Submitted:
28 July 2025
Posted:
30 July 2025
You are already at the latest version
Abstract
In this comprehensive review, we explore the full spectrum of the most common incidentally found bone lesion in children and adolescents. Non-ossifying fibromas (NOFs) are benign, self-limiting bone lesions that represent a significant proportion of skeletal abnormalities in this population. Beginning with their first description by Sontag and Pyle and the subsequent histological characterization by Jaffe and Lichtenstein, we trace the historical evolution of understanding NOFs, including early theories on etiology, while outlining key epidemiologic, histopathologic, and advanced imaging findings. Furthermore, we discuss diagnostic criteria, management strategies, and emerging molecular insights for NOFs, emphasizing their clinical significance. By integrating historical perspectives, current diagnostic protocols, and emerging discoveries, this paper seeks to assist clinicians in optimizing diagnostic and treatment proto-cols to prevent unnecessary interventions through a comprehensive understanding of NOFs.
Keywords:
Non-ossifying fibroma
; Benign bone lesion
; Pediatric Orthopedics
1. Introduction
Did you know the Fels Longitudinal Study – one of the most comprehensive, serial imaging studies on skeletal growth in children - established in the first half of the 20th century led to the characterization of the most common bone tumor in children? The only reference to this interesting factoid was found in a single hard copy of a journal in an east coast medical school, spurring the authors to refresh the pediatric medical community on this very common and often over analyzed entity [1]. The incidental discovery of a bone lesion on imaging in a child is a common clinical scenario that frequently causes anxiety for patients, families, and referring physicians. The primary challenge lies in differentiating benign, self-limiting entities from aggressive or malignant pathologies that require urgent intervention. Non-ossifying fibromas represent the most common of these benign lesions. While NOFs are well-known, recent discoveries in their molecular pathogenesis, evolving risk-stratification for pathologic fractures, and controversies in management warrant a contemporary and comprehensive review. This article will synthesize the historical understanding of NOFs with modern molecular insights, critically evaluate diagnostic and management algorithms, and identify key areas for future research.
Non-ossifying fibromas (NOFs) are benign fibrous bone lesions that are among the most common skeletal abnormalities observed in children and adolescents. Historically they were known as fibrous cortical defect or metaphyseal fibrous defect. This came from an original thought that these were on the spectrum of “chronic fibrous osteomyelitis”. [14] Described in a paper by Phemister in 1929, it was noted as incidentally found fibrous lesions that were asymptomatic. They were thought to be in the spectrum of chronic osteomyelitis. Phemister speculated “it is impossible to state when the lesion had developed, but it was apparently in a quiescent state and had it been left alone, would probably no disturbance in the future.” [14]. Cultures of these lesions were found to be negative but it was speculated that they were formed of “organisms of low virulence belong to the pyogenic group.” [14] The consensus over the 11 cases discussed in the article was that it was likely streptococcus viridans and reflected osteomyelitis. This nomenclature has persisted in radiology reports and clinical literature.
First formally described by Sontag and Pyle in 1941 as "pseudocysts," these lesions were identified through radiographic observations in a longitudinal skeletal growth study as mentioned above [1]. Their work was particularly significant due to the scale of their study, which was one of the first to comprehensively track skeletal development over time, and their novel use of radiographic technology to identify such lesions systematically. They observed an incidence of 53% in boys and 22% in girls. Later, Jaffe and Lichtenstein (1942) applied the term "nonosteogenic fibroma of bone" and provided a detailed histological and radiographic description, emphasizing the characteristic spindle-shaped fibroblasts and hemosiderin deposits [2]. Their meticulous differentiation between NOFs and other bone lesions had a lasting impact on diagnostic practices, particularly when it comes to identifying benign versus malignant features in skeletal abnormalities. This historical context underscores the evolving understanding of NOFs, from early descriptive studies to modern diagnostic and management approaches. Often asymptomatic and discovered incidentally, NOFs have a characteristic radiographic appearance and a self-limiting natural history. Despite their benign nature, large or symptomatic lesions may require intervention to prevent complications such as pathologic fracture. Up to 60% of referrals to orthopedic oncology practices are for benign bone lesions, with NOFs comprising a significant proportion [2,3]. This high referral rate likely stems from the challenge of differentiating benign from malignant bone lesions in initial assessments, coupled with the frequent incidental discovery of NOFs during imaging for unrelated issues. Despite their benign nature, their distinctive radiographic appearance can mimic more serious conditions, prompting further evaluation to rule out malignancy. This article provides a comprehensive review of NOFs, with an emphasis on their clinical presentation, diagnosis, and management.
2. Etiology
The exact etiology of NOFs is not fully understood. Early studies, such as those by Sontag and Pyle in 1941, conjectured that these lesions might arise from an abnormal developmental process during skeletal growth. They described NOFs as "pseudocysts," suggesting a misstep in ossification that results in fibroblastic proliferation within the metaphyseal regions of long bones. Jaffe and Lichtenstein (1942) expanded upon this by identifying hallmark histological features, including whorled connective tissue stroma and hemosiderin deposits, which they believed were indicative of a benign lesion with distinct biological behavior. In 1986, Ritschl and Karnel discussed that the NOFs only occur in areas where tendons insert into the perichondrium of the epiphyseal plate. [15] These metaphyseal areas where tendinous insertions lead to lesions is well known in the so called “tug lesions” such as the cortical desmoids of distal medial femur from traction of the adductor magnus. While tug lesions can be noted in all ages, NOFs existence in pediatric patients only implies the role of the physeal plate. Other noted tendinous relationships maybe include the pes anserinus in the proximal tibia and the interosseous membrane in the distal tibia. However, some non-ossifying fibromas are not attached to the physeal plate and can originate from non-fibrous or tendinous location. Thus, the etiology of NOFs is not clearcut from histology and radiology alone. However, these early insights laid the groundwork for contemporary investigations into the cellular and molecular mechanisms of NOFs.
Despite the foundational work by Sontag, Pyle, Jaffe, and Lichtenstein, the specific factors driving lesion formation and regression remain unknown. In 2018, DNA sequencing demonstrated activating mutations in genes involved in the MAP-Kinase signaling pathway such as KRAS, FGFR1, and NF1. In particular, the natural regression of NOF lesions is not fully understood but suspected to be related to a transient phenomenon or insufficient activation of the MAP-Kinase pathway to sustain continuous tumor growth [4]. The term “RASopathies” has been used to label a conglomerate of genetic conditions or syndromes, like NF1 and Noonan syndrome, driven by mutations in genes involved in the Ras/MAPK signaling pathway. For this reason, adding NOF to the family of RASopathies has been proposed [5].
