Submitted:
26 June 2025
Posted:
27 June 2025
You are already at the latest version
Abstract
The fungal kingdom, with an estimated five million species, has undergone extensive diversification over the past billion years and now occupies a wide array of ecological niches from terrestrial to aquatic ecosystems. To thrive in such diverse environments, fungi must exhibit finely tuned physiological and morphological responses orchestrated by conserved molecular pathways. Increasing evidence suggests that aquaporins (AQPs) play a key role in mediating these adaptive responses, particularly under varying abiotic and biotic stresses conditions. However, despite notable advances in recent decades, the precise functional roles of AQPs within fungal kingdom remains largely unresolved in the field of cell biology. AQPs are transmembrane proteins belonging to the major intrinsic proteins (MIPs) superfamily, which is characterized by remarkable sequence and structural diversity. Beyond their established function in facilitating water transport, MIPs mediated the bidirectional diffusion of a range of small inorganic and organic solutes, ions, and gases across cellular membranes. In fungi, MIPs are classified into three main subfamilies: orthodox (i.e., classical) AQPs, aquaglyceroporins (AQGP), and X-intrinsic proteins (XIPs). This review provides a concise summary of the fundamental structural and functional characteristics of fungal aquaporins, including their structure, classification, and known physiological roles. While the majority of current literature has focused on the AQP and AQGP subfamilies, this review also aims to offer a comprehensive and original overview of the relatively understudied X-intrinsic protein subfamily, highlighting its potential implication in fungal biology.
Keywords:
Aquaporin
; Aquaglyceroporin
; X-Intrinsic Protein (XIP)
; Evolution
; Deep divergence
; Phylogenetic
; 3D modeling
1. General Considerations on MIPs
Membranes, whether plasmamembranes, endomembranes, or those delimiting organelles such as chloroplasts or mitochondria, serve as semi-permeable barriers that separate various cellular systems (cytosol and organelles) from their respective external environments (apoplasm or cytosol). Due to their hydrophobic nature, membranes establish the physical and chemical boundaries of cell or subcellular structures within water-soluble environments. Consequently, these systems must rely on a vast and highly complex network of transmembrane protein transporters to regulate the controlled influx and efflux of polar molecules and ions. The maintenance of distinct organic and hydro-mineral compositions between the extracellular fluid and the cytosol is essential for cellular stability and survival. In this context, aquaporins (AQPs) figure as major actors [1].
AQPs are members of the major intrinsic proteins (MIPs) superfamily and facilitate the passive movement of water and small, neutral solutes across biological membranes. They belong to an ancient family of water channel proteins that are found across all kingdoms of life (Archaea, Bacteria, Fungi, Plantae, Animalia, Protozoa, Chromista, and virus), underscoring their essential role in a plethora of fundamental biological processes [2,3]. The discovery of aquaporins has provided valuable insights into the molecular mechanisms underlying solute transmembrane transport, encompassing both natural and synthetic substances (e.g., drugs).
Structurally, AQP proteins typically assemble into homo- and heterotetrameric arrangements, with each protomer acting as an independent pore [4,5]. These tetrameric configurations are regarded as a crucial regulatory mechanism for the permeability of each AQP protomer [6]. Each protomer is composed of six transmembrane (TM) α-helices (H1–H6), connected by five loops (denoted LA through LE), with both the N- and C-terminal regions localized in the cytosol, as presented for the fungus Trichoderma atroviride in Figure 1. The helices are symmetrically organized into two vestibules, forming a central conduction pore that governs transport activity. The pore spans approximately 55 Å in height, from the intra- to the extra-cellular space. The three-dimensional arrangement of these secondary structure elements thus imparts the characteristic hourglass shape to aquaporins.
Inside the channel, solute permeation is facilitated by a series of polar interactions between the solute molecules and specific amino acids, whose side chains are embedded within the central channel. Two primary constriction sites within the pore are involved in its selectivity. The LB and LE loops each contain a short α-helix, forming a seventh pseudo-transmembrane helix, and are connected by highly conserved Asparagine-Proline-Alanine (NPA) motifs [7]. These loops are partially located within the membrane. The asparagine (N) side chains, oriented towards the pore’s interior, are situated at the ends of two half-helices, and their antiparallel dipoles create an electrostatic barrier in this region, which is essential for proton exclusion.
A second structural feature common to aquaporins is the aromatic/arginine (ar/R) constriction residue quartet [8,9]. This quartet comprises four amino acid residues arranged within the same spatial plane, with two residues located in the H2 and H5 helices, and two in loop E (LE1 and LE2). Forming the narrowest part of the pore, this constriction plays a crucial role in the substrate selectivity. It acts as a size-exclusion barrier, blocking the transport of bulkier substrates, while simultaneously providing key hydrogen bonds and van der Waals contacts that stabilize the solutes and/or water molecules being transported [8,10]. In this regard, the selectivity filter residues of water-specific aquaporins typically form a smaller (∼2.8 Å) and more hydrophilic pore, whereas those in glycerol transporters form a larger (∼3.4 Å) and more hydrophobic pore [11,12].
Natural substitutions within the ar/R selectivity filter and NPA motifs are believed to play a critical role in determining the broad substrate specificity of AQPs. Additionally, residues surrounding these key sites may vary, substantially influencing the overall size and hydrophobicity of the pore. Molecules transported through aquaporins pass via a network of hydrogen bonds, which are formed with the carbonyl groups of the B and E loops, as well as with the side chains of specific residues. While the aforementioned motifs are highly conserved across all aquaporins, their local chemical environment –defined by the chemical groups of amino acids that are spatially proximal, though not necessarily adjacent in the linear protein sequence- determines the selectivity and efficiency of solute transport. These atomic interactions and chemical groups underpin the functional variability of aquaporins, which, in part, explains the complexity and multifactorial nature of AQP gating, as observed in various cell systems [13,14].
In addition, various motifs involved in co- and post-translational modifications (such as N-glycosylation, deamidation, N-terminal acetylation, methylation, ubiquitination, and phosphorylation), along with external factors such as pH, temperature, solute gradients, pressure, and membrane tension, all contribute to shaping the functional core of the channels. These factors, along with trafficking processes (where AQPs are transported from intracellular storage sites to the plasma membrane) and interactions with protein or minerals partners (e.g., cadmium, calcium, etc.), govern the selective transport of water, neutral solutes, ions, small uncharged molecules, and gases across cellular membranes. Notably, structural variations in the α-helices, loops, and the N- and C-termini of AQPs appear to confer substantial functional diversity [15].
Thus, two “prototype” members of the MIP superfamily can be distinguished: “orthodox“ or “classical” aquaporins (i.e., aquaporins, stricto sensu), which mediate the rapid and selective flux of water across biological membranes and play important roles in the osmoregulation of cells and organisms, and aquaglyceroporins (AQGPs, or AGPs), which facilitate the transmembrane transport of small uncharged molecules such as polyols, H2O2, urea, arsenite, and others, in addition to or independent water (depending on the specific member). These AQGPs are involved in osmoregulation, nutrient uptake, and potentially other physiological processes. However, within both groups, a significant diversity of members and dedicated subfamilies exists. In animals, aquaporins are categorized into four major groups: Aqp4-like classical aquaporins (Aqp), Aqp8-like aqua-ammoniaporin, Aqp12-like unorthodox aquaporins, and Glp aquaglyceroporins [15]. In Viridiplantae, based on sequence homology and potential subcellular localization, the diversity of MIPs is organized into eight subfamilies. Four of these subfamilies are well characterized: PIP (Plasma Membrane Intrinsic Protein), TIP (Tonoplast Intrinsic Protein), NIP (Nodulin 26-like Intrinsic Protein), and SIP (Small and Basic Intrinsic Protein) [16,17]. A fifth subfamily, XIP (Uncharacterized X-Intrinsic Protein), was identified in some bryophytes and dicots [18,19]. Three additional subfamilies -GIP (GlpF-Like Intrinsic Protein), HIP (Hybrid Intrinsic Protein), and LIP (Large Intrinsic Proteins)- have been described in more basal plant groups. GIP and HIP were identified in the bryophyte Physcomitrella patens [18] and the lycophyte Selaginella moellendorffii [20], respectively, while LIP has been found exclusively in some algae, such as the Ochrophytes [21]. In bacteria, approximately 10% of studied genomes encode MIPs, which fall into two groups: AQP and AQGP (or Glp) [2,22] [. However, the increasing power of phylogenetic models and the availability of annotated genomes from various taxa are revealing a complex evolutionary history of both Archaeal and bacterial aquaporins, categorized into four main types: AqpZ, AqpN, AqpM, and GlpF [15,23]. Finally, as the focus of this special issue, fungal MIPs have been classified into three major subfamilies: the fungal “orthodox” aquaporins (AQPs), the fungal aquaglyceroporins (AQGPs), and the fungal uncategorized (X)-intrinsic proteins (XIPs) [23,24,25].
Although AQPs are remarkably conserved in their core structural architecture and function as permeases for a variety of solutes essential for life, they paradoxically exhibit considerable diversity in the residues that shape them. Numerous future studies will be need to support and refine our understanding of the evolutionary dynamics of AQPs across the major clades of life. However, it is already striking that the evolution of most major AQPs groups closely correlates with the diversification of the biosphere, as reflected in the tree of life. There are, however, notable exceptions, such as the XIPs, which appear to be shared exclusively by a few plant and fungal groups. This subfamily warrant particular attention, as its original nomenclature suggests that it remains poorly studied and the diversity of its members is still largely unexplored.
2. MIP Fungal Diversity and Function
Fungi constitute one of the five kingdoms of the Eucaryota life domain and are likely the most species-rich group of eukaryotic organisms after insects [26]. A recent reassessment of global fungal diversity suggests that the total number of fungal species may range from 2.2 to 5 million worldwide [27]. According to widely accepted classifications and alternative simplifications [28,29,30,31], more than 200 fungal orders are classified into 19 phyla, which are divided into six major groups: the subkingdoms Dikarya (comprising the phyla Ascomycota and Basidiomycota) and Chytridiomyceta (comprising the phyla Chytridiomycota, Monoblepharidomycota, and Neocallimastigomycota), the phyla Mucoromycota, Zoopagomycota, and Blastocladiomycota, and the major group Opisthosporidia (comprising the phyla Aphelidiomycota, Cryptomycota/Rozellomycota, and Microsporidia) (Figure 2). Among the relatively small fraction of fungal species currently described, 2,683 annotated/reference genomes are available through the JGI’s MicoCosm portal [32], and 5,028 through the NCBI portal, covering the major phyla that structure the fungal kingdom (April 2025).
