Submitted:
07 July 2025
Posted:
08 July 2025
Read the latest preprint version here
Abstract
Glioblastoma, the most frequent malignant primary brain tumor in adults, presents a significant challenge in cancer treatment, with a mean survival of merely 12-15 months and a 5-year survival rate of less than 2%. Despite extensive research and current treatments, involving maximal surgical excision, radiation, and chemotherapy mainly with temozolomide (TMZ), recurrence remains nearly universal. This is attributable to its infiltrative nature and tumor cells may be found far from the surgical resection cav-ity. To address the critical need for improved diagnostic and therapeutic strategies for glioblastoma multiform (GBM), we have developed an innovative theranostic molecule, AGT-7, based on tetrofosmin, a compound used in nuclear medicine bioimaging, and TMZ. AGT-7 was specifically designed to enable simultaneous tumor imaging and tar-geted chemotherapy. Its diagnostic component was inspired by the ability of tetrofosmin to selectively accumulate in glioma mitochondria, driven by its positive charge and the negative membrane potential of these organelles. A chelating moiety allows for radio-labeling with technetium-99m (⁹⁹ᵐTc), enabling precise SPECT imaging. The therapeutic arm comprises a tethered TMZ moiety, delivering localized cytotoxic activity. In vitro studies demonstrated that AGT-7 exerts potent cytotoxic effects in GBM cell lines (T98 and U87), with superior therapeutic efficacy compared to TMZ alone. Toxicity assess-ment in zebrafish embryos confirmed a favorable safety profile. Furthermore, biodis-tribution studies in CFW mice showed that [⁹⁹ᵐTc]Tc-AGT-7 exhibited approximately tenfold lower heart uptake than [⁹⁹ᵐTc]Tc-tetrofosmin, a standard cardiac imaging agent, indicating a significant reduction in off-target cardiac localization. This reduced heart uptake is particularly advantageous, as it minimizes potential cardiotoxicity and en-hances the suitability of AGT-7 as a glioma-targeted theranostic agent.
Keywords:
Glioblastoma
; Tetrofosmin
; Temozolamide
; Theranostics
; Technetium-99m
1. Introduction
Glioblastoma, accounting for approximately 50% of adult malignant brain tumors, stands out as one of the most lethal CNS diseases. Despite its relatively low incidence rate (approximately 5/100,000), the tumor’s intrinsic aggressiveness is associated with a median survival period of 15 months after initial diagnosis [1]. Compounds targeted to treat GBM face challenges due to its highly infiltrative growth, rapid proliferation, tumor heterogeneity, distinct interactions with the brain microenvironment, and the presence of the blood-brain barrier (BBB). The BBB complicates the development of treatments for intracranial malignancies by limiting access to only lipophilic and low molecular weight (less than 400 Da) drugs [2]. It is well known that surgical resection alone leads to median survival of about 5 months. Radiotherapy alone can increase survival to 10-12 months, whereas addition of the alkylating agent temozolomide (TMZ) in addition to radiation increases survival to a median of 15 months. Nowadays, the initial phase of treatment involves surgical excision, when possible, followed by a combination of radiotherapy and concurrent and adjuvant chemotherapy. TMZ is the most commonly utilized chemotherapy drug, often employed as a primary treatment option. The United States Food and Drug Administration (USFDA) granted approval to TMZ, also recognized under the brand name Temodal, in 2005 as a second-generation DNA alkylating agent. This oral medication is well-tolerated, and exhibits effective penetration of the blood-brain barrier, contributing to its therapeutic efficacy [3]. There are particular protocols that specify the dosage and how it should be administered with radiation therapy, which has an average duration of six months [4]. Also, studies have demonstrated that administering TMZ over a longer period of time has some beneficial outcomes [5]. However, problems arising from the administration of the drug are hematologic toxicity and nausea, although the most important of all of these is resistance to its activity [4]. Understanding the mechanism of drug resistance is pivotal, and a closer examination of TMZ mechanisms of action sheds light on this phenomenon.
Initially inert, TMZ operates as a prodrug. Upon hydrolysis, it transforms into active metabolite methyltriazen-1-yl imidazole-4-carboxamide (MTIC), activating the production of the reactive DNA methylating species methyl hydrazine. Of note, this hydrolysis occurs at normal pH (pH>7) without requiring liver metabolism. Subsequently, genomic DNA undergoes methylation at specific sites: N7 (>70%), O6 (6%) of guanine, and N3 (9%) of adenine. While methylation at guanine's O6 position (O6-MeG) is infrequent, it holds significant importance due to its potential to induce mismatched base errors during DNA replication, resulting in cytotoxicity. This comprehensive understanding of the mechanism of TMZ action provides insights into the intricate dynamics involved in drug resistance [3,6]. The robust DNA repair capabilities observed in GBM cells enable the reversal of TMZ-induced methylation effects, leading to resistance. Three primary DNA repair pathways contribute significantly to TMZ resistance: Mismatch Repair (MMR), O6-methylguanine-DNA methyltransferase (MGMT), and Base Excision Repair (BER) via the poly (ADP)-ribose polymerase (PARP) pathway. Notably, MGMT assumes a pivotal role in counteracting the cytotoxic effects of TMZ by proficiently repairing O6-methylguanine (O6-MeG) damage resulting from methylation. Understanding these repair mechanisms is essential in unraveling the intricate dynamics of TMZ resistance in glioblastoma multiforme (GBM) [6]. Extensive research efforts have been dedicated to dealing with the challenges associated with GBM and its resistance to treatments such as TMZ. These efforts demonstrate a commitment to advancing our understanding of the intricate mechanisms at operating and developing strategies to overcome the formidable obstacles posed by this aggressive form of cancer. Significant progress in research shows a shared commitment to improving treatments and ultimately improves outcomes for individuals facing the complex challenges of GBM [1,7,8].
Although TMZ maintains a key role in treating gliomas, the need for precise imaging agents to monitor treatment response and disease progression has driven interest in radiopharmaceuticals. Technetium-99m tetrofosmin, also known by its brand name MyoviewTM, was introduced in 1993 by Kelley et al [9]. The aim of their research was to develop a novel class of radiopharmaceuticals intended for application in Myocardial Perfusion Imaging (MPI). Since then, [99mTc]-tetrofosmin has been the standard of excellence for myocardial perfusion imaging in clinical practice. Because it is patient-safe, cost-efficient and produces good images, it is widely employed [10]. Researchers have also employed [99mTc]-tetrofosmin in SPECT/CT imaging investigations, including Stacy et al [11]. They discovered that [99mTc]-tetrofosmin helps to detect reduced blood circulation in cases of lower body vascular blockage, particularly in regions such as the calf muscles. This diagnostic ability is useful for evaluating diseases such as peripheral arterial disease.
Tetrofosmin (TF) is a compound that acts as a ligand which can complex 99mTc. The molecule formed as a result is a positively charged complex that functions as a hybrid of TF and technetium (99mTc). The term "lipophilic di-oxo-monocation complex" refers to the formation of a central technetium atom by means of two identical di-phosphine ligands [12]. The uptake of 99mTc-tetrofosmin is closely associated with energy-dependent transport pathways, cellular ion homeostasis, and mitochondrial membrane potentials due to its positive charge and lipophilic nature [10].
99mTc-tetrofosmin has also been used for brain tumor imaging. It can enter viable cells through passive transport, due to its positively charged chemical nature. In previous studies, 99mTc-tetrofosmin has been found to be helpful for distinguishing between glioma recurrence and necrosis induced by radiochemotherapy, differentiate low from high-grade tumor, assess response to treatment and overall prognosis in glioblastoma patients [13,14]. Furthermore, 99mTc-tetrofosmin uptake in glioblastoma cells has been correlated with the sensitivity of the cells to TMZ, making it a promising tool for assessing early response to treatment in patients with high-grade gliomas [15]. Due to the positive charge of the complex, its uptake is strongly related to both cell membrane and mitochondrial potentials. Furthermore, 99mTc-tetrofosmin proved superior to 99mTc-MIBI for brain tumor imaging since its uptake is less dependent on multidrug resistance phenotype of glioblastoma, both in vivo and in vitro [20552283, 25436147]. In a study by Arbab et al., 99mTc-tetrofosmin accumulated inside the mitochondria of HBL-2 cells to only a low extent, in comparison to 99mTc-MIBI [16].
In recent years, targeting intracellular organelles, such as mitochondria, has evolved as a promising therapeutic approach for the treatment of different malignancies. Mitochondria are generally characterized as the energy powerhouse of the cell and they take part in several cellular processes [17]. Moreover, they are susceptible to oxidative damage, and can lead damaged cells to apoptosis [18]. Hence, targeting mitochondria with photosensitizers has gained the interest of several scientific groups. However, a strong issue that remains is the premature leakage of photosensitizers before reaching mitochondria. With the development of nanocarriers, this issue has been at least partially resolved. Recently, brain endothelial cell-derived extracellular vesicles were used to encapsulate a positively charged lipophilic molecule that targets mitochondria and a photosensitizer. This complex effectively inhibited GBM cell growth in vivo, without causing systemic toxicity [19].
For SPECT or PET imaging applications, short-lived gamma- or positron-emitting radionuclides such as technetium-99m (99mTc), gallium-68 (68Ga), iodine-123 (123I), indium-111 (111In), fluorine- 18 (18F), and copper-64 (64Cu), are extensively used for radiolabeling various molecules. Due to excellent nuclear characteristics such as the emission of low-energy gamma rays (140 keV) and short half-life (t1/2 = 6.02 h), as well as its convenient availability from 99Mo/99mTc generators, 99mTc is the most commonly used radionuclide in diagnostic imaging [20]. A very efficient way of radiolabeling compounds with 99mTc was first introduced by Alberto et al in 1998 and involves the [99mTc][Tc(H2O)3(CO)3]+ carbonyl core. This method enables short radiolabeling procedures without compound modification with bifunctional chelators, under mild conditions, and has been widely used in the development of new radiopharmaceuticals [21].
There is an unmet clinical need for the development of innovative compounds that combine both a diagnostic entity and a therapeutic payload, while offering reduced potential side effects compared to existing compounds within the same theranostic class. Along these lines, the overall objective of the present work is the development of an innovative theranostic molecule that provides simultaneous imaging and therapy of glioblastoma. The diagnostic component is based on a TF derivative radiolabeled with 99mTc, while the payload comprises TMZ attached to the TF derivative, which also serves as the targeting agent.
Our designed compound bears technetium affinity similar to that of TF. This molecule is engineered to selectively localize within glioma cells by targeting overexpressed mitochondria. Additionally, it functions as a carrier for the cytotoxic agent TMZ, combining targeted imaging with effective therapeutic capacity.
