaccines. Clin. Immunol. 2017, 183, 99–108. [Google Scholar] [CrossRef]
- Lua, L.H.L.; Connors, N.K.; Sainsbury, F.; Chuan, Y.P.; Wibowo, N.; Middelberg, A.P.J. Bioengineering virus-like particles as vaccines. Biotechnol. Bioeng. 2014, 111, 425–440. [Google Scholar] [CrossRef]
- da Silva, A.J.; Zangirolami, T.C.; Novo-Mansur, M.T.M.; Giordano, R.d.C.; Martins, E.A.L. Live bacterial vaccine vectors: An overview. Braz. J. Microbiol. 2014, 45, 1117–1129. [Google Scholar] [CrossRef]
- Humphreys, I.R.; Sebastian, S. Novel viral vectors in infectious diseases. Immunology 2018, 153, 1–9. [Google Scholar] [CrossRef]
- Ertl, H.C. Viral vectors as vaccine carriers. Curr. Opin. Virol. 2016, 21, 1–8. [Google Scholar] [CrossRef]
- Lambricht, L.; Lopes, A.; Kos, S.; Sersa, G.; Préat, V.; Vandermeulen, G. Clinical potential of electroporation for gene therapy and DNA vaccine delivery. Expert Opin. Drug Deliv. 2015, 13, 295–310. [Google Scholar] [CrossRef]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef]
- Alameh, M-G; Weissman, D; Pardi, N. Messenger RNA-based Vaccines against Infectious Diseases. Springer, Berlin/Heidelberg, Germany, 2020,1-35.
- Andries, O.; Mc Cafferty, S.; De Smedt, S.C.; Weiss, R.; Sanders, N.N.; Kitada, T. N1-methylpseudouridine-incorporated mRNA outperforms pseudouridine-incorporated mRNA by providing enhanced protein expression and reduced immunogenicity in mammalian cell lines and mice. J. Control. Release 2015, 217, 337–344. [Google Scholar] [CrossRef]
- Chaudhary, N; Weissman, D; Whitehead, KA. mRNA Vaccines for Infectious Diseases. Principles, Delivery and Clinical Translation. Nat Rev Drug Discov 2021, 20, 817–838.
- Magini, D.; Giovani, C.; Mangiavacchi, S.; Maccari, S.; Cecchi, R.; Ulmer, J.B.; De Gregorio, E.; Geall, A.J.; Brazzoli, M.; Bertholet, S.; et al. Self-Amplifying mRNA Vaccines Expressing Multiple Conserved Influenza Antigens Confer Protection against Homologous and Heterosubtypic Viral Challenge. PLOS ONE 2016, 11, e0161193. [Google Scholar] [CrossRef]
- Kristensen, L.S.; Andersen, M.S.; Stagsted, L.V.W.; Ebbesen, K.K.; Hansen, T.B.; Kjems, J. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 2019, 20, 675–691. [Google Scholar] [CrossRef] [PubMed]
- Alameh, M-G; Tombacz, I; Bettini, E; Lederer, K; Sittplangkoon, C; Wilmore, JR; Gaudette, BT; Soliman, OY; Pine, M; Hicks, P, et al. Lipid Nanoparticles Enhance the Efficacy of mRNA and Protein Subunit Vaccines by Inducing Robust T Follicular Helper Cell and Humoral Responses. Immunity 2021, 54, 2877–2892.
- Verdegaal, E.M.E.; de Miranda, N.F.C.C.; Visser, M.; Harryvan, T.; van Buuren, M.M.; Andersen, R.S.; Hadrup, S.R.; van der Minne, C.E.; Schotte, R.; Spits, H.; et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature 2016, 536, 91–95. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.-P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat. Commun. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Hammerich, L; Binder, A; Brody, JD. In situ Vaccination: Cancer Immunotherapy both Personalized and Off-the-shelf. Mol Oncol 2015, 9, 1966–1981. [CrossRef]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel, E.E., III; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Oweida, A.; Hararah, M.K.; Phan, A.; Binder, D.; Bhatia, S.; Lennon, S.; Bukkapatnam, S.; Van Court, B.; Uyanga, N.; Darragh, L.; et al. Resistance to Radiotherapy and PD-L1 Blockade Is Mediated by TIM-3 Upregulation and Regulatory T-Cell Infiltration. Clin. Cancer Res. 2018, 24, 5368–5380. [Google Scholar] [CrossRef]
- Smith, C.C.; Selitsky, S.R.; Chai, S.; Armistead, P.M.; Vincent, B.G.; Serody, J.S. Alternative tumour-specific antigens. Nat. Rev. Cancer 2019, 19, 465–478. [Google Scholar] [CrossRef]
- Roudko, V.; Bozkus, C.C.; Orfanelli, T.; McClain, C.B.; Carr, C.; O’dOnnell, T.; Chakraborty, L.; Samstein, R.; Huang, K.-L.; Blank, S.V.; et al. Shared Immunogenic Poly-Epitope Frameshift Mutations in Microsatellite Unstable Tumors. Cell 2020, 183, 1634–1649.e17. [Google Scholar] [CrossRef]
- Engelhard, V.H.; Obeng, R.C.; Cummings, K.L.; Petroni, G.R.; Ambakhutwala, A.L.; A Chianese-Bullock, K.; Smith, K.T.; Lulu, A.; Varhegyi, N.; E Smolkin, M.; et al. MHC-restricted phosphopeptide antigens: preclinical validation and first-in-humans clinical trial in participants with high-risk melanoma. J. Immunother. Cancer 2020, 8, e000262. [Google Scholar] [CrossRef]
- Brentville, V.; Vankemmelbeke, M.; Metheringham, R.; Durrant, L. Post-translational modifications such as citrullination are excellent targets for cancer therapy. Semin. Immunol. 2020, 47, 101393. [Google Scholar] [CrossRef]
- Yarchoan, M.; Albacker, L.A.; Hopkins, A.C.; Montesion, M.; Murugesan, K.; Vithayathil, T.T.; Zaidi, N.; Azad, N.S.; Laheru, D.A.; Frampton, G.M.; et al. PD-L1 expression and tumor mutational burden are independent biomarkers in most cancers. J. Clin. Investig. 2019, 4. [Google Scholar] [CrossRef]
- Bulk, J.v.D.; Verdegaal, E.M.E.; Ruano, D.; Ijsselsteijn, M.E.; Visser, M.; van der Breggen, R.; Duhen, T.; van der Ploeg, M.; de Vries, N.L.; Oosting, J.; et al. Neoantigen-specific immunity in low mutation burden colorectal cancers of the consensus molecular subtype 4. Genome Med. 2019, 11, 1–15. [Google Scholar] [CrossRef]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Van der Burg, SH; Arens, R; Ossendorp, F; van Hall, T; Mielief, CJ. Vaccines foe Established Cancer: Overcomin the Challenges Posed by Immune Evasion. Nat Rev Cancer 2016, 16, 219–233.
- Melief, CJ, van Hall, T; Arens, R; Ossendorp, F; van der Burg, SH. Therapeutic Cancer Vaccines. J Clin Invest 2015, 125, 3401–3412.
- Baharom, F.; Ramirez-Valdez, R.A.; Tobin, K.K.S.; Yamane, H.; Dutertre, C.-A.; Khalilnezhad, A.; Reynoso, G.V.; Coble, V.L.; Lynn, G.M.; Mulè, M.P.; et al. Intravenous nanoparticle vaccination generates stem-like TCF1+ neoantigen-specific CD8+ T cells. Nat. Immunol. 2020, 22, 41–52. [Google Scholar] [CrossRef] [PubMed]
- van der Maaden, K.; Heuts, J.; Camps, M.; Pontier, M.; van Scheltinga, A.T.; Jiskoot, W.; Ossendorp, F.; Bouwstra, J. Hollow microneedle-mediated micro-injections of a liposomal HPV E743–63 synthetic long peptide vaccine for efficient induction of cytotoxic and T-helper responses. J. Control. Release 2018, 269, 347–354. [Google Scholar] [CrossRef]
- Li, A.W.; Sobral, M.C.; Badrinath, S.; Choi, Y.; Graveline, A.; Stafford, A.G.; Weaver, J.C.; Dellacherie, M.O.; Shih, T.-Y.; Ali, O.A.; et al. A facile approach to enhance antigen response for personalized cancer vaccination. Nat. Mater. 2018, 17, 528–534. [Google Scholar] [CrossRef]
- Meshii, N.; Takahashi, G.; Okunaga, S.; Hamada, M.; Iwai, S.; Takasu, A.; Ogawa, Y.; Yura, Y. Enhancement of systemic tumor immunity for squamous cell carcinoma cells by an oncolytic herpes simplex virus. Cancer Gene Ther. 2013, 20, 493–498. [Google Scholar] [CrossRef]
- Cerullo, V; Diaconu, J; Romano, V; Hirvinen, M; Ugolini, M; Escutenaire, S; Holm, S_L; Kipar, A; Kanerva, A; Hemminki, A. An Oncolytic Adenovirus Enhanced for Toll-like Receptor 9 Stimulation Increases Antitumor Clearance. Mol Ther 2012, 20, 2076–2086. [CrossRef]
- Motoyoshi, Y.; Kaminoda, K.; Saitoh, O.; Hamasaki, K.; Nakao, K.; Ishii, N.; Nagayama, Y.; Eguchi, K. Different mechanisms for anti-tumor effects of low- and high-dose cyclophosphamide. Oncol. Rep. 2006, 16, 141–146. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Kangasniemi, L.; Rajecki, M.; Escutenaire, S.; Koski, A.; Romano, V.; Rouvinen, N.; Tuuminen, T.; Laasonen, L.; et al. Immunological Effects of Low-dose Cyclophosphamide in Cancer Patients Treated With Oncolytic Adenovirus. Mol. Ther. 2011, 19, 1737–1746. [Google Scholar] [CrossRef] [PubMed]
- Banissi, C.; Ghiringhelli, F.; Chen, L.; Carpentier, A.F. Treg depletion with a low-dose metronomic temozolomide regimen in a rat glioma model. Cancer Immunol. Immunother. 2009, 58, 1627–1634. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized Oncolytic Virotherapy Overcomes Systemic Tumor Resistance to Immune Checkpoint Blockade Immunotherapy. Sci. Transl. Med. 2014, 6, 226ra32–226ra32. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Cai, Y.; Jiang, Y.; He, X.; Wei, Y.; Yu, Y.; Tian, X. Vaccine adjuvants: mechanisms and platforms. Signal Transduct. Target. Ther. 2023, 8, 1–24. [Google Scholar] [CrossRef]
- Coffman, R.L.; Sher, A.; Seder, R.A. Vaccine Adjuvants: Putting Innate Immunity to Work. Immunity 2010, 33, 492–503. [Google Scholar] [CrossRef]
- Xie, C.; Yao, R.; Xia, X. The advances of adjuvants in mRNA vaccines. npj Vaccines 2023, 8, 1–6. [Google Scholar] [CrossRef]
- Parkin, D.M. The global health burden of infection-associated cancers in the year 2002. Int. J. Cancer 2006, 118, 3030–3044. [Google Scholar] [CrossRef]
- Morales-Sánchez, A.; Fuentes-Pananá, E.M. Human Viruses and Cancer. Viruses 2014, 6, 4047–4079. [Google Scholar] [CrossRef]
- De Martel, C; Georges, D; Bray, F’ Ferlay, J; Clifford, GM. Global Burden of Cancer Attributable to Infections in 2018: A Worldwide Incidence Analysis. Lancet Glob Health 2020, 8, e180–e190.
