Submitted:
12 July 2025
Posted:
14 July 2025
You are already at the latest version
Abstract
Algorithms are essential to the modern transformation of biomedicine, enabling the analysis of large and complex biological datasets through computational models, machine learning, and network analysis. These indirect methods allow for quick hypothesis generation, discovering potential interactions, and gaining insights into biological systems that would be difficult to achieve with experimental methods alone. However, relying only on computational predictions can lead to misinterpretation and false positives. Therefore, we must combine algorithms with rigorous experimental validation to ensure scientific accuracy and reliability. Improvements in algorithm efficiency, data integration, and validation strategies continually strengthen their robustness, making them vital tools in systems biology. Combining algorithms with experimentation creates a dynamic cycle of hypothesis testing and refinement, leading to deeper insights into biological functions and advancements in diagnostics and therapeutics. We illustrate, in the text, what happens when preventive checks on data reliability are missing. Using STRING and similar resources, we show how to apply Popper's Falsification Principle to interactomics, for falsification, not mere exploration.
Keywords:
algorithms
; interactomics
; experimental methods
; big data
; computational models
; omics data
; drug repurposing
; text mining
; STRING
; principle of falsification in science
1. The Acquisition of Current Scientific Knowledge in Biomedicine
In the rapidly growing field of biomedicine, computational algorithms and high-throughput data analysis have become essential tools for generating hypotheses and understanding system-level processes [1]. However, rigorous experimental validation should complement these powerful methods, not replace them [2]. The strength of computational approaches lies in their ability to analyze large datasets, identify potential interactions, and suggest new directions for research [3]. Yet, without experimental confirmation, such findings remain speculative and vulnerable to false positives, oversights, or misinterpretations.
Recognizing current limitations in data quality, database curation, and interpretive frameworks is crucial. Advances are continuously enhancing the reliability and predictive accuracy of computational models. Combining these tools with careful laboratory validation grounds our hypotheses in biological reality, reducing the risk of mistaking correlation for causation or over-interpreting indirect associations. The future of biomedicine relies on a collaborative cycle: using computational methods to narrow down hypotheses efficiently, then rigorously testing them experimentally to verify their biological relevance [4]. This balanced strategy maximizes the strengths of both approaches—quick hypothesis generation and solid validation—ultimately producing more trustworthy, detailed, and impactful scientific knowledge.
Today's scientific understanding of biomedicine comes from both direct experimental methods and indirect algorithmic analyses [5]. Each approach has its strengths, limitations, and complexities in interpretation. To discuss algorithms effectively, we also need to consider the object being studied and the experimental data that support it. The more quantitative information available about the object, the more likely the algorithm will produce significant and reliable results. This is especially relevant when working with Big Data, a key aspect of systems biology [6]. It is important for professionals in the field, whether they use experimental or computational methods, to understand the advantages and disadvantages of both, as they can sometimes overlap and cause confusion.
Systems Biology is revolutionizing our understanding of biology [7]. Its experimental and computational methods shed light on the complex and causal layers of metabolism, where groups of molecules, especially proteins, work together. Until now, defining these molecular processes and their interactions has been challenging because they occur at a microscopic level that is difficult to study. Many researchers still adopt a reductionist approach, often producing causal explanations that are inaccurate or not supported by their data [8]. As a result, current literature lacks comprehensive in vivo data showing the chronological order and specific conditions under which these functional relationships occur. While in vitro studies offer valuable insights, they may not fully represent the complexities of protein behavior within a living system. For example, many interactions between proteins remain poorly understood, and further research using advanced methods is necessary to explain the "where," "how," and "when" of these interactions in a physiological context. To understand the functional roles of protein interactions, it is essential to define these parameters clearly, as current literature often does not adequately address them [9]. Recognizing this context is important because proteins can behave differently depending on their cellular environment and other molecules.
Many studies provide valuable initial insights, but they often fail to fully capture the complexities of protein interactions within living organisms [10]. By acknowledging these complexities and the limitations of our current knowledge in biomedicine, we can present a more nuanced understanding of our findings. A "nuanced understanding" goes beyond surface-level knowledge, allowing us to recognize and appreciate the intricacies and subtleties of an issue. There is a significant need for more comprehensive research that combines advanced experimental and computational techniques. This approach would bridge the gap between our current understanding and the complexities of biological interactions. It is essential in scientific discourse to recognize the limitations of our knowledge [11]. This humility encourages further inquiry and exploration and motivates researchers to seek more nuanced answers, rather than oversimplifying complex interactions.
2. The Principles Behind It
Today, biology is a data-driven science [12]. Systems biology uses massive, complex data models that are impossible for humans to analyze. In this, computer science, with its algorithms and large storage capacity, assists us. This help, however, progressively detaches the researcher from genuine scientific inquiry, potentially leading to negligence of essential research methodologies. For centuries, science has served as the foundation of human knowledge and relies on the direct observation of natural phenomena [13]. Observation generates the "data," the numerical representation of what we observe [14]. Information technology has gradually transformed the words provided into information.
Scientific literature is the most crucial source of human knowledge, generated by the scientific community through research. When researchers develop scientific projects, they select and collect sources of information that allow them to gather fragments of knowledge and rework them in planning hypotheses. However, this process is highly personal, carried out with a unique and specific criterion for each researcher, based on their scientific project and the argument they aim to pursue. When conducting research, the researcher focuses more on describing the topic than measuring it, aiming to develop a logical explanation for the "why" behind the results in that area of human knowledge [15]. In doing so, they place less emphasis on statistics and structured data, primarily using textual information to gain a deeper understanding of scientific motivations and behaviors driven by human emotions. This process involves evaluating opinions, viewpoints, and attributes, presenting significantly fewer concrete numbers in graphs or tables.
Researchers mainly perform this work today through text mining, or text data mining, which automatically extracts information from electronically published scientific publications. Text mining uses advanced deep learning algorithms to extract information. This process involves knowledge discovery in databases, information extraction, and data mining. Public archives contain large amounts of data related to biological systems, and even more data exists in semi-structured form in the literature [16]. Therefore, we believe that even experts manually populate databases in a curated manner. However, manual curation of literature and its inclusion in databases depend on the human characteristics of the editors [17]. Different editors may interpret the text of an article differently, mainly because they need to tailor it to the specific purposes of the curated database. The number of published articles, which is growing exponentially, makes it practically impossible to examine all of them completely and accurately [18]. Undoubtedly, the enormous number of scientific papers significantly affects researchers' ability to generate meaningful and testable hypotheses. It creates a bottleneck in scientific research processes. However, text mining quickly extracts scientific information, which helps develop new scientific hypotheses more easily [19]. Therefore, it is potentially an effective system for systematically reviewing a topic through direct and immediate observation.
But one detail eludes us. There is a significant difference between scientific information and data [20]. We represent observations with data: concrete and quantitative information, verifiable by coded reference quantities, which consistently produce similar results. We use a direct experimental approach to solve problems, applying scientific principles for data collection. This is the scientific method [12]. It relies on observation and experimentation, measurement, generating results through generalization (induction), and confirming them through multiple tests. Information is the transmission or reception of a notion or news collected or communicated for practical or immediate use, considered valuable or essential for the humanity that receives it [21]. This method employs algorithmic techniques to analyze extensive data and information. This method is regarded as an indirect analysis approach.
Scientists need to analyze scientific data, an objective, experimental, and repeatable representation of reality expressed through quantitative symbols. This data characterizes observable phenomena. Conversely, processing this data (the output) yields information that, through communication, creates knowledge about a specific subject. The formulation of a hypothesis, based on a scientific theory, occurs before experimental observation. The resulting experimental data serves as a verification process to confirm or refute the hypothesis. By using text mining, which relies on computer algorithms, we can extract scientific information, as algorithms evaluate the information as either true or false. To be considered scientifically valid, experimental data or some means of experimental interpretation must support information. This is where the concept of metadata becomes important [22]. Metadata, a term from computer science, refers to a set of information that describes the characteristics and accuracy of data. In databases, the reliability of this metadata often depends heavily on the work of curators [23]. For information to be statistically significant, it needs to be highly accurate [24]. Precision, in this context, refers to how consistent or close the results are when multiple pieces of information about the same object agree.
Precision refers to the likelihood of randomly selected information being a true positive [25]. We calculate this metric by dividing the number of true positives by the sum of true positives and false positives. In simpler terms, we can understand precision as the probability of accurately identifying a positive outcome. To compute precision, we divide the number of true positives by the total number of items expected, which is the sum of true positives and false negatives. When evaluating information in the literature, we often find that its precision is low, showing a high level of speculation [26]. Speculation involves curators thoughtfully considering a theoretical area of investigation and study, guiding their selection of material (information) to include in databases. It is important to remember that curators work for the databases they curate. As a result, curators often skip information standardization and normalization processes. This lack of standardization limits our ability to fully understand the extent of experimentally proven structure/function relationships (metadata) [27].
A systematic analysis of quantitative data (metadata) structured for statistical purposes can yield conclusive results and help us achieve our goals. This analysis allows for drawing conclusions and making informed decisions regarding a proposed course of action (i.e., the hypothesis we aim to prove). Methodologically, it is the most viable way to confirm or refute a predefined hypothesis. Researchers often rework and synthesize theories and concepts that others have already developed, using information sources to plan their hypotheses [28]. In this context, text mining and databases are among the most commonly used methods for documenting and planning scientific projects. However, they come with significant challenges. The ongoing risk is that we may contaminate the foundations of human knowledge with distorted or meaningless information.
2. Direct Experimental Method
Direct experimental methods for generating scientific knowledge rely on observable and repeatable laboratory techniques. These techniques include structural and biochemical assays, X-ray crystallography, cryo-electron microscopy (cryo-EM), mass spectrometry, and co-immunoprecipitation. The primary goal of these methods is to clarify specific relationships between structure and function, molecular interactions, and mechanistic insights within controlled environments. By providing measurable and directly observable data, these methods have historically formed the foundation of scientific knowledge, offering the most concrete validation of hypotheses. For example, we can directly measure the strength of protein interactions and visualize their combined shapes.
