
Submitted:
27 July 2025
Posted:
28 July 2025
You are already at the latest version
Abstract
Diabetes insipidus (DI) in newborn is an extremely rare condition, simultaneously the age of presentation is a strong suggestion of genetic background of disease. The differ-ential diagnosis should include arginine vasopressin deficiency (AVD) and arginine vasopressin resistance (AVR). Some novel diagnostic tools such as copeptin evaluation and genetic tests are vital for the early diagnosis. Case report: We present the case of a 1-month-old boy with polyuria observed since birth. Laboratory tests showed persis-tent hypernatremia, elevated plasma and low urine osmolality. An attempt at oral administration of desmopressin had no effect, additionally copeptin level was in-creased. A genetic study (NGS of the AVP, AVPR2 and AQP2 genes) was taken and a new pathogenic variant in the AVPR2 gene (hemizygous c.157del) was detected. After the genetic test result was obtained, treatment with hydrochlorothiazide was started. The patient is now 3 months old, developing normally, weight and height are normal. Conclusions: A newborn with DI should be subjected to extensive multidisciplinary diagnostics including endocrine and renal causes. Copeptin evaluation and prompt genetic diagnosis allows for early diagnosis and implementation of appropriate treat-ment.
Keywords:
diabetes insipidus
; arginine vasopressin deficiency
; arginine vasopressin resistance
; neonate
; pathogenic variant
1. Introduction
Diabetes insipidus (DI) in a newborn is an extremely rare condition, simultaneously the age of presentation is a strong suggestion of genetic background of disease. Two diagnostic directions should be taken into account in such a case: arginine vasopressin deficiency (AVD) or arginine vasopressin resistance (AVR). Inherited AVD affects only 1% of AVD and the majority of cases is acquired condition. The frequency of hereditary AVR is estimated for 1 per 1,000,000 and the cases are associated with pathogenic variants of arginine vasopressin receptor 2, AVPR2 (90% of cases) or in aquaporin 2, AQP2 (10% of cases) [1]. A comparison between the frequency of AVD and AVR indicated, that in newborn statistically more probable is AVR than inherited AVD. Nevertheless the investigation always starts with the evaluation of the symptoms of DI and the response to the desmopressin (DDAVP).
Arginine vasopressin resistance (AVR), previously known as nephrogenic diabetes insipidus (NDI), is defined as a decreased sensitivity of kidneys to arginine vasopressin (AVP) resulting in an inability to concentrate urine. Congenital cases of AVR are less frequent than the acquired ones, which could be secondary to intake of certain drugs, such as lithium, as well to electrolyte disturbances (hypokalemia, hypercalcemia) and other renal or systemic diseases such as renal sarcoidosis, sickle cell anemia or polycystic kidney disease [2,3]. Primary congenital AVR is known to be caused by pathogenic variants of AVPR2 or AQP2 genes that encode the type 2 receptor to arginine vasopressin and aquaporin-2 protein, respectively. The loss-of-function mutations in the AVPR2 gene located on the X chromosome are believed to be responsible for about 90%-98% of hereditary AVR cases with an estimated incidence of approximately 8-8.8 per million male live births [3,4]. The remaining cases result from pathogenic variants of the AQP2 gene located on chromosome 12, which may be inherited in either an autosomal recessive or dominant manner depending on the variant. The dominant form is less common and typically associated with a milder clinical presentation [3]. Symptoms typically appear during the first year of life with a median age of 0.6 years, according to the study by Lopez-Garcia et al. [5]. These include polyuria and polydipsia, although they may be easily overlooked in infants. Additional symptoms more likely to alert parents are poor feeding, vomiting, fever, growth faltering, constipation, and visible signs of dehydration [4,5].
In this case report, we present a 1-month-old boy with symptomatic AVR caused by a previously not reported pathogenic variant of the AVPR2 gene.
2. Case Presentation
2.1. Patient Presentation and Clinical Assessment
We present a case of a male child of non-consanguineous, healthy parents, born from the first pregnancy, in the 36th week of gestation +3 days, by spontaneous delivery, assessed for 10 Apgar points. The pregnancy was complicated by gestational diabetes, well controlled with diet, and hypothyroidism, treated with L-thyroxin oral supplementation. The child’s birth weight was 2830 g, length 48 cm, and head circumference 33 cm. The boy was breastfed, sucked effectively, but constant weight loss was observed from birth. From the first days of life, parents noticed very abundant diaper wetting. On the 12th day of life, the body weight was 2590g, the estimated diuresis was 5-7 mL/kg/hour. Laboratory tests revealed hypernatremia of 158mmol/L, increased serum osmolality, and significantly reduced urine osmolality, without other urinary abnormalities. Capillary blood acid-base balance was normal (pH-7.34, BE (-3.1)). After intravenous rehydration, sodium concentration decreased to 147mmol/l, and serum osmolality decreased to values slightly above the norm (298 mOsm/kg H2O). The boy was discharged home, where he was breastfed on demand for 8 to 12 feedings a day; he was still gaining weight poorly, and he was soaking diapers profusely.
