
Submitted:
01 August 2025
Posted:
05 August 2025
You are already at the latest version
Abstract
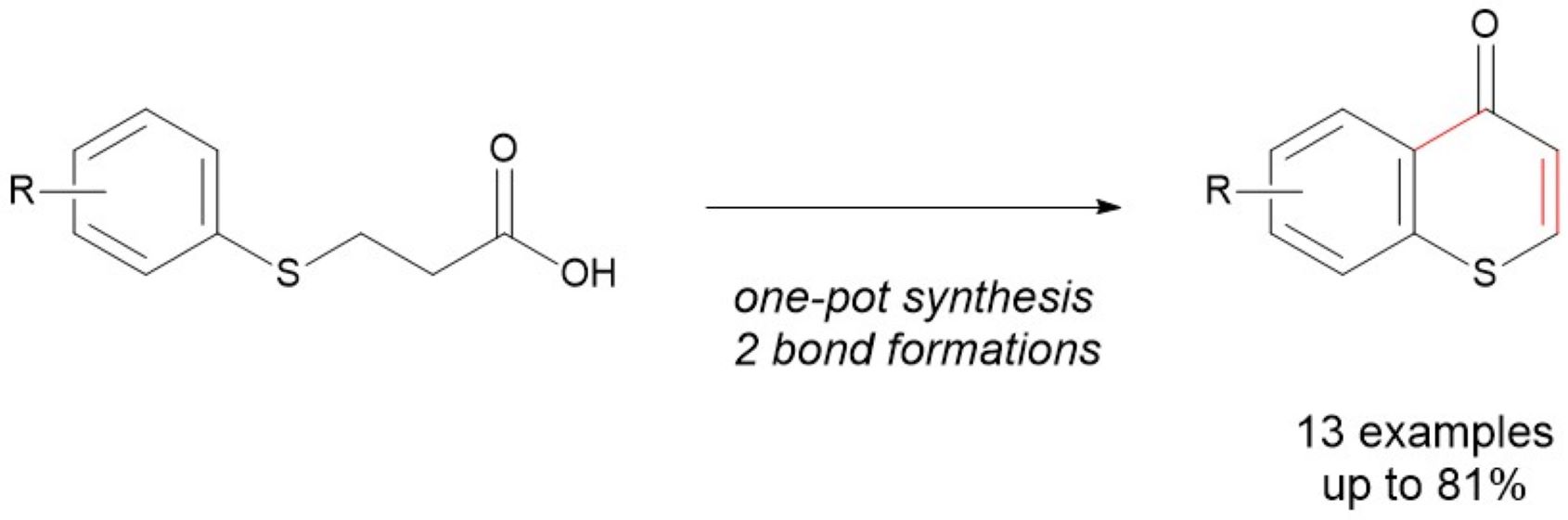
Thiochromones are known to possess useful optical properties and rich bioactivities, including antioxidant, antimicrobial, and anticancer properties. They are known to inhibit tumor cell growth, induce apoptosis, and have antiplatelet aggregation effects. Thiochromones are also used as synthons and precursors in organic synthesis for bioactive agents. Although many synthetic approaches to oxygen containing counterparts, chromones, have been reported, research on the synthesis of thiochromones are scarce. The synthesis of thiochromones can be challenging due to the inherent nature of sulfur, including its multiple oxidation states and tendency to form diverse bonding patterns. Here we report the one-pot synthesis of thiochromone, where two transformations of the starting material, 3-(arylthiol)propanoic acid, are performed within a single reaction vessel, eliminating the need for intermediate purification step. This one-pot reaction worked well with a variety of substrates with both electron withdrawing and donating groups on the aromatic ring of 3-(arylthiol)propanoic acids to give thiochromone with good yields (up to 81%). This approach offers advantages like time and cost savings, increased efficiency, and reduced waste. This synthetic approach will allow access to a broader scope of thiochromones due to the readily available thiophenols. The thiochromones can be utilized for additional synthetic applications for the synthesis of thioflavanones and 2-alkylthiochroman-4-ones, an important class of sulfur-heterocycles with rich biological activities.
Keywords:
thiochromone
; 3-(arylthiol)propanoic acids
; thiochroman-4-one
; one-pot synthesis
1. Introduction
Chromones are an important class of heterocycles and known as privileged scaffolds [1,2,3] in medicinal chemistry due to their wide range of biological activities including antioxidants [4], antiprotozoal [5] or anticancer agents [6,7,8]. Synthetic approaches to chromones have been extensively reported [9]. Thiochromones [10] are the sulfur analogs of chromone, in which the O-1 atom is replaced by a sulfur atom. The sulfur containing heterocyclic compounds benzothiopyrans or thiochromones stand out as having promising biological activities due to their structural relationship with chromones (benzopyrans), which are widely known as privileged scaffolds in medicinal chemistry [1,2,3,4,5,6,7,8]. However, the sulfur containing thiochromones have been significantly less explored presumably due to the reduced reactivity of the thiopyrone moiety as the result of the replacement of oxygen by sulfur atom. Thiochromones are known to possess useful optical properties and rich bioactivities, including antioxidant, antimicrobial, and anticancer properties. They are known to inhibit tumor cell growth, induce apoptosis, and have antiplatelet aggregation effects [11,12]. Thiochromones are also used as synthons and precursors in organic synthesis for useful sulfur heterocycles and other bioactive agents [13]. In the past several years, we have reported the conjugate addition of organometallic reagents to thiochromones in the synthesis of thioflavanones, thiochroman-4-ones with additional synthetic applications [14,15,16,17]. Although many synthetic approaches to oxygen containing counterparts, chromones, have been reported, [9] research on the synthesis of thiochromones are scarce. The synthesis of thiochromones can be challenging due to the inherent nature of sulfur, including its multiple oxidation states and tendency to form diverse bonding patterns. One of the earliest methods for the preparation of 2,3-unsubstituted thiochromone involved the bromination of thiochroman-4-one and subsequent dehydrohalogenation to give the desired thiochromone (Figure 1, A, a) [18].
Another method utilized a 3-component synthesis of 2,3-unsubstituted thiochromones from o-haloaroyl chlorides, trimethylsilylacetylene, and sodium sulfide nonahydrate with modest yields (35-39%, Figure 1, A, b) [19,20]. Another synthetic approach to thiochromone involved a key intermediate (Z)-3-arylthioacrylic acids, which were synthesized from aryl halides, sodium sulfide pentahydrate, and propiolic acid. The subsequent Friedel-Crafts acylation reaction of (Z)-3-arylthioacrylic acids under treatment with sulfuric acid at 100 °c to give thiochromones in good yields (51-80%, Figure 1, A, c) [21]. However, accessing a broad scope of thiochromones, a class of sulfur-containing heterocycles, remains a significant synthetic challenge. Here we report the one-pot synthesis [22] of thiochromone (Figure 1, B), where two transformations of the starting material, 3-(arylthiol)propanoic acid, are performed within a single reaction vessel, eliminating the need for intermediate purification step. This approach offers advantages like time and cost savings, increased efficiency, and reduced waste.