- Molina, M.A.; Steenbergen, R.D.; Pumpe, A.; Kenyon, A.N.; Melchers, W.J. HPV integration and cervical cancer: a failed evolutionary viral trait. Trends Mol. Med. 2024, 30, 890–902. [Google Scholar] [CrossRef]
- WHO Vaccine Position Papers. World Health Prganization. Geneva (www.who.int/teams/immunization-vaccines-and biologicals/policies/position papers, accessed August 2022).
- Cancer Genome Atlas Research Network, Albert Einstein College of Medicine; Analytic Biological services, Barretos Cancer Hospital, Baylor College of Medicine, City of Hope; Buck Institute of research on Aging; Canada’s Michael Smith Genome Science Centre; Harvard Medical School; Helen F Graham Cancer & Research Institute at Christiana Care Health Services; Hudson Alpha Institute for Biotechnology, IL Sbio, LLC, Indiana University School of Medicine; Institute of Human Virology, Institute for System Biology, International Genomics Consortium, Leidos Biomedical, Massachusetts General Hospital; McDonnell Denome Institute at Washington University, Medical College of Wisconsin, et al. Integrated Genomic and Molecular Characterization of Cervical Cancer. Nature 2017,543,378-384. Doi:10.1038/nature21386.
- Ren, S.; Gaykalova, D.A.; Guo, T.; Favorov, A.V.; Fertig, E.J.; Tamayo, P.; Callejas-Valera, J.L.; Allevato, M.; Gilardi, M.; Santos, J.; et al. HPV E2, E4, E5 drive alternative carcinogenic pathways in HPV positive cancers. Oncogene 2020, 39, 6327–6339. [Google Scholar] [CrossRef]
- Maehama, T.; Nishio, M.; Otani, J.; Mak, T.W.; Suzuki, A. The role of Hippo-YAP signaling in squamous cell carcinomas. Cancer Sci. 2020, 112, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Bossler, F.; Hoppe-Seyler, K.; Hoppe-Seyler, F. PI3K/AKT/mTOR Signaling Regulates the Virus/Host Cell Crosstalk in HPV-Positive Cervical Cancer Cells. Int. J. Mol. Sci. 2019, 20, 2188. [Google Scholar] [CrossRef] [PubMed]
- von Knebel Doeberitz, M.; Prigge, E.-S. Role of DNA methylation in HPV associated lesions. Papillomavirus Res. 2019, 7, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Saco, A; Mills, AM; Park, KJ; Focchi, GRA; Carrhilho, C; Regauer, S; Kong, CS. Squamous Cell Carcinoma. HPV-associated, of the Uterine Cervix. In: WHO Classification of Female Genital Tumours, 5th edition, 2020, pp.347-349; Herrington, CS; Ordi, J (eds). Lyons, France, IARC.
- Saco, A; Mills, AM; Park, KJ; Focchi, GRA; Carrhilho, C; Regauer, S; Kong, CS. Squamous Cell Carcinoma, HPV-independent, of the Uterine Cervix. In: WHO Classification of Female Genital Tumours, 5th edition, 2020, p.350. Herrington, CS; Ordi, J (eds). Lyons, France, IARC.
- Lam, E.W.; Chan, J.Y.-W.; Chan, A.B.; Ng, C.S.; Lo, S.T.; Lam, V.S.; Chan, M.M.; Ngai, C.M.; Vlantis, A.; Ma, R.K.; et al. Prevalence, Clinicopathological Characteristics, and Outcome of Human Papillomavirus–Associated Oropharyngeal Cancer in Southern Chinese Patients. Cancer Epidemiol. Biomark. Prev. 2016, 25, 165–173. [Google Scholar] [CrossRef]
- Lei, J; Ploner, A; Elfstrom, M; Wang, J; Roth, A; Fang, F’ Sundstrom, K; Dillner, J. HPV Vaccination and the Risk of Invasive Cervical Cancer. N Eng J Med 2020, 383, 1340–1348. [CrossRef]
- Falcaro, M.; Soldan, K.; Ndlela, B.; Sasieni, P. Effect of the HPV vaccination programme on incidence of cervical cancer and grade 3 cervical intraepithelial neoplasia by socioeconomic deprivation in England: population based observational study. BMJ 2024, 385, e077341. [Google Scholar] [CrossRef]
- Suksanpaisan, L; Xu, T; Tesfay, MZ; Bomidi, C; Hamm, S; Vandergaast, R; Jenks, N; Steele, MB; Ota-Setlik, A; Akhtar, H, et al. Preclinical Development of Oncolytic Immunotherapy for Treatment of HPV-positive Cancers. Mol Ther Oncolytics 2018, 10, 1–13.
- Bushara, O.; Krogh, K.; Weinberg, S.E.; Finkelman, B.S.; Sun, L.; Liao, J.; Yang, G.-Y. Human Immunodeficiency Virus Infection Promotes Human Papillomavirus-Mediated Anal Squamous Carcinogenesis: An Immunologic and Pathobiologic Review. Pathobiology 2021, 89, 1–12. [Google Scholar] [CrossRef]
- Liu, G.; Sharma, M.; Tan, N.; Barnabas, R.V. HIV-positive women have higher risk of human papilloma virus infection, precancerous lesions, and cervical cancer. AIDS 2018, 32, 795–808. [Google Scholar] [CrossRef]
- Palefsky, J.M. Human papillomavirus-associated anal and cervical cancers in HIV-infected individuals. Curr. Opin. HIV AIDS 2017, 12, 26–30. [Google Scholar] [CrossRef] [PubMed]
- Lien, K.; Mayer, W.; Herrera, R.; Padilla, N.T.; Cai, X.; Lin, V.; Pholcharoenchit, R.; Palefsky, J.; Tugizov, S.M.; Sinclair, A. HIV-1 Proteins gp120 and Tat Promote Epithelial-Mesenchymal Transition and Invasiveness of HPV-Positive and HPV-Negative Neoplastic Genital and Oral Epithelial Cells. Microbiol. Spectr. 2022, 10, e0362222. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef] [PubMed]
- Gordon, K.J.; Blobe, G.C. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim. et Biophys. Acta (BBA) - Mol. Basis Dis. 2008, 1782, 197–228. [Google Scholar] [CrossRef]
- Glauser, D.A.; Schlegel, W. Sequential actions of ERK1/2 on the AP-1 transcription factor allow temporal integration of metabolic signals in pancreatic β cells. FASEB J. 2007, 21, 3240–3249. [Google Scholar] [CrossRef]
- Di Mascio, M.; Srinivasula, S.; Bhattacharjee, A.; Cheng, L.; Martiniova, L.; Herscovitch, P.; Lertora, J.; Kiesewetter, D. Antiretroviral Tissue Kinetics: In Vivo Imaging Using Positron Emission Tomography. Antimicrob. Agents Chemother. 2009, 53, 4086–4095. [Google Scholar] [CrossRef]
- Boomgarden, A.C.; Upadhyay, C. Progress and Challenges in HIV-1 Vaccine Research: A Comprehensive Overview. Vaccines 2025, 13, 148. [Google Scholar] [CrossRef]
- Silver, M.I.; Paul, P.; Sowjanya, P.; Ramakrishna, G.; Vedantham, H.; Kalpana, B.; Shah, K.V.; Gravitt, P.E. Shedding of Epstein-Barr Virus and Cytomegalovirus from the Genital Tract of Women in a Periurban Community in Andhra Pradesh, India. J. Clin. Microbiol. 2011, 49, 2435–2439. [Google Scholar] [CrossRef]
- Kahla, S.; Oueslati, S.; Achour, M.; Kochbati, L.; Chanoufi, M.B.; Maalej, M.; Oueslati, R. Correlation between ebv co-infection and HPV16 genome integrity in Tunisian cervical cancer patients. Braz. J. Microbiol. 2012, 43, 744–753. [Google Scholar] [CrossRef]
- de Lima, M.A.P.; Neto, P.J.N.; Lima, L.P.M.; Júnior, J.G.; Junior, A.G.T.; Teodoro, I.P.P.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr virus (EBV) and cervical carcinoma: A meta-analysis. Gynecol. Oncol. 2018, 148, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Blanco, R.; Carrillo-Beltrán, D.; Osorio, J.C.; Calaf, G.M.; Aguayo, F. Role of Epstein-Barr Virus and Human Papillomavirus Coinfection in Cervical Cancer: Epidemiology, Mechanisms and Perspectives. Pathogens 2020, 9, 685. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, F.; Rasizadeh, R.; Sharaflou, S.; Aghbash, P.S.; Shamekh, A.; Jafari-Sales, A.; Baghi, H.B. Coinfection of EBV with other pathogens: a narrative review. Front. A J. Women Stud. 2024, 4, 1482329. [Google Scholar] [CrossRef]
- Dillner, J; Lenner, P; Lehtinen, M; Eklundl, C; Heino, P; Wiklund, F; Hallmans, G; Stendahl, U. A Population-based Seroepidemiological Study of Cervical Cancer. Cancer Res 1994,54,134-141.
- Rodrigues, FR; Miranda, NL; Fonseca, EC; Pires, ARC; Dias, EP. Investigation of the LMPEBV and Co-infection by HPV in Genital Lesions of Patients Infected or Not by HIV. J Bras Patol Med Lab 2010, 46, 415–420.
- Kaiser, C.; Laux, G.; Eick, D.; Jochner, N.; Bornkamm, G.W.; Kempkes, B. The Proto-Oncogene c- myc Is a Direct Target Gene of Epstein-Barr Virus Nuclear Antigen 2. J. Virol. 1999, 73, 4481–4484. [Google Scholar] [CrossRef]
- Sausen, D.G.; Basith, A.; Muqeemuddin, S. EBV and Lymphomagenesis. Cancers 2023, 15, 2133. [Google Scholar] [CrossRef]
- Cui, X.; Snapper, C.M. Epstein Barr Virus: Development of Vaccines and Immune Cell Therapy for EBV-Associated Diseases. Front. Immunol. 2021, 12, 734471. [Google Scholar] [CrossRef]
- Glover, A.; Shannon-Lowe, C. From pathobiology to targeted treatment in Epstein Barr virus related T cell and Natural Killer cell lymphoproliferative diseases. Ann. Lymphoma 2021, 5, 31–31. [Google Scholar] [CrossRef]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med Microbiol. Immunol. 2018, 208, 573–583. [Google Scholar] [CrossRef]
- Lurain, K.A.; Ramaswami, R.; Krug, L.T.; Whitby, D.; Ziegelbauer, J.M.; Wang, H.-W.; Yarchoan, R.; Forrest, G.N. HIV-associated cancers and lymphoproliferative disorders caused by Kaposi sarcoma herpesvirus and Epstein-Barr virus. Clin. Microbiol. Rev. 2024, 37, e0002223. [Google Scholar] [CrossRef]
- Nash, A.; Ryan, E.J. The oncogenic gamma herpesviruses Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV) hijack retinoic acid-inducible gene I (RIG-I) facilitating both viral and tumour immune evasion. Tumour Virus Res. 2022, 14, 200246. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Liu, Y.; Wang, C.; Gan, R. Signaling pathways of EBV-induced oncogenesis. Cancer Cell Int. 2021, 21, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Cross, JR; Postigo, A; Blight, K; Downward, J. Viral Pro-survival Proteins Block Separate Stages in Bax Inactivation but Changes in Mitochondrial Ultrastructure Still Occur. Cell death Differ 2008, 15, 997.
