
Submitted:
21 July 2025
Posted:
24 July 2025
You are already at the latest version
Abstract
Rivaroxaban, a direct oral anticoagulant (DOAC), is a widely used drug for the prevention and treatment of thromboembolic disorders. While its fixed dosing regimen and predictable anticoagulation profile improve clinical usability, significant inter-individual variability in pharmacokinetics (PK) and pharmacodynamics (PD) exists, particularly in patients with altered physiology or comorbidities. This study established an open database of rivaroxaban PK and PD data from 14 clinical studies. The data were used to develop and validate a physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) model of rivaroxaban that captures its absorption, distribution, metabolism, excretion, and coagulation effects. The model incorporates the effects of dose, food intake, hepatic and renal function, and allows simulation of activated Factor X (Factor Xa) inhibition, prothrombin time (PT), and activated partial thromboplastin time (aPTT). Simulations accurately reproduced observed plasma concentration-time and coagulation profiles across a range of doses, fed/fasting conditions, as well as with different degrees of cirrhosis and renal impairment. This mechanistic PBPK/PD model provides a framework for exploring variability in rivaroxaban response across diverse clinical scenarios and supports its use in personalized dosing strategies, clinical trial design, and regulatory evaluation.
Keywords:
rivaroxaban
; direct oral anticoagulant
; DOAC
; PBPK/PD modeling
; pharmacokinetics
; pharmacodynamics
1. Introduction
Direct oral anticoagulants (DOACs) have transformed the prevention and treatment of thromboembolic disorders, offering predictable pharmacokinetics, and fewer drug–food interactions. One of the most widely prescribed DOACs is rivaroxaban (brand name: Xarelto), a selective, direct inhibitor of activated coagulation factor X (Factor Xa). By binding directly to factor Xa, rivaroxaban inhibits both its free and clot-bound form, reducing thrombin generation and fibrin clot formation [1,2]. Rivaroxaban is administered orally, which increases its efficacy and patient compliance, therefore eliminating the need for constant monitoring that is necessary with vitamin K antagonists (VKAs). These characteristics make rivaroxaban particularly effective for managing conditions that may require lifelong treatment, such as stroke consequences and systemic embolism in patients with non-valvular atrial fibrillation. It also prevents recurrent deep vein thrombosis or pulmonary embolism, especially in patients with permanent risk factors or unprovoked venous thromboembolism [3,4].
Rivaroxaban is rapidly absorbed after oral administration and normally reaches peak plasma concentrations within 2 to 4 hours. It exhibits high bioavailability, between 80% to 100% for a 10 mg dose, independent of food intake. Interestingly, the patient’s fasting state plays a more important role when increasing the dose. In particular, to achieve the same bioavailability of 80% to 100% for a 15 mg and 20 mg dose, it is necessary to take rivaroxaban with food [5,6,7]. On the contrary, the bioavailability for a dose of 15 mg to 20 mg can drop to around 66% if taken while fasting [8]. Rivaroxaban is highly protein-bound (fraction unbound is equal to 5–8%) and has a moderate volume of distribution (around 50 L). It is eliminated via two different pathways: approximately one-third is excreted unchanged by the kidneys, while the rest is metabolized in the liver via CYP3A4/A5 and CYP2J2, and the inactive metabolites are subsequently excreted through biliary and renal routes [9].
Two key laboratory assays used to evaluate coagulation are prothrombin time (PT) and activated partial thromboplastin time (aPTT), which assess the intrinsic and extrinsic coagulation pathways, respectively [10]. Both PT and aPTT are prolonged by rivaroxaban in a dose-dependent manner [11,12,13,14], reflecting its anticoagulant effect, which follows an Emax model of Factor Xa inhibition [15].
Despite its predictable pharmacologic profile, significant inter-individual variability in drug exposure remains a clinical concern. Factors such as renal and hepatic functional state can, for instance, influence rivaroxaban pharmacokinetics and pharmacodynamics. In particular, in patients with moderate hepatic impairment (Child-Pugh B), total body clearance of rivaroxaban is decreased, leading to an increase in systemic exposure. This is accompanied by enhanced pharmacodynamic effects, such as greater inhibition of Factor Xa, raising the risk of bleeding complications [16]. Renal impairment also affects rivaroxaban pharmacokinetics and pharmacodynamics as the drug is partially eliminated by the kidneys [17,18]. Therefore, reduced renal function leads to a higher plasma concentration and increased bleeding risk. It is recommended to adjust the dose in patients with moderate renal impairment (creatinine clearance 30–49 mL/min), and rivaroxaban should be used with caution in these individuals, especially if they are taking other medications that may further increase rivaroxaban levels (i.e. potent CYP3A4 and/or P-gp inhibitors, such as ketoconazole) [19,20].
Understanding the influence of renal and hepatic function, age, weight, dose, and food interaction on rivaroxaban PK/PD is essential for optimizing therapy, especially in vulnerable populations. To better account for the variability caused by these factors, physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) modeling has emerged as a powerful approach. PBPK/PD models combine drug characteristics with patient-specific physiological data and can be employed to simulate how rivaroxaban behaves under different clinical conditions, providing insights for supporting individualized dosing strategies.
The aim of this work was to study rivaroxaban pharmacokinetics and pharmacodynamics using PBPK/PD modeling. By addressing key factors influencing intra- and inter-individual variability we aim to better understand how altered metabolism and excretion impact drug behaviour. Additionally, we will investigate the role of dosing regimens and fasting states in modulating rivaroxaban’s action. The outcomes of this research will contribute to the development of predictive models and support precision medicine approaches to optimize rivaroxaban therapy across diverse patient populations.
2. Materials and Methods
2.1. Systematic Literature Research
A systematic literature review was conducted for clinical pharmacokinetic and pharmacodynamic data on rivaroxaban (Figure 1). The keywords rivaroxaban AND pharmacokinetics were used to search the PubMed and PKPDAI databases. This search, conducted on February 3rd, 2025, yielded a wide range of studies, including single and multiple dose pharmacokinetic profiles, pharmacodynamic data, as well as population-specific data (renal and hepatic impairment, and food effect). To determine study eligibility, the following criteria were applied: Clinical trials reporting plasma concentration-time data, dose levels, and elimination parameters in adult humans were included. Preclinical studies, pediatric trials, or studies lacking primary pharmacokinetic endpoints were excluded.
2.2. Data Curation
Relevant pharmacokinetic and pharmacodynamic data were extracted from selected studies and curated in the PK-DB database [21]. Information on demographics (age, sex, weight), clinical condition (renal/hepatic status), rivaroxaban dose, co-administration with other medications, and fasting state was recorded. Data from figures were digitized using WebPlotDigitizer [22], and all numerical entries were standardized according to PK-DB’s data schema [21]. Pharmacokinetic parameters such as peak plasma concentration (Cmax), time to reach peak plasma concentration (Tmax), half-life (T1/2), and area under the curve ((AUC0–∞)) were included.
2.3. Model
A whole-body PBPK/PD model of rivaroxaban was developed in SBML format using sbmlutils [23], with all simulations performed using sbmlsim [24] and libRoadRunner [25,26]. Visualization was performed with cy3sbml [27]. The model structure includes absorption (oral input), distribution across major organ compartments, hepatic metabolism, renal and hepatic excretion, and coagulation dynamics.
Hepatic Impairment. Hepatic clearance was scaled using the parameter f_cirrhosis, where 0 represents normal function. Simulated impairments followed Child-Pugh classifications where 0.40 corresponded to mild, 0.70 to moderate and 0.81 to severe degree of cirrhosis [28].
Renal Impairment. Renal elimination was adjusted using KI__f_renal_function, based on estimated glomerular filtration rate. The model simulated normal (1.0), mild (0.5), moderate (0.35), and severe (0.2) renal impairment, consistent with KDIGO staging [29].
Food Effect. Food effect was controlled through GU__F_riv_abs, which represents the fraction of drug absorbed in the gastrointestinal tract. This parameter was scaled to account for the impact of food on rivaroxaban’s bioavailability. The model assumes a 18% reduction in absorption under fasted conditions (0.82) compared to food (1.0), consistent with clinical observations of reduced bioavailability when the drug is taken without food [5].
Coagulation Dynamics. The pharmacodynamic model describes rivaroxaban’s anticoagulant effects by simulating its inhibition of Factor Xa. This interaction is represented using an Emax model, with rivaroxaban venous plasma concentration (Cve_riv) as input and fractional Factor Xa activity as output. The resulting inhibition cascade affects downstream coagulation markers, including prothrombin time (PT) and activated partial thromboplastin time (aPTT). These are modeled using similar Emax relationships, capturing both the absolute clotting times and their fold-changes relative to baseline values ( and ).