2. Results and Discussions
The starting 3-(arylphenylthio)propanoic acids 1 were prepared according to procedure previously reported in the literature [23,24]. Arylthiols were purchased from commercial source and used as received without further purification. A 250 mL flask was charged with a stirrer bar, aqueous NaOH (25 mL, 1.0 M) and aqueous Na2CO3 (25 mL, 1.0 M). To the above solution, arylthiols (50 mmol) was then added as solution in 30 mL EtOH followed by the addition of 3-chloropropanoic acid (51 mmol) as an aqueous solution in 20 mL water. The resultant reaction mixture was stirred at room temperature for 2 hrs, and then heated to reflux in an oil bath for overnight (12 hrs). The resultant mixture was then cooled down to room temperature, EtOH was then evaporated under rotovap, and then the aqueous phase was acidified to with conc. HCl (6.0 M) to pH 1~2. The solution was diluted with H2O (30 mL), and extracted with CH2Cl2 for three times (3 X 40 mL), and the organic layers were combined, dried (Na2SO4), filtered, and concentrated to get crude product. It was purified by flash column chromatography (ethyl acetate/hexanes, 5% to 30%) to furnish the desired 3-(arylthio)propanoic acid 1 in good chemical yield (Figure 2, 80-93%).
We started our investigation with 3-(4-MeOphenylthiol)propanoic acid 1a and concentrated sulfuric acid. Upon treatment of 3-(4-MeOphenylthiol)propanoic acid 1a in DCM (1.0 mL, added to help dissolve solid starting material before cooling down in ice bath and the addition of concentrated sulfuric acid) with excess concentrated sulfuric acid (95-97%, 0.5 mL) at 0 oC using ice bath, no desired thiochromone 3a was observed after warming up to room temperature and stirred for 12 hrs. Instead, it was found that demethylated thiochroman-4-one was attained in modest yield (Table 1, entry 1). Then excess amount of concentrated phosphoric acid (85%, 0.5 mL) was added to 3-(4-MeOphenylthiol)propanoic acid 1a in DCM (1.0 mL, added to help dissolve solid starting material before cooling down in ice bath and the addition of concentrated phosphoric acid), similar result was attained as no desired thiochromone 3a was observed (Table 1, entry 2). Similarly, demethylated thiochroman-4-one was observed. These results indicated that strong acids such as concentrated sulfuric acid and phosphoric acid are not tolerant with methoxy (MeO-) substituent on the 3-(4-MeOphenylthiol)propanoic acid 1a. So, a weaker acid polyphosphoric acid (PPA) was then deployed instead. DCM was added to help dissolve the solid starting material and very viscous PPA so that the magnetic stir bar can stir. The start material 3-(4-MeOphenylthiol)propanoic acid 1a is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well using the magnetic a stir bar (the magnetic stir bar would not stir due to the high viscosity of PPA and solid starting material). So, 1.0 mL of DCM was used to dissolve the starting material 3-(4-MeOphenylthiol)propanoic acid initially at RT before cooling in an ice bath and the addition of very viscous PPA at 0 oC. When excess amount of polyphosphoric acid (PPA, 0.5 mL) was added under similar conditions (0 oC using ice bath), no reaction was observed after warming to room temperature and stirred for 12 hrs as we only recovered the starting material (Table 1, entry 3). When the reaction temperature is above the boiling point of DCM (Table 1, entries 4-8), 1.0 mL of DCM was used to dissolve the starting material 3-(4-MeOphenylthiol)propanoic acid initially at room temperature and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the mixture is heated to the boiling point of DCM (40 oC). Subsequently, when the reaction was heated to 50 oC for 2 hrs with excess polyphosphoric acid (PPA, 0.5 mL), the corresponding thiochroman-4-one 2a was attained in low yield (entry 4, 20%) but no desired thiochromone 3a was observed. Slightly higher yield of thiochromone-4-one 2a was attained when the reaction was heated to 50 oC for 4 hrs with excess polyphosphoric acid (PPA, 0.5 mL) (entry 5, 25%, no desired thiochromone 3a). Although no desired thiochromone 3a was observed, the yield of thiochromone-4-one 2a was significantly increased when the reaction mixture was heated at 50 oC for extended period of time (entry 6, 12 hrs). Finally, we were delighted to see the formation of thiochromone 3a when the reaction temperature was further increased to 100 °C (entry 7). The highest yield can be attained with 3-(4-MeOphenylthiol)propanoic acid and polyphosphoric acid (PPA) at 100 °C for extended period of time (entry 8)
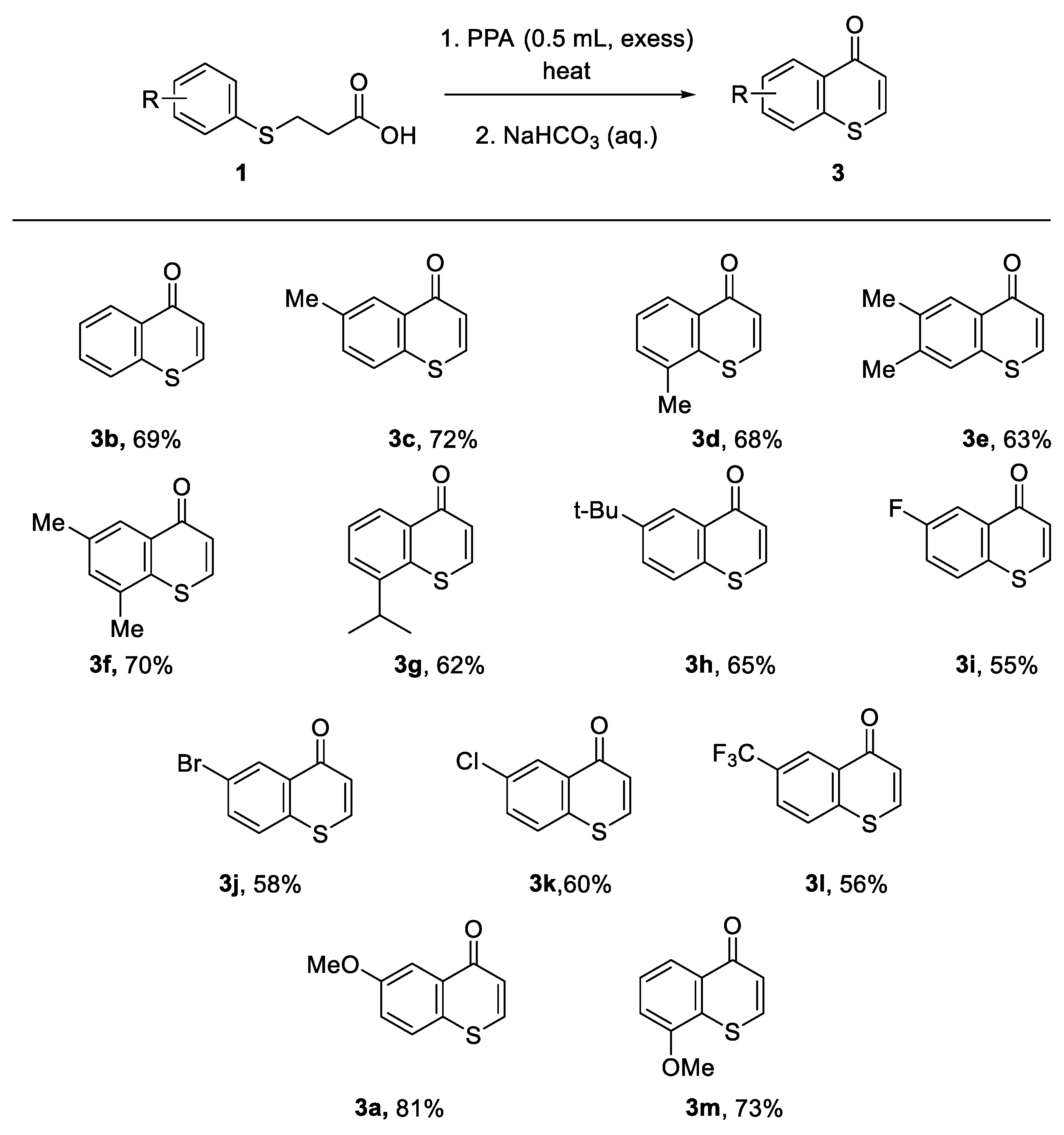
Having found the optimal reaction condition for the one-pot synthesis of thiochromone, we next turned our attention to explore the scope of this reaction. A variety of 3-(arylthiol)propanoic acids with both electron withdrawing and donating group were investigated. It was found that 3-(arylthiol)propanoic acids with both electron withdrawing and donating group on the aromatic ring undergo the one-pot reaction to afford thiochromones 3a-3m with 55–81% yields (Scheme 1). 3-(Phenylthiol)propanoic acid worked well under the optimal one-pot synthesis reaction condition to give thiochromone 3b in good yield. 3-(Arylthiol)propanoic acids bearing simple alkyl substituents on aromatic ring, such as methyl group, reacted well to afford 3c-f in 63–72% yields (Scheme 1). Steric hindrance is not a factor as slightly bulky substituents such as i-Pr worked well to furnish the desired thiochromone 3g in 62% yield. Bulky t-butyl group is also tolerated to afford the corresponding 3h in good yield (Scheme 1, 65%). 3-(Arylthiol)propanoic acids with halides F, Br, and Cl also work well under this one-pot reaction condition but with slightly lower chemical yields (Scheme 1, 55–60%). Strong electron-withdrawing group trifluoromethyl also worked with slightly lower chemical yield (Scheme 1, 3l, 56%). 3-(Arylthiol)propanoic acids with electron-donating groups, such as MeO-, also work well to afford thiochromones 3a (81%) and 3m (73%) in good yields (Scheme 1).