In contrast to the remarkable diversity of fungal species evolving in extraordinarily varied biotopes, the diversity of fungal MIPs appears to be more limited than that observed in animals and plants. Fungal genomes typically contain an average of 4.4 MIP members, with a range of 1 to 20 (Figure 2). Notably, the highest numbers of MIP members are found in four clades of filamentous fungi: two in Basidiomycota (Pucciniomycotina and Dacrymycetes), and two in Ascomycota (Dothideomycetes and Leotiomycetes). Basal fungal lineages (according to the classification of [31] exhibit a low number of MIPs, with some species possessing only a single member: Aphelidiomycota, Cryptomycota, Microsporidia, and Neocallimastigomycetes. However, it remains challenging to determine whether their single member should be classified as an AQP or an AQGP based solely on sequence alignment, phylogeny analysis, and the presence of certain predicted protein motifs. Functional studies using heterologous expression systems are necessary to resolve this issue.
For clades with multiple MIP members, they are classified into three main subfamilies of fungal MIPs: fungal aquaporins (AQPS, which correspond to water channels, including both “orthodox” and “facultative” types), fungal aquaglyceroporins (AQGPs, which facilitate the transmembrane transport of small uncharged molecules, and includes Fps-like proteins with a conserved regulatory region in the N-terminus, Yfl054p-like proteins with a very long N-terminal extension, and “Other” aquaglyceroporins with significant sequences divergence that do not fit into the Fps-like or Yfl054p-like categories), and the unique fungal X-intrinsic proteins (XIPs) (Figure 3a; Supplementary Figure S1). This classification of fungal MIPs is now widely accepted and comprehensively detailed in several studies [22,24,25,33,34,35,36].
It is important to emphasize that the number of AQPs and AQGPs can vary significantly between closely related species, plausibly due to overlapping functions as bifunctional water/solute channels. This is exemplified by the AQPs identified in the parasites Plasmodium falciparum [37] and Toxoplasma gondii [38]. Similarly, in organisms exhibiting more than five aquaporins, the number of each subtype differs: Trichoderma atroviride contains three AQPs, three AQGPs, and one XIP [25], while Periconia macrospinosa has two AQGPs, six AQPs, and one XIP, with very low sequence identity among subfamilies (Figure 3b). However, it should be stressed that, in comparison with the extensive data available on plant and animal AQPs, the knowledge regarding the structural topologies, regulatory mechanisms, permease capacities, and biological functions of fungal AQPs remains limited. Examples of fungal AQPs, such as those from Saccharomyces cerevisiae [33,39,40,41], Laccaria bicolor [34,42], Terfezia claveryi [43,44], Tuber melanosporum [45], Glomus intraradices [46], Aspergillus glaucus [35], Botrytis cinerea [47], Cenococcum geophilum [48], Trichoderma atroviride [25,49], and Aspergillus niger [50], illustrate some of the structural and biochemical peculiarities of fungal AQPs, as well as their functional significance in various physiological processes of hyphal cells and their interactions with the biotic and/or abiotic environment.
As we have seen, AQPs are involved in a wide range of processes, acting as key regulators of material and gas exchange, hydro-mineral homeostasis, and cellular signaling. On the other hand, beyond their ecological significance, fungi possess considerable industrial potential across various sectors, including agriculture, pollution control, agri-food, and pharmaceuticals. Then, exploring the diversity of fungal aquaporins represent a highly promising avenue for both fundamental and applied research, opening up new opportunities for the scientific community.
3. Case Study of Fungal XIPs – State of Art
3.1. State of Art
The diversity of fungal aquaporins is relatively well understood; however, the XIP subfamily remains largely unexplored. The second objective of this review is to investigate the origin and diversification of X-intrinsic proteins within the evolutionary history of the MIP superfamily in fungi. XIPs constitute the most recently identified subfamily of AQPs, discovered in the era of high-throughput ‘omics’ technologies. Initially discovered in certain non-vascular plant lineages [18], XIPs have also been characterized in some protozoa [18] and fungal species [34,51]. However, this subfamily remains quite distinct within the MIP family, as it is entirely absent from the genome of animals and insects sequenced to date. Similarly, several fungal clades (such as yeasts), Oomycetes, and some plant groups including Coniferophytes [52], Brassicaceae [53], and Monocotyledons [54], also lack the XIP subfamily, which, in plants, has been attributed to functional redundancy [55].
XIPs are localized on the plasma membrane of plant cells [19,56]. Phylogenetic analyses suggest that they are closely related to orthodox AQPs, including those of fungi [49,50]. XIPs exhibit high transport activity for small solutes such as H2O2 and glycerol, although they show variable activity for water, as demonstrated in heterologous expression experiments using Xenopus oocytes or yeast models [19,56]. The biological functions of XIPs have primarily been characterized in plants [19,56,57,58,59,60,61]. Like other MIPs, XIPs are hypothesized to function as significant cellular checkpoints, regulating permeability and osmolarity; however, their biological significance remains largely speculative.
This knowledge gap is even more pronounced in the context of fungi. To date, the most comprehensive structural and functional studies on fungal XIPs come from two studies: one on the Sordariomycetes species, Trichoderma atroviride [49], a beneficial fungus for sustainable and eco-friendly agriculture, and another on, the Eurotiomycetes species, Aspergillus niger, a major species in the chemical industry (microbial fermentations) and in medicine (as both a food contaminant and a potential human pathogen) [50]. In Trichoderma atroviride, the XIP subfamily consists of a single XIP member, which plays a notable role in hyphal development and chlamydosporogenesis. However, despite its involvement in these processes, and plausibly due to compensating effect with other MIP counterparts, mutants lacking XIP do not show significant changes in mycoparasitic activity against three plant pathogens: Botrytis cinerea, Fusarium graminearum, and Rhizoctonia solani. In A. niger, two XIP genes, so-called AQPC and AQPF, are significantly transcribed in response to hydrogen peroxide. However, only AQPF appears to play a role in facilitating the transport this molecule across cellular membranes.
Beyond these studies, several other notable investigations have examined the MIPs of fungal species that possess XIPs in their genomes, such as Tuber melanosporum [45], Cenococcum geophilum [48], and Botrytis cinerea [47]. Paradoxically, none of these studies have specifically addressed the biochemical and/or functional properties of XIPs, with this subfamily consistently overlooked.
3.2. Diversity of the Fungal XIPs – Evolution and Topology
The diversity of fungal XIPs has been examined within the context of the fungal kingdom’s exceptionally rich and complex evolutionary history. Given the limited availability of information on XIPs in the current literature, we assembled a dataset of 950 full-length XIP sequences by mining two major public genomic repositories: JGI (http://genome.jgi-psf.org/) and NCBI (http://www.ncbi.nlm.nih.gov/). This was achieved through a combination of BLAST searches (BLASTp and tBLASTn) and keyword-based queries (i.e., “KOG0223, Aquaporin”, and “PF00230, Major intrinsic protein”), as described in Supplemental file S1. A comprehensive list of sequences, along with their corresponding species and phylum-level classifications, is provided in Supplementary Table S1.
Based on extensive phylogenetic analyses and multiple sequence alignments (methods detailed in Supplemental File S1), eight principal conclusions were drawn (Table 1, and Figure 2 and Figure 4):
1) According to currently available sequenced genomes, XIPs appear to be absent from several fungal lineages. Specifically, no XIP sequences were identified in the Kickxellomycotina (XXIII), Mucoromycotina (XX), Mortierellomycotina (XIX), Taphrinomycotina (XVII), Wallmiomycetes (VI), and Ustilaginomycotina (II) (Figure 2). Similarly, XIPs are entirely absent from all lineages within the Opisthosporidia subdivision (including Aphelidiomycota (XXX), Cryptomycota (XXIX), and Microsporidia (XXVIII)), as well as from all subdivisions of the Chytridiomycota (XXV to XXVII) and the Blastocladiomycotina (XXIV). These basal clades, generally regarded as the earliest-diverging lineages of the fungal kingdom, are predominantly composed of unicellular organisms and are characterized by the production of both motile and non-motile sporangiospores [62,63].
2) In their review of fungal AQPs, Nehls and Dietz [35] reported the presence of a putative XIP sequence in a species belonging to the phylum Microsporidia. Although the specific origin of this sequence remains unspecified, its mention is nonetheless significant, as it highlights the limitations inherent to bioinformatic approaches commonly used to characterize specific subfamilies. Unless this observation stems from an annotation error or the inadvertent incorporation of foreign genetic material into public databases -an event we have not yet been able to confirm- our analyses suggest that Microsporidia do not possess any genuine XIP members (Supplementary Figure S2 and Supplementary Table S1). tBLASTn searches and keyword-based queries conducted in both the JGI and NCBI databases consistently retrieve aquaporin sequences that are not related to the XIP subfamily. Furthermore, the protein translations of these candidate sequences exhibit very low sequence identity with known XIPs (approximately 22%), and phylogenetic analyses -including XIP sequences from both closely and distantly related fungal species- do not support the hypothesis that Microsporidia harbor members of this aquaporin subfamily. This conclusion appears to extend to all members of the Chytridiomycetes and Opisthosporidia as well. Continued efforts to identify new species of basal fungi, particularly those inhabiting underexploited environments, combined with expanded genome sequencing initiatives, will be crucial for enriching and potentially refining our understanding of aquaporin diversity and evolution within these early-diverging fungal lineages.
3) The abundance of XIPs is markedly lower compared to that of the AQP and AQGP subfamilies. In the vast majority of fungal species, the XIP subfamily is represented by a single gene copy, a trend particularly evident in six clades: three within Agaricomycotina (II, IV, and V), as well as Orbiliomycetes (VIII), Arthoniomycetes (XI), and Saccharomycotina (XVI). This limited diversity among fungal XIPs is reminiscent of the low number of XIP members observed in plants, as previously reported by Verma et al. [23], and may reflect evolutionary constraints or functional specificity that limit the expansion of this subfamily.
4) A striking concordance emerges between the phylogenetic distribution of XIPs and the currently established fungal phylogeny, suggesting an evolutionary trajectory in which most XIP members have diversified in a lineage-specific manner across multiple distinct fungal clades (Figure 4; Supplementary Figure S3). This distribution pattern parallels the divergence of major fungal phyla, implying that XIPs may have undergone multiple and independent expansion events throughout fungal evolutionary history. Notably, such lineage-specific expansions of XIP genes in fungi closely mirror those documented in plants [19,54], further supporting the hypothesis of convergent evolutionary strategies among eukaryotes adapting to diverse environmental niches.
