Submitted:
30 June 2025
Posted:
01 July 2025
You are already at the latest version
Abstract
Liquid organic hydrogen carrier (LOHC) compounds represent a promising option for hydrogen storage. Beyond the use of pure hydrocarbons, the incorporation of oxygen atoms offers a way to modify thermodynamic properties and potentially improve suitability for hydrogen storage. This study investigates the effect of oxygen functionalization in aromatic ethers and lactones on the reaction equilibrium of reversible hydrogenation. To address this question, reaction enthalpies and entropies are calculated using both experimental and theoretically determined pure substance data. The equilibrium position shift of the hydrogenation of furan derivatives has been shown to follow a similar trend to that of their hydrocarbon counterparts upon the addition of aromatic rings. This shift is, however, more pronounced in the case of the furan-based systems. The effect is reflected in increasing Gibbs reaction energies during the dehydrogenation process. Both the formation of lactones and the addition of a second ring to the furan core leads to a further increase in the Gibbs reaction energy. The highest value is observed for dibenzofuran, with a Gibbs reaction energy of 36.6 kJ∙mol-1 at 500 K. These findings indicate that, from a thermodynamic perspective, hydrogen release is feasible at temperatures below 500 K, which is an important feature for the potential application as a hydrogen storage system.
Keywords:
hydrogen storage
; LOHC
; enthalpies of phase transitions
; enthalpy of formation
; vapor pressure
; structure-property correlation
; quantum-chemical calculations
1. Introduction
LOHC systems with oxygen functionality have been demonstrated to provide very favourable hydrogen release performance at low temperatures and very attractive properties for potential technical applications [1]. For example, it has been reported that benzophenone can be selectively and completely hydrogenated to di-cyclohexyl-methanol over Ru/Al2O3 catalysts at a hydrogen pressure of 50 bar and temperatures between 90 to 180 °C. Hydrogen is released from di-cyclohexyl methanol at a temperature of 250 °C using Pt-based catalysts in order to recover benzophenone. This reversible process exhibits an excellent theoretical hydrogen storage capacity of 7.2 mass percent [2]. This successful example has motivated further investigations of oxygen-containing LOHC systems [3]. The effects of oxygen functionality on the thermodynamic properties of hydrogenation/dehydrogenation reactions were evaluated for LOHC systems based on methoxy-benzene, diphenyl ether, benzyl phenyl ether, dibenzyl ether, and methoxy-naphthalenes. The respective data were derived and compared with the data for the similarly structured hydrogen carriers based on benzene, diphenyl-methane, 1,2-diphenyl-ethane, 1,3-diphenyl-propane, and naphthalene [3]. It was observed that for methoxy-benzene and methoxy-naphthalenes, a decrease in reaction enthalpy and Gibbs free energy for the dehydrogenation reactions can be observed, leading to more favourable reaction conditions. However, in systems based on other aromatic ethers, the thermodynamic properties of the reaction are very similar to those of the analogous system without an oxygen function within the LOHC. These new observations contributed to a better understanding of the thermodynamic properties of LOHC systems. The investigation of the thermodynamic properties of hydrogenation/dehydrogenation reactions in LOHC systems with oxygen functions would need to be expanded to evaluate this interesting structure-property relationship for the screening of molecules suitable for practical applications.
In order to simplify the evaluation and comparison of thermodynamic data for LOHC systems, it makes sense to select a few indicators that relate to physical and chemical properties and are relevant for comparison and practical implementation. The most obvious of them are the hydrogen storage capacity (e.g. in % mass) and the equilibrium temperature, :
where is the standard molar reaction enthalpy and the is the standard molar reaction entropy. This temperature is considered the potential optimum reaction temperature for a hydrogenation/dehydrogenation process. Generally, the temperature should be as low as possible. The reaction enthalpy values are also considered a valuable indicator, which should be as low as possible, since lower values reduce the temperature level needed for dehydrogenation. An example for the application of the indicators is shown in Figure 1, where the oxygen functionality is directly bound to the benzene ring and compared with similarly structured hydrocarbon systems.
Obviously, introducing oxygen into the aromatic system can only reduce the hydrogen storage capacity, but in return, the value in the methoxy-benzene/methoxy-cyclohexane system drops to 554 K compared to 569 K in the benzene/cyclohexane system. An analogous trend can be observed in the reaction enthalpy in the methoxy-benzene/methoxy-cyclohexane system compared to the benzene/cyclohexane system (see Figure 1).
From a structural point of view, the oxygen functionality cannot only be bound to the aromatic ring of a promising LOHC structures as shown in Figure 1, but also as part of bicyclic molecules in which the oxygen is part of a five-membered ring (e.g. benzofuran, dibenzofuran, indanol, indanone, etc.) or a six-membered ring (e.g. chromene, isochromene, tetralones, tetralols, etc.). The strain on the corresponding rings can certainly influence the thermodynamic properties of these auspicious LOHC compounds. Therefore, the current study focuses on the question of whether the introduction of oxygen functionality into aromatic bi- and tricyclic molecules with a five-membered ring (see Figure 2) improves the thermodynamic properties of hydrogen storage compared to similarly shaped LOHC systems without oxygen functionality.
It is well established that the hydrogenation/dehydrogenation of LOHC systems is a chemically reversible process. However, optimisation of the experimental reaction conditions is essential to shift the equilibrium towards sufficient hydrogen release at moderate temperatures. In this work, the available thermodynamic data for the hydrogen-lean (HL) counterparts of the LOHC systems (see Figure 2) is compiled and evaluated using empirical and quantum chemical methods. Since experimental data for partially and fully hydrogenated aromatics (see Figure 2) are practically non-existent in the literature, the thermodynamic data for these hydrogen-rich (HR) counterparts of the LOHC systems were derived using specially developed empirical correlations and methods as well as high-level quantum chemical calculations and finally are recommended for chemical engineering calculations.
2. Approach
As a rule for our considerations, the hydrogenation/dehydrogenation reactions of the LOHC system take place in the liquid phase. Unfortunately, the enthalpies of formation of the HL and HR counterparts of the LOHC systems are not always found in the literature, or the available data originate from a single measurement and therefore need to be validated. Admittedly, modern high-level quantum-chemical (QC) calculations are capable of providing reliable gas-phase enthalpies of formation, (g, 298.15 K), and absolute entropies, (g, 298.15 K) with chemical accuracy [4].
In a Step I, high-level QC calculations were performed to determine the gas phase enthalpies of formation and absolute entropies of the compounds involved in the hydrogenation/dehydrogenation reaction.
In a Step II, the combination of experimental vapor pressures and empirical methods enabled the assessment of reliable vaporisation enthalpies (298.15 K), and vaporisation entropies, (298.15 K), which were required to derive the enthalpies of formation in the liquid phase, (liq, 298.15 K), according to Eq. (2):
and the liquid phase absolute entropies, (liq, 298.15 K) according to Eq. (3):
In a Step III, the (liq, 298.15 K) and (liq, 298. 15 K) values were used to derive the (liq, 298.15 K) and (liq, 298. 15 K) and used to calculate the Gibbs free energies (liq, 298.15 K), according to Eq. (4):
and finally to calculate the equilibrium temperatures for the hydrogenation/dehydrogenation reactions with the compounds given in Figure 2 and to perform a thermodynamic analysis of the LOHC system of interest. These three steps comprise the general roadmap for the present work, which focuses on evaluating the effects and practical potential of introducing oxygen functionality into aromatic ether-based LOHC systems compared to similarly structured aromatics.
3. Results and Discussion
3.1. Step I: Quantum-Chemical Calculations of the Gas-Phase Enthalpies of Formation
Aromatic ethers (see Figure 2) are generally rigid molecules, but aliphatic ethers (the partially or completely hydrogenated products of the species shown in Figure 2) are flexible molecules and exist in the gas phase as a mixture of conformers. A careful conformational analysis was performed using CREST software [5]. The G3MP2 method implemented in the Gaussian 16 software [6] was used to calculate the energies E0 and total enthalpies H298 of the most stable conformers. The calculated H298 enthalpies were converted to the standard molar enthalpies of formation based on conventional atomisation (AT) procedure [7] (e.g., for 2,3-benzofuran):
C8H6O → 8×C + 6×H + 1×O
The results of the quantum chemical G3MP2 calculations for the hydrogen-lean and hydrogen-rich counterparts of the LOHC systems are summarised in Table S1 (column 3).
The AT reaction is the choice with the least bias compared to other isodesmic, isogyric, homodesmotic, etc. reactions for the conversion of the H298 enthalpies to the standard molar enthalpies of formation[7]. A major advantage of the AT reaction is the fact that the enthalpies of the atoms are precisely quantified. In contrast, the enthalpies of formation of the various molecules involved in the aforementioned reactions are only known with significant uncertainties. However, it has also been shown in references [7] and [8] that the QC results derived from G3MP2 modelling and the AT reactions,(g, AT), exhibit a small but noticeable deviation from values derived experimentally. Yet, utilizing a set of similarly structured molecules with reliably known experimental enthalpies of formation, (g)exp, (see Table S1) allows for a correction of QC and experimental results. Thus, the (g, AT)-values are correlated with the experimental values. The following equation:
was used to get the “corrected” atomisation results for the aromatic ethers (see Figure 2) and their hydrogenated products. The enthalpies calculated with the corrected atomisation reactions are shown in Table 1, column 2, and compared with the QC results from other studies (see Table 1, columns 3 and 4).
Notario et al. [9] calculated the enthalpies of formation of all aromatic ethers shown in Figure 2 using the G3MP2//B3LYP method and the atomisation reaction. The original atomisation enthalpies were corrected as shown in Table S2, and the resulting “corrected” enthalpies of formation (see Table 1, column 3) agree with our G3MP2 calculations (see Table 1, column 2).
Sousa et al. [10] calculated the enthalpies of formation of 2-cumarone, 3-cumarone and phthalide using the G3MP2 method and using 10 different well-balanced reactions, which are shown in Figure S2. It should be noted that the selection of the appropriate reactions cannot be considered optimal, as the fluctuations in the resulting (g)G3MP2 for each aromatic lactone were unacceptably large (e.g., for phthalide, they ranged between -210.0 and -230.6 kJ·mol-1). Surprisingly, despite this variation, the values averaged by Sousa et al. [10] (see Table 1, column 4) agree well with our G3MP2 calculations (see Table 1, column 2) and with the “corrected” results of Notario et al. [9] (see Table 1, column 3). The weighted average value (g)QC, which was determined from the data in columns 2, 3 and 4 (Table 1, column 5) using the uncertainty as a weighting factor, was recommended as reliable and compared with the available experimental results evaluated in this work and presented in Table 2, column 5.
With the exception of 2-coumaranone, the quantum chemical and experimental gas phase enthalpies of formation of the compounds listed in Table 2 agree well within the uncertainties specified for these values. In order to find an explanation for the observed discrepancy, the experimental details of the combustion experiments with 2-coumaranone were analysed as described in the original paper [10], as follows. The combustion experiments with 2-coumaranone and 3-coumaranone were carried out using a micro-bomb calorimeter with sample masses of approximately 20 mg. The samples of 2-coumaranone were burned in pellet form, which were enclosed in Melinex bags due to the high volatility of the compound. Moreover, incomplete combustion was observed for 2-coumaranone. Even when an appropriate correction for the formation of carbon soot was made by authors, the reliability of the experimental enthalpy of formation for this compound remains questionable.
The good agreement between the quantum chemical and experimental gas phase enthalpies of formation of the compounds listed in Table 2 can be regarded as mutual validation of both data sets, and these data have been recommended for further thermodynamic calculations.
3.2. Step II: Evaluation of the Thermodynamic Functions of Vaporisation
3.2.1. Absolute Vapor Pressures and Vaporisation Enthalpies
The primary vapor pressure-temperature dependencies for aromatic ethers and lactones as well as their hydrogenated counterparts were compiled from the literature and uniformly fitted using a three-parameter equation (see ESI for details). From these data, the standard molar enthalpies of vaporisation at the corresponding temperatures Tav (average temperature of the measurement interval) were derived and adjusted to the reference temperature T = 298.15 K for comparison. The resulting (298.15 K)-values for the compounds of interest are summarised and compared in Table 3.
The (298.15 K)–values for 2,3-benzofuran, 2,3-dihydrobenzofuran and 1,3-dihydroisobenzofuran (phthalane), which were determined using various techniques, are very consistent, and a weighted average value was determined for each compound and recommended for thermochemical calculations.
For 2-coumaranone, 3-coumaranone, and phthalide, only the sublimation enthalpies were measured directly [10,20] using drop calorimetry. The corresponding enthalpies of vaporisation, (298.15 K), were derived in this work using fusion enthalpies (see Table S3). These values still need to be validated, especially for phthalide, where (298.15 K) = 71.5±4.6 kJ⋅mol-1 (Table 3) deviates significantly from the value of (298.15 K) = 66.9±2.2 kJ⋅mol-1 (see Table 3) derived from the Antoine equation coefficients given in the comprehensive compilation by Stephenson and Malanowski [21].
