Submitted:
27 July 2025
Posted:
28 July 2025
You are already at the latest version
Abstract
Renal cell carcinoma (RCC) is a biologically diverse malignancy with a rising global incidence and a high propensity for metastasis, particularly in its clear cell subtype. While traditional treatments have centered on surgery and targeted therapies, immunotherapy has emerged as a transformative approach in the management of advanced RCC. This review explores the evolution of immunotherapy in RCC, from early cytokine-based treatments to the advent of immune checkpoint inhibitors (ICIs) and their integration with tyrosine kinase inhibitors (TKIs). We detail the immunologic mechanisms underpinning these therapies, the rationale from preclinical models, and the pivotal clinical trials that redefined standard of care. Challenges such as immune resistance, tumor heterogeneity, and immune escape mechanisms are critically analyzed, highlighting tumor-intrinsic and microenvironmental factors. Lastly, we examine ongoing innovations including novel immune checkpoints, cytokine modulators, CAR-T therapies, and neoantigen-based vaccines, offering a forward-looking perspective on enhancing immunotherapeutic efficacy and personalization in RCC management.
Keywords:
renal cell carcinoma
; immunotherapy
; immune checkpoint inhibitors
; tyrosine kinase inhibitors
; PD-1
; CLTA-4
; belzutifan
; advanced RCC
1. Introduction
Renal cell carcinoma (RCC) arises from the renal tubular epithelium and accounts for over 80% of all kidney cancers [1]. In 2024, it is estimated that 81,610 new cases of RCC will be diagnosed, representing 4.1% of all new cancer cases. Additionally, the projected number of deaths due to RCC in the same year is 14,390, which constitutes 2.4% of all cancer-related deaths [2]. The primary risk factors for RCC include tobacco use, obesity, hypertension, Von Hippel–Lindau syndrome, acquired cystic kidney disease, and genetic predisposition [3]. The higher incidence of RCC observed in Europe and North America compared to Asia and Africa is largely attributed to the greater accessibility and utilization of advanced diagnostic imaging in Western countries [4].
Clear cell RCC (ccRCC) is the most common subtype, accounting for approximately 80% of cases, while the papillary subtype represents around 15%. Papillary RCC (pRCC) is the second most common histologic subtype of RCC after ccRCC , followed by less common subtypes such as chromophobe, translocation, unclassified, collecting duct carcinoma, and medullary RCC [5]. Each subtype varies in genetic profile, biological characteristics, and clinical behavior. Both ccRCC and pRCC originate from the proximal tubule epithelium, while chromophobe and collecting duct RCC arise from the distal tubular system. Medullary RCC originates from the renal papillae or calyceal epithelium. Importantly, medullary and collecting duct carcinomas are often found in younger patients and typically follow a more aggressive clinical course [6]. Additional subtypes include translocation-associated RCC, typically affecting younger patients and characterized by gene fusions involving TFE3 or TFEB, as well as unclassified RCC, which represents tumors that do not fit established morphologic or molecular criteria [7]. Furthermore, sarcomatoid differentiation is not considered a distinct histologic entity but rather a high-grade transformation that can occur within any RCC subtype, most frequently in ccRCC and other non-clear cell variants, and is associated with poor prognosis [8].
Various treatment modalities have been developed to inhibit tumor progression and metastasis in RCC. Current treatment strategies for RCC include surgical interventions such as nephrectomy, systemic therapies including ICI and targeted agents, and localized approaches such as thermal or cryoablation [9]. Radiation therapy is typically reserved for palliation in select metastatic or symptomatic cases [9]. Although many RCC cases are resectable at diagnosis, a significant proportion of patients present with advanced or metastatic disease, where nephrectomy may not be beneficial or feasible [10]. Furthermore, it is important to recognize that metastatic progression can occur in over one-third of patients, even after the surgical removal of the primary tumor [6].
Immunotherapy has become a cornerstone of modern cancer treatment, with increasing evidence supporting its role in improving long-term survival and disease control in RCC. Among the most studied and widely used immunotherapies are ICIs, which target specific transmembrane proteins that cancer cells exploit to evade immune surveillance. By inhibiting these proteins, ICIs restore the immune system’s ability to recognize and attack tumors, thereby preventing disease progression and metastasis [9].
Despite their efficacy, tumors may develop resistance to ICI, limiting the durability of therapeutic benefit [11]. A major challenge in ICI is tumor heterogeneity, wherein genetic and molecular diversity within tumors leads to variable responses to treatment. As a result, specific drugs may only be effective against certain tumor subpopulations, complicating the treatment landscape [12].
To address these challenges, recent research suggests that the combination of various immunotherapeutic approaches or their integration with non-immunotherapeutic treatments may yield more effective solutions. This synergistic strategy holds the potential to enhance treatment efficacy, counteract resistance mechanisms, and extend the benefits of immunotherapy to a broader range of cancer types [9,13].
2. Mechanism of Action of Immunotherapy: A Detailed Overview
Immunotherapy represents a diverse and transformative approach to treating diseases by enhancing or regulating the immune system’s ability to recognize and respond to harmful agents. It plays a pivotal role in addressing conditions such as allergies and cancer by modifying immune responses to achieve therapeutic benefits. Depending on the type of immunotherapy, specific mechanisms are employed to either suppress or amplify immune activity, ensuring that the body maintains a balanced and effective defense mechanism against allergens or malignant cells.
2.1. Allergen-Specific Immunotherapy (AIT)
AIT is a well-established approach used to treat IgE-mediated allergic diseases by promoting immune tolerance to specific allergens. The goal of AIT is to shift the immune system from an allergic state to one of desensitization, thus reducing the severity of allergic reactions over time. This process involves several key immunological mechanisms as outlined below.
2.1.1. Early Desensitization
AIT induces early desensitization by rapidly modifying the reactivity of mast cells and basophils, the primary effector cells responsible for allergic reactions. When allergens bind to IgE, they typically trigger degranulation, releasing histamine and other inflammatory mediators that cause symptoms such as itching, swelling, and airway constriction. Through repeated allergen exposure in controlled doses, AIT reduces the sensitivity of these cells, thereby decreasing the intensity of immediate allergic responses. Over time, mast cells and basophils become less reactive, leading to long-term symptom relief [15].
2.1.2. Regulatory T and B Cells (Treg & Breg)
AIT also enhances the activity of regulatory T (Treg) and regulatory B (Breg) cells, which play a crucial role in immune tolerance. These cells work by suppressing the activity of effector T cells, particularly Th2 cells, which are responsible for driving allergic responses. Tregs secrete anti-inflammatory cytokines such as IL-10 and Transforming Growth Factor β (TGF-β), which help shift the immune system away from an allergic state and toward a more tolerant, non-reactive state. Bregs contribute to this process by modulating antibody production and promoting an anti-inflammatory environment, further supporting immune tolerance [15,16].
2.1.3. Antibody Modulation
AIT alters antibody responses to allergens by decreasing the production of allergen-specific IgE, the antibody responsible for immediate hypersensitivity reactions, while increasing IgG4 levels. IgG4 serves as a blocking antibody that competes with IgE for allergen binding, thereby preventing IgE from triggering allergic reactions. This antibody shift is a critical mechanism by which AIT reduces allergy symptoms and provides long-term protection against allergen exposure [17].
2.1.4. Cellular Changes and Inflammation Reduction
AIT reduces the migration and activation of key inflammatory cells, including eosinophils, basophils, and mast cells, within allergic tissues. This suppression of cellular activity leads to decreased tissue inflammation, ultimately alleviating allergic symptoms. The recruitment of these immune cells to affected tissues is diminished over time, leading to a long-lasting reduction in allergic reactions, even after the completion of immunotherapy treatment [14,15,16,17].
2.2. Cancer Immunotherapy
Cancer immunotherapy enhances the body's ability to detect and eliminate malignant cells by targeting and modulating various components of the immune system. Unlike traditional cancer treatments such as cytotoxic chemotherapy and radiation, which directly kill cancer cells, immunotherapy focuses on augmenting the immune system’s capacity to recognize and eliminate tumor cells more effectively. The major mechanisms of action involved in cancer immunotherapy include the following:
2.2.1. Immune Checkpoint Inhibition
Cancer cells evade immune detection by exploiting immune checkpoint pathways that regulate T cell activation. Two primary immune checkpoint molecules, CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) and PD-1 (programmed death-1), act as immune brakes that dampen T cell responses, allowing tumors to escape immune attack. PD-L1 (programmed death-ligand 1), the principal ligand for PD-1, is often overexpressed on tumor cells and contributes to T cell exhaustion in the tumor microenvironment. Checkpoint inhibitors, such as anti-PD-1, anti-PD-L1, and anti-CTLA-4 antibodies, block these inhibitory signals, thereby reactivating T cells and enhancing their ability to attack cancer cells. This strategy has led to remarkable improvements in cancer treatment, particularly in melanoma, non-small cell lung cancer, and RCC, among others [18,19].
2.2.2. Adoptive Cell Therapy (ACT)
ACT is an advanced immunotherapy approach that involves extracting a patient’s T cells, genetically modifying them to enhance their tumor-targeting ability, and reinfusing them into the patient. One of the most effective forms of ACT is chimeric antigen receptor T-cell (CAR-T) therapy, where T cells are engineered to express synthetic receptors that recognize specific cancer antigens. CAR-T cells demonstrate potent tumor-killing ability and have been particularly successful in treating hematologic malignancies such as various subtypes of leukemia and lymphoma [20].
2.2.3. Cancer Vaccines
Cancer vaccines stimulate the immune system to recognize and destroy cancer cells by presenting tumor-associated antigens. These vaccines can be designed using dendritic cells, peptides, or viral vectors to train the immune system to mount an effective response against malignancies. Unlike preventive vaccines for infectious diseases, therapeutic cancer vaccines aim to enhance immune surveillance and eliminate existing cancer cells. Ongoing research is focused on developing personalized cancer vaccines tailored to an individual’s tumor-specific mutations [19].