- Fanidi, A.; Hancock, D.C.; Littlewood, T.D. Suppression of c-Myc-Induced Apoptosis by the Epstein-Barr Virus Gene Product BHRF1. J. Virol. 1998, 72, 8392–8395. [Google Scholar] [CrossRef]
- Chan, JKC; Peterson, BF; Bray, F; Lee, AWM; Rajadurai, P; Lo, KW. Nasopharyngeal Carcinoma. In: WHO Classification of Head and Neck Tumours 5th edition, 2023.
- Wei, K.-R.; Zheng, R.-S.; Zhang, S.-W.; Liang, Z.-H.; Li, Z.-M.; Chen, W.-Q. Nasopharyngeal carcinoma incidence and mortality in China, 2013. Chin. J. Cancer 2017, 36, 1–8. [Google Scholar] [CrossRef]
- Lin, J.-C.; Wang, W.-Y.; Chen, K.Y.; Wei, Y.-H.; Liang, W.-M.; Jan, J.-S.; Jiang, R.-S. Quantification of Plasma Epstein–Barr Virus DNA in Patients with Advanced Nasopharyngeal Carcinoma. New Engl. J. Med. 2004, 350, 2461–2470. [Google Scholar] [CrossRef]
- Su, W.-H.; Hildesheim, A.; Chang, Y.-S. Human Leukocyte Antigens and Epstein–Barr Virus-Associated Nasopharyngeal Carcinoma: Old Associations Offer New Clues into the Role of Immunity in Infection-Associated Cancers. Front. Oncol. 2013, 3, 299. [Google Scholar] [CrossRef]
- Bei, J.-X.; Li, Y.; Jia, W.-H.; Feng, B.-J.; Zhou, G.; Chen, L.-Z.; Feng, Q.-S.; Low, H.-Q.; Zhang, H.; He, F.; et al. A genome-wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat. Genet. 2010, 42, 599–603. [Google Scholar] [CrossRef]
- Bruce, JP; To, KF; Lui, VEY; Chung, GTY; Chan, Y; Tsang, CM; Yip, KY; Ma, BBY; Woo, JKS; Hui, EP, et al. Whole-Genome Profiling of Nasopharyngeal Carcinoma Reveals Viral-Host Co-operation in Inflammatory NF-KB Activation and Immune Escape. Nat Commun 2021, 12, 4193. [CrossRef]
- Carneiro, F; Fukayama, M; Grabsch, HI; Yasui, W. Gastric Adenocarcinoma. In: WHO Classification of Digestive system Tumours, 5th edition, 2019, p.91. Lyons, France, IARC.
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Shimokuri, K.; Kobayashi, Y.; Sasaki, S.; Nakamura, M.; Yanai, H.; Sakai, K.; Suehiro, Y.; et al. Clinical Importance of Epstein–Barr Virus-Associated Gastric Cancer. Cancers 2018, 10, 167. [Google Scholar] [CrossRef]
- Saito, M.; Kono, K. Landscape of EBV-positive gastric cancer. Gastric Cancer 2021, 24, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Cho, H.J.; Seo, J.; Park, K.B.; Kwon, Y.H.; Bae, H.I.; Na Seo, A.; Kim, M. Genetic landscape and PD-L1 expression in Epstein–Barr virus-associated gastric cancer according to the histological pattern. Sci. Rep. 2023, 13, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Salnikov, M.Y.; MacNeil, K.M.; Mymryk, J.S. The viral etiology of EBV-associated gastric cancers contributes to their unique pathology, clinical outcomes, treatment responses and immune landscape. Front. Immunol. 2024, 15, 1358511. [Google Scholar] [CrossRef] [PubMed]
- Saito, R; Abe, H; Kunita, A; Yamashita, H; Setoo, Y; Fukayama, M. Overexpression of PD-L1 in Cancer Cells and PD-L1+ Immune Cells in Epstein-Barr Virus-associated Gastric Cancer, the Prognostic Implications. Mod Pathol 2017, 30, 427–439. [CrossRef]
- Chou, T; Wong, P; Chang, Y. Lymphoepithelial Carcinoma of the Lung.In: WHO Classification of Thoracic Tumours, 5th edition, 2021, pp.94-96. Travis, WD; Chan, JKC (eds). Lyons, France, IARC.
- The, Y; Kao, H; Lee,K; Wu, M; Ho, H; Chou, T. Epstein-Barr Virus-associated pulmonary Carcinoma: Proposing an Alternative Term and Expanding the Histologic Spectrum of Lymphoepithelioma-like Carcinoma of the Lung. Am J Surg Pathol 2019, 43,211-219. [CrossRef]
- Chang, Y.-L.; Wu, C.-T.; Shih, J.-Y.; Lee, Y.-C. New Aspects in Clinicopathologic and Oncogene Studies of 23 Pulmonary Lymphoepithelioma-Like Carcinomas. Am. J. Surg. Pathol. 2002, 26, 715–723. [Google Scholar] [CrossRef]
- Zhao, L.; Ding, J.-Y.; Tao, Y.-L.; Zhu, K.; Chen, G. Detection of Epstein–Barr virus infection in thymic epithelial tumors by nested PCR and Epstein–Barr-encoded RNA ISH. Infect. Agents Cancer 2023, 18, 1–8. [Google Scholar] [CrossRef]
- Wu, T.-C.; Kuo, T.-T. Study of Epstein-Barr virus early RNA1 (EBER1) expression by in situ hybridization in thymic epithelial tumors of Chinese patients in Taiwan. Hum. Pathol. 1993, 24, 235–238. [Google Scholar] [CrossRef]
- Zhang, G.; Yu, Z.; Shen, G.; Chai, Y.; Liang, C. Association between Epstein-Barr virus and Thymic epithelial tumors: a systematic review. Infect. Agents Cancer 2019, 14, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Chan, JKC, Chalabreysse, L; Mukai, K, Tateyama, H. Lymphoepithelial Carcinoma of the Thymus. In: WHO Classification of Thoracic Tumours, 5th edition, 2021, pp. 361-363. Lyons, France, IARC.
- Huang, J.; Ahmad, U.; Antonicelli, A.; Catlin, A.C.; Fang, W.; Gomez, D.; Loehrer, P.; Lucchi, M.; Marom, E.; Nicholson, A.; et al. Development of the International Thymic Malignancy Interest Group International Database: An Unprecedented Resource for the Study of a Rare Group of Tumors. J. Thorac. Oncol. 2014, 9, 1573–1578. [Google Scholar] [CrossRef] [PubMed]
- Sayed, S; Leoncini, L; Siebert, R; Ferry, JA; Meideiros, LJ; Cheuk, W, Klapper, W; d’Amore, ESG; Naresh, KN; Dave, SS, et al. Burkitt Lymphoma. In: WHO Classification of Hematolymphoid Tumours, 5th edition, 2024.
- Johnston, W.; Erdmann, F.; Newton, R.; Steliarova-Foucher, E.; Schüz, J.; Roman, E. Childhood cancer: Estimating regional and global incidence. Cancer Epidemiology 2021, 71, 101662. [Google Scholar] [CrossRef] [PubMed]
- Leoncini, L. Epstein–Barr virus positivity as a defining pathogenetic feature of Burkitt lymphoma subtypes. Br. J. Haematol. 2021, 196, 468–470. [Google Scholar] [CrossRef]
- Grande, B.M.; Gerhard, D.S.; Jiang, A.; Griner, N.B.; Abramson, J.S.; Alexander, T.B.; Allen, H.; Ayers, L.W.; Bethony, J.M.; Bhatia, K.; et al. Genome-wide discovery of somatic coding and noncoding mutations in pediatric endemic and sporadic Burkitt lymphoma. Blood 2019, 133, 1313–1324. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Deroubaix, S.; Feldhahn, N.; Oliveira, T.Y.; Callen, E.; Wang, Q.; Jankovic, M.; Silva, I.T.; Rommel, P.C.; Bosque, D.; et al. Plasmodium Infection Promotes Genomic Instability and AID-Dependent B Cell Lymphoma. Cell 2015, 162, 727–737. [Google Scholar] [CrossRef]
- Ramaswami, R; Chia, G; Pria, AD; Pinato, DJ; Parker, K; Nelson, M; Bower, M. Evolution of HIV-associated Lymphoma over 3 Decades. J Acquir Immune Defic Synd 2016,72,177-183. [CrossRef]
- Mundo, L.; Del Porro, L.; Granai, M.; Siciliano, M.C.; Mancini, V.; Santi, R.; Marcar, L.; Vrzalikova, K.; Vergoni, F.; Di Stefano, G.; et al. Correction: Frequent traces of EBV infection in Hodgkin and non-Hodgkin lymphomas classified as EBV-negative by routine methods: expanding the landscape of EBV-related lymphomas. Mod. Pathol. 2020, 33, 2637–2637. [Google Scholar] [CrossRef]
- Ng, C.S.; Qin, J. New Facets of Hematolymphoid Eponymic Diseases. Lymphatics 2025, 3, 9. [Google Scholar] [CrossRef]
- Alsharif, R.; Dunleavy, K. Burkitt Lymphoma and Other High-Grade B-Cell Lymphomas with or without MYC, BCL2, and/or BCL6 Rearrangements. Hematol. Clin. North Am. 2019, 33, 587–596. [Google Scholar] [CrossRef]
- Borges, AM; Delabie, J; Vielh, P; d’Amore, ESG; Hebeda, KM; Diepstra, A; Naresh, KM; Garcia, JF; Tamarn, J; Laskar, S, et al. Classic Hodgkin Lymphoma. In: WHO Classification of Haematolymphoid Tumours, 5th edition, 2024.
- Kanzler, H.; Küppers, R.; Hansmann, M.L.; Rajewsky, K. Hodgkin and Reed-Sternberg cells in Hodgkin's disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells. J. Exp. Med. 1996, 184, 1495–1505. [Google Scholar] [CrossRef] [PubMed]
- Harabuchi, Y; Imai, S; Wakashima, J; Hirao, M; Katanra, A; Osato, T; Kon, S. Nasal T-cell Lymphoma Causally Associated with Epstein-Barr Virus: Clinicopathologic, Phenotypic and Genotypic Studies. Cancer 1996, 77, 2137–2149.
