
Submitted:
16 July 2025
Posted:
21 July 2025
You are already at the latest version
Preprints on COVID-19 and SARS-CoV-2
Abstract
SARS-CoV and MERS-CoV are two coronaviruses that received great attention due to their high pathogenicity and mortality rates in human populations. In this study, we compared their evolutionary dynamics to provide a One Health perspective of their different results of disease control. The phylogenetic network of SARS-CoVs showed that human isolates gathered into a “super-spreader” cluster, and were distinct from civet isolates. In contrast, dromedary camel- and human-isolated MERS-CoVs were clustered together. Thus, most clades of MERS-CoV can infect humans, and MERS-CoVs seem easier to spill over from animal-to-human interface. Additionally, the civet can be easily controlled, while the intermediate host (dromedary camels) of MERS-CoV is important livestock, so it is impossible to eliminate all animals. This further leads to difficulties in disease control of MERS. Although MERS-CoVs are endemic to dromedary camels in both the Middle East and Africa, human infections are mainly linked to the Middle East. The nucleotide sequences of the MERS-CoV receptor gene--dipeptidyl peptidase 4 (DPP4) from 30 Egyptians, 36 Sudanese, and 34 Saudi Arabians showed little difference. These findings suggest that the observed disparities in MERS prevalence between populations in the Middle East and Africa may be attributed more to inadequate disease surveillance and the limited camel-to-human transmission of clade C MERS-CoV in Africa, rather than variations in DPP4 gene.
Keywords:
SARS-CoV
; MERS-CoV
; evolutionary dynamics
; DPP4
1. Introduction
Coronaviruses (CoVs), a large family of single-stranded RNA viruses, cause respiratory, hepatic, gastrointestinal, and neurologic diseases of varying severity in a wide range of animal species, including humans [1]. Although some CoVs have been known for decades, the potential threat of these viruses to global health security was not fully realized until the outbreaks of Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) [2,3,4,5]. In 2017, a novel HKU2-related bat coronavirus, swine acute diarrhea syndrome coronavirus (SADS-CoV), caused the death of 24,693 piglets [6,7]. Especially, the outbreak of a new SARS-like coronavirus (SARS-CoV-2) in Wuhan at the end of 2019 further raises concern about these coronaviruses [8,9].
SARS-CoV first emerged in November 2002 in Guangdong province of Southern China and then rapidly spread to 29 countries and regions, infecting over 8,000 individuals with a death toll of nearly 800 [2]. The ultimate reservoir host for SARS-CoVs appears to be horseshoe bats (Rhinolophus sp.) [10], where the palm civet (Paguma larvata), sold for food at markets in southern China, act as an important intermediate amplifying host [11,12]. Fortunately, since the control of this SARS outbreak, it has not occurred again since 2005.
Ten years after SARS, MERS emerged in 2012 [13]. While dromedary camels (Camelus dromedarius) are thought to be the intermediate host for MERS-CoV, bats are widely considered to be the ultimate source of this virus [14,15,16]. Angiotensin-converting enzyme 2 (ACE2) acts as the receptor for SARS-CoV, while MERS-CoV uses dipeptidyl peptidase-4 (DPP4) [17]. Receptor genes play a vital role in how coronaviruses adapt. A recombination event in the ancestral receptor binding domain of MERS-CoV altered its receptor usage, allowing new variants to gain the ability to infect humans [18,19]. It is important to note that the MERSr-CoV sublineage, which includes NeoCoV and PDF-2180, has evolved convergently to use ACE2 by modifying the receptor-binding domain (RBD) of its spike protein. This adaptation allows the sublineage to attach to both bat and human ACE2, highlighting its potential risk for zoonotic transmission [20,21]. SARS-CoV and MERS-CoV share several important common features, such as, they both have an intermediate host, bats are their natural reservoir. While SARS was controlled, MERS continues to be a global public health concern [22,23,24]. To examine differences in the epidemic patterns of these two viruses, here in this study, we collected all available sequences to compare the different evolutionary characteristics of SARS-CoV and MERS-CoV.
The MERS-CoV is endemic in dromedary camels in the Middle East and Africa. However, all of the human infection cases occurred in the Middle East, and cases that occurred outside of the Middle East involved travelers from this region [16], while African infections have so far not been reported. This raises the question whether genetic differences between Africans and Arabians lead to differences in their susceptibility? Given that DPP4 functions as the cell surface receptor facilitating the cell entry of MERS-CoV [25], and considering that MERS-CoV adaptive evolution is linked to its capacity for human infection [18,19], this study involved the sequencing and comparative analysis of DPP4 alleles from a cohort comprising 30 individuals from Egyptians, 36 from Sudanese, and 34 from Saudi Arabians.
2. Materials and Methods
2.1. Source of Virus Genomes and Sequence Treatment
All published genomic sequences for MERS-CoV and MERSr-CoV (total 734), SARS-CoV and SARSr-CoV (total 274) were obtained from GenBank (Supplementary Table S1). SARS-CoV and SARSr-CoV sequences were separated into human, bat, and carnivore groups, based on the host source of the virus. MERS-CoV and MERSr-CoV strains were similarly separated into human, dromedary camel, and bat groups. The complete ORF1ab, S (spike), E (envelope), M (matrix), and N (nucleocapsid) gene sequences were extracted from each genome and separately aligned using MAFFT v7.505 [26]. Nucleotide diversity (π) of these genes in each population was calculated using MEGA v 11.0.13 [27].
To detect possible recombination signals in coronavirus genomes, seven different detection methods were used: RDP, GENECONV, BootScan, Chimaera, MaxChi, SiScan, and 3Seq, all implemented through the Recombination Detection Program (RDP) software version 4.101 [28]. To minimize the chances of false positives, only recombination events confirmed by four or more independent methods were taken into account. Additionally, statistical significance was assessed using a p-value threshold of ≤ 0.05, which was adjusted for multiple comparisons using Bonferroni correction. We analyzed the selective pressure acting on the protein-coding S, E, M, and N genes. Nonsynonymous to synonymous substitution ratios (dN/dS) were calculated using MEGA v 11.0.13 [27]. To quantify levels of selection and population expansion, Tajima’s D and Fu & Li’ D* tests were performed using DnaSP v6.12.03 [29]. Models M8 (beta&w) and M8a (beta&ws=1) from PAML 4.9 [30] were applied to identify sites that potentially experienced positive selection, where the null model M8a was compared to model M8 that allows positive selection. When likelihood ratio tests for these models were significant (P < 0.01), amino acid residues that showed posterior probabilities (PP) >95% under a Bayes Empirical Bayes (BEB) analysis [31] were regarded as being under positive selection. Simultaneously, the methodologies of Single-Likelihood Ancestor Counting (SLAC) [32], Fixed Effects Likelihood (FEL) [32], Fast Unconstrained Bayesian AppRoximation (FUBAR) [33], and Mixed Effects Model of Evolution (MEME) [34] were utilized with default settings to analyze positive selection through the DataMonkey online platform (http://www.datamonkey.org). To reduce the influence of confounding factors on selection analysis, sequences showing possible recombination signals, as detected by the RDP4 software, were removed from consideration. Results that were supported by at least three of the mentioned algorithms across these five methods were considered reliable.
