Submitted:
07 July 2025
Posted:
08 July 2025
You are already at the latest version
Abstract
Background: Factor XI (FXI) deficiency, or hemophilia C, is a rare bleeding disorder resulting from reduced levels or dysfunctional FXI protein due to mutations in the F11 gene. Aim of the Study: This study investigated the correlation between FXI activity levels, F11 genotype and bleeding phenotypes Methods: Clinical and genetic characteristics of 93 individuals from southern Italy diagnosed with congenital FXI deficiency, including 39 index cases and their relatives were evaluated. FXI:C plasma levels were measured. Sanger sequencing of F11 was performed and pathogenicity of variants identified was assessed using in silico tools. Results: FXI activity levels ranged widely (1–69%), with most cases being heterozygous and showing moderate deficiency. Only 12 individuals had severe FXI deficiency, typically associated with homozygosity or compound heterozygosity. Bleeding symptoms varied from mild to severe and occurred in 31% of subjects, though only a minority of those with severe deficiency experienced spontaneous or surgery-related bleeding. Sanger sequencing revealed 24 distinct F11 gene variants, predominantly missense mutations, with three novel variants (p.Val89*, p.Leu306Pro, and p.Trp515Gly). Common mutations included p.Glu135* and p.Glu315Lys. Variants were distributed across the gene, with no domain-specific clustering. No clear genotype-phenotype correlation was observed). Conclusions: FXI levels alone did not reliably predict bleeding risk, highlighting the influence of additional factors such as age, gender, and clinical history. The study reinforces the allelic and clinical heterogeneity of FXI deficiency and the limited utility of FXI:C levels alone for predicting bleeding severity. Comprehensive risk assessment models integrating clinical and molecular data are necessary for optimal patient management. Further research is needed to clarify the complex genotype–phenotype relationships in FXI deficiency.
Keywords:
FXI deficiency
; gene variant
; levels
; bleeding
; phenotype
1. Introduction
Factor XI (FXI) deficiency, also known as hemophilia C, is a rare bleeding disorder that affects individuals due to reduced levels or impaired function of FXI, a coagulation factor that plays a crucial role in the intrinsic pathway. [1] This deficiency is considered rare, affecting a small proportion of the population. FXI deficiency is most commonly found in individuals of Ashkenazi Jewish descent, with a prevalence of 1 in 1,000 individuals. [2] The pathogenesis of FXI deficiency lies in genetic mutations affecting the F11 gene, which is located on chromosome 4 and encodes the FXI protein. F11 variants can lead to a large array of alterations in the protein synthesis or functionality, resulting in reduced or dysfunctional FXI. [3] Any disruption in the molecular processes involved in the synthesis, post-translational modification, or function of FXI can contribute to deficiency. [4] This may involve abnormalities in the protein folding, transportation, or interaction with other clotting factors. The inheritance pattern of severe FXI deficiency is autosomal recessive. This means that an individual has to carry two alleles with a pathogenic variant, usually inherited one from each parent, to manifest the severe form of the disorder. Heterozygotes, individuals with a normal and a mutated copy of the gene, typically do not show symptoms or manifest an injury-related bleeding phenotype but can transmit the variant allele to their offspring.
FXI deficiency exhibits considerable genetic heterogeneity and a high number of variants has been identified in the F11 gene. These variations may include missense mutations, nonsense mutations, deletions, or insertions, each contributing to the varied clinical manifestations observed in affected individuals. The type and location of these gene variants can influence the severity of the deficiency and the associated bleeding tendencies.
Bleeding in FXI deficient patients can range from mild to severe, with symptoms including prolonged bleeding after injury or surgery, nosebleeds, and easy bruising. [5] However, the severity of bleeding can vary greatly among individuals with FXI deficiency, making it difficult to predict the risk of bleeding in each patient. [6]
Understanding the predictability of bleeding in patients with FXI deficiency is crucial for effective patient management and the development of appropriate treatment strategies. [7]
FXI protein structure is a homodimer, with each subunit containing four apple domains (A1 to A4) and a trypsin-like catalytic domain. F11 mutations have been identified in all domains suggesting that distinct structure-function relationships of different domains of the FXI could play a role leading to the clinical phenotype. [1]
In this study, we have evaluated the incidence and characteristics of gene variants affecting F11 in a cohort of patients with FXI deficiency recruited in reference hospitals in southern Italy.
2. Materials and Methods
2.1. Case Index and Relatives
A total of 93 individuals belonging to independent families and additional unrelated index patients with congenital FXI deficiency were recruited at four reference centers for thrombotic and hemorrhagic disorders. The inclusion criteria encompassed patients with a congenital FXI deficiency, characterized by a factor XI activity of <70 IU/dL. Exclusion criteria included acquired FXI deficiency, liver failure, and consumptive coagulopathy. Severe FXI deficiency was defined as an activity level of < 20 IU/dL. [8] Partial FXI deficiency was diagnosed among individuals who had FXI activity levels of 20 to <70 IU/dL. Data were collected from medical records, including clinical, biological, and therapeutic data at the time of diagnosis and subsequent evolution. Clinical and genetic investigations were performed in accordance with the Helsinki declaration and based on written informed consent for clinical and genetic testing. Written informed consent was requested from their legal representatives for subjects under 18 years. All data presented in the manuscript were properly anonymized. The study was approved by the local ethical committee. (protocol code 3261/CE/20).