- Wang, H.; Fu, B.-B.; Gale, R.P.; Liang, Y. NK-/T-cell lymphomas. Leukemia 2021, 35, 2460–2468. [Google Scholar] [CrossRef] [PubMed]
- Au, W.-Y.; Pang, A.; Choy, C.; Chim, C.-S.; Kwong, Y.-L. Quantification of circulating Epstein-Barr virus (EBV) DNA in the diagnosis and monitoring of natural killer cell and EBV-positive lymphomas in immunocompetent patients. Blood 2004, 104, 243–249. [Google Scholar] [CrossRef]
- Wai, C.M.M.; Chen, S.; Phyu, T.; Fan, S.; Leong, S.M.; Zheng, W.; Low, L.C.Y.; Choo, S.-N.; Lee, C.-K.; Chung, T.-H.; et al. Immune pathway upregulation and lower genomic instability distinguish EBV-positive nodal T/NK-cell lymphoma from ENKTL and PTCL-NOS. Haematologica 2022, 107, 1864–1879. [Google Scholar] [CrossRef]
- Cohen, J.I.; Kimura, H.; Nakamura, S.; Ko, Y.-H.; Jaffe, E.S. Epstein–Barr virus-associated lymphoproliferative disease in non-immunocompromised hosts: a status report and summary of an international meeting, 8–9 September 2008. Ann. Oncol. 2009, 20, 1472–1482. [Google Scholar] [CrossRef]
- Tang, Y.-T.; Wang, D.; Luo, H.; Xiao, M.; Zhou, H.-S.; Liu, D.; Ling, S.-P.; Wang, N.; Hu, X.-L.; Luo, Y.; et al. Aggressive NK-cell leukemia: clinical subtypes, molecular features, and treatment outcomes. Blood Cancer J. 2017, 7, 1–5. [Google Scholar] [CrossRef]
- Jiang, X.-N.; Zhang, Y.; Xue, T.; Chen, J.-Y.; Chan, A.C.F.; Cheuk, W.F.; Chan, J.K.F.; Li, X.-Q. New Clinicopathologic Scenarios of EBV+ Inflammatory Follicular Dendritic Cell Sarcoma. Am. J. Surg. Pathol. 2020, 45, 765–772. [Google Scholar] [CrossRef]
- Cheuk, W.M.; Chan, J.K.C.M.; Shek, T.W.H.M.; Chang, J.H.; Tsou, M.-H.; Yuen, N.W.F.M.; Ng, W.-F.M.; Chan, A.C.L.M.; Prat, J. Inflammatory Pseudotumor-Like Follicular Dendritic Cell Tumor. Am. J. Surg. Pathol. 2001, 25, 721–731. [Google Scholar] [CrossRef]
- Li, Y.; Yang, X.; Tao, L.; Zeng, W.B.; Zuo, M.; Li, S.; Wu, L.B.; Lin, Y.; Zhang, Z.; Yun, J.; et al. Challenges in the Diagnosis of Epstein-Barr Virus-positive Inflammatory Follicular Dendritic Cell Sarcoma. Am. J. Surg. Pathol. 2022, 47, 476–489. [Google Scholar] [CrossRef]
- Natkunam, Y; Bhagat, G; Chadbum, A; Naresh, KN; Chan, J; Michelow, P; Satou, A; Satom Y; Bower, M; Gratzinger, D, et al. EBV-mucocutaneous Ulcer. In: WHO Classification of Haematolymphoid Tumours, 5th edition, 2024. deJong, D; Siebert, R; Alaggio, R; Coupland, SE (eds). Lyoms, France, IARC (available online from https://tumourclassification.iarc.who.int/chapters/63).
- Anagnostopoulos, I; Siebert, R; Nicholson, AG; Deckert, M. Lymphomatoid Granulomatosis. In: WHO Classification of Haematolymphoid Tumours, 5th edition, 2024. Oh, G; de jong, D (eds). Lyons, France, IARC (available online from https://tumourclassification.iarc.who.int/chapters/63).
- Anagnostopoulis, I; Medeiros, J; Klapper, W; de Jong, D; Miles, RR; Lenz, G; Asano, N; Chapman, JR; Steidi, C. EBV-positive Diffuse Large B-cell Lymphoma. In: WHO Classification of Haematolymphoid Tumours, 5th edition, 2024. Oh, G; Chan, J (eds). Lyons, france, IARC (available online from https://tumourclassification.iarc.who.int/chapters/63.).
- Montes-Morento, S; Leoncini, L; Miranda, R; Louissaint, A, Jr; sengar, M. Plasmablastic Lymphoma. In: WHO Classification of Haematolymphoid Tumours, 5th edition, 2024. Oh, G; de Jong, D; Dave, SS (eds).Lyons, France, IARC (available online from https://tumour classification.iarc.who.int/chapters/63).
- Cui, X.; Cao, Z.; Ishikawa, Y.; Cui, S.; Imadome, K.-I.; Snapper, C.M. Immunization with Epstein–Barr Virus Core Fusion Machinery Envelope Proteins Elicit High Titers of Neutralizing Activities and Protect Humanized Mice from Lethal Dose EBV Challenge. Vaccines 2021, 9, 285. [Google Scholar] [CrossRef] [PubMed]
- Moutschen, M.; Léonard, P.; Sokal, E.M.; Smets, F.; Haumont, M.; Mazzu, P.; Bollen, A.; Denamur, F.; Peeters, P.; Dubin, G.; et al. Phase I/II studies to evaluate safety and immunogenicity of a recombinant gp350 Epstein–Barr virus vaccine in healthy adults. Vaccine 2007, 25, 4697–4705. [Google Scholar] [CrossRef] [PubMed]
- Draper, S.J.; Angov, E.; Horii, T.; Miller, L.H.; Srinivasan, P.; Theisen, M.; Biswas, S. Recent advances in recombinant protein-based malaria vaccines. Vaccine 2015, 33, 7433–7443. [Google Scholar] [CrossRef]
- Zhong, L.; Zhao, Q.; Zeng, M.-S.; Zhang, X. Prophylactic vaccines against Epstein–Barr virus. Lancet 2024, 404, 845–845. [Google Scholar] [CrossRef]
- Li, P.; Meng, Z.; Zhou, Z.; Zhong, Z.; Kang, M. Therapeutic vaccines for Epstein–Barr virus: a way forward. Lancet 2024, 403, 2779–2780. [Google Scholar] [CrossRef]
- Chang Y; Cesarman, E; Pessin, MS; Lee,F; Culpepper, J; Knowles, DM; Moore, PS. Identification of Herpes-like DNA Sequences in AIDS-associated Kaposi’s Sarcoma. Science 1994, 266, 1865–1869.
- Ablashi, D.V.; Chatlynne, L.G.; Whitman, J.J.E.; Cesarman, E. Spectrum of Kaposi's Sarcoma-Associated Herpesvirus, or Human Herpesvirus 8, Diseases. Clin. Microbiol. Rev. 2002, 15, 439–464. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Prim. 2019, 5, 1–21. [Google Scholar] [CrossRef]
- Gantts,S; i Casper, C. Human Herpesvirus 8-associated Neoplasms: The Roles of Viral Replication and Antiviral Treatment. Curr Opin. Infect. Dis. 2011, 24, 295–301. [Google Scholar] [CrossRef]
- Suda, T; Katano, H; Delso, G; Kakiuchi, C; Nakamura, T; Shiota, T; Higashihara, M; Mori, S. HHV-8 Infection Status of AIDS-unrelated and AIDS-associated Multicentric Castleman Disease. Pathol. Int. 2001, 51, 671–679. [Google Scholar] [CrossRef]
- Chadbum, A; Said, JW; Du, M; Vega, F. KSHV/HHV8-positive Germinotropic Lymohoproliferative Disorder. In: WHO Classification of Haematolymphoid Tumours, 5th Edition, 2024.
- Douglas, Jl; Gustin, JK; Moses, AV; Dezube, BJ; Pantanowitz, L. Kaposi Sarcoma Pathogenesis: A Triad of Viral Infection, Oncogenesis and Chronic Inflammation. Transl Biomed 2010,1,172.
- Radu, O; Pantanowitz, L. Kpaosi Sarcoma. Arch. Pathol. Lab. Med. 2013, 137, 289–294. [Google Scholar] [CrossRef]
- Guillet, S.; Gérard, L.; Meignin, V.; Agbalika, F.; Cuccini, W.; Denis, B.; Katlama, C.; Galicier, L.; Oksenhendler, E. Classic and extracavitary primary effusion lymphoma in 51 HIV-infected patients from a single institution. Am. J. Hematol. 2015, 91, 233–237. [Google Scholar] [CrossRef]
- Bruce-Brand, C.; Rigby, J. Kaposi Sarcoma With Intravascular Primary Effusion Lymphoma in the Skin: A Potential Pitfall in HHV8 Immunohistochemistry Interpretation. Int. J. Surg. Pathol. 2020, 28, 868–871. [Google Scholar] [CrossRef]
- Said, JW; Chadbum, A; Medeiros, LJ; Bacon, CM; Cesarman, E; Michelow, P; Du, M; Bower, M. Primary Effusion Lymphoma. In: WHO Classification of Haematolymphoid Tumours, 5th Edition, 2024.
- Rossi, G.; Cozzi, I.; Della Starza, I.; De Novi, L.A.; De Propris, M.S.; Gaeta, A.; Petrucci, L.; Pulsoni, A.; Pulvirenti, F.; Ascoli, V. Human herpesvirus-8–positive primary effusion lymphoma in HIV-negative patients: Single institution case series with a multidisciplinary characterization. Cancer Cytopathol. 2020, 129, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Das, D.K. Serous effusions in malignant lymphomas: A review. Diagn. Cytopathol. 2006, 34, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Lurain, K.; Polizzotto, M.N.; Aleman, K.; Bhutani, M.; Wyvill, K.M.; Gonçalves, P.H.; Ramaswami, R.; Marshall, V.A.; Miley, W.; Steinberg, S.M.; et al. Viral, immunologic, and clinical features of primary effusion lymphoma. Blood 2019, 133, 1753–1761. [Google Scholar] [CrossRef] [PubMed]
- Hong, Y.-K.; Foreman, K.; Shin, J.W.; Hirakawa, S.; Curry, C.L.; Sage, D.R.; Libermann, T.; Dezube, B.J.; Fingeroth, J.D.; Detmar, M. Lymphatic reprogramming of blood vascular endothelium by Kaposi sarcoma–associated herpesvirus. Nat. Genet. 2004, 36, 683–685. [Google Scholar] [CrossRef]
- Dittmer, D.P.; Damania, B. Kaposi sarcoma associated herpesvirus pathogenesis (KSHV)—an update. Curr. Opin. Virol. 2013, 3, 238–244. [Google Scholar] [CrossRef]
- Mariggiò, G.; Koch, S.; Schulz, T.F. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160275. [Google Scholar] [CrossRef]
- Pantanowitz, L.; Moses, A.V.; Dezube, B.J. The inflammatory component of Kaposi sarcoma. Exp. Mol. Pathol. 2009, 87, 163–165. [Google Scholar] [CrossRef]
- Kaposi Idiopathisches multiples Pigmentsarkom der Haut. Arch. Dermatol. Res. 1872, 4, 265–273. [CrossRef]
- Bigi, R.; Landis, J.T.; An, H.; Caro-Vegas, C.; Raab-Traub, N.; Dittmer, D.P. Epstein–Barr virus enhances genome maintenance of Kaposi sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. 2018, 115, 201810128–E11387. [Google Scholar] [CrossRef]
- Cesarmann, E; Chang, Y; Moore, Y; Said, JW; Knowles, DM. Kaposi’s Sarcoma-associated Herpesvirus-like DNA Sequences in AIDS-related Body Cavity-based Lymphoma. N Eng J Med 1995,4,1186-1191. Doi:10.1056/NEJM199505043321802.
- Vega, F; Chadbum, A; Said, JW; Cesarmann, E; Du, M; Bower, M. KSH/HHV8-positive Diffuse Large B-cell Lymphoma. In; WHO Classification of Haematolymphoid Tumours, 5th Edition, 2024. De Jong, D; Naresh, KN; Chan, J (eds). Lyons, France, IARC (available online from https://tumourclassification.iarc.who.int/chapters/63).
