
Submitted:
15 July 2025
Posted:
16 July 2025
You are already at the latest version
Abstract
Heat shock proteins (HSPs) are highly conserved molecular chaperones that play a key role in maintaining protein homeostasis, or proteostasis, especially under stressful envi-ronmental conditions such as hyperthermia, hypoxia, or the presence of reactive oxygen species. In pancreatic cancer, the expression of many HSP isoforms is dysregulated, con-tributing to the activation of mechanisms that promote tumor development, including proliferation, invasion, angiogenesis, treatment resistance, and cancer cachexia syndrome. HSPs are significant diagnostic and prognostic biomarkers. Some of them, such as HSP27, HSP70, and HSP90, have been shown to correlate with treatment response and patient survival. Others, including HSPA2 and HSPB6, may indicate an increased risk of disease recurrence. These proteins also represent promising therapeutic targets. Preclinical and clinical studies suggest that inhibiting HSP activity and associated signaling pathways may inhibit tumor growth and increase treatment efficacy. These therapeutic effects include inducing apoptosis, autophagy, and ferroptosis, as well as sensitizing cancer cells to chemotherapy and immunotherapy. This article summarizes the current knowledge about the role of HSPs in pancreatic cancer biology, their significance as biomarkers, and their potential therapeutic applications in treating pancreatic ductal adenocarcinoma (PDAC). Most studies conducted so far have been preclinical, and due to the promising results, further clinical investigation is warranted.
Keywords:
heat shock proteins
; pancreatic cancer
; clinical value
1. Introduction
HSPs are a large family of conserved proteins found in both prokaryotic and eukaryotic cells [1]. Their key functions include helping to fold newly synthesized polypeptides, refolding denatured or unstable proteins, facilitating intracellular transport, and preventing the formation of toxic protein aggregates [2]. This is why they are referred to as "molecular chaperones" [3]. The functional diversity of heat shock proteins arises from their capacity to perform multiple roles, including those of foldases, holdases, sequestrases, aggregases, and disaggregases [4].
Under normal conditions, the level of HSP synthesis is similar to that of other proteins. However, in response to cellular stress, such as elevated temperature, hypoxia, infection, oxidative stress, DNA damage, or the accumulation of improperly folded proteins, there is a significant increase in HSP expression [1,5]. This mechanism depends on the activation of transcription factors from the HSF family. After oligomerization, these factors bind to heat shock elements (HSE) located within the promoter regions of HSP genes [3]. Dysregulation of HSP expression occurs in many cancers. These proteins have been shown to regulate the proliferation, apoptosis, invasion, and metastasis of cancer cells. They also play a role in developing resistance to chemotherapy and radiotherapy [6].
HSPs are classified by molecular weight into six main families: HSP100, HSP90, HSP70, HSP60, HSP40, and HSP20, which is the low molecular weight HSP [1,3]. These groups each perform specific functions that affect cellular homeostasis and tumor transformation processes differently.
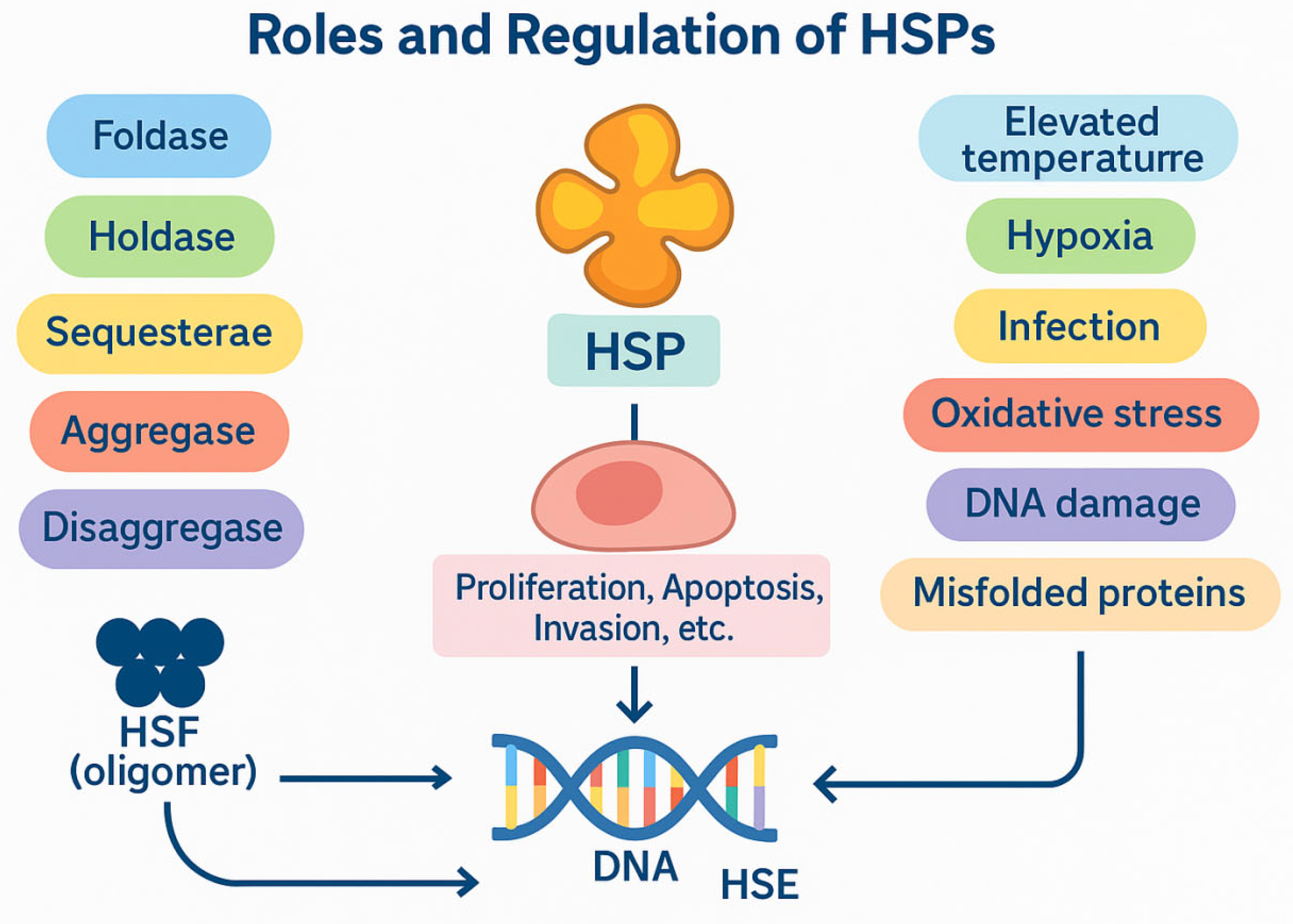
Figure 1.
Roles and regulation of Heat Shock Proteins (HSPs).
2. Aims
This study aims to critically analyze the role of heat shock proteins in diagnosing, predicting, and treating pancreatic cancer, with a focus on their potential use as biomarkers and therapeutic targets. The current state of knowledge regarding the significance of individual HSP families in the pathogenesis of pancreatic ductal cancer was discussed. Experimental data and clinical observations were considered to highlight the molecular mechanisms of HSP action and their role in disease progression, treatment response, and therapeutic resistance. We also analyzed therapeutic strategies based on HSP inhibition and their potential application in cancer treatment.
3. The Biological and Prognostic Significance of HSP Families in Pancreatic Ductal Adenocarcinoma (PDAC)
3.1. Low-Molecular-Weight Heat Shock Protein (lmHSPs)
Low-molecular-weight heat shock proteins (lmHSPs), including HSP27, HSPB2, HSPB6, and αA-crystallin, play a complex role in the biology of pancreatic cancer by exhibiting both pro- and anti-tumorigenic effects. Studies conducted on pancreatic cancer cell lines and cancer-associated fibroblasts (CAFs) have shown that increased HSPB6 expression in CAFs may contribute to the modulation of the tumor stroma. Moreover, analysis of data from the Clinical Proteomic Tumor Analysis Consortium (CPTAC) revealed that high HSPB6 expression correlates with longer overall survival in patients with PDAC, indicating its potential as a prognostic marker [7].