2.2. Coagulation Tests
Peripheral venous blood was obtained from all patients using siliconized glass tubes containing sodium citrate anticoagulant. The upper layer of poor-platelet plasma was utilized for routine coagulation screening after centrifugation at 3000 xg for 10 min. The prothrombin time (PT), activated partial thromboplastin time (APTT), fibrinogen, and FXI:C were detected using SynthASil and FXI-deficient plasma, both from Werfen on the ACL top automatic analyzer (Werfen, Barcelona, Spain). The detection limits of the FXI:C assay was <1 U /dL. Reference intervals were obtained dosing plasma of 100 healthy blood donors from local healthy population.
2.3. Genetic Investigation
According to the manufacturer’s instructions, leukocytes from participants were utilized for extracting genomic DNA via QIAamp DNA Blood Kits (GIAGEN, Hilden, Germany). All 15 exons of the F11 gene, along with exon/intron boundary regions, were amplified using specific primers and PCR products were then sequenced using BigDye Terminator v.3.1 (Thermo Fisher Scientific, Waltham, MA, USA), according to standard protocols. [9] PCR products were purified with PCR multiwell plates 96 well plate for PCR 96 (Merck Millipore ldt – Tullagreen, Ireland). The purification protocol involves adding 55 μL H2O to the PCR product and transferring it to the plate, leave the plate at room temperature on the vacuum pump for 8 min. Afterwards add 26 μL H2O and let the plate shake for 10 min at 500rpm. At the end we obtained the purified product. Then, the sequencing reaction was performed by BigDye Terminator v3.1 Cycle Sequencing Kit and were analyzed on an ABI 3130xl DNA Sequencer (Applied Biosystems, Nor-walk, USA). The sequencing reactions mixtures conteined 2 μL of BigDie Terminator v1.1, 3.1 5X sequencing buffer; 1.2 μL of the same primer used in PCR previously di-luted 1:100; 0.5 μL of BigDie Terminator v3.1 Cycle Sequencing RR-100; 0.5 μL of purified PCR in a final volume of 10 μL H2O. Sequencing files were processed using Sequence Analysis Software (Applied Biosystems, Norwalk, USA) and were aligned and analyzed using Sequencer 4.7 Software. Prioritized Variant was validated in probands by Sanger sequencing and then studied in additional affected family members when DNA was available to perform segregation analysis.
2.4. In Silico Analysis of Pathogenicity
Population data were obtained from the Genome Aggregation Database (gnomAD; https://gnomad.broadinstitute.org/). ClinVar (https://www.ncbi.nlm.nih.gov/clinvar accessed on 1 June 2025), Franklin by genoox (https://franklin.genoox.com/clinical-db/home on accessed on 1 June 2025), EAHAD coagulation Factor XI UCL Factor XI Gene (F11) (https://dbs.eahad.org/FXI), (https://www.factorxi.org/torifxi_reference.html.php), and The Human Gene Mutation Database (https://www.hgmd.cf.ac.uk/ac/) were used as tools to sum up actual knowledge about the variants.
To investigate the putative pathogenic effect of new unreported F11 gene variants established available bioinformatics tools were used (MISSENSE3D: https://missense3d.bc.ic.ac.uk/).
To explore the potential deleterious impact of the new missense variations DynaMut2 (https://biosig.lab.uq.edu.au/dynamuth2) was then utilized by using the AlfaFold prediction structure (AF-P03951-F1) of a human FXI. DynaMut2 is a web server, which can evaluate the effects of mutations on the vibrational entropy variations induced and modifications in protein dynamics and stability, as well as to analyze and visualize protein dynamics by sampling conformations.
3. Results
3.1. Clinical Features of Cases with FXI Deficiency
In the present study was investigated a cohort of 93 individuals (37 men and 56 women) with a diagnosis of FXI deficiency from 39 unrelated families recruited in four southern Italian hospitals over a 15-year period (2007- 2022). Mean age at the enrollment was 31.6 years (range: 1-82). The large majority of individuals (81/93: 87%) showed a moderate FXI deficiency, suggestive of heterozygous defects, but 12 cases had severe FXI deficiency compatible with homozygosity or compound heterozygosity (Figure 1). Mean FXI activity levels were 35.1 IU/dL (range: 1-69).
Most of 39 index cases were referred for abnormal values during preoperative screening and check-ups (n= 21, 54%). The remaining index cases were referred for bleeding symptoms. Among the cases where abnormal bleeding occurred (n=18, 46%), eight resulted from bleeding after surgery or trauma. In addition, four women presenting with a personal history of menorrhagia were diagnosed with a FXI deficiency, whereas four cases suffered from recurrent nosebleed and two from spontaneous ecchymoses. Among the nine index cases presenting with severe FXI deficiency, three bleed: an 8-yrs old young female had bleeding after surgery; a 24-yrs old woman showed a history of menorrhagia; and a 49-yrs old woman who had repeated hemorrhagic episodes after surgery or operative dentistry procedures.
Fifty-four relatives with reduced FXI levels were identified. All relatives but four showed a moderate FXI deficiency. Among them, nine out of 50 with a moderate deficiency (18%) and two out of four with a severe deficiency (50%) suffered from bleeding after surgery or trauma (n=8), recurrent nosebleed (n=2), or menorrhagia (n=1).
3.2. Molecular Characterization
Sanger sequencing of F11 identified 24 potential causative variants. All were single nucleotide variations (SNVs) mainly causing missense changes (N = 17). In addition, three nonsense variants, three variants affecting splicing, and a small deletion were detected. As expected, variations dispersed over the entire F11 gene with no evidence of clustering at specific coding and non-coding regions (Table 1) but primarily involved residues in the Apple 2, Apple 4, and serine protease domains (Figure 1). Bleeding tendency was equally distributed among subjects carrying variants in different FXI protein domains e no significant genotype-phenotype correlation was detected.
