
Submitted:
18 July 2025
Posted:
21 July 2025
You are already at the latest version
Abstract
The intracellular topography of RNA molecules, encompassing ribonucleotides with biochemical modifications, such as N6-methyladenosine(m6A), 5-methylcytosine (m5C) and isomerization of uridine to pseudouridine (Ψ), as well as of the non-coding RNA molecules, are currently studied within the frame of the epigenome. Circulating RNA molecules in the intracellular space, that have incorporated information by carrying specific modifications, depend on the balanced activity and correct subcellular installation of their modifying enzymes, the “writers”, “readers” and “erasers”. Modifications are critical for RNA translocation from nucleus to cytoplasm, stability and translation efficiency and other, still uncovered functions. Moreover, intracellular movement of non-coding RNA molecules depends on membrane transporters, capable of recognizing signal sequences and RNA recognition-binding proteins, that can facilitate their transport to different intracellular locations, guiding the establishment of interconnection possibilities with different macromolecular networks. The potential of long non-coding RNAs to form multilayer molecular connections, as well as the differential topology of micro-RNAs in cell nucleus over cytoplasm have been recognized by several studies. The study of the intracellular compartmentalization of these molecules has recently become feasible thanks to technological progress, however, a wealth of information has not yet been produced that would lead to safe conclusions, regarding non-coding RNA contribution to the early steps of pathogenesis and disease progression of hematological malignancies. Both the bone marrow, as the main hematopoietic tissue, and the lymphoid tissues are composed of cells with high potential of reaction to signals affecting the epigenome and initiating cascade pathways in response. Independently or in combination with the coexistent driver genetic mutations, especially of enzymes involved in epigenomic surveillance, intracellular microenvironmental alterations within the cell nuclear, cytoplasmic and mitochondrial compartments, can lead to disorganization of hematopoietic stem cells’ epigenome, promoting the generation of hematological malignancies. In this review, we discuss the various intracellular processes that, when disrupted, may result in the ectopic placement of RNA molecules, either inducing specific modifications or non-coding molecules, promoting hematological malignant phenotypes. The crosstalk between mitochondrial and nuclear genomes and the complex regulatory effects of mis-localized RNA molecules is highlighted. This research approach may constitute a field for new, more specifically targeted therapies in hematology, based on the RNA technology.
Keywords:
non-coding RNAs
; intracellular topography
; hematological malignancies
; RNA modification
; abnormal signalling
Introduction
A variety of covalent bonded modifications are mainly prevalent following biosynthesis of protein and RNA molecules, rather than of DNA. These chemical modifications represent an efficient way of post-transcriptional / post-translational regulation of the function of macromolecules and of an additional control of gene expression profiles, providing a high degree of adaptation to the continuously changing cellular micro- and macro-environment [1]. Chemical modification of the macromolecules has been extensively highlighted and studied over the last decades, since when Conrad Waddington (1905–1975) introduced the term “epigenetic mechanisms” to describe it. Thereafter epigenetic signatures have gradually been uncovered and have revealed their potential, as disease-causing and disease-modifying mechanisms [2]. The field of RNA epitranscriptomics has been delayed being thoroughly studied, due to lack of highly sensitive quantitative and qualitative detection techniques. Although RNA modifications were described over 50 years ago, only recent studies have uncovered that their presence and binding partners are dysregulated in cancer, creating unique opportunities for new therapeutic approaches. Currently, more than 170 post-transcriptional modifications have been identified in the cellular RNA, apart from the 5’ cap and 3’ poly-A tail, and more than 350 proteins involved in these modifications have been included in the MODOMICS database [3]. Modifications can occur via the addition of simple or complex chemical groups to the ribonucleosides. Other than chemical modifications have also been detected, such as isomerization (e.g., uridine to pseudouridine [Ψ]), oxidation (e.g., 5-methylcytosine to 5-methylcytidine and 5-hydroxymethylcytosine to 5-hydroxymethylcytidine), reduction (e.g., uridine to dihydrouridine), and substitution (e.g., uridine to 4-thiouridine). They are spread among all kinds of RNA and are apparent in nuclear, cytoplasmic and mitochondrial RNAs [3,4]. RNA modifications and the associated modifying enzymes or binding proteins are known to be implicated in homeostasis of Hematopoietic Stem cells (HSCs), providing quiescence balance, self-renewal and differentiation capacity and consequently, instructing cell fate decisions. When RNA modifications are disrupted, the situation appears to favor the development of clonal hematologic malignancies. Alterations in RNA splicing and RNA editing during HSC aging, due to defective RNA modification pathways contribute to increased myeloid lineage skewing and activation of inflammation-responsive transcription factors [5]. These facts underscore the importance of epitranscriptomic mechanisms in the acquisition of the HSC age-related abnormal phenotypes.
Also, non-coding (nc) RNAs represent another group οf epigenetic players, which are strongly associated with the development of clonal / neoplastic hematological disorders. Here, we provide an in-depth overview of how subcellular mis-localization of ncRNAs, mainly microRNAs (miRs) and long non-coding RNAs (lncRNAs), may stimulate their differentiated activity and their participation in alternative intra-molecular roots, and how these mechanisms may contribute to the generation of hematological malignancies.
The precise subcellular localization of all the macromolecules is crucial for the integrity and specificity of their function. Altered localization, caused by specific mutation profiles, stress signals, or structural nuclear defects, can modulate their regulatory roles. Of the most profound and well-studied paradigms in hematology is the AML subtype, carrying mutations in the NPM1 gene, that direct nucleophosmin1 molecule to translocate in the cytoplasm, possibly misrouting other molecules and altering various gene regulatory networks in both compartments [6]. Under physiological conditions NPM1 phosphoprotein shuttles between nucleus and cytoplasm, chaperoning ribosomal proteins and core histones between subcellular compartments, but NPM1 mutations trigger its favorable localization in the cytoplasmic cell compartment [7,8,9]. Other examples implicating ncRNAs, such as hsa-miR-223, hsa-miR-690 [10,11,12], lncRNA-HOTAIRM1, lncRNA-HOTTIP, etc [13,14,15], which are discussed in detail in separate sections of this review, provide clear evidence of how ncRNA mis-localization directly or indirectly contributes to disease pathogenesis.
NcRNAs in the extracellular matrix constitute mostly signaling molecules, transported to other areas of the same or different tissues and are not within purposes of this review. On the contrary, we address issues raised from the altered compartmentalization of endo-cellular ncRNA molecules and defected pathways related to m6A, m5C and Ψ modified RNAs and to the underlying mechanisms, discussing also the potential effects in cells’ fate within the hematopoietic compartment [16,17,18,19,20,21,22,23,24]. The composition of post-transcriptional modifications in RNA transcripts can determine their decay, versus stabilization/increased translation; due to the subcellular localization of associated RNA modifying-enzymes they can potentially impinge different aspects of transcript metabolism and finally, modified expression/activity of ‘reader’ proteins, can serve as the final arbiters of the fate of specifically decorated transcripts [25,26,27,28,29,30]. We speculate that these functional variabilities may, at least in part, explain some experimental observations combining altered RNA modification and topology with tumorigenesis in hematology.
A. Subcellular Dynamics of Chemically Modified RNA Molecules and Their Modifying Enzymes in hematologic malignancies
N6-methyladenosine (m6A) is one of the most abundant RNA modifications in mammalian cells. Additional well-documented marks in RNA molecules include the 5-methylcytosine (m5C) and isomerization of uridine to pseudouridine (Ψ), which have been reported to associate with hematologic malignancies [16]. 2′-O-Methylation in only briefly mentioned, since there are very few references implicating the alternative topology of RNA molecules carrying this modification in hematologic malignancies [31,32].
I. N6-Methyladenosine RNA Modification
m6A is a methylation modification, located on the sixth nitrogen atom of RNA adenine and is the most prevalent and well-studied internal modification in eukaryotic mRNA and ncRNAs, affecting various biological processes, including chromatin conformation and consequent regulation of gene expression, RNA processing and half-life, translation, etc. [17,33,34,35]. m6A occurs both in the nuclear and cytoplasmic cell compartment and is mainly distributed in the internal gene exons and the 3′ untranslated regions, with a significant enrichment just upstream of the stop codon of messenger RNAs and of long non-coding RNAs [18].
In the nucleus m6A is primarily added to immature RNA molecules during transcription, acting as functional signal for the downstream mRNA maturation. m6A mark, as well as the modifying enzymes, associated with m6A RNA are implicated in maturation processes, including splicing (and alternative splicing) [25], function of the spliceosome component U6 [26], RNA export to the cytoplasm through specified transporters and RNA stability to hydrolysis [19]. Among ncRNA species, m6A methylation of micro-RNA (miRNA) primary transcripts has been documented as a requirement for their efficient recognition and enzymatic processing to mature miRNA molecules [36].
In the cytoplasm, m6A modification influences translation efficiency and mRNA decay [35,37]. Many studies support that m6A contributes to surveillance pathways, to selectively degrade faulty transcripts, by enhancing and sensitizing the nonsense-mediated mRNA decay (NMD)-mediated clearance from cell [38]. NMD is a quality control mechanism that selectively degrades mRNAs containing premature stop codons (nonsense mutations), to prevent the production of truncated, potentially harmful proteins. m6A mark recruits UPF1, a critical NMD factor, to enhance the decay of transcripts with premature stop codons, and the same accounts for the recruitment of m6A ‘reader’ protein YTHDF2, leading to accelerating degradation of those transcripts [27,39].
Delving into the molecular mechanisms and pathways influenced by m6A RNA modification may provide insights into potential therapeutic targets and novel prognostic biomarkers for hematologic malignancies. m6A RNA modification has been reported as an important factor assisting the balance between self-renewal and differentiation of the Hematopoietic Stem Cells (HSCs). Dysregulation of m6A modifying enzymes, such as ‘writers’ of m6A-RNA mark (m6A methyltransferases METTL3, METTL14, WTAP, RBM15, VIRMA, and ZC3H13, etc), ‘erasers’ (m6A demethylases FTO and ALKBH5) and ‘readers’ (m6A binding proteins YTHDF1/2/3, YTHDC1/2, HNRNPs, IGF2BP, eIF3, etc.) influence HSCs’ gene expression programs. Dysregulated expression profiles of several modifying enzymes often lead to malignant phenotypes within the hematopoietic compartment [28,40,41,42]. Relevance of m6A modification in various studies on hematological malignancies underscores the importance of proper endo-cellular localization of m6A-modifying enzymes in maintaining normal hematopoietic homeostasis [20,43]. Aberrantly m6A RNA modified molecules contribute to immune evasion and resistance mechanisms, commonly observed in B-cell lymphomas, by altering the stability and translational output of key immune and oncogenic transcripts. Upon activating signals, m6A demethylase ALKBH5 and the lncRNA-treRNA1 (translation regulatory lnc-RNA1) translocate within the nucleus, where they form a functional complex with the RNA helicase DDX46. The ALKBH5/lnc-treRNA1/DDX46 nuclear complex facilitates the removal of m6A modifications from key B-Cell Receptor (BCR)-related transcripts, thereby providing fine-tuning to BCR expression levels. Disruption of this axis leads to impaired transcript processing and diminished BCR-related gene expression, essential for B-cell functionality [44]. Furthermore, m6A demethylase ALKBH5, frequently enhanced in human Diffuse large B-cell lymphoma (DLBCL, the most common and aggressive form of non-Hodgkin lymphoma) samples, directly demethylates BCR signature transcripts, hijacking the post-transcriptional/translational regulation of gene expression, crucial for normal B-cell development and lymphomagenesis [45] (Figure 1A).
The m6A demethylases FTO (fat-mass and obesity-associated protein) and ALKBH5 have distinct subcellular localization and largely non-overlapping targets. Their activity is modulated by intermediary metabolism, affecting RNA demethylation processes. The dynamic nature of epitranscriptome, is in part influenced by the subcellular movement of FTO, which shuttles between the cytosol and the nucleus, in contrast to the more stably nuclear localization of ALKBH5 [29,46]. Studies in AML and B-cell lymphomas have demonstrated that FTO, a multifunctional RNA demethylase, is capable of removing several types of methyl groups (including m6A) from diverse RNA classes. FTO localization patterns across the nucleus or cytoplasm enables selective RNA demethylation. Nuclear FTO preferentially targets m6A in pre-mRNAs and snRNAs, whereas cytoplasmic FTO accesses mainly cap-adjacent m6Aₘ on mature cytoplasmic mRNAs. As a result, FTO knockdown increases m6A, affecting RNA function in the different cell compartments and further implying an emerging pattern for oncogenic transformation, via subcellular mis-localization of FTO [29]. ALKBH5 activity on the other hand, is regulated by post-translational modifications, such as Ubiquitination. A recent study reports that USP36, a deubiquitinating enzyme, is associated with and promotes ALKBH5 stabilization, thus contributing to tumorigenesis [47]; towards the same direction, ALKBH5 deficiency was suggested to represent an inhibiting factor for tumor proliferation in AML [48].