The Ras/MAPK pathway is critical in normal cell growth, proliferation and differentiation, particularly in osteogenesis. The pathway generally starts with a bioactive peptide binding to a cell surface receptor. This leads to the activation of receptor-linked tyrosine kinase. Downstream MAP (mitogen-active protein) kinase is activated leading to activation of a transcription factor and then cell division. This is a well-regulated step that is integrated into the cell cycle and is speculated to contribute to a restriction point in cell proliferation. It plays a complex role in osteogenesis where increased Ras-MAPK signaling can antagonize bone formation but can also promote Runx2/Cbfa1 activity and osteogenic gene expression [16]. With the rise of Ras/MAPK targeting therapeutics, this duality is particularly important to elucidate.
In NOFs, KRAS, FGFR1 and NF1 represent activating mutations leading to constitutive, ligand-independent signaling down the MAPK pathway, which promotes fibroblastic proliferation over normal ossification. KRAS (Kirsten rat sarcoma viral oncogene homolog) is a known oncogene in variety of neoplastic conditions including non-small cell lung cancer, colorectal cancer and pancreatic cancer. In terms of benign bone tumors, KRAS mutations are found in brown tumors, a rare self-limiting lesion associated with hyperparathyroidism [4]. FGFR1 (Fibroblast growth factor receptor 1) is a receptor tyrosine kinase that receives signals from fibroblast growth factors. It is known to be critical in normal embryonic bone development as well as a key mutation in many genetic syndromes and skeletal dysplasia. However, patients with NOFs typically do not manifest clinical features of skeletal deformities, it reflects that FGFR1 is a somatic and not a germline mutation [4]. NF1 (neurofibromatosis 1) gene encodes for neurofibromin which is a negative regulator of the Ras protein. In contrast to the KRAS and FGFR1 mutations, the NF1 mutation appears to be germline. Patients with NOFs with the NF1 mutation also have neurofibromatosis type 1.
Spontaneous regression of NOFs has been theorized that the mutations noted provide a proliferative stimulus but not strong enough for malignant transformation or sustained growth. Thus, there is an eventual “burnout” or regression, leading to apoptosis as the bone matures and the microenvironment changes. This has not been fully elucidated but is rather theorized given the scarcity of reports in adults with non-ossifying fibromas. In contrast, those with NOFs due to germline mutations in NF1 may not have regression due to persistent stimulus in NF1 patients.
RASopathies represent a spectrum of syndromes that involve mutations in the RAS/MAPK pathway. They are noted for general craniofacial dysmorphology, cardiovascular abnormalities, musculoskeletal abnormalities, cutaneous manifestations. Neurocognitive impairment and increased risk of benign and malignant tumors. Patients with NOFs do not typically have these other clinical manifestations despite the common pathway to mutations. This again proposes that these mutations are not germline and somatic and sporadic without sufficient stimulus to lead to further proliferation.
Jaffe-Campanacci syndrome reflects a true RASopathy that includes non-ossifying fibromas. The syndrome is characterized by multiple NOFs of the long bones and jaw and café-au-lait patches. Some patients are noted to have neurocognitive impairment along with cardiovascular malformations, hypogonadism, cryptorchidism and ocular abnormalities [4]. There is discussion about whether Jaffe-Campanacci syndrome is a distinct entity from NF1 or a variant. Jaffe-Campanacci syndrome does not manifest with the neurofibromas of NF1. Both syndromes are allelic, meaning that they both have NF1 mutations with different presentations. Jaffe-Campanacci syndrome has noted polyostotic NOFs that can lead to bone development issues and potential pathologic fractures. This represents the strong clinical-genetic relationship between RAS/MAPK pathway and NOFs.
3. Epidemiology
NOFs are most commonly diagnosed in individuals aged 5 to 20 years, with a peak incidence during adolescence when skeletal growth is most active. They are observed in up to 30-40% of children on routine radiographs, often as incidental findings due to their asymptomatic nature [6]. The true incidence of the lesions is unknown because patients are mostly asymptomatic. Thus, radiographic prevalence is estimated to be 30-40% and used as an estimate for clinical population.
The lesions are slightly more prevalent in males than females, which was first noted by Sontag and Pyle in 1941. These authors studied serial radiographs of 200 children obtained by the Samuel S. Fels Research institute as part of a natural history skeletal growth study. They noted that NOFs are generally around 1-3cm in diameter and located most frequently in the metaphysis of long bones, particularly the medial condyle and posterior aspect of the femur. Lesions most commonly appear in the distal femur (57 of 107 cases), proximal tibia (25 of 107 cases), and distal tibia (19 of 107 cases). They are not observed in sites lacking tendinous attachments near the peri-chondrium of epiphyseal cartilage, such as the femoral neck. [6] The average age of first appearance was 46 months, with maximum size being reached by approximately 50 months. Most lesions persisted for around 2.5 years (29 months). In this population, there was an incidence of 53% in boys and 22% in girls [1].
There have been no large-scale studies that have tried to capture the prevalence of NOFs. Given that these tumors often do not require treatment, there appears to be limited interest in updating this data. The seminal study from Sontag and Pyle in 1941 remains the most cited article discussing the prevalence of NOFs in children. No studies have been noted to document the persistence of these tumors in the adult population.
As mentioned, the distal femur and proximal tibia are the two most common sites for NOFs. This is likely the result of these physeal plates being the most active in the body, increasing the probability of somatic mutations leading to development of NOFs. The distal femur is also proposed as a common location due to the relative weakness of the posterior cortex and the presence of multiple vascular channels [15]. These areas also represent areas of high mechanical stress as well. Goldin et al in 2017 noted that in 68 NOFs in 60 patients noted tendinous insertions in 93% of distal femoral lesions [15].
4. Clinical Presentation
4.1. Asymptomatic Patient
The most common presentation for patients with NOFs is an incidental finding in an asymptomatic patient. Often the typical case will be a patient who underwent a trauma or has pain, and the tumor is noted incidentally on radiograph. Patients are often then referred to pediatric orthopaedic surgeons or orthopaedic oncologists for further characterization of the lesions. The physician will often reassure the patient and the family and will potentially recommend surveillance of the lesion, depending on location or size. As most tumors will spontaneously regress, long-term surveillance is not often necessary. However, if the lesion remains stable over at least 6-12 months, surveillance is usually ceased. The surveillance course is dependent on the patient’s age, especially if they are close to the age of skeletal maturity, size and location of the lesion, and the context of how the lesion was noted.