As expected, most of cases were heterozygotes (79/93: 88%), whereas homozygous, and compound heterozygous were four (4/93: 1%) and ten (10/93: 10%), respectively (Table 1). Heterozygotes presented approximately four-fold higher mean plasma FXI:C levels than those recorded in homozygotes or compound heterozygotes, being 39 IU/dL (range: 18-79) and 10.6 IU/dL (range: 1-36), respectively. Among the 25 FXI defective cases presenting with a hemorrhagic phenotype, two were compound heterozygotes and all others were heterozygotes.
In index cases, the most common F11 gene variants identified were p.Glu135* (aka Jewish mutation type II; 26%, 12/46), p.Glu315Lys (15%, 7/46), and p.Phe301Leu (aka Jewish mutation type III; 13%, 6/46).
Of the 24 different FXI gene variants identified, 21, 20, and 18 have been previously reported in subjects with a FXI deficiency and were included by June 1, 2025 in the EAHAD coagulation Factor XI variant, in the UCL Factor XI Gene (F11), and in the HGMD databases, respectively.
In the gnomAD v4.1.0 database, 15 of these variants had a germline classification and were reported in the ClinVar archive as pathogenic (n=7), likely pathogenic (n=3), with conflicting interpretations (n=3), or of uncertain significance (n=2).
Among gene variants of uncertain significance, the p.Asp34His substitution has been suggested to interfere with chain folding. [10] The p.Arg162Cys substitution replaces a polar residue Arginine containing an electrically charged side chain with a sulfur-containing neutral and slightly polar amino acid, Cysteine. Among gene variants with conflicting interpretations, the substitution of a Threonine with a Methionine at residue #141 results in the introduction of a hydrophobic sulfur atom and the loss of hydrophilic hydroxyl group.[11] The p.Thr150Met substitution replaces a hydrophilic residue with a hydrophobic one. Finally, the p.Glu565Lys substitution has been suggested to affect with F11 mRNA splicing and induce the exon 13 skipping. [12,13]
In the gnomAD v4.1.0 database, the p.Cys136Arg and the p.Arg497Gln variants were present at a very low frequency (1.86e-6 and 9.30e-6, respectively). The first missense variation results in the substitution of a positively charged amino acid for a neutral one containing a sulfur atom. The latter causes the substitution of an Arginine for a Glutamine, which has no ionizable side chain.
Two variants with unknown implication on gene transcription were located in exon-intron boundaries. The first was the c.595+3A>G transition and occurred in the intron 6. In silico investigation of the effect of the substitution predicted a loss of efficiency of the splicing machinery (score of the donor site changing from 0.98 to 0.56) and causing the skipping of the affected exon 7. The second was the c.1717-2A>G transition and occurred in the intron 14. In silico investigation of the effect of the substitution predicted a deleterious effect with the suppression of the acceptor site (score changing from 0.98 to 0.0) causing the skipping of the affected exon 14.
The c.G325A (p.Ala109Thr) variant has been reported to interfere with the physiological donor splice site, resulting in the skipping of exon 4. [14]
Among the remaining FXI gene variants identified, the p.Ala561Asp was identified together with the p.Glu315Lys in a women and her daughter. In both cases, FXI plasma levels were comparable (33,6 IU/dL and 36,3 IU/dL). Inheritance of both variants suggested that they are in linkage and the moderately reduction of FXI plasma levels indicated the lack of an important effect of the p.Ala561Asp variant on the protein functionality. The p.Trp515Gly replaces a large aromatic and hydrophobic residue with a small, non-polar, and hydrophilic amino acid. The substitution was predicted to produce a pathogenic effect (suggested classification: Varsome: PP3; Franklin: Likely pathogenic).
The remaining three FXI gene variants (p.Val89*, p.Leu306Pro, and p.Trp515Gly) were new and previously unreported. The p:Val89* variant causes the appearance of a stop codon and is predicted to severely affect protein expression. In keeping with this, the index case showed reduced plasma FXI levels (42.3%). Analysis of the p.Leu306Pro variant using computational prediction tools (Table 2) showed an extremely low frequency in gnomAD population databases (PM2: Pathogenic Moderate) and supported a deleterious effect on the gene (PP3: Pathogenic Moderate). In addition, the missense variant occurred in a gene with a low rate of benign missense mutations and for which missense mutation is a common mechanism of a disease (PP2: Pathogenic Supporting). The p.Trp515Gly replaces a large aromatic and hydrophobic residue with a small, non-polar, and hydrophilic amino acid. The substitution was predicted to produce a pathogenic effect (suggested classification: Varsome: pathogenic supporting; Franklin: Likely pathogenic). The in silico analysis using the MISSENSE3D tool detected structural damage in both missense variant. p. Leu306Pro substitution triggers disallowed phi/psi alert. The phi/psi angles are in favoured region for the wild-type residue but outlier region for the mutant residue. The p.Trp515Gly substitution disrupts all H-bonds formed by a buried TRP residue (RSA 6.6%). In addition The it leads to the expansion of cavity volume by 113.832 Å^3.
For each genetic variant, the following information is shown: variant allele frequency according to gnomAD exome/genome database, DyneMut2_ (ΔΔGStability), Predicted Stability Change, effect on Protein Structure, ACMG annotation, ACMG Supporting Criteria. VUS: variant of uncertain significance.
To analyze the movement and flexibility of the mutant p.Leu306Pro and p.Trp515Gly proteins, we used the AlfaFold prediction structure (AF-P03951-F1) of the human Coagulation factor XI as a template and DynaMut2, a web server that combines Normal Mode Analysis (NMA) methods. DynaMut2 predicted a significant effect of both p.Leu306Pro and p.trp515Gly missense variations on protein stability with a Predicted Stability Change (ΔΔGStability) of -0.32 kcal/mol and-2.94 kcal/mol respectively, indicating a destabilizing effect. In this tool, ΔΔG ≥ 0 is considered stabilizing, and ΔΔG < 0 is considered destabilizing (Table 2). The modeling identifies in both model modifications in a mutant structure, allowing the formation of new extra bonds between the mutated protein and nearby residues or the loss of wild type bonds (Figure 2).