After one week of home observation, follow-up tests again revealed hypernatremia (154 mmol/L), hyperchloremia (120 mol/L), and normal potassium concentration (4.3 mmol/L). Capillary blood acid-base balance was again normal (pH-7.363, BE (-3.5)), plasma osmolality was substantially increased (311 mOsm/kg H2O), and urinalysis revealed specific gravity of 1002. The boy was admitted to the hospital, to pediatric endocrinology department. The general condition on admission was good; his blood pressure was 88/48 mm Hg. He presented with constant polyuria of 6-8 mL/kg/hour. Physical examination showed slight signs of dehydration (dry mucous membranes) and umbilical hernia; otherwise, there were no significant deviations. Laboratory tests showed: normal complete blood count, normal glycemia and liver and kidney function parameters, high serum osmolality (323 mOsm/kg H2O) with significantly reduced urinary osmolality (73 mOsm/kg H2O), and very high plasma renin (1001 mU/L) and aldosterone (206 ng/dL) concentrations. Imaging tests - transfontanel and abdominal ultrasound were normal. Magnetic resonance imaging of hypothalamo- pituitary region described a poorly separated posterior pituitary lobe. Pending the result of copeptin and the genetic test, suspecting AVD and hoping to reduce polyuria, the boy was started on oral desmopressin (lyophilisate) in increasing doses: in the following days, 3.125 µg (1/8 tablet a 25 µg, Noqturina, Ferring GmbH, Kiel, Germany), then 9.375 µg (3/8 tablet a 25 µg Noqturina) and 15 µg (1/4 tablet a 60 µg, Minirin melt, Ferring GmbH, Kiel, Germany) (freeze-dried freshly divided with a scalpel). Administration of desmopressin did not alter serum and urine osmolality, and increased diuresis persisted. At the first test, the boy had an extended pause in feedings to 3 hours, and during this time, the osmolality (values) of serum and sodium level (154 mmol/L) increased. After 10 days from the start of diagnostics, the result of copeptin was 271 pmol/l, which was collected at an osmolality of 310 mOsm/kg H2O (normal value: <28.2 pmol/l with an osmolality of 296-300 mOsm/kg H2O, according to adult reference producer’s values). The increased copeptin concentration and no effect after administration of desmopressin indicated renal insipidus. The genetic tests detected a potentially pathogenic variant in the AVPR2 gene.
Afterwards, the boy remains under nephrological care. He has been receiving hydrochlorothiazide in increasing doses since the age of 2 months (1mg/kg of body weight/day). The boy is fed exclusively with breast milk and supplemented with water, and he gains weight well. The fluid supply is about 200mL/kg/day; polyuria of 5-7 mL/kg/hour still persists. In follow-up examinations, sodium and potassium concentrations are normal (141 mmol/L and 5.0 mmol/L, respectively), as are serum osmolality, urine specific gravity remains low: 1003, similar to urine osmolality.
2.1. Genetic Diagnosis
METHODS
Genomic DNA of the patient and his mother was extracted from whole blood using NucleoSpin Dx Blood (Macherey-Nagel). Purity of DNA was checked using spectrophotometric method on Nanodrop (Thermo Fisher Scientific). The DNA was quantified using fluorometric-based method on Quantus with QuantiFluor ONE dsDNA System (Promega). Integrity of genomic DNA was determined with Genomic DNA Reagents Analysis Kit on TapeStation System (Agilent). Library of the proband DNA was prepared with AmpliSeq for Illumina On-Demand Panel Custom (Illumina) and the testing was performed with Next Generation Sequencing method on the MiSeq System (Illumina).Detected variant was verified with Sanger sequencing (Figure 1) and analyzed with Mutation Surveyor software (Softgenetics).