In pancreatic cancer cell line models, HSP27 has been shown to regulate the expression of Snail, E-cadherin, and ERCC1, thereby potentially contributing to the development of resistance to gemcitabine. Lower levels of HSP27 are associated with poorer prognosis, while elevated expression correlates with improved responses to chemotherapy, suggesting its role as a predictive biomarker of treatment efficacy [6,8].
Deng et al. (2010) demonstrated that the small heat shock protein αA-crystallin is physiologically expressed in normal pancreatic tissue and acts as a negative regulator of pancreatic tumorigenesis. Their study revealed a significant downregulation of αA-crystallin in pancreatic tumor tissue compared to adjacent non-neoplastic tissue. Functional assays further confirmed that overexpression of αA-crystallin in pancreatic cancer cells reduced tumorigenic potential, whereas its silencing enhanced tumorigenicity [9].
3.2. HSP40 (DnaJ Family)
HSPs from the HSP40 family, also referred to as DnaJ proteins, play a crucial role in regulating the function of HSP70 chaperones and are increasingly recognized as contributors to PDAC progression by modulating cancer cell survival, metabolic reprogramming, and apoptosis.
Liu et al. demonstrated that DnaJB11, a co-chaperone of HSPA5 (BiP, Grp78), is overexpressed in pancreatic cancer cells. Their study, which was primarily based on in vitro assays and in vivo mouse xenograft models implanted with human PDAC cells, showed that increased DnaJB11 levels promote tumor growth and inhibit apoptosis, suggesting a tumor-supportive role for this co-chaperone in the PDAC microenvironment [10].
Similarly, Roth et al. provided further evidence for the involvement of HSP40 family members in PDAC pathogenesis. Their research, conducted on established PDAC cell lines, revealed that elevated expression of DnaJA1 enhances the Warburg effect, upregulates anti-apoptotic Bcl-2 protein levels, and reduces apoptotic signaling, thereby promoting tumor cell survival and invasiveness [11]. These findings, although limited to preclinical models, highlight the potential of HSP40 proteins as modulators of cancer metabolism and apoptosis in pancreatic cancer.
3.3. HSP60 (Chaperonins)
HSP60, a mitochondrial chaperonin, plays a multifaceted role in PDAC by regulating protein homeostasis, cellular metabolism, and apoptotic signaling. Elevated HSP60 expression has been observed in PDAC tissues and is associated with enhanced tumor cell proliferation and resistance to cell death. Mechanistically, HSP60 supports mitochondrial integrity and oxidative phosphorylation, contributing to cancer cell survival under metabolic stress. Additionally, HSP60 may inhibit apoptotic pathways by stabilizing anti-apoptotic proteins and preventing cytochrome c release, thereby promoting PDAC progression and chemoresistance [12].
3.4. HSP70
HSP70 plays a critical role in the progression of pancreatic cancer, affecting both tumor biology and the systemic condition of patients. In a study conducted on pancreatic cancer cell lines, Liumei et al. demonstrated that elevated HSP70 expression activates the NF-κB signaling pathway and promotes epithelial–mesenchymal transition (EMT), thereby enhancing the proliferation, migration, and invasiveness of cancer cells [13]. In contrast, Zhai et al., using tumor samples derived from pancreatic cancer patients, confirmed that increased expression of the HSPA2 gene—also present in stromal components—correlates with a more aggressive clinical course [14]. HSP70, which is released in extracellular vesicles by PDAC cells, has been shown to activate the p38β MAPK catabolic cascade, contributing to muscle wasting and the development of cancer cachexia. This mechanism was demonstrated in study [15] using patient-derived pancreatic cancer cells, while its systemic effects were further confirmed in vivo in a murine model of cancer cachexia [16]. Notably, HSP70 has been identified as an independent prognostic factor for both overall and progression-free survival. Studies by Xiong et al. indicate that analyzing HSP70 and VEGF levels simultaneously may improve the accuracy of predicting responses to chemotherapy and radiotherapy; a decrease in these levels after treatment was associated with a more favorable prognosis [13]. These findings were obtained in clinical studies conducted in patients.
3.5. HSP90
HSP90 represents a family of proteins comprising isoforms localized in distinct cellular compartments, including GRP94 in the endoplasmic reticulum, TRAP1 in mitochondria, and HSP90α and HSP90β in the cytosol [20,21]. Studies using murine pancreatic cancer cell lines have shown a significant upregulation of HSP90 expression in PDAC cells [17].
Clinical studies have identified HSP90 as a potential prognostic biomarker in humans. Elevated HSP90 levels were associated with a threefold increased risk of mortality, particularly in patients with a history of acute pancreatitis, independently of other clinical variables [18].
HSP90 also holds promise in imaging diagnostics. Its expression can be visualized using positron emission tomography (PET) tracers such as ⁶⁴Cu-Di-San A1 and ¹⁸F-PEGylated San A. However, these tracers may yield false-positive signals in areas of inflammation. To improve specificity, a novel PET probe, ¹⁸F-NOTA-Dimer-San A, was developed and validated in murine models, allowing for precise detection of HSP90 expression in malignant tissues while distinguishing it from inflammatory lesions [19,20,21].
A summary of the role of HSPs in pathogenesis and biology in PDAC is presented in Table 1.
4. The Role of HSP in Treating Pancreatic Cancer
HSPs, including HSP27, HSP47, HSP60, HSP70, and HSP90, are essential molecular chaperones involved in the maintenance of proteostasis under physiological and pathological conditions. In PDAC, their overexpression is closely associated with aggressive tumor behavior, therapeutic resistance, and immune evasion. Elevated levels of these chaperones correlate with advanced disease stages, poor prognosis, and decreased responsiveness to standard therapies, thus highlighting their potential as actionable molecular targets. The therapeutic efficacy of HSP inhibition appears to be dependent on the tumor’s specific genetic background. For example, mortalin (HSPA9), a mitochondrial HSP70 family member, is significantly upregulated in KRAS-mutated PDAC. Silencing mortalin in such contexts induces apoptosis and increases mitochondrial membrane permeability. Preclinical investigations have demonstrated that JG-231, a hydrophilic derivative of the HSP70 inhibitor MKT-077, effectively suppresses tumor growth in PDAC models harboring the KRASG12C mutation, suggesting a promising therapeutic approach tailored to molecular tumor profiles [39].
4.1. HSP27: Marker of Resistance and Therapeutic Target
HSP27 contributes to PDAC progression and metastasis via activation of the β-catenin/MMP-3 axis. Retrospective clinical studies indicate that elevated HSP27 expression correlates with advanced tumor stage and poor prognosis [40]. These associations, however, have not been clinically validated in interventional trials. Preclinical models—primarily in vitro and mouse xenografts—demonstrate that HSP27 knockdown using siRNA or small-molecule inhibitors such as OGX-427 enhances FOLFIRINOX efficacy and mitigates phosphorylation-related resistance mechanisms [41]. Additionally, gemcitabine-induced accumulation of methylglyoxal (MG) has been shown to trigger HSP27 expression as a cytoprotective response [42,43]. Compounds like triptolide, AHCC, and melatonin have been reported to downregulate HSP27 and restore apoptosis in PDAC cells, with melatonin exerting its effects via inhibition of NF-κB and STAT3 signaling, as demonstrated in experimental in vitro studies [44,45].
In the study by Drexler et al., lower HSP27 expression was associated with shorter overall survival (OS). Furthermore, high expression was associated with a better response to the gemcitabine regimen in patients with resectable, non-metastatic disease [46].