Thiochromones have become increasingly valuable synthons and vital precursors in organic synthesis for useful sulfur heterocycles and other bioactive agents [13]. In the past several years, we have reported the conjugate addition of organometallic reagents to thiochromones in the synthesis of thioflavanones and 2-alkylthiochroman-4-ones demonstrating the synthetic applications of thiochromones [14,15,16,17]. For example, thiochromones have been reported to be used as the vital precursor for the synthesis of a variety of thioflavanones [17], an important class of sulfur-containing heterocycles with rich biological activities including the abilities to significantly inhibit cellular proliferation with weak cytotoxicity and induce apoptosis in human breast cancer cells [25,26]. We have also previously reported the synthesis of a large scope of 2-alkylthiochroman-4-one using thiochromones as the vital precursors [14,16]. Here are some of the synthetic applications of thiochromones for the synthesis of a variety of thioflavanones and 2-alkylthiochroman-4-ones that have been reported by our research group [Scheme 2]. A variety of 2-alkylthiochroman-4-ones 4a-i with both electron-donating and -withdrawing on the aromatic ring can be synthesized in high chemical yields using thiochromones 3 (Scheme 2, 75-86%). A number of thioflavanones 5a-i with both electron-donating and -withdrawing on the aromatic ring can be synthesized from thiochromones 3 in high chemical yields (Scheme 2, 77-89%).
3. Materials and Methods
3.1. General Methods
The 1H and 13C-NMR spectra were recorded on a BRUKER AscendTM 400 NMR spectrometer, operating at 400 MHz for 1H and 100 MHz for 13C. Samples for NMR spectra were dissolved in deuterated chloroform (with TMS). Analytical thin layer chromatography (TLC) was performed on silica gel plates, 60 µ mesh with F254 indicator. Visualization was accomplished by UV light (254 nm), and/or a 10% ethanol solution of phosphomolybdic acid and/or KMnO4 stain prepared by dissolving 1.5 g KMnO4, 10 g potassium carbonate, and 1.25 mL 10% sodium hydroxide in 200 mL water. Flash chromatography was performed with 200–400 µ silica gel.
3.2. Materials
All glassware was flamed-dried under high vacuum and purged with argon to cool to room temperature. Chemicals and solvents were obtained from commercial sources and used without further purification unless stated otherwise. TMSCl was distilled from CaH2 under a positive N2 atmosphere. Grignard reagents were purchased from Sigma Aldrich or prepared from the corresponding bromocompounds. Anhydrous tetrahydrofuran (THF) was purchased from Sigma Aldrich (Sigma-Aldrich, St. Louis, MO, USA). Low-temperature baths were prepared using dry ice-isopropanol slush bath mixtures. All 1,4-conjugate addition reactions with Grignard reagents were conducted under a positive dry argon atmosphere in anhydrous solvents in flasks fitted with a rubber septum.
3.3. General Procedures
3.3.1. General Procedures A
A round bottom flask with a stir bar was charged with 3-(arylthiol)propanoic acids (1, 1.0 mmol, 1.0 equivalent, Scheme 1), were added dichloromethane (1.0 mL) and polyphosphoric acid (PPA, 0.5 mL, excess). DCM was added to help dissolve the solid starting material and very viscous PPA. The start material 3-(arylthiol)propanoic acids is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well. So, 1.0 mL of DCM was used to dissolve the starting material 3-(arylthiol)propanoic acids initially at RT and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the reaction mixture was heated to the boiling point of DCM (40 oC). The result mixture was then heated to 100 °C (oil bath temperature) and the reaction was monitored by TLC. Once the TLC monitoring showed complete consumption of starting material, the reaction mixture was allowed to cool down to room temperature. An aqueous saturated NaHCO3 solution was then added dropwise (5.0 mL), and the resultant mixture was allowed to stir for 2 hours at room temperature. It was then extracted with dichloromethane (3 X 15.0 mL). The organic layers were combined, dried with Na2SO4, filtered, and evaporated by vacuum to give crude product. The crude product was purified by column chromatography on silica gel with a mixture of hexanes/ethyl acetate as eluent to give the product thiochromone 3 (Scheme 1) in 55-81% yield.
3.3.2. General Procedures B: Synthetic Applications of Thiochromones - Conjugate Addition Reactions of Grignard Reagents (RMgX or ArMgX; X = Cl or Br) to Thiochromones Catalyzed by CuCN∙2LiCl (0.2 eq)
CuCN (9.0 mg, 0.1 mmol, 0.2 equivalent) was added to flame-dried LiCl (8.5 mg, 0.2 mmol, 0.4 equivalent) in a 50 mL round bottom flask under argon. Then THF (1.0 mL) was added and the resultant mixture was stirred for 10 min at room temperature and then cooled to 0 °C in an ice bath followed by the addition of a Grignard reagent (0.75 mmol, 1.5 equivalent). The resultant solution was stirred for an additional 30 min at 0 °C under argon. Then thiochromones were added as a mixture with TMSCl in THF [0.5 mmol mixed with TMSCl (1.0 mmol) in THF 1.0 mL)]. After the resultant reaction mixture was allowed to warm up to room temperature during overnight stirring (12 hrs), the reaction mixture was quenched with saturated aqueous NH4Cl (10.0 mL) and extracted with ethyl acetate (3 × 10.0 mL). The combined organic phase was subsequently washed with brine (15.0 mL), dried (Na2SO4), filtered, concentrated in vacuo, and purified by flash column chromatography (silica gel, 0 - 2% ethyl acetate in hexane, v/v) to give pure compounds.
