Submitted:
28 June 2025
Posted:
30 June 2025
You are already at the latest version
Abstract
Chronic kidney disease (CKD) has now reached epidemic proportions in many parts of the world primarily due to the high incidence of diabetes and hypertension. By 2040, CKD is predicted to be the fifth leading cause of years of life lost. Developing strategies to prevent CKD and to reduce its progression to kidney failure is thus of great public health significance. Hypertension is known to be both a cause and a consequence of kidney damage and an eminently modifiable risk factor. An increased risk for hypertension, especially among women, has been linked to chronic exposure to the ubiquitous food contaminant cadmium (Cd). The mechanism is unclear but is likely to involve its action on the proximal tubular cells (PTCs) of the kidney, where Cd accumulates. Here it leads to chronic tubular injury and a sustained drop in the estimated glomerular filtration rate (eGFR), a common sequela of ischemic acute tubular necrosis, acute and chronic tubulointerstitial inflammation, all of which hinders glomerular filtration. The present review discusses exposure levels of Cd that have been associated with an increased risk of hypertension, albuminuria, and eGFR ≤ 60 mL/min/1.73 m2 (low eGFR) in environmentally exposed people. It highlights the potential role for 20-hydroxyeicosatetraenoic acid (20-HETE), the second messenger produced in kidneys, as the contributing factor to gender differentiated effects of Cd-induced hypertension. Use of GFR loss and albumin excretion in toxicological risk calculation, and derivation of Cd exposure limits, instead of β2-microglobulin (β2M) excretion at a rate of 300 µg/g creatinine, are recommended.
Keywords:
albuminuria
; blood pressure
; cadmium
; glomerular filtration rate
; hypertension
; 20-hydroxyeicosatetraenoic acid
; kidney disease
1. Introduction
Exposure to the metal cadmium (Cd) is widespread globally because it is ubiquitously present as a contaminant in nearly all food types, notably the staple food group [1]. Over half of the world’s population consume rice as a staple food, which forms half or more of total oral Cd exposure levels in many regions. A recent finding that 15% of arable soils around the world is loaded with toxic metals, Cd included, raises concern on dietary Cd exposure guidelines and its permissible levels in foods [2]. The European Food Safety Authority (EFSA) set the maximum levels (ML) in rice at 0.2 mg/kg [3,4], while the Codex’s ML for Cd in rice is as high as 0.4 mg/kg dry grain weight [http://www.fao.org/fileadmin/user_upload/livestockgov/documents/1_CXS_193e.pdf] (accessed 20 June 2025).
Cd has a high toxicity potential; its electronegativity is close to zinc and its ionic radius is within the same range as calcium, thus it can enter the body through the metal transporters and pathways for essential metals iron [5], zinc [6], copper [7] and calcium [8,9]. Moreover, there is no excretory route for Cd [10,11]. Most acquired Cd are stored, especially by the proximal tubular cells (PTCs) of the kidney, and it appears in urine when these cells are injured or die [1,12]. Thus, Cd excretion can be viewed as a manifestation of current tissue injury and reduction of present and future exposure cannot mitigate injury in progress [1,12]. A half-life of Cd in the body ranges from 7.4 to 30 years; the lower the body burden, the longer the half-life of Cd [13,14,15].
Cells with high metabolic rates, like PTCs are particularly susceptible to Cd effects on mitochondria [16,17,18]. There, it reduces the synthesis of adenosine triphosphate (ATP), disrupts the electron transport chain, and promotes the formation of reactive oxygen species (ROS), leading to mitochondrial injury, and a release of mitochondrial DNA. In response, the nuclear factor-kappaB (NF-кB) and the DNA-sensing mechanism, cyclic GMP-AMP synthase–stimulator of interferon genes (cGAS-STING), signaling pathways are activated and proinflammatory cytokines are released, followed by premature cell death, inflammation and tubulointerstitial fibrosis [1]. Also, Cd cytotoxicity targets the nucleus, lysosome, and endoplasmic reticulum [19,20,21]. Many other molecular mechanisms have been postulated to explain the nephrotoxicity of Cd [22,23,24].
The present review is focused on hypertension in people chronically exposed to Cd, most likely in a normal diet It is aimed to illustrate the gender differentiated effects of Cd on blood pressure control by kidneys. It is hypothesized that the eicosanoid 20-hydroxyeicosatetraenoic acid (20-HETE), produced by PTCs, is a mediator of Cd-induced hypertension in female gender. Inverse relationships between estimated glomerular filtration rate (GFR) and blood pressure increment following exposure to Cd are presented. It highlights the potential mechanisms that Cd increases excretion of both albumin and β2-microglobulin (β2M), while causing a reduction in eGFR. Use of Cd-induced eGFR reduction, as opposed to β2M excretion at a rate above 300 µg/g creatinine, to define critical Cd exposure levels is discussed.
2. Food as a Modifiable Cd Exposure Route
For non-occupationally exposed populations, foods represent the main source of Cd. This section briefly discusses food safety monitoring programs, known as total diet studies and market basket surveys. The vulnerable groups of people who have enhanced Cd uptake rates are briefly discussed.
2.1. Exposure Limits
Reliable data on the concentration of Cd in various food items and estimated dietary exposure levels can be found in reports of total diet studies [25,26,27,28]. Because Cd is notoriously known for its toxicity to kidneys and bones, exposure limits, namely the tolerable intake level, the minimal risk level, the toxicological reference value (TRV), and the reference dose (RfD) have been determined for these targets [29]. Like the ML values of Cd in rice, there is no consensus on exposure limits nor thresholds among the existing exposure guidelines. Notably, however, most countries adopt Cd exposure at 0.83 μg/kg bw/day (58 μg/day for a 70 kg person), and Cd excretion rate at 5.24 µg/g creatinine as an exposure threshold level. These Cd exposure guidelines were determined by the Joint Food and Agriculture Organization and World Health Organization Expert Committee on Food Additives and Contaminants (JECFA) [https://apps.who.int/iris/handle/10665/44521] (accessed on 20 June 2025).
2.2. Populations At-Risk for Dietary Cd Exposure and Adverse Health Effects
Young children are at risk because enhanced intestinal absorption of metals, and a larger amount of food intake relative to body size and thus a larger dose of food-borne toxicants [30,31]. An excessive Cd exposure early in life has been evident from the U.S. study; the mean and 90th percentile Cd exposures for children 1–6 years, estimated from total diet study data from 2018 to 2020 and food consumption data from 2015 to 2018, were 0.43 and 0.71 μg/kg bw/day, respectively. The food groups contributing most to Cd exposure were grains/baking, dairy and fruit and grains/baking and vegetables, respectively [28]. These dietary Cd exposure levels exceeded the TRV for oral Cd exposure ranging between 0.21 and 0.36 μg/kg bw/day [32]. These dietary exposure levels, however, were within the JECFA’s acceptable exposure level of 0.83 μg/kg bw/day. The discrepancies in existing Cd exposure limits need to be addressed.
Excessive Cd exposures early in life may adversely affect the kidney. This has been observed in a prospective cohort study of rural Bangladeshi children, where Cd exposure was inversely associated with eGFR and reduced kidney volume, particularly in girls [33]. The eGFR values in Mexican children, aged 8–12 years, varied inversely with eGFR [34].
2.3. A Broad Range of Adverse Health Effects of Cd
Systematic reviews and meta-analyses suggested that risk for diabetes [35], cardiovascular disease (CVD) [36], chronic kidney disease (CKD) [37] and hypertension [38] rose with blood Cd levels and/or urinary Cd excretion rates, in a dose-dependent manner, as did mortality from any cause and CVD [39]. Furthermore, Dutch prospective cohort study has shown that Cd promoted the progressive deterioration of eGFR in patients with diabetics [40]. A fall of eGFR at a high rate in Swiss cohort participants has been linked with Cd exposure [41]. Notably, in the mentioned meta-analyses, effects of Cd were found to be independent of traditional risk factors, like adiposity. In a review of Cd and diabetes Cd exposure was inversely associated with BMI and other measures of obesity in children, adolescents, and adults [42].
It can be inferred from the above findings that a significant proportion of people has been affected from their exposure to Cd in a normal diet. This calls into questions whether safe exposure levels exist for a cumulative toxicant like Cd. The existing permissible dietary exposure levels and exposure thresholds were outdated as was the utility of excretion of β2-microglobulin (β2M) at a rate of 300 µg/g creatinine, termed, β2-microglobulinuria (β2M-uria), as an indicator of the nephrotoxicity of Cd [29]. A further discussion on use of eGFR decline instead of β2M-uria in determining critical exposure level for Cd is provided in Section 5.
3. Cd Exposure and Hypertension
This section describes definition, and prevalence of hypertension together with CKD diagnosis and staging and the cardiovascular–kidney–metabolic (CKM) syndrome, proposed by the American Heart Association in viewing of the high prevalence of metabolic and kidney disease. Cd exposure levels that may increase risk for hypertension are summarized. High blood pressure as a mediator of Cd-induced albuminuria is discussed.
3.1. Hypertension Prevalence
Hypertension is defined as systolic blood pressure (SBP) and/or diastolic blood pressure (DBP) ≥ 140/90 mm Hg [43,44]. In most economically developed countries, at least, one third of their adult population live with primary or essential hypertension with notable gender differences in incidence and treatment outcomes [45,46].
In the U.S. prospective cohort study, the Health and Retirement Study (1992−2014), hypertension occurred in 41% of cohort participants over a median observation period of 7.8 years, and an estimated incident hypertension was 45.3 per 1000 person-years [47]. A cross-sectional study from France (the CONSTANCES study) reported a higher prevalence of hypertension in men (37.3%) than women (23.2%) with body mass index (BMI) being a strong determinant, especially in women [48]. In rural Bangladesh, hypertension was found to be more prevalent in women (8.9%) than men (4.5%), and the highest prevalence (21.3%) was found in women aged ≥ 60 years [49]. Risk factors for hypertension in rural residents of Bangladesh included age, education, current tobacco use, BMI, and hyperglycemia [49].
3.2. Resistance Hypertension: An Emerging Challenge
Use of antihypertensive medications, which include diuretics, dihydropyridine calcium channel blockers and renin-angiotensin system inhibitors fails to achieve target blood pressure control in 10-15% of hypertensive patients [50,51]. The condition is designated as resistant hypertension. Risk factors for resistant hypertension are age, high blood pressure, obesity, salt consumption, chronic kidney disease (CKD), and diabetes mellitus (DM) [50,51]. Notably, hypertension and DM are leading causes of end-stage kidney disease and kidney damage is a pathological sign common among those with hypertension and DM. Those who had hypertension onset at <35, <45 and ≥ 45 years of age had respectively, 2.52-fold, 1.59- fold, and 1.54-fold higher odds for CKD [52].
As enlisted in Table 1, the diagnosis and staging of CKD are based on eGFR reduction and albuminuria [53].
Lower eGFR and higher albumin excretion rates both are predictors of death from any cause and CVD, independent of each other and of traditional risk factors for CVD [54]. Because of the high prevalence of metabolic and kidney disease, the American Heart Association has put forward the CKM syndrome, and sex-specific, race-free risk equations: PREVENT (AHA Predicting Risk of CVD Events) [55,56,57].
3.3. Cd as a Risk Factor for Hypertension and CKD: Epidemiolical Data
Table 2 provides Cd exposure levels that have been found to be associated with risk for hypertension and other adverse effects in the environmentally exposed populations.
In a repeated measurements of exposure to metals and kidney injury biomarkers [62], Yin et al. (2024) applied the pathway enrichment analysis to identify the molecular entities that may account for an association between an increased risk for CKD and exposure to toxic metals; Cd, Cr and Pb. They connected metal-induced CKD onset to the oxidative stress pathway and a decrease in the expression of the transcription factor NFE2 like BZIP transcription factor 2 (NFE2L2), also known as nuclear recetor factor 2 (NRF2).
3.4. Albuminuria in Cd-Exposed People
3.4.1. Tubular Handling of Albumin
Albumin is synthesized in the liver and secreted into the circulation 10–15 g per day [65,66]. It undergoes catabolism in muscle, the liver, and kidneys, which balances synthesis, and homeostasis is maintained. Normal plasma albumin concentration is between 3.5 g/dL and 5 g/dL, and half-life in plasma is 19 days [65,66]. With a molecular weight of 66 kDa and negative charge, albumin is not filtered by glomeruli. By means of transcytosis through endothelial cells and podocyte foot processes, 1–10 g of albumin enters the urinary space each day [65,66,67,68].