4.2. HSP47: Modulator of Tumor Microenvironment
HSP47 facilitates extracellular matrix (ECM) remodeling, creating a physical barrier that impedes drug penetration. Its inhibition improves gemcitabine sensitivity, indicating its relevance in targeting the tumor microenvironment [47,48]. These findings are based on experimental in vitro and in vivo studies using PDAC cell lines and mouse xenograft models. Han et al. presented a strategy to increase drug delivery to the tumor site. In their study, they used a tumor microenvironment-responsive nanosystem based on PEGylated polyethylenimine-coated gold nanoparticles to deliver all-trans retinoic acid (ATRA) and siRNA targeting heat shock protein 47. This influences activated pancreatic stellate cells (PSCs) and inhibits extracellular matrix hyperplasia [49]. To date, no clinical trials in humans have evaluated therapeutic strategies directly targeting HSP47 in pancreatic cancer.
4.3. HSP60: Regulator of Mitochondrial Metabolism and Tumor Immunogenicity
HSP60 has been implicated in the progression of PDAC by promoting cancer cell proliferation, migration, and tumorigenic potential. Zhou et al. demonstrated that HSP60 expression is significantly elevated in PDAC tissues and correlates with tumor progression. Analysis of patient-derived pancreatic cancer cells revealed that HSP60 exerts its oncogenic effects through stabilization of mitochondrial oxidative phosphorylation (OXPHOS) and modulation of the HSP60/OXPHOS/Erk1/2 signaling axis. This pathway supports tumor cell survival by maintaining mitochondrial function and sustaining Erk1/2 phosphorylation. Inhibition of OXPHOS—either by genetic silencing of HSP60 or pharmacologically via metformin—leads to reduced Erk1/2 activation, induction of apoptosis, and cell cycle arrest [12]. Additionally, HSP60 interacts with anti-apoptotic proteins such as Bcl-xL, survivin, and clusterin, further enhancing its role in resistance to cell death. Functional studies confirmed that HSP60 knockdown suppresses PDAC cell proliferation and invasiveness, whereas its overexpression accelerates tumor progression. Additionally, thermal stress induced by local hyperthermia in the range of 39–43 °C leads to increased surface expression of HSP60 and HSP70 on cancer cells, which enhances their antigenic profile. This promotes antigen presentation by dendritic cells and augments antitumor immune responses through interferon-gamma (IFN-γ) secretion by activated T cells [50]. These findings are based primarily on experimental in vitro and in vivo models. Although local hyperthermia is used clinically as an adjunct to cancer therapy, there is currently no direct clinical evidence confirming that this approach enhances HSP-mediated immunogenicity or IFN-γ-driven immune responses in patients.
4.4. HSP70: Multifaceted Therapeutic Target
HSP70 is markedly overexpressed in pancreatic PDAC and is closely associated with increased tumor proliferation, apoptosis resistance, invasiveness, and poor prognosis [51]. Due to its multifunctional role in cancer progression, HSP70 has emerged as a promising therapeutic target. Pharmacological inhibition of HSP70 suppresses tumor growth and enhances the efficacy of chemotherapy and immunotherapy. Small-molecule inhibitors such as PES, MKT-077, VER-155008, and Ap-4-139B have demonstrated preclinical efficacy, with the latter showing synergistic antitumor activity when combined with hydroxychloroquine in murine models of metastatic PDAC [51].
Natural compounds including ursenolide and maslinic acid preferentially target glucose-deprived PDAC cells by inhibiting HSPA5 (GRP78) and GRP94, inducing endoplasmic reticulum stress in vitro [59]. Additionally, maslinic acid suppresses HSPA8 and promotes autophagy, although its overexpression may limit treatment efficacy, underscoring the need for combination strategies targeting multiple HSP70 isoforms [52].
Beyond cytoprotection, HSP70 inhibition elicits immunomodulatory effects by activating dendritic cells and enhancing antitumor immune responses, as demonstrated in in vitro and in vivo models [53]. HSP70 also stabilizes the oncogenic mutant p53 R175H protein, and its inhibition promotes degradation of this variant, attenuating tumor progression [54]. Moreover, HSPA5 facilitates ferroptosis in PDAC cells via EP300-mediated acetylation, with HDAC6 acting as a negative regulator—suggesting potential synergy through dual inhibition [55].
Combination therapies targeting HSP70 show promise. Leja-Szpak et al. demonstrated that gemcitabine combined with melatonin or AFMK enhances apoptotic signaling in PANC-1 cells more effectively than monotherapy by downregulating HSP70 and cIAP-2 [56]. Similarly, radiofrequency ablation (RFA) increases HSP70 expression and activates the AKT–mTOR axis, promoting survival; however, its combination with mTOR inhibitors achieves a synergistic antitumor effect in murine PDAC models [57]. These findings are based on preclinical studies; although RFA is used clinically, the described molecular effects remain unconfirmed in humans.
HSP70 also contributes to PDAC metastasis through interaction with its co-chaperone STIP1, which stabilizes the HSP70–HSP90 complex and activates the FAK/AKT/MMP signaling pathway. High STIP1 expression correlates with poor prognosis, and its inhibition reduces migration and invasion in experimental models [58]. Additionally, miR-634 acts as a tumor suppressor by targeting HSPA2, inhibiting epithelial-to-mesenchymal transition and extracellular matrix degradation. Clinical sample analysis supports an inverse correlation between miR-634 and HSPA2 levels, while in vitro assays confirm that miR-634 restoration suppresses malignant traits [58]. These findings remain limited to preclinical studies and have not yet been validated in clinical trials.
The glucose-regulated proteins (GRPs) are Ca2+-binding chaperone proteins with protective properties whose transcription is induced in response to several stimuli that disrupt ER structure and function, related to HSP70. In Park's study, the novel therapeutic agents PST-A and PST-B exhibited selective cytotoxicity against PANC-1 pancreatic cancer cells under glucose deprivation. This was attributed to the inhibition of glucose-regulated protein 78 (GRP78), a heat shock protein (HSP) that protects pancreatic cancer cells [59]. Another novel strategy was presented in the study by Tang et al. Secalonic acid D was found to inhibit the Akt signaling pathway and affect the induction of glucose-regulated protein 78 (GRP78) under glucose-starved conditions, resulting in a cytotoxic effect on human pancreatic carcinoma PANC-1 cells [60].