3.4. Synthesis
HRMS data for compounds 3a, 3d, 3f, 3g, 3m, and 4c were analyzed by TOF MS. Compounds 3b, 3c, 3e, and 3h-l have been fully characterized and reported [21]. Compounds 4a, 4b, 4d-i, and 5a-i have been fully characterized and reported by our group [14,16,17]. (The NMR spectra and Mass spectra for new compounds are included in Supplementary materials)
3.4.1. Synthesis of 6-Methoxylthiochromone (3a)
Employing General Procedure A and using 3-(4-methoxylphenyl)propanoic acids (212 mg, 1.0 mol), after purification by flash column chromatography (silica, 5-10% Ethyl acetate : hexanes, v/v) gave 3a (155 mg, 81%). 1H NMR (400 MHz, CDCl3): δ 3.92 (s, 3H), 7.13 (dd, J = 0.8, 10.4 Hz, 1 H), 7.26 (dd, J = 2.8, 8.8 Hz, 1 H), 7.55 (d, J = 8.8 Hz, 1 H), 7.91 (d, J = 10.4 Hz, 1 H), 7.99 (d, J = 2.8 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ 56.1, 108.7, 123.5, 123.7, 128.3, 131.3, 132.4, 141.3, 160.4, 179.0; HRMS (EI-ion trap) m/z: [M]+ calcd. for C10H8O2S, 192.0245; found 192.0241.
3.4.2. Synthesis of 8-Methylthiochromone (3d)
Employing General Procedure A and using 3-(2-methylphenyl)propanoic acids (196 mg, 1.0 mol), after purification by flash column chromatography (silica, 5-10% Ethyl acetate : hexanes, v/v) gave 3d (119 mg, 68%). 1H NMR (400 MHz, CDCl3): δ 2.30 (s, 3H), 6.85 (d, J = 10.4 Hz, 1 H), 7.19-7.28 (m , 2H), 7.64 (d, J = 10.4 Hz, 1 H), 8.21 (ddd, J = 0.8, 2.4, 7.2 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ 19.7, 125.6, 126.7, 127.4, 132.6, 132.8, 135.0, 137.4, 137.8, 180.3; HRMS (EI-ion trap) m/z: [M]+ calcd. for C10H8OS, 176.0296; found 176.0293.
3.4.3. Synthesis of 6, 8-Dimethylthiochromone (3f)
Employing General Procedure A and using 3-(2,4-dimethylphenyl)propanoic acids (210 mg, 1.0 mol), after purification by flash column chromatography (silica, 5-10% Ethyl acetate : hexanes, v/v) gave 3f (133 mg, 70%). 1H NMR (400 MHz, CDCl3): δ 2.38 (s, 3H), 2.42 (s, 3H), 6.97 (d, J = 10.4 Hz, 1 H), 7.24 (s , 1H), 7.78 (d, J = 10.4 Hz, 1 H), 8.18 (s, 1 H); 13C NMR (100 MHz, CDCl3): δ 19.6, 21.3, 125.5, 126.4, 132.6, 134.2, 134.4, 134.8, 137.3, 137.5, 180.4; HRMS (EI-ion trap) m/z: [M]+ calcd. for C11H10OS, 190.0452; found 190.0455.
3.4.4. Synthesis of 8-Isopropylthiochromone (3g)
Employing General Procedure A and using 3-(2-methylphenyl)propanoic acids (224 mg, 1.0 mol), after purification by flash column chromatography (silica, 5-10% Ethyl acetate : hexanes, v/v) gave 3g (126 mg, 62%). 1H NMR (400 MHz, CDCl3): δ 1.38 (s, 3H), 1.40 (s, 3H), 3.43 (septet, J = 6.8 Hz, 1H), 7.04 (d, J = 10.4 Hz, 1 H), 7.55 (t, J = 8 Hz, 1 H), 7.61 (dd, J = 1.6, 7.2 Hz, 1 H), 7.88 (d, J = 10.4 Hz, 1 H), 8.48 (dd, J = 1.6, 8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ 23.2, 30.5, 124.5, 127.0, 128.1, 128.7, 132.2, 136.7, 139.4, 145.8, 180.5; HRMS (EI-ion trap) m/z: [M]+ calcd. for C12H12OS, 204.0609; found 204.0611.
3.4.5. Synthesis of 8-Methoxylthiochromone (3m)
Employing General Procedure A and using 3-(2-methoxylphenyl)propanoic acids (212 mg, 1.0 mol), after purification by flash column chromatography (silica, 5-10% Ethyl acetate : hexanes, v/v) gave 3m (140 mg, 73%). 1H NMR (400 MHz, CDCl3): δ 4.00 (s, 3H), 7.06-7.18 (m, 2 H), 7.50 (t, J = 8.0 Hz, 1 H), 7.92 (d, J = 10.4 Hz, 1 H), 8.16 (dd, J = 0.8, 8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3): δ 56.7, 111.7, 120.7, 125.2, 128.2, 128.7, 133.0, 139.6, 155.1, 179.8; HRMS (EI-ion trap) m/z: [M]+ calcd. for C10H8O2S, 192.0245; found 192.0249.
3.4.6. Synthesis of 2-Cyclopropylthiochroman-4-one (4c)
Employing General Procedure B, using cyclobutylmagnesium bromide (1.0 M, 0.75 mL, 0.75 mmol) and thiochromone (81 mg, 0.5 mmol), after purification by flash column chromatography (silica gel, 0–2% ethyl acetate: hexanes, v/v), gave a light-yellow solid 4c (89 mg, 82%): IR (neat) 3055 (w), 3001 (w), 2922 (w), 1672 (s), 1591 (s), 1455 (w), 1433 (s), 1393 (w), 1283 (s), 1228 (m), 1153 (w), 1067 (w), 1022 (w), 954 (w) cm−1; 1H NMR (400 MHz, CDCl3) δ 1.65-1.85 (m, 4 H),1.98-2.10 (m, 2H), 2.41-2.53 (M, 1H), 2.62 (dd, J = 10.4, 16.4 Hz, 1 H), 2.90 (dd, J = 3.2, 16.4 Hz, 1 H), 3.34 (dt, J = 3.2, 10.4 Hz, 1 H), 7.06 (ddd, J = 1.2, 7.2, 9.2 Hz, 1H), 7.17 (ddd, J = 0.4, 1.2, 8.0 Hz, 1H), 7.28 (ddd, J = 1.2, 6.8, 8.0 Hz, 1H), 7.99 (ddd, J = 0.4, 1.6, 8.0 Hz, 1H); 13C NMR (100 MHz, CDCl3) δ 17.6, 26.4, 26.6, 38.7, 43.2, 47.3, 124.8, 127.7, 128.9, 130.8, 133.4, 141.6, 194.5; HRMS (EI-ion trap) m/z: [M + 1]+ calcd for C13H15OS, 219.0844; found 219.0840.