Most albumin that reaches tubular lumen is reabsorbed via the high-capacity fluid phase endocytosis and is returned to blood supply via transcytosis [67,68,69]. This albumin reuptake mechanism occurs in S1, S2 and S3 segments of tubules. A relatively small proportion of albumin is reabsorbed via the high affinity, low-capacity (megalin/cubilin) mediated endocytosis, followed by lysosomal degradation [67,68,69]. This mechanism occurs in S1 segment of tubules. Reuptake of albumin and β2M through megalin/cubilin-mediated endocytosis, expressed on the apical membrane of PTCs, is depicted in Figure 1.
Given that β2M-uria is used as a criterion to judge the toxicity of Cd, the most frequently reported sign of the nephrotoxicity of Cd is β2M-uria, detailed in Section 5. The pathogenesis of albuminuria in Cd-exposed people has rarely been investigated. Herein, studies examining albumin excretion in two Cd exposure scenarios are recapitulated.
3.4.2. Moderate-to-High Exposure to Cd
For 519 study subjects with low-to-moderate exposure (geometric mean for Cd excretion at a rate of 5.44 µg/g creatinine), estimated fractional reductions in albumin and β2M reabsorption were 18 and 21%, assuming the glomerular sieving coefficients for albumin and β2M of 10−4 and 0.01, respectively [70]. Cd may adversely, affect megalin, a component involved in the uptake of both albumin and β2M [70,71]. In theory, change in fractional clearance of any filtered protein is a result of filtration and reuptake [69].
As Figure 1 depicts, “toxic” unbound Cd can be released as albumin is degraded. Free (unbound) Cd can reach the inner membrane of mitochondria [72,73], where it causes excessive ROS production, membrane lipid peroxidation, and mitochondrial DNA leakage. As a result, the DNA-sensing mechanism (cGAS-STING) and NF-кB signaling pathways are stimulated and proinflammatory cytokines are released, and followed by cell death.
Initially, the stimulator of interferon genes (STING) was found to mediate innate immunity via a DNA-sensing pathway. Its involvement in ferroptosis-induced tubular cell death was demonstrated using an experimental model of ischemic kidney injury [74]. Intriguingly, the potential contribution of the immune system to modulate blood pressure variability and renal injury has emerged [75] as is the role for proximal tubule endocytosis in renal fibrosis development [76].
3.4.3. Low-to-Moderate Exposure to Cd
Among 641 study subjects with low-to-moderate exposure, evident from geometric mean for Cd excretion of 1.11 µg/g creatinine, hypertension was the most prevalent (39.8%), followed by albuminuria (16.5%) and a low eGFR (4.8%) [77]. It is inferred from a simple mediation model analysis that albumin excretion rate in this group is causally related to an increase in SBP, which accompanies Cd-induced eGFR reductions. Direct relationship between albumin excretion rate and blood pressures is shown in Figure 2.
Albumin excretion rate varied directly with SBP and DBP (Figure 2a, b). Those with SBP ≥ 130 mm Hg plus low eGFR had 17.2% and 21.2% higher albumin excretion rates, compared to those with eGFR 61–90 and ≥ 90 mL/min/1.73 m2, respectively (Figure 2c). Similarly, those with high DBP and low eGFR had 19.0% and 22.5% higher albumin excretion rates, compared to those with eGFR 61–90 and ≥ 90 mL/min/1.73 m2, respectively (Figure 2d). Thus, there was an increase in albumin excretion in those with low eGFR without hypertension (SBP/DBP of 130/80 mm Hg).
The above observations were in line with data from the SPRINT Trial, where participants who had low eGFR and 16 mm Hg higher SBP than mean SBP were found to have an albumin excretion rate rise by 16% [78]. Proteinuria/albuminuria are predictors of continued progressive decline of eGFR [79,80,81,82].
3.5. The Kidney and Gender Differences in Cd-Induced Hypertension
3.5.1. Cd and Kidney’s Role in Blood Pressure Regulation
The essential role of the kidney in long-term blood pressure control was first demonstrated in 1970s, when the transplant of a kidney from a hypertensive rat to a normotensive host raised blood pressure, and the transplant of a normal kidney to hypertensive host lowered blood pressure [83,84,85]. The close relationships between renal perfusion pressure, urine flow and sodium excretion led to the Guyton’s theory that hypertension develops when the pressure-natriuresis response is shifted to higher pressure levels [86]. Thus, the regulation of sodium transport in kidneys by blood pressure is central to Guyton's model of blood pressure control [87]. Many mediators of the pressure-natriuresis response have been investigated such as 20-HETE [88,89], renal sympathetic activity [90], and the renin-angiotensin-aldosterone system (RAAS) [91].
Herein, role of 20-HETE in regulating salt balance in the kidney is discussed together with its potential involvement in Cd-induced hypertension. This eicosanoid is produced from arachidonic acid via ɷ-hydroxylation reaction, catalyzed by CYP4A11 and CYP4F2 enzymes [92]. Evidence that links these enzymes to hypertension comes from human gene mutation research and sodium retention and hypertension development in Dahl salt-sensitive rats is caused by 20-HETE deficiency [88].
Below is a hypothetical model for Cd-intoxicated kidney tubular cell (Figure 3).
3.5.2. The Increment of Tubular Avidity for Filtered Sodium After Cd Exposure
In 1990’s, the synthesis and excretion of 20-HETE by human kidneys were observed [95,96]. Later, CYP4F2 and CYP4A11 were found to be responsible for the synthesis of 20-HETEin the kidneys from arachidonic acid [90]. CYP4F2 was found to be more prominently expressed in glomeruli and proximal tubular cells than the distal tubules [97]. In comparison, CYP4A11 was faintly expressed in the proximal and distal tubules, but not in glomeruli. Notably, the zinc influx transporter, ZIP8, known to mediate Cd uptake, was more prominently expressed in the distal than the proximal tubules, but not in the glomeruli [98]. The Na/K-ATPase is expressed in the distal tubule DT in the highest density, especially in the basolateral membrane
The activity of Na/K-ATPase pump was highly sensitive to oxidative damage [94,99], and the oxidized Na/K-ATPase is subjected to degradation by the proteasome and endo-lysosomal protease [100]. This decreases cellular Na/K-ATPase content and sodium transport activity. In the cortical proximal tubular cell, where relatively higher Cd accumulation levels than the distal tubules, an increased CYP4F2 expression was noted [101,102,103]. However, because of low abundance of Na/K-ATPase due to Cd-induced oxidative damage, the ability for 20-HETE to inhibit salt re-absorption is impaired.
Low CYP4A11 protein expression in the distal tubules in those with high Cd body burden, as suggested by an inverse correlation between kidney Cd levels and CYP4A11 protein abundance [103] suggests that only low-level 20-HETE production in this nephron region, where 20-HETE reduces sodium reabsorption through inhibition of Na+-K+-2Cl− and 70pS K+ transport. Low 20-HETE levels in the distal tubule, thus, lead to a greater re-uptake of salt and water into systemic circulation. Hypertension develops after prolonged volume overload, increasing sodium retention and shifting the natriuretic response to higher pressures. Rats with Cd-induced hypertension showed increased sodium retention and reduced sodium excretion [104,105].
3.5.3. Gender Differences in Cd-Induced Hypertension
The female preponderance of Cd-associated hypertension has been observed across populations, and it has been attributed to sex hormones. Japanese and Swedish women studies showed that serum estradiol levels inversely associated with Cd excretion rate [106,107]. Another study on postmenopausal Japanese women observed a 28% increase in serum testosterone, when Cd excretion rates rose with Cd excretion rate [108].
In U.S. NHANES 2013-2016 dataset, hyperandrogenemia was associated with exposure to Cd and/or Pb in women aged ≥ 35 years [109]. Hyperandrogenemia was defined by free androgen index, the ratio of total testosterone to sex hormone-binding globulin ≥ 5 %. A 20% rise in risk for hyperandrogenemia was noted, comparing blood Cd in the top to the bottom tertile, was found to be associated with exposure to Cd. Inflammation associated with exposure to Cd was suggested to mediate the effect observed.
Clinical and experimental data suggested 20-HETE to mediate androgen effect on blood pressure [109,110,111,112]. The potential role for 20-HETE in hypertension comes from a study on Thai women, where there was a positive correlation between SBP and urine 20-HETE [113]. A rise of 20-HETE excretion from the bottom to the top tertile was associated with 6 mm Hg higher SBP. Furthermore, urinary 20-HETE concentrations ≥ 469 pg/mL were associated with a 1.9-fold, 4.36-fold and 1.53-fold increases in odds for hypertension. However, due to a cross-sectional study design, further research is required to examine whether 20-HETE is a cause or a consequence of hypertension development following Cd exposure.
4. Critical Exposure Levels of Cd
This section highlights the critical Cd excretion rate, obtained by applying advanced benchmark dose (BMD) methodology. Special emphasis is given to the BMD modeling of dose-effect datasets from population samples which could be the representative of the general populations with low environmental Cd exposures. It recapitulates results from a recent investigation, suggesting the use of eGFR, as opposed to β2M excretion rate, in Cd toxicological risk assessment. Also, it reiterates the imprecisions in measuring Cd exposure and its associated adverse effects that can obscure the relationship between eGFR and Cd excretion. In some instance an effect of Cd is nullified, the phenomena described in the literature as “reverse causality”.
4.1. Benchmark Dose Limit (BMDL)
The benchmark dose (BMD) of any exposure indicator is estimated through fitting the entire exposure–effect dataset to mathematical dose-response models with a pre-defined specific effect size, referred to as the benchmark response (BMR) [114]. Difference between the lower bound (BMDL) and upper bound (BMDU) of the 95% confidence interval (CI) of BMD reflects the statistical uncertainties in the BMD estimates. A narrow difference indicates a high degree of certainty of estimated figures. The BMD estimates deem unreliable when BMDU/BMDL ratio ≥ 100.
Assessment of relative goodness of fit of different models is through the Akaike information criterion (AIC). Relative model weights provide an additional insight into the shape and steepness of the slope [115,116]. The mathematical dose–response models often used are inverse exponential, natural logarithmic, exponential, and Hill models [117,118].
The benchmark dose limit (BMDL) value of any exposure indicator is referred to as the lower 95% confidence bound of BMD, computed at 5% BMR. This BMDL value has become a replacement of the no-observed-adverse-effect level (NOAEL) and it can represent a critical exposure level [114].
4.2. JECFA and EFSA Dietary Cd Exposure Guidelines and Thresholds
Assuming lifetime exposure of 2 g of Cd and β2M excretion at a rate above 300 µg/g creatinine as a critical effect, JECFA found a Cd exposure level of 0.83 µg/kg bw/day to be the exposure level carrying an unappreciable health risk, and Cd excretion rate at 5.24 µg/g creatinine was designated as Cd exposure threshold. With an application of the same β2M endpoint, the European Food Safety Authority (EFSA) found dietary Cd exposure at 0.36 μg/kg bw/day to be the reference dose (RfD) and Cd excretion rate at 1 µg/g creatinine was assigned as the threshold level after a safety margin was included to account for interindividual differences in Cd exposure levels [3,4].
The RfD value for human Cd exposure, estimated from a conventional dosing experiment was 0.2 μg/kg bw/day [119]. This empirical Cd exposure limit is lower than the figures derived by JECFA and EFSA. In such a conventional study, pigs were fed with four dose levels of Cd; 0, 0.5, 2, 8, and 32 mg Cd/kg feed, for 100 days [119], and it was found that a Cd dose producing abnormal β2M excretion was the highest, abnormal excretion of retinol binding protein (RBP) occurred at the lowest feeding dose. These findings indicate β2M excretion was not an early warning sign of Cd nephrotoxicity and its use as a basis to estimate exposure guideline represents a conceptual flaw.
4.3. Falling eGFR as an Early Warning Sign of Cd Nephrotoxicity
Although a declining eGFR is clinically relevant (Table 1), this eGFR endpoint has rarely been used in evaluating Cd effects on kidney. A dearth of research into the eGFR and albuminuria endpoints could be attributed a non-association between Cd exposure and eGFR reported in two meta-analyses, leading to an erroneous conclusion that Cd had no effect on eGFR nor were there progressive reductions in eGFR toward kidney failure among Cd-exposed people [120,121].
Table 3 provide results of logistic regression analyses, evaluating effects of doubling Cd excretion rate on the prevalence odds for CKD along with other independent variables.