4.2. Symptomatic Patient
This scenario represents an uncommon presentation but has been reported in the literature. These patients often complain of a dull ache or pain, especially with weight-bearing of the long bone. Patients will also complain of increasing pain with physical activity and loss of exercise endurance. They may also complain of swelling at the site of the tumor. These symptoms reflect direct compression of surrounding tissue from the weakening of the bone tissue or the presence of microfractures. If the pain becomes persistent this could reflect that the lesion is at risk for a pathologic fracture. These patients require close surveillance to avoid pathologic fracture through the lesion and may need prophylactic fixation.
4.3. Pathologic Fracture
A pathologic fracture is a rare complication of NOFs but represents a significant clinical event. They are often low energy fractures that occur without significant trauma or injury. Patients can complain of antecedent pain prior to fracture, but this is not universal in presentation. While it is common to have a minimally displaced pathologic fracture, deformity and displacement is possible.
4.4. Pathologic Fracture
A pathologic fracture is a rare complication of NOFs but represents a significant clinical event. They are often low energy fractures that occur without significant trauma or injury. Patients can complain of antecedent pain prior to fracture, but this is not universal in presentation. While it is common to have a minimally displaced pathologic fracture, deformity and displacement is possible.
5. Differential Diagnosis

Table 1.
Differential diagnosis of common bone lesions that can be compared to NOFs.
| Lesion | Typical Age | Location | Radiographic Features | Histology Key Feature |
|---|---|---|---|---|
| Non-ossifying Fibroma | 5-20 yrs | Metaphysis (eccentric) | Lytic, scalloped, sclerotic rim, bubbly appearance | Storiform spindle cells, hemosiderin |
| Fibrous Dysplasia | <30 yrs | Metaphysis/Diaphysis (central) | Ground-glass, "Rind" sign, Shepherd’s Crook deformity | "Chinese character" woven bone |
| Aneurysmal Bone Cyst | <20 yrs | Metaphysis (central) | Expansile, lytic, fluid-fluid levels on MRI | Blood-filled spaces, giant cells |
| Simple Bone Cyst (UBC) | <20 yrs | Metaphysis (central) | Lytic, well-defined, "fallen leaf" sign after fracture | Thin fibrous lining, clear fluid |
| Giant Cell Tumor | 20-40 yrs | Epiphysis (abuts joint) | Lytic, expansile, non-sclerotic margin | Numerous osteoclast-like giant cells |
6. Radiographic and Advanced Imaging
6.1. Radiographic Features
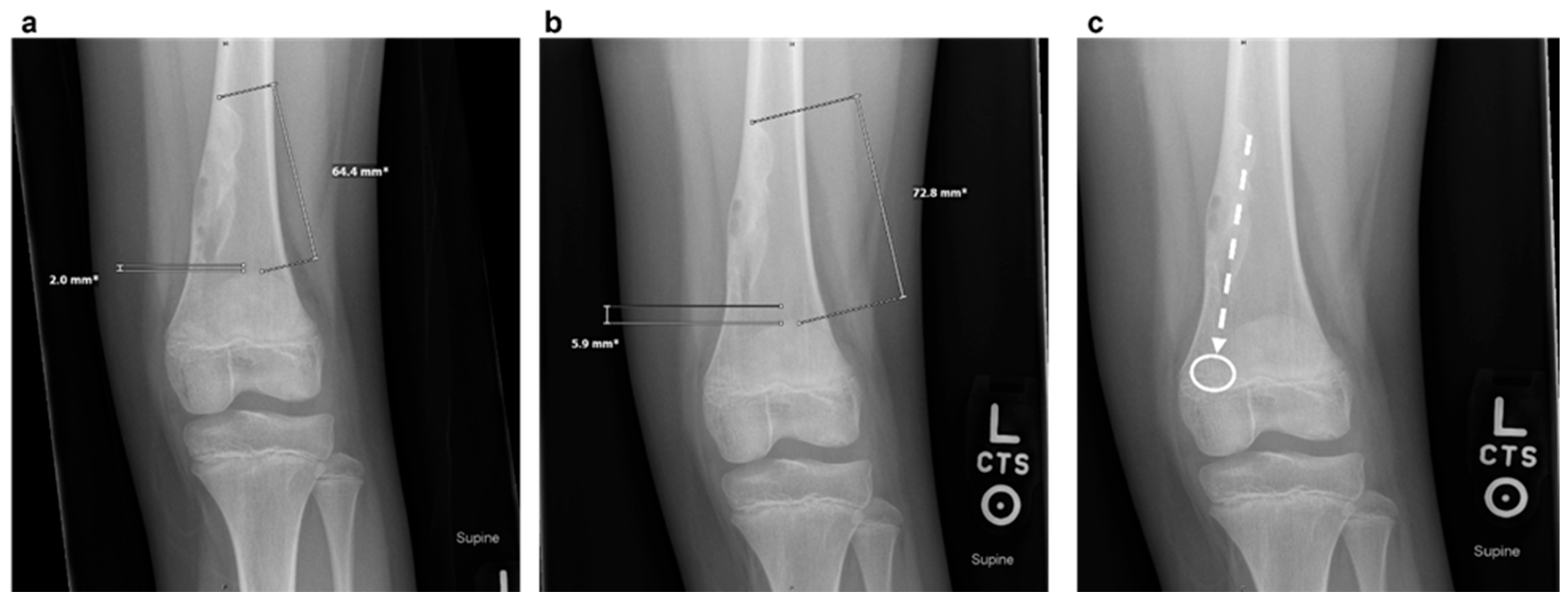
Plain radiographs typically show a well-demarcated, lytic lesion located eccentrically within the metaphysis of the affected bone. Over time, as the child grows, NOFs tend to migrate away from the growth plate towards the diaphysis due to bone remodeling [6]. Unlike enchondromas, which often present centrally within the medullary cavity and exhibit a chondroid matrix, NOFs are uniquely characterized by their eccentric location, elongated shape fibrous matrix and sclerotic border. This differentiation underscores the diagnostic importance of radiographic positioning and lesion morphology. As the defect migrates away from the tendon insertion, an imaginary line can often be drawn along the lesion’s long axis, pointing toward the soft tissue insertion from which the defect presumably arises. This is illustrated in Figure 1.