2.2.4. Cytokine Therapy
Cytokines, such as interleukin-2 (IL-2), interferon-alpha (IFN-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF), are essential mediators of the immune response against tumors. IL-2 therapy promotes the expansion and activation of cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, enhancing their tumor-killing capabilities. IFN-α and GM-CSF contribute to anti-tumor immunity by stimulating antigen presentation and increasing immune cell infiltration into tumors. Although historically used in metastatic RCC (mRCC), cytokine therapies such as IL-2 and IFN-α have largely fallen out of favor due to significant toxicity and limited efficacy. Their use has been supplanted by more effective and better-tolerated agents such as immune checkpoint inhibitors and VEGF-targeted therapies [1].
3. Preclinical Studies That Led to Immunotherapy
Preclinical research in immunotherapy for RCC has played a central role in shaping today’s treatment landscape. ccRCC, the most common histologic subtype, is recognized as an immunogenic tumor, largely due to its tendency to express PD-L1 and foster an immunosuppressive tumor microenvironment. This may help explain why some RCC tumors could evade immune detection despite being infiltrated by T cells [21].
In mouse models, blockade of the PD-1/PD-L1 and CTLA-4 pathways was shown to restore T-cell activity, enhance tumor killing, and delay disease progression, findings that directly supported early clinical trials of ICIs like nivolumab [22]. Another key insight from preclinical models was the immune-modulating effect of VEGF inhibitors. The clinical validation of immune checkpoint blockade began with the landmark CheckMate 025 trial, in which PD-1 inhibition with single-agent nivolumab significantly improved overall survival (OS) compared to everolimus in previously treated mRCC (median OS 25.0 vs. 19.6 months; HR 0.73), leading to the first FDA approval of nivolumab in RCC as a second-line therapy [23]. This success catalyzed further investigation into combination regimens for the first-line setting. Agents like sunitinib were shown not only to block angiogenesis but also to reduce myeloid-derived suppressor cells (MDSC) and improve T-cell infiltration, laying the groundwork for successful ICI-TKI combinations such as pembrolizumab + axitinib (KEYNOTE-426) and nivolumab + cabozantinib (CheckMate 9ER) [24,25].
In addition to checkpoint blockade, recent preclinical work has explored novel strategies such as neoantigen-based vaccines. A phase I trial demonstrated that personalized cancer vaccines targeting RCC-specific neoantigens could generate robust T-cell responses and potentially prevent tumor recurrence [26]. Meanwhile, studies have identified mechanisms of resistance to current therapies, including upregulation of alternative checkpoints like LAG-3 and TIM-3. To address this, dual ICI and cytokine-based approaches are under investigation to counteract immune escape and broaden response [27]. These preclinical insights have provided critical biological rationale for many of the combination regimens now tested in clinical trials and continue to guide the next generation of RCC immunotherapies.
4. Overview of Clinical Trials in RCC
The evolution of immunotherapy in RCC has shifted the treatment landscape from non-specific immune stimulation to precision-driven checkpoint blockade, offering durable outcomes for many patients.
4.1. Early Cytokine Therapies in RCC
The first major efforts in immunotherapy for RCC emerged from the observation that RCC was largely resistant to chemotherapy and radiotherapy, yet occasionally showed spontaneous regression, suggesting immune sensitivity(28). Notably, some patients with metastatic disease who underwent nephrectomy experienced spontaneous resolution of pulmonary metastases. This phenomenon was attributed to the removal of the so-called "immunologic sink," wherein the primary tumor was thought to suppress systemic anti-tumor immune responses, and its removal allowed for immune reactivation [29].
These insights led to the development of cytokine-based therapies, notably IL-2 and IFN-α [30]. High-dose IL-2, approved by the FDA in 1992, became the first immunotherapy for mRCC. IL-2 plays a critical role in activating lymphokine-activated killer (LAK) cells, which are immune cells derived from natural killer (NK) and T cells following several days of IL-2 stimulation. These LAK cells, which are part of the activated peripheral blood mononuclear cell (PBMC) population, exhibit potent cytotoxic activity against a broad spectrum of tumors while sparing normal tissues [31,32]. Early trials, including a pivotal study by Fyfe et al., demonstrated durable complete responses in approximately 7% of patients (17 out of 255) with mRCC treated with high-dose IL-2 However, its use was limited by high toxicity, including vascular leak syndrome, hypotension, and multi-organ failure, confining its administration to select patients at specialized centers [30]. Similarly, IFN-α was evaluated in multiple studies and showed modest benefits in progression-free survival (PFS) and (OS), however, its clinical impact was tempered by flu-like symptoms, fatigue, and psychiatric side effects [33].
4.2. Landmark Trials and the Turning Point in RCC Immunotherapy
The transition from cytokine therapy to ICIs marked a paradigm shift in RCC treatment. During this transitional period, targeted therapies particularly TKIs directed against the VEGF and mTOR pathways emerged as the dominant treatment approach, replacing cytokine therapy as the standard of care in advanced RCC [34]. Preclinical studies had identified PD-1/PD-L1 and CTLA-4 pathways as key mechanisms of immune escape in various solid tumors, including RCC [22]. This prompted the initiation of clinical trials targeting these checkpoints. The CheckMate 025 trial was the first major trial to demonstrate the clinical potential of PD-1 inhibition in RCC. In this phase III study, nivolumab significantly improved OS (25.0 vs. 19.6 months; HR 0.73; p = 0.002) compared to everolimus in previously treated patients with mRCC, establishing immune checkpoint blockade as a standard second-line therapy [23]. Importantly, the safety profile of nivolumab was more favorable than that of cytokines or mTOR inhibitors, with fewer grade 3–4 adverse events [23].
4.3. Advancements with ICI and Combinations
Following the success of CheckMate 025, efforts turned to earlier lines of therapy. The CheckMate 214 trial evaluated nivolumab + ipilimumab, combining PD-1 and CTLA-4 blockade, in treatment-naïve mRCC patients. In those with intermediate- and poor-risk disease, the combination showed superior efficacy over sunitinib. Median OS was not reached in the combination group versus 26.0 months with sunitinib (HR 0.63), and median PFS was 11.6 months vs. 8.3 months respectively (HR 0.73). The objective response rate (ORR) was also higher (42% vs. 27%). This trial established dual checkpoint inhibition as a viable first-line option in a biomarker-unselected population [35]. Updated 8-year follow-up data presented at ASCO 2024 confirmed the durability of this benefit, reporting a sustained OS hazard ratio of 0.75 in the same subgroup, along with a higher complete response rate of 11% compared to 2% with sunitinib [36].
Subsequently, ICI-based combinations with TKIs were investigated. KEYNOTE-426, a pivotal phase III trial, evaluated the efficacy of pembrolizumab, a PD-1 inhibitor, in combination with axitinib, a VEGFR tyrosine kinase inhibitor, compared to sunitinib in treatment-naïve patients with advanced RCC. A total of 861 patients were randomized to receive either pembrolizumab plus axitinib or sunitinib monotherapy. The combination demonstrated a 47% lower risk of death (hazard ratio for death, 0.53; 95% CI, 0.38–0.74; p < 0.0001), and a median PFS of 15.1 months compared to 11.1 months with sunitinib (HR 0.69; 95% CI, 0.57–0.84). The ORR was also higher in the pembrolizumab–axitinib arm (59.3%) than with sunitinib (35.7%), including a complete response rate of 10% versus 3.5%, respectively. These benefits were consistent across the International Metastatic RCC Database Consortium (IMDC) risk groups and PD-L1 expression subgroups [24]. Updated 5-year follow-up data presented at ASCO 2023 confirmed the durability of benefit, with a sustained OS advantage (HR 0.67; 95% CI, 0.52–0.84) and a 5-year OS rate of 41.9% versus 31.7%, favoring the pembrolizumab–axitinib combination [37].
This was followed by CheckMate 9ER, a pivotal phase III trial comparing the combination of nivolumab, a PD-1 inhibitor, and cabozantinib, a VEGFR tyrosine kinase inhibitor, against sunitinib monotherapy in treatment-naïve patients with advanced RCC. A total of 651 patients were randomized to receive either nivolumab–cabozantinib (n=323) or sunitinib (n=328). The combination regimen resulted in a significant improvement in PFS, with a median of 16.6 months versus 8.3 months for sunitinib (HR 0.51; 95% CI, 0.41–0.64; p < 0.001). Improvements were also seen in OS, with a 12-month OS rate of 85.7% for the combination arm versus 75.6% for sunitinib (HR 0.60; 98.89% CI, 0.40–0.89; p = 0.001). The ORR was notably higher with nivolumab–cabozantinib (55.7%) than with sunitinib (27.1%), including complete response rates of 8.0% and 4.6%, respectively. These clinical benefits were consistent across subgroups stratified by IMDC risk status and PD-L1 expression. Grade ≥3 treatment-related adverse events were reported in 60.6% of patients in the combination arm, compared to 50.9% in the sunitinib group [25]. Based on these findings, the combination of nivolumab and cabozantinib was approved as a first-line treatment option for advanced RCC.
The CLEAR trial further expanded first-line treatment options by evaluating lenvatinib, a multikinase inhibitor, in combination with pembrolizumab versus sunitinib in advanced RCC. In this phase III trial, 1,069 treatment-naïve patients were randomized to receive either lenvatinib–pembrolizumab, lenvatinib–everolimus, or sunitinib. The lenvatinib–pembrolizumab arm demonstrated a significant improvement in PFS, with a median of 23.9 months compared to 9.2 months with sunitinib (HR 0.39; 95% CI, 0.32–0.49; p < 0.001). OS was also significantly improved (HR 0.66; 95% CI, 0.49–0.88; p = 0.005), and the ORR was 71.0%, including a complete response rate of 16.1%, compared to 36.1% and 4.2% with sunitinib, respectively. These results established lenvatinib–pembrolizumab as another highly effective first-line regimen for patients with advanced RCC, particularly for those with high disease burden or rapid progression [38].