All independent variables incorporated in models A and B were identical with an exception for Cd excretion rates. In model A, Cd excretion (ECd) was normalized to creatinine excretion (Ecr), denoted as as ECd/Ecr. This practice is to correct for different urine volumes or dilutions among study subjects in case of a single time (spot) urine sampling. In model B, ECd was normalized to creatinine clearance (Ccr), denoted as ECd/Ccr. This Ccr normalization corrects for differences in urine dilution and number of functioning nephrons, simultaneously.
In model A, age, BMI ≥ 24 kg/m2 and ECd/Ecr were associated with increased odds for CKD. There was a 1.98-fold increase in CKD risk per doubling ECd/Ecr. In model B, hypertension was found to be associated risk for CKD along with age, BMI ≥ 24 kg/m2 and ECd/Ccr. Odds for CKD rose 3.13-fold and 2.6-fold per doubling ECd/Ccr in those who had hypertension.
In summary, an effect size of Cd on risk for CKD was reduced by 63%, compared to Ccr-normalized dataset. An effect of hypertension on risk for CKD was completely obscure in Ecr-adjusted dataset. These data clearly demonstrate an impact of Ecr-adjustment. Ecr is influenced by muscle mass which vary among people and gender (universally higher in males than females). Adjusting ECd with Ecr adds variance to the data sets which can diminish or nullify an effect size [123]. Normalizing ECd to Ccr corrects for differences in urine dilution and number of functioning nephrons, simultaneously and it is unaffected by muscle mass [124]. Ccr-normalization is computed by an equation, which does not require timed urine collection [124].
4.4. Use of β2M Excretion as Indicator of Cd Effect on Proximal Tubules?
The protein β2M is the light-chain component of class I histocompatibility complexes, expressed on the surface of most nucleated cells and is released into blood circulation [125,126]. With the molecular weight of 11,800 Daltons, β2M is filtered totally with plasma by the glomeruli, reabsorbed by PTCs in S1 segment of tubules through the endocytosis, mediated by megalin/cubilin receptor system, and is degraded in the lysosome (Figure 4). Unlike albumin, which is taken up, and returned to the circulation, there is little evidence that retrieved β2M is re-entered blood supply.
Excretion of proteins with low-molecular weights, like β2M, α1-microglobulin (α1M), and retinol binding protein (RBP) have long been used as a sign of Cd-induced tubular dysfunction [127]. Such use relies on the promise that they are completely filtered by glomeruli and reuptake by kidney tubules. In theory, excretion of these proteins is a function of their production, glomerular filtration, re-uptake and degradation by PTCs.
A thorough dissection of the determinants of β2M homeostasis has shown for the first time that fractional tubular degradation of filtered β2M (FrTDβ2M) should be used to assess the impact of Cd on PTCs [128]. There was a large interindividual variation in the flux of β2M from cells into plasma as such β2M excretion was minimally related to tubular degradation of β2M, and it could not a reliable measure of proximal tubular re-absorptive function [128].
The fact that excretion of β2M, especially among those with low-dose exposures does not reflect Cd effect on PTCs is evident from BMD modeling of data from 799 participants of Thai cohort of non-diabetics, mean age 49.2 years, (range: 18-87) [122]. Based in the geometric mean of Cd excretion of 2.15 µg/g creatinine (1.82 µg/L filtrate), this group can be considered to represent low-to-moderate exposure observed in most populations. The BMDL values of Cd excretion were estimated from changes in eGFR concurrently with changes in β2M excretion rate. The BMR was set at 5% for both endpoints.
Figure 5 provides results of correlation analysis, while Figure 6 shows outputs from the PROAST software for BMD computation (https://proastweb.rivm.nl) (accessed on 25 June 2025).
As Figure 5 depicts, strength of the correlation of eGFR vs. Cd excretion rate and β2M excretion rate vs. eGFR were higher when ECd and Eβ2M were normalized to Ccr, compared to Ecr-adjusted datasets.
As Figure 6 depicts, Cd excretion at a rate of 0.17 µg/g creatinine was found to be a critical Cd exposure level (BMDL), when a decrease in eGFR was a toxic endpoint. The BMDU/BMDL ratio for the eGFR endpoint was 16.9, meaning the high degree of certainty of the estimated BMDL value. However, when an increase in β2M excretion was used as an endpoint, the BMDL value of Cd excretion rate could not be determined because of a high degree of uncertainty around the BMD estimates (BMDU/BMDL= 182).
This finding on β2M excretion endpoint can be interpreted to suggest that an increase in excretion of β2M in people exposed to a low-dose Cd is minimally related to Cd excretion. It appears, therefore that there is little basis to use β2M excretion to derive exposure guidelines. The β2M-nuria, defined as β2M excretion at a rate of 300 µg/g creatinine observed most frequently at Cd excretion rates higher than 1 µg/g creatinine and eGFR ≤ 60 mL/min/1.73 m2 is a severe consequence of nephron destruction attributable to the nephrotoxicity of Cd and hypertension (Table 3). Cd excretion rate as little as 0.17 µg/g creatinine could serve as a basis to compute an exposure level below which kidney functional integrity is preserved.
A small increase in excretion of β2M has been found to be associated with increased risk of hypertension independently of albuminuria in the general Japanese population [129,130]. Two studies conducted on Cd-exposed subjects suggested β2M excretion to be a risk factor for hypertension, especially among those with diabetes [131,132]. These findings are consistent with a notion that β2M may be involved in blood pressure control [133,134,135]. An elevated plasma β2M has been found to be associated with increases in prevalent and incident hypertension in the Framingham Heart Study [135].
5. Conclusions
The prevalence of people living with hypertension, diabetes type 2, CVD, CKD, and death from CVD all increased with blood Cd concentration and/or excretion rate of Cd, indicators of the presence of Cd in the human body [35,36,37,38,39]. These disease prevalence increments are independent of the traditional risk factors, obesity, and tobacco consumption. These data together with current scientific understanding of the nephrotoxicity of Cd at cellular and molecular levels are indisputable. Exposure to Cd at the levels presently found in a normal diet has posed significant public health threat and toxic levels of Cd can be found in a significant proportion of the general population. Notably, however, the impact of dietary Cd exposure on health has been vastly underappreciated. Now is the time to recognize the health threat of Cd and the need to address key issues of toxic risk assessment criteria.
CKD in its early stage is largely asymptomatic. This makes its early detection difficult and the initiation of early treatment, which can significantly prevent progression to kidney failure, limited. In low-to-moderate Cd exposure scenarios, albuminuria is at least 3-time more prevalent than a low eGFR. Thus, it is suggested that an increase in albumin excretion can be used to find those with early CKD. Proteinuria also can be used for such purposes because the BMDL value of Cd excretion is as little as 0.05 µg/g creatinine [136].
ACR within the normal range was independently associated with a higher risk of hypertension in the general population [137]. Comparing the lowest category of ACR, the risk was increases 1.75-folld in the highest ACR group. Hypertension risk associated with ACR increment was particularly high in women; the risk for hypertension was 2.47-fold higher, compared to those with lowest ACR [137] An ACR as low as 7 mg/g creatinine was a predictor of incident CKD within 10 years [138]. An ACR ≥10 mg/g creatinine may increase mortality from all causes and CVD [139].
For the eGFR endpoint, the BMDL value of Cd excretion rate is 0.17 µg/g creatinine. In contrast, the BMDL value of Cd excretion rate could not be identified for the β2M endpoint. A large interindividual in plasma β2M concentrations can explain the failure of such attempt to define the BMDL value of Cd excretion based on the β2M endpoint (Figure 6). Given that a declining eGFR is clinically relevant, the Cd excretion at a rate of 0.17 µg/g creatinine could serve as a basis to compute a meaningful exposure limit for Cd. This figure is 3.2 % of the Cd exposure threshold level at 5.24 µg/g creatinine, the JECFA derived from the β2-microglobulinuria endpoint.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions are included in this review. Further inquiries can be directed to the author.
Acknowledgments
The author thanks David A. Vesey for his assistance with access to the literature and administrative supports. The author also thanks Aleksandar Cirovic for his assistance with designing Figures 1,3 and 4. The work was supported with resources from the Centre for Kidney Disease Research, Translational Research Institute, and the Department of Kidney and Transplant Services, Princess Alexandra Hospital, QLD, Australia.
Conflicts of Interest
The author declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| ACR | albumin-to-creatinine ratio |
| β2M | β2-microglobulin |
| BMDL | Benchmark dose limit |
| BMR | Benchmark dose response |
| Cd | Cadmium |
| Cr | Chromium |
| CKM | Cardiovascular–kidney–metabolic syndrome |
| Ccr | Creatinine clearance |
| CYP4A11 | Cytochrome P450 4A11 |
| CYP4F2 | Cytochrome P450 4F2 |
| cGAS-STING | cyclic GMP-AMP synthase–stimulator of interferon genes |
| DMT1 | Divalent metal transporter1 |
| EFSA | European Food Safety Authority |
| eGFR | estimated glomerular filtration rate |
| JECFA | Joint Food and Agriculture Organization and World Health Organization Expert Committee on Food Additives and Contaminants |
| 20-HETE | 20-hydroxyeicosatetraenoic acid |
| NAG | N-acetyl-β-D-glucosaminidase |
| NFE2L2 | NFE2 like BZIP transcription factor 2 |
| NF-кB | nuclear factor-kappaB |
| TRV | Toxicological Reference Value |
| ZIP8 | Zrt- and Irt-related protein 8 |
References
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Phelps, K.R. Estimation of health risks associated with dietary cadmium exposure. Arch. Toxicol. 2023, 97, 329–358. [Google Scholar] [CrossRef]
- Hou, D.; Jia, X.; Wang, L.; McGrath, S.P.; Zhu, Y.G.; Hu, Q.; Zhao, F.J.; Bank, M.S.; O'Connor, D.; Nriagu, J. Global soil pollution by toxic metals threatens agriculture and human health. Science 2025, 388, 316–321. [Google Scholar] [CrossRef]
- European Food Safety Authority (EFSA). Scientific opinion on cadmium in food. EFSA J. 2009, 980, 1–139. [Google Scholar]
- European Food Safety Authority (EFSA). Statement on tolerable weekly intake for cadmium. EFSA J. 2011, 9, 1975. [Google Scholar]