The haplotype network for the S gene sequences from MERS-CoV and SARS-CoV genomes were constructed respectively using PopART version 1.7 [35].
2.2. DNA Extraction, PCR, and Sequencing DPP4 Gene
Blood samples from 30 Egyptians, 36 Sudanese, and 34 Saudi Arabians were collected. The study was approved by the research ethics committee of King Abdulaziz University. Genomic DNA was extracted by the standard phenol/chloroform method. To amplify the DPP4 gene, a total of 14 pairs of primers were designed (Supplementary Table S2). The primers produced a total gene length of 13,474 base pairs, including all exon regions and 10,208 base pairs of intronic sequences, accounting for 16.4% of the complete DPP4 gene, which is 81,971 base pairs long. PCR was performed in a 50 µL volume containing 25 µL of 2×PrimeSTARTM GC Buffer, 0.25mM dNTPs, 0.2µM each primer, 1.5 U PrimeSTARTM HS DNA Polymerase (TaKaRa Biosystems, Dalian, China), and 100ng genomic DNA. The PCR amplification profile was 95°C for 5 min, followed by 35 cycles of 98oC for 10s, 53oC for 15s, and 72°C for 1 min, with a final extension for 10 min at 72°C. PCR products were visualized on 1.0% agarose gels, purified on spin columns (Watson Biotechnologies Inc., Shanghai, China), and directly sequenced for both strands using a BigDyeTM Terminator Cycle Sequence Kit (ABI Applied Biosystems 3730, USA) according to the manufacturer’s manual. DNA sequences were edited using DNAstar software (DNASTAR Inc., Madison, WI, USA), with the newly determined sequences deposited into GenBank (Accession numbers: MK670823-MK670922).
3. Results
3.1. The Evolutionary Characteristics of SARS-CoVs
Of the SARS-CoV gene examined, the S gene in bats has the highest genetic diversity. The nucleotide diversity of all genes (ORF1ab, S, E, M, and N) from human and carnivore isolates are significantly lower than those of the bat isolates (Figure 1A). Except for the E and M genes, the genetic diversity of the remaining genes (ORF1ab, S, and N) isolated from carnivores are higher than those from human isolates (Table 1).
To test for neutrality in the evolution of the SARS-CoV sequences, we conducted Tajima’s D and Fu & Li’s D* tests. Significant negative Tajima’s D and Fu & Li’s D* values are indications of deviation from neutrality that suggest selective sweeps and/or population expansions. Values for Tajima’s D and Fu & Li’s D* calculated from human isolated ORF1, S, M, and N genes were significantly less than zero. The dN/dS values for S genes from carnivores and the human M genes were greater than 1. Site model tests revealed that the bat, carnivore and human S genes had undergone positive selection, with a series of positive selected amino acid sites detected. Notably, bat SARS-CoV lineages revealed adaptive evolution signatures at N protein residues 8, 22, 25, 81, 268, 410, 540 (Table 2).
The haplotype network based on the S gene of SARS-CoVs showed that the human-isolated SARS-CoV sequences were isolated mainly in 2003 and were particularly concentrated (Figure 2). 47 of 144 human SARS-CoV sequences in the analysis were related to the central haplotype (the biggest blue cycle in Figure 2). Those strains that cause super transmission (HZS2-C, HZS2-D, Sin2500, Sin2677, Sin2748, CUHK-Su10, CUHK-AG01, CUHK-AG03, CUHK-AG03) were all in this haplotype. Therefore, most human infection cases were due to this cluster. Bat-isolated SARS-CoVs showed much more divergence than the human isolates, while the carnivore SARS-CoV sequences form an independent clade that is very close to the human isolates.
3.2. The Evolutionary Characteristics of MERS-CoV
Similar to SARS-CoVs, the nucleotide diversity of all genes from the human and dromedary camel isolates are significantly lower than those from the bat isolates (Figure 1B). The nucleotide diversity of the human isolate E genes is much lower than that from the dromedary camel isolates, while the other genes had similar nucleotide diversity in these two groups. The E gene is the shortest gene of MERS-CoV and is only 249 bp. Therefore, any mutations may lead to the sharp increase of nucleotide diversity. It is possible that the significant difference of nucleotide diversity of E gene between human- and dromedary camel isolates may not be reliable. Values for Fu & Li’s D* calculated for all genes from the human and dromedary camel isolates were significantly less than zero. The dN/dS value for the E gene from dromedary camel was greater than 1 (Table 1). Camel-derived variants exhibited adaptive changes at S protein positions 26, 28, 424, 459, 723 and 1,224, Furthermore, camel MERS-CoV lineages displayed evidence of positive selection at position 69 in the M protein and residue 3,198 in the N protein. Bat-associated strains exhibited positive selection adaptations at N protein residues 200, 328, 389, and 424, while no positive selection sites were found in human strains (Table 3).
A haplotype network was constructed using the MERS-CoV S gene sequences . A total of 308 haplotypes were identified in these sequences. As shown in Figure 3, the dromedary camel MERS-CoVs from the Middle East was divided into multiple clusters, most of which were shared by dromedary camels and humans. The MERS-CoVs from dromedary camels in Africa formed a distinct single cluster (clade C) that is different from the MERS-CoVs present in humans and camels from other areas (clades A and B).
3.3. The Evolutionary Characteristics of MERS-CoV
DPP4 exon and partial intron sequences, in 14 fragments, were obtained from 30 Egyptians, 36 Sudanese, and 34 Saudi Arabian and were sequenced. We identified 4 exon haplotypes and 6 intron haplotypes in these sequences. The central haplotype for both the exon and intron sequences were shared by the Egyptians, Sudanese, and Saudi Arabians (Figure 4). The genetic diversity of the exons and introns were 0.0~0.3% and 0.0~0.1%, respectively, indicating that there was a very limited difference in the DPP4 sequences between Arabs and Africans.
4. Discussion
Bats are the reservoir hosts for both SARS-CoV and MERS-CoV, which then use palm civets and dromedary camels as intermediary host before dissemination to humans [36,37]. Although they both have the same reservoir host, and use intermediary hosts before dissemination to humans, SARS was controlled quickly while MERS continues to have sporadic human infections.
