
Submitted:
25 June 2025
Posted:
26 June 2025
You are already at the latest version
Abstract
Skeletal morphogenesis is a highly complicated interaction cascade of molecular cues, with the Ectodysplasin-A (EDA) pathway emerging as a key player in this biological process. This review focuses on the molecular complexity of the EDA pathway's role in shaping the diverse skeletal architectures observed across vertebrates. At the molecular level, we first discuss the signaling cascades initiated by EDA and briefly explore its impact on skeletal development. Insights into the transcriptional regulation and downstream effectors activated by EDA provide a greater understanding of its influence on skeletal formation. Beyond its standalone role in skeletogenesis, the review mainly focuses on the dynamic cross-talk between the EDA pathway and other important skeletogenic/morphogenic pathways. The multi-layered interplay with signaling networks, such as BMP, Hedgehog, Wnt, and FGF, highlights the integration of this pathway into broader molecular process governing skeletal morphogenesis. The physiological role of EDA in skeletal tissues appears highly context-dependent, varying with the interacting pathway, cell type, and developmental stage. We explore instances where EDA acts as a conductor, harmonizing its effects with those of other pathways to achieve distinct outcomes in skeletal diversity. By summarizing the interactions of EDA and their associated physiological roles, we provide a comprehensive perspective on the EDA-dependent molecular underpinnings of skeletal diversity, offering new and valuable insights for future research and potential applications in skeletal biology.
Keywords:
Ectodysplasin-A
; EDA pathway
; skeletogenesis
; Bone
; Cartilage
; Tooth
1. Introduction
Skeletogenesis, the formation of bones and other skeletal structures in animals, is a highly complicated process that involves a multitude of molecular players. One such emerging key player is Ectodysplasin (EDA), a molecule crucial for shaping the outer layers of an organism’s body [1,2,3]. Dysfunctions in the EDA gene can lead to X-linked hypohidrotic ectodermal dysplasia (XLHED), a condition characterized by missing teeth, sparse hair, and sweat gland problems [4,5,6]. Studies on animal models, such as Tabby mice with EDA mutations, as well as EDA-deficient fish models, have revealed abnormalities in bone shape and density, particularly in the facial region and appendages, prompting further investigation into EDA’s role in craniofacial and dermal skeletogenesis [7,8]. Despite some understanding of EDA’s function, many aspects of its involvement in bone and cartilage development remain elusive, highlighting the need for deeper exploration. Hence, we believe that the time has come to gather all the relevant recent findings about the EDA pathway and its molecular interactions with other known skeletogenic pathways to provide a comprehensive picture of how integrating EDA signaling in skeletogenesis can guide future investigations in the field of skeletal biology. In this review, we focus on the complex molecular mechanisms underlying the role of EDA in skeletogenesis, with a specific focus on fish and mammals. Firstly, we highlight the signaling pathways and molecular processes through which EDA influences bone and cartilage formation. Secondly, we explore various crosstalk between the EDA pathway and other skeletogenic signaling pathways, such as BMP, Hedgehog, Wnt, and FGF pathways, that are well-known to affect skeletal development and morphogenesis across vertebrates [6,9,10,11]. The overarching idea is to better understand how these pathways collectively regulate skeletal development. Through this pathway-based framework of molecular interactions, we discuss empirical studies that have investigated the influence of EDA on skeletal development, morphogenesis, regeneration, and adaptation. In summary, this review endeavors to provide a timely ad comprehensive understanding of the role of EDA in shaping skeletal structures in animals. Our synthesis will contribute valuable insights to the fields of developmental, evolutionary, and regenerative biology, with potential implications for the development of novel therapeutic strategies targeting skeletal-related disorders.
2. Role of the EDA Pathway in Skeletal Formation
Ectodysplasin-A (EDA) is a type II transmembrane protein that belongs to the tumor necrosis factor (TNF) ligand superfamily and plays a central role in the development of ectodermal derivatives. It is synthesized as a membrane-bound precursor that undergoes proteolytic cleavage by furin-like convertases, releasing a soluble, biologically active form [4,12]. The mature EDA protein contains a conserved C-terminal TNF homology domain, which facilitates trimerization and receptor binding. Alternative splicing of the EDA gene (on chromosome Xq12-q13.1 in humans) gives rise to at least two major isoforms in humans; EDA-A1 and EDA-A2, which differ by the presence of a short amino acid insertion [2,4]. This difference confers selective binding to distinct receptors: EDA-A1 binds exclusively to the EDA receptor (EDAR), while EDA-A2 interacts with the X-linked ectodysplasin-A2 receptor (XEDAR). Ligand-receptor engagement activates downstream signaling cascades, most notably the nuclear factor kappa B (NF-κB) pathway, through adaptor proteins such as EDARADD and TRAF6 [13,14,15]. These biochemical features define EDA as a critical morphogen with tightly regulated post-translational processing, isoform-specific receptor affinity, and signal transduction capacity, all of which contribute to its spatial and temporal specificity during vertebrate development [16]. Mutations in this pathway disrupt epithelial-mesenchymal interactions, causing X-linked hypohidrotic ectodermal dysplasia (XLHED), characterized by hypodontia, hypohidrosis, and hypotrichosis [5]. Over the past decades, most previous studies have focused on EDA’s regulatory role in epithelial development, including teeth and hair [6]. Meanwhile, the observation of mandibular abnormalities in EDA-deficient mice (Tabby mice) and reduced bone density in calvarial bones has prompted researchers to further investigate the role of the EDA pathway in skeletal development, especially during craniofacial skeletogenesis [7]. Nonetheless, many details regarding the molecular mechanisms of EDA in both typical and atypical skeletal development remain unclear.
Skeletal cell differentiation: Mutations in the Eda gene lead to X-linked hypohidrotic ectodermal dysplasia (XLHED), characterized by abnormalities in ectodermal appendages and sometimes mesodermal features like craniofacial dysmorphism [8]. Study of Eda1-deficient mice has revealed a crucial role for EDA signaling on bone development through affecting both osteoblast and osteoclast differentiation, as it is activity was found to be essential in Osterix (Osx)+ osteoblasts and Edar-positive osteoclasts [17,18]. The Eda1-deficient mice exhibited osteopetrosis-like changes with reduced marrow space and mature osteoclastic differentiation, as well as impaired osteoclast function, both indicating altered bone homeostasis [17]. The same study showed that EDA treatment restores these effects in osteoclasts (through Nfatc1 translocation and NF-κB activity), indicating its potential therapeutic implications [17]. Early postnatal EDA treatment in Eda1-deficient mice normalized vertebral bone density in adults, indicating the requirement of EDA1 for normal osteogenesis in later life stages as well [18]. A similar finding about the osteogenic activity of EDA signaling (through EDA treatment) has been already reported in study of osteosarcoma in human cells [19]. Compared to bone, less is known about the involvement of EDA signaling in cartilage development and morphogenesis; however, initial studies have found roles for the EDA pathway in the chondrogenic activities of fish and mammals [7,20,21,22,23]. In zebrafish, for instance, research has demonstrated that EDA signaling regulates the differentiation of skeletal progenitor cells into chondrocytes, a key process in cartilage development [21,24,25]. Activation of EDA signaling has been shown to promote chondrocyte proliferation and extracellular matrix synthesis, which are essential for cartilage formation and morphogenesis [21]. Similarly, studies in mice have highlighted the importance of EDA signaling in chondrogenesis and cartilage development. EDA signaling has been correlated with the expression of genes associated with chondrocyte differentiation, suggesting a potential influence on cartilage formation and growth [7]. Moreover, clinical studies have identified mutations in genes encoding components of the EDA pathway in patients with skeletal dysplasias, further implicating the pathway in cartilage-related disorders [23].
Skeletal regeneration: The ectodysplasin A (Eda) pathway plays role in skeletal tissue regeneration across various vertebrate species. Extensive research in fish and mammalian models, has elucidated the molecular mechanisms underlying Eda-mediated osteogenesis and chondrogenesis. For instance, it has been demonstrated that Eda signaling regulates the differentiation of skeletal progenitor cells into osteoblasts and chondrocytes through activation of the NF-κB pathway [21]. Furthermore, investigations in mammalian models, including mice and humans, have provided valuable insights into the role of the Eda pathway in skeletal tissue regeneration [9]. This study showed that Eda signaling controls the expression of genes involved in osteoblast and chondrocyte differentiation in mice, while clinical studies identified mutations in genes encoding Eda pathway components in patients with skeletal dysplasias with impaired regenerative capacities in these tissues [9,26,27]. Moreover, recent studies have highlighted the therapeutic potential of targeting the Eda pathway for bone and cartilage regeneration [7]. It is demonstrated that activation of Eda signaling promotes bone regeneration by enhancing the proliferation and differentiation of osteoblasts in mouse models. Similarly, it has been shown that Eda signaling plays roles in cartilage repair and Eda treatment significantly improved cartilage regeneration by enhancing chondrocyte proliferation and extracellular matrix synthesis [7,20]. Furthermore, molecular studies have revealed the multi-layered regulatory network of the Eda pathway in skeletal tissue regeneration. Overall, these findings reveal the necessity of further research on role of the Eda pathway in skeletal tissue regeneration and highlight its therapeutic potential for treating bone and cartilage-related disorders and injuries.
Dermal skeletogenesis: The dermal skeleton, comprising the external morphology of adult fish, encompasses various elements such as the skull’s dermocranium, opercular lateral bones, scales, fin rays, teeth, and gill rakers. Unlike the endochondral ossification process in which osteoblasts deposit organic matrix over a chondrogenic scaffold, dermal skeletal elements arise from direct mineralization of a collagenous matrix deposited by dermal fibroblasts, closely associated with the epidermis [28]. Development and patterning of dermal elements are akin to epidermal appendages and are regulated by reciprocal signaling between epithelium and mesenchyme [29]. Most dermal skeletal elements in teleosts do not form during larval development but rather through juvenile metamorphosis, with variations playing a significant role in fish population adaptations to diverse environments, and mutations in genes like ectodysplasin (eda) affecting these elements, suggesting an ancestral role of Eda signaling in dermal skeleton formation and patterning [21]. Throughout vertebrate evolution, from fish to tetrapods, there has been a transition in dermal structures, with lateral bones, scales, dermal plates, and fin rays either reduced or lost, accompanied by the evolution of specialized keratinized integumentary appendages [30]. The diversity of form in extant bony fishes involves modifications in the size, shape, and number of scales, fin rays, cranial dermal bones, and teeth. Mutations in genes like ectodysplasin (eda) and edar affecting the Eda signaling pathway, crucial for hair and teeth formation in mammals, reveal the importance of this pathway in fish skeletal morphogenesis [9]. Loss of Eda signaling in zebrafish mirrors human hereditary disease hypohidrotic ectodermal dysplasia (HED) phenotypes, highlighting zebrafish mutants as genetic models of this disease [9,21]. The activation of Eda signaling in fish epidermis contributes to the formation of an epidermal placode, resembling early development in other vertebrate integumentary appendages. Variations in the expressivity of dominant alleles sensitive to background modifiers and organ-specific responses to reduction of Eda signaling suggest these alleles as potential drivers of morphological variation in evolution [21,31,32,33]. Furthermore, other major pathway such as Wnt signaling can act as an upstream regulator of EDA during the morphogenesis of dermal bones such as scales and armor plates [31,33].
3. Cross-Talk Between EDA and Major Skeletogenic Pathways
3.1. Transforming Growth Factor-Beta Signaling Pathways
The TGF-β superfamily, a group of structurally related polypeptides conserved across the animal kingdom, includes members synthesized as large precursors that undergo proteolytic cleavage, releasing mature and active forms (e.g., BMPs) or mature and latent forms (e.g., TGF-β) [34]. Secreted TGF-βs bind to transmembrane receptors, regulated by various factors and transmit signals through intracellular SMAD proteins, regulating target genes and influencing biological processes, including ECM synthesis and skeletal remodeling [34,35,36]. TGF-β subfamily members and their receptors contribute to the development and morphogenesis of various skeletal structures [11,37,38]. TGF-β1, a ubiquitously expressed member, plays a key role in skeletogenesis, influencing skeletal metabolism and the balance between bone formation and resorption [39,40]. TGF-β pathways’ effects on skeletal morphogenesis involve modulation of extracellular matrix (ECM) production, as well as regulation of major skeletogenic factors including BMPs, Runx2, RANK, OPG, and twist1 [37,39,41].
In mammals, it has been shown that EDA signal can act upstream of several components of TGF-β pathway during embryonic development, for example, in the regulation of the expression of Bambi, a pseudoreceptor related to the Tgf-β superfamily type I receptors, thereby inhibiting the activity of Tgf-β signaling [20]. Another layer of potential cross-talk between EDA and TGF-β pathways can be through regulation of the Smad7 transcription factor by EDA activation [20,42]. Inhibitory cross-talk between EDA and TGF-β pathways has also been reported during tooth development; however, the detailed molecular mediators of these interactions have remained unexplored [43]. In fish, regulatory interactions between components of EDA and TGF-β pathways have been predicted during dermal bone (scale) regeneration [24]. During craniofacial skeletal development in both mammals and fish, EDA through its downstream effector, NF-KB, regulates palatal morphogenesis and TGF-β signal is also an essential player in this process, acting upstream of NF-KB [44]. This may indicate the competitive interactions between the two signals in regulating shared downstream effectors during skeletogenesis. Overall, the physiological output of EDA and TGF-β interaction in skeletal tissues appears to be involved in developmental growth mechanisms.
3.2. Bone Morphogenetic Protein Signaling Pathways
Bone morphogenetic proteins (BMPs) are part of the TGF-β superfamily and signal through specific BMP receptors (BMPRs), activating SMAD proteins [45,46]. Signaling modulation occurs through extracellular and intracellular BMP antagonists, differential SMAD regulation, inhibitors, and negative feedback loops [45,47]. In vertebrates, BMPs contribute to diverse skeletal structures; Bmp4 signaling, for instance, influences tooth and neural crest-derived skeletal development [48,49]. During early vertebrate development, ectodermal BMP signals interact with other morphogens to establish gene expression domains [50]. BMP signaling regulates musculoskeletal cell differentiation and chondrogenesis. Inhibiting BMP signaling affects bone and cartilage formation differently across developmental stages, with high BMP levels promoting chondrogenesis over osteogenesis. BMP signals regulate numerous TFs, including essential skeletogenic factors, orchestrating gene expression networks in a dose-dependent manner [51].
The regulatory crosstalk between the EDA/EDAR and BMP pathways plays a crucial role in skeletogenesis and the development of skeletal tissues. Research indicates that during fin formation in medaka, the activation of the EDA/EDAR signaling is necessary for osteoblast differentiation and typically precedes the expression of BMP2b, suggesting a sequential interplay during fin skeletogenesis [52]. This EDA–BMP interaction in the fin appears to involve only the dermal (but not endochondral) bone structures; namely, the fin rays [52]. This pattern is echoed in zebrafish, where EDA/EDAR signaling is also essential for BMP2b expression and subsequent dermal bone formation during scale and fin development [21]. Moreover, EDA-A2 has been implicated in bone formation, possibly through a synergistic interaction with BMP-4, which together activate caspase-3 mediated osteoblast differentiation [19]. In endochondral-derived bone tissue, BMP pathways regulate major transcription factors involved in the EDA pathway, such as Nfatc1, where BMP-2 enhances osteoblast proliferation and differentiation by inducing Nfatc1 expression through SMAD1/5 binding to its promoter [53]. Moreover, during tooth development, EDA-A1 is known to inhibit BMP4 expression via NF-kB-dependent induction of Ccn2/CTGF, a BMP inhibitor, thus inhibiting ameloblast differentiation and tooth formation [20,54]. These studies collectively highlight a potential complex interplay between EDA/EDAR and BMP signaling in regulating various aspects of skeletal development (summarized in Figure 1A). Thus, the physiological output of EDA and BMP interactions in skeletal tissues is likely primarily involved in developmental growth and patterning.