- Casper, C.; Corey, L.; Cohen, J.I.; Damania, B.; Gershon, A.A.; Kaslow, D.C.; Krug, L.T.; Martin, J.; Mbulaiteye, S.M.; Mocarski, E.S.; et al. KSHV (HHV8) vaccine: promises and potential pitfalls for a new anti-cancer vaccine. npj Vaccines 2022, 7, 1–10. [Google Scholar] [CrossRef]
- Chandran, B. Early Events in Kaposi's Sarcoma-Associated Herpesvirus Infection of Target Cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Mulama, D.H.; Mutsvunguma, L.Z.; Totonchy, J.; Ye, P.; Foley, J.; Escalante, G.M.; Rodriguez, E.; Nabiee, R.; Muniraju, M.; Wussow, F.; et al. A multivalent Kaposi sarcoma-associated herpesvirus-like particle vaccine capable of eliciting high titers of neutralizing antibodies in immunized rabbits. Vaccine 2019, 37, 4184–4194. [Google Scholar] [CrossRef] [PubMed]
- Muniraju, M.; Mutsvunguma, L.Z.; Foley, J.; Escalante, G.M.; Rodriguez, E.; Nabiee, R.; Totonchy, J.; Mulama, D.H.; Nyagol, J.; Wussow, F.; et al. Kaposi Sarcoma-Associated Herpesvirus Glycoprotein H Is Indispensable for Infection of Epithelial, Endothelial, and Fibroblast Cell Types. J. Virol. 2019, 93. [Google Scholar] [CrossRef] [PubMed]
- Kwun, H.J.; da Silva, S.R.; Qin, H.; Ferris, R.L.; Tan, R.; Chang, Y.; Moore, P.S. The central repeat domain 1 of Kaposi's sarcoma-associated herpesvirus (KSHV) latency associated-nuclear antigen 1 (LANA1) prevents cis MHC class I peptide presentation. Virology 2011, 412, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Sugden, A.U.; Hayes, M.; Sugden, B. How Epstein–Barr Virus and Kaposi’s Sarcoma-Associated Herpesvirus Are Maintained Together to Transform the Same B-Cell. Viruses 2021, 13, 1478. [Google Scholar] [CrossRef]
- Böni, M.; Rieble, L.; Münz, C. Co-Infection of the Epstein–Barr Virus and the Kaposi Sarcoma-Associated Herpesvirus. Viruses 2022, 14, 2709. [Google Scholar] [CrossRef]
- Julius, P.; Kang, G.; Siyumbwa, S.; Musumali, J.; Tso, F.Y.; Ngalamika, O.; Kaile, T.; Maate, F.; Moonga, P.; West, J.T.; et al. Co-infection and co-localization of Kaposi sarcoma-associated herpesvirus and Epstein-Barr virus in HIV-associated Kaposi sarcoma: a case report. Front. Cell. Infect. Microbiol. 2023, 13, 1270935. [Google Scholar] [CrossRef]
- King, SR. HIV: Virology and Mechanisms of Disease. Ann. Emerg. Med. 1994, 24, 3. [Google Scholar] [CrossRef]
- Chinen, J.; Shearer, W.T. Molecular virology and immunology of HIV infection. J. Allergy Clin. Immunol. 2002, 110, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Kaposi’s Sarcoma and Pneucystitis Pneumonia Among Homosexual Men – New York City and California. MMWR Morb Mortal Wkly Rep 1981, 30, 305–308.
- Yarchoan, R.; Uldrick, T.S. HIV-Associated Cancers and Related Diseases. N. Engl. J. Med. 2018, 378, 1029–1041. [Google Scholar] [CrossRef]
- Isaguliants, M.; Bayurova, E.; Avdoshina, D.; Kondrashova, A.; Chiodi, F.; Palefsky, J.M. Oncogenic Effects of HIV-1 Proteins, Mechanisms Behind. Cancers 2021, 13, 305. [Google Scholar] [CrossRef]
- Omar, A.; Marques, N.; Crawford, N. Cancer and HIV: The Molecular Mechanisms of the Deadly Duo. Cancers 2024, 16, 546. [Google Scholar] [CrossRef]
- Lurain, K.A.; Ramaswami, R.; Krug, L.T.; Whitby, D.; Ziegelbauer, J.M.; Wang, H.-W.; Yarchoan, R.; Forrest, G.N. HIV-associated cancers and lymphoproliferative disorders caused by Kaposi sarcoma herpesvirus and Epstein-Barr virus. Clin. Microbiol. Rev. 2024, 37, e0002223. [Google Scholar] [CrossRef]
- Zhao, L.-H.; Liu, X.; Yan, H.-X.; Li, W.-Y.; Zeng, X.; Yang, Y.; Zhao, J.; Liu, S.P.; Zhuang, X.-H.; Lin, C.; et al. Genomic and oncogenic preference of HBV integration in hepatocellular carcinoma. Nat. Commun. 2016, 7, 12992. [Google Scholar] [CrossRef]
- Nevola, R.; Beccia, D.; Rosato, V.; Ruocco, R.; Mastrocinque, D.; Villani, A.; Perillo, P.; Imbriani, S.; Femine, A.D.; Criscuolo, L.; et al. HBV Infection and Host Interactions: The Role in Viral Persistence and Oncogenesis. Int. J. Mol. Sci. 2023, 24, 7651. [Google Scholar] [CrossRef]
- Zhang, M; Chen, H; Liu, H; Tang, H. The Impact of Integrated Hepatitis B Virus DNA on Oncogenesis and Anti-viral Therapy. Biomarkers Res. 2024, 12, 84. [Google Scholar] [CrossRef]
- Flores, J.E.; Thompson, A.J.; Ryan, M.; Howell, J. The Global Impact of Hepatitis B Vaccination on Hepatocellular Carcinoma. Vaccines 2022, 10, 793. [Google Scholar] [CrossRef] [PubMed]
- Forner, A; Llovet, JM; Brnix, J. Hepatocellular Carcinoma. Lancet 2012, 379, 1245–1255. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.R.; Barnes, E.; Cox, A.L. Approaches, Progress, and Challenges to Hepatitis C Vaccine Development. Gastroenterology 2019, 156, 418–430. [Google Scholar] [CrossRef] [PubMed]
- Virzì, A.; Suarez, A.A.R.; Baumert, T.F.; Lupberger, J. Oncogenic Signaling Induced by HCV Infection. Viruses 2018, 10, 538. [Google Scholar] [CrossRef]
- Fiehn, F.; Beisel, C.; Binder, M. Hepatitis C virus and hepatocellular carcinoma: carcinogenesis in the era of direct-acting antivirals. Curr. Opin. Virol. 2024, 67, 101423. [Google Scholar] [CrossRef]
- Martineau, C.-A.; Rivard, N.; Bisaillon, M. From viruses to cancer: exploring the role of the hepatitis C virus NS3 protein in carcinogenesis. Infect. Agents Cancer 2024, 19, 1–14. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Yoon, S.K.; Lencioni, R. The Etiology of Hepatocellular Carcinoma and Consequences for Treatment. Oncol. 2010, 15, 14–22. [Google Scholar] [CrossRef]
- Torbenson, M; Ng, I; Park, YN; Roncalli, M; Sakamato, M. Hepatocellular Carcinoma. In: WHO Classification of Digestive System Tumours, 5th Edition, 2019, p.229-239. Shirmacher, P (eds). Lyons, France, IARC.
- Burt, A.D.; Alves, V.; Bedossa, P.; Clouston, A.; Guido, M.; Hübscher, S.; Kakar, S.; Ng, I.; Park, Y.N.; Reeves, H.; et al. Data set for the reporting of intrahepatic cholangiocarcinoma, perihilar cholangiocarcinoma and hepatocellular carcinoma: recommendations from the International Collaboration on Cancer Reporting (ICCR). Histopathology 2018, 73, 369–385. [Google Scholar] [CrossRef]
- Romano, L.; Paladini, S.; Galli, C.; Raimondo, G.; Pollicino, T.; Zanetti, A.R. Hepatitis B Vaccination. Hum. Vaccin. Immunother. 2015, 11, 53–57. [Google Scholar] [CrossRef]
- Vesikari, T.; Finn, A.; van Damme, P.; Leroux-Roels, I.; Leroux-Roels, G.; Segall, N.; Toma, A.; Vallieres, G.; Aronson, R.; Reich, D.; et al. Immunogenicity and Safety of a 3-Antigen Hepatitis B Vaccine vs a Single-Antigen Hepatitis B Vaccine. JAMA Netw. Open 2021, 4, e2128652–e2128652. [Google Scholar] [CrossRef]
- Chang, M.-H.; You, S.-L.; Chen, C.-J.; Liu, C.-J.; Lee, C.-M.; Lin, S.-M.; Chu, H.-C.; Wu, T.-C.; Yang, S.-S.; Kuo, H.-S.; et al. Decreased Incidence of Hepatocellular Carcinoma in Hepatitis B Vaccinees: A 20-Year Follow-up Study. JNCI J. Natl. Cancer Inst. 2009, 101, 1348–1355. [Google Scholar] [CrossRef]
- Chen, T.-W. Paths toward hepatitis B immunization in South Korea and Taiwan. Clin. Exp. Vaccine Res. 2013, 2, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Fu, Y.; Allain, J.-P.; Li, C. Chronic and occult hepatitis B virus infections in the vaccinated Chinese population. Ann. Blood 2016, 2, 4–4. [Google Scholar] [CrossRef]
- Cao, M.; Fan, J.; Lu, L.; Fan, C.; Wang, Y.; Chen, T.; Zhang, S.; Yu, Y.; Xia, C.; Lu, J.; et al. Long term outcome of prevention of liver cancer by hepatitis B vaccine: Results from an RCT with 37 years. Cancer Lett. 2022, 536, 215652. [Google Scholar] [CrossRef]
- Wong, NS; Chan, DPC, Poon, CM; Chan, CP; Lau, LHW; Yeoh, EK; Lee, SS. Hepatitis B Burden and Population Immunity in a High Endemicity City – a Geographically Random Household Epidemiology Study for Evaluating Achievability of Elimination. Epidemiol Infect 2022, 151, e22, 1–11.