3.4.7. Synthesis of 8-Methyl-2-n-butylthiochroman-4-one (4f) [16]
Employing General Procedure B and using 8-methylthiochromone (88 mg, 0.50 mmol) and n-BuMgCl (2.8 M, 0.28 mL, 0.75 mmol), after purification by flash column chromatography (silica gel, 0–2% ethyl acetate:hexanes, v/v) gave light yellow oil 4f (96 mg, 82%); IR (neat) 3061 (w), 2955 (m) 2926 (s), 2857 (m), 1676 (s), 1583 (m), 1449 (m), 1401 (m), 1379 (w), 1295 (m), 1279 (m), 1248 (w), 1056 (w), 1000 (w), 841 (w) 784 (w), 722 (w) cm−1; 1H-NMR (400 MHz, CDCl3) δ 0.73 (t, J = 7.2 Hz, 3H), 1.11–1.21 (m, 2H), 1.22–1.35 (m, 2H), 1.51–1.60 (m, 2H), 2.12 (s, 3H), 2.58 (dd, J = 11.6, 16.0 Hz, 1H), 2.83 (dd, J = 2.8, 16.0 Hz, 1H), 3.21–3.29 (m, 1H), 6.88 (t, J = 7.6 Hz, 1H), 7.09 (qd, J = 0.8, 8.0 Hz, 1H), 7.78 (qd, J = 0.4, 8.0 Hz, 1H); 13C-NMR (100 MHz, CDCl3) δ 13.9, 20.1, 22.4, 28.8, 34.4, 40.7, 45.7, 123.9, 126.6, 130.8, 134.5, 135.4, 141.3, 195.2; HRMS (EI-ion trap) m/z: [M]+ calcd. for C14H18OS, 234.1078; found 234.1075.
3.4.8. Synthesis of 6-Isopropyl-2-phenylthiochroman-4-one (5g) [14]
Employing General Procedure B, using phenylmagnesium bromide (2.8 M, 0.28 mL, 0.75 mmol) and thiochromone (101 mg, 0.50 mmol), after purification by flash column chromatography (silica gel, 0–2% ethyl acetate: hexanes, v/v), gave a yellow solid 5g (109 mg, 77%): mp 78–79 °C; IR (neat) 3056 (w), 2965 (s), 2927 (m), 1673 (s), 1576 (m), 1489 (w), 1451 (w), 1411 (m), 1286 (w), 1262 (s), 1147 (w), 1973 (w), 1042 (w), 780 (w), 766 (w), 731 (w), 698 (w) cm−1; 1H NMR (400 MHz, CDCl3) δ 1.24 (d, J = 6.8 Hz, 3 H), 1.31 (d, J = 6.8 Hz, 3 H), 3.23 (dd, J = 2.8, 16.4 Hz, 1 H), 3.26–3.34 (m, 1H), 3.35 (dd, J = 13.6, 16.4 Hz, 1 H), 4.66 (dd, J = 2.8, 13.6 Hz, 1 H), 7.25 (dd, J = 7.6, 15.2 Hz, 1H), 7.34–7.51 (m, 6H), 8.09 (dd, J = 1.6, 8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 22.7, 23.1, 30.3, 44.9, 46.3, 124.8, 127.2, 127.5, 128.5, 129.0, 130.3, 130.9, 138.7, 140.4,, 145.8, 195.1; HRMS (EI-ion trap) m/z: [M + 1]+ calcd for C18H19OS, 283.1157; found 283.1147.
3.4.9. Synthesis of 8-Methoxy-2-phenylthiochroman-4-one (5i) [14]
Employing General Procedure B, using phenylmagnesium bromide (2.8 M, 0.28 mL, 0.75 mmol) and thiochromone (95 mg, 0.50 mmol), after purification by flash column chromatography (silica gel, 0–2% ethyl acetate: hexanes, v/v), gave a light-yellow solid 5i (109 mg, 81%): mp 147–148 °C; IR (neat) 3017 (w), 2972 (W), 2936 (w), 1673 (s), 1579 (m), 1559 (m), 1451 (m), 1420 (m), 1316 (m), 1254 (s), 1155 (w), 1055 (w), 1031 (s), 789 (m), 770 (m), 712 (w), 697 (m) cm−1; 1H NMR (400 MHz, CDCl3) δ 3.08 (dd, J = 2.8, 16.0 Hz, 1 H), 3.21 (dd, J = 13.6, 16 Hz, 1 H), 3.82 (s, 3 H), 4.54 (dd, J = 2.8, 13.6 Hz, 1 H), 6.91 (dd, J = 1.2, 8.0 Hz, 1 H), 7.09 (t, J = 8.0 Hz, 1 H), 7.22–7.32 (m, 3H), 7.33–7.38 (m, 2H), 7.71 (dd, J = 1.2, 8.0 Hz, 1 H); 13C NMR (100 MHz, CDCl3) δ 44.4, 46.0, 56.3, 114.2, 121.1, 124.8, 127.5, 128.5, 129.0, 131.3, 131.9, 138.7, 155.1, 194.6; HRMS (EI-ion trap) m/z: [M + 1]+ calcd for C16H15O2S, 271.0793; found 271.0794.
4. Conclusions
In conclusion, we successfully developed the one-pot synthesis of thiochromones from 3-(arylthiol)propanoic acids. This reaction was shown to work well with a broad range of substrates with both electron withdrawing and donating groups on the aromatic ring of 3-(arylthiol)propanoic acids. 3-(arylthiol)propanoic acids with both electron withdrawing and donating group on the aromatic ring undergo the one-pot reaction to afford thiochromones with good chemical yields (56-81% in one-pot synthesis). With this one-pot approach, two transformations were performed within a single reaction vessel, eliminating the need for intermediate purification step. This one-pot approach offers advantages like time and cost savings, increased efficiency, and reduced waste. This synthetic approach will allow access to a broader scope of thiochromones due to the readily available thiophenols. The thiochromones can be utilized for additional synthetic applications for the synthesis of thioflavanones and 2-alkylthiochroman-4-ones, an important class of sulfur-heterocycles with rich biological activities.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
For research articles with several authors, a short paragraph specifying their individual contributions must be provided. The following statements should be used “Conceptualization, F. Guo.; methodology, F. Guo, H. A. H.; validation, K. S. S., M. Y. G., T. D. S., H. A. H.. and F.G.; investigation, K. S. S., M. Y. G., T. D. S., H. A. H.. and F.G.; data curation, K. S. S., M. Y. G., T. D. S., H. A. H.. and F.G.; writing—F. Guo; supervision, F. Guo; project administration, F. Guo; funding acquisition, F. Guo. All authors have read and agreed to the published version of the manuscript.” Please turn to the CRediT taxonomy for the term explanation. Authorship must be limited to those who have contributed substantially to the work reported.
Funding
We thank the NIH National Institute of General Medical Sciences (R16GM145541) for generous financial support. We would also like to thank the National Science Foundation (Award no. 2433178) for financial support.
Data Availability Statement
Data is available via supporting information.
Acknowledgments
We thank Dr. Marcus Wright from the Chemistry Department, Wake Forest University, Winston-Salem for access to NMR facility and assistance in attaining NMR spectra.
Conflicts of Interest
Declare conflicts of interest or state “The authors declare no conflicts of interest.”.
References
- Costa, M.; Dias, T.A.; Brito, A.; Proença, F. Biological importance of structurally diversified chromenes. Eur. J. Med. Chem. 2016, 123, 487–507.
- Keri, R.S.; Budagumpi, S.; Pai, R.K.; Balakrishna, R.G. Chromones as a privileged scaffold in drug discovery: A review. Eur. J. Med. Chem. 2014, 78, 340–374.
- Welsch, M.E.; Snyder, S.A.; Stockwell, B.R. Privileged scaffolds for library design and drug discovery. Curr. Opin. Chem. Biol. 2010, 14, 347–361.