ALKBH5 and FTO are also (αKG) alpha-ketoglutarate-dependent enzymes, potentially inhibited by the high accumulation of D-2-HG (D-2-hydroxyglutarate), a natural metabolite that is aberrantly produced by IDH1/2 (isocitrate dehydrogenases 1/2), proteins frequently found mutated in primary AML. D-2-HG competitively inhibits the activity of αKG-dependent dioxygenases, leading to a m6A hypermethylation profile in AML [49,50]. Along with αKG, other Krebs’ cycle metabolites can also alter the activity of m6A demethylases FTO/ALKBH5 [51,52]. FTO and ALKBH5 are shown to be enriched or depleted in the nucleus, when O-GlcNAcylation (bonding of a single O-linked N-acetylglucosamine moiety to a serine or threonine residue) is elevated or suppressed respectively, suggesting that deposition of O-GlcNAc may also influence the epitranscriptome. O-GlcNAcylation is a ubiquitous post-translational protein modification, influencing the subcellular compartmentalization of several proteins and its elevation within the cell is regulated by MYC expression and αKG exposure. The exact protein network intervening in this mechanism merits further elucidation [51]. Transcription factor MYC binds to the IDH2 promoter to transcriptionally activate it, leading to increased αKG cellular pool. Along with IDH2, MYC also regulates various enzymes that positively influence anapleorotic mitochondrial metabolism, glycolysis and the transport of glucose and glutamine through cell membrane, suggesting its critical role on cancer-associated metabolic reprogramming and representing an important mechanism of nuclear-mitochondrial crosstalk [53]. Data from AML show that IDH1/2 mutations lead to increased D-2-HG production, which competes with αKG resulting in αKG depletion and inhibition of ALKBH2 and FTO activity (Figure 1B). Also, other αKG-dependent enzymes, such as DNA demethylases TET-1 and -3, under circumstances of decreased αKG tolerate adverse effects, compromising their activity [49,50,51,54].
In Acute Lymphoblastic Leukemia (ALL) either adult or pediatric type, as well as in Multiple Myeloma (MM), altered m6A modification patterns have been reported to affect malignant cell survival and proliferation [55,56,57,58]. In AML, the m6A ‘reader’ YTHDC1 has been found elevated in the nucleus and forms nuclear speckles and super enhancer condensates. m6A chemical marks on RNA molecules is a significant requirement for YTHDC1, to form the nuclear YTHDC1-m6A condensates (nYACs), presenting a nest for protecting m6A-mRNAs from the PAXT-complex (Poly(A) Tail eXosome Targeting complex) and the related exosome-associated RNA degradation. Protection and stabilization of m6A transcripts accelerates cell survival and proliferation [59] (Figure 1B).
Inhibitors targeting mainly m6A RNA demethylases FTO and ALKBH5, are in constant investigation as they represent promising targets to fine-tuning of cells’ RNA epigenome. FTO/ALKBH5 inhibitors related to hematology are presented in Table 1.
II. 5-Methylcytosine (m5C) RNA Modification
5-Methylcytosine (m5C) RNA modification refers to a post-transcriptional chemical addition of a methyl group to the fifth carbon of cytosine residues. m5C modification occurs in all (already known) kinds of RNA molecules (mRNAs, rRNAs, tRNAs and regulatory ncRNAs), distributed in non-random patterns. Its presence is detected in all endo-cellular compartments, including nucleus, cytoplasm and mitochondria [21,22]. mRNA sequences are enriched with m5C modification within 5’ and 3’UTRs (untranslated regions), prevailing mainly near the translational start codon, primarily affecting protein synthesis rates, and as a consequence, the overall gene expression profiles [65]. Enzymes Involved in m5C RNA modification are categorized as ‘writers’ with distinguished members the NSUN1-7 (NOP2/Sun RNA Methyltransferase) family and TRDMT1 (TRNA Aspartic Acid Methyltransferase 1 or DNMT2), which preferentially methylates the tRNA cytosines [66,67]. NSUN2 modifies cytoplasmic tRNAs, mediating their cleavage and stability, particularly under stress conditions. It also methylates non-coding RNAs, affecting their maturation process [68,69]. Another category is comprised of the m5C ‘erasers’, that remove methyl groups from RNA, reversing the m5C modification. TET (Tet Methylcytosine Dioxygenase) proteins belong to this family, primarily known for their role in DNA demethylation however, they are also implicated in RNA demethylation [70]. Finally, the ‘readers’ are proteins that recognize and bind to RNA methylated cytosine residues, influencing downstream processes. Among them ALYREF recognizes and binds to m5C modified RNAs, mediating their nuclear export. Depletion of this m5C ‘reader’ protein causes nuclear retention of m5C methylated transcripts [30]. Additionally, YBX1 (Y-Box Binding Protein 1) ‘reader’ protein interacts with m5C modified RNAs to regulate their translation and stability, influencing various aspects of RNA metabolism. This m5C-binding protein ensures the expansion of HSCs, by stabilizing m5C modified mRNAs essential for cell proliferation. Disruption in YBX1 function can impair HSC maintenance and differentiation [23].
The relevance of m5C RNA modification in hematological malignancies is an area of active research, with emerging evidence highlighting its impact on the development and progression of these disorders. TET2-deficient HSCs manifest global increase in chromatin accessibility, genome instability and finally favoring the development of myeloid malignancies [64,71]. Beyond its established function in DNA demethylation, TET2 in the nucleus oxidizes m5C in RNA, especially in retrotransposon-derived RNAs. This oxidation prevents the binding of MBD6 (methyl-CpG-binding domain protein) to chromatin-associated retrotransposon RNA (carrying m5C modifications), which further recruits deubiquitinases, that remove mono-ubiquitination from histone H2A at lysine 119 (H2AK119ub), leading to an “open chromatin” conformation. Briefly, TET2 oxidizes m5C and antagonizes this MBD6-dependent H2AK119ub deubiquitination. Loss of TET2 activity leads to globally open chromatin conformation, promoting increased gene transcription in HSCs, that contributes to leukemic transformation, explaining the paradox of the highly demethylated (instead of methylated) DNA status in TET2 deficient HSCs [64] (Figure 1C). Confirming to the above is the observation that TET2 mutations impacting its expression levels occur very frequently (about 15%) in patients with various myeloid neoplasias [72]. Expression levels of m5C regulators may serve as biomarkers for AML prognosis and treatment response, while NSUN2, NSUN5, and TET2 inhibitors are suggested to act as potential modulators, in an effort to restore normal HSCs’ methylation patterns, by reversing the malignant phenotype [73].
Mitochondrial dysfunction is a hallmark of several hematological malignancies, and mitochondrial RNA modifications contribute to the regulation of proper mitochondrial function. A recent study focuses on the mitochondrial RNA methyltransferase METTL17, which catalyzes m5C modifications on mitochondrial RNAs (mainly mt-ribosomal RNA), enhancing translation efficiency and supporting the assembly of oxidative phosphorylation (OXPHOS) complexes. Elevated METTL17 expression in AML cells leads to increased OXPHOS activity, shifting metabolism from glycolysis to oxidative phosphorylation, therefore satisfying the high energy demands of proliferating leukemic cells. METTL17 (Knock Out) KO cells have exhibited significant inhibition of AML cell proliferation, induced cell cycle arrest and concurrent reduced OXPHOS [74].
Although most functional studies implicate m5C RNA modification in tumorigenesis, design of antagonists or inhibitors of modifying enzymes responsible for m5C incorporation in RNA molecules and recognition, has not yet yielded results in clinical hematology.
III. Pseudouridine (Ψ) RNA Modification
Pseudouridine (Ψ) is one of the most abundant RNA modifications, displaying chemical properties distinct from the naturally occurring nucleotides. It is found in various types of RNA, including transfer (tRNA), ribosomal (rRNA), small-nuclear (snRNA) and messenger (mRNA). The modification involves the isomerization of uridine to pseudouridine, catalyzed by either RNA-independent or RNA-dependent pseudouridylation [75]. RNA-independent pseudouridylation is performed by stand-alone pseudouridine synthases (PUSs), based on structural or sequence recognition motifs of their target RNA substrates. On the other hand, RNA-dependent pseudouridylation relies on a complex involving a Box H/ACA RNA, along with four core proteins: the Ψ synthase dyskerin (DKC1), glycine-arginine-rich protein 1 (GAR1), non-histone protein 2 (NHP2), and nucleolar protein 10 (NOP10), which form a box H/ACA small ribonucleoprotein (RNP) [24,76]. PUSs are widespread within all cell compartments, including nucleus, cytoplasm and mitochondria, and their relocalization upon stimuli, can influence the RNA substrate pool available for pseudouridylation [77]. Based on its highly conserved nature and abundance within cell, Ψ is believed to be functionally very important, since it enhances the stability of RNA molecules, by improving their base-pairing capabilities and structural rigidity.
This modification has very early been recognized as a critical feature for the tRNAs folding, located in nearly all tRNAs within the TΨC stem–loop, while other, less frequent Ψ sites are also found at different tRNA positions [78,79]. At the three-dimensional level of the ribosomal subunits interactive surface, Ψ is highly concentrated in functionally important sites [80,81]. The same accounts for Ψ locations within spliceosomal snRNAs U1, U2, and U5, all of which participate in spliceosome assembly and pre-mRNA splicing reactions [82]. SnRNA U2 is extensively modified, featuring 14 Ψs in conserved residues, which contribute to snRNP and spliceosome assembly during the splicing process. SHQ1, a critical factor for H/ACA snoRNP assembly, is overexpressed in T-cell acute lymphoblastic leukemia (T-ALL), directly activated via the oncogenic NOTCH1. Elevated SHQ1 facilitates U2 snRNA pseudouridylation and efficient global pre-mRNA splicing, therefore, supporting T-cell leukemogenesis [83]. Pseudouridylation of snRNAs (U2, U4, U5) ensures correct spliceosome function. Loss of DKC1 or snoRNA-guided pseudouridylation impairs splicing fidelity, possibly leading to aberrant splicing of tumor suppressors or oncogenes, exon skipping or intron retention, all known as crucial features in hematological cancers, especially in MDS with splicing factor mutations (e.g., SF3B1, SRSF2).
Artificially enriched with Ψ mRNA, encoding erythropoietin, has been shown to increase its translation efficiency [84]. Pseudouridylation has also been detected in several miRNAs, including members of let-7 family, evolutionary conserved microRNAs that mediates post-transcriptional gene silencing, aiming to regulate/attenuate various normal biological processes, but also induce tumor suppression. Ψ is embedded in pre-miRNAs by PUS1 and/or TruB1, promoting their maturation [85,86]. In addition, pseudouridylation of various mitochondrial tRNAs (mt-tRNACys, mt-tRNASer and mt-tRNATyr), induced by the P175fs mutation of PUS1 reduces the mitochondrial protein synthesis, contributing to mitochondrial dysfunction and inducing among other disorders the syndrome of primary acquired, refractory sideroblastic anemia syndrome (RARS) [87].
Pseudouridine‘s contribution to hematological malignancies has been documented by several studies. HSCs from patients with myelodysplastic syndrome (MDS) are highly concentrated with a specific subset of tRNA-derived fragments (tRFs), termed mini tRFs with a characteristic 5′ terminal oligoguanine tail (mTOGs), which frequently undergoes pseudouridylation (Ψ), enhancing their stability and function. Pseudouridylated mTOGs interact with the RNA-binding protein PABPC1. This interaction disrupts the recruitment of the translational co-activator PAIP1 (Poly(A)-binding protein-interacting protein 1), leading to selective repression of mRNAs with pyrimidine-enriched sequences (PES) in their 5′ untranslated regions (UTRs), including 5′ terminal oligopyrimidine tracts (TOP). These mRNAs typically encode components of the protein synthesis machinery. In high-risk MDS patients, dysregulation of mTOGs results in increased translation of pyrimidine-enriched sequences (PES)-containing mRNAs, contributing to aberrant protein synthesis and impaired differentiation of HSCs. This dysregulation is clinically associated with progression to acute myeloid leukemia (AML) and reduced patient survival [88].
Abnormal hematopoiesis associated with DKC1 mutations may also arise from aberrant rRNA pseudouridylation. Profiled snoRNAs from patients with X-linked dyskeratosis congenita (X-DC), a multisystem disorder characterized by multiple DKC1 mutations and bone marrow failure, uncovered highly heterogeneous snoRNA landscapes in CD34+ cells. Ψ levels at specific uridine residues were correlated with snoRNA levels, that guide pseudouridylation at those sites. Reduced rRNA pseudouridylation can lead to defective ribosome biogenesis and impaired translation of mRNAs, especially IRES-containing transcripts (e.g., p53, c-MYC), promoting skewing protein synthesis. Wild-type DKC1 overexpression could largely rescued normal snoRNA levels and restored faithful CD34+ progenitor differentiation into mature blood lineage-specific cells [89].
Conclusively, defective pseudouridylation disrupts RNA metabolism at multiple levels: translation, splicing, ribosome function, and all these perturbations can initiate or aggravate the course of several hematological malignancies.
IV. 2′-O-Methylation RNA Modification
This RNA modification involves the methylation of the 2′-O position of the ribose sugar in RNA, catalyzed by the Fibrillarin and NOP56 complex. It influences RNA stability, splicing, and protein synthesis. Nucleophosmin 1 (NPM1) participates in rRNA 2′-O-methylation by direct binding to several C/D box small nucleolar RNAs and to the methyltransferase fibrillarin. The fibrillarin–NPM1–small nucleolar RNA complex actively methylates rRNA. NPM1 loss of function, a very frequent event in AML, results in altered rRNA 2′-O-methylation and dysregulated translation [31].