- Garrick, M.D.; Dolan, K.G.; Horbinski, C.; Ghio, A.J.; Higgins, D.; Porubcin, M.; Moore, E.G.; Hainsworth, L.N.; Umbreit, J.N.; Conrad, M.E.; et al. DMT1: A mammalian transporter for multiple metals. Biometals 2003, 16, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Aydemir, T.B.; Cousins, R.J. The multiple faces of the metal transporter ZIP14 (SLC39A14). J. Nutr. 2018, 148, 174–184. [Google Scholar] [CrossRef]
- Ohta, H.; Ohba, K. Involvement of metal transporters in the intestinal uptake of cadmium. J. Toxicol. Sci. 2020, 45, 539–548. [Google Scholar] [CrossRef]
- Kovacs, G.; Danko, T.; Bergeron, M.J.; Balazs, B.; Suzuki, Y.; Zsembery, A.; Hediger, M.A. Heavy metal cations permeate the TRPV6 epithelial cation channel. Cell Calcium 2011, 49, 43–55. [Google Scholar] [CrossRef]
- Kovacs, G.; Montalbetti, N.; Franz, M.C.; Graeter, S.; Simonin, A.; Hediger, M.A. Human TRPV5 and TRPV6: Key players in cadmium and zinc toxicity. Cell Calcium 2013, 54, 276–286. [Google Scholar] [CrossRef]
- Mitchell, C.J.; Shawki, A.; Ganz, T.; Nemeth, E.; Mackenzie, B. Functional properties of human ferroportin, a cellular iron exporter reactive also with cobalt and zinc. Am. J. Physiol. Cell Physiol. 2014, 306, C450–C459. [Google Scholar] [CrossRef]
- Hoch, E.; Lin, W.; Chai, J.; Hershfinkel, M.; Fu, D.; Sekler, I. Histidine pairing at the metal transport site of mammalian ZnT transporters controls Zn2+ over Cd2+ selectivity. Proc. Natl. Acad. Sci. USA 2012, 109, 7202–7207. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Vesey, D.A.; Ruangyuttikarn, W.; Nishijo, M.; Gobe, G.C.; Phelps, K.R. The Source and Pathophysiologic Significance of Excreted Cadmium. Toxics 2019, 7, 55. [Google Scholar] [CrossRef]
- Elinder, C.G.; Lind, B.; Kjellström, T.; Linnman, L.; Friberg, L. Cadmium in kidney cortex, liver, and pancreas from Swedish autopsies. Estimation of biological half time in kidney cortex, considering calorie intake and smoking habits. Arch. Environ. Health 1976, 31, 292–302. [Google Scholar] [CrossRef]
- Suwazono, Y.; Kido, T.; Nakagawa, H.; Nishijo, M.; Honda, R.; Kobayashi, E.; Dochi, M.; Nogawa, K. Biological half-life of cadmium in the urine of inhabitants after cessation of cadmium exposure. Biomarkers 2009, 14, 77–81. [Google Scholar] [CrossRef]
- Ishizaki, M.; Suwazono, Y.; Kido, T.; Nishijo, M.; Honda, R.; Kobayashi, E.; Nogawa, K.; Nakagawa, H. Estimation of biological half-life of urinary cadmium in inhabitants after cessation of environmental cadmium pollution using a mixed linear model. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2015, 32, 1273–1276. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: Physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef] [PubMed]
- Branca, J.J.V.; Pacini, A.; Gulisano, M.; Taddei, N.; Fiorillo, C.; Becatti, M. Cadmium-induced cytotoxicity: Effects on mitochondrial electron transport chain. Front. Cell Dev. Biol. 2020, 8, 604377. [Google Scholar] [CrossRef]
- Lv, Y.T.; Liu, T.B.; Li, Y.; Wang, Z.Y.; Lian, C.Y.; Wang, L. HO-1 activation contributes to cadmium-induced ferroptosis in renal tubular epithelial cells via increasing the labile iron pool and promoting mitochondrial ROS generation. Chem Biol Interact. 2024, 399, 111152. [Google Scholar] [CrossRef]
- Lee, W.K.; Thévenod, F. Cell organelles as targets of mammalian cadmium toxicity. Arch. Toxicol. 2020, 94, 1017–1049. [Google Scholar] [CrossRef]
- Li, K.; Guo, C.; Ruan, J.; Ning, B.; Wong, C.K.-C.; Shi, H.; Gu, J. Cadmium disrupted ER Ca2+ homeostasis by inhibiting SERCA2 expression and activity to induce apoptosis in renal proximal tubular cells. Int. J. Mol. Sci. 2023, 24, 5979. [Google Scholar] [CrossRef]
- Liu, F.; Li, Z.F.; Wang, Z.Y.; Wang, L. Role of subcellular calcium redistribution in regulating apoptosis and autophagy in cadmium-exposed primary rat proximal tubular cells. J. Inorg. Biochem. 2016, 164, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Dong, P.F.; Liu, T.B.; Chen, K.; Li, D.; Li, Y.; Lian, C.Y.; Wang, Z.Y.; Wang, L. Cadmium targeting transcription factor EB to inhibit autophagy-lysosome function contributes to acute kidney injury. J. Adv Res. 2025, 72, 653–669. [Google Scholar] [CrossRef]
- Guo, Y.Y.; Liang, N.N.; Zhang, X.Y.; Ren, Y.H.; Wu, W.Z.; Liu, Z.B.; He, Y.Z.; Zhang, Y.H.; Huang, Y.C.; Zhang, T.; et al. Mitochondrial GPX4 acetylation is involved in cadmium-induced renal cell ferroptosis. Redox Biol. 2024, 73, 103179. [Google Scholar] [CrossRef]
- Zhang, K.; Long, M.; Dong, W.; Li, J.; Wang, X.; Liu, W.; Huang, Q.; Ping, Y.; Zou, H.; Song, R.; et al. Cadmium Induces Kidney Iron Deficiency and Chronic Kidney Injury by Interfering with the Iron Metabolism in Rats. Int. J. Mol. Sci. 2024, 25, 763. [Google Scholar] [CrossRef] [PubMed]
- Suomi, J.; Valsta, L.; Tuominen, P. Dietary Heavy Metal Exposure among Finnish Adults in 2007 and in 2012. Int. J. Environ. Res. Public Health. 2021, 18, 10581. [Google Scholar] [CrossRef]
- Watanabe, T.; Kataoka, Y.; Hayashi, K.; Matsuda, R.; Uneyama, C. Dietary exposure of the Japanese general population to elements: Total diet study 2013–2018. Food Saf. 2022, 10, 83–101. [Google Scholar] [CrossRef]
- Vasco, E.; Dias, M.G.; Oliveira, L. The first harmonised total diet study in Portugal: Arsenic, cadmium and lead exposure assessment. Chemosphere 2025, 372, 144003. [Google Scholar] [CrossRef] [PubMed]
- Hoffman-Pennesi, D.; Winfield, S.; Gavelek, A.; Santillana Farakos, S.M.; Spungen, J. Infants’ and young children's dietary exposures to lead and cadmium: FDA total diet study 2018-2020. Food Addit. Contam. Part A Chem. Anal. Control. Expo. Risk Assess. 2024, 41, 1454–1479. [Google Scholar] [CrossRef]
- Satarug, S. Environmental Cadmium Exposure Limits and Thresholds for Adverse Effects on Kidneys. J. Environ. Expose. Assess. [CrossRef]
- Vogt, R.; Bennett, D.; Cassady, D.; Frost, J.; Ritz, B.; Hertz-Picciotto, I. Cancer and non-cancer health effects from food contaminant exposures for children and adults in California: a risk assessment. Environ Health 2012, 11, 83. [Google Scholar] [CrossRef]
- Flannery, B.M.; Schaefer, H.R.; Middleton, K.B. A scoping review of infant and children health effects associated with cadmium exposure. Regul. Toxicol. Pharmacol. 2022, 131, 105155. [Google Scholar] [CrossRef]
- Schaefer, H.R.; Flannery, B.M.; Crosby, L.M.; Pouillot, R.; Farakos, S.M.S.; Van Doren, J.M.; Dennis, S.; Fitzpatrick, S.; Middleton, K. Reassessment of the cadmium toxicological reference value for use in human health assessments of foods. Regul. Toxicol. Pharmacol. 2023, 144, 105487. [Google Scholar] [CrossRef] [PubMed]
- Skröder, H.; Hawkesworth, S.; Kippler, M.; El Arifeen, S.; Wagatsuma, Y.; Moore, S.E.; Vahter, M. Kidney function and blood pressure in preschool-aged children exposed to cadmium and arsenic--potential alleviation by selenium. Environ. Res. 2015, 140, 205–213. [Google Scholar] [CrossRef]
- Rodríguez-López, E.; Tamayo-Ortiz, M.; Ariza, A.C.; Ortiz-Panozo, E.; Deierlein, A.L.; Pantic, I.; Tolentino, M.C.; Estra-da-Gutiérrez, G.; Parra-Hernández, S.; Espejel-Núñez, A.; et al. Early-Life Dietary Cadmium Exposure and Kidney Function in 9-Year-Old Children from the PROGRESS Cohort. Toxics 2020, 8, 83. [Google Scholar] [CrossRef]
- Filippini, T.; Wise, L.A.; Vinceti, M. Cadmium exposure and risk of diabetes and prediabetes: A systematic review and dose-response meta-analysis. Environ. Int. 2022, 158, 106920. [Google Scholar] [CrossRef]
- Verzelloni, P.; Urbano, T.; Wise, L.A.; Vinceti, M.; Filippini, T. Cadmium exposure and cardiovascular disease risk: A systematic review and dose-response meta-analysis. Environ, Pollut. 2024, 345, 123462. [Google Scholar] [CrossRef] [PubMed]
- Doccioli, C.; Sera, F.; Francavilla, A.; Cupisti, A.; Biggeri, A. Association of cadmium environmental exposure with chronic kidney disease: A systematic review and meta-analysis. Sci. Total Environ. 2024, 906, 167165. [Google Scholar] [CrossRef]
- Verzelloni, P.; Giuliano, V.; Wise, L.A.; Urbano, T.; Baraldi, C.; Vinceti, M.; Filippini, T. Cadmium exposure and risk of hypertension: A systematic review and dose-response meta-analysis. Environ. Res. 2024, 263 Pt 1, 120014. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Su, W.; Li, N.; Song, Q.; Wang, H.; Liang, Q.; Li, Y.; Lowe, S.; Bentley, R.; Zhou. Z.; et al. Association of urinary or blood heavy metals and mortality from all causes, cardiovascular disease, and cancer in the general population: a systematic review and meta-analysis of cohort studies. Environ. Sci. Pollut. Res. Int. 2022, 29, 67483–67503. [Google Scholar] [CrossRef]
- Oosterwijk, M.M.; Hagedoorn, I.J.M.; Maatman, R.G.H.J.; Bakker, S.J.L.; Navis, G.; Laverman, G.D. Cadmium, active smoking and renal function deterioration in patients with type 2 diabetes. Nephrol. Dial. Transplant. 2023, 38, 876–883. [Google Scholar] [CrossRef]
- Xie, S.; Perrais, M.; Golshayan, D.; Wuerzner, G.; Vaucher, J.; Thomas, A.; Marques-Vidal, P. Association between urinary heavy metal/trace element concentrations and kidney function: A prospective study. Clin. Kidney J. 2024, 18, sfae378. [Google Scholar] [CrossRef]
- Satarug, S. Is Environmental Cadmium Exposure Causally Related to Diabetes and Obesity? Cells 2024, 13, 83. [Google Scholar] [CrossRef] [PubMed]
- Unger, T.; Borghi, C.; Charchar, F.; Khan, N.A.; Poulter, N.R.; Prabhakaran, D.; Ramirez, A.; Schlaich, M.; Stergiou, G.S.; Tomaszewski, M.; Wainford, R.D.; Williams, B.; Schutte, A.E. 2020 International Society of Hypertension Global Hypertension Practice Guidelines. Hypertension 2020, 75, 1334–1357. [Google Scholar] [CrossRef]
- Murray, C.J.L.; Aravkin, A.Y.; Zheng, P.; Abbafati, C.; Abbas, K.M.; Abbasi-Kangevari, M.; Abd-Allah, F.; Abdelalim, A.; Abdollahi, M.; Abdollahpour, I.; et al. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar] [CrossRef] [PubMed]
- Reckelhoff, J.F. Mechanisms of sex and gender differences in hypertension. J. Hum. Hypertens. 2023, 37, 596–601. [Google Scholar] [CrossRef]
- Connelly, P.J.; Currie, G.; Delles, C. Sex differences in the prevalence, outcomes and management of hypertension. Curr. Hypertens. Rep. 2022, 24, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Neufcourt, L.; Zins, M.; Berkman, L.F.; Grimaud, O. Socioeconomic disparities and risk of hypertension among older Americans: the Health and Retirement Study. J. Hypertens. 2021, 39, 2497–2505. [Google Scholar] [CrossRef] [PubMed]
- Neufcourt, L.; Deguen, S.; Bayat, S.; Paillard, F.; Zins, M.; Grimaud, O. Geographical variations in the prevalence of hypertension in France: Cross-sectional analysis of the CONSTANCES cohort. Eur. J. Prev. Cardiol. 2019, 26, 1242–1251. [Google Scholar] [CrossRef]
- Islam, J.Y.; Zaman, M.M.; Ahmed, J.U.; Choudhury, S.R.; Khan, H.; Zissan, T. Sex differences in prevalence and determinants of hypertension among adults: a cross-sectional survey of one rural village in Bangladesh. BMJ Open 2020, 10, e037546. [Google Scholar] [CrossRef]
- Calhoun, D.A.; Jones, D.; Textor, S.; Goff, D.C.; Murphy, T.P.; Toto, R.D.; White, A.; Cushman, W.C.; White, W.; et al. Resistant hypertension: diagnosis, evaluation, and treatment. A scientific statement from the American Heart Association Professional Education Committee of the Council for High Blood Pressure Research. Hypertension 2008, 51, 1403–1419. [Google Scholar] [CrossRef]