Scientists often use simplified systems for experiments, which may not fully represent the complexity of biological processes in living organisms. Proteins can interact differently depending on various cellular environments [30]. This variability makes it harder to characterize interactions clearly and highlights the need for more advanced and comprehensive study methods. Technical and financial limitations can restrict these methods, making system-level application difficult. Structural methods can be sensitive to ambiguous data, and phenomena such as epigenetic changes and post-translational modifications (PTMs) are tough to observe directly in vitro [31]. For example, in studying viral-human interactions, high-resolution techniques like cryo-electron microscopy (cryo-EM) can visualize viral protein binding; however, they might miss transient or context-dependent interactions that are crucial for understanding pathogenicity in vivo [32]. Misinterpreting or overextending findings from these models can lead to inaccurate conclusions when applied more broadly. The costs and long timelines needed to get these results further add to the challenges.
3. Indirect Algorithmic Analysis
Systems biology uses algorithmic methods to analyze large amounts of data from different omics fields, such as genomics, proteomics, transcriptomics, and interactomics, as well as biological interaction databases [33]. By applying machine learning, network analysis, and statistical modeling, these methods aim to find patterns, correlations, and possible causative links between biological entities without direct experimental validation [34]. Algorithmic approaches help researchers process big datasets, identify relationships within complex biological systems, and predict previously unknown interactions or functions [35]. This is especially helpful when analyzing thousands of genes, proteins, or pathways—information that would be difficult, if not impossible, to gather through direct experiments alone. These algorithmic predictions often depend on multiple data sources of varying quality, and inaccuracies, including assumptions from literature, can introduce systemic biases [36]. As a result, these analyses are indirect and often show observational correlations. Data biases or model-specific artifacts, which might be mistaken for real biological phenomena, can also influence these analyses. It's important to remember that correlation does not imply causation. For example, in interactome studies, a computational model might predict interactions between two proteins based on their co-expression or literature-reported associations. Without validation through direct experiments, such predictions can be misleading, potentially representing temporary or non-functional interactions. These predictions might overstate the importance of specific pathways or suggest incorrect biological functions.
In scientific research, particularly in biology, "indirect" refers to insights and conclusions not derived from direct observation or experimentation on the biological system. Computational analysis of existing data yields these insights, enabling researchers to make inferences or predictions.
Reliance on data and models: Algorithms process data such as gene sequences, protein structures, and gene expression levels that experiments, observations, or simulations. They then apply mathematical models, statistical methods, and computational logic to identify patterns, correlations, and make predictions [37].
Inference, not direct observation: The algorithm does not "see" a protein interacting with a DNA molecule in a test tube, nor does it directly measure a metabolic rate in a cell [38]. Instead, it might infer a protein-DNA interaction based on sequence homology, structural predictions, or co-expression patterns across large datasets.
Need for experimental validation: Since the analyses are indirect, their results are essentially hypotheses or predictions that require further direct experimental testing. For example, an algorithm may predict a new drug target. Still, scientists must then test this prediction in the lab through biochemical assays, cell cultures, or animal models to verify its practical effectiveness.
Abstraction and representation: We represent the biological system abstractly in the computer using numerical values, graphs, networks, or other data structures. The algorithm operates on these representations, not the biological reality itself [39].
Complementary, not replacement: Indirect algorithmic approaches are potent for generating hypotheses, prioritizing experiments, and identifying potential areas of interest within massive datasets [40]. However, they complement, not replace, direct experimental work, which provides the ultimate ground truth.
By using indirect algorithmic analysis, scientists can analyze and interpret large amounts of biological data, which helps guide their discoveries that are later confirmed through direct experimental investigations. Even though experiments are still the best way to verify how things work in biology, modern biology also uses computer models and new methods to study complex systems. These models can generate testable hypotheses that inform experimental design and help prioritize targets more confidently. Their effectiveness relies on transparent methodologies, high-quality data inputs, and ongoing refinement through iterative feedback from experiments.
4. The Role of the Experimental Method in the Era of Big Data
Experimental validation remains essential as systems biology increasingly depends on high-throughput data and algorithmic analysis. Algorithms can identify promising targets or pathways, but direct methods are necessary to verify the functional importance of these predictions [41]. We employ experimental techniques to assess the reliability of algorithmically inferred data. For example, co-immunoprecipitation or functional assays can confirm predicted interactions, helping to reduce false positives caused by computational assumptions. A balanced approach where computational predictions generate hypotheses that are then tested through direct experiments results in a stronger scientific foundation. For instance, computational predictions of protein-protein interactions in viral-host systems require thorough in vitro or in vivo validation to establish biological relevance in disease processes, such as those seen in COVID-19 pathology [42].
5. Dubious Interpretations in Literature: Examples of Failures Because of Overreliance on Computational Predictions
Many scientific articles use indirect methods to establish complex biological relationships, sometimes leading to conclusions that seem robust but lack experimental support. Key examples include, A) Protein-Protein Interaction Networks (PPINs), where large databases like BioGRID and STRING aggregate data from diverse studies, often mixing high-confidence experimental interactions with inferred ones [43]. Mixing the data misrepresents transient interactions or context-dependent associations as stable or universal relationships, thus creating "hairball" networks that obscure actual functional relevance [44]. A "hairball," in network visualization, describes a visually confusing graph with so many densely connected nodes and edges it is hard to understand. This complex network of connections makes it hard to uncover patterns, insights, and specific relationships within the data; B) Omics Data Analysis and Biomarker Discovery. Here, high-throughput omics analyses often identify potential biomarkers or therapeutic targets based on differential expression. However, a lack of biochemical validation reveals many of these biomarkers as false positives [45]. For instance, biomarkers for cancer diagnostics discovered solely through transcriptomic analysis frequently fail clinical validation, as they may reflect tissue-specific noise rather than disease-specific changes [46]; C) Drug Repurposing and Target Discovery. Computational methods to identify drug targets often rely on correlations in expression patterns or inferred pathway links without functional testing [47]. This approach led to initial suggestions that hydroxychloroquine might inhibit SARS-CoV-2 based on indirect interactions, which proved inaccurate in clinical trials [48].
6. Achieving Reliable Scientific Knowledge Through Balanced Approaches
A realistic approach recognizes that both experimental and computational methods are invaluable, but one should not rely solely on either. The synergy between computational predictions and experimental validation fosters a cycle of hypothesis generation and testing that leads to more grounded, reliable knowledge. Ideally, predictions from Systems Biology should iteratively cycle through hypothesis, testing, and refinement [49]. This might mean shifting from one-off validations to dynamic updates as models improve or as new experimental findings emerge. Also, experimental resources should focus on validating highly affected predictions to balance exploratory science and practical verification. Finally, to enhance interpretative clarity, scientific publications should include confidence levels of computational predictions, validation statuses, and any limitations specific to the datasets or methods used [50]. The lower the confidence level, the larger the interaction until it becomes a hairball. In conclusion, as we grapple with Big Data in biology, computational methods offer an unprecedented ability to generate hypotheses. Still, they cannot replace direct experimental validation's foundational, interpretative clarity. Each approach enriches our understanding when balanced by the other, ensuring that scientific knowledge does not remain a hairball [51].
What do algorithms do to determine the efficiency in the calculation of interactomes?
Interactome computational algorithms analyze the interactions of proteins and other biomolecules within cells. We can assess the effectiveness of these algorithms in several ways: accuracy, computational speed, scalability, resource use, robustness, and interoperability [52]. An efficient algorithm should have high accuracy, minimize false positives and negatives, and process information quickly, even with extensive datasets. The accuracy of interactome calculation algorithms is essential to ensure that predictions are reliable and valuable for biomedical research. Here's how experimental data contributes to improving accuracy:
Model validation: While experimental validation remains a cornerstone of biomedical research, the developing field of systems biology increasingly shows that well-designed computational models can serve as powerful hypothesis-generating tools, often predicting interactions, pathways, or targets that are experimentally testable even before laboratory validation [53]. These models, built on integrative, multi-omics data, can guide targeted experiments, saving time and resources, and enhancing the iterative cycle of discovery. We must confirm these predictions experimentally to establish their biological relevance and accuracy. Experimental data is used to validate predictive models of algorithms [54]. Researchers can identify and correct discrepancies by comparing the algorithm's predictions with experimental results.
Error reduction: Experimental data helps identify and reduce errors in predictive models. For example, researchers can revise and improve the model if an algorithm predicts an interaction that is not observed experimentally [55].
Parameter improvement: Experimental data provide valuable information about biological parameters, such as binding affinities and protein concentrations [56]. This information can optimize algorithm parameters, improving their accuracy.
Adaptation to real data: Experimental data represent the conditions under which protein interactions occur [57]. Using this data, we can adapt algorithms to reflect real-world biological situations better, increasing the' accuracy of predictions.
Cross-validation: Researchers can use experimental data to perform cross-validation techniques, dividing the data into training and test sets [58]. This helps assess the algorithm's accuracy of unseen data and prevents overfitting.
Experimental data is essential to ensure that interactome computation algorithms are accurate and reliable. This data allows predictive models to be validated, optimized, and adapted, thus improving their usefulness in biomedical research. In addition, an algorithm must work effectively with small and large volumes of data, efficiently using computing resources, such as memory and processor power. Optimized algorithms minimize resource use without compromising performance. An algorithm should possess the ability to maintain high performance even in the presence of noisy or incomplete data [59]. Finally, it should be robust enough to withstand data anomalies.
7. Computer and Automatic Methods in Biology
In our era, data availability has grown exponentially thanks to computer technologies and text mining. It is undeniable that computer science and data mining algorithms have revolutionized the way researchers can analyze and interpret large amounts of biological data [60,61]. This has led to increased scientific productivity and opened new avenues for the discovery and understanding of complex biological phenomena. However, this growing dependence on computer science distracts researchers from the fundamental core of scientific research, the correct method, and in-depth understanding of biological phenomena. Automated text analysis may cause a loss of quality and precision in extracted information, because algorithm interpretations are susceptible to human variability and the specific context of each database [62]. The increasing emphasis on textual information can sometimes neglect quantitative and structured data, which is essential for verifying and validating scientific hypotheses [63]. Without a solid basis of experimental and quantitative data, the conclusions, and hypotheses planned can be fragile and subject to misinterpretation.