5) In contrast to AQPs and AQGPs members, XIPs display an uneven distribution across fungal clades, a pattern that persists even at finer taxonomic resolutions. For example, the class Sordariomycetes (XIV) contains the highest number of identified XIPs, totaling 271 sequences. However, these sequences are not uniformly distributed across all subclasses. XIP members have been identified in several orders belonging to four subclasses: Diaporthomycetidae, Sordariomycetidae, Xylariomycetidae, and Hypocreomycetidae (Supplementary Figures S4 and S5). To date, no XIP sequences have been detected within the subclasses Savoryellomycetidae and Lulworthiomycetidae. This observation highlights the uneven retention of XIPs across fungal lineages and raises questions about the evolutionary and ecological factors underlying the differential presence of this subfamily among closely related taxonomic groups.
6) Fungal XIPs, based on their nucleotide sequences, are phylogenetically grouped into three well-supported clades (Figure 4, Supplementary Figure S3). The first (Clusters A) consists exclusively of members of the Pucciniomycotina (I); the second (Clusters B) comprises a subset of divergent sequences from the Agaricomycetes (III), and the third (Clusters C), which encompasses 96% of the retrieved members, forms the largest group. Notably, this major clade is sub-partitioned into two distinct branches, hereafter referred to as Clusters C1 and C2. This bipartite structure, described here for the first time, is consistently observed in fungal species that harbor multiple XIP paralogs. Despite a relatively low average sequence similarity across the four clusters, approximately 39%, all proteins within both clusters retain conserved molecular signatures characteristic of the XIP family [25,51]. This phylogenetic organization underscores an unexpected level of diversity and divergence within the fungal XIP subfamily. Importantly, the taxonomic distribution across the two clusters is markedly uneven. Cluster C2 contains the most divergent sequences, spanning a broad range of fungal phyla, from basal lineages (XVIII, XXI, and XXII) to more recently evolved groups such as the Dikarya. In contrast, Cluster C1 is restricted to the Dikarya and includes representatives from only a subset of its constituent phyla. Within the Dikarya, certain major phyla are represented exclusively in one cluster: for example, Pucciniomycotina, Pezizomycetes, and Lecanoromycetes in Cluster C2, and Dacrymycetes, Orbiliomycetes, and Leotiomycetes in Cluster C1. Intriguingly, some sequences (highlighted with an asterisk in Figure 4) do not cluster with their expected fungal clades, but instead appear among distantly related fungal groups. This applies to several phyla (e.g., Eurotiomycetes (IX), Dothideomycetes (X), Sordariomycetes (XIV) and Leotiomycetes (XIII)); however, this pattern is particularly noticeable in Tremellomycetes (V), which fails to form a distinct cluster. These atypical phylogenetic placements suggest plausible horizontal gene transfer (HGT) between fungal species, a phenomenon previously reported for other fungal proteins [64,65]. HGT, coupled with the stable integration of the transferred genetic material, may confer adaptive advantages to the host, including enhanced environmental responsiveness and the acquisition of novel traits and functions [66,67]. This hypothesis warrants further dedicated investigation.
Revisiting the example of the Sordariomycetes (XIV), the two identified XIP clusters reflect the four major lineage-specific subdivisions within this class: i) Diaporthomycetidae (orders: Diaporthales, Togniniales, Magnaporthales, and Ophiostomatales), ii) Sordariomycetidae (orders: Sordariales and Chaetosphaeriales), iii) Xylariomycetidae (order: Xylariales), and iv) Hypocreomycetidae (orders: Glomerellales, Microascales, and Hypocreales) (Supplementary Figure S4). While Cluster C2 includes representatives from all these taxonomic groups, Cluster C1 is restricted to sequences from Diaporthomycetidae and Hypocreomycetidae. Notably, two sequences from Apiospora montagnei (order: Xylariales) fall within Cluster C2, and display a highly degree of similarity (62% identity), likely reflecting a recent gene duplication event. In contrast, Trichoderma virens (order: Hypocreales) possesses two highly divergent XIP homologs (33% identity) that segregate into both Clusters C1 and C2, suggesting the presence of two ancestral “XIP archetypes“ prior to the diversification of the Sordariomycetes lineage. This pattern appears to be broadly representative of the major fungal phyla distributed across the two clusters.
Collectively, these findings support the hypothesis that the four XIP clusters originated from ancient out-paralogs that arose in the common ancestor of the Dikarya, followed by lineage-specific losses of one cluster over evolutionary time. To the best of our knowledge, this is the first report of such a dichotomous organization among fungal XIPs, which, intriguingly, closely mirrors the phylogenetic structuring reported for plant XIPs within the Viridiplantae [19,54]. This similarity reinforces the notion of a conserved evolutionary trajectory across two distantly related eukaryotic lineages: fungi and plants.
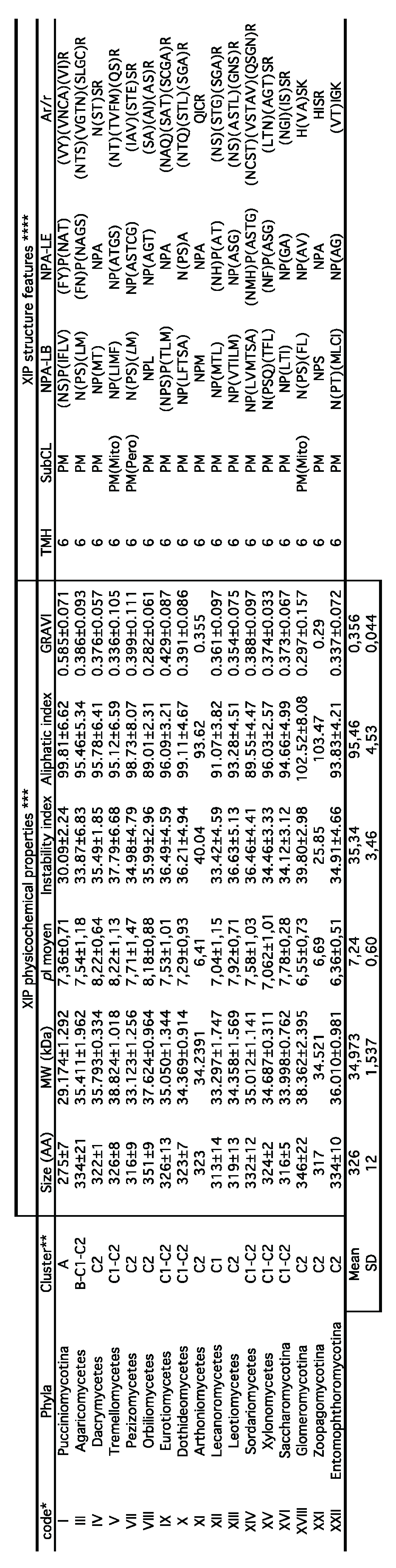
7) The amino acid length of fungal XIPs ranges from 274 to 362 residues, with an average of 326, corresponding to theoretical molecular weights between 29.17 and 38.82 kDa (mean: 34.97) (Table 1). The isoelectric points (pI) of these proteins span a range from 6.41 to 8.22, suggesting functional constraints potentially linked to regulatory mechanisms. These include conserved regulatory motifs in the C-terminal regions and in several electrostatic loops, which may be involved in post-translational modifications such as methylation, phosphorylation, and trafficking, as well as interactions with regulatory partners. Such modifications, known to vary among MIP families and subfamilies, underscore the multifunctional and dynamic regulation of solute permeability mediated by AQPs; this phenomenon is-documented in plants and animals [68,69], but still insufficiently characterized in filamentous fungi. Additionally, XIPs exhibit favorable biochemical properties, including high intrinsic stability (instability index <40), and thermal resilience, as indicated by an average aliphatic index of 95. These features are characteristic of canonical MIP protomers. In silico predictions of subcellular localization predominantly place XIPs in the plasma membranes; however, a marginal presence in other cellular compartments -such as the mitochondria, endoplasmic reticulum, and peroxisomes- cannot be excluded. In this context, the positive GRAVY (Grand Average of Hydropathy) index of 0.389 further supports their hydrophobic nature, a fundamental trait for aquaporins that facilitates efficient transmembrane transport of solutes [70].
8) As expected for members of the MIP superfamily, the protomeric architecture of all fungal XIPs is highly conserved and adopts the canonical hourglass configuration. This structure comprises six transmembrane helices (TMH1 to TMH6), connected by five inter-helical loops (Loop A to E), with both the N- and C-terminal regions oriented toward the cytosolic side (Figure 3C and Table 1). Centrally located within the pore are two half-helices, derived from loops B and E, each capped by conserved “NPA” motifs, along with the ar/R selectivity filter. The latter is composed of specific hydrophobic amino acids whose side chains line the pore and form a narrow constriction at the extracellular entrance. Although this ar/R filter appears to be somewhat less hydrophobic that its counterparts in dicotyledonous plants and moss [51], it is likely optimized for the transmembrane passage of relatively large and/or hydrophobic solutes, such as glycerol, urea, ammonia (NH3), lactic and boric acids, polyols, and various metalloids or ions including arsenic (As), antimony (Sb), zinc (Zn2+), aluminum (Al3+), silicon (Si), and selenium (Se). Notably, there is considerable variability in the amino acid residues lining the channel both between and within fungal phyla, which may reflect adaptations to distinct physiological environments and suggest divergent substrate specificities of XIPs across fungal lineages.
3.3. Fungal XIPs: A Relatively Recent Chapter in the Evolution of Fungal AQP
The most ancient fungal phyla in which genomes contain at least one XIP-encoding gene belong to Zoopagomycota, specifically within the subphyla Zoopagomycotina and Entomophthoromycotina. Zoopagomycota represent the most basal lineage among zygomycetous fungi and are notably associated with three major phenotypic transitions in the fungal evolutionary history: successful adaptation to terrestrial environments (terrestrialization), the emergence of zygospores and novel sexual reproductive structures, and the acquisition of the spindle pole body (SPB) [63]. These findings support the hypothesis that fungal XIPs emerged more recently than the two other major aquaporin subgroups –AQPs and AQGPs-, as previously suggested by [24]. This emergence is estimated to have occurred approximately 450 million years ago (Mya), potentially coinciding with or shortly following the divergence of the Mucoromycota and Dikarya subkingdoms, which occurred between 350 and 400 Mya. It is therefore plausible to propose that fungal XIPs originated from a common ancestral gene that duplicated early in the evolutionary history of the fungal kingdom, followed by a significant diversification burst, particularly within the Dikarya. This evolutionary window overlaps with the Silurian and Devonian geological periods, a critical epoch in the history of eukaryotic biodiversity. This period is marked by increased terrestrialization events, the emergence and complexification of multicellular body plans (exemplified in fungi by the development of non-flagellated, multicellular thalli composed of apically growing and branching hyphae, whose development and spatial organization exhibit characteristic analogous to fractal geometry), the expansion of terrestrial biodiversity, and the plateauing of atmospheric O2 concentrations at approximately 21% [71,72,73]. Interestingly, the timing of complex innovation in the fungal multicellularity coincides with the explosive diversification of terrestrial animals and plants, suggesting a possible co-evolutionary relationship between fungi and Embryophyta (land plants), which also experienced a major radiation around 430-450 Mya [74,75].