2′-O-methylation of rRNAs, guided by snoRNAs and executed by fibrillarin (FBL), is dynamically remodeled in AML, and specific methylation patterns on the ribosome surface correlate with leukemia stem cell (LSC) signatures. Enhanced 2′-O-methylation is reported to rewire translation towards amino-acid transporter mRNAs enriched in optimal codons, increasing intracellular amino acid levels and supporting the LSC phenotype [32].
B. Consequences from Altered Subcellular Topology of Non-Coding (nc)RNA Molecules
The regulatory potential of many ncRNAs has been revealed at specific subcellular location. Nevertheless, many ncRNAs are discretely distributed in different cellular compartments, and the related biological significance remains largely unclear.
I. Intracellular Distribution and Function of Micro-RNAs (miRs)
Among well-documented functions of mature miRs (21-23 nt) in the cytoplasm is the targeting of mRNAs molecules, mainly at the 3’ UTR, leading to their degradation. To accomplish their aggressive function against mRNAs, miRs are tethered with the catalytic Argonaute (Ago) proteins and are assembled into the RISC (RNA-induced silencing) complex [90]. Pre-miRNAs with a hairpin 2D construction are transported to the cytoplasm, through their nuclear transport receptor exportin-5 and another karyopherin, the CRM1, which mediates their nuclear-cytoplasmic shuttling [91]. Importin 8, a member of the karyopherin β transporters on the nuclear envelope, has been reported to participate and facilitate miRs’ relocation within cell nucleus [92]. Despite their clearly defined role in the cytoplasm, numerous mature miRs have also been reported to selectively relocate within cell nucleus, assigned with gene expression regulatory roles. These miRs encompass variations of a 6-nucleotide motif [93] responsible, but not sufficient, for their reimport in the nucleus. These motifs have been evident in the 5p-arm of the let-7 family members [94].
Studies that differentiate the intracellular localization of miRs between the nucleus and the cytoplasm are relatively few. A basic reason for that is the important methodological difficulties, arising from RNA extraction procedures from different cell fractions: nucleus, cytoplasm and mitochondria. Pure RNA extraction preventing cross-contaminations is challenging, mostly related to insufficient gradient methods, that result in incomplete separation and consequent overlap between organelle fractions. Also, RNases can act during processing, especially if cell compartments aren't promptly stabilized, leading to massive RNA degradation. Nevertheless, well-defined functions of nuclear miRs is the regulation of other miR’s biogenesis [95]. Some studies have expanded miRs’ silencing mechanism in the nucleus and suggest that this is assisted by the GW182 family proteins (components of miR-induced silencing complexes) and by Ago proteins that translocate in nuclear foci, to target apart from miRs and nuclear long non-coding RNAs (lncRNAs), such as MALAT-1[96]. Analysis of subcellular nuclear and cytoplasmic compartments of neural stem cells derived from the human embryonic cell line WA09, revealed miRs with consistently stronger standard scores in the nuclear fraction, namely hsa-miR-30b/c, hsa-miR-374a/b and hsa-miR-19a/b, as well as different miR species significantly enriched in the cytoplasmic fraction, such as hsa-miR-20a/b, hsa-miR-17, hsa-miR-106a/b, hsa-miR-93, ect [97].
In the hematopoietic compartment hsa-miR-223, stands for as a well-documented double functioning molecule. When this miR is localized in the nucleus silences the NF1A (Nuclear Factor I 1A) promoter via recruitment of the polycomb repressive complex (PcG), responsible for the trimethylation of H3K27. In the cytoplasm of myeloid progenitors, hsa-miR-223 targets for degradation the NF1A-mRNA during granulopoiesis [98,99]. Conclusively, hsa-miR-233 is devoted to silence NF1A factor, having a dual role in both, the nucleus and the cytoplasm of myeloid progenitors. Also, hsa-miR-223 and C/EBPa (CCAAT Enhancer Binding Protein alpha) regulate the expression of E2F1 in AML. E2F1 upregulates hsa-miR-223 transcription via promoter binding, and cytoplasmic hsa-miR-223 suppresses E2F1-mRNA. In AML cells, this feedback loop is disrupted, resulting in the aberrant expression levels of E2F1 and hsa-miR-223, contributing to uncontrolled cell proliferation [100]. Several other miRs: hsa-miR-690, hsa-miR-706, hsa-miR-709 have been reported to be localized within the nuclear compartment, regulating or fine-tuning other miR species’ expression patterns [101]. Hsa-miR-690 is enriched in the nucleus of myeloid cells and upon its overexpression attenuates the expression of C/EBPα protein, involved in the development of granulocyte-monocyte progenitors, and affecting their subsequent terminal differentiation [102]. Loss of C/EBPα leads to increased myeloid proliferation [103].
Loss or down-regulation of HOX regulatory miRs, such as hsa-miR-204 and hsa-miR-128a and overexpression of Homeobox (HOX) genes (specifically HOXA and HOXB), have been suggested as potential disease-specific signatures in AML patients, simultaneously carrying mutations in the nucleophosmin 1 (NPM1) gene. At steady-state, NPM1 usually resides in the nucleolus, whereas only a minimal fraction shuttles between the other cell compartments [7]. NPM1 mutations in AML result in the translocalization of NPM1 protein into the cytoplasm, representing the most common (50–60%) genetic alteration in AML-NK (Normal Karyotype) and in about one-third of all AML cases [8,9]. Quantification of the precise proportion of NPM1 protein, localized to the cytoplasm of NPM1-mutated cells is challenging, and the biological relevance of how NPM1 mutant cells are prone to develop acute leukemia, by acting at the chromatin level, in cooperation with miR species (hsa-miR-204 and hsa-miR-128a) as potential partners, remains unclear. This active network can include lncRNAs, which is discussed in the lncRNA section.
Studies in MDS and AML have shown substantial discrepancies in miR expression profiles, and a potential etiologic factor for these discrepancies can be the different cell sources, used for RNA extraction. Unsorted total BM cells, BM mononuclear cells or selected CD34+ BM cells have been used as a primary source for RNA isolation and miR detection, with each investigative approach having both, advantages and disadvantages. MiRs secreted into the extracellular fluids and imported into exosomes are also subject of investigations, mainly for biomarkers’ identification. Analyses of the unsorted BM cells can deliver more information about the microenvironment and the impaired erythroid differentiation, than about the more homogeneously isolated CD34+ BM cell population, which captures in more detail the clonal, MDS-specific blast cell population and demonstrates the profound, intrinsic changes of miR expression profiles. However, significant differences in miR expression have been reported between healthy controls, MDS and AML, independent of the cell source [104]. The most significantly down- or up-regulated miRs found in patients with MDS and AML are listed in Table 2. These observations support the idea for the existing absolute specialization in the creation of molecular networks/interactions within functionally different cells, even in the same tissue.
Mutations in genes encoding spliceosome components occur in more than 50% of MDS patients with SF3B1 (Splicing Factor 3b Subunit 1), SRSF2 (Serine and Arginine Rich Splicing Factor 2), U2AF1 (U2 Small Nuclear RNA Auxiliary Factor 1) and ZRSR2 (Zinc Finger CCCH-Type, RNA Binding Motif And Serine/Arginine Rich 2) as the most frequently mutated [105,106]. Aberrant splicing as a consequence of the spliceosome mutations have shown to affect along with mRNA transcripts and miR expression and their post-transcriptional maturation. In particular, pri-miRNAs encoded within introns of coding genes, termed as miRtrons, are processed by the nuclear splicing machinery like typical introns, to form intra-nuclear stable hairpins with a shorter stem feature before they are exported to the cytoplasm [107]. Downregulation of the tumor suppressive miRs such as let-7 family, miR-423 and miR-103a have been documented in MDS patients carrying spliceosome mutations [108], which can be associated with impaired splicing events in the nucleus.
Among mitochondrial ncRNAs, the mitochondrial microRNAs (mt-miRs) are transcribed directly from the mitochondrial genome as their nuclear counterparts and regulate mitochondrial gene expression post-transcriptionally [109]. Along with mt-miRs, nuclear derived miR species are imported into the mitochondria, to modulate the expression levels of mRNAs originating from the mitochondrial genome. One of the key proteins implicated in this complex transport process is Argonaute2 (Ago2) [110] however, mitochondrial miR-derived species and hematologic malignancies still remain unconnected.
Another category of small nuclear ncRNAs, the Piwi-interacting RNAs, have been framed as regulatory molecules in hematology. In DLBCL, upregulation of the piRNA-30473 has been shown to induce increased expression of methylation factor WTAP (Wilms' tumor 1-associating protein) in the nucleus and increased m6A levels mainly in (hexokinase2) HK2-mRNA, contributing to the high glycolytic rates and promoting tumorigenesis [111].
II. Abundance of Long Non-Coding RNAs (lncRNAs) in the Subcellular Compartments
Long non-coding RNAs (lncRNAs) are long transcripts (mainly ≥200nt) that do not code for proteins, but play roles in regulating gene expression, modulating chromatin dynamics by recruiting histone-modifying and chromatin-remodeling complexes and RNA splicing. Recent evidence reveals that some lncRNAs actually harbor small open reading frames (sORFs), that are actively translated into functional micropeptides, but their actual relevance in promoting malignant phenotypes remains uncovered [112]. LncRNAs associated with chromatin remodeling are highlighted as important regulators of normal and malignant hematopoiesis [113]. Further, to their chromatin remodeling properties, concerns have arisen from lncRNAs’ transportation to different locations within the cell and about the consequences in cell homeostasis and performance.
Distribution of the lncRNAs among the different endo-cellular clusters has been determined by cell fractionation, followed by RNA-seq and has been shown to be proportionally higher in the cytoplasmic ribosome-enriched clusters, than in the nuclear clusters. TUG1-lncRNA, which is involved in the upregulation of the HOX family and other growth-control genes in the nucleus, has also been detected in cytosolic cell fractions containing five or six ribosomes. Furthermore, DANCR- and H19- lncRNAs have shown clear enrichment in the cytoplasm over nucleus, providing evidence for other, still unrecognized functions within the cell, and more specifically lncRNA implication in the regulation of translation process [114]. However, there is a debate concerning the precise lncRNA endo-cellular compartmentalization, since other groups propose that they preferentially localize and function within the cell nucleus [115].
The cytoplasmic translocation of the NPM1 mutants in AML patients is accompanied by upregulation of HOXA/B genes and alters the hematopoietic cells’ genome topology, as previously mentioned [9]. In this complex microenvironment specific lncRNAs, deriving from HOXA/B loci (on chromosomes 7 and 17 respectively), are also implicated. The anterior HOXB derived lncRNA-HOXBLINC (Gene ID: 128462380), is activated in AML and promotes HSC transcription signatures, by exploiting an active 3D interactome to recruit the MLL1/Setd1a complex and deposit H3K4me3 marks on each of the anterior HOXB genes for their activation [116]. LncRNA-HOTAIRM1 is hosted within HOXA genomic cluster, between HOXA1 and HOXA2 genes. Its significantly higher expression in AML patients, carrying NPM1 mutations, attributes to lncRNA-HOTAIRM1 a great prognostic impact [117]. LncRNA-HOTTIP, which is transcribed from the distal HOXA locus, recruits the mixed lineage leukemia MLL family of histone methyltransferases (MLL1/WDR5 complex) to activate targeted genes in cis [118]. Given that both, LncRNA-HOTTIP and NPM1 interact with CTCF (CCCTC-binding factor) a critical factor responsible for chromatin organization and for anchoring CTCF-flanked Topologically Associated Domains (TADs) [14,15], renders particularly interesting and important to determine deeper mechanisms of action for NPM1 mutants and mis-localization of NPM1 associations within the evolutionary process of leukemogenesis (Figure 2A).
In AML with mutated NPM1, more evidence exists for the implication of lncRNA translocations. The lncRNA-LONA shifts from the cytoplasm to the nucleus, alongside the mutant NPM1 export towards the opposite direction. Within the nucleus, lncRNA-LONA alters myeloid differentiation by regulating tissue-specific mRNAs. By this mechanism promotes leukemogenesis, while reduces chemotherapy response in vivo [119]. The involvement of lncRNAs in m6A RNA modification pathways has been also identified in AML. LncRNA-UCA1 has been reported to contribute to AML progression, by upregulating the expression of CXCR4 (C-X-C Motif Chemokine Receptor 4) and CYP1B1 (Cytochrome P450 Family 1 Subfamily B Member 1), through binding and stabilization of the m6A methyltransferase METTL14. This interaction affects m6A levels and influences localization and function of METTL14 in AML cells [120]. Another study has demonstrated that the lncRNA-MALAT-1 binds to METTL14, promoting m6A modification of ZEB1-mRNA (Zinc Finger E-Box Binding Homeobox 1). This interaction enhances the aggressiveness of AML, by influencing the localization and function of METTL14 intranuclearly [121] (Figure 2B).