2. Results
2.1. Rational Design of AGT-7 as a Multifunctional Compound Bearing a Therapeutic and Diagnostic Modality
Figure 1.
The architecture of AGT-7 as a glioma theranostic compound.
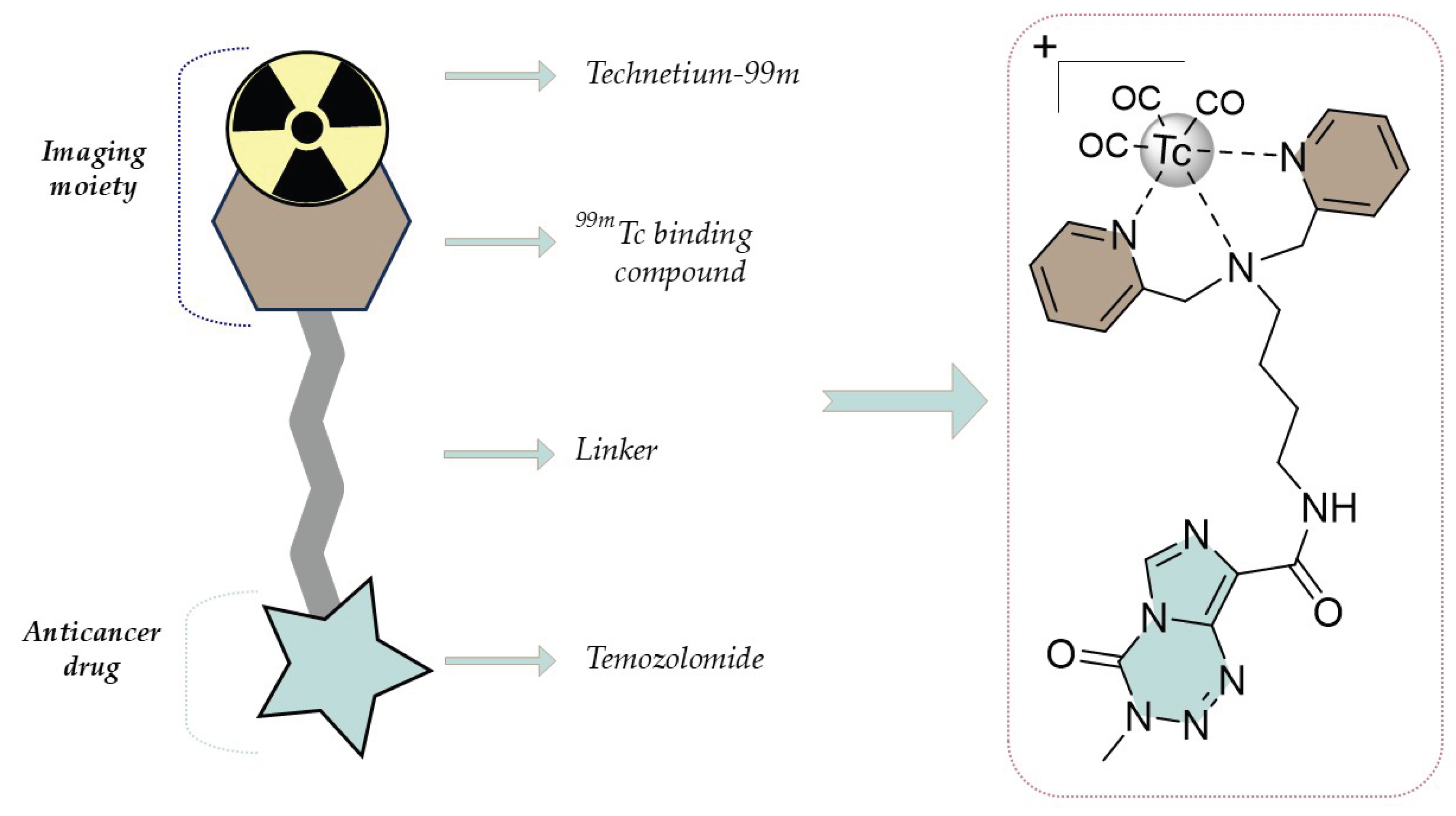
Three key pillars guided the development of the compound AGT-7: imaging, targeting, and treatment. Our objective was to harness 99mTc as an imaging agent, prompting the incorporation of a moiety specifically designed to chelate it. However, a critical aspect of our approach involved targeting the overexpressed mitochondria in glioblastoma cells. Mitochondria, enriched in glioma cells, play a pivotal role in the altered cellular metabolism characteristic of these tumors. Numerous vital cellular functions are significantly influenced by mitochondria with the most important being programmed cell death (apoptosis), which is controlled by permeabilization of the mitochondrial outer membrane (MOMP) [22]. Mitochondrial dysfunction is a key characteristic of neoplasia. Due to substantial changes in the mitochondrial genome, gliomas have impaired mitochondrial function resulting in altered morphology and unusual bioenergetics[23]. Recognizing this, AGT-7 was rationally engineered to exploit the overabundance of mitochondria. The mitochondrial membrane potential (∆Ψm) is present on both sides of the Inner Mitochondrial Membrane (IMM) and is typically negative, ranging from 180 to 200 mV [24]. Due to this membrane potential, compounds conjugated with lipophilic cations can accumulate within mitochondria inside cells [25]. We, therefore, strategically designed the compound to bear a positive charge to enhance its glioma targeting efficacy. Moreover, we aimed to utilize TMZ as a therapeutic agent, given its well-established efficacy in treating patients with brain tumors [5]. The specific design could maximize the therapeutic potential while minimizing off-target effects on healthy cells.
2.2. Synthetic Route for AGT-7
The synthetic route of AGT-7 begins with the formation of the chelating substituent through a double reductive amination with the linker molecule. This is followed by the removal of the BOC protecting groups, leading to the release of the secondary amine. The latter then reacts with TMZ-COOH via an amide bond, yielding the final product (Scheme 1).
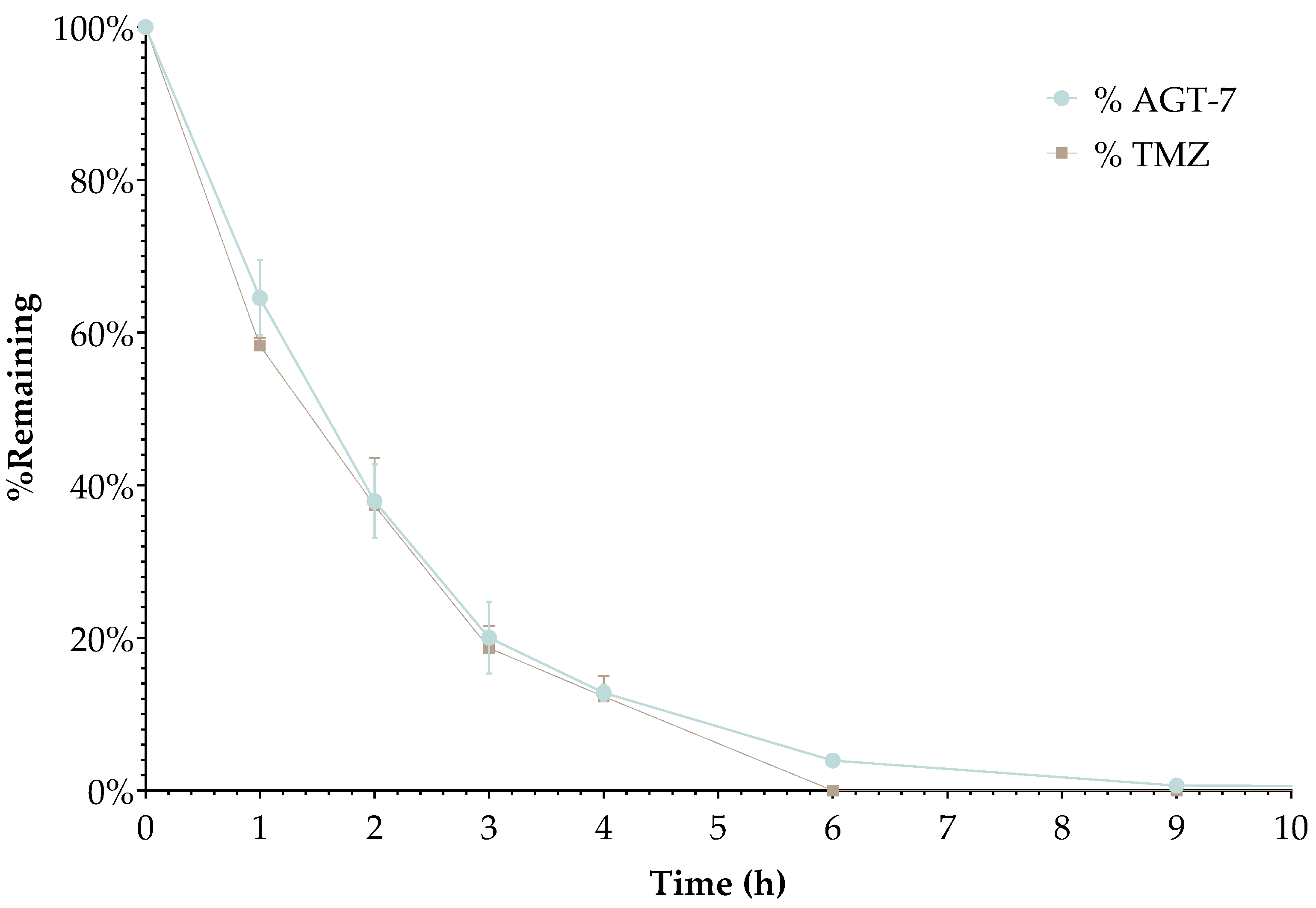
2.3. LC-MS Based Plasma Stability of AGT-7
AGT-7 demonstrated a plasma stability profile very similar to that of the parent drug, TMZ. Throughout the 9-hour incubation period, only minimal stability differences were observed. This slight variation is likely due to the compound being a structural analog of TMZ, which could clarify similar behaviour in plasma. Overall, the results suggest that the compound retains the stability profile of TMZ.
Figure 2.
Time-dependent stability of AGT-7 compared to TMZ in human plasma. Plasma samples’ final concentration asjusted to 1 μM (2.5% DMSO, v/v), and incubated at 37 °C, and tested at various time points (0, 1, 2, 3, 4, 6, 9, and 24 h). Each point represents the mean analyte concentration from three independent replicates.
Figure 2.