GENETIC ANALYSIS
The analysis revealed a hemizygous variant in the AVPR2 gene (NM_000054.7:c.157del (p.Leu53Ter)) in the boy and a heterozygous variant in the patient’s mother. The detected variant is not registered in the dbSNP, ClinVar, HGMD-Public or other available databases containing information about mutations in the human genome. There are no information about population variant frequency.
OMIM database indicates that mutations in the AVPR2 gene (OMIM *300538) are connected to X-linked nephrogenic diabetes insipidus, NDI1 (OMIM #304800). Loss of function of the AVPR2 gene is known mechanism of the disease. Detected variant id located in 2 of 3 exons coding the gene, in the transmembrane domain. The expected effect of the change is leucine at position 53 to a STOP codon and shortening of the protein. It is predicted that the transcript with a premature STOP codon will be eliminated by NMD (Nonsense-Mediated mRNA Decay).
Clinical interpretation of the variant using a classification of the American College of Medical Genetics and Genomics guidelines is likely pathogenic (PVS1, PM2).
The gene AVPR2 encodes the vasopressin receptor, type 2, also known as the V2 receptor, which belongs to the seven-transmembrane-domain G protein-coupled receptor (GPCR) superfamily, and couples to Gs thus stimulating adenylate cyclase. The AVPR2 gene (located on Xq28) encodes a protein of 371 amino acids protein characterized by six transmembrane domains connected by five loops and intracellular N- and C- termini (Figure 2). When the function of this gene is lost, the disease Nephrogenic Diabetes Insipidus (NDI) results, whereas gain-of-function mutations cause nephrogenic syndrome of inappropriate antidiuresis (NSIAD; https://doi.org/10.1016/bs.vh.2019.08.012) . The V2 receptor is expressed in the kidney tubule, predominantly in the distal convoluted tubule and collecting ducts, where its primary property is to respond to the pituitary hormone arginine vasopressin (AVP) by stimulating mechanisms that concentrate the urine and maintain water homeostasis in the organism. X-linked NDI associated with variants in the AVPR2 gene mostly affects males, with variable penetrance in heterozygous women, depending on the presence of inactivated X bias.
3. Discussion
We presented the case of a boy who had symptoms of diabetes insipidus (polyuria, hypernatremia, high plasma osmolality with low urine osmolality) since birth. Molecular studies revealed a new, previously undescribed pathogenic variant in the AVPR2 gene, producing an inactive protein responsible for the child’s phenotype. Efficient cooperation between endocrinologists, nephrologists, and geneticists allowed for making an early diagnosis and initiating treatment.
Until 2022, the traditional terms for central diabetes insipidus caused by vasopressin deficiency and nephrogenic diabetes insipidus, resulting from a lack of sensitivity of the renal tubules to vasopressin, were used. In 2022, the Working Group for Renaming Diabetes Insipidus changed the names of these diseases to arginine vasopressin deficiency (AVD) and arginine vasopressin resistance (AVR) to name these two entities according to their true pathophysiologic mechanism [6].
Children with AVR usually have a normal pregnancy, and polyuria and consequently polyhydramnios are not typical symptoms of the disease [7]. According to large registries, the first symptoms usually become evident later than in our patient, around 3-4 months of age [5,8]. Although infants are generally more prone to dehydration, in the case of children with AVR, life-threatening dehydration usually occurs only after switching to solid foods, due to their greater osmotic load compared to mother’s milk or milk-replacing formulas. It is worth emphasizing that our patient presented severe polyuria with typical biochemical disorders from the first days of life.
Our patient presented typical biochemical disorders: high plasma osmolality caused by hypernatremia, simultaneous low (always lower than plasma!) urine osmolality, and polyuria. No other biochemical disorders were found in the patient that could have suggested other tubulopathies (e.g., renal tubular acidosis or Bartter syndrome); potassium, calcium, and magnesium levels were normal, and the patient had normal calcium-phosphate metabolism [7,9].
According to the 2025 international expert consensus statement, the detection of inappropriately diluted urine (i.e., urinary osmolality <200 mOsm/kg H2O), combined with high–normal or elevated serum sodium, is pathognomonic for the diagnosis of diabetes insipidus (nephrogenic or central) and warrants early genetic testing [7]. Possible central, renal and other causes of polyuria in neonates and infants are presented in Table 1
Among patients with genetically determined AVR, 90% of patients have a pathogenic variant in the aquaporin receptor 2 gene (AVPR2) - the gene is located on the long arm of the X chromosome (Xq28) (OMIM #304800), hence symptoms occur in boys, though some women may also develop the disease as a consequence of X-inactivation of the chromosome carrying the normal variant. The remaining 10% have pathogenic variants in the AQP2 gene encoding aquaporin 2 - in these patients (of both sexes), there may be autosomal recessive (OMIM #222000) or dominant (OMIM #125800) inheritance, and symptoms may be milder than in the X-linked form [7,10]. Of note, many acquired conditions may cause renal tubule resistance to AVP. These conditions are primarily seen in older children and adults (Table 2).