- Park, S.; Shin, J.; Ihm, S.H.; Kim, K.I.; Kim, H.L.; Kim, H.C.; Lee, E.M.; Lee, J.H.; Ahn, S.Y.; Cho, E.J.; et al. Resistant hypertension: consensus document from the Korean society of hypertension. Clin. Hypertens. 2023, 29, 30. [Google Scholar] [CrossRef]
- Qiu, L.; Wu, B. The relationship between the age of onset of hypertension and chronic kidney disease: a cross-sectional study of the American population. Front. Cardiovasc. Med. 2024, 11, 1426953. [Google Scholar] [CrossRef] [PubMed]
- Bloch, M.J.; Basile, J.N. Review of recent literature in hypertension: Updated clinical practice guidelines for chronic kidney disease now include albuminuria in the classification system. J. Clin. Hypertens. 2013, 15, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Mulè, G.; Castiglia, A.; Cusumano, C.; Scaduto, E.; Geraci, G.; Altieri, D.; Di Natale, E.; Cacciatore, O.; Cerasola, G.; Cottone, S. Subclinical Kidney Damage in Hypertensive Patients: A Renal Window Opened on the Cardiovascular System. Focus on Microalbuminuria. Adv. Exp. Med. Biol. 2017, 956, 279–306. [Google Scholar]
- Khan, S.S.; Coresh, J.; Pencina, M.J.; Ndumele, C.E.; Rangaswami, J.; Chow, S.L.; Palaniappan, L.P.; Sperling, L.S.; Virani, S.S.; Ho, J.E.; et al. Novel Prediction Equations for Absolute Risk Assessment of Total Cardiovascular Disease Incorporating Cardiovascular-Kidney-Metabolic Health: A Scientific Statement From the American Heart Association. Circulation 2023, 148, 1982–2004. [Google Scholar] [CrossRef]
- Massy, Z.A.; Drueke, T.B. Combination of Cardiovascular, Kidney, and Metabolic Diseases in a Syndrome Named Cardiovascular-Kidney-Metabolic, With New Risk Prediction Equations. Kidney Int. Rep. 2024, 9, 2608–2618. [Google Scholar] [CrossRef] [PubMed]
- Javaid, A.; Hariri, E.; Ozkan, B.; Lang, K.; Khan, S.S.; Rangaswami, J.; Stone, N.J.; Blumenthal, R.S.; Ndumele, C.E. Cardiovascular-Kidney-Metabolic (CKM) Syndrome: A Case-Based Narrative Review. Am. J. Med. Open 2025, 13, 100089. [Google Scholar] [CrossRef]
- Chen, H.; Zou, Y.; Leng, X.; Huang, F.; Huang, R.; Wijayabahu, A.; Chen, X.; Xu, Y. Associations of blood lead, cadmium, and mercury with resistant hypertension among adults in NHANES, 1999-2018. Environ. Health Prev. Med. 2023, 28, 66. [Google Scholar] [CrossRef]
- Liu, L.; Xu, A.; Cheung, B.M.Y. Associations Between Lead and Cadmium Exposure and Subclinical Cardiovascular Disease in U.S. Adults. Cardiovasc Toxicol. 2025, 25, 282–293. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Ma, Z.; Dang, Y.; Yang, Y.; Cao, S.; Ouyang, C.; Shi, X.; Pan, J.; Hu, X. Associations of urinary and blood cadmium concentrations with all-cause mortality in US adults with chronic kidney disease: A prospective cohort study. Environ. Sci. Pollut. Res. Int. 2023, 30, 61659–61671. [Google Scholar] [CrossRef]
- Yin, G.; Zhao, S.; Zhao, M.; Xu, J.; Ge, X.; Wu, J.; Zhou, Y.; Liu, X.; Wei, L.; Xu, Q. Complex interplay of heavy metals and renal injury: New perspectives from longitudinal epidemiological evidence. Ecotoxicol. Environ. Saf. 2024, 278, 116424. [Google Scholar] [CrossRef]
- Yin, G.; Xin, M.; Zhao, S.; Zhao, M.; Xu, J.; Chen, X.; Xu, Q. Heavy metals and elderly kidney health: A multidimensional study through Enviro-target Mendelian Randomization. Ecotoxicol. Environ. Saf. 2024, 281, 116659. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Lee, S.; Jang, M.; Kim, K. Effect of combined exposure to lead, mercury, and cadmium on hypertension: the 2008-2013 Korean National Health and Nutrition Examination Surveys. Int. J. Occup. Med. Environ. Health 2025, 203648. [Google Scholar] [CrossRef] [PubMed]
- Yeon, J.; Kang, S.; Park, J.; Ahn, J.H.; Cho, E.A.; Lee, S.H.; Ryu, K.H.; Shim, J.G. Association between blood cadmium levels and the risk of chronic kidney disease in Korea, based on the Korea National Health and Nutrition Examination Survey 2016-2017. Asian Biomed. (Res Rev News) 2025, 19, 60–66. [Google Scholar] [CrossRef]
- Gburek, J.; Konopska, B.; Gołąb, K. Renal handling of albumin-from early findings to current concepts. Int. J. Mol. Sci. 2021, 22, 5809. [Google Scholar] [CrossRef]
- Benzing, T.; Salant, D. Insights into glomerular filtration and albuminuria. N. Engl. J. Med. 2021, 384, 1437–1446. [Google Scholar] [CrossRef]
- Molitoris, B.A.; Sandoval, R.M.; Yadav, S.P.S.; Wagner, M.C. Albumin uptake and processing by the proximal tubule: Physiological, pathological, and therapeutic implications. Physiol. Rev. 2022, 102, 1625–1667. [Google Scholar] [CrossRef]
- Eshbach, M.L.; Weisz, O.A. Receptor-mediated endocytosis in the proximal tubule. Annu. Rev. Physiol. 2017, 79, 425–448. [Google Scholar] [CrossRef] [PubMed]
- Comper, W.D.; Vuchkova, J.; McCarthy, K.J. New insights into proteinuria/albuminuria. Front. Physiol. 2022, 13, 991756. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Gobe, G.C.; Phelps, K.R. The pathogenesis of albuminuria in cadmium nephropathy. Curr. Res. Toxicol. 2023, 6, 100140. [Google Scholar] [CrossRef]
- Nielsen, R.; Christensen, E.I.; Birn, H. Megalin and cubilin in proximal tubule protein reabsorption: from experimental models to human disease. Kidney Int. 2016, 89, 58–67. [Google Scholar] [CrossRef]
- Lee, W.K.; Thévenod, F. Cell organelles as targets of mammalian cadmium toxicity. Arch. Toxicol. 2020, 94, 1017–1049. [Google Scholar] [CrossRef] [PubMed]
- Thévenod, F.; Lee, W.K.; Garrick, M.D. Iron and cadmium entry into renal mitochondria: Physiological and toxicological implications. Front. Cell Dev. Biol. 2020, 8, 848. [Google Scholar] [CrossRef]
- Jin, L.; Yu, B.; Wang, H.; Shi, L.; Yang, J.; Wu, L.; Gao, C.; Pan, H.; Han, F.; Lin, W.; et al. STING promotes ferroptosis through NCOA4-dependent ferritinophagy in acute kidney injury. Free Radic. Biol. Med. 2023, 208, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Dasinger, J.H.; Abais-Battad, J.M.; McCrorey, M.K.; Van Beusecum, J.P. Recent advances on immunity and hypertension: the new cells on the kidney block. Am. J. Physiol. Renal Physiol. 2025, 328, F301–F315. [Google Scholar] [CrossRef]
- Chen, M.; Gu, X. Emerging roles of proximal tubular endocytosis in renal fibrosis. Front. Cell Dev. Biol. 2023, 11, 1235716. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S.; Yimthiang, S.; Khamphaya, T.; Pouyfung, P.; Vesey, D.A.; Buha Đorđević, A. Albuminuria in People Chronically Exposed to Low-Dose Cadmium Is Linked to Rising Blood Pressure Levels. Toxics 2025, 13, 81. [Google Scholar] [CrossRef]
- Ikeme, J.C.; Katz, R.; Muiru, A.N.; Estrella, M.M.; Scherzer, R.; Garimella, P.S.; Hallan, S.I.; Peralta, C.A.; Ix, J.H.; Shlipak, M.G. Clinical Risk Factors For Kidney Tubule Biomarker Abnormalities Among Hypertensive Adults With Reduced eGFR in the SPRINT Trial. Am. J. Hypertens. 2022, 35, 1006–1013. [Google Scholar] [CrossRef]
- Liu, D.; Lv, L.L. New Understanding on the Role of Proteinuria in Progression of Chronic Kidney Disease. Adv. Exp. Med. Biol. 2019, 1165, 487–500. [Google Scholar]
- Sharma, S.; Smyth, B. From Proteinuria to Fibrosis: An Update on Pathophysiology and Treatment Options. Kidney Blood Press. Res. 2021, 46, 411–420. [Google Scholar] [CrossRef]
- Makhammajanov, Z.; Gaipov, A.; Myngbay, A.; Bukasov, R.; Aljofan, M.; Kanbay, M. Tubular toxicity of proteinuria and the progression of chronic kidney disease. Nephrol. Dial. Transplant. 2024, 39, 589–599. [Google Scholar] [CrossRef]
- Faivre, A.; Verissimo, T.; de Seigneux, S. Proteinuria and tubular cells: Plasticity and toxicity. Acta Physiol. (Oxf) 2025, 241, e14263. [Google Scholar] [CrossRef]
- Dahl, L.K.; Heine, M.; Thompson, K. 1972. Genetic influence of renal homografts on the blood pressure of rats from different strains. Proc. Soc. Exp. Biol. Med. 1972, 140, 852–856. [Google Scholar] [CrossRef]
- Dahl, L.K.; Heine, M.; Thompson, K. 1974. Genetic influence of the kidneys on blood pressure: evidence from chronic renal homografts in rats with opposite predispositions to hypertension. Circ. Res. 1974, 34, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.K.; Heine, M. Primary role of renal homografts in setting chronic blood pressure levels in rats. Circ. Res. 1975, 36, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Guyton, A.C. 1991. Blood pressure control-special role of the kidneys and body fluids. Science 1991, 252, 1813–1816. [Google Scholar] [CrossRef] [PubMed]
- Baek, E.J.; Kim, S. Current Understanding of Pressure Natriuresis. Electrolyte Blood Press. 2021, 19, 38–45. [Google Scholar] [CrossRef]
- Roman, R.J. 20-HETE and Hypertension. Hypertension 2024, 81, 2012–2015. [Google Scholar] [CrossRef]
- Froogh, G.; Garcia, V.; Schwartzman, M.L. The CYP/20-HETE/GPR75 axis in hypertension. Adv. Pharmacol. 2022, 94, 1–25. [Google Scholar]
- Díaz-Morales, N.; Baranda-Alonso, E.M.; Martínez-Salgado, C.; López-Hernández, F.J. Renal sympathetic activity: A key modulator of pressure natriuresis in hypertension. Biochem. Pharmacol. 2023, 208, 115386. [Google Scholar] [CrossRef]
- Leite, A.P.O.; Li, X.C.; Nwia, S.M.; Hassan, R.; Zhuo, J.L. Angiotensin II and AT1a Receptors in the Proximal Tubules of the Kidney: New Roles in Blood Pressure Control and Hypertension. Int. J. Mol. Sci. 2022, 23, 2402. [Google Scholar] [CrossRef]
- Lasker, J.M.; Chen, W.B.; Wolf, I.; Bloswick, B.P.; Wilson, P.D.; Powell, P.K. Formation of 20-hydroxyeicosatetraenoic acid, a vasoactive and natriuretic eicosanoid, in human kidney. Role of CYP4F2 and CYP4A11. J. Biol. Chem. 2000, 275, 4118–4126. [Google Scholar] [CrossRef] [PubMed]
- Gonick, H.C. Pathophysiology of human proximal tubular transport defects. Klin. Wochenschr. 1982, 60, 1201–1211. [Google Scholar] [CrossRef] [PubMed]
- Palmer, L.G.; Schnermann, J. Integrated control of Na transport along the nephron. Clin. J. Am. Soc. Nephrol. 2015, 10, 676–687. [Google Scholar] [CrossRef] [PubMed]
- Schwartzman, M.L.; Martasek, P.; Rios, A.R.; Levere, R.D.; Solangi, K.; Goodman, A.L.; Abraham, N.G. Cytochrome P450-dependent arachidonic acid metabolism in human kidney. Kidney Int. 1990, 37, 94–99. [Google Scholar] [CrossRef]
- Prakash, C.; Zhang, J.Y.; Falck, J.R.; Chauman, K.; Blair, A. 20-Hydroxyeicosatetraenoic acid is excreted as a glucuronide conjugate in human urine. Biochem. Biophys. Res. Comm. 1992, 158, 728–733. [Google Scholar] [CrossRef]
- Boonprasert, K.; Satarug, S.; Morais, C.; Gobe, G.C.; Johnson, D.W.; Na-Bangchang, K.; Vesey, D.A. The stress response of human proximal tubule cells to cadmium involves up-regulation of haemoxygenase 1 and metallothionein but not cytochrome P450 enzymes. Toxicol. Lett. 2016, 249, 5–14. [Google Scholar] [CrossRef]
- Ajjimaporn, A.; Botsford, T.; Garrett, S.H.; Sens, M.A.; Zhou, X.D.; Dunlevy, J.R.; Sens, D.A.; Somji, S. ZIP8 expression in human proximal tubule cells, human urothelial cells transformed by Cd+2 and As+3 and in specimens of normal human urothelium and urothelial cancer. Cancer Cell Int. 12, 16.