Time-dependent stability of AGT-7 compared to TMZ in human plasma. Plasma samples’ final concentration asjusted to 1 μM (2.5% DMSO, v/v), and incubated at 37 °C, and tested at various time points (0, 1, 2, 3, 4, 6, 9, and 24 h). Each point represents the mean analyte concentration from three independent replicates.
2.4. Cytotoxic Evaluation of AGT-7 in Glioma Cell Lines
To explore the effect of AGT-7 on the viability of both GBM cell lines, T98 and U87 cells were cultured with escalating concentrations of the compounds for 72 h and afterward the Trypan Blue Exclusion Assay was carried out. The results demonstrate a more pronounced decrease in cell viability at higher concentrations of the compound, indicating a concentration-dependent inhibitory effect on both cell lines. Specifically, the IC50 values of AGT-7 found at 72 h post treatment were 27 μM for the T98 cell line and 30 μM for the U87 cell line (Figure 3). In comparison, the IC₅₀ values for temozolomide were 143 μM in T98 cells and 50 μM in U87 cells, indicating a markedly higher potency of AGT-7, particularly in the TMZ-resistant T98 line.
2.5. In vivo Toxicity Evaluation and LD₅₀ Evaluation of AGT-7 in Zebrafish Embryos
Prior to advancing the biological evaluation of AGT-7, it was essential to assess its in vivo acute toxicity. To this end, we employed a standardized acute exposure assay using zebrafish (Danio rerio) embryos as a vertebrate model system. The exposure lasted 96 hours post-fertilization (hpf), following the guidelines established by the OECD (2010) for fish embryo toxicity testing. As illustrated in Figure 4, AGT-7 exhibited a dose-dependent increase in mortality, with an LD₅₀ value of 46.81 μM, and LD₂₅ and LD₇₅ values of 35.54 μM and 61.64 μM, respectively. Notably, as shown in Figure 4, no significant mortality was observed at concentrations up to 30 μM—a range that corresponds to the IC₅₀ values observed in glioblastoma T98 (27 μM) and U87 (30 μM) cell lines. This alignment between therapeutically effective concentrations and sub-lethal doses in vivo underscores a favorable safety profile for AGT-7 and supports its further preclinical development.
2.6. Radiolabeling and In Vitro Stability
Radiolabeling of [99mTc]Tc-TF was achieved after addition of freshly-eluted [99mTc]TcO4- to a kit formulation of Tetrofosmin (Myoview®). Radiochemical purity was assessed by radio-TLC and was >97% (Rf ~ 0.6). Colloid formation was <2%, while free pertechnetate was ~1% (Figure 5).
Radiolabeling of AGT-7 with the [99mTc][Tc(CO)3(OH2)3]+ precursor was accomplished after 45 min at 40°C. Radiochemical yields > 95% were achieved, and the radiotracer was used without further purification (Figure 6). The semi aqua ion [99mTc][Tc(CO)3(OH2)3]+ has three available coordination sites and the metal center is primarily found in the oxidative state +1, so after radiolabeling, the water molecules were substituted with the three nitrogen atoms, thus forming the 99mTc complex. The radiolabeled compound remained stable at RT for at least 24 h. [99mTc]Tc-AGT-7 was also stable in serum up to 24h post-radiolabeling (Figure 7).
2.7. Lipophilicity Studies—Determination of Partition Coefficient
Lipophilicity studies were performed for the radiotracer [99mTc]Tc-AGT-7. The lipophilicity of [99mTc]Tc-TF has previously been reported but was repeated in the present study, for reasons of direct comparison. The partition coefficients (P) were determined using the shake-flask method and are expressed as logP. While both examined compounds exhibited lipophilic behavior, higher lipophilicity values were observed for [99mTc]Tc-TF, which is also reflected in the results of the biodistribution studies shown below, where heart uptake of [99mTc]Tc-AGT-7 was significantly lower than that of [99mTc]Tc-TF.

Table 1.
Partition Coefficient of [99mTc]Tc-TF, TMZ and [99mTc]Tc-AGT-7.
| LogP | Theoretical | Experimental |
|---|---|---|
| [99mTc]Tc-TF | 3.25 | 2.41 ± 1.03 |
| TMZ | -0.84 | |
| [99mTc]Tc-AGT-7 | -0.99 | 0.71 ± 0.20 |
2.8. Ex Vivo Biodistribution Evaluation of [⁹⁹ᵐTc]Tc-AGT-7 Compared to [⁹⁹ᵐTc]Tc-TF
The ex vivo biodistribution of [99mTc]Tc-AGT-7 in comparison to [99mTc]Tc-TF was evaluated in normal CFW mice at 1 h and 24 h post-intravenous injection via the tail vein (Figure 8).
Table 2.
Biodistribution data for [99mTc]Tc-AGT-7 and [99mTc]Tc-Tetrofosmin at 1 and 24h post-injection.
Table 2.
Biodistribution data for [99mTc]Tc-AGT-7 and [99mTc]Tc-Tetrofosmin at 1 and 24h post-injection.
| 1h | 24h | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [99mTc]Tc-AGT-7 | [99mTc]Tc-Tetrofosmin | [99mTc]Tc-AGT-7 | [99mTc]Tc-Tetrofosmin | |||||||||
| Blood | 3.78 | ± | 0.81 | 0.74 | ± | 0.39 | 0.72 | ± | 0.16 | 0.02 | ± | 0.01 |
| Liver | 8.37 | ± | 1.57 | 10.20 | ± | 1.19 | 8.19 | ± | 3.11 | 0.27 | ± | 0.07 |
| Heart | 1.70 | ± | 0.32 | 21.20 | ± | 3.15 | 0.76 | ± | 0.19 | 0.63 | ± | 0.22 |
| Kidney | 25.36 | ± | 15.26 | 32.94 | ± | 0.94 | 7.32 | ± | 1.77 | 0.54 | ± | 0.22 |
| Stomach | 1.59 | ± | 0.22 | 5.39 | ± | 1.60 | 0.37 | ± | 0.01 | 0.11 | ± | 0.03 |
| Intestine | 4.01 | ± | 1.87 | 8.37 | ± | 1.25 | 0.56 | ± | 0.08 | 0.23 | ± | 0.07 |
| Spleen | 1.39 | ± | 0.31 | 1.39 | ± | 0.13 | 0.44 | ± | 0.13 | 0.06 | ± | 0.01 |
| Muscle | 0.30 | ± | 0.15 | 2.46 | ± | 0.70 | 0.13 | ± | 0.04 | 0.50 | ± | 0.07 |
| Lung | 5.49 | ± | 1.00 | 1.36 | ± | 0.54 | 1.57 | ± | 0.15 | 0.06 | ± | 0.03 |
| Bone | 0.48 | ± | 0.14 | 2.22 | ± | 1.05 | 0.18 | ± | 0.07 | 0.20 | ± | 0.05 |
| Pancreas | 1.39 | ± | 0.33 | 4.37 | ± | 0.30 | 0.48 | ± | 0.17 | 0.35 | ± | 0.16 |
| Brain | 0.13 | ± | 0.02 | 0.17 | ± | 0.02 | 0.04 | ± | 0.02 | 0.01 | ± | 0.00 |
3. Discussion
Our aim was to develop a theranostic compound, AGT-7 that selectively targets glioblastoma, while avoiding undesirable off-target localization, especially in the myocardium, a known accumulation site for [⁹⁹ᵐTc]Tc-TF. To verify this we evaluated the biodistribution profile of the novel agent [⁹⁹ᵐTc]Tc-AGT-7 and compare it with [⁹⁹ᵐTc]Tc-tetrofosmin ([⁹⁹ᵐTc]Tc-TF) -a well-established cardiac imaging agent that, according to our previous work, also exhibits homing to glioma tissue [26].
As shown by our data, the biodistribution patterns of [⁹⁹ᵐTc]Tc-AGT-7 and [⁹⁹ᵐTc]Tc-TF differ significantly, primarily due to their distinct physicochemical characteristics. The most notable distinction was observed in heart uptake: while [⁹⁹ᵐTc]Tc-TF exhibited very high myocardial accumulation (>20% ID/g), [⁹⁹ᵐTc]Tc-AGT-7 displayed approximately tenfold lower uptake. This contrast reflects the higher lipophilicity of [⁹⁹ᵐTc]Tc-TF, enabling its strong cardiac retention, whereas the more moderate lipophilicity/hydrophilicity balance of [⁹⁹ᵐTc]Tc-AGT-7 reduces cardiac sequestration. The reduced heart uptake of [⁹⁹ᵐTc]Tc-AGT-7 is especially desired, as excessive myocardial accumulation would impair its effectiveness and safety as a glioma theranostic agent. Both tracers showed elevated liver uptake at 1 hour post-injection, consistent with their lipophilicity. Both compounds exhibit similar permeability across the ΒΒΒ. However, the slightly more hydrophilic nature of [⁹⁹ᵐTc]Tc-AGT-7 led to higher renal clearance at 24 hours post-injection, suggesting a more favorable excretion pathway. Additionally, low stomach uptake for [⁹⁹ᵐTc]Tc-AGT-7 indicates in vivo radiochemical stability. These findings are consistent with previous literature from other research groups [27,28]. Throughout the 24-hour study period, [⁹⁹ᵐTc]Tc-AGT-7 exhibited low uptake in all other evaluated organs, reinforcing its target specificity and capitalizing its potential as a safe and effective theranostic tool for glioblastoma.
4. Materials and Methods
Scheme 2.
Synthesis of the tert-butyl (4-(bis(pyridin-2-ylmethyl)amino)butyl)carbamate.
