Submitted:
21 July 2025
Posted:
22 July 2025
You are already at the latest version
Abstract
Almost every cell of a multicellular organism is in contact with the extracellular matrix (ECM), which provides the shape and mechanic stability of tissue, organs and the entire body. At the molecular level, cells contact the ECM via integrins. Integrins are transmembrane cell adhesion molecules that connect the ECM and the cytoskeleton, which they bind with their extracellular and intracellular domains, respectively. Cysteine residues are abundant in both integrin subunits, α and β. If pairwise oxidized into disulfide bridges, they stabilize the folding and molecular structure of the integrin. However, despite the oxidative environment of the extracellular space, not all pairs of cysteines in the extracellular integrin domains are permanently engaged in disulfide bridges. Rather, the reversible and temporary closure to cystine bridge of these cysteine pairs by oxidation or their reductive cleavage can cause major conformational changes within the integrin, thereby changing ligand binding affinity and altering cellular functions, such as adhesion and migration. Recent years have characterized several oxidoreductases and thiol isomerases, which target such allosteric disulfide bridges. This outlines much better, albeit not fully comprehensively the role that such thiol switches play in redox regulation of integrins. The platelet integrin, αIIIbβ3, is the best examined example so far. Mostly referring to this integrin, this review will provide insights into the thiol switch-based redox regulation of integrins, the known effects of their allosteric disulfide bridge on conformational changes and cell functions, as well as on the machinery of redox-modifying enzymes that contribute to the redox regulation of cell contacts with the ECM.
Keywords:
allosteric disulfide bonds
; integrin
; redox regulation
; conformational signaling
1. Introduction
Cysteine residues are the most conserved amino acid in proteins, even more conserved than tryptophane [1]. This is presumably due to its structure-stabilizing effect on proteins. Two cysteine that are spatially close-by can be oxidized to form a disulfide bond. This covalent bond is among the strongest bonds that can stabilize the tertiary and sometimes quarternary structure, if formed intra- and intercatenarily, respectively. This stabilizing effect on protein structure is important and worthwhile to be evolutionarily preserved [1], especially if reductive cleavage of a cysteine bridge would make a protein lose its structure and concomitantly its function. However, not every pair of vicinal cysteines serves such a structurally indispensable role. In this review light is shed on cell adhesion mediating molecules, especially of integrins, in which, along with several structure-stabilizing cysteine bridges, also cysteine pairs exist that, due to their reversible formation and cleavage of a disulfide bond, act as a thiol switches and regulate protein function.
A redox pair, such as cysteines and cysteine, only undergoes a redox reaction, if a convenient oxidant or reductant with an appropriate redox potential thermodynamically allows an electron transfer and is kinetically able to react in reasonable time periods [2]. Therefore, the oxidative formation of a disulfide bridge or its reductive cleavage depends on its environment, including redox-active compounds and thiol group-modifying enzymes [3]. Moreover, in order to allow redox regulation, this environment has to be altered by the cell. Whereas the cytoplasm of a cell remains largely at a low, hence reducing redox potential due to the GSH/GSSG and cysteine/cysteine buffer with high molar fractions of the reduced species [4,5], certain compartments within the cells, such as mitochondria, endoplasmic reticulum (ER), Golgi organelle and transfer vesicles, as well as the extracellular space have a higher redox potential [6,7,8]. Moreover, in addition to the access of oxidizing molecular oxygen, certain cells can produce highly oxidative species, such as reactive oxygen species (ROS) and reactive nitrogen species (RNS) upon certain stimuli, that increase the redox potential locally and temporally [9,10,11]. As an example, vascular smooth muscle cells transiently produce a pulse of ROS locally and thus form redox hot spots at sites of adhesion [12]. NADPH oxidase (Nox) isoforms, preferentially Nox4, are recruited to these local cellular protrusions, where also integrins gather to form adhesive contacts with the ECM. Conversely, these cell-produced oxidants cause disulfide formation within integrins, especially at their allosteric thiol switches, which will be highlighted in this review.
2. Not All Cysteine Pairs in Proteins Are Equal
Disulfide bridges within proteins and their formation have been considered as a path-determining step in protein folding and as a stabilizing element for the tertiary and quaternary structure of proteins, especially of those in the extracellular space, where the redox potential generally is higher than in the cytoplasm, due to the higher oxygen tension and the low concentration of redox buffers, such as glutathione [3]. Thus, they have been seen as static elements of protein structure, called structural cysteines. However, labeling of extracellular proteins with thiol-selective mass spectrometric probes, which differ in their isotope composition, before and after reductive cleavage of disulfide bridges (differential cysteine alkylation), revealed that a remarkable fractions of vicinal cysteine residues, albeit in a distance of the two thiol groups close enough to form a disulfide bridge (0.28 nm), were not oxidized to a disulfide bridge. Moreover, the fraction of disulfide-linked and reduced cysteine residues varied for each cysteine pair in different sites of these proteins [13]. Interestingly, a wide investigation of the protein structure data base revealed that many proteins contain such pairs of cysteines, which would form labile and mechanically stressed disulfide bridges and are prone to be reversible and temporal [14]. In addition, upon formation and cleaving, these disulfide bridges are able to induce conformational changes within the protein and hence were called allosteric disulfide bonds [15,16]. Such cysteine pairs are mostly located at or close to the protein surface and thus are accessible to redox-modifying enzymes.
Many extracellular proteins bear such cysteine pairs, some of which are not necessarily oxidized into a linking disulfide bridge, or some of which can be temporarily crosslinked by oxidation and cleaved by reduction [15,17,18,19]. Such allosteric disulfide bridges can be found in several of the coagulation factors and other blood proteins [20], such as fibrinogen, α2 macroglobulin [21], von Willebrand factor (vWF), Histidin-rich glycoprotein (HRG) [17], and in several extracellular domains of hemostasis-relevant membrane proteins, such as tissue factor [22,23], vWF receptor GPIbα [24] and integrins [25].
Along with the finding of allosteric cysteine pairs in extracellular proteins, several redox-modifying enzymes, originally thought to be localized within the cells and the lumen of vesicular organelles, such as ER and Golgi, have been detected in the extra- and pericellular space, among them protein disulfide isomerase (PDI), several thiol isomerases, thioredoxins, peroxiredoxin-4 (Prx4) and quiescin–sulfhydryl oxidase-1 (QSOX1) [3,26,27]. They serve functions in the redox regulation of these proteins, both under physiological and pathological conditions, such in normal thrombus formation [28] and cancer-induced thrombosis [29].
3. Adhesion of Platelets and Cells Are Mediated by Members of the Cysteine-Rich Integrin Receptor Family
One of the best example of how cellular adhesion is regulated by the redox properties of the environment is platelet aggregation at injured blood vessels, the initial step of hemostasis that is followed by coagulation and thrombus formation [29,30,31,32]. Platelets are anucleated cell fragments pinched off from megakaryocytes in the blood marrow and floating in the blood stream for a life time of 8-10 days [33]. After stimulation by soluble procoagulant agents, such as purine nucleotides or proteases, they become activated with morphologic changes from a discoid to protrusion-rich stellate shape, cytoskeletal rearrangement, degranulation of their vesicles, and activation of the fibrin-binding platelet integrin αIIbβ3 [33]. Another stimulus for platelet activation is the contact with accessible extracellular matrix molecules, such as collagens from the subendothelial layers of blood vessels [34]. Six different integrin receptors are expressed on the platelet surface [35]. With its 80.000 copies per platelet, integrin αIIbβ3 outnumbers any other integrin receptor, hence also called the platelet integrin. After being activated, the integrin αIIbβ3 binds fibrin bundles that have been formed as a result of blood coagulation, resulting in a firm fibrin-platelet clot, as well as in stabilization and contraction of the thrombus [36]. Its binding activity is regulated not only by ligand occupancy, but also by intracellular adaptor molecules [37] and extracellular redox-modifying agents and enzymes [28,30].
Along with 23 other members, the integrin αIIbβ3 belongs to the integrin family, all of which consist of two non-covalently associated and glycoconjugates bearing subunits, α and β [38,39]. Eighteen α and 8 β subunits, most of them specified by a number (α1 through α11, β1 through β8) or acronyms (αV for the vitronectin-binding integrins, and αIIb for the platelet glycoprotein IIb), heterodimerize within the endoplasmic reticulum during their biosynthesis and form receptors on the cell surface [40,41]. The integrins containing the β1, β3, and β4 subunit recognize proteins of the extracellular matrix (ECM) [42,43], thereby anchoring cells to the extracellular scaffold and transmitting mechanical forces and environmental cues between the cells and the ECM scaffold [44]. According to their ECM ligands, subgroups are defined as collagen-binding (α1β1, α2β1, α10β1, and α11β1) and laminin-binding integrins (α3β1, α6β1, α6β4, and α7β1). They recognize their ligands by an array of amino acids presented in a characteristic three-dimensional tertiary structure, such as the triple helix of collagens and the globular domain of laminins [45,46]. In contrast, RGD-dependent integrins bind their ligands via the characteristic tripeptide motif, Asp-Gly-Arg (RGD in the one-letter-code), such as the fibronectin receptor α5β1, as well as the integrins α8β1, α9β1, αV-subunit containing integrins and the platelet integrin αIIbβ3 [40,41,45]. The integrin containing the β2 integrin subunit, αLβ2, αMβ2, αXβ2, and αDβ2, are expressed on leukocytes and are often grouped with other immunologically relevant integrins (α4β1, α4β7, and αEβ4). They mediate cohesive cell-cell contacts with endothelial cells during lymphocyte patrolling and homing or with E-cadherin of epithelial cells during immune surveillance. Some of them are also involved in pathogen defense as they essentially contribute in phagocytosis of complement-opsonized pathogens [47,48].
Integrin-mediated signaling in platelets is a well-studied example as of how cells sense and regulate adhesive interactions with the ECM via integrins [25,33,49]. On quiescent thrombocytes, the platelet integrin αIIbβ3 is surface-exposed but does not bind to its ligand, fibrinogen, which is abundant in the blood plasma. Upon damage of the vessel wall, ECM proteins, such as collagen fibrils, become accessible and are recognized by thrombocytes via different receptors, among them the collagen-binding integrin α2β1 [50,51]. This triggers an outside-in signaling cascade within the platelets resulting in release of additional platelet-activating substances, cytoskeletal rearrangement, and inside-out activation of the platelet integrin αIIbβ3 via association of cytoplasmic integrin-associating proteins, such as kindlins [33,37,49,52]. Only activated αIIbβ3 binds to fibrinogen and its coagulation-converted product, fibrin. Via the αIIbβ3 integrin-fibrin axis, thrombocytes adhere to the fibrin network and keep the fibrin network under stabilizing tension, thus closing the bleeding wound [53].
Upon ligand binding, integrins coral into clusters and recruit additional adaptor and signaling proteins to their cytoplasmic domains [54,55,56]. Thus, in an adhesion-dependent manner, new adhesive supramolecular complexes, called adhesomes are formed, among which focal contacts and focal adhesion (plaques) in the cell periphery during cell spreading and migration can be distinguished from fibrillary adhesions underneath the cell soma [57,58]. They all contain distinct signaling and adaptor proteins and cytoskeleton-linking molecules, that enables the integrins in the adhesome to transmit mechanical forces and convey signals between cells and their environment [44,56]. Noteworthy, not only the integrins but also integrin associating adaptor molecules within focal adhesions [59,60] and several ECM proteins are subject to redox modifications [14,47,48,61].
4. Structure; Domains and Disulfide Pattern of Integrins
Integrins are characterized by a high number of cysteine residues, which are distributed throughout the extracellular domains of both subunits. While the integrin β subunits bear more than 50 cysteine residues, the α subunit still has a high number of about 20 cysteine residues. Not all of them are engaged in disulfide bridges, and the fraction of disulfide engagement varies for each cysteine pair [25].
Both subunits are type I membrane proteins with a large N-terminal ectodomains, which is anchored by a single pass transmembrane domain and ends with a short C-terminal cytoplasmic domain of about 20-50 and 15-65 residues in the long α and β subunit, respectively (Figure 1). Only the cytoplasmic tail of the β4 chain extends to more than 1000 amino acids and contains four fibronectin type II domains arranged in two tandem pairs. The cytoplasmic domains harbor binding sites for various adaptor and signaling molecules that are relevant for integrin-mediated signal transduction and mechanical force transmission [56,58,62]. Reversible phosphorylation of tyrosine residues within the integrin β cytoplasmic tail regulates the interaction with several of these adaptor and signalling molecules [63,64,65]. In non-activated integrins, the transmembrane domains of both subunits heterodimerize via several hydrophobic interactions and a interchain salt bridge between membrane-proximal cytoplasmic residues [66,67], whereas integrin activation make the two different transmembrane domains separate from each other [68,69].
The molecular structure of the cysteine residues-bearing ectodomains of the integrin αVβ3 was the first to be resolved by crystallographic X-ray diffraction analysis in 2001 [70], followed by the ectodomains of other αV-integrins, α5β1 and β2 integrins, and eventually of the entire αVβ3 and αIIbβ3 heterodimer [71,72,73,74]. Both integrin subunits consist of several domains. The most N-terminal domain of the mature integrin α subunit is the propeller domain with seven blades with four-β-strands each and a center pore [70]. Between the second and third blade, an additional A-domain is inserted in about half of the integrin α subunits, especially in the subfamilies of collagen-binding and β2-integrins (Figure 1). As a magnesium ion serves as complexing bridge for binding the ECM ligand, the ligand binding crevice, located on top of the A-domain, is referred to as metal ion-dependent binding site (MIDAS) [75,76,77]. In addition to the Mg2+ ion, two Ca2+ ions are complexed in close vicinity to the Mg2+ ion and shape the so-called adjacent metal ion binding site (ADMIDAS) and synergistic metal binding site (SyMBS) [78]. In those integrin α subunits that lack an A domain, the propeller binds to the ECM ligand directs. The last four blades of the propeller domain also complex Ca2+ ions. However, these metal ion binding sites likely serve structural purposes as they are located on the bottom face of the propeller domain opposite to the ligand binding site. The propeller domain and, if present, the A-domain of the α subunit, together with the A-domain of the β subunit, shapes the integrin headpiece. Therein, the propeller domain is the rather inflexible structural foundation. C-terminally adjacent to the head domain, the leg/stalk of the integrin α subunit is formed by β-strand-rich thigh domain, and two calf-domains, calf 1 and 2. In integrin α subunits that lack an αA-domain, the calf 2 domain usually is proteolytically cleaved into the N-terminal heavy and C-terminal light chains, which remained covalently cross-linked via a disulfide bridge (Figure 1). Exceptionally, integrin α4 is cleaved within the thigh domain.
In the ectodomain of the integrin β subunit, the peptide chain starts with the N-terminal plexin-semaphorin-integrin (PSI) domain, passes through the hybrid domain to the βA-domain, and passes a second time through the hydrid domain connecting the βA domain with the EGF1-domain (Figure 1). The βA-domain is inserted between the first and second peptide chain passage through the hybrid domain. The A-domains of both integrin subunits have a homologous fold and complex a divalent cation on their top. Interestingly, a conserved basic amino acid side chain, R261, of the βA-domain points into the central pore of the propeller domain of the integrin α subunit [70]. In addition, hydrophobic side chains, especially ones of phenylalanine residues, also interacts between the propeller and A domains of both subunits and thus jointly form the integrin head domain [79].
The stalk of the integrin β subunit is shaped by 4 EGF domains, each of which contain three or four disulfide bridges, and a membrane-proximal β tail domain (βTD), which despite its distance from the ligand binding head domain also influences ligand binding activity by interacting with the βA-domain in one of the integrin conformations [80,81]. Moreover, the conformation changes that occur during integrin activation and ligand binding involves the distance and orientation of the integrin stalk/leg domains [82].
The location and pair-wise distances of cysteines residues and the resulting disulfide pattern within both integrin subunit had been a thrilling conundrum. Early protein chemical and mass spectrometric analysis mapped most of the 28 and 9 potential disulfide bridges within the integrin β and α subunits, respectively, of the platelet integrin [83,84,85]. However, data from crystallographic protein structure determination showed some discrepancies on the location of some disulfide bonds [74]. These discrepancies might be due to conformational differences, as crystallography revealed the structure of only one of different integrin conformations.
In the primary sequences of both ectodomains of integrin αIIbβ3, cysteines are comparatively abundant with around 20 to 35 and between 50 and 60 residues in the α and β subunit, respectively [85]. Within the α-chain, cysteines are localized in the first three blades of the propeller domain (C56-C65, C107-130, C 146-C167, numbering refers to the αIIB subunit), however in non-homologous strands or linker sequences within each blade, and likely serve structural functions. The thigh domain and calf 1 domain contain four and two cysteine residues, respectively, engaged in two (C475-C484, C490-C545) and one (C674-C687) disulfide bridges, respectively (Figure 2A). These two domains flank the α subunit hinge region, which contains a pair of cysteines (C602-C608) and complexes one divalent metal cation. The disulfide pattern between the four cysteines (C826, C880, C885, and C890) within the calf 2 domain differ between the crystallographic and protein-chemical analyses [74,83]. Being oxidizable by hydrogen peroxide, these cysteines are likely not fully engaged in disulfide bridges [12,13,25]. Interestingly, the most C-terminal cysteine within the heavy chain (C826) is covalently crosslinked with one of the other three cysteines of the calf2 domain, located within the light chain. This disulfide bridge holds the heavy and light chain of the integrin α subunit together and hence is of structural relevance (Figure 1).