No consistent data sets regarding vapor pressure measurements are available for the majority of the hydrogenated aromatic ethers. Still, the experimental boiling point (BP) data of the desired aliphatic cyclic ethers at different reduced pressures have been reported before (see a compilation in Table S4). These data may be helpful in obtaining the missing thermodynamic information. These boiling point data usually originate from distillation experiments on reaction mixtures after synthesis and are usually only utilized to identify the compounds. In such measurements, uncalibrated manometers are usually used to measure the pressures and the temperatures are given within the range of a few degrees. However, in our previous work (e.g. for methyl-biphenyls [26]) it has been demonstrated that reasonable trends can be obtained even from such rough data. This general conclusion is also applicable for these aromatic ethers [3]. We therefore carefully collected the available boiling points at reduced pressures for 2,3-benzofuran, 2,3-dihydrobenzofuran, 2-coumaranone, 3-coumaranone phthalide, and their hydrogenated products, and used these data to calculate the vaporisation enthalpies (298.15 K) and vaporisation entropies (298.15 K) required for the thermodynamic analysis of the hydrogenation/dehydrogenation reactions relevant to this work. The enthalpies of vaporisation derived from the approximation of the boiling points at reduced pressures selected in Table S4 are given in Table 3 as BP.
3.2.2. Validation of Vaporisation Enthalpies by Structure-Property Correlations
Structure-property relationships are a central pillar of physical-organic chemistry. Provided that new data are consistent with already known trends, they can be recommended for further applications. If this is not the case, the measurements should be repeated. Thus, before using (298.15 K)-values derived from single experiments with conventional methods or from BP data, it is advisable to evaluate the values with structure-property correlations. For instance, correlations between enthalpy of vaporisation and normal boiling points (Tb) enable a simple plausibility test.
For example, in our recent work [25], we established a reliable (298.15 K) - Tb correlation for a large series of aliphatic ethers:
We used this correlation to estimate the (298.15 K)-values for octahydro-benzofuran and octahydro-isobenzofuran and the results (shown in Table 3 as Tb) agreed well with the enthalpies of vaporisation derived from the BP approximation.
A valuable option called the “centrepiece approach”, which is closely related to group additivity (GA) methods, has been developed in a series of our recent work on the correlation of structures and thermodynamic properties [27]. The “centerpiece approach” is an empirical method for assessing the thermochemical properties (e.g. enthalpies of vaporisation, enthalpies of formation, etc.) of organic molecules with improved accuracy and efficiency. It involves selecting a well-characterised “centerpiece” molecule, typically a structurally similar compound with reliable properties, as a starting point. The target molecule is then analysed by calculating the relative differences in thermochemical properties between it and the “centrepiece molecule” (more details see in ESI). This differential approach helps to reduce systematic errors and improve the reliability of predictions, especially for large or complex organic molecules. Examples of calculations using the “centerpiece approach” for LOHC pairs are shown in Figure 3.
Indeed, to calculate the enthalpy of vaporisation of 2,3-dihydrobenzofuran, it is logical to start with indane as the “centerpiece" molecule that is structurally most similar to 2,3-dihydrobenzofuran. To assess the enthalpy contribution for the exchange of the CH2 fragment in the five-membered ring of indane with oxygen, the difference between the enthalpy of vaporisation of tetrahydrofuran and cyclopentane of ΔO = 3.5 kJ⋅mol-1 was calculated (see Figure 3, left) and assumed to be equal to the value for the indane/2,3-dihydrobenzofuran pair. The latter contribution of 3.5 kJ⋅mol-1 was added to the reliable enthalpy of vaporisation of indane, (298.15 K) = 49.0±0.5 kJ⋅mol-1 [Table S6], and the empirical CP value of 52.5 kJ⋅mol-1 derived in this way agrees well with the experimental result of (298.15 K) = 53.2±0.2 kJ⋅mol-1 [Table 3].
The same “centerpiece" molecule indane was used to estimate the vaporisation enthalpies of 8H-benzofuran or 8H-isobenzofuran as fully hydrogenated counterparts of 2,3-dihydrobenzofuran and 1,3-dihydro-isobenzofuran (phthalan) (see Figure 3, right). In this case, it was assumed that the difference in the enthalpy of vaporisation of indane and 8H-indene (CAS 3296-50-2) is Δ(6+5) = -4.2 kJ⋅mol-1 and it is equal for all types of combinations of 6+5-membered rings. Based on this assumption and using the vaporisation enthalpies of 2,3-dihydrobenzofuran and phthalan given in Table 3, the missing vaporisation enthalpies for 8H-benzofuran or 8H-isobenzofuran were estimated. The latter results agree very well with the values determined using other methods (see Table 3).
In this work, the “centerpiece approach" was used to estimate the (298.15 K)-values for all partially and fully hydrogenated molecules in Figure 2. The “stepwise” estimates are documented in Figures S3 to S10, and the final results are summarised in Table 3 with the notation CP (“centrepiece”).
The bicyclic and polycyclic fully hydrogenated compounds shown in Figure 2 are general “hydrogen-rich” products of reversible hydrogenation/dehydrogenation reactions with LOHC. Admittedly, thermochemical data for this type of organic molecule are almost completely lacking in the literature. In this context, it is important to test various structure-property relationships in order to obtain reliable estimates for the vaporisation enthalpies of such cyclic molecules.
Another highly valuable tool for organic physical chemistry are group additivity (GA) methods. This approach somehow follows the principal idea of a Lego box, from which a property of a molecule is assembled from small building blocks, which contribute well-established numerical values to the overall value. Nowadays, a comprehensive set of GA methods is available for the major classes of organic compounds [28,29].
The group contributions used in this work to predict thermodynamic properties are listed in Table S7. However, it is known that conventional GA methods can have difficulties with cyclic molecules. To overcome this disadvantage, we have developed some additional cyclic increments, e.g. the methylene fragment in the five-membered aliphatic ring [CH2]5 and the six-membered aliphatic ring [CH2]6 or the oxygen atom [O]5 incorporated into the five-membered ring. The numerical values for these new increments are also given in Table S7 and have significantly improved the prediction quality, as shown in Figure S3 using the example of 8H-benzofuran. The sum of the required increments for 8H-benzofuran yields an estimated value of (298.15 K) = 48.9±1.5 kJ⋅mol-1 [Figure S3], which, within the specified uncertainties, is hardly distinguishable from the result of the BP method (298.15 K) = 48.8±1.9 kJ⋅mol-1 [Table 3]. The vaporisation enthalpies estimated using the group-additivity contributions from Table S7 are given as GA values in Table 3.
An additional option to overcome the difficulties with prediction of vaporisation enthalpies of cyclic molecule is to combine the “centerpiece approach” with the group-additivity contributions from Table S7. Such an example is shown in Figure 4 for calculation of the enthalpy of vaporisation, (298.15 K) of 8H-benzofuran using data for 8H-indene.
The reliable vaporisation enthalpy, (298.15 K) = 44.8±1.3 kJ⋅mol-1 [Table S6], available for 8H-indene is a suitable “centerpiece” molecule in which replacing the cyclic hydrocarbon contribution [CH2]5 with the cyclic oxygen contribution [O]5 given in Table S7 directly leads to the desired vaporisation enthalpy of 8H-benzofuran, as shown in Figure 4. The value (298.15 K) = 48.4±1.5 kJ⋅mol-1 [Table 3], estimated in this way is consistent with other results collected for this molecule in Table 3.
The values derived by CP and GA are summarised in Table 3. For all substances, the enthalpies of vaporisation (298.15 K) evaluated in this way agree well with values obtained from other methods. To obtain more reliable data, the weighted average values were derived for all species considered in this work (see Table 3). These data were used to calculate the enthalpies of formation for the liquid phase in Section 3.
3.2.3. Validation of Vaporisation Enthalpies Using Correlation with the Kovats Indices Jx
A further means of validation are Kovats retention indices, Jx. These can be measured by gas chromatography (GC) [30]. They reflect the intensity of interaction between a molecule moving in the gas phase and the stationary phase in GC. It can be directly related to the enthalpy of evaporation. The (298.15 K)-values within structurally parent series of molecules usually correlate linearly with the Kovats indices [31]. The Kovats indices for furan derivatives on the non-polar column DB-5 (see Table 4) were taken from literature [32].
The (298.15 K)-values collected in Table 4 show a clear linear correlation with their corresponding Jx-values:
In column 6 of Table 4 it can be seen that the deviations between the experimental values and those calculated according to Eq. (8) values are mostly below 1.0 kJ·mol-1. The "empirical" enthalpies of vaporisation derived by applying Eq. (8) (see Table 4, column 5) are referred to as Jx-values in Table 3 and agree well with results obtained by other methods.
3.2.4. Standard Molar Vaporisation Entropies
At the reference temperature T = 298.15 K, the standard molar entropies in the liquid phase and in the gas phase are connected via the standard molar entropy of vaporisation, (Eq. 3). In this work, the (298.15 K)-values have been derived from the intercept of the temperature dependence of vapor pressures as presented in Table S4. The results for (298.15 K) are shown in Table 5, column 2. The gas-phase absolute entropies were calculated using the G3MP2 method (see Table 5, column 3. The liquid phase entropies, estimated according to Eq. (3) are shown in Table 5, column 5.
It is important to note that the absolute entropies in the liquid phase, (liq), calculated indirectly according to Eq. (3) (see Table 5, column 5) based on quantum chemical calculations (Table 5, column 3) agree well with the experimental (liq)-values, which were mostly measured using cryogenic adiabatic calorimetry (Table 5, column 4). This agreement confirms the reliability of the sophisticated quantum chemical methods for (g) calculations.
3.3. Step III: Thermodynamic Analysis of the LOHC Systems Based on Aromatic Ethers
3.3.1. Reaction Enthalpies, Entropies and Gibbs Energies of the LOHC Hydrogenation
Hess's Law was used to derive the standard molar enthalpies of chemical reactions, , from the standard molar enthalpies of formation, , of the reactants:
Since the -values for cyclic aliphatic molecules of the hydrogen-rich LOHC counterparts are not available in the literature, they were calculated using the G3MP2 method (see Table S1). This method has proven to be reliable for the alkyl-cyclohexane series in our recent work [47]. The gas phase enthalpies of formation of perhydrogenated aromatic ethers were converted into the required liquid phase enthalpies of formation using the vaporisation enthalpies, (298.15 K), evaluated in Table 3. The (liq, 298.15 K)-values, derived according to Eq. (2) are given in Table S9 for the HL compounds and in Table S10 for the HR compounds and can now be used as input variables for Hess's Law according to Eq. (9).
The liquid phase reaction enthalpies, (liq), of the full hydrogenation of aromatic ethers are given in Table S11 and the liquid phase reaction enthalpies, (liq), of the partial hydrogenation of aromatic ethers are given in Table S12.
The reaction entropies of the hydrogenation of aromatic ethers were calculated according to Eq. (10), using the standard molar entropies of the reactants from Table 5 as follows:
The resulting total liquid phase reaction entropies, (liq, 298.15 K), of the hydrogenation reactions with LOHC candidates shown in Figure 2, a compiled in Table S13.
The Gibbs energies for liquid phase for the hydrogenation reactions are reported in Table S13. The calculations have initially been carried out at the reference temperature T = 298 K. Utilizing the standard molar isobaric heat capacities of the reactants, the thermodynamic feasibility of the reaction can be evaluated at any desired temperature aid of Kirchhoff's law and the van’t-Hoff equation. The change of reaction enthalpies and entropies as a function of temperature is determined by these thermodynamic relations
where is the change in standard molar heat capacity during the reaction. It is calculated analogously to Hess´s Law from the standard molar heat capacities, , of the reactants and products with the stoichiometric coefficients as weighting factors (see Table S5). These values are depending on temperature and are not available for all potential LOHC structures (this particularly holds for the hydrogenated forms). Nevertheless, even though the -values for the individual species might be large, the –values for reactions are generally quite small and change little over temperature as the effects of reactants and products cancel each other. Therefore, in this work the -values at 298.15 K have been utilized for the adjustment of (liq, T) and (liq, T) to T = 500 K, as well as for calculation of (500 K) values, which are shown in Table S13.
3.3.2. Comparison of the Thermodynamics of Hydrogenation Reactions with and Without Oxygen Functionality
Detailed information on the calculations leading to the thermodynamic parameters of the hydrogenation/dehydrogenation reactions is provided in the electronic supplementary information (Tables S9 to S13). The final results for the enthalpies of the liquid phase reaction enthalpies, , reaction entropies, , Gibbs energies, , and equilibrium temperatures, , are summarised in Table 6 for discussion and comparison.
As a preliminary assessment, the thermodynamic suitability of a compound as a hydrogen storage material is often evaluated based on the reaction enthalpies of hydrogenation at 298.15 K. The relevant benchmark in this context is approximately –63 kJ∙mol-1, which is characteristic of commercial hydrogen storage materials. However, from a thermodynamic perspective, the key parameter is the Gibbs reaction energy. This value should be as large as possible to enable hydrogen release at temperatures below 500 K. The data relevant for comparing the suitability of the different compounds as hydrogen storage materials are shown in Table 6.
3.3.3. Comparison of Reactions Involving Five-Membered Aliphatic Rings
First of all, it is interesting to compare the thermodynamic characteristics of the hydrogenation reactions for 1,3-cyclopentadiene and furan (see Figure 5)
In this system, there is a substantial difference in the hydrogenation reaction enthalpies nearly 30 kJ∙mol-1/H2. In both cases, the full hydrogenation reactions remain strongly exergonic even at 500 K (-88.4 kJ∙mol-1 for 1,3-cyclopentadiene and -34.0 kJ∙mol-1 for furan. As a result, these compounds enable stable hydrogen storage in terms of neglectable dehydrogenation; however, the energy required for hydrogen release would be unacceptably high. It is noteworthy that the hydrogenation steps in 1,3-cyclopentadiene exhibit approximately the same reaction enthalpy, whereas a pronounced difference is observed in the case of furan. Interestingly, the first hydrogenation step of furan is associated with a remarkably low reaction enthalpy of -40.7 kJ∙mol-1/H2. The selective and reversible hydrogenation of a single double bond could therefore represent a largely unexplored approach to hydrogen storage.