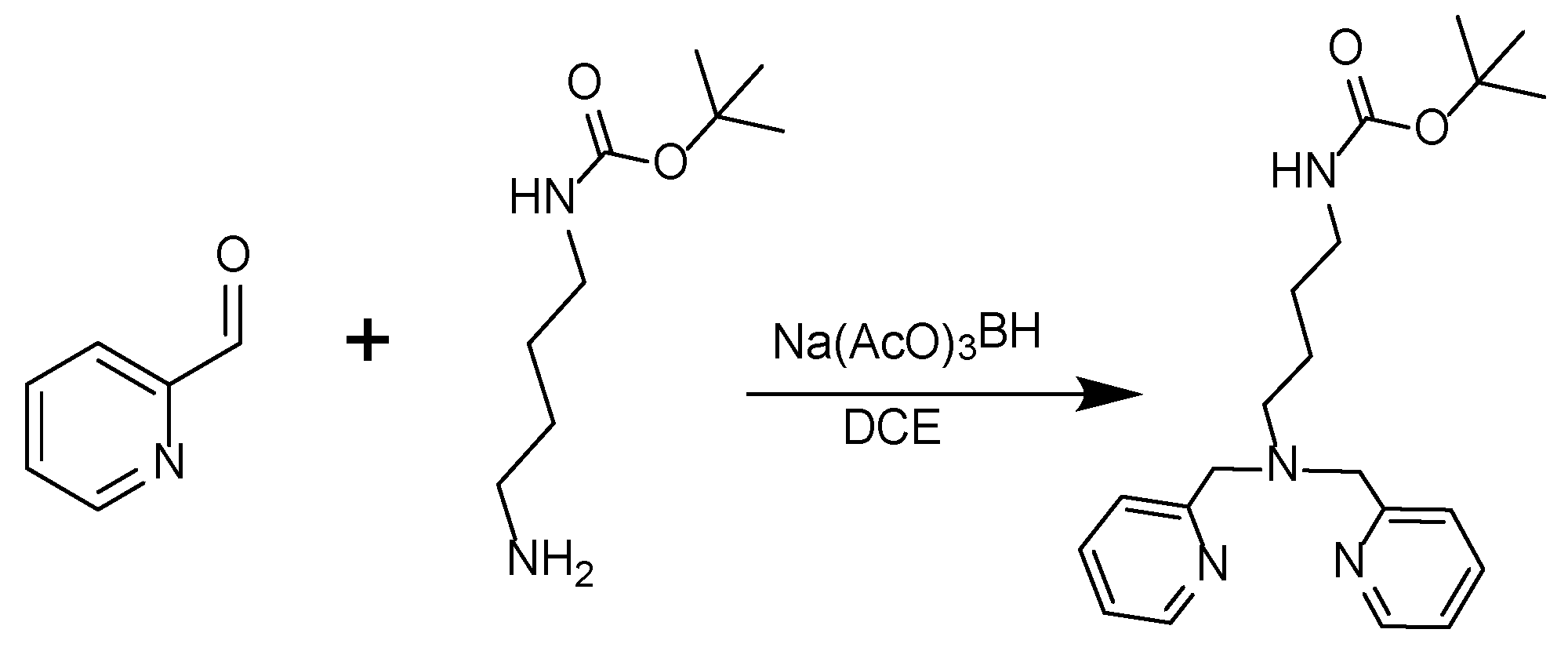
2-Pyridinecarboxyaldehyde (334 μl, 3.51 mmol), tert-Butyl N-(4-aminobutyl)carbamate (305 μl, 1.59 mmol) and 1,2-Dichloroethane (DCE, 15 ml) were added in a two neck round bottom flask. The flask was purged with N2 and the mixture was stirred in an ice bath for 10 mins. Then Sodium triacetoxyborohydride Na(AcO)3BH (845 mg, 3.98 mmol) was added and the reaction was further stirred for 10 mins in the ice bath and 3.5 h in room temperature. The reaction was quenched with 10 ml H2O and stirred for 1.5 h. The pH of the mixture was adjusted to 10 with the addition of drops of a 2.5 M NaOH solution and then extracted with CH2Cl2 (3 x 10 ml). The organic layers were collected, dried with Na2SO4, and filtered. The solvent was removed under reduced pressure in the rotary evaporator. The crude mixture was purified with silica gel column chromatography starting from 100% CH2Cl2 up to 5% MeOH/CH2Cl2 as eluent. The collected fraction containing the product was concentrated to dryness under reduced pressure in the rotary evaporator to afford a dark yellow oil (511.628 mg, yield = 86.8%). 1H-NMR (250 MHz, CDCl3-d, δ ppm): 8.51 (ddd, J = 4.9, 1.9, 0.9 Hz, 2H), 7.63 (td, J = 7.6, 1.8 Hz, 2H), 7.48 (dt, J = 7.9, 1.2 Hz, 2H), 7.13 (ddd, J = 7.5, 4.9, 1.3 Hz, 2H), 4.78 (s, 1H), 3.79 (s, 4H), 3.04 (q, J = 6.5 Hz, 2H), 2.54 (t, J = 6.9 Hz, 2H), 1.60 – 1.48 (m, 2H), 1.42 (s, 11H).13C-NMR (63 MHz, DMSO-d6, δ ppm): 159.66, 156.14, 148.90, 136.46, 123.04, 121.98, 60.33, 53.91, 40.50, 28.42, 27.84, 24.17.
Synthesis of N1,N1-bis(pyridin-2-ylmethyl)butane-1,4-diamine [29]:
Scheme 3.
Synthesis of the N1,N1-bis(pyridin-2-ylmethyl)butane-1,4-diamine.
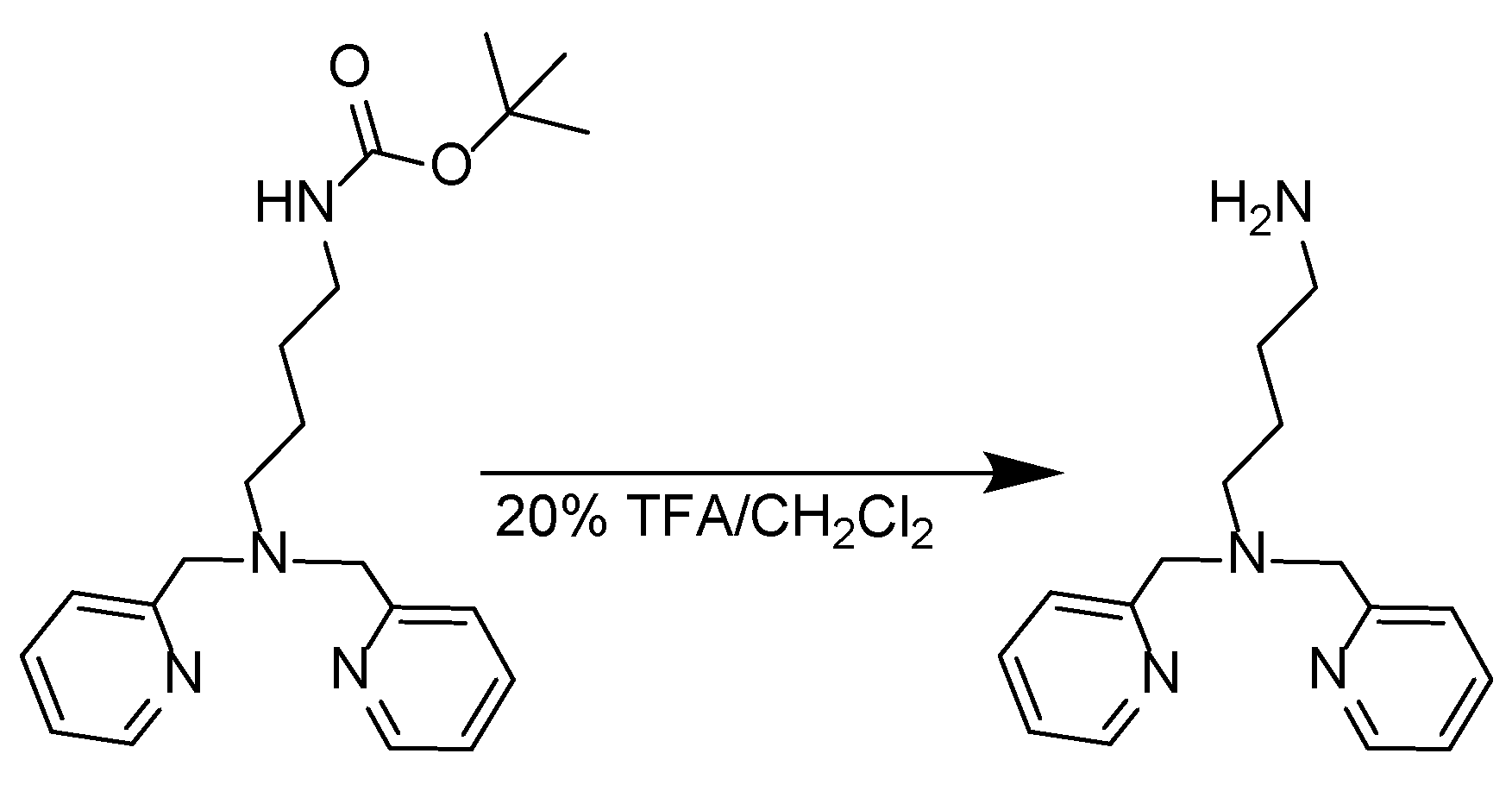
Tert-butyl (4-(bis(pyridin-2-ylmethyl)amino)butyl)carbamate (500 mg, 1.35 mmol) was transferred in a round bottom flask with 10 ml of 20% TFA/CH2Cl2. The mixture was stirred at room temperature for 2 h. The solvent was removed under high vacuum to afford a red-brown oil. The oil was dissolved in CH2Cl2 (20 ml) and washed with H2O whose pH was adjusted to 10 with the addition of drops of 2.5 M NaOH solution. The organic layer was then collected and the aqueous layer was extracted again with CH2Cl2 (2 × 10 mL). The organic layers were collected, dried with Na2SO4, and filtered. The solvent was removed under reduced pressure in the rotary evaporator to afford an amber-colored oil (308.583 mg, yield = 84.6%). 1H-NMR (250 MHz, CDCl3-d, δ ppm): 8.54 – 8.44 (m, 2H), 7.62 (td, J = 7.6, 1.8 Hz, 2H), 7.50 (d, J = 7.8 Hz, 2H), 7.17 – 7.05 (m, 2H), 3.79 (s, 4H), 2.56 (dt, J = 17.2, 7.1 Hz, 4H), 1.65 – 1.50 (m, 2H), 1.47 – 1.37 (m, 2H), 1.38 – 1.28 (m, 2H). 13C-NMR (63 MHz, DMSO-d6, δ ppm): 159.97, 148.94, 136.31, 122.83, 121.85, 60.46, 54.20, 42.02, 31.47, 24.42 .
Synthesis of AGT-7.
Scheme 4.
Synthesis of the AGT-7.