4. Discussion
Circulating FXI levels play a significant role in determining the bleeding tendencies of individuals with FXI deficiency. Several studies have demonstrated a correlation between lower FXI levels and an increased risk of bleeding events. Lower levels of FXI are associated with an increased risk of bleeding, while higher levels may provide some degree of protection against bleeding episodes. Patients with FXI levels below 30 IU/dL have been more likely found to experience spontaneous bleeding. [15] In addition, FXI levels have been reported that can serve as a reliable predictor of bleeding severity in FXI-deficient patients. [16] We report a higher bleeding prevalence among subjects with a severe FXI deficiency. These findings highlight the importance of monitoring FXI levels to better understand bleeding predictability in this population. However, it is important to note that FXI levels alone do not fully predict the severity of bleeding in FXI-deficient patients. In the current context, even in severe cases of FXI deficiency, the bleeding tendency is generally mild or moderate, while spontaneous bleeding is infrequent. We confirm a high variability in bleeding tendency, which was barely associated with FXI activity. In fact, 29 out of 93 individuals showed bleeding episodes and only 5 out of these had shown a severe deficiency.
Potentially pathogenic variants were identified in all subjects presenting with FXI deficiency. A total of 24 different F11 gene variations were identified and all were a single nucleotide variant. Most of them were previously reported while three were described for the first time (p.Val89*, and p.Leu306Pro, and p.Trp515Gly). Pathogenic variants were equally distributed throughout the entire F11 gene. In keeping with other studies in Italian patients, [17,18] some F11 gene variants were prevalent, i.e. p.Glu135* (aka Jewish mutation type II). All these results demonstrate the high allelic heterogeneity of FXI deficiency.
The novel p.Ala561Asp variant was found both in a young woman and in her daughter in association with the p.Glu315Lys variation. This finding suggests that the two variants form a haplotype and segregate in cis with each other. The p.Glu315Lys variant is reported in the ClinVar archive as pathogenic. In addition, in the present investigation heterozygotes showed FXI levels similar to those observed in previous studies. All these findings suggest that the p.Ala561Asp variant in cis does not apparently influence FXI plasma values or the clinical phenotype. However, it occurs in a highly conserved sequence - Cysteine-Alanine-Glycine - among serine proteases. Missense alterations involving Cysteine-Alanine-Glycine residues of the F11 gene or other serine proteases of the coagulation system have been described. [19]
The most common F11 gene variants identified were p.Glu135* and p.Glu315Lys, recorded in 21 (14 heterozygotes, four compound heterozygotes and three homozygotes) and 17 individuals (15 heterozygotes, including those also carrying the p.Ala561Asp variant, one compound heterozygote and one homozygote), respectively. Overall, a high variability in the expression of the clinical phenotype was observed among heterozygotes with one of these two variants. In fact, 19 out of 29 individuals were asymptomatic and only four of the 19 women suffered from menorrhagia. There are currently limited data on bleeding complications in women with FXI deficiency. In our cohort of women (25 probands and 28 relatives) we recorded 5 menorrhagia (9.4%). This prevalence did not differ from that recorded in apparently normal women, in which up to one forth may experience some form of heavy menstrual bleeding. [20] Overall, all these data indicated that bleeding tendency is equally distributed among subjects carrying variants in different domains e no significant genotype-phenotype correlation was detected.
Other factors, such as age, gender, and the presence of comorbidities, can also influence the bleeding tendencies in these individuals. [1] Risk stratification models have been developed to enhance the accuracy of bleeding risk assessment in FXI-deficient patients. These models take into account various clinical parameters, including FXI levels, to provide a more comprehensive evaluation of bleeding tendencies. By integrating FXI levels with other relevant factors, such as age, bleeding history, and comorbidities, these models offer a more accurate prediction of bleeding risk. The ISTH bleeding assessment tool and the EN-RBD bleeding score are examples of risk prediction models that have been widely used in clinical practice to guide management decisions for FXI-deficient patients. [7,21]
5. Limitations
The present study has some limitations because longitudinal functional data are lacking. Due to the sample size, we did not explore and thus cannot rule out whether the relationship between FXI deficiency and outcomes differ by sex. In addition, drugs and comorbidities might have somewhat interfered with FXI levels though many potential cofounders were excluded. However, the present study confirms that FXI activity levels poorly correlate with bleeding phenotypes.
6. Conclusion
The data from the present study confirm the wide heterogeneity of clinical and molecular findings in subjects with FXI deficiency and a weak correlation between FXI plasma levels and clinical outcome in FXI-deficient patients. Further studies are needed to better define the genotype–phenotype relationship in subjects with FXI deficiency.
Author Contributions
Conceptualization, R.S., G.DA., M.DA., and M.M.; methodology, R.S., F.M., D.B., and M.M.; validation, GL.T., D.B., P.DB., and M.M.; formal analysis, GL.T.; resources, R.S., E.G., and M.M.; data curation, R.S., G.L., G.DA., P.DB., and M.M.; writing—original draft preparation, R.S., GL.T., and D.B.; writing—review and editing, D.B., F.M., G.L., and M.M.; visualization, G.DA., P.G.; supervision, R.S., P.G., and M.M.; project administration, R.S., and M.M. All authors have read and agreed to the published version of the manuscript.