Genetic diversity of all genes from human- and carnivore-isolated SARS-CoVs was much lower than that in the bat-isolated SARS-CoVs (Figure 1A). It seems that only a group of the viruses could be transmitted through carnivores, and then able to infect humans. This suggests that most SARS-CoVs might be highly incompatible with humans and palm civets. Tajima’s D and Fu & Li’s D* values revealed that population expansion occurred in the human isolates. This is in accord with the rapid expansion of human infections. Although a total of 8,096 human infection cases were reported, they were not scattered in the network, but rather occurred around a “super-spreader” strain. Only the “super-spreader” can spread, further illustrating that most SARS-CoVs were incompatible with human transmission. Although bats were the reservoir host for SARS-CoV [38], carnivore SARS-CoVs were much closer to the human isolates. This indicates that without the intermediary host of carnivores, bat-derived SARS-CoVs likely cannot directly infect humans. Therefore, although SARS-CoVs are still circulating in bats [39], management of carnivores to prevent infection allowed quick control of SARS. However, as bats remain as a reservoirs for coronavirus, cross-species transmission of viruses from bats still poses a threat to human and animal health [6].
Compared with SARS, MERS has been a much longer epidemic. Specific antibodies against MERS-CoV have been detected in the serum of dromedary camels from Africa and Arabian Peninsula collected since 1992 [40,41], suggesting that MERS-CoV has had a long co-evolutionary time to develop a bat-dromedary camel-human transmission route. The genetic diversity of all genes, except the E gene, were similar from both human- and dromedary camel-isolated MERS-CoV (Figure 1B). In addition, most clades shared both dromedary camel and human isolates (Figure 3), which is consistent with previous studies[42,43,44,45], indicating that most clades of MERS-CoV can infect humans. This suggests that MERS-CoV has a better ability to spread from dromedary camels to humans than SARS-CoV. Moreover, since the carnivore-intermediate amplifying host of SARS-CoV was easy to control by closing wild animal markets, the dromedary camel, as an important livestock species in Arab countries playing key roles in transportation, food, fabric (wool), and entertainment, could not be so easily controlled [46]. It is likely impossible to eliminate all dromedary camels from Arab countries. Serological surveys have also showed that there is a high prevalence of MERS-CoV-neutralizing antibodies in dromedary camels [47,48], therefore, unlike SARS-CoV, the spread of MERS-CoV has a stable natural reservoir and thus, its epidemic may last longer. Eradication of MERS-CoVs from the dromedary camels is the primary condition needed for the control of this disease in the Arabian Peninsula.
To study the genetic basis of the host jumps, a series of adaptive evolutionary analyses were conducted. The S gene from SARS-CoVs in bat, carnivore and human isolates, and the S gene of MERS-CoVs in dromedary camels showed evidence of experiencing positive selection (Table 2 and Table 3). Although SARS-Cov and MERS-Cov use different receptors [49,50,51], their S proteins form spikes on the surface of CoVs particles which are involved in receptor recognition and are important for viral entry into host cells, thus can alter viral tropism [2,52]. Adaptive evolution of the S protein should help viruses recognize receptors in the new hosts, a conclusion supported by previous studies [12,19,53].
Although MERS-CoV is endemic to dromedaries in Africa [45,54,55,56,57,58,59]. MERS-CoV infection in humans in African countries, including East Africa (Kenya, Sudan), West Africa (Ghana, Nigeria), and North Africa (Morocco, Tunisia, Egypt) was very rare [60]. The major determinant of viral species-tropism is host cell entry level, which is mediated by the MERS-CoV spike protein binding to dipeptidyl peptidase 4 (DPP4) on host cells [50,51]. Therefore, we compared sequences of DPP4 alleles from Arabs and North Africans to determine whether difference in the DPP4 sequences might explain differences in susceptibilities. However, no differences were found in the DPP4 alleles found in these two groups of people (Figure 4). The difference in reported susceptibility of Middle East and African people might be due to poor disease surveillance in Africans [60,61], or the low camel-to-human transmission of calde C MERS-CoV in Africa [60], rather than the difference in DPP4 gene. In addition to the diversity of DPP4 genes, cytokine-related genes (such as TNF-α and IL-6), HLA (human leukocyte antigen), and innate immune receptor genes may also regulate susceptibility to coronaviruses [62,63,64]. For instance, the alleles HLA-DQA1*01:03 and DQB1*06:01 are commonly found in Asian populations, particularly in southern China, and are linked to a higher risk of severe SARS-CoV-2 infection [65,66]. HLA-DRB111:01 and DQB102:02 have been identified in the Saudi Arabian population and are linked to mild cases of MERS [67]. Future studies should investigate other genetic factors to explore their possible connections to differences in MERS infection rates across populations in the Middle East and Africa. In summary, our study compares the different evolutionary characteristics between SARS-CoV and MERS-CoV. Most SARS-CoVs are incompatible with humans, with most human infections by SARS-CoVs being mainly caused by “super-spreaders”. The intermediate host (civets) for the SARS-CoV epidemic is an exotic animal, which was easy to control to reduce new human infections. In contrast, all clades of MERS-CoVs appear to be able to infect humans. In addition, the intermediate host (dromedary camel) of the MERS-CoV epidemic is an important livestock species for Arab countries. What is more, a high percentage of dromedary camels have been exposed to MERS-CoV, which could potentially lead to human infection. Both of these factors lead to difficulties in disease control. It is important to note that DPP4 protein sequences do not show differences between Arabs and Africans; thus, there is no genetic evidence that supports the proposal that Africans are less likely to be infected by MERS-CoV. These findings indicate that African countries should strengthen their monitoring of MERS-CoV, with an expanded scope of surveillance, especially for those with frequent contact with dromedary camels, to determine the true extent of MERS-CoV infections in Africa.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
YS designed the experiments; JM and XS performed the research; JSMS, NHH, MMMA, MJS, and OGA conducted surveillance; MJS did the phylogenetic networks; JP and XY provided a technical help; DMI edited the paper; YS and JM wrote the paper. All authors have read and agreed to the published version of the manuscript.
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Acknowledgments
This work was supported by Guangdong Provincial Key R&D Program (2022B1111040001), Guangdong Major Project of Basic and Applied Basic Research (2020B0301030007), the National Natural Science Foundation of China (U24A20363), and Major Project of Guangzhou National Laboratory (GZNL2023A01001).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Cui, J.; Li, F.; Shi, Z.-L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Song, Z.; Xu, Y.; Bao, L.; Zhang, L.; Yu, P.; Qu, Y.; Zhu, H.; Zhao, W.; Han, Y.; Qin, C. From SARS to MERS, Thrusting Coronaviruses into the Spotlight. Viruses 2019, 11, 59. [Google Scholar] [CrossRef]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.-R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.M.; et al. Identification of a Novel Coronavirus in Patients with Severe Acute Respiratory Syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.M.E.; Fouchier, R.A.M. Isolation of a Novel Coronavirus from a Man with Pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yáng, X.-L.; Shi, W.-F.; Zhang, W.; Zhu, Y.; Zhang, Y.-W.; Xie, Q.-M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Gong, L. , et al., A new bat-HKU2-like coronavirus in swine, China, 2017. Emerg Infect Dis, 2017. 23(9).