In biomedical sciences, computer methods have become increasingly common and have opened new opportunities for analyzing biological data. However, it is important to understand that computer science does not fully replace the experimental method, but complements and enhances its potential [64]. Laboratory experiments provide concrete and repeatable data that are essential for validating scientific hypotheses. Although computer methods can aid in data collection and analysis, it is often necessary to confirm and deepen these findings through direct experience [65]. The exponential growth of data and the widespread use of computer tools can pose risks of distortion, misinterpretation, or even manipulation of scientific information [66]. We can no longer consistently rely on a complete understanding of the published literature. Because the exponential trend is unlikely to slow down, automatic information retrieval tools, such as text mining, have quickly emerged.
8. The Contribution of AI
The often-mentioned artificial intelligences, while powerful tools for data analysis and information processing, also rely on the quality of the data and information they receive [67]. Responsible training and use of artificial intelligences are essential. This includes critically evaluating input data, validating sources and methods used in analyses, and ensuring transparency in the decision-making process of artificial intelligences. If data becomes contaminated or distorted, artificial intelligences may produce equally unreliable conclusions and answers [68].
Artificial intelligence can help address scientific knowledge pollution [69] in several ways. Programmers can use AI to identify and filter unreliable or distorted data sources, ensuring quality data for scientific analysis. We can train AI systems to evaluate key scientific sources and spot biases or discrepancies in data and conclusions.
Using anomaly detection algorithms [70,71], an AI can identify discrepancies or anomalies in scientific data that may indicate knowledge pollution. It can continuously monitor the quality of scientific databases, reporting anomalies or significant changes in archived data. It can assist the peer review process by detecting potential weaknesses in a scientific study's methods or findings. The reproducibility of the authors' method, assessed through the data presented, could enable quick intervention for complex calculations. This should serve as a supplementary tool for reviewers, not a replacement. Artificial intelligence can also help develop advanced analytical tools, enhancing the quality and reliability of scientific data interpretation [72]. Properly trained AI can help reduce pollution in scientific knowledge by providing tools and techniques that effectively identify, filter, and analyze data. It can also promote transparency and openness in science by enabling data sharing and dissemination of results, ensuring access to trustworthy information.
9. How to Mitigate Pollution of Scientific Knowledge and Enhance the Effectiveness of Computational Algorithms
To preserve the integrity and reliability of scientific knowledge, especially in complex fields like systems biology, it is crucial to implement rigorous data collection, curation, and verification procedures. These measures include:
- Replication of experiments: Repeating studies under varying conditions to confirm findings.
- Use of Adequate Controls: Ensuring experimental designs account for confounding factors.
- Transparency: Sharing raw data, protocols, and analysis methods openly for peer validation.
- Promotion of Data Sharing: Encouraging researchers to deposit datasets in accessible repositories facilitates cross-verification.
- Role of Algorithms in Interaction Networks: Computational algorithms are central to constructing and refining interactomes and comprehensive maps of molecular interactions. We can evaluate their performance based on several key criteria:
- Accuracy: Correctly predicting true biological interactions while minimizing false positives [73].
- Computational Speed: Rapid processing is vital given the vast size of biological datasets.
- Scalability: Algorithms should handle increasing data volumes without performance loss [74].
- Resource Efficiency: Optimizing memory and processing power to facilitate workable analysis [75].
- Robustness: Maintaining performance despite noisy, incomplete, or conflicting data [76].
It is crucial to recognize that ongoing advancements in algorithms, validation protocols, and data integration platforms are actively addressing many current limitations related to data quality and interpretative ambiguities [77]. For example, advanced machine learning techniques and improved bioinformatics pipelines now include multiple levels of data authentication, cross-validation, and confidence scoring, which help reduce false positives and assess prediction reliability more systematically [78]. These improvements are gradually strengthening the robustness of computational predictions, although challenges still exist.
10. Practical Approaches
Here are some practical tips for using various control tools.
STRING Database and Confidence Scoring: STRING (https://string-db.org/) combines data from various sources, including experimental data, curated databases, text mining, co-expression, and gene neighborhood, to predict interactions [79,80]. Algorithms assign confidence scores to these interactions; for instance, a high-confidence cutoff (e.g., 0.900) filters out less reliable data but may also exclude meaningful yet less-studied interactions. Finding the right balance is essential; overly strict filters can oversimplify the extensive networks ("hairballs') and hide biological truths.
Model Validation Using Experimental Data: Algorithms frequently produce hypotheses about protein interactions that researchers need to validate through experiments. For instance, co-immunoprecipitation assays confirm predicted interactions [81]. Without such validation, predictions may include false positives, interactions suggested by algorithms but not occurring biologically, leading to misleading "hairball" networks that are difficult to interpret.
Algorithmic Challenges [82]:
- Dealing with Noisy Data: Biological datasets often contain experimental artifacts or context-dependent interactions; algorithms must distinguish genuine signals.
- Handling Incomplete Data: We do not know all interactions. Algorithms should predict missing links without overfitting known data.
- Resource Management: Developers must optimize advanced algorithms, such as graph-based machine learning models, for efficient operation on multi-terabyte datasets.
Biological datasets often contain experimental artifacts, noise, or context-dependent interactions, which can challenge the accuracy of computational models [83]. However, new algorithms employing ensemble methods, Bayesian approaches, and integrative platforms can distinguish accurate biological signals from such confounders, improving the reliability of interaction predictions [84].
10. Advances and Future Directions
Machine learning and artificial intelligence developments have introduced more sophisticated algorithms capable of integrating diverse datasets for more accurate interaction prediction. For example, deep learning models trained on experimentally validated interaction datasets can uncover subtle patterns that traditional methods miss. They remain dependent on high-quality input data and transparent methodologies to ensure reliability. Computational algorithms are powerful tools for elucidating biological networks, but their success hinges on rigorous validation, careful parameterization, and transparent reporting. Ongoing improvements in algorithm design, combined with experimental validation, are essential to advancing trustworthy systems biology research [85].
We should adopt several preventive measures to maintain the integrity and reliability of scientific knowledge and reduce the risk of pollution. Establishing thorough procedures for collecting, curating, and verifying scientific data is essential. This might include replicating experiments, using proper controls, and ensuring data collection and analysis transparency. We should encourage scientists to share their data, methods, and results for verification and replication by other researchers [86]. This fosters openness and reproducibility of scientific findings. We should also teach researchers the best data handling and research practices. This includes paying more attention to statistics and data analysis, and recognizing potential biases and distortions. However, scientific institutions and funding agencies must implement oversight and monitoring mechanisms to ensure researchers adhere to ethical and scientific standards [87]. Encouraging collaboration among researchers and integrating peer review as a key part of the scientific process can help find and fix errors or inconsistencies in results. Ongoing investment in developing advanced tools and methods for data analysis and interpretation can improve the quality and trustworthiness of scientific research.
Implementing these measures requires a long-term commitment from the scientific community, academic institutions, funders, and government agencies. It is a gradual process that requires time, resources, and coordination, but it is essential to ensure that scientific knowledge remains robust, reliable, and at the service of human progress. However, we need simple protocols for efficiently conducting necessary validity checks, primarily on the data used for planning scientific projects.
11. The Need for a Control Tool
Let us illustrate with a practical example from interactomics, a field that heavily depends on experimental data. Datasets on protein-protein interactions are manually curated and derived from the literature [88]. The systems biology community uses them as the gold standard, and they establish practices to extract parameters for mechanistic models from the literature. Sometimes, data extraction faces limitations because some journals do not grant access to the full text because of copyright restrictions, allowing only access to abstracts and titles. This restricts the use of data and information that must be based on experimental results. The frequent presence of datasets on highly studied proteins creates a large body of knowledge [89]. However, statistical analysis often dismisses data from less-studied proteins as insignificant background noise, leading to their exclusion [90].
11.1. Interactomics as a Tool for Controlling Metabolic Analyses
Recently, researchers published a metabolomic study [91] in the prestigious Nature Medicine. This study showed that blood contains metabolites from the breakdown of niacin (vitamin B3), mainly when people absorb excess vitamin through dietary supplements. These metabolites strongly link to heart attacks, strokes, and other adverse cardiac events. This suggests that niacin supplementation, which is very common among humans, may require a controlled clinical and quantitative approach. The researchers, therefore, issued a clear warning to the medical community. The two metabolites, identified by mass spectrometry as N1-methyl-2-pyridone-5-carboxamide (2PY) and N1-methyl-4-pyridone-3-carboxamide (4PY), were associated with the expression of a soluble vascular adhesion protein (sVCAM-1). Plasma levels of the two metabolites, end products of excess niacin metabolism, were linked to an increased risk of cardiovascular disease (MACE) at 3 years in two validation cohorts, in which a genetic variant (rs10496731) appeared to be significantly associated with sVCAM-1 levels. The authors also found that 4PY, but not 2PY, was directly responsible for inducing VCAM-1 expression in the vascular endothelium in mice, suggesting an inflammation-dependent molecular causal mechanism related to cardiovascular risk. In functional assays, a physiological level of 4PY induced the expression of VCAM-1 mRNA and proteins on human endothelial cells. Nicotinamide-riboside and nicotinamide mononucleotide also increased plasma levels of 2PY and 4PY. These observations have raised many concerns about using niacin-containing supplements in inflammatory processes. The researchers' efforts were significant, as was the experimental design they followed.
A simple way to verify this information is to use interactomics. Interactomics is an analytical tool and a true epistemological filter that helps distinguish between speculative hypotheses and solid experimental data [92]. A set of principles, methods, and assumptions that shape scientific data analysis and, indirectly, the development of reliable knowledge governs it. It acts as a set of algorithmic lenses that guide us in selecting, organizing, and interpreting information, influencing what we consider valid and meaningful [93].