- Csepanyi, E.; Szabados-Furjesi, P.; Kiss-Szikszai, A.; Frensemeier, L.M.; Karst, U.; Lekli, I.; Haines, D.D.; Tosaki, A.; Bak, I. Antioxidant properties and oxidative transformation of different chromone derivatives. Molecules 2017, 22, 588.
- Presley, C.C.; Valenciano, A.L.; Fernández-Murga, M.L.; Du, Y.; Shanaiah, N.; Cassera, M.B.; Goetz, M.; Clement, J.A.; Kingston, D.G.I. Antiplasmodial chromanes and chromenes from the monotypic plant species Koeberlinia spinosa. J. Nat. Prod. 2018, 81, 475-483.
- Payen, L.; Honorat, M.; Guitton, J.; Gauthier, C.; Bouard, C.; Lecerf-Schmidt, F.; Peres, B.; Terreux, R.; Gervot, H.; Rioufol, C.; et al. MBL-II-141, a chromone derivative, enhances irinotecan (CPT-11) anticancer efficiency in ABCG2-positive xenografts. Oncotarget 2014, 5, 11957–11970.
- China Raju, B.; Nageswara Rao, R.; Suman, P.; Yogeeswari, P.; Sriram, D.; Shaik, T.B.; Kalivendi, S.V. Synthesis, structure-activity relationship of novel substituted 4H-chromen-1,2,3,4-tetrahydropyrimidine-5-carboxylates as potential anti-mycobacterial and anticancer agents. Bioorg. Med. Chem. Lett. 2011, 21, 2855–2859.
- Shaw, A.Y.; Chang, C.Y.; Liau, H.H.; Lu, P.J.; Chen, H.L.; Yang, C.N.; Li, H.Y. Synthesis of 2-styrylchromones as a novel class of antiproliferative agents targeting carcinoma cells. Eur. J. Med. Chem. 2009, 44, 2552–2562.
- Li, N.-G.; Shi, Z.-H.; Tang, Y.-P.; Ma, H.-Y.; Yang, J.-P.; Li, B.-Q.; Wang, Z.-J.; Song, S.-L.; Duan, J.-A. Synthetic strategies in the construction of chromones. J. Heterocycl. Chem. 2010, 47, 785-799.
- Sosnovskikh, V. Y. Synthesis and chemical properties of thiochromone and its 3-subsituted derivatives. Chemistry of Heterocyclic compounds 2016, 52, 427-440.
- Wang, H.-K.; Bastow, K. F.; Cosentino, L. M.; Lee, K.-H. J. Med. Chem. 1996, 39, 1975.
- Schneller, S. W. Adv. Heterocycl. Chem. 1975, 18, 79.
- Guo, F.; Young, J.A.; Perez, M.S.; Hankerson, H.A.; Chavez, A.M. Progress on the Cu-Catalyzed 1,4-Conjugate Addition to Thiochromones. Catalysts 2023, 13, 713.
- Bellinger, T.J.; Harvin, T.; Pickens-Flynn, T.; Austin, N.; Whitaker, S.H.; Tang Yuk Tutein, M.L.C.; Hukins, D.T.; Deese, N.; Guo, F. Conjugate Addition of Grignard Reagents to Thiochromones Catalyzed by Copper Salts: A Unified Approach to Both 2-Alkylthiochroman-4-One and Thioflavanone. Molecules 2020, 25, 2128.
- In Lee, J. Synthetic Approaches to 2-Alkylthiochroman-4-ones and Thioflavanones. Bull. Korean Chem. Soc. 2021, 42, 852–862.
- Bass, S.A.; Parker, D.M.; Bellinger, T.J.; Eaton, A.S.; Dibble, A.S.; Koroma, K.L.; Sekyi, S.A.; Pollard, D.A.; Guo, F. Development of Conjugate Addition of Lithium Dialkylcuprates to Thiochromones: Synthesis of 2-Alkylthiochroman-4-ones and Additional Synthetic Applications. Molecules 2018, 23, 1728.
- Guo, F.; Jeffries, M.C.; Graves, B.N.; Graham, S.A.; Pollard, D.A.; Pang, G.; Chen, H.Y. A rapid entry into thioflavanones via conjugate additions of diarylcuprates to thiochromones. Tetrahedron 2017, 73, 5745–5750.
- Nakazumi, H.; Endo, T.; Nakaue, T.; Kitao, T. J. Heterocycl. Chem. 1985, 22, 89.
- Willy, B.; Müller, T. J. J. A novel consecutive three-component Coupling-Addition-SNAr (CASNAR) synthesis of 4H-thiochromen-4-ones, Synlett. 2009, 1255-1260.
- Willy, B.; Frank, W.; Müller, T. J. J. Microwave-assisted three-component coupling-addition-SNAr (CASNAR) sequences to annelated 4H-thiopyran-4-ones. Org. Biomol. Chem. 2010, 8, 90.
- Palani, T.; Park, K.; Song, K. H.; Lee, S. Palladium-catalyzed synthesis of (Z)-3-arylthioacrylic acids and thiochromenones, Adv. Synth. Catal. 2013, 355, 1160.
- Hayashi, Y. Pot economy and one-pot synthesis, Chem. Sci., 2016, 7, 866–880.
- Giles, P. R.; Marson, C. M. Aust. J. Chem. 1992, 45, 439-443.
- Jia, W.; Y.-J.; Li, W.; Liu, Y.; Zhang, D.-J.; Zhang, P.; Gong, P. Bioorg. Med. Chem. 2009, 17, 4569-4574.
- Choi, E.J., Lee, J.I., Kim, G.H. Int. J. Mol. Med., 2012, 29, 252-256.
- Song, Y.-L., Wu, F., Zhang, C.-C., Liang, G.-C., Zhou, G., Yu, J.-J. Bioorg Med Chem Lett, 2015, 25, 259-261.
Figure 1.
Synthesis of Thiochromones.