Following the 2025 consensus, we performed a genetic test on the patient as soon as possible using the next-generation sequencing method for three genes: AVP, AVPR2, and AQP2 [7]. We found a new, previously undescribed pathogenic variant of the receptor 2 gene in the patient under examination. The variant (c.157del) causes shortening and complete inactivation of the protein. The complete inactivity of the receptor, in the absence of a second correct copy of the gene, explains the severe phenotype in the boy observed, unlike in most patients with AVR, from the first days of life [10].
The recommendations emphasize that other tests differentiating polyuria should be performed in the case of a long wait for a genetic test, the impossibility of performing it, or a negative result of genetic studies [7]. Copeptin is secreted together with AVP in equimolar amounts. In contrast to AVP, it is characterized by stability. The concentration of copeptin has already been assessed in various clinical settings, including children with nocturnal enuresis [17], primary hypertension [18], and chronic kidney disease [19]. In adults, the cut-off point is assumed to be 21.4 pmol/L, without such standards for children. In a patient with polyuria, the concentration of copeptin > 21.4 pmol/L indicates insensitivity of the tubule to vasopressin [20]. It is worth emphasizing that the concentration of copeptin in our patient was about ten times higher than the cut-off point, which is further evidence of a peripheral background of polyuria.
The tests assessing the hypothalamic-renal system are the water deprivation and DDAVP tests. DDAVP is a synthetic analogue of AVP that does not affect the V1 receptor. The drug has been widely used in the treatment of AVR and primary nocturnal enuresis for many years. The DDAVP test should be performed carefully, ensuring full control over the child’s condition. In the case of a normal tubular response to DDAVP, there may be sudden water retention and a rapid decrease in serum sodium, which may have adverse consequences for the central nervous system [7]. In our patient, while waiting for the genetic test result, a DDAVP test was performed. A commercially available oral preparation was administered in gradually increasing doses, in controlled, hospital conditions. We did not observe any side effects, the child’s condition did not change, and there was no effect on biochemical disorders, which is another confirmation of the diagnosis. We did not perform a water deprivation test for obvious reasons, such as patient safety.
Treating a child with AVR is difficult and requires very good cooperation with the family. The period of infancy and early childhood is particularly dangerous because the patient does not articulate his or her needs regarding fluids. In addition, due to the body’s higher physiological content of water (70% vs. 60%), dehydration occurs more easily at this age. In the youngest children, it is recommended to flush with water, and if intravenous hydration is necessary, unlike in the general population, a 5% glucose solution is recommended to avoid additional sodium load [21]. For infants, breast milk or a traditional formula milk substitute is recommended. According to literature data, about one-fourth of children with AVR may require tube feeding or gastrostomy in early childhood [5,8,22]. Our patient is gaining adequate weight on mixed feeding. We will closely monitor his further development.
In patients with persistent hypernatremia and a tendency to dehydration, also in infancy, it is recommended to include drugs that limit diuresis - thiazide/thiazide-like diuretics and cyclooxygenase inhibitors. The mechanism of action of thiazide diuretics is unclear. It is postulated that these drugs cause a small intravascular depletion, which causes increased reabsorption of sodium and water in the initial parts of the tubules, as a result of which less water enters the collecting tubules and less urine is produced. Patients treated with thiazide diuretics are at risk of developing biochemical complications: fluctuations in sodium concentration to hyponatremia, hypokalemia, hypercalcemia, and hyperuricemia. In our patient, we used hydrochlorothiazide as wafers in gradually increased doses starting from 0,5mg/kg/day. Non-pharmacological treatment together with pharmacological treatment allowed for the maintenance of normonatremia, proper hydration, and nutrition status in our patient despite persistent polyuria.
Large international registries assessing long-term prognosis in patients with AVR have shown that the most commonly used drugs in this group of patients are thiazide/thiazide-like diuretics and potassium-sparing diuretics (approx. 70% of patients), less frequently NSAIDs (less than 50% of patients). Over time, there is an improvement in height and weight expressed in SDS. Most adults have normal height, but an increased prevalence of obesity was noted. Urological complications (hydronephrosis, bladder dysfunction with residual urine volume) occur in approximately 40% of patients. Chronic kidney disease in stage 2 or higher was revealed in as many as 32% of children and half of the adults [5,8].