The paradox of scientific computing is that the more data we have, the more we risk building castles on sand if we don't check the quality of the foundations [94]. Automated analysis can generate fascinating networks and correlations, but we cannot attribute certain biological functions without experimental confirmation. The algorithm can suggest, but it cannot replace the scientific method. Experimental validation remains at the heart of molecular biology. Without it, we risk confusing correlation with causality and hypothesis with truth.
One of the best and most well-known interactome tools is STRING. STRING is a biological network analysis system that collects physical and functional biological interactions and uses seven different sources of data and information [62,63]. Using a low confidence score and all seven open channels on STRING [95], we get a statistically significant interactome after enrichment; this interactome likely includes all the metabolic implications for the proteins (nodes) found in the literature. However, if we increase the confidence score to 0.900 and close the text mining and annotated database channels, we can isolate the most significant data. However, we could also drastically reduce the number of connected nodes and the number of edges. In that case, we would not confirm most metabolic relationships.
Incorporating interactomics as a verification tool, rather than just for exploration, might initially seem unusual. However, using STRING and similar systems to test the robustness of hypotheses before developing experimental models can be very helpful. Let's examine how to apply STRING's "algorithmic lenses" to validate the findings reported in the article published in Nature Medicine.
In their research, the authors performed a transcriptomic analysis of human endothelial cells exposed to 2PY or 4PY. Their aim was to identify the genes induced and associated with VCAM1 at the physiological level of the metabolites. Bioinformatic analyses of differentially expressed genes showed an enrichment of various transcriptional regulation and signaling pathways. Along with VCAM1, this included genes such as BEST1, WNT, AKT1, MAPK3 (refer to Figures 5 and 8 of the article), and ACMSD, which control the de novo synthesis of NAD.
We used these genes as functional seeds in our interactomics analysis to identify the functional relationships they have within the entire human proteome. To do this, we applied an average confidence score of 0.400, kept all seven active interaction sources open (Text mining, Experiments, Databases, Co-expression, Neighborhood, Gene Fusion, and Co-occurrence), and included 500 first-order proteins (direct interactions) and 500 second-order proteins (indirect interactions) in the analysis. In Figure 1, we show the interactome generated by STRING.
The interactome, calculated by STRING using data from over 10,000 publications on PubMed (freely available), is highly compact, featuring 1,006 nodes and 74864 edges. It has an average node degree of 149 and a p-value less than 1.0e-16. Among the most significant hub nodes are VCAM1, AKT1, and MAPK3, with 391, 757, and 574 interactions, respectively. The interactome includes 9,107 terms across 15 functional categories. The most significant Biological Processes (see Figure 2) include all the "functional concerns" identified by the authors.
We observe a link between VCAM1 and AKT1 and cardiovascular conditions (DOID:1287 Cardiovascular system disease), supported by 110 of 493 relevant genes, a signal value of 2.28, and a p-value of 6.37e-32. By focusing on the interactome centered on VCAM1 using STRING's specific functionality, we can identify VCAM1's unique interactome, highlighting the functional relationships within the human proteome (see Figure 3).
The interactome has confidence 0.400, and all seven channels are open. We get a compact graph of 972 nodes, 80,262 edges, 9,051 functional terms, again calculated from PubMed's 10,000 publications. However, if we pass the confidence score to 0.900 to select the most significant and reliable data, eliminating Textmining and Databases, we have (Figure 4) only one node, VCAM1.
Applying strict criteria (confidence score ≥ 0.900 and excluding text mining and databases), the STRING analysis shows that VCAM-1 has no interactions within the human proteome. This finding, which conflicts with the current functional consensus, forces us to reassess the translational validity of the proposed model. The exportable TVS file from STRING displays the key information. In the interactome, VCAM-1 shows 471 functional interactions. Table 1 shows the framework of experimentally validated functional interactions.
PubMed lacks vital experimental data on VCAM1 interactions across the human proteome. Filling this gap is essential for better understanding the functional relationships discussed by the authors of the Nature paper and their implications for human health. It is concerning that, without reliable experimental data on any of the 471 VCAM 1 interactions, the information from annotated databases and text mining has a confidence score of nearly 0.900. All this raises important questions about the trustworthiness of these findings and their impact on our understanding of VCAM1 functions. Therefore, we cannot definitively assign a function to VCAM1 in humans at this stage. Understanding its role depends on identifying its interacting proteins. The cellular systems used as models are often based on hypotheses rather than validated experimental evidence. Scientific research accuracy and functional analyses directly depend on the reliability of the information used. The exponential growth of data from IT and text mining creates a significant challenge, especially today. Computer science and data mining algorithms have revolutionized how researchers analyze and interpret large biological datasets. This progress has boosted scientific productivity and opened new avenues for exploring complex biological phenomena. However, over-reliance on technology risks distracting researchers from core scientific principles, emphasizing proper methods and a deep understanding of biological processes. Automated text analysis can sometimes compromise data quality and accuracy, as human factors and the specific context of each database influence algorithm results. The growing focus on textual data may lead to neglecting quantitative and structured data, which is crucial for testing and validating hypotheses. Without a solid foundation of experimental and quantitative data, conclusions, and assumptions risk becoming weak and misleading.
The Nature Medicine study is impressive in its breadth and rigor, but the use of non-experimentally validated databases for VCAM-1 compromised the robustness of the conclusions. The real problem is that, to plan the development of their project, the authors used a set of "information" stored in different databases, without carrying out any specific verification to verify whether the data of the physical/functional interactions related to VCAM-1 had an experimental, significant, and specific origin. 4PY induces VCAM-1 expression in vitro and mouse models. However, the observed effect does not guarantee a defined biological role for VCAM-1 in humans without knowing its interaction network. A protein's function arises from its molecular context; without interactions, it has no assignable function. Functional analysis vs. molecular analysis is asymmetric. Researchers investigate various aspects of biochemical and biological interactions. Functional analysis emphasizes biological functions, while molecular analysis focuses on chemical and physical characteristics of molecules. The methods and tools used in molecular analysis differ from those used in functional analysis, leading to distinct approaches in technique and interpretation. Interactomics underscores this disparity in biological interactions, where the functions and molecular characteristics do not always align.
In this context, STRING is a testing platform that improves the confidence score while filtering out non-experimental sources. It isolates VCAM-1 and shows that there are no confirmed interactions, which raises a methodological concern. The paradox of scientific computing is that, despite having more data, we risk building theories on unstable foundations if we do not evaluate the data quality. Automated analysis can generate interesting networks and correlations, but we cannot accurately assign specific biological functions without experimental validation.
VCAM-1 becomes a prime example: we cannot determine its functions if we do not understand its interactions. The definition of VCAM-1's role depends on a methodological approach based on interactions researchers have not yet confirmed experimentally. While text mining algorithms can provide suggestions, they cannot replace the scientific method. Experimental validation is essential to molecular biology; without it, we risk conflating correlation with causation and hypothesis with fact.
Perhaps our major challenge today is incorporating interactomics as a verification tool rather than solely for exploration. Using STRING and similar resources to test the robustness of hypotheses before creating experimental models could develop into a valuable scientific practice.
12. Recommendations and Practical Frameworks for Ensuring Balance
Because of limited resources, researchers can use some practical approaches.
- Prioritize High-Confidence Predictions for Validation: Use computational scoring systems (like confidence scores in STRING) to select the most promising hypotheses for experimental validation. Focus on interactions or targets with high confidence, reducing costs associated with testing less likely candidates.
- Sequential Validation Strategy:
- Step 1: Use in silico methods to generate hypotheses [97].
- Step 2: Apply secondary computational filters (e.g., cross-validation across datasets, orthogonal methods) to refine predictions [98].
- Step 3: Experimentally validate only the top-tier predictions, such as through targeted assays or minimal confirmatory experiments.
- 3.
- Use Collaborations and Shared Resources: Partner with institutions or consortia that can offer access to specialized experimental platforms or datasets, which help to lessen individual resource burdens. Taking part in shared repositories or consortium initiatives can make validation more cost-effective.
- 4.
- Implement a Hypothesis-Driven Approach [99]: Restrict computational analysis to well-defined hypotheses rather than broad exploratory searches. This focused approach minimizes unnecessary experiments and maximizes resource efficiency.
- 5.
- Emphasize Open Science and Data Sharing: Share datasets and validation results transparently. This prevents redundant efforts, helps refine models collectively, and speeds up validation efforts [100].
- 6.
- Use Computational Validation as a Filter, Not an Ultimate Authority: Recognize computational predictions as hypotheses rather than conclusions. Establish a workflow incorporating initial predictions, systematic prioritization, and targeted experimental validation. This layered approach optimizes resource expenditure.
- 7.
- Incorporate training on data interpretation and biases. Researchers should receive training on data interpretation and biases. To avoid misinterpreting computational results, they must understand algorithms' limitations and potential biases and interpret findings critically.
A balanced strategy incorporates computational methods to refine hypotheses, uses objective confidence metrics, collaborates to share validation responsibilities, and prioritizes predictions that are highly affected and confident for experimental testing. This approach significantly strengthens scientific rigor while effectively addressing resource limitations.
13. Interactomics as a Criterion for Scientific Validation
Many studies identify metabolites or proteins, or genes related to disease states. Although the experimental design is often robust, the molecular model does not rely on experimentally confirmed protein interaction data.
Interactomic verification protocol: from hypothesis to confirmation
1. Biological Goal Definition:
- Identify the target protein (e.g., VCAM-1) and pathological/biological context (e.g., vascular inflammation). Determine if the proposed function is observed or merely hypothesized.
2. Extracting interactions from STRING
First phase (exploratory):
Low confidence score (≥0.150).
All active channels (including text mining and predictive sources).
Aim: to identify potential relationships and functional enrichments.
Second phase (confirmatory):
Confidence score, high (≥0.900).
Experimental channels only (exclude text mining and annotated databases).
Aim: to validate the physicochemical relationships observed.