Table 1.
Notable Clinical Trials Related to RCC Immunotherapy.
| Trial Name | Trial ID | Phase | Objective | Results |
|---|---|---|---|---|
| KEYNOTE-426 [24] | NCT02853318 | III | Pembrolizumab + Axitinib vs Sunitinib in first-line advanced RCC | Median PFS: 15.1 vs 11.1 months (HR 0.69; p<0.001); OS: HR 0.53 (p<0.0001); ORR: 59.3% vs 35.7% |
| CheckMate 9ER [25] | NCT03141177 | III | Nivolumab + Cabozantinib vs Sunitinib in first-line metastatic RCC | Median PFS: 16.6 vs 8.3 months (HR 0.51; p<0.001); OS at 12 months: 85.7% vs 75.6% (HR 0.60; p=0.001); ORR: 55.7% vs 27.1% |
| CheckMate 214 [35] | NCT02231749 | III | Nivolumab + Ipilimumab vs Sunitinib in treatment-naive metastatic RCC | OS: HR 0.63 (p<0.001); ORR: 42% vs 27%; more benefit in intermediate/poor-risk patients |
| JAVELIN Renal 101 [39] | NCT02684006 | III | Avelumab + Axitinib vs Sunitinib in first-line advanced RCC | Median PFS: 13.8 vs 8.4 months (HR 0.69; p<0.001); OS not significantly improved; ORR higher in combination arm |
| CheckMate 025 [23] | NCT01668784 | III | Nivolumab vs Everolimus in previously treated metastatic RCC | OS: 25.0 vs 19.6 months (HR 0.73; p=0.002); better safety profile |
| CLEAR Trial [38] | NCT02811861 | III | Lenvatinib + Pembrolizumab vs Sunitinib in first-line advanced RCC | Median PFS: 23.9 vs 9.2 mo (HR 0.39; p<0.001); OS: HR 0.66 (p<0.001); ORR: 71% vs 36% |
| Phase I Trial of Personalized Cancer Vaccines in RCC | NCT03472238 | I | Personalized cancer vaccines targeting RCC-specific neoantigens | Robust T-cell responses observed; early-phase safety and immunogenicity data encouraging |
| Combination of Dual Checkpoint Inhibition (PD-1/PD-L1 + LAG-3/TIM-3) | NCT03871297 | I/II | PD-1/PD-L1 + LAG-3/TIM-3 inhibitors in resistant advanced RCC | Ongoing trial; aims to overcome resistance with novel checkpoint blockade combinations |
5. Standard of Care:
5.1. Localized and Locally Advanced RCC
Localized RCC, encompassing stages I and II, is primarily managed surgically, with the choice of procedure guided by tumor size, location, and patient-specific factors. For tumors ≤4 cm (T1a), partial nephrectomy is the preferred approach, aiming to preserve renal function while achieving oncologic control. Tumors measuring 4–7 cm (T1b) may still be amenable to partial nephrectomy, depending on their anatomical location and complexity; however, radical nephrectomy is often considered for larger or centrally located masses. Stage II tumors (>7 cm, T2) confined to the kidney are typically treated with radical nephrectomy [40].
Stage III RCC, classified as locally advanced, is defined by tumor extension into major veins (T3), perinephric tissues, or regional lymph node involvement (N1). It is primarily treated with radical nephrectomy. Lymph node dissection is selectively performed in patients with radiologically or intraoperatively suspicious lymphadenopathy, but is not routinely indicated in the absence of nodal involvement [40].
Postoperative management decisions are influenced by the risk of recurrence. The KEYNOTE-564 trial demonstrated that adjuvant pembrolizumab significantly improved disease-free survival and overall survival in patients with clear cell RCC at high risk of recurrence following nephrectomy, defined as those patients with stage II RCC with grade 4 or sarcomatoid features, stage III RCC, regional lymph node involvement (N+), or oligometastatic disease following metastatectomy (M1 NED) [41]. This led to FDA approval of pembrolizumab for adjuvant treatment in these patients. The S-TRAC trial demonstrated improved disease-free survival (DFS) with adjuvant sunitinib in high-risk clear cell RCC, but no benefit in OS, and its use remains limited due to toxicity [42]. Several other adjuvant strategies have failed to demonstrate benefit, including atezolizumab in the IMmotion010 trial, nivolumab plus ipilimumab in CheckMate 914, and axitinib in the ATLAS trial, which was terminated early due to futility [43,44,45].
Table 2.
Adjuvant Treatment Studies:.
| Trial | Therapy | Population | Outcome | Result |
|---|---|---|---|---|
| S-TRAC [42] | Sunitinib | High-risk (≥pT3 and/or N+) | Improved DFS; no OS benefit | Positive (DFS) |
| KEYNOTE-564 [41] | Pembrolizumab | Intermediate-high/high-risk | Improved DFS and OS | Positive |
| CheckMate 914 [35] | Nivolumab + Ipilimumab | High-risk | No DFS benefit | Negative |
| IMmotion010 [43] | Atezolizumab | High-risk | No DFS benefit | Negative |
| ATLAS [45] | Axitinib | High-risk | Trial stopped early; no benefit | Negative |
5.2. Advanced/Metastatic Disease:
Stage IV RCC, characterized by the presence of distant metastases, is managed primarily with systemic therapy. The current standard-of-care first-line treatment options fall into four major therapeutic classes: ICI combined with TKI, dual ICI therapy, TKI monotherapy (less commonly), and participation in clinical trials [40]. Selection among these regimens is largely guided by risk stratification based on the IMDC criteria. The IMDC model incorporates six clinical and laboratory factors: time from diagnosis to initiation of systemic therapy less than one year, Karnofsky performance status below 80%, anemia, hypercalcemia, neutrophilia, and thrombocytosis. Patients are categorized as favorable risk (0 risk factors), intermediate risk (1–2 factors), or poor risk (3 or more factors), and these classifications inform both prognosis and treatment selection [40,46].
For favorable-risk patients, ICI–TKI combinations are the preferred first-line approach. Three landmark trials have established these regimens: KEYNOTE-426, which evaluated pembrolizumab plus axitinib, CheckMate 9ER, which assessed nivolumab plus cabozantinib, and CLEAR, which assessed pembrolizumab plus Lenvatinib. All three combinations demonstrated superior OS, PFS, and ORR compared to sunitinib in previously untreated advanced RCC, including patients with favorable-risk disease [24,25,38]. These regimens are now widely adopted in this setting.
In patients with intermediate- or poor-risk disease, both ICI–TKI combinations and dual checkpoint blockade are approved options. The CheckMate 214 trial evaluated the combination of nivolumab and ipilimumab, demonstrating a significant OS benefit and a higher rate of durable complete responses compared to sunitinib in intermediate- and poor-risk groups(35). This dual ICI regimen remains a standard first-line option in patients, especially those with sarcomatoid histology or those for whom treatment-free intervals are a clinical goal. ICI–TKI combinations also remain appropriate alternatives in this population [24,25].
In the second-line setting, treatment selection is guided by the patient’s prior exposure and tolerability to first-line therapy. With the increasing adoption of ICI-based regimens as the standard of care in the frontline setting, the use of nivolumab monotherapy in the second-line therapy has become relatively uncommon. Most patients who do not receive ICI up front typically have a contraindication, such as a history of solid organ transplantation or uncontrolled autoimmune or rheumatologic disease, that precludes immunotherapy altogether [40,47]. In such cases, sequential use of VEGF-targeted TKIs remains the primary approach [40].
For patients previously treated with ICI-TKI combinations, second-line options include cabozantinib, lenvatinib plus everolimus, or other VEGF inhibitors [40,48,49]. For the small subset of patients who received frontline VEGF monotherapy, ICIs such as nivolumab may still be considered, as supported by results from the CheckMate 025 trial, which demonstrated improved OS compared to everolimus [23].
In the third-line setting and beyond, therapeutic decision-making is individualized and largely influenced by prior lines of treatment, disease burden, and patient comorbidities. Agents such as tivozanib, axitinib, and lenvatinib plus everolimus remain viable options if not previously utilized [40,48,50,51]. Belzutifan, a hypoxia-inducible factor 2-alpha (HIF-2α) inhibitor that targets the VHL–HIF pathway has demonstrated promising efficacy in patients with VHL-associated RCC [52].The phase II LITESPARK-005 trial examined belzutifan in the ccRCC setting, and found that belzutifan significantly improved PFS compared to everolimus in previously treated patients with advanced ccRCC, supporting its potential role as a non-immunologic, molecularly targeted therapy in later lines [53]. As the therapeutic landscape continues to evolve, clinical trial participation remains strongly encouraged to expand treatment options and refine sequencing strategies.
6. ICI Resistance and Escape Mechanisms
Like other treatments, resistance to ICI remains a major clinical challenge. Once resistance develops, current standard second- and third-line therapies offer limited benefits in terms of PFS and OS [48,53]. To understand resistance, it is important to first review the basic steps of the anti-tumor immune response triggered by ICI. The process begins with dendritic cells presenting tumor-associated antigens to T lymphocytes, enabling recognition of cancer cells. These activated T cells then migrate into the tumor microenvironment, where they are further activated and secrete cytokines. These cytokines recruit additional immune cells, such as macrophages and dendritic cells, to sustain the immune attack. Finally, the immune system forms memory T cells that help maintain long-term surveillance against the tumor [54,55].