- Shoukry, N.H. Hepatitis C vaccines, antibodies, and T cells. Front. Immunol. 2018, 9, 1480. [Google Scholar] [CrossRef]
- Czarnota, A.; Raszplewicz, A.; Sławińska, A.; Bieńkowska-Szewczyk, K.; Grzyb, K. Minicircle-based vaccine induces potent T-cell and antibody responses against hepatitis C virus. Sci. Rep. 2024, 14, 1–11. [Google Scholar] [CrossRef]
- Garcia-Samaniego, J.; Rodriguez, M.; Berenguer, J.; Rodriguez-Rosado, R.; Carbo, J.; Asensi, V.; Soriano, V. Hepatocellular carcinoma in HIV-infected patients with chronic hepatitis C. Am. J. Gastroenterol. 2001, 96, 179–183. [Google Scholar] [CrossRef]
- Gjærde, L.I.; Shepherd, L.; Jablonowska, E.; Lazzarin, A.; Rougemont, M.; Darling, K.; Battegay, M.; Braun, D.; Martel-Laferriere, V.; Lundgren, J.D.; et al. Trends in Incidences and Risk Factors for Hepatocellular Carcinoma and Other Liver Events in HIV and Hepatitis C Virus–coinfected Individuals From 2001 to 2014: A Multicohort Study. Clin. Infect. Dis. 2016, 63, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Awadh, A.A.; Alharthi, A.A.; Alghamdi, B.A.; Alghamdi, S.T.; Baqays, M.K.; Binrabaa, I.S.; Malli, I.A. Coinfection of Hepatitis B and C Viruses and Risk of Hepatocellular Carcinoma: Systematic Review and Meta-analysis. J. Glob. Infect. Dis. 2024, 16, 127–134. [Google Scholar] [CrossRef] [PubMed]
- Zampino, R.; Pisaturo, M.A.; Cirillo, G.; Marrone, A.; Macera, M.; Rinaldi, L.; Stanzione, M.; Durante-Mangoni, E.; Gentile, I.; Sagnelli, E.; et al. Hepatocellular carcinoma in chronic HBV-HCV co-infection is correlated to fibrosis and disease duration. Ann. Hepatol. 2015, 14, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Maqsood, Q.; Sumrin, A.; Iqbal, M.; Younas, S.; Hussain, N.; Mahnoor, M.; Wajid, A. Hepatitis C virus/Hepatitis B virus coinfection: Current prospectives. Antivir. Ther. 2023, 28. [Google Scholar] [CrossRef]
- Mehershanhi, S.; Haider, A.; Kandhi, S.; Sun, H.; Patel, H. Prevalence of Hepatocellular Carcinoma in HIV Patients Co-infected or Triple Infected With Hepatitis B and Hepatitis C in a Community Hospital in South Bronx. Cureus 2022, 14, e26089. [Google Scholar] [CrossRef]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal Integration of a Polyomavirus in Human Merkel Cell Carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef]
- Spurgeon, M.E.; Lambert, P.F. Merkel cell polyomavirus: A newly discovered human virus with oncogenic potential. Virology 2013, 435, 118–130. [Google Scholar] [CrossRef]
- Silling, S.; Kreuter, A.; Gambichler, T.; Meyer, T.; Stockfleth, E.; Wieland, U. Epidemiology of Merkel Cell Polyomavirus Infection and Merkel Cell Carcinoma. Cancers 2022, 14, 6176. [Google Scholar] [CrossRef]
- Yang, J.F.; You, J. Merkel cell polyomavirus and associated Merkel cell carcinoma. Tumour Virus Res. 2021, 13, 200232. [Google Scholar] [CrossRef]
- DeCaprio, J.A. Merkel cell polyomavirus and Merkel cell carcinoma. Philos. Trans. R. Soc. B: Biol. Sci. 2017, 372, 20160276. [Google Scholar] [CrossRef]
- Zhou, X.; Yin, C.; Lin, Z.; Yan, Z.; Wang, J. Merkel Cell Polyomavirus Co-Infection in HIV/AIDS Individuals: Clinical Diagnosis, Consequences and Treatments. Pathogens 2025, 14, 134. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.-Y.; Lai, Y.-R.; Cheng, P.-S.; Yu, W.-W.; Wang, R.C.; Shen, W.-L.; Chuang, S.-S. Merkel cell carcinoma in Taiwan: a subset is chronic arsenicism-related, and the Merkel cell polyomavirus-negative cases are pathologically distinct from virus-related cases with a poorer outcome. Pathology 2024, 57, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Jiang, S.; He, Y.; Jin, X.; Zhao, G.; Wang, B. Development of a therapeutic vaccine targeting Merkel cell polyomavirus capsid protein VP1 against Merkel cell carcinoma. npj Vaccines 2021, 6, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Frey, A.; Pevzy, C.; olino, K.; Ishizwka, J. Development of an mRNA Therapeutic Vaccine for Virally Driven Merkel Cell Carcinoma. J. Clin. Oncol. 2024, 42, s2637. [Google Scholar] [CrossRef]
- Gambichler, T.; Schrama, D.; Käpynen, R.; Weyer-Fahlbusch, S.S.; Becker, J.C.; Susok, L.; Kreppel, F.; Abu Rached, N. Current Progress in Vaccines against Merkel Cell Carcinoma: A Narrative Review and Update. Vaccines 2024, 12, 533. [Google Scholar] [CrossRef]
- Rosean, C.B.; Leyder, E.C.; Hamilton, J.; Carter, J.J.; Galloway, D.A.; Koelle, D.M.; Nghiem, P.; Heiland, T. LAMP1 targeting of the large T antigen of Merkel cell polyomavirus results in potent CD4 T cell responses and tumor inhibition. Front. Immunol. 2023, 14, 1253568. [Google Scholar] [CrossRef]
- Zeng, Q.; Gomez, B.P.; Viscidi, R.P.; Peng, S.; He, L.; Ma, B.; Wu, T.-C.; Hung, C.-F. Development of a DNA vaccine targeting Merkel cell polyomavirus. Vaccine 2011, 30, 1322–1329. [Google Scholar] [CrossRef]
- A Engels, E.; Frisch, M.; Goedert, J.J.; Biggar, R.J.; Miller, R.W. Merkel cell carcinoma and HIV infection. Lancet 2002, 359, 497–498. [Google Scholar] [CrossRef]
- Izikson, L.; Nornhold, E.; Iyer, J.G.; Nghiem, P.; Zeitouni, N.C. Merkel cell carcinoma associated with HIV: review of 14 patients. AIDS 2011, 25, 119–121. [Google Scholar] [CrossRef]
- Brites, C.; Grassi, M.F.; Quaresma, J.A.S.; Ishak, R.; Vallinoto, A.C.R. Pathogenesis of HTLV-1 infection and progression biomarkers: An overview. Braz. J. Infect. Dis. 2021, 25, 101594. [Google Scholar] [CrossRef]
- Forlani, G.; Shallak, M.; Accolla, R.S.; Romanelli, M.G. HTLV-1 Infection and Pathogenesis: New Insights from Cellular and Animal Models. Int. J. Mol. Sci. 2021, 22, 8001. [Google Scholar] [CrossRef]
- Hirons, A.; Khoury, G.; Purcell, D.F.J. Human T-cell lymphotropic virus type-1: a lifelong persistent infection, yet never truly silent. Lancet Infect. Dis. 2021, 21, e2–e10. [Google Scholar] [CrossRef] [PubMed]
- GonçaLves, D.U.; Proietti, F.A.; Ribas, J.G.R.; AraújO, M.G.; Pinheiro, S.R.; Guedes, A.C.; Carneiro-Proietti, A.B.F. Epidemiology, Treatment, and Prevention of Human T-Cell Leukemia Virus Type 1-Associated Diseases. Clin. Microbiol. Rev. 2010, 23, 577–589. [Google Scholar] [CrossRef]
- Bangham, C.R.M. HTLV-1 persistence and the oncogenesis of adult T cell leukemia/lymphoma. Blood 2023, 141, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Letafati, A.; Bahari, M.; Ardekani, O.S.; Jazi, N.N.; Nikzad, A.; Norouzi, F.; Mahdavi, B.; Aboofazeli, A.; Mozhgani, S.-H. HTLV-1 vaccination Landscape: Current developments and challenges. Vaccine: X 2024, 19, 100525. [Google Scholar] [CrossRef] [PubMed]
- Attygalle, A.D.; Karube, K.; Jeon, Y.K.; Cheuk, W.; Bhagat, G.; Chan, J.K.C.; Naresh, K.N. The fifth edition of the WHO classification of mature T cell, NK cell and stroma-derived neoplasms. J. Clin. Pathol. 2025, 78, 217–232. [Google Scholar] [CrossRef]
- Suehiro, Y.; Hasegawa, A.; Iino, T.; Sasada, A.; Watanabe, N.; Matsuoka, M.; Takamori, A.; Tanosaki, R.; Utsunomiya, A.; Choi, I.; et al. Clinical outcomes of a novel therapeutic vaccine with Tax peptide-pulsed dendritic cells for adult T cell leukaemia/lymphoma in a pilot study. Br. J. Haematol. 2015, 169, 356–367. [Google Scholar] [CrossRef]
- McFee, R.B. SARS2 Human Coronavirus (COVID-19, SARSCoV2). Dis. Mon. 2020, 66, 10163. [Google Scholar] [CrossRef]
- Hu, B.; Guo, H.; Zhou, P.; Shi, Z.-L. Characteristics of SARS-CoV-2 and COVID-19. Nat. Rev. Microbiol. 2021, 19, 141–154. [Google Scholar] [CrossRef]
- Policard, M.; Jain, S.; Rego, S.; Dakshanamurthy, S. Immune characterization and profiles of SARS-CoV-2 infected patients reveals potential host therapeutic targets and SARS-CoV-2 oncogenesis mechanism. Virus Res. 2021, 301, 198464–198464. [Google Scholar] [CrossRef]
- Jahankhani, K.; Ahangari, F.; Adcock, I.M.; Mortaz, E. Possible cancer-causing capacity of COVID-19: Is SARS-CoV-2 an oncogenic agent? Biochimie 2023, 213, 130–138. [Google Scholar] [CrossRef]
- Stingi, A.; Crillo, L. SARS-Cov-2 Infection and Cancer. Bio. Essays. 2021, 43, 2000289. [Google Scholar] [CrossRef] [PubMed]
- López-Romero, R.; Nambo-Lucio, M.d.J.; Salcedo-Carrillo, E.; Hernández-Cueto, M.d.L.Á.; Salcedo, M. El gran desafío de la latencia de SARS-CoV-2: el testículo como reservorio. Gac. medica de Mex. 2020, 156, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Goubran, H.; Stakiw, J.; Seghatchian, J.; Ragab, G.; Burnouf, T. SARS-CoV-2 and cancer: the intriguing and informative cross-talk. Transfus. Apher. Sci. 2022, 61, 103488–103488. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; De Giglio, M.A.R.; Roviello, G.N. Deciphering the Relationship between SARS-CoV-2 and Cancer. Int. J. Mol. Sci. 2023, 24, 7803. [Google Scholar] [CrossRef]
- De Souza, L.M.; Ismail, M.; Elaskandrany, M.-A.; Fratella-Calabrese, A.; Grossman, I.R. Primary Hepatic EBV-DLBCL Lymphoma in the Setting of COVID-19 Infection. ACG Case Rep. J. 2024, 11, e01276. [Google Scholar] [CrossRef]
- Pietroluongo, E.; Luciano, A.; Peddio, A.; Buonaiuto, R.; Caltavituro, A.; Servetto, A.; De Angelis, C.; Arpino, G.; Palmieri, G.; Veneziani, B.M.; et al. Exploring the interplay between Kaposi's sarcoma and SARS-CoV-2 infection: A case series and systematic review. J. Med Virol. 2024, 96, e29849. [Google Scholar] [CrossRef]
- Pasi, F; Calveri, MM; Pizzarelli, G; Calabresse, A; Andreoli, M; Bongiovanni, I; Cattaneo, C; Rignanese, G. Oncolytic Effect of SARS-Cov-2 in a Patient with NK Lymphoma. Acta Biomed 2020,91,e2020047.
- Challenor, S; Tucker, D. SARS-Cov-2 Induced Remission of Hodgkin Lymphoma. Br J Haematol 2021, 192, 415.
- Ohadi, L.; Hosseinzadeh, F.; Dadkhahfar, S.; Nasiri, S. Oncolytic effect of SARS-CoV-2 in a patient with mycosis fungoides: A case report. Clin. Case Rep. 2022, 10, e05682. [Google Scholar] [CrossRef]
- Han, X.; Xu, P.; Ye, Q. Analysis of COVID-19 vaccines: Types, thoughts, and application. J. Clin. Lab. Anal. 2021, 35, e23937. [Google Scholar] [CrossRef]
- Panagiotakopoulos, L. Use of 2025-2026 Covid-19 Vaccines-Work Group Considerations. CDC Advisory Committee on Immunization Practices, April 15, 2025.
Figure 1.
CIN3 and NKSCC of cervix. F/40, vaginal bleeding. (A) CIN3 with glandular involvement H&E x100. (B) CIN3 H&Ex400. (C) NKSCC with co-existing CIN3 (lower right) H&E x100. (D) SCC with CINs (left) H&E x200. (E) SCC with inflammation H&Ex400. (F) Positive p16 immunostaining, strong in carcinoma cells and moderate in CIN3 cells x100.
Figure 1.
CIN3 and NKSCC of cervix. F/40, vaginal bleeding. (A) CIN3 with glandular involvement H&E x100. (B) CIN3 H&Ex400. (C) NKSCC with co-existing CIN3 (lower right) H&E x100. (D) SCC with CINs (left) H&E x200. (E) SCC with inflammation H&Ex400. (F) Positive p16 immunostaining, strong in carcinoma cells and moderate in CIN3 cells x100.