Note: STRING shows how the interactome varies significantly depending on the source and confidence score. At lower scores and with active text mining, metabolic networks consistent with the hypothesis appear; however, with more stringent criteria, the identified gene, metabolite, or protein lacks documented interactions. This creates an epistemological paradox: We only assume the protein's biological relevance in permissive computational, not experimental, environments.
3. Interactome topology analysis
Verify:
Key nodes (hubs) and connectivity.
Enriched pathways (Reactome, KEGG).
P-value of significance for the global network.
4. Cross-check with other sources
BioGRID, IntAct, Human Protein Atlas.
PubMed: search for experimental articles (not just reviews).
Proteome and phosphoproteome in relevant cell/tissue models.
The casual use of mixed databases can cause the creation of biological models lacking a real molecular basis. Accepting interactions predicted or derived from text mining without physical-chemical verification risks producing an "illusory biology" effect, where nodes appear functional. This virtual connection alone can cause such an effect.
5. Translational validation
Re-contextualize interactions in biological models:
Mouse models, human cell lines, and primary tissues.
To test the effect of metabolites on proteins with confirmed interactions.
6. Epistemological revision
Classify your data into three categories:
Confirmed Experimental
Predicted computational
Unverifiable
Discuss the impact of each on the validity of the hypothesis.
7 Methodological proposals
Clearly distinguish the sources of interaction in experimental designs.
Use interactomics as a robustness test: If a protein lacks confirmed interactions, we cannot accurately attribute its function. Re-evaluate the translational validity of cellular models: the induced effect (e.g., VCAM-1 expression) does not imply that the protein has a functional endogenous role.
8. Final Considerations
Interactomics is a valuable exploration tool and an essential epistemological criterion [101]. In network science, a key challenge is not only integrating data and information from various experiments involving genes or proteins, but also ensuring the accuracy of these integrations [102]. Validated experimental networks are vital for correctly assigning protein functions; biological hypotheses remain speculative without them. Preserving the integrity of the scientific method is crucial, especially in the era of big data. This structured approach, tailored to needs, could help build robust and verifiable biological models.
So, we use STRING and similar sources as tools for falsification, not just exploration. The proposal is to introduce a "molecular verifiability index" in the design of experiments. This aims to distinguish functional induction from biological relevance. Computational data is an invaluable resource. However, even the most elegant model is unacceptable without experimental verification. Recognizing the limitations of predictive interactomics allows us to improve real-world data further and develop more robust, humane, and scientific hypotheses.
14. The Principle of Falsification
When we speak of a "tool of falsification," we refer to a fundamental concept of the philosophy of science, in particular the thought of Karl Popper [103]. For Popper, a scientific theory is only scientific if one can falsify it; that is, if one can test it to disprove it [104]. In computational biology, researchers often use STRING and similar platforms to explore biological networks; they select proteins, expand nodes, search for interactions, and build models. This approach is practical, but it has a risk: it can generate speculative networks, also built on predictive sources, text mining, and databases, which do not have an experimental basis. When using STRING with stringent parameters, for example, a confidence score of ≥0.900 and only experimental sources, we use the tool falsified. Not to discover what might be true, but to check if real data supported something. If a protein like VCAM-1 does not show confirmed interactions, we cannot attribute a specific molecular function to it. In this sense, STRING becomes a scientific test bed: not to build hypotheses, but to test them severely.
The principle of falsification in scientific review is essential for evaluation. We should not penalize bold hypothesis creators; we must make clear distinctions between data and deductions. The boundary between observed data and interpreted data is where critical thinking truly emerges, based on three methodological principles:
1. Epistemological clarity: This principle requires interaction.
2. Conscious use of databases: We should use databases as tools for control, not just for justification.
3. Distinction between induced effects and endogenous functions: The induced effect responds to external factors, while the endogenous function describes a dynamic within the system.
Popper was well aware of all this. He expressed it in these words: "Clinical observations, like all other observations, are interpretations in the light of theories; and for this reason alone, they are apt to seem to support those theories in the light of which they were interpreted. But real support can be obtained only from observations undertaken as tests (by "attempted refutations"); and for this purpose, criteria of refutation have to be laid down beforehand; it must be agreed which observable situations, if actually observed, mean that the theory is refuted [103]."
References
- Yang, X., Huang, K., Yang, D., Zhao, W., Zhou, X. "Biomedical big data technologies, applications, and challenges for precision medicine: a review." Global Challenges 8.1 (2024): 2300163. [CrossRef]
- Jafari, M., Guan, Y., Wedge, D.C., Ansari-Pour, N. "Re-evaluating experimental validation in the Big Data Era: a conceptual argument." Genome Biology 22 (2021): 1-6. [CrossRef]
- Iqbal, R., Doctor, F., More, B., Mahmud, S., & Yousuf, U. (2020). Big Data analytics and Computational Intelligence for Cyber–Physical Systems: Recent trends and state of the art applications. Future Generation Computer Systems, 105, 766-778. [CrossRef]
- Martin-Sanchez, F., Iakovidis, I., Nørager, S., Maojo, V., de Groen, P., Van der Lei, J., Jones, T., Abraham-Fuchs, K., Apweiler, R., Babic, A., et al. (2004). Synergy between medical informatics and bioinformatics: facilitating genomic medicine for future health care. Journal of biomedical informatics, 37(1), 30-42. [CrossRef]
- Preim, Bernhard, and Charl P. Botha. Visual computing for medicine: theory, algorithms, and applications. Second Edition (2014), Elsevier. ISBN: 978-0-12-415873-3.
- Altaf-Ul-Amin, M., Afendi, F. M., Kiboi, S. K., & Kanaya, S. (2014). Systems biology in the context of big data and networks. BioMed research international, 2014(1), 428570. [CrossRef]
- Kirschner, Marc W. "The meaning of systems biology." Cell 121.4 (2005): 503-504. [CrossRef]
- Kaiser, Marie I. Reductive explanation in the biological sciences. (2015) History, Philosophy and Theory of the Life Sciences (HPTL), Cham: Springer International Publishing. [CrossRef]
- Colonna, G. Overcoming Barriers in Cancer Biology Research: Current Limitations and Solutions. Cancers 2025, 17, 2102. [CrossRef]
- Van Segbroeck, S.; De Jong, S.; Nowé, A.; Santos, F.C.; Lenaerts, T. Learning to coordinate in complex networks. Adapt. Behav. (2010), 18, 416–427. [CrossRef]
- Ioannidis, John PA. "Limitations are not properly acknowledged in the scientific literature." Journal of clinical epidemiology 60.4 (2007): 324-329. [CrossRef]
- Strasser, B. J. (2012). Data-driven sciences: From wonder cabinets to electronic databases. Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences, 43(1), 85-87. [CrossRef]
- Windelband, Wilhelm. "History and natural science." Theory & Psychology 8.1 (1998): 5-22. [CrossRef]
- Stoelinga, M. T., Hobbs, P. V., Mass, C. F., Locatelli, J. D., Colle, B. A., Houze Jr, R. A., Rangno, A.L., Bond, N.A., Smull, B.F., Rasmussen, R.M., et al. (2003). Improvement of microphysical parameterization through observational verification experiment. Bulletin of the American Meteorological Society, 84(12), 1807-1826. [CrossRef]
- McGregor, S. L. (2017). Understanding and evaluating research: A critical guide. Sage Publications.
- Mullins, Simon, and Sean A. Spence. "Re-examining thought insertion: Semi-structured literature review and conceptual analysis." The British Journal of Psychiatry 182.4 (2003): 293-298. [CrossRef]
- Dahdul, Wasila M., et al. "Evolutionary characters, phenotypes and ontologies: curating data from the systematic biology literature." PLoS One 5.5 (2010): e10708. ttps://doi.org/10.1371/journal.pone.0010708.
- Szalay, Alexander, and Jim Gray. "Science in an exponential world." Nature 440.7083 (2006): 413-414. http://research.microsoft.com/towards2020science.
- Salloum, S.A., Al-Emran, M., Monem, A.A., Shaalan, K. (2018). Using Text Mining Techniques for Extracting Information from Research Articles. In: Shaalan, K., Hassanien, A., Tolba, F. (eds) Intelligent Natural Language Processing: Trends and Applications. Studies in Computational Intelligence, vol 740. Springer, Cham. [CrossRef]
- Leonelli, Sabina, "Scientific Research and Big Data", The Stanford Encyclopedia of Philosophy (Summer 2020 Edition), Edward N. Zalta (ed.). https://plato.stanford.edu/archives/sum2020/entries/science-big-data/.
- Capurro, Rafael. "Past, present, and future of the concept of information." TripleC: communication, capitalism & critique. open access journal for a global sustainable information society 7.2 (2009): 125-141. [CrossRef]
- Mayernik, Matthew S. "Metadata." KO Knowledge Organization 47.8 (2021): 696-713. [CrossRef]
- Buneman, P., Cheney, J., Tan, W. C., & Vansummeren, S. (2008, June). Curated databases. In Proceedings of the twenty-seventh ACM SIGMOD-SIGACT-SIGART symposium on Principles of database systems (pp. 1-12). [CrossRef]
- Lemson, Joris, and Willem P. de Boode. "Importance of using the correct statistics." Archives of Disease in Childhood. Fetal and Neonatal Edition (2024). [CrossRef]
- Zhang, Peng, and Wanhua Su. "Statistical inference on recall, precision and average precision under random selection." 2012 9th international conference on fuzzy systems and knowledge discovery. IEEE, 2012. pp. 1348-1352. [CrossRef]
- Haase, Marco, Yvonne Seiler Zimmermann, and Heinz Zimmermann. "The impact of speculation on commodity futures markets–A review of the findings of 100 empirical studies." Journal of Commodity Markets 3.1 (2016): 1-15. [CrossRef]
- Zheng, H., Porebski, P. J., Grabowski, M., Cooper, D. R., & Minor, W. (2017). Databases, repositories, and other data resources in structural biology. Protein Crystallography: Methods and Protocols, 643-665. Methods in Molecular Biology ((MIMB, volume 1607)). [CrossRef]
- Storey, M. A. (2006). Theories, tools and research methods in program comprehension: past, present and future. Software Quality Journal, 14, 187-208. [CrossRef]
- Hepburn, Brian and Hanne Andersen, "Scientific Method", The Stanford Encyclopedia of Philosophy (Summer 2021 Edition), Edward N. Zalta (ed.). ISSN 1095-5054 https://plato.stanford.edu/archives/sum2021/entries/scientific-method/.