6.2. Staging
The most widely accepted staging system, first described by Ritschl et al. and later expanded upon, is as follows:
- Stage A: Small, eccentric, cortically based lesions near the epiphysis with smooth, round borders.
- Stage B: Lesions become polycystic with clear but thin sclerotic borders, increasing in size and exhibiting variable distances from the epiphysis. The most significant growth typically occurs during this stage, transitioning from Stage A or within Stage B.
- Stage C: Lesions demonstrate increased sclerosis and reduced growth potential.
- Stage D: Complete and homogeneous sclerosis is observed, with no further growth.
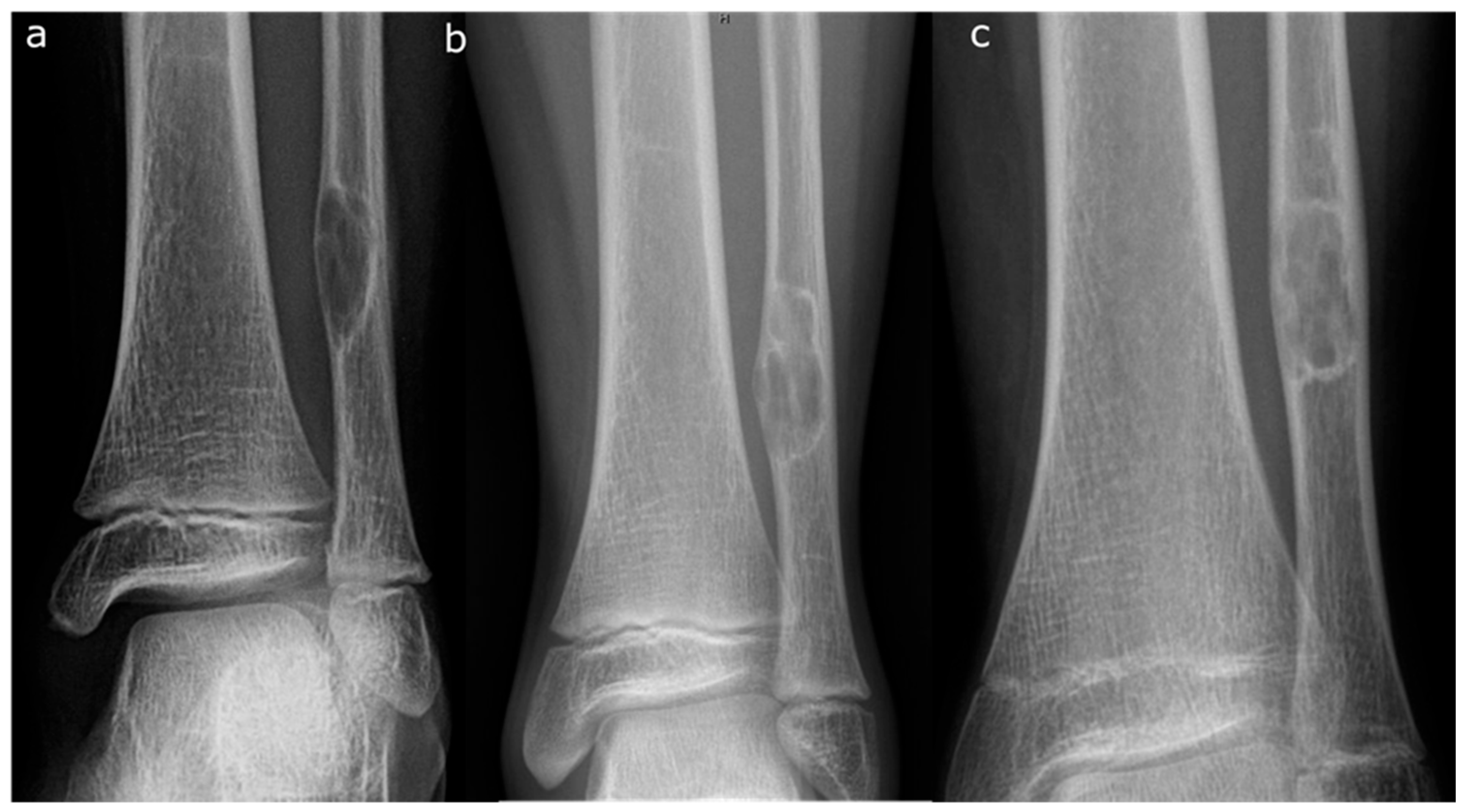
Figure 2.
(a) A 8-year-old girl at the first presentation. AP radiograph showing a lytic lesion of the left distal fibula in stage A. (b) Two years later the lesion was polycyclic in shape with sclerotic borders (stage B). (c) At the age of 12 years there was evidence of ossification beginning at the diaphysis representing a stage C lesion.
Figure 2.
(a) A 8-year-old girl at the first presentation. AP radiograph showing a lytic lesion of the left distal fibula in stage A. (b) Two years later the lesion was polycyclic in shape with sclerotic borders (stage B). (c) At the age of 12 years there was evidence of ossification beginning at the diaphysis representing a stage C lesion.
6.3. Advanced Imaging
Advanced imaging modalities can provide additional information in atypical cases by offering enhanced visualization of lesion characteristics and their surrounding anatomy. It is not routine to order routine imaging in most cases, and it is reserved for unusual cases. For example, computed tomography (CT) is particularly useful in assessing cortical thinning and evaluating fracture risk, which can guide decisions regarding the necessity of activity restrictions or surgical intervention. Magnetic resonance imaging (MRI), with its superior soft tissue contrast, is most indicated in cases where the differentiation of NOFs from other pathologies, such as fibrous dysplasia or malignancy, is challenging. The decision to pursue advanced imaging often depends on the lesion’s size, location, and the presence of symptoms, as these factors significantly influence management strategies. In some cases, advanced imaging findings, like the "Pac-Man sign", described below, may prompt closer surveillance or preemptive treatment to mitigate fracture risk. The “Syndesmosis sign” is positive when the syndesmosis can be seen inserting into a distal tibia lesion. While both signs are better seen on MRI, they can also be visualized on CT in some cases. Clinicians could benefit from clear guidelines for when to employ advanced imaging modalities in order to optimize patient care and minimize healthcare costs.