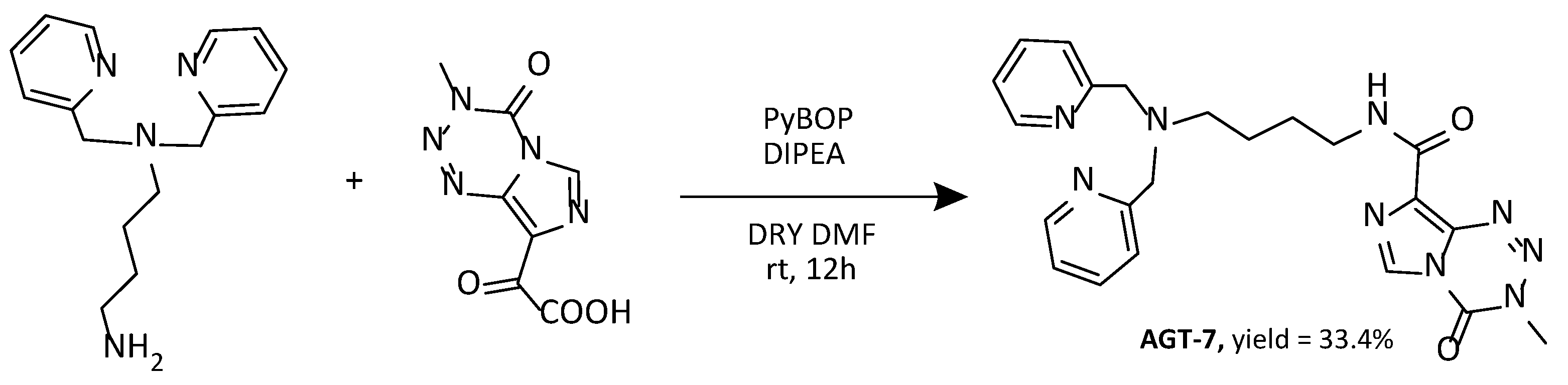
In a round bottom flask PyBOP (benzotriazol-1-yloxytripyrrolidinophosphonium hexafluorophosphate, 90.9 mg, 0.174 mmol), N,N-Diisopropylethylamine (DIPEA, 26 μl, 0.149 mmol) and TMZ-COOH (28.42 mg, 0.145 mmol) were added with dry DMF (2 ml). The flask was purged with N2. The reaction was stirred under N2 for 30 mins in room temperature. Then N1,N1-bis(pyridin-2-ylmethyl)butane-1,4-diamine (35.8 mg, 0.132mmol) was added and the reaction was stirred for 12 h. The solvent was evaporated under high vacuum. The crude mixture was purified with High Performance Column Chromatography, HPLC (A: H2O + 0,1% TFA, B: ACN + 0,1% TFA, from 70% A: 30% Β to 35% A: 65% Β 20 mL/min, 20 mins, 254 nm) to obtain a white solid (18.356 mg, yield = 33.4%). 1H NMR (500 MHz, DMSO-d6) δ: 8.85 (s, 1H, H-6’), 8.66 – 8.61 (m, 2H, H-6), 8.54 (t, J = 6.0 Hz, 1H, NH-13), 7.88 (td, J = 7.7, 1.8 Hz, 2H, H-4), 7.52 (d, J = 7.6, 2H, H-5), 7.44 (dd, J = 7.6, 5.0 Hz, 2H, H-3), 4.53 (d, J = 9.0 Hz, 4H, H-1), 3.88 (s, 3H, H-3’), 3.28 (q, J = 6.8 Hz, 2H, H-10), 3.23 – 3.17 (m, 2H, H-12), 1.80 (p, J = 7.9 Hz, 2H, H-9), 1.53 (p, J = 7.3 Hz, 2H, H-11). 13C NMR (126 MHz, DMSO-d6) δ: 159.60 (C-14), 151.2(C-2),149.14(C-6), 139.09(C-8’), 137.45(C-4), 134.17(C-4’), 130.27(C-8’a), 128.22(C-6’), 124.53(C-3), 123.71(C-5), 53.57(C-9), 39.41(C-8’), 37.57(C-12), 35.93(C-3’), 26.08(C-11), 20.96(C-10).
Chemicals.
All chemicals and reagents used in this study were of analytical grade. Sodium triacetoxyborohydride (Na(AcO)3BH) and 2-Pyridinecarboxaldehyde was purchased from Thermo Fisher Scientific. N-Boc-1,4-butanediamine and 3-Methyl-4-oxo-3,4dihydroimidazo[5,1-d][1,2,3,5]tetrazine-8-carboxylic acid (TMZ-COOH) were purchased from Fluorochem.
The solvents employed in the synthesis of the compounds, including Dichloromethane (CH2Cl2), 1,2-Dichloroethane, Methanol (MeOH), Trifluoroacetic acid (TFA) were procured from Sigma-Aldrich. Additionally, HPLC-grade Acetonitrile (ACN) was obtained from Avantor.
For column chromatography, 0.040–0.063 mm (230–400 mesh) silica gel was used. Thin layer chromatography (TLC) with pre-coated Merck silica gel 60 F254 plates was performed to check the reaction progress. Then, UV light exposure was used to visualize the TLC plates.
Characterization.
Bruker Avance FT-NMR spectrometers were utilized for the experiments: a 250 MHz instrument for recording 1H and 13C NMR spectra and a 500 MHz instrument for 1H NMR spectra and 2D 1H/13C - NMR experiments.
The Xevo G2 Q-TOF mass spectrometer was operated in positive electrospray ionization (ESI) mode for direct infusion analysis, employing full-scan MS across a mass range of 50–1000 m/z. The following optimized source conditions were applied to achieve maximum ion intensity: capillary voltage of 3.5 kV, sample cone voltage of 120 V, source temperature set to 120 °C, desolvation temperature of 250 °C, cone gas flow rate of 100 L h⁻¹, and desolvation gas (N₂) flow rate of 600 L h⁻¹. These settings ensured enhanced sensitivity and ionization efficiency during the analysis.
Product purity was further evaluated by an Agilent analytical HPLC system equipped with an InfinityLab Poroshell 120 EC-C18 column (4.6x150 mm) and a diode array detector (DAD). Detection was carried out at a wavelength of 254 nm. The analysis occupied under a gradient solvent system, from A (2%) and B (98%) to A (100%) and B (0%), where solvent A consisted of ACN with 0.1% trifluoroacetic acid, and solvent B was H2O with 0.1% trifluoroacetic acid. This was followed by a 3-minute phase with A (100%) and B (0%), and finally, the system reverted to the initial conditions of A (2%) and B (98%) for an additional 2 minutes. The flow rate was maintained at 1.0 mL/min over a 20-minute period. The peak corresponding to AGT-7 appeared at 5.15 minutes, indicating a purity of 95.3%.
Plasma Stability
Sample Preparation
Plasma samples were diluted 1:1 (v/v) with phosphate buffer (pH 7.4). For each analyte, three independent samples were prepared by spiking 50μL of a 40 μM stock solution in dimethyl sulfoxide (DMSO) into 1950μL of diluted plasma, resulting in a final analyte concentration of 1 μM and a DMSO content of 2.5% (v/v) in plasma [30,31]. The mixtures were vortexed vigorously and incubated at 37°C on a thermoshaker (Kisker Biotech, Steinfurt, Germany) at 600 rpm. Reactions were terminated at 0, 1, 2, 3, 4, 6, 9, and 24 hours by transferring 150 μL of the incubated plasma into 450 μL of ice-cold acetonitrile. After vortexing, samples were stored at −20°C for 30 min, centrifuged, and 200 μL of the supernatant was diluted with 600 μL of water. The resulting solution was filtered through a 0.22μm nylon syringe filter (Target Analysis, Thessaloniki, Greece) into 2 mL glass vials prior to analysis [32].
Chromatographic Analysis
Chromatographic separation was achieved using a Waters ACQUITY i-Class Plus UPLC system (Waters, Manchester, UK) equipped with a temperature-controlled autosampler (6 °C) and an Waters ACQUITY UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm particle size) maintained at 35°C. The mobile phase consisted of (A) 0.1% formic acid in water and (B) 0.05% formic acid in acetonitrile, delivered constantly at 0.2 mL/min. The gradient program was optimized as follows: 1% B (0.00–3.00 min), linear ramp to 100% B (3.00–10.00 min), hold at 100% B (10.00–12.00 min), return to initial conditions (12.00–12.01 min), and re-equilibration at 1% B (12.01–15.00 min). A 5 μL injection volume was employed for all analyses.
Mass Spectrometric Detection
The UPLC system was coupled to a Xevo G2-XS QToF mass spectrometer (Waters) via an electrospray ionization (ESI) source operated in positive ion mode. Key ion source parameters included a capillary voltage of 1.0 kV, cone voltage of 20 V, source temperature of 120°C, and desolvation temperature of 550°C, with cone and desolvation gas flows set to 20 L/h and 1000 L/h (nitrogen), respectively. Mass accuracy was ensured using a LockSpray system with leucine-enkephalin (5 ng/mL in 50% acetonitrile/0.1% formic acid; 10 μL/min) as the lock mass (reference ion: [M+H]⁺ at m/z 556.2771).
Data Acquisition and Processing
Full-scan MS data were acquired in MSE mode with two alternating collision energy functions: low energy (4 eV) for precursor ions and ramped high energy (25–45 eV) for fragment ions. Scans covered m/z 100–900 at a rate of 0.3 s/scan. System control and data acquisition were managed using MassLynx v4.2 (SCN 1029), while UNIFI v1.9.4.053 facilitated data processing, including adduct identification ([M+H]⁺, [M+Na]⁺, [M+H₂]²⁺) and relative quantitation.
Evaluation of cytotoxicity in cancer cell lines
The sensitivity of T98 and U87 cells to the compound was assessed using the Trypan Blue Exclusion Test. 20,000 cells were seeded onto 12-well plates and incubated in a humidified atmosphere. After 24 hours, cells were treated with the compound at concentrations of 20-100 μM for 72 hours followed by trypan blue exclusion analysis. The results were derived as the mean of three independent experiments.
Flow Cytometric Analysis of DNA Cell Cycle
For DNA cell cycle analysis, 20,000 cells were seeded in 12-well plates, and after 24 h were treated with the compound at IC50 concentration for a further 72 h. Subsequently, cells were washed with PBS solution, harvested by incubation with trypsin, and held at 37 °C for 20 min with a PI working solution (50 g/mL PI, 20 mg/mL RNase A, and 0.1% Triton X-100). PI fluorescence data were collected using a flow cytometer (Omnicyt, Cytognos S.L.) and were analyzed using the GraphPad Prism version 6 software and MedCalc software (Trial version).
Zebrafish maintenance, breeding and toxicity tests
Zebrafish Housing and Husbandry
Adult zebrafish specimens of the wild-type strain (AB) were maintained in a colony room, in a recirculated system, at 28 ± 1 °C, pH 6.5–7.5, water conductivity of 500 ± 50 μS cm−1 with a 14-h light/10-h dark photoperiod (lights on at 8:00 a.m.). Feeding of the fish was performed twice a day with zebrafish feed (Zebrafeed, Sparos) following common practices. Sexually mature zebrafish (at least three months old) were used for spawning.