- Keskin, O., Gursoy, A., Ma, B., & Nussinov, R. (2008). Principles of protein− protein interactions: what are the preferred ways for proteins to interact? Chemical reviews, 108(4), 1225-1244. [CrossRef]
- Ramazi, S., & Zahiri, J. (2021). Post-translational modifications in proteins: resources, tools and prediction methods. Database, 2021, baab012. [CrossRef]
- Gordon, D. E., Hiatt, J., Bouhaddou, M., Rezelj, V. V., Ulferts, S., Braberg, H., Jureka, A., Obernier, K., Guo, J.Z., Batra, J., et al. (2020). Comparative host-coronavirus protein interaction networks reveal pan-viral disease mechanisms. Science, 370(6521), eabe9403. [CrossRef]
- Tolani, P., Gupta, S., Yadav, K., Aggarwal, S., & Yadav, A. K. (2021). Big data, integrative omics and network biology. Advances in protein chemistry and structural biology, 127, 127-160. [CrossRef]
- Lecca, P. (2021). Machine learning for causal inference in biological networks: perspectives of this challenge. Frontiers in Bioinformatics, 1, 746712. [CrossRef]
- Koyutürk, M. (2010). Algorithmic and analytical methods in network biology. Wiley Interdisciplinary Reviews: Systems Biology and Medicine, 2(3), 277-292. [CrossRef]
- Arab, Ali. "Knowledge discovery: from correlation to causation." (2024). (Thesis) Ph.D., Simon Fraser University, Computing Science Theses. Identifier etd23245.
- Ratner, Bruce. Statistical and machine-learning data mining: Techniques for better predictive modeling and analysis of big data. Chapman and Hall/CRC, 2017. [CrossRef]
- Miteva, Yana V., Hanna G. Budayeva, and Ileana M. Cristea. "Proteomics-based methods for discovery, quantification, and validation of protein–protein interactions." Analytical chemistry 85.2 (2013): 749-768. [CrossRef]
- Cardelli, L. (2005). Abstract machines of systems biology. In Transactions on Computational Systems Biology III (pp. 145-168). Berlin, Heidelberg: Springer Berlin Heidelberg. [CrossRef]
- Sarker, I. H. (2021). Machine learning: Algorithms, real-world applications and research directions. SN computer science, 2(3), 160. [CrossRef]
- Agamah, F. E., Mazandu, G. K., Hassan, R., Bope, C. D., Thomford, N. E., Ghansah, A., & Chimusa, E. R. (2020). Computational/in silico methods in drug target and lead prediction. Briefings in bioinformatics, 21(5), 1663-1675. [CrossRef]
- Messina, F., Giombini, E., Agrati, C., Vairo, F., Ascoli Bartoli, T., Al Moghazi, S., Piacentini, M., Locatelli, F., Kobinger, G., Maeurer, M., et al. (2020). COVID-19: viral–host interactome analyzed by network based-approach model to study pathogenesis of SARS-CoV-2 infection. Journal of translational medicine, 18, 1-10. [CrossRef]
- Gaudelet, T. (2021). Integration of multi-scale protein interactions for biomedical data analysis (Doctoral dissertation), UCL (University College, London)). ttps://discovery.ucl.ac.uk/id/eprint/10125012.
- Röttjers, Lisa, and Karoline Faust. "From hairballs to hypotheses–biological insights from microbial networks." FEMS microbiology reviews 42.6 (2018): 761-780. [CrossRef]
- Ioannidis, John PA. "Biomarker failures." Clinical chemistry 59.1 (2013): 202-204. [CrossRef]
- Goossens, N., Nakagawa, S., Sun, X., & Hoshida, Y. (2015). Cancer biomarker discovery and validation. Translational cancer research, 4(3), 256. [CrossRef]
- Dimitrakopoulos, Christos M., and Niko Beerenwinkel. "Computational approaches for the identification of cancer genes and pathways." Wiley Interdisciplinary Reviews: Systems Biology and Medicine 9.1 (2017): e1364. [CrossRef]
- Rakedzon, S., Khoury, Y., Rozenberg, G., & Neuberger, A. (2020). Hydroxychloroquine and coronavirus disease 2019: A systematic review of a scientific failure. Rambam Maimonides medical journal, 11(3), e0025.. [CrossRef]
- O'malley, Maureen A., and Orkun S. Soyer. "The roles of integration in molecular systems biology." Studies in History and Philosophy of Science Part C: Studies in History and Philosophy of Biological and Biomedical Sciences 43.1 (2012): 58-68. [CrossRef]
- Luo, W., Phung, D., Tran, T., Gupta, S., Rana, S., Karmakar, C., Shilton, A., Yearwood, J., Dimitrova, N., Ho, T.B., et al. (2016). Guidelines for developing and reporting machine learning predictive models in biomedical research: a multidisciplinary view. Journal of medical Internet research, 18(12), e323. [CrossRef]
- Jaeger, J. (2017). The Importance of Being Dynamic: Systems Biology Beyond the Hairball. In: Green, S. (eds) Philosophy of Systems Biology. History, Philosophy and Theory of the Life Sciences, vol 20. Springer, Cham. [CrossRef]
- Brohee, Sylvain, and Jacques Van Helden. "Evaluation of clustering algorithms for protein-protein interaction networks." BMC bioinformatics 7 (2006): 1-19. [CrossRef]
- Sornette, D., Davis, A. B., Ide, K., Vixie, K. R., Pisarenko, V., & Kamm, J. R. (2007). Algorithm for model validation: Theory and applications. Proceedings of the National Academy of Sciences, 104(16), 6562-6567. [CrossRef]
- Dougherty, Edward R., and Ulisses Braga-Neto. "Epistemology of computational biology: mathematical models and experimental prediction as the basis of their validity." Journal of Biological Systems 14.01 (2006): 65-90. [CrossRef]
- Ahady Dolatsara, H., Chen, Y. J., Leonard, R. D., Megahed, F. M., & Jones-Farmer, L. A. (2023). Explaining predictive model performance: An experimental study of data preparation and model choice. Big Data, 11(3), 199-214. [CrossRef]
- Kairys, V., Baranauskiene, L., Kazlauskiene, M., Matulis, D., & Kazlauskas, E. (2019). Binding affinity in drug design: experimental and computational techniques. Expert opinion on drug discovery, 14(8), 755-768. [CrossRef]
- Kopac T. Leveraging Artificial Intelligence and Machine Learning for Characterizing Protein Corona, Nanobiological Interactions, and Advancing Drug Discovery. Bioengineering (Basel). 2025 Mar 18;12(3):312. [CrossRef] [PubMed] [PubMed Central]
- Refaeilzadeh, P., Tang, L., & Liu, H. (2009). Cross-validation. In Encyclopedia of database systems (pp. 532-538). Springer, Boston, MA. [CrossRef]
- Adadi, A. (2021). A survey on data-efficient algorithms in big data era. Journal of Big Data, 8(1), 24. [CrossRef]
- Mahmud, M., Kaiser, M. S., McGinnity, T. M., & Hussain, A. (2021). Deep learning in mining biological data. Cognitive computation, 13(1), 1-33. [CrossRef]
- Altman, R. B., Bergman, C. M., Blake, J., Blaschke, C., Cohen, A., Gannon, F., ... & Valencia, A. (2008). Text mining for biology-the way forward: opinions from leading scientists. Genome biology, 9, 1-15. [CrossRef]
- Agarwal, S., Godbole, S., Punjani, D., & Roy, S. (2007, October). How much noise is too much: A study in automatic text classification. In Seventh IEEE International Conference on Data Mining (ICDM 2007) (pp. 3-12). IEEE. [CrossRef]
- Kar, A. K., & Dwivedi, Y. K. (2020). Theory building with big data-driven research–Moving away from the "What" towards the "Why". International Journal of Information Management, 54, 102205. [CrossRef]
- Johanson, A., & Hasselbring, W. (2018). Software engineering for computational science: Past, present, future. Computing in Science & Engineering, 20(2), 90-109. [CrossRef]
- Tracy, Sarah J. Qualitative research methods: Collecting evidence, crafting analysis, communicating impact. Third Edition. John Wiley & Sons, 2024. ePUB ISBN: 9781119988670.
- Aldoseri, A., Al-Khalifa, K. N., & Hamouda, A. M. (2023). Re-thinking data strategy and integration for artificial intelligence: concepts, opportunities, and challenges. Applied Sciences, 13(12), 7082. [CrossRef]
- Chelly Dagdia, Z., Avdeyev, P. & Bayzid, M.S. Biological computation and computational biology: survey, challenges, and discussion. Artif Intell Rev 54, 4169–4235 (2021). [CrossRef]
- Kaur, D., Uslu, S., Rittichier, K. J., & Durresi, A. (2022). Trustworthy artificial intelligence: a review. ACM computing surveys (CSUR), 55(2), 1-38. [CrossRef]
- Nickles, Thomas J. "Knowledge Pollution." NSF Award 95.9530174 (1996): 30174. https://www.nsf.gov/awardsearch/showAward?AWD_ID=9530174.