Resistance to ICIs in RCC can be broadly categorized as primary, adaptive, or acquired. Primary resistance refers to a lack of clinical benefit from immunotherapy despite adequate drug exposure and no apparent pharmacologic failure. It is generally defined as disease progression or lack of response (per RECIST 1.1 criteria) at the first radiographic evaluation, usually within 8–12 weeks of initiating ICI therapy [54,55,56]. Mechanisms include an inherently non-immunogenic tumor microenvironment, low tumor mutational burden, or absence of T-cell infiltration (also known as “cold tumors”) [55,57].
Adaptive resistance occurs when tumors initially trigger immune activation, but subsequently upregulate immune checkpoint molecules (e.g., PD-L1) or immunosuppressive pathways in response to immune pressure. This dynamic immune evasion allows tumors to escape destruction after initial immune recognition [58].
Acquired resistance describes cases where a patient initially responds to immunotherapy but later experiences disease progression after a period of tumor control. Mechanisms may include loss of neoantigen expression, immune editing, or T-cell exhaustion [59].
These resistance patterns may stem from tumor-intrinsic factors (e.g., genomic mutations, antigen presentation defects), host-related factors (e.g., T-cell repertoire, HLA type), or extrinsic influences within the tumor microenvironment (e.g., immunosuppressive cytokines, Tregs) [59].
6.1. Tumor-Related Factors for Resistance to ICI:
Tumor-related mechanisms of ICI resistance are multifaceted and begin at the level of antigen recognition. Effective immune surveillance requires that dendritic cells present tumor-associated antigens to T cells, a process that can be disrupted when tumors are poorly differentiated or undergo dedifferentiation. Such changes may lead to loss or mutation of critical cell surface molecules, including major histocompatibility complex class I (MHC-I) and β2-microglobulin, impairing antigen presentation. Mutations or dysregulation in signaling pathways, such as the interferon (IFN), MAPK, and JAK/STAT pathways, can further diminish tumor visibility to the immune system [60]. Stress within the endoplasmic reticulum also interferes with antigen processing, while mutations in mitochondrial cytochrome proteins may disrupt apoptotic signaling. Aberrant expression of PD-L1, whether upregulated as an immune evasion tactic or downregulated in certain resistant phenotypes, influences tumor susceptibility to checkpoint blockade. Additionally, epigenetic reprogramming can alter the expression of immune-related proteins even in the absence of genetic mutations. The tumor microenvironment itself can become immunosuppressive through the secretion of inhibitory metabolites and cytokines, creating a barrier to effective T cell infiltration and activation. Resistance to apoptosis further enables tumor cells to evade immune destruction. Epithelial-to-mesenchymal transition (EMT), a key process in tumor progression, has also been implicated in immunotherapy resistance through downregulation of immune checkpoints like PD-L1, ultimately contributing to immune evasion and therapeutic failure [60,61,62].
6.2. External factors in the Tumor Microenvironment that lead to resistance to ICI:
When tumor-infiltrating lymphocytes (TILs) penetrate the tumor microenvironment, some may differentiate into Tregs, which suppress the antitumor immune response and promote tumor progression [63]. This immunosuppressive shift is further influenced by alterations in cytokine signaling. Elevated levels of immunosuppressive cytokines such as TGF-β and IL-2 facilitate Treg development and inhibit cytotoxic T cell function [64,65]. Chemokines, which orchestrate the recruitment of immune cells, also play a dual role in tumor biology. While chemokines such as CXCL9 and CXCL10 are associated with antitumor immunity through the recruitment of effector T cells, others like CCL2 and IL-8 exhibit pro-tumorigenic effects [66,67,68]. These pro-tumor chemokines promote the infiltration of MDSCs, which in turn suppress cytotoxic T lymphocytes and secrete tumor-promoting growth factors such as TGF-β, fibroblast growth factor (FGF), platelet-derived growth factor (PDGF), and epidermal growth factor (EGF), ultimately enhancing tumor growth and immune evasion [69].
VEGF secreted by tumor cells can lead to abnormal blood vessel formation, leading to stress in the TME because of the increased oxygen requirement by the tumor, leading to hypoxia and acidosis [70]. Hypoxia contributes to an immunosuppressive tumor microenvironment by inducing HIF-1α in both tumor cells and MDSCs, a heterogeneous population of immature myeloid cells that suppress T cell responses, resulting in increased PD-L1 expression, T cell depletion, and resistance to ICIs [71]. Additionally, HIF-1α upregulates VEGF expression through a positive feedback loop, further promoting immune evasion and tumor progression [72]. VEGF can also bind to dendritic cells (DC) and stop its maturation process, thereby impairing antigen presentation and subsequent T cell activation [73]. Abnormal blood vessel formation causes difficulty in infiltration of T cells because of the alterations in adhesion molecules like vascular cell adhesion molecule (VCAM), P selectin, E selectin, Platelet-endothelial cell adhesion molecule-1 (PECAM-1), CD31, and CD99 [74]. Furthermore, these changes in vasculature can inhibit penetrance to the tumor itself [75].
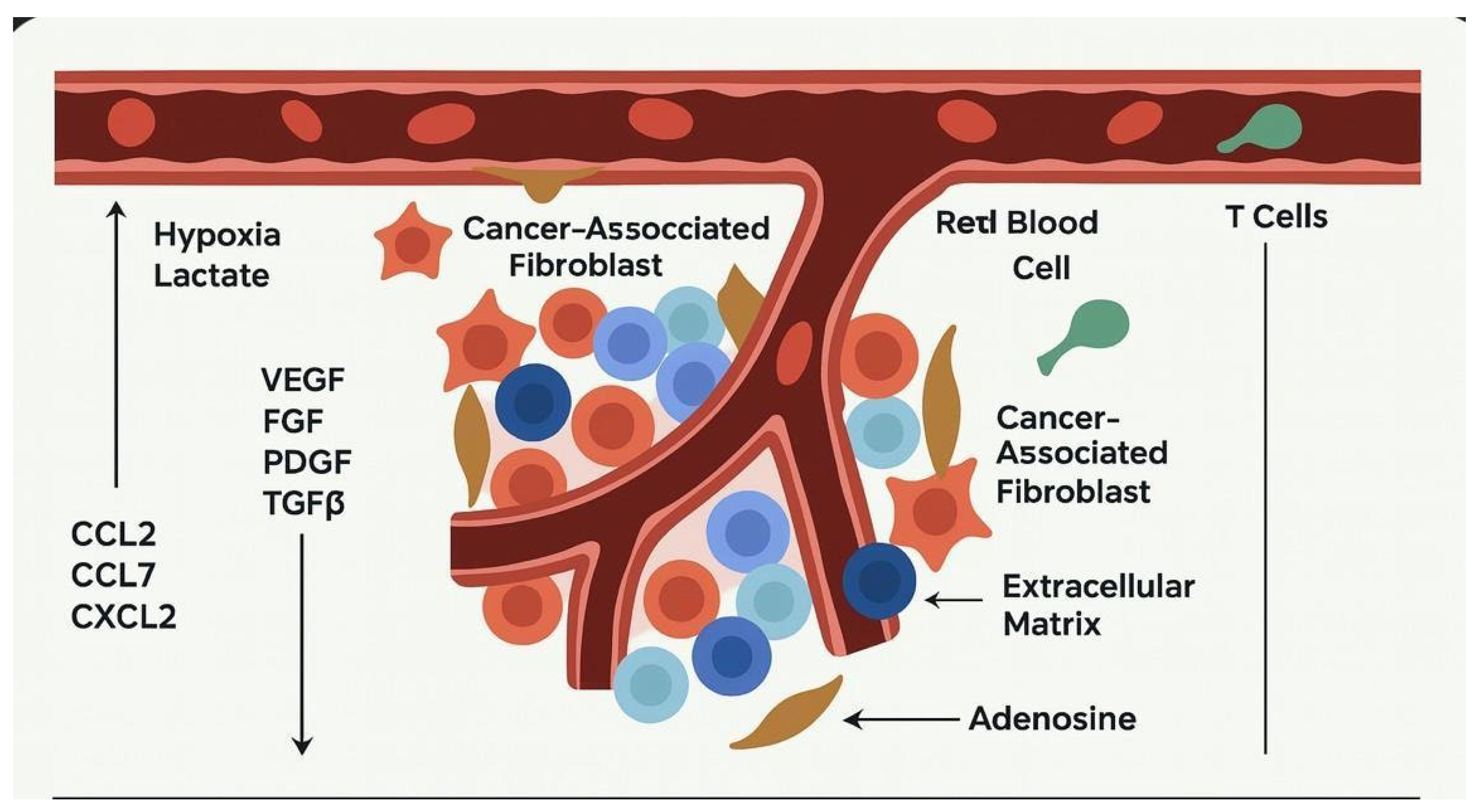
Figure 1.
This figure illustrates how cancer-associated fibroblasts (CAFs) actively shape the tumor microenvironment to support cancer progression. CAFs promote angiogenesis by secreting pro-angiogenic factors such as VEGF, FGF, PDGF, and TGF-β, facilitating the formation of new blood vessels to supply nutrients to the tumor. They also remodel the extracellular matrix (ECM), creating a dense stromal barrier that not only supports tumor cell invasion but also impedes the infiltration of immune cells. In response to tumor hypoxia and metabolic stress, CAFs contribute to an immunosuppressive milieu through the release of cytokines (such as CCL2, CCL7, and CXCL2) and immunomodulatory metabolites like adenosine. These secretions inhibit the recruitment and function of cytotoxic T cells, thereby allowing the tumor to evade immune surveillance. Collectively, these mechanisms orchestrated by CAFs enable tumors to grow, invade surrounding tissues, and resist immune-mediated destruction.
Figure 1.