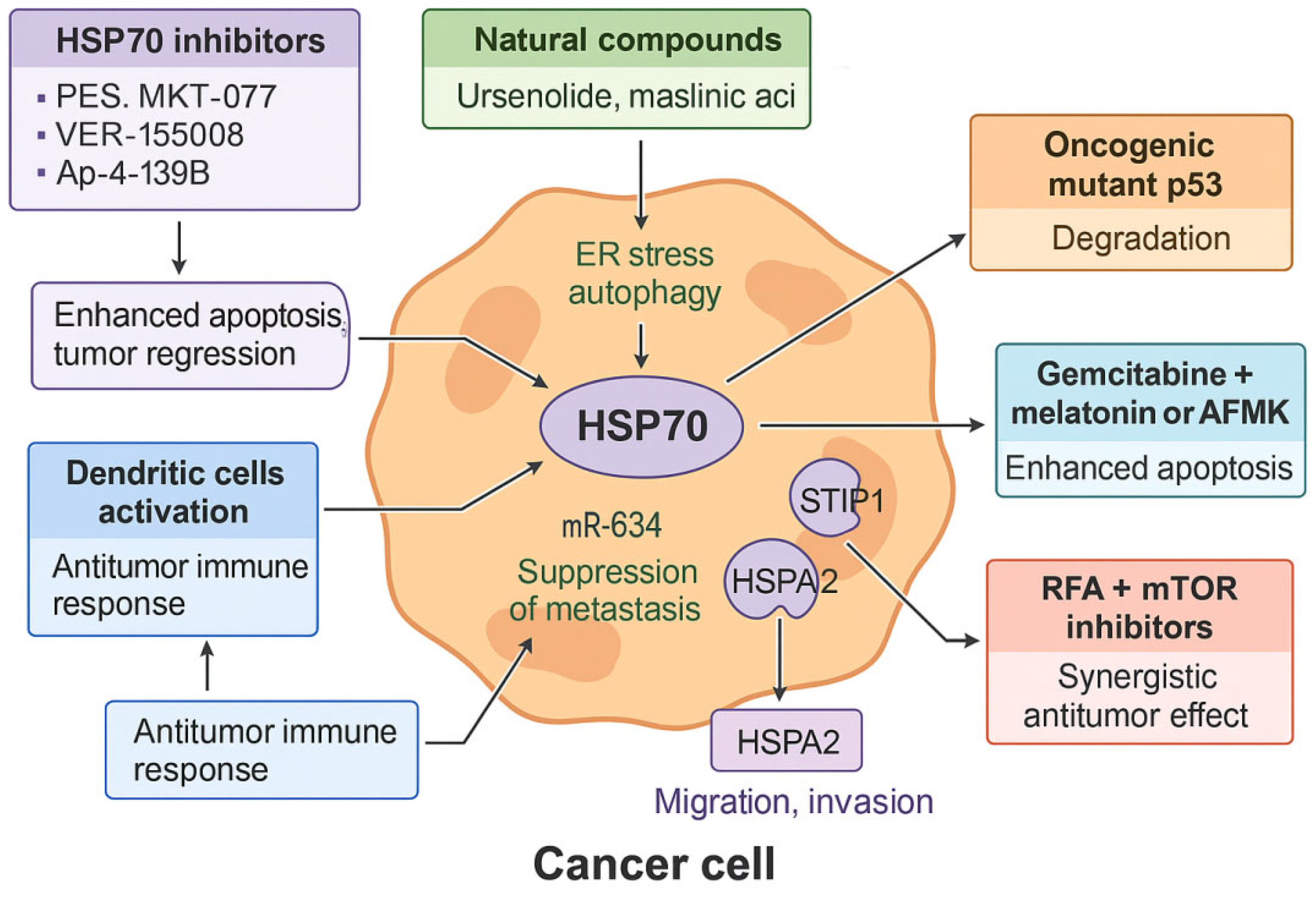
Figure 2.
The role of HSP70 in PDAC and therapeutic intervention strategies.
4.5. HSP90: Central Regulator of Oncogenic Stability
HSP90 is a central molecular chaperone that stabilizes a wide array of oncogenic client proteins, including EGFR, HER2, VEGF, phosphorylated STAT3, Src, and IGF-1Rβ. Through this activity, it supports key hallmarks of PDAC, such as sustained proliferation, invasion, angiogenesis, and resistance to therapy [61,62,63,64]. Pharmacological inhibition of the HSP90 ATPase domain using agents such as ganetespib, 17-AAG (tanespimycin), and AUY922 (luminespib) promotes proteasomal degradation of these client proteins, enhances sensitivity to chemotherapy and radiotherapy, and disrupts DNA repair mechanisms by downregulating ATM, ATR, RAD51, and DNA-PK [64]. Although these compounds have shown potent antitumor activity in preclinical models, early-phase clinical trials in PDAC patients—such as those evaluating 17-AAG in combination with gemcitabine—have yielded mixed results, and further clinical validation is needed to confirm therapeutic efficacy.
A secreted isoform, HSP90α, also contributes to PDAC progression via paracrine mechanisms. It binds to the LRP1 (CD91) receptor, activating the AKT signaling cascade and inducing epithelial-to-mesenchymal transition (EMT). In preclinical studies, neutralization of extracellular HSP90α using the monoclonal antibody HH01 reversed EMT and suppressed metastatic potential [65,66]. Moreover, small-molecule inhibitors that disrupt the HSP90–Cdc37 interaction (e.g., x6506 and x1540) have been shown to inhibit ERK and AKT signaling in KRAS-mutated PDAC cells [67].
HSP90 also cooperates with HSP70 to regulate the stability and membrane localization of SLC6A14, an amino acid transporter frequently overexpressed in PDAC. Inhibition of HSP90 destabilizes SLC6A14 and reduces amino acid uptake, while combination with SLC6A14 antagonists such as α-methyl-tryptophan enhances antitumor effects in vivo [68].
Resistance to therapy in PDAC is often driven by compensatory activation of survival pathways. Thiadiazole-based HSP90 inhibitors have been shown to overcome such resistance by destabilizing oncogenic proteins and inhibiting the PI3K/AKT/mTOR axis. Co-administration with MEK inhibitors results in robust tumor growth inhibition and prolonged survival in murine models [69,70].
Within the immunosuppressive tumor microenvironment, HSP90 plays an additional role by stabilizing STAT1 and promoting IFN-γ-induced upregulation of immune checkpoint molecules such as PD-L1 and immunomodulatory enzymes including IDO1. Pharmacological inhibition of HSP90 using agents such as luminespib, ganetespib, SNX-2112, or XL888 decreases the expression of these immunosuppressive markers and enhances the efficacy of immune checkpoint blockade, including anti–PD-1 therapies, in preclinical PDAC models [71,72].
Lastly, iron oxide nanoparticles (DIO-NPs) have been shown to trigger oxidative stress in PDAC cells, leading to upregulation of HSP70 and HSP90 as part of a cytoprotective response. This stress adaptation can be effectively counteracted by co-treatment with HSP inhibitors, thereby amplifying the overall antitumor effect both in vitro and in vivo [73].
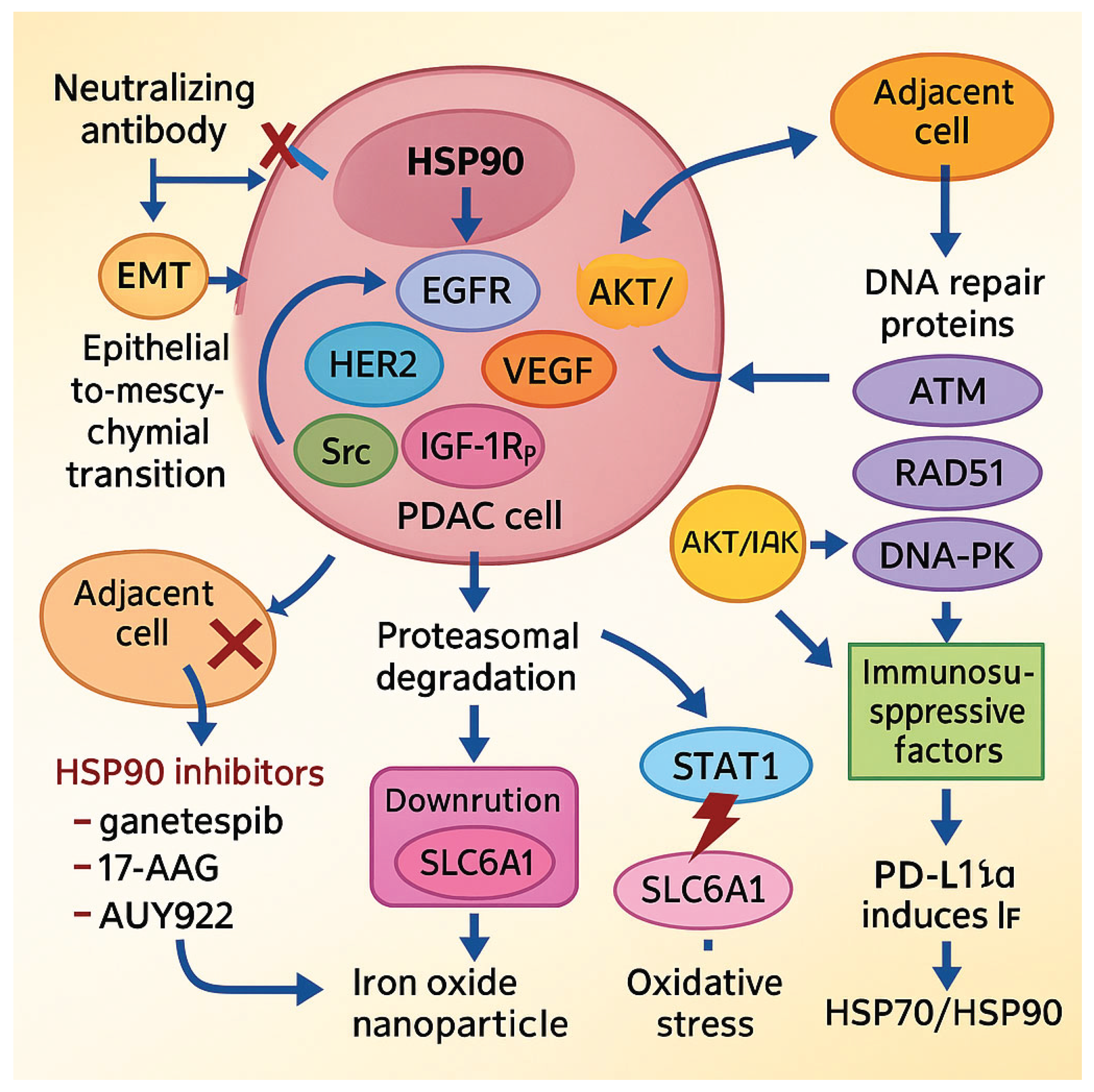
Figure 3.