3.3. Hedgehog Signaling Pathway
One of the earliest basic genetic pathways for animal development to be discovered was hedgehog (Hh) signaling, which has been thoroughly investigated in a number of model species [55]. Desert hedgehog (dhh), Sonic hedgehog (shh), and Indian hedgehog (ihh) are the three main kinds of Hh proteins that are encoded by genes in vertebrates that follow whole genome duplication (WGD) and functional diversification. The transmembrane protein called Dispatched in the generating cells releases the activated forms of Hh proteins, which are then bound by Ptch1 and Ptch2 receptors on target cells that are sensitive. Upon binding Hh, Ptch releases Smoothened (Smo), an additional membrane protein that links with the Gli family of transcription factors to control target gene transcription [56]. During the development of skeletal structures and as an early initiator of cartilage cell differentiation, the functions of Hh signaling components have been intensively researched [57,58]. Shh plays a critical role as an intermediary in the formation of skeletal structures. Shh derived from the endoderm assures the continued existence of neural crest cells in the craniofacial skeleton [59]. Ihh, an additional Hh ligand that plays a variety of functions in skeletogenesis, modulates the palatogenesis process, and promotes chondrocyte differentiation and proliferation, osteoblastogenesis, and ossification [58,60,61].
The regulatory crosstalk between the Hh and EDA pathways plays a crucial role in skeletal development and skeletogenesis across various species. In zebrafish and medaka, EDA/EDAR signaling notably influences the expression of Sonic hedgehog (shh), which is essential for the anterior-posterior patterning and bone formation in paired fins [21,52]. Specifically, mutations in the eda gene can lead to a total absence of shh expression, inhibiting osteoblast proliferation and differentiation, and consequently impairing fin formation and regeneration [52]. Similarly, in zebrafish scales and cichlid fish, EDA-mediated activation of shh is necessary for bone formation and the differentiation of osteoblast-like cells [33,62], with variations in shh expression suggested as a mechanism for adaptive morphological divergence [33]. In cichlids, this signaling synergy extends to the development of jaw cartilage [63]. In mice, during early tooth development, Shh is induced by Eda signaling, promoting the growth of the dental bud [2,20]. Moreover, in pathological contexts, such as jaw bone diseases, Shh can drive Nfatc1 expression and activity, promoting osteoclast proliferation and differentiation, which leads to bone resorption [64], while Indian hedgehog (Ihh) activation in endochondral bone tissue (trabecular and cortical bone) can inhibit Nfatc1 expression and osteoclastogenesis, favoring osteoblast differentiation and bone formation [65]. This evidence highlights a complex interplay between EDA and Hh pathways, highlighting their potential indirect interactions through transcriptional regulation mechanisms that significantly impact bone and cartilage development and regeneration (Figure 1B). Thus at physiological level, EDA-Hh interactions might be not only play role during skeletal development and patterning but also under pathological conditions in these tissues.
3.4. Wnt/β-Catenin Signaling Pathway
Wnts, which are a family of secreted glycoproteins, are essential for key processes such as embryonic growth and morphological development, as they activate numerous pathways for signal transduction. Due to their critical role in skeletogenesis, potential for therapeutic skeletal regeneration and master modulatory role via links with multiple morphogenic channels [66,67]. Modest variations in the strength, periodicity, and duration of Wnt signals influence developmental skeletogenesis, bone remodeling, and regeneration [66,68]. Bone mineral density is correlated with polymorphisms in Wnt pathway components. Within the canonical pathway, Wnts establish a binding interaction with a transmembrane receptor of the Frizzled (FZD) family in its extracellular domain. Facilitating the binding process are co-receptors including LRPs (LRP5/6). A sequence of molecular events is initiated by the Wnt/LRPs/FZD complex, culminating in the establishment of a gene regulatory complex in the nucleus between β-catenin and additional factors (Lef and TCFs). β-catenin/Lef/TCF combinations are capable of modulating the expression of an extensive array of target genes. Prominent osteogenic and chondrogenic factors, including Runx2 and Ihh, are included among these [69]. Furthermore, Wnt/β-catenin signaling has the ability to modulate the transcription of key factors involved in bone homeostasis and remodeling, RANKL and OPG [66,70]. The activity of the Wnt/β-catenin signaling pathway can be hindered by various skeletogenic factors and can also be regulated by transmembrane inhibitors (Kremen1/2, Ror2, and Ryk) and secreted Wnt antagonists (Dkks, Sfrps, Wif1, and Sost) [71]. The transition from epithelial-to-mesenchymal of the cranial neural crest cells, as well as their breakdown and translocation into distinctive cranial regions, are all dependent on canonical Wnt signaling [72].
The regulatory crosstalk between the Wnt and EDA pathways is among the most studied EDA crosstalk in the skeletal system and it influences various aspects of bone, cartilage, and tooth development, homeostasis and morphogenesis across species. During tooth development, EDA is known to enhance the expression of WNT10A and WNT10B via NF-κB signaling, promoting the differentiation of odontoblasts and ameloblasts [54]. Similarly, the interaction between these pathways is essential for craniofacial skeletal patterning, with evidence showing that Wnt signaling acts upstream of EDA during bone formation, although it inhibits osteoblast differentiation [23,73]. This upstream regulatory effect is also observed in palatogenesis, where Pax9-induced upregulation of Wnt signaling via suppression of DKK, an inhibitor of Wnt, leads to the induction of EDA signaling and bone formation [74]. In zebrafish, the cooperation between Wnt and Eda/NF-κB signaling facilitates a signaling wave essential for scale bone morphogenesis [62,75]. This cooperative relationship is also significant in cichlid and stickleback fishes, where Wnt/Lef1 signaling is suggested as an upstream regulator of EDA during the morphogenesis of dermal bones such as scales and armor plates [31,33].
In studies related to endochondral skeletal tissues, the interaction between EDA and Wnt signaling may occur indirectly through EDA’s major downstream effectors, NF-κB and NFATc1. For instance, NF-κB can induce RSPO2 expression during inflammation, activating Wnt signaling and subsequently inhibiting cartilage formation, while also participating in a negative feedback loop that may be essential for late-stage chondrogenesis [76,77,78]. In this negative feedback loop, RSPO2 mediated activation of Wnt signal can later block Nf-kb activity. It is important to note that the Nf-kb mediated induction of RSPO2 can be triggered by both inflammation and mechanical overload [76]. The Wnt pathway activity is required for the late stage of chondrocyte differentiation (chondrocyte hypertrophy) while Nf-kb induces chondrocyte apoptosis; therefore this negative feedback might be required for promoting the late stage of chondrocyte differentiation without entering apoptosis [77,78]. Moreover, Wnt signaling plays a crucial role as a major inhibitor of NFATc1, therefore controlling osteoclast differentiation. This inhibition is mediated through both canonical (GSK3β/β-catenin) and non-canonical (WNT4, WNT16, WNT5A, WNT3A) pathways, highlighting the multifaceted nature of Wnt signaling in bone resorption and remodeling [79,80,81,82]. Taken together, these findings highlight the complex and integral interactions between the Wnt and EDA pathways in craniofacial skeletal tissues, and potentially in endochondral skeletal tissues, as well (Figure 1C). At the physiological level, their interactions may be essential for skeletal tissue homeostasis, growth, and remodeling.
3.5. Notch Signaling Pathway
Notch proteins are transmembrane receptors that are exceptionally conserved. They comprise three domains: extracellular, transmembrane, and intracellular [83]. By interacting with the extracellular domain of Notch, the canonical ligands Jagged and delta-like (Dll) facilitate the unloading of the intracellular domain. When inducing the expression of target genes, including Hes and Hey, the liberated domain assembles with its transcriptional regulator CSL (RBPjk) [83]. A multitude of biological processes, especially those involving the determination of cell fate, are remarkably governed by the canonical Notch signal, which is an incredibly simple molecular cascade. In addition to early somitogenesis, skeletal growth, and bone remodeling, the pathway is implicated in various facets of skeletal development [84,85,86]. Chondrocyte and osteoblast differentiation are initially inhibited by Notch signaling, which subsequently initiates chondrogenesis [87]. By differentially regulating genes such as RANKL and OPG, the pathway also influences osteoclastogenesis throughout lineage commitment and maturation [84,85,88]. Notch signaling hinders the differentiation of chondrocytes as well as osteoblasts at various phases [85,89].
The regulatory crosstalk between the Notch and EDA pathways has been found to play significant role in certain processes involving the development and differentiation of skeletal tissues across various species. For instance, a recent study in stickleback fish has shown that mutations in Eda along with alterations in Notch signaling components, specifically Dld and Egfl6, impact the development of lateral plates by influencing osteoblast differentiation in these structures [90]. Similarly, in pufferfish, the development of dermal spines, a scale derivative, is regulated by the interaction between Eda signaling and Notch3 expression [91]. In mammalian bone marrow, the interaction between Notch signaling and EDA pathways manifests differently; for example, the overexpression of Jagged1 and Notch2 enhances the transcription of NFATc1, promoting osteoclast differentiation [92]. Conversely, in a mammalian jaw bone, suppression of Notch3 leads to reduced expression of its downstream targets (Jag1 and Hey1), which in turn increases NFATc1 expression and thereby promotes osteoclast differentiation [93].
In endochondrally derived skeletal tissues, the EDA-Notch regulatory connection extends to the inhibition of osteoblast differentiation where Notch1 and their signaling through RBPjk are found to suppress osteoblast activity by inhibiting NFATc1 [94,95]. Furthermore, under inflammatory conditions in bone, the opposing roles of RBPjk and NFATc1 regulate miR-182, which plays a stimulatory role in osteoclast differentiation and inflammatory bone resorption [96]. In cartilage, activation of Notch1/2 signaling leads to the suppression of NFATc1 and inhibition of early-stage chondrocyte differentiation, illustrating a divergent role in cartilage compared to bone [97]. Collectively, these studies highlight the complex interactions between Notch and EDA pathways in skeletal development, emphasizing their pivotal roles in regulating cell differentiation and tissue formation (Figure 1D). Hence, EDA-Notch interactions may play a crucial physiological role in regulating skeletal homeostasis by modulating osteoblast and osteoclast differentiation, ensuring balanced bone formation and resorption in both endochondral and craniofacial skeletal tissues.
4. Cross-Talk Between EDA and Growth Factor-Mediated Signals
4.1. Fibroblast Growth Factor Signaling Pathway
A large family of primarily paracrine ligands known as fibroblast growth factors (FGFs) activates numerous conserved signaling pathways. FGF signaling is fundamental at various phases of vertebrate development and is involved in a vast array of biological processes [98]. FGFs generate signals via FGF receptors (FGFRS), which comprise a family of tyrosine kinases. Due to alternative splicing that is strictly regulated, the quantity of FGFR isoforms in vertebrates significantly surpasses the quantity of genes that encode them [99]. During development, FGFs and FGFR isoforms exhibit discrete spatiotemporal expression patterns, and their function disruption has been linked to an assortment of developmental and morphological abnormalities [100,101,102,103,104]. Various aspects of endochondral and intramembranous bone development, chondrogenesis, and bone mechanical sensing are regulated by FGF-regulated pathways [100,105,106].
FGF signaling pathway is an immediate target of EDA during tooth development and Fgf20 is an essential downstream effector of Eda and affects Eda-regulated characteristics of tooth morphogenesis, including the number, size and shape of teeth [107]. Studies in ectodermal tissues also proposed Fgf20 as a key mediator and a direct target of EDA signaling transduction [108,109], and interestingly, the absence of Fgf20 also leads to the suppression of EDAR expression and, consequently, also EDA signaling [6]. These findings suggest a synergistic and reciprocal crosstalk between EDA and FGF signals at transcriptional levels in these tissues. In zebrafish, activation of both EDA and FGF signals are essential for osteoblastogenesis during scale development, however, their direct interaction in this process remained unexplored [110]. In cichlid fish, a positive expression correlation between eda and fgf20 has been shown during morphogenesis of posterior scales [33]. An indirect interaction between EDA and FGF signals is indicated during zebrafish scale development through the Wnt signaling pathway [62], thus their crosstalk in scale osteoblasts might not be the result of a direct transcriptional activation. On the contrary, a study of endochondrally-driven mammalian osteoblasts has shown that activation of FGF signaling through FGF2 can also suppress NF-kB signaling and promote bone formation [111]. Compared to bone and tooth, less is known about potential crosstalk between EDA and FGF signals in cartilage, although a recent study in endochondral mammalian chondrocytes has found that FGF8 induces the expression of ECM components through induction of NF-κB transcriptional activity [112]. Taken together, these observations indicate the presence of direct crosstalk in the tooth and indirect crosstalk in bone and cartilage between EDA and FGF signals (Figure 2A). The interactions appear to be synergistic and reciprocal, with broader implications for the physiological role of their coordination in skeletal development and morphogenesis.
4.2. Insulin-Like Growth Factor Signaling Pathway
Insulin-like growth factors (IGFs), initially identified in the musculoskeletal system, mediate growth and differentiation [113]. They activate an evolutionarily conserved signaling cascade involving IGFs, IGF receptors, IGF binding proteins (IGFBPs), and IGFBP proteases. IGFs bind to activated receptors, initiating gene regulatory signals via the MAPK and PI3K-AKT pathways. IGFBPs, regulating IGF bioavailability, impact various facets of IGF function. Present in all tissues, IGFs play crucial roles in homeostasis, embryonic/postnatal development, and tissue survival [114,115,116]. Predominant in bones, IGFs promote mineralization, differentiation, and formation but exert a multifaceted impact on bone [117]. IGF-mediated signaling regulates chondrocyte proliferation, differentiation, and apoptosis [118].