3.3.4. Comparison of Reactions Involving Aromatic Ethers with Attached Five-Membered Aliphatic Ring
The condensation of an aromatic ring onto the 1,3-cyclopentadiene or furan backbone significantly alters the enthalpy difference for hydrogenation (see Figure 6). The difference in reaction enthalpies of the full hydrogenation is reduced to approximately 10 kJ∙mol-1/H2 between benzofuran and indene. Moreover, the hydrogenation enthalpies of the benzofuran systems decrease to roughly −62 kJ∙mol-1/H2, which is comparable to the typical values observed for commercial hydrogen carriers. Partial hydrogenation is not advantageous for these compounds. In indene, benzofuran, and isobenzofuran, the hydrogenation of the first double bond is significantly more exothermic than subsequent steps. According to the Gibbs free energy of reaction, dehydrogenation becomes thermodynamically favourable for 8H-benzofuran at temperatures above approximately 500 K. Thermodynamically, this represents a significant advantage for its application as a hydrogen storage material.
3.3.5. Comparison of Reactions Involving a Five-Membered Aliphatic Ring Fused to Two Benzene Rings
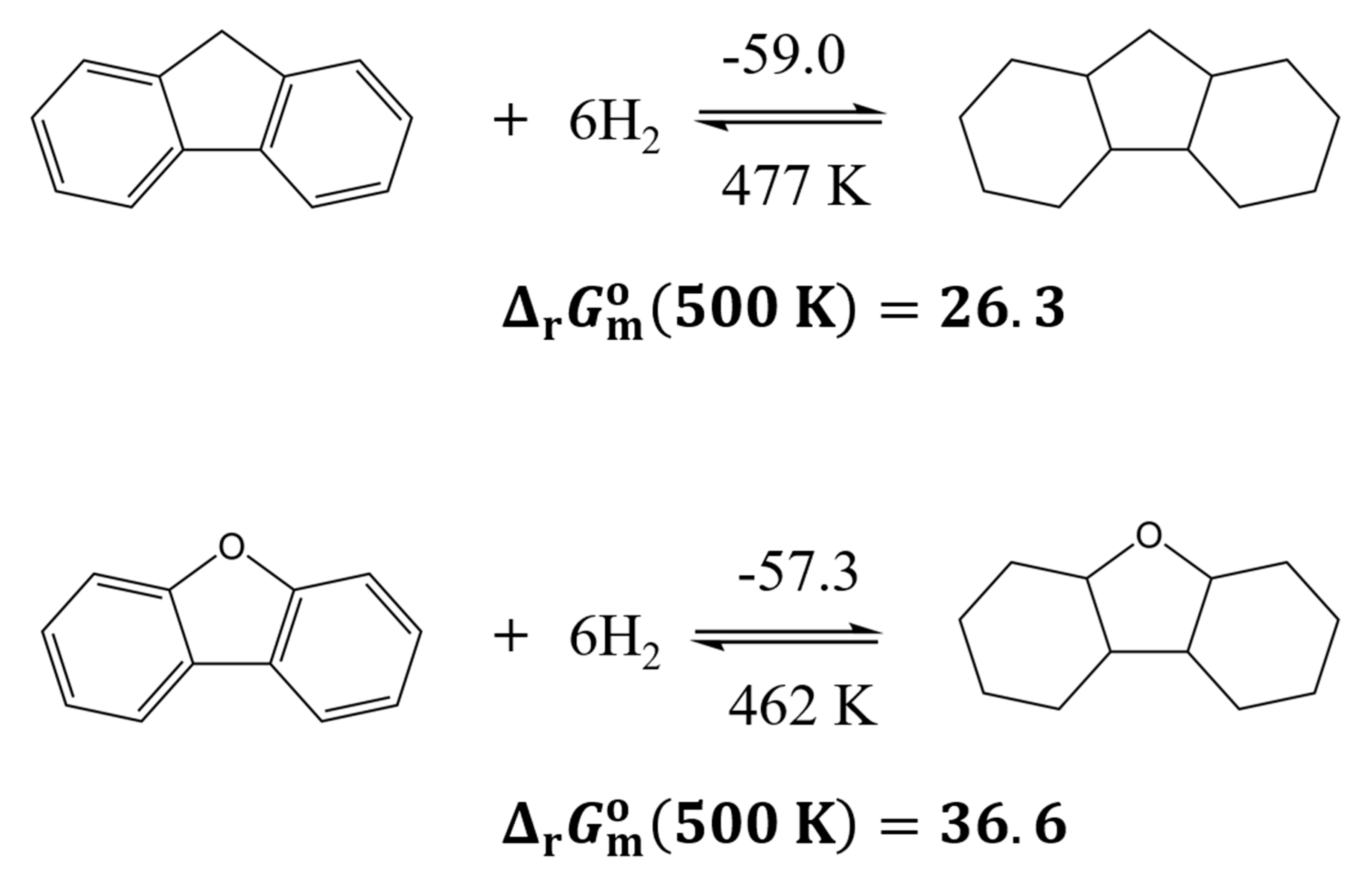
There is no longer a significant difference in the reaction enthalpies of fluorene and dibenzofuran hydrogenation, with both values ranging between approximately (-57 to -59) kJ∙mol-1/H2. The condensation of an additional aromatic ring further reduces the enthalpy gap between the pure hydrocarbon and the furan derivative, as already seen in the benzofuran systems. Furthermore, the Gibbs free reaction energies for hydrogenation at 500 K are strongly positive, with values of -24.6 kJ∙mol-1 for fluorene and -36.6 kJ∙mol-1 for dibenzofuran. This indicates that high hydrogen yields during dehydrogenation can already be expected at temperatures below 500 K.
Figure 7.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for fluorene and dibenzofuran. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
Figure 7.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for fluorene and dibenzofuran. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
3.3.6. Could the Lactone Ring Improve Hydrogen Storage Thermodynamics?
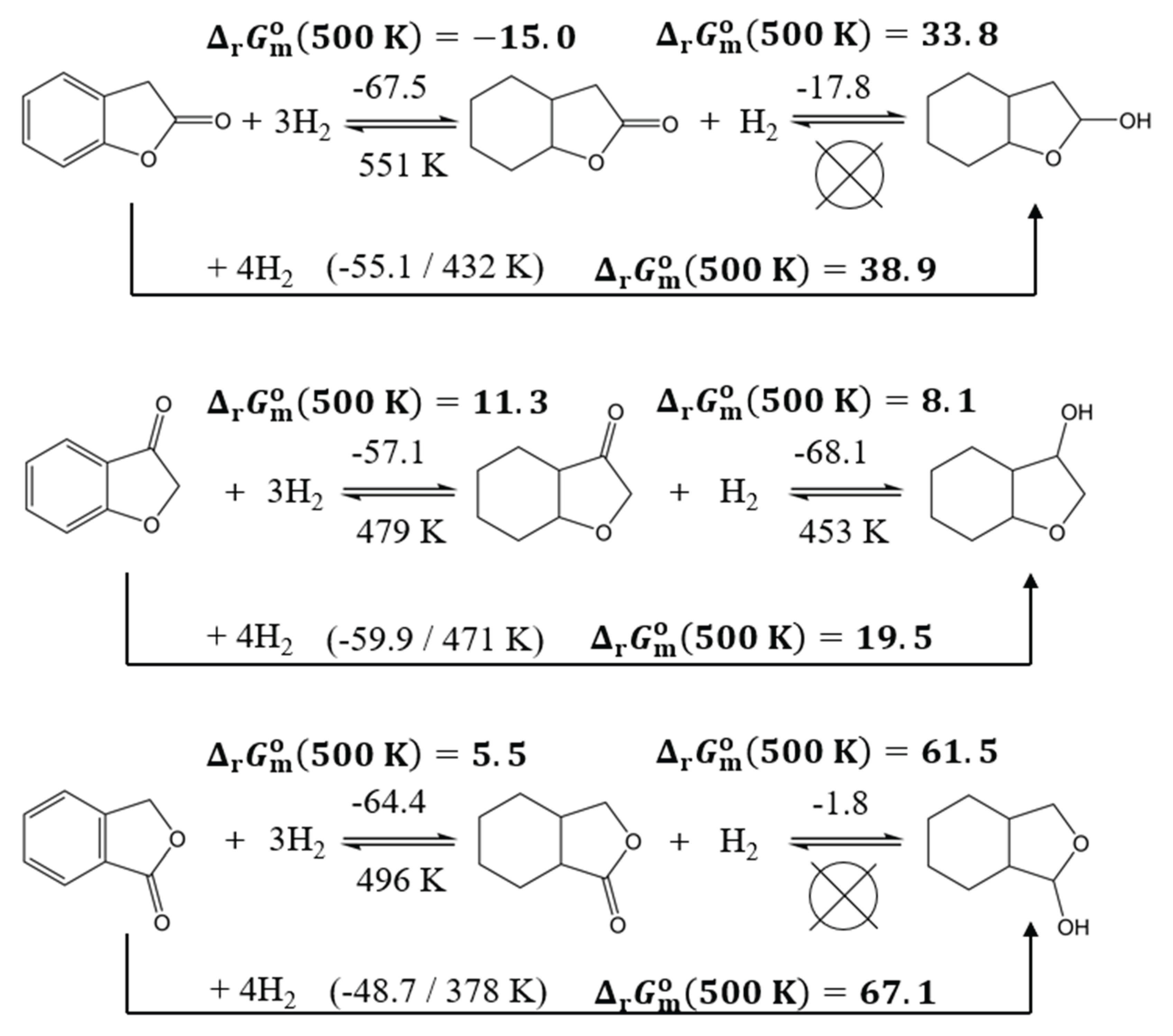
In comparison to benzofurans, lactones exhibit a lower reaction enthalpy for the full hydrogenation because of the possible hydrogenation of the keto group to a hydroxy group. Regarding hydrogenation of the aromatic ring, the reaction enthalpy for 2-coumaranone (-67.5 kJ∙mol-1/H2) and phthalide (-64.4 kJ∙mol-1/H2) is not favourable in comparison to commercial LOHCs. Moreover, the absolute values of the hydrogenation enthalpies of the keto group are so low that long-term hydrogen storage is not feasible.
The most promising candidate among the lactones is 3-coumaranone with a complete hydrogenation reaction enthalpy -59.9 kJ∙mol-1/H2 and possible dehydrogenation below 500 K. In addition, the hydrogenation enthalpies of the aromatic ring (-57.1 kJ∙mol-1) and the keto group (-68.1 kJ∙mol-1) are of similar magnitude.
Figure 8.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for aromatic lactones. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
Figure 8.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for aromatic lactones. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
3.3.7. Thermodynamic Analysis of the Reversible Hydrogenation/Dehydrogenation Process
Generally, hydrogen release from the hydrogen-rich form of a LOHC is thermodynamically more challenging than the hydrogen charging reaction [48]. Suitability as a hydrogen storage material is assessed based on the magnitude of the Gibbs reaction energy at 500 K, with higher values indicating greater potential. When comparing the Gibbs reaction enthalpies in Table 6, it is important to consider the differing values associated with the various degrees of hydrogenation. For lactones, in particular, a significant difference is observed between the hydrogenation of the aromatic ring and that of the hydroxyl group. Although 2-coumaranone, 3-coumaranone, and phthalide exhibit very high values, these are primarily due to the hydrogenation of the keto group. Hydrogenation of the aromatic ring is more favorable than in established hydrogen carriers only in the case of 3-coumaranone.
The addition of aromatic rings to the furan backbone leads to increasingly favorable properties for dehydrogenation. The benzofuran derivatives containing one aromatic ring already exhibit Gibbs reaction enthalpies comparable to those of typical hydrogen carriers. The condensation of an additional ring further amplifies this effect. Alternatively, a lactone may form in place of the second ring. However, dibenzofuran possesses the most favourable Gibbs reaction energy of the compounds investigated in this work. For this reason, lactone formation is comparatively less advantageous in comparison to the addition of the second aromatic ring.
4. Conclusions
This study demonstrates that the incorporation of oxygen functionalities into bi- and tricyclic aromatic compounds containing five-membered rings significantly influences their thermodynamic properties as liquid organic hydrogen carriers (LOHCs). The combined use of experimental vaporisation data and high-level quantum chemical calculations allowed reliable estimation of reaction enthalpies and entropies, confirming that optimal balances of these parameters could enable effective hydrogen release at moderate temperatures below 500 K. Thermodynamic evaluation based on Gibbs reaction energies at 500 K reveals that dibenzofuran exhibits the most favourable characteristics, with a Gibbs energy of 36.6 kJ·mol⁻¹, indicating high potential for efficient hydrogen storage and release. In contrast, lactone formation offers comparatively less advantage for hydrogen storage applications. In summary, aromatic furan derivatives with extended ring systems, particularly low melting dibenzofuran derivatives, emerge as promising candidates for LOHC materials.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Acknowledgments
This project has been funded by the Free State of Bavaria through the project "Oxo-LOHC-Autotherme und ultratiefe Wasserstoff-Freisetzung aus LOHC-Systeme - Oxo-LOHC“ (grant number: 84-6665a2/201/11). AAS acknowledges gratefully the Committee on Science and Higher Education of the Government of St. Petersburg. This work was partly supported by the Ministry of Science and Higher Education of the Russian Federation (Project No. FSSE-2023-0003) under the state assignment of Samara State Technical University.