The collection of zebrafish eggs was performed at the beginning of the 14-h light phase following the mating procedure that took place overnight. After inspecting them, the unfertilized eggs and those that showed developmental disorders were removed. The dechorionation process of the eggs followed at 24 h post-fertilization (hpf).
The dechorionated embryos were placed in 24-well culture plates (2 embryos per well, 1.5 mL of solution per well) and each experiment was performed in triplicate. Preliminary tests were performed to evaluate the range of 0%-100% mortality. DMSO (0.1 %) was used as dissolver and as well as vehicle control (non-exposed). In the current study, seven different concentrations of AGT-7 were tested (0.0, 10.0, 20.0, 40.0, 60.0, 80.0 and 100.0 μΜ). In total, 881 embryos were studied, of which 96 belonged to the non-exposed group. Six (6) embryos of the non-exposed group were found to be dead. Each experiment lasted 96 hours post-fertilization time (hpf) and began at 24 hpf, approximately at 26-somite point, according to Kimmel et al. [33,34].
Zebrafish Toxicity Testing
It is essential to apply an acute test that uses short-term exposure (96hpf) to the AGT-7 and then to determine the concentration that is lethal to 50% of zebrafish embryos. This is a valuable indicator of acute fish toxicity. According to OECD, 2010 indications of death of an embryo include coagulation of the embryo, lack of somite formation, non-detachment of the tail and/or lack of heartbeat.
Lethal Dose (LD50) Determination
Toxicity assays (LD50 values) and confidence limits (LD25 and LD75) were calculated based on cumulative mortality at the end of the experiment (96 hpf). The LD50 values were assessed using Regression Probit analysis (the chi-square test, Pearson goodness of fit test, and 95% confidence interval).
Radiolabeling of Myoview
Technetium-99m (99mTc) is a gamma emitter with a photon energy of 140 keV, which requires radiation protection precautions during handling to reduce the risk of harm. All work associated with radiolabeling procedures was conducted in a licensed radiochemistry facility, where such experiments could be safely conducted.
All reagents and solvents had a purity >95% and were used without further purification. Acetonitrile (>99.5%) was purchased from Carlo Erba (Val-de-Reuil, France). Dimethylsulfoxide (>99.5%) was purchased from Aldrich Chemical (St. Louis, MO, USA). Human serum was purchased from Sigma Aldrich (St. Louis, MO, USA). Trifluoroacetic acid (>99%) was purchased from Alfa Aesar (Loughborough, UK). 99mTc was eluted as Na[99mTc]TcO4 from a commercial 99Mo/99mTc generator (Mallinckrodt Medical B.V., Hazelwood, MO, USA). Commercially available tetrofosmin (Myoview, GE Healthcare) was used in our experiments, for reasons of comparison to the novel theranostic agent.
Analyses and separation as well as purification processes were performed by High-Performance Liquid Chromatography (HPLC) using a Waters 600 Controller pump, a Waters 996 Photodiode Array detector (set at 220 nm for all experiments) and a γ-RAM radioactivity detector to measure radioactive flow on a NUCLEOSIL 100-5 C18 (Macherey-Nagel, Dueren, Germany). All HPLC solvents were filtered through 0.22 mm membrane filters (Millipore, Milford, MA, USA) before use. Instant thin layer chromatography - silica gel (ITLC-SG) 60 sheets (5 × 10 cm) (Merck, Darmstadt, Germany) were used for the determination of radiolabeling purity of [99mTc]Tc-TF (Myoview). These were developed in Acetone:Dichloromethane 35:65 and then measured with a Radio-TLC Scanner (Scan-Ram, LabLogic, Sheffield, UK). Radioactivity measurements were conducted in a dose calibrator (Capintec, Ramsey, NJ, USA).
The Myoview Tetrofosmin kit for radiopharmaceutical preparation contains 230 micrograms of tetrofosmin (active ingredient), 0.03mg stannous chloride dehydrate, 0.32mg disodium sulphosalicylate, 1.0mg sodium D-gluconate and 1.8mg sodium hydrogen carbonate as a freeze-dried mixture under nitrogen. Labeling is performed by direct addition of freshly-eluted sodium pertechnetate (4 mL, 20 mCi) and incubating for 15 min at room temperature. Radiolabeling yield was assessed by ITLC-SG, with a mixture of 35:65 acetone:dichloromethane as the mobile phase. In this solvent system, free [99mTc]Tc-pertechnetate runs to the top piece of the strip, [99mTc]Tc-tetrofosmin runs to the centre piece of the strip, while reduced, hydrolysed 99mTc and any hydrophilic complex impurities remain at the origin in the bottom piece of the strip. [99mTc]Tc-Tetrofosmin should be used within 12 hours of preparation.
Preparation of the precursor [99mTc][Tc(H2O)3(CO)3]+
The labeling precursor [99mTc][Tc(H2O)3(CO)3]+was prepared as described in the literature [35]. Briefly, a vial containing 4 mg Na2CO3, 20 mg sodium tartrate and 7 mg NaBH4 was sealed and CO gas was purged for 2 min prior to addition of 1 mL Na[99mTc]TcO4 eluate obtained from a 99Mo/99mTc generator. The vial was heated at 115 ◦C for 30 min and, at the end of the reaction, was left to cool at room temperature. Finally, the precursor was brought to pH 6.5–7 through the addition of HCl 1 M. The formation of the precursor [99mTc][Tc(H2O)3(CO)3]+ was determined by reverse-phase HPLC (RP-HPLC) on a C18-RP column by applying a linear gradient system from 0% to 100% B over 30 min at a flow rate of 1 mL/min, where solvent A was H2O/0.1% TFA and solvent B was MeOH/0.1% TFA, at a flow rate of 1 mL/min. The radioactivity of the precursor was measured using a dose calibrator.
Radiolabeling with [99mTc][Tc(H2O)3(CO)3]+
Radiolabeling of AGT-7 was accomplished via the 99mTc carbonyl precursor (0.8–2 mCi) at 40 °C for 45 min. Radiochemical yield was assessed by HPLC, as mentioned above.
In Vitro Stability Studies
In vitro stability of radiolabeled AGT-7 was evaluated at room temperature (bench stability at RT) and in the presence of serum at 37 °C up to 24 h post-radiolabeling. Serum stability is performed in order to assess the in vivo stability of [99mTc]Tc-AGT-7. Stable binding of the radioisotope onto the novel compound is very important in order to ensure that freely-circulating radioisotope after in vivo administration will be minimal, thus avoiding excess background noise during imaging. Serum stability studies do not always ensure in vivo stability of the radiolabeled compounds under investigation, however they provide a good indication of their in vivo fate. Thus, stability of the radiolabeled samples in the presence of human serum is tested before assessing the biological behavior in animal models.
Human Serum Stability
For human serum stability assessment, 50 μL of [99mTc]Tc-AGT-7 were challenged against 450 μL human serum. The samples were incubated at 37 ◦C for 2 h and 24 h. Afterwards, 100 μL of each mixture were treated with 200 μL ethanol and centrifuged at 450× g for 10 min to precipitate serum proteins. The supernatants were removed and analyzed by HPLC, as mentioned above. The experiment was performed thrice.
Lipophilicity Studies
Lipophilicity studies were performed in order to determine the lipophilic behavior of [99mTc]Tc-Tetrofosmin and [99mTc]Tc-AGT-7. The lipophilicity of the radiolabeled complexes was determined by calculating the partition coefficient (P) with the shake-flask method. Briefly, in a centrifuge tube, 1 mL of 1-octanol and 1 mL PBS (0.01 M, pH 7.4) were mixed with 1–2 μCi of each of the radiolabeled complexes. The samples were vortexed for 1 min and the radioactivity of the aliquots (200 μL) of each phase was counted in a gamma counter. The partition coefficient was calculated according to the following equation:
The results were expressed as logP. The procedure was repeated three times.
Ex Vivo Biodistribution Studies
For the ex vivo biodistribution studies, normal Carworth Farms White (CFW) mice of both genders were used. The mice were housed in individually ventilated cages (IVC) under constant temperature (22 ± 2 °C) and humidity (45 – 50%) and a 12 h light/dark cycle, with free access to food and water. The animals were obtained from the breeding facilities of the Institute of Biosciences and Applications, NCSR “Demokritos”. The experimental animal facility is registered according to Greek Presidential Decree 56/2013 (Reg. Number: EL 25 BIO 022), in accordance with European Directive 2010/63, which is harmonized with national legislation, on the protection of animals used for scientific purposes. All applicable national guidelines for the care and use of animals were followed. The study protocol was approved by the Department of Agriculture and Veterinary Service of the Prefecture of Athens (Protocol Number 634365/27-7-21). These studies have been further approved by our institutional ethics committee, and the procedures followed are in accordance with institutional guidelines. Intravenous injections were performed using insulin syringes BD Micro-Fine 1 mL (29G). The animals were euthanized in a chamber saturated with isofluorane vapors (isoflurane 1000 mg/g, Iso-Vet, Chanelle Pharma). The radioactivity of samples and syringes was measured using a dose calibrator (Capintec, Ramsey, New Jersey), while a Cobra II automatic gamma counter (Canberra, Packard, Schwadorf, Austria), was used to measure the radioactivity of the samples from the ex vivo biodistribution studies.
The ex vivo biodistribution of [99mTc]Tc-AGT-7 was investigated at 1 and 24 h post-injection and compared to the ex vivo biodistribution of [99mTc]Tc-TF. The ex vivo behavior of the two radiolabeled complexes was evaluated in mixed-gender Carworth Farms White (CFW) mice, 6–8 weeks old, weighing 20–30 g. The radiotracers were intravenously injected via the tail vein (100 μL/~100 μCi). At 1 and 24 h post-administration, the animals were euthanized by isoflurane inhalation and the major organs and tissues of interest (blood, liver, heart, kidneys, stomach, intestines, spleen, muscle, lung, bone, pancreas and brain) were collected, weighed and measured in an automatic gamma counter. For the calculation of the injected dose in each animal, a standard solution was prepared, while the radioactivity remaining in the tail was subtracted. The syringes containing the radiolabeled samples were measured before and after injection, to determine the precise dose administered to each mouse. All measurements were corrected for background and radioactive decay. All the distribution data were calculated as the percentage of injected dose per gram (%ID/g).