Funding
The author recognizes the financial contribution of European Union– NextGenerationUE as part of PNRR MUR – M4C2 – Investimento 1.3 - Public Call “Partenariati Estesi” - D.D. n. 341/2022.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Ethics Committee) of Policlinico Foggia University-Hospital (protocol code 3261/CE/20).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Barg AA, Livnat T, Kenet G. Factor XI deficiency: phenotypic age-related considerations and clinical approach towards bleeding risk assessment. Blood. 2024;143:1455-1464. [CrossRef]
- Asakai R, Chung DW, Davie EW, Seligsohn U. Factor XI deficiency in Ashkenazi Jews in Israel. N Engl J Med. 1991;325:153-158. [CrossRef]
- Asselta R, Paraboschi EM, Rimoldi V, Menegatti M, Peyvandi F, Salomon O, Duga S. Exploring the global landscape of genetic variation in coagulation factor XI deficiency. Blood. 2017;130:e1-e6. [CrossRef]
- Peyvandi F, Palla R, Menegatti M, Siboni SM, Halimeh S, Faeser B, et al. European Network of Rare Bleeding Disorders Group. Coagulation factor activity and clinical bleeding severity in rare bleeding disorders: results from the European Network of Rare Bleeding Disorders. J Thromb Haemost. 2012;10:615-621. [CrossRef]
- Saes JL, Verhagen MJA, Meijer K, Cnossen MH, Schutgens REG, Peters M, et al. Bleeding severity in patients with rare bleeding disorders: real-life data from the RBiN study. Blood Adv. 2020;4:5025-5034. [CrossRef]
- Moellmer SA, Puy C, McCarty OJT. Biology of factor XI. Blood. 2024;143:1445-1454.
- Palla R, Siboni SM, Menegatti M, Musallam KM, Peyvandi F; European Network of Rare Bleeding Disorders EN-RBD group. European Network of Rare Bleeding Disorders (EN-RBD) group. Establishment of a bleeding score as a diagnostic tool for patients with rare bleeding disorders. Thromb Res. 2016;148:128-134. [CrossRef]
- Duga S, Salomon O. Congenital factor XI deficiency: an update. Semin Thromb Hemost. 2013;39:621-631. [CrossRef]
- D'Andrea, G.; Colaizzo, D.; Vecchione, G.; Grandone, E.; Di Minno, G.; Margaglione, M.; GLAnzmann's Thrombasthenia Ita- 489 lian Team (GLATIT). Glanzmann's thrombasthenia: identification of 19 new mutations in 30 patients. Thromb. Haemost. 2002, 490 87, 1034-42. [CrossRef]
- Pugh RE, McVey JH, Tuddenham EG, Hancock JF. Six point mutations that cause factor XI deficiency. Blood. 1995;85:1509-16. [CrossRef]
- Castaman G, Giacomelli SH, Dragani A, Iuliani O, Duga S, Rodeghiero F. Severe factor XI deficiency in the Abruzzo region of Italy is associated to different FXI gene mutations. Haematologica. 2008;93:957-8. [CrossRef]
- Saunders RB, Shiltagh N, Gomez K, Mellars G, Cooper C, Perry DJ, et al. Structural analysis of eight novel and 112 previously reported missense mutations in the interactive FXI mutation database reveals new insight on FXI deficiency. Thromb Haemost. 2009;102:287-301. [CrossRef]
- Esteban J, de la Morena-Barrio ME, Salloum-Asfar S, Padilla J, Miñano A, Roldán V, Soria JM, Vidal F, Corral J, Vicente V. High incidence of FXI deficiency in a Spanish town caused by 11 different mutations and the first duplication of F11: Results from the Yecla study. Haemophilia. 2017;23:e488-e496. [CrossRef]
- Guella I, Soldà G, Spena S, Asselta R, Ghiotto R, Tenchini ML et al. Molecular characterization of two novel mutations causing factor XI deficiency: a splicing defect and a missense mutation responsible for a CRM+defect. Thromb Haemost 2008; 99: 523-530. [CrossRef]
- Smith SB, Gailani D. Update on the physiology and pathology of factor IX activation by factor XIa. Expert Rev Hematol. 2008;1:87-98. [CrossRef]
- Sharman Moser S, Chodick G, Ni YG, Chalothorn D, Wang MD, Shuldiner AR, Morton L, Salomon O, Jalbert JJ. The Association between Factor XI Deficiency and the Risk of Bleeding, Cardiovascular, and Venous Thromboembolic Events. Thromb Haemost. 2022;122:808-817. [CrossRef]
- Zadra G, Asselta R, Tenchini ML, Castaman G, Seligsohn U, Mannucci PM et al. Simultaneous genotyping of coagulation factor XI type II and type III mutations by multiplex real-time polymerase chain reaction to determine their prevalence in healthy and factor XI-deficient Italians. Haematologica 2008;93: 715-721. [CrossRef]
- Castaman G, Giacomelli SH, Caccia S, Riccardi F, Rossetti G, Dragani A et al. The spectrum of factor XI deficiency in Italy. Haemophilia 2014; 20: 106-113. [CrossRef]
- O'Connell NM, Saunders RE, Lee CA, Perry DJ, Perkins SJ. Structural interpretation of 42 mutations causing factor XI deficiency using homology modeling. J Thromb Haemost 2005; 3: 127-138. [CrossRef]
- Harlow SD, Campbell OM. Epidemiology of menstrual disorders in developing countries: a systematic review. BJOG. 2004; 111:6-16. [CrossRef]
- Fasulo MR, Biguzzi E, Abbattista M, Stufano F, Pagliari MT, Mancini I, et al. The ISTH Bleeding Assessment Tool and the risk of future bleeding. J Thromb Haemost. 2018;16:125- 130. [CrossRef]
- Tiscia G, Favuzzi G, Chinni E, Colaizzo D, Fischetti L, Intrieri M, et al. Factor VII deficiency: a novel missense variant and genotype-phenotype correlation in patients from Southern Italy. Hum Genome Var. 2017;4:17048. [CrossRef]
- Barcellona D, Favuzzi G, Vannini ML, Piras SM, Ruberto MF, Grandone E, Marongiu F. A Sardinian Family with Factor XI Deficiency. Hamostaseologie. 2019;39:398-403. [CrossRef]
Figure 1.