5. Conclusions
We presented a case of an infant with polyuria occurring from the first days of life and indicated that such a situation requires urgent medical intervention. First, serum sodium concentration, plasma osmolality, and urine should be assessed to confirm or exclude diabetes insipidus. At the next stage, distinguishing between arginine vasopressin resistance and deficiency is necessary. In this process the vital role play the copeptin evaluation and genetic tests. In the described boy, we detected a new pathogenic variant of the AVPR2 gene, resulting in early-onset AVR managed with a careful non-pharmacological and pharmacological treatment. Cooperation between specialist clinicians and the genetic laboratory is crucial for early diagnosis and implementation of proper treatment.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, A.K. and A.S.; methodology, D.C. and M.D.; software, A.L.; validation, A.K., P.S. and B.P.; formal analysis, P.S.; investigation, A.K.,B.P.; resources, A.K., P.S. A.S.; data curation, A.S., A.K..; writing—original draft preparation, P.S.,A.K.; writing—review and editing, A.K.; visualization, D.C., M.D.; supervision, A.K.; project administration, A.K.,P.S.; All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Bioethics Committee of Medical University of Warsaw (AKBE/218/25 date of approval 09.07.2025)
Informed Consent Statement
Informed consent was obtained from parents of the patient involved in the study. Written informed consent for publication has been obtained from parents.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Angelousi A, Alexandraki KI, Mytareli C, Grossman AB, Kaltsas G. New developments and concepts in the diagnosis and management of diabetes insipidus (AVP-deficiency and resistance). J Neuroendocrinol. 2023; 35(1):e13233. [CrossRef]
- Christ-Crain, M.; Bichet, D.G.; Fenske, W.K.; Goldman, M.B.; Rittig, S.; Verbalis, J.G. Verkman, A.S. Diabetes insipidus. Nat Rev Dis Primers. 2019, 5, 54.
- Arthus, M.F.; Lonergan, M.; Crumley, M.J.; Naumova, A.K.; Morin, D.; LA, D.E.M.; Kaplan, B.S.; Robertson, G.L.; Sasaki, S.; Morgan, K., et al. Report of 33 novel AVPR2 mutations and analysis of 117 families with X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2000, 11, 1044–1054.
- Hureaux, M.Vargas-Poussou, R. Genetic basis of nephrogenic diabetes insipidus. Mol Cell Endocrinol. 2023, 560, 111825.
- Lopez-Garcia, S.C.; Downie, M.L.; Kim, J.S.; Boyer, O.; Walsh, S.B.; Nijenhuis, T.; Papizh, S.; Yadav, P.; Reynolds, B.C.; Decramer, S., et al. Treatment and long-term outcome in primary nephrogenic diabetes insipidus. Nephrol Dial Transplant 2020.
- Arima, H.; Cheetham, T.; Christ-Crain, M.; Cooper, D.; Drummond, J.; Gurnell, M.; Levy, M.; McCormack, A.; Newell-Price, J.; Verbalis, J.G., et al. Changing the Name of Diabetes Insipidus: A Position Statement of the Working Group for Renaming Diabetes Insipidus. J Clin Endocrinol Metab. 2022, 108, 1–3.
- Levtchenko, E.; Ariceta, G.; Arguedas Flores, O.; Bichet, D.G.; Bockenhauer, D.; Emma, F.; Hoorn, E.J.; Koster-Kamphuis, L.; Nijenhuis, T.; Trepiccione, F., et al. International expert consensus statement on the diagnosis and management of congenital nephrogenic diabetes insipidus (arginine vasopressin resistance). Nat Rev Nephrol. 2025, 21, 83–96.
- D’Alessandri-Silva, C.; Carpenter, M.; Ayoob, R.; Barcia, J.; Chishti, A.; Constantinescu, A.; Dell, K.M.; Goodwin, J.; Hashmat, S.; Iragorri, S., et al. Diagnosis, Treatment, and Outcomes in Children With Congenital Nephrogenic Diabetes Insipidus: A Pediatric Nephrology Research Consortium Study. Front Pediatr. 2019, 7, 550.
- Duicu, C.; Pitea, A.M.; Săsăran, O.M.; Cozea, I.; Man, L. Bănescu, C. Nephrogenic diabetes insipidus in children (Review). Exp Ther Med. 2021, 22, 746.