During follicular hair formation, EDA acts upstream of IGF signaling indirectly through Shh-dependent transcriptional regulation of IGFBP-5, a major IGF inhibitor in various tissues [119]. Moreover, IGFBP-3 can modulate EDA transcription through a Wnt/β-catenin-dependent mechanism during tooth mineralization development [120]. To date, no study has been conducted to investigate direct crosstalk between EDA and IGF signals in any skeletal cells, even though direct regulatory interactions between NF-kB and IGF-1 mediated signals have been found in both bone and cartilage under normal and pathological conditions [121,122,123]. During cartilage inflammation, IGF-1 directly inhibits NF-kB transcriptional activity, leading to promotion of chondrogenesis and cartilage repair [122]. During endochondral ossification, the promoting effects of NF-kB transcriptional activity are mediated through activation of IGF-1 signaling pathway [121]. Also, during mechanical stress in the endochondral growth plate, the biomechanical signals induce NF-κB transcriptional activation and IGF-1 signal appears to act downstream of NF-κB signal in this condition; however, the detailed regulatory connection has not been studied [124]. Moreover, NF-κB signal-dependent bone resorption deficiency affects tooth root development from failure of releasing of IGF-1 from bone matrix through osteoclasts and IGF-1 inhibition in root odontoblasts [125]. These results suggest that NF-κB signal can act upstream of the IGF-1 signaling pathway and activate it in both osteoclasts and odontoblasts. Taken together, these observations indicate various crosstalk between EDA and IGF signals in skeletal cells. The regulatory nature of these can be very different, as IGF-1 inhibits NF-κB in regenerating cartilage, whereas NF-κB signal stimulates IGF-1 activity during bone and cartilage growth, tooth root development, cartilage mechanical overloading and bone resorption (Figure 2B). A broader physiological aspect of their interaction may be their coordinated role in skeletal pathobiological processes.
4.3. Signals Mediated by MAPKs
The conserved family of serine/threonine kinases, mitogen-activated protein kinases (MAPKs), plays a crucial role in transducing external signals into cells via membrane receptors [126]. Major regulatory cascades, including extracellular signal-regulated kinase (ERK), c-Jun NH2-terminal kinase (JNK), and p38 MAPK, are incorporated within MAPKs. Despite growth factors being primary activators, each MAPK cascade mediates unique cellular signals related to apoptosis, stress, differentiation, and growth [127,128,129]. Ap-1 complex members (c-Jun and c-Fos heterodimer) regulate gene expression during osteoblast differentiation and are targeted by distinct MAPKs. Crucially, MAPK cascade activation is involved in mesodermal derivatives, skeleton, and dentition formation. Bone mechanotransduction is facilitated through Ap-1 transcriptional activity induction via JNK and ERK [130].
The essential role of p38 MAPK signaling pathway during tooth morphogenesis and enamel secretion has been shown to be independent of EDA signaling [131]. During ectodermal differentiation, however, Edar mediated signal has been found to directly (but modestly) activate MAPK/JNK [132]. Though no study has ever investigated presence of direct crosstalk between MAPK and EDA signaling pathways, it is well known that activated NF-κB acts as potent inhibitor of JNK by which cell survival versus cell death in various tissues may be balanced [133]. Conversely, activated MAPK pathway (including JNK signal) enhances osteoclast differentiation through induction of NF-κB transcriptional activity [134]. Interestingly, a recent study has shown that activated NF-κB can also induce osteoclast differentiation through activation of MAPK signal components (ERK, JNK and p38) [135]. These suggest that a synergistic activation of NF-κB and MAPK signals is required for bone remodeling and both signals can act upstream of each other during this process. In endochondral-type chondrocytes, p38 MAPK signaling acts directly at upstream of NF-κB, and enhanced p38 activity induces NF-κB signal, leading to promotion of both chondrocyte differentiation and inflammation [136]. Similarly, mechanical pressure in bone activates p38 MAPK signaling and in turn enhanced p38 activity again induces NF-κB signal leading to osteogenesis osteoblast differentiation [137]. Taken together, these studies indicate potential crosstalk between MAPK and EDA signaling pathways in various skeletogenic processes, which is most likely mediated through synergistic activation of NF-κB dependent signals (Figure 2C). At a broader physiological level, their interaction might be a potential future research topic in processes involving skeletal remodeling under normal or pathological conditions.
5. Cross-Talk Between EDA and Signals Mediated by Nuclear Receptors
5.1. Retinoic Acid Signaling Pathway
Retinoic Acid (RA), among the initial vertebrate morphogens [138], plays diverse roles in developmental patterning. Its inactive precursor, Vitamin A (retinol), is vital for growth, development, and tissue maintenance [139,140]. RA rapidly diffuses and activates specific heterodimeric nuclear receptors, primarily RXRα/RAR (α, β, and γ), influencing RA-responsive gene expression through distinct sequences. In vertebrates, RA signaling coordinates early skeletal morphogenesis and anterior-posterior patterning during embryogenesis by regulating homeobox gene expression [138]. Dysregulation of RA synthesis and signaling, including variations in RA receptor-encoding genes and enzyme-encoding genes (Rdh10 and Raldh3), can induce various skeletal abnormalities [141,142,143]. RA-metabolizing enzymes, linked to skeletal development, impact spatiotemporal RA levels post-synthesis.
Coordinated activation of both RA and EDA signals are essential for tooth development and morphogenesis across vertebrates; however, the detailed molecular interactions between the two pathways remained unexplored during this process [144]. In several metabolic diseases, RXRα has been found to bind to EDA promoter and induced EDA transcription [6]. In human skin, EDAR has been found to be induced by activation of RA signaling pathway [145]. Such a direct transcriptional regulation of EDA or EDAR by RA pathway has not been reported in skeletal cells. However, during endochondral skeletogenesis, RA mediated suppression of NF-κB transcriptional activity is proposed as a molecular mechanism underlying chondrogenic effects of RA during cartilage regeneration [146]. On the contrary, all-trans-retinoic acid (ATRA), an active vitamin A compound binding to RXRα/RAR and activating their regulatory function, inhibits osteogenesis by enhancing NF-κB transcriptional activity [147]. Strikingly, a recent study in zebrafish has demonstrated that NF-κB signaling acts upstream of RA signaling during osteoblast dedifferentiation which is essential for skeletal regeneration [148]. This is done through suppression of the RA-degrading enzyme cyp26b1 by activated NF-κB signaling. Previously, in mammals, it has been shown that NF-κB impair osteoblastogenesis through inhibition of VDR and RXR function [149]. These observations indicate potential direct and indirect reciprocal crosstalk between RA and EDA signals, which may involve both inhibitory and stimulatory effects, during skeletogenesis (Figure 3A). Furthermore, their interaction may represent a promising avenue for future research into the mechanisms underlying skeletal regeneration at a broader physiological level.
5.2. Aryl Hydrocarbon Signaling Pathway
The Aryl hydrocarbon/Dioxin receptor (Ahr), a member of the bHLH-PAS family of heterodimeric TFs, initially identified for mediating a signaling pathway, is implicated in various skeletogenic phases [150]. At developmental level, for instance, Ahr loss-of-function mutation can cause craniofacial and skeletal phenotypes with elongation along the anterior-posterior axis [151,152]. Activated Ahr pathway may influence skeleton formation via various skeletogenic factors, dependent on time, dose, and ligand. The direct regulatory link between Ahr and EDA pathways in the skeletal system is only reported in zebrafish during craniofacial and fin skeletal development [153]. This connection is proposed to be through Ahr2 and Edar, by which Ahr2 seems to act at upstream of Edar inducing its expression during skeletal development [153]. Another likely scenario can be EDA regulation through interaction between NF-κB and cyp1a1, the main target of Ahr pathway [153], via competition with NF-κB in transcriptional regulation of cyp1a1 [154]. In mammals, Ahr mediated transcriptional induction of Edar is only demonstrated in liver cells [155]. During inflammatory responses in different tissues, it is already known that NF-κB induces Ahr expression by directly binding to the Ahr promoter [156]. Moreover, the Ahr pathway modulates osteoclast differentiation and bone remodeling through regulation of NF-κB nuclear translocation or competing with its transcriptional activity in osteoclasts [154]. Interestingly, the Ahr pathway can also enhance osteoclastogenesis through crosstalk with NF-κB signal [154]. The differentiation of osteoblasts is also hindered by the Ahr pathway through a complex mechanism involving NF-κB signal modulation [156,157]. In cartilage tissue, activation of Ahr pathway by its endogenous ligands can reduce inflammation in chondrocytes by blocking NF-κB signal [158]. These findings implicate both inhibitory and stimulatory regulatory connections between the Ahr pathway and NF-κB mediated signals in various aspects of skeletogenesis (Figure 3B). Although the molecular mechanisms underlying potential direct crosstalk between Ahr and EDA signaling pathways in skeletal cells remain poorly understood, existing findings suggest that their interaction may be important for coordinating skeletal remodeling and regeneration at a broader physiological level.
5.3. Glucocorticoid Signaling Pathway
Derived from steroids, glucocorticoids (GCs) bind to glucocorticoid receptors (GR) present in virtually all tissues [159]. GCs traverse cell membranes, modulating transcription via nuclear GR post-conversion to an active state. Ligand-bound GR regulates gene transcription positively or negatively by interacting with other TFs. GR-mediated signaling is implicated in various aspects of skeletogenesis and morphological adaptation of skeletal structures [160,161]. GC-induced osteoporosis results from GR pathway interactions with signals regulating skeletal cell processes [161]. Essential GR signaling elements respond to environmental and cellular stresses [162]. Maternal GR transcripts in zebrafish embryos are crucial for early skeletal development [163]. Elevated GC levels during growth can subtly manifest craniofacial and vertebral skeletal abnormalities and during adulthood cause various skeletal pathologies [164,165,166,167]. Major genes involved in ECM biogenesis, including ctsk, dcn, and mmp2/9/13, are among the direct downstream effectors of the GR pathway during skeletogenesis [165,168]. Differential regulation of these genes during growth results in distinctive morphological modifications to skeletal structures.
Multiple components of GR and EDA signaling pathways are required for hair, skin and tooth development and morphogenesis [169,170]. In mice, the effects of induced activation of the GR pathway (through GR overexpression) is mediated by reduction of NF-κB transcriptional activity and interference with NF-κB function, which is the result of significant decrease in NF-κB binding activity in tooth epithelium [170]. The same study also suggests that the GR signaling pathway can interfere with the NF-κB function at multiple levels during tooth development [170]. In mice, GR signaling can inhibit normal osteoblastogenesis in the absence of NF-κB activity indicating independency of GR signal during this process [171]. However, under inflammatory conditions, GR signaling appears to interfere NF-κB mediated signals in bone via hindering NF-κB transcriptional activity or directly regulating its downstream target genes [172]. Similarly, in articular cartilage, GR signaling has been found as a strong suppressor of NF-κB transcriptional activity and function in cartilage under inflammatory condition [173]. These findings indicate inhibitory effects of GR-dependent signaling on the EDA pathway (through suppression of NF-κB activity) during normal tooth development, with a potentially broad physiological role in pathological conditions affecting bone and cartilage (Figure 3C).
5.4. Estrogen Signaling Pathway
Oestrogens, often known as estrogens, are hormones derived from precursor molecules with androgenic properties. While initially identified as sex hormones, they have the ability to impact a range of developmental and physiological processes, such as the creation and regeneration of the skeletal system [174,175]. Given the prevalence of sexual dimorphism, which is ultimately influenced by sex-hormone signaling, this is not unexpected. In skeletal cells, oestrogens transmit signals through two types of receptors: ER-alpha/-beta, which are estrogen receptors regulated by ligands [174], and receptors connected with G-proteins (e.g., GPR-30 and GPER1) [176,177,178]. The mediators of estrogen signals are present in chondrocytes and are involved in the process of chondrogenesis [178,179]. The impact of estrogen on the multiplication of chondrocytes and the development of cartilage varies among different species [180,181,182]. High levels of estrogen during zebrafish development can completely interfere with the construction of craniofacial and trunk skeletal structures [183,184].
In zebrafish, the expression of edar and esr2a (estrogen receptor 2a) are both up-regulated in similar areas of the epidermis at initiator sites of ectodermal/dermal appendage where developing scales appear [185]. In mammalian epithelial cells, selective activation of estrogen receptor-β (Er-a) induces Edar transcription [186]. Nonetheless, there has been no investigation into the potential direct crosstalk between EDA and estrogen signaling pathways in skeletal tissues. This is particularly surprising because estrogen signaling has been well known as a potent repressor of NF-κB activity in various skeletal cells such as osteoblasts, osteoclasts and chondrocytes [187,188,189]. Estrogen signaling can also inhibit osteoclast differentiation through transcriptional repression of NFATc1, the master regulator of osteoclastogenesis [190]. Furthermore, NFATc1 has been found to promote osteoblast differentiation by suppressing Er-a transcription [191]. In this study, NFATc1 was found to act as an upstream transcriptional inhibitor of the Er-a gene by directly binding to its promoter [191]. However, a recent study has found unexpectedly that a direct cooperative interaction between Er-a and NFATc1 leads to suppression of WNT5B transcription and consequently to promotion of osteoblast differentiation in human [192]. Based on these observations, EDA and estrogen signaling pathways might have indirect regulatory interactions in skeletal tissues through competitive modulation of NF-κB and NFATc1 activities; such crosstalk of EDA and estrogen signaling pathways would be more likely to have inhibitory outcomes (Figure 3D). At a physiological systems level, the interaction between EDA and estrogen signaling may be important for maintaining skeletal tissue homeostasis and should be explored in future studies.
6. Cross-Talk Between EDA and Calcium Dependent Pathways
6.1. Signaling Pathways Mediated by Nuclear Factor of Activated T-Cells
The NFAT (nuclear factor of activated T-cells) signaling pathway plays a role in various aspects of skeletal development and morphogenesis, orchestrating processes such as osteoblastogenesis, osteoclastogenesis as well as skeletal remodeling, inflammation and homeostasis [193,194,195]. This pathway, which is induced by calcium through the Orai1 calcium channel and STIM1 calcium sensor, is involved in transducing extracellular signals into intracellular responses, ultimately regulating gene expression and cellular functions essential for skeletal development. NFAT pathway activation promotes the differentiation of mesenchymal stem cells into osteoblasts [196]. Studies have shown that NFATc1, a key transcription factor downstream of calcineurin, plays a central role in this process by regulating the expression of osteogenic genes such as Runx2 and Osterix [196]. The NFAT pathway also plays a crucial role in regulating the differentiation and function of osteoclasts [197]. NFATc1 is a master regulator of osteoclastogenesis, and its activation promotes osteoclast differentiation by inducing the expression of genes essential for osteoclast formation and activity, including TRAP and Cathepsin K [197]. Moreover, NFATc1 regulates the expression of genes involved in osteoclast fusion and bone resorption, thereby contributing to bone remodeling [193]. NFATc1 regulates the balance between osteoblast and osteoclast activities, thereby modulating bone remodeling processes such as bone formation, resorption, and turnover. Dysregulation of NFATc1 activity can disrupt this balance, leading to pathological conditions such as osteoporosis and osteopetrosis [193,198]. Interestingly, the conservation of the NFAT pathway between mammals and fish makes it an interesting target for comparative molecular studies of bone remodeling across vertebrates [199]. Moreover, the NFAT pathway is involved in regulating bone and cartilage inflammation. NFATc1 activation in immune cells, such as macrophages and T cells, promotes the production of pro-inflammatory cytokines and mediators, which can contribute to bone and cartilage inflammation [197]. Finally, NFAT pathway plays a critical role in maintaining skeletal homeostasis by regulating the expression of genes involved in bone and cartilage metabolism, mineralization, and turnover [200]. NFATc1 activation modulates the activity of osteoblasts and osteoclasts in response to various extracellular signals, thereby ensuring proper skeletal development, growth, and maintenance throughout life [201].