References
- Teichmann, D.; Arlt, W.; Wasserscheid, P.; Freymann, R. A Future Energy Supply Based on Liquid Organic Hydrogen Carriers (LOHC). Energy Environ. Sci. 2011, 4, 2767. [CrossRef]
- Zakgeym, D.; Hofmann, J.D.; Maurer, L.A.; Auer, F.; Müller, K.; Wolf, M.; Wasserscheid, P. Better through Oxygen Functionality? The Benzophenone/Dicyclohexylmethanol LOHC-System. Sustain. Energy Fuels 2023, 7, 1213–1222. [CrossRef]
- Verevkin, S.P.; Samarov, A.A.; Vostrikov, S. V. Does the Oxygen Functionality Really Improve the Thermodynamics of Reversible Hydrogen Storage with Liquid Organic Hydrogen Carriers? Oxygen 2024, 4, 266–284. [CrossRef]
- Ghahremanpour, M.M.; van Maaren, P.J.; Ditz, J.C.; Lindh, R.; van der Spoel, D. Large-Scale Calculations of Gas Phase Thermochemistry: Enthalpy of Formation, Standard Entropy, and Heat Capacity. J. Chem. Phys. 2016, 145, 114305. [CrossRef]
- Pracht, P.; Bohle, F.; Grimme, S. Automated Exploration of the Low-Energy Chemical Space with Fast Quantum Chemical Methods. Phys. Chem. Chem. Phys. 2020, 22, 7169–7192. [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Revision C.01 Gaussian, Inc., Wallingford CT. 2016.
- Wheeler, S.E.; Houk, K.N.; Schleyer, P. v. R.; Allen, W.D. A Hierarchy of Homodesmotic Reactions for Thermochemistry. J. Am. Chem. Soc. 2009, 131, 2547–2560. [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Notario, R.; Roux, M.V.; Chickos, J.S.; Liebman, J.F. Rediscovering the Wheel. Thermochemical Analysis of Energetics of the Aromatic Diazines. J. Phys. Chem. Lett. 2012, 3, 3454–3459. [CrossRef]
- Notario, R.; Victoria Roux, M.; Castaño, O. The Enthalpy of Formation of Dibenzofuran and Some Related Oxygen-Containing Heterocycles in the Gas Phase. A GAUSSIAN-3 Theoretical Study. Phys. Chem. Chem. Phys. 2001, 3, 3717–3721. [CrossRef]
- Sousa, C.C.S.; Matos, M.A.R.; Santos, L.M.N.B.F.; Morais, V.M.F. Reprint of: Energetics of 2- and 3-Coumaranone Isomers: A Combined Calorimetric and Computational Study. J. Chem. Thermodyn. 2014, 73, 283–289. [CrossRef]
- Steele, W.V.; Chirico, R.D. Thermodynamics and the Hydrodeoxygenation of 2,3-Benzofuran; 1990;
- Verevkin, S.P.; Emel’yanenko, V.N.; Pimerzin, A.A.; Vishnevskaya, E.E. Thermodynamic Analysis of Strain in Heteroatom Derivatives of Indene. J. Phys. Chem. A 2011, 115, 12271–12279. [CrossRef]
- Verevkin, S.P.; Emel’yanenko, V.N.; Pimerzin, A.A.; Vishnevskaya, E.E. Thermodynamic Analysis of Strain in the Five-Membered Oxygen and Nitrogen Heterocyclic Compounds. J. Phys. Chem. A 2011, 115, 1992–2004. [CrossRef]
- Steele, W. V.; Chirico, R.D.; Knipmeyer, S.E.; Nguyen, A.; Smith, N.K.; Tasker, I.R. Thermodynamic Properties and Ideal-Gas Enthalpies of Formation for Cyclohexene, Phthalan (2,5-Dihydrobenzo-3,4-Furan), Isoxazole, Octylamine, Dioctylamine, Trioctylamine, Phenyl Isocyanate, and 1,4,5,6-Tetrahydropyrimidine. J. Chem. Eng. Data 1996, 41, 1269–1284. [CrossRef]
- Chirico, R..; Gammon, B..; Knipmeyer, S..; Nguyen, A.; Strube, M..; Tsonopoulos, C.; Steele, W.. The Thermodynamic Properties of Dibenzofuran. J. Chem. Thermodyn. 1990, 22, 1075–1096. [CrossRef]
- Verevkin, S.P. Enthalpy of Sublimation of Dibenzofuran: A Redetermination. Phys. Chem. Chem. Phys. 2003, 5, 710–712. [CrossRef]
- Rakus, K.; Verevkin, S.P.; Schätzer, J.; Beckhaus, H.; Rüchardt, C. Thermolabile Hydrocarbons, 33. Thermochemistry and Thermal Decomposition of 9,9′-Bifluorenyl and 9,9′-Dimethyl-9,9′-bifluorenyl – The Stabilization Energy of 9-Fluorenyl Radicals. Chem. Ber. 1994, 127, 1095–1103. [CrossRef]
- Verevkin, S.P. Vapor Pressure Measurements on Fluorene and Methyl-Fluorenes. Fluid Phase Equilib. 2004, 225, 145–152. [CrossRef]
- Tavernier, P. Donnees Thermochimiques Relatives Aux Constituants Des Poudres. Mem. Poudres 1956, 301–327.
- Freitas, V.L.S.; Santos, C.P.F.; Ribeiro da Silva, M.D.M.C.; Ribeiro da Silva, M.A.V. The Effect of Ketone Groups on the Energetic Properties of Phthalan Derivatives. J. Chem. Thermodyn. 2016, 96, 74–81. [CrossRef]
- Stephenson, R.M.; Malanowski, S. Handbook of the Thermodynamics of Organic Compounds; Springer Netherlands: Dordrecht, 1987; ISBN 978-94-010-7923-5.
- Gobble, C.; Chickos, J.S. The Vapor Pressure and Vaporization Enthalpy of R-(+)-Menthofuran, a Hepatotoxin Metabolically Derived from the Abortifacient Terpene, (R)-(+)-Pulegone by Correlation Gas Chromatography. J. Chem. Thermodyn. 2016, 98, 135–139. [CrossRef]
- Stull, D.R. Vapor Pressure of Pure Substances. Organic and Inorganic Compounds. Ind. Eng. Chem. 1947, 39, 517–540. [CrossRef]
- Verevkin, S.P. Weaving a Web of Reliable Thermochemistry around Lignin Building Blocks: Phenol, Benzaldehyde, and Anisole. J. Therm. Anal. Calorim. 2022, 147, 6073–6085. [CrossRef]
- Verevkin, S.P.; Samarov, A.A.; Vostrikov, S. V. Thermodynamics of Reversible Hydrogen Storage: Are Methoxy-Substituted Biphenyls Better through Oxygen Functionality? Hydrogen 2023, 4, 862–880. [CrossRef]
- Samarov, A.A.; Verevkin, S.P. Hydrogen Storage Technologies: Methyl-Substituted Biphenyls as an Auspicious Alternative to Conventional Liquid Organic Hydrogen Carriers (LOHC). J. Chem. Thermodyn. 2022, 165, 106648. [CrossRef]
- Verevkin, S.P.; Andreeva, I. V.; Zherikova, K. V.; Pimerzin, A.A. Prediction of Thermodynamic Properties: Centerpiece Approach—How Do We Avoid Confusion and Get Reliable Results? J. Therm. Anal. Calorim. 2022, 147, 8525–8534. [CrossRef]
- Benson, S.W. Thermochemical Kinetics, 2nd Ed. John Wiley & Sons, New York-London-Sydney-Toronto. 1976, 1–320.
- Verevkin, S.P.; Emel’yanenko, V.N.; Diky, V.; Muzny, C.D.; Chirico, R.D.; Frenkel, M. New Group-Contribution Approach to Thermochemical Properties of Organic Compounds: Hydrocarbons and Oxygen-Containing Compounds. J. Phys. Chem. Ref. Data 2013, 42, 033102. [CrossRef]
- Kováts, E. Gas-chromatographische Charakterisierung Organischer Verbindungen. Teil 1: Retentionsindices Aliphatischer Halogenide, Alkohole, Aldehyde Und Ketone. Helv. Chim. Acta 1958, 41, 1915–1932. [CrossRef]
- Verevkin, S.P. Vapour Pressures and Enthalpies of Vaporization of a Series of the Linear N-Alkyl-Benzenes. J. Chem. Thermodyn. 2006, 38, 1111–1123. [CrossRef]
- NIST Chemistry WebBook. Available Online: Https://Webbook.Nist.Gov/Chemistry/ (Accessed on 15.06.2025).
- Majer, V.; Svoboda, V. Enthalpies of Vaporization of Organic Compounds: A Critical Review and Data Compilation, Blackwell Scientific Publications, Oxford; 1985;
- Steele, W. V.; Chirico, R.D.; Knipmeyer, S.E.; Nguyen, A. Measurements of Vapor Pressure, Heat Capacity, and Density along the Saturation Line for Cyclopropane Carboxylic Acid, N , N -Diethylethanolamine, 2,3-Dihydrofuran, 5-Hexen-2-One, Perfluorobutanoic Acid, and 2-Phenylpropionaldehyde. J. Chem. Eng. Data 2002, 47, 715–724. [CrossRef]
- Steele, W. V.; Chirico, R.D. Thermodynamics and the Hydrodeoxygenation of 2,3-Benzofuran NIPER-457. Publ. by DOE Foss. Energy, Bartlesv. Proj. Off. Available from NTIS, Rep. No. DE-90000218 1990.
- Chirico, R.D.; Nguyen, A.; Steele, W.V.; Strube, M.M.; Hossenlopp, I.A.; Gammon, B.E. Thermochemical and Thermophysical Properties of Organic Compounds Derived from Fossil Substances. Chemical Thermodynamic Properties of Organic Oxygen Compounds Found in Fossil Materials. NIPER Rep. 1986, 135, 42.
- Sasse, K.; Jose, J.; Merlin, J.-C. A Static Apparatus for Measurement of Low Vapor Pressures. Experimental Results on High Molecular-Weight Hydrocarbons. Fluid Phase Equilib. 1988, 42, 287–304. [CrossRef]
- Guthrie, G.B.; Scott, D.W.; Hubbard, W.N.; Katz, C.; McCullough, J.P.; Gross, M.E.; Williamson, K.D.; Waddington, G. Thermodynamic Properties of Furan. J. Am. Chem. Soc. 1952, 74, 4662–4669. [CrossRef]
- Parsana, V.M.; Parikh, S.; Ziniya, K.; Dave, H.; Gadhiya, P.; Joshi, K.; Gandhi, D.; Vlugt, T.J.H.; Ramdin, M. Isobaric Vapor–Liquid Equilibrium Data for Tetrahydrofuran + Acetic Acid and Tetrahydrofuran + Trichloroethylene Mixtures. J. Chem. Eng. Data 2023, 68, 349–357. [CrossRef]
- Lebedev, B.V.; Lityagov, V.Y. Calorimetric Study of Tetrahydrofuran and Its Polymerization in the Temperature Range 0–400°K. Vysok. Soedin. 1977, A19, 2283–2290.
- Furuyama, S.; Golden, D.M.; Benson, S.W. Thermochemistry of Cyclopentene and Cyclopentadiene from Studies of Gas-Phase Equilibria. J. Chem. Thermodyn. 1970, 2, 161–169. [CrossRef]
- Beckett, C.W.; Freeman, N.K.; Pitzer, K.S. The Thermodynamic Properties and Molecular Structure of Cyclopentene and Cyclohexene 1. J. Am. Chem. Soc. 1948, 70, 4227–4230. [CrossRef]
- Huffman, H.M.; Eaton, M.; Oliver, G.D. The Heat Capacities, Heats of Transition, Heats of Fusion and Entropies of Cyclopentene and Cyclohexene. J. Am. Chem. Soc. 1948, 70, 2911–2914. [CrossRef]
- Douslin, D.R.; Huffman, H.M. The Heat Capacities, Heats of Transition, Heats of Fusion and Entropies of Cyclopentane, Methylcyclopentane and Methylcyclohexane 1. J. Am. Chem. Soc. 1946, 68, 173–176. [CrossRef]
- Stull, D.R.; Sinke, G.C.; McDonald, R.A.; Hatton, W.E.; Hildebrand, D.L. Thermodynamic Properties of Indane and Indene. Pure Appl. Chem. 1961, 2, 315–322. [CrossRef]
- Finke, H..; McCullough, J..; Messerly, J..; Osborn, A.; Douslin, D.. Cis- and Trans-Hexahydroindan. Chemical Thermodynamic Properties and Isomerization Equilibrium. J. Chem. Thermodyn. 1972, 4, 477–494. [CrossRef]
- Verevkin, S.P.; Samarov, A.A.; Vostrikov, S. V.; Wasserscheid, P.; Müller, K. Comprehensive Thermodynamic Study of Alkyl-Cyclohexanes as Liquid Organic Hydrogen Carriers Motifs. Hydrogen 2023, 4, 42–59. [CrossRef]
- Müller, K.; Völkl, J.; Arlt, W. Thermodynamic Evaluation of Potential Organic Hydrogen Carriers. Energy Technol. 2013, 1, 20–24. [CrossRef]
Figure 1.