The role of HSP90 in PDAC progression and potential therapeutic strategies.
5. Summary
This paper provides a comprehensive overview of current knowledge regarding the importance of heat shock proteins (HSPs) in pancreatic cancer pathogenesis, diagnosis, and treatment, with a focus on their potential as biomarkers and therapeutic targets. Numerous scientific reports confirm HSP participation in fundamental carcinogenic processes, including tumor growth, invasion, metastasis, tumor microenvironment remodeling, apoptosis avoidance, cachexia development, and systemic treatment and radiotherapy resistance. Interestingly, some HSP isoforms, such as HSPB2, exhibit anti-cancer properties, e.g., activating the p53 protein and limiting PDAC cell proliferation. From a translational perspective, HSPs show significant potential as diagnostic and prognostic biomarkers in pancreatic cancer. For example, increased HSPB6 expression correlates with a more favorable prognosis in patients with pancreatic ductal adenocarcinoma. In contrast, decreased HSP27 levels are associated with unfavorable clinical parameters, such as poor histopathological differentiation, more frequent liver metastases, and shorter survival after tumor resection. Simultaneously assessing HSP70 and VEGF levels is a promising method for predicting treatment response. HSPA2 and HSP90, on the other hand, have been identified as unfavorable prognostic factors associated with a higher risk of disease recurrence and shorter overall survival. Additionally, HSP90 is used in molecular imaging as a target for radiolabeled ligands in positron emission tomography (PET); however, its clinical use is limited due to its rapid metabolism and elimination by the hepatobiliary system.
Heat shock proteins are important therapeutic targets for treating pancreatic cancer. Preclinical studies have demonstrated that inhibiting these proteins can make cancer cells more susceptible to chemotherapy, modulate the immune response by affecting PD-L1 and IDO1 expression, and induce direct cytotoxic effects. Substances such as melatonin, metformin, AFMK, ganetespib, and JG-231 demonstrate antitumor activity in PDAC models by inducing apoptosis, autophagy, and ferroptosis and by inhibiting epithelial-mesenchymal transformation processes and amino acid metabolism. Recently, there has been growing interest in combination therapy strategies, which combine HSP inhibitors with MEK and mTOR pathway inhibitors or epigenetic drugs. This approach can significantly increase therapy efficacy by affecting multiple tumor resistance mechanisms simultaneously. Although preclinical results are promising, many HSP-targeted therapies require clinical validation. Further, well-designed randomized studies are required. Nevertheless, mounting evidence suggests that heat shock proteins are an essential component of tumor biology and may play a pivotal role in future therapeutic strategies for pancreatic cancer.
Author Contributions
Conceptualization, J.K.; methodology J.K., Ł.M., I.G.G.; validation, J.K. and I.G.G. formal analysis, J.K.; investigation, J.S., W.Ż., M.S., M.Str. N.J. resources, Ł.M..; data curation, J.K.; writing—original draft preparation, J.K., J.S., W.Ż., M.S., M. Str, N.J., J.W., A.G.M. writing—review and editing, J.W. and J.K.; visualization, E.CH.; supervision, J.K. and I.GG; project administration, J.K.; funding acquisition, I.G.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
This research has received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hu C, Yang J, Qi Z, Wu H, Wang B, Zou F, et al. Heat shock proteins: Biological functions, pathological roles, and therapeutic opportunities. MedComm. 2022 Sep 1;3(3). [CrossRef]
- Kakkar V, Meister-Broekema M, Minoia M, Carra S, Kampinga HH. Barcoding heat shock proteins to human diseases: Looking beyond the heat shock response. DMM Dis Model Mech. 2014;7(4):421–34. [CrossRef]
- Singh MK, Shin Y, Ju S, Han S, Choe W, Yoon KS, et al. Heat Shock Response and Heat Shock Proteins: Current Understanding and Future Opportunities in Human Diseases. Int J Mol Sci. 2024 Apr 10;25(8). [CrossRef]
- Melikov A, Biocev PN. Review Article Heat Shock Protein Network: the Mode of Action, the Role in Protein Folding and Human Pathologies (HSP / protein folding / chaperone / aggregation / neurodegenerative disease / cancer).