2.4. Parameter Optimization
Both PK and PD model parameters were optimized using a subset of the curated clinical data. The fitted parameters of the PK model consisted of gastrointestinal dissolution rate (GU__Ka_dis_riv), absorption rate (GU__RIVABS_k), hepatic metabolic rate (LI__RIV2RX_Vmax), and renal elimination rate (KI__RIVEX_k). For the PD model, Emax and EC50 values for Factor Xa inhibition (Emax_Xa, EC50_riv_Xa), prothrombin time (Emax_PT, EC50_riv_PT), and activated partial thromboplastin time (Emax_aPTT, EC50_riv_aPTT) were optimized. Optimization was performed across multiple dose levels and under both fed and fasted conditions. Multiple local optimization runs () were conducted to first optimize the PK parameters and subsequently the PD parameters. Model performance was evaluated using independent datasets not used for calibration.
3. Results
3.1. Rivaroxaban Database
An open database of pharmacokinetic and pharmacodynamic data from 14 clinical studies was established, including diverse populations, dosing regimens and physiological conditions (Table 1). The data was used to develop the rivaroxaban PBPK/PD model.
3.2. Computational Model
A whole-body PBPK/PD model of rivaroxaban was created that includes key factors that determine intra- and inter-individual variability (Figure 2). The model includes the organs involved in the pharmacokinetics of rivaroxaban (gastrointestinal tract, liver, and kidney), connected through the systemic circulation, as well as the main pharmacodynamic outputs (activated Factor Xa inhibition, prothrombin time, and activated partial thromboplastin time). The main patient-specific factors that are known to affect the pharmacokinetics and pharmacodynamics of rivaroxaban are included in the model. Specifically, four key factors were included: dose dependency, liver impairment, renal impairment, and food effect.
3.3. Dose Dependency
The model confirmed the dose dependency of rivaroxaban pharmacokinetic and pharmacodynamic profiles in a wide range of oral doses (1-100 mg), as shown in Figure 3. Simulated plasma concentration-time profiles matched observed data from multiple studies, both with single and multiple doses, demonstrating increases in Cmax and (AUC0–∞) with increasing doses. More specifically, the (AUC0–∞) increased proportionally from 1 to 20 μmolh/L as the dose increased from 1 to 100 mg. The sum of rivaroxaban metabolites also exhibited a proportional, if not as sharp, increase, going up to about 4 μmol/L when the dose reached 100 mg. The half-life remained constant across all doses. The simulations matched the data from Graff2007 [14], Jiang2010 [12], Kubitza2005a [11] and Zhao2009 [13]. The urinary data from Jiang2010 [12] and Zhao2009 [13] were also consistently in good agreement with the simulations, which exhibited higher levels of rivaroxaban in the urine at higher doses. Additionally, pharmacodynamic responses are also scaled with the dose, showing higher values of maximum prothrombin time (up to 34 for a 100 mg dose), activated partial thromboplastin time (up to 43 ), and activated factor X inhibition (up to 65%) for higher doses.
3.4. Food Effect
The model reproduced the known effects of food on rivaroxaban pharmacokinetics and pharmacodynamics (Figure 4), with enhanced oral bioavailability and increased coagulation effects when the drug is co-administered with food. Following administration with food, and the subsequent increase in the fraction of the drug that is absorbed (from about 0.82 to 1.0), the (AUC0–∞)0–∞ of both rivaroxaban and its metabolites increased by approximately 20% compared to the fasted state. Despite the changes in exposure, the half-life of rivaroxaban remained relatively unchanged, indicating that the observed increases were primarily driven by increased bioavailability, rather than alterations in clearance. Pharmacodynamically, food intake amplified rivaroxaban’s anticoagulant effects. The maximum prothrombin time increased from 15.4 to 15.9 seconds, and maximum aPTT rose from 38.2 to 38.9 seconds. The peak Factor Xa inhibition increased from 0.30 to 0.34, reflecting the higher systemic exposure to active drug under fed conditions. These findings align with the clinical data from Kubitza2006 [6] and Liu2024 [7], as well as with label recommendations to administer rivaroxaban with food, especially for doses greater than 15 mg.
3.5. Hepatic Impairment
Simulation of rivaroxaban pharmacokinetics and pharmacodynamics under increasing degrees of hepatic impairment showed a consistent increase in rivaroxaban exposure (Figure 5). (AUC0–∞) values increased with worsening liver function from approximately 2 to 2.5 μmolh/L. In contrast, rivaroxaban metabolites displayed a decreasing trend, with (AUC0–∞) falling from 0.5 to 0 μmolh/L, which is consistent with the reduced hepatic metabolic activity in the case of cirrhosis. The simulated half-life only increased minimally with the progression of liver cirrhosis.
On the pharmacodynamic side, prothrombin time (PT) increased from 15.5 to 18.5 , activated partial thromboplastin time (aPTT) from 39 to 44 , and Factor Xa inhibition from 0.32 to 0.44 across the hepatic impairment scale. These results align with the higher exposure that leads to increased anticoagulation effects.
3.6. Renal Impairment
Figure 6 shows the effect of renal function on the pharmacokinetics and pharmacodynamics of rivaroxaban. With decreasing renal function, systemic exposure to rivaroxaban and its metabolites increases. Specifically, the model predicts an increase in rivaroxaban (AUC0–∞) from 1 to 3 μmolh/L, and in metabolite (AUC0–∞) from 0 to 5 μmolh/L.
These trends reflect enhanced drug accumulation associated with worse renal clearance. The minor increase of half-life (from 4.35 to 4.36 for rivaroxaban and from 4.35 to 4.42 for metabolites) also supports a marginal decrease in the drug excretion. All this is aligned with the underlying physiology and with clinical findings from Greenblatt2018 [18] and Moore2014 [17], which show greater exposure with worsening renal function.
4. Discussion
In this study, we developed and evaluated a whole-body physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) model of rivaroxaban, a direct oral anticoagulant commonly prescribed for the prevention and treatment of thromboembolic events. The model was constructed using a comprehensive, literature-derived database of clinical pharmacokinetic and pharmacodynamic data, enabling simulation of plasma concentrations, drug exposure metrics ((AUC0–∞), Cmax, half-life), and PD characteristics (Factor Xa inhibition, PT, aPTT) across a range of physiological conditions. This work demonstrates the potential of PBPK/PD modeling to support precision dosing and address questions that could arise in real-world clinical scenarios, even in the absence of complete empirical datasets.
The curated database used in this project not only allowed robust model calibration and validation but also laid the foundation for future clinical decision support systems (CDSS) and digital twin applications. By aggregating and harmonizing clinical trial data, including various dosing regimens, patient populations, and intervention contexts, the database allows flexible reuse for both academic and clinical purposes. Importantly, it serves as a scalable resource that can be linked to other pharmacological and physiological models in systems pharmacology frameworks.
Our dose dependency simulations successfully reproduced the expected increases in rivaroxaban pharmacokinetic and pharmacodynamic response across a range of doses. These results are consistent with clinical findings and support the model’s capacity to simulate rivaroxaban pharmacology under varying therapeutic scenarios.
In terms of food effects, the model correctly captured the qualitative trend of increased rivaroxaban exposure when administered with food, in alignment with known clinical data. Further improvements could include making the food effect itself dose-dependent. Notably, the fact that food has a greater impact on rivaroxaban bioavailability at higher doses (particularly above 15 mg) [34] is an effect that was not fully captured. Incorporating this knowledge, e.g. by modifying intestinal permeability or gastric emptying rates in a dose-dependent manner, could enhance the model’s translational relevance, especially for clinical scenarios that require the administration of high doses of rivaroxaban.
As expected, simulations of hepatic impairment demonstrated increases in rivaroxaban exposure and pharmacodynamic effect with progression of liver dysfunction. This is consistent with the known role of hepatic metabolism and hepatic excretion in rivaroxaban clearance, and with clinical observations [16]. The model also predicted a progressive decline in the formation of rivaroxaban metabolites, which is biologically plausible given the reduced metabolic capacity in cirrhosis. Additionally, Factor Xa inhibition, PT, and aPTT all increased correspondingly, further validating the model’s ability to accurately reproduce liver impairment.
Renal impairment simulations likewise reflected physiologically consistent trends. Exposure metrics such as (AUC0–∞) increased with worsening renal function, reflecting increased drug accumulation due to reduced renal clearance. While the model slightly underestimated the extent of drug retention seen in severe renal impairment in some studies [17], it was nonetheless able to replicate concentration–time profiles observed in plasma and urine. In particular, although the detailed pharmacodynamic timecourses under renal impairment conditions are lacking, the data reported by Moore2014 showed increases in (AUC0–∞) for the PD curve and in Emax in individuals with impaired renal function. The model accurately represented this trends by predicting higher values in Factor Xa inhibition, prolonged PT, and elevated aPTT when the renal function is impaired, which is consistent with higher systemic exposure that would be expected from impaired clearance. This demonstrates one of the major strengths of PBPK/PD modeling: the ability to bridge data gaps and make physiologically grounded predictions even where detailed empirical data are lacking.