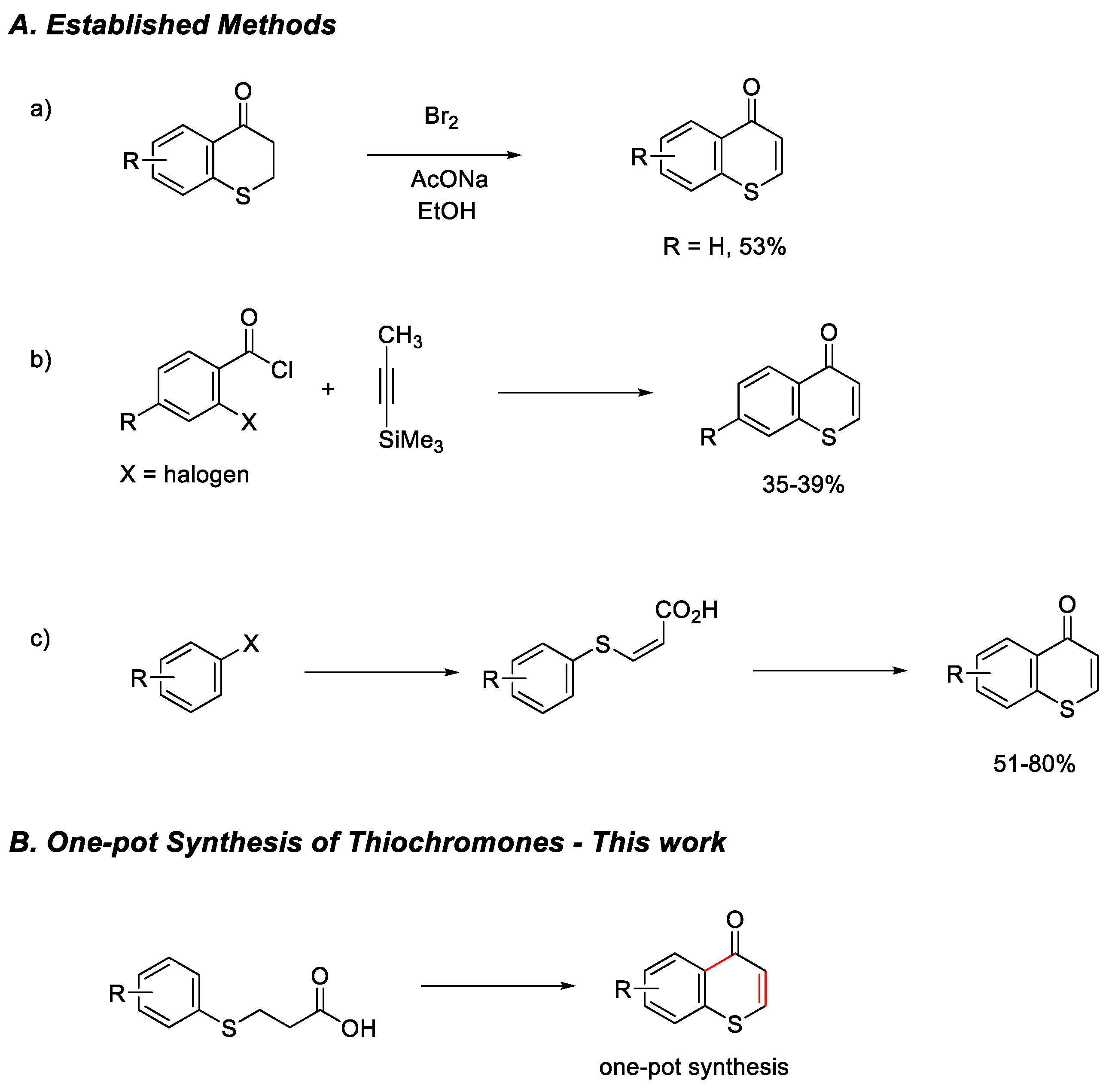
Figure 2.
Preparation of 3-(arylphenylthio)propanoic acid.
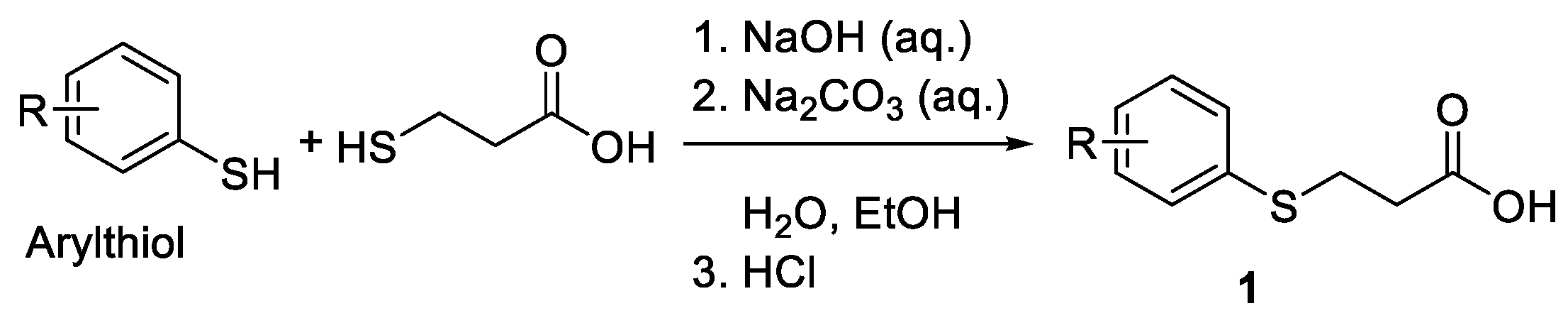
Scheme 1.
The scope of one-pot reaction of 3-(Arylthiol)propanoic acids. DCM was added to help dissolve the solid starting material and very viscous PPA. The start material 3-(arylthiol)propanoic acids is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well. So, 1.0 mL of DCM was used to dissolve the starting material 3-(arylthiol)propanoic acids initially at RT and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the mixture is heated to the boiling point of DCM (40 oC). Yields are based on isolated products by column chromatography.
Scheme 1.
The scope of one-pot reaction of 3-(Arylthiol)propanoic acids. DCM was added to help dissolve the solid starting material and very viscous PPA. The start material 3-(arylthiol)propanoic acids is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well. So, 1.0 mL of DCM was used to dissolve the starting material 3-(arylthiol)propanoic acids initially at RT and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the mixture is heated to the boiling point of DCM (40 oC). Yields are based on isolated products by column chromatography.
Scheme 2.
Synthetic applications of thiochromones - synthesis of thioflavanones and 2-alkylthiochroman-4-ones. a. All the reactions were performed using 1.5 equiv of RMgX (X = Cl, Br), or PhMgBr in the presence of TMSCl. b. MeMgBr, PhMgBr, n-BuMgCl were commercially available. Other Grignard reagents were prepared from corresponding bromocompounds in THF and used as a THF solution. c. Yields are based on isolated products by column chromatography.
Scheme 2.
Synthetic applications of thiochromones - synthesis of thioflavanones and 2-alkylthiochroman-4-ones. a. All the reactions were performed using 1.5 equiv of RMgX (X = Cl, Br), or PhMgBr in the presence of TMSCl. b. MeMgBr, PhMgBr, n-BuMgCl were commercially available. Other Grignard reagents were prepared from corresponding bromocompounds in THF and used as a THF solution. c. Yields are based on isolated products by column chromatography.