The translocation of non-coding RNAs (ncRNAs) between the nucleus and mitochondria is essential for regulating mitochondrial function and mitochondrial gene expression. Among lnc-RNAs identified in both, nuclear and mitochondrial territories, nuclear lnc-RMRP has been identified as a component of the nuclear RNase MRP complex, mediating the processing of the short mature 5.8S rRNA [122]. In addition, lnc-RMRP in the cell nucleus interacts with telomerase, to form a complex with the RNA-dependent RNA polymerase, capable of synthesizing dsRNA precursors, which are processed by DICER1 into siRNAs [123]. Two RNA-binding proteins: HuR (human antigen R) and GRSF1 (G-rich RNA sequence-binding factor 1) are responsible for the mobilization of the nuclear encoded lncRNA-RMRP into the mitochondrial matrix. As a component of the mitochondrial RNase MRP, lnc-RMRP is important for mitochondrial DNA replication and RNA processing [124].
Another nucleus-derived lncRNA, linc-p21, has been shown to increase HIF-1a (hypoxia-inducible factor-1) stability with high specificity, and consequently to promote glycolysis, affecting also mitochondrial OXPHOS [125]. Inhibition of the lncRNA-HOTAIR, a Hox Transcript Antisense Intergenic RNA, encoded from the nuclear genome and imported into mitochondria, has also been reported to be associated with a remarkable decrease of the UQCRQ (Ubiquinol-Cytochrome C Reductase, Complex III Subunit VII), inducing deficiency of complex III, and impairment of the mitochondrial respiratory chain [126]. These studies highlight an as yet undiscovered field of interplay between endonuclearly expressed lnc-RNAs and mitochondrial regulation. In hematological malignancies, dysregulation of this transport system can contribute to altered cellular metabolism, apoptosis, and abnormal proliferation, affecting disease progression and treatment response, hence requires deeper investigation.
LncRNAs are also transcribed from mitochondrial DNA and additionally, some nuclear lncRNAs are transported into the mitochondrial matrix and influence mitochondrial gene expression and function. Mitochondrial long non-coding RNAs (mt-lncRNAs), transcribed from the mitochondrial genome, have emerged as key players in various cellular processes and have been implicated in the pathogenesis of several cancers. Their function within the mitochondrial compartment is not only restricted on metabolism reprogramming, but is also extended in mitochondria-associated apoptosis, mitochondrial genome expression regulation and stress signal transmission [127,128,129,130]. A circular non-coding RNA, the mc-COX2 is produced from the COX2 locus on the L-strand of mitochondrial DNA and has been found at high levels in the plasma exosomes of patients with chronic lymphocytic leukemia (CLL), affecting disease recurrence and progression [131]. Another mitochondrial lncRNA, the SncmtRNA, derives from mitochondrial 16S rRNA sequences and is consistently overexpressed in proliferating cells—both normal and malignant—but is absent in non-dividing cells, indicating its role in cell division. Two antisense lncRNAs, ASncmtRNA-1 and ASncmtRNA-2, with similar hairpin structures to SncmtRNA, are expressed only in normal proliferating cells and are downregulated in tumor cells, suggesting a potential tumor-suppressor function. Subcellular localization studies, using in situ hybridization, confocal and electron microscopy, have revealed that SncmtRNA localizes predominantly to the nucleus, and more precisely in the nucleoli and heterochromatin, in both, normal and tumor cells. However, in tumor tissues, SncmtRNA shows variable localization, being nuclear in some cases and cytoplasmic in others, possibly reflecting its involvement in epigenetic regulation during malignant transformation. In contrast, ASncmtRNAs localize to the nucleus in normal cells, but are notably absent in tumor tissues [129,132]. Conclusively, downregulation of SncmtRNA and the upregulation of ASncmtRNA-2 suggest a pivotal role of these transcripts in cell replicative senescence. ASncmtRNA-2 is linked to the production of mitochondrial encoded miRs hsa-miR-4485-3p and hsa-miR-1973, with miR-4485 showing a significant increase. Overexpression of these mtmiRs in cells results in delayed progression through the G1 and G2/M phases of the cell cycle. These findings highlight a functional role for mitochondrial lncRNA ASncmtRNA-2 in generating regulatory miRs that influence cell cycle control [130].
Conclusions and Future Perspectives
There is clear evidence based on investigations, although still very limited, that non-coding RNA molecules localized into different membrane separated cell compartments are prone to cooperate with different macromolecule networks, therefore, exhibiting alternate functions. "Genetic" over "epigenetic" events are considered the first and most important drivers for several hematological malignancies. However, epigenetic alterations are documented to play early and sometimes initiating pathogenetic roles, especially in age-related clonal hematopoiesis and pre-leukemic states. The debate between these two pathogenetic mechanisms, as driver factors for tumorigenesis, is still under consideration and under continuous investigation. Nevertheless, "genetic" and "epigenetic" lesions are strictly interconnected; mutations in genes encoding modification enzymes, that control the epigenome blur the line between "genetic" and "epigenetic" events. Current technologies are increasingly capable of unraveling the spatial organization of RNA modifications and ncRNAs within cellular compartments, and these approaches are proving highly useful for understanding how alterations in RNA biology may contribute to tumorigenesis.
This review highlights the endo-cellular defected pathways, referring to aberrant topology of altered RNA modifications, mainly m6A, m5C and Ψ, presenting an overview on factors triggering this phenomenon, and also the downstream pathways that are affected and stimulate the development of hematological malignancies. Mis-localized modified RNAs have been shown to be conditionally produced by relevant malfunctional modifying enzymes, affecting RNA maturation, stabilization and interference to chromatin conformation (Figure 1). These insights point to RNAs and their modifying enzymes as potential therapeutic targets in various hematological malignancies. Efforts for testing and utilization of new drugs, targeting RNA modifying enzymes are ongoing, and a few of them are already in the early stages of clinical trials (Table 1).
Further to RNA modifications implicated in hematology, miRs and lncRNAs, of the most recognized and studied ncRNA molecules, have emerged as critical epigenetic factors, capable for disrupting chromatin structure and gene expression levels, by misregulating either pre- or post-transcriptionally tumor suppressors or oncogenes, and alter RNA/protein interactions critical for cell integrity (Figure 2). With advances in delivery systems, RNA biology, and epigenetic understanding, ncRNA-based therapies are likely to become a part of future treatment options for several hematological malignancies. Particularly promising is their potential to effectively target malignant stem cells, overcome drug resistance, and act synergistically with the existing therapies, targeting other dysregulated cellular pathways. Few attempts have entered clinical trials, such as the MRG-106 (Cobomarsen), an inhibitor of hsa-miR-155 tested in cutaneous T-cell lymphoma (CTCL) and other lymphomas however, development was paused due to company restructuring [133]. Currently, there are some RNA-based therapies in clinical trials representing a growing and highly promising class of novel agents but have not yet received approval from the Regulatory Organizations as treatment for hematological malignancies. Several of them are currently tested in clinical trials, specifically for blood cancers, including antisense oligonucleotides (ASOs), small interfering RNAs (siRNAs), RNA aptamers etc. As single-cell RNA-seq, epitranscriptomics, and AI-driven drug design evolve, new possibilities arise for the prediction of RNA structure-function interactions and the tumor-specific RNA profiles, which will enable a “smart” RNA drug design, addressing many of the unmet needs for the effective treatment of hematological malignancies.
Author Contributions
A.S. (Argyro Sgourou) with A.S. (Argiris Symeonidis) and DC conceived the context and perspectives. ID, AC and DC reviewed the related literature and A.S. (Argyro Sgourou) drafted the manuscript. A.S. (Argiris Symeonidis) revised, A.S. (Argyro Sgourou) and DC finalized the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
Irene Dereki was supported by a three year-fellowship provided from the Hellenic Open University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kouroukli, O.; Symeonidis, A.; Foukas, P.; Maragkou, M.-K.; Kourea, E.P. Bone Marrow Immune Microenvironment in Myelodysplastic Syndromes. Cancers (Basel) 2022, 14, 5656. [CrossRef]
- Godfrey, K.M.; Lillycrop, K.A.; Burdge, G.C.; Gluckman, P.D.; Hanson, M.A. Epigenetic Mechanisms and the Mismatch Concept of the Developmental Origins of Health and Disease. Pediatr Res 2007, 61.
- Boccaletto, P.; MacHnicka, M.A.; Purta, E.; Pitkowski, P.; Baginski, B.; Wirecki, T.K.; De Crécy-Lagard, V.; Ross, R.; Limbach, P.A.; Kotter, A.; et al. MODOMICS: A Database of RNA Modification Pathways. 2017 Update. Nucleic Acids Res 2018, 46, D303–D307. [CrossRef]
- Rebelo-Guiomar, P.; Powell, C.A.; Van Haute, L.; Minczuk, M. The Mammalian Mitochondrial Epitranscriptome. Biochim Biophys Acta Gene Regul Mech 2019, 1862, 429–446.
- van der Werf, I.; Sneifer, J.; Jamieson, C. RNA Modifications Shape Hematopoietic Stem Cell Aging: Beyond the Code. FEBS Lett 2024.
- Khan, I.; Amin, M.A.; Eklund, E.A.; Gartel, A.L. Regulation of HOX Gene Expression in AML. Blood Cancer J 2024, 14, 42. [CrossRef]
- Correction for Garzon et al., Distinctive MicroRNA Signature of Acute Myeloid Leukemia Bearing Cytoplasmic Mutated Nucleophosmin. Proceedings of the National Academy of Sciences 2024, 121. [CrossRef]
- Falini, B.; Mecucci, C.; Tiacci, E.; Alcalay, M.; Rosati, R.; Pasqualucci, L.; Starza, R. La; Diverio, D.; Colombo, E.; Santucci, A.; et al. Cytoplasmic Nucleophosmin in Acute Myelogenous Leukemia with a Normal Karyotype; 2005;
- Falini, B. NPM1-Mutated Acute Myeloid Leukemia: From Bench to Bedside. https://doi.org/10.1182/blood.2019004226/1747787/blood.2019004226.pdf. [CrossRef]
- Zardo, G.; Ciolfi, A.; Vian, L.; Starnes, L.M.; Billi, M.; Racanicchi, S.; Maresca, C.; Fazi, F.; Travaglini, L.; Noguera, N.; et al. Polycombs and MicroRNA-223 Regulate Human Granulopoiesis by Transcriptional Control of Target Gene Expression. Blood 2012, 119, 4034–4046. [CrossRef]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A Minicircuitry Comprised of MicroRNA-223 and Transcription Factors NFI-A and C/EBPα Regulates Human Granulopoiesis. Cell 2005, 123, 819–831. [CrossRef]
- Rasko, J.E.J.; Wong, J.J.L. Nuclear MicroRNAs in Normal Hemopoiesis and Cancer. J Hematol Oncol 2017, 10.
- Díaz-Beyá, M.; Brunet, S.; Nomdedéu, J.; Pratcorona, M.; Cordeiro, A.; Gallardo, D.; Escoda, L.; Heras, I.; Ribera, J.M.; Duarte, R.; et al. The LincRNA HOTAIRM1, Located in the HOXA Genomic Region, Is Expressed in Acute Myeloid Leukemia, Impacts Prognosis in Patients in the Intermediate-Risk Cytogenetic Category, and Is Associated with a Distinctive MicroRNA Signature; Vol. 6;.
- Yusufzai, T.M.; Tagami, H.; Nakatani, Y.; Felsenfeld, G. CTCF Tethers an Insulator to Subnuclear Sites, Suggesting Shared Insulator Mechanisms across Species; 2004; Vol. 13;.
- Luo, H.; Zhu, G.; Xu, J.; Lai, Q.; Yan, B.; Guo, Y.; Fung, T.K.; Zeisig, B.B.; Cui, Y.; Zha, J.; et al. HOTTIP LncRNA Promotes Hematopoietic Stem Cell Self-Renewal Leading to AML-like Disease in Mice. Cancer Cell 2019, 36, 645-659.e8. [CrossRef]
- Qing, Y.; Su, R.; Chen, J. RNA Modifications in Hematopoietic Malignancies: A New Research Frontier. Blood 2021, 138, 637–648. [CrossRef]
- Meyer, K.D.; Jaffrey, S.R. The Dynamic Epitranscriptome: N6-Methyladenosine and Gene Expression Control. Nat Rev Mol Cell Biol 2014, 15, 313–326.
- Dominissini, D.; Moshitch-Moshkovitz, S.; Schwartz, S.; Salmon-Divon, M.; Ungar, L.; Osenberg, S.; Cesarkas, K.; Jacob-Hirsch, J.; Amariglio, N.; Kupiec, M.; et al. Topology of the Human and Mouse M6A RNA Methylomes Revealed by M6A-Seq. Nature 2012, 485, 201–206. [CrossRef]
- Ke, S.; Pandya-Jones, A.; Saito, Y.; Fak, J.J.; Vågbø, C.B.; Geula, S.; Hanna, J.H.; Black, D.L.; Darnell, J.E.; Darnell, R.B. M 6 A MRNA Modifications Are Deposited in Nascent Pre-MRNA and Are Not Required for Splicing but Do Specify Cytoplasmic Turnover. 2017. [CrossRef]
- Zhao, Y.; Peng, H. The Role of N6-Methyladenosine (M6A) Methylation Modifications in Hematological Malignancies. Cancers (Basel) 2022, 14.