Among the calcium-dependent signaling pathways, EDA signal has the most direct and well-characterized crosstalk with the NFAT pathway [17,202]. This is due to the fact that NFATc1 (the major transcription factor in the NFAT pathway) is also one of the most prominent downstream transcriptional targets (after NF-κB) of the EDA signaling pathway in skeletal tissues and EDA treatment can potently induce transcription of both NFATc1 and NF-κB in both craniofacial and endochondral skeletal cells [17,202]. However, a recent study in mice has shown that enhancement of NF-κB signal can impair osteoclastogenesis through direct inhibition of NFATc1 activity as well [203]. Previously, NF-κB was always considered as a potent transcriptional inducer of NFATc1 during osteoclast differentiation [201]. This contrasting evidence may indicate the presence of an unknown negative feedback loop in which NF-κB signal limits NFATc1 activity in skeletal cells, but further functional studies are required to validate this hypothesis. Although the possibility for the presence of such a feedback is proposed in osteoclasts under inflammatory condition, the potential involvement of EDA signal in this remained elusive [204]. Considering the extensive role of NFAT pathway in various aspects of skeletogenesis (Figure 4A), it is no surprise that future studies might reveal interference of EDA signal in all these processes, particularly skeletal development, through modulation of NFAT pathway.
6.2. Parathyroid Hormone Signaling Pathway
Parathyroid hormone (PTH) and parathyroid hormone-related peptide (PTHrP) are closely related proteins secreted by distinct cell types. PTH, from parathyroid glands, regulates calcium and phosphate levels in the bloodstream. PTHrP, with RNA-splicing variations, plays critical roles in growth and maturation as paracrine/autocrine hormones [205]. They can attach to different or overlapping receptors, triggering diverse signaling pathways, including elevated Ca2+, activation of enzymes like PKA and PLC, and modulation of pathways like MAPK [205]. The PTH/PTHrP pathways regulate osteoblastogenesis and bone formation [206,207]. PTHrP signaling influences Sox9 and Runx2, key proteins for cartilage and bone formation and controls RANKL and Ap-1 activity in skeletal cells.
In mammals, it has been already shown that PTHrP is a direct downstream transcriptional target of Eda/Edar/NF-κB in epithelial cells in skin and mammary glands and its expression is induced by EDA treatment during mammary gland morphogenesis [208]. Interestingly, the impairment in bone remodeling and osteoclast differentiation in HED patients with EDA mutation is attributed to reduced PTH function since the expression of PTH is decreased in their craniofacial skeletal tissues [23]. This indicates that EDA signal may act upstream of the PTH pathway during skeletogenesis; however the molecular mechanisms underlying such a regulatory connection have not been further explored. Because PTH has crosstalk with Wnt and FGF pathways in skeletal tissues, it is also likely that the regulatory connection between EDA and PTH signals is indirect and mediated through these pathways. Such a scenario is worth investigating since Eda/Edar/NF-κB pathway targets both PTHrP and Wnt10 in epithelial cells where they exhibit similar expression pattern [209]. Notably, cooperative regulatory interactions between EDA, PTHrP, Wnt and FGF signaling pathways have been already demonstrated during tooth formation in mammals [210]. Taken together, these findings imply on presence of a stimulatory regulatory connection between EDA and PTH/PTHrP pathways in skeletal tissues which might be indirect through Wnt and FGF pathways (Figure 4B). Although limited data are available to interpret the roles of EDA and PTH/PTHrP pathways in the general physiology of the skeletal system, mechanistically their interaction is likely important for key skeletal developmental processes.
6.3. Calmodulin Signaling Pathway
Calcium (Ca2+) is a ubiquitous signaling molecule regulating Ca2+-binding factors and associated cascades. It plays a vital role in cellular processes and is integral to skeletal biology [211,212]. Stimulated cells experience a rapid Ca2+ release through voltage-sensitive channels, increasing cytoplasmic Ca2+ that binds to calmodulin (CaM), a conserved protein. CaM, binding calcium ions, activates proteins like CaM kinases (CaMK) and Calcineurin (Cn), which are crucial for skeleton formation. Ca2+/CaM signals regulate bone processes, influencing osteoblast and osteoclast differentiation and proliferation [212,213]. They also affect chondrocyte differentiation, mechanotransduction signals, and interact with MAPKs, CREB, NFAT. The Ca2+/CaM signal interacts with the BMP pathway, influencing skeletal development [211,214,215]. Differentially regulated components of the Ca2+/CaM pathway contribute to skeletal variation in closely related species [214,215].
Similar to EDA signal, the pathway mediated by Ca2+/CaM is known to be essential during tooth development and morphogenesis [216] and recently it has been shown that mutations in components of both pathway can result in similar dental deformities in human [217]. Nevertheless, there is no study investigating the possibility of direct crosstalk between EDA and Ca2+/CaM signaling pathways. However, it is demonstrated that during endochondral ossification, a CaM kinase (CaMKII) can inhibit chondrogenic differentiation of progenitor cells through activation of the two major downstream transcription factor targets of EDA signal (NF-κB and NFATc1) [218]. On the other hand, activation of another CaM kinase (CaMKIV) has been found to induce osteoclast differentiation again through enhancement of NF-kB and NFATc1 activity [219]. Calcineurin (Cn) is another major target protein that is activated by Ca2+/CaM signal, and strikingly, activation of Cn inhibits osteoblast proliferation and differentiation through direct dephosphorylation of NFATc1 [212]. Despite the absence of demonstrated direct crosstalk between EDA and Ca2+/CaM signaling pathways, the regulation of key targets of the EDA signal by components of the Ca2+/CaM signal in skeletal cells (Figure 4C) warrants further investigation into potential direct interactions during skeletogenesis. Overall, based on the findings discussed above, it is conceivable that interactions between EDA and Ca2+/CaM could become a future topic in skeletal physiology, particularly in processes related to skeletal remodeling.
6.4. Endothelin Signaling Pathway
Endothelins (Edns), initially produced as inert proteins, undergo complex enzymatic processes and are secreted by cells in response to stimulation [220]. Edns act in both paracrine and autocrine manners, binding to transmembrane receptors (Ednrs) that initiate downstream signaling cascades, increasing Ca2+ levels and activating MAPK and PI3K-AKT pathways. The endothelin signaling pathway is a critical regulator in skeletal biology, influencing both bone and cartilage development [221]. Endothelins, a family of peptides, interact with endothelin receptors to modulate cellular processes such as proliferation, differentiation, and matrix production. In bone, endothelin signaling supports osteoblast activity and bone matrix deposition, contributing to skeletal growth and remodeling. In cartilage, it plays a role in chondrocyte proliferation and the maintenance of cartilage structure, essential for joint function and integrity [222]. Dysregulation of endothelin signaling is associated with skeletal abnormalities and disorders such as osteoarthritis, emphasizing its importance in maintaining healthy bone and cartilage dynamics.
In the skin of EDA-deficient mice, Edn1 has been found to be a transcriptional target of EDA signaling and Edn1 expression is reduced in the absence of active EDA signal in keratinocytes [223]. Although, EDA and Edn/Ednr signaling pathways have been shown to be essential for jaw skeletogenesis and tooth development [224], surprisingly, to date no study has investigated their potential regulatory interaction during skeletal development and morphogenesis. A major target of Edn/Ednr during craniofacial skeletal development is Nr2f, and activation of Edn signal strongly suppresses Nr2f transcription in developing skeleton (mainly upper jaw) [225]. Nr2f encodes a crucial transcription factor regulating various aspects of bone and cartilage development and remodeling [226]. Strikingly, Nr2f is a very potent suppressor of NF-κB transcription in skeletal cells, particularly during osteoclastogenesis and bone remodeling [226]. Therefore, it is conceivable that the activated Edn/Ednr signal could enhance the effects of the EDA/NF-κB signal in bone by repressing Nr2f transcription, but confirmation of such an indirect regulatory synergy between these pathways in skeletal tissues requires experimental validation. Finally, the activation of both EDA and Edn/Ednr/Grem2 signaling pathways are essential during tooth development, indicating their potential cooperative/synergistic interactions in skeletal development [227]. Based on these findings, the interaction between EDA and Edn/Ednr may play an important role in skeletal tissue remodeling, particularly in processes involving skeletal cell apoptosis (Figure 4D).
7. Potential Cross-Talk Between EDA and Non-Canonical Microenvironment-Responsive Pathways Affecting Skeletogenesis
7.1. Serotonin Signaling Pathway
The serotonin signaling pathway plays a multifaceted role in skeletal development and maintenance, acting both locally within bone and cartilage and systemically through its endocrine effects [228]. Serotonin, a neurotransmitter, exists in two distinct pools: central (produced in the brain) and peripheral (produced in the gut), with the latter being particularly influential in bone biology. Peripheral serotonin can inhibit bone formation by acting on osteoblasts, while central serotonin has been shown to promote bone mass accrual through neural signaling pathways. In cartilage, serotonin regulates chondrocyte proliferation and matrix production, contributing to proper cartilage formation and maintenance [229,230].
In zebrafish, the development of skin and scales both involve activation of serotonin and EDA signaling pathways, but the potential molecular crosstalk between these pathways has not been investigated [231]. Currently, no research has explored the potential direct molecular interaction between serotonin and EDA signaling pathways in skeletal tissues. However, studies of EDA-deficient mice indicated that EDA mediated regulation of osteoclast differentiation is tightly linked to activation of RANKL (Eda1/Edar/NF-κB axis) [17], a major factor controlling osteoclastogenesis, and interestingly, in endochondral bone, serotonin signal also exerts its stimulatory effects on osteoclastogenesis through activation of NF-κB and NFATc1 [232]. This may indicate synergistic effects of serotonin and EDA signaling pathways on bone resorption and remodeling through NF-κB and NFATc1. Moreover, the adverse effects of excessive activation of 5-HT2B receptor on endochondral bone appeared to be linked with transcriptional dysregulation of NF-κB, the main transcription factor in EDA signaling [233]. These also suggest a potential competitive regulatory effect of serotonin and EDA signaling pathways on NF-κB transcription in skeletal cells. In addition, NF-κB function is implicated in tryptophan metabolism, which is required for serotonin synthesis, and reciprocally, serotonin signal mediates its effects on inflammatory responses through regulation of NF-κB transcription [234]. Finally, a complex indirect regulatory link between activity of EDA signaling and expression of tryptophan hydroxylase (TPH), encoding the main enzyme in serotonin synthesis, has been reported in cartilage [235]. Taken together, these findings suggest potential indirect synergistic (through RANKL transcription and tryptophan metabolism) or competitive (through NF-κB transcription) regulatory connections between serotonin and EDA signaling pathways which requires further validations in skeletal tissues (Figure 5A). From a systemic physiological perspective, their interactions may be important for advancing our understanding of skeletal tissue maintenance.
7.2. Integrin Signaling Pathway
Integrins are transmembrane receptors that facilitate ECM interactions, playing a pivotal role in skeletal development and maintenance. In bone biology, integrins such as α1β1 and α2β1 mediate osteoblast adhesion to collagen, influencing bone formation and remodeling processes [236,237]. In cartilage, integrins like α5β1 and αVβ3 are expressed on chondrocytes and interact with ECM components, regulating cell adhesion, mechanotransduction, and matrix production [238]. These integrin-mediated interactions are essential for maintaining cartilage integrity and function. Dysregulation of integrin signaling has been implicated in skeletal disorders, including osteoarthritis, where altered integrin expression contributes to disease progression.
There have been no investigations examining the possible direct molecular interplay between integrin and EDA signaling pathways in skeletal tissue. In epithelial cells, however, EDA signaling pathway modulates the affinity of adhesion receptors such as integrins, thus affecting integrin-mediated cell-matrix morphogenesis in these cells [239]. In the skin of EDA-deficient mice, the direct physical interactions between the extracellular domain of EDA and matrix elements like integrins are lost suggesting that the EDA mediated skin morphogenesis is exerted through direct physical interactions with integrin molecules [240]. A network-based analysis of molecular players during scale development and morphogenesis also revealed extensive regulatory connections between integrins and EDA signaling components [241]. Similarly, potential regulatory connections between the activity of EDA and integrin-mediated signals have been reported in human cartilage. It is important to note that NF-kB signaling directly modulate integrin-β1 expression in bone and acts upstream of integrin signaling [242]. On the other hand, activation of certain integrin signals can activate NF-kB mediated signals in osteoblasts in endochondral bone upon mechanical stimulation [243], but the potential involvement of EDA in this regulatory connection has remained unclear. Given these observations, it is conceivable to propose a hypothetical model in which EDA activates integrin signaling through direct physical interaction with integrin molecules during skeletogenesis (Figure 5B), emphasizing the need for further investigations to explore this possibility. Moreover, based on the current cellular findings, the interaction between EDA and integrin signaling is likely to play a broader physiological role in skeletal mechanobiology and apoptosis-related processes.
7.3. Nitric Oxide Signaling Pathway
Initially, nitric oxide (NO) signaling was discovered to regulate endochondral ossification and later was found to be involved in skeletal cell differentiation and mechanical adaptation [244,245,246]. To date, there is no study investigating the possibility of direct molecular crosstalk between NO and EDA signaling pathways in any skeletal tissue; however, NF-κB, the main transcription factor mediating EDA signal, has been shown to directly act upstream of the nitric oxide synthase II encoding gene (NOS2) and increases its transcription in the brain [247]. Moreover, in several ectodermal-derived tissues, NF-κB activates the transcription of iNOS, another major gene encoding inducible nitric oxide synthase [248]. In articular cartilage, NF-κB activates the transcription of iNOS in chondrocytes in response to inflammatory stress [249]. A later study in endochondral cartilage has shown that the mechanotransduction signal mediated by NF-κB in cartilage might involve activation of NOS signal in addition to other mechanosensing signals [250]. Furthermore, the inhibition of NF-κB signal is essential during endochondral cartilage repair and this mechanism appears to also involve activation of NO signaling [251]; however, further studies are required to elucidate the molecular interaction between NF-κB and NO signals in cartilage regeneration/repair. In endochondral bone, NO signaling (through NOS2) is required for mediating the mechanical-induced responses and inhibition of NO signaling reduces osteoblast proliferation and increases their differentiation [252]. Interestingly, this mechanism in bone is also mediated by NF-κB at the transcriptional level [252]. These findings indicate the necessity of future research to explore potential direct molecular crosstalk between components of EDA and NO signaling pathways in skeletal system since they both share the same transcription factor (NF-κB) in these processes (Figure 5B). Furthermore, their interactions may be important in broader physiological contexts, particularly in studies of mechanical sensing and tissue regeneration within skeletal systems.
Conclusion
The Ectodysplasin-A (EDA) signaling pathway is frequently studied in the context of dermal skeletal development, particularly in craniofacial bone and cartilage, where it significantly influences the formation and diversity of skeletal structures in vertebrates. Its interactions with other critical skeletogenic and morphogenic pathways that we synthesize in this review highlight the incredibly complex network of molecular cross-talk governing skeletal development and morphogenesis (see summary Table 1). By integrating signals from multiple pathways, EDA may contribute to the precise regulation of cellular processes essential for various aspects of skeletal physiology (see summary Table 2) – making it a coordinator of sculptors. Understanding these complex relationships enhances our comprehension of skeletal biology and highlights potential avenues for therapeutic intervention in skeletal disorders, such as craniofacial skeletal anomalies in humans. Future research focusing on EDA’s multifaceted roles and its synergistic actions with other signaling networks holds promise for advancing skeletal biology and regenerative medicine.