Within the β-subunit, 8 cysteines are located within the N-terminal PSI-domains [86]. Six of them form intradomain disulfide links (C5-C23, C16-C38, and C26-C49, numbering of the β3 subunit), whereas the second cysteine, C13, forms a long-range disulfide bond with a cysteine, C435, situated more than 400 residues C-terminally within the first EGF-domain [70,74,86,87] (Figure 2B). Both βA and hybrid domain contain four cysteines each, which, as cysteine bonds (C177-C184 and C232-273 in the βA-domain), stabilize the globular and rod-like structures, respectively, of these domains, but may also serve regulatory function (Figure 2C). Two disulfide bridge, C374-C386 and C433-443 of the hybrid domain crosslink the peptide chain on its second passage through the hybrid domain. Within the four EGF-domains, especially the last two of them, the cysteines share a common folding pattern in the order 1-5, 2-4, 3-6, and 7-8 [88,89]. Being connected with its first cysteine, C435, to the PSI domain via the long-range disulfide bond, C13-C435, the EGF1 domain exceptionally has only three intradomain cystine bridges. Its last cysteine, C472, forms a interdomain disulfide bridge, C473-C503, to the first cysteine, C503, of the EGF2 domain, thereby spanning the domain-connecting loop between EGF1 and EGF2 (Figure 2D), which is the very flexible hinge region of the integrin β subunit [70,74,89]. This is in conspicuous contrast to the common disulfide pattern of EGF-domains [90,91]. The 10 cysteines of the βTD form are engaged in well-defined intradomain disulfide bridges.
All of those previous studies assumed that a pair of cysteine residues must be engaged within a disulfide bridge in an all-or-none fashion. However, more recent differential alkylation studies, in which thiol groups in integrins were labelled with two isotopically different labelled 2-iodo-N-phenylacetamide (IPA) before and after reductive cleavage of disulfide bridges [13,25,92], revealed that the pairs of cysteines in integrins are not completely oxidized into cysteine bridges and, in fact, that each pairs of cysteines have a distinct ratio of engaged or disengaged disulfide bridge [25], explaining some of the differences in disulfide patterns within integrins of previous studies. Additional studies revealed that cysteine pairs are more prone to redox-mediated disulfide engagement, when being exposed on the protein surface, indicating their accessibility to redox-modifying agents or enzymes [25]. The analysis of thousands of cysteine bonds in protein structure data bases revealed that in proteins, cysteine bonds can take 20 different conformations depending on the 5 dihedral angles of the five rotatable single bonds between the αC-atoms of two cysteine residues engaged in such a cysteine bond [14-16] (Figure 3). Three conformers are the main representatives within the group of allosteric disulfide bonds, which are called /+ RH hook, -LH hook, and -RH staple [15](Figure 3 bottom panel). Together, they make about 80% of the allosteric disulfide bonds, while their distribution frequency within several thousands of disulfide bonds, irrespective of their structural or regulatory role, only run to 7.20 %, 7,48 %, and 6.44 %, respectively, in total about 20 % [16]. The -RH staple conformation has an exceptionally high dihedral strain energy, a measure for the mechanical tension that they withstand to hold the peptide chains in the tertiary structure of the protein. High dihedral strain energy values also indicate instability of the disulfide bond and its tendency to be cleaved. Thus, a group of redox-labile cysteines also in the integrin αIIbβ3 integrin could be identified, which are likely to be reversible oxidized in disulfide bridges and again reduced into free thiol groups, depending on external factors [15,25]. These novel insights have opened a new field of research, whether and how the formation and cleavage of these particular allosteric disulfide bridges is regulated in a redox-dependent manner, as well as what consequences the oxidation and reduction of cysteines and disulfide bridges, respectively, may have for regulation of integrin activation.
5. The Machinery That Modifies Integrins on the Cell Surface and Redox-Regulate Platelet Adhesion and Deadhesion
Formation and remodeling of disulfide bonds is well studied for intracellular proteins in the cytoplasma, a compartment with a comparatively low redox potential due to the redox buffers of cysteine and glutathione with a surplus of their reducing forms, cysteine and glutathione (GSH), as compared to their oxidized counterparts, cystine and oxidized, homodimerized glutathione (GSSG). This reducing redox environment is maintained by a high concentration of NADPH∙H+, which constantly reduces thioredoxin or glutaredoxin via thioredoxin reductase and glutaredoxin reductase. However, extracellular proteins are synthesized into the endoplasmic reticulum (ER), further processed and modified in the Golgi compartments, and transported and secreted in transport vesicles into the extracellular space or, in case of membrane proteins, onto the extracellular face of the cell membrane. Already at their first stage of production within the ER, they embrace an oxidative environment with a higher redox potential, which is necessary for a proper formation of disulfide bridges in a process called oxidative protein folding. This would require a high amount of cystine, GSSG or NADP+. However, the concentrations of such oxidants are scarce in the ER and oxidative protein folding relies on the oxidative power of molecular oxygen and its derivative hydrogen peroxide (H2O2). The predominant enzyme for the electron withdrawal from two cysteine residues in proteins is the endoplasmic reticulum oxidoreductin 1 (Ero1), which comes in two isoforms, Ero 1α and Ero 1β. It transfers the electrons via an flavin adenine dinucleotide (FAD) to molecular oxygen thereby forming H2O2 [93]. A similar reaction is performed by the enzyme quiescin–sulfhydryl oxidase (QSOX), which also comes in two isoforms, QSOX1 and QSOX2 [27]. Hydrogen peroxide, produced by either Ero1 or QSOX, has even a higher oxidizing power and can be used by peroxiredin-4 (Prx4) to assist oxidative folding of secretory proteins by forming a disulfide bond via a temporary cysteine sulfenic acid and water [27]. Interestingly, Prx4 is the only member of the six-member family of peroxiredoxins, whose genes encode a signal sequence directing its secretory translation into the ER.
The oxidoreductases, Ero1 and QSOX, do not introduce disulfide bonds directly into the final target protein, but mainly use protein disulfide isomerases (PDI) as intermediate disulfide carriers that eventually oxidize the vicinal cysteine residues into a cystine bridge, while being reduced into dithiol forms that are recycled and reoxidized by Ero1 and QSOX [28] (Figure 4). A function similar to the one of PDI is fulfilled by so-called endoplasmic reticulum proteins (ERps), such as ERp5, ERp46, ERp72 and ERp57, the latter also known for its important role in the calreticulin-calcineurin pathway of ER stress and apoptosis [94]. PDIs and ERps form a large family of thiol isomerases with about 20 members. Their common feature is thioredoxin-homologous domain (referred to as a or a’ domain) with the typical CGHC motif which is usually present in tandem within the thiol isomerase, either connected next to each other (e.g., within ERp5, ERp72, and ERp46, the latter even in a triplet repeat) or flanking two connecting domains (commonly called b or b’ domain) that form the substrate protein binding site (e.g., in PDI, and ERp57). Bearing an oxidized disulfide bridge, they approach the two spatially close cysteine residues of the substrate protein. These two vicinal cysteines will be oxidized and thus a disulfide bridge is formed, while the CGHC motif of the thiol isomerase is reduced into two free thiol group by the end of the reaction.
Most of the thiol isomerases and their redox modifying partners carry the ER retention peptide sequence, KDEL. However, in recent years, it has become more and more evident, that at least six thiol isomerases are secreted, or leaking, into the peri- and extracellular space of the cell [26,28]. It is not clear yet, how they can fulfill their oxidative function of forming disulfide bridges, as, outside of the ER, they are separated from a constant supply of oxidants provided e.g. by ERo1. However, the extracellular form of QSOX1 may substitute for Ero1 in the peri- and extracellular environment [27]. The importance for the extracellular thiol isomerases, which are stored in vesicles by platelets and by endothelial cells and secreted in case of vessel damage, nowadays summarized as vascular thiol isomerase, has become more and more evident in the last few years. They play essential roles in platelet activation and blood coagulation. Moreover, they have even been identified as pharmacological targets in thrombosis [30,31,95,96].
So far, six vascular thiol isomerase have been identified to be secreted by platelets and endothelial cells into the extra- and pericellular space upon vessel damage [28,97,98]. Being their targets, the integrins αIIbβ3 and αVβ3 on platelets and endothelial cells, respectively, play pivotal roles in thrombus formation and stabilization. Several studies have highlighted the role of the secreted vascular thiol isomerases in coagulation and thrombus formation (reviewed in [3,28,30,31,99,100]. However, only a few ones showed a direct interaction of a particular thiol isomerase with the platelet integrin or integrin αVβ3 at the protein-chemical level [25,30]. The six thiol isomerases are PDI, ERp57, ERp72, ERp5, ERp46, and TMX1.
PDI has a thiol isomerase activity on the platelet integrin [101]. This prototypic thiol isomerase, encoded by the gene prolyl-4-hydroxylase-β (P4HB), consists of two thioredoxin-like domains, a and a’, with a typical CGHC motif that flank two homologous domains, b and b’, the latter of which binds the substrate protein. A linker sequence between the b’ and a’ domains bestows flexibility of the active site. Affecting platelet aggregation and adhesion to fibrin, PDI itself was identified to be redox-regulated. H2O2 and endothelial cell-derived NO cause sulfenylation and nitrosylation, respectively, of thiol groups within PDI, thereby reducing fibrin binding of platelet integrin [95,102].
ERp57 (PDIA3) has a structure similar to PDI. It is an essential component of the oxidative folding machinery of the ER, and connects protein folding with the calreticulin-calcineurin pathway [103]. Although its role of disulfide formation in platelet integrin has not exactly delineated, the interaction of integrins with calreticulin in the ER makes its close association with ERp57 very likely. However, the extracellular role of ERp57 on platelet integrin regulation at the molecular level has remained elusive, although deficiency of ERp57 activity on platelets reduces thrombus formation drastically [104].
In addition to the basic a-b-b’-a’ domain structure, ERp72 (PDIA4) has an additional thioredoxin domain with a CGHC motif, named a°, that precedes the a-domain at its N-terminus. As an extracellular thiol isomerase, ERp72 oxidizes the long-range disulfide bridge (C654-C711, numbering of the αM subunit, homologous to C490-C545 of the αIIB subunit) within the thigh domain of αM subunit of the integrin αMβ2 (Mac1) on neutrophils. This interaction promotes integrin activation and neutrophil attachment to its ligand, the intercellular adhesion molecule-1 (ICAM1) on endothelial cells [105]. The homologous cysteine bond, C490-C545, within the αIIB subunit, is also redox-regulated and mostly not engaged in a disulfide bond [106]. However, the corresponding thiol isomerase for the platelet integrin has not been identified yet.
ERp5 (PDIA6) has the extra a° domain in common with ERp72, but lacks the b’ and a’ domains. A platelet-specific ERp5 knockout shows increased secretion of other thiol isomerases in platelets and an increased platelet adhesion to collagen, presumably via the integrin α2β1 [107]. This suggests an attenuating effect of ERp5 on integrins. In fact, at the molecular level, ERp5 reductively cleaves the disulfide bridge C177-C184 within the A-domain of the integrin β3 subunit of the platelet integrin causing its release from its fibrin ligand [108]. Interestingly, this occurs preferentially, after the platelet integrin has bound fibrin and transmits forces, which likely induce a conformation change within the integrin thereby making it more accessible to ERp5.
In contrast, ERp46 (TXNDC5) activates the platelet integrin by cleaving the cysteine bridge C473-C503 within the hinge region of the integrin β3 subunit [109]. Of structural interest, ERp46 lacks any conventional substrate-binding b-domain and basically consists only of 3 thioredoxin-like domains with CGHC motif [28].
The sixth member of the secreted thiol isomerase, the thioredoxin-related transmembrane protein-1 (TMX1) stands apart from the others, with respect to both structure and function [110]. TMX1 is a type I transmembrane protein, which contains one thioredoxin-like a-domain with a less homologous CPAC motif in its extracellular domain [31]. Because of its membrane anchorage, its action is limited to the pericellular space. Nevertheless, TMX1 is able to oxidize vicinal thiol groups within the platelet integrins, thereby inhibited integrin-mediated platelet adhesion and migration [111]. As a negative regulator of integrin action, it counteracts the action of the other secreted thiol isomerases [31].
6. The Allosteric Disulfide Bridges and Their Sites Within Integrins
Which of the numerous disulfide bridges within integrins are redox-modified by the extracellular thiol isomerase? For the integrin αIIbβ3, they are listed in a recent review by Pijning et al. [25], including those ones that are of structural relevance. The latter are the majority of disulfide bridges and are required for the correct folding and function. In fact, their mutations prevent the expression or function of a properly working integrin αIIbβ3. It may not be expressed or may have too low or too high an activity, resulting in bleeding disorders and constitutive activation with thrombosis, respectively, generally known as Glanzman thrombasthenia [25]. In most of these cases, cysteine residues within the EGF-domains of the β3 subunits are affected. Also, the four cysteines within the calf-2 domain of the integrin α7 subunit serve a structural role, among them the cysteine pair that crosslink the heavy and light chain of the proteolytically cleaved integrin α subunit, as their replacement for alanine drastically reduced or abolished its expression [12,112]
First evidence of an allosteric disulfide bridges within the platelet integrin that serve regulatory functions redox-dependently were reported 25 years ago [113]. Some years later, the fact that the oxidizing “zero spacer” crosslinker, such as phenylarsenic acid (PAO), form a disulfide bridge between such spatially vicinal cysteine residues proved that a pair of such two cysteine thiol groups at a close distance are not necessarily engaged in a disulfide bridge in the native protein, and that their induced formation with PAO would do so and cause functional consequences [114]. Since then, potential thiol switches have been mapped at different sites within both subunits of integrin heterodimers. They are listed in Table 1.
The PSI domain contains a tandem array of two CXXC motifs, C13QQC16 and C23AWC26, which are similarly found in members of the PDI family (Figure 2B). The hypothesis, that this integrin might not only be a substrate for redox modifying enzymes, but also might have PDI functions itself were positively tested in protein-chemical assays [115,116]. In addition, antibodies directed against the cysteine-containing region of the PSI domain blocked fibrin binding to integrin αIIbβ3 and reduced platelet aggregation [116]. However, the PSI domain only shows low homology of the intervening amino acids to the typical CGHC-sequence of the thioredoxin and it does not resemble the thioredoxin-fold [117]. Moreover, in the crystal structure of the integrin αIIbβ3 in its bent conformation, the two cysteines in both CXXC motifs are neither reduced nor engaged with each other. Interesting the first cysteine, C13 of the first CXXC motif forms a long-range interdomain disulfide bond to cysteine C435 of the first EFG domain [25,74].
The A-domain of the β3 domain bears two cysteine pairs, C177-C184 and C232-C273 (Figure 2C). The former one acts like a thiol switch, which upon ligand binding becomes under mechanical stress and due to its surface exposure becomes more easily reduced by the extracellular thiol isomerase ERp5 [108]. This finding is extremely interesting, as it links mechanical force transmission via integrin-ligand interaction with the accessibility and susceptibility of the cysteine bridge that leads to disengagement of two cysteine residues and enforced detachment of the platelet from fibrin [108]. Also within the integrin β2 subunit, the two homologous disulfide bonds, C169-C176 and C224-C264, of the A domain of β2 in Mac-1 were identified to be allosteric disulfide bridges [118]. The first one is homologous to C177-C184 within the integrin β3 subunit. Reduction of the disulfide bonds within the β2 A-domain converts Mac1 (integrin αMβ2) into a less active conformation and consequentially to disengagement of Mac1 from its ligand, ICAM-1, especially under shear forces of the blood stream. As PDI associates with Mac-1 at the trailing edge of neutrophils, PDI activity increases motility of neutrophils on the endothelial cell layer of capillaries and thus promotes extravasation of neutrophils [118].
The highest number of disulfide bridges are located in the 4 EGF-repeats forming the leg of the integrin β subunit (Figure 2D). Two of these numerous disulfide bridges are of allosteric nature, C13-C435 and C437-C457. The first one connects the PSI domain (C13) with the N-terminal face of the EGF1 domain, while the second allosteric disulfide bridge, C473-C503, connects the C-terminal end of EGF1 domain to the N-terminal end of the EGF2 domain, spanning the hinge region of the β3 subunit between the domains, EGF1 and EGF2. Although not entirely assigned to a redox-regulatory role, the first intradomain cystine bond of the EGF1 domain, C437-C457, is in close vicinity to the interdomain cysteine bridge between the PSI and EGF1-domain. The EGF1 and EGF2 domains flank the hinge sequence, around which the integrin head domain, together with the EGF1-domain, can pivot against the lower leg, consisting of the EGF2 through EGF4-domains. Right within this hinge region, there is the disulfide bond, C473-C503, which is highly surface exposed (Figure 2D) and is in a mechanically strained conformation [25]. It is reductively cleaved by the platelet thiol isomerase ERp46 [109]. Blocking of ERp46 with antibodies or knockout of ERp46 reduces binding of integrin αIIbβ3 to fibrin, platelet activation and thrombus formation.

Table 1.
Potential allosteric disulfide bonds in integrin α IIBβ3, and homologously in other integrins.
Table 1.