- Trixl, L.; Lusser, A. The Dynamic RNA Modification 5-methylcytosine and Its Emerging Role as an Epitranscriptomic Mark. WIREs RNA 2019, 10. [CrossRef]
- Squires, J.E.; Patel, H.R.; Nousch, M.; Sibbritt, T.; Humphreys, D.T.; Parker, B.J.; Suter, C.M.; Preiss, T. Widespread Occurrence of 5-Methylcytosine in Human Coding and Non-Coding RNA. Nucleic Acids Res 2012, 40, 5023–5033. [CrossRef]
- Liu, F.; Wang, M.; Gao, S.; Song, G.; Liu, M.; Li, Y.; Sun, P.; Lai, W.; Wang, H.; Yang, Y.G.; et al. RNA M5C Methylation Mediated by Ybx1 Ensures Hematopoietic Stem and Progenitor Cell Expansion. Cell Rep 2025, 44. [CrossRef]
- Cerneckis, J.; Cui, Q.; He, C.; Yi, C.; Shi, Y. Decoding Pseudouridine: An Emerging Target for Therapeutic Development. Trends Pharmacol Sci 2022, 43, 522–535.
- Xiao, W.; Adhikari, S.; Dahal, U.; Chen, Y.S.; Hao, Y.J.; Sun, B.F.; Sun, H.Y.; Li, A.; Ping, X.L.; Lai, W.Y.; et al. Nuclear M6A Reader YTHDC1 Regulates MRNA Splicing. Mol Cell 2016, 61, 507–519. [CrossRef]
- Pendleton, K.E.; Chen, B.; Liu, K.; Hunter, O. V.; Xie, Y.; Tu, B.P.; Conrad, N.K. The U6 SnRNA M6A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell 2017, 169, 824-835.e14. [CrossRef]
- Zaccara, S.; Jaffrey, S.R. A Unified Model for the Function of YTHDF Proteins in Regulating M6A-Modified MRNA. Cell 2020, 181, 1582-1595.e18. [CrossRef]
- Li, Z.; Weng, H.; Su, R.; Weng, X.; Zuo, Z.; Li, C.; Huang, H.; Nachtergaele, S.; Dong, L.; Hu, C.; et al. FTO Plays an Oncogenic Role in Acute Myeloid Leukemia as a N6-Methyladenosine RNA Demethylase. Cancer Cell 2017, 31, 127–141. [CrossRef]
- Wei, J.; Liu, F.; Lu, Z.; Fei, Q.; Ai, Y.; He, P.C.; Shi, H.; Cui, X.; Su, R.; Klungland, A.; et al. Differential m 6 A, m 6 A m , and m 1 A Demethylation Mediated by FTO in the Cell Nucleus and Cytoplasm. Mol Cell 2018, 71, 973-985.e5. [CrossRef]
- Yang, X.; Yang, Y.; Sun, B.F.; Chen, Y.S.; Xu, J.W.; Lai, W.Y.; Li, A.; Wang, X.; Bhattarai, D.P.; Xiao, W.; et al. 5-Methylcytosine Promotes MRNA Export-NSUN2 as the Methyltransferase and ALYREF as an m 5 C Reader. Cell Res 2017, 27, 606–625. [CrossRef]
- Nachmani, D.; Bothmer, A.H.; Grisendi, S.; Mele, A.; Bothmer, D.; Lee, J.D.; Monteleone, E.; Cheng, K.; Zhang, Y.; Bester, A.C.; et al. Germline NPM1 Mutations Lead to Altered RRNA 2′-O-Methylation and Cause Dyskeratosis Congenita. Nat Genet 2019, 51, 1518–1529. [CrossRef]
- Zhou, F.; Aroua, N.; Liu, Y.; Rohde, C.; Cheng, J.; Wirth, A.K.; Fijalkowska, D.; Göllner, S.; Lotze, M.; Yun, H.; et al. A Dynamic RRNA Ribomethylome Drives Stemness in Acute Myeloid Leukemia. Cancer Discov 2023, 13, 332–347. [CrossRef]
- Liu, J.; Dou, X.; Chen, C.; Chen, C.; Liu, C.; Michelle Xu, M.; Zhao, S.; Shen, B.; Gao, Y.; Han, D.; et al. N6-Methyladenosine of Chromosome-Associated Regulatory RNA Regulates Chromatin State and Transcription. Science (1979) 2020, 367, 580–586. [CrossRef]
- Huang, J.; Yin, P. Structural Insights into N6-Methyladenosine (M6A) Modification in the Transcriptome. Genomics Proteomics Bioinformatics 2018, 16, 85–98.
- Wang, X.; Zhao, B.S.; Roundtree, I.A.; Lu, Z.; Han, D.; Ma, H.; Weng, X.; Chen, K.; Shi, H.; He, C. N6-Methyladenosine Modulates Messenger RNA Translation Efficiency. Cell 2015, 161, 1388–1399. [CrossRef]
- Alarcón, C.R.; Lee, H.; Goodarzi, H.; Halberg, N.; Tavazoie, S.F. N6-Methyladenosine Marks Primary MicroRNAs for Processing. Nature 2015, 519, 482–485. [CrossRef]
- Meyer, K.D.; Patil, D.P.; Zhou, J.; Zinoviev, A.; Skabkin, M.A.; Elemento, O.; Pestova, T. V.; Qian, S.B.; Jaffrey, S.R. 5′ UTR M6A Promotes Cap-Independent Translation. Cell 2015, 163, 999–1010. [CrossRef]
- Hoek, T.A.; Khuperkar, D.; Lindeboom, R.G.H.; Sonneveld, S.; Verhagen, B.M.P.; Boersma, S.; Vermeulen, M.; Tanenbaum, M.E. Single-Molecule Imaging Uncovers Rules Governing Nonsense-Mediated MRNA Decay. Mol Cell 2019, 75, 324-339.e11. [CrossRef]
- Du, H.; Zhao, Y.; He, J.; Zhang, Y.; Xi, H.; Liu, M.; Ma, J.; Wu, L. YTHDF2 Destabilizes m 6 A-Containing RNA through Direct Recruitment of the CCR4-NOT Deadenylase Complex. Nat Commun 2016, 7. [CrossRef]
- Lv, J.; Zhang, Y.; Gao, S.; Zhang, C.; Chen, Y.; Li, W.; Yang, Y.G.; Zhou, Q.; Liu, F. Endothelial-Specific M6A Modulates Mouse Hematopoietic Stem and Progenitor Cell Development via Notch Signaling. Cell Res 2018, 28, 249–252.
- Gao, Y.; Vasic, R.; Song, Y.; Teng, R.; Liu, C.; Gbyli, R.; Biancon, G.; Nelakanti, R.; Lobben, K.; Kudo, E.; et al. M6A Modification Prevents Formation of Endogenous Double-Stranded RNAs and Deleterious Innate Immune Responses during Hematopoietic Development. Immunity 2020, 52, 1007-1021.e8. [CrossRef]
- Weng, H.; Huang, H.; Wu, H.; Qin, X.; Zhao, B.S.; Dong, L.; Shi, H.; Skibbe, J.; Shen, C.; Hu, C.; et al. METTL14 Inhibits Hematopoietic Stem/Progenitor Differentiation and Promotes Leukemogenesis via MRNA M6A Modification. Cell Stem Cell 2018, 22, 191-205.e9. [CrossRef]
- Weng, H.; Huang, H.; Chen, J. N6-Methyladenosine RNA Modification in Normal and Malignant Hematopoiesis. In Advances in Experimental Medicine and Biology; Springer, 2023; Vol. 1442, pp. 105–123.
- Kapadia, B.; Roychowdhury, A.; Kayastha, F.; Lee, W.S.; Nanaji, N.; Windle, J.; Gartenhaus, R. M6A Eraser ALKBH5/TreRNA1/DDX46 Axis Regulates BCR Expression. Neoplasia (United States) 2025, 62. [CrossRef]
- Song, W.; Fei, F.; Qiao, F.; Weng, Z.; Yang, Y.; Cao, B.; Yue, J.; Xu, J.; Zheng, M.; Li, J. ALKBH5-Mediated N6-Methyladenosine Modification of TRERNA1 Promotes DLBCL Proliferation via P21 Downregulation. Cell Death Discov 2022, 8. [CrossRef]
- Jaafar, C.; Aguiar, R.C.T. Dynamic Multilayered Control of M6A RNA Demethylase Activity. Proc Natl Acad Sci U S A 2024, 121. [CrossRef]
- Chang, G.; Xie, G.S.; Ma, L.; Li, L.; Richard, H.T. USP36 Promotes Tumorigenesis and Drug Sensitivity of Glioblastoma by Deubiquitinating and Stabilizing ALKBH5. Neuro Oncol 2023, 25, 841–853. [CrossRef]
- Shen, C.; Sheng, Y.; Zhu, A.C.; Robinson, S.; Jiang, X.; Dong, L.; Chen, H.; Su, R.; Yin, Z.; Li, W.; et al. RNA Demethylase ALKBH5 Selectively Promotes Tumorigenesis and Cancer Stem Cell Self-Renewal in Acute Myeloid Leukemia. Cell Stem Cell 2020, 27, 64-80.e9. [CrossRef]
- Elkashef, S.M.; Lin, A.P.; Myers, J.; Sill, H.; Jiang, D.; Dahia, P.L.M.; Aguiar, R.C.T. IDH Mutation, Competitive Inhibition of FTO, and RNA Methylation. Cancer Cell 2017, 31, 619–620.
- Qing, Y.; Dong, L.; Gao, L.; Li, C.; Li, Y.; Han, L.; Prince, E.; Tan, B.; Deng, X.; Wetzel, C.; et al. R-2-Hydroxyglutarate Attenuates Aerobic Glycolysis in Leukemia by Targeting the FTO/M6A/PFKP/LDHB Axis. Mol Cell 2021, 81, 922-939.e9. [CrossRef]
- Lin, A.P.; Qiu, Z.; Ethiraj, P.; Sasi, B.; Jaafar, C.; Rakheja, D.; Aguiar, R.C.T. MYC, Mitochondrial Metabolism and O-GlcNAcylation Converge to Modulate the Activity and Subcellular Localization of DNA and RNA Demethylases. Leukemia 2022, 36, 1150–1159. [CrossRef]
- Qiu, Z.J.; Lin, A.P.; Jiang, S.; Elkashef, S.M.; Myers, J.; Srikantan, S.; Sasi, B.; Cao, J.Z.; Godley, L.A.; Rakheja, D.; et al. MYC Regulation of D2HGDH and L2HGDH Influences the Epigenome and Epitranscriptome. Cell Chem Biol 2020, 27, 538-550.e7. [CrossRef]
- Stine, Z.E.; Walton, Z.E.; Altman, B.J.; Hsieh, A.L.; Dang, C. V. MYC, Metabolism, and Cancer. Cancer Discov 2015, 5, 1024–1039.
- Cerchione, C.; Romano, A.; Daver, N.; DiNardo, C.; Jabbour, E.J.; Konopleva, M.; Ravandi-Kashani, F.; Kadia, T.; Martelli, M.P.; Isidori, A.; et al. IDH1/IDH2 Inhibition in Acute Myeloid Leukemia. Front Oncol 2021, 11.
- Wang, Y.; Zeng, H. min; Xue, Y. juan; Lu, A. dong; Jia, Y. ping; Zuo, Y. xi; Zhang, L. ping The Gene Expression Level of M6A Catalytic Enzymes Is Increased in ETV6/RUNX1-Positive Acute Lymphoblastic Leukemia. Int J Lab Hematol 2021, 43, e89–e91.