The comparison of reaction enthalpies, ( per H2, given in kJ·mol-1), equilibrium temperatures (Teq in K), gravimetric capacities (in %mass), and Gibbs energies ( in kJ·mol-1) of hydrogenation reactions of C,H and C,H,O containing LOHC.
Figure 1.
The comparison of reaction enthalpies, ( per H2, given in kJ·mol-1), equilibrium temperatures (Teq in K), gravimetric capacities (in %mass), and Gibbs energies ( in kJ·mol-1) of hydrogenation reactions of C,H and C,H,O containing LOHC.
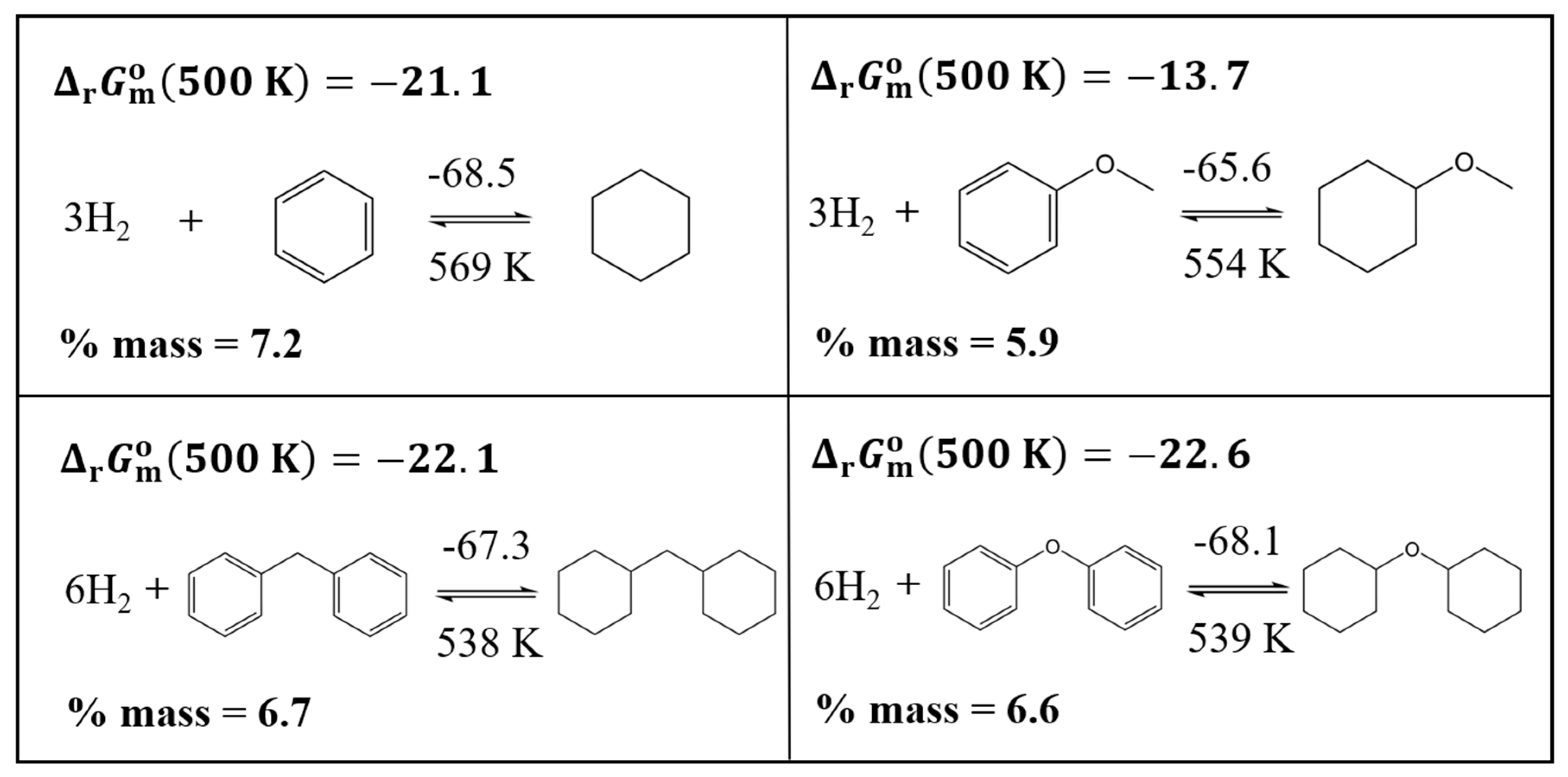
Figure 2.
Structures of aromatic and aliphatic compounds with oxygen functionalities studied in this work.
Figure 2.
Structures of aromatic and aliphatic compounds with oxygen functionalities studied in this work.
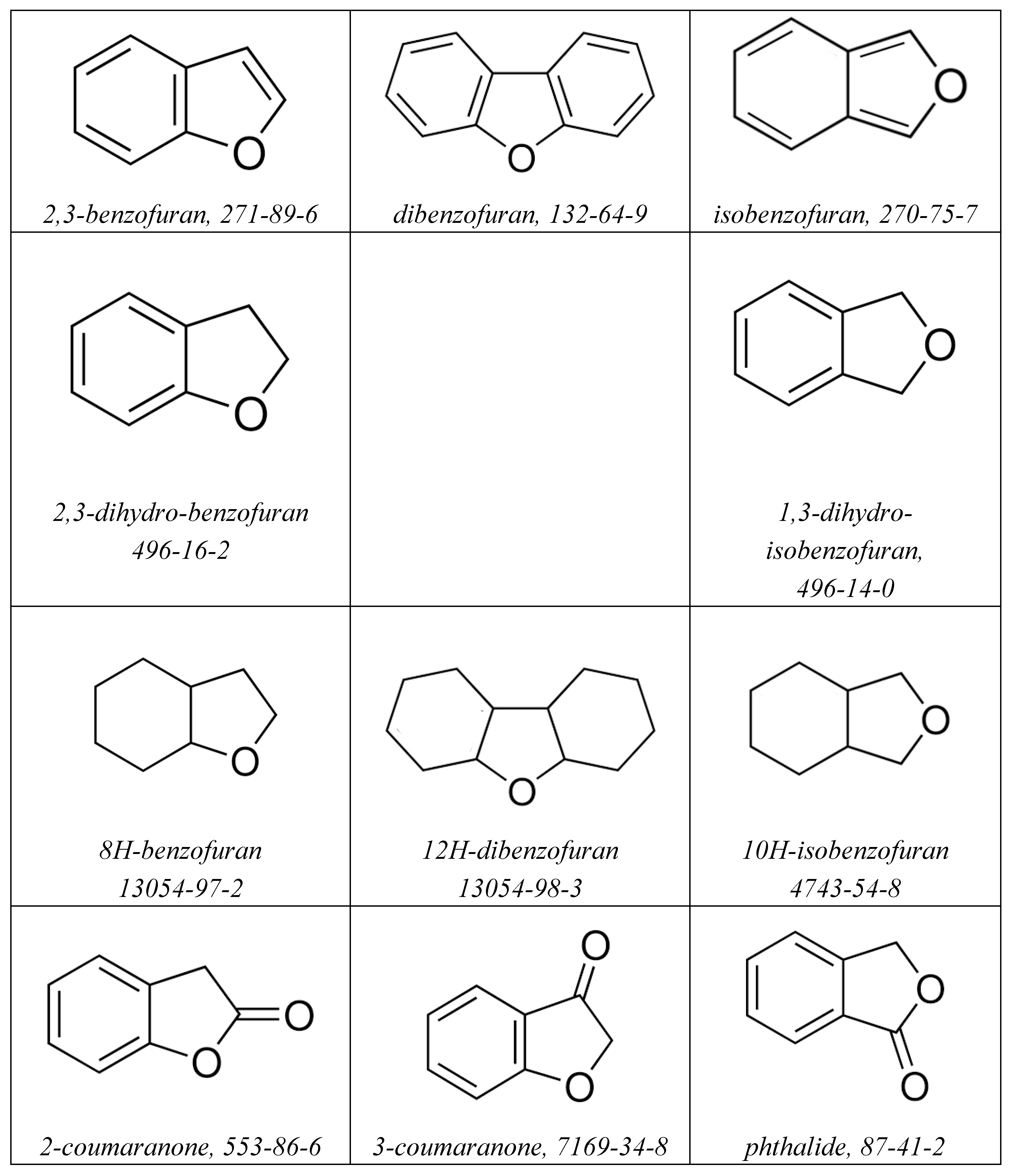
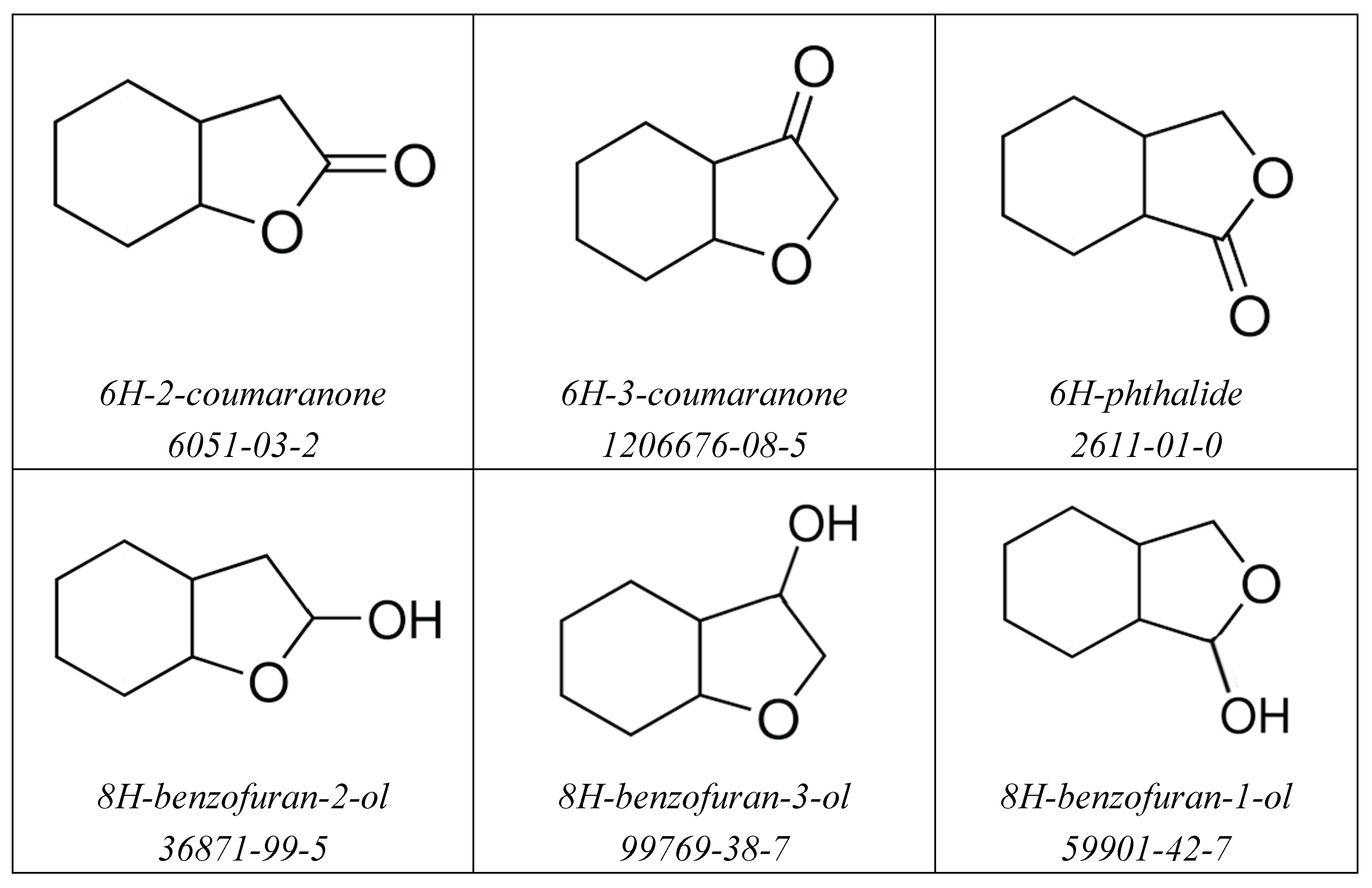
Figure 3.
Calculating the enthalpy of vaporisation, (298.15 K)/ kJ⋅mol-1 of 2,3-dihydrobenzofuran (left) and 8H-benzofuran or 8H-isobenzofuran (right) using the “centerpiece approach” (CP). The numerical values of enthalpies of vaporisation required for calculations are given in Table S6.
Figure 3.
Calculating the enthalpy of vaporisation, (298.15 K)/ kJ⋅mol-1 of 2,3-dihydrobenzofuran (left) and 8H-benzofuran or 8H-isobenzofuran (right) using the “centerpiece approach” (CP). The numerical values of enthalpies of vaporisation required for calculations are given in Table S6.