-
Magnetic Resonance Imaging (MRI): MRI can help differentiate NOFs from other lesions such as fibrous dysplasia or malignancies [7]. On MRI, NOFs are typically T1-hypointense and T2-hyperintense. A peripheral, low-signal rim on all sequences corresponds to the sclerotic border seen on radiographs. Post-contrast sequences usually show minimal, peripheral enhancement, whereas more aggressive lesions often demonstrate avid, diffuse enhancement. In 2021, Baghdadi et al. identified advanced imaging features, such as the "Pac-Man sign” and the “syndesmosis sign” that may indicate increased fracture risk in distal tibia NOFs [8].
- o
- Pac-Man Sign - Proliferation of bone anterior and posterior to the syndesmosis results in a shape which resembles the video game character “Pac-Man”. This sign was found to be highly specific (95%) but not very sensitive (47%) for predicting pathologic fracture. (Figure 3)
- o
- Syndesmosis Sign - Advanced imaging shows the syndesmosis inserting into the distal tibia lesion. This sign was found to be highly sensitive (94%) but less specific (48%) for predicting fracture risk.
- Computed Tomography (CT): CT can be useful for evaluating degree of cortical thinning, which is important when assessing fracture risk [9]. CT is superior for delineating the precise cortical integrity. It allows for quantitative measurement of the cross-sectional area occupied by the lesion, which is a key factor in biomechanical models predicting fracture risk. The signs described above can also be visualized on CT scan. Figure 3 demonstrates a “Pac-Man Sign” on CT of a 13-year-old boy who did have a fracture through an NOF.
- Bone Scintigraphy: Demonstrates mild uptake, reflecting the lesion’s low metabolic activity.
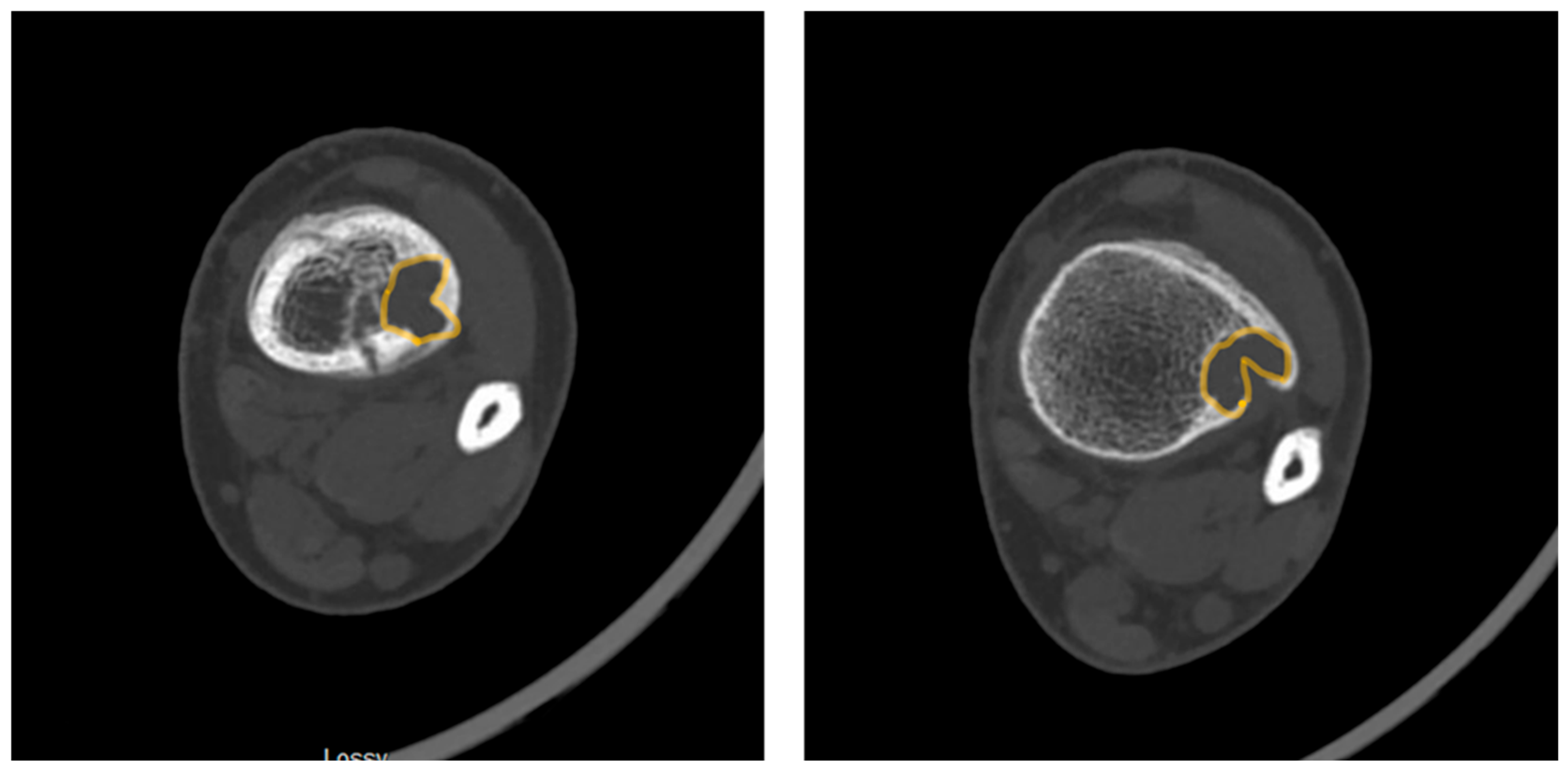
Figure 3.
Axial computed tomography slices demonstrating the “Pac-Man Sign” in a patient with a pathologic fracture through a distal tibia NOF.
Figure 3.
Axial computed tomography slices demonstrating the “Pac-Man Sign” in a patient with a pathologic fracture through a distal tibia NOF.