- Liu, X.; Huang, L.; Huang, K.; Yang, L.; Yang, X.; Luo, A.; Cai, M.; Wu, X.; Liu, X.; Yan, Y.; et al. Novel Associations Between METTL3 Gene Polymorphisms and Pediatric Acute Lymphoblastic Leukemia: A Five-Center Case-Control Study. Front Oncol 2021, 11. [CrossRef]
- Song, S.; Fan, G.; Li, Q.; Su, Q.; Zhang, X.; Xue, X.; Wang, Z.; Qian, C.; Jin, Z.; Li, B.; et al. IDH2 Contributes to Tumorigenesis and Poor Prognosis by Regulating M6A RNA Methylation in Multiple Myeloma. Oncogene 2021, 40, 5393–5402. [CrossRef]
- Qu, J.; Hou, Y.; Chen, Q.; Chen, J.; Li, Y.; Zhang, E.; Gu, H.; Xu, R.; Liu, Y.; Cao, W.; et al. RNA Demethylase ALKBH5 Promotes Tumorigenesis in Multiple Myeloma via TRAF1-Mediated Activation of NF-ΚB and MAPK Signaling Pathways. Oncogene 2022, 41, 400–413. [CrossRef]
- Cheng, Y.; Xie, W.; Pickering, B.F.; Chu, K.L.; Savino, A.M.; Yang, X.; Luo, H.; Nguyen, D.T.; Mo, S.; Barin, E.; et al. N6-Methyladenosine on MRNA Facilitates a Phase-Separated Nuclear Body That Suppresses Myeloid Leukemic Differentiation. Cancer Cell 2021, 39, 958-972.e8. [CrossRef]
- Huang, Y.; Su, R.; Sheng, Y.; Dong, L.; Dong, Z.; Xu, H.; Ni, T.; Zhang, Z.S.; Zhang, T.; Li, C.; et al. Small-Molecule Targeting of Oncogenic FTO Demethylase in Acute Myeloid Leukemia. Cancer Cell 2019, 35, 677-691.e10. [CrossRef]
- Cao, K.; Du, Y.; Bao, X.; Han, M.; Su, R.; Pang, J.; Liu, S.; Shi, Z.; Yan, F.; Feng, S. Glutathione-Bioimprinted Nanoparticles Targeting of N6-methyladenosine FTO Demethylase as a Strategy against Leukemic Stem Cells. Small 2022, 18. [CrossRef]
- Selberg, S.; Seli, N.; Kankuri, E.; Karelson, M. Rational Design of Novel Anticancer Small-Molecule RNA M6A Demethylase ALKBH5 Inhibitors. ACS Omega 2021, 6, 13310–13320. [CrossRef]
- Wang, Y.-Z.; Li, H.-Y.; Zhang, Y.; Jiang, R.-X.; Xu, J.; Gu, J.; Jiang, Z.; Jiang, Z.-Y.; You, Q.-D.; Guo, X.-K. Discovery of Pyrazolo[1,5-a]Pyrimidine Derivative as a Novel and Selective ALKBH5 Inhibitor for the Treatment of AML. J Med Chem 2023, 66, 15944–15959. [CrossRef]
- Zou, Z.; Dou, X.; Li, Y.; Zhang, Z.; Wang, J.; Gao, B.; Xiao, Y.; Wang, Y.; Zhao, L.; Sun, C.; et al. RNA M5C Oxidation by TET2 Regulates Chromatin State and Leukaemogenesis. Nature 2024. [CrossRef]
- Amort, T.; Rieder, D.; Wille, A.; Khokhlova-Cubberley, D.; Riml, C.; Trixl, L.; Jia, X.Y.; Micura, R.; Lusser, A. Distinct 5-Methylcytosine Profiles in Poly(A) RNA from Mouse Embryonic Stem Cells and Brain. Genome Biol 2017, 18. [CrossRef]
- Bohnsack, K.E.; Höbartner, C.; Bohnsack, M.T. Eukaryotic 5-Methylcytosine (M 5 C) RNA Methyltransferases: Mechanisms, Cellular Functions, and Links to Disease. Genes (Basel) 2019, 10.
- Goll, M.G.; Kirpekar, F.; Maggert, K.A.; Yoder, J.A.; Hsieh, C.-L.; Zhang, X.; Golic, K.G.; Jacobsen, S.E.; Bestor, T.H. Methylation of TRNA Asp by the DNA Methyltransferase Homolog Dnmt2;
- Blanco, S.; Dietmann, S.; Flores, J. V; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant Methylation of t RNA s Links Cellular Stress to Neuro-developmental Disorders . EMBO J 2014, 33, 2020–2039. [CrossRef]
- Hussain, S.; Sajini, A.A.; Blanco, S.; Dietmann, S.; Lombard, P.; Sugimoto, Y.; Paramor, M.; Gleeson, J.G.; Odom, D.T.; Ule, J.; et al. NSun2-Mediated Cytosine-5 Methylation of Vault Noncoding RNA Determines Its Processing into Regulatory Small RNAs. Cell Rep 2013, 4, 255–261. [CrossRef]
- Fu, L.; Guerrero, C.R.; Zhong, N.; Amato, N.J.; Liu, Y.; Liu, S.; Cai, Q.; Ji, D.; Jin, S.G.; Niedernhofer, L.J.; et al. Tet-Mediated Formation of 5-Hydroxymethylcytosine in RNA. J Am Chem Soc 2014, 136, 11582–11585. [CrossRef]
- Moran-Crusio, K.; Reavie, L.; Shih, A.; Abdel-Wahab, O.; Ndiaye-Lobry, D.; Lobry, C.; Figueroa, M.E.; Vasanthakumar, A.; Patel, J.; Zhao, X.; et al. Tet2 Loss Leads to Increased Hematopoietic Stem Cell Self-Renewal and Myeloid Transformation. Cancer Cell 2011, 20, 11–24. [CrossRef]
- Delhommeau, F.; Dupont, S.; Valle, V. Della; James, C.; Trannoy, S.; Massé, A.; Kosmider, O.; Le Couedic, J.-P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in Myeloid Cancers. New England Journal of Medicine 2009, 360, 2289–2301. [CrossRef]
- Ding, Y.; Bajpai, A.K.; Wu, F.; Lu, W.; Xu, L.; Mao, J.; Li, Q.; Pan, Q.; Lu, L.; Wang, X. 5-Methylcytosine RNA Modification Regulators-Based Patterns and Features of Immune Microenvironment in Acute Myeloid Leukemia. Aging 2024. [CrossRef]
- Wang, X.; Zhang, H.; Dong, L.; Zhang, X.; Xue, M.; Ren, L.; Bi, H.; Ghoda, L.Y.; Wunderlich, M.; Mulloy, J.C.; et al. Mitochondrial RNA Methylation By METTL17 Rewires Metabolism and Regulates Retrograde Mitochondrial-Nuclear Communication in AML. Blood 2024, 144, 625–625. [CrossRef]
- Ge, J.; Yu, Y.T. RNA Pseudouridylation: New Insights into an Old Modification. Trends Biochem Sci 2013, 38, 210–218.
- Mcmahon, M.; Contreras, A.; Ruggero, D. Small RNAs with Big Implications: New Insights into H/ACA SnoRNA Function and Their Role in Human Disease. Wiley Interdiscip Rev RNA 2015, 6, 173–189.
- Schwartz, S.; Bernstein, D.A.; Mumbach, M.R.; Jovanovic, M.; Herbst, R.H.; León-Ricardo, B.X.; Engreitz, J.M.; Guttman, M.; Satija, R.; Lander, E.S.; et al. Transcriptome-Wide Mapping Reveals Widespread Dynamic-Regulated Pseudouridylation of NcRNA and MRNA. Cell 2014, 159, 148–162. [CrossRef]
- Grosjean, H.; Sprinzl, M.; Steinberg, S. Posttranscriptiona||y Modified Nucleosides in Transfer RNA: Their Locations and Frequencies; 1995; Vol. 77;.
- Motorin, Y.; Helm, M. TRNA Stabilization by Modified Nucleotides. Biochemistry 2010, 49, 4934–4944.
- Liang, X.H.; Liu, Q.; Fournier, M.J. Loss of RRNA Modifications in the Decoding Center of the Ribosome Impairs Translation and Strongly Delays Pre-RRNA Processing. RNA 2009, 15, 1716–1728. [CrossRef]
- Piekna-Przybylska, D.; Przybylski, P.; Baudin-Baillieu, A.; Rousset, J.P.; Fournier, M.J. Ribosome Performance Is Enhanced by a Rich Cluster of Pseudouridines in the A-Site Finger Region of the Large Subunit. Journal of Biological Chemistry 2008, 283, 26026–26036. [CrossRef]
- Yu, A.T.; Ge, J.; Yu, Y.T. Pseudouridines in Spliceosomal SnRNAs. Protein Cell 2011, 2, 712–725.
- Su, H.; Hu, J.; Huang, L.; Yang, Y.; Thenoz, M.; Kuchmiy, A.; Hu, Y.; Li, P.; Feng, H.; Zhou, Y.; et al. SHQ1 Regulation of RNA Splicing Is Required for T-Lymphoblastic Leukemia Cell Survival. Nat Commun 2018, 9. [CrossRef]
- Karikó, K.; Muramatsu, H.; Keller, J.M.; Weissman, D. Increased Erythropoiesis in Mice Injected with Submicrogram Quantities of Pseudouridine-Containing MRNA Encoding Erythropoietin. Molecular Therapy 2012, 20, 948–953. [CrossRef]
- Kurimoto, R.; Chiba, T.; Ito, Y.; Matsushima, T.; Yano, Y.; Miyata, K.; Yashiro, Y.; Suzuki, T.; Tomita, K.; Asahara, H. The TRNA Pseudouridine Synthase TruB1 Regulates the Maturation of Let-7 MiRNA. EMBO J 2020, 39. [CrossRef]
- Herridge, R.P.; Dolata, J.; Migliori, V.; de Santis Alves, C.; Borges, F.; Schorn, A.J.; van Ex, F.; Lin, A.; Bajczyk, M.; Parent, J.S.; et al. Pseudouridine Guides Germline Small RNA Transport and Epigenetic Inheritance. Nat Struct Mol Biol 2024. [CrossRef]
- Wang, B.; Shi, D.; Yang, S.; Lian, Y.; Li, H.; Cao, M.; He, Y.; Zhang, L.; Qiu, C.; Liu, T.; et al. Mitochondrial TRNA Pseudouridylation Governs Erythropoiesis. Blood 2024, 144, 657–671. [CrossRef]
- Guzzi, N.; Muthukumar, S.; Cieśla, M.; Todisco, G.; Ngoc, P.C.T.; Madej, M.; Munita, R.; Fazio, S.; Ekström, S.; Mortera-Blanco, T.; et al. Pseudouridine-Modified TRNA Fragments Repress Aberrant Protein Synthesis and Predict Leukaemic Progression in Myelodysplastic Syndrome. Nat Cell Biol 2022, 24, 299–306. [CrossRef]
- Bellodi, C.; McMahon, M.; Contreras, A.; Juliano, D.; Kopmar, N.; Nakamura, T.; Maltby, D.; Burlingame, A.; Savage, S.A.; Shimamura, A.; et al. H/ACA Small RNA Dysfunctions in Disease Reveal Key Roles for Noncoding RNA Modifications in Hematopoietic Stem Cell Differentiation. Cell Rep 2013, 3, 1493–1502. [CrossRef]
- Iwakawa, H.; Tomari, Y. Life of RISC: Formation, Action, and Degradation of RNA-Induced Silencing Complex. Mol Cell 2022, 82, 30–43. [CrossRef]
- Castanotto, D.; Lingeman, R.; Riggs, A.D.; Rossi, J.J. CRM1 Mediates Nuclear-Cytoplasmic Shuttling of Mature MicroRNAs. Proceedings of the National Academy of Sciences 2009, 106, 21655–21659. [CrossRef]
- Wei, Y.; Li, L.; Wang, D.; Zhang, C.-Y.; Zen, K. Importin 8 Regulates the Transport of Mature MicroRNAs into the Cell Nucleus. Journal of Biological Chemistry 2014, 289, 10270–10275. [CrossRef]
- Hwang, H.-W.; Wentzel, E.A.; Mendell, J.T. A Hexanucleotide Element Directs MicroRNA Nuclear Import. Science (1979) 2007, 315, 97–100. [CrossRef]
- Turunen, T.A.; Roberts, T.C.; Laitinen, P.; Väänänen, M.-A.; Korhonen, P.; Malm, T.; Ylä-Herttuala, S.; Turunen, M.P. Changes in Nuclear and Cytoplasmic MicroRNA Distribution in Response to Hypoxic Stress. Sci Rep 2019, 9, 10332. [CrossRef]
- Zisoulis, D.G.; Kai, Z.S.; Chang, R.K.; Pasquinelli, A.E. Autoregulation of MicroRNA Biogenesis by Let-7 and Argonaute. Nature 2012, 486, 541–544. [CrossRef]
- Nishi, K.; Nishi, A.; Nagasawa, T.; Ui-Tei, K. Human TNRC6A Is an Argonaute-Navigator Protein for MicroRNA-Mediated Gene Silencing in the Nucleus. RNA 2013, 19, 17–35. [CrossRef]
- Jeffries, C.D.; Fried, H.M.; Perkins, D.O. Nuclear and Cytoplasmic Localization of Neural Stem Cell MicroRNAs. RNA 2011, 17, 675–686. [CrossRef]
- Zardo, G.; Ciolfi, A.; Vian, L.; Starnes, L.M.; Billi, M.; Racanicchi, S.; Maresca, C.; Fazi, F.; Travaglini, L.; Noguera, N.; et al. Polycombs and MicroRNA-223 Regulate Human Granulopoiesis by Transcriptional Control of Target Gene Expression. Blood 2012, 119, 4034–4046. [CrossRef]
- Fazi, F.; Rosa, A.; Fatica, A.; Gelmetti, V.; De Marchis, M.L.; Nervi, C.; Bozzoni, I. A Minicircuitry Comprised of MicroRNA-223 and Transcription Factors NFI-A and C/EBPα Regulates Human Granulopoiesis. Cell 2005, 123, 819–831. [CrossRef]