This figure illustrates how cancer-associated fibroblasts (CAFs) actively shape the tumor microenvironment to support cancer progression. CAFs promote angiogenesis by secreting pro-angiogenic factors such as VEGF, FGF, PDGF, and TGF-β, facilitating the formation of new blood vessels to supply nutrients to the tumor. They also remodel the extracellular matrix (ECM), creating a dense stromal barrier that not only supports tumor cell invasion but also impedes the infiltration of immune cells. In response to tumor hypoxia and metabolic stress, CAFs contribute to an immunosuppressive milieu through the release of cytokines (such as CCL2, CCL7, and CXCL2) and immunomodulatory metabolites like adenosine. These secretions inhibit the recruitment and function of cytotoxic T cells, thereby allowing the tumor to evade immune surveillance. Collectively, these mechanisms orchestrated by CAFs enable tumors to grow, invade surrounding tissues, and resist immune-mediated destruction.
6.3. Alterations in Cell Metabolites and effects on Immune response:
High lactate levels caused by hypoxia result in the expression of CD44 and hyaluronic-associated metastasis [76]. Tumor cells increase CD38 expression to produce adenosine, leading to tumor metastasis and causing increased expression of PD-1 on the T cells and impaired signalling [77]. Additionally, tumor cells and MDSCs utilize tryptophan and form an immunosuppressive metabolite kynurenine(78). Tumor cell esterification of cholesterol can lead to maturation of inhibitory T cells, and lead to deactivation DCs [76].
Table 3.
Mechanisms of Resistance to Immunotherapy in RCC.
| Category | Factors/Mechanisms | Description |
| 1. Tumor-Related Factors | Poorly Differentiated Tumors/De-differentiation | Tumor cells with poor differentiation or de-differentiation exhibit reduced antigen recognition, hindering T-cell recognition. |
| Loss or Mutation of MHC-I and β2 Microglobulin | Tumor cells may lose MHC-I expression, impairing antigen presentation to T cells. | |
| Altered Signaling Pathways (INF, MAP kinase, JAK-STAT) | Changes in signaling pathways can impact immune recognition by tumor cells. | |
| Endoplasmic Reticulum Stress/Mutation in Mitochondrial Cytochrome Proteins | Stress or mutations may affect antigen processing and presentation, preventing effective immune response. | |
| Epigenetic Reprogramming of Tumor Cell Proteins | Tumor cells may alter protein expression without genetic mutations, further hindering immune recognition. | |
| Resistance to Apoptosis | Tumor cells may evade apoptosis, which prevents immune-mediated cell death. | |
| Epithelial-Mesenchymal Transition (EMT) | EMT may downregulate PD-L1, reducing responsiveness to PD-1 blockade therapy, and contributing to therapy resistance. | |
| 2. External Factors in Tumor Microenvironment (TME) | TIL Differentiation into Tregs | TIL can differentiate into Tregs, which suppress immune responses and promote tumor growth. |
| Cytokine Alterations (e.g., TGF-β, IL-2) | Cytokines like TGF-β can suppress immune responses, promoting resistance to ICI therapies. | |
| Pro-Tumor Chemokines (e.g., CCL-2, IL-8) | Pro-tumor chemokines recruit MDSCs that suppress T-cell cytotoxicity and promote tumor growth. | |
| MDSCs and Secretion of Growth Factors (TGF-β, FGF, PDGF, EGF) | MDSCs produce growth factors like TGF-β, FGF, and PDGF, supporting tumor growth and immune suppression. | |
| VEGF-Induced Hypoxia and Acidosis | VEGF promotes abnormal blood vessel formation, leading to hypoxia and acidosis, which create an immunosuppressive environment. Hypoxia also upregulates PD-L1 expression, leading to immune evasion. | |
| Altered Blood Vessel Adhesion Molecules (VCAM, P-selectin, PECAM-1) | Abnormal vasculature hinders T-cell infiltration into tumors, reducing the effectiveness of ICI. | |
| 3. Alterations in Cell Metabolites | High Lactate Levels and CD44 Expression | Lactate production by tumor cells induces CD44 expression, facilitating metastasis and immune resistance. |
| Increased CD38 Expression and Adenosine Production | Increased CD38 expression in tumor cells promotes adenosine production, which impairs T-cell function and enhances immune evasion. | |
| Tryptophan Degradation and Immunosuppressive Metabolite (Kynurenine) | Tumor cells and MDSCs degrade tryptophan, forming kynurenine, which suppresses T-cell function and promotes immune evasion. | |
| Cholesterol Esterification and Inhibition of DC Maturation | Tumor cells modify cholesterol metabolism, leading to impaired DC maturation and reduced immune response. |
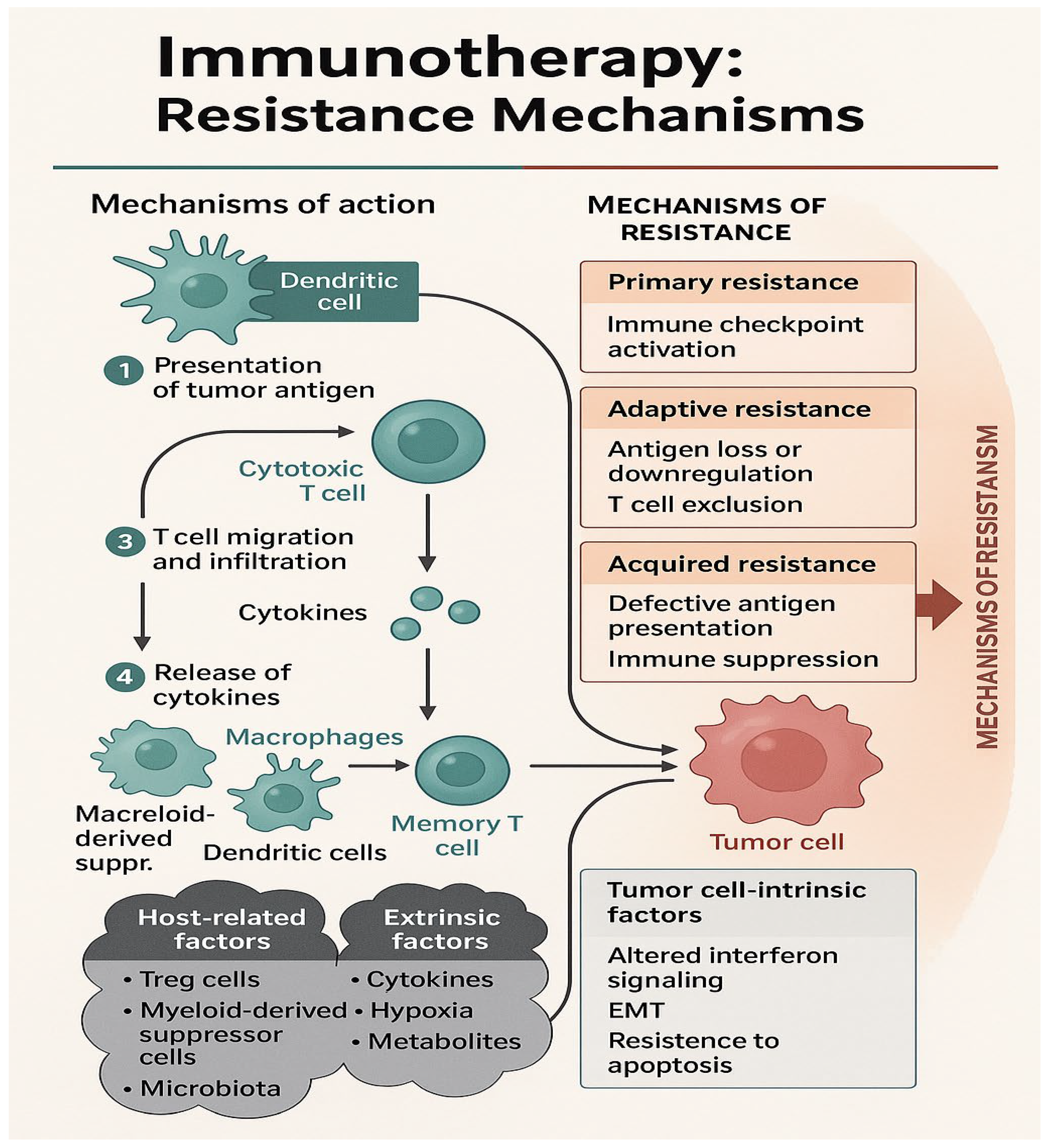
Figure 2.
This schematic illustrates the key steps of immunotherapy (IO) action and the multifaceted mechanisms by which resistance can arise. The left panel outlines the canonical cancer-immunity cycle, including: (1) presentation of tumor antigens by dendritic cells to cytotoxic T cells, (2) T-cell migration and tumor infiltration, (3) cytokine release, and (4) recruitment of additional immune cells such as macrophages and dendritic cells, ultimately leading to memory T-cell formation and tumor cell death. The right panel classifies IO resistance into three categories: primary (pre-existing immune checkpoint activation), adaptive (tumor-mediated antigen loss or T-cell exclusion), and acquired resistance (loss of antigen presentation and immune suppression). Resistance arises from various sources, including tumor-intrinsic factors (e.g., altered interferon signaling, epithelial-mesenchymal transition [EMT], and resistance to apoptosis), host-related factors (e.g., T regulatory cells, myeloid-derived suppressor cells, and microbiota), and extrinsic factors (e.g., cytokines, hypoxia, and immunosuppressive metabolites). Together, these mechanisms create a hostile tumor microenvironment that impairs the efficacy of immunotherapy.
Figure 2.