Looking forward, the model offers numerous opportunities for expansion. Demographic and physiological factors such as age, sex, and body weight have well-documented effects on rivaroxaban pharmacokinetics [30,35] and could be incorporated to support population-level simulations or individualized dosing. In particular, future model versions could differentiate between lean body mass and adipose tissue in order to more accurately capture distribution volumes and clearance dynamics in obese or underweight individuals. This distinction could be especially important given the moderate lipophilicity of rivaroxaban [36] and the increasing clinical emphasis on dosing optimization in diverse body types.
Additional refinements may include better representation of transporter proteins (e.g., P-gp) and enzyme polymorphisms (e.g., CYP3A4/5), as well as simulation of drug–drug interactions and adherence scenarios. Ultimately, the integration of this model into digital twin platforms could enable real-time therapeutic monitoring and decision-making support, particularly for vulnerable populations such as the elderly or patients with organ dysfunction.
5. Conclusions
In this study, we developed a whole-body physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) model of rivaroxaban to simulate its behaviour under varying physiological conditions and treatment scenarios. Using the established clinical database, we successfully reproduced key pharmacokinetic and pharmacodynamic outcomes across different doses, food intake states, and levels of hepatic and renal function. This work demonstrates the value of PBPK/PD modeling in understanding complex drug behaviour, supporting personalized dosing strategies, and informing clinical decision-making. The database and model together form a strong foundation for future integration into digital twin platforms and clinical decision support systems.
Author Contributions
Conceptualization, M.K..; methodology, M.K.; software, M.K.; validation, M.K.; formal analysis, E.C.; investigation, E.C.; resources, M.K.; data curation, E.C., M.K., M.M.; writing—original draft preparation, E.C.; writing—review and editing, M.K., E.C., M.M.; visualization, M.K.; supervision, M.K., M.M.; project administration, M.K.; funding acquisition, M.K., E.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Erasmus Mundus Joint Master Degree (EMJMD) program Be in Precision Medicine. MK was supported by the BMBF grant number 031L0304B and by the DFG grant number 436883643 and 465194077. MM was supported by the BMBF grant number 031L0304B.
Data Availability Statement
All curated pharmacokinetic data are publicly available in the PK-DB database (https://pk-db.com). The model and all associated materials (simulation scripts, parameters, and documentation) are publicly available in SBML format under a CC-BY 4.0 license at https://github.com/matthiaskoenig/rivaroxaban-model, version 0.7.0 [37].
Acknowledgments
This work was supported by the BMBF-funded de.NBI Cloud within the German Network for Bioinformatics Infrastructure (de.NBI) (031A537B, 031A533A, 031A538A, 031A533B, 031A535A, 031A537C, 031A534A, 031A532B).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Samama, M.M. The Mechanism of Action of Rivaroxaban – an Oral, Direct Factor Xa Inhibitor – Compared with Other Anticoagulants. Thrombosis Research 2011, 127, 497–504. [Google Scholar] [CrossRef]
- Orfeo, T.; Butenas, S.; Brummel-Ziedins, K.E.; Gissel, M.; Mann, K.G. Anticoagulation by Factor Xa Inhibitors. Journal of thrombosis and haemostasis : JTH 2010, 8, 1745–1753. [Google Scholar] [CrossRef]
- Kvasnicka, T.; Malikova, I.; Zenahlikova, Z.; Kettnerova, K.; Brzezkova, R.; Zima, T.; Ulrych, J.; Briza, J.; Netuka, I.; Kvasnicka, J. Rivaroxaban - Metabolism, Pharmacologic Properties and Drug Interactions. Current Drug Metabolism 2017, 18, 636–642. [Google Scholar] [CrossRef]
- Tittl, L.; Marten, S.; Naue, C.; Beyer-Westendorf, J. 5-Year Outcomes from Rivaroxaban Therapy in Atrial Fibrillation: Results from the Dresden NOAC Registry. Thrombosis Research 2021, 202, 24–30. [Google Scholar] [CrossRef]
- Ding, S.; Wang, L.; Xie, L.; Zhou, S.; Chen, J.; Zhao, Y.; Deng, W.; Liu, Y.; Zhang, H.; Shao, F. Bioequivalence Study of 2 Formulations of Rivaroxaban, a Narrow-Therapeutic-Index Drug, in Healthy Chinese Subjects Under Fasting and Fed Conditions. Clinical pharmacology in drug development 2020, 9, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Becka, M.; Zuehlsdorf, M.; Mueck, W. Effect of Food, an Antacid, and the H2 Antagonist Ranitidine on the Absorption of BAY 59-7939 (Rivaroxaban), an Oral, Direct Factor Xa Inhibitor, in Healthy Subjects. Journal of clinical pharmacology 2006, 46, 549–558. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Xie, Q.; Zhao, X.; Tan, Y.; Wang, W.; Cao, Y.; Wei, X.; Mu, G.; Zhang, H.; Zhou, S.; et al. The Pharmacogenetic Variability Associated with the Pharmacokinetics and Pharmacodynamics of Rivaroxaban in Healthy Chinese Subjects: A National Multicenter Exploratory Study. Clinical therapeutics 2024, 46, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Konicki, R.; Weiner, D.; Herbert Patterson, J.; Gonzalez, D.; Kashuba, A.; Cao, Y.C.; Gehi, A.K.; Watkins, P.; Powell, J.R. Rivaroxaban Precision Dosing Strategy for Real-World Atrial Fibrillation Patients. Clinical and translational science 2020, 13, 777–784. [Google Scholar] [CrossRef]
- Pereira Portela, C.; Gautier, L.A.; Zermatten, M.G.; Fraga, M.; Moradpour, D.; Bertaggia Calderara, D.; Aliotta, A.; Veuthey, L.; De Gottardi, A.; Stirnimann, G.; et al. Direct Oral Anticoagulants in Cirrhosis: Rationale and Current Evidence. JHEP Reports 2024, 6, 101116. [Google Scholar] [CrossRef]
- Suchman, A.L.; Griner, P.F. Diagnostic Uses of the Activated Partial Thromboplastin Time and Prothrombin Time. Annals of Internal Medicine 1986, 104, 810–816. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Voith, B.; Zuehlsdorf, M.; Wensing, G. Safety, Pharmacodynamics, and Pharmacokinetics of Single Doses of BAY 59-7939, an Oral, Direct Factor Xa Inhibitor. Clinical pharmacology and therapeutics 2005, 78, 412–421. [Google Scholar] [CrossRef]
- Jiang, J.; Hu, Y.; Zhang, J.; Yang, J.; Mueck, W.; Kubitza, D.; Bauer, R.J.; Meng, L.; Hu, P. Safety, Pharmacokinetics and Pharmacodynamics of Single Doses of Rivaroxaban - an Oral, Direct Factor Xa Inhibitor - in Elderly Chinese Subjects. Thrombosis and haemostasis 2010, 103, 234–241. [Google Scholar] [CrossRef]
- Zhao, X.; Sun, P.; Zhou, Y.; Liu, Y.; Zhang, H.; Mueck, W.; Kubitza, D.; Bauer, R.J.; Zhang, H.; Cui, Y. Safety, Pharmacokinetics and Pharmacodynamics of Single/Multiple Doses of the Oral, Direct Factor Xa Inhibitor Rivaroxaban in Healthy Chinese Subjects. British journal of clinical pharmacology 2009, 68, 77–88. [Google Scholar] [CrossRef]
- Graff, J.; von Hentig, N.; Misselwitz, F.; Kubitza, D.; Becka, M.; Breddin, H.K.; Harder, S. Effects of the Oral, Direct Factor Xa Inhibitor Rivaroxaban on Platelet-Induced Thrombin Generation and Prothrombinase Activity. Journal of clinical pharmacology 2007, 47, 1398–1407. [Google Scholar] [CrossRef]
- Mueck, W.; Stampfuss, J.; Kubitza, D.; Becka, M. Clinical Pharmacokinetic and Pharmacodynamic Profile of Rivaroxaban. Clinical Pharmacokinetics 2014, 53, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Roth, A.; Becka, M.; Alatrach, A.; Halabi, A.; Hinrichsen, H.; Mueck, W. Effect of Hepatic Impairment on the Pharmacokinetics and Pharmacodynamics of a Single Dose of Rivaroxaban, an Oral, Direct Factor Xa Inhibitor. British journal of clinical pharmacology 2013, 76, 89–98. [Google Scholar] [CrossRef]
- Moore, K.T.; Vaidyanathan, S.; Natarajan, J.; Ariyawansa, J.; Haskell, L.; Turner, K.C. An Open-Label Study to Estimate the Effect of Steady-State Erythromycin on the Pharmacokinetics, Pharmacodynamics, and Safety of a Single Dose of Rivaroxaban in Subjects with Renal Impairment and Normal Renal Function. Journal of clinical pharmacology 2014, 54, 1407–1420. [Google Scholar] [CrossRef]
- Greenblatt, D.J.; Patel, M.; Harmatz, J.S.; Nicholson, W.T.; Rubino, C.M.; Chow, C.R. Impaired Rivaroxaban Clearance in Mild Renal Insufficiency With Verapamil Coadministration: Potential Implications for Bleeding Risk and Dose Selection. Journal of clinical pharmacology 2018, 58, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Ashton, V.; Kerolus-Georgi, S.; Moore, K.T. The Pharmacology, Efficacy, and Safety of Rivaroxaban in Renally Impaired Patient Populations. Journal of clinical pharmacology 2021, 61, 1010–1026. [Google Scholar] [CrossRef] [PubMed]
- Mueck, W.; Kubitza, D.; Becka, M. Co-Administration of Rivaroxaban with Drugs That Share Its Elimination Pathways: Pharmacokinetic Effects in Healthy Subjects. British journal of clinical pharmacology 2013, 76, 455–466. [Google Scholar] [CrossRef]
- Grzegorzewski, J.; Brandhorst, J.; Green, K.; Eleftheriadou, D.; Duport, Y.; Barthorscht, F.; Köller, A.; Ke, D.Y.J.; De Angelis, S.; König, M. PK-DB: Pharmacokinetics Database for Individualized and Stratified Computational Modeling. Nucleic Acids Research 2021, 49, D1358–D1364. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A. WebPlotDigitizer, 2024.