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.F. A novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats Are Natural Reservoirs of SARS-Like Coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef]
- Guan, Y.J.; Zheng, B.J.; He, Y.Q.; Liu, X.L.; Zhuang, Z.X.; Cheung, C.L.; Luo, S.W.; Li, P.H.; Zhang, L.J.; Butt, K.M.; et al. Isolation and Characterization of Viruses Related to the SARS Coronavirus from Animals in Southern China. Science 2003, 302, 276–278. [Google Scholar] [CrossRef]
- Song, H.-D.; Tu, C.-C.; Zhang, G.-W.; Wang, S.-Y.; Zheng, K.; Lei, L.-C.; Chen, Q.-X.; Gao, Y.-W.; Zhou, H.-Q.; Xiang, H.; et al. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. USA 2005, 102, 2430–2435. [Google Scholar] [CrossRef]
- de Wit, E. , et al., SARS and MERS: recent insights into emerging coronaviruses. Nat Rev Micro, 2016. 14(8): p. 523-34.
- Sabir, J.S.M.; Lam, T.T.-Y.; Ahmed, M.M.M.; Li, L.; Shen, Y.; Abo-Aba, S.E.M.; Qureshi, M.I.; Abu-Zeid, M.; Zhang, Y.; Khiyami, M.A.; et al. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science 2015, 351, 81–84. [Google Scholar] [CrossRef]
- Anthony, S.J.; Gilardi, K.; Menachery, V.D.; Goldstein, T.; Ssebide, B.; Mbabazi, R.; Navarrete-Macias, I.; Liang, E.; Wells, H.; Hicks, A.; et al. Further Evidence for Bats as the Evolutionary Source of Middle East Respiratory Syndrome Coronavirus. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Fehr, A.R.; Channappanavar, R.; Perlman, S. Middle East Respiratory Syndrome: Emergence of a Pathogenic Human Coronavirus. Annu. Rev. Med. 2017, 68, 387–399. [Google Scholar] [CrossRef] [PubMed]
- Millet, J.K.; A Jaimes, J.; Whittaker, G.R. Molecular diversity of coronavirus host cell entry receptors. FEMS Microbiol. Rev. 2020, 45. [Google Scholar] [CrossRef]
- Tolentino, J.E.; Lytras, S.; Ito, J.; Holmes, E.C.; Sato, K. Recombination as an evolutionary driver of MERS-related coronavirus emergence. Lancet Infect. Dis. 2024, 24, e546–e546. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, L.; Gu, X. Evolutionary Dynamics of MERS-CoV: Potential Recombination, Positive Selection and Transmission. Sci. Rep. 2016, 6, 25049–25049. [Google Scholar] [CrossRef]
- Tolentino, J.E.; Lytras, S.; Ito, J.; Sato, K. Recombination analysis on the receptor switching event of MERS-CoV and its close relatives: implications for the emergence of MERS-CoV. Virol. J. 2024, 21, 1–11. [Google Scholar] [CrossRef]
- Ma, C.-B.; Liu, C.; Park, Y.-J.; Tang, J.; Chen, J.; Xiong, Q.; Lee, J.; Stewart, C.; Asarnow, D.; Brown, J.; et al. Multiple independent acquisitions of ACE2 usage in MERS-related coronaviruses. Cell 2025, 188, 1693–1710.e18. [Google Scholar] [CrossRef]
- Jiang, S.; Wu, F. Global surveillance and countermeasures for ACE2-using MERS-related coronaviruses with spillover risk. Cell 2025, 188, 1465–1468. [Google Scholar] [CrossRef]
- Peeri, N.C. , et al., The SARS, MERS and novel coronavirus (COVID-19) epidemics, the newest and biggest global health threats: what lessons have we learned? Int J Epidemiol, 2020. 49(3): p. 717-726.
- Rabaan, A.A. , et al., MERS-CoV: epidemiology, molecular dynamics, therapeutics, and future challenges. Ann Clin Microbiol Antimicrob, 2021. 20(1): p. 8.
- Raj, V.S.; Mou, H.; Smits, S.L.; Dekkers, D.H.; Müller, M.A.; Dijkman, R.; Muth, D.; Demmers, J.A.; Zaki, A.; Fouchier, R.A.; et al. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 2013, 495, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Toh, H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics 2010, 26, 1899–1900. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.P.; Murrell, B.; Golden, M.; Khoosal, A.; Muhire, B. RDP4: Detection and analysis of recombination patterns in virus genomes. Virus Evol. 2015, 1, vev003. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes Empirical Bayes Inference of Amino Acid Sites Under Positive Selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef]
- Murrell, B.; Moola, S.; Mabona, A.; Weighill, T.; Sheward, D.; Pond, S.L.K.; Scheffler, K. FUBAR: A Fast, Unconstrained Bayesian AppRoximation for Inferring Selection. Mol. Biol. Evol. 2013, 30, 1196–1205. [Google Scholar] [CrossRef]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Pond, S.L.K.; Malik, H.S. Detecting Individual Sites Subject to Episodic Diversifying Selection. PLOS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef]
- Ge, X.-Y.; Li, J.-L.; Yang, X.-L.; Chmura, A.A.; Zhu, G.; Epstein, J.H.; Mazet, J.K.; Hu, B.; Zhang, W.; Peng, C.; et al. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature 2013, 503, 535–538. [Google Scholar] [CrossRef]
- Fan, Y.; Zhao, K.; Shi, Z.-L.; Zhou, P. Bat Coronaviruses in China. Viruses 2019, 11, 210. [Google Scholar] [CrossRef]
- Alagaili, A.N. , et al., Middle East respiratory syndrome coronavirus infection in dromedary camels in Saudi Arabia. MBio, 2014. 5(2): p. e00884-14.
- Corman, V.M. , et al., Antibodies against MERS coronavirus in dromedary camels, Kenya, 1992-2013. Emerg Infect Dis, 2014. 20(8): p. 1319-22.