Potential allosteric disulfide bonds in integrin α IIBβ3, and homologously in other integrins.
| Disulfide bond | Domainlocalization | Homologous sites in other integrins | αC-αC Distance [nm] | Disulfide strain energy [kJ/mol]a | Solvent accessibility [Å2]a | Stereochemical conformation | Redox-modifying enzyme(and effect) | References |
|---|---|---|---|---|---|---|---|---|
| C490-C545 | αIIB thigh | C654-C711 in αM |
0.42 | 19.2 | 6.59 | -RH staple | ERp72 on integrin αMβ2on neutrophils (promoting adhesion) | [25,119] |
| C602-C608 | αIIB hinge | C589-C594 in α4 C606-C611 in murine α7 (X2 splice variant) | 0.41 | 9.1 | 2.2 | -LH hook | Reductive cleavage in α4 and disulfide bond formation in α7 promotes ligand binding | [112,120,121] |
| C177-C184 | β3 A | C169-C176 in β2 A | 0.56 | 17.7 | 0.17 | -/+RH hook | Erp5 (attenuating; reduction of disulfide bond under tension) | [108] |
| C232-C273 | β3 A | C224-C264 in β2 A | 0.52 | 10.0 | 8.56 | -RH hook | PDI (attenuating binding affinity) | [118] |
| C13-C435 | interdomain β3 PSI-β3 EGF1 |
0.66 | 15.7 | 1.70 | +/-LH spiral | unknown | ||
| C473-C503 | hinge between EGF1 and EGF2 | 0.68 | 48.4 | 27.37 | -/+RH hook | ERp46 (reductive cleavage, activating integrin) | [109] | |
| C437-C457 | EGF1 of β3 | C494-C526 in β7 | 0.54 | 17.2 | 19.90 | -/+RH hook | in αVβ3 and in α4β7 (in the latter, reductive cleavage activates integrin) | [88,90,121,122] |
| C523-C544 | EGF3 of β3 (at interface with EGF2) | 0.41 | 17.3 | 3.36 | -LH hook | Putatively ERp57 (inhibiting in αIIbβ3, but not in αVβ3) | [88,90,122] |
a, taken from [25].
Also highly surface-exposed and under mechanical strain is the disulfide bond C523-C544, the first disulfide bridge within EGF3, which is located at the interdomain face towards EGF2 within αIIbβ3[25]. The replacement of this cysteine pair for redox inactive serine residues resulted in a constitutively active platelet integrin, whereas the same mutation was ineffective within the integrin αVβ3, albeit containing the same β subunit [90,123]. A systematic replacements of additional cysteine residues within the EGF domains were carried out, but as the outcomes depended also on the combination with other mutated cysteine residues, the interpretation of the mutational effects have not yet provided a clear picture as of which cysteines, apart from the above named ones, are of structural importance or have a activity-regulating, allosteric effect on integrins. The thiol isomerase ERp57 have been hypothesized to be active at these EGF sites [88,90,122].
The integrin α subunits possess less cysteine residues than the integrin β subunits. However, exposure of recombinant α7β1 integrin ectodomain to hydrogen peroxide, a typical member of the reactive oxygen species (ROS), revealed thiol oxidation products of cysteines other than disulfide bridges only within the integrin α7 but not β1 subunit, which were mapped to the hinge and calf2 domain [12]. They suggested free thiol groups. Similarly, differential cysteine alkylation studies on platelet integrin revealed that the long range cysteine bond, C490-C545, within the thigh domain of the αIIB subunit is not formed in one of three cases, but remains in the reduced form [13,25]. It spans between two β strands of the thigh domain and thus appears to be of structural importance. Nevertheless, mutation of these two cysteine into non-crosslinkable alanine residues made the cell spread less and recruit them into adhesion complexes [106,119]. As these supramolecular adhesive structures are located preferentially underneath the nuclei, not in the cell periphery, they appear to be fibrillar adhesions that mediate firm and static cell adhesion rather than focal adhesions plaques of lamellipodia that are involved in cell migration [38,55,56]. Interestingly, different behavior of such mutated integrin was not caused by an altered reaction of the ectodomains but by the reduced association with adaptor protein-2 (AP2) towards the cytoplasmic domain, which is relevant for clathrin-dependent integrin recycling. Its mutation-related reduction of AP2 association failed to renew focal adhesions in the periphery of cells, along with reduced lamellipodia formation and thus decreased spreading onto an adhesive substratum [106,119]. As a consequence, abundant surface integrins recruited into fibrillar adhesions. Likewise, the homologous cysteine bond, Cys654–Cys711, within the thigh domain of the αM subunit of integrin αMβ2 (Mac1) is cleaved by ERp72, thereby promoting neutrophil granulocytes to adhere to their cognate ligand on the endothelial cells [105].
Although not experimentally addressed in the platelet integrin αIIbβ3, the cysteine pair within the hinge region between the thigh and calf1 domain, which is highly conserved throughout all integrin α subunits, forms an important allosteric disulfide bond. This was proven for the α4 and α7 subunits of the integrin α4β7 and α7β1 integrins, respectively [112,121]. The cysteine bridge of the integrin α7 subunit serves a s a thiol switch, as its formation by hydrogen peroxide increases its affinity towards its ligand laminin-111, induces a structural conversion from its bent to the more active elongated conformation, and promotes migration of integrin α7β1 bearing cells on laminin-111 [112]. The oxidant H2O2 is formed at the adhesion sites of cells by NADPH-oxidase 4 (NOX4) and thus activates the integrin for cell adhesion [12]. Chemical crosslinkage of the two free thiol groups of the cysteine pair with a homobifunctional crosslinker resulted in a similar integrin activation and provided evidence, that the formation of the disulfide bridge within the α subunit hinge region primed the shaping of a chelator site for a divalent cation, most likely Ca2+ ion, that is essential for ligand binding [120]. The paramount importance of this thiol switch was likewise proven for the integrin α4 subunit, which was done as a double mutant along with mutating the cysteine bridge in the β7 subunit spanning the hinge region between EGF1-and EGF2 [121]. The latter is also conserved within the different integrin β subunits and is homologous to the cysteine bond C437-C457 within the β3 subunit of the platelet integrin. However, in contrast to our work [112,120], Zhang et al. (2013) showed that a reductant, dithiothreitol (DTT), rather than the oxidizing H2O2, increased ligand binding of α4β7 integrin [121]. Conspiciously, each of the hinge regions of both α and β integrin subunit, located between the thigh and calf-1 and between the EGF1 with its adjacent PSI and EGF2 domains, respectively, contain a redox-sensitive cysteine pair. As the hinge region is spatially distant from the ligand binding head domain of integrins, these two thiol switches are of clear allosteric nature. Their influence on ligand binding and integrin activation must be conveyed via conformational changes.
7. The Formation and Cleavage of Allosteric Disulfide Bonds in Integrins Cause Conformational Changes
By forming a disulfide bridge, the distance between the αC-atoms, and thus of the protein backbone chain, is fixed in a distance range of 0.43 to 0.65 nm [16]. How can this covalent fixation across such a short distance lead to a signal? A conceivable way to amplify this subtle structural change of a sub-nanometer scaled bond is the use of a lever structure within the protein. Indeed, such amplification is achieved by mechanical movements within the protein, such as piston-like shifts of secondary structure elements, viz. α-helices, against each other in combination with rotational movements of tertiary structural elements and domains around hinges within the integrin. Molecular structure analysis have revealed such a piston shift of two α-helices in the A domain of the integrin α subunit [75,76]. Moreover, a rotational movement, in which the head piece together with the upper legs pivots against the lower leg domains, amplifies the distances. The pivot is the integrin hinge region, also known as knee region, which is flanked by the thigh and calf1 domains, and by the EGF1 and EGF2 domains within the integrin α and β subunits, respectively [74,124] (Figure 1). Conspiciously, the redox-regulated disulfide bridges of integrins are located in these conformationally relevant domains, the A-domain of the α subunit and the hinge regions of both α and β subunit, including the domains adjacent to the hinge regions. The hinge regions are not part of the ECM ligand binding site, which is mapped to the A-domain of the integrin α subunit and, in case of αA-domain-lacking integrins, to the α subunit propeller and the β subunit A-domain [56,125].
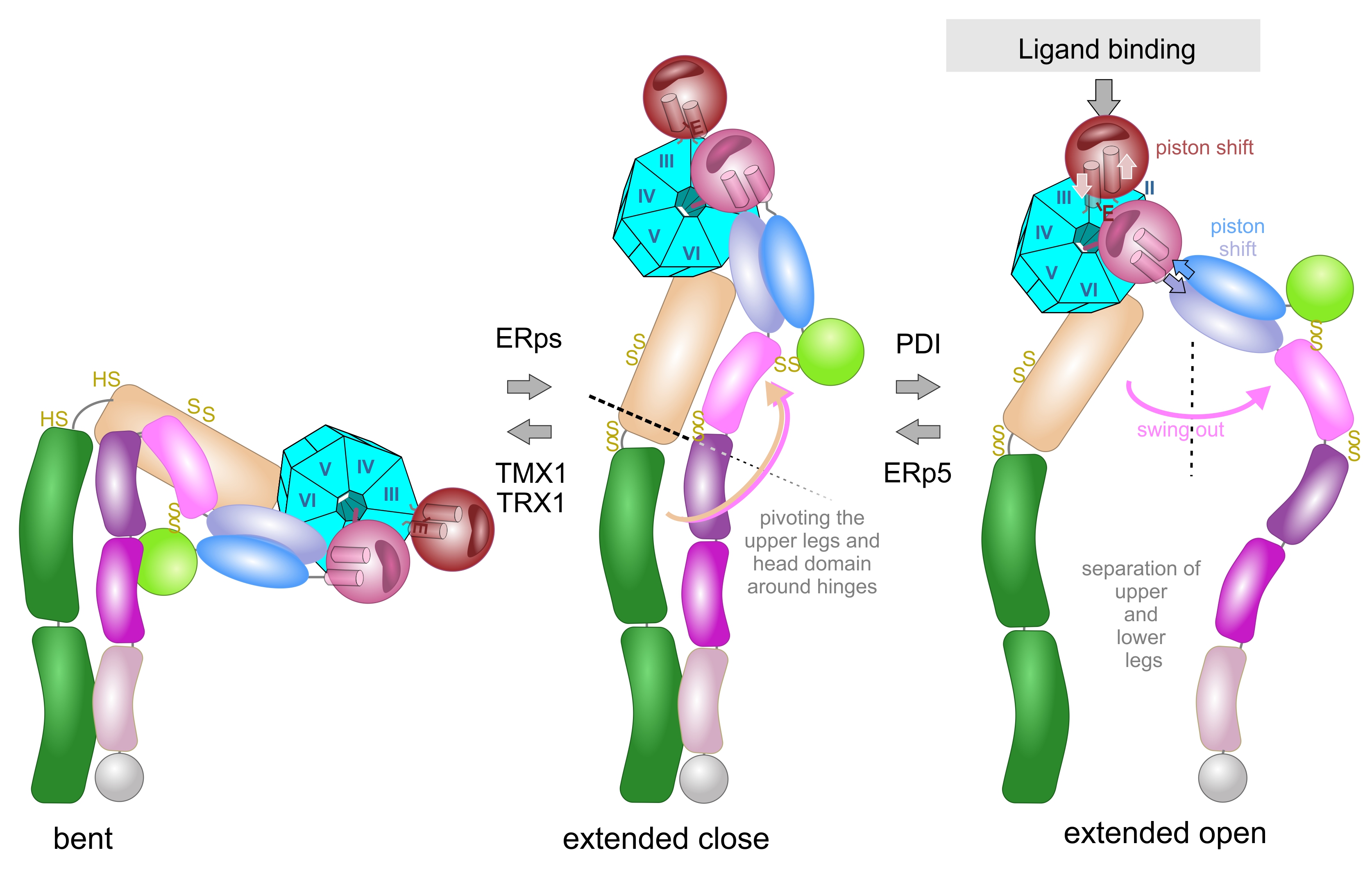
The rotational movement around the hinge regions is an essential part in the extension model, in which the integrin changes between a bent and extended conformation (Figure 5). In a reversible manner, the bent conformation, in which the ligand binding head domain points towards the cell membrane, converts into the extended conformation, in which the entire integrin ectodomains takes an upright shape with the ligand binding site pointing away from the cell membrane towards the ECM [124,125,126,127]. Electron microscopy, combined with single particle analysis, and small angle x-ray scattering proved the extension of the integrin upon activation in vitro [124,128,129] and on the cell surface [130]. Additionally, conformation-dependent antibodies helped to define specific integrin conformations [131]. During extension, contacts between the upper and lower leg domains, as well as between the headpiece and the lower leg domains are loosened, especially the intra-β-chain interactions between the βA and βTD domains [80] and the inter-subunit contacts between the membrane-proximal calf 2 and EGF4 domains [132].
During the extension of the integrin, the hinge regions of both subunit still remain together, so that the bent conformation converts into the so called extended-close conformation with an increased binding affinity to its ECM ligand. Subsequently, ligand binding triggers another conformational change of the integrin ectodomain, in which the knees/hinge regions of both integrin subunits separate. This so-called swing out movement converts the extended close conformation into the extended open one with the latter having the highest affinity to its ECM ligand [82,133,134]. Interestingly the conformations differ thermodynamically, with the free enthalpy rising strongly upon integrin extension and slightly from the extended close to the extended open conformations rendering integrin activation an energy-demanding process [135].
Ligand binding triggers the second conformational change, the swing out movement of the upper leg regions (Figure 5). In the αA-containing integrins, the ligand, such as collagen, binds via the MIDAS to the top of the αA-domain. This binding crevice is opposite to the N- and C-terminal linker sequences of the αA-domain, with which it is connected to the propeller domain. The major structural consequences are unfolding of an additional α-helic C, a shift of the ADMIDAS metal ion close to the binding crevice and a piston movement of α-helix 7 against α-helix 1 by about 1 nm [56,76,133]. As a consequence of the ligand-induced piston shift of the α-helix 7, a glutamate residue in its C-terminally adjacent linker-region approaches the MIDAS of the neighboring βA-domain and serves as an “internal” ligand [136]. During this process, the residue R261 on the side face of the βA domain holds it in place towards the propeller domain, by poking its side chain deeply into the central pore with the propeller domain (Figure 6). This interaction likely contributes to the stable platform of the head piece, consisting of the βA-domain and propeller domain as well as αA-domain, if present. From the head piece, both flexible legs eradiate. Whereas in αA-domain-containing integrins, the emerging glutamate residue of the αA domain serves as an internal ligand for the βA-domain, this role is taken by an acidic amino acid side chain of the ECM ligand in αA-domain-lacking integrins. For example, this can be the aspartate side chain of the RGD-motif in fibrin [77,137]. Then, this likely leads to a piston shift between α-helices 1 and 7, similar to the one within the A domain of the integrin α subunit [138].
How do the cysteines within the A-domains potentially influence the conformational changes? In the A-domain of the integrin α subunit, the two cysteines are too far apart, separated by the central β-sheet and hardly accessible from the protein surface to be oxidized into a disulfide bridge. In contrast, the two disulfide bridges within the βA domain, C177-C184 and C232-C273, are redox-regulated and located at conformationally relevant sites. Whereas C177-C184 is located to the top of the βA domain, close to the ligand-binding crevice, but not directly participating in the loops that shape the metal-binding sites (MIDAS and ADMIDAS), the second disulfide bridge, C232-C273 of βA domain is situated in a loop structure between α-helix 4 and β-strand D, which also harbors the R261 residue that anchors the βA domain with the propeller domain [70,77]. It can be envisioned, albeit not proven yet, that opening the disulfide bridge C232-273 results in a loosening of this interdomain static platform of the βA-domain with the propeller domain, thereby affecting the mechanics of secondary structure movements.
The piston shifts within the A-domains that both are associated to the propeller domain within the head piece is conveyed towards the hinge domain via the rod-like hybrid domain. As the βA domain is inserted into the hybrid domain, there are two peptide strands connecting the two domains. Along the N→C direction, the peptide chain enters into the helix 1 of the αA domain from the hybrid domain, while it leads out from helix 7 of the αA domain back into the hybrid domain. The piston shift of helices 1 and 7 via these two connections is transmitted into a rotational movement of the hybrid domain, which swings out like a lever which is even enlarged by the adjacent EGF1-domain [139].
The EGF1 domain contains two allosteric disulfide bridges, which are located at either end of the domain. The first one, C13-C435, connects the PSI domain with the EGF1, while the latter, C475-C503, links EGF1 to EGF2 in the hinge region [134,139,140]. Hence it can easily imagined that both disulfide bridges influence the swing out movement of the integrin upper legs from the head piece.
The hinge domains of both subunits play a role not only in the extension but also in the swing out movement, which eventually separates the knee regions from both integrin subunits. The thiol switch within the α subunit hinge region is of dominant importance in this conformational conversion [112,120,127]. The swing out movement goes along with the disengagement of the interfaces between the two integrin subunits. In the bent and extended-close conformation, before the swing out, the upper legs of both subunits still remain close together with several molecular interactions [73], as the thigh domain interacts with the hybrid and EGF1 domain. Also, the calf1 and 2 domains form interfaces with the EGF-domains 3 and 4 (Figure 6). It might be noteworthy, that the disulfide bridge C523-C544 of EGF3 domain of β3 subunit comes into vicinity of the disulfide bridge C674-C687 of the calf1 domain of the αIIb subunit. Likewise, the thigh domain and the hinge domain of the integrin α subunit also contain redox-regulated disulfide bridges, C490-545 and C602-C608, respectively. They are close to the EGF2 domain of the β-subunit that loses its contact with the α-subunit thigh domain upon separation of the upper legs [73,133,140].