References
- C.Y. Cui, D. Schlessinger, EDA Signaling and Skin Appendage Development, Cell Cycle 5 (2006) 2477–2483. [CrossRef]
- T. Mustonen, M. Ilmonen, M. Pummila, A.T. Kangas, J. Laurikkala, R. Jaatinen, J. Pispa, O. Gaide, P. Schneider, I. Thesleff, M.L. Mikkola, Ectodysplasin A1 promotes placodal cell fate during early morphogenesis of ectodermal appendages, Development 131 (2004) 4907–4919. [CrossRef]
- A. Sadier, L. Viriot, S. Pantalacci, V. Laudet, The ectodysplasin pathway: From diseases to adaptations, Trends Genet. 30 (2014) 24–31. [CrossRef]
- P. Schneider, S.L. Street, O. Gaide, S. Hertig, A. Tardivel, J. Tschopp, L. Runkel, K. Alevizopoulos, B.M. Ferguson, J. Zonana, Mutations leading to X-linked hypohidrotic ectodermal dysplasia affect three major functional domains in the tumor necrosis factor family member ectodysplasin-A, J. Biol. Chem. 276 (2001) 18819–18827. [CrossRef]
- C. Cluzeau, S. Hadj-Rabia, M. Jambou, S. Mansour, P. Guigue, S. Masmoudi, E. Bal, N. Chassaing, M.C. Vincent, G. Viot, F. Clauss, M.C. Manière, S. Toupenay, M. Le Merrer, S. Lyonnet, V. Cormier-Daire, J. Amiel, L. Faivre, Y. De Prost, A. Munnich, J.P. Bonnefont, C. Bodemer, A. Smahi, Only four genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases, Hum. Mutat. 32 (2011) 70–72. [CrossRef]
- Z. Cai, X. Deng, J. Jia, D. Wang, G. Yuan, Ectodysplasin A/Ectodysplasin A Receptor System and Their Roles in Multiple Diseases, Front. Physiol. 12 (2021). [CrossRef]
- L. Xing, Y. Liu, J. Wu, C. Song, B. Jiang, Spatial and Temporal Expression of Ectodysplasin-A Signaling Pathway Members During Mandibular Condylar Development in Postnatal Mice 71 (2023) 631-642. [CrossRef]
- O. Montonen, S. Ezer, U.K. Saarialho-Kere, R. Herva, M.-L. Karjalainen-Lindsberg, I. Kaitila, D. Schlessinger, A.K. Srivastava, I. Thesleff, J. Kere, Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity, Int. J. Mol. Sci. 2022, Vol. 23, Page 12189 23 (2022) 12189. [CrossRef]
- M.L. Mikkola, I. Thesleff, Ectodysplasin signaling in development, Cytokine Growth Factor Rev. 14 (2003) 211–224. [CrossRef]
- E.P. Ahi, Signalling pathways in trophic skeletal development and morphogenesis: Insights from studies on teleost fish, Dev. Biol. 420 (2016) 11–31. [CrossRef]
- S. Dash, P.A. Trainor, The development, patterning and evolution of neural crest cell differentiation into cartilage and bone, Bone 137 (2020) 115409. [CrossRef]
- O. Elomaa, K. O. Elomaa, K. Pulkkinen, U. Hannelius, M. Mikkola, U. Saarialho-Kere, J. Kere, Ectodysplasin is released by proteolytic shedding and binds to the EDAR protein, Hum. Mol. Genet. 10 (2001) 953–962. [CrossRef]
- K. Verhelst, S. Gardam, A. Borghi, M. Kreike, I. Carpentier, R. Beyaert, XEDAR activates the non-canonical NF-κB pathway, Biochem. Biophys. Res. Commun. 465 (2015) 275–280. [CrossRef]
- H. Fujikawa, M. Farooq, A. Fujimoto, M. Ito, Y. Shimomura, Functional studies for the TRAF6 mutation associated with hypohidrotic ectodermal dysplasia, Br. J. Dermatol. 168 (2013) 629–633. [CrossRef]
- A. Morlon, A. Munnich, A. Smahi, TAB2, TRAF6 and TAK1 are involved in NF-κB activation induced by the TNF-receptor, Edar and its adaptator Edaradd, Hum. Mol. Genet. 14 (2005) 3751–3757. [CrossRef]
- T. Mustonen, J. Pispa, M.L. Mikkola, M. Pummila, A.T. Kangas, L. Pakkasjärvi, R. Jaatinen, I. Thesleff, Stimulation of ectodermal organ development by Ectodysplasin-A1, Dev. Biol. 259 (2003) 123–136. [CrossRef]
- C. Schweikl, S. Maier-Wohlfart, H. Schneider, J. Park, Ectodysplasin A1 Deficiency Leads to Osteopetrosis-like Changes in Bones of the Skull Associated with Diminished Osteoclastic Activity, Int. J. Mol. Sci. 2022, Vol. 23, Page 12189 23 (2022) 12189. [CrossRef]
- C.S. Kossel, M. Wahlbuhl, S. Schuepbach-Mallepell, J. Park, C. Kowalczyk-Quintas, M. Seeling, K. von der Mark, P. Schneider, H. Schneider, Correction of Vertebral Bone Development in Ectodysplasin A1-Deficient Mice by Prenatal Treatment With a Replacement Protein, Front. Genet. 12 (2021) 709736. [CrossRef]
- B. Chang, V. Punj, M. Shindo, P.M. Chaudhary, Adenoviral-mediated gene transfer of ectodysplasin-A2 results in induction of apoptosis and cell-cycle arrest in osteosarcoma cell lines, Cancer Gene Ther. 2007 1411 14 (2007) 927–933. [CrossRef]
- M. Pummila, I. Fliniaux, R. Jaatinen, M.J. James, J. Laurikkala, P. Schneider, I. Thesleff, M.L. Mikkola, Ectodysplasin has a dual role in ectodermal organogenesis: inhibition of Bmp activity and induction of Shh expression, Development 134 (2007) 117–125. [CrossRef]
- M.P. Harris, N. Rohner, H. Schwarz, S. Perathoner, P. Konstantinidis, C. Nüsslein-Volhard, Zebrafish eda and edar Mutants Reveal Conserved and Ancestral Roles of Ectodysplasin Signaling in Vertebrates, PLoS Genet. 4 (2008) e1000206. [CrossRef]
- A. Williams, E.C.Y. Wang, L. Thurner, C.J. Liu, Review: Novel Insights Into Tumor Necrosis Factor Receptor, Death Receptor 3, and Progranulin Pathways in Arthritis and Bone Remodeling, Arthritis Rheumatol. (Hoboken, N.J.) 68 (2016) 2845. [CrossRef]
- F. Clauss, M.C. Manière, F. Obry, E. Waltmann, S. Hadj-Rabia, C. Bodemer, Y. Alembik, H. Lesot, M. Schmittbuhl, Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): a review, J. Dent. Res. 87 (2008) 1089–1099. [CrossRef]
- R.A. Costa, D.M. Power, Skin and scale regeneration after mechanical damage in a teleost, Mol. Immunol. 95 (2018) 73–82. [CrossRef]
- F. Tonelli, J.W. Bek, R. Besio, A. De Clercq, L. Leoni, P. Salmon, P.J. Coucke, A. Willaert, A. Forlino, Zebrafish: A Resourceful Vertebrate Model to Investigate Skeletal Disorders, Front. Endocrinol. (Lausanne). 11 (2020) 555577. [CrossRef]
- J. Kere, A.K. Srivastava, O. Montonen, J. Zonana, N. Thomas, B. Ferguson, F. Munoz, D. Morgan, A. Clarke, P. Baybayan, E.Y. Chen, S. Ezer, U. Saarialho-Kere, A. De La Chapelle, D. Schlessinger, X–linked anhidrotic (hypohidrotic) ectodermal dysplasia is caused by mutation in a novel transmembrane protein, Nat. Genet. 1996 134 13 (1996) 409–416. [CrossRef]
- A.W. Monreal, B.M. Ferguson, D.J. Headon, S.L. Street, P.A. Overbeek, J. Zonana, Mutations in the human homologue of mouse dl cause autosomal recessive and dominant hypohidrotic ectodermal dysplasia, Nat. Genet. 1999 224 22 (1999) 366–369. [CrossRef]
- J.Y. Sire, A. Huysseune, Formation of dermal skeletal and dental tissues in fish: A comparative and evolutionary approach, Biol. Rev. Camb. Philos. Soc. 78 (2003). [CrossRef]
- P.C.J. Donoghue, I.J. Sansom, Origin and early evolution of vertebrate skeletonization, Microsc. Res. Tech. 59 (2002) 352–372. [CrossRef]
- T.W.P. Wood, T. Nakamura, Problems in Fish-to-Tetrapod Transition: Genetic Expeditions Into Old Specimens, Front. Cell Dev. Biol. 6 (2018). [CrossRef]
- N.M. O’brown, B.R. Summers, F.C. Jones, S.D. Brady, D.M. Kingsley, A recurrent regulatory change underlying altered expression and Wnt response of the stickleback armor plates gene EDA, Elife 2015 (2015). [CrossRef]
- T.G. Laurentino, N. Boileau, F. Ronco, D. Berner, The ectodysplasin-A receptor is a candidate gene for lateral plate number variation in stickleback fish., G3 (Bethesda). 12 (2022) jkac077–jkac077. [CrossRef]
- M. Wagner, S. Bračun, A. Duenser, C. Sturmbauer, W. Gessl, E.P. Ahi, Expression variations in ectodysplasin-A gene (eda) may contribute to morphological divergence of scales in haplochromine cichlids, BMC Ecol. Evol. 22 (2022) 28. [CrossRef]
- M. de Caestecker, The transforming growth factor-β superfamily of receptors, Cytokine Growth Factor Rev. 15 (2004) 1–11. [CrossRef]
- J.G. Kim, Y.A. Rim, J.H. Ju, The Role of Transforming Growth Factor Beta in Joint Homeostasis and Cartilage Regeneration, Tissue Eng. Part C. Methods 28 (2022) 570–587. [CrossRef]
- E.P. Ahi, Regulation of Skeletogenic Pathways by m6A RNA Modification: A Comprehensive Review, Calcif. Tissue Int. 2025 1161 116 (2025) 1–23. [CrossRef]
- A.S. Patil, R.B. Sable, R.M. Kothari, An update on transforming growth factor-β (TGF-β): Sources, types, functions and clinical applicability for cartilage/bone healing, J. Cell. Physiol. 226 (2011) 3094–3103. [CrossRef]
- A. Machiya, S. Tsukamoto, S. Ohte, M. Kuratani, N. Suda, T. Katagiri, Smad4-dependent transforming growth factor-β family signaling regulates the differentiation of dental epithelial cells in adult mouse incisors, Bone 137 (2020) 115456. [CrossRef]
- W. Wang, D. Rigueur, K.M. Lyons, TGFβ as a gatekeeper of BMP action in the developing growth plate, Bone 137 (2020) 115439. [CrossRef]
- L. Li, S. Xiang, B. Wang, H. Lin, S. Kihara, H. Sun, P.G. Alexander, R.S. Tuan, TGF-β1 plays a protective role in glucocorticoid-induced dystrophic calcification, Bone 136 (2020) 115355. [CrossRef]
- E.C. Ekholm, L. Ravanti, V.M. Kähäri, P. Paavolainen, R.P.K. Penttinen, Expression of extracellular matrix genes: transforming growth factor (TGF)-β1 and ras in tibial fracture healing of lathyritic rats, Bone 27 (2000) 551–557. [CrossRef]
- A.G. Li, M.I. Koster, X.J. Wang, Roles of TGFβ signaling in epidermal/appendage development, Cytokine Growth Factor Rev. 14 (2003) 99–111. [CrossRef]
- M. Bei, Molecular genetics of tooth development, Curr. Opin. Genet. Dev. 19 (2009) 504–510. [CrossRef]
- S. Trakanant, J. Nihara, M. Kawasaki, F. Meguro, A. Yamada, K. Kawasaki, I. Saito, M. Takeyasu, A. Ohazama, Molecular mechanisms in palatal rugae development, J. Oral Biosci. 62 (2020) 30–35. [CrossRef]
- G.R. Gipson, E.J. Goebel, K.N. Hart, E.C. Kappes, C. Kattamuri, J.C. McCoy, T.B. Thompson, Structural perspective of BMP ligands and signaling, Bone 140 (2020) 115549. [CrossRef]
- T.K. Sampath, A.H. Reddi, Discovery of bone morphogenetic proteins – A historical perspective, Bone 140 (2020) 115548. [CrossRef]
- G. Sanchez-Duffhues, E. Williams, M.J. Goumans, C.H. Heldin, P. ten Dijke, Bone morphogenetic protein receptors: Structure, function and targeting by selective small molecule kinase inhibitors, Bone 138 (2020) 115472. [CrossRef]
- J. Gluhak-Heinrich, D. Guo, W. Yang, M.A. Harris, A. Lichtler, B. Kream, J. Zhang, J.Q. Feng, L.C. Smith, P. Dechow, S.E. Harris, New roles and mechanism of action of BMP4 in postnatal tooth cytodifferentiation, Bone 46 (2010) 1533–1545. [CrossRef]
- C. da Silva Madaleno, J. Jatzlau, P. Knaus, BMP signalling in a mechanical context – Implications for bone biology, Bone 137 (2020) 115416. [CrossRef]
- D.M. Medeiros, J.G. Crump, New perspectives on pharyngeal dorsoventral patterning in development and evolution of the vertebrate jaw., Dev. Biol. 371 (2012) 121–35. [CrossRef]
- T. Katagiri, T. Watabe, Bone Morphogenetic Proteins, Cold Spring Harb. Perspect. Biol. 8 (2016) a021899. [CrossRef]
- Y. Iida, K. Hibiya, K. Inohaya, A. Kudo, Eda/Edar signaling guides fin ray formation with preceding osteoblast differentiation, as revealed by analyses of the medaka all-fin less mutant afl, Dev. Dyn. 243 (2014) 765–777. [CrossRef]
- C.C. Mandal, F. Das, S. Ganapathy, S.E. Harris, G.G. Choudhury, N. Ghosh-Choudhury, Bone morphogenetic protein-2 (BMP-2) activates NFATc1 transcription factor via an autoregulatory loop involving Smad/Akt/Ca2+ signaling, J. Biol. Chem. 291 (2016) 1148–1161. [CrossRef]
- W. Shen, Y. Wang, Y. Liu, H. Liu, H. Zhao, G. Zhang, M.L. Snead, D. Han, H. Feng, Functional Study of Ectodysplasin-A Mutations Causing Non-Syndromic Tooth Agenesis, PLoS One 11 (2016) e0154884. [CrossRef]
- H.W.A. Ehlen, L.A. Buelens, A. Vortkamp, Hedgehog signaling in skeletal development, Birth Defects Res. Part C Embryo Today Rev. 78 (2006) 267–279. [CrossRef]
- D. Huangfu, K. V Anderson, Signaling from Smo to Ci/Gli: conservation and divergence of Hedgehog pathways from Drosophila to vertebrates., Development 133 (2006) 3–14. [CrossRef]
- S. Ohba, Hedgehog Signaling in Skeletal Development: Roles of Indian Hedgehog and the Mode of Its Action, Int. J. Mol. Sci. 2020, Vol. 21, Page 6665 21 (2020) 6665. [CrossRef]
- J. Yang, P. Andre, L. Ye, Y.Z. Yang, The Hedgehog signalling pathway in bone formation, Int. J. Oral Sci. 2015 72 7 (2015) 73–79. [CrossRef]
- J. Xu, P.P.R. Iyyanar, Y. Lan, R. Jiang, Sonic hedgehog signaling in craniofacial development, Differentiation 133 (2023) 60–76. [CrossRef]
- A. Marumoto, R. Milani, R.A. da Silva, C.J. da Costa Fernandes, J.M. Granjeiro, C. V. Ferreira, M.P. Peppelenbosch, W.F. Zambuzzi, Phosphoproteome analysis reveals a critical role for hedgehog signalling in osteoblast morphological transitions, Bone 103 (2017) 55–63. [CrossRef]