Distribution of single nucleotide variants along protein domains of FXI.
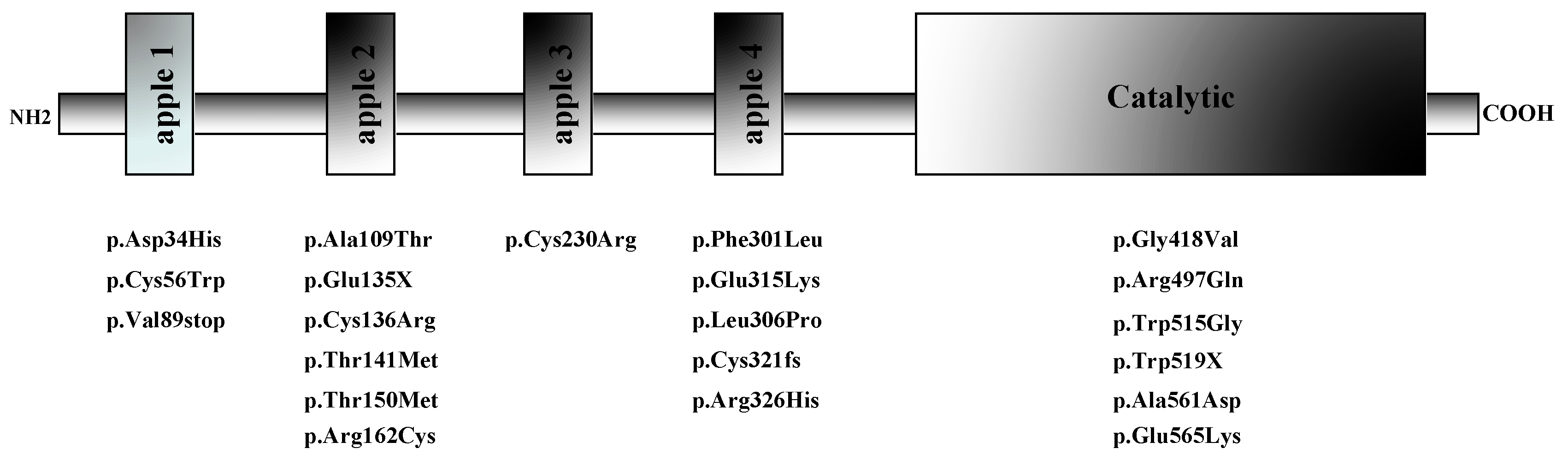
Figure 2.
Predicted structures of the wild-type (A left: 306L; B left 515W) and variant protein (A right:306P; B right: 306G) are compared using the DynaMut2 online server.
Figure 2.
Predicted structures of the wild-type (A left: 306L; B left 515W) and variant protein (A right:306P; B right: 306G) are compared using the DynaMut2 online server.
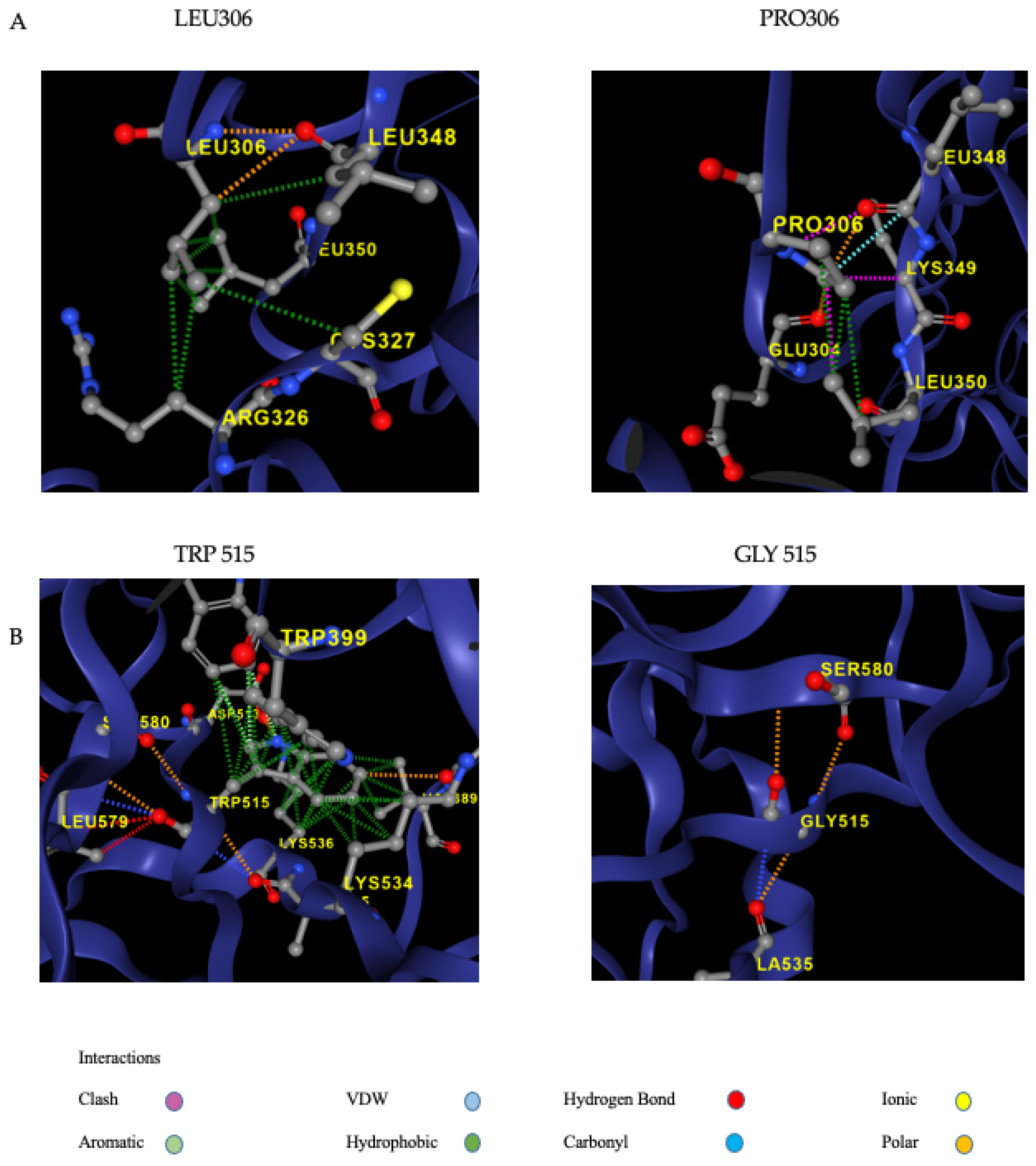

Table 1.