As a consequence of the swing out movement, the separated stalks push the two transmembrane and cytoplasmic domains of both integrin subunits apart, enabling the conformational signaling to be conveyed and translated into the association of adaptor and signaling molecules to the cytoplasmic tails. Of pivotal importance, the conformational conversion correlates with the biological functions of the integrin that act as anchoring receptor and signalling relais within the cell membrane [44,66,133].
8. From Disulfide Bridge-Induced Conformational Changes to Cellular Consequences of Integrin-ECM Contacts
The oxidative formation and reductive cleavage of disulfide bridges within integrins cause conformational changes. This alters their affinity towards the respective ECM ligands. In addition, ECM occupancy of integrins also induces clustering of integrins [51], or if integrin clusters exist already before ligand binding, eventuallyto recruitment of adaptor and signaling molecules to the separated integrin cytoplasmic tails. Thus, a new adhesive cell organelle termed adhesome is formed [58,62,141]. In this highly ordered stratified structure, cytoskeletal adaptor molecules and their regulator proteins connect the cytoplasmic integrin tails with the actin fiber network and its motor proteins to the ECM [142].Thus, adhesomes serve as both mechanical anchorage points and signaling hubs.
However, it must be stated that redox-active compounds and redox-modifying enzymes are not the only factors that affect integrin binding to their ECM ligands or influence integrin association with intracellular adaptor and signaling molecules, but also other parameters, such as concentration of divalent cations, tensile forces and intracellular adaptor proteins, are also valid effectors of integrins’ activity. Divalent metal ions may serve both structural and regulatory functions within integrins [78,143]. Some of the divalent cations, such as the ones on the bottom face of the propeller domain likely keep up a structure, whereas the divalent cations within the A-domains are essential to bridge the ECM with the integrin during binding [78,143]. Within the α subunit hinge region, the divalent cation binding site depends on the formation of the disulfide bridge [120]. Tensile mechanical forces that are transmitted via the integrins across the cell membrane also influence integrin activity [56,144,145]. In addition, the rate of force exertion is important, as integrin form a catch bond to their extracellular ligand. It also has an effect on integrin conformation [146,147]. Moreover, the adhesome structure and composition changes with the load and duration of the integrin-mediated forces [56,148]. Also regulating integrin activity, intracellular adaptor proteins, such as kindlin and talin, activate integrins during inside-out signaling and thus trigger cell attachment [149,150]. Upon binding to the cytoplasmic tail of the integrin β-subunit, kindlin and talin keep integrins in the extended open conformation with high ECM ligand-binding activity [37,149]. Their association with integrins depend on small G-proteins, the corresponding guanine nucleotide exchange factors (GEFs), and GTPase-activating proteins (GAPs) [150,151,152,153].
Besides the fact that several different factors influence integrins, it must also be stated that integrins are not the only components of the supramolecular adhesome complex that are redox-regulated [59,60]. Therefore, redox regulation of integrins via their thiol switches has to be seen as part of a complex regulatory and signaling network that orchestrate cell adhesion, migration and other integrin-relation cellular functions, such as anchorage-dependent growth and survival, as well as differentiation [25,154,155].
Being anucleated cell fragments, platelets and their hemostatic function to stop blood leakage through injured blood vessels have been more intensively studied for redox regulation of integrin-mediated functions. A common target for vascular thiol isomerases is integrin αIIbβ3, the fibrin(ogen) receptor, which is by far the most abundant integrin receptor with 80.000 copies per platelet [35]. Among the numerous stimuli of platelet activation, the contact of platelets with immobilized ECM components, such as collagen, indicative of vessel damage, activates integrin α2β1, the sole collagen-binding integrin on platelets, in a redox-dependent manner [156]. Via inside-out signaling, activated platelets convert platelet integrin αIIbβ3 into a highly active receptor for fibrin. Fibrin is the end product of the coagulation cascade and its network stabilizes the newly formed thrombus. Both platelet integrin and fibrin are redox-regulated molecules, along with several factors of the coagulation cascade, such as the tissue factor [21,49,157]. Particular disulfide bridges within these molecules are the molecular redox switches, which are targets for cognate vascular thiol reductases [30,96,97].
Redox regulation is not only restricted to the anucleated platelets, but also occurs in numerous cells with different integrins. In addition to αIIbβ3, disulfide-based redox regulation has also been described for the collagen-binding integrins, α2β1 and α11β1 [156,158], for the laminin-binding integrin α7β1 [12,112,120], and for the immunlogically relevant integrins, α4β1, α4β7 αMβ2, and αLβ2, on leukocytes [118,121,159,160,161,162,163]. In most of these cases, PDI or other thiol isomerases are involved, and even physical association of them with the respective integrin was proven [30,104,158]. They influence the ECM ligand binding affinity positively [30,59,104,154,158] or negatively [108]. Mostly, the thiol isomerases use an oxidant, e.g., molecular oxygen or H2O2, to form a disulfide bridge. Exceptionally, the thiol isomerase TMX1 reduces disulfide bridges, but has so far only seen operative on the platelet integrin [111]. Also, reduced thioredoxin-1 can reduce α7β1-mediated cell migration by cleaving the α hinge thiol switch [112]. Albeit found in the extracellular space [3,164], thioredoxin-1, however, would need a constant supply of NADPH∙H+ in the pericellular space to be reduced by thioredoxin reductase and to maintain a redox regulation-supporting electron transport chain.
By altering integrin conformation, a disulfide-based redox regulation of integrin enables the cells to regulate or fine-tune their capabilities to transmit forces and to migrate. In order to adhere and spread, cell have to exert forces onto their ECM substrate. The binding of integrins to their ligands withstand decent forces [165,166,167,168,169,170]. Moreover, maximum force load transmitted by integrins is regulated by the redox modification of their allosteric disulfide bridges [57,118,144,171]. Vice versa, mechanical force mediated conformation changes of integrin may make particular disulfide bonds more accessible to thiol isomerase and thus influence their redox modification [108]. The mechanical forces transmitted via the integrins may not only serve firm cellular adhesion, but may also be a way, how cells set the ECM under tension, a biophysical parameter that influences behavior of other cells in tissues via integrin-mediated mechanosensing [38,172].
Cell migration is a multistep process, which includes integrin engagement with the ECM ligand and force transmission at the cell front, but also detachment of the cell and retraction at its rear end and recycling of disengaged integrins from the rear end to the front end of the cell [54,173,174,175]. Detachment of cell due to a redox-mediated decrease of integrin binding affinity have been shown for the integrin αMβ2 on neutrophil granulocytes at their rear end, thereby increasing their migratory extravasation [118]. Also, redox-modification of integrins within their thigh domain by thiol isomerases affects integrin recycling. Consequentially, a longer retainment of integrins changes its location from migration-supporting focal adhesions to fibrillary adhesions, a different adhesome type, which favors firm cell adhesion [25,106]
Although the platelet integrin and their thrombotic events have been in the focus for redox regulation of cell adhesion, all integrin-related cellular functions, also on nucleated cells, are redox-regulated resulting in the fact that redox regulation of integrins and their adhesome structure orchestrate various (patho)physiological processess, such as blood coagulation, vascular remodeling, mechanosensing, endothelial function, immune responses, inflammation, tumor progression and metastasis [3,59,176], which we only start to understand now.
Author Contributions
writing—original draft preparation, review and editing; and visualization, were done by JAE.
Funding
This research was funded by Deutsche Forschungsgemeinschaft (DFG), grant numbers EB177/17-1 and EB177/19-1, the latter as project P9 of the Clinical Research Unit (CRU) 342. The APC was also funded by Deutsche Forschungsgemeinschaft (DFG).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wong, J.W.; Hogg, P.J. Analysis of disulfide bonds in protein structures. J Thromb Haemost 2010. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.G.; Nardi, A.N.; Amadei, A.; D'Abramo, M. Theoretical Modeling of Redox Potentials of Biomolecules. Molecules 2022, 27. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, L.Y.; Oliveira, P.V.S.; Laurindo, F.R.M. Peri/Epicellular Thiol Oxidoreductases as Mediators of Extracellular Redox Signaling. Antioxid Redox Signal 2020, 33, 280–307. [Google Scholar] [CrossRef]
- Liu, Y.; Hyde, A.S.; Simpson, M.A.; Barycki, J.J. Emerging regulatory paradigms in glutathione metabolism. Adv Cancer Res 2014, 122, 69–101. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.C. Regulation of glutathione synthesis. Mol Aspects Med 2009, 30, 42–59. [Google Scholar] [CrossRef]
- Poole, L.B. The basics of thiols and cysteines in redox biology and chemistry. Free Radic Biol Med 2015, 80, 148–157. [Google Scholar] [CrossRef]
- Jezek, P.; Dlaskova, A.; Engstova, H.; Spackova, J.; Tauber, J.; Pruchova, P.; Kloppel, E.; Mozheitova, O.; Jaburek, M. Mitochondrial Physiology of Cellular Redox Regulations. Physiol Res 2024, 73, S217–S242. [Google Scholar] [CrossRef]
- Kervella, M.; Bertile, F.; Bouillaud, F.; Criscuolo, F. The cell origin of reactive oxygen species and its implication for evolutionary trade-offs. Open Biol 2025, 15, 240312. [Google Scholar] [CrossRef]
- Eble, J.A.; de Rezende, F.F. Redox-relevant aspects of the extracellular matrix and its cellular contacts via integrins. Antioxid Redox Signal 2014, 20, 1977–1993. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat Rev Mol Cell Biol 2020. [Google Scholar] [CrossRef]
- Bhopale, V.M.; Yang, M.; Yu, K.; Thom, S.R. Factors Associated with Nitric Oxide-mediated beta2 Integrin Inhibition of Neutrophils. J Biol Chem 2015, 290, 17474–17484. [Google Scholar] [CrossRef]
- de Rezende, F.F.; Martins Lima, A.; Niland, S.; Wittig, I.; Heide, H.; Schroder, K.; Eble, J.A. Integrin alpha7beta1 is a redox-regulated target of hydrogen peroxide in vascular smooth muscle cell adhesion. Free Radic Biol Med 2012, 53, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Pijning, A.E.; Butera, D.; Hogg, P.J. Not one, but many forms of thrombosis proteins. J Thromb Haemost 2022, 20, 285–292. [Google Scholar] [CrossRef]
- Pijning, A.E.; Chiu, J.; Yeo, R.X.; Wong, J.W.H.; Hogg, P.J. Identification of allosteric disulfides from labile bonds in X-ray structures. R Soc Open Sci 2018, 5, 171058. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J.; Hogg, P.J. Allosteric disulfides: Sophisticated molecular structures enabling flexible protein regulation. J Biol Chem 2019, 294, 2949–2960. [Google Scholar] [CrossRef]
- Schmidt, B.; Ho, L.; Hogg, P.J. Allosteric disulfide bonds. Biochemistry 2006, 45, 7429–7433. [Google Scholar] [CrossRef]
- Lv, K.; Chen, S.; Xu, X.; Chiu, J.; Wang, H.J.; Han, Y.; Yang, X.; Bowley, S.R.; Wang, H.; Tang, Z.; et al. Protein disulfide isomerase cleaves allosteric disulfides in histidine-rich glycoprotein to regulate thrombosis. Nat Commun 2024, 15, 3129. [Google Scholar] [CrossRef]
- Azimi, I.; Wong, J.W.; Hogg, P.J. Control of mature protein function by allosteric disulfide bonds. Antioxid Redox Signal 2011, 14, 113–126. [Google Scholar] [CrossRef]
- Hogg, P.J. Contribution of allosteric disulfide bonds to regulation of hemostasis. J Thromb Haemost 2009, 7 Suppl 1, 13–16. [Google Scholar] [CrossRef]
- Butera, D.; Cook, K.M.; Chiu, J.; Wong, J.W.; Hogg, P.J. Control of blood proteins by functional disulfide bonds. Blood 2014, 123, 2000–2007. [Google Scholar] [CrossRef]
- Butera, D.; Hogg, P.J. Fibrinogen function achieved through multiple covalent states. Nat Commun 2020, 11, 5468. [Google Scholar] [CrossRef]
- Zhou, B.; Hogg, P.J.; Grater, F. One-Way Allosteric Communication between the Two Disulfide Bonds in Tissue Factor. Biophys J 2017, 112, 78–86. [Google Scholar] [CrossRef]
- Chen, V.M.; Hogg, P.J. Encryption and decryption of tissue factor. J Thromb Haemost 2013, 11 Suppl 1, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, K.; Jeong, S.Y.; Chiu, J.; Xiong, B.; Petukhov, P.A.; Dai, X.; Li, X.; Andrews, R.K.; Du, X.; et al. Platelet Protein Disulfide Isomerase Promotes Glycoprotein Ibalpha-Mediated Platelet-Neutrophil Interactions Under Thromboinflammatory Conditions. Circulation 2019, 139, 1300–1319. [Google Scholar] [CrossRef] [PubMed]
- Pijning, A.E.; Hogg, P.J. Disulfide bond control of platelet alphaIIbbeta3 integrin. Thromb Res 2025, 250, 109320. [Google Scholar] [CrossRef] [PubMed]
- Lorenzen, I.; Eble, J.A.; Hanschmann, E.M. Thiol switches in membrane proteins - Extracellular redox regulation in cell biology. Biol Chem 2020. [Google Scholar] [CrossRef]
- Kodali, V.K.; Thorpe, C. Oxidative protein folding and the Quiescin-sulfhydryl oxidase family of flavoproteins. Antioxid Redox Signal 2010, 13, 1217–1230. [Google Scholar] [CrossRef]
- Schulman, S.; Bendapudi, P.; Sharda, A.; Chen, V.; Bellido-Martin, L.; Jasuja, R.; Furie, B.C.; Flaumenhaft, R.; Furie, B. Extracellular Thiol Isomerases and Their Role in Thrombus Formation. Antioxid Redox Signal 2016, 24, 1–15. [Google Scholar] [CrossRef]
- Stopa, J.D.; Zwicker, J.I. The intersection of protein disulfide isomerase and cancer associated thrombosis. Thromb Res 2018, 164 Suppl 1, S130–S135. [Google Scholar] [CrossRef]
- Essex, D.W.; Wang, L. Recent advances in vascular thiol isomerases and redox systems in platelet function and thrombosis. J Thromb Haemost 2024, 22, 1806–1818. [Google Scholar] [CrossRef]
- Wu, Y.; Essex, D.W. Vascular thiol isomerases in thrombosis: The yin and yang. J Thromb Haemost 2020, 18, 2790–2800. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.D.; Reddy, E.C.; Moran, N.; O'Neill, S. Regulation of platelet activity in a changing redox environment. Antioxid Redox Signal 2014, 20, 2074–2089. [Google Scholar] [CrossRef] [PubMed]
- van der Meijden, P.E.J.; Heemskerk, J.W.M. Platelet biology and functions: new concepts and clinical perspectives. Nat Rev Cardiol 2019, 16, 166–179. [Google Scholar] [CrossRef] [PubMed]
- Eble, J.A.; Niland, S. The extracellular matrix of blood vessels. Current pharmaceutical design 2009, 15, 1385–1400. [Google Scholar] [CrossRef]
- Janus-Bell, E.; Mangin, P.H. The relative importance of platelet integrins in hemostasis, thrombosis and beyond. Haematologica 2023, 108, 1734–1747. [Google Scholar] [CrossRef]