- Zuo WF, Pang Q, Zhu X, Yang QQ, Zhao Q, He G, et al. Heat shock proteins as hallmarks of cancer: insights from molecular mechanisms to therapeutic strategies. J Hematol Oncol. 2024 Dec 1;17(1):81. [CrossRef]
- Yun CW, Kim HJ, Lim JH, Lee SH. Heat shock proteins: Agents of cancer development and therapeutic targets in anti-cancer therapy. Cells. 2020 Jan 1;9(1). [CrossRef]
- Lau R, Yu L, Roumeliotis TI, Stewart A, Pickard L, Riisanes R, et al. Unbiased differential proteomic profiling between cancer-associated fibroblasts and cancer cell lines. J Proteomics. 2023 Sep 30;288. [CrossRef]
- Zhang S, Zhang XQ, Huang SL, Chen M, Shen SS, Ding XW, et al. The effects of HSP27 on gemcitabine-resistant pancreatic cancer cell line through snail. Pancreas. 2015 Oct 1;44(7):1121–9. [CrossRef]
- Deng M, Chen PC, Xie S, Zhao J, Gong L, Liu J, et al. The small heat shock protein αA-crystallin is expressed in pancreas and acts as a negative regulator of carcinogenesis. Biochim Biophys Acta - Mol Basis Dis. 2010 Jul 1;1802(7–8):621–31. [CrossRef]
- Liu P, Zu F, Chen H, Yin X, Tan X. Exosomal DNAJB11 promotes the development of pancreatic cancer by modulating the EGFR/MAPK pathway. Cell Mol Biol Lett. 2022 Dec 1;27(1):1–20. [CrossRef]
- Roth HE, Bhinderwala F, Franco R, Zhou Y, Powers R. DNAJA1 Dysregulates Metabolism Promoting an Antiapoptotic Phenotype in Pancreatic Ductal Adenocarcinoma. J Proteome Res. 2021 Aug 6;20(8):3925–39. [CrossRef]
- Zhou C, Sun H, Zheng C, Gao J, Fu Q, Hu N, et al. Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis 2018 92. 2018 Feb 7;9(2):1–14. [CrossRef]
- Xiong L, Li D, Xiao G, Tan S, Xu L, Wang G. HSP70 promotes pancreatic cancer cell epithelial-mesenchymal transformation and growth via the NF-κB signaling pathway. Pancreas. 2024 Feb 1;54(2). [CrossRef]
- Zhai LL, Qiao PP, Sun YS, Ju TF, Tang ZG. Tumorigenic and immunological roles of Heat shock protein A2 in pancreatic cancer: a bioinformatics analysis. Rev Assoc Med Bras. 2022;68(4):470–5. [CrossRef]
- Wu HY, Trevino JG, Fang BL, Riner AN, Vudatha V, Zhang GH, et al. Patient-Derived Pancreatic Cancer Cells Induce C2C12 Myotube Atrophy by Releasing Hsp70 and Hsp90. Cells. 2022 Sep 1;11(17). [CrossRef]
- Yang J, Zhang Z, Zhang Y, Ni X, Zhang G, Cui X, et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology. 2019 Feb 1;156(3):722-734.e6. [CrossRef]
- Zhang Y, Ware MB, Zaidi MY, Ruggieri AN, Olson BM, Komar H, et al. Heat shock protein-90 inhibition alters activation of pancreatic stellate cells and enhances the efficacy of PD-1 blockade in pancreatic cancer. Mol Cancer Ther. 2021 Jan 1;20(1):150–60. [CrossRef]
- Gulla A, Strupas K, Chun M, Wanglong Q, Su G. Heat Shock Protein 90: Target Molecular Regulator in Acute Pancreatitis and Pancreatic Ductal Adenocarcinoma. HPB. 2023;25:S440. [CrossRef]
- Wang X, Zhang J, Wu H, Li Y, Conti PS, Chen K. PET imaging of Hsp90 expression in pancreatic cancer using a new 64Cu-labeled dimeric Sansalvamide A decapeptide. Amino Acids. 2018 Jul 1;50(7):897–907. [CrossRef]
- Wang X, Han Z, Zhang J, Chen M, Meng W. Development and Preclinical Evaluation of 18F-Labeled PEGylated Sansalvamide A Decapeptide for Noninvasive Evaluation of Hsp90 Status in Pancreas Cancer. Mol Pharm. 2024 Oct 7;21(10):5238–46. [CrossRef]
- Wang X, Zhang J, Han Z, Ma L, Li Y. 18F-labeled Dimer-Sansalvamide A Cyclodecapeptide: A Novel Diagnostic Probe to Discriminate Pancreatic Cancer from Inflammation in a Nude Mice Model. J Cancer. 2022;13(6):1848–58. [CrossRef]
- Richter K, Haslbeck M, Buchner J. The Heat Shock Response: Life on the Verge of Death. Mol Cell. 2010 Oct;40(2):253–66. [CrossRef]
- Wu Y, Zhao J, Tian Y, Jin H. Cellular functions of heat shock protein 20 (HSPB6) in cancer: A review. Cell Signal. 2023 Dec 1;112:110928. [CrossRef]
- Zhang L, Yang L, Du K. Exosomal HSPB1, interacting with FUS protein, suppresses hypoxia-induced ferroptosis in pancreatic cancer by stabilizing Nrf2 mRNA and repressing P450. J Cell Mol Med. 2024 May 1;28(9):e18209. [CrossRef]
- Yu Z, Wang H, Fang Y, Lu L, Li M, Yan B, et al. Molecular chaperone HspB2 inhibited pancreatic cancer cell proliferation via activating p53 downstream gene RPRM, BAI1, and TSAP6. J Cell Biochem. 2020 Mar 1;121(3):2318–29. [CrossRef]
- Yun CW, Kim HJ, Lim JH, Lee SH. Heat shock proteins: Agents of cancer development and therapeutic targets in anti-cancer therapy. Cells. 2020 Jan 1;9(1). [CrossRef]
- Montresor S, Pigazzini ML, Baskaran S, Sleiman M, Adhikari G, Basilicata L, et al. HSP110 is a modulator of amyloid beta (Aβ) aggregation and proteotoxicity. J Neurochem. 2024 Jan 1;169(1). [CrossRef]
- Evans CG, Chang L, Gestwicki JE. Heat shock protein 70 (Hsp70) as an emerging drug target. J Med Chem. 2010 Jun 24;53(12):4585–602. [CrossRef]
- Sha G, Jiang Z, Zhang W, Jiang C, Wang D, Tang D. The multifunction of HSP70 in cancer: Guardian or traitor to the survival of tumor cells and the next potential therapeutic target. Int Immunopharmacol. 2023 Sep 1;122. [CrossRef]
- Youness RA, Gohar A, Kiriacos CJ, El-Shazly M. Heat Shock Proteins: Central Players in Oncological and Immuno-Oncological Tracks. Adv Exp Med Biol. 2023;1409:193–203. [CrossRef]
- Zuo WF, Pang Q, Zhu X, Yang QQ, Zhao Q, He G, et al. Heat shock proteins as hallmarks of cancer: insights from molecular mechanisms to therapeutic strategies. J Hematol Oncol. 2024 Dec 1;17(1). [CrossRef]
- Nawaz MS, Fournier-Viger P, Nawaz S, Gan W, He Y. FSP4HSP: Frequent sequential patterns for the improved classification of heat shock proteins, their families, and sub-types. Int J Biol Macromol. 2024 Oct 1;277(Pt 1). [CrossRef]
- Peng YF, Lin H, Liu DC, Zhu XY, Huang N, Wei YX, et al. Heat shock protein 90 inhibitor ameliorates pancreatic fibrosis by degradation of transforming growth factor-β receptor. Cell Signal. 2021 Aug 1;84. [CrossRef]
- Neckers L, Workman P. Hsp90 molecular chaperone inhibitors: Are we there yet? Clin Cancer Res. 2012 Jan 1;18(1):64–76. [CrossRef]
- Liu B, Chen Z, Li Z, Zhao X, Zhang W, Zhang A, et al. Hsp90α promotes chemoresistance in pancreatic cancer by regulating Keap1-Nrf2 axis and inhibiting ferroptosis. Acta Biochim Biophys Sin (Shanghai). 2024 Feb 1;57(2). [CrossRef]
- Zhang J, Li H, Liu Y, Zhao K, Wei S, Sugarman ET, et al. Targeting HSP90 as a Novel Therapy for Cancer: Mechanistic Insights and Translational Relevance. Cells 2022, Vol 11, Page 2778. 2022 Sep 6;11(18):2778. [CrossRef]
- Malkeyeva D, Kiseleva E V., Fedorova SA. Heat shock proteins in protein folding and reactivation. Vavilov J Genet Breed. 2025 Mar 2;29(1):7–14. [CrossRef]
- Lee G, Kim RS, Lee SB, Lee S, Tsai FTF. Deciphering the mechanism and function of Hsp100 unfoldases from protein structure. Biochem Soc Trans. 2022 Dec 1;50(6):1725–36. [CrossRef]
- Wu PK, Hong SK, Starenki D, Oshima K, Shao H, Gestwicki JE, et al. Mortalin/HSPA9 targeting selectively induces KRAS tumor cell death by perturbing mitochondrial membrane permeability. Oncogene. 2020 May 21;39(21):4257–70. [CrossRef]
- Fang Z, Liang W, Luo L. HSP27 promotes epithelial-mesenchymal transition through activation of the β-catenin/MMP3 pathway in pancreatic ductal adenocarcinoma cells. Transl Cancer Res. 2019;8(4):1268–78. [CrossRef]