7. Pathology
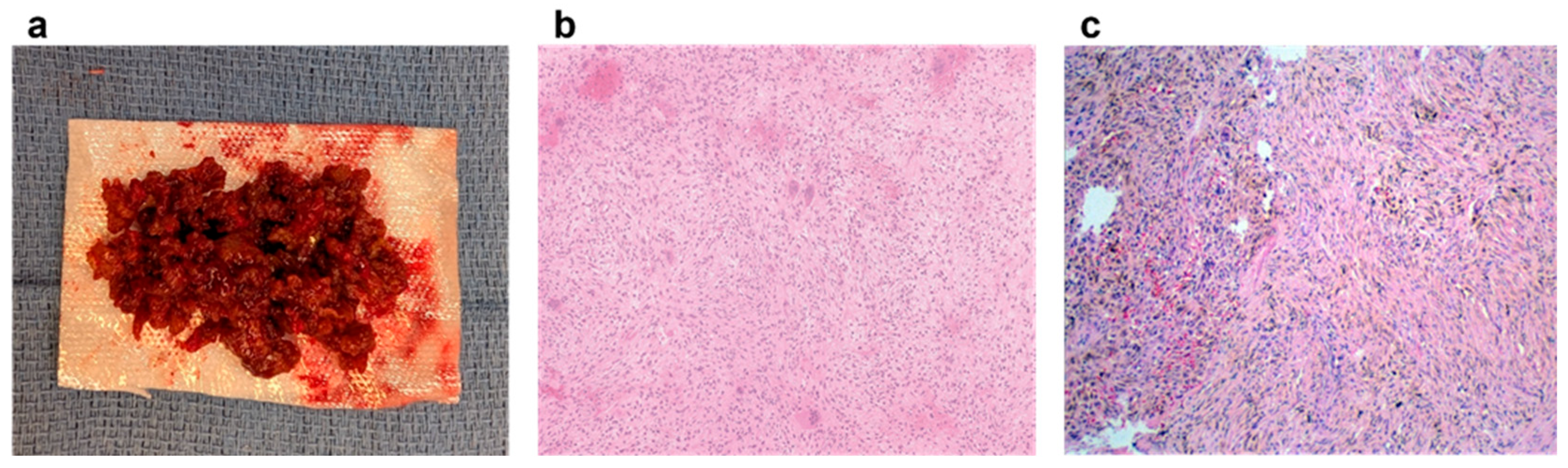
On gross examination, the curetted tissue is typically rubbery and firm, with a characteristic brownish-yellow or reddish-brown color due to the high content of hemosiderin-laden macrophages. Histologically, NOFs consist of spindle-shaped fibroblasts arranged in a whorled or storiform pattern, a hallmark feature first noted by Jaffe and Lichtenstein in 1942. This distinctive arrangement, along with the absence of cellular atypia or mitotic figures, supports the benign nature of NOFs. Additionally, the presence of hemosiderin deposits and scattered multinucleated giant cells further underscores their unique histological profile, differentiating them from other bone lesions such as fibrous dysplasia or malignant tumors. Jaffe and Lichtenstein also observed that these lesions lack bone formation, a finding that further distinguishes NOFs from other bony pathologies. The hemosiderin accounts for the brownish coloration observed grossly. Variability in cellularity, vascularity, and hemosiderin content may be observed, depending on the lesion’s stage. Figure 4 demonstrates gross pathology and histology of a curetted lesion.
While diagnosis is nearly always made on standard H&E staining, in challenging cases, IHC can be used to rule out other spindle cell lesions. NOF cells are typically negative for markers like S100 and desmin. Given the presence of multinucleated giant cells, H3.3G34W stains can be used to differentiate from giant cell tumors of bone. This is a stain against the G34W mutation in histone 3F3A, and mutations in the G34 position are present in 96% of giant cell tumors of bones. This mutation is not expressed in NOFs or other giant cell rich histologic mimics.
Figure 4.
Gross specimen (a) of a non-ossifying fibroma which has been curetted out of a patient. Representative histopathology showing mutli-nucleated giant cells (b) and hemosiderin (c) with both examples demonstrating a characteristic the whorled stromal pattern4.
Figure 4.
Gross specimen (a) of a non-ossifying fibroma which has been curetted out of a patient. Representative histopathology showing mutli-nucleated giant cells (b) and hemosiderin (c) with both examples demonstrating a characteristic the whorled stromal pattern4.
8. Non-Operative Management
8.1. Observation and Monitoring
Most NOFs are asymptomatic and require no intervention, as their natural history involves spontaneous resolution during skeletal maturation. Observation is sufficient in these cases because NOFs are self-limiting and rarely lead to complications. Periodic radiographic monitoring is typically recommended to assess lesion size and ensure resolution or stabilization over time. Most lesions spontaneously regress as the individual reaches skeletal maturity [3,6]. In a retrospective review of over 1300 NOFs, Patel and Damron found that fewer than 3.5% of patients fractured or required surgery, suggesting that serial surveillance may be unnecessary for small, asymptomatic lesions [10]. Historically, large asymptomatic lesions prompted frequent radiographic surveillance and activity modification. This retrospective study challenges that intensive surveillance may represent unnecessary use of resources and radiation exposure in many cases.
8.2. Activity Modification
In cases of lesions presenting a higher fracture risk, activity restriction may be advised. High-impact activities, such as contact sports, are generally discouraged until the lesion stabilizes beyond Stage B [11].
8.3. Closed Reduction and Casting of Pathologic Fractures
In 1997, Easley et al examined the necessity of prophylactic treatment of impending pathologic fractures in NOFs. At the time, the approach supported by the orthopedic community was to treat NOFs prophylactically with curettage and bone grafting if they demonstrated more than 50% cortical involvement on AP and lateral radiographs [12]. The authors anecdotally noted many large NOFs were found incidentally and did not ultimately fracture before self-resolving. They used this clinical experience to question the status quo of the era. In their investigation, the authors noted that nearly 60% of patients with “large” NOFs did not go on to fracture. While 9/22 patients did incur a fracture, all fractures were traumatic in nature without prodromal pain and 4 of the fractures occurred distant to the lesion within the same long bone. All fractures were treated successfully with closed reduction and cast immobilization.
9. Operative Intervention
9.1. Indications
Surgical intervention is reserved for symptomatic lesions, those causing significant pain, or lesions at high risk for fracture based on size and location. Factors such as cortical thinning (<1-2mm), involvement of more than 50% of the bone diameter on two orthonal views, lesion size (greater than 3-4 cm in major weight-bearing bone), presence of Pac-Man sign in the distal tibia, and advanced lesion staging (e.g., Ritschl Stage B) inform the decision for surgery [9,11].