- Durrant, T.N.; van den Bosch, M.T.; Hers, I. Integrin alpha(IIb)beta(3) outside-in signaling. Blood 2017, 130, 1607–1619. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fassler, R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Katoh, K. Integrin and Its Associated Proteins as a Mediator for Mechano-Signal Transduction. Biomolecules 2025, 15. [Google Scholar] [CrossRef]
- Arnaout, M.A. The Integrin Receptors: From Discovery to Structure to Medicines. Immunol Rev 2025, 329, e13433. [Google Scholar] [CrossRef]
- Humphries, J.D.; Byron, A.; Humphries, M.J. Integrin ligands at a glance. J Cell Sci 2006, 119, 3901–3903. [Google Scholar] [CrossRef]
- Barczyk, M.; Carracedo, S.; Gullberg, D. Integrins. Cell Tissue Res 2010, 339, 269–280. [Google Scholar] [CrossRef]
- Singh, B.; Fleury, C.; Jalalvand, F.; Riesbeck, K. Human pathogens utilize host extracellular matrix proteins laminin and collagen for adhesion and invasion of the host. FEMS Microbiol Rev 2012, 36, 1122–1180. [Google Scholar] [CrossRef]
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv Drug Deliv Rev 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Humphries, J.D.; Chastney, M.R.; Askari, J.A.; Humphries, M.J. Signal transduction via integrin adhesion complexes. Curr Opin Cell Biol 2018, 56, 14–21. [Google Scholar] [CrossRef]
- Eble, J.A. The molecular basis of integrin-extracellular matrix interactions. Osteoarthritis and cartilage / OARS, Osteoarthritis Research Society 2011, 9 Suppl A, S131–140. [Google Scholar]
- Hohenester, E. Structural biology of laminins. Essays Biochem 2019, 63, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Zhi, K.; Hu, L.; Fan, Z. The Activation and Regulation of beta2 Integrins in Phagocytes and Phagocytosis. Front Immunol 2021, 12, 633639. [Google Scholar] [CrossRef]
- Fagerholm, S.C.; Guenther, C.; Llort Asens, M.; Savinko, T.; Uotila, L.M. Beta2-Integrins and Interacting Proteins in Leukocyte Trafficking, Immune Suppression, and Immunodeficiency Disease. Front Immunol 2019, 10, 254. [Google Scholar] [CrossRef]
- Periayah, M.H.; Halim, A.S.; Mat Saad, A.Z. Mechanism Action of Platelets and Crucial Blood Coagulation Pathways in Hemostasis. Int J Hematol Oncol Stem Cell Res 2017, 11, 319–327. [Google Scholar]
- Madamanchi, A.; Santoro, S.A.; Zutter, M.M. alpha2beta1 Integrin. Adv Exp Med Biol 2014, 819, 41–60. [Google Scholar] [CrossRef]
- Lima, A.M.; Wegner, S.V.; Martins Cavaco, A.C.; Estevao-Costa, M.I.; Sanz-Soler, R.; Niland, S.; Nosov, G.; Klingauf, J.; Spatz, J.P.; Eble, J.A. The spatial molecular pattern of integrin recognition sites and their immobilization to colloidal nanobeads determine alpha2beta1 integrin-dependent platelet activation. Biomaterials 2018, 167, 107–120. [Google Scholar] [CrossRef]
- Li, Z.; Shao, R.; Xin, H.; Zhu, Y.; Jiang, S.; Wu, J.; Yan, H.; Jia, T.; Ge, M.; Shi, X. Paxillin and Kindlin: Research Progress and Biological Functions. Biomolecules 2025, 15. [Google Scholar] [CrossRef] [PubMed]
- Versteeg, H.H.; Heemskerk, J.W.; Levi, M.; Reitsma, P.H. New fundamentals in hemostasis. Physiol Rev 2013, 93, 327–358. [Google Scholar] [CrossRef] [PubMed]
- Case, L.B.; Waterman, C.M. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol 2015, 17, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Geiger, B.; Boujemaa-Paterski, R.; Winograd-Katz, S.E.; Balan Venghateri, J.; Chung, W.L.; Medalia, O. The Actin Network Interfacing Diverse Integrin-Mediated Adhesions. Biomolecules 2023, 13. [Google Scholar] [CrossRef]
- Bachmann, M.; Kukkurainen, S.; Hytonen, V.P.; Wehrle-Haller, B. Cell Adhesion by Integrins. Physiol Rev 2019, 99, 1655–1699. [Google Scholar] [CrossRef]
- Revach, O.Y.; Grosheva, I.; Geiger, B. Biomechanical regulation of focal adhesion and invadopodia formation. J Cell Sci 2020, 133. [Google Scholar] [CrossRef]
- Horton, E.R.; Humphries, J.D.; James, J.; Jones, M.C.; Askari, J.A.; Humphries, M.J. The integrin adhesome network at a glance. J Cell Sci 2016, 129, 4159–4163. [Google Scholar] [CrossRef]
- Matrullo, G.; Filomeni, G.; Rizza, S. Redox regulation of focal adhesions. Redox Biol 2025, 80, 103514. [Google Scholar] [CrossRef]
- Meissner, J.; Rezaei, M.; Siepe, I.; Ackermann, D.; Konig, S.; Eble, J.A. Redox proteomics reveals an interdependence of redox modification and location of adhesome proteins in NGF-treated PC12 cells. Free Radic Biol Med 2021, 164, 341–353. [Google Scholar] [CrossRef]
- Grosche, J.; Meissner, J.; Eble, J.A. More than a syllable in fib-ROS-is: The role of ROS on the fibrotic extracellular matrix and on cellular contacts. Mol Aspects Med 2018, 63, 30–46. [Google Scholar] [CrossRef]
- Horton, E.R.; Byron, A.; Askari, J.A.; Ng, D.H.J.; Millon-Fremillon, A.; Robertson, J.; Koper, E.J.; Paul, N.R.; Warwood, S.; Knight, D.; et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 2015, 17, 1577–1587. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Calderwood, D.A.; Ginsberg, M.H. Integrin cytoplasmic domain-binding proteins. J Cell Sci 2000, 113 ( Pt 20) Pt 20, 3563–3571. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, D.A.; Fujioka, Y.; de Pereda, J.M.; Garcia-Alvarez, B.; Nakamoto, T.; Margolis, B.; McGlade, C.J.; Liddington, R.C.; Ginsberg, M.H. Integrin beta cytoplasmic domain interactions with phosphotyrosine-binding domains: a structural prototype for diversity in integrin signaling. Proc Natl Acad Sci U S A 2003, 100, 2272–2277. [Google Scholar] [CrossRef] [PubMed]
- Horton, E.R.; Humphries, J.D.; Stutchbury, B.; Jacquemet, G.; Ballestrem, C.; Barry, S.T.; Humphries, M.J. Modulation of FAK and Src adhesion signaling occurs independently of adhesion complex composition. J Cell Biol 2016, 212, 349–364. [Google Scholar] [CrossRef]
- Arnaout, M.A.; Mahalingam, B.; Xiong, J.P. Integrin structure, allostery, and bidirectional signaling. Annu Rev Cell Dev Biol 2005, 21, 381–410. [Google Scholar] [CrossRef]
- Luo, B.H.; Carman, C.V.; Takagi, J.; Springer, T.A. Disrupting integrin transmembrane domain heterodimerization increases ligand binding affinity, not valency or clustering. Proc Natl Acad Sci U S A 2005, 102, 3679–3684. [Google Scholar] [CrossRef]
- Zhu, J.; Luo, B.H.; Barth, P.; Schonbrun, J.; Baker, D.; Springer, T.A. The structure of a receptor with two associating transmembrane domains on the cell surface: integrin alphaIIbbeta3. Mol Cell 2009, 34, 234–249. [Google Scholar] [CrossRef]
- Zhu, J.; Carman, C.V.; Kim, M.; Shimaoka, M.; Springer, T.A.; Luo, B.H. Requirement of alpha and beta subunit transmembrane helix separation for integrin outside-in signaling. Blood 2007, 110, 2475–2483. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Diefenbach, B.; Zhang, R.; Dunker, R.; Scott, D.L.; Joachimiak, A.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin αVβ3. Science 2001, 294, 339–345. [Google Scholar] [CrossRef]
- Nagae, M.; Re, S.; Mihara, E.; Nogi, T.; Sugita, Y.; Takagi, J. Crystal structure of α5β1 integrin ectodomain: atomic details of the fibronectin receptor. J. Cell Biol. 2012, 197, 131–140. [Google Scholar] [CrossRef]
- Xie, C.; Zhu, J.; Chen, X.; Mi, L.; Nishida, N.; Springer, T.A. Structure of an integrin with an αI domain, complement receptor type 4. EMBO J. 2010, 29, 666–679. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.P.; Mahalingham, B.; Alonso, J.L.; Borrelli, L.A.; Rui, X.; Anand, S.; Hyman, B.T.; Rysiok, T.; Muller-Pompalla, D.; Goodman, S.L.; et al. Crystal structure of the complete integrin alphaVbeta3 ectodomain plus an alpha/beta transmembrane fragment. J Cell Biol 2009, 186, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Luo, B.H.; Xiao, T.; Zhang, C.; Nishida, N.; Springer, T.A. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell 2008, 32, 849–861. [Google Scholar] [CrossRef] [PubMed]
- Emsley, J.; King, S.L.; Bergelson, J.M.; Liddington, R.C. Crystal structure of the I domain from integrin α2β1. J. Biol. Chem. 1997, 272, 28512–28517. [Google Scholar] [CrossRef]
- Emsley, J.; Knight, C.G.; Farndale, R.W.; Barnes, M.J.; Liddington, R.C. Structural basis of collagen recognition by integrin alpha2beta1. Cell 2000, 101, 47–56. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Zhang, R.; Joachimiak, A.; Frech, M.; Goodman, S.L.; Arnaout, M.A. Crystal structure of the extracellular segment of integrin alpha Vbeta3 in complex with an Arg-Gly-Asp ligand. Science 2002, 296, 151–155. [Google Scholar] [CrossRef]
- Zhang, K.; Chen, J. The regulation of integrin function by divalent cations. Cell Adh Migr 2012, 6, 20–29. [Google Scholar] [CrossRef]
- Gupta, V.; Alonso, J.L.; Sugimori, T.; Essafi, M.; Xiong, J.P.; Arnaout, M.A. Role of the beta-subunit arginine/lysine finger in integrin heterodimer formation and function. J Immunol 2008, 180, 1713–1718. [Google Scholar] [CrossRef]
- Gupta, V.; Gylling, A.; Alonso, J.L.; Sugimori, T.; Ianakiev, P.; Xiong, J.P.; Arnaout, M.A. The beta-tail domain (betaTD) regulates physiologic ligand binding to integrin CD11b/CD18. Blood 2007, 109, 3513–3520. [Google Scholar] [CrossRef]
- Zhu, J.; Boylan, B.; Luo, B.H.; Newman, P.J.; Springer, T.A. Tests of the extension and deadbolt models of integrin activation. J Biol Chem 2007, 282, 11914–11920. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.H.; Carman, C.V.; Springer, T.A. Structural basis of integrin regulation and signaling. Annu Rev Immunol 2007, 25, 619–647. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Henschen, A.; Gonzalez-Rodriguez, J. Complete localization of the intrachain disulphide bonds and the N-glycosylation points in the alpha-subunit of human platelet glycoprotein IIb. Biochem J 1989, 261, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Henschen, A.; Gonzalez-Rodriguez, J. Assignment of disulphide bonds in human platelet GPIIIa. A disulphide pattern for the beta-subunits of the integrin family. Biochem J 1991, 274 ( Pt 1), 63–71. [Google Scholar] [CrossRef]
- Krokhin, O.V.; Cheng, K.; Sousa, S.L.; Ens, W.; Standing, K.G.; Wilkins, J.A. Mass spectrometric based mapping of the disulfide bonding patterns of integrin α chains. Biochemistry 2003, 42, 12950–12959. [Google Scholar] [CrossRef]
- Xiong, J.P.; Stehle, T.; Goodman, S.L.; Arnaout, M.A. A novel adaptation of the integrin PSI domain revealed from its crystal structure. J Biol Chem 2004, 279, 40252–40254. [Google Scholar] [CrossRef]
- Shi, M.; Sundramurthy, K.; Liu, B.; Tan, S.M.; Law, S.K.; Lescar, J. The crystal structure of the plexin-semaphorin-integrin domain/hybrid domain/I-EGF1 segment from the human integrin beta2 subunit at 1.8-A resolution. J Biol Chem 2005, 280, 30586–30593. [Google Scholar] [CrossRef]
- Mor-Cohen, R. Disulfide Bonds as Regulators of Integrin Function in Thrombosis and Hemostasis. Antioxid Redox Signal 2016, 24, 16–31. [Google Scholar] [CrossRef]
- Takagi, J.; Beglova, N.; Yalamanchili, P.; Blacklow, S.C.; Springer, T.A. Definition of the EGF-like, closely interacting modules that bear activation epitopes in integrin β subunits. Proc. Natl. Acad. Sci. USA 2001, 98, 11175–11180. [Google Scholar] [CrossRef]
- Mor-Cohen, R.; Rosenberg, N.; Einav, Y.; Zelzion, E.; Landau, M.; Mansour, W.; Averbukh, Y.; Seligsohn, U. Unique disulfide bonds in epidermal growth factor (EGF) domains of beta3 affect structure and function of alphaIIbbeta3 and alphavbeta3 integrins in different manner. J Biol Chem 2012, 287, 8879–8891. [Google Scholar] [CrossRef]
- Wouters, M.A.; Rigoutsos, I.; Chu, C.K.; Feng, L.L.; Sparrow, D.B.; Dunwoodie, S.L. Evolution of distinct EGF domains with specific functions. Protein Sci. 2005, 14, 1091–1103. [Google Scholar] [CrossRef] [PubMed]
- Chiu, J. Measurement of redox states of the beta3 integrin disulfide bonds by Mass Spectrometry. Bio Protoc 2019, 9, e3156. [Google Scholar] [CrossRef] [PubMed]
- Zito, E. ERO1: A protein disulfide oxidase and H2O2 producer. Free Radic Biol Med 2015, 83, 299–304. [Google Scholar] [CrossRef] [PubMed]
- Makio, T.; Chen, J.; Simmen, T. ER stress as a sentinel mechanism for ER Ca(2+) homeostasis. Cell Calcium 2024, 124, 102961. [Google Scholar] [CrossRef]
- Bekendam, R.H.; Iyu, D.; Passam, F.; Stopa, J.D.; De Ceunynck, K.; Muse, O.; Bendapudi, P.K.; Garnier, C.L.; Gopal, S.; Crescence, L.; et al. Protein disulfide isomerase regulation by nitric oxide maintains vascular quiescence and controls thrombus formation. J Thromb Haemost 2018, 16, 2322–2335. [Google Scholar] [CrossRef]
- Tanaka, L.Y.; Laurindo, F.R.M. Vascular remodeling: A redox-modulated mechanism of vessel caliber regulation. Free Radic Biol Med 2017, 109, 11–21. [Google Scholar] [CrossRef]
- Chiu, J.; Passam, F.; Butera, D.; Hogg, P.J. Protein Disulfide Isomerase in Thrombosis. Semin Thromb Hemost 2015, 41, 765–773. [Google Scholar] [CrossRef]
- Soares Moretti, A.I.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch Biochem Biophys 2017, 617, 106–119. [Google Scholar] [CrossRef]
- Furie, B.; Flaumenhaft, R. Thiol isomerases in thrombus formation. Circ Res 2014, 114, 1162–1173. [Google Scholar] [CrossRef]
- Gaspar, R.S.; Gibbins, J.M. Thiol Isomerases Orchestrate Thrombosis and Hemostasis. Antioxid Redox Signal 2021, 35, 1116–1133. [Google Scholar] [CrossRef]
- Manickam, N.; Sun, X.; Li, M.; Gazitt, Y.; Essex, D.W. Protein disulphide isomerase in platelet function. Br J Haematol 2008, 140, 223–229. [Google Scholar] [CrossRef]
- Yang, M.; Chiu, J.; Scartelli, C.; Ponzar, N.; Patel, S.; Patel, A.; Ferreira, R.B.; Keyes, R.F.; Carroll, K.S.; Pozzi, N.; et al. Sulfenylation links oxidative stress to protein disulfide isomerase oxidase activity and thrombus formation. J Thromb Haemost 2023, 21, 2137–2150. [Google Scholar] [CrossRef]
- Groenendyk, J.; Lynch, J.; Michalak, M. Calreticulin, Ca2+, and calcineurin - signaling from the endoplasmic reticulum. Mol Cells 2004, 17, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Essex, D.W.; Wu, Y. Multiple protein disulfide isomerases support thrombosis. Curr Opin Hematol 2018, 25, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, X.; Wang, H.J.; Chen, Y.C.; Chen, Y.; Chiu, J.; Li, L.; Wang, L.; Wang, J.; Tang, Z.; et al. Endoplasmic Reticulum Protein 72 Regulates Integrin Mac-1 Activity to Influence Neutrophil Recruitment. Arterioscler Thromb Vasc Biol 2024, 44, e82–e98. [Google Scholar] [CrossRef] [PubMed]
- Pijning, A.E.; Blyth, M.T.; Coote, M.L.; Passam, F.; Chiu, J.; Hogg, P.J. An alternate covalent form of platelet alphaIIbbeta3 integrin that resides in focal adhesions and has altered function. Blood 2021, 138, 1359–1372. [Google Scholar] [CrossRef]
- Lay, A.J.; Dupuy, A.; Hagimola, L.; Tieng, J.; Larance, M.; Zhang, Y.; Yang, J.; Kong, Y.; Chiu, J.; Gray, E.; et al. Endoplasmic reticulum protein 5 attenuates platelet endoplasmic reticulum stress and secretion in a mouse model. Blood Adv 2023, 7, 1650–1665. [Google Scholar] [CrossRef]
- Passam, F.; Chiu, J.; Ju, L.; Pijning, A.; Jahan, Z.; Mor-Cohen, R.; Yeheskel, A.; Kolsek, K.; Tharichen, L.; Aponte-Santamaria, C.; et al. Mechano-redox control of integrin de-adhesion. Elife 2018, 7. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, Y.; Rauova, L.; Koma, G.; Wang, L.; Poncz, M.; Li, H.; Liu, T.; Fong, K.P.; Bennett, J.S.; et al. A novel role for endoplasmic reticulum protein 46 (ERp46) in platelet function and arterial thrombosis in mice. Blood 2022, 139, 2050–2065. [Google Scholar] [CrossRef]
- Hogg, P.J. TMX1: a new vascular thiol isomerase. Blood 2019, 133, 188–190. [Google Scholar] [CrossRef]
- Zhao, Z.; Wu, Y.; Zhou, J.; Chen, F.; Yang, A.; Essex, D.W. The transmembrane protein disulfide isomerase TMX1 negatively regulates platelet responses. Blood 2019, 133, 246–251. [Google Scholar] [CrossRef]