5. Conclusions
In conclusion, we have successfully developed AGT-7, a theranostic compound based on the glioma tumor-homing properties of Tetrofosmin (MyoviewTM), designed to integrate both diagnostic and therapeutic functionalities. AGT-7 utilizes a chelating core for 99mTc radiolabeling, tethered to the cytotoxic agent TMZ. Cytotoxicity evaluations in glioma cell lines demonstrated significantly enhanced efficacy for AGT-7 compared to TMZ alone. AGT-7 exhibited IC50 values of 27 μM in T98 cells and 30 μM in U87 cells, compared to respective values of 143 μM and 50 μM for TMZ, highlighting its superior cytotoxic potential. In vivo toxicity assessments in zebrafish revealed dose-dependent effects, with mortality rates increasing at concentrations higher than the IC50 values in glioma cells, suggesting a favorable therapeutic window for clinical evaluation. Radiolabeling of AGT-7 with [99mTc][Tc(CO)3(OH2)3]+ was achieved efficiently, with radiochemical yields exceeding 95% after 45 minutes at 40°C. The radiolabeled compound demonstrated stability at room temperature and in serum for at least 24 hours post-radiolabeling. Ex vivo biodistribution studies revealed distinct differences in organ uptake between [99mTc]Tc-AGT-7 and [99mTc]Tc-TF. Notably, [99mTc]Tc-AGT-7 exhibited tenfold lower cardiac uptake compared to [99mTc]Tc-TF, a well-established heart imaging agent, which demonstrated over 20% ID/g heart uptake. This reduced cardiac uptake, attributed to the lower lipophilicity of AGT-7, underscores its suitability as a glioma theranostic agent. These findings highlight the therapeutic potential of AGT-7 as a dual-functional agent for glioblastoma, warranting further preclinical evaluation.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Data curation, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Evangelia-Alexandra Salvanou, Maria Giannakopoulou, Maximos Leonardos, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Funding acquisition, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Maria Giannakopoulou, Vasiliki Zoi, Andreas Fotopoulos, Chrissa Sioka, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Investigation, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Evangelia-Alexandra Salvanou, Maria Giannakopoulou and Maximos Leonardos; Project administration, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Resources, Andreas Fotopoulos, Ioannis Leonardos, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Supervision, Timothy Crook, Ioannis Leonardos, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Validation, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Evangelia-Alexandra Salvanou, Maria Giannakopoulou and Maximos Leonardos; Visualization, George A. Alexiou, Penelope Bouziotis and Andreas Tzakos; Writing – original draft, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Evangelia-Alexandra Salvanou, Maria Giannakopoulou, Penelope Bouziotis and Andreas Tzakos; Writing – review & editing, Stavroula Kyrkou, Vasileios-Panagiotis Bistas, Penelope Bouziotis and Andreas Tzakos.
Funding
This research was funded by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH—CREATE—INNOVATE (project code: TAEDK-06189/ Τ2EDK-0326, Acronym: Glioblastoma).
Institutional Review Board Statement
The animal study protocol was approved by the Department of Agriculture and Veterinary Service of the Prefecture of Athens (protocol number: 634365; date of approval: 27 July 2021).
Acknowledgments
The authors would like to thank Nikolaos A. Parisis for performing the mass spectrometry analyses and for his valuable assistance with the plasma stability experiments and Dr. Stamatis S. Passadis for participating at the onset on this study. Additionally the authors kindly acknowledge Nektarios Pirmettis for preparation of the technetium precursor used in this study and Stavros Xanthopoulos for excellent technical execution of the biodistribution studies.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Jiapaer, S.; Furuta, T.; Tanaka, S.; Kitabayashi, T.; Nakada, M. , Potential Strategies Overcoming the Temozolomide Resistance for Glioblastoma. Neurol Med Chir (Tokyo) 2018, 58, 405–421. [Google Scholar] [CrossRef] [PubMed]
- Pandey, P.K.; Sharma, A.K.; Gupta, U. , Blood brain barrier: An overview on strategies in drug delivery, realistic in vitro modeling and in vivo live tracking. Tissue Barriers 2016, 4, e1129476. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, D.T.; Chow, F.; Yew, A.; Kim, W.; Cremer, N.; Yang, I. , Temozolomide and Other Potential Agents for the Treatment of Glioblastoma Multiforme. Neurosurgery Clinics of North America 2012, 23, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Chua, J.; Nafziger, E.; Leung, D.; Practice, E.-B. Current Oncology. Reports 2019, 21, 30. [Google Scholar] [CrossRef]
- Jia, J.L.; Alshamsan, B.; Ng, T.L. , Temozolomide Chronotherapy in Glioma: A Systematic Review. Curr Oncol 2023, 30, 1893–1902. [Google Scholar] [CrossRef]
- Teraiya, M.; Perreault, H.; Chen, V.C. , An overview of glioblastoma multiforme and temozolomide resistance: can LC-MS-based proteomics reveal the fundamental mechanism of temozolomide resistance? Front Oncol 2023, 13, 1166207. [Google Scholar] [CrossRef]
- Cruz, J.V.; Batista, C.; Afonso, B.D.; Alexandre-Moreira, M.S.; Dubois, L.G.; Pontes, B.; Neto, V.M.; Mendes, F.D., Obstacles to Glioblastoma Treatment Two Decades after Temozolomide. Cancers, 2022.
- Li, H.-Y.; Feng, Y.-H.; Lin, C.-L.; Hsu, T.-I. , Mitochondrial Mechanisms in Temozolomide Resistance: Unraveling the Complex Interplay and Therapeutic Strategies in Glioblastoma. Mitochondrion 2024, 75, 101836. [Google Scholar] [CrossRef]
- Kelly, J.D.; Forster, A.M.; Higley, B.; Archer, C.M.; Booker, F.S.; Canning, L.R.; Chiu, K.W.; Edwards, B.; Gill, H.K.; McPartlin, M. , et al., Technetium-99m-tetrofosmin as a new radiopharmaceutical for myocardial perfusion imaging. J Nucl Med 1993, 34, 222–227. [Google Scholar]
- D’Arceuil, H. , Technetium-99m tetrofosmin: Use for myocardial perfusion imaging in the detection of coronary artery disease. Reports in Medical Imaging 2010, 2010. [Google Scholar] [CrossRef]
- Stacy, M.R.; Zhou, W.; Sinusas, A.J. , Radiotracer imaging of peripheral vascular disease. J Nucl Med 2013, 54, 2104–2110. [Google Scholar] [CrossRef]
- Boschi, A.; Uccelli, L.; Marvelli, L.; Cittanti, C.; Giganti, M.; Martini, P., Technetium-99m Radiopharmaceuticals for Ideal Myocardial Perfusion Imaging: Lost and Found Opportunities. Molecules, 2022.
- Fotopoulos, A.D.; Kyritsis, A.P.; Tsiouris, S.; Al-Boucharali, J.; Papadopoulos, A.; Voulgaris, S.; Alexiou, G.A. , Characterization of intracranial space-occupying lesions by ⁹⁹(m)Tc-Tetrofosmin SPECT. J Neurooncol 2011, 101, 83–89. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Fotopoulos, A.D.; Papadopoulos, A.; Kyritsis, A.P.; Polyzoidis, K.S.; Tsiouris, S. , Evaluation of brain tumor recurrence by 99mTc-tetrofosmin SPECT: a prospective pilot study. Annals of Nuclear Medicine 2007, 21, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Xourgia, X.; Gerogianni, P.; Vartholomatos, E.; Kalef-Ezra, J.A.; Fotopoulos, A.D.; Kyritsis, A.P. , <sup>99m</sup>Tc-Tetrofosmin Uptake Correlates with the Sensitivity of Glioblastoma Cell Lines to Temozolomide. World J Nucl Med 2017, 16, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Arbab, A.S.; Koizumi, K.; Toyama, K.; Araki, T. ; Uptake of technetium-99m-tetrofosmin, technetium-99m-MIBI and thallium-201 in tumor cell lines. J Nucl Med 1996, 37, 1551–1556. [Google Scholar]
- Weinberg, S.E.; Chandel, N.S. , Targeting mitochondria metabolism for cancer therapy. Nature Chemical Biology 2015, 11, 9–15. [Google Scholar] [CrossRef]
- Murphy, M.P. , How mitochondria produce reactive oxygen species. Biochem J 2009, 417, 1–13. [Google Scholar] [CrossRef]
- Cao, T.G.N.; Kang, J.H.; Kang, S.J.; Hoang, Q.T.; Kang, H.C.; Rhee, W.J.; Zhang, Y.S.; Ko, Y.T.; Shim, M.S. , Brain endothelial cell-derived extracellular vesicles with a mitochondria-targeting photosensitizer effectively treat glioblastoma by hijacking the blood‒brain barrier. Acta Pharmaceutica Sinica B 2023, 13, 3834–3848. [Google Scholar] [CrossRef]
- Dilworth, J.R.; Parrott, S.J. , The biomedical chemistry of technetium and rhenium. Chemical Society Reviews 1998, 27, 43–55. [Google Scholar] [CrossRef]
- Alberto, R.A.; Schibli, R.; Egli, A.; Schubiger, A.P.; Abram, U.; Kaden, T. , A Novel Organometallic Aqua Complex of Technetium for the Labeling of Biomolecules: Synthesis of [99mTc(OH2)3(CO)3]+ from [99mTcO4]- in Aqueous Solution and Its Reaction with a Bifunctional Ligand. Journal of the American Chemical Society 1998, 120, 7987–7988. [Google Scholar] [CrossRef]
- Lopez, J.; Tait, S.W.G. , Mitochondrial apoptosis: killing cancer using the enemy within. British Journal of Cancer 2015, 112, 957–962. [Google Scholar] [CrossRef]
- Lu, R.O.; Ho, W.S.; Dysfunction, M. ; Macrophage, and Microglia in Brain Cancer. Frontiers in Cell and Developmental Biology 2021, 8. [Google Scholar] [CrossRef]
- Smith, R.A.; Hartley, R.C.; Cochemé, H.M.; Murphy, M.P. ; Mitochondrial pharmacology. Trends Pharmacol Sci 2012, 33, 341–352. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. , Targeting lipophilic cations to mitochondria. Biochim Biophys Acta 2008, 1777, 1028–1031. [Google Scholar] [CrossRef] [PubMed]
- Alexiou, G.A.; Tsiouris, S.; Goussia, A.; Papadopoulos, A.; Kyritsis, A.P.; Polyzoidis, K.S.; Fotopoulos, A.D. , Evaluation of glioma proliferation by 99mTc-Tetrofosmin. Neuro-Oncology 2008, 10, 104–105. [Google Scholar] [CrossRef]
- Makrypidi, K.; Kiritsis, C.; Roupa, I.; Triantopoulou, S.; Shegani, A.; Paravatou-Petsotas, M.; Chiotellis, A.; Pelecanou, M.; Papadopoulos, M.; Pirmettis, I. , Evaluation of Rhenium and Technetium-99m Complexes Bearing Quinazoline Derivatives as Potential EGFR Agents. Molecules 2023, 28. [Google Scholar] [CrossRef]
- Bouziotis, P.; Gourni, E.; Patsis, G.; Psimadas, D.; Zikos, C.; Fani, M.; Xanthopoulos, S.; Loudos, G.; Paravatou-Petsotas, M.; Livaniou, E.; Varvarigou, A.D.; Pirmettis, I.; Papadopoulos, M. Radiochemical and radiobiological assessment of a pyridyl-S-cysteine functionalized bombesin derivative labeled with the (99m)Tc(CO)3(+) core. Bioorg Med Chem 2013, 21, 6699–6707. [Google Scholar] [CrossRef]
- Kasten, B.B.; Ma, X.; Cheng, K.; Bu, L.; Slocumb, W.S.; Hayes, T.R.; Trabue, S.; Cheng, Z.; Benny, P.D. , Isothiocyanate-Functionalized Bifunctional Chelates and fac-[MI(CO)3]+ (M = Re, 99mTc) Complexes for Targeting uPAR in Prostate Cancer. Bioconjugate Chemistry 2016, 27, 130–142. [Google Scholar] [CrossRef]
- Di, L.; Kerns, E.H.; Chapter; Di, L.; Kerns, E.H.; Press, A.; Boston, 2016, pp. 387–392.