9.2. Techniques
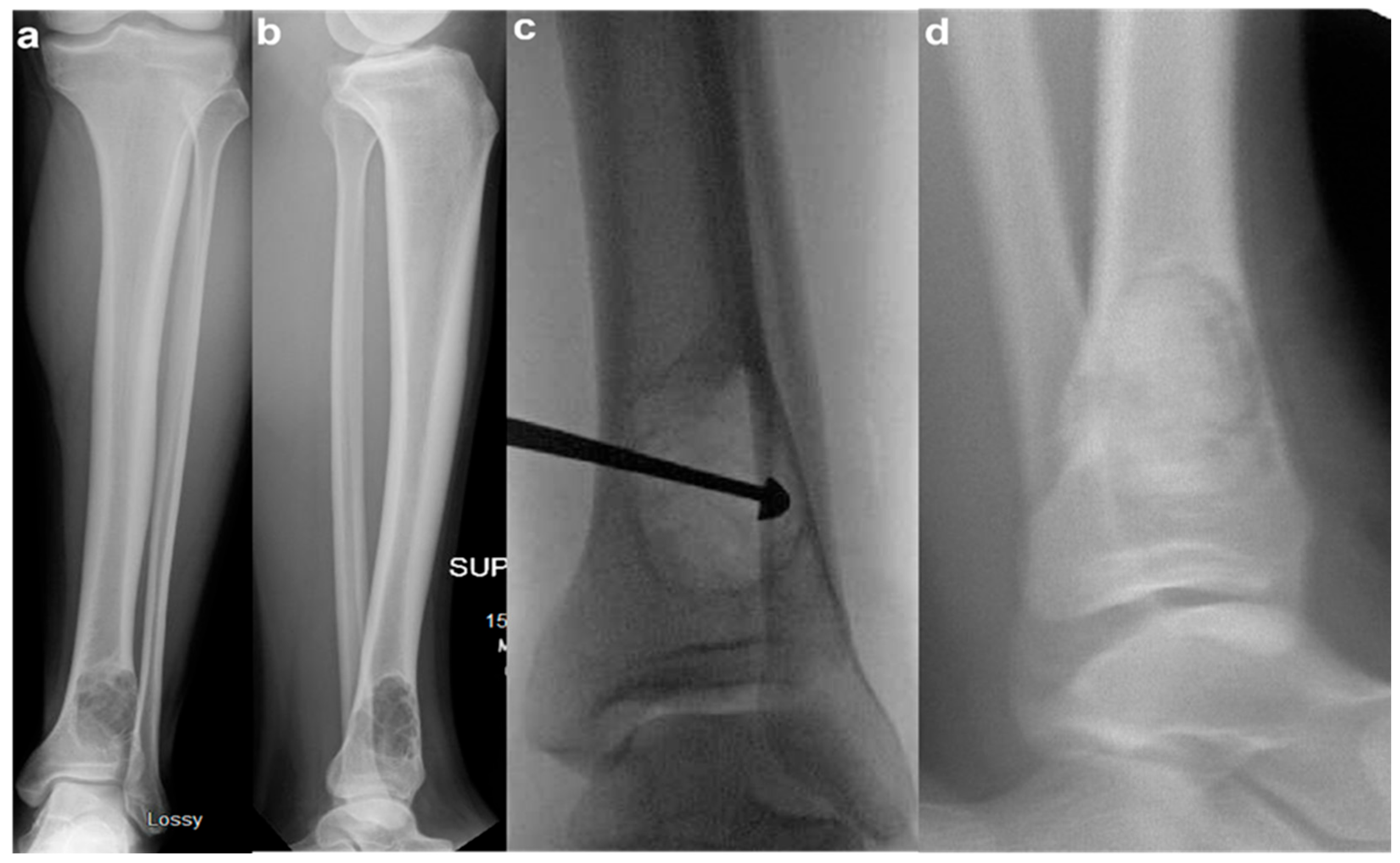
- Curettage and Bone Grafting: This is by far the most performed procedure. After curettage, the cavity is filled with autograft, allograft, or synthetic bone substitute [12]. While autograft is theoretically biologically superior, the donor site morbidity makes allograft the most used graft choice. In addition, the relatively high success rate and low risk of recurrence, make the risks of an additional incision with autograft prohibitive. Synthetic substitutes such as calcium sulfate and calcium phosphate can provide structural support to allow earlier weight bearing. Again, the risks of cementation, in the setting of a benign disease with good operative results, makes this a less common choice. Adjuvants are typically not used in the curettage stage for NOFs as they are in giant cell tumors of bone or aneurysmal bone cysts. NOFs are not locally aggressive, and the use of phenol or argon beam coagulation are not typically indicated to reduce recurrence. Phenol and argon have risk of local tissue damage, making them uncommonly used in the setting of NOFs. Figure 5 demonstrates pre-, intra-, and post-operative radiographic images of a 17-year-old boy with a large symptomatic distal tibia NOF who underwent a curettage and bone grafting procedure.
- Internal Fixation: For structural support in large lesions or fractures, internal fixation with plates or intramedullary nails may be necessary.
Figure 5.
Pre-operative AP (a) and lateral (b) tibia films of a 17-year-old boy with a large, symptomatic left distal tibia NOF. Intra-operative fluoroscopy (c) demonstrating an AP radiograph with a curette in place. Post-operative lateral radiograph (d) demonstrating bone grafting in place.
Figure 5.
Pre-operative AP (a) and lateral (b) tibia films of a 17-year-old boy with a large, symptomatic left distal tibia NOF. Intra-operative fluoroscopy (c) demonstrating an AP radiograph with a curette in place. Post-operative lateral radiograph (d) demonstrating bone grafting in place.
Surgical treatment typically results in excellent outcomes, with low recurrence rates, cited under 5% [13]. Complications are rare but may include infection, graft resorption, or refracture [13]. Andreacchio et al. (1997) reported favorable outcomes in surgically treated patients, particularly those with symptomatic or high-risk lesions [13]. In this case series, 9 patients with symptomatic NOFs of the lower extremity were treated with curettage, phenol treatment, and bone grafting with calcium sulfate pellets. All but one patient in this series had a Ritschl Type B lesion and there were no long-term complications or sequalae related to the lesion or operative intervention [12,13]. Recurrence after surgical intervention is always due to incomplete curettage of the lesion.