- Azhar, E.I.; El-Kafrawy, S.A.; Farraj, S.A.; Hassan, A.M.; Al-Saeed, M.S.; Hashem, A.M.; Madani, T.A. Evidence for Camel-to-Human Transmission of MERS Coronavirus. N. Engl. J. Med. 2014, 370, 2499–2505. [Google Scholar] [CrossRef] [PubMed]
- Dudas, G.; Carvalho, L.M.; Rambaut, A.; Bedford, T. MERS-CoV spillover at the camel-human interface. eLife 2018, 7. [Google Scholar] [CrossRef]
- Kandeil, A.; Gomaa, M.; Nageh, A.; Shehata, M.M.; Kayed, A.E.; Sabir, J.S.M.; Abiadh, A.; Jrijer, J.; Amr, Z.; Said, M.A.; et al. Middle East Respiratory Syndrome Coronavirus (MERS-CoV) in Dromedary Camels in Africa and Middle East. Viruses 2019, 11, 717. [Google Scholar] [CrossRef] [PubMed]
- Reusken, C.B.; Raj, V.S.; Koopmans, M.P.; Haagmans, B.L. Cross host transmission in the emergence of MERS coronavirus. Curr. Opin. Virol. 2016, 16, 55–62. [Google Scholar] [CrossRef]
- Shen, X.; Sabir, J.S.M.; Irwin, D.M.; Shen, Y. Vaccine against Middle East respiratory syndrome coronavirus. Lancet Infect. Dis. 2019, 19, 1053–1054. [Google Scholar] [CrossRef]
- Reusken, C.B.; Haagmans, B.L.; A Müller, M.; Gutierrez, C.; Godeke, G.-J.; Meyer, B.; Muth, D.; Raj, V.S.; Vries, L.S.-D.; Corman, V.M.; et al. Middle East respiratory syndrome coronavirus neutralising serum antibodies in dromedary camels: a comparative serological study. Lancet Infect. Dis. 2013, 13, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Meyer, B.; Müller, M.A.; Corman, V.M.; Reusken, C.B.; Ritz, D.; Godeke, G.-J.; Lattwein, E.; Kallies, S.; Siemens, A.; van Beek, J.; et al. Antibodies against MERS Coronavirus in Dromedary Camels, United Arab Emirates, 2003 and 2013. Emerg. Infect. Dis. 2014, 20, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Moore, M.J.; Vasilieva, N.; Sui, J.; Wong, S.K.; Berne, M.A.; Somasundaran, M.; Sullivan, J.L.; Luzuriaga, K.; Greenough, T.C.; et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 2003, 426, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Cao, D.; Zhang, Y.; Ma, J.; Qi, J.; Wang, Q.; Lu, G.; Wu, Y.; Yan, J.; Shi, Y.; et al. Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat. Commun. 2017, 8, 15092. [Google Scholar] [CrossRef]
- Millet, J.K.; E Goldstein, M.; Labitt, R.N.; Hsu, H.-L.; Daniel, S.; Whittaker, G.R. A camel-derived MERS-CoV with a variant spike protein cleavage site and distinct fusion activation properties. Emerg. Microbes Infect. 2016, 5, 1–9. [Google Scholar] [CrossRef]
- Hulswit, R.J.G.; De Haan, C.A.M.; Bosch, B.J. Coronavirus Spike Protein and Tropism Changes. Adv. Virus Res. 2016, 96, 29–57. [Google Scholar] [CrossRef]
- Consortium, C.S.M.E. , Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science, 2004. 303(5664): p. 1666-9.
- Chu, D.K.; Poon, L.L.; Gomaa, M.M.; Shehata, M.M.; Perera, R.A.; Abu Zeid, D.; El Rifay, A.S.; Siu, L.Y.; Guan, Y.; Webby, R.J.; et al. MERS Coronaviruses in Dromedary Camels, Egypt. Emerg. Infect. Dis. 2014, 20, 1049–1053. [Google Scholar] [CrossRef]
- Chu, D.K.; O Oladipo, J.; Perera, R.A.; A Kuranga, S.; Chan, S.M.; Poon, L.L.; Peiris, M. Middle East respiratory syndrome coronavirus (MERS-CoV) in dromedary camels in Nigeria, 2015. Eurosurveillance 2015, 20. [Google Scholar] [CrossRef]
- Miguel, E.; Chevalier, V.; Ayelet, G.; Ben Bencheikh, M.N.; Boussini, H.; Chu, D.K.; El Berbri, I.; Fassi-Fihri, O.; Faye, B.; Fekadu, G.; et al. Risk factors for MERS coronavirus infection in dromedary camels in Burkina Faso, Ethiopia, and Morocco, 2015. Eurosurveillance 2017, 22, 15–24. [Google Scholar] [CrossRef]
- Chu, D.K.W.; Hui, K.P.Y.; Perera, R.A.P.M.; Miguel, E.; Niemeyer, D.; Zhao, J.; Channappanavar, R.; Dudas, G.; Oladipo, J.O.; Traoré, A.; et al. MERS coronaviruses from camels in Africa exhibit region-dependent genetic diversity. Proc. Natl. Acad. Sci. 2018, 115, 3144–3149. [Google Scholar] [CrossRef]
- Kiambi, S.; Corman, V.M.; Sitawa, R.; Githinji, J.; Ngoci, J.; Ozomata, A.S.; Gardner, E.; von Dobschuetz, S.; Morzaria, S.; Kimutai, J.; et al. Detection of distinct MERS-Coronavirus strains in dromedary camels from Kenya, 2017. Emerg. Microbes Infect. 2018, 7, 1–4. [Google Scholar] [CrossRef]
- Ommeh, S.; Zhang, W.; Zohaib, A.; Chen, J.; Zhang, H.; Hu, B.; Ge, X.-Y.; Yang, X.-L.; Masika, M.; Obanda, V.; et al. Genetic Evidence of Middle East Respiratory Syndrome Coronavirus (MERS-Cov) and Widespread Seroprevalence among Camels in Kenya. Virol. Sin. 2018, 33, 484–492. [Google Scholar] [CrossRef]
- Karani, A.; Ombok, C.; Situma, S.; Breiman, R.; Mureithi, M.; Jaoko, W.; Njenga, M.K.; Ngere, I. Low-Level Zoonotic Transmission of Clade C MERS-CoV in Africa: Insights from Scoping Review and Cohort Studies in Hospital and Community Settings. Viruses 2025, 17, 125. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E. MERS coronavirus: diagnostics, epidemiology and transmission. Virol. J. 2015, 12, 1–21. [Google Scholar] [CrossRef]
- Barlan, A.; Zhao, J.; Sarkar, M.K.; Li, K.; McCray, P.B.; Perlman, S.; Gallagher, T.; Dermody, T.S. Receptor Variation and Susceptibility to Middle East Respiratory Syndrome Coronavirus Infection. J. Virol. 2014, 88, 4953–4961. [Google Scholar] [CrossRef]
- Sims, A.C.; Schäfer, A.; Okuda, K.; Leist, S.R.; Kocher, J.F.; Cockrell, A.S.; Hawkins, P.E.; Furusho, M.; Jensen, K.L.; Kyle, J.E.; et al. Dysregulation of lung epithelial cell homeostasis and immunity contributes to Middle East respiratory syndrome coronavirus disease severity. mSphere 2025, 10, e0095124. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Bhattacharya, M.; Das, A.; Saha, A. Regulation of miRNA in Cytokine Storm (CS) of COVID-19 and Other Viral Infection: An Exhaustive Review. Rev. Med Virol. 2025, 35, e70026. [Google Scholar] [CrossRef] [PubMed]
- Dobrijević, Z.; Gligorijević, N.; Šunderić, M.; Penezić, A.; Miljuš, G.; Tomić, S.; Nedić, O. The association of human leucocyte antigen (HLA) alleles with COVID-19 severity: A systematic review and meta-analysis. Rev. Med Virol. 2022, 33, e2378. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K. , et al., HLA-DQA1*01:03 and DQB1*06:01 are risk factors for severe COVID-19 pneumonia. HLA, 2024. 104(1): p. e15609.