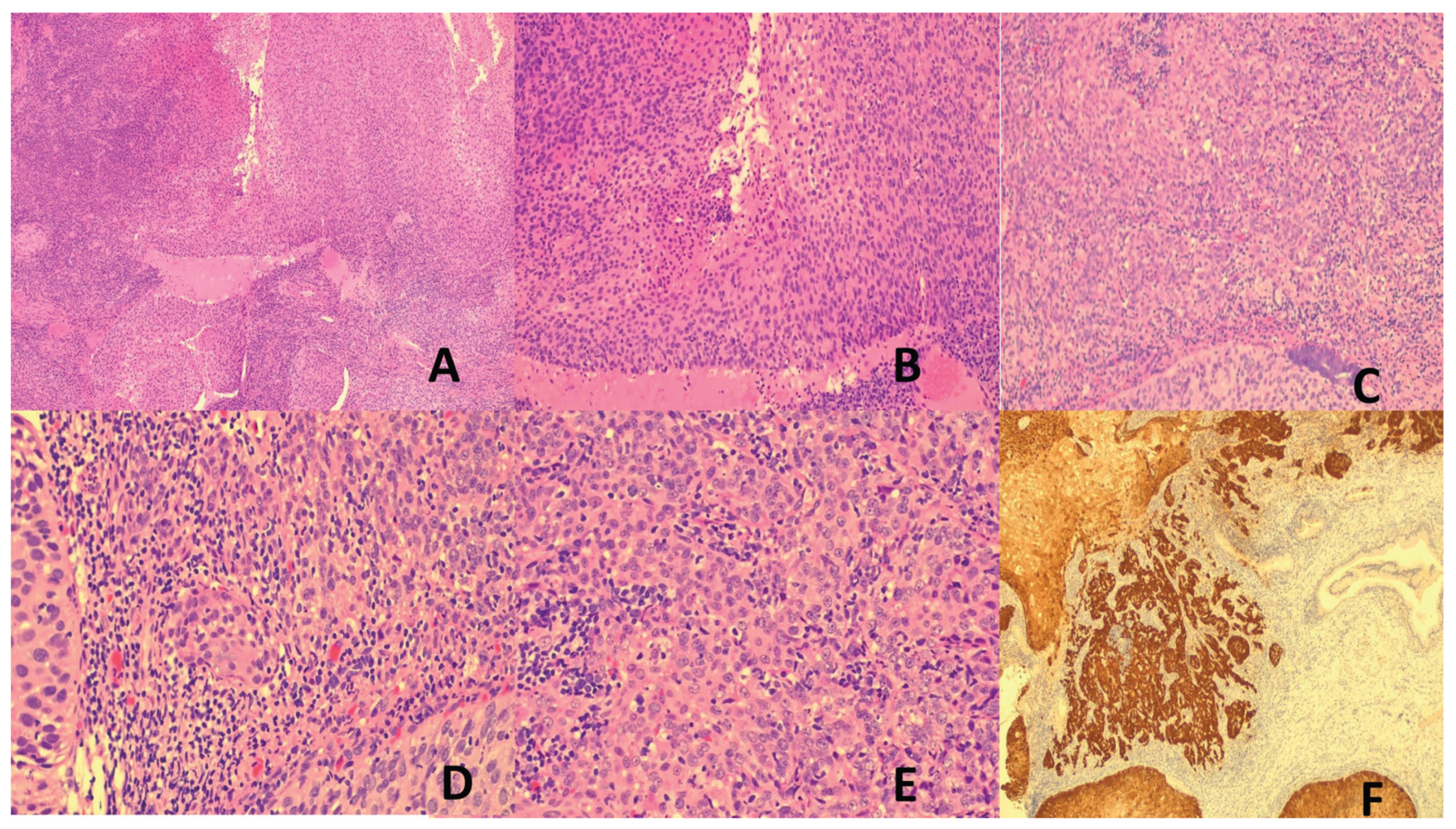
Figure 2.
(A) to (C) NPC, M/40, post-nasal bleeding. (A) Syncytial epithelioid and spindle tumor cells H&E x100. (B) Syncytial sheets of plump spindle tumor cells H&E x400. (C) EBER-positive tumor cells. (D) to (G) LEC of lung, M/56, right lung nodule. (D) Spindle tumor cells H&E x100. (E) Sheets of plump spindle and epithelioid tumor cells H&E x400. (F) EBER-positive tumor cells x100.
Figure 2.
(A) to (C) NPC, M/40, post-nasal bleeding. (A) Syncytial epithelioid and spindle tumor cells H&E x100. (B) Syncytial sheets of plump spindle tumor cells H&E x400. (C) EBER-positive tumor cells. (D) to (G) LEC of lung, M/56, right lung nodule. (D) Spindle tumor cells H&E x100. (E) Sheets of plump spindle and epithelioid tumor cells H&E x400. (F) EBER-positive tumor cells x100.
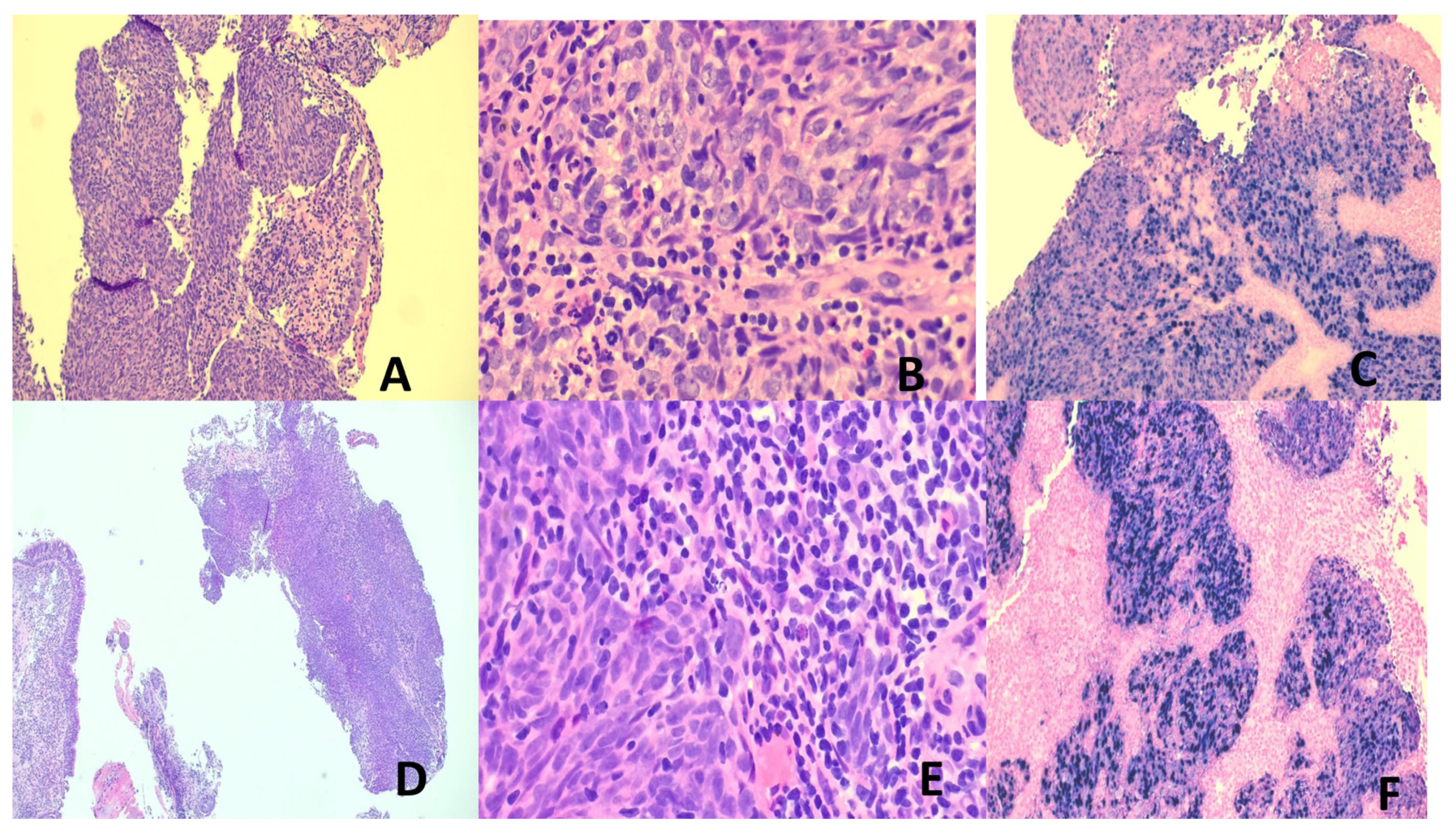
Figure 3.
(A-C) Burkitt Lymphoma. M/74, jejunal tumor. (A) Mural invasion H&E x100. (B) Medium-sized “squared” tumor cells and “starry sky” appearance H&E x 400. (C) CD20-positive x100. (D) to (F) Classic Hodgkin Lymphoma, mixed cellularity type. M/41, large cervical lymph node. (D) & (E) Hodgkin and Reed-Sternberg cells in mixed cell backdrop H&E x100 (D) & x400(E). (F) CD30 highlights neoplastic cells. x200.
Figure 3.
(A-C) Burkitt Lymphoma. M/74, jejunal tumor. (A) Mural invasion H&E x100. (B) Medium-sized “squared” tumor cells and “starry sky” appearance H&E x 400. (C) CD20-positive x100. (D) to (F) Classic Hodgkin Lymphoma, mixed cellularity type. M/41, large cervical lymph node. (D) & (E) Hodgkin and Reed-Sternberg cells in mixed cell backdrop H&E x100 (D) & x400(E). (F) CD30 highlights neoplastic cells. x200.