- Pulikkan, J.A.; Dengler, V.; Peramangalam, P.S.; Peer Zada, A.A.; Müller-Tidow, C.; Bohlander, S.K.; Tenen, D.G.; Behre, G. Cell-Cycle Regulator E2F1 and MicroRNA-223 Comprise an Autoregulatory Negative Feedback Loop in Acute Myeloid Leukemia. Blood 2010, 115, 1768–1778. [CrossRef]
- Rasko, J.E.J.; Wong, J.J.-L. Nuclear MicroRNAs in Normal Hemopoiesis and Cancer. J Hematol Oncol 2017, 10, 8. [CrossRef]
- Hegde, V.L.; Tomar, S.; Jackson, A.; Rao, R.; Yang, X.; Singh, U.P.; Singh, N.P.; Nagarkatti, P.S.; Nagarkatti, M. Distinct MicroRNA Expression Profile and Targeted Biological Pathways in Functional Myeloid-Derived Suppressor Cells Induced by Δ9-Tetrahydrocannabinol in Vivo. Journal of Biological Chemistry 2013, 288, 36810–36826. [CrossRef]
- Porse, B.T.; Bryder, D.; Theilgaard-Mönch, K.; Hasemann, M.S.; Anderson, K.; Damgaard, I.; Jacobsen, S.E.W.; Nerlov, C. Loss of C/EBPα Cell Cycle Control Increases Myeloid Progenitor Proliferation and Transforms the Neutrophil Granulocyte Lineage. Journal of Experimental Medicine 2005, 202, 85–96. [CrossRef]
- Bauer, M.; Vaxevanis, C.; Heimer, N.; Al-Ali, H.K.; Jaekel, N.; Bachmann, M.; Wickenhauser, C.; Seliger, B. Expression, Regulation and Function of MicroRNA as Important Players in the Transition of MDS to Secondary AML and Their Cross Talk to RNA-Binding Proteins. Int J Mol Sci 2020, 21, 7140. [CrossRef]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of Genetic Lesions in 944 Patients with Myelodysplastic Syndromes. Leukemia 2014, 28, 241–247. [CrossRef]
- Papaemmanuil, E.; Cazzola, M.; Boultwood, J.; Malcovati, L.; Vyas, P.; Bowen, D.; Pellagatti, A.; Wainscoat, J.S.; Hellstrom-Lindberg, E.; Gambacorti-Passerini, C.; et al. Somatic SF3B1 Mutation in Myelodysplasia with Ring Sideroblasts . New England Journal of Medicine 2011, 365, 1384–1395. [CrossRef]
- Berezikov, E.; Chung, W.-J.; Willis, J.; Cuppen, E.; Lai, E.C. Mammalian Mirtron Genes. Mol Cell 2007, 28, 328–336. [CrossRef]
- Aslan, D.; Garde, C.; Katrine Nygaard, M.; Søgaard Helbo, A.; Dimopoulos, K.; Werner Hansen, J.; Tang Severinsen, M.; Bach Treppendahl, M.; Dissing Sjø, L.; Grønbaek, K.; et al. Tumor Suppressor MicroRNAs Are Downregulated in Myelodysplastic Syndrome with Spliceosome Mutations; 2016;
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The Human Mitochondrial Transcriptome. Cell 2011, 146, 645–658. [CrossRef]
- Luo, L.; An, X.; Xiao, Y.; Sun, X.; Li, S.; Wang, Y.; Sun, W.; Yu, D. Mitochondrial-Related MicroRNAs and Their Roles in Cellular Senescence. Front Physiol 2024, 14. [CrossRef]
- Han, H.; Fan, G.; Song, S.; Jiang, Y.; Qian, C.; Zhang, W.; Su, Q.; Xue, X.; Zhuang, W.; Li, B. PiRNA-30473 Contributes to Tumorigenesis and Poor Prognosis by Regulating M6A RNA Methylation in DLBCL. Blood 2021, 137, 1603–1614. [CrossRef]
- Li, J.; Qu, L.; Sang, L.; Wu, X.; Jiang, A.; Liu, J.; Lin, A. Micropeptides Translated from Putative Long Noncoding RNAs. Acta Biochim Biophys Sin (Shanghai) 2022. [CrossRef]
- Qiu, Y.; Xu, M.; Huang, S. Long Noncoding RNAs: Emerging Regulators of Normal and Malignant Hematopoiesis. Blood 2021, 138, 2327–2336. [CrossRef]
- van Heesch, S.; van Iterson, M.; Jacobi, J.; Boymans, S.; Essers, P.B.; de Bruijn, E.; Hao, W.; MacInnes, A.W.; Cuppen, E.; Simonis, M. Extensive Localization of Long Noncoding RNAs to the Cytosol and Mono- and Polyribosomal Complexes. Genome Biol 2014, 15, R6. [CrossRef]
- Djebali, S.; Davis, C.A.; Merkel, A.; Dobin, A.; Lassmann, T.; Mortazavi, A.; Tanzer, A.; Lagarde, J.; Lin, W.; Schlesinger, F.; et al. Landscape of Transcription in Human Cells. Nature 2012, 489, 101–108. [CrossRef]
- Zhu, G.; Luo, H.; Feng, Y.; Guryanova, O.A.; Xu, J.; Chen, S.; Lai, Q.; Sharma, A.; Xu, B.; Zhao, Z.; et al. HOXBLINC Long Non-Coding RNA Activation Promotes Leukemogenesis in NPM1-Mutant Acute Myeloid Leukemia. Nat Commun 2021, 12. [CrossRef]
- Díaz-Beyá, M.; Brunet, S.; Nomdedéu, J.; Pratcorona, M.; Cordeiro, A.; Gallardo, D.; Escoda, L.; Tormo, M.; Heras, I.; Ribera, J.M.; et al. The LincRNA HOTAIRM1, Located in the HOXA Genomic Region, Is Expressed in Acute Myeloid Leukemia, Impacts Prognosis in Patients in the Intermediate-Risk Cytogenetic Category, and Is Associated with a Distinctive MicroRNA Signature. Oncotarget 2015, 6, 31613–31627. [CrossRef]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A Long Noncoding RNA Maintains Active Chromatin to Coordinate Homeotic Gene Expression. Nature 2011, 472, 120–126. [CrossRef]
- Gourvest, M.; De Clara, E.; Wu, H.-C.; Touriol, C.; Meggetto, F.; De Thé, H.; Pyronnet, S.; Brousset, P.; Bousquet, M. A Novel Leukemic Route of Mutant NPM1 through Nuclear Import of the Overexpressed Long Noncoding RNA LONA. Leukemia 2021, 35, 2784–2798. [CrossRef]
- Li, J.; Li, Z.; Bai, X.; Chen, X.; Wang, M.; Wu, Y.; Wu, H. LncRNA UCA1 Promotes the Progression of AML by Upregulating the Expression of CXCR4 and CYP1B1 by Affecting the Stability of METTL14. J Oncol 2022, 2022, 1–13. [CrossRef]
- Jin, J.; Fu, L.; Hong, P.; Feng, W. MALAT-1 Regulates the AML Progression by Promoting the M6A Modification of ZEB1. Acta Biochim Pol 2023. [CrossRef]
- Schmitt, M.E.; Clayton, D.A. Nuclear RNase MRP Is Required for Correct Processing of Pre-5.8S RRNA in Saccharomyces Cerevisiae. Mol Cell Biol 1993, 13, 7935–7941. [CrossRef]
- Maida, Y.; Yasukawa, M.; Furuuchi, M.; Lassmann, T.; Possemato, R.; Okamoto, N.; Kasim, V.; Hayashizaki, Y.; Hahn, W.C.; Masutomi, K. An RNA-Dependent RNA Polymerase Formed by TERT and the RMRP RNA. Nature 2009, 461, 230–235. [CrossRef]
- Noh, J.H.; Kim, K.M.; Abdelmohsen, K.; Yoon, J.-H.; Panda, A.C.; Munk, R.; Kim, J.; Curtis, J.; Moad, C.A.; Wohler, C.M.; et al. HuR and GRSF1 Modulate the Nuclear Export and Mitochondrial Localization of the LncRNA RMRP. Genes Dev 2016, 30, 1224–1239. [CrossRef]
- Yang, F.; Zhang, H.; Mei, Y.; Wu, M. Reciprocal Regulation of HIF-1α and LincRNA-P21 Modulates the Warburg Effect. Mol Cell 2014, 53, 88–100. [CrossRef]
- Zheng, P.; Xiong, Q.; Wu, Y.; Chen, Y.; Chen, Z.; Fleming, J.; Gao, D.; Bi, L.; Ge, F. Quantitative Proteomics Analysis Reveals Novel Insights into Mechanisms of Action of Long Noncoding RNA Hox Transcript Antisense Intergenic RNA (HOTAIR) in HeLa Cells*. Molecular & Cellular Proteomics 2015, 14, 1447–1463. [CrossRef]
- Zhao, Y.; Sun, L.; Wang, R.R.; Hu, J.-F.; Cui, J. The Effects of Mitochondria-Associated Long Noncoding RNAs in Cancer Mitochondria: New Players in an Old Arena. Crit Rev Oncol Hematol 2018, 131, 76–82. [CrossRef]
- Rackham, O.; Shearwood, A.-M.J.; Mercer, T.R.; Davies, S.M.K.; Mattick, J.S.; Filipovska, A. Long Noncoding RNAs Are Generated from the Mitochondrial Genome and Regulated by Nuclear-Encoded Proteins. RNA 2011, 17, 2085–2093. [CrossRef]
- Landerer, E.; Villegas, J.; Burzio, V.A.; Oliveira, L.; Villota, C.; Lopez, C.; Restovic, F.; Martinez, R.; Castillo, O.; Burzio, L.O. Nuclear Localization of the Mitochondrial NcRNAs in Normal and Cancer Cells. Cellular Oncology 2011, 34, 297–305. [CrossRef]
- Bianchessi, V.; Badi, I.; Bertolotti, M.; Nigro, P.; D’Alessandra, Y.; Capogrossi, M.C.; Zanobini, M.; Pompilio, G.; Raucci, A.; Lauri, A. The Mitochondrial LncRNA ASncmtRNA-2 Is Induced in Aging and Replicative Senescence in Endothelial Cells. J Mol Cell Cardiol 2015, 81, 62–70. [CrossRef]
- Wu, Z.; Sun, H.; Wang, C.; Liu, W.; Liu, M.; Zhu, Y.; Xu, W.; Jin, H.; Li, J. Mitochondrial Genome-Derived CircRNA Mc-COX2 Functions as an Oncogene in Chronic Lymphocytic Leukemia. Mol Ther Nucleic Acids 2020, 20, 801–811. [CrossRef]
- Burzio, V.A.; Villota, C.; Villegas, J.; Landerer, E.; Boccardo, E.; Villa, L.L.; Martínez, R.; Lopez, C.; Gaete, F.; Toro, V.; et al. Expression of a Family of Noncoding Mitochondrial RNAs Distinguishes Normal from Cancer Cells. Proceedings of the National Academy of Sciences 2009, 106, 9430–9434. [CrossRef]
- Querfeld, C.; Foss, F.M.; Kim, Y.H.; Pinter-Brown, L.; William, B.M.; Porcu, P.; Pacheco, T.; Haverkos, B.M.; DeSimone, J.; Guitart, J.; et al. Phase 1 Trial of Cobomarsen, an Inhibitor of Mir-155, in Cutaneous T Cell Lymphoma. Blood 2018, 132, 2903–2903. [CrossRef]
Figure 1.
Hematologic malignancies subtypes with defective m6A and 5mC RNA modification pathways. A. In B-cell lymphomas m6A demethylase ALKBH5, lncRNA-treRNA1 and the RNA helicase DDX46, form a functional complex in the cell nucleus, affecting the removal of m6A modifications from key BCR-related transcripts with severe consequences to the B-cell functionality [44]. Also, ALKBH5 is frequently enhanced in human DLBCL and demethylates BCR signature transcripts, guiding lymphomagenesis [45]. B. In AML ALKBH5 and FTO are αKG-dependent enzymes and are inhibited by the high accumulation of D-2-HG an alternative metabolite produced by IDH1/2 mutants, leading to a m6A hypermethylation profile [49,50]. MYC transcriptionally activates the IDH2 promoter along with various other enzymes’ promoters that positively influence anapleorotic mitochondrial metabolism, glycolysis and the glucose transport, leading to increased αKG cellular pool, suggesting its critical role in the nuclear-mitochondrion crosstalk [53]. Increased levels of the m6A ‘reader’ YTHDC1 in AML, leads to the formation of nuclear speckles, protecting m6A-mRNAs from degradation. Protection and stabilization of m6A transcripts accelerates cells’ survival and proliferation [59]. C. In AML with mutated TET2, oxidation of m5C especially in RNAs derived from retrotransposons fails. Remaining m5C marks in RNA recruits MBD6 and deubiquitinases that remove mono-ubiquitination from H2AK119ub, leading to open chromatin status, increased transcription of retrotransposons and genomic instability, all hallmarks of cells’ cancer state [64].
Figure 1.