Within this evolutionary and ecological context, the expansion of the XIP family appears to have paralleled the early colonization of terrestrial habitats by emerging eukaryotic biodiversity. Nonetheless, despite these broadly similar evolutionary trajectories, the expansion and diversification of the XIP subfamily in fungi appears to have been more constrained than in plants. While land plant genomes frequently encode multiple XIP isoforms [19,54], fungal genomes typically harbor only a single XIP candidate per species. This pattern suggests that, although the XIP archetype has been maintained across both fungal and plant lineages, its evolutionary expansion has followed markedly divergent trajectories. These differences likely reflect distinct functional roles and regulatory constraints within the respective cellular architectures and ecological niches of fungi and plants.
3.4. Fungal XIP – Outstanding Questions
Despite the high sequence diversity among XIP homologs, accumulating evidence supports the hypothesis that XIPs have a deep evolutionary origin [76]. This is substantiated by the presence of key conserved residues and a set of synapomorphic amino acids identified in XIPs across a broad spectrum of eukaryotic lineages, including Angiosperms, Bryophytes, Dyctyostelium (Amoebozoa), Chlorarachniophyte algae (Rhizaria), a Stramenopile (diatom), and fungi [22,51]. Although absent in many Protists, Oomycetes, and Metazoans, available evidence suggests that an ancestral XIP-like protein was already present in certain early eukaryotic ancestors. This ancestral archetype likely underwent extensive diversification, potentially driven by ancient burst of gene duplication, which, according to the most recent estimates of the eukaryotic molecular clock, may have occurred over 2 billion years ago [77,78].
Within the fungal kingdom, the phylogenetic topology of the XIP subgroup is paraphyletic, encompassing members with varying degrees of relatedness. This paraphyly implies the emergence of a structural and functional diversification of fungal XIPs, potentially shaped by distinct ecological and physiological pressures, such as nutritional strategies, habitat specificity, or biotic interactions. The retention of XIP genes in some fungal lineages, alongside their absence in others, highlights unresolved questions concerning the selective forces that may underpin their maintenance or loss. Elucidating the roles of XIPs in fungal growth, development, and solute homeostasis could provide key insights into their evolutionary significance. Notably, only a limited number of fungal species within each phylum possess identifiable XIP orthologs.
Given the current state of limited knowledge, no clear functional rationale has yet emerged to explain the selective presence of the XIP family in certain fungal species. This uncertainty gives rise to several key questions:
- Does the expansion and relative conservation of the XIP subgroup within the Dikarya, together with its apparent correlation to the terrestrialization of the fungal kingdom, point to a specialized function for these proteins in transporting solutes essential to the full developmental cycle of these fungi?
- Could the concomitant presence of XIP homologs in both fungal and green plant lineages be associated with their parallel evolutionary success of in terrestrial ecosystems? This prompts broader questions regarding the potential roles of XIPs as an evolutionary link between these two kingdoms, whose ecological interdependence is both diverse and deep. Despite their distinct phylogenetic classifications, remarkably, XIP proteins in plants and fungi share striking structural similarities, hinting at a plausible shared ancestral origin.
- Do the two divergent XIP archetypes observed across fungal species reflect a degree of functional specialization, potentially involving distinct solute transport profiles and regulatory mechanisms, or are they associated with tissue-specific expression patterns in multicellular organisms, indicative of sub-functionalization?
Expanding the scope of phylogenetic analyses to include a broader range of XIP sequences should improve the resolution of these outstanding questions. More broadly, unlike MIP families in animals and plants, fungal MIP repertories are typically composed of a relatively small number of isoforms. This trend is particularly pronounced for the XIP group, which is generally represented by a single isoform in the majority of fungal species. Additionally, the spectrum of substrates transported by fungal MIPs -especially XIPs- remains largely uncharacterized. This raises the possibility that the limited number of fungal MIP channels may have evolved multifunctional capabilities, thereby compensating for reduced isoform diversity through functional versatility adapted to the wide array of ecological niches that fungi inhabit.
Collectively, these observations offer novel perspectives on the classification and evolutionary history of fungal XIPs, and lay the groundwork for future investigations into their functional and ecological relevance.
4. Conclusion
The extensive body of research on aquaporins (AQPs) has yielded highly detailed insights into their diversity, topology, function, regulatory mechanisms, transported solutes and evolutionary trajectories across biological kingdoms. While AQPs in plant and animal are the most thoroughly studied, largely due to their economic and biomedical relevance, substantial knowledge gaps persist regarding fungal AQPs, particularly those belonging to less common subfamilies such as XIPs. A key current challenge is to achieve a comprehensive understanding of how fungal AQPs, AQGPs, and XIPs contribute to fundamental biological processes, including growth, development, defense mechanisms, and overall fitness under fluctuating environmental conditions. This entails elucidating their roles in diverse fungal lifestyle strategies and their involvement in the establishment and evolution of specialized interactions with surrounding organisms, including animals, insects, and plants.
The number of functionally characterized fungal aquaporins remains limited, making it difficult to draw generalizable conclusions. However, in light of the animal models and the evolutionary complexity of fungal cell biology, it is reasonable to anticipate that in fungal genomes encoding multiple potentially expressible AQP isoforms, these proteins exhibit specialized expression patterns and subcellular localization, depending on developmental stage and specific cellular structures. Physiologically, AQPs allow for the rapid modulation of water permeability in response to osmotic stress, particularly during the influx and efflux of solutes between the cytosol and the extracellular environment. Likewise, AQPs facilitate the uptake of key metabolic precursors and nutrients (e.g., glycerol, nitrogen, gases, etc.), as well as the expulsion of potentially toxic metabolic byproducts, such as methylglyoxal or ammonia. In addition, from a therapeutic perspective, fungal AQPs have already emerged as promising drug targets and potential gateways for antifungal compounds [79]. The development of high-affinity and highly specific inhibitors targeting these channels remains a critical avenue for future research, with potential applications in the treatment of severe cryptogamic diseases affecting both plants and animals.
In brief, a major research priority will be to predict and characterize the diverse ecological functions of MIPs across the fungal kingdom, guided by large-scale structural and phylogenetic analyses. Integrating multidisciplinary approaches, from molecular biology and structural bioinformatics to ecology and biotechnology, will be essential for advancing our understanding of the physiological roles of AQP subclasses and their contributions to fungal biology.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org. Supplementary Files S1 - The materials and methods used to generate the new data that supplement this review. Supplementary Figure S1. Phylogenetic analysis of full-length aquaporin nucleotide sequences from one species of each major fungal phylum detailed on JGI's MycoCosm portal. The assignment of the AQP, AQGP, Yfl054-like, Fps-like, “Other” AQGP, and XIP subgroups is predicted based on the characterized heterologous AQP sequences of Trichoderma atroviride [25] and Aspergillus niger [50] (highlighted by one or two asterisks, resp.). Species, and related sequences are listed in Supplementary Table S1. Tree Inference was done on the maximum-likelihood (1,000 bootstrap replicates), using FastTree program and based on a MAFFT alignment, and edited with ITOL V7. Supplementary Figure S2. Phylogenetic distribution of all MIPs extracted from the Chytridiomycota and Opisthosporidia phyla (MycoCosm portal of JGI), with one XIP member from each phylum that possesses one. These XIP are those used in the phylogeny presented in Figure 3a. All the MIP sequences of the basal phyla are available in Supplementary Table S2, and the XIP sequences in Supplementary Table S1. The assignment of the AQP and AQGP subgroups is putatively predicted based on an alignment that includes the characterized orthodox AQP, AQGP, Yfl054-like, Fps-like, “Other” AQGP, and XIP sequences of Trichoderma atroviride (highlighted by an asterisk) [25]. Sequences for which it is uncertain to assign a subgroup without further biochemical and bioinformatic analysis are named MIP and colored in purple. Tree Inference was done on the maximum-likelihood (1,000 bootstrap replicates). Supplementary Figure S3. Detailed view of the maximum likelihood (1,000 bootstrap replicates) tree based on 950 full-length fungal XIP nucleotide sequences, presented in Figure 4. The scale bar represents the number of amino acid substitutions per site. The complete fungal names corresponding to the sequences are provided in Supplementary Table S1. Supplementary Figure S4. Maximum-likelihood phylogenetic tree of 271 nucleotide sequences from Sordariomycetes. The bootstrap values indicated at the nodes are based on 1,000 bootstrap replicates. Branch values lower than 50% are hidden. The tree scale denotes the evolutionary distance expressed at the number of nucleotide substitutions per site. Clusters C1 and C2 correspond to the primary clusters illustrated in Figure 4. Accession numbers and sequences of XIP are provided in Supplementary Table S1. Supplementary Figure S5. Modified representation of Sordariomycetes Maximum Likelihood phylogenetic tree from 345 taxa (100 bootstraps), based on Hongsanan et al [80]. Orders containing fungal species with identified XIP sequences in their genomes are highlighted in red. Branch lengths are not proportional to genetic distances.
Author Contributions
Conceptualization, J.-S.V.; Methodology, B.F., G.B., M.S., M.F.; Software, GB, B.F., J.-S.V.; Validation, B.F., J.-S.V; Formal analysis, GB, B.F., P;R.D., J.-S.V.; Investigation, B.F., J.-S.V.; Writing – original draft, J.-S.V.; Writing – review & editing, B.F., G.B., M.S., M.F., P.R.D., J.-S.V; Project Administration, J.-S.V ; Funding Acquisition, J.-S.V.
All authors contributed to review the whole manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no funding.
Data availability
All data generated or analyzed during this study are included in the supplementary information files.