- Makita, N.; Manaka, K.; Sato, J. Iiri, T. V2 vasopressin receptor mutations. Vitam Horm. 2020, 113, 79–99.
- Newell-Price, J.; Drummond, J.B.; Gurnell, M.; Levy, M.; McCormack, A.; Cooper, D.; Wass, J.; Christ-Crain, M. Verbalis, J.G. Approach to the Patient With Suspected Hypotonic Polyuria. J Clin Endocrinol Metab. 2025, 110, e506–e514.
- Lopes, A.; Campos, A.C.; Marques Simões, J. Jordão, A. Lithium-Induced Arginine Vasopressin Resistance (AVP-R): A Case of Chronic Exposure to Lithium. Cureus. 2023, 15, e41677. [Google Scholar]
- Williams, A.A.; Pir Muhammad, A.I.; Ruchi, R. Ali, R. Managing Ifosfamide-Induced Arginine Vasopressin Resistance: Diagnostic and Treatment Strategies. Cureus. 2025, 17, e81236. [Google Scholar] [PubMed]
- Wadehra, A.Ghandour, M. Foscarnet-Associated Nephrogenic Diabetes Insipidus. Am J Ther. 2022, 29, e750–e751.
- Krishnamurthy, A.; Bhattacharya, S.; Lathia, T.; Kantroo, V.; Kalra, S. Dutta, D. Anticancer Medications and Sodium Dysmetabolism. Eur Endocrinol. 2020, 16, 122–130. [Google Scholar] [PubMed]
- Downie, M.L.; Lopez Garcia, S.C.; Kleta, R. Bockenhauer, D. Inherited Tubulopathies of the Kidney: Insights from Genetics. Clin J Am Soc Nephrol. 2021, 16, 620–630. [Google Scholar] [PubMed]
- Szymanik-Grzelak, H.; Daniel, M.U.; Skrzypczyk, P.; Kotuła, I. Pańczyk-Tomaszewska, M. Is copeptin a reliable biomarker of primary monosymptomatic nocturnal enuresis? Cent Eur J Immunol. 2019, 44, 38–44. [Google Scholar] [PubMed]
- Tenderenda-Banasiuk, E.; Wasilewska, A.; Filonowicz, R.; Jakubowska, U. Waszkiewicz-Stojda, M. Serum copeptin levels in adolescents with primary hypertension. Pediatr Nephrol. 2014, 29, 423–429. [Google Scholar] [PubMed]
- Skrzypczyk, P.; Okarska-Napierała, M.; Górska, E.; Stelmaszczyk-Emmel, A. Pańczyk-Tomaszewska, M. [Copeptin in children with chronic kidney disease]. Pol Merkur Lekarski. 2018, 44, 165–170. [Google Scholar] [PubMed]
- Morgenthaler, N.G.; Struck, J.; Alonso, C. Bergmann, A. Assay for the measurement of copeptin, a stable peptide derived from the precursor of vasopressin. Clin Chem. 2006, 52, 112–119. [Google Scholar] [PubMed]
- Moritz, M.L. Ayus, J.C. Maintenance Intravenous Fluids in Acutely Ill Patients. N Engl J Med. 2015, 373, 1350–1360. [Google Scholar] [PubMed]
- Sharma, S.; Ashton, E.; Iancu, D.; Arthus, M.F.; Hayes, W.; Van’t Hoff, W.; Kleta, R.; Bichet, D.G. Bockenhauer, D. Long-term outcome in inherited nephrogenic diabetes insipidus. Clin Kidney J. 2019, 12, 180–187. [Google Scholar] [PubMed]
Figure 1.
Result of arginine vasopressin receptor 2 (AVPR2) sequencing. The sequence chromatogram of AVPR2 in the patient and mother.
Figure 1.
Result of arginine vasopressin receptor 2 (AVPR2) sequencing. The sequence chromatogram of AVPR2 in the patient and mother.