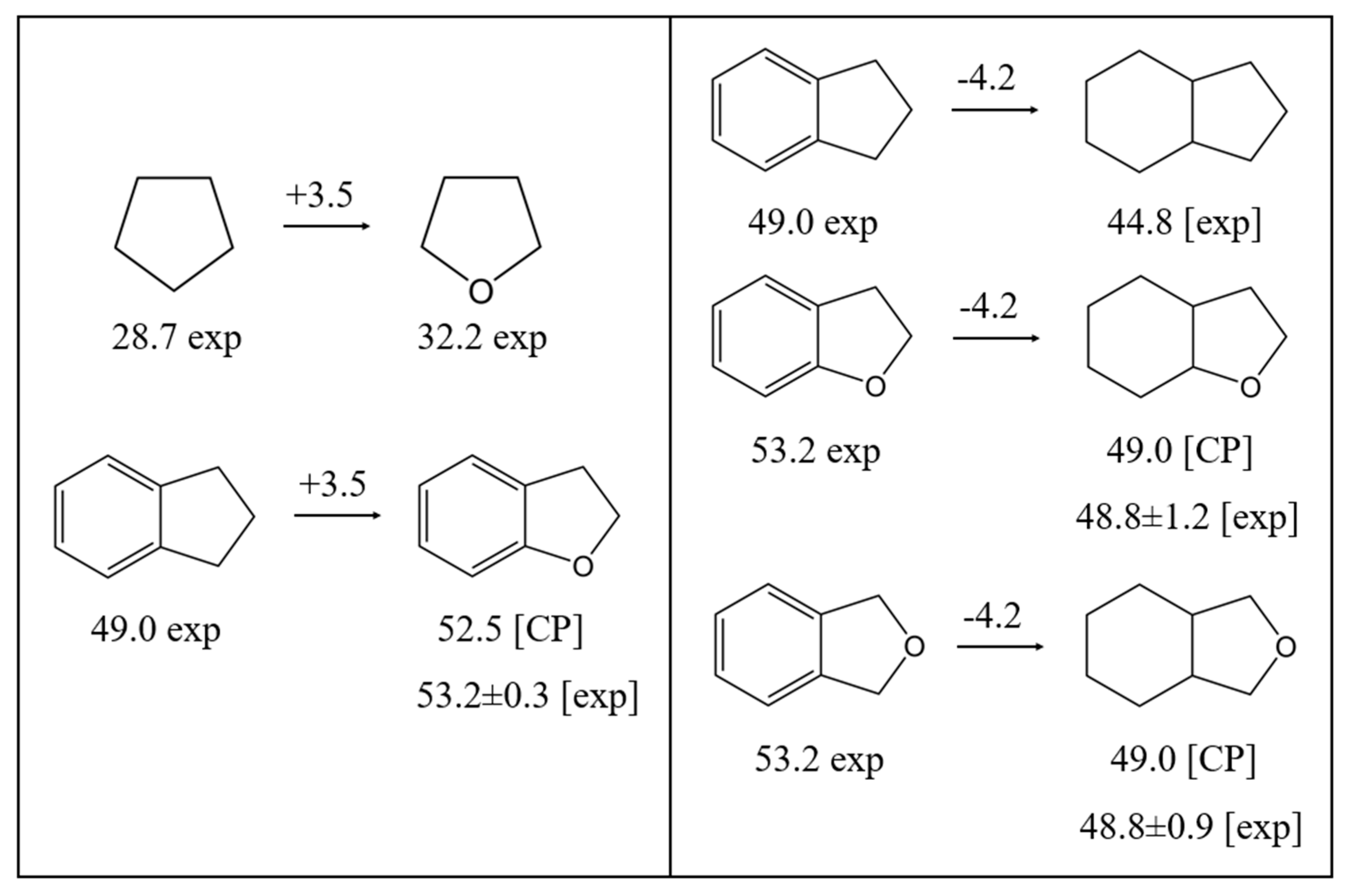
Figure 4.
Calculating the enthalpy of vaporisation, (298.15 K)/ kJ⋅mol-1 of 8H-benzofuran from 8H-indene using the combination of the centerpiece approach” (CP) and the GA method. The numerical values of enthalpies of vaporisation required for calculations are given in Tables S6 and S7.
Figure 4.
Calculating the enthalpy of vaporisation, (298.15 K)/ kJ⋅mol-1 of 8H-benzofuran from 8H-indene using the combination of the centerpiece approach” (CP) and the GA method. The numerical values of enthalpies of vaporisation required for calculations are given in Tables S6 and S7.
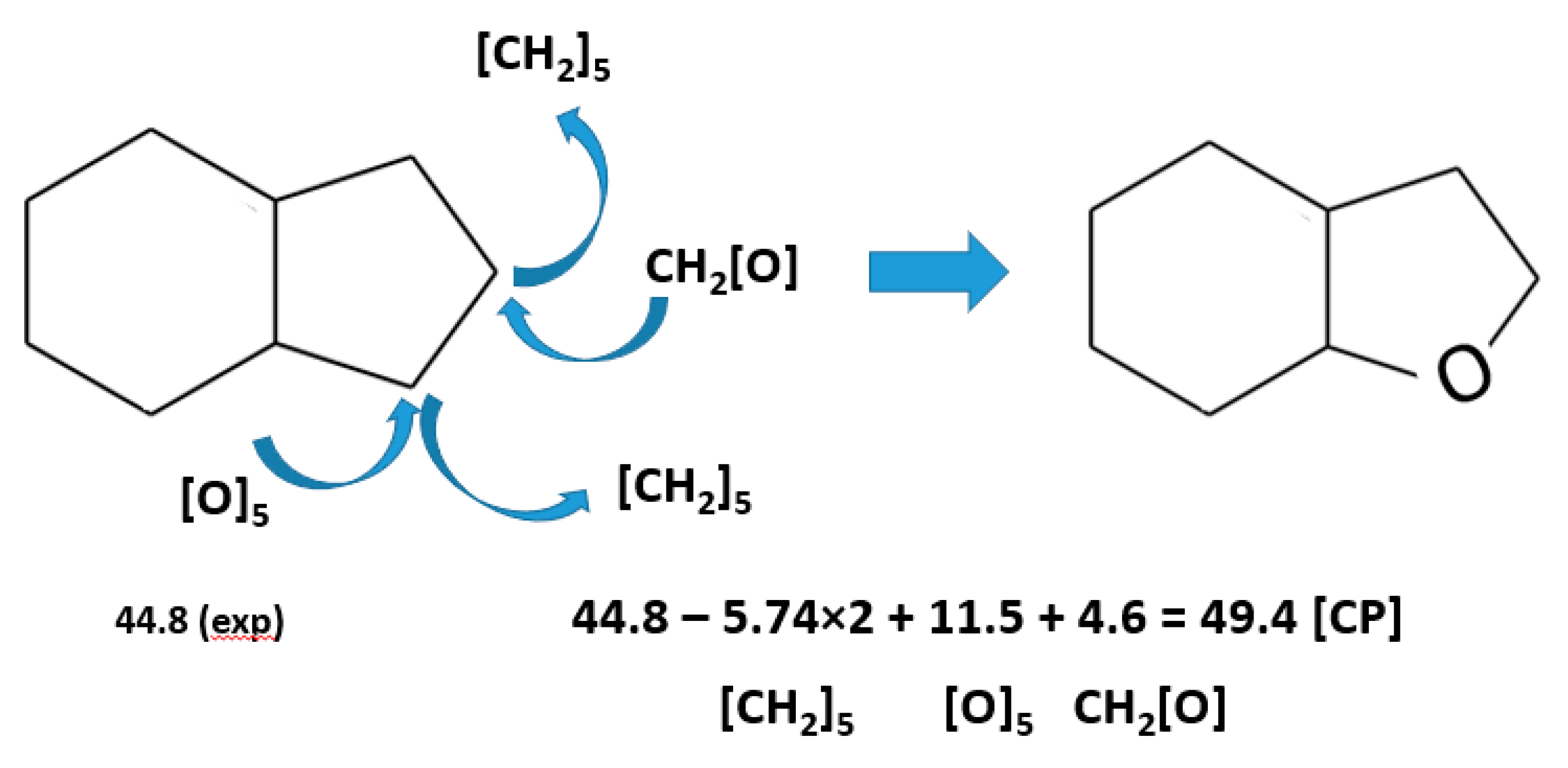
Figure 5.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for 1,3-cyclopentadiene and furan. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
Figure 5.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for 1,3-cyclopentadiene and furan. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
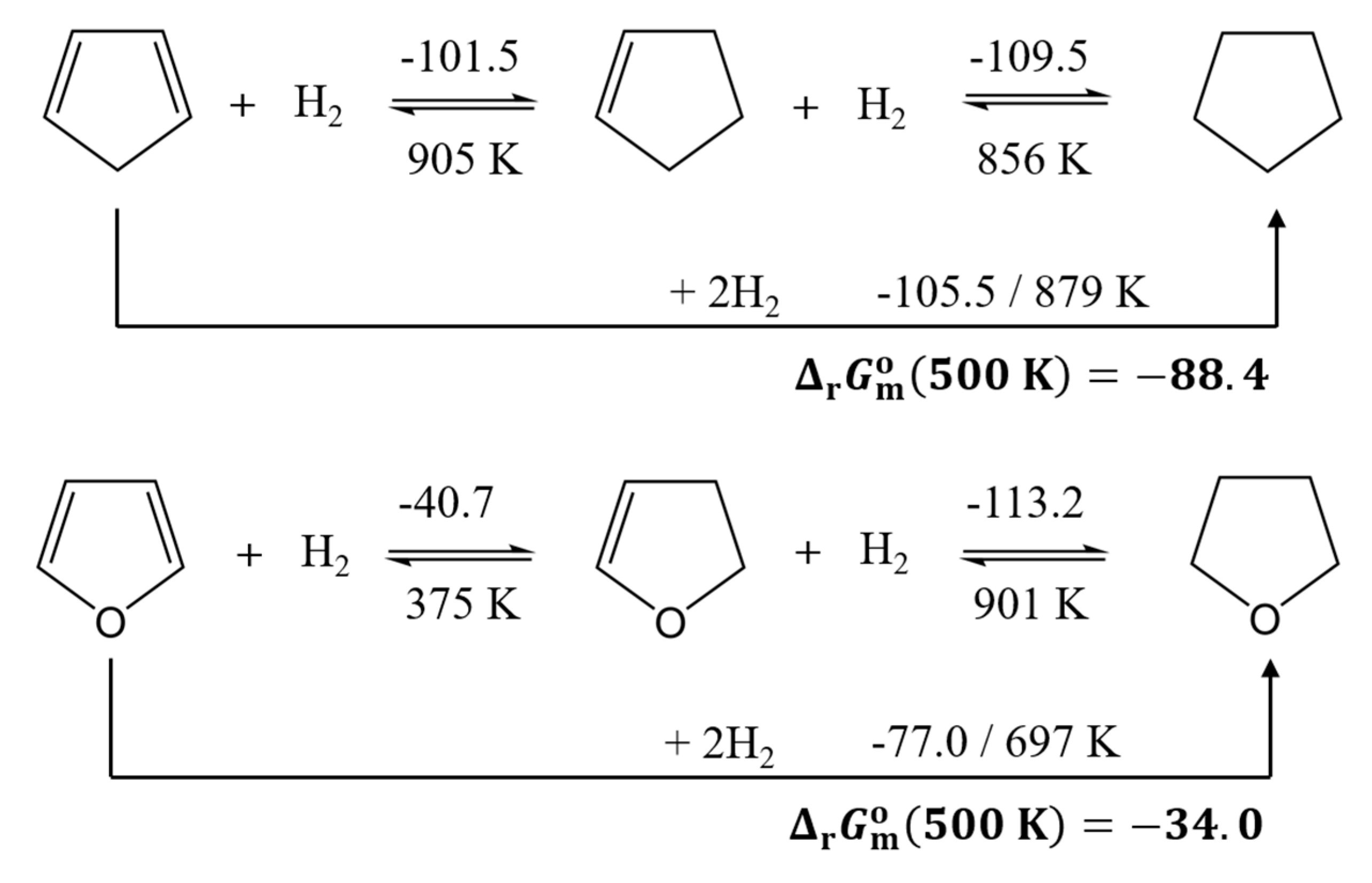
Figure 6.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for benzofuran derivatives. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
Figure 6.
Comparison of the thermodynamic characteristics of the hydrogenation reactions for benzofuran derivatives. The numerical values for reaction enthalpies, (298 K), are given above arrows (in kJ∙mol-1/H2) and the -values are given bellow arrows in K. Gibbs reaction energies of full hydrogenation, (500 K) are given in kJ∙mol-1.