Acknowledgments
The authors express their gratitude to all contributors who have made fungal genome data publicly available through the databases of the Joint Genome Institute (JGI) and the National Center for Biotechnology Information (NCBI). We are grateful to the Mésocentre Clermont-Auvergne, and the plateforme AuBi of the University Clermont Auvergne for providing help, computing, and storage resources. Computations have been performed on the supercomputer facilities of the Mésocentre Clermont-Auvergne of the University Clermont Auvergne. We are indebted to the anonymous reviewers for their helpful suggestions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Agre, P.; Sasaki, S.; Chrispeels, M.J. Aquaporins: a family of water channel proteins. American Journal of Physics 1993, 265, F461. [Google Scholar] [CrossRef] [PubMed]
- Zardoya, R. Phylogeny and evolution of the major intrinsic protein family. Biology of the Cell 2005, 97, 397–414. [Google Scholar] [CrossRef] [PubMed]
- Gazzarrini, S.; Kang, M.; Epimashko, S.; Van Etten, J.L.; Dainty, J.; Thiel, G.; et al. Chlorella virus MT325 encodes water and potassium channels that interact synergistically. Proceedings of the National Academy of Sciences of USA 2006, 103, 5355–5360. [Google Scholar] [CrossRef] [PubMed]
- Fujiyoshi, Y.; Mitsuoka, K.; de Groot, BL.; Philippsen, A.; Grubmüller, H.; Agre, P.; Engel, A. Structure and function of water channels. Current Opinion in Structural Biology 2002, 12, 509–515. [Google Scholar] [CrossRef]
- Ozu, M.; Galizia, L.; Acuña, C.; Amodeo, G. Aquaporins: More Than Functional Monomers in a Tetrameric Arrangement. Cells 2018, 7, 209. [Google Scholar] [CrossRef]
- Luu, D.T.; Maurel, C. Aquaporins in a challenging environment: molecular gears for adjusting plant water status. Plant, Cell & Environment 2005, 28, 85–96. [Google Scholar]
- Tajkhorshid, E.; Nollert, P.; Jensen, M.; Miercke, L.; O’Connell, J.; Stroud, R.M.; Schulten, K. Control of the selectivity of the aquaporin water channel family by global orientation tuning. Science 2002, 296, 525–530. [Google Scholar] [CrossRef]
- Sui, H.; Han, B.G.; Lee, J.K.; Walian, P.; Jap, B.K. Structural basis of water specific transport through the AQP1 water channel. Nature 2001, 414, 872–878. [Google Scholar] [CrossRef]
- Wallace, I.S.; Roberts, D.M. Homology modeling of representative subfamilies of Arabidopsis major intrinsic proteins. Classification based on the aromatic/arginine selectivity filter. Plant Physiology 2004, 135, 1059–1068. [Google Scholar] [CrossRef]
- Fu, D.; Libson, A.; Miercke, L.J. : Weitzman, C.; Nollert, P.; Krucinski, J.; Stroud, R.M. Structure of a glycerol-conducting channel and the basis for its selectivity. Science 2000, 290, 481–486. [Google Scholar] [CrossRef]
- Wang, Y.; Schulten, K.; Tajkhorshid, E. What makes an aquaporin a glycerol channel? A comparative study of AqpZ and GlpF. Structure 2005, 13, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Wu, B.; Beitz, E. Aquaporins with selectivity for unconventional permeants. Cellular and Molecular Life Sciences 2007, 64, 2413–2421. [Google Scholar] [CrossRef] [PubMed]
- Saboe, P. O,. Rapisarda, C,. Kaptan, S,. Hsiao, Y.S,. Summers, S.R,. De Zorzi, R,. et al. Role of Pore-Lining Residues in Defining the Rate of Water Conduction by Aquaporin-0. Biophysical Journal 2017, 112, 953–965. [Google Scholar] [CrossRef] [PubMed]
- Pluhackova, K.; Schittny, V.; Bürkner, P.C.; Siligan, C.; Horner, A. Multiple pore lining residues modulate water permeability of GlpF. Protein Science 2022, 31, e4431. [Google Scholar] [CrossRef]
- 15 Finn, R.N.; Cerdà, J. Evolution and functional diversity of aquaporins. Biol. Bull. 2015, 229, 6–23. [Google Scholar] [CrossRef]
- Johanson, U.; Karlsson, M.; Johansson, I.; Gustavsson, S.; Sjovall, S.; Fraysse, L.; Weig, A.R.; Kjellbom, P. The complete set of genes encoding major intrinsic proteins in Arabidopsis provides a framework for a new nomenclature for major intrinsic proteins in plants. Plant Physiology 2001, 126, 358–1369. [Google Scholar] [CrossRef]
- Maurel, C. Plant aquaporins: Novel functions and regulation properties. FEBS Letters 2007, 581, 2227–2236. [Google Scholar] [CrossRef]
- Danielson, J.Å.H.; Johanson, U. Unexpected complexity of the aquaporin gene family in the moss Physcomitrella patens. BMC Plant Biology 2008, 8, 1–15. [Google Scholar] [CrossRef]
- Lopez, D.; Bronner, G.; Brunel, N.; Auguin, D.; Bourgerie, S.; Brignolas, F.; et al. Insights into Populus XIP aquaporins: evolutionary expansion, protein functionality, and environmental regulation. Journal of Experimental Botany 2012, 65, 2217–2230. [Google Scholar] [CrossRef]
- Anderberg, H.I.; Kjellbom, P.; Johanson, U. Annotation of Selaginella moellendorffii major intrinsic proteins and the evolution of the protein family in terrestrial plants. Front. Plant Sciences 2012, 3, 1–14. [Google Scholar] [CrossRef]
- Khabudaev, K.V.; Petrova, D.P.; Grachev, M.A.; Likhoshway, Y.V. A new subfamily LIP of the major intrinsic proteins. BMC Genomics 2014, 15, 173. [Google Scholar] [CrossRef] [PubMed]
- Abascal, F.; Irisarri, I.; Zardoya, R. Diversity and evolution of membrane intrinsic proteins. Biochimica et Biophysica Acta 2014, 1840, 1468–1481. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.K.; Prabh, N.D.; Sankararamakrishnan, R. New subfamilies of major intrinsic proteins in fungi suggest novel transport properties in fungal channels: Implications for the host-fungal interactions. BMC Ecology and Evolution 2014, 14, 173. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Cooke, J.E.K.; Zwiazek, J.J. Phylogenetic analysis of fungal aquaporins provide insight into their possible role in water transport of mycorrhizal associations. Botany 2013, 91, 495–504. [Google Scholar] [CrossRef]
- Ben Amira, M.; Mom, R.; Lopez, D.; Chaar, H.; Khouaja, A.; Pujade-Renaud, V.; Fumanal, B.; et al. MIP diversity from Trichoderma: Structural considerations and transcriptional modulation during mycoparasitic association with Fusarium solani olive trees. PLoS ONE 2018, 13, e0193760. [Google Scholar] [CrossRef]
- Purvis, A.; Hector, A. Getting the measure of biodiversity. Nature 2000, 405, 212–219. [Google Scholar] [CrossRef]
- Hawksworth, DL.; Luecking, R. Fungal diversity revisited: 2.2 to 3.8 million species. Microbiol Spectrum 2017, 5, FUNK-0052-2016. [Google Scholar] [CrossRef]
- James, T.Y.; Stajich, J.E.; Hittinger, C.T.; Rokas, A. Toward a Fully Resolved Fungal Tree of Life. Annual Review of Microbiology 2020, 74, 291–313. [Google Scholar] [CrossRef]
- Wijayawardene, N.N.; Hyde, K.D.; Al-Ani, L.; Tedersoo, L.; Haelewaters, D.; Rajeshkumar, K.C.; et al. Outline of Fungi and fungus-like taxa. Mycosphere 2020, 11, 1060–1456. [Google Scholar] [CrossRef]
- Baldrian, P.; Větrovský, T.; Lepinay, C.; Kohout, P. High-throughput sequencing view on the magnitude of global fungal diversity. Fungal Diversity 2021, 114, 539–547. [Google Scholar] [CrossRef]
- Li, Y.; Steenwyk, J.L.; Chang, Y.; Wang, Y.; James, T.Y.; Stajich, J.E.; et al. A genome-scale phylogeny of the kingdom Fungi. Curr Biol. 2021, 31, 1653–1665. [Google Scholar] [CrossRef] [PubMed]
- Grigoriev, I.V.; Nikitin, R.; Haridas, S.; Kuo, A.; Ohm, R.; Otillar, R.; et al. MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Research 2014, 42, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, N.; Filipsson, C.; Becit, E.; Brive, L.; Hohmann, S. Aquaporins in yeasts and filamentous fungi. Biology of the Cell 2005, 97, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Dietz, S.; Bülow, J.V.; Beitz, E.; Nehls, U. The aquaporin gene family of the ectomycorrhizal fungus Laccaria bicolor: Lessons for symbiotic functions. New Phytologist 2011, 190, 927–940. [Google Scholar] [CrossRef]
- Nehls, U.; Dietz, S. Fungal aquaporins: Cellular functions and ecophysiological perspectives. Applied Microbiology and Biotechnology 2014, 98, 8835–8851. [Google Scholar] [CrossRef]
- Xu, H.; Zwiazek, JJ. Fungal Aquaporins in Ectomycorrhizal Root Water Transport. Frontiers in Plant Science 2020, 11, 302. [Google Scholar] [CrossRef]
- Hansen, M.; Kun, J.F.; Schultz, J.E.; Beitz, E. A single, bi-functional aquaglyceroporin in blood-stage Plasmodium falciparum malaria parasites. Journal of Biological Chemistry 2002, 277, 4874–4882. [Google Scholar] [CrossRef]
- Pavlovic-Djuranovic, S.; Schultz, J.E.; Beitz, E. A single aquaporin gene encodes a water/glycerol/urea facilitator in Toxoplasma gondii with similarity to plant tonoplast intrinsic proteins. FEBS Letters 2003, 555, 500–504. [Google Scholar] [CrossRef]
- Laize, V.; Tacnet, F.; Ripoche, P.; Hohmann, S. Polymorphism of Saccharomyces cerevisiae aquaporins. Yeast 2000, 16, 897–903. [Google Scholar] [CrossRef]
- Sidoux-Walter, F.; Pettersson, N.; Hohmann, S. The Saccharomyces cerevisiae aquaporin Aqy1 is involved in sporulation. Proceedings of the National Academy of Sciences, USA 2004, 101, 17422–17427. [Google Scholar] [CrossRef]
- Pettersson, N.; Hagstrom, J.; Bill, RM.; Hohmann, S. Expression of heterologous aquaporins for functional analysis in Saccharomyces cerevisiae. Current Genetics 2006, 50, 247–255. [Google Scholar] [CrossRef] [PubMed]
- Xu, H. Major Intrinsic Proteins of Laccaria Bicolor: Characterization, Transcript Profiling and Functions in Ectomycorrhizal Associations with Picea glauca. Ph.D. thesis, University of Alberta, Edmonton. 2015, pp 99.