Hematologic malignancies subtypes with defective m6A and 5mC RNA modification pathways. A. In B-cell lymphomas m6A demethylase ALKBH5, lncRNA-treRNA1 and the RNA helicase DDX46, form a functional complex in the cell nucleus, affecting the removal of m6A modifications from key BCR-related transcripts with severe consequences to the B-cell functionality [44]. Also, ALKBH5 is frequently enhanced in human DLBCL and demethylates BCR signature transcripts, guiding lymphomagenesis [45]. B. In AML ALKBH5 and FTO are αKG-dependent enzymes and are inhibited by the high accumulation of D-2-HG an alternative metabolite produced by IDH1/2 mutants, leading to a m6A hypermethylation profile [49,50]. MYC transcriptionally activates the IDH2 promoter along with various other enzymes’ promoters that positively influence anapleorotic mitochondrial metabolism, glycolysis and the glucose transport, leading to increased αKG cellular pool, suggesting its critical role in the nuclear-mitochondrion crosstalk [53]. Increased levels of the m6A ‘reader’ YTHDC1 in AML, leads to the formation of nuclear speckles, protecting m6A-mRNAs from degradation. Protection and stabilization of m6A transcripts accelerates cells’ survival and proliferation [59]. C. In AML with mutated TET2, oxidation of m5C especially in RNAs derived from retrotransposons fails. Remaining m5C marks in RNA recruits MBD6 and deubiquitinases that remove mono-ubiquitination from H2AK119ub, leading to open chromatin status, increased transcription of retrotransposons and genomic instability, all hallmarks of cells’ cancer state [64].
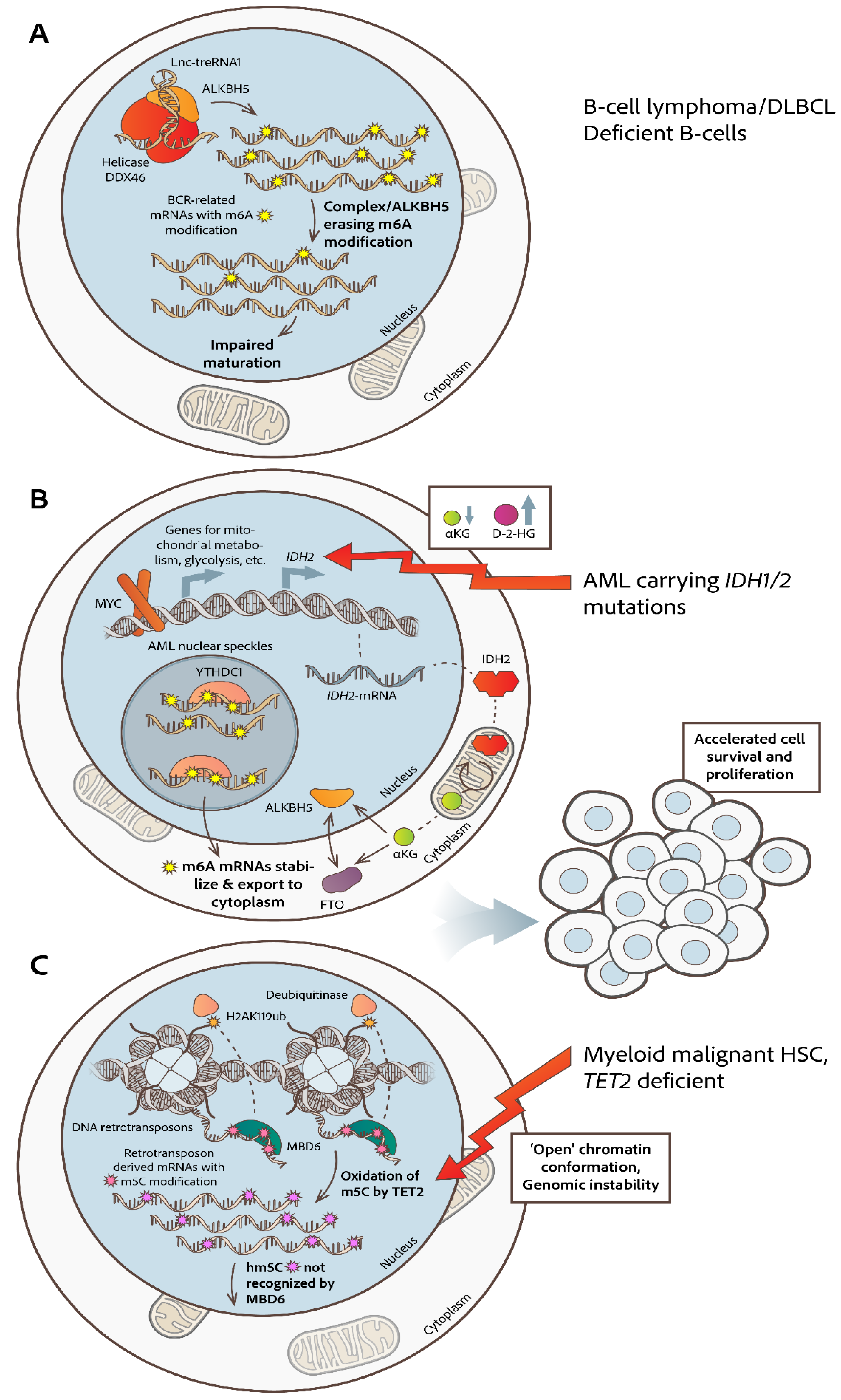
Figure 2.
Implicated lncRNAs in malignancies of myeloid origin. A. In AML carrying mutated NPM1, there is upregulation of the HOXA/B loci (on chromosomes 7 and 17 respectively) and of lncRNA-HOTAIRM1, lncRNA-HOTTIP and lncRNA-HOXBLINC (highlighted with light green color), which are encoded from the same loci, but are reversely transcribed [9]. Altered lncRNAs’ expression profiles are implicated in several dysregulated pathways. B. (Left) NPM1 mutated AML produces specific leukemia stem cell signatures by remodeling 2′-O-methylation of rRNAs, guided by snoRNAs and executed by fibrillarin (FBL). (Right) LncRNA-UCA1 contributes to AML progression, by upregulating the expression of CXCR4 and CYP1B1 and by promoting stabilization of the m6A methyltransferase METTL14. This interaction affects m6A levels and influences localization and function of METTL14 in AML cells [120]. Also, lncRNA-MALAT-1 binds to METTL14, promoting m6A modification of ZEB1-mRNA and influencing the localization of METTL14 intranuclearly [121].
Figure 2.
Implicated lncRNAs in malignancies of myeloid origin. A. In AML carrying mutated NPM1, there is upregulation of the HOXA/B loci (on chromosomes 7 and 17 respectively) and of lncRNA-HOTAIRM1, lncRNA-HOTTIP and lncRNA-HOXBLINC (highlighted with light green color), which are encoded from the same loci, but are reversely transcribed [9]. Altered lncRNAs’ expression profiles are implicated in several dysregulated pathways. B. (Left) NPM1 mutated AML produces specific leukemia stem cell signatures by remodeling 2′-O-methylation of rRNAs, guided by snoRNAs and executed by fibrillarin (FBL). (Right) LncRNA-UCA1 contributes to AML progression, by upregulating the expression of CXCR4 and CYP1B1 and by promoting stabilization of the m6A methyltransferase METTL14. This interaction affects m6A levels and influences localization and function of METTL14 in AML cells [120]. Also, lncRNA-MALAT-1 binds to METTL14, promoting m6A modification of ZEB1-mRNA and influencing the localization of METTL14 intranuclearly [121].
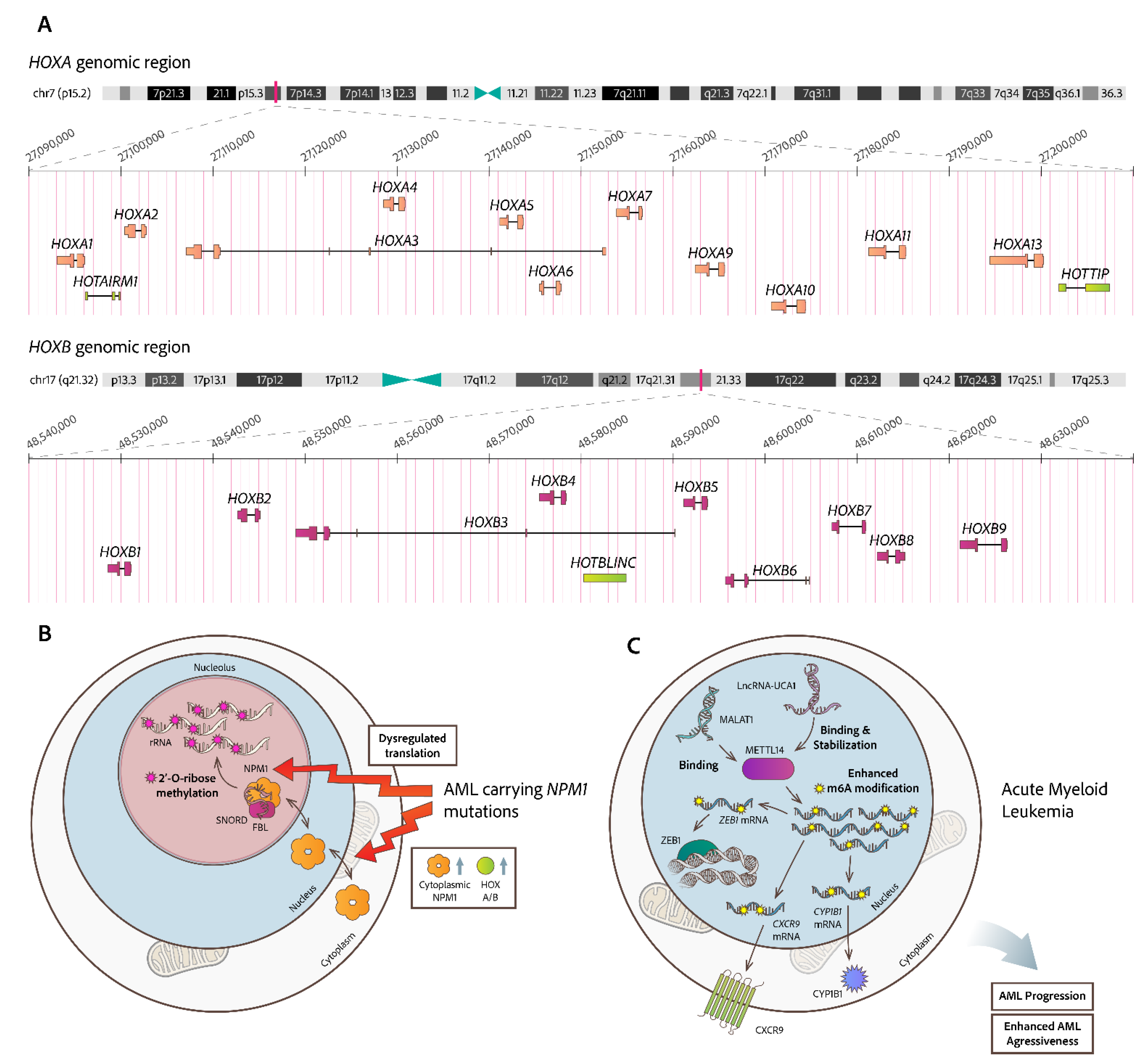

Table 1.
FTO/ALKBH5 inhibitors as therapeutic options for hematologic malignancies.
| Inhibitor | target | disease | REF |
|---|---|---|---|
| R-2 hydroxyglutarate (FTO inhibitor) | inhibits aerobic glycolysis without affecting HSCs | sensitive leukemia cells | [50] |
| FB23 and FB23-2 (FTO inhibitor) |
Directly bind to FTO and inhibit he progression of human AML cells | AML | [60] |
| glutathione (GSH) bioimprinted nanocomposite material, GNPIPP12MA, loaded with FTO inhibitors | GNPIPP12MA targets the FTO/m6A pathway, synergistically enhancing anti-leukemia effects by depleting GSH | leukemia stem cells | [61] |
| Zantrene (FTO inhibitor) |
Targeted inhibitor of FTO | AML | Phase I clinical trial (NCT05456269) |
| 2-(2-hydroxyethylsulfanyl)acetic acid (RD3) and 4-[(methyl)amino]-3,6-dioxo (RD6) (ALKBH5 inhibitors) | Decreased cell viability at low concentrations | Leukemia cell lines | [62] |
| Pyrazolo[1,5-a]pyrimidine (DO-2728a) (ALKBH5 inhibitor) | Enhances m6A modification abundance and impedes cell cycle progression | AML | [63] |
Table 2.
Intracellular miRs contributing to MDS and AML hematologic neoplasias. The cell type source is bone marrow mononuclear cells, BMMCs. MiR level comparisons are against physiological controls or samples from patients with aplastic anemia (AA).
Table 2.
Intracellular miRs contributing to MDS and AML hematologic neoplasias. The cell type source is bone marrow mononuclear cells, BMMCs. MiR level comparisons are against physiological controls or samples from patients with aplastic anemia (AA).
| MiRNA | Regulation in MDS/AML vs healthy or AA (aplastic anemia) |
|---|---|
| hsa-miR-130a-3p | Significantly higher in de novo MDS vs AA |
| hsa-miR-221-3p | Significantly higher in de novo MDS vs AA |
| hsa-miR-126-3p | Significantly higher in de novo MDS vs AA |
| hsa-miR-27b-3p | Significantly higher in de novo MDS vs AA |
| hsa-miR-196b-5p | Significantly higher in de novo MDS vs AA |
| hsa-let-7e-5p | Upregulated in de novo MDS vs AA |
| hsa-miR-181c-5p | Associated with progression to sAML |
| hsa-miR-155 | Downregulated in MDS (prognostic value) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