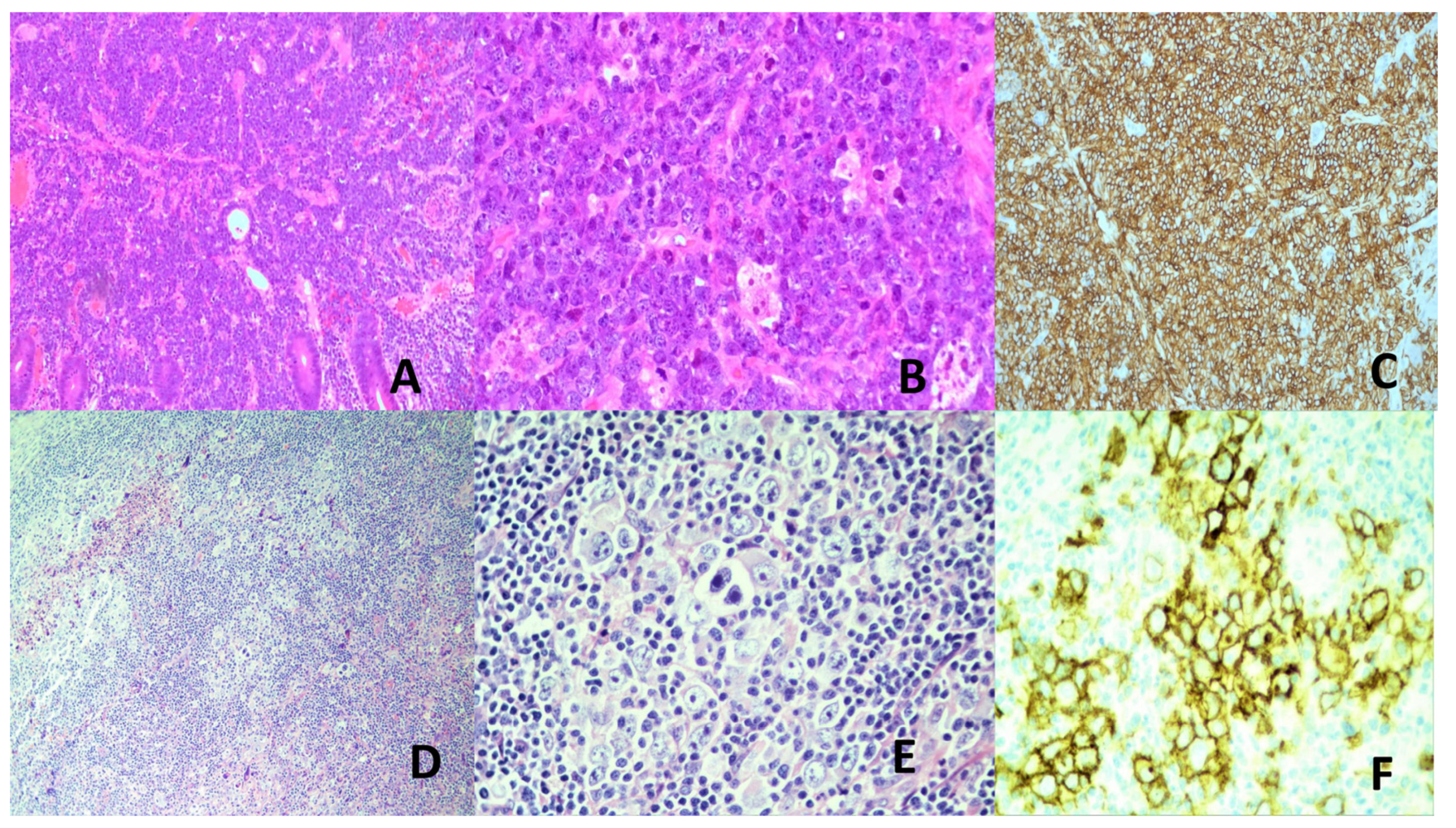
Figure 4.
ENNKTL of nose. M/44, nasal tumor. (A) & (B) Mixed lymphoid infiltrates H&E x100(A), x200(B). (C–E) Glandular epithelium invasion H&E x100(C), x200(D & E). (F) Vascular invasion with necrosis H&E x200. (G) CD56 positivity x100. (H) EBER positive tumor cells x100.
Figure 4.
ENNKTL of nose. M/44, nasal tumor. (A) & (B) Mixed lymphoid infiltrates H&E x100(A), x200(B). (C–E) Glandular epithelium invasion H&E x100(C), x200(D & E). (F) Vascular invasion with necrosis H&E x200. (G) CD56 positivity x100. (H) EBER positive tumor cells x100.
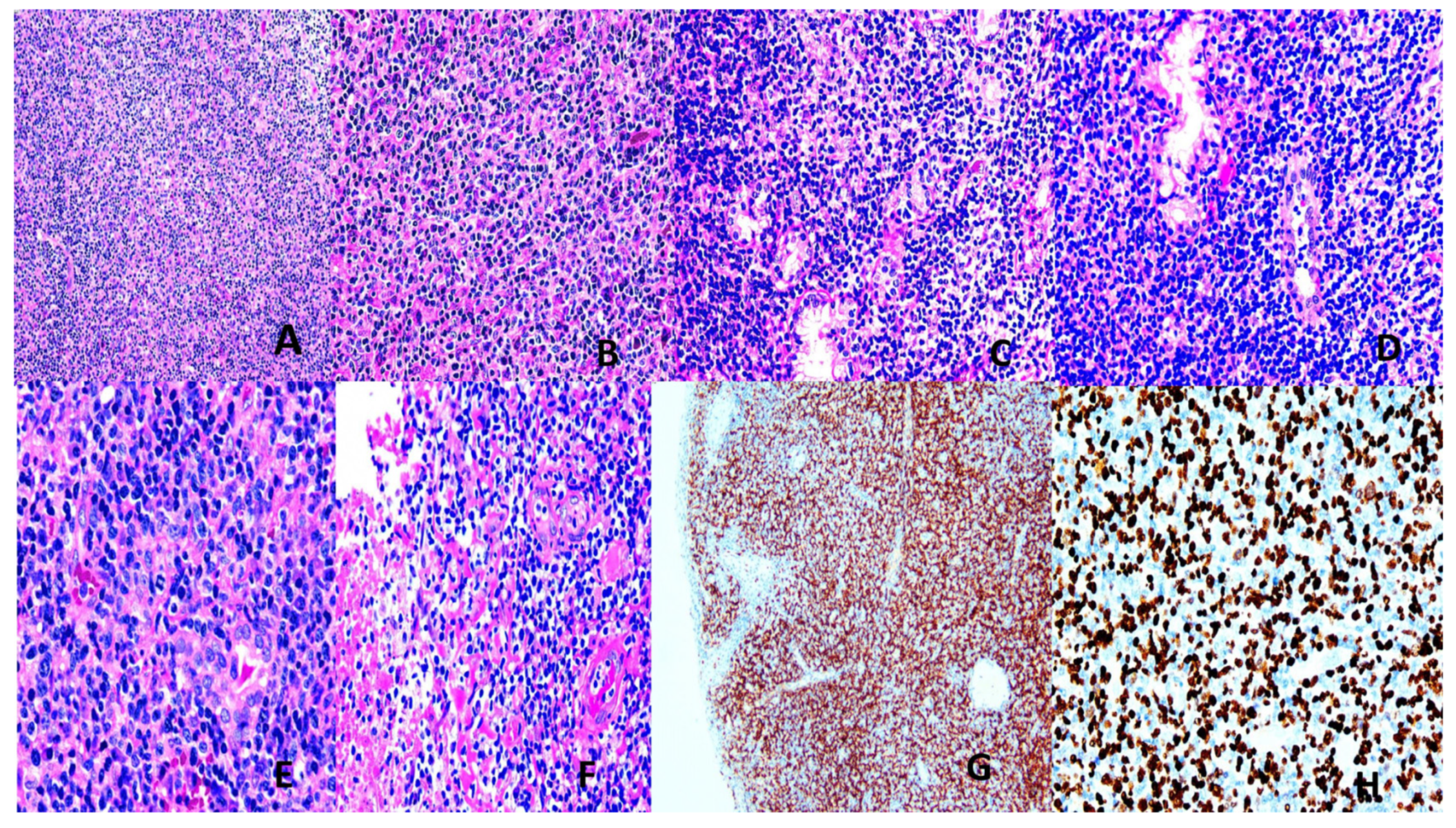
Figure 5.
(A–D) Kaposi sarcoma. M/38, HIV-positive, multiple skin lesions. (A) Spindle tumor cells H&E x100. (B) Red cells in slits and spaces among spindle tumor cells H&E x400. (C) ERG-positive tumor cells. x100 (D) LANA-positive tumor cells x100. (E,F) Hepatocellular carcinoma. M/61, liver tumor. (E) Pseudoglandular pattern and many extracellular globules H&E x200. (F) Macrotrabecular pattern and fatty change in tumor cells H&E x200.
Figure 5.
(A–D) Kaposi sarcoma. M/38, HIV-positive, multiple skin lesions. (A) Spindle tumor cells H&E x100. (B) Red cells in slits and spaces among spindle tumor cells H&E x400. (C) ERG-positive tumor cells. x100 (D) LANA-positive tumor cells x100. (E,F) Hepatocellular carcinoma. M/61, liver tumor. (E) Pseudoglandular pattern and many extracellular globules H&E x200. (F) Macrotrabecular pattern and fatty change in tumor cells H&E x200.
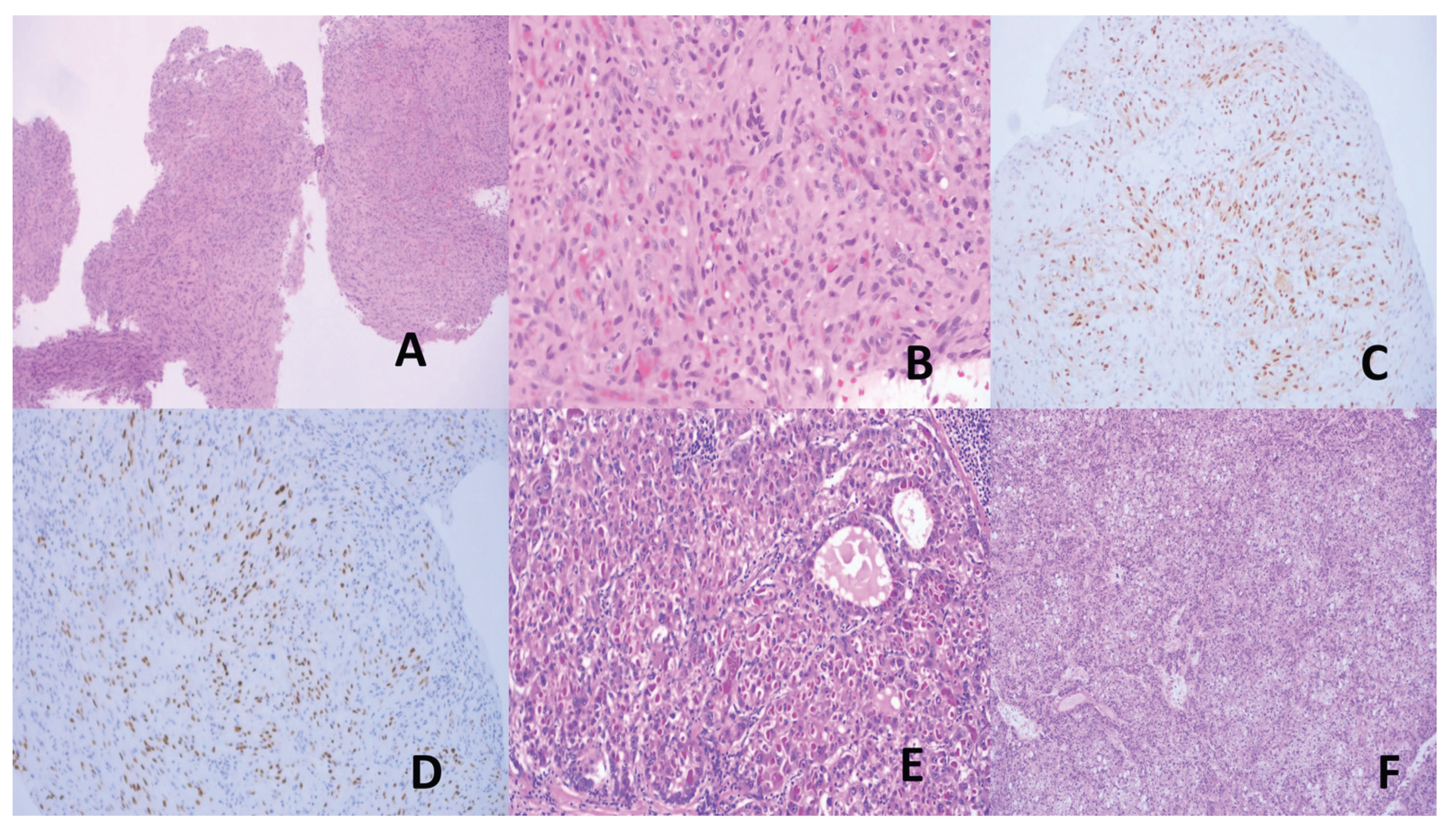
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