- Hajeer, A.H.; Balkhy, H.; Johani, S.; Yousef, M.Z.; Arabi, Y. Association of human leukocyte antigen class II alleles with severe Middle East respiratory syndrome-coronavirus infection. Ann. Thorac. Med. 2016, 11, 211–3. [Google Scholar] [CrossRef]
Figure 1.
Nucleotide diversity in the ORF1ab, S, E, M, and N genes of SARS-CoV and MERS-CoV isolated from bats, carnivores (for SARS-CoV) or camels (for MERS-CoV), and human hosts. (A) SARS-CoVs. (B) MERS-CoVs.
Figure 1.
Nucleotide diversity in the ORF1ab, S, E, M, and N genes of SARS-CoV and MERS-CoV isolated from bats, carnivores (for SARS-CoV) or camels (for MERS-CoV), and human hosts. (A) SARS-CoVs. (B) MERS-CoVs.
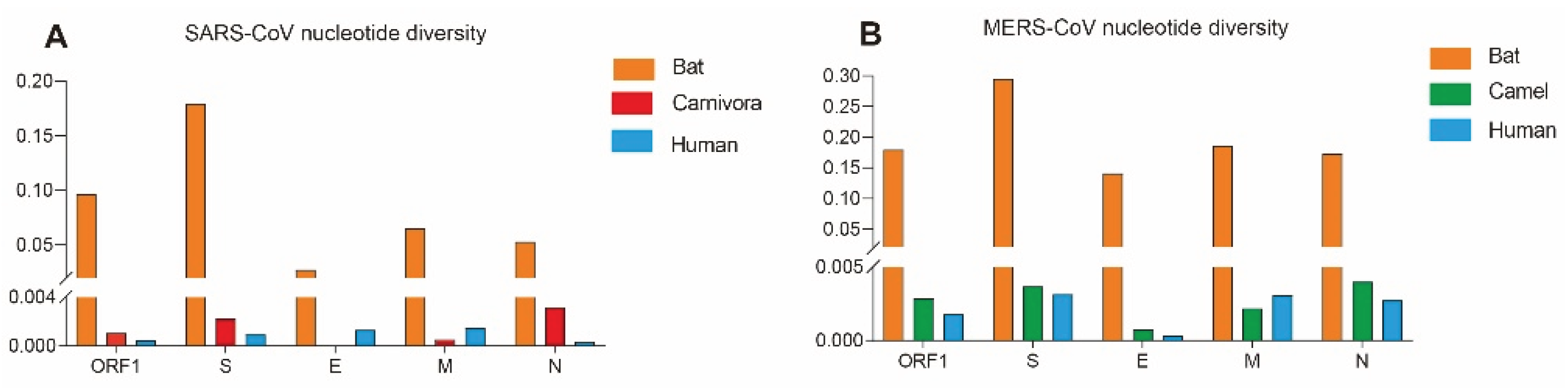
Figure 2.
Haplotype network of the S gene sequences from SARS-CoV and SARSr-CoV genomes.
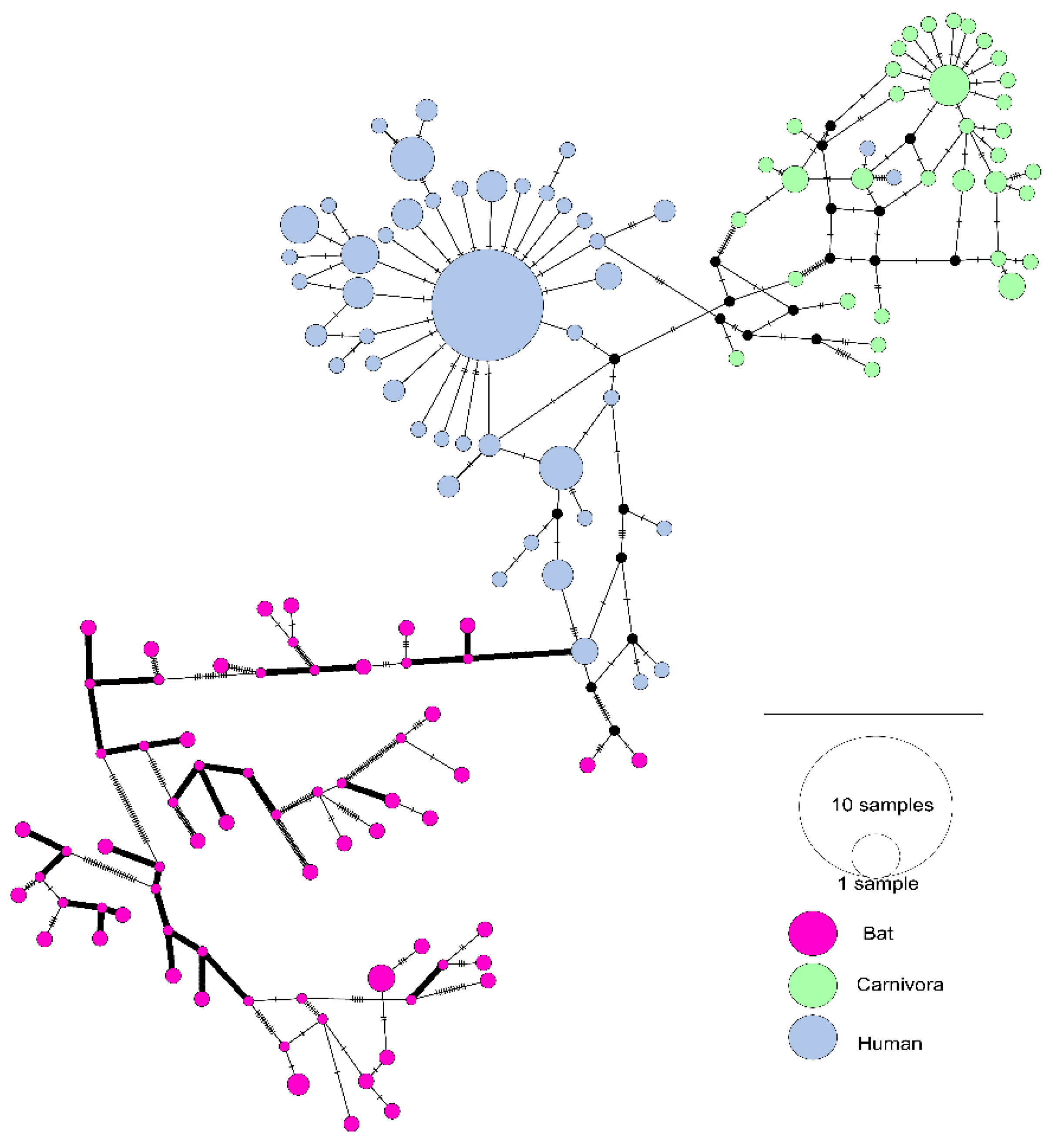
Figure 3.
Haplotype network of the S gene sequences from MERS-CoV and MERSr-CoV genomes.