- Subramanya, A.R.; Ellison, D.H. Distal convoluted tubule. Clin J Am Soc Nephrol. 2014, 9, 2147–2163. [Google Scholar] [CrossRef]
- Thevenod, F.; Friedmann, J.M. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+/K+-ATPase through proteasomal and endo-/lysosomal proteolytic pathways. FASEB J. 1999, 13, 1751–1761. [Google Scholar] [CrossRef]
- Baker, J.R.; Satarug, S.; Urbenjapol, S.; Edwards, R.J.; Williams, D.J.; Moore, M.R.; Reilly, P.E. Associations between human liver and kidney cadmium content and immunochemically detected CYP4A11 apoprotein. Biochem. Pharmacol. 2002, 63, 693–696. [Google Scholar] [CrossRef]
- Baker, J.R.; Satarug, S.; Edwards, R.J.; Moore, M.R.; Williams, D.J.; Reilly, P.E. Potential for early involvement of CYP isoforms in aspects of human cadmium toxicity. Toxicol. Lett. 2003, 137, 85–93. [Google Scholar] [CrossRef]
- Baker, J.R.; Edwards, R.J.; Lasker, J.M.; Moore, M.R.; Satarug, S. Renal and hepatic accumulation of cadmium and lead in the expression of CYP4F2 and CYP2E1. Toxicol. Lett. 2005, 159, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Perry, H.M., Jr.; Erlanger, M.W. Sodium retention in rats with cadmium-induced hypertension. Sci. Total Environ. 1981, 22, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Pena, A.; Iturri, S.J. 1993. Cadmium as hypertensive agent. Effect on ion excretion in rats. Comp. Biochem. Physiol. C 1993, 106, 315–319. [Google Scholar]
- Ali, I.; Engström, A.; Vahter, M.; Skerfving, S.; Lundh, T.; Lidfeldt, J.; Samsioe, G.; Halldin, K.; Åkesson, A. Associations between cadmium exposure and circulating levels of sex hormones in postmenopausal women. Environ. Res. 2014, 134, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Nagata, C.; Konishi, K.; Goto, Y.; Tamura, T.; Wada, K.; Hayashi, M.; Takeda, N.; Yasuda, K. Associations of urinary cadmium with circulating sex hormone levels in pre- and postmenopausal Japanese women. Environ. Res. 2016, 150, 82–87. [Google Scholar] [CrossRef]
- Nagata, C.; Nagao, Y.; Shibuya, C.; Kashiki, Y.; Shimizu, H. Urinary cadmium and serum levels of estrogens and androgens in postmenopausal Japanese women. Cancer Epidemiol. Biomark. Prev. 2005, 14, 705–708. [Google Scholar] [CrossRef]
- Lu, Y.; Geng, L.; Zhou, D.; Sun, Y.; Xu, H.; Du, X.; Xu, Q.; Chen, M. Toxic metals and trace elements, markers of inflammation, and hyperandrogenemia in women and testosterone deficiency in men: Associations and potential mediating factors. Ecotoxicol. Environ. Saf. 2025, 299, 118352. [Google Scholar] [CrossRef]
- Wu, C.C.; Schwartzman, M.L. The role of 20-HETE in androgen-mediated hypertension. Prostaglandins Other Lipid Mediat. 2011, 96, 45–53. [Google Scholar] [CrossRef]
- Dalmasso, C.; Maranon, R.; Patil, C.; Moulana, M.; Romero, D.G.; Reckelhoff, J.F. 20-HETE and CYP4A2 ω-hydroxylase contribute to the elevated blood pressure in hyperandrogenemic female rats. Am. J. Physiol. Renal Physiol. 2016, 311, F71–F77. [Google Scholar] [CrossRef]
- Pandey, V.; Garcia, V.; Gilani, A.; Mishra, P.; Zhang, F.F.; Paudyal, M.P.; Falck, J.R.; Nasjletti, A.; Wang, W.H.; Schwartzman, M.L. The Blood Pressure-Lowering Effect of 20-HETE Blockade in Cyp4a14(-/-) Mice Is Associated with Natriuresis. J. Pharmacol. Exp. Ther. 2017, 363, 412–418. [Google Scholar] [CrossRef]
- Boonprasert, K.; Vesey, D.A.; Gobe, G.C.; Ruenweerayut, R.; Johnson, D.W.; Na-Bangchang, K.; Satarug, S. Is renal tubular cadmium toxicity clinically relevant? Clin. Kidney J. 2018, 11, 681–687. [Google Scholar] [CrossRef]
- EFSA Scientific Committee Update: Use of the benchmark dose approach in risk assessment. EFSA J. 2017, 15, 4658.
- Slob, W.; Setzer, R.W. Shape and steepness of toxicological dose-response relationships of continuous endpoints. Crit. Rev. Toxicol. 2014, 44, 270–297. [Google Scholar] [CrossRef] [PubMed]
- Slob, W. A general theory of effect size, and its consequences for defining the benchmark response (BMR) for continuous endpoints. Crit. Rev. Toxicol. 2017, 47, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Filipsson, A.F.; Sand, S.; Nilsson, J.; Victorin, K. The benchmark dose method--review of available models, and recommendations for application in health risk assessment. Crit. Rev. Toxicol. 2003, 33, 505–542. [Google Scholar]
- Sand, S.J.; von Rosen, D.; Filipsson, A.F. Benchmark calculations in risk assessment using continuous dose-response information: the influence of variance and the determination of a cut-off value. Risk Anal. 2003, 23, 1059–1068. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Wei, S.; Wei, Y.; Guo, B.; Yang, M.; Zhao, D.; Liu, X.; Cai, X. The reference dose for subchronic exposure of pigs to cadmium leading to early renal damage by benchmark dose method. Toxicol. Sci. 2012, 128, 524–531. [Google Scholar] [CrossRef]
- Byber, K.; Lison, D.; Verougstraete, V.; Dressel, H.; Hotz, P. Cadmium or cadmium compounds and chronic kidney disease in workers and the general population: A systematic review. Crit. Rev. Toxicol. 2016, 46, 191–240. [Google Scholar] [CrossRef]
- Jalili, C.; Kazemi, M.; Cheng, H.; Mohammadi, H.; Babaei, A.; Taheri, E.; Moradi, S. Associations between exposure to heavy metals and the risk of chronic kidney disease: A systematic review and meta-analysis. Crit. Rev. Toxicol. 2021, 51, 165–182. [Google Scholar] [CrossRef]
- Đorđević, A.B.; Vesey, D.A.; Satarug, S. Cadmium-induced nephrotoxicity assessed by benchmark dose calculations in two exposure-effect datasets. Discover Toxicol. [CrossRef]
- Grandjean, P.; Budtz-Jørgensen, E. Total imprecision of exposure biomarkers: Implications for calculating exposure limits. Am. J. Ind. Med. 2007, 50, 712–719. [Google Scholar] [CrossRef]
- Phelps, K.R.; Gosmanova, E.O. A generic method for analysis of plasma concentrations. Clin. Nephrol. 2020, 94, 43–49. [Google Scholar] [CrossRef]
- Argyropoulos, C.P.; Chen, S.S.; Ng, Y.-H.; Roumelioti, M.-E.; Shaffi, K.; Singh, P.P.; Tzamaloukas, A.H. Rediscovering Beta-2 Microglobulin as a Biomarker across the Spectrum of Kidney Diseases. Front. Med. 2017, 4, 73. [Google Scholar] [CrossRef] [PubMed]
- Sivanathan, P.C.; Ooi, K.S.; Mohammad Haniff, M.A.S.; Ahmadipour, M.; Dee, C.F.; Mokhtar, N.M.; Hamzah, A.A.; Chang, E.Y. Lifting the Veil: Characteristics, Clinical Significance, and Application of β-2-Microglobulin as Biomarkers and Its Detection with Biosensors. ACS Biomater. Sci. Eng. 2022, 8, 3142–3161. [Google Scholar] [CrossRef]
- Satarug, S.; Phelps, K.R. Cadmium exposure and toxicity. In: Bagchi D, Bagchi M, eds: Metal Toxicology Handbook, Ch 14, pp 219-272. New York, CRC Press, 2021.
- Phelps, K.R.; Yimthiang, S.; Pouyfung, P.; Khamphaya, T.; Vesey, D.A.; Satarug, S. Homeostasis of β2-Microglobulin in Diabetics and Non-Diabetics with Modest Cadmium Intoxication. [CrossRef]
- Mashima, Y.; Konta, T.; Kudo, K.; Takasaki, S.; Ichikawa, K.; Suzuki, K.; Shibata, Y.; Watanabe, T.; Kato, T.; Kawata, S.; et al. Increases in urinary albumin and beta2-microglobulin are independently associated with blood pressure in the Japanese general population: The Takahata Study. Hypertens. Res. 2011, 34, 831–835. [Google Scholar] [CrossRef]
- Kaneda, M.; Wai, K.M.; Kanda, A.; Ando, M.; Murashita, K.; Nakaji, S.; Ihara, K. Low Level of Serum Cadmium in Relation to Blood Pressures Among Japanese General Population. Biol. Trace Elem. Res. 2022, 200, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Yimthiang, S.; Pouyfung, P.; Khamphaya, T.; Vesey, D.A.; Gobe, G.C.; Satarug, S. Evidence Linking Cadmium Exposure and β2-Microglobulin to Increased Risk of Hypertension in Diabetes Type 2. Toxics 2023, 11, 516. [Google Scholar] [CrossRef] [PubMed]
- Satarug, S. Urinary β2-Microglobulin Predicts the Risk of Hypertension in Populations Chronically Exposed to Environmental Cadmium. J. Xenobiot. 2025, 15, 49. [Google Scholar] [CrossRef]
- Huan, T.; Meng, Q.; Saleh, M.A.; Norlander, A.E.; Joehanes, R.; Zhu, J.; Chen, B.H.; Zhang, B.; Johnson, A.D.; Ying, S.; et al. Integrative network analysis reveals molecular mechanisms of blood pressure regulation. Mol. Syst. Biol. 2015, 11, 799. [Google Scholar] [CrossRef]
- Alexander, M.R.; Hank, S.; Dale, B.L.; Himmel, L.; Zhong, X.; Smart, C.D.; Fehrenbach, D.J.; Chen, Y.; Prabakaran, N.; Tirado, B.; et al. A single nucleotide polymorphism in SH2B3/LNK promotes hypertension development and renal damage. Circ. Res. 2022, 131, 731–747. [Google Scholar] [CrossRef]
- Keefe, J.A.; Hwang, S.J.; Huan, T.; Mendelson, M.; Yao, C.; Courchesne, P.; Saleh, M.A.; Madhur, M.S.; Levy, D. Evidence for a causal role of the SH2B3-β2M axis in blood pressure regulation. Hypertension 2019, 73, 497–503. [Google Scholar] [CrossRef]
- Satarug, S.; Vesey, D.A.; Đorđević, A.B. The NOAEL equivalent for the cumulative body burden of cadmium: Focus on proteinuria as an endpoint. J. Environ. Expo. Assess. 2024, 3, 26. [Google Scholar] [CrossRef]
- Ren, F.; Li, M.; Xu, H.; Qin, X.; Teng, Y. Urine albumin-to-creatinine ratio within the normal range and risk of hypertension in the general population: A meta-analysis. J. Clin. Hypertens. 2021, 23, 1284–1290. [Google Scholar] [CrossRef] [PubMed]
- Okubo, A.; Nakashima, A.; Doi, S.; Doi, T.; Ueno, T.; Maeda, K.; Tamura, R.; Yamane, K.; Masaki, T. High-normal albuminuria is strongly associated with incident chronic kidney disease in a nondiabetic population with normal range of albuminuria and normal kidney function. Clin. Exp. Nephrol. 2020, 24, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Song, W.; Zhou, Y.; Gao, Y.; Wang, Y.; Wang, Y.; Liu, Y.; Deng, L.; Liao, Y.; Wu, B.; et al. Elevated urine albumin creatinine ratio increases cardiovascular mortality in coronary artery disease patients with or without type 2 diabetes mellitus: A multicenter retrospective study. Cardiovasc. Diabetol. 2023, 22, 203. [Google Scholar] [CrossRef]
Figure 1.
Albumin and β2-microglobulin reuptake by the proximal tubular cells (PTCs). Albumin reaches tubular lumen through endothelial cells and podocyte foot processes [65,66], and nearly all is reabsorbed and returned to blood circulation [67,68]. A relatively small proportion of albumin is reabsorbed by megalin/cubilin-mediated endocytosis and it undergoes lysosomal degradation. In Cd-intoxicated PTC, “toxic” unbound Cd is released as albumin is degraded.