- Elouataoui, W. (2024). AI-Driven frameworks for enhancing data quality in big data ecosystems: Error_detection, correction, and metadata integration. arXiv preprint arXiv:2405.03870. [CrossRef]
- Samariya, D., & Thakkar, A. (2023). A comprehensive survey of anomaly detection algorithms. Annals of Data Science, 10(3), 829-850. [CrossRef]
- Paramesha, M., Rane, N. L., & Rane, J. (2024). Big data analytics, artificial intelligence, machine learning, internet of things, and blockchain for enhanced business intelligence. Partners Universal Multidisciplinary Research Journal, 1(2), 110-133. [CrossRef]
- Forstmeier, W., Wagenmakers, E. J., & Parker, T. H. (2017). Detecting and avoiding likely false-positive findings–a practical guide. Biological Reviews, 92(4), 1941-1968. [CrossRef]
- E. M. Hassib, A. I. El-Desouky, E. -S. M. El-Kenawy and S. M. El-Ghamrawy, "An Imbalanced Big Data Mining Framework for Improving Optimization Algorithms Performance," in IEEE Access, vol. 7, pp. 170774-170795, 2019. [CrossRef]
- Chedid, W., Yu, C., & Lee, B. (2005). Power analysis and optimization techniques for energy efficient computer systems. Advances in Computers, 63, 129-164. [CrossRef]
- Hamidzadeh, J., & Moradi, M. (2020). Enhancing data analysis: uncertainty-resistance method for handling incomplete data. Applied Intelligence, 50(1), 74-86. [CrossRef]
- Jiao, Y., & Du, P. (2016). Performance measures in evaluating machine learning based bioinformatics predictors for classifications. Quantitative Biology, 4, 320-330. [CrossRef]
- Lei, Jing. "Cross-validation with confidence." Journal of the American Statistical Association 115.532 (2020): 1978-1997. [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein–protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2020, 49, D605–D612, Erratum in: Nucleic Acids Res. 2021, 49, 10800.. [CrossRef].
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein–protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2022, 51, D638–D646. [CrossRef] [PubMed].
- Lin, J. S., & Lai, E. M. (2017). Protein–protein interactions: co-immunoprecipitation. Bacterial protein secretion systems: Methods and protocols, 211-219. [CrossRef]
- Galas, D. J., Nykter, M., Carter, G. W., Price, N. D., & Shmulevich, I. (2010). Biological information as set-based complexity. IEEE Transactions on Information Theory, 56(2), 667-677. [CrossRef]
- Kholodenko, B., Yaffe, M. B., & Kolch, W. (2012). Computational approaches for analyzing information flow in biological networks. Science signaling, 5(220), re1-re1. [CrossRef]
- Li, Y., Wu, F. X., & Ngom, A. (2018). A review on machine learning principles for multi-view biological data integration. Briefings in bioinformatics, 19(2), 325-340. [CrossRef]
- Sokouti, B., & Amjad, E. (2025). Validation strategies in systems biology research. In Systems Biology and In-Depth Applications for Unlocking Diseases (pp. 183-190). Academic Press. [CrossRef]
- Boué, S., Byrne, M., Hayes, A. W., Hoeng, J., & Peitsch, M. C. (2018). Embracing transparency through data sharing. International Journal of Toxicology, 37(6), 466-471. [CrossRef]
- Roberts, M. K., Fisher, D. M., Parker, L. E., Darnell, D., Sugarman, J., Carrithers, J., Weinfurt, K., Jurkovich, G., Zatzick, D. (2020). Ethical and regulatory concerns in pragmatic clinical trial monitoring and oversight. Ethics & Human Research, 42(5), 29-37. [CrossRef]
- Cannataro, Mario, Pietro H. Guzzi, and Pierangelo Veltri. "Protein-to-protein interactions: Technologies, databases, and algorithms." ACM Computing Surveys (CSUR) 43.1 (2010): 1-36. [CrossRef]
- Leung, M. K., Delong, A., Alipanahi, B., & Frey, B. J. (2015). Machine learning in genomic medicine: a review of computational problems and data sets. Proceedings of the IEEE, 104(1), 176-197. [CrossRef]
- Lualdi, Marta, and Mauro Fasano. "Statistical analysis of proteomics data: a review on feature selection." Journal of proteomics 198 (2019): 18-26. [CrossRef]
- Ferrell, M., Wang, Z., Anderson, J.T. et al. A terminal metabolite of niacin promotes vascular inflammation and contributes to cardiovascular disease risk. Nat Med 30, 424–434 (2024). [CrossRef]
- Karunakaran, Kalyani Bindu. Interactome-based framework to translate disease genetic data into biological and clinical insights. Diss. University of Reading, UK, 2024. School of Chemistry, Food & Pharmacy. [CrossRef]
- Singh, Vikram, and Vikram Singh. "Inferring interaction networks from transcriptomic data: methods and applications." Transcriptome Data Analysis. New York, NY: Springer US, 2024. 11-37. [CrossRef]
- MacKenzie, Donald. Mechanizing proof: computing, risk, and trust. MIT Press, London, 2004. ISBN 0-262-13393-8.
- Mering, C. V., Huynen, M., Jaeggi, D., Schmidt, S., Bork, P., & Snel, B. (2003). STRING: a database of predicted functional associations between proteins. Nucleic acids research, 31(1), 258-261. [CrossRef]
- Colonna, G. Understanding the SARS-CoV-2–Human Liver Interactome Using a Comprehensive Analysis of the Individual Virus–Host Interactions. Livers 2024, 4, 209–239. [CrossRef]
- Klinke, David J. "In silico model-based inference: A contemporary approach for hypothesis testing in network biology." Biotechnology progress 30.6 (2014): 1247-1261. [CrossRef]
- Roberts, D. R., Bahn, V., Ciuti, S., Boyce, M. S., Elith, J., Guillera-Arroita, G., Hauenstein, S., Lahoz-Monfort, J.J., Schroeder, B., Thuiller, W., et al. (2017). Cross-validation strategies for data with temporal, spatial, hierarchical, or phylogenetic structure. Ecography, 40(8), 913-929. [CrossRef]
- Kalinichenko, L. A., Kovalev, D. Y. E., Kovaleva, D. A., & Malkov, O. Y. E. (2015). Methods and tools for hypothesis-driven research support: A survey. Informatics and its Applications, 9(1), 28-54. [CrossRef]
- Abele-Brehm, A. E., Gollwitzer, M., Steinberg, U., & Schönbrodt, F. D. (2019). Attitudes toward open science and public data sharing. Social Psychology. Vol 50, No4, Res. Report. [CrossRef]
- Campaner, Raffaella. "Complexity and Integration." Explaining Disease: Philosophical Reflections on Medical Research and Clinical Practice. European Studies in Philosophy of Science. Cham: Springer International Publishing, 2022. 65-88. [CrossRef]
- Sharma, A., Colonna, G. System-Wide Pollution of Biomedical Data: Consequence of the Search for Hub Genes of Hepatocellular Carcinoma Without Spatiotemporal Consideration. Mol Diagn Ther 25, 9–27 (2021). [CrossRef]
- Karl Popper, Conjectures and Refutations, London: Routledge and Keagan Paul, 1963, pp. 33-39; from Theodore Schick, ed., Readings in the Philosophy of Science, Mountain View, CA: Mayfield Publishing Company, 2000, pp. 9-13. Excerpt from Royal Institute of Technology (KTH), Sweden. https://www.kth.se/social/files/57da705ef276540c0f789308/KPopper_Falsification.pdf.
- Popper, Karl. "Science: Conjectures and refutations." Arguing about Science. Routledge, 2012. 15-43. Taylor & Francis Group. [CrossRef]
Figure 1.
Interactome calculated with functional seeds taken from [91].
Figure 1.
Interactome calculated with functional seeds taken from [91].
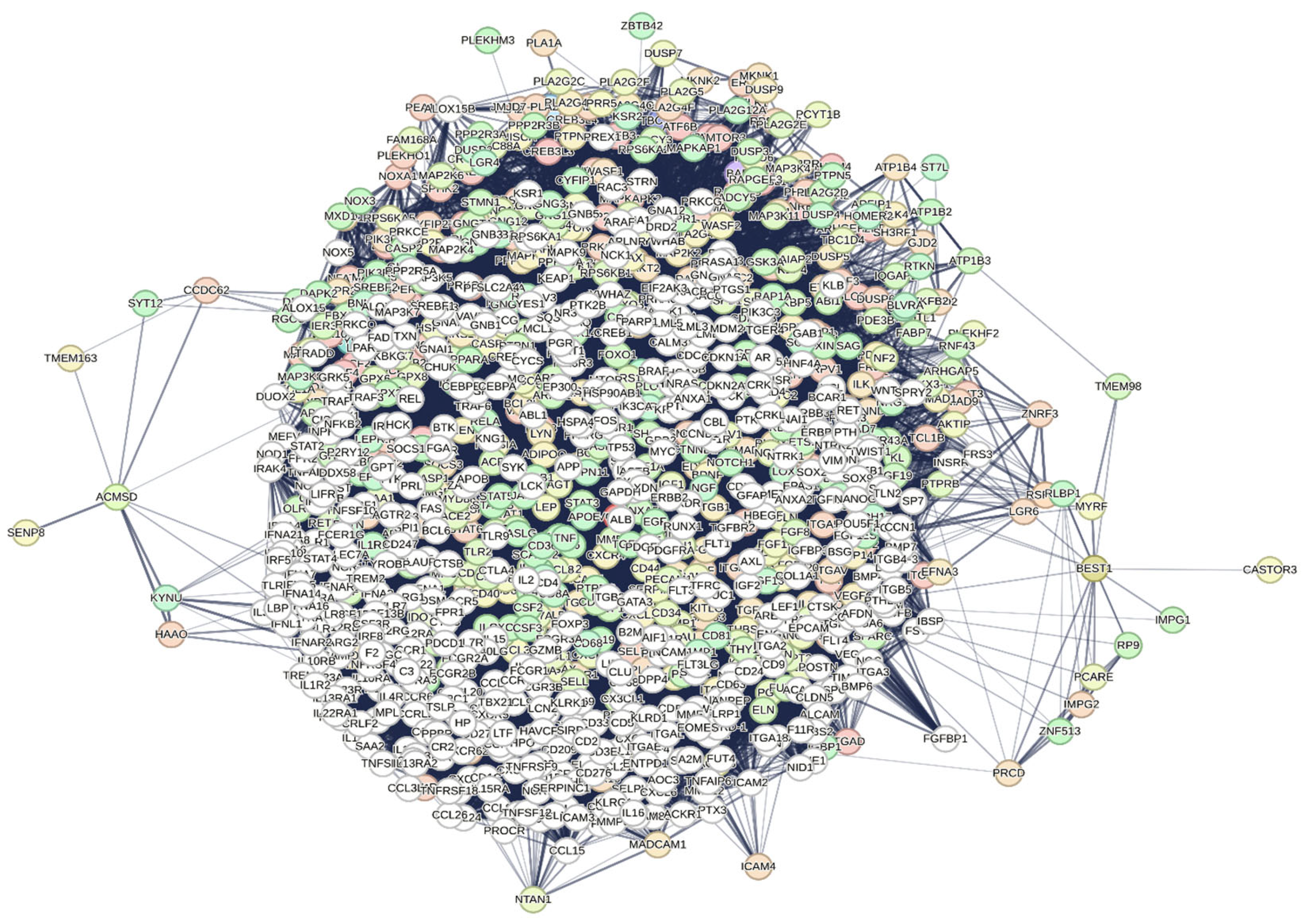
Figure 2.