- König, M. Sbmlutils: Python Utilities for SBML. Zenodo, 2024. [CrossRef]
- König, M. Sbmlsim: SBML Simulation Made Easy. [object Object], 2021. [CrossRef]
- Welsh, C.; Xu, J.; Smith, L.; König, M.; Choi, K.; Sauro, H.M. libRoadRunner 2.0: A High Performance SBML Simulation and Analysis Library. Bioinformatics 2023, 39, btac770. [Google Scholar] [CrossRef]
- Somogyi, E.T.; Bouteiller, J.M.; Glazier, J.A.; König, M.; Medley, J.K.; Swat, M.H.; Sauro, H.M. libRoadRunner: A High Performance SBML Simulation and Analysis Library. Bioinformatics 2015, 31, 3315–3321. [Google Scholar] [CrossRef]
- König, M.; Dräger, A.; Holzhütter, H.G. CySBML: A Cytoscape Plugin for SBML. Bioinformatics 2012, 28, 2402–2403. [Google Scholar] [CrossRef]
- Köller, A.; Grzegorzewski, J.; König, M. Physiologically Based Modeling of the Effect of Physiological and Anthropometric Variability on Indocyanine Green Based Liver Function Tests. Frontiers in Physiology 2021, 12, 757293. [Google Scholar] [CrossRef]
- Stevens, P.E.; Ahmed, S.B.; Carrero, J.J.; Foster, B.; Francis, A.; Hall, R.K.; Herrington, W.G.; Hill, G.; Inker, L.A.; Kazancıoğlu, R.; et al. KDIGO 2024 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease. Kidney International 2024, 105, S117–S314. [Google Scholar] [CrossRef]
- Alalawneh, M.; Awaisu, A.; Abdallah, I.; Elewa, H.; Danjuma, M.; Matar, K.M.; ElKashlan, A.M.; Elshayep, Y.; Ibrahim, F.; Rachid, O. Pharmacokinetics of Single-Dose Rivaroxaban under Fed State in Obese vs. Non-Obese Subjects: An Open-Label Controlled Clinical Trial (RIVOBESE-PK). Clinical and translational science 2024, 17, e13853. [Google Scholar] [CrossRef]
- Genis-Najera, L.; Sañudo-Maury, M.E.; Moquete, T. A Single-blind, Randomized, Single-dose, Two-sequence, Two-period, Crossover Study to Assess the Bioequivalence between Two Oral Tablet Formulations of Rivaroxaban 20 Mg in Healthy Mexican Volunteers. Clinical pharmacology in drug development 2022, 11, 826–831. [Google Scholar] [CrossRef] [PubMed]
- Kubitza, D.; Becka, M.; Wensing, G.; Voith, B.; Zuehlsdorf, M. Safety, Pharmacodynamics, and Pharmacokinetics of BAY 59-7939–an Oral, Direct Factor Xa Inhibitor–after Multiple Dosing in Healthy Male Subjects. European Journal of Clinical Pharmacology 2005, 61, 873–880. [Google Scholar] [CrossRef] [PubMed]
- Weinz, C.; Schwarz, T.; Kubitza, D.; Mueck, W.; Lang, D. Metabolism and Excretion of Rivaroxaban, an Oral, Direct Factor Xa Inhibitor, in Rats, Dogs, and Humans. Drug metabolism and disposition: the biological fate of chemicals 2009, 37, 1056–1064. [Google Scholar] [CrossRef]
- Kubitza, D.; Becka, M.; Roth, A.; Mueck, W. Dose-Escalation Study of the Pharmacokinetics and Pharmacodynamics of Rivaroxaban in Healthy Elderly Subjects. Current medical research and opinion 2008, 24, 2757–2765. [Google Scholar] [CrossRef] [PubMed]
- Speed, V.; Green, B.; Roberts, L.N.; Woolcombe, S.; Bartoli-Abdou, J.; Barsam, S.; Byrne, R.; Gee, E.; Czuprynska, J.; Brown, A.; et al. Fixed Dose Rivaroxaban Can Be Used in Extremes of Bodyweight: A Population Pharmacokinetic Analysis. Journal of thrombosis and haemostasis : JTH 2020, 18, 2296–2307. [Google Scholar] [CrossRef] [PubMed]
- Remko, M. Molecular Structure, Lipophilicity, Solubility, Absorption, and Polar Surface Area of Novel Anticoagulant Agents. Journal of Molecular Structure: THEOCHEM 2009, 916, 76–85. [Google Scholar] [CrossRef]
- Casabianca, E.; König, M. Physiologically Based Pharmacokinetic/Pharmacodynamic (PBPK/PD) Model of Rivaroxaban. Zenodo, 2025. [CrossRef]
Figure 1.
PRISMA flow diagram of the literature search and study selection process.
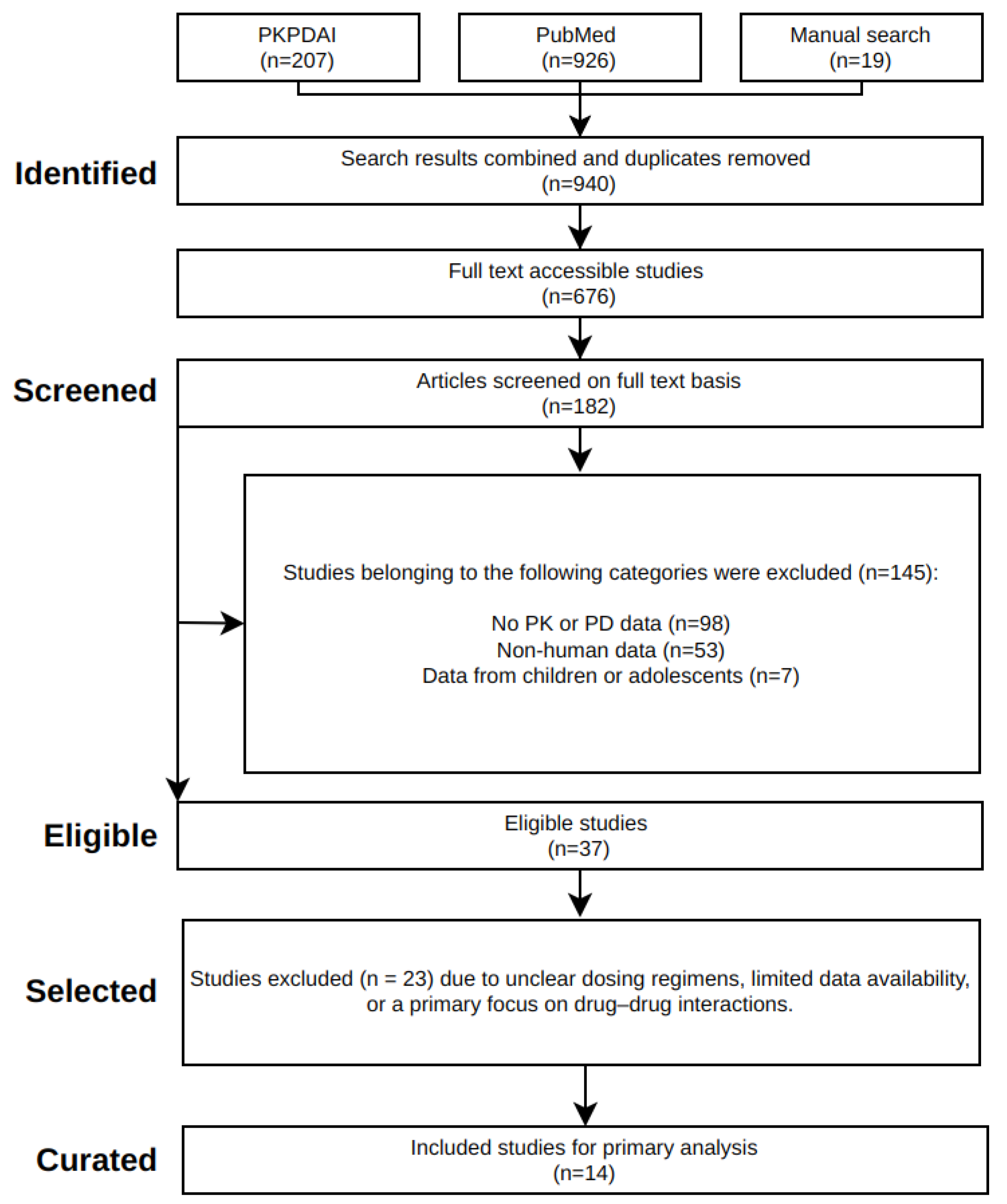
Figure 2.
Physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) model of rivaroxaban. A) Whole-body model illustrating oral administration of rivaroxaban (RIV), its distribution via systemic circulation, and the key organs involved in its absorption, metabolism, and elimination—including the gastrointestinal tract, liver, and kidneys. B) Gastrointestinal model depicting the absorption of RIV and highlighting the food and dose dependency in the percentage of absorption. C) Pharmacodynamic model capturing inhibition of activated Factor Xa by rivaroxaban, with downstream effects on clot formation and consequent prolongation of PT and aPTT. D) Hepatic model showing metabolism of rivaroxaban, mediated by CYP3A4, CYP3A5 and CYPJ2 to produce its metabolites RX. E) Renal model illustrating the contribution of renal excretion to total clearance. F) Key factors influencing rivaroxaban PK and PD profiles accounted for in the model.
Figure 2.
Physiologically based pharmacokinetic/pharmacodynamic (PBPK/PD) model of rivaroxaban. A) Whole-body model illustrating oral administration of rivaroxaban (RIV), its distribution via systemic circulation, and the key organs involved in its absorption, metabolism, and elimination—including the gastrointestinal tract, liver, and kidneys. B) Gastrointestinal model depicting the absorption of RIV and highlighting the food and dose dependency in the percentage of absorption. C) Pharmacodynamic model capturing inhibition of activated Factor Xa by rivaroxaban, with downstream effects on clot formation and consequent prolongation of PT and aPTT. D) Hepatic model showing metabolism of rivaroxaban, mediated by CYP3A4, CYP3A5 and CYPJ2 to produce its metabolites RX. E) Renal model illustrating the contribution of renal excretion to total clearance. F) Key factors influencing rivaroxaban PK and PD profiles accounted for in the model.
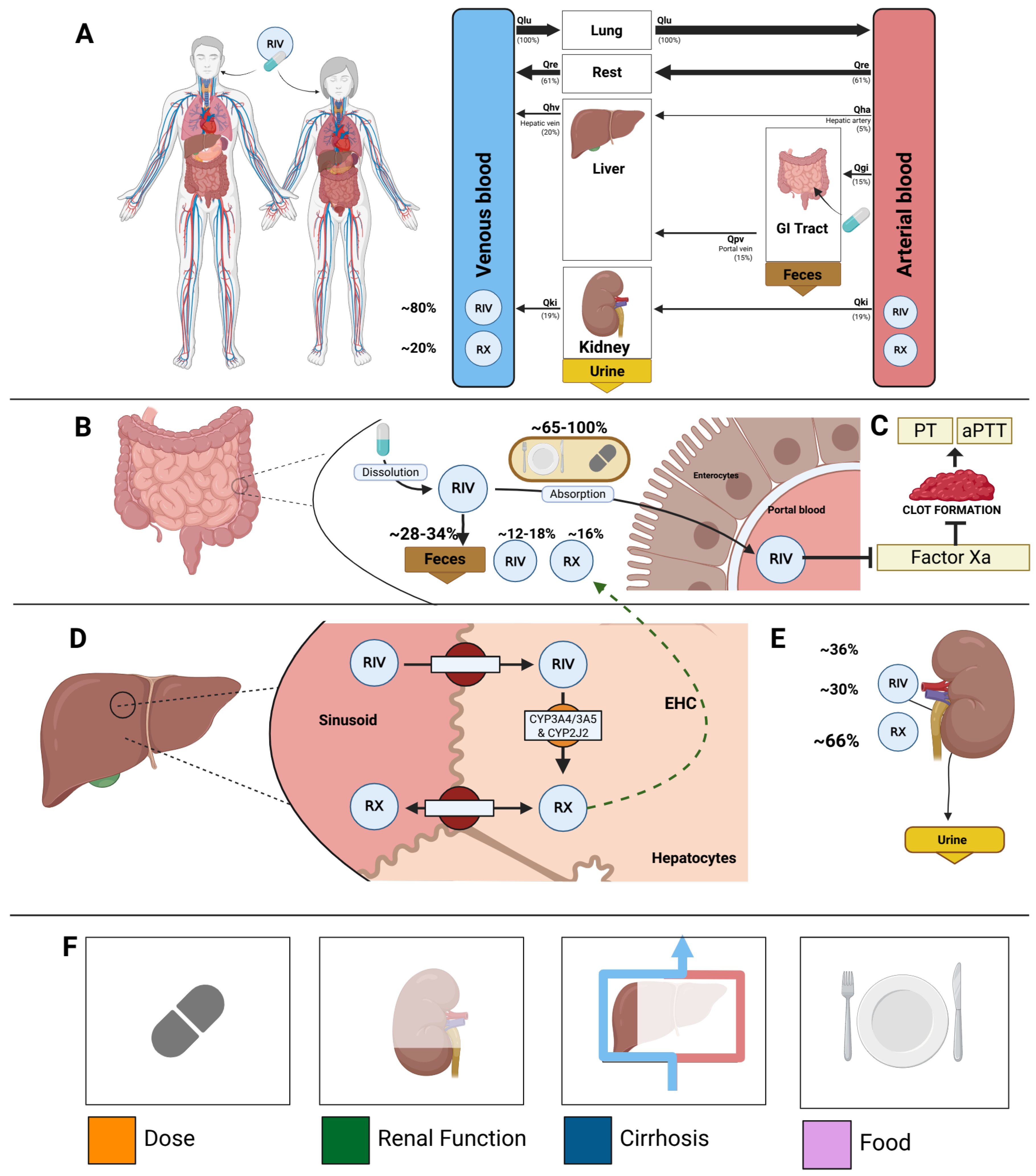
Figure 3.
Dose-dependent pharmacokinetics and pharmacodynamics of rivaroxaban. A) Illustration of the
rivaroxaban oral dose range (1–100 mg) evaluated in the simulations. B) Simulated PK and PD timecourses
depending on administered dose. C) Summary of simulated PK parameters across the dose range for rivaroxaban
and the sum of rivaroxaban metabolites. D) Summary of simulated maximum PD parameters across the dose range.
E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles of rivaroxaban
across various doses in selected studies (Graff2007 [14], Jiang2010 [12], Kubitza2005a [11], Kubitza2005 [32],
Zhao2009 [13]). F) Simulated (solid lines) versus observed (symbols) pharmacodynamic profiles of rivaroxaban
across various doses in selected studies (Graff2007 [14], Jiang2010 [12], Kubitza2005a [11], Kubitza2005 [32],
Zhao2009 [13]).
Figure 3.
Dose-dependent pharmacokinetics and pharmacodynamics of rivaroxaban. A) Illustration of the
rivaroxaban oral dose range (1–100 mg) evaluated in the simulations. B) Simulated PK and PD timecourses
depending on administered dose. C) Summary of simulated PK parameters across the dose range for rivaroxaban
and the sum of rivaroxaban metabolites. D) Summary of simulated maximum PD parameters across the dose range.
E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles of rivaroxaban
across various doses in selected studies (Graff2007 [14], Jiang2010 [12], Kubitza2005a [11], Kubitza2005 [32],
Zhao2009 [13]). F) Simulated (solid lines) versus observed (symbols) pharmacodynamic profiles of rivaroxaban
across various doses in selected studies (Graff2007 [14], Jiang2010 [12], Kubitza2005a [11], Kubitza2005 [32],
Zhao2009 [13]).