- Navarro-Ródenas, A.; Ruíz-Lozano, J.M.; Kaldenhoff, R.; Morte, A. The aquaporin TcAQP1 of the desert truffle Terfezia claveryi is a membrane pore for water and CO2 transport. Molecular Plant-Microbe Interactions 2012, 25, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Ródenas, A.; Bárzana, G.; Nicolás, E.; Carra, A.; Schubert, A.; Morte, A. Expression analysis of aquaporins from desert truffle mycorrhizal symbiosis reveals a fine-tuned regulation under drought. Molecular Plant-Microbe Interactions 2013, 26, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Hacquard, S.; Tisserant, E.; Brun, A.; Lege, V.; Martin, F.; Kohler, A. Laser microdissection and microarray analysis of Tuber melanosporum ectomycorrhizas reveal functional heterogeneity between mantle and Hartig net compartments. Environmental Microbiology 2013, 15, 1853–1869. [Google Scholar] [CrossRef]
- Li, T.; Hu, Y.J.; Hao, Z.P.; Li, H.; Wang, Y.S.; Chen, B.D. First cloning and characterization of two functional aquaporin genes from an arbuscular mycorrhizal fungus Glomus intraradices. New Phytologist 2013, 197, 617–630. [Google Scholar] [CrossRef]
- An, B.; Li, B.; Li, H.; Zhang, Z.; Qin, G.; Tian, S. Aquaporin-8 regulates cellular development and reactive oxygen species production, a critical component of virulence in Botrytis cinerea. New Phytol. 2016, 209, 1668–1680. [Google Scholar] [CrossRef]
- Peter, M.; Kohler, A.; Ohm, R.A.; Kuo, A.; Krützmann, J.; Morin, E.; et al. Ectomycorrhizal ecology is imprinted in the genome of the dominant symbiotic fungus Cenococcum geophilum. Naure Communication 2016, 7, 1–15. [Google Scholar] [CrossRef]
- Ben Amira, M.; Faize, M.; Karlsson, M.; Dubey, M.; Frąc, M.; Panek, J.; et al. Fungal X-Intrinsic Protein aquaporin from Trichoderma atroviride: structural and functional considerations. Biomolecules 2021, 11, 338. [Google Scholar] [CrossRef]
- Laothanachareon, T.; Asin-Garcia, E.; Volkers, R.J.M.; Tamayo-Ramos, J.A.; Martins dos Santos, V.A.P.; Schaap, P.J. Identification of Aspergillus niger Aquaporins Involved in Hydrogen Peroxide Signaling. Journal of Fungi 2023, 9, 499. [Google Scholar] [CrossRef]
- Gupta, A.B.; Sankararamakrishnan, R. Genome-wide analysis of major intrinsic proteins in the tree plant Populus trichocarpa: characterization of XIP subfamily of aquaporins from evolutionary perspective. BMC Plant Biology 2009, 9, 134. [Google Scholar] [CrossRef]
- Laur, J.; Hacke, U.G. Exploring Picea glauca aquaporins in the context of needle water uptake and xylem refilling. New Phytologist 2014, 203, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Diehn, T.A.; Pommerrenig, B.; Bernhardt, N.; Hartmann, A.; Bienert, G.P. Genome-wide identification of aquaporin encoding genes in Brassica oleracea and their phylogenetic sequence comparison to Brassica crops and Arabidopsis. Front. Plant Sciences 2015, 6, 166. [Google Scholar] [CrossRef] [PubMed]
- Venkatesh, J.; Yu, J.W.; Gaston, D.; et al. Molecular evolution and functional divergence of X-intrinsic protein genes in plants. Mol. Genet. Genomics 2015, 290, 443–460. [Google Scholar] [CrossRef] [PubMed]
- Maurel, C.; Boursiac, Y.; Luu, D.T.; Santoni, V.; Shahzad, Z.; Verdoucq, L. Aquaporins in Plants. Physiological Reviews 2015, 95, 1321–1358. [Google Scholar] [CrossRef]
- Bienert, G.P.; Bienert, M.D.; Jahn, T.P.; Boutry, M.; Chaumont, F. Solanaceae XIPs are plasma membrane aquaporins that facilitate the transport of many uncharged substrates. Plant J. 2011, 66, 306–317. [Google Scholar] [CrossRef]
- Zhang, DY.; Ali, Z.; Wang, CB.; Xu, L.; Yi, JX.; Xu, ZL.; et al. Genome-wide sequence characterization and expression analysis of major intrinsic proteins in Soybean (Glycine max L.). PLoS ONE 2013, 8, e56312. [Google Scholar] [CrossRef]
- Reuscher, S.; Akiyama, M.; Mori, C.; Aoki, K.; Shibata, D.; Shiratake, K. Genome-wide identification and expression analysis of aquaporins in tomato. PLoS ONE 2013, 8, e79052R. [Google Scholar] [CrossRef]
- Venkatesh, J.; Yu, J.W.; Park, S.W. Genome-wide analysis and expression profiling of the Solanum tuberosum aquaporins. Plant Physiology and Biochemistry 2013, 73, 392–404. [Google Scholar] [CrossRef]
- Yue, C.; Cao, H.; Wang, L.; Zhou, Y.; Hao, X.; Zeng, J.; Wang, X.; Yang, Y. Molecular cloning and expression analysis of tea plant aquaporin (AQP) gene family. Plant Physiology and Biochemistry 2014, 83, 65–76R. [Google Scholar] [CrossRef]
- de Paula Santos Martins, C.; Pedrosa, A.M.; Du, D.; Gonçalves, L.P.; Yu, Q.; Gmitter, F.G., Jr.; et al. Genome-Wide Characterization and Expression Analysis of Major Intrinsic Proteins during Abiotic and Biotic Stresses in Sweet Orange (Citrus sinensis L. Osb.). PLoS ONE 2015, 10, e0138786. [Google Scholar]
- Berbee, M.L.; James, T.Y.; Strullu-Derrien, C. Early diverging fungi: diversity and impact at the dawn of terrestrial life. Annual Review of Microbiology 2017, 71, 41–60. [Google Scholar] [CrossRef] [PubMed]
- Voigt, K.; James, T.Y.; Kirk, P.M.; et al. Early-diverging fungal phyla: taxonomy, species concept, ecology, distribution, anthropogenic impact, and novel phylogenetic proposals. Fungal Diversity 2021, 109, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, V.D.A.; Sukno, S.A.; Thon, M.R. Identification of horizontally transferred genes in the genus Colletotrichum reveals a steady tempo of bacterial to fungal gene transfer. BMC Genomics 2015, 16, 2. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Sun, S.; Ge, W.; Zhao, L.; Hou, B.; Wang, K.; et al. Horizontal gene transfer of Fhb7 from fungus underlies Fusarium head blight resistance in wheat. Science 2020, 368, eaba5435. [Google Scholar] [CrossRef]
- Tiwari, P.; Bae, H. Horizontal Gene Transfer and Endophytes: An Implication for the Acquisition of Novel Traits. Plants 2020, 9, 305. [Google Scholar] [CrossRef]
- Liu, F.; Wang, S.; Cheewangkoon, R.; Zhao, R. Uneven distribution of prokaryote-derived horizontal gene transfer in fungi: a lifestyle-dependent phenomenon. mBio 2025, 16, e02855-24. [Google Scholar] [CrossRef]
- Chaumont, F.; Moshelion, M.; Daniels, M.J. Regulation of plant aquaporin activity. Biology of the Cell 2005, 97, 749–764. [Google Scholar] [CrossRef]
- Day, R.E.; Kitchen, P.; Owen, D.S.; Bland, C.; Marshall, L.; Conner, A.C.; Bill, R.M.; Conner, M.T. Human aquaporins: regulators of transcellular water flow. Biochimica et Biophysica Acta 2014, 1840, 1492–1506. [Google Scholar] [CrossRef]
- De Groot, B.L.; Grubmüller, H. Water permeation across biological membranes: Mechanism and dynamics of aquaporin-1 and GlpF. Science 2001, 294, 2353–2357. [Google Scholar] [CrossRef]
- Hanson, K.; et al. Fungi use efficient algorithms for the exploration of microfluidic networks. Small 2006, 2, 1212–1220. [Google Scholar] [CrossRef]
- Nagy, L.G.; Kovács, G.M.; Krizsán, K. Complex multicellularity in fungi: evolutionary convergence, single origin, or both? Biological Reviews 2018, 93, 1778–1794. [Google Scholar] [CrossRef] [PubMed]
- László, G. Nagy, Torda Varga, Árpád Csernetics, Máté Virágh. Fungi took a unique evolutionary route to multicellularity: Seven key challenges for fungal multicellular life. Fungal Biology Reviews 2020, 34, 151–169. [Google Scholar]
- Silberfeld, T.; Leigh, J.W.; Verbruggen, H.; Cruaud, C.; de Reviers, B.; Rousseau, F. A multi-locus time-calibrated phylogeny of the brown algae (Heterokonta, Ochrophyta, Phaeophyceae): Investigating the evolutionary nature of the “brown algal crown radiation”. Molecular Phylogenetics and Evolution 2010, 56, 659–674. [Google Scholar] [CrossRef]
- Lutzoni, F.; Nowak, M.D.; Alfaro, M.E.; Reeb, V.; Miadlikowska, J.; Krug, M.; et al. Contemporaneous radiations of fungi and plants linked to symbiosis. Nature Communication 2018, 9, 5451. [Google Scholar] [CrossRef] [PubMed]
- Irisarri, I.; Lorente-Martínez, H.; Strassert, J.F.H.; Zardoya, A.A.R.; San Mauro, D.; de Vries, J. Early Diversification of Membrane Intrinsic Proteins (MIPs) in Eukaryotes. Genome Biology and Evolution 2024, 16, evae164. [Google Scholar] [CrossRef]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G.B. The new tree of eukaryotes. Trends in Ecology & Evolution 2020, 35, 43–55. [Google Scholar] [CrossRef]
- Strassert, J.F.H.; Irisarri, I.; Williams, T.A.; Burki, F. A molecular timescale for eukaryote evolution with implications for the origin of red algal- derived plastids. Nature Communication 2021, 12, 1879. [Google Scholar] [CrossRef]
- Song, J.; Mak, E.; Wu, B.; Beitz, E. Parasite aquaporins: current developments in drug facilitation and resistance. Biochimica et Biophysica Acta 2014, 1840, 1566–1573. [Google Scholar] [CrossRef]
- Hongsanan, S.; Maharachchikumbura, S.S.N.; Hyde, K.D.; Samarakoon, M.C.; et al. An updated phylogeny of Sordariomycetes based on phylogenetic and molecular clock evidence. Fungal Diversity 2017, 84, 25–41. [Google Scholar] [CrossRef]
- Altschul, S.; Madden, T.; Schaffer, A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D. Gapped BLASt and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Research 1997, 25, 3389–3401. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Sonnhammer, E.L.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. In Proceedings of the Sixth International Conference on Intelligent Systems for Molecular Biology, Montreal, QC, Canada, 28 June–1 July 1998; AAAI Press: Menlo Park, CA, USA, 1998; pp. 175–182. [Google Scholar]
- Hirokawa, T.; Boon Chieng, S.; Mitaku, S. SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 1998, 14, 378–379. [Google Scholar] [CrossRef] [PubMed]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Hanák, O.; Sehnal, D.; Banáš, P.; Navratilova, V.; Jaiswal, D.; et al. MOLE online 2.0: Interactive web-based analysis of biomacromolecular channels. Nucleic Acids Research 2012, 40, W222–W227. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Molecular Biology and Evolution 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.; Wong, T.; et al. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Research 2019, 47, W256–CW259. [Google Scholar] [CrossRef]
Figure 1.