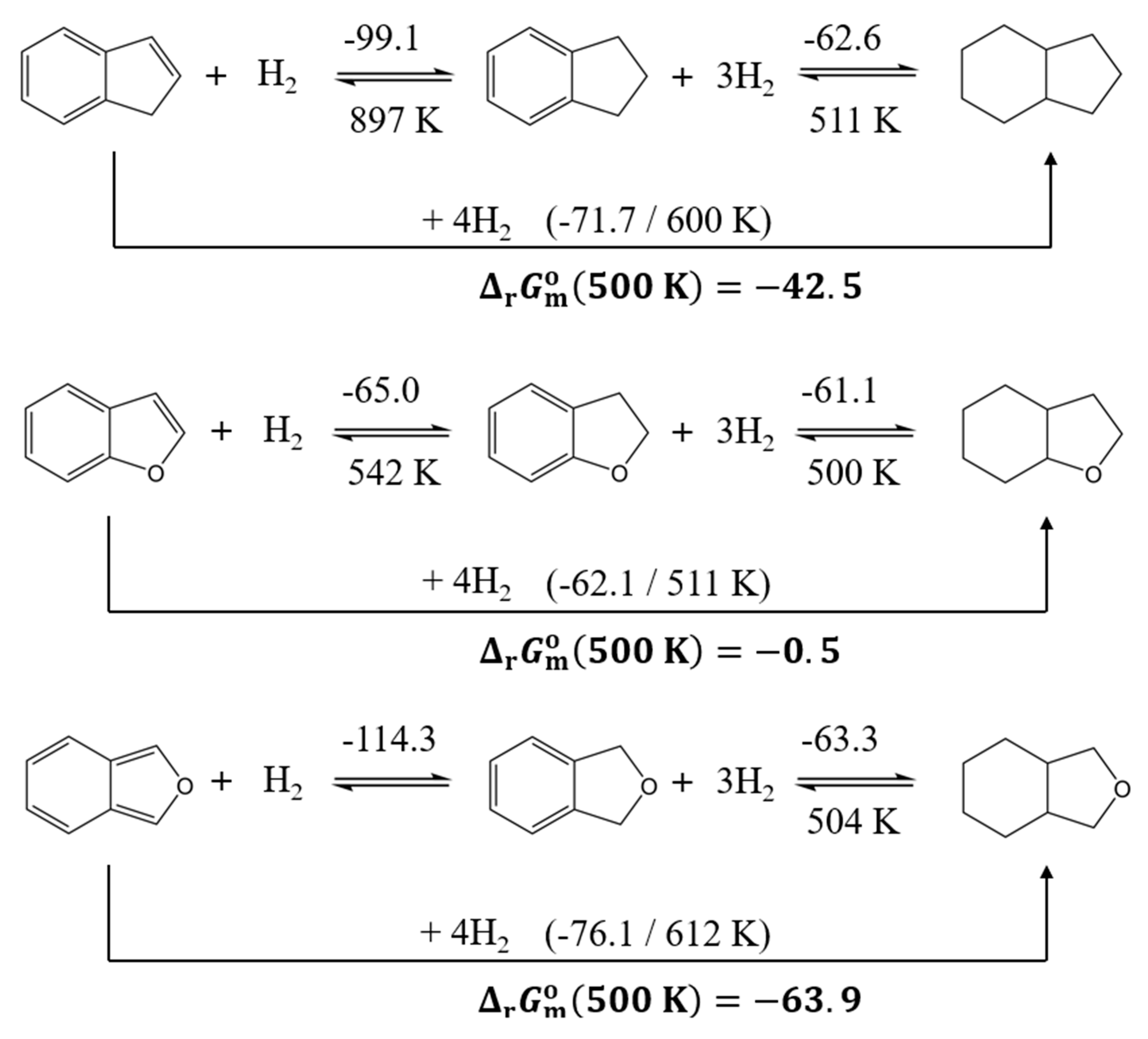

Table 1.
Comparison of the quantum-chemical and experimental enthalpies of formation of the HL compounds (at T = 298.15 K and p° = 0.1 MPa, in kJ·mol-1) a.
Table 1.
Comparison of the quantum-chemical and experimental enthalpies of formation of the HL compounds (at T = 298.15 K and p° = 0.1 MPa, in kJ·mol-1) a.
| Compound |
(g) b G3MP2/AT |
(g) c G3MP2//B3LYP |
(g) d G3MP2 |
(g)QC e |
(g) f (exp) |
|---|---|---|---|---|---|
| 2,3-benzofuran | 17.2±4.1 | 16.6±4.0 | 16.9±2.9 | 13.9±0.7 | |
| 2,3-dihydrobenzofuran | -47.1±4.1 | -45.5±4.0 | -46.3±2.9 | -46.7±0.9 | |
| isobenzofuran | 79.3±4.1 | 79.3±4.1 | |||
| 1,3-dihydro-isobenzofuran | -26.3±4.1 | -26.0±4.0 | -26.1±2.9 | -30.6±1.2 | |
| dibenzofuran | 50.1±4.1 | 48.9±4.0 | 49.5±2.9 | 52.9±0.7 | |
| 2-coumaranone | -222.7±4.1 | -217.5±2.8 | -218.1±3.4 | -218.7±1.9 | (-210.8±4.0) |
| 3-coumaranone | -166.3±4.1 | -162.1±2.6 | -162.3±3.4 | -162.9±1.9 | -168.8±2.4 |
| phthalide | -224.1±4.1 | -220.4±3.4 | -220.7±4.4 | -221.4±2.3 | -220.6±2.8 |
a Uncertainties in this table are expressed as two times the standard deviation. b From Table S1, column 5, calculated in this work by G3MP2 and the atomization reaction, corrected according to Eq. (6). c From Table S2, column 5, calculated by Notario et al. [9] using G3MP2//B3LYP method and the “corrected” atomization reaction. d Calculated by Sousa et al. [10] using G3MP2 method and the well-balanced reactions shown in Figure S2. e The weighted average value determined from the data in columns 2 to 4 (using uncertainty as the weighting factor). f The available experimental results, evaluated in this work and shown in Table 2, column 5. The value in parentheses seems to be in error.
Table 2.
Comparison of experimental and theoretical thermochemical data for the hydrogen lean (HL) compounds (at T = 298.15 K, p° = 0.1 MPa, in kJ·mol-1)a.
Table 2.
Comparison of experimental and theoretical thermochemical data for the hydrogen lean (HL) compounds (at T = 298.15 K, p° = 0.1 MPa, in kJ·mol-1)a.
| Compounds | (liq or cr) | b | (g)expc | (g)QCd |
|---|---|---|---|---|
| 2,3-benzofuran [271-89-6] (liq) | -34.8±0.7 [11] | |||
| -35.3±1.6 [12] | ||||
| -34.9±0.6 | 48.4±0.4 | 13.9±0.7 | 16.9±2.9 | |
| 2,3-dihydro-benzofuran [496-16-2] (liq) | -99.8±0.7 [11] | |||
| -100.6±1.9 [13] | ||||
| -99.9±0.8 | 53.2±0.3 | -46.7±0.9 | -46.3±2.9 | |
| 1,3-dihydro-isobenzofuran [496-14-0] (liq) | -83.8±0.9 [14] | 53.2±0.8 | -30.6±1.2 | -26.1±2.9 |
| dibenzofuran [132-64-9] (cr) | -29.1±0.6 [15] | 82.0±0.3 [16] | 52.9±0.7 | 49.5±2.9 |
| fluorene [86-73-7] (cr) | 89.9±1.4 [17] | 86.1±0.1 [18] | 176.0±1.4 | - |
| 2-coumaranone [553-86-6] (cr) | (-292.4±3.7) [10] | 81.6±1.4 f | (-210.8±4.0) | -218.7±1.9 |
| 3-coumaranone [7169-34-8] (cr) | -254.9±2.0 [10] | 86.1±1.4 g | -168.8±2.4 | -162.9±1.9 |
| phthalide [87-41-2] (cr) | (-366±10) [19] | |||
| -312.4±1.5 [20] | 92.0±2.2h | -220.6±2.8 | -221.4±2.3 |
a Uncertainties correspond to expanded uncertainties of the mean (0.95 level of confidence). The values highlighted in bold were used for further thermochemical calculations. The values in brackets were considered unreliable. b The evaluated data from Table 3. c Sum of columns 2 and 3. d The averaged quantum-chemical results from Table 1, column 6. e For 2-coumaranone, the sublimation enthalpy, (298.15 K) = 81.6±1.4 kJ·mol-1 was calculated as a sum of the enthalpy of vaporisation (298.15 K) = 64.8±1.0 kJ·mol-1 [Table 3] and the enthalpy of fusion (298.15 K) = 16.8±1.1 kJ·mol-1 [Table S3]. f For 3-coumaranone, the sublimation enthalpy, (298.15 K) = 86.1±1.4 kJ·mol-1 was calculated as the average from (298.15 K) = 85.8±1.7 kJ·mol-1 [10] and the sublimation enthalpy (298.15 K) = 86.8±2.8 kJ·mol-1 calculated as the sum of enthalpy of vaporisation (298.15 K) = 68.8±2.3 kJ·mol-1 [Table 3] and the enthalpy of fusion (298.15 K) = 18.0±1.6 kJ·mol-1 [Table S3]. g For phthalide, the sublimation enthalpy, (298.15 K) = 92.0±2.2 kJ·mol-1 was calculated as the average from (298.15 K) = 88.4±4.4 kJ·mol-1 [20] and the sublimation enthalpy (298.15 K) = 93.2±2.8] kJ·mol-1 calculated as the sum of the enthalpy of vaporisation (298.15 K) = 76.3±2.5 kJ·mol-1 [Table 3] and the enthalpy of fusion (298.15 K) = 16.9±1.2 kJ·mol-1 [Table S3].
Table 3.
Compilation of the standard molar enthalpies of vaporisation of HL and HR counterparts of LOHC systems (in kJ⋅mol-1).
Table 3.
Compilation of the standard molar enthalpies of vaporisation of HL and HR counterparts of LOHC systems (in kJ⋅mol-1).
| Compounds | Methoda |
T-range/ K |
Tav |
298.15 Kb |
|
|---|---|---|---|---|---|
| 2,3-benzofuran [271-89-6] | n/a | 323-403 | 45.0±1.5 | 47.9±1.6 | [21] |
| E | 273.2-488.1 | 45.1±0.1 | 48.0±0.6 | [11] | |
| T | 278.7-313.4 | 49.0±0.4 | 48.8±0.6 | [12] | |
| 48.4±0.4c | average | ||||
| Jx | 49.4±1.0 | Table 4 | |||
| 2,3-dihydro-benzofuran [496-16-2] | E | 285.0-503.2 | 48.7±0.1 | 53.0±0.9 | [11] |
| T | 278.7-333.0 | 52.7±0.2 | 53.2±0.4 | [13] | |
| BP | 341-465 | 47.8±0.8 | 53.2±1.3 | Table S4 | |
| 53.2±0.3c | |||||
| Jx | 52.7±1.0 | Table 4 | |||
| CP | 52.5±1.5 | Figure 3 | |||
| 1,3-dihydro-isobenzofuran [496-14-0] | E | 285.0-503.2 | 48.9±0.1 | 53.3±0.9 | [14] |
| phthalan | CGC | 298 | 52.7±2.9 | [22] | |
| BP | 318-465 | 48.0±1.1 | 53.1±1.5 | Table S4 | |
| 53.2±0.8c | average | ||||
| Jx | 52.6±1.0 | Table 4 | |||
| 8H-octahydro-benzofuran | BP | 337-446 | 44.5±0.8 | 48.8±1.9 | Table S4 |
| [13054-97-2] | Tb | 48.8±1.5 | Eq. 7 | ||
| 48.8±1.2c | average | ||||
| CP | 49.0±1.5 | Figure 3 | |||
| CP | 48.3±1.5 | Figure S3 | |||
| CP | 48.4±1.5 | Figure 3 | |||
| GA | 48.9±1.5 | Figure S3 | |||
| 8H-octahydro-isobenzofuran | BP | 324-453 | 43.8±0.8 | 48.1±1.2 | Table S4 |
| [4743-54-8] | Tb | 49.9±1.5 | Eq. 7 | ||
| 48.8±0.9c | average | ||||
| CP | 48.8±1.5 | Figure 3 | |||
| CP | 48.3±1.5 | Figure 3 | |||
| CP | 48.3±1.5 | Figure S4 | |||
| GA | 48.8±1.5 | Figure S4 | |||
| 12H-dodecahydro-dibenzofuran | BP | 381-532 | 52.2±1.4 | 63.8±2.7 | Table S4 |
| [13054-98-3] | |||||
| 2(3H)-benzofuranone [553-86-6] | PhT | 298 | 61.9±2.0 | Table S3 | |
| 2-coumaranone | BP | 384-521 | 57.0±1.8 | 65.3±2.4 | Table S4 |
| Jx | 65.7±1.5 | Table 6-2 | |||
| Jx | 65.7±2.0 | Table 6-2 | |||
| CP | 67.3±1.5 | Figure S5 | |||
| CP | 66.2±1.5 | Figure S5 | |||
| 65.7±0.7 | average | ||||
| benzofuran-3(2H)-one [7169-34-8] | PhT | 298 | 67.8±2.3 | Table S3 | |
| 3-coumaranone | BP | 425-551 | 60.3±1.5 | 68.8±2.3 | Table S4 |
| CP | 66.7±1.5 | Figure S6 | |||
| CP | 67.3±1.5 | Figure S6 | |||
| CP | 65.5±1.5 | Figure S6 | |||
| 66.9±0.8c | average | ||||
| 2-benzofuran-1(3H)-one [87-41-2] | n/a | 368.7-563.0 | 58.7±1.5 | (66.9±2.2) | [23] |
| phthalide | PhT | 298 | 71.5±4.6 | Table S3 | |
| BP | 346-563 | 69.0±2.0 | 76.3±2.5 | Table S4 | |
| CP | 74.7±1.5 | Figure S7 | |||
| CP | 75.4±1.5 | Figure S7 | |||
| CP | 73.6±1.5 | Figure S7 | |||
| 74.7±0.8c | average | ||||
| 6H-hexahydro-2-coumaranone | BP | 327-537 | 56.3±1.7 | 64.3±2.3 | Table S4 |
| [6051-03-2] | |||||
| 6H-hexahydro-3-coumaranone | BP | 353-626 | 54.2±0.7 | 65.2±2.3 | Table S4 |
| [1206676-08-5] | CP | 62.5±1.5 | Figure S8 | ||
| CP | 63.0±1.5 | Figure S8 | |||
| CP | 62.2±1.5 | Figure S8 | |||
| 62.9±0.8c | average | ||||
| 6H-hexahydro-phthalide | BP | 337-503 | 72.3±0.8 | 77.6±1.3 | Table S4 |
| [2611-01-0] | |||||
| octahydro-benzofuran-2-ol | CP | 66.5±2.0 | Figure S9 | ||
| [36871-99-5] | CP | 67.2±2.0 | Figure S9 | ||
| 66.9±1.4c | average | ||||
| 8H-octahydro-benzofuran-3-ol | BP | 354-513 | 63.8±1.6 | 73.2±2.5 | Table S4 |
| [99769-38-7] | CP | 73.0±2.0 | Figure S9 | ||
| CP | 73.5±2.0 | Figure S9 | |||
| 73.2±1.2c | average | ||||
| 8H-octahydro-isobenzofuran-1-ol | CP | 66.7±2.0 | Figure S9 | ||
| [59901-42-7] | CP | 66.5±2.0 | Figure S9 | ||
| 66.6±1.4c | average | ||||
| methoxy-benzene [100-66-3] | 46.4±0.2 | [24] | |||
| methoxy-cyclohexane [931-56-6] | 43.0±0.5 | [25] |
a Methods: n/a = method is not available; T = transpiration method; E = ebulliometry; Tb = from correlation with the normal boiling temperatures; PhT = from consistency of phase transitions (see Table S3); BP = estimated from boiling points at different pressures (see Table S4); Jx = calculated from correlation with the Kovats retention indices; CP = calculated according to the centerpiece approach (see text). b Vapor pressures available in the literature were treated using Eqs. (S2) and (S3) with help of heat capacity differences from Table S5 to calculate the enthalpies of vaporisation at 298.15 K. Uncertainties of the vaporisation enthalpies U() are the expanded uncertainties (0.95 level of confidence). They include uncertainties from the fitting equation and uncertainties from temperature adjustment to T = 298.15 K. Uncertainties in the temperature adjustment of vaporisation enthalpies to the reference temperature T= 298.15 K are estimated to account with 20% to the total adjustment. c Weighted mean value (uncertainties were taken as the weighting factor). Values given in bold are recommended for further thermochemical calculations.