Clinical and molecular findings of the families investigated.
| Case index # | Sex | Age at the presentation | FXI activity (IU/dL ) | Variant 1 | Variant 2 | Symptoms |
| 1 | F | 21 | 51 | p.Asp34His | Asymptomatic | |
| 1-1 | F | 26 | 47 | p.Asp34His | Asymptomatic | |
| 1-2 | F | 49 | 55 | p.Asp34His | Asymptomatic | |
| 2 | M | 58 | 43 | p.Cys56Trp | Bleeding after surgery | |
| 2-1 | M | 32 | 38 | p.Cys56Trp | Asymptomatic | |
| 2-2 | F | 25 | 38 | p.Cys56Trp | Asymptomatic | |
| 3 | F | 56 | 42 | p.Val89stop | Repeated bleeding after surgery | |
| 4 | M | 35 | 32 | C.325+1G>A | Epixastis | |
| 5 (a) | F | 1 | 1 | p.Ala109Thr | C.325+1G>A | Asymptomatic |
| 5-1 | M | 31 | 48 | C.325+1G>A | Asymptomatic | |
| 5-2 | F | 4 | 41 | p.Ala109Thr | Asymptomatic | |
| 5-3 | F | 29 | 35 | p.Ala109Thr | Asymptomatic | |
| 6 (a) | M | 32 | 39 | p.Glu135X | Epixastis | |
| 7 (a) | F | 60 | 44 | p.Glu135X | Repeated bleeding after surgery, Menorrhagia | |
| 8 (a) | F | 26 | 38 | p.Glu135X | Asymptomatic | |
| 8-1 | F | 37 | 34 | p.Glu135X | Asymptomatic | |
| 9 | M | 16 | 28 | p.Glu135X | Asymptomatic | |
| 9-1 | F | 13 | 25 | p.Glu135X | Asymptomatic | |
| 9-2 | M | 43 | 48 | p.Glu135X | bleeding after surgery or trauma | |
| 9-3 | F | 46 | 33 | p.Glu135X | Asymptomatic | |
| 10 | F | 7 | 4 | p.Glu135X | p.Cys321fs | Asymptomatic |
| 10-1 | F | 34 | 69 | p.Cys321fs | Asymptomatic | |
| 10-2 | F | 5 | 3 | p.Glu135X | p.Cys321fs | Asymptomatic |
| 10-3 | F | 3 | 39 | p.Glu135X | Asymptomatic | |
| 11 (b) | F | 47 | 1 | p.Glu135X | Repeated bleeding after surgery or trauma | |
| 11-1 | M | 76 | 50 | p.Glu135X | Asymptomatic | |
| 11-2 | F | 74 | 76 | p.Glu135X | Asymptomatic | |
| 11-3 | M | 45 | 1 | p.Glu135X | Repeated bleeding after surgery or trauma | |
| 11-4 | F | 33 | 52 | p.Glu135X | Asymptomatic | |
| 11-5 | M | 41 | 2 | p.Glu135X | Repeated bleeding after surgery or trauma | |
| 12 (a) | F | 55 | 1 | p.Glu135X | p.Cys136Arg | Asymptomatic |
| 12-1 | F | 57 | 2 | p.Glu135X | p.Cys136Arg | Asymptomatic |
| 13 | F | 26 | 40 | p.Glu135X | Menorrhagia | |
| 14 | F | 41 | 60 | p.Glu135X | Menorrhagia | |
| 15 | M | 10 | 22 | p-Thr141Met | Asymptomatic | |
| 15-1 | M | 54 | 35 | p-Thr141Met | Asymptomatic | |
| 16 (a) | M | 7 | 34 | p.Thr150Met | Epistaxis | |
| 16-1 | M | 38 | 52 | p.Thr150Met | Epistaxis | |
| 16-2 | M | 46 | 41 | p.Thr150Met | Asymptomatic | |
| 16-3 | M | 21 | 34 | p.Thr150Met | Epistaxis | |
| 17 (a) | F | 6 | 47 | p.Arg162Cys | Asymptomatic | |
| 17-1 | F | 6 | 43 | p.Arg162Cys | Asymptomatic | |
| 17-2 | F | 36 | 45 | p.Arg162Cys | Asymptomatic | |
| 18 (a) | M | 20 | 34 | p.Cys230Arg | Bleeding after surgery | |
| 18-1 | F | 50 | 34 | p.Cys230Arg | Asymptomatic | |
| 19 | M | 41 | 44 | p.Phe301Leu | Repeated bleeding after surgery | |
| 20 | F | 5 | 46 | p.Phe301Leu | Asymptomatic | |
| 21 (a) | F | 8 | 6 | p.Phe301Leu | p.Trp519X | Bleeding after surgery |
| 21-1 | F | 39 | 40 | p.Phe301Leu | Asymptomatic | |
| 22 (a) | F | 14 | 4 | p.Phe301Leu | c.595+3A>G | Asymptomatic |