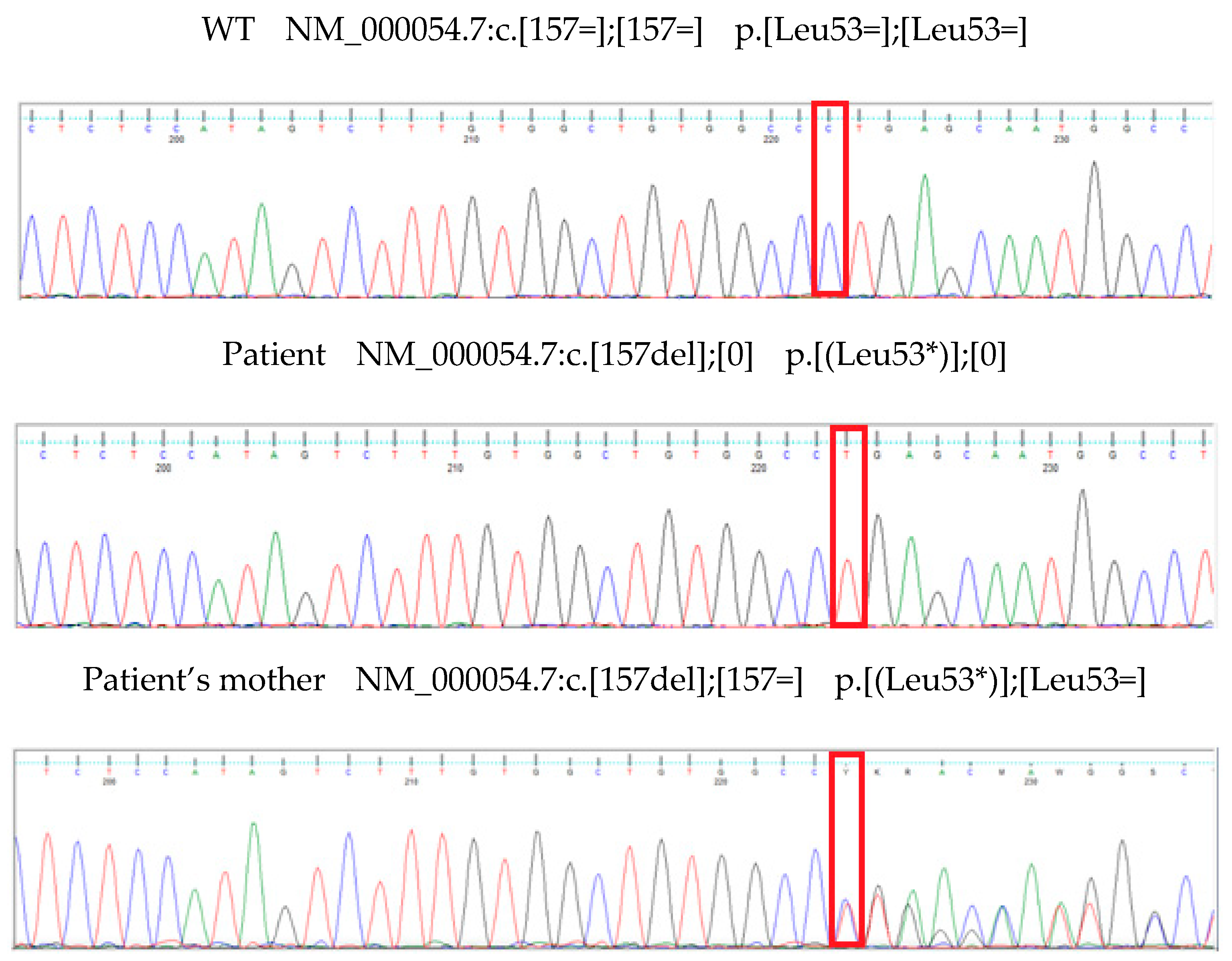
Figure 2.
Predicted structural model of arginine vasopressin receptor 2 (AVPR2) protein. The simulation performed using web tool I-TASSER Protein Structure & Functional Predictions (https://doi.org/10.1038/s41587-025-02654-4).
Figure 2.
Predicted structural model of arginine vasopressin receptor 2 (AVPR2) protein. The simulation performed using web tool I-TASSER Protein Structure & Functional Predictions (https://doi.org/10.1038/s41587-025-02654-4).