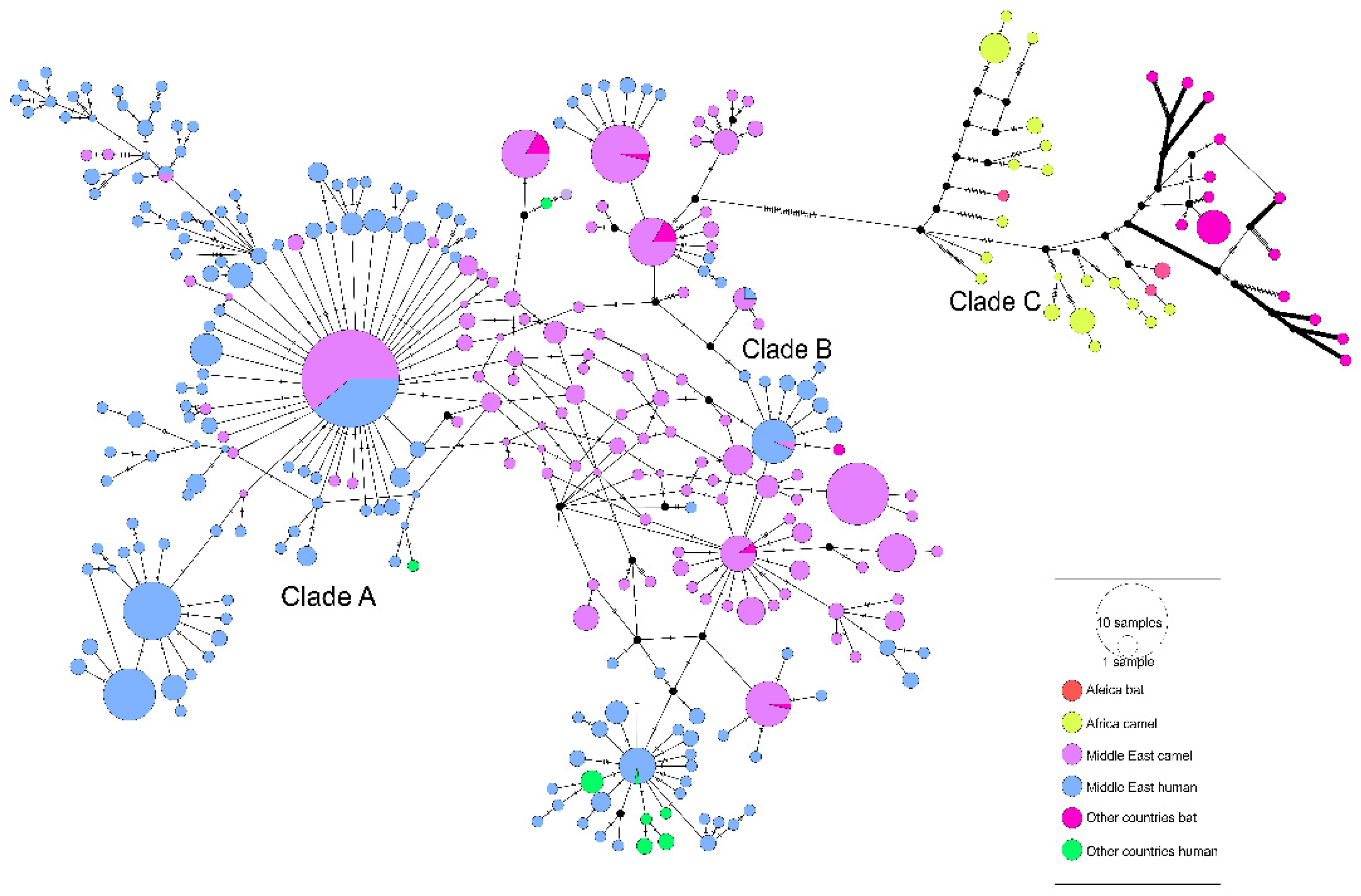
Figure 4.
Haplotype networks of exon and intron regions of DPP4 gene. (A) Intron region. (B) Exons.
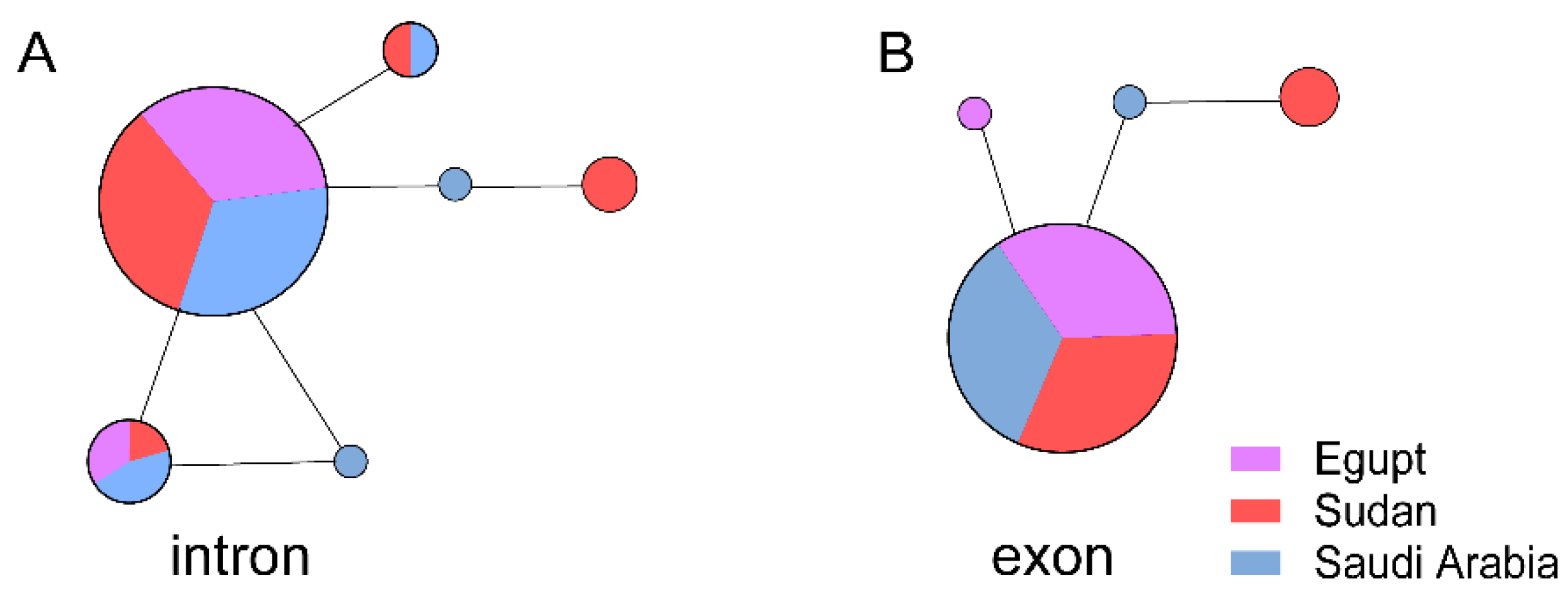

Table 1.