- Bergerhausen, L.; Grosche, J.; Meissner, J.; Hecker, C.; Caliandro, M.F.; Westerhausen, C.; Kamenac, A.; Rezaei, M.; Morgelin, M.; Poschmann, G.; et al. Extracellular Redox Regulation of α7β1 Integrin-Mediated Cell Migration Is Signaled via a Dominant Thiol-Switch. Antioxidants (Basel) 2020, 9, 227–250. [Google Scholar] [CrossRef]
- Yan, B.; Smith, J.W. A redox site involved in integrin activation. J Biol Chem 2000, 275, 39964–39972. [Google Scholar] [CrossRef]
- Manickam, N.; Ahmad, S.S.; Essex, D.W. Vicinal thiols are required for activation of the alphaIIbbeta3 platelet integrin. J Thromb Haemost 2011, 9, 1207–1215. [Google Scholar] [CrossRef]
- MacKeigan, D.T.; Ni, T.; Shen, C.; Stratton, T.W.; Ma, W.; Zhu, G.; Bhoria, P.; Ni, H. Updated Understanding of Platelets in Thrombosis and Hemostasis: The Roles of Integrin PSI Domains and their Potential as Therapeutic Targets. Cardiovasc Hematol Disord Drug Targets 2020, 20, 260–273. [Google Scholar] [CrossRef]
- Zhu, G.; Zhang, Q.; Reddy, E.C.; Carrim, N.; Chen, Y.; Xu, X.R.; Xu, M.; Wang, Y.; Hou, Y.; Ma, L.; et al. The integrin PSI domain has an endogenous thiol isomerase function and is a novel target for antiplatelet therapy. Blood 2017, 129, 1840–1854. [Google Scholar] [CrossRef]
- Weichsel, A.; Gasdaska, J.R.; Powis, G.; Montfort, W.R. Crystal structures of reduced, oxidized, and mutated human thioredoxins: evidence for a regulatory homodimer. Structure 1996, 4, 735–751. [Google Scholar] [CrossRef]
- Dupuy, A.; Aponte-Santamaria, C.; Yeheskel, A.; Hortle, E.; Oehlers, S.H.; Grater, F.; Hogg, P.J.; Passam, F.H.; Chiu, J. Mechano-Redox Control of Mac-1 De-Adhesion by PDI Promotes Directional Movement Under Flow. Circ Res 2023, 132, e151–e168. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Shi, P.; Chen, X.; Zhang, L.; Liu, J.; Fan, X.; Luo, X. Clathrin-mediated integrin alphaIIbbeta3 trafficking controls platelet spreading. Platelets 2018, 29, 610–621. [Google Scholar] [CrossRef] [PubMed]
- Caliandro, M.F.; Schmalbein, F.; Todesca, L.M.; Morgelin, M.; Rezaei, M.; Meissner, J.; Siepe, I.; Grosche, J.; Schwab, A.; Eble, J.A. A redox-dependent thiol-switch and a Ca2+ binding site within the hinge region hierarchically depend on each other in α7β1 integrin regulation. Free Radic Biol Med 2022. [Google Scholar] [CrossRef]
- Zhang, K.; Pan, Y.; Qi, J.; Yue, J.; Zhang, M.; Xu, C.; Li, G.; Chen, J. Disruption of disulfide restriction at integrin knees induces activation and ligand-independent signaling of α4β7. J Cell Sci 2013, 126, 5030–5041. [Google Scholar] [CrossRef] [PubMed]
- Mor-Cohen, R.; Rosenberg, N.; Averbukh, Y.; Seligsohn, U.; Lahav, J. Disulfide bond exchanges in integrins alphaIIbbeta3 and alphavbeta3 are required for activation and post-ligation signaling during clot retraction. Thromb Res 2014, 133, 826–836. [Google Scholar] [CrossRef] [PubMed]
- Leader, A.; Mor-Cohen, R.; Ram, R.; Sheptovitsky, V.; Seligsohn, U.; Rosenberg, N.; Lahav, J. The role of protein disulfide isomerase in the post-ligation phase of β3 integrin-dependent cell adhesion. Thromb Res 2015, 136, 1259–1265. [Google Scholar] [CrossRef] [PubMed]
- Adair, B.D.; Xiong, J.P.; Alonso, J.L.; Hyman, B.T.; Arnaout, M.A. EM structure of the ectodomain of integrin CD11b/CD18 and localization of its ligand-binding site relative to the plasma membrane. PLoS One 2013, 8, e57951. [Google Scholar] [CrossRef]
- Xiao, T.; Takagi, J.; Coller, B.S.; Wang, J.H.; Springer, T.A. Structural basis for allostery in integrins and binding to fibrinogen-mimetic therapeutics. Nature 2004, 432, 59–67. [Google Scholar] [CrossRef]
- Kamata, T.; Handa, M.; Ito, S.; Sato, Y.; Ohtani, T.; Kawai, Y.; Ikeda, Y. Structural requirements for activation in αIIbβ3 integrin. J. Biol. Chem. 2010, 285, 38428–38437. [Google Scholar] [CrossRef]
- Xie, C.; Shimaoka, M.; Xiao, T.; Schwab, P.; Klickstein, L.B.; Springer, T.A. The integrin alpha-subunit leg extends at a Ca2+-dependent epitope in the thigh/genu interface upon activation. Proc Natl Acad Sci U S A 2004, 101, 15422–15427. [Google Scholar] [CrossRef]
- Adair, B.D.; Xiong, J.P.; Maddock, C.; Goodman, S.L.; Arnaout, M.A.; Yeager, M. Three-dimensional EM structure of the ectodomain of integrin {alpha}V{beta}3 in a complex with fibronectin. J Cell Biol 2005, 168, 1109–1118. [Google Scholar] [CrossRef]
- Eng, E.T.; Smagghe, B.J.; Walz, T.; Springer, T.A. Intact alphaIIbbeta3 integrin is extended after activation as measured by solution X-ray scattering and electron microscopy. J Biol Chem 2011, 286, 35218–35226. [Google Scholar] [CrossRef]
- Moore, T.I.; Aaron, J.; Chew, T.L.; Springer, T.A. Measuring Integrin Conformational Change on the Cell Surface with Super-Resolution Microscopy. Cell Rep 2018, 22, 1903–1912. [Google Scholar] [CrossRef]
- Byron, A.; Humphries, J.D.; Askari, J.A.; Craig, S.E.; Mould, A.P.; Humphries, M.J. Anti-integrin monoclonal antibodies. J Cell Sci 2009, 122, 4009–4011. [Google Scholar] [CrossRef]
- Wang, J.H.; Fu, G.; Luo, B.H. Dissociation of the α-subunit calf-2 domain and the β-subunit I-EGF4 domain in integrin activation and signaling. Biochem. 2010, 49, 10158–10165. [Google Scholar] [CrossRef]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr Opin Cell Biol 2006, 18, 579–586. [Google Scholar] [CrossRef]
- Springer, T.A.; Dustin, M.L. Integrin inside-out signaling and the immunological synapse. Curr Opin Cell Biol 2012, 24, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Su, Y.; Xia, W.; Qin, Y.; Humphries, M.J.; Vestweber, D.; Cabanas, C.; Lu, C.; Springer, T.A. Conformational equilibria and intrinsic affinities define integrin activation. EMBO J 2017, 36, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Shimaoka, M.; Salas, A.; Takagi, J.; Springer, T.A. Intersubunit signal transmission in integrins by a receptor-like interaction with a pull spring. Proc Natl Acad Sci U S A 2004, 101, 2906–2911. [Google Scholar] [CrossRef] [PubMed]
- Springer, T.A.; Zhu, J.; Xiao, T. Structural basis for distinctive recognition of fibrinogen gammaC peptide by the platelet integrin alphaIIbbeta3. J Cell Biol 2008, 182, 791–800. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Li, N.; Shu, X.; Zhou, L.; Lu, S.; Chen, S.; Mao, D.; Long, M. Salt bridge interactions within the beta2 integrin alpha7 helix mediate force-induced binding and shear resistance ability. FEBS J 2017. [Google Scholar] [CrossRef]
- Liddington, R.C.; Ginsberg, M.H. Integrin activation takes shape. J Cell Biol 2002, 158, 833–839. [Google Scholar] [CrossRef]
- Zhu, J.; Zhu, J.; Springer, T.A. Complete integrin headpiece opening in eight steps. J Cell Biol 2013, 201, 1053–1068. [Google Scholar] [CrossRef]
- Winograd-Katz, S.E.; Fassler, R.; Geiger, B.; Legate, K.R. The integrin adhesome: from genes and proteins to human disease. Nat Rev Mol Cell Biol 2014, 15, 273–288. [Google Scholar] [CrossRef]
- Kanchanawong, P.; Shtengel, G.; Pasapera, A.M.; Ramko, E.B.; Davidson, M.W.; Hess, H.F.; Waterman, C.M. Nanoscale architecture of integrin-based cell adhesions. Nature 2010, 468, 580–584. [Google Scholar] [CrossRef]
- Xia, W.; Springer, T.A. Metal ion and ligand binding of integrin alpha5beta1. Proc Natl Acad Sci U S A 2014, 111, 17863–17868. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Zhou, L.; Zhang, Y.; Lu, S.; Long, M. Mechanochemical modeling of neutrophil migration based on four signaling layers, integrin dynamics, and substrate stiffness. Biomech Model Mechanobiol 2018, 17, 1611–1630. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Springer, T.A. Integrin extension enables ultrasensitive regulation by cytoskeletal force. Proc Natl Acad Sci U S A 2017, 114, 4685–4690. [Google Scholar] [CrossRef] [PubMed]
- Kong, F.; Garcia, A.J.; Mould, A.P.; Humphries, M.J.; Zhu, C. Demonstration of catch bonds between an integrin and its ligand. J Cell Biol 2009, 185, 1275–1284. [Google Scholar] [CrossRef]
- Zhang, H.; Yang, M.; Kim, S.H.; Li, I.T.S. Integrin force loading rate in mechanobiology: From model to molecular measurement. QRB Discov 2025, 6, e9. [Google Scholar] [CrossRef]
- Zaidel-Bar, R.; Milo, R.; Kam, Z.; Geiger, B. A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci 2007, 120, 137–148. [Google Scholar] [CrossRef]
- Rognoni, E.; Ruppert, R.; Fassler, R. The kindlin family: functions, signaling properties and implications for human disease. J Cell Sci 2016, 129, 17–27. [Google Scholar] [CrossRef]
- Shattil, S.J.; Kim, C.; Ginsberg, M.H. The final steps in integrin activation: the end game. Nat. Rev. Mol. Cell Biol. 2010, 11, 288–300. [Google Scholar] [CrossRef]
- Hota, P.K.; Buck, M. Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functions. Cell Mol Life Sci 2012, 69, 3765–3805. [Google Scholar] [CrossRef]
- Bromberger, T.; Zhu, L.; Klapproth, S.; Qin, J.; Moser, M. Rap1 and membrane lipids cooperatively recruit talin to trigger integrin activation. J Cell Sci 2019, 132. [Google Scholar] [CrossRef]
- Zhu, L.; Yang, J.; Bromberger, T.; Holly, A.; Lu, F.; Liu, H.; Sun, K.; Klapproth, S.; Hirbawi, J.; Byzova, T.V.; et al. Structure of Rap1b bound to talin reveals a pathway for triggering integrin activation. Nat Commun 2017, 8, 1744. [Google Scholar] [CrossRef]
- Rosenberg, N.; Mor-Cohen, R.; Sheptovitsky, V.H.; Romanenco, O.; Hess, O.; Lahav, J. Integrin-mediated cell adhesion requires extracellular disulfide exchange regulated by protein disulfide isomerase. Exp Cell Res 2019, 381, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Jones, M.C.; Askari, J.A.; Humphries, J.D.; Humphries, M.J. Cell adhesion is regulated by CDK1 during the cell cycle. J Cell Biol 2018, 217, 3203–3218. [Google Scholar] [CrossRef] [PubMed]
- Lahav, J.; Wijnen, E.M.; Hess, O.; Hamaia, S.W.; Griffiths, D.; Makris, M.; Knight, C.G.; Essex, D.W.; Farndale, R.W. Enzymatically catalyzed disulfide exchange is required for platelet adhesion to collagen via integrin α2β1. Blood 2003, 102, 2085–2092. [Google Scholar] [CrossRef] [PubMed]
- Zelaya, H.; Rothmeier, A.S.; Ruf, W. Tissue factor at the crossroad of coagulation and cell signaling. J Thromb Haemost 2018, 16, 1941–1952. [Google Scholar] [CrossRef]
- Popielarski, M.; Ponamarczuk, H.; Stasiak, M.; Michalec, L.; Bednarek, R.; Studzian, M.; Pulaski, L.; Swiatkowska, M. The role of Protein Disulfide Isomerase and thiol bonds modifications in activation of integrin subunit alpha11. Biochem Biophys Res Commun 2018, 495, 1635–1641. [Google Scholar] [CrossRef]
- Blouin, E.; Halbwachs-Mecarelli, L.; Rieu, P. Redox regulation of β2-integrin CD11b/CD18 activation. Eur J Immunol 1999, 29, 3419–3431. [Google Scholar] [CrossRef]
- Liu, S.Y.; Tsai, M.Y.; Chuang, K.P.; Huang, Y.F.; Shieh, C.C. Ligand binding of leukocyte integrin very late antigen-4 involves exposure of sulfhydryl groups and is subject to redox modulation. Eur J Immunol 2008, 38, 410–423. [Google Scholar] [CrossRef]
- Hahm, E.; Li, J.; Kim, K.; Huh, S.; Rogelj, S.; Cho, J. Extracellular protein disulfide isomerase regulates ligand-binding activity of alphaMbeta2 integrin and neutrophil recruitment during vascular inflammation. Blood 2013, 121, 3789–3800. [Google Scholar] [CrossRef]
- Laragione, T.; Bonetto, V.; Casoni, F.; Massignan, T.; Bianchi, G.; Gianazza, E.; Ghezzi, P. Redox regulation of surface protein thiols: identification of integrin alpha-4 as a molecular target by using redox proteomics. Proc Natl Acad Sci U S A 2003, 100, 14737–14741. [Google Scholar] [CrossRef]
- Cho, J. Protein disulfide isomerase in thrombosis and vascular inflammation. J Thromb Haemost 2013, 11, 2084–2091. [Google Scholar] [CrossRef]
- Hanschmann, E.M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, glutaredoxins, and peroxiredoxins--molecular mechanisms and health significance: from cofactors to antioxidants to redox signaling. Antioxid Redox Signal 2013, 19, 1539–1605. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.; Kanchanawong, P. Nanoscale mechano-adaption of integrin-based cell adhesions: New tools and techniques lead the way. Curr Opin Cell Biol 2025, 94, 102509. [Google Scholar] [CrossRef] [PubMed]
- Berdiaki, A.; Neagu, M.; Tzanakakis, P.; Spyridaki, I.; Perez, S.; Nikitovic, D. Extracellular Matrix Components and Mechanosensing Pathways in Health and Disease. Biomolecules 2024, 14. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, M.L.; Graham, B.T.; Pabon, L.M.; Han, S.J.; Murry, C.E.; Sniadecki, N.J. Measuring the contractile forces of human induced pluripotent stem cell-derived cardiomyocytes with arrays of microposts. J Biomech Eng 2014, 136, 051005. [Google Scholar] [CrossRef]
- Boye, K.; Ligezowska, A.; Eble, J.A.; Hoffmann, B.; Klosgen, B.; Merkel, R. Two barriers or not? Dynamic force spectroscopy on the integrin α7β1 invasin complex. Biophys J 2013, 105, 2771–2780. [Google Scholar] [CrossRef]
- Niland, S.; Westerhausen, C.; Schneider, S.W.; Eckes, B.; Schneider, M.F.; Eble, J.A. Biofunctionalization of a generic collagenous triple helix with the α2β1 integrin binding site allows molecular force measurements. The international journal of biochemistry & cell biology 2011, 43, 721–731. [Google Scholar] [CrossRef]
- Saraswathibhatla, A.; Indana, D.; Chaudhuri, O. Cell-extracellular matrix mechanotransduction in 3D. Nat Rev Mol Cell Biol 2023, 24, 495–516. [Google Scholar] [CrossRef]
- Dupuy, A.; Ju, L.A.; Chiu, J.; Passam, F.H. Mechano-Redox Control of Integrins in Thromboinflammation. Antioxid Redox Signal 2022, 37, 1072–1093. [Google Scholar] [CrossRef]
- Mathieu, M.; Isomursu, A.; Ivaska, J. Positive and negative durotaxis - mechanisms and emerging concepts. J Cell Sci 2024, 137. [Google Scholar] [CrossRef]
- Larocque, G.; Royle, S.J. Integrating intracellular nanovesicles into integrin trafficking pathways and beyond. Cell Mol Life Sci 2022, 79, 335. [Google Scholar] [CrossRef]
- Mana, G.; Valdembri, D.; Serini, G. Conformationally active integrin endocytosis and traffic: why, where, when and how? Biochem Soc Trans 2020, 48, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Shafaq-Zadah, M.; Gomes-Santos, C.S.; Bardin, S.; Maiuri, P.; Maurin, M.; Iranzo, J.; Gautreau, A.; Lamaze, C.; Caswell, P.; Goud, B.; et al. Persistent cell migration and adhesion rely on retrograde transport of beta(1) integrin. Nat Cell Biol 2016, 18, 54–64. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.M.; Muthuramalingam, K.; Cho, M. Redox Regulation of NOX Isoforms on FAK((Y397))/SRC((Y416)) Phosphorylation Driven Epithelial-to-Mesenchymal Transition in Malignant Cervical Epithelial Cells. Cells 2020, 9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Domain structure of integrins that do (i) or do not (ii) contain an A-domain in their α subunit. 24 integrins are known so far, all of which consists of two subunits, α and β. Half of the 18 different α subunits carry an A-domain inserted between blades II and III within the propeller domain. The domain names of both integrin subunits are labelled. The domains comprise several functional units: the head piece, the upper and lower legs. The head piece binds the ECM ligand and consists of the propeller domain and, if present the α subunit A-domain (αA), and the A-domain of the β-subunit (βA), which is anchored to the propeller domain via the side chain of a basic amino acid residue. The domains of the β subunit are: plexin-semaphorin-integrin (PSI), hybrid, four epidermal growth factor-homology (EGF), βtail-domain (βTD) and, like in the α subunit, a single membrane-spanning transmembrane (TM) and cytoplasmic domain (CD). The calf-2 domain of the integrin α subunits, which lack an A-domain, is poteolytically processed and cleaved into a heavy and light chain that are hold together via an intercatenary disulfide bridge.