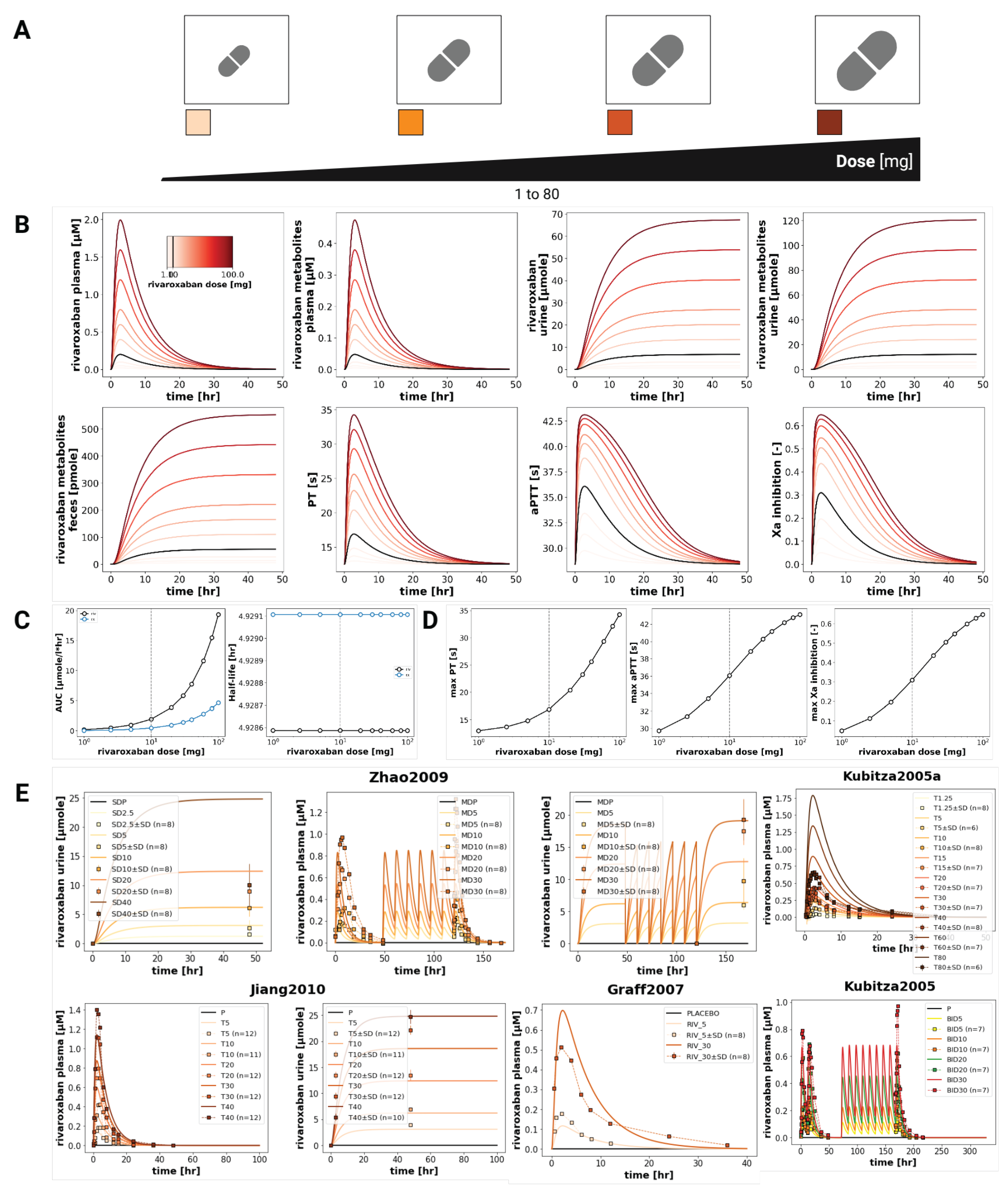
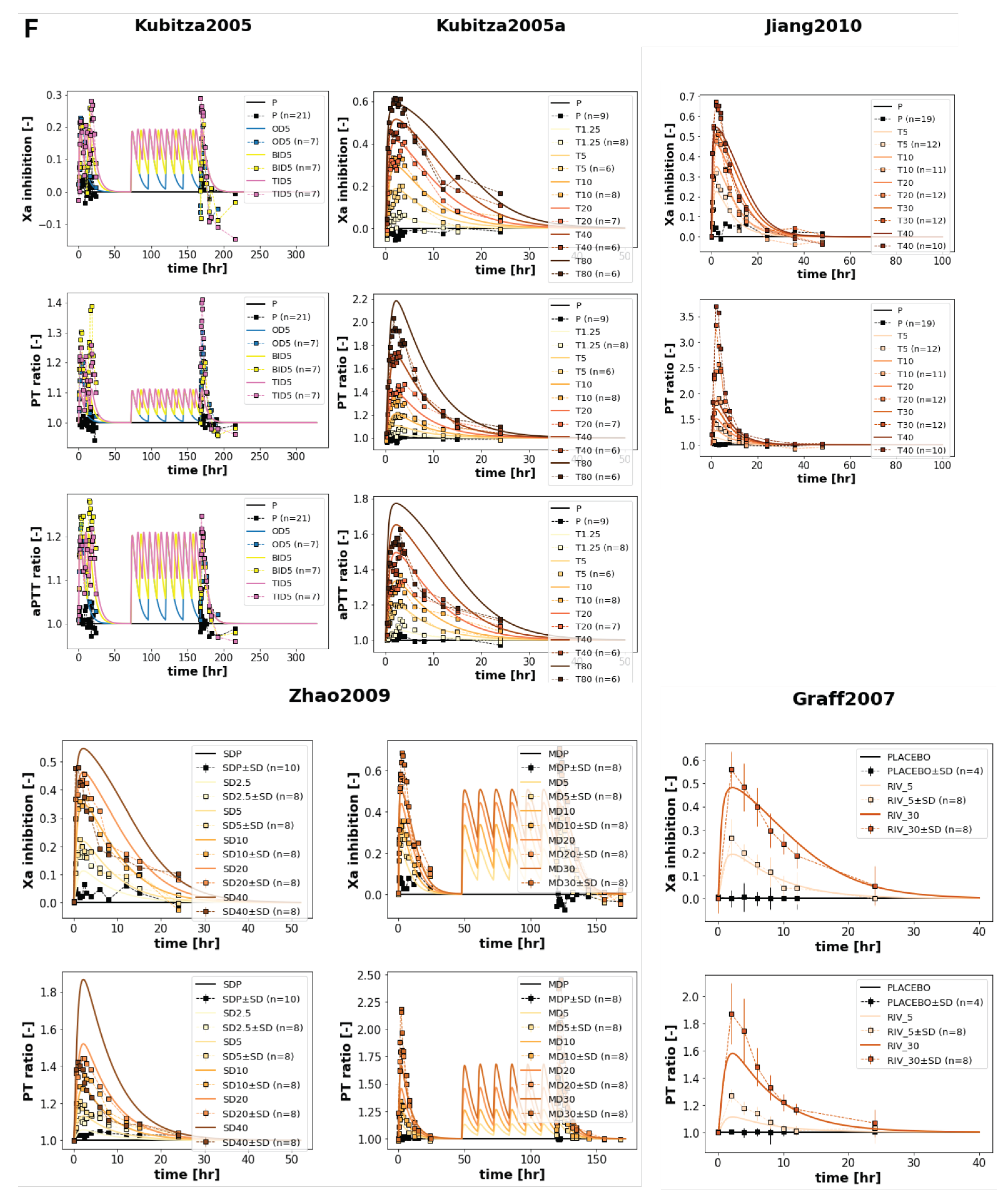
Figure 4.
Food-dependent pharmacokinetics and pharmacodynamics of rivaroxaban. A) Illustration of the
absorbed fraction of rivaroxaban between 1% and 100% (82% in the fasted state to 100% in the fed state) evaluated
in the simulations. B) Simulated PK and PD timecourses depending on fasting state. C) Summary of simulated
PK parameters depending on fasting state for rivaroxaban and the sum of rivaroxaban metabolites. D) Summary
of simulated PD parameters depending on fasting state. E) Simulated (solid lines) versus observed (symbols)
plasma and urine concentration-time profiles across fed and fasted conditions in Ding2020 [5], Kubitza2006 [6]
and Liu2024 [7]. F) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time
profiles and pharmacodynamic timecourses of rivaroxaban across fed and fasted conditions in Liu2024 [7] and
Kubitza2006 [6].
Figure 4.