- Grierson PM, Dodhiawala PB, Cheng Y, Chen THP, Khawar IA, Wei Q, et al. The MK2/Hsp27 axis is a major survival mechanism for pancreatic ductal adenocarcinoma under genotoxic stress. Sci Transl Med. 2021 Dec 1;13(622). [CrossRef]
- Crake R, Gasmi I, Dehaye J, Lardinois F, Peiffer R, Maloujahmoum N, et al. Resistance to Gemcitabine in Pancreatic Cancer Is Connected to Methylglyoxal Stress and Heat Shock Response. Cells. 2023 May 1;12(10). [CrossRef]
- Tamtaji OR, Mirhosseini N, Reiter RJ, Behnamfar M, Asemi Z. Melatonin and pancreatic cancer: Current knowledge and future perspectives. J Cell Physiol. 2019 May 1;234(5):5372–8. [CrossRef]
- Hussain MS, Mujwar S, Babu MA, Goyal K, Chellappan DK, Negi P, et al. Pharmacological, computational, and mechanistic insights into triptolide’s role in targeting drug-resistant cancers. Naunyn Schmiedebergs Arch Pharmacol. 2025 Jun 1;398(6). [CrossRef]
- Kuhara K, Tokuda K, Kitagawa T, Baron B, Tokunaga M, Harada K, et al. CUB Domain-containing Protein 1 (CDCP1) is down-regulated by active hexose-correlated compound in human pancreatic cancer cells. Anticancer Res. 2018 Nov 1;38(11):6107–11. [CrossRef]
- Drexler R, Wagner KC, Küchler M, Feyerabend B, Kleine M, Oldhafer KJ. Significance of unphosphorylated and phosphorylated heat shock protein 27 as a prognostic biomarker in pancreatic ductal adenocarcinoma. J Cancer Res Clin Oncol. 2020 May 1;146(5):1125–37. [CrossRef]
- Yoneda A, Minomi K, Tamura Y. Heat shock protein 47 confers chemoresistance on pancreatic cancer cells by interacting with calreticulin and IRE1α. Cancer Sci. 2021 Jul 1;112(7):2803–20. [CrossRef]
- Duarte BDP, Bonatto D. The heat shock protein 47 as a potential biomarker and a therapeutic agent in cancer research. J Cancer Res Clin Oncol. 2018 Dec 1;144(12):2319–28. [CrossRef]
- Han X, Li Y, Xu Y, Zhao X, Zhang Y, Yang X, et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat Commun 2018 91. 2018 Aug 23;9(1):1–18. [CrossRef]
- Mahmood J, Shukla HD, Soman S, Samanta S, Singh P, Kamlapurkar S, et al. Immunotherapy, Radiotherapy, and Hyperthermia: A Combined Therapeutic Approach in Pancreatic Cancer Treatment. Cancers 2018, Vol 10, Page 469. 2018 Nov 28;10(12):469. [CrossRef]
- Ferretti GDS, Quaas CE, Bertolini I, Zuccotti A, Saatci O, Kashatus JA, et al. HSP70-mediated mitochondrial dynamics and autophagy represent a novel vulnerability in pancreatic cancer. Cell Death Differ 2024 317. 2024 May 28;31(7):881–96. [CrossRef]
- Tian Y, Xu H, Farooq AA, Nie B, Chen X, Su S, et al. Maslinic acid induces autophagy by down-regulating HSPA8 in pancreatic cancer cells. Phyther Res. 2018 Jul 1;32(7):1320–31. [CrossRef]
- Giri B, Sharma P, Jain T, Ferrantella A, Vaish U, Mehra S, et al. Hsp70 modulates immune response in pancreatic cancer through dendritic cells. Oncoimmunology. 2021;10(1). [CrossRef]
- Polireddy K, Singh K, Pruski M, Jones NC, Manisundaram N V., Ponnela P, et al. Mutant p53 R175H promotes cancer initiation in the pancreas by stabilizing HSP70. Cancer Lett. 2019 Jul 1;453:122–30. [CrossRef]
- Zhu S, Zhang Q, Sun X, Zeh HJ, Lotze MT, Kang R, et al. HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 2017 Apr 15;77(8):2064–77. [CrossRef]
- Leja-Szpak A, Nawrot-Porąbka K, Góralska M, Jastrzębska M, Link-Lenczowski P, Bonior J, et al. Melatonin and its metabolite N1-acetyl-N2-formyl-5-methoxykynuramine (afmk) enhance chemosensitivity to gemcitabine in pancreatic carcinoma cells (PANC-1). Pharmacol Reports. 2018 Dec 1;70(6):1079–88. [CrossRef]
- Zhang F, Wu G, Sun H, Ding J, Xia F, Li X, et al. Radiofrequency ablation of hepatocellular carcinoma in elderly patients fitting the Milan criteria: A single centre with 13 years experience. Int J Hyperth. 2014;30(7):471–9. [CrossRef]
- Jing Y, Liang W, Liu J, Zhang L, Wei J, Zhu Y, et al. Stress-induced phosphoprotein 1 promotes pancreatic cancer progression through activation of the FAK/AKT/MMP signaling axis. Pathol Res Pract. 2019 Nov 1;215(11). [CrossRef]
- Park HR. Pancastatin A and B Have Selective Cytotoxicity on Glucose-Deprived PANC-1Human Pancreatic Cancer Cells. J Microbiol Biotechnol. 2020 May 28;30(5):733–8. [CrossRef]
- Tang R, Kimishima A, Setiawan A, Arai M. Secalonic acid D as a selective cytotoxic substance on the cancer cells adapted to nutrient starvation. J Nat Med. 2020 Mar 1;74(2):495–500. [CrossRef]
- Xue N, Lai F, Du T, Ji M, Liu D, Yan C, et al. Chaperone-mediated autophagy degradation of IGF-1Rβ induced by NVP-AUY922 in pancreatic cancer. Cell Mol Life Sci. 2019 Sep 1;76(17):3433–47. [CrossRef]
- Rochani AK, Balasubramanian S, Girija AR, Maekawa T, Kaushal G, Sakthi Kumar D. Heat Shock Protein 90 (Hsp90)-Inhibitor-Luminespib-Loaded-Protein-Based Nanoformulation for Cancer Therapy. Polym 2020, Vol 12, Page 1798. 2020 Aug 11;12(8):1798. [CrossRef]
- Nagaraju GP, Zakka KM, Landry JC, Shaib WL, Lesinski GB, El-Rayes BF. Inhibition of HSP90 overcomes resistance to chemotherapy and radiotherapy in pancreatic cancer. Int J Cancer. 2019 Sep 15;145(6):1529–37. [CrossRef]
- Mehta RK, Pal S, Kondapi K, Sitto M, Dewar C, Devasia T, et al. Low-Dose Hsp90 Inhibitor Selectively Radiosensitizes HNSCC and Pancreatic Xenografts. Clin Cancer Res. 2020 Oct 1;26(19):5246–57. [CrossRef]
- Fan CS, Chen LL, Hsu TA, Chen CC, Chua KV, Li CP, et al. Endothelial-mesenchymal transition harnesses HSP90α-secreting M2-macrophages to exacerbate pancreatic ductal adenocarcinoma. J Hematol Oncol. 2019 Dec 17;12(1):1–15. [CrossRef]
- Zhao ZX, Li S, Liu LX. Thymoquinone affects hypoxia-inducible factor-1α expression in pancreatic cancer cells via HSP90 and PI3K/AKT/mTOR pathways. World J Gastroenterol. 2024 Jun 7;30(21):2793–816. [CrossRef]
- Siddiqui FA, Parkkola H, Vukic V, Oetken-lindholm C, Jaiswal A, Kiriazis A, et al. Novel Small Molecule Hsp90/Cdc37 Interface Inhibitors Indirectly Target K-Ras-Signaling. Cancers 2021, Vol 13, Page 927. 2021 Feb 23;13(4):927. [CrossRef]
- Nałęcz KA. Amino Acid Transporter SLC6A14 (ATB0,+) – A Target in Combined Anti-cancer Therapy. Front Cell Dev Biol. 2020 Oct 21;8:594464. [CrossRef]
- Gulla A, Kazlauskas E, Liang H, Strupas K, Petrauskas V, Matulis D, et al. Heat Shock Protein 90 Inhibitor Effects on Pancreatic Cancer Cell Cultures. Pancreas. 2021 Apr 1;50(4):625–32. [CrossRef]
- Grbovic-Huezo O, Pitter KL, Lecomte N, Saglimbeni J, Askan G, Holm M, et al. Unbiased in vivo preclinical evaluation of anticancer drugs identifies effective therapy for the treatment of pancreatic adenocarcinoma. Proc Natl Acad Sci U S A. 2020 Dec 1;117(48):30670–8. [CrossRef]
- Liu J, Kang R, Kroemer G, Tang D. Targeting HSP90 sensitizes pancreas carcinoma to PD-1 blockade. Oncoimmunology. 2022 Dec 31;11(1). [CrossRef]
- Tang D, Kang R. HSP90 as an emerging barrier to immune checkpoint blockade therapy. Oncoscience. 2022 Apr 22;9:20–2. [CrossRef]
- Balas M, Predoi D, Burtea C, Dinischiotu A. New Insights into the Biological Response Triggered by Dextran-Coated Maghemite Nanoparticles in Pancreatic Cancer Cells and Their Potential for Theranostic Applications. Int J Mol Sci 2023, Vol 24, Page 3307. 2023 Feb 7;24(4):3307. [CrossRef]
- Gao S, Pu N, Yin H, Li J, Chen Q, Yang M, et al. Radiofrequency ablation in combination with an mTOR inhibitor restrains pancreatic cancer growth induced by intrinsic HSP70. Ther Adv Med Oncol. 2020;12. [CrossRef]

Table 1.