This schematic illustrates the key steps of immunotherapy (IO) action and the multifaceted mechanisms by which resistance can arise. The left panel outlines the canonical cancer-immunity cycle, including: (1) presentation of tumor antigens by dendritic cells to cytotoxic T cells, (2) T-cell migration and tumor infiltration, (3) cytokine release, and (4) recruitment of additional immune cells such as macrophages and dendritic cells, ultimately leading to memory T-cell formation and tumor cell death. The right panel classifies IO resistance into three categories: primary (pre-existing immune checkpoint activation), adaptive (tumor-mediated antigen loss or T-cell exclusion), and acquired resistance (loss of antigen presentation and immune suppression). Resistance arises from various sources, including tumor-intrinsic factors (e.g., altered interferon signaling, epithelial-mesenchymal transition [EMT], and resistance to apoptosis), host-related factors (e.g., T regulatory cells, myeloid-derived suppressor cells, and microbiota), and extrinsic factors (e.g., cytokines, hypoxia, and immunosuppressive metabolites). Together, these mechanisms create a hostile tumor microenvironment that impairs the efficacy of immunotherapy.
7. Future of IO in RCC
Several ICI–TKI combinations are approved for patients across all IMDC risk groups, along with dual checkpoint blockade with nivolumab and ipilimumab for intermediate- and poor-risk groups. Ongoing research is focused on refining existing strategies and introducing novel immunotherapeutic approaches. Among emerging agents, LAG-3 + PD-1 inhibitors are under investigation for PD-L1 low tumors, while additional immune checkpoints such as TIGIT, TIM-3, KIR3DL3, and PSGL-1 are also being explored as potential targets [79,80,81]. Cytokine-based therapies, including CD8-IL-2 fusion proteins and decoy-resistant IL-18, aim to rejuvenate exhausted T-cell pools and have shown promising in vitro responses [82,83]. Personalized mRNA-based cancer vaccines using tumor-specific neoantigens have demonstrated encouraging recurrence-free survival beyond 40 months in resected stage III and IV RCC in early-phase trials [26]. Cell-based therapies such as CAR-T targeting CD70 (e.g., CTX130 and ALLO-316) have also shown potential in early studies of advanced or refractory RCC [84,85]. Utilizing the gut microbiome, the addition of Clostridium Butyricum improves the response from immunotherapy, showing significantly longer PFS in early phase clinical trials [86].
Importantly, several recent and ongoing clinical trials may shift practice. The Phase III LITESPARK-005 trial led to the FDA approval of belzutifan in 2023 after demonstrating a higher ORR (22% vs. 4%) and a significantly reduced risk of progression or death compared to everolimus (HR 0.75; p = 0.0008) in pretreated ccRCC [53]. Rechallenge strategies have thus far not shown benefit, with both the CONTACT-03 study [87] (IO rechallenge with atezolizumab) and the TiNivo-2 study [88] (IO rechallenge with nivolumab) showing lack of benefit. Adaptive trial designs, such as the ongoing PEDIGREE trial, tailor post-induction therapy based on early responses to combination treatment (nivolumab and ipilimumab), allowing for the potential early addition of cabozantinib, aiming to improve outcomes through a more personalized approach [89]. Additionally, the PROBE trial (NCT04510597) is evaluating the role of cytoreductive nephrectomy in patients with metastatic disease receiving IO-based systemic therapy, the results of which are eagerly anticipated [90].
8. Conclusions
Immunotherapy has revolutionized the treatment landscape of RCC, offering durable responses and improved survival outcomes, particularly in advanced and metastatic settings. While checkpoint inhibitors and combination regimens have become integral to standard care, challenges such as resistance, immune evasion, and tumor heterogeneity continue to limit long-term efficacy. Ongoing research into novel immune targets, personalized vaccines, CAR-T therapies, and modulation of the tumor microenvironment holds promise for enhancing and extending the benefits of immunotherapy. Continued innovation and a deeper understanding of RCC immunobiology will be key to developing more effective, personalized, and durable treatment strategies in the future.
Author Contributions
Conceptualization, S.P. and W.P.S.; methodology, S.P.; software, A.D.; validation, S.P., A.D., .; formal analysis, S.P.; investigation, S.P.; resources, S.P and A.D.; data curation, S.P and A.D.; writing—original draft preparation, S.P.; A.D and N.M writing—review and editing, S.P , A.D, and W.P.S.; visualization, S.P.; supervision, W.P.S.; project administration, S.P. W.P.S ; funding acquisition, W.P.S. All authors have read and agreed to the published version of the manuscript.
Funding
“This research received no external funding” or “This research was funded by William Paul Skelton IV and “The APC was funded byWilliam Paul Skelton IV and MDPI as it is an invited review article”.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Ethics Statement
This study did not involve experiments on humans or animals. For any retrospective data reviewed, all patient information was de-identified, and the study was conducted in accordance with institutional guidelines and the Declaration of Helsinki. Ethical approval was not required for this analysis.
References
- Rini, B.I.; Campbell, S.C.; Escudier, B. Renal cell carcinoma. The Lancet. 2009, 373, 1119–32. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA Cancer J Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Woolf, N. Pathology: Basic and Systemic. London, UK: W.B. Saunders; 1998. 699–702 p.
- Lara, PN, Jonasch E, editors. Kidney Cancer: Principles and Practice [Internet]. Berlin, Heidelberg: Springer Berlin Heidelberg; 2012 [cited 2025 Apr 3]. Available from: https://link.springer.com/10.1007/978-3-642-21858-3.
- Cheville, J.C.; Lohse, C.M.; Zincke, H.; Weaver, A.L.; Blute, M.L. Comparisons of Outcome and Prognostic Features Among Histologic Subtypes of Renal Cell Carcinoma: Am J Surg Pathol. 2003, 27, 612–24.
- Banasik, J.L. Pathophysiology. 7th ed. St. Louis: Elsevier; 2021.
- Athanazio, D.A.; Amorim, L.S.; Da Cunha, I.W.; Leite, K.R.M.; Da Paz, A.R.; De Paula Xavier Gomes, R.; et al. Classification of renal cell tumors – current concepts and use of ancillary tests: recommendations of the Brazilian Society of Pathology. Surg Exp Pathol. 2021, 4, 4. [Google Scholar] [CrossRef]
- Cheville, J.C.; Lohse, C.M.; Zincke, H.; Weaver, A.L.; Leibovich, B.C.; Frank, I.; et al. Sarcomatoid Renal Cell Carcinoma: An Examination of Underlying Histologic Subtype and an Analysis of Associations With Patient Outcome. Am J Surg Pathol. 2004, 28, 435–41. [Google Scholar] [CrossRef]
- Ross, K.; Jones, R.J. Immune checkpoint inhibitors in renal cell carcinoma. Clin Sci Lond Engl 1979, 131, 2627–42. [Google Scholar] [CrossRef] [PubMed]
- SEERProgram, S.E.E.R. 2024 [cited 2025 May 11]. Kidney and Renal Pelvis Cancer- Cancer Stat Facts. Available from: https://seer.cancer.gov/statfacts/html/kidrp.html.
- Moreira, M.; Pobel, C.; Epaillard, N.; Simonaggio, A.; Oudard, S.; Vano, Y.A. Resistance to cancer immunotherapy in metastatic renal cell carcinoma. Cancer Drug Resist Alhambra Calif. 2020, 3, 454–71. [Google Scholar]
- Beksac, A.T.; Paulucci, D.J.; Blum, K.A.; Yadav, S.S.; Sfakianos, J.P.; Badani, K.K. Heterogeneity in renal cell carcinoma. Urol Oncol Semin Orig Investig. 2017, 35, 507–15. [Google Scholar] [CrossRef] [PubMed]
- Sammarco, E.; Manfredi, F.; Nuzzo, A.; Ferrari, M.; Bonato, A.; Salfi, A.; et al. Immune Checkpoint Inhibitor Rechallenge in Renal Cell Carcinoma: Current Evidence and Future Directions. Cancers. 2023, 15, 3172. [Google Scholar] [CrossRef] [PubMed]
- Akdis, M.; Akdis, C.A. Mechanisms of allergen-specific immunotherapy: Multiple suppressor factors at work in immune tolerance to allergens. J Allergy Clin Immunol. 2014, 133, 621–31. [Google Scholar] [CrossRef]
- Akdis, C.A.; Akdis, M. Mechanisms of allergen-specific immunotherapy. J Allergy Clin Immunol. 2011, 127, 18–27. [Google Scholar] [CrossRef]
- Till, S.J.; Francis, J.N.; Nouri-Aria, K.; Durham, S.R. Mechanisms of immunotherapy. J Allergy Clin Immunol. 2004, 113, 1025–34. [Google Scholar] [CrossRef] [PubMed]
- Głobińska, A.; Boonpiyathad, T.; Satitsuksanoa, P.; Kleuskens, M.; Van De Veen, W.; Sokolowska, M.; et al. Mechanisms of allergen-specific immunotherapy. Ann Allergy Asthma Immunol. 2018, 121, 306–12. [Google Scholar] [CrossRef]
- Barbari, C.; Fontaine, T.; Parajuli, P.; Lamichhane, N.; Jakubski, S.; Lamichhane, P.; et al. Immunotherapies and Combination Strategies for Immuno-Oncology. Int J Mol Sci. 2020, 21, 5009. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Hu, T.; Zhou, J.; Gu, X.; Chen, S.; Qi, Q.; et al. Overview of tumor immunotherapy based on approved drugs. Life Sci. 2024, 340, 122419. [Google Scholar] [CrossRef]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol. 2020, 20, 651–68. [Google Scholar] [CrossRef] [PubMed]
- Monjaras-Avila, C.U.; Lorenzo-Leal, A.C.; Luque-Badillo, A.C.; D’Costa, N.; Chavez-Muñoz, C.; Bach, H. The Tumor Immune Microenvironment in Clear Cell Renal Cell Carcinoma. Int J Mol Sci. 2023, 24, 7946. [Google Scholar] [CrossRef]
- Sharma, P.; Goswami, S.; Raychaudhuri, D.; Siddiqui, B.A.; Singh, P.; Nagarajan, A.; et al. Immune checkpoint therapy—current perspectives and future directions. Cell. 2023, 186, 1652–69. [Google Scholar] [CrossRef]
- Motzer, R.J.; Escudier, B.; McDermott, D.F.; George, S.; Hammers, H.J.; Srinivas, S.; et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015, 373, 1803–13. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019, 380, 1116–27. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2021, 384, 829–41. [Google Scholar] [CrossRef]
- Braun, D.A.; Moranzoni, G.; Chea, V.; McGregor, B.A.; Blass, E.; Tu, C.R.; et al. A neoantigen vaccine generates antitumour immunity in renal cell carcinoma. Nature. 2025, 639, 474–82. [Google Scholar] [CrossRef] [PubMed]
- Andrews, L.P.; Yano, H.; Vignali, D.A.A. Inhibitory receptors and ligands beyond PD-1, PD-L1 and CTLA-4: breakthroughs or backups. Nat Immunol. 2019, 20, 1425–34. [Google Scholar] [CrossRef]
- Janiszewska, A.D.; Poletajew, S.; Wasiutyński, A. Reviews Spontaneous regression of renal cell carcinoma. Współczesna Onkol. 2013, 2, 123–7. [Google Scholar] [CrossRef]
- Maruschke, M. Spontaneous regression of renal cell carcinoma: Reality or myth? World J Clin Urol. 2014, 3, 201. [Google Scholar] [CrossRef]
- Fyfe, G.; Fisher, R.I.; Rosenberg, S.A.; Sznol, M.; Parkinson, D.R.; Louie, A.C. Results of treatment of 255 patients with metastatic renal cell carcinoma who received high-dose recombinant interleukin-2 therapy. J Clin Oncol. 1995, 13, 688–96. [Google Scholar] [CrossRef]
- Grimm, E.A.; Mazumder, A.; Zhang, H.Z.; Rosenberg, S.A. Lymphokine-activated killer cell phenomenon. Lysis of natural killer-resistant fresh solid tumor cells by interleukin 2-activated autologous human peripheral blood lymphocytes. J Exp Med. 1982, 155, 1823–41. [Google Scholar]
- West, E.J.; Scott, K.J.; Jennings, V.A.; Melcher, A.A. Immune activation by combination human lymphokine-activated killer and dendritic cell therapy. Br J Cancer. 2011, 105, 787–95. [Google Scholar] [CrossRef] [PubMed]
- Interferon-alpha and survival in metastatic renal carcinoma: early results of a randomised controlled trial. Medical Research Council Renal Cancer Collaborators. Lancet Lond Engl. 1999, 353, 14–7.