Food-dependent pharmacokinetics and pharmacodynamics of rivaroxaban. A) Illustration of the
absorbed fraction of rivaroxaban between 1% and 100% (82% in the fasted state to 100% in the fed state) evaluated
in the simulations. B) Simulated PK and PD timecourses depending on fasting state. C) Summary of simulated
PK parameters depending on fasting state for rivaroxaban and the sum of rivaroxaban metabolites. D) Summary
of simulated PD parameters depending on fasting state. E) Simulated (solid lines) versus observed (symbols)
plasma and urine concentration-time profiles across fed and fasted conditions in Ding2020 [5], Kubitza2006 [6]
and Liu2024 [7]. F) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time
profiles and pharmacodynamic timecourses of rivaroxaban across fed and fasted conditions in Liu2024 [7] and
Kubitza2006 [6].
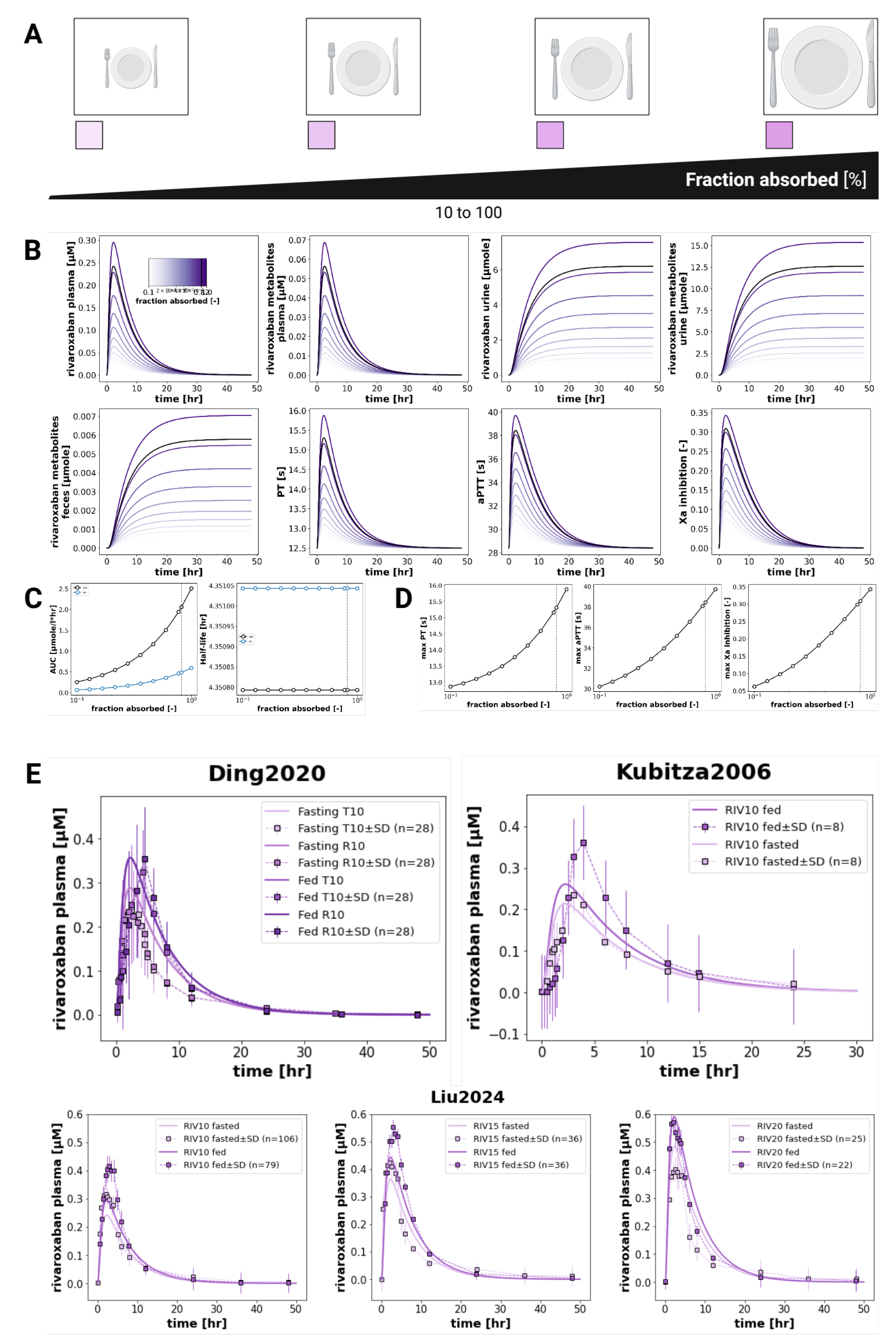
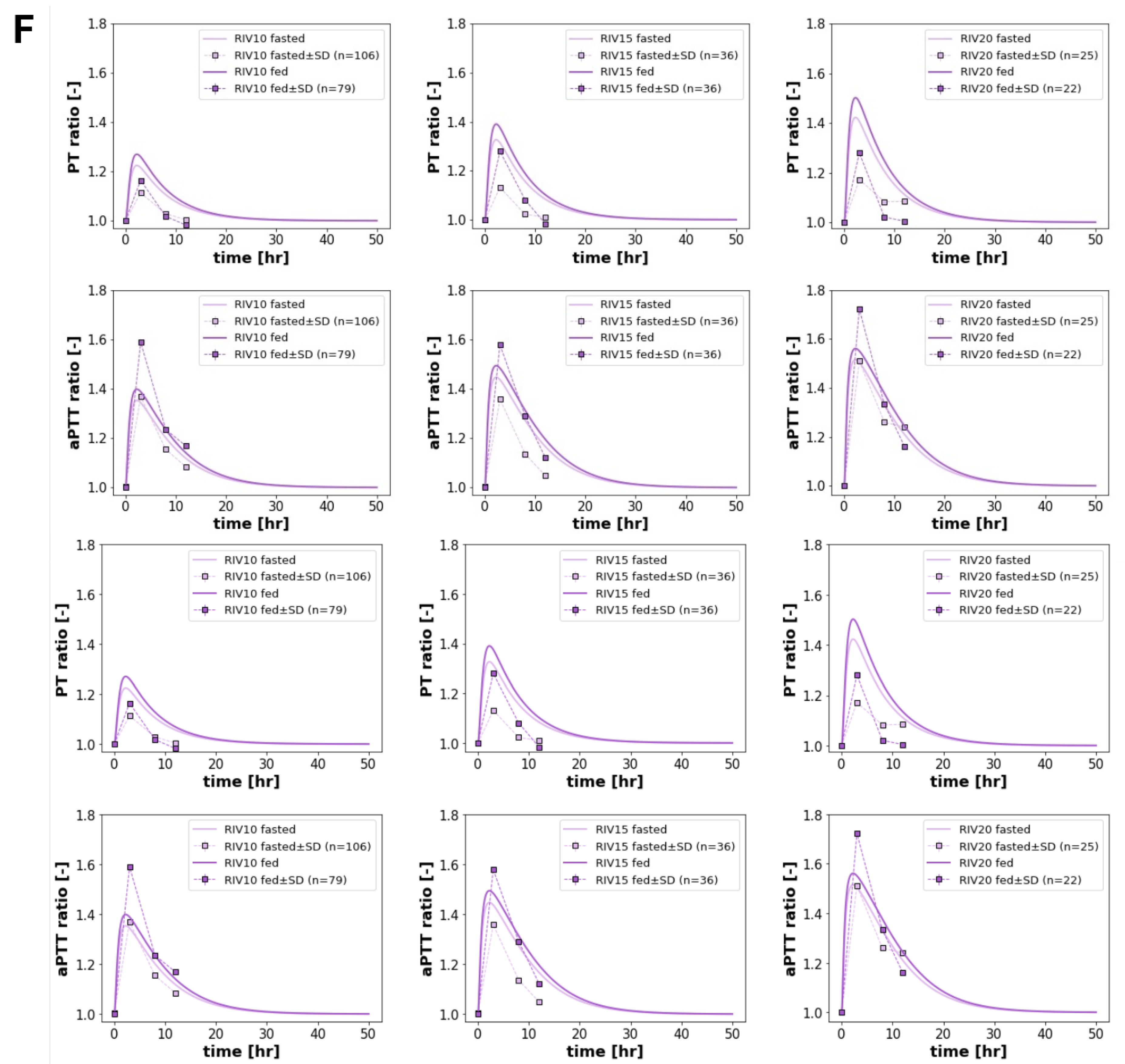
Figure 5.
Effect of hepatic impairment on rivaroxaban pharmacokinetic and pharmacodynamic profiles. A) Illustration of the level of cirrhosis evaluated in the simulations. B) Simulated PK and PD timecourses across different levels of cirrhosis. C) Summary of simulated PK parameters across different levels of cirrhosis for rivaroxaban and the sum of rivaroxaban metabolites. D Summary of simulated maximum PD parameters across different levels of cirrhosis. E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles of rivaroxaban for different degrees of cirrhosis in Kubitza2013 [16]. F) Simulated (solid lines) versus observed (symbols) pharmacodynamic timecourses for different degrees of cirrhosis in Kubitza2013 [16].