- Di, L.; Kerns, E.H.; Hong, Y.; Chen, H. , Development and application of high throughput plasma stability assay for drug discovery. International Journal of Pharmaceutics 2005, 297, 110–119. [Google Scholar] [CrossRef]
- Parisis, N.A.; Bousdouni, P.; Kandyliari, A.; Spyridaki, M.-H.; Koutsogianni, A.D.; Telli, C.; Tsilidis, K.K.; Koutelidakis, A.E.; Tzakos, A.G. , Development and Validation of a Simple and Cost-Effective LC-MS/MS Method for the Quantitation of the Gut-Derived Metabolite Trimethylamine N-Oxide in Human Plasma of Healthy and Hyperlipidemic Volunteers. Molecules, 2025.
- Kimmel, C.B.; Patterson, J.; Kimmel, R.O. , The development and behavioral characteristics of the startle response in the zebra fish. Developmental Psychobiology 1974, 7, 47–60. [Google Scholar] [CrossRef]
- OECD, Short Guidance on the Treshold Approach for Acute Fish Toxicity, 2010.
- Schibli, R.; La Bella, R.; Alberto, R.; Garcia-Garayoa, E.; Ortner, K.; Abram, U.; Schubiger, P.A. , Influence of the Denticity of Ligand Systems on the in Vitro and in Vivo Behavior of 99mTc(I)−Tricarbonyl Complexes: A Hint for the Future Functionalization of Biomolecules. Bioconjugate Chemistry 2000, 11, 345–351. [Google Scholar] [CrossRef]
Scheme 1.
Synthetic route of the AGT-7, (i) Na(AcO)3BH - adding at 0 oC, DCE, rt, 3.5 hours, then H2O for 1.5 hours, (ii) 20% TFA/CH2Cl2, rt, 2 hours, (iii) PyBOP, DIPEA, dry DMF, rt, 12 hours.
Scheme 1.
Synthetic route of the AGT-7, (i) Na(AcO)3BH - adding at 0 oC, DCE, rt, 3.5 hours, then H2O for 1.5 hours, (ii) 20% TFA/CH2Cl2, rt, 2 hours, (iii) PyBOP, DIPEA, dry DMF, rt, 12 hours.
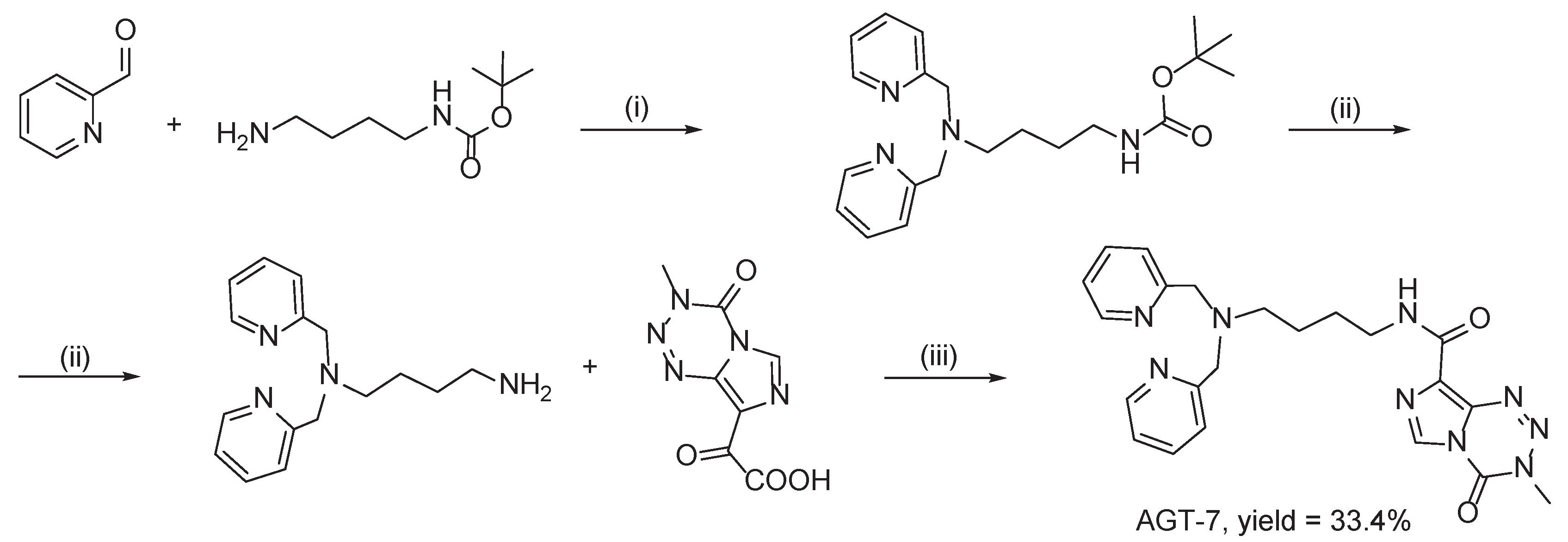
Figure 3.
Cytotoxic effect of AGT-7 and TMZ on T98 (A) and U87 cell lines (B) 72 hours after treatment. The results are the average of three independent experiments and are standardized to untreated cells. The IC50 values were calculated using the GraphPad Prism Version 6 non-linear regression analysis model.
Figure 3.
Cytotoxic effect of AGT-7 and TMZ on T98 (A) and U87 cell lines (B) 72 hours after treatment. The results are the average of three independent experiments and are standardized to untreated cells. The IC50 values were calculated using the GraphPad Prism Version 6 non-linear regression analysis model.
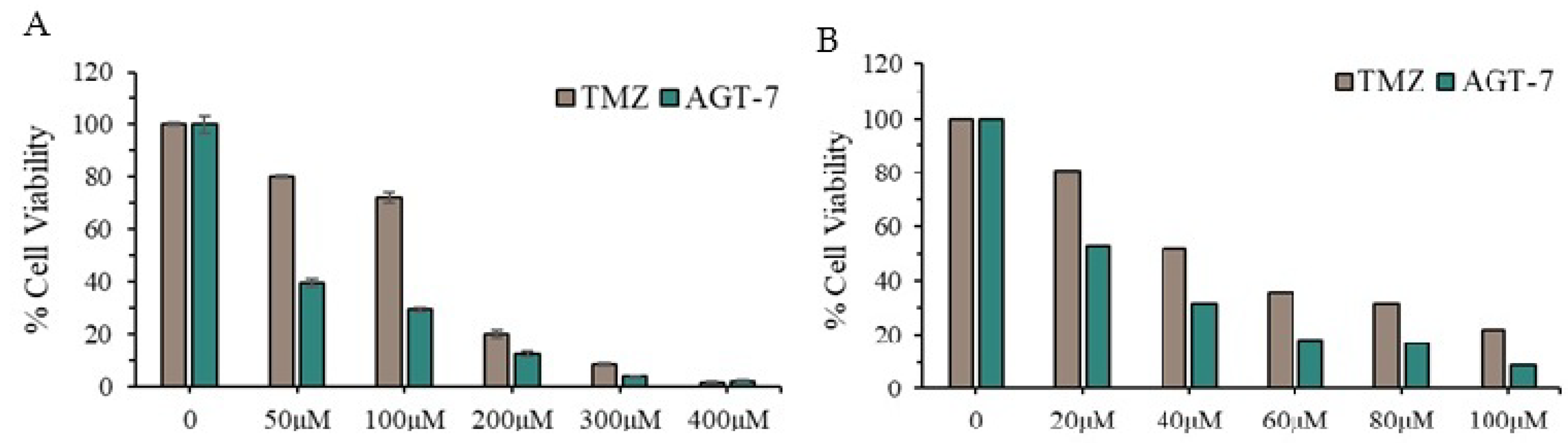
Figure 4.
Concentration response of AGT-7 toxicity for 96 hpf zebrafish embryos.
Figure 5.
Representative radio-TLC graph of [99mTc]Tc-Tetrofosmin.
Figure 6.
Representative HPLC graph of [99mTc]Tc-AGT-7. The retention times are ~4-6 min for the [99mTc][Tc(CO)3(OH2)3]+ precursor and ~3 min for [99mTc][TcO4]-. The main peak corresponds to [99mTc]Tc-AGT-7.
Figure 6.
Representative HPLC graph of [99mTc]Tc-AGT-7. The retention times are ~4-6 min for the [99mTc][Tc(CO)3(OH2)3]+ precursor and ~3 min for [99mTc][TcO4]-. The main peak corresponds to [99mTc]Tc-AGT-7.
Figure 7.
In vitro stability of [99mTc]Tc-AGT-7 in RT and serum up to 24 h.
Figure 8.
Ex vivo biodistribution results of [99mTc]Tc-AGT-7 and [99mTc]-TF, expressed as %IA/g (n = 3).
Figure 8.
Ex vivo biodistribution results of [99mTc]Tc-AGT-7 and [99mTc]-TF, expressed as %IA/g (n = 3).
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