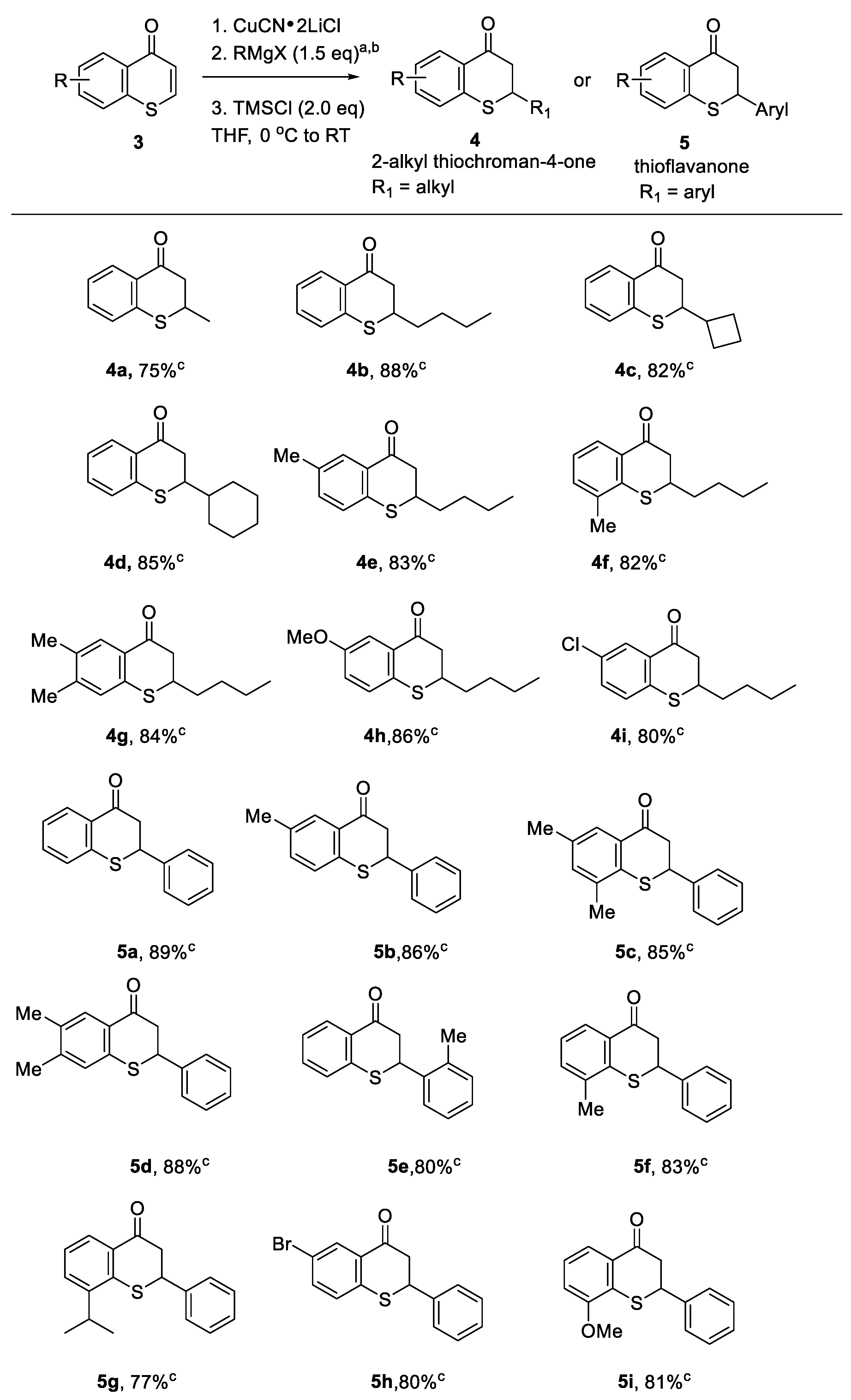

Table 1.
Reactions of with 3-(4-MeOphenylthiol)propanoic acid. a. DCM was added to help dissolve the solid starting material and very viscous PPA. The start material 3-(4-MeOphenylthiol)propanoic acid is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well. So, 1.0 mL of DCM was used to dissolve the starting material 3-(4-MeOphenylthiol)propanoic acid initially at RT and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the mixture is heated to the boiling point of DCM (40 oC). b. Excess amount of acids were used (0.5 mL acids for 1.0 mmol of starting material. c. Yields are based on isolated products by column chromatography.
Table 1.
Reactions of with 3-(4-MeOphenylthiol)propanoic acid. a. DCM was added to help dissolve the solid starting material and very viscous PPA. The start material 3-(4-MeOphenylthiol)propanoic acid is solid and PPA is very viscous at room temperature, which makes it very difficult to mix them well. So, 1.0 mL of DCM was used to dissolve the starting material 3-(4-MeOphenylthiol)propanoic acid initially at RT and to mix well with very viscous PPA. The DCM was then distilled and collected in a collecting flask when the mixture is heated to the boiling point of DCM (40 oC). b. Excess amount of acids were used (0.5 mL acids for 1.0 mmol of starting material. c. Yields are based on isolated products by column chromatography.
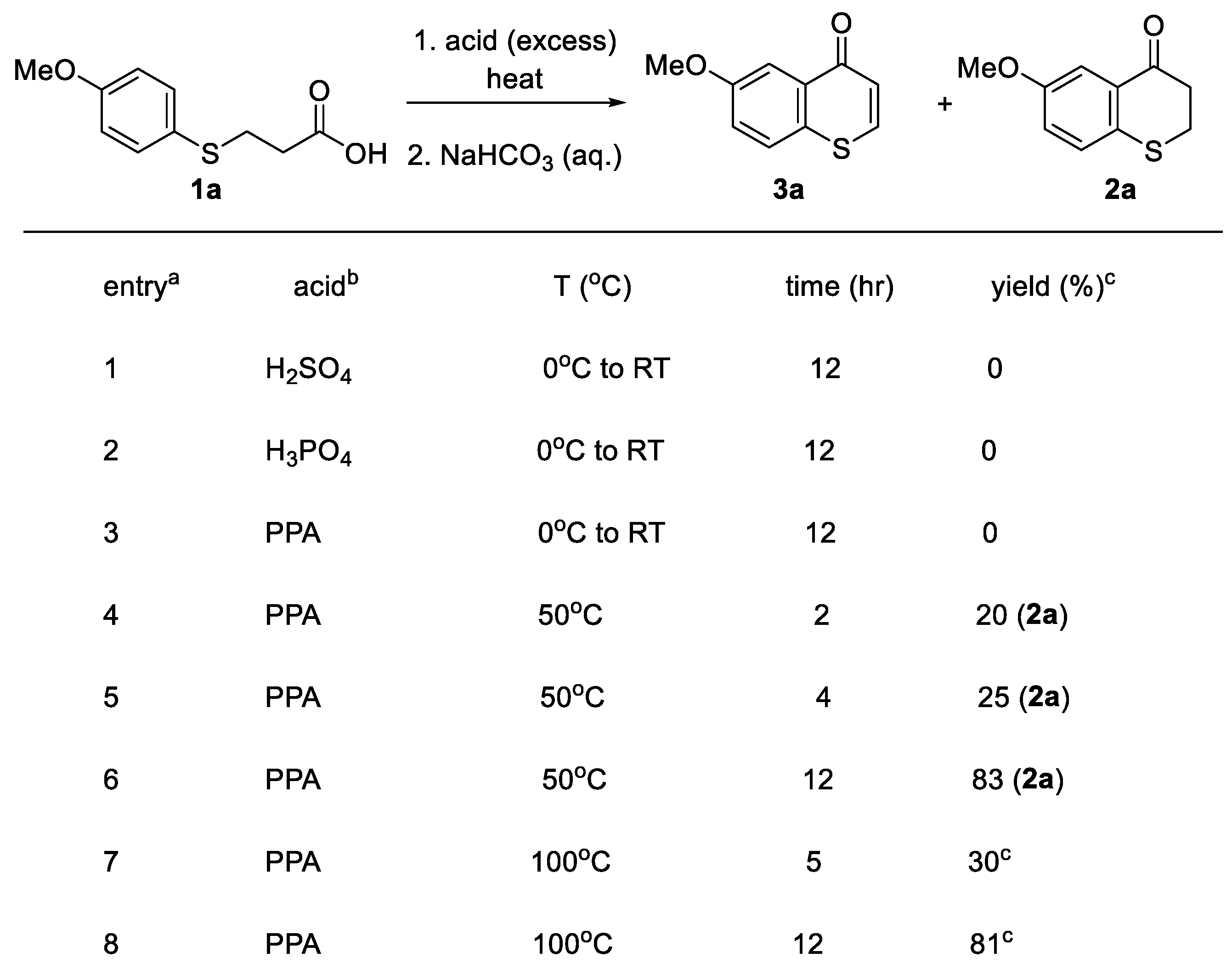 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