Figure 1.
Domain structure of integrins that do (i) or do not (ii) contain an A-domain in their α subunit. 24 integrins are known so far, all of which consists of two subunits, α and β. Half of the 18 different α subunits carry an A-domain inserted between blades II and III within the propeller domain. The domain names of both integrin subunits are labelled. The domains comprise several functional units: the head piece, the upper and lower legs. The head piece binds the ECM ligand and consists of the propeller domain and, if present the α subunit A-domain (αA), and the A-domain of the β-subunit (βA), which is anchored to the propeller domain via the side chain of a basic amino acid residue. The domains of the β subunit are: plexin-semaphorin-integrin (PSI), hybrid, four epidermal growth factor-homology (EGF), βtail-domain (βTD) and, like in the α subunit, a single membrane-spanning transmembrane (TM) and cytoplasmic domain (CD). The calf-2 domain of the integrin α subunits, which lack an A-domain, is poteolytically processed and cleaved into a heavy and light chain that are hold together via an intercatenary disulfide bridge.

Figure 2.
Location of allosteric disulfide bonds within the integrin domains. (A) The two thiol switches within the thigh (wheat) and hinge (red) domains of the integrin α subunit. Although the two cysteines, C490 and C545, in the thigh domain is mostly reduced [25] and would allow flexibility, both residues are located in two neighboring strands within one β sheet thus being in an apparently structurally fixed position without much conformational flexibility. Nevertheless, this cysteine pair is highly accessible on the surface of the entire protein. The thiol switch, C602-C608, located in the hinge domain of the α subunit that flanks the upper leg and the calf1-domain of the lower leg (dark green), is of dominant importance as it determines the conformational change of the integrin ectodomains between the bent and extended conformation. The hinge sequence embraces residues that together with residues of the calf1 domain complex a Ca2+ ion. (B) PSI domain of the integrin β domain (green) with its long distance interdomain disulfide bridge C13-C435 to the EGF1 domain (magenta). Although the cysteines, C13 and C16, of the PSI show a sequence homology to the CXXC motif of thiol isomerases, both cysteines are not engaged with themselves, but with other cysteines within the EGF1-domain and the PSI domain. (C) Side views of βA domain (raspberry) with its two redox-relevant pairs of cysteine, C177-C184 and C232-C273. The Mg2+ ion (grey sphere) and two Ca2+ ions (darkblue spheres) mark the top of the A-domain and are complexed by residues in the loops connecting the β strands of the central β-sheet with each other or with the α-helices that flank the central β-sheet on either side. The βA-domain is firmly anchored with the integrin α subunit via its R261 residue, that deeply intrudes into the central pore of propeller domain (cyan). Visible from one side (i) are the first and last α-helices, 1 and 7, that are closest connected to the peptide chain that enters from (navy blue) and leaves into (lilac blue) the respective peptide chains of the hybrid domain, respectively. The redox-regulated thiol switch, C177-C184 (encircled in red), is located in the second loop between β-strands A and B, on top of the A-domain in close vicinity to the divalent cation-binding site. Visible after a 180° rotation around the long axis of the A domain (ii), the cysteine bridge, C232-C273 (encircled in red), is located in a loop that is directly connected to the R261 residue, which mediates the firm interaction of propeller domain and βA domain within the integrin headpiece. (D) The disulfide-rich EGF-domains, EGF1-EGF4, of the integrin β subunit (in different hues of magenta). While EGF1 is part of the upper leg, together with the hybrid domain (navy blue and lilac blue) in the upper part of the image, the domains EGF2, EGF3 and EGF4 form the lower leg in bottom part of the panel. The hinge region of the β subunit connects EGF1 with EGF2, contains another redox-regulated disulfide bridge, C475-C503 (encircled in red), and is close to the PSI domain with its long range disulfide bridge to the EGF1 domain (see also (B)). The molecular structure was rendered with Pymol Molecular Graphics System software, version 2.3.1, using the protein data file 3FCS.pdb of RCSB Protein Data BANK.
Figure 2.
Location of allosteric disulfide bonds within the integrin domains. (A) The two thiol switches within the thigh (wheat) and hinge (red) domains of the integrin α subunit. Although the two cysteines, C490 and C545, in the thigh domain is mostly reduced [25] and would allow flexibility, both residues are located in two neighboring strands within one β sheet thus being in an apparently structurally fixed position without much conformational flexibility. Nevertheless, this cysteine pair is highly accessible on the surface of the entire protein. The thiol switch, C602-C608, located in the hinge domain of the α subunit that flanks the upper leg and the calf1-domain of the lower leg (dark green), is of dominant importance as it determines the conformational change of the integrin ectodomains between the bent and extended conformation. The hinge sequence embraces residues that together with residues of the calf1 domain complex a Ca2+ ion. (B) PSI domain of the integrin β domain (green) with its long distance interdomain disulfide bridge C13-C435 to the EGF1 domain (magenta). Although the cysteines, C13 and C16, of the PSI show a sequence homology to the CXXC motif of thiol isomerases, both cysteines are not engaged with themselves, but with other cysteines within the EGF1-domain and the PSI domain. (C) Side views of βA domain (raspberry) with its two redox-relevant pairs of cysteine, C177-C184 and C232-C273. The Mg2+ ion (grey sphere) and two Ca2+ ions (darkblue spheres) mark the top of the A-domain and are complexed by residues in the loops connecting the β strands of the central β-sheet with each other or with the α-helices that flank the central β-sheet on either side. The βA-domain is firmly anchored with the integrin α subunit via its R261 residue, that deeply intrudes into the central pore of propeller domain (cyan). Visible from one side (i) are the first and last α-helices, 1 and 7, that are closest connected to the peptide chain that enters from (navy blue) and leaves into (lilac blue) the respective peptide chains of the hybrid domain, respectively. The redox-regulated thiol switch, C177-C184 (encircled in red), is located in the second loop between β-strands A and B, on top of the A-domain in close vicinity to the divalent cation-binding site. Visible after a 180° rotation around the long axis of the A domain (ii), the cysteine bridge, C232-C273 (encircled in red), is located in a loop that is directly connected to the R261 residue, which mediates the firm interaction of propeller domain and βA domain within the integrin headpiece. (D) The disulfide-rich EGF-domains, EGF1-EGF4, of the integrin β subunit (in different hues of magenta). While EGF1 is part of the upper leg, together with the hybrid domain (navy blue and lilac blue) in the upper part of the image, the domains EGF2, EGF3 and EGF4 form the lower leg in bottom part of the panel. The hinge region of the β subunit connects EGF1 with EGF2, contains another redox-regulated disulfide bridge, C475-C503 (encircled in red), and is close to the PSI domain with its long range disulfide bridge to the EGF1 domain (see also (B)). The molecular structure was rendered with Pymol Molecular Graphics System software, version 2.3.1, using the protein data file 3FCS.pdb of RCSB Protein Data BANK.
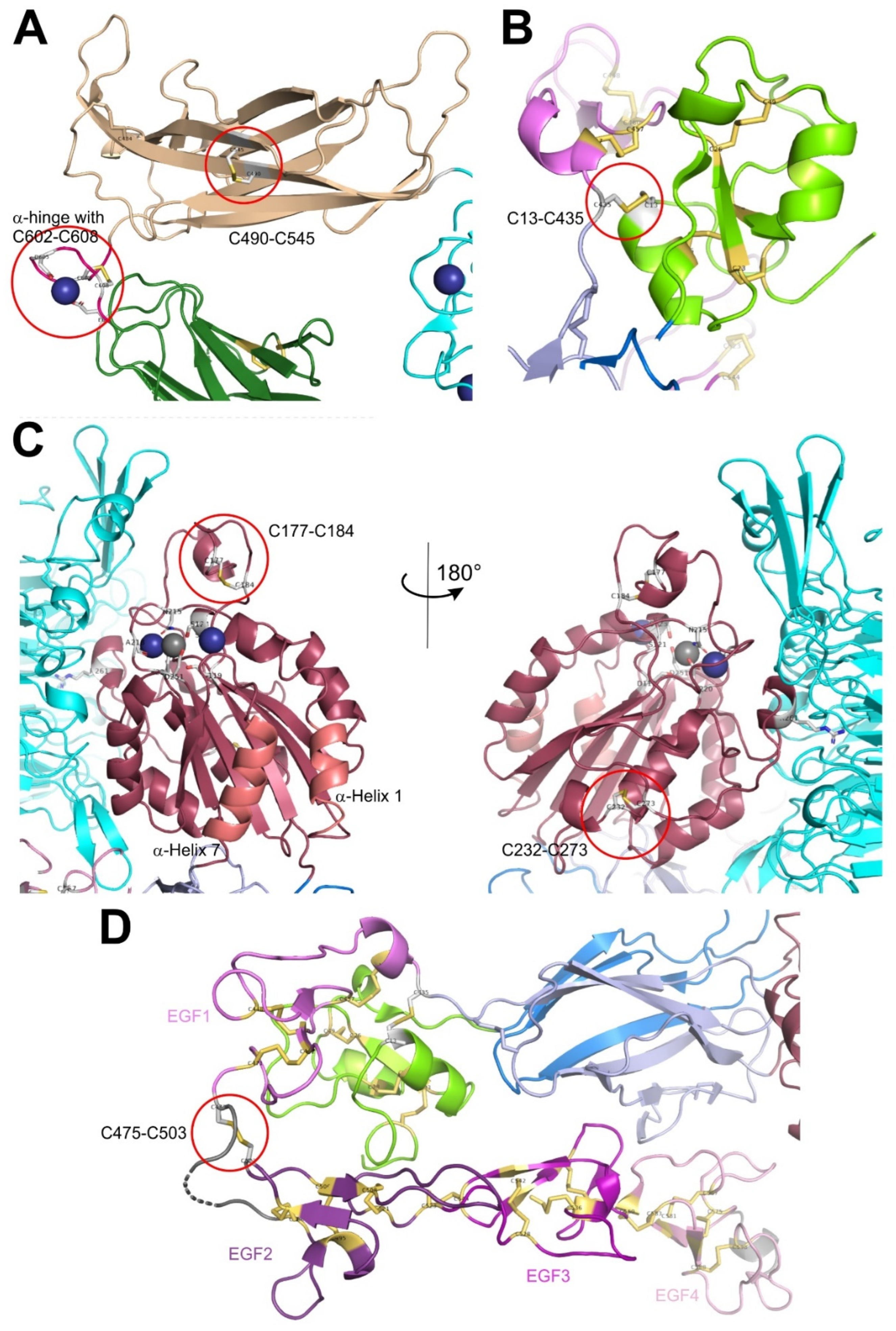
Figure 3.
Stereochemistry of conformers of disulfide bond-linked peptide chains and the most prominent conformations of allosteric disulfide bridges. Two cysteine-containing peptide chains can form a disulfide bond, marked in yellow. Like the four freely rotatable single bonds between the αC- and βC-atoms and between the βC-atoms and the sulfur atoms (yellow), the disulfide bond (highlighted in yellow) between the sulfur atoms can also rotate around its single bond. These five bonds are indicated in rainbow colors and the dihedral angles, that these bonds may take are labelled by ϕ1 through ϕ5. For clarity, the conformation, in which all dihedral angles lead to an anti-periplanar conformation is shown in the upper panel, which is likely to have the lowest conformational energy. The second lowest conformational energy level is taken by conformers in syn gauche conformations, which are reached from the anti-periplanar conformation by turning the dihedral angles, ϕ1, ϕ2, ϕ3, ϕ4, and ϕ5, around 120°, 120°, 97°, 120°, 120°, either clockwise (+) or counterclockwise (-), resulting in two possible conformers for each dihedral angles. As compared to the distribution of more than 13,000 disulfide bond conformation in protein data banks [15], allosteric disulfide bonds show a significant over-representation of three typical conformers, -/+RH hook (+,+,+,-,-), -LH hook ((-,+,-,-,-), and -RH (-,-,+,-,-), with the rotational orientation of the five dihedral angles given in parentheses. The dihedral strain energies, that represents the mechanical tension on the disulfide bond, are in the ranges of 12.8-14.7 kJ/mol, 15.1-17.1 kJ/mol, and 17.3-18.8 kJ/mol, respectively, for these three conformers, with the latter two being above average of all disulfide conformations of 14.5-15.0 kJ/mol [16]. The image were rendered with GeoGebra Classics software, version 6.0.892.0.
Figure 3.
Stereochemistry of conformers of disulfide bond-linked peptide chains and the most prominent conformations of allosteric disulfide bridges. Two cysteine-containing peptide chains can form a disulfide bond, marked in yellow. Like the four freely rotatable single bonds between the αC- and βC-atoms and between the βC-atoms and the sulfur atoms (yellow), the disulfide bond (highlighted in yellow) between the sulfur atoms can also rotate around its single bond. These five bonds are indicated in rainbow colors and the dihedral angles, that these bonds may take are labelled by ϕ1 through ϕ5. For clarity, the conformation, in which all dihedral angles lead to an anti-periplanar conformation is shown in the upper panel, which is likely to have the lowest conformational energy. The second lowest conformational energy level is taken by conformers in syn gauche conformations, which are reached from the anti-periplanar conformation by turning the dihedral angles, ϕ1, ϕ2, ϕ3, ϕ4, and ϕ5, around 120°, 120°, 97°, 120°, 120°, either clockwise (+) or counterclockwise (-), resulting in two possible conformers for each dihedral angles. As compared to the distribution of more than 13,000 disulfide bond conformation in protein data banks [15], allosteric disulfide bonds show a significant over-representation of three typical conformers, -/+RH hook (+,+,+,-,-), -LH hook ((-,+,-,-,-), and -RH (-,-,+,-,-), with the rotational orientation of the five dihedral angles given in parentheses. The dihedral strain energies, that represents the mechanical tension on the disulfide bond, are in the ranges of 12.8-14.7 kJ/mol, 15.1-17.1 kJ/mol, and 17.3-18.8 kJ/mol, respectively, for these three conformers, with the latter two being above average of all disulfide conformations of 14.5-15.0 kJ/mol [16]. The image were rendered with GeoGebra Classics software, version 6.0.892.0.
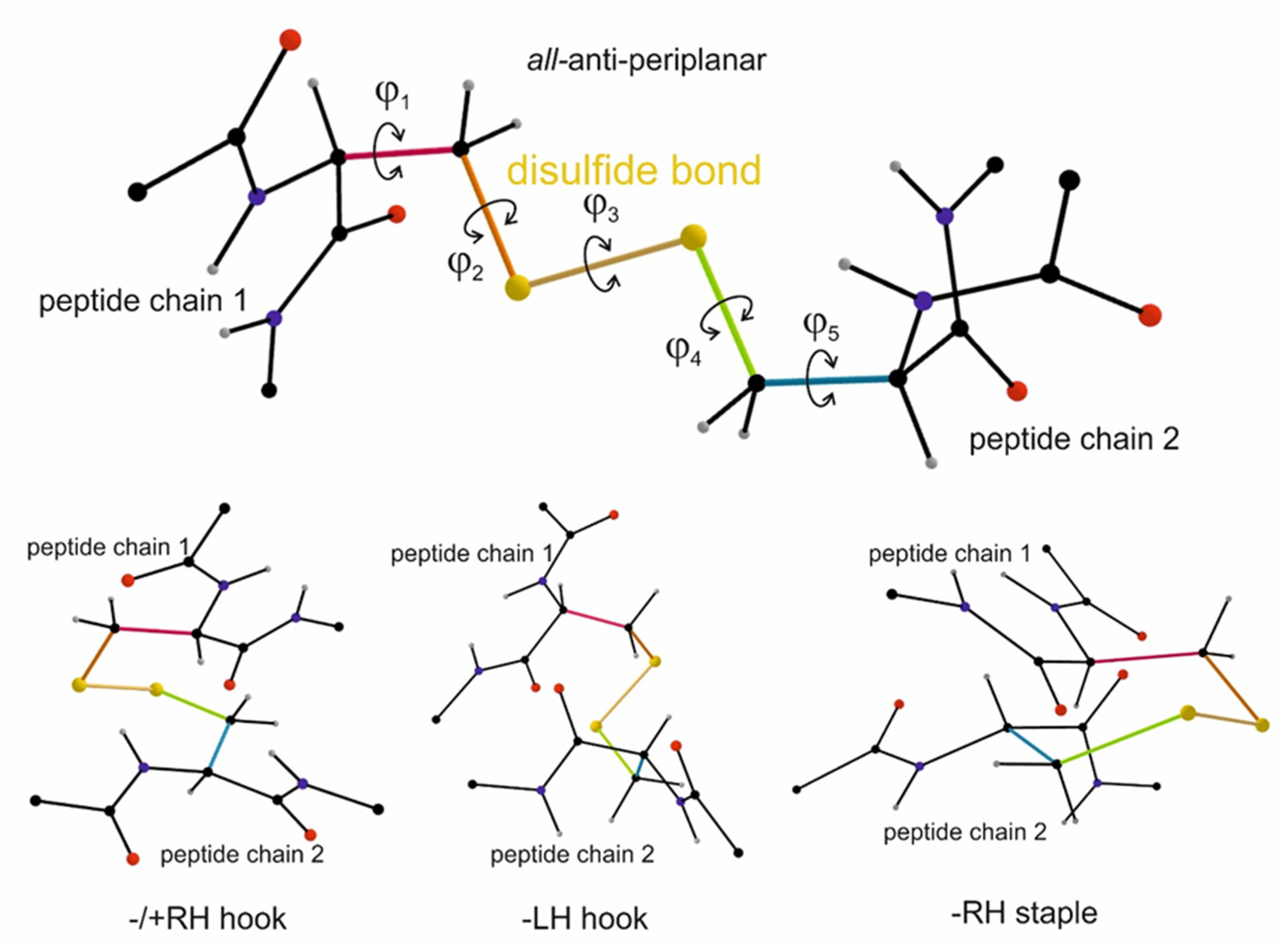
Figure 4.