- C. Kan, L. Chen, Y. Hu, N. Ding, Y. Li, T.L. McGuire, H. Lu, J.A. Kessler, L. Kan, Gli1-labeled adult mesenchymal stem/progenitor cells and hedgehog signaling contribute to endochondral heterotopic ossification, Bone 109 (2018) 71–79. [CrossRef]
- A.J. Aman, A.N. Fulbright, D.M. Parichy, Wnt/β-catenin regulates an ancient signaling network during zebrafish scale development, Elife 7 (2018). [CrossRef]
- S.-W. Cho, J.C. van Rijssel, F. Witte, M.A.G. de Bakker, M.K. Richardson, The sonic hedgehog signaling pathway and the development of pharyngeal arch Derivatives in Haplochromis piceatus, a Lake Victoria cichlid, J. Oral Biosci. 57 (2015) 148–156. [CrossRef]
- T. Shimo, K. Matsumoto, K. Takabatake, E. Aoyama, Y. Takebe, S. Ibaragi, T. Okui, N. Kurio, H. Takada, K. Obata, P. Pang, M. Iwamoto, H. Nagatsuka, A. Sasaki, The Role of Sonic Hedgehog Signaling in Osteoclastogenesis and Jaw Bone Destruction, PLoS One 11 (2016) e0151731. [CrossRef]
- L. Zhang, Y. Yang, Z. Liao, Q. Liu, X. Lei, M. Li, Saijilafu, Z. Zhang, D. Hong, M. Zhu, B. Li, H. Yang, J. Chen, Genetic and pharmacological activation of Hedgehog signaling inhibits osteoclastogenesis and attenuates titanium particle-induced osteolysis partly through suppressing the JNK/c-Fos-NFATc1 cascade, Theranostics 10 (2020) 6638. [CrossRef]
- D. Schupbach, M. Comeau-Gauthier, E. Harvey, G. Merle, Wnt modulation in bone healing, Bone 138 (2020) 115491. [CrossRef]
- T.A. Burgers, B.O. Williams, Regulation of Wnt/β-catenin signaling within and from osteocytes, Bone 54 (2013) 244–249. [CrossRef]
- D.G. Monroe, M.E. McGee-Lawrence, M.J. Oursler, J.J. Westendorf, Update on Wnt signaling in bone cell biology and bone disease, Gene 492 (2012) 1–18. [CrossRef]
- T. Oichi, S. Otsuru, Y. Usami, M. Enomoto-Iwamoto, M. Iwamoto, Wnt signaling in chondroprogenitors during long bone development and growth, Bone 137 (2020) 115368. [CrossRef]
- R.B. Choi, A.G. Robling, The Wnt pathway: An important control mechanism in bone’s response to mechanical loading, Bone 153 (2021) 116087. [CrossRef]
- L. Hu, W. Chen, A. Qian, Y.P. Li, Wnt/β-catenin signaling components and mechanisms in bone formation, homeostasis, and disease, Bone Res. 2024 121 12 (2024) 1–33. [CrossRef]
- P.J. Niziolek, T.L. Farmer, Y. Cui, C.H. Turner, M.L. Warman, A.G. Robling, High-bone-mass-producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes, Bone 49 (2011) 1010–1019. [CrossRef]
- D. yu Bao, Y. Yang, X. Tong, H. yan Qin, Activation of wnt/β-catenin signaling pathway down regulated osteogenic differentiation of bone marrow-derived stem cells in an anhidrotic ectodermal dysplasia patient with EDA/EDAR/EDARADD mutation, Heliyon 10 (2024). [CrossRef]
- H.J. Won, J.W. Kim, H.S. Won, J.O. Shin, Gene Regulatory Networks and Signaling Pathways in Palatogenesis and Cleft Palate: A Comprehensive Review, Cells 2023, Vol. 12, Page 1954 12 (2023) 1954. [CrossRef]
- M.N. Evanitsky, S. Di Talia, An active traveling wave of Eda/NF-κB signaling controls the timing and hexagonal pattern of skin appendages in zebrafish, Dev. 150 (2023). [CrossRef]
- F. Liu, Y. Zhao, Y. Pei, F. Lian, H. Lin, Role of the NF-kB signalling pathway in heterotopic ossification: biological and therapeutic significance, Cell Commun. Signal. 2024 221 22 (2024) 1–26. [CrossRef]
- E. Horváth, Á. Sólyom, J. Székely, E.E. Nagy, H. Popoviciu, Inflammatory and Metabolic Signaling Interfaces of the Hypertrophic and Senescent Chondrocyte Phenotypes Associated with Osteoarthritis, Int. J. Mol. Sci. 2023, Vol. 24, Page 16468 24 (2023) 16468. [CrossRef]
- K.S. Alharbi, O. Afzal, A.S.A. Altamimi, W.H. Almalki, I. Kazmi, F.A. Al-Abbasi, S.I. Alzarea, H.A. Makeen, M. Albratty, Potential role of nutraceuticals via targeting a Wnt/β-catenin and NF-κB pathway in treatment of osteoarthritis, J. Food Biochem. 46 (2022). [CrossRef]
- Y.Q. Liu, Z.L. Hong, L. Bin Zhan, H.Y. Chu, X.Z. Zhang, G.H. Li, Wedelolactone enhances osteoblastogenesis by regulating Wnt/β-catenin signaling pathway but suppresses osteoclastogenesis by NF-κB/c-fos/NFATc1 pathway, Sci. Reports 2016 61 6 (2016) 1–12. [CrossRef]
- M.M. Weivoda, M. Ruan, C.M. Hachfeld, L. Pederson, A. Howe, R.A. Davey, J.D. Zajac, Y. Kobayashi, B.O. Williams, J.J. Westendorf, S. Khosla, M.J. Oursler, Wnt Signaling Inhibits Osteoclast Differentiation by Activating Canonical and Noncanonical cAMP/PKA Pathways, J. Bone Miner. Res. 31 (2016) 65–75. [CrossRef]
- Y. Kobayashi, S. Uehara, M. Koide, N. Takahashi, The regulation of osteoclast differentiation by Wnt signals, Bonekey Rep. 4 (2015) 713. [CrossRef]
- N. Takahashi, Regulatory Mechanism of Osteoclastogenesis by RANKL and Wnt Signals, Front. Biosci. 16 (2011) 21. [CrossRef]
- S.J. Bray, Notch signalling: a simple pathway becomes complex., Nat. Rev. Mol. Cell Biol. 7 (2006) 678–89. [CrossRef]
- J. Yu, E. Canalis, Notch and the regulation of osteoclast differentiation and function, Bone 138 (2020) 115474. [CrossRef]
- T.J. Mead, K.E. Yutzey, Notch signaling and the developing skeleton., Adv. Exp. Med. Biol. 727 (2012) 114–30. [CrossRef]
- A. Kamalakar, J.M. McKinney, D. Salinas Duron, A.M. Amanso, S.A. Ballestas, H. Drissi, N.J. Willett, P. Bhattaram, A.J. García, L.B. Wood, S.L. Goudy, JAGGED1 stimulates cranial neural crest cell osteoblast commitment pathways and bone regeneration independent of canonical NOTCH signaling, Bone 143 (2021) 115657. [CrossRef]
- R.A. Oldershaw, T.E. Hardingham, Notch signaling during chondrogenesis of human bone marrow stem cells, Bone 46 (2010) 286–293. [CrossRef]
- E. Canalis, L. Schilling, E. Denker, C. Stoddard, J. Yu, A NOTCH2 pathogenic variant and HES1 regulate osteoclastogenesis in induced pluripotent stem cells, Bone 191 (2025) 117334. [CrossRef]
- F. Engin, B. Lee, NOTCHing the bone: Insights into multi-functionality, Bone 46 (2010) 274–280. [CrossRef]
- C.E. Rodríguez-Ramírez, M. Hiltbrunner, V. Saladin, S. Walker, A. Urrutia, C.L. Peichel, Molecular mechanisms of Eda-mediated adaptation to freshwater in threespine stickleback, Mol. Ecol. 00 (2023) 1–19. [CrossRef]
- T. Shono, A.P. Thiery, R.L. Cooper, D. Kurokawa, R. Britz, M. Okabe, G.J. Fraser, Evolution and Developmental Diversity of Skin Spines in Pufferfishes, IScience 19 (2019) 1248–1259. [CrossRef]
- H. Fukushima, A. Nakao, F. Okamoto, M. Shin, H. Kajiya, S. Sakano, A. Bigas, E. Jimi, K. Okabe, The association of Notch2 and NF-kappaB accelerates RANKL-induced osteoclastogenesis., Mol. Cell. Biol. 28 (2008) 6402–12. [CrossRef]
- X.W. Dou, W. Park, S. Lee, Q.Z. Zhang, L.R. Carrasco, A.D. Le, Loss of Notch3 Signaling Enhances Osteogenesis of Mesenchymal Stem Cells from Mandibular Torus, J. Dent. Res. 96 (2017) 347–354. [CrossRef]
- S. Zanotti, A. Smerdel-Ramoya, E. Canalis, Reciprocal regulation of Notch and nuclear factor of activated T-cells (NFAT) c1 transactivation in osteoblasts, J. Biol. Chem. 286 (2011) 4576–4588. [CrossRef]
- X. Tu, J. Chen, J. Lim, C.M. Karner, S.Y. Lee, J. Heisig, C. Wiese, K. Surendran, R. Kopan, M. Gessler, F. Long, Physiological Notch Signaling Maintains Bone Homeostasis via RBPjk and Hey Upstream of NFATc1, PLOS Genet. 8 (2012) e1002577. [CrossRef]
- K. Inoue, X. Hu, B. Zhao, Regulatory network mediated by RBP-J/NFATc1-miR182 controls inflammatory bone resorption, FASEB J. 34 (2020) 2392. [CrossRef]
- S. Zanotti, E. Canalis, Notch regulation of bone development and remodeling and related skeletal disorders., Calcif. Tissue Int. 90 (2012) 69–75. [CrossRef]
- L. Dailey, D. Ambrosetti, A. Mansukhani, C. Basilico, Mechanisms underlying differential responses to FGF signaling., Cytokine Growth Factor Rev. 16 (2005) 233–47. [CrossRef]
- M.A. Lemmon, J. Schlessinger, Cell signaling by receptor tyrosine kinases., Cell 141 (2010) 1117–34. [CrossRef]
- X. Du, Y. Xie, C.J. Xian, L. Chen, Role of FGFs/FGFRs in skeletal development and bone regeneration., J. Cell. Physiol. 227 (2012) 3731–43. [CrossRef]
- D.M. Ornitz, P.J. Marie, Fibroblast growth factor signaling in skeletal development and disease, Genes Dev. 29 (2015) 1463–1486. [CrossRef]
- A. Duenser, P. Singh, L.A. Lecaudey, C. Sturmbauer, R.C. Albertson, W. Gessl, E.P. Ahi, Conserved Molecular Players Involved in Human Nose Morphogenesis Underlie Evolution of the Exaggerated Snout Phenotype in Cichlids, Genome Biol. Evol. 15 (2023). [CrossRef]
- L.A. Lecaudey, P. Singh, C. Sturmbauer, A. Duenser, W. Gessl, E.P. Ahi, Transcriptomics unravels molecular players shaping dorsal lip hypertrophy in the vacuum cleaner cichlid, Gnathochromis permaxillaris, BMC Genomics 22 (2021) 506. [CrossRef]
- L.A. Lecaudey, C. Sturmbauer, P. Singh, E.P. Ahi, Molecular mechanisms underlying nuchal hump formation in dolphin cichlid, Cyrtocara moorii, Sci. Rep. (2019). [CrossRef]
- J.E. Lazarus, A. Hegde, A.C. Andrade, O. Nilsson, J. Baron, Fibroblast growth factor expression in the postnatal growth plate, Bone 40 (2007) 577–586. [CrossRef]
- Y. Xie, A. Zinkle, L. Chen, M. Mohammadi, Fibroblast growth factor signalling in osteoarthritis and cartilage repair, Nat. Rev. Rheumatol. 2020 1610 16 (2020) 547–564. [CrossRef]
- O. Häärä, E. Harjunmaa, P.H. Lindfors, S.H. Huh, I. Fliniaux, T. Åberg, J. Jernvall, D.M. Ornitz, M.L. Mikkola, I. Thesleff, Ectodysplasin regulates activator-inhibitor balance in murine tooth development through fgf20 signaling, Dev. 139 (2012) 3189–3199. [CrossRef]
- S.H. Huh, K. Närhi, P.H. Lindfors, O. Häärä, L. Yang, D.M. Ornitz, M.L. Mikkola, Fgf20 governs formation of primary and secondary dermal condensations in developing hair follicles, Genes Dev. 27 (2013) 450–458. [CrossRef]
- D. Dhouailly, The avian ectodermal default competence to make feathers, Dev. Biol. 508 (2024) 64–76. [CrossRef]
- M. Iwasaki, J. Kuroda, K. Kawakami, H. Wada, Epidermal regulation of bone morphogenesis through the development and regeneration of osteoblasts in the zebrafish scale, Dev. Biol. 437 (2018) 105–119. [CrossRef]
- A. Suzuki, G. Sugiyama, Y. Ohyama, W. Kumamaru, T. Yamada, Y. Mori, Regulation of NF-kB Signalling Through the PR55β-RelA Interaction in Osteoblasts, In Vivo (Brooklyn). 34 (2020) 601. [CrossRef]
- H. Huang, J. Xie, J. Wei, S. Xu, D. Zhang, X. Zhou, Fibroblast growth factor 8 (FGF8) up-regulates gelatinase expression in chondrocytes through nuclear factor-κB p65, J. Bone Miner. Metab. 41 (2023) 17–28. [CrossRef]
- D.D. Bikle, C. Tahimic, W. Chang, Y. Wang, A. Philippou, E.R. Barton, Role of IGF-I signaling in muscle bone interactions, Bone 80 (2015) 79–88. [CrossRef]
- R.G. Maki, Small is beautiful: insulin-like growth factors and their role in growth, development, and cancer., J. Clin. Oncol. 28 (2010) 4985–95. [CrossRef]
- A. Esposito, M. Klüppel, B.M. Wilson, S.R.K. Meka, A. Spagnoli, CXCR4 mediates the effects of IGF-1R signaling in rodent bone homeostasis and fracture repair, Bone 166 (2023) 116600. [CrossRef]
- M.H.C. Sheng, X.D. Zhou, L.F. Bonewald, D.J. Baylink, K.H.W. Lau, Disruption of the insulin-like growth factor-1 gene in osteocytes impairs developmental bone growth in mice, Bone 52 (2013) 133–144. [CrossRef]
- K. Fulzele, T.L. Clemens, Novel functions for insulin in bone, Bone 50 (2012) 452–456. [CrossRef]
- X. Ruan, X. Jin, F. Sun, J. Pi, Y. Jinghu, X. Lin, N. Zhang, G. Chen, IGF signaling pathway in bone and cartilage development, homeostasis, and disease, FASEB J. 38 (2024) e70031. [CrossRef]
- B. Hammerschmidt, T. Schlake, Localization of Shh expression by Wnt and Eda affects axial polarity and shape of hairs, Dev. Biol. 305 (2007) 246–261. [CrossRef]
- M.D. Zhang, J. Zheng, S. Wu, H. Chen, L. Xiang, Dynamic expression of IGFBP3 modulate dual actions of mineralization micro-environment during tooth development via Wnt/beta-catenin signaling pathway, Biol. Direct 18 (2023) 1–16. [CrossRef]
- F. De Luca, Regulatory role of NF-κB in growth plate chondrogenesis and its functional interaction with Growth Hormone, Mol. Cell. Endocrinol. 514 (2020) 110916. [CrossRef]
- M.A. Hossain, A. Adithan, M.J. Alam, S.R. Kopalli, B. Kim, C.W. Kang, K.C. Hwang, J.H. Kim, IGF-1 Facilitates Cartilage Reconstruction by Regulating PI3K/AKT, MAPK, and NF-kB Signaling in Rabbit Osteoarthritis, J. Inflamm. Res. 14 (2021) 3555. [CrossRef]
- Y. Wang, D.D. Bikle, W. Chang, Autocrine and Paracrine Actions of IGF-I Signaling in Skeletal Development, Bone Res. 2013 11 1 (2013) 249–259. [CrossRef]