Significant GO functions from the interactome of Figure 1.
Figure 2.
Significant GO functions from the interactome of Figure 1.
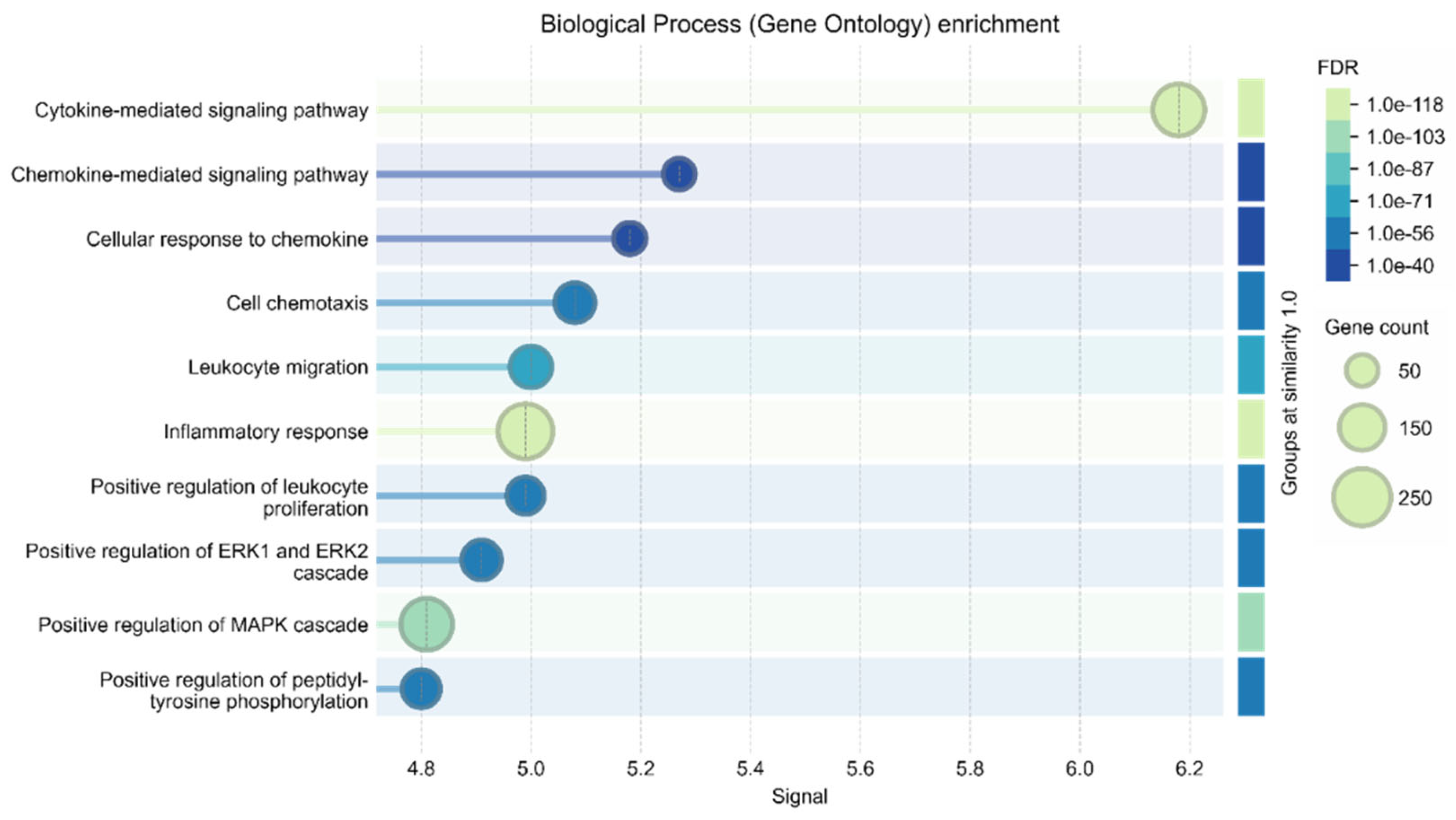
Figure 3.
Interactome related to VCAM1 and its specific functional relationships in the human proteome. We extracted this interactome from Figure 1. STRING has recalculated this new interactome using the "recenter on the node" function.
Figure 3.
Interactome related to VCAM1 and its specific functional relationships in the human proteome. We extracted this interactome from Figure 1. STRING has recalculated this new interactome using the "recenter on the node" function.
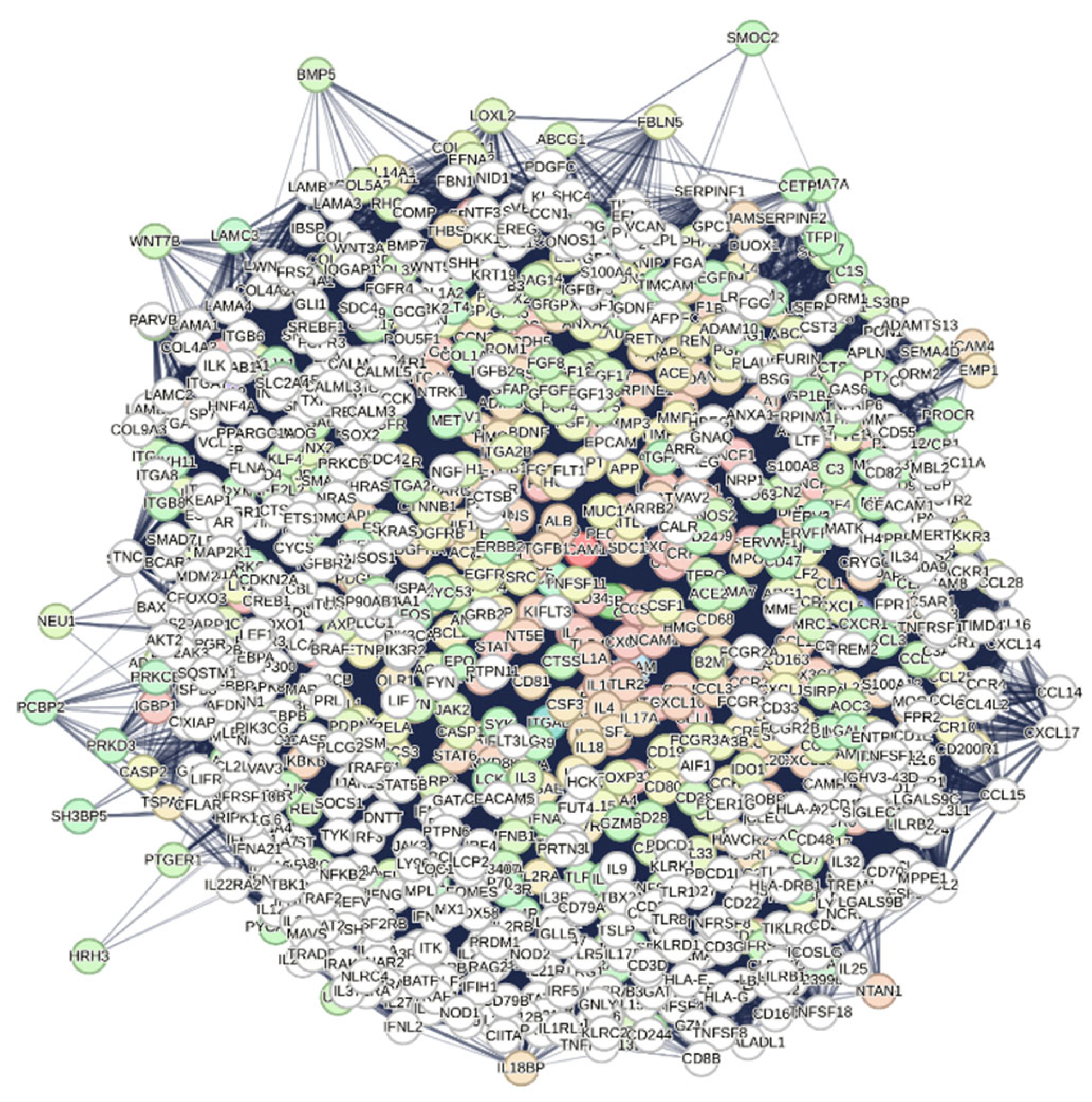
Figure 4.
The result of the interactomic analysis made with stringent parameters.


Table 1.
Experimental validations of VCAM1 interactions.
| Confidence score | Experimental validation | % |
| 0.900 | 0 | 0 |
| 0.800 | 0 | 0 |
| 0.700 | 0 | 0 |
| 0.600 | 0 | 0 |
| 0.500 | 0 | 0 |
| 0.400 | 2 | 0.42 |
| 0.300 | 7 | 1.48 |
| 0.200 | 1 | 0.02 |
| 0.100 | 81 | 17.19 |
| 0<x<0.1 | 108 | 22.92 |
| 0 | 272 | 57.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