Structure of the AQP protomer (Orthodox AQP; Triat6990) from Trichoderma atroviride. A Primary structure of Triat6990. TMH, transmembrane α-helices domain. The green asterisks correspond to the amino acid tetrad constituting the Ar/R domain. In orange, the two pseudo TMH (LE and LB) including each the NPA motif. B Schematic representation of the secondary structures. C Schematic representation of the folding of the 6 secondary structures (TMH1-6) and the 5 electrostatic loops (Loops A-E) into a tertiary structure, an aquaporin protomer. D Predicted 3D models of the protomer, in both lateral (left) and top views (apoplastic side, right). Models were generated by using the PHYRE2.0 Protein Fold Recognition server, using the Normal mode modeling based on alignment to experimentally solved protein structures. The modeling of TriatAQP-6990 was performed by comparing it with the c5bn2A template (AQY1 from Saccharomycas cerevisiae), with 241 residues (i.e., 80% of the sequence) modeled with 100% confidence. X, Y and Z model dimensions were expressed in Angströms (Å). The two NPA motifs are indicated by red asterisks. E Physicochemical properties (radius, charge, and hydrophobicity) of Triat6990 pore structure, and details of the amino acid residues involved in the Ar/R (F64, H184, T193, and Arg199) and the two NPA motifs and their positions within the protomer. Only the Arginines (N1 from NPA1 in Loop A, and N2 from NPA2 in Loop E), which are positioned antiparallel and involved in the selectivity and inversion of each water molecule as it passes through the channel, are depicted. The NPA motifs fold into half-membrane-spanning helices that create a dipole moment and prevent proton (H+) entry into the cell. The Ar/R selectivity filters act as size-exclusion filters for water and small molecules. Each schema and models is visualized by rainbow colour from N to C termini. The 3D model, physicochemical properties, and the predicted water column were calculated using Mole 2.5 software.
Figure 1.
Structure of the AQP protomer (Orthodox AQP; Triat6990) from Trichoderma atroviride. A Primary structure of Triat6990. TMH, transmembrane α-helices domain. The green asterisks correspond to the amino acid tetrad constituting the Ar/R domain. In orange, the two pseudo TMH (LE and LB) including each the NPA motif. B Schematic representation of the secondary structures. C Schematic representation of the folding of the 6 secondary structures (TMH1-6) and the 5 electrostatic loops (Loops A-E) into a tertiary structure, an aquaporin protomer. D Predicted 3D models of the protomer, in both lateral (left) and top views (apoplastic side, right). Models were generated by using the PHYRE2.0 Protein Fold Recognition server, using the Normal mode modeling based on alignment to experimentally solved protein structures. The modeling of TriatAQP-6990 was performed by comparing it with the c5bn2A template (AQY1 from Saccharomycas cerevisiae), with 241 residues (i.e., 80% of the sequence) modeled with 100% confidence. X, Y and Z model dimensions were expressed in Angströms (Å). The two NPA motifs are indicated by red asterisks. E Physicochemical properties (radius, charge, and hydrophobicity) of Triat6990 pore structure, and details of the amino acid residues involved in the Ar/R (F64, H184, T193, and Arg199) and the two NPA motifs and their positions within the protomer. Only the Arginines (N1 from NPA1 in Loop A, and N2 from NPA2 in Loop E), which are positioned antiparallel and involved in the selectivity and inversion of each water molecule as it passes through the channel, are depicted. The NPA motifs fold into half-membrane-spanning helices that create a dipole moment and prevent proton (H+) entry into the cell. The Ar/R selectivity filters act as size-exclusion filters for water and small molecules. Each schema and models is visualized by rainbow colour from N to C termini. The 3D model, physicochemical properties, and the predicted water column were calculated using Mole 2.5 software.
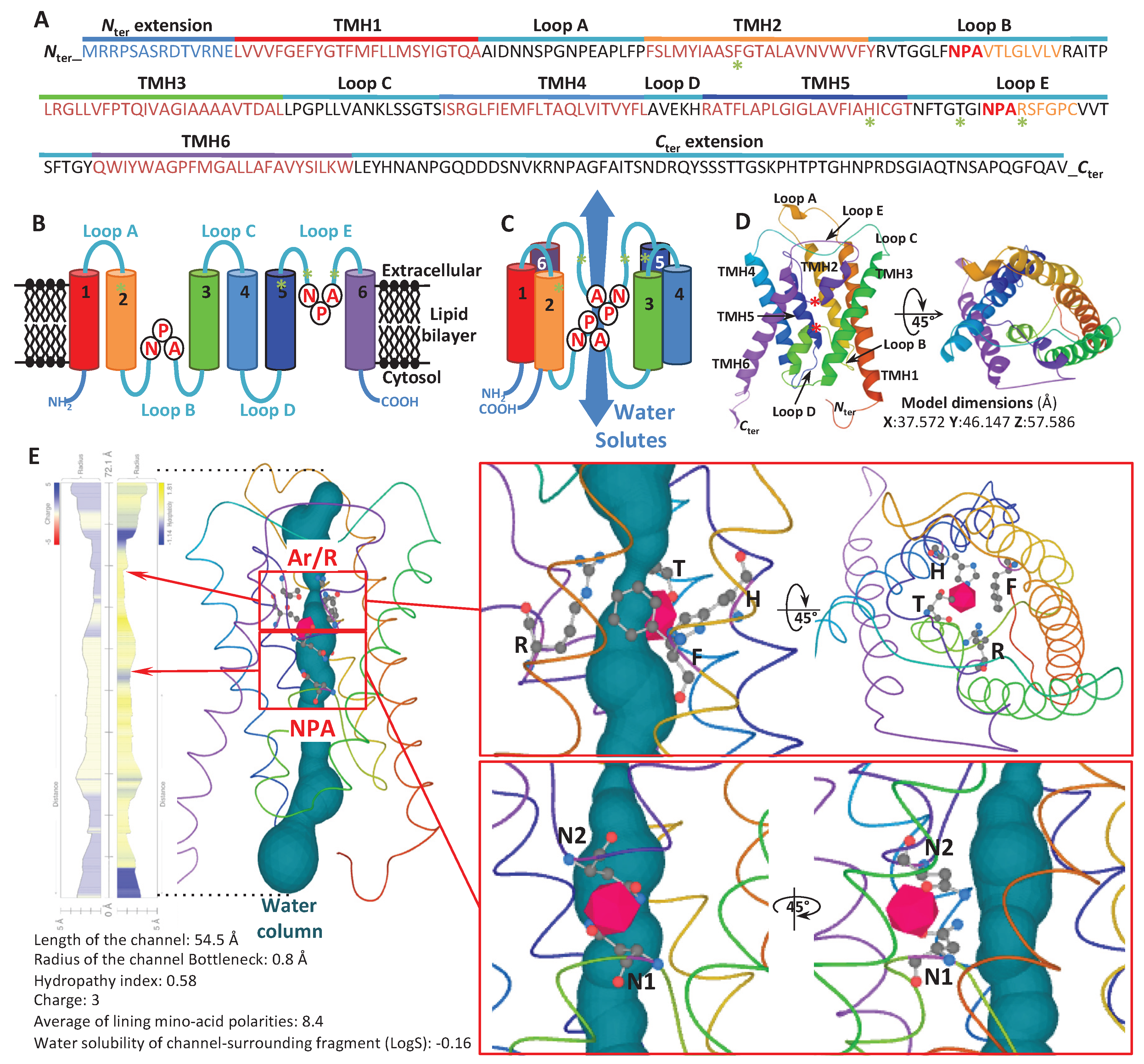
Figure 2.
Diversity and distribution of fungal aquaporins within the major phyla of the fungal kingdom. *Code assigned to each phylum and used in the phylogenetic analyses for this study. **Average number of aquaporins, without subfamily discrimination. ***Percentage of species sharing the XIP subfamily. The distribution of major phyla was derived from the data displayed in JGI's MycoCosm portal. The phyla Arthonomycetes and Aphelidiomycota were added. All the data were extracted from the information available on the JGI public platform.
Figure 2.
Diversity and distribution of fungal aquaporins within the major phyla of the fungal kingdom. *Code assigned to each phylum and used in the phylogenetic analyses for this study. **Average number of aquaporins, without subfamily discrimination. ***Percentage of species sharing the XIP subfamily. The distribution of major phyla was derived from the data displayed in JGI's MycoCosm portal. The phyla Arthonomycetes and Aphelidiomycota were added. All the data were extracted from the information available on the JGI public platform.

Figure 3.
Structure and diversity of fungal aquaporins. A Maximum-likelihood (1,000 bootstrap replicates) phylogenetic tree of 188 full-length aquaporin protein sequences from one species of each major fungal phylum detailed on JGI's MycoCosm portal. Details of the phylogenetic tree are given in Supplementary Figure S1. Species, and related sequences are listed in Supplementary Table S1. The nomenclature of each MIP group was established based on the classification adopted for Trichoderma atroviride [49] and Aspergillus niger [50] B Protein identity percentage between the nine aquaporins present in the Periconia macrospinosa genome, calculated using “Align Sequences Protein BLAST” software on the NCBI resource. C Monomeric lateral of the water path (white column) through the "Orthodox" AQP, Fsp-like, AQGP, and XIP aquaporins, including the predicted aromatic-arginine (ar/R) constrictions, which are detailed in the right view. The 3D model, physicochemical properties, and the predicted water column were calculated using Mole 2.5 software.
Figure 3.
Structure and diversity of fungal aquaporins. A Maximum-likelihood (1,000 bootstrap replicates) phylogenetic tree of 188 full-length aquaporin protein sequences from one species of each major fungal phylum detailed on JGI's MycoCosm portal. Details of the phylogenetic tree are given in Supplementary Figure S1. Species, and related sequences are listed in Supplementary Table S1. The nomenclature of each MIP group was established based on the classification adopted for Trichoderma atroviride [49] and Aspergillus niger [50] B Protein identity percentage between the nine aquaporins present in the Periconia macrospinosa genome, calculated using “Align Sequences Protein BLAST” software on the NCBI resource. C Monomeric lateral of the water path (white column) through the "Orthodox" AQP, Fsp-like, AQGP, and XIP aquaporins, including the predicted aromatic-arginine (ar/R) constrictions, which are detailed in the right view. The 3D model, physicochemical properties, and the predicted water column were calculated using Mole 2.5 software.