- M. Anghelina, D. Sjostrom, P. Perera, J. Nam, T. Knobloch, S. Agarwal, Regulation of biomechanical signals by NF-κB transcription factors in chondrocytes, Biorheology 45 (2008) 245. [CrossRef]
- H. Huang, J. Wang, Y. Zhang, G. Zhu, Y.P. Li, J. Ping, W. Chen, Bone resorption deficiency affects tooth root development in RANKL mutant mice due to attenuated IGF-1 signaling in radicular odontoblasts, Bone 114 (2018) 161–171. [CrossRef]
- M. Qi, E.A. Elion, MAP kinase pathways., J. Cell Sci. 118 (2005) 3569–72. [CrossRef]
- B.E. Bobick, W.M. Kulyk, Regulation of cartilage formation and maturation by mitogen-activated protein kinase signaling, Birth Defects Res. Part C Embryo Today Rev. 84 (2008) 131–154. [CrossRef]
- M.B. Greenblatt, J.H. Shim, L.H. Glimcher, Mitogen-activated protein kinase pathways in osteoblasts, Annu. Rev. Cell Dev. Biol. 29 (2013) 63–79. [CrossRef]
- T.W. Tai, F.C. Su, C.Y. Chen, I.M. Jou, C.F. Lin, Activation of p38 MAPK-regulated Bcl-xL signaling increases survival against zoledronic acid-induced apoptosis in osteoclast precursors, Bone 67 (2014) 166–174. [CrossRef]
- D.J. Papachristou, P. Pirttiniemi, T. Kantomaa, A.G. Papavassiliou, E.K. Basdra, JNK/ERK-AP-1/Runx2 induction “paves the way” to cartilage load-ignited chondroblastic differentiation., Histochem. Cell Biol. 124 (2005) 215–23. [CrossRef]
- M.B. Greenblatt, J.M. Kim, H. Oh, K.H. Park, M.K. Choo, Y. Sano, C.E. Tye, Z. Skobe, R.J. Davis, J.M. Park, M. Bei, L.H. Glimcher, J.H. Shim, P38α MAPK is required for tooth morphogenesis and enamel secretion, J. Biol. Chem. 290 (2015) 284–295. [CrossRef]
- A. Kumar, M.T. Eby, S. Sinha, A. Jasmin, P.M. Chaudhary, The Ectodermal Dysplasia Receptor Activates the Nuclear Factor-κB, JNK, and Cell Death Pathways and Binds to Ectodysplasin A, J. Biol. Chem. 276 (2001) 2668–2677. [CrossRef]
- S. Papa, C. Bubici, F. Zazzeroni, C.G. Pham, C. Kuntzen, J.R. Knabb, K. Dean, G. Franzoso, The NF-κB-mediated control of the JNK cascade in the antagonism of programmed cell death in health and disease, Cell Death Differ. 2006 135 13 (2006) 712–729. [CrossRef]
- S. Abbas, J.C. Clohisy, Y. Abu-Amer, Mitogen-activated protein (MAP) kinases mediate PMMA-induction of osteoclasts, J. Orthop. Res. 21 (2003) 1041–1048. [CrossRef]
- M.T. Su, K. Ono, D. Kezuka, S. Miyamoto, Y. Mori, T. Takai, Fibronectin-LILRB4/gp49B interaction negatively regulates osteoclastogenesis through inhibition of RANKL-induced TRAF6/TAK1/NF–kB/MAPK signaling, Int. Immunol. 35 (2023) 135–145. [CrossRef]
- V. Ulivi, P. Giannoni, C. Gentili, R. Cancedda, F. Descalzi, p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes, J. Cell. Biochem. 104 (2008) 1393–1406. [CrossRef]
- L. Wang, J.Y. Li, X.Z. Zhang, L. Liu, Z.M. Wan, R.X. Li, Y. Guo, Involvement of p38MAPK/NF-κB signaling pathways in osteoblasts differentiation in response to mechanical stretch, Ann. Biomed. Eng. 40 (2012) 1884–1894. [CrossRef]
- K. Niederreither, P. Dollé, Retinoic acid in development: towards an integrated view., Nat. Rev. Genet. 9 (2008) 541–53. [CrossRef]
- M. Pacifici, Retinoid roles and action in skeletal development and growth provide the rationale for an ongoing heterotopic ossification prevention trial, Bone 109 (2018) 267–275. [CrossRef]
- M. Theodosiou, V. Laudet, M. Schubert, From carrot to clinic: an overview of the retinoic acid signaling pathway, Cell. Mol. Life Sci. 67 (2010) 1423–1445. [CrossRef]
- E. Van Beek, C. Löwik, M. Karperien, S. Papapoulos, Independent pathways in the modulation of osteoclastic resorption by intermediates of the mevalonate biosynthetic pathway: The role of the retinoic acid receptor, Bone 38 (2006) 167–171. [CrossRef]
- A.M. Ashique, S.R. May, M.A. Kane, A.E. Folias, K. Phamluong, Y. Choe, J.L. Napoli, A.S. Peterson, Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling., Genesis 50 (2012) 415–23. [CrossRef]
- T. Yu, M. Chen, J. Wen, J. Liu, K. Li, L. Jin, J. Yue, Z. Yang, J. Xi, The effects of all-trans retinoic acid on prednisolone-induced osteoporosis in zebrafish larvae, Bone 189 (2024) 117261. [CrossRef]
- A. Sadier, W.R. Jackman, V. Laudet, Y. Gibert, The Vertebrate Tooth Row: Is It Initiated by a Single Organizing Tooth?, BioEssays 42 (2020) 1900229. [CrossRef]
- D. Kim, R. Chen, M. Sheu, N. Kim, S. Kim, N. Islam, E.M. Wier, G. Wang, A. Li, A. Park, W. Son, B. Evans, V. Yu, V.P. Prizmic, E. Oh, Z. Wang, J. Yu, W. Huang, N.K. Archer, Z. Hu, N. Clemetson, A.M. Nelson, A. Chien, G.A. Okoye, L.S. Miller, G. Ghiaur, S. Kang, J.W. Jones, M.A. Kane, L.A. Garza, Noncoding dsRNA induces retinoic acid synthesis to stimulate hair follicle regeneration via TLR3, Nat. Commun. 2019 101 10 (2019) 1–13. [CrossRef]
- T. Shimo, H. Takebe, T. Okui, Y. Kunisada, S. Ibaragi, K. Obata, N. Kurio, K. Shamsoon, S. Fujii, A. Hosoya, K. Irie, A. Sasaki, M. Iwamoto, Expression and Role of IL-1β Signaling in Chondrocytes Associated with Retinoid Signaling during Fracture Healing, Int. J. Mol. Sci. 2020, Vol. 21, Page 2365 21 (2020) 2365. [CrossRef]
- L. Guo, Y. Zhang, H. Liu, Q. Cheng, S. Yang, D. Yang, All-trans retinoic acid inhibits the osteogenesis of periodontal ligament stem cells by promoting IL-1β production via NF-κB signaling, Int. Immunopharmacol. 108 (2022). [CrossRef]
- R. Mishra, I. Sehring, M. Cederlund, M. Mulaw, G. Weidinger, NF-κB Signaling Negatively Regulates Osteoblast Dedifferentiation during Zebrafish Bone Regeneration, Dev. Cell 52 (2020) 167-182.e7. [CrossRef]
- P.K. Farmer, X. He, M.L. Schmitz, J. Rubin, M.S. Nanes, Inhibitory effect of NF-κB on 1,25-dihydroxyvitamin d3 and retinoid X receptor function, Am. J. Physiol. - Endocrinol. Metab. 279 (2000). [CrossRef]
- D.W. Alhamad, H. Bensreti, J. Dorn, W.D. Hill, M.W. Hamrick, M.E. McGee-Lawrence, Aryl hydrocarbon receptor (AhR)-mediated signaling as a critical regulator of skeletal cell biology, J. Mol. Endocrinol. 69 (2022) R109–R124. [CrossRef]
- B.C. Goodale, J.K. La Du, W.H. Bisson, D.B. Janszen, K.M. Waters, R.L. Tanguay, AHR2 Mutant Reveals Functional Diversity of Aryl Hydrocarbon Receptors in Zebrafish, PLoS One 7 (2012) e29346. [CrossRef]
- E.P. Ahi, S.S. Steinhäuser, A. Pálsson, S.R. Franzdóttir, S.S. Snorrason, V.H. Maier, Z.O. Jónsson, Differential expression of the aryl hydrocarbon receptor pathway associates with craniofacial polymorphism in sympatric Arctic charr, Evodevo 6 (2015) 27. [CrossRef]
- J.P. Souder, D.A. Gorelick, ahr2, But Not ahr1a or ahr1b, Is Required for Craniofacial and Fin Development and TCDD-dependent Cardiotoxicity in Zebrafish, Toxicol. Sci. 170 (2019) 25–44. [CrossRef]
- R. Park, S. Madhavaram, J.D. Ji, The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function, Cells 2020, Vol. 9, Page 2294 9 (2020) 2294. [CrossRef]
- N. Chen, Q. Shan, Y. Qi, W. Liu, X. Tan, J. Gu, Transcriptome analysis in normal human liver cells exposed to 2, 3, 3′, 4, 4′, 5 - Hexachlorobiphenyl (PCB 156), Chemosphere 239 (2020) 124747. [CrossRef]
- C.F.A. Vogel, E.M. Khan, P.S.C. Leung, M.E. Gershwin, W.L.W. Chang, D. Wu, T. Haarmann-Stemmann, A. Hoffmann, M.S. Denison, Cross-talk between Aryl Hydrocarbon Receptor and the Inflammatory Response: A ROLE FOR NUCLEAR FACTOR-κB *, J. Biol. Chem. 289 (2014) 1866–1875. [CrossRef]
- Q. Ye, X. Xi, D. Fan, X. Cao, Q. Wang, X. Wang, M. Zhang, B. Wang, Q. Tao, C. Xiao, Polycyclic aromatic hydrocarbons in bone homeostasis, Biomed. Pharmacother. 146 (2022) 112547. [CrossRef]
- H. Zhuang, X. Ren, F. Jiang, P. Zhou, Indole-3-propionic acid alleviates chondrocytes inflammation and osteoarthritis via the AhR/NF-κB axis, Mol. Med. 29 (2023) 1–13. [CrossRef]
- G.P. Chrousos, T. Kino, Glucocorticoid Signaling in the Cell, Ann. N. Y. Acad. Sci. 1179 (2009) 153–166. [CrossRef]
- M.S. Cooper, M.J. Seibel, H. Zhou, Glucocorticoids, bone and energy metabolism, Bone 82 (2016) 64–68. [CrossRef]
- W. Yao, W. Dai, J.X. Jiang, N.E. Lane, Glucocorticoids and osteocyte autophagy, Bone 54 (2013) 279–284. [CrossRef]
- A.J. Galliher-Beckley, J.G. Williams, J.A. Cidlowski, Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling., Mol. Cell. Biol. 31 (2011) 4663–75. [CrossRef]
- S. Pikulkaew, F. Benato, A. Celeghin, C. Zucal, T. Skobo, L. Colombo, L. Dalla Valle, The knockdown of maternal glucocorticoid receptor mRNA alters embryo development in zebrafish., Dev. Dyn. 240 (2011) 874–89. [CrossRef]
- J.M. Hillegass, C.M. Villano, K.R. Cooper, L.A. White, Glucocorticoids Alter Craniofacial Development and Increase Expression and Activity of Matrix Metalloproteinases in Developing Zebrafish (Danio rerio), Toxicol. Sci. 102 (2008) 413–424. [CrossRef]
- E.P. Ahi, K.H. Kapralova, A. Pálsson, V.H. Maier, J. Gudbrandsson, S.S. Snorrason, Z.O. Jónsson, S.R. Franzdóttir, Transcriptional dynamics of a conserved gene expression network associated with craniofacial divergence in Arctic charr, Evodevo 5 (2014). [CrossRef]
- G. Adami, D. Gatti, M. Rossini, A. Giollo, M. Gatti, F. Bertoldo, E. Bertoldo, A.S. Mudano, K.G. Saag, O. Viapiana, A. Fassio, Risk of fracture in women with glucocorticoid requiring diseases is independent from glucocorticoid use: An analysis on a nation-wide database, Bone 179 (2024) 116958. [CrossRef]
- D.E. Robinson, E.M. Dennison, C. Cooper, T.P. van Staa, W.G. Dixon, A review of the methods used to define glucocorticoid exposure and risk attribution when investigating the risk of fracture in a rheumatoid arthritis population, Bone 90 (2016) 107–115. [CrossRef]
- K. Harrison, L. Loundagin, B. Hiebert, A. Panahifar, N. Zhu, D. Marchiori, T. Arnason, K. Swekla, P. Pivonka, D. Cooper, Glucocorticoids disrupt longitudinal advance of cortical bone basic multicellular units in the rabbit distal tibia, Bone 187 (2024) 117171. [CrossRef]
- M. Nakamura, M.R. Schneider, R. Schmidt-Ullrich, R. Paus, Mutant laboratory mice with abnormalities in hair follicle morphogenesis, cycling, and/or structure: An update, J. Dermatol. Sci. 69 (2013) 6–29. [CrossRef]
- J.L. Cascallana, A. Bravo, E. Donet, H. Leis, M.F. Lara, J.M. Paramio, J.L. Jorcano, P. Pérez, Ectoderm-Targeted Overexpression of the Glucocorticoid Receptor Induces Hypohidrotic Ectodermal Dysplasia, Endocrinology 146 (2005) 2629–2638. [CrossRef]
- A. Rauch, S. Seitz, U. Baschant, A.F. Schilling, A. Illing, B. Stride, M. Kirilov, V. Mandic, A. Takacz, R. Schmidt-Ullrich, S. Ostermay, T. Schinke, R. Spanbroek, M.M. Zaiss, P.E. Angel, U.H. Lerner, J.P. David, H.M. Reichardt, M. Amling, G. Schütz, J.P. Tuckermann, Glucocorticoids suppress bone formation by attenuating osteoblast differentiation via the monomeric glucocorticoid receptor, Cell Metab. 11 (2010) 517–531. [CrossRef]
- B. Frenkel, W. White, J. Tuckermann, Glucocorticoid-Induced osteoporosis, Adv. Exp. Med. Biol. 872 (2015) 179–215. [CrossRef]
- J.A. Roman-Blas, S.A. Jimenez, NF-κB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis, Osteoarthr. Cartil. 14 (2006) 839–848. [CrossRef]
- A.B. Khalid, S.A. Krum, Estrogen receptors alpha and beta in bone, Bone 87 (2016) 130–135. [CrossRef]
- V. Shi, E.F. Morgan, Estrogen and estrogen receptors mediate the mechanobiology of bone disease and repair, Bone 188 (2024) 117220. [CrossRef]
- F.A. Syed, U. IL Mödder, D.G. Fraser, T.C. Spelsberg, C.J. Rosen, A. Krust, P. Chambon, J.L. Jameson, S. Khosla, Skeletal Effects of Estrogen Are Mediated by Opposing Actions of Classical and Nonclassical Estrogen Receptor Pathways, J. Bone Miner. Res. 20 (2005) 1992–2001. [CrossRef]
- Y. Feng, H. Wang, S. Xu, J. Huang, Q. Pei, Z. Wang, The detection of Gper1 as an important gene promoting jawbone regeneration in the context of estrogen deficiency, Bone 180 (2024) 116990. [CrossRef]
- L.B. Tankó, B.-C. Søndergaard, S. Oestergaard, M.A. Karsdal, C. Christiansen, An update review of cellular mechanisms conferring the indirect and direct effects of estrogen on articular cartilage., Climacteric 11 (2008) 4–16. [CrossRef]
- C.E. Metzger, P. Olayooye, L.Y. Tak, O. Culpepper, A.N. LaPlant, P. Jalaie, P.M. Andoh, W. Bandara, O.N. Reul, A.A. Tomaschke, R.K. Surowiec, Estrogen deficiency induces changes in bone matrix bound water that do not closely correspond with bone turnover, Bone 186 (2024) 117173. [CrossRef]
- K.E. Warner, J.J. Jenkins, Effects of 17alpha-ethinylestradiol and bisphenol A on vertebral development in the fathead minnow (Pimephales promelas)., Environ. Toxicol. Chem. 26 (2007) 732–7. Available online: http://www.ncbi.nlm.nih.gov/pubmed/17447558 (accessed on 14 September 2015).