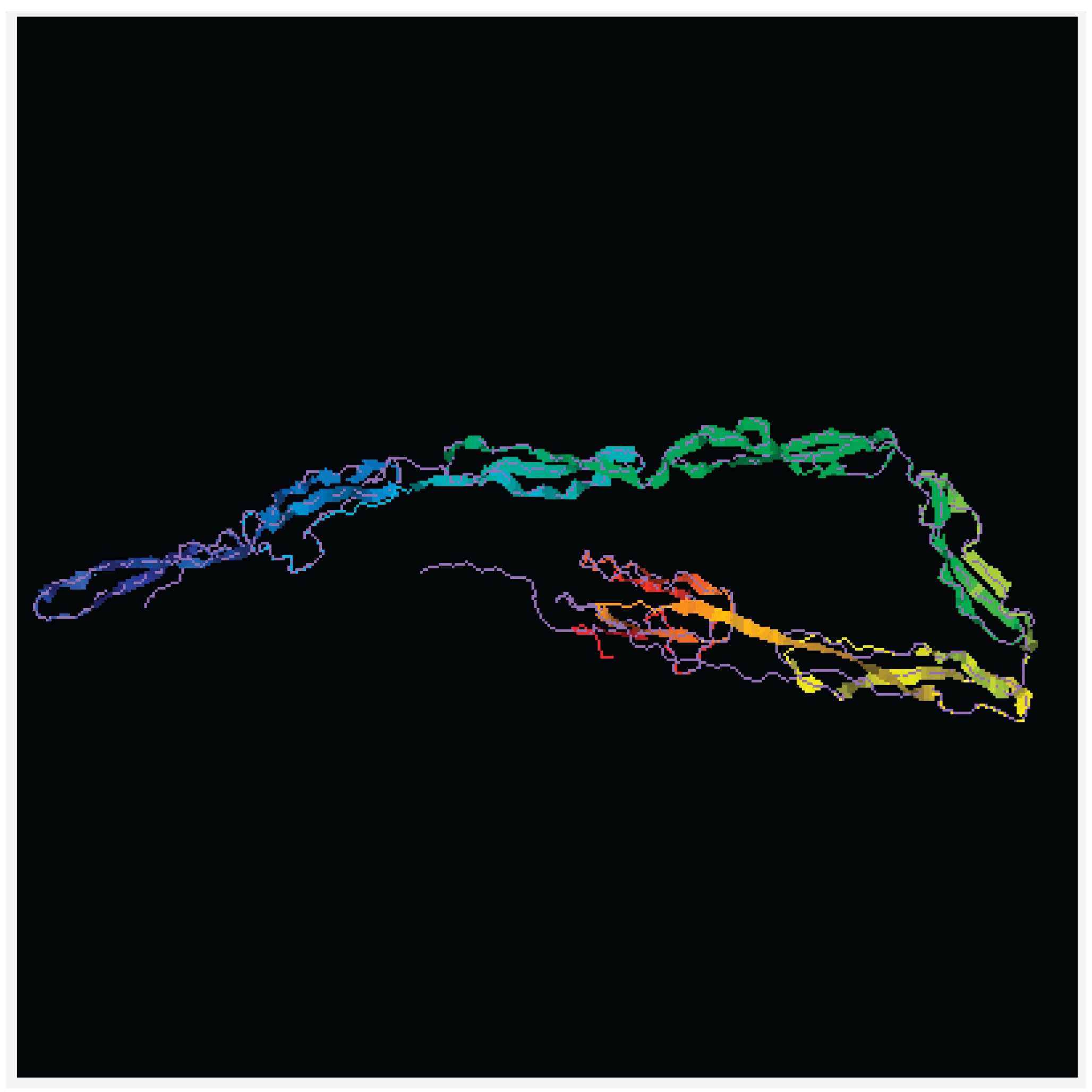

Table 1.
Central, renal and other possible causes of polyuria and polydipsia in infants and neonates.
Table 1.
Central, renal and other possible causes of polyuria and polydipsia in infants and neonates.
| Central etiology | Renal etiology | Other |
| Arginine vasopressin deficiency (formerly: central diabetes insipidus) | Arginine vasopressin resistance (formerly: nephrogenic diabetes insipidus) | Congenital adrenal hyperplasia with salt wasting |
| Cerebral salt wasting syndrome | Chronic kidney disease of different etiologies (especially congenital anomalies of the kidney and urinary tract, nephronophthisis, ciliopathies) | Diabetes mellitus |
| Bartter syndrome (especially types 1, 2, and 5) | Hypercalcemia (e.g., vitamin D intoxication, idiopathic infantile hypercalcemia) | |
| Congenital or acquired Fanconi syndrome | Hypokalemia (e.g., familial hyperaldosteronism) | |
| Distal renal tubular acidosis | ||
| Apparent mineralocorticoid excess | ||
Table 2.
Possible causes of arginine vasopressin resistance (AVR) according to [7,9,11,12,13,14,15,16].
| Congenital | Acquired |
|
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