- Motzer, R.J.; Hutson, T.E.; Tomczak, P.; Michaelson, M.D.; Bukowski, R.M.; Rixe, O.; et al. Sunitinib versus Interferon Alfa in Metastatic Renal-Cell Carcinoma. N Engl J Med. 2007, 356, 115–24. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med. 2018, 378, 1277–90. [Google Scholar] [CrossRef]
- Tannir, N.M.; Escudier, B.; McDermott, D.F.; Burotto, M.; Choueiri, T.K.; Hammers, H.J.; et al. Nivolumab plus ipilimumab (NIVO+IPI) vs sunitinib (SUN) for first-line treatment of advanced renal cell carcinoma (aRCC): Long-term follow-up data from the phase 3 CheckMate 214 trial. J Clin Oncol. 2024, 42, 363–363. [Google Scholar] [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Waddell, T.; Nosov, D.; et al. Pembrolizumab plus axitinib versus sunitinib as first-line therapy for advanced clear cell renal cell carcinoma: 5-year analysis of KEYNOTE-426. J Clin Oncol. 2023, 41, LBA4501–LBA4501. [Google Scholar] [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. N Engl J Med. 2021, 384, 1289–300. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. N Engl J Med. 2019, 380, 1103–15. [Google Scholar] [CrossRef]
- National Comprehensive Cancer Network. NCCN.org. 2024. NCCN Clinical Practice Guidelines in Oncology: Kidney Cancer, v2.2024. Available from: https://www.nccn.org/professionals/physician_gls/pdf/kidney.pdf.
- Choueiri, T.K.; Tomczak, P.; Park, S.H.; Venugopal, B.; Ferguson, T.; Symeonides, S.N.; et al. Overall Survival with Adjuvant Pembrolizumab in Renal-Cell Carcinoma. N Engl J Med. 2024, 390, 1359–71. [Google Scholar] [CrossRef] [PubMed]
- Ravaud, A.; Motzer, R.J.; Pandha, H.S.; George, D.J.; Pantuck, A.J.; Patel, A.; et al. Adjuvant Sunitinib in High-Risk Renal-Cell Carcinoma after Nephrectomy. N Engl J Med. 2016, 375, 2246–54. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Uzzo, R.; Karam, J.A.; Master, V.A.; Donskov, F.; Suarez, C.; et al. Adjuvant atezolizumab versus placebo for patients with renal cell carcinoma at increased risk of recurrence following resection (IMmotion010): a multicentre, randomised, double-blind, phase 3 trial. The Lancet. 2022, 400, 1103–16. [Google Scholar] [CrossRef] [PubMed]
- Motzer, R.J.; Bex, A.; Russo, P.; Tomita, Y.; Cutuli, H.J.; Rojas, C.; et al. Adjuvant Nivolumab for Localized Renal Cell Carcinoma at High Risk of Recurrence After Nephrectomy: Part B of the Randomized, Placebo-Controlled, Phase III CheckMate 914 Trial. J Clin Oncol. 2025, 43, 189–200. [Google Scholar] [CrossRef]
- Gross-Goupil, M.; Kwon, T.G.; Eto, M.; Ye, D.; Miyake, H.; Seo, S.I.; et al. Axitinib versus placebo as an adjuvant treatment of renal cell carcinoma: results from the phase III, randomized ATLAS trial. Ann Oncol. 2018, 29, 2371–8. [Google Scholar] [CrossRef]
- Heng, D.Y.C.; Xie, W.; Regan, M.M.; Warren, M.A.; Golshayan, A.R.; Sahi, C.; et al. Prognostic Factors for Overall Survival in Patients With Metastatic Renal Cell Carcinoma Treated With Vascular Endothelial Growth Factor–Targeted Agents: Results From a Large, Multicenter Study. J Clin Oncol. 2009, 27, 5794–9. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Lacchetti, C.; Schneider, B.J.; Atkins, M.B.; Brassil, K.J.; Caterino, J.M.; et al. Management of Immune-Related Adverse Events in Patients Treated With Immune Checkpoint Inhibitor Therapy: American Society of Clinical Oncology Clinical Practice Guideline. J Clin Oncol. 2018, 36, 1714–68. [Google Scholar] [CrossRef]
- Motzer, R.J.; Hutson, T.E.; Glen, H.; Michaelson, M.D.; Molina, A.; Eisen, T.; et al. Lenvatinib, everolimus, and the combination in patients with metastatic renal cell carcinoma: a randomised, phase 2, open-label, multicentre trial. Lancet Oncol. 2015, 16, 1473–82. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Escudier, B.; Powles, T.; Mainwaring, P.N.; Rini, B.I.; Donskov, F.; et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med. 2015, 373, 1814–23. [Google Scholar] [CrossRef]
- Rini, B.I.; Pal, S.K.; Escudier, B.J.; Atkins, M.B.; Hutson, T.E.; Porta, C.; et al. Tivozanib versus sorafenib in patients with advanced renal cell carcinoma (TIVO-3): a phase 3, multicentre, randomised, controlled, open-label study. Lancet Oncol. 2020, 21, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Rini, B.I.; Wilding, G.; Hudes, G.; Stadler, W.M.; Kim, S.; Tarazi, J.; et al. Phase II Study of Axitinib in Sorafenib-Refractory Metastatic Renal Cell Carcinoma. J Clin Oncol. 2009, 27, 4462–8. [Google Scholar] [CrossRef]
- Jonasch, E.; Donskov, F.; Iliopoulos, O.; Rathmell, W.K.; Narayan, V.K.; Maughan, B.L.; et al. Belzutifan for Renal Cell Carcinoma in von Hippel–Lindau Disease. N Engl J Med. 2021, 385, 2036–46. [Google Scholar] [CrossRef] [PubMed]
- Choueiri, T.K.; Powles, T.; Peltola, K.; De Velasco, G.; Burotto, M.; Suarez, C.; et al. Belzutifan versus Everolimus for Advanced Renal-Cell Carcinoma. N Engl J Med. 2024, 391, 710–21. [Google Scholar] [CrossRef] [PubMed]
- Said, S.S.; Ibrahim, W.N. Cancer Resistance to Immunotherapy: Comprehensive Insights with Future Perspectives. Pharmaceutics. 2023, 15, 1143. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology Meets Immunology: The Cancer-Immunity Cycle. Immunity. 2013, 39, 1–10. [Google Scholar] [CrossRef]
- Makhov, P.; Joshi, S.; Ghatalia, P.; Kutikov, A.; Uzzo, R.G.; Kolenko, V.M. Resistance to Systemic Therapies in Clear Cell Renal Cell Carcinoma: Mechanisms and Management Strategies. Mol Cancer Ther. 2018, 17, 1355–64. [Google Scholar] [CrossRef]
- Binnewies, M.; Roberts, E.W.; Kersten, K.; Chan, V.; Fearon, D.F.; Merad, M.; et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med. 2018, 24, 541–50. [Google Scholar] [CrossRef] [PubMed]
- Topalian, S.L.; Drake, C.G.; Pardoll, D.M. Immune Checkpoint Blockade: A Common Denominator Approach to Cancer Therapy. Cancer Cell. 2015, 27, 450–61. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Hu-Lieskovan, S.; Wargo, J.A.; Ribas, A. Primary, Adaptive, and Acquired Resistance to Cancer Immunotherapy. Cell. 2017, 168, 707–23. [Google Scholar] [CrossRef] [PubMed]
- Sprent, J. Antigen-Presenting Cells: Professionals and amateurs. Curr Biol. 1995, 5, 1095–7. [Google Scholar] [CrossRef]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science. 2015, 348, 69–74. [Google Scholar] [CrossRef]
- Harryvan, T.J.; De Lange, S.; Hawinkels, L.J.A.C.; Verdegaal, E.M.E. The ABCs of Antigen Presentation by Stromal Non-Professional Antigen-Presenting Cells. Int J Mol Sci. 2021, 23, 137. [Google Scholar] [CrossRef]
- Lin, B.; Du, L.; Li, H.; Zhu, X.; Cui, L.; Li, X. Tumor-infiltrating lymphocytes: Warriors fight against tumors powerfully. Biomed Pharmacother. 2020, 132, 110873. [Google Scholar] [CrossRef]