Figure 1.
Albumin and β2-microglobulin reuptake by the proximal tubular cells (PTCs). Albumin reaches tubular lumen through endothelial cells and podocyte foot processes [65,66], and nearly all is reabsorbed and returned to blood circulation [67,68]. A relatively small proportion of albumin is reabsorbed by megalin/cubilin-mediated endocytosis and it undergoes lysosomal degradation. In Cd-intoxicated PTC, “toxic” unbound Cd is released as albumin is degraded.
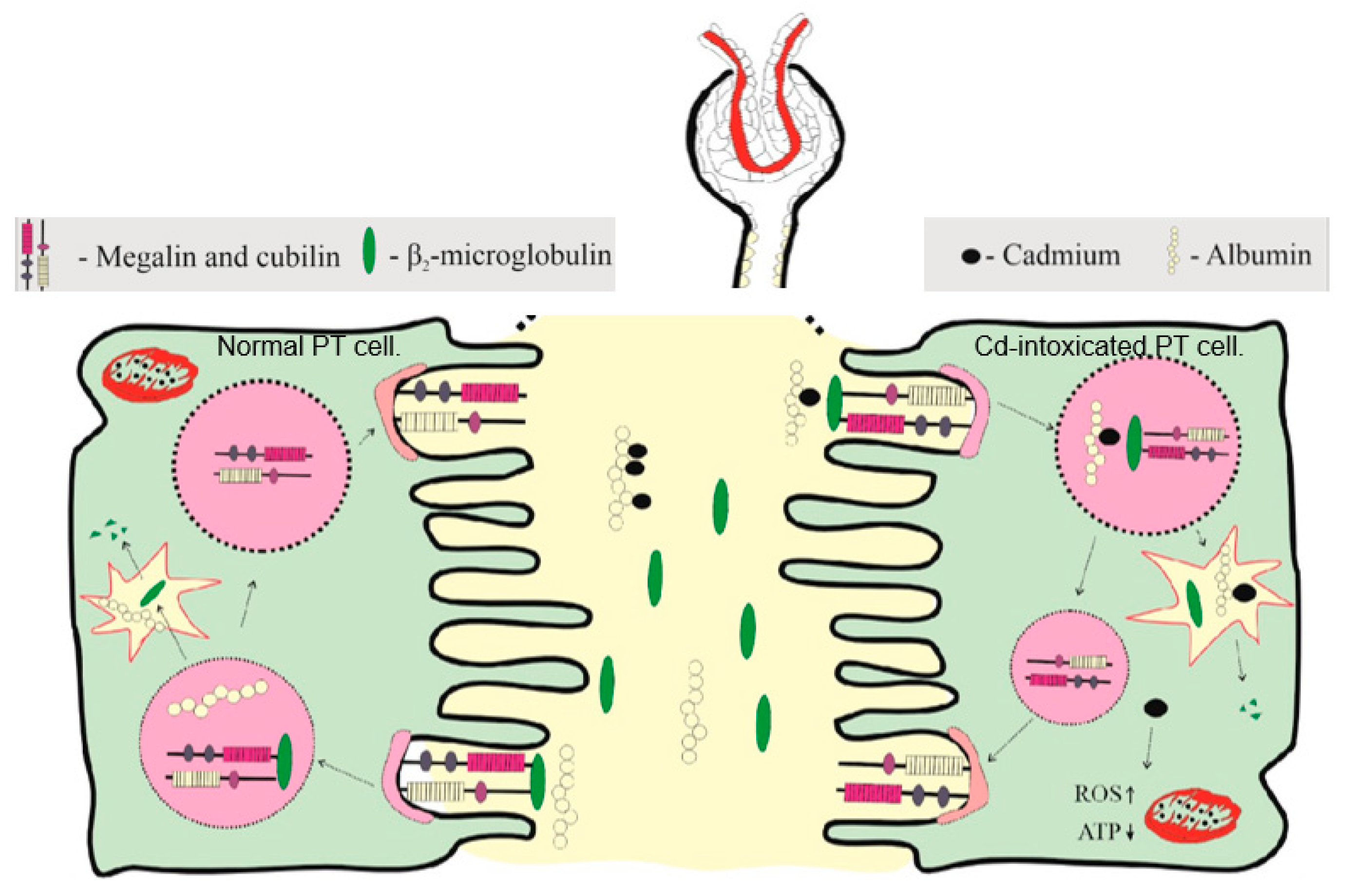
Figure 2.
Effects of blood pressure and eGFR on albumin excretion. Scatterplots relate albumin excretion to SBP (a) and DBP (b) across eGFR groups; ≤ 60, 61–90, and ≥ 90 mL/min/1.73 m2. Mean albumin excretion rate in eGFR subgroups with SBP (c) and DBP above medians (d). The median SBP/DBP was 130/80 80 mmHg. All mean values were adjusted for covariates and interactions.
Figure 2.
Effects of blood pressure and eGFR on albumin excretion. Scatterplots relate albumin excretion to SBP (a) and DBP (b) across eGFR groups; ≤ 60, 61–90, and ≥ 90 mL/min/1.73 m2. Mean albumin excretion rate in eGFR subgroups with SBP (c) and DBP above medians (d). The median SBP/DBP was 130/80 80 mmHg. All mean values were adjusted for covariates and interactions.
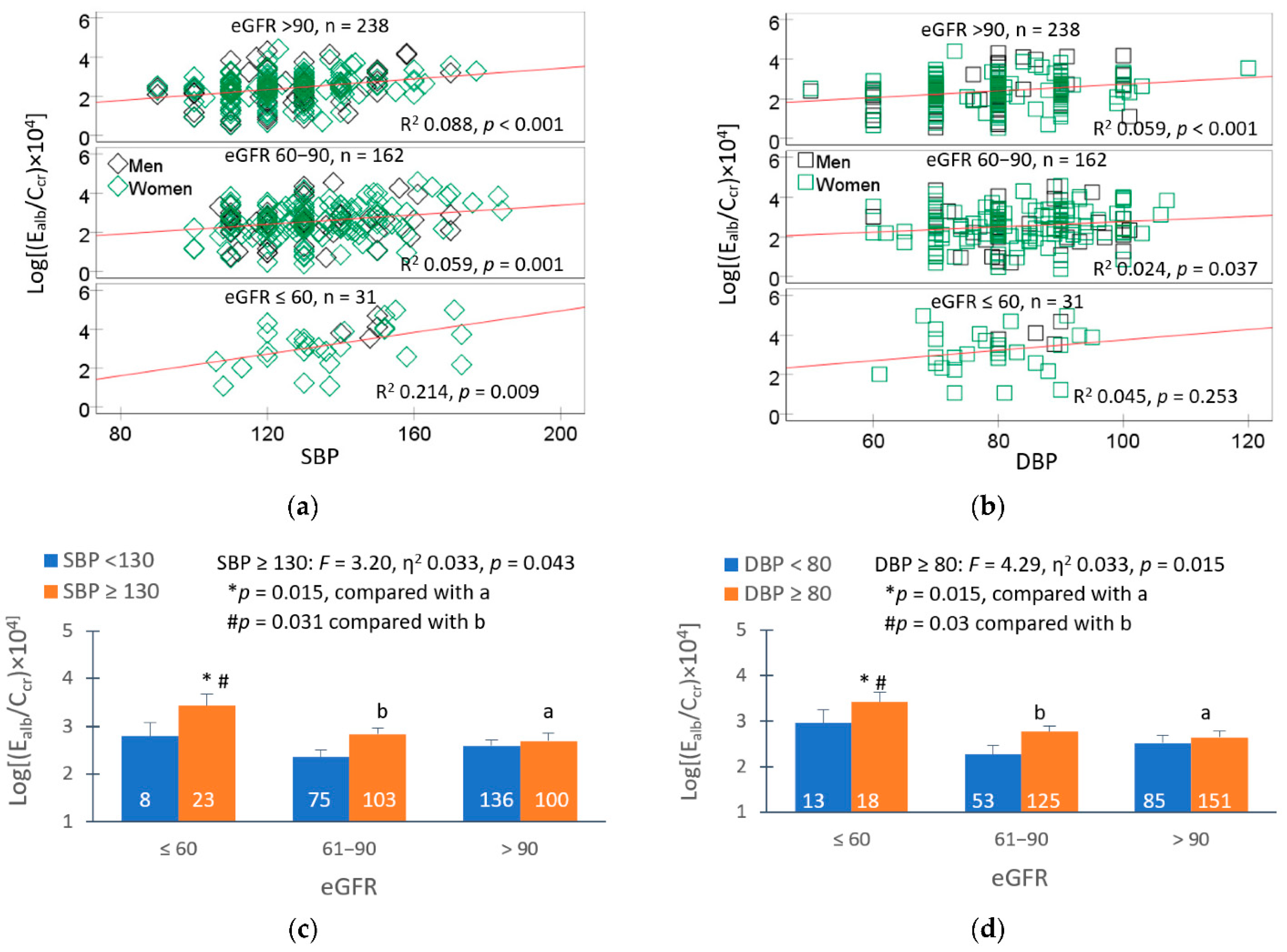
Figure 3.
The kidney tubular cell as Cd target of toxicity. Cd enters tubular cells and reaches mitochondria through receptors and transporters for essential metals. It targets also lysosome, endoplasmic reticulum, and the nucleus. It induces expression of CYP4F2, which produces 20-HETE. It enhances mitochondrial production of ROS which can damage Na/K-ATPase and apical Na-cotransporters. The electrochemical Na gradient generated by the Na/K ATPase pump promotes co-transport of Na with other filtered substances, notably glucose [93,94].
Figure 3.
The kidney tubular cell as Cd target of toxicity. Cd enters tubular cells and reaches mitochondria through receptors and transporters for essential metals. It targets also lysosome, endoplasmic reticulum, and the nucleus. It induces expression of CYP4F2, which produces 20-HETE. It enhances mitochondrial production of ROS which can damage Na/K-ATPase and apical Na-cotransporters. The electrochemical Na gradient generated by the Na/K ATPase pump promotes co-transport of Na with other filtered substances, notably glucose [93,94].

Figure 4.
Fate of β2-microglobulin in the body. The protein β2M is expressed on the surface of most nucleated cells and is shed into blood stream. Being a low-molecular-weight protein, β2M is filtered completely by the glomeruli, retrieved by PTCs, and subjected to lysosomal degradation.
Figure 4.
Fate of β2-microglobulin in the body. The protein β2M is expressed on the surface of most nucleated cells and is shed into blood stream. Being a low-molecular-weight protein, β2M is filtered completely by the glomeruli, retrieved by PTCs, and subjected to lysosomal degradation.
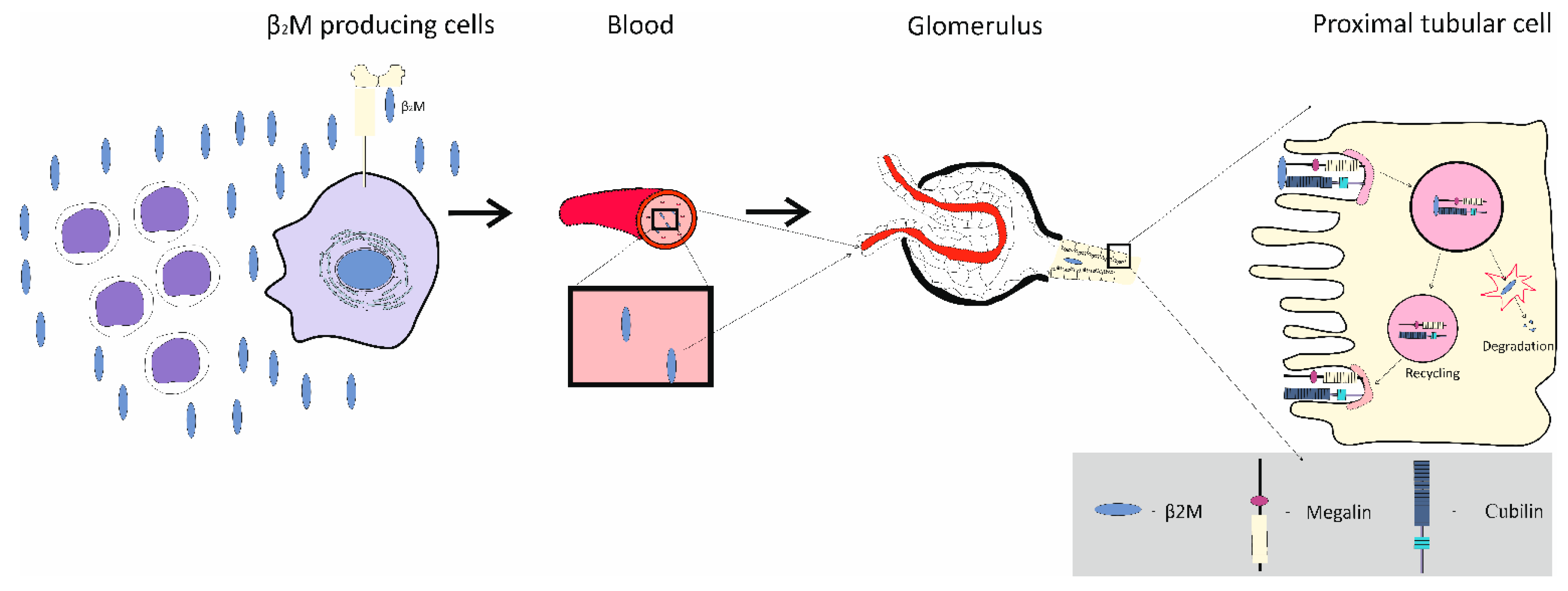
Figure 5.