Figure 5.
Effect of hepatic impairment on rivaroxaban pharmacokinetic and pharmacodynamic profiles. A) Illustration of the level of cirrhosis evaluated in the simulations. B) Simulated PK and PD timecourses across different levels of cirrhosis. C) Summary of simulated PK parameters across different levels of cirrhosis for rivaroxaban and the sum of rivaroxaban metabolites. D Summary of simulated maximum PD parameters across different levels of cirrhosis. E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles of rivaroxaban for different degrees of cirrhosis in Kubitza2013 [16]. F) Simulated (solid lines) versus observed (symbols) pharmacodynamic timecourses for different degrees of cirrhosis in Kubitza2013 [16].
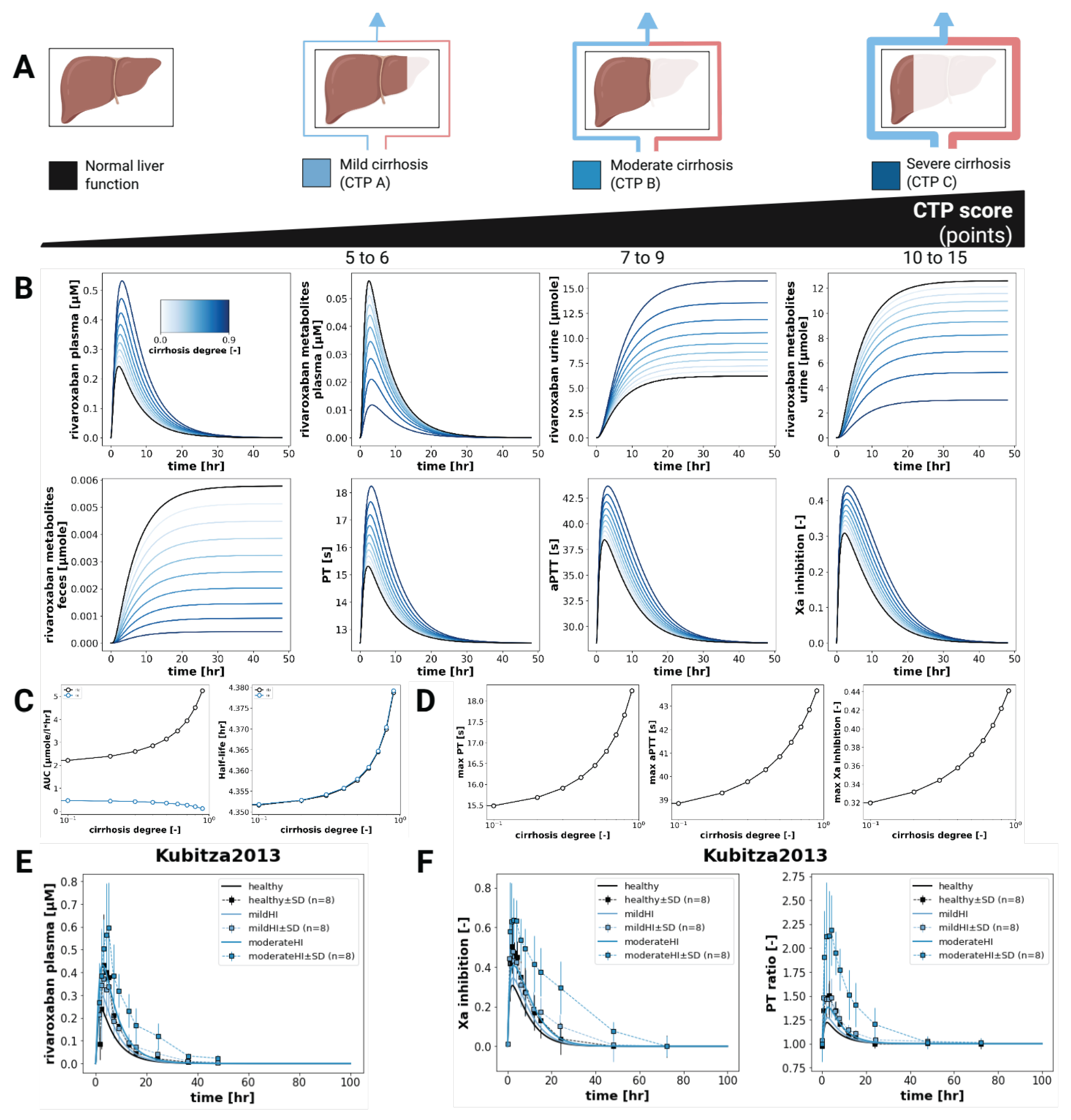
Figure 6.
Effect of renal impairment on rivaroxaban pharmacokinetics and pharmacodynamics. A) Illustration of the level of renal impairment evaluated in the simulations. B) Simulated PK and PD timecourses depending on the degree of renal function. C) Summary of simulated PK parameters across different levels of renal impairment for rivaroxaban and the sum of rivaroxaban metabolites. D) Summary of simulated maximum PD parameters for different levels of renal impairment. E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles for different degrees of renal function in selected studies (Greenblatt2018 [18] and Moore2014 [17].)
Figure 6.
Effect of renal impairment on rivaroxaban pharmacokinetics and pharmacodynamics. A) Illustration of the level of renal impairment evaluated in the simulations. B) Simulated PK and PD timecourses depending on the degree of renal function. C) Summary of simulated PK parameters across different levels of renal impairment for rivaroxaban and the sum of rivaroxaban metabolites. D) Summary of simulated maximum PD parameters for different levels of renal impairment. E) Simulated (solid lines) versus observed (symbols) plasma and urine concentration-time profiles for different degrees of renal function in selected studies (Greenblatt2018 [18] and Moore2014 [17].)

Table 1.
Overview of rivaroxaban studies. This table summarizes the included studies with key identifiers (Study, PK-DB ID), dosing regimen (Dosing), dose (Dose), and participant characteristics: H (healthy), RI (renal impairment), and HI (hepatic impairment). Fasting status is indicated as FA (fasted) or FE (fed). Pharmacokinetic data include RP (plasma rivaroxaban), RU (urine rivaroxaban), and RF (feces rivaroxaban); pharmacodynamic data include Xa (Factor Xa inhibition), PT (prothrombin time), and aP (activated partial thromboplastin time). S study reference.
Table 1.
Overview of rivaroxaban studies. This table summarizes the included studies with key identifiers (Study, PK-DB ID), dosing regimen (Dosing), dose (Dose), and participant characteristics: H (healthy), RI (renal impairment), and HI (hepatic impairment). Fasting status is indicated as FA (fasted) or FE (fed). Pharmacokinetic data include RP (plasma rivaroxaban), RU (urine rivaroxaban), and RF (feces rivaroxaban); pharmacodynamic data include Xa (Factor Xa inhibition), PT (prothrombin time), and aP (activated partial thromboplastin time). S study reference.
| Study | PK-DB | Dosing | Dose [mg] | H | RI | HI | FA | FE | RP | RU | RF | Xa | PT | aP | S |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Alalawneh2024 | PKDB00983 | single | 20 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [30] | |||||
| Ding2020 | PKDB00972 | single | 10 | ✓ | ✓ | ✓ | ✓ | [5] | |||||||
| GenisNajera2022 | PKDB00973 | single | 20 | ✓ | ✓ | ✓ | [31] | ||||||||
| Graff2007 | PKDB00984 | single | 5, 30 | ✓ | ✓ | ✓ | ✓ | ✓ | [14] | ||||||
| Greenblatt2018 | PKDB00985 | single | 20 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [18] | ||||
| Jiang2010 | PKDB00986 | single | 5, 10, 20, 30, 40 | ✓ | ✓ | ✓ | ✓ | ✓ | [12] | ||||||
| Kubitza2005 | PKDB00987 | multi | 5, 10, 20, 30 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [32] | |||||
| Kubitza2005a | PKDB00988 | single, multi | 1.25, 5, 10, 20, 40, 80 | ✓ | ✓ | ✓ | ✓ | ✓ | [11] | ||||||
| Kubitza2006 | PKDB00989 | single, multi | 5, 20 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [6] | |||||
| Kubitza2013 | PKDB00990 | single | 10 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [16] | |||||
| Liu2024 | PKDB00991 | single | 10, 15, 20 | ✓ | ✓ | ✓ | ✓ | ✓ | [7] | ||||||
| Moore2014 | PKDB00992 | single | 10 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [17] | |||||
| Weinz2009 | PKDB00993 | single | 10 | ✓ | ✓ | ✓ | ✓ | ✓ | [33] | ||||||
| Zhao2009 | PKDB00994 | single, multi | 2.5, 5, 10, 20, 40 | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | [13] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