Genetic diversity and neutrality analyses of the S, E, M, and N genes from SARS-CoV and MERS-CoV.
Table 1.
Genetic diversity and neutrality analyses of the S, E, M, and N genes from SARS-CoV and MERS-CoV.
| Gene |
Host |
Nucleotide diversity (π) |
Neutrality analyses | ||
|---|---|---|---|---|---|
| Fu and Li's D* | Fu and Li's D* | ||||
| SARS-CoV | ORF1 | Human | 0.00048 | -2.63679** | -5.05802** |
| Bat | 0.09634 | -0.72289 | -0.85435 | ||
| Carnivora | 0.00107 | -1.45552 | -1.05296 | ||
| Human | 0.00094 | -2.29683** | -4.31580** | ||
| S | Bat | 0.17954 | 0.35301 | -0.36182 | |
| Carnivora | 0.00225 | -0.10083 | -1.05012 | ||
| Human | 0.00132 | -1.63734* | -0.82508 | ||
| E | Bat | 0.02742 | -1.05697 | -1.11301 | |
| Carnivora | / | / | / | ||
| Human | 0.00151 | -1.99699* | -3.68341** | ||
| M | Bat | 0.06514 | -1.22163 | -1.01881 | |
| Carnivora | 0.00054 | -1.03789 | -0.50381 | ||
| Human | 0.00037 | -2.23097 ** | -4.79424** | ||
| N | Bat | 0.053134 | -1.43029 | -1.77074** | |
| Carnivora | 0.00320 | -0.63115 | 0.04240 | ||
| Human | 0.00048 | -2.63679** | -5.05802** | ||
| MERS-CoV | ORF1 | Human | 0.00180 | -2.32084** | -8.53056** |
| Camel | 0.00286 | -2.03977* | -6.02846** | ||
| Bat | 0.17953 | -0.04466 | 0.68164 | ||
| Human | 0.00316 | -2.23642 ** | -7.86620** | ||
| S | Camel | 0.00369 | -2.12122** | -5.43913** | |
| Bat | 0.29554 | -0.13075 | 0.44654 | ||
| Human | 0.00033 | -2.06853* | -5.60248** | ||
| E | Camel | 0.00078 | -1.89892* | -3.04304* | |
| Bat | 0.14117 | 0.35028 | 0.68165 | ||
| Human | 0.00307 | -1.65090 | -5.28654** | ||
| M | Camel | 0.00219 | -2.08234* | -3.89494** | |
| Bat | 0.18639 | 0.06353 | 0.68401 | ||
| Human | 0.00276 | -2.15013** | -4.22054** | ||
| N | Camel | 0.00401 | -2.10318* | -4.26757** | |
| Bat | 0.17372 | -0.07776 | 0.54725 | ||
Note:*P≤0.05; **0.05>P >0.001; ***P≤0.001.
Table 2.
Selective pressure analyses of the S, E, M, and N genes from SARS-CoV.
| Gene | Positive selection pressure sites identified by different methods | |||||
|---|---|---|---|---|---|---|
| M8 vs. M8a | FEL | SLAC | FUBAR | MEME | PSC* | |
| Bat S | 5,8,20,132,133,135,136,138,154,182,195,411,412,422,439,481,503,589,689 | 166,540,596,633 | None | 451,540,648 | 7,20,24,43,81,82,83,89,91,101,130,156,157,160,166,174,178,185,190,207,210,231,242,252,314,410,411,451,454,464,465,477,501,502,509,516,540,562,596,633,648,689,693,753,876,935,1117,1250,1253 | 540 |
| Bat E | None | None | None | None | None | - |
| Bat M | 14 | 97 | None | None | 97 | - |
| Bat N | 8,22,268,410 | 8,22,25,34,81,121,268,410 | None | 8,25,81,410 | 8,22,25,34,81,121,196,268,297,408, 410 |
8,22,25,81,268,410 |
| Carnivora S | 77,108,113,139,147,194,227,239,243,244,261,294,336,344,360,461,470,477,478,556,575,578,605,607,611,630,642,645,648,663,699,701,741,752,763,776,819,837,842,892,898,1050,1078,1161,1217, | 77,147,227,479, 609,743,894,1080 |
None | 147,227,244,344,360,440,462,479,480,609,613,743,1052,1080,1219 | 147,227,479,609, |
147,227,462, 479,609 |
| Carnivoral E | None | None | None | None | None | - |
| Carnivora M | None | None | None | None | None | - |
| Carnivora N | 384 | None | None | 384 | None | - |
| Human S | 2,5,12,49,75,77,78,138,139,144,147,238,243,310,343,349,352,359,383,424,435,441,462,471,479,486,500,576,599,604,607,608,612,622,651,664,665,742,764,777,793,855,859,860,862,1000,1131,1147,1162,1168,1182,1207,1222,1246 | None | None | 12,138,311,608,609,1148,1163,1208 | 138 | 138 |
| Human E | 5,6,23,29 | None | None | None | None | - |
| Human M | 5,11,27,38,68,73,81,86,91,99,113, 119,154,210 |
None | None | 11 | None | - |
| Human N | None | None | None | None | None | - |
* Note: Positively Selected Codons: only the codons identified by at least three of the five methods were considered to be positively selected codons.“ - ” indicates that no positive selection site was detected in this gene.
Table 3.
Selective pressure analyses of the S, E, M, and N genes from MERS-CoV.
| Gene | Positive selection pressure sites identified by different methods | |||||
|---|---|---|---|---|---|---|
| M8 vs. M8a | FEL | SLAC | FUBAR | MEME | PSCa | |
| Bat S | None | 3,7,25,225,328,625,731,777 | None | 219 | 3,7,25,145,199,222,225,232,235,239,591,687,713,731,772,777,794,795,908,966,1277,1293 |
- |
| Bat E | None | None | None | None | None | - |
| Bat M | None | None | None | None | 96 | - |
| Bat N | None | 200,328,378,389,394,402, 403,424,435 | None | 200,328,389,398,424 | 111,200,210,328,367,378,389,402,403,406, 424,431 | 200,328,389,424 |
| Camel S | 26,459,465,612,723,1193,1224 | 26,28,424,459,723,1224 | 26 | 26,28,158,390,424,710,723,1193,1224 | 26,28,424,459,723,1224 | 26,28,424,459,723,1224 |
| Camel E | None | None | None | None | None | - |
| Camel M | None | 69,111,155 | None | 8,69,82 | 69 | 69 |
| Camel N | None | 3,198 | None | 3,198 | 3,198 | 3,198 |
| Human S | None | 424 | None | 26,91,95,301,424,507,509,534,914,1158 | 1020 | - |
| Human E | None | None | None | None | None | - |
| Human M | None | None | None | 15,20,69 | None | - |
| Human N | None | None | None | 8,126,300 | None | - |
| * Note: Positively Selected Codons: only the codons identified by at least three of the five methods were considered to be positively selected codons.“-” indicates that no positive selection site was detected in this gene. | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