- Malek, T.R.; Castro, I. Interleukin-2 Receptor Signaling: At the Interface between Tolerance and Immunity. Immunity. 2010, 33, 153–65. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.L.; Flavell, R.A. TRANSFORMING GROWTH FACTOR-β REGULATION OF IMMUNE RESPONSES. Annu Rev Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Tokunaga, R.; Zhang, W.; Naseem, M.; Puccini, A.; Berger, M.D.; Soni, S.; et al. CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation – A target for novel cancer therapy. Cancer Treat Rev. 2018, 63, 40–7. [Google Scholar] [CrossRef]
- Xu, M.; Wang, Y.; Xia, R.; Wei, Y.; Wei, X. Role of the CCL2-CCR2 signalling axis in cancer: Mechanisms and therapeutic targeting. Cell Prolif. 2021, 54, e13115. [Google Scholar] [CrossRef]
- David, J.; Dominguez, C.; Hamilton, D.; Palena, C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines. 2016, 4, 22. [Google Scholar] [CrossRef] [PubMed]
- Oberholtzer, N.; Quinn, K.M.; Chakraborty, P.; Mehrotra, S. New Developments in T Cell Immunometabolism and Implications for Cancer Immunotherapy. Cells. 2022, 11, 708. [Google Scholar] [CrossRef]
- Aguiar, R.B.D.; Moraes, J.Z.D. Exploring the Immunological Mechanisms Underlying the Anti-vascular Endothelial Growth Factor Activity in Tumors. Front Immunol. 2019, 10, 1023. [Google Scholar] [CrossRef]
- Al-Fahdawi, M.Q.; Al-Doghachi, F.A.J.; Abdullah, Q.K.; Hammad, R.T.; Rasedee, A.; Ibrahim, W.N.; et al. Oxidative stress cytotoxicity induced by platinum-doped magnesia nanoparticles in cancer cells. Biomed Pharmacother. 2021, 138, 111483. [Google Scholar] [CrossRef]
- Hu, K.; Babapoor-Farrokhran, S.; Rodrigues, M.; Deshpande, M.; Puchner, B.; Kashiwabuchi, F.; et al. Hypoxia-inducible factor 1 upregulation of both VEGF and ANGPTL4 is required to promote the angiogenic phenotype in uveal melanoma. Oncotarget. 2016, 7, 7816–28. [Google Scholar] [CrossRef]
- Chen, W.; Shen, L.; Jiang, J.; Zhang, L.; Zhang, Z.; Pan, J.; et al. Antiangiogenic therapy reverses the immunosuppressive breast cancer microenvironment. Biomark Res. 2021, 9, 59. [Google Scholar] [CrossRef] [PubMed]
- Harjunpää, H.; Llort Asens, M.; Guenther, C.; Fagerholm, S.C. Cell Adhesion Molecules and Their Roles and Regulation in the Immune and Tumor Microenvironment. Front Immunol. 2019, 10, 1078. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, L.C.; Ikenberg, K.; Cetintas, T.; Kapaklikaya, K.; Hutmacher, C.; Detmar, M. Tumor-Associated Lymphatic Vessels Upregulate PDL1 to Inhibit T-Cell Activation. Front Immunol [Internet]. 2017 Feb 3 [cited 2025 Apr 3];8. Available from: http://journal.frontiersin.org/article/10.3389/fimmu.2017.00066/full.
- Bai, R.; Chen, N.; Li, L.; Du, N.; Bai, L.; Lv, Z.; et al. Mechanisms of Cancer Resistance to Immunotherapy. Front Oncol. 2020, 10, 1290. [Google Scholar] [CrossRef]
- Vigano, S.; Alatzoglou, D.; Irving, M.; Ménétrier-Caux, C.; Caux, C.; Romero, P.; et al. Targeting Adenosine in Cancer Immunotherapy to Enhance T-Cell Function. Front Immunol. 2019, 10, 925. [Google Scholar] [CrossRef]
- Lamplugh, Z.; Fan, Y. Vascular Microenvironment, Tumor Immunity and Immunotherapy. Front Immunol. 2021, 12, 811485. [Google Scholar] [CrossRef]
- Takamatsu, K.; Tanaka, N.; Matsumoto, K.; Kosaka, T.; Mizuno, R.; Oya, M. Uncovering LAG-3 related tumor immunology in renal cell carcinoma and pan-cancer evaluation. J Clin Oncol. 2023, 41, e16517–e16517. [Google Scholar] [CrossRef]
- Chocarro, L.; Bocanegra, A.; Blanco, E.; Fernández-Rubio, L.; Arasanz, H.; Echaide, M.; et al. Cutting-Edge: Preclinical and Clinical Development of the First Approved Lag-3 Inhibitor. Cells. 2022, 11, 2351. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, K.; Tanaka, N.; Hakozaki, K.; Takahashi, R.; Teranishi, Y.; Murakami, T.; et al. Profiling the inhibitory receptors LAG-3, TIM-3, and TIGIT in renal cell carcinoma reveals malignancy. Nat Commun. 2021, 12, 5547. [Google Scholar] [CrossRef]
- Kaptein, P.; Slingerland, N.; Metoikidou, C.; Prinz, F.; Brokamp, S.; Machuca-Ostos, M.; et al. CD8-Targeted IL2 Unleashes Tumor-Specific Immunity in Human Cancer Tissue by Reviving the Dysfunctional T-cell Pool. Cancer Discov. 2024, 14, 1226–51. [Google Scholar] [CrossRef]
- Schoenfeld, D.A.; Djureinovic, D.; Su, D.G.; Zhang, L.; Lu, B.Y.; Kamga, L.; et al. Decoy-resistant IL-18 reshapes the tumor microenvironment and enhances rejection by anti–CTLA-4 in renal cell carcinoma. JCI Insight. 2025, 10, e184545. [Google Scholar] [CrossRef]
- Pal, S.K.; Tran, B.; Haanen, J.B.A.G.; Hurwitz, M.E.; Sacher, A.; Tannir, N.M.; et al. CD70-Targeted Allogeneic CAR T-Cell Therapy for Advanced Clear Cell Renal Cell Carcinoma. Cancer Discov. 2024, 14, 1176–89. [Google Scholar] [CrossRef] [PubMed]
- Furlow, B. CAR T-cell therapy ALL-316 targets CD70 in clear cell renal cell carcinoma (ccRCC) [Internet]. Available from: https://www.cancertherapyadvisor.com/reports/car-t-cell-therapy-allo-316-ccrcc/.
- Dizman, N.; Meza, L.; Bergerot, P.; Alcantara, M.; Dorff, T.; Lyou, Y.; et al. Nivolumab plus ipilimumab with or without live bacterial supplementation in metastatic renal cell carcinoma: a randomized phase 1 trial. Nat Med. 2022, 28, 704–12. [Google Scholar] [CrossRef] [PubMed]
- Pal, S.K.; Albiges, L.; Tomczak, P.; Suárez, C.; Voss, M.H.; De Velasco, G.; et al. Atezolizumab plus cabozantinib versus cabozantinib monotherapy for patients with renal cell carcinoma after progression with previous immune checkpoint inhibitor treatment (CONTACT-03): a multicentre, randomised, open-label, phase 3 trial. The Lancet. 2023, 402, 185–95. [Google Scholar] [CrossRef]
- Choueiri, T.K.; Albiges, L.; Barthélémy, P.; Iacovelli, R.; Emambux, S.; Molina-Cerrillo, J.; et al. Tivozanib plus nivolumab versus tivozanib monotherapy in patients with renal cell carcinoma following an immune checkpoint inhibitor: results of the phase 3 TiNivo-2 Study. The Lancet. 2024, 404, 1309–20. [Google Scholar] [CrossRef]
- Zhang, T.; Ballman, K.V.; McGregor, B.A.; Moon, H.; Matrana, M.R.; Alter, R.S.; et al. Ipilimumab and nivolumab in patients with metastatic clear cell renal cell carcinoma (mccRCC) treated on the phase 3 PDIGREE (Alliance A031704) trial: Results from Step 1 analysis. J Clin Oncol. 2025, 43, 4516–4516. [Google Scholar] [CrossRef]
- Vaishampayan, U.N.; Tangen, C.; Tripathi, A.; Shuch, B.M.; Pal, S.K.; Barata, P.C.; et al. SWOG S1931 (PROBE): Phase III randomized trial of immune checkpoint inhibitor (ICI) combination regimen with or without cytoreductive nephrectomy (CN) in advanced renal cancer. J Clin Oncol. 2022, 40, TPS402–TPS402. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