| 22-1 | F | 43 | 85 | c.595+3A>G | Asymptomatic | |
| 22-2 | M | 49 | 51 | p.Phe301Leu | Asymptomatic | |
| 23 | M | 15 | 40 | p.Leu306Pro | Asymptomatic | |
| 23-1 | F | 11 | 45 | p.Leu306Pro | Asymptomatic | |
| 23-2 | F | 49 | 38 | p.Leu306Pro | Asymptomatic | |
| 24 | F | 3 | 34 | p.Glu315Lys | p.Ala561Asp | Asymptomatic |
| 24-1 | F | 29 | 36 | p.Glu315Lys | p.Ala561Asp | Asymptomatic |
| 25 | F | 37 | 28 | p.Glu315Lys | Menorrhagia | |
| 25-1 | F | 36 | 33 | p.Glu315Lys | Bleeding after surgery | |
| 25-2 | M | 66 | 18 | p.Glu315Lys | Bleeding after surgery | |
| 26 (a) | F | 24 | 7 | p.Glu315Lys | p.Trp519X | Menorrhagia |
| 26-1 | F | 22 | 29 | p.Glu315Lys | Asymptomatic | |
| 26-2 | M | 60 | 49 | p.Trp519X | Asymptomatic | |
| 26-3 | F | 46 | 39 | p.Glu315Lys | Repeated bleeding after trauma | |
| 27 | M | 16 | 34 | p.Glu315Lys | Asymptomatic | |
| 27-1 | M | 45 | 27 | p.Glu315Lys | Asymptomatic | |
| 28 | M | 10 | 31 | p.Glu315Lys | Asymptomatic | |
| 28-1 | F | 49 | 37 | p.Glu315Lys | Menorrhagia | |
| 29 | M | 4 | p.Glu315Lys | Asymptomatic | ||
| 30 | F | 15 | 36 | p.Glu315Lys | Asymptomatic | |
| 30-1 | M | 38 | 28 | p.Glu315Lys | Asymptomatic | |
| 31 | M | 6 | 41 | p.Glu315Lys | Asymptomatic | |
| 31-1 | M | 40 | 40 | p.Glu315Lys | Asymptomatic | |
| 32 (a) | F | 18 | 38 | p.Arg326His | Asymptomatic | |
| 32-2 | F | 44 | 62 | p.Arg326His | Asymptomatic | |
| 33 (a) | F | 29 | 43 | p.Gly418Val | Spontaneous ecchymoses | |
| 33-1 | F | 52 | 45 | p.Gly418Val | Repeated bleeding after trauma | |
| 34 | M | 10 | 21 | p.Arg497Gln | Asymptomatic | |
| 35 (a) | F | 40 | 14 | p.Trp515Gly | Asymptomatic | |
| 35-1 | M | 17 | 30 | p.Trp515Gly | Asymptomatic | |
| 35-2 | F | 15 | 44 | p.Trp515Gly | Asymptomatic | |
| 35-3 | F | 43 | 27 | p.Trp515Gly | Asymptomatic | |
| 35-4 | M | 46 | 36 | p.Trp515Gly | Asymptomatic | |
| 35-6 | F | 69 | 36 | p.Trp515Gly | Asymptomatic | |
| 36 | F | 82 | 40 | P.Glu565Lys | Repeated bleeding after surgery | |
| 37 | F | 18 | 29 | P.Glu565Lys | Spontaneous ecchymoses | |
| 37-1 | M | 57 | 50 | P.Glu565Lys | Asymptomatic | |
| 38 | M | 10 | 30 | P.Glu565Lys | Asymptomatic | |
| 38-1 | M | 6 | 28 | P.Glu565Lys | Asymptomatic | |
| 38-2 | M | 36 | 28 | P.Glu565Lys | Repeated bleeding after surgery - Epistaxis | |
| 38-3 | F | 63 | 39 | P.Glu565Lys | Asymptomatic | |
| 39 | M | 21 | 48 | c.1717-2A>G | Epistaxis | |
| 39-1 | F | 24 | 42 | c.1717-2A>G | Asymptomatic |
Table 2.
Information on the new F11 gene variants identified.
| GENE | Protein Variation | Frequencies | ClinVar | DyneMut2_ (ΔΔGStability) | Predicted Stability Change | Effect on Protein Structure | ACMG | ACMG Supporting Criteria |
| FXI | p.Leu306Pro | Exomes: not found genomes: not found |
No data available | -0,32 kcal/mol | destabilising | disallowed phi/psi | VUS | PM2,PP3,PP2 |
| FXI | p.Trp515Gly | exomes: not found genomes: not found (cov: 31.9) |
No data available | -2,94 kcal/mol | destabilising |
-disrupts all H-bonds
-expansion of cavity |
VUS | PM2,PP3,PP2 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