The role of HSPs in pancreatic cancer pathogenesis.
| HSP Family | Pathological Role in PDAC | References |
| lmHSPs | Ferroptosis inhibition, promoting chemioresistance via Snail/E-cadherin/ERCC1 (HSP27); tumor suppression via p53 (HSPB2) | [8,22,23,24,25] |
| HSP40 (DnaJ Family) | Promoting PDAC development via BiP/GRP78 (DnaJB11); apoptosis inhibition, promoting invasiveness, enhancing Warburg effect and Bcl-2 expression (DnaJA1) | [10,11,26] |
| HSP60 (Chaperonins) | Apoptosis inhibition via HSP60/OXPHOS/Erk1/2 pathway; overexpression correlates with PDAC severity | [3,12,22] |
| HSP70 | Promoting EMT via NF-κB; development of cachexia via p38βMAPK; overexpression in tumor cells and CAFs (HSPA2) | [3,5,13,14,15,16,27,28,29,30] |
| HSP90 | Ferroptosis resistance via Nrf2\GPx; mutant p53 stabilization; inducing invassiveness via MMP2/9 activtion; promoting EMT and immune evasion | [31,32,33,34,35,36] |
| Large HSPs (HSP100) | Unknown | [31,32,37,38] |
Table 2.
The importance of HSPs as diagnostic and prognostic markers in pancreatic cancer.
| Marker (HSP) | Diagnostic/Prognostic Relevance | Methods/Models | References |
| HSPB6 | Overexpressed in cancer associated fibroblasts (CAFs); associated with improved overall survival in patients with PDAC; prognostic marker in PDAC | Mass spectrometry analysis of cancer-associated fibroblasts and cancer cell lines (Clinical Proteomic Tumor Analysis Consortium) | [7] |
| HSPB1 (HSP27) | Lower expression linked to poor overall survival in patients with PDAC after resection and liver metastases; higher expression associated with a better response to gemcitabine in the resected, non-metastasisedpatients group | Immunoreactive score (IRS), post-resection PDAC patient data (Dexter et al.) | [46] |
| HSP90 | High levels indicate poor prognosis; in PET imaging the expression of this protein enables monitoring and early detection of pancreatic cancer; | PET radiotracers, mouse model, immunochemistry (Wang et al.); pathologic data (Gamboa et al.); mice and rat models (Kacar et al.) | [18,19,20,21] |
Table 3.
Selected Experimental and Pharmacological Strategies Targeting HSP in Pancreatic Cancer Models.
Table 3.
Selected Experimental and Pharmacological Strategies Targeting HSP in Pancreatic Cancer Models.
| Strategy/Compound | Mechanism of Action | Targeted HSPs | References |
| Triptolide (TPL) | HSF1 inhibition and caspase-3, caspase-9 degradation promotes apoptosis and leads to increased tumor sensitivity to chemotherapy | HSP27, HSP70, HSP90 | [39] |
| Active hexose-correlated compound (AHCC) | Gemcytabine/methylglyoxal pathway leads to overexpression of HSP27, which is downregulated by AHCC inducing apoptosis and preventing resistance to chemotherapy | HSP27 | [45] |
| siRNA + ATRA delivered by PEGylated polyethylenimine-coated gold nanoparticles | HSP47-specific mRNA degradation by siRNA prevents ECM proliferation and increases gemcytabine sensitivity | HSP47 | [49] |
| AK-778,Col003,Pirfenidon | Direct inhibition of HSP47 inhibits tumor growth and increases gemcytabine sensitivity | HSP47 | [48] |
| Local hyperthermia | Increases tumor antigenicity and drug penetration by enhancing HSP70 and HSP60 expression; HSP70 promotes anti-tumor immune response, while HSP60 activates T cells and IFN-γ secretion | HSP60, HSP70 | [50] |
| Metformin + aminoguanidine | GLO-1 inhibition interferes with methylglyoxal/HSP27/HSP70 pathway increasing PDAC sensitivity to gemcytabine | HSP27, HSP70 | [42] |
| Melatonin | HSP27, HSP60, HSP70, HSP90 and HSP100 downregulation via NF-κB and STAT3 inhibition promotes apoptosis and increases tumor sensitivity to chemotherapy | HSP27, HSP60, HSP70, HSP90, HSP100 | [45] |
| Melatonin, AFMK | Suppression of HSP70 and cIAP-2 enhances gemcitabine efficacy and promotes apoptosis | HSP70 | [56] |
| Ap-4-139B + Hydroxychloroquine | Selective HSP70 inhibition induces mitochondrial swelling and activates the apoptotic pathway; combination with hydroxychloroquine (autophagy inhibitor) enhances antitumor efficacy | HSP70 | [51] |
| Pancastatin A and B | GRP78 (HSPA5) inhibition during glucose deprivation. | HSP70 | [59] |
| Xanthone derivative of secalonic acid D | AKT signaling pathway inhibition under glucose-starved condition and GRP78 (HSPA5) downregulation leads to cytotoxic activity on PANC-1 | HSP70 | [60] |
| Maslinic acid | Proliferation inhibition and inducing autophagy in PANC-28 through HSPA8 downregulation | HSP70 | [52] |
| DIO-NPs + HSP Inhibitors | DIO-NPs induce cellular stress leading to increased HSP70/HSP90 expression; combination with HSP inhibitors may impair survival mechanisms of PDAC and enhance therapy efficacy | HSP70, HSP90 | [73] |
| RFA + mTOR Inhibitors | Inhibition of RFA-induced via HSP70 AKT/mTOR pathway leads to suppression of proliferation and enhanced therapeutic response | HSP70 | [74] |
| JG-231 | Mortalin (HSPA9, GRP75) inhibition in K-RasG12C mutation PDAC increases the permeability of the mitochondrial membrane and promotes apoptosis | HSP70 | [39] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