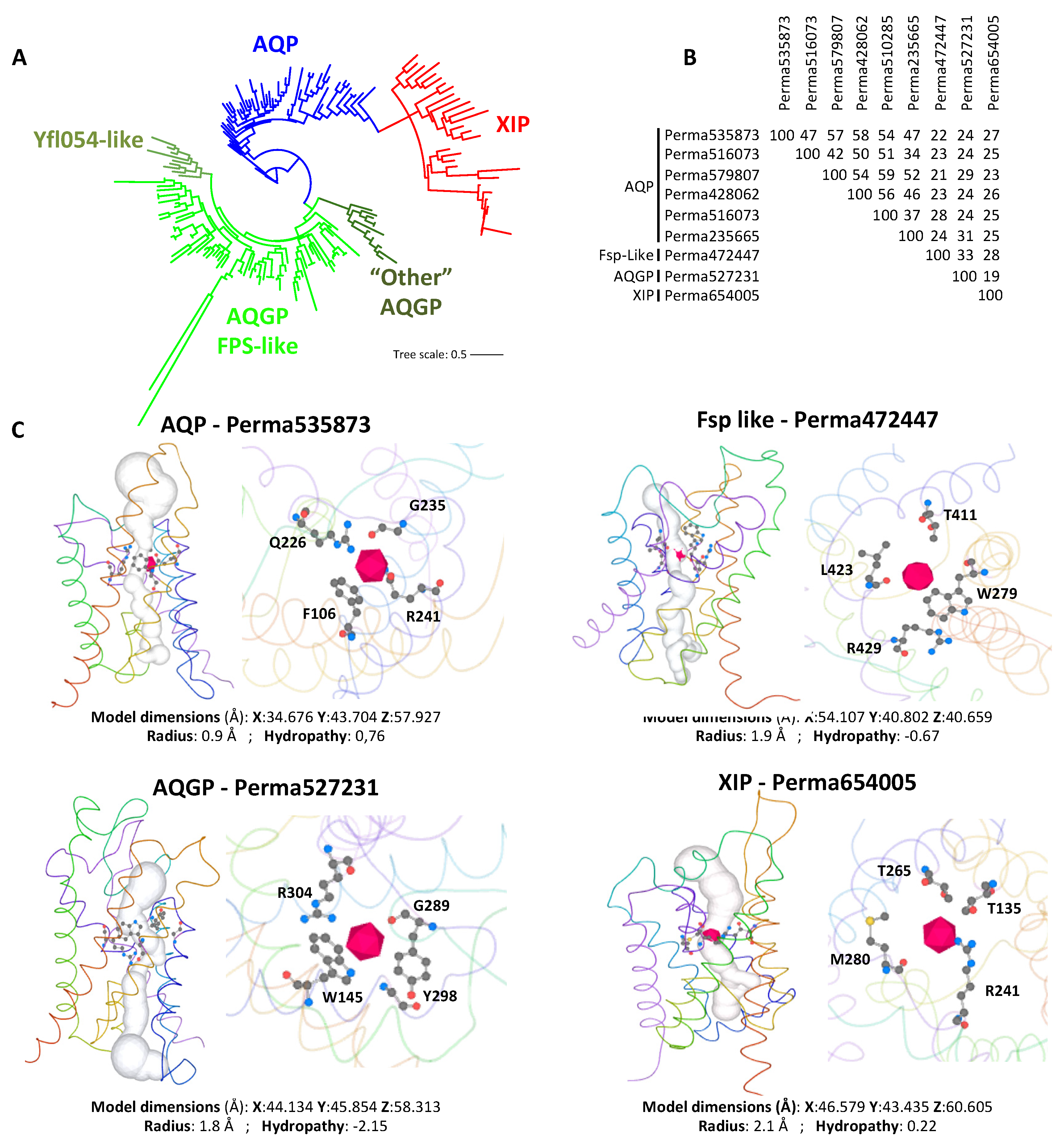
Figure 4.
Overview of XIP diversity in fungi. Maximum-likelihood (1,000 bootstrap replicates) phylogenetic tree of 950 full-length fungal XIP nucleotide sequences. Only selected significant bootstrap values are displayed; the complete set is provided in Supplementary Figure S3. The scale bar indicates the number of amino acid substitutions per site. The fungal names are provided in Supplementary Table S1 and Supplementary Figure S3. * Indicates sequences with atypical phylogenetic placements that do not cluster within their expected fungal clade, but instead group with distantly related fungal groups. § Indicates sequences originating from basal fungal phyla: Glomeromycotina (XVIII), Zoopagomycotina (XXI), and Entomophthoromycotina (XXII).
Figure 4.
Overview of XIP diversity in fungi. Maximum-likelihood (1,000 bootstrap replicates) phylogenetic tree of 950 full-length fungal XIP nucleotide sequences. Only selected significant bootstrap values are displayed; the complete set is provided in Supplementary Figure S3. The scale bar indicates the number of amino acid substitutions per site. The fungal names are provided in Supplementary Table S1 and Supplementary Figure S3. * Indicates sequences with atypical phylogenetic placements that do not cluster within their expected fungal clade, but instead group with distantly related fungal groups. § Indicates sequences originating from basal fungal phyla: Glomeromycotina (XVIII), Zoopagomycotina (XXI), and Entomophthoromycotina (XXII).
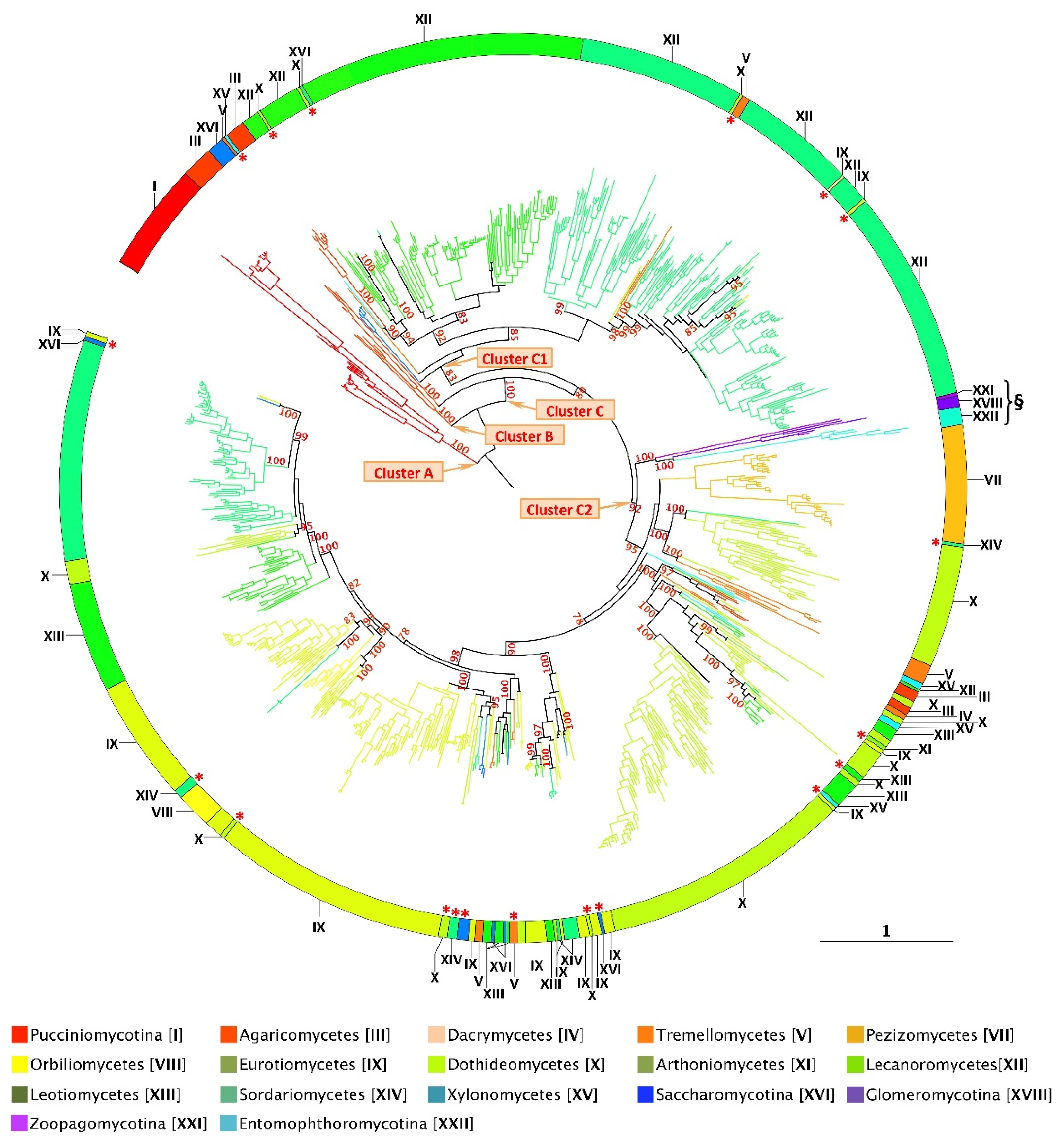

Table 1.
Protein characteristics of the fungal XIP sequences identified through the JGI and NCBI databases. * Code related to the phylogenetic distribution of the major fungal phyla. ** Clusters were determined based on the phylogenetic distribution of the 950 XIP sequences. *** Physicochemical properties were predicted using the ProtParam tool (Expasy portal): AA, number of amino acids; MW, molecular weight; pI, isoelectric point, GRAVI, grand average of hydropathicity. **** Structural features were predicted through sequence alignment. Transmembrane helices (TMH) were predicted using TMHMM, and subcellular localization (SubCL) was assessed using WoLF PSORT.
Table 1.
Protein characteristics of the fungal XIP sequences identified through the JGI and NCBI databases. * Code related to the phylogenetic distribution of the major fungal phyla. ** Clusters were determined based on the phylogenetic distribution of the 950 XIP sequences. *** Physicochemical properties were predicted using the ProtParam tool (Expasy portal): AA, number of amino acids; MW, molecular weight; pI, isoelectric point, GRAVI, grand average of hydropathicity. **** Structural features were predicted through sequence alignment. Transmembrane helices (TMH) were predicted using TMHMM, and subcellular localization (SubCL) was assessed using WoLF PSORT.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