Table 4.
Correlation of vaporisation enthalpies, (298.15 K)/ kJ.mol-1 of furan derivatives with their Kovats indices (Jx).
Table 4.
Correlation of vaporisation enthalpies, (298.15 K)/ kJ.mol-1 of furan derivatives with their Kovats indices (Jx).
| CAS | Compound | Jxa | (298.15 K)exp b | (298.15 K)calcc | d |
|---|---|---|---|---|---|
| 109-99-9 | tetrahydrofuran | 611 | 32.6±0.2 [33] | 33.1 | -0.5 |
| 110-00-9 | furan | 468 | 27.7±0.2 [33] | 27.8 | -0.1 |
| 1191-99-7 | 2,3-dihydrofuran | 564 | 31.3±0.3 [34] | 31.3 | 0.0 |
| 1708-29-8 | 2,5-dihydrofuran | 614 | 32.6±0.2 [33] | 33.2 | -0.6 |
| 534-22-5 | 2-methyl-furan | 603 | 32.7±0.2 [33] | 32.7 | 0.0 |
| 625-86-5 | 2,5-dimethyl-furan | 729 | 37.7±0.2 [33] | 37.4 | 0.3 |
| 3208-16-0 | 2-ethyl-furan | 720 | 37.1±0.2 [33] | 37.1 | 0.0 |
| 271-89-6 | 2,3-benzofuran | 1054 | 48.8±0.3 [12] | 49.4 | -0.6 |
| 496-16-2 | 2,3-dihydrobenzofuran | 1144 | 53.2±0.4 [13] | 52.7 | 0.5 |
| 496-14-0 | phthalan | 1142e | 53.7±0.4 [14] | 52.6 | 1.1 |
| 4265-25-2 | 2-methyl-benzofuran | 1158 | 53.3±1.5 [35] | 53.2 | 0.1 |
| 132-64-9 | dibenzofuran | 1537 | 66.3±0.6 [16] | 67.2 | -0.9 |
a Kovats indices from Table S8, column 5. b Experimental data evaluated in Table 5. c Calculated using equation (298.15 K) = 10.46 + 0.0369×Jx with R² = 0.9981, with the assessed expanded uncertainty of ±1.0 kJ.mol-1 (0.95 level of confidence, k = 2). d Difference between column 3 and 4 in this table. e From Table S5.
Table 5.
Compilation of the standard molar entropies of vaporisation, , and the absolute standard molar entropies, (g or liq), of HL and HR compounds. (All values at T = 298.15 K in J·K-1·mol-1).
Table 5.
Compilation of the standard molar entropies of vaporisation, , and the absolute standard molar entropies, (g or liq), of HL and HR compounds. (All values at T = 298.15 K in J·K-1·mol-1).
| Compound | a | (g) b | (liq) c | (liq) d |
|---|---|---|---|---|
| 2,3-benzofuran [271-89-6] | 111.3±0.1 [11] | 328.3 | 215.6 [36] | 217.0 |
| isobenzofuran [270-75-7] | 102.4±8.0 g | 330.7 | 228.3 | |
| 2,3-dihydro-benzofuran [496-16-2] | 119.2±3.0 | 344.1 | 226.4 [36] | 224.9 |
| 1,3-dihydro-isobenzofuran [496-14-0] | 119.2±0.1 [14] | 357.5 | 238.3 | |
| dibenzofuran [132-64-9] | 128.4±2.0 [16] | 384.0 | 248.5 e | 255.6 |
| fluorene [86-73-7] | 136.7±0.4 [37] | 392.0 | 257.8 f | 255.3 |
| 2-coumaranone [553-86-6] | 132.4±3.0 | 353.8 | 221.4 | |
| 3-coumaranone [7169-34-8] | 132.2±5.1 | 355.8 | 223.6 | |
| phthalide [87-41-2] | 144.5±0.2 | 353.6 | 209.1 | |
| furan [110-00-9] | 93.4±0.2 [38] | 272.7 | 176.7 [38] | 179.3 |
| 2,3-dihydro-furan [1191-99-7] | 95.8±0.1 [34] | 294.7 | 198.9 | |
| tetrahydrofuran [109-99-9] | 96.5±0.2 [39] | 301.7 | 203.9 [40] | 205.2 |
| 8H-benzofuran [13054-97-2] | 113.1±2.6 | 365.5 | 252.4 | |
| 8H-isobenzofuran [4743-54-8] | 110.7±1.4 | 364.2 | 253.5 | |
| 12H-dibenzofuran [13054-98-3] | 135.5±4.6 | 430.7 | 295.2 | |
| 12H-fluorene | 136.0±2.7 | 434.7 | 298.7 | |
| 6H-2-coumaranone [6051-03-2] | 127.6±5.6 | 373.9 | 246.3 | |
| 6H-3-coumaronone [1206676-08-5] | 119.8±2.4 | 377.9 | 258.1 | |
| 6H-phthalide [2611-01-0] | 162.5±2.9 | 374.1 | 211.6 | |
| 8H-benzofuran-2-ol [36871-99-5] | 152.0±8.0 g | 385.9 | 233.9 | |
| 8H-benzofuran-3-ol [99769-38-7] | 151.9±5.5 | 390.3 | 238.4 | |
| 8H-isobenzofuran-1-ol [59901-42-7] | 172.0±8.0 g | 387.8 | 215.8 | |
| 1,3-cyclopentadiene [542-92-7] | 274.5 [41] | 182.7 [40] | ||
| cyclopentene [142-29-0] | 289.7 [42] | 201.3 [43] | ||
| cyclopentane [287-92-3] | 292.9 | 204.1 [44] | ||
| indene [95-13-6] | 115.4±0.6 [13] | 335.9 | 214.2 [45] | 220.5 |
| indane [496-11-7] | 110.8±0.8 [13] | 345.9 | 234.4 [45] | 235.1 |
| trans-8H-indene [3296-50-2] | 106.2±0.1 | 371.6 | 258.9 [46] | 265.4 |
a From the vapor pressure values compiled in Table S4 or from the vapor pressure-temperature dependencies given in the corresponding original literature. b Calculated with the G3MP2 method. c The experimental data available in the literature. d Calculated according to Eq. (3) using the entries from columns 2 and 3 of this table. e Estimated based on the following experimental data from the literature: (liq) = (cr) + = 196.2 [15] + 52.3 [15] = 248.5 J·K-1·mol-1. f Estimated based on the following experimental data from the literature: (liq) = (cr) + = 207.3 [46] + 50.5 [46] = 257.8 J·K-1·mol-1. f The assessed value.
Table 6.
The liquid phase reaction enthalpies, , reaction entropies, , Gibbs energies, , and equilibrium temperatures, , of the full hydrogenation reactions of aromatic ethers given in Figure 2, (p°=0.1 MPa).
Table 6.
The liquid phase reaction enthalpies, , reaction entropies, , Gibbs energies, , and equilibrium temperatures, , of the full hydrogenation reactions of aromatic ethers given in Figure 2, (p°=0.1 MPa).
| compound | (298 K)/H2 d | (298 K)/H2 | (298 K)/H2 b | (500 K)/H2 b | (298 K) | (298 K) | (298 K) b | (500 K) b | |
|---|---|---|---|---|---|---|---|---|---|
| kJ·mol-1 | J·K-1·mol-1 | kJ·mol-1 | kJ·mol-1 | kJ·mol-1 | J·K-1·mol-1 | kJ·mol-1 | kJ·mol-1 | K | |
| (aliphatic)5 | |||||||||
| 1,3-cyclopentadiene | -105.5 | -120.0 | -69.7 | -44.2 | -211.0 | -240.0 | -139.4 | -88.4 | 879 |
| furan | -77.0 | -117.1 | -42.0 | -17.0 | -153.9 | -234.2 | -84.1 | -34.0 | 697 |
| (aromatic)6 + (aliphatic)5 |
|||||||||
| indene | -71.7 | -119.5 | -72.1 | -10.6 | -286.8 | -478.1 | -144.3 | -42.5 | 600 |
| 2,3-benzofuran | -62.1 | -121.5 | -25.8 | -0.1 | -248.2 | -486.0 | -103.3 | -0.5 | 511 |
| 2,3-dihydrobenzofuran | -61.1 | -122.0 | -24.7 | 1.2 | -183.2 | -366.1 | -74.0 | 3.5 | 500 |
| isobenzofuran | -76.1 | -124.4 | -39.0 | -16.0 | -304.3 | -497.6 | -155.9 | -63.9 | 612 |
| 1,3-dihydro-isobenzofuran | -63.3 | -125.6 | -25.9 | -2.6 | -190.0 | -376.9 | -77.6 | -7.7 | 504 |
| (aromatic)6 + (aliphatic)5 +(aromatic)6 |
|||||||||
| fluorene | -59.0 | -123.9 | -22.1 | 4.1 | -354.2 | -743.3 | -132.6 | 24.6 | 477 |
| dibenzofuran | -57.3 | -124.1 | -20.3 | 6.1 | -343.7 | -744.6 | -121.7 | 36.6 | 462 |
| (aromatic)6 + (lactone)5 | |||||||||
| indene | -71.7 | -119.5 | -72.1 | -10.6 | -286.8 | -478.1 | -144.3 | -42.5 | 600 |
| 2-coumaranone | -55.1 | -127.6 | -17.0 | 9.7 | -220.2 | -510.3 | -68.1 | 38.9 | 432 |
| 3-coumaranone | -59.9 | -127.0 | -22.0 | 4.9 | -239.4 | -508.0 | -87.9 | 19.5 | 471 |
| phthalide | -48.7 | -129.0 | -10.3 | 16.8 | -194.9 | -516.1 | -41.0 | 67.1 | 378 |
| open-chained ethers | |||||||||
| benzene | -68.5 | -120.3 | -32.6 | -7.0 | -205.4 | -360.9 | -97.8 | -21.1 | 569 |
| methoxy-benzene | -65.6 | -118.9 | -30.1 | -4.6 | -196.8 | -356.8 | -90.4 | -13.7 | 552 |
| diphenyl ether | -68.1 | -126.2 | -30.5 | -3.8 | -408.5 | -757.3 | -182.7 | -22.6 | 539 |
a Liquid-phase enthalpies of formation of of aromatic ethers and hydrocarbons (hydrogen-lean (HL) counterparts of the LOHC system) from Table S9. b Liquid-phase enthalpies of formation of perhydrogenated aromatic ethers and hydrocarbons (hydrogen-rich (HR) counterparts of the LOHC system from Table S10. c Calculated according to the Hess´s Law applied to the reactions of aromatic ethers given in Figure 2. d Reaction enthalpy per mole H2.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