Redox chain for oxidative folding of proteins and the formation of disulfide bonds in secreted target proteins. This pathway occurs during the secretary pathway of extracellular proteins or of ectodomains of membrane proteins, such as integrin ectodomains. However, as several of these redox-modifying enzymes are also secreted in the pericellular space, it is likely that this oxidative redox chain to form disulfide bridges also takes place in the environment of cells. Alternative pathways starting with either molecular oxygen (via ERo1) or hydrogen peroxide (via peroxiredoxin-4, PRDX4) are shown in the bottom part. The Ero1-based redox circle can hypothetically be substituted for by the redox enzyme QSOX1, which is a membrane-anchored redox enzyme that is secreted onto the cell surface.
Figure 4.
Redox chain for oxidative folding of proteins and the formation of disulfide bonds in secreted target proteins. This pathway occurs during the secretary pathway of extracellular proteins or of ectodomains of membrane proteins, such as integrin ectodomains. However, as several of these redox-modifying enzymes are also secreted in the pericellular space, it is likely that this oxidative redox chain to form disulfide bridges also takes place in the environment of cells. Alternative pathways starting with either molecular oxygen (via ERo1) or hydrogen peroxide (via peroxiredoxin-4, PRDX4) are shown in the bottom part. The Ero1-based redox circle can hypothetically be substituted for by the redox enzyme QSOX1, which is a membrane-anchored redox enzyme that is secreted onto the cell surface.
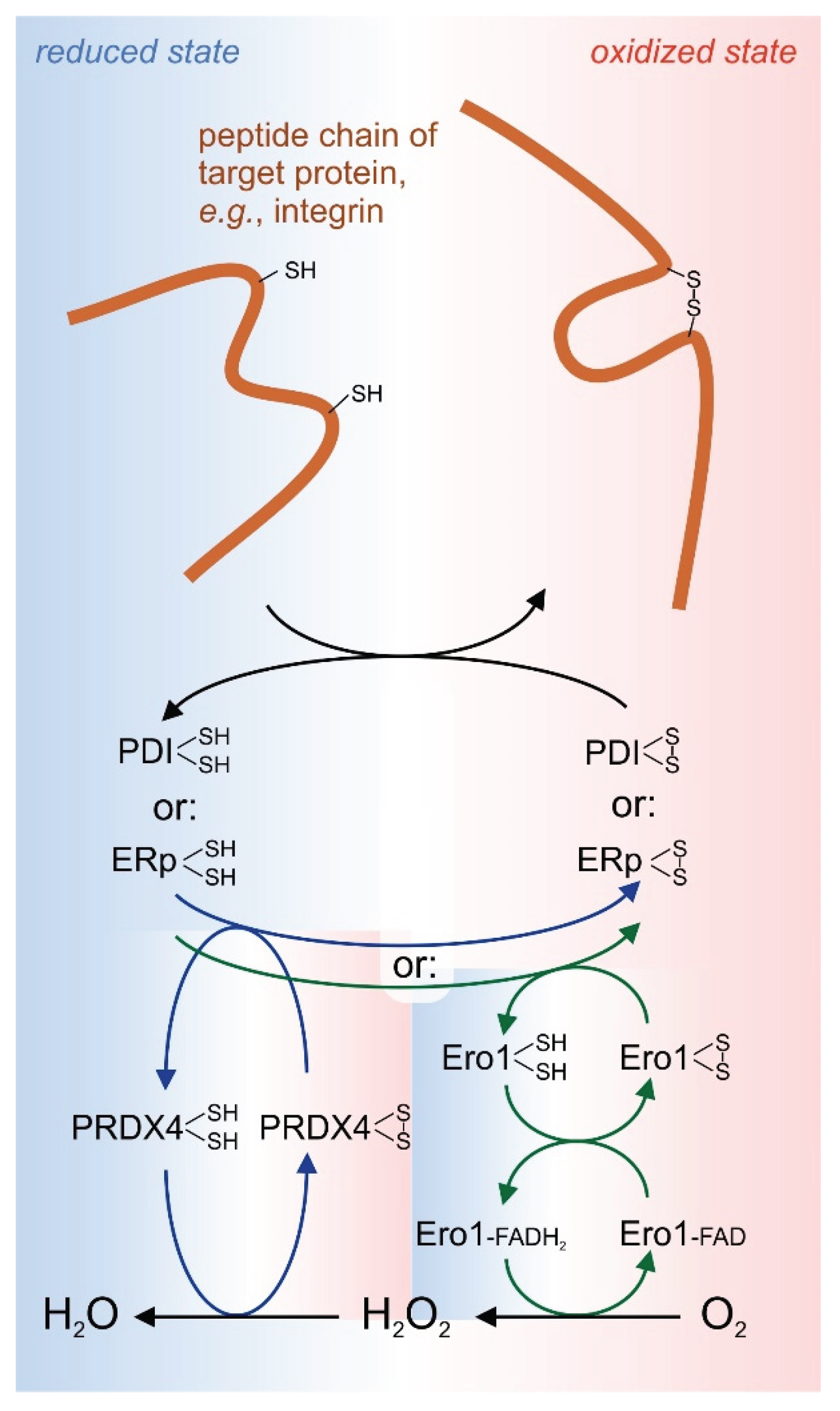
Figure 5.
Conformational changes within the integrin ectodomains structure and the potential involvement of allosteric disulfide bonds. A two step model of conformational changes of an integrin are shown. The first step is the extension of the ectodomain from a bent to an extended close conformation by jointly rotating the head piece and upper leg regions around the hinges away from the lower legs. The second molecular movement is the swing out movement and separation of the upper legs of both subunits. This eventually separates the transmembrane and cytoplasmic domains. These conformational changes convey the signal of ECM ligand binding to the cytoplasm during outside-in signaling. The involvement of redox-modifying enzymes on the conformational changes are indicated. TRX1, thioredoxin1.
Figure 5.
Conformational changes within the integrin ectodomains structure and the potential involvement of allosteric disulfide bonds. A two step model of conformational changes of an integrin are shown. The first step is the extension of the ectodomain from a bent to an extended close conformation by jointly rotating the head piece and upper leg regions around the hinges away from the lower legs. The second molecular movement is the swing out movement and separation of the upper legs of both subunits. This eventually separates the transmembrane and cytoplasmic domains. These conformational changes convey the signal of ECM ligand binding to the cytoplasm during outside-in signaling. The involvement of redox-modifying enzymes on the conformational changes are indicated. TRX1, thioredoxin1.
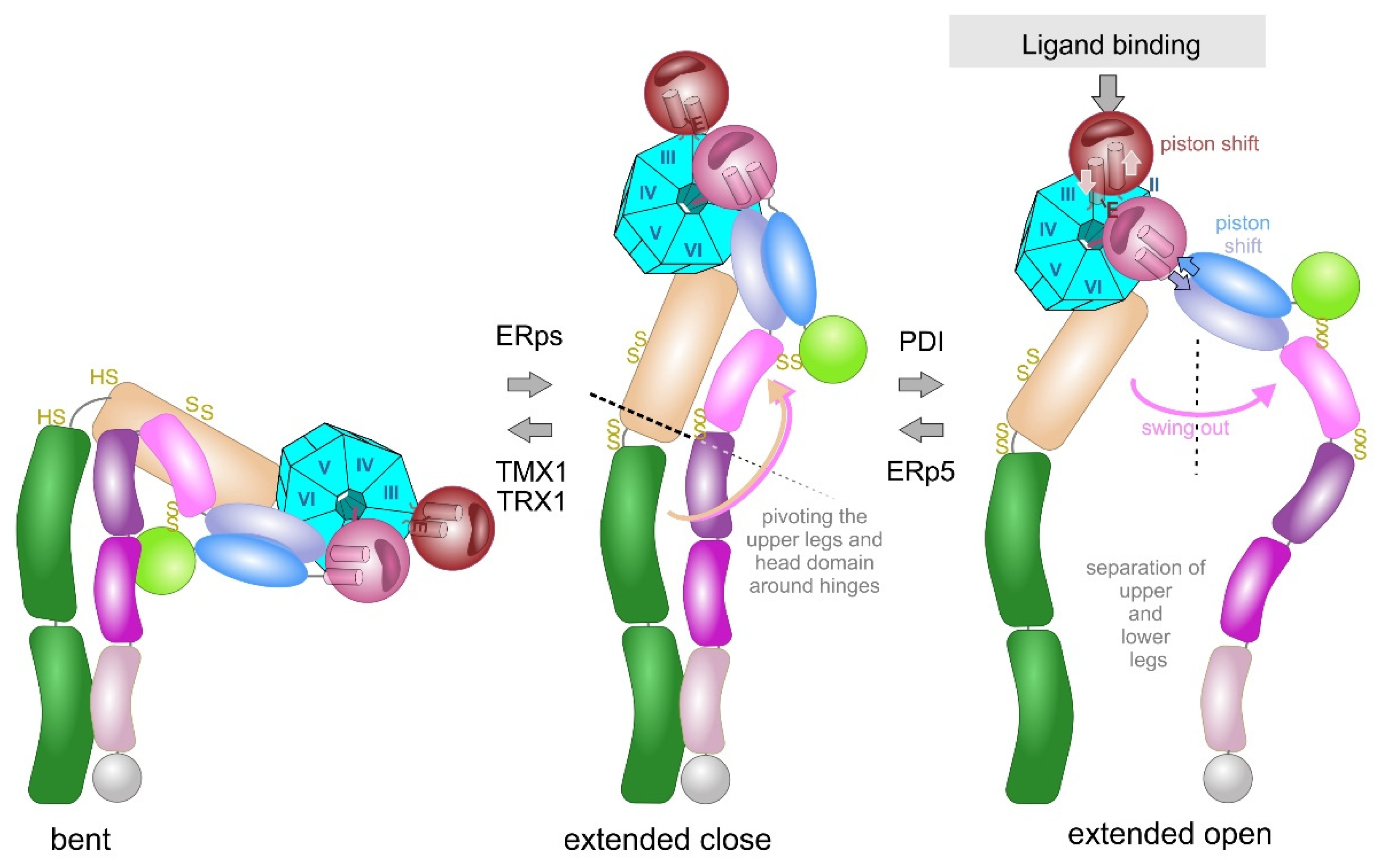
Figure 6.
Interfaces between the two integrin subunits in a bookview presentation. The stable interaction between the two subunits is the linkage of a basic amino acid side chain (R261 in β3) that serves as a knob pointing into the central pore of the propeller domain (encircled in green). This tight intersubunit interaction provides a structurally robust platform for the integrin headpiece. Other interactions between the two subunits occur in the hinges, which are the essential pivots for the ectodomains extension movement. During the swing out movement, the interactions between the calf1 and EGF2/3 and hybrid/PSI domain of the upper legs are loosened. Conspiciously, allosteric disulfide bonds are mapped to these interfaces within the hinges (C602-C608 in α subunit hinge and C473-C503 of EGF1 and EGF2 in ß subunit) and upper leg regions (C490-C545 in thigh domain and disulfide bonds in PSI, EGF1, and in EGF3) (encircled in red). The molecular structure was rendered with Pymol Molecular Graphics System software, version 2.3.1, using the protein data file 3FCS.pdb of RCSB Protein Data Bank.
Figure 6.
Interfaces between the two integrin subunits in a bookview presentation. The stable interaction between the two subunits is the linkage of a basic amino acid side chain (R261 in β3) that serves as a knob pointing into the central pore of the propeller domain (encircled in green). This tight intersubunit interaction provides a structurally robust platform for the integrin headpiece. Other interactions between the two subunits occur in the hinges, which are the essential pivots for the ectodomains extension movement. During the swing out movement, the interactions between the calf1 and EGF2/3 and hybrid/PSI domain of the upper legs are loosened. Conspiciously, allosteric disulfide bonds are mapped to these interfaces within the hinges (C602-C608 in α subunit hinge and C473-C503 of EGF1 and EGF2 in ß subunit) and upper leg regions (C490-C545 in thigh domain and disulfide bonds in PSI, EGF1, and in EGF3) (encircled in red). The molecular structure was rendered with Pymol Molecular Graphics System software, version 2.3.1, using the protein data file 3FCS.pdb of RCSB Protein Data Bank.
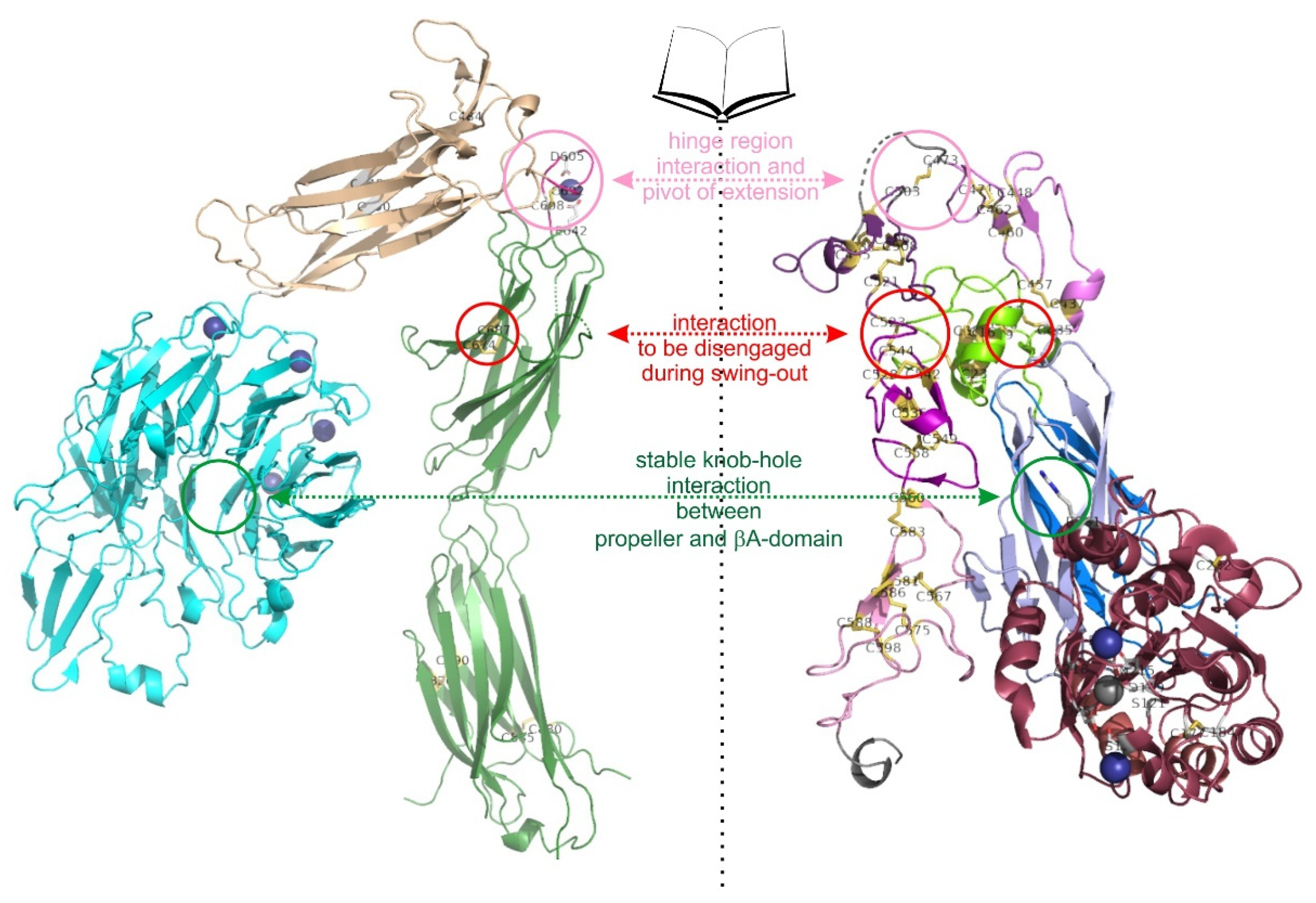
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