Inverse relationship of eGFR vs. ECd and Eβ2M vs ECd. Scatterplots relate eGFR to ECd/Ecr (A) and ECd/Ccr (B). Scatterplots relate Eβ2M/Ecr (C) and Eβ2M/Ccr (D) to eGFR.
Figure 5.
Inverse relationship of eGFR vs. ECd and Eβ2M vs ECd. Scatterplots relate eGFR to ECd/Ecr (A) and ECd/Ccr (B). Scatterplots relate Eβ2M/Ecr (C) and Eβ2M/Ccr (D) to eGFR.

Figure 6.
BMDL values of Cd excretion computed from two endpoints. Bootstrap dose-effect averaging for Eβ2M/Ecr−ECd/Ecr (A) eGFR−ECd/Ecr datasets (D). The mathematical dose-response models and relative fitting weights for Eβ2M/Ecr −ECd/Ecr (B) and eGFR −ECd/Ecr datasets (E). BMDL/BMDU values of ECd/Ecr for the β2M excretion (C) and falling eGFR (F) endpoints.
Figure 6.
BMDL values of Cd excretion computed from two endpoints. Bootstrap dose-effect averaging for Eβ2M/Ecr−ECd/Ecr (A) eGFR−ECd/Ecr datasets (D). The mathematical dose-response models and relative fitting weights for Eβ2M/Ecr −ECd/Ecr (B) and eGFR −ECd/Ecr datasets (E). BMDL/BMDU values of ECd/Ecr for the β2M excretion (C) and falling eGFR (F) endpoints.
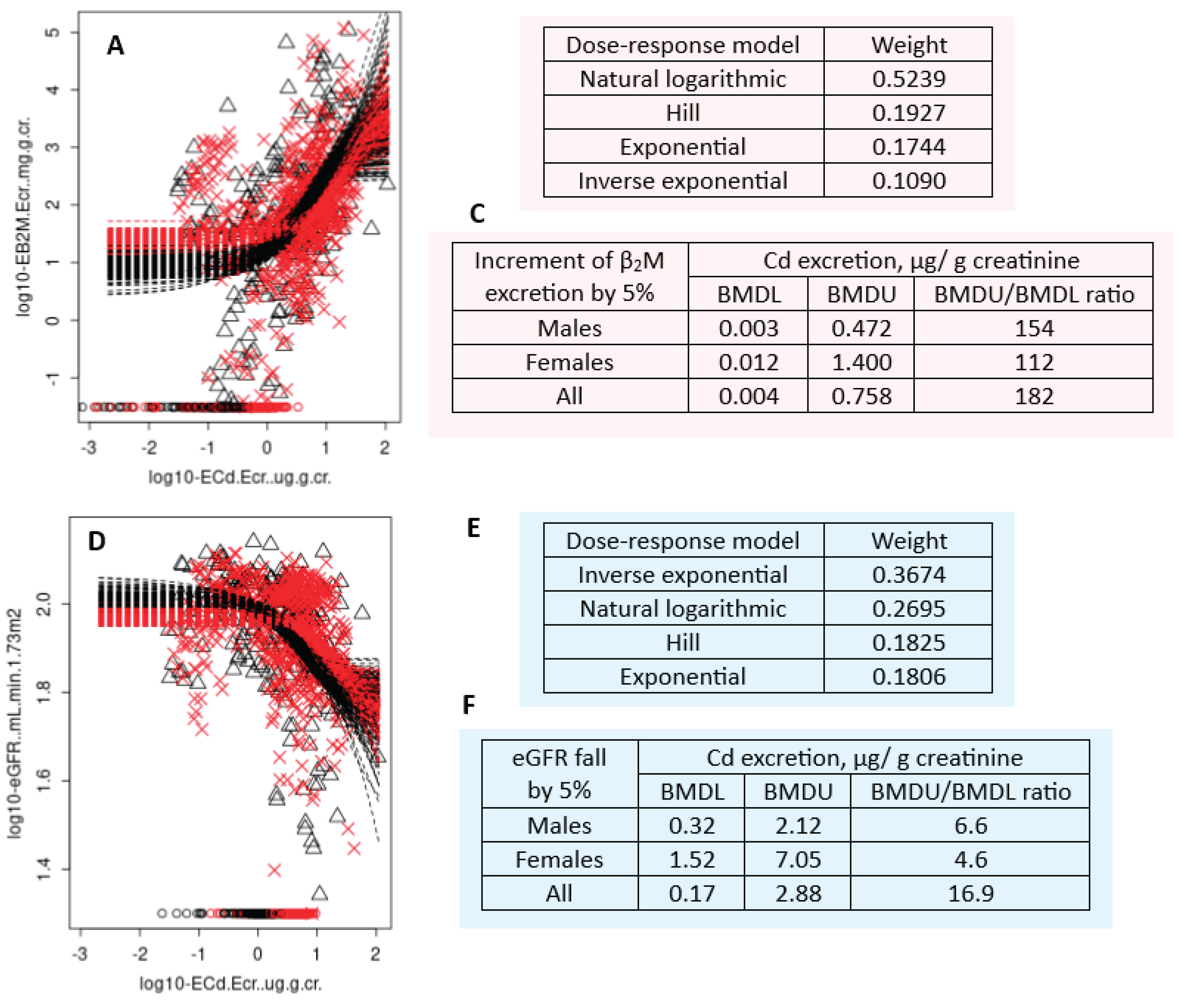

Table 1.
Staging of CKD according to eGFR and albuminuria.
| GFR domain | Albuminuria domain |
|---|---|
| G1: Normal or high eGFR ≥ 90 mL/min/1.73 m2 |
A1: Normal to mildly increased AER < 30 mg/d or ACR <30 mg albumin/g creatinine |
| G2: Mildly decreased eGFR 60–89 mL/min/1.73 m2 |
A2: Moderately increased AER 30–300 mg/d or ACR 30–300 mg/g creatinine |
| G3a: Mildly to moderately decreased eGFR 45–59 mL/min/1.73 m2 G3b: Moderately to severely decreased eGFR 30–44 mL/min/1.73 m2 |
A3: Severely increased AER > 300 mg/d or ACR >300 mg/g creatinine |
| G4: Severely decreased eGFR 15–29 mL/min/1.73 m2 |
|
| G5: Kidney failure eGFR <15 mL/min/1.73 m2 |
eGFR, estimated glomerular filtration rate; AER, albumin excretion rate; ACR, albumin-to-creatinine ratio.
Table 2.
Blood and urine Cd levels associated with adverse health effects in different populations.
| Study Population | Findings | Reference |
|---|---|---|
| NHANES, 1999-2018 n = 38,281, 3.7% resistance hypertension, 27.6% hypertension. |
Risk for resistant hypertension rose 30 and 35% comparing blood Cd in quartiles 3 and 4, with blood Cd in the bottom quartile, respectively. | Chen et al. 2023 [58] |
| NHANES 1999-2004, n = 10,197, ≥ 20 years |
Risk for plasma levels of cardiac troponin (cTnT) ≥ 19 ng/L and of N-terminal pro b-type natriuretic peptide (NT-proBNP) ≥ 125 pg/mL rose 33 and 39% at blood Cd concentration ≥ 1.0 μg/L | Liu et al. 2025 [59] |
| NHANES 1999–2014 CKD cohort, n =1825, Follow-up period, 6.8 years |
Risk for all-cause mortality rose 75 and 59% at Cd excretion rates ≥ 0.60 μg/g creatinine, and blood Cd concentrations ≥ 0.70 μg/L, respectively. | Zhang et al. 2023 [60] |
| Northeast China n = 384, four-time repeated measurements, 2016-2021 |
Cd and Cr produced synergistic effects on NAG excretion, albuminuria, and ACR Cd and Pb produced synergistic effects on NAG excretion and ACR. |
Yin et al. 2024 [61] |
| Jinzhou, Liaoning, China, n = 529, three-time repeated measurements of Cd, Cr and Pb excretion rates and effects on kidneys. 2016–2021. |
Baseline median values for urine Cd, Cr, and Pb were 2.41, 3.96 and 2.49 μg/L, respectively. Baseline median values for urine NAG, β2M, Alb, ACR, and eGFR were 8.86 U/L, 790 µg/L, 24.4 mg/L, 21.2 mg/g creatinine, and 102 mL/min/1.73 m², respectively, A combined exposure to Cd, Cr and Pb caused more extensive injury to kidneys than did each individual metal. |
Yin et al. 2024 [62] |
| Korean NHANES 2008-2013 n = 40,328, GM for blood Pb and blood Cd in males (females) were 2.5 (1.84) µg/dL and 0.88 (1.04) µg/L. |
Increment in risk for hypertension by 29, 47 and 78% were associated with Pb, Cd and a combined Cd and Pb exposure, respectively. | Kim et al. 2025 [63] |
| Korean NHANES 2016-2017 n = 4,222, ≥ 30 years, 5.1% had CKD. Mean blood Cd 1.2 µg/L. |
A 2.70-fold increase in risk for CKD was associated blood Cd in those who had hypertension. A 2.40-fold increase in risk for CKD was associated with blood Cd in non-diabetics. |
Yeon et al. 2025 [64] |
NHANES, national health and nutrition examination survey; CKD, chronic kidney disease Cd, cadmium; Cr, chromium; Pb, lead; NAG, N-acetyl-β-D-glucosaminidase; ACR, albumin-to-creatinine ratio; GM, geometric mean.
Table 3.
Independent effect of Cd on prevalence odds for chronic kidney disease (low eGFR).
| a CKD | |||||
|---|---|---|---|---|---|
| Model A | POR | 95% CI | p | ||
| Lower | Upper | ||||
| Age | 1.173 | 1.128 | 1.220 | <0.001 | |
| Log2[(ECd/Ecr) × 103], µg/g creatinine | 1.981 | 1.500 | 2.615 | <0.001 | |
| Gender | 1.135 | 0.533 | 2.415 | 0.743 | |
| Hypertension | 1.933 | 0.965 | 3.874 | 0.063 | |
| Smoking | 1.140 | 0.529 | 2.456 | 0.738 | |
| BMI, kg/m2 | |||||
| 12–18 | Referent | ||||
| 19–23 | 1.150 | 0.441 | 3.002 | 0.775 | |
| ≥ 24 | 4.002 | 1.351 | 11.86 | 0.012 | |
| Model B | POR | Lower | Upper | p | |
| Age | 1.168 | 1.119 | 1.219 | <0.001 | |
| Log2[(ECd/Ccr) × 105], µg/L filtrate | 3.132 | 2.249 | 4.361 | <0.001 | |
| Gender | 0.719 | 0.315 | 1.643 | 0.434 | |
| Hypertension | 2.656 | 1.231 | 5.727 | 0.013 | |
| Smoking | 1.103 | 0.487 | 2.495 | 0.815 | |
| BMI, kg/m2 | |||||
| 12–18 | Referent | ||||
| 19–23 | 1.134 | 0.403 | 3.189 | 0.812 | |
| ≥ 24 | 4.784 | 1.468 | 15.59 | 0.009 | |
a CKD was defined as eGFR ≤ 60 mL/min/1.73 m2. ECd was normalized to Ecr and Ccr in models A and B, respectively. Data were from 691 subjects without diabetes and workplace exposure to Cd (420 females, 271 males), aged 18–87 years [122].
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