9. Future Directions
As NOFs represent a benign process that often does not require intervention, research in the field is limited. However, further research could be targeted on its origins and treatment. While the role of the MAPK pathway is established, the upstream triggers for these somatic mutations remain unknown. Why do they occur in specific locations and at specific ages? Further elucidation of the MAPK pathway may help unlock its role in other etiologies as well. While the risk of pathologic fractures is low, there are real sequalae from when they do occur. Future research should focus on refining fracture risk prediction. While size criteria exist, the development of patient-specific biomechanical models using Finite Element Analysis (FEA) based on CT data could provide a more accurate, individualized risk assessment that could be applied broadly. For syndromic cases with multiple, problematic lesions (Jaffe-Campanacci), could MAPK inhibitors one day play a role? This remains a speculative but logical area for future investigation.
10. Conclusions
Non-ossifying fibroma has journeyed from being considered a simple developmental ’defect’ to being understood as a true, genetically driven benign neoplasm of the RAS/MAPK pathway. They are benign, self-limiting bone lesions most found in children and adolescents. For the clinician, its classic radiographic features remain the cornerstone of diagnosis, allowing for confident observation in the vast majority of cases and preventing unnecessary interventions. The primary challenge remains the identification of the small subset of lesions at high risk for pathologic fractures. While surgical treatment is highly effective when indicated, future efforts should focus on refining our risk stratification models to further optimize patient care. The story of the NOF is a perfect example of how molecular discovery continues to reshape our understanding of even the most common orthopedic pathologies
Author Contributions
All authors contributed to the article’s conception, literature search, drafting, review, and editing. All authors read and approved of the final manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
This review article synthesizes existing literature and does not create or analyze new data. Data sharing is not applicable to this review article.
Acknowledgments
Not applicable.
Conflicts of Interest
The authors declare no conflicts of interest. None of the following authors or any immediate family member has received anything of value from, has stock, or stock options held in a commercial company or institution related directly or indirectly to the subject of this article: KW, JBS, NT, DB. RLR: Committee member: Children’s Oncology Group (COG); Editorial Board: Journal of Clinical Medicine and Journal of Surgical Oncology; Paid consultant Balmoral Medical, Onkos Surgical, SpringWorks, and Zimmer; Paid speaker: Deciphera, OncLive, SpringWorks, and Zimmer.
References
- Sontag L, Pyle S. The appearance and nature of cyst-like areas in the distal femoral metaphyses of children. Am J Roentgenol. 1941;46(185).
- Jaffe HL, Lichtenstein L. Non-osteogenic fibroma of bone. Am J Pathol. 1942;18(2):205-221.
- Collier CD, Nelson GB, Conry KT, Kosmas C, Getty PJ, Liu RW. The Natural History of Benign Bone Tumors of the Extremities in Asymptomatic Children: A Longitudinal Radiographic Study. JBJS. 2021;103(7):575. [CrossRef]
- Baumhoer D, Kovac M, Sperveslage J, et al. Activating mutations in the MAP-kinase pathway define non-ossifying fibroma of bone. J Pathol. 2019;248(1):116-122. [CrossRef]
- Bovée JV, Hogendoorn PC. Non-ossifying fibroma: A RAS-MAPK driven benign bone neoplasm. J Pathol. 2019;248(2):127-130. [CrossRef]
- Ritschl P, Karnel F, Hajek P. Fibrous metaphyseal defects — determination of their origin and natural history using a radiomorphological study. Skeletal Radiol. 1988;17(1):8-15. [CrossRef]
- Rammanohar J, Zhang C, Thahir A, Krkovic M. Imaging of Non-ossifying Fibromas: A Case Series. Cureus. 13(3):e14102. [CrossRef]
- Baghdadi S, Nguyen JC, Arkader A. Nonossifying Fibroma of the Distal Tibia: Predictors of Fracture and Management Algorithm. J Pediatr Orthop. 2021;41(8):e671. [CrossRef]
- Goldin AN, Muzykewicz DA, Mubarak SJ. Nonossifying Fibromas: A Computed Tomography–based Criteria to Predict Fracture Risk. J Pediatr Orthop. 2020;40(2):e149-e154. [CrossRef]
- Patel RR, Damron TA. The Role of Surveillance in Predicting Fracture in Pediatric Patients With Incidentally Discovered Nonossifying Fibromas and Fibrous Cortical Defects: Is It Worth It? J Pediatr Orthop. 2024;44(6):395. [CrossRef]
- Herget GW, Mauer D, Krauß T, et al. Non-ossifying fibroma: natural history with an emphasis on a stage-related growth, fracture risk and the need for follow-up. BMC Musculoskelet Disord. 2016;17:147. [CrossRef]
- Arata MA, Peterson HA, Dahlin DC. Pathological fractures through non-ossifying fibromas. Review of the Mayo Clinic experience. JBJS. 1981;63(6):980.
- Andreacchio A, Alberghina F, Testa G, Canavese F. Surgical treatment for symptomatic non-ossifying fibromas of the lower extremity with calcium sulfate grafts in skeletally immature patients. Eur J Orthop Surg Traumatol. 2018;28(2):291-297. [CrossRef]
- Phemister DB. CHRONIC FIBROUS OSTEOMYELITIS. Ann Surg. 1929;90(4):756-764.
- Goldin A, Muzykewicz DA, Dwek J, Mubarak SJ. The aetiology of the non-ossifying fibroma of the distal femur and its relationship to the surrounding soft tissues. J Child Orthop. 2017;11(5):373-379. [CrossRef]
- Schindeler A, Little DG. Ras-MAPK signaling in osteogenic differentiation: friend or foe? J Bone Miner Res. 2006 Sep;21(9):1331-8. PMID: 16939391. [CrossRef]
Figure 1.
Images (a) and (b) demonstrate migration of the non-ossifying fibroma in a 9-year-old girl over a 6-month time span. Image (c) demonstrates the major axis of the non-ossifying fibroma with an imaginary line (dashed arrow) pointing toward the gastrocnemius insertion (white circle) as is a typical migration pattern for non-ossifying fibromas.
Figure 1.
Images (a) and (b) demonstrate migration of the non-ossifying fibroma in a 9-year-old girl over a 6-month time span. Image (c) demonstrates the major axis of the non-ossifying fibroma with an imaginary line (dashed arrow) pointing toward the gastrocnemius insertion (white circle) as is a typical migration pattern for non-ossifying fibromas.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