- E. Pashay Ahi, B.S. Walker, C.S. Lassiter, Z.O. Jónsson, Investigation of the effects of estrogen on skeletal gene expression during zebrafish larval head development, PeerJ 4 (2016) e1878. [CrossRef]
- P. Singh, E.P. Ahi, C. Sturmbauer, Gene coexpression networks reveal molecular interactions underlying cichlid jaw modularity, BMC Ecol. Evol. 21 (2021) 1–17. [CrossRef]
- S. Fushimi, N. Wada, T. Nohno, M. Tomita, K. Saijoh, S. Sunami, H. Katsuyama, 17beta-Estradiol inhibits chondrogenesis in the skull development of zebrafish embryos., Aquat. Toxicol. 95 (2009) 292–8. [CrossRef]
- E. Pashay Ahi, B.S. Walker, C.S. Lassiter, Z.O. Jónsson, Investigation of the effects of estrogen on skeletal gene expression during zebrafish larval head development., PeerJ 4 (2016) e1878. [CrossRef]
- A. Tingaud-Sequeira, J. Forgue, M. André, P.J. Babin, Epidermal transient down-regulation of retinol-binding protein 4 and mirror expression of apolipoprotein Eb and estrogen receptor 2a during zebrafish fin and scale development, Dev. Dyn. 235 (2006) 3071–3079. [CrossRef]
- S. Paruthiyil, A. Cvoro, X. Zhao, Z. Wu, Y. Sui, R.E. Staub, S. Baggett, C.B. Herber, C. Griffin, M. Tagliaferri, H.A. Harris, I. Cohen, L.F. Bjeldanes, T.P. Speed, F. Schaufele, D.C. Leitman, Drug and Cell Type-Specific Regulation of Genes with Different Classes of Estrogen Receptor β-Selective Agonists, PLoS One 4 (2009) e6271. [CrossRef]
- M.E. Quaedackers, C.E. Van Den Brink, S. Wissink, R.H.M.M. Schreurs, J.Å. Gustafsson, P.T. Van Der Saag, B. Van Der Burg, 4-Hydroxytamoxifen Trans-Represses NuclearFactor-κB Activity in Human Osteoblastic U2-OS Cells through EstrogenReceptor (ER)α, and Not through ERβ, Endocrinology 142 (2001) 1156–1166. [CrossRef]
- M. Martin-Millan, M. Almeida, E. Ambrogini, L. Han, H. Zhao, R.S. Weinstein, R.L. Jilka, C.A. O’Brien, S.C. Manolagas, The Estrogen Receptor-α in Osteoclasts Mediates the Protective Effects of Estrogens on Cancellous But Not Cortical Bone, Mol. Endocrinol. 24 (2010) 323–334. [CrossRef]
- F.C. Liu, C.C. Wang, J.W. Lu, C.H. Lee, S.C. Chen, Y.J. Ho, Y.J. Peng, Chondroprotective Effects of Genistein against Osteoarthritis Induced Joint Inflammation, Nutr. 2019, Vol. 11, Page 1180 11 (2019) 1180. [CrossRef]
- H. Allison, L.M. McNamara, Inhibition of osteoclastogenesis by mechanically stimulated osteoblasts is attenuated during estrogen deficiency, Am. J. Physiol. - Cell Physiol. 317 (2019) C969–C982. [CrossRef]
- L. Penolazzi, M. Zennaro, E. Lambertini, E. Tavanti, E. Torreggiani, R. Gambari, R. Piva, Induction of Estrogen Receptor α Expression with Decoy Oligonucleotide Targeted to NFATc1 Binding Sites in Osteoblasts, Mol. Pharmacol. 71 (2007) 1457–1462. [CrossRef]
- S. Suthon, J. Lin, R.S. Perkins, J.R. Crockarell, G.A. Miranda-Carboni, S.A. Krum, Estrogen receptor alpha and NFATc1 bind to a bone mineral density-associated SNP to repress WNT5B in osteoblasts, Am. J. Hum. Genet. 109 (2022) 97. [CrossRef]
- D. Sitara, A.O. Aliprantis, Transcriptional regulation of bone and joint remodeling by NFAT, Immunol. Rev. 233 (2010) 286–300. [CrossRef]
- H. Zheng, Y. Liu, Y. Deng, Y. Li, S. Liu, Y. Yang, Y. Qiu, B. Li, W. Sheng, J. Liu, C. Peng, W. Wang, H. Yu, Recent advances of NFATc1 in rheumatoid arthritis-related bone destruction: mechanisms and potential therapeutic targets, Mol. Med. 2024 301 30 (2024) 1–23. [CrossRef]
- H. min Kim, L. He, S. Lee, C. Park, D.H. Kim, H.J. Han, J. Han, J. Hwang, H. Cha-Molstad, K.H. Lee, S.K. Ko, J.H. Jang, I.J. Ryoo, J. Blenis, H.G. Lee, J.S. Ahn, Y.T. Kwon, N.K. Soung, B.Y. Kim, Inhibition of osteoclasts differentiation by CDC2-induced NFATc1 phosphorylation, Bone 131 (2020) 115153. [CrossRef]
- R. Ren, J. Guo, Y. Chen, Y. Zhang, L. Chen, W. Xiong, The role of Ca2+ /Calcineurin/NFAT signalling pathway in osteoblastogenesis, Cell Prolif. 54 (2021). [CrossRef]
- J.H. Kim, N. Kim, Regulation of NFATc1 in Osteoclast Differentiation, J. Bone Metab. 21 (2014) 233. [CrossRef]
- J. Wang, B.M. Gardner, Q. Lu, M. Rodova, B.G. Woodbury, J.G. Yost, K.F. Roby, D.M. Pinson, O. Tawfik, H.C. Anderson, Transcription factor Nfat1 deficiency causes osteoarthritis through dysfunction of adult articular chondrocytes, J. Pathol. 219 (2009) 163–172. [CrossRef]
- C.M. Park, H.M. Kim, D.H. Kim, H.J. Han, H. Noh, J.H. Jang, S.H. Park, H.J. Chae, S.W. Chae, E.K. Ryu, S. Lee, K. Liu, H. Liu, J.S. Ahn, Y.O. Kim, B.Y. Kim, N.K. Soung, Ginsenoside Re Inhibits Osteoclast Differentiation in Mouse Bone Marrow-Derived Macrophages and Zebrafish Scale Model, Mol. Cells 39 (2016) 855. [CrossRef]
- M. Asagiri, K. Sato, T. Usami, S. Ochi, H. Nishina, H. Yoshida, I. Morita, E.F. Wagner, T.W. Mak, E. Serfling, H. Takayanagi, Autoamplification of NFATc1 expression determines its essential role in bone homeostasis, J. Exp. Med. 202 (2005) 1261. [CrossRef]
- Y. Abu-Amer, NF-κB signaling and bone resorption, Osteoporos. Int. 2013 249 24 (2013) 2377–2386. [CrossRef]
- M. Noordijk, J.L. Davideau, S. Eap, O. Huck, F. Fioretti, J.F. Stoltz, W. Bacon, N. Benkirane-Jessel, F. Clauss, Bone defects and future regenerative nanomedicine approach using stem cells in the mutant Tabby mouse model, Biomed. Mater. Eng. 25 (2015) S111–S119. [CrossRef]
- Z. Li, K. Chen, Q. Yu, Y. Li, S. Tong, R. Xu, R. Hu, Y. Zhang, W. Xu, Suppression of NFATc1 through NF-kB/PI3K signaling pathway by Oleandrin to inhibit osteoclastogenesis and bone resorption, Eng. Regen. (2024). [CrossRef]
- J. Yang, R. Tang, J. Yi, Y. Chen, X. Li, T. Yu, J. Fei, Diallyl disulfide alleviates inflammatory osteolysis by suppressing osteoclastogenesis via NF-κB–NFATc1 signal pathway, FASEB J. 33 (2019) 7261. [CrossRef]
- R.C. Gensure, T.J. Gardella, H. Jüppner, Parathyroid hormone and parathyroid hormone-related peptide, and their receptors, Biochem. Biophys. Res. Commun. 328 (2005) 666–678. [CrossRef]
- M. Wang, A.R. Nasiri, A.E. Broadus, S.M. Tommasini, Periosteal PTHrP Regulates Cortical Bone Remodeling During Fracture Healing, Bone 81 (2015) 104–111. [CrossRef]
- T. John Martin, Parathyroid hormone-related protein, its regulation of cartilage and bone development, and role in treating bone diseases, Physiol. Rev. 96 (2016) 831–871. [CrossRef]
- M. Voutilainen, P.H. Lindfors, S. Lefebvre, L. Ahtiainen, I. Fliniaux, E. Rysti, M. Murtoniemi, P. Schneider, R. Schmidt-Ullrich, M.L. Mikkola, Ectodysplasin regulates hormone-independent mammary ductal morphogenesis via NF-κB, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 5744–5749. [CrossRef]
- P. Liu, Y. Li, W. Wang, Y. Bai, H. Jia, Z. Yuan, Z. Yang, Role and mechanisms of the NF-ĸB signaling pathway in various developmental processes, Biomed. Pharmacother. 153 (2022) 113513. [CrossRef]
- P. Rao, J. jing, Y. Fan, C. Zhou, Spatiotemporal cellular dynamics and molecular regulation of tooth root ontogeny, Int. J. Oral Sci. 2023 151 15 (2023) 1–12. [CrossRef]
- C. Matta, R. Zakany, Calcium signalling in chondrogenesis: implications for cartilage repair., Front. Biosci. (Schol. Ed). 5 (2013) 305–24. Available online: http://www.ncbi.nlm.nih.gov/pubmed/23277053 (accessed on 8 September 2015).
- M. Zayzafoon, Calcium/calmodulin signaling controls osteoblast growth and differentiation, J. Cell. Biochem. 97 (2006) 56–70. [CrossRef]
- E.C. Seales, K.J. Micoli, J.M. McDonald, Calmodulin is a critical regulator of osteoclastic differentiation, function, and survival, J. Cell. Biochem. 97 (2006) 45–55. [CrossRef]
- A. Abzhanov, W.P. Kuo, C. Hartmann, B.R. Grant, P.R. Grant, C.J. Tabin, The calmodulin pathway and evolution of elongated beak morphology in Darwin’s finches., Nature 442 (2006) 563–7. [CrossRef]
- K.J. Parsons, R.C. Albertson, Roles for Bmp4 and CaM1 in Shaping the Jaw: Evo-Devo and Beyond, Annu. Rev. Genet. 43 (2009) 369–388. [CrossRef]
- Mikkola, The role of growth factors in tooth development, Int. Rev. Cytol. 217 (2002) 93–135. [CrossRef]
- H. Park, K. Hosomichi, Y. Il Kim, Y. Hikita, A. Tajima, T. Yamaguchi, Comprehensive Genetic Exploration of Fused Teeth by Whole Exome Sequencing, Appl. Sci. 2022, Vol. 12, Page 11899 12 (2022) 11899. [CrossRef]
- F. Qu, Z. Zhao, B. Yuan, W. Qi, C. Li, X. Shen, C. Liu, H. Li, G. Zhao, J. Wang, Q. Guo, Y. Liu, CaMKII plays a part in the chondrogenesis of bone marrow-derived mesenchymal stem cells, Int. J. Clin. Exp. Pathol. 8 (2015) 5981. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC4503202/ (accessed on 12 February 2024).
- Y.-H. Choi, E.-J. Ann, J.-H. Yoon, J.-S. Mo, M.-Y. Kim, H.-S. Park, Calcium/calmodulin-dependent protein kinase IV (CaMKIV) enhances osteoclast differentiation via the up-regulation of Notch1 protein stability, Biochim. Biophys. Acta - Mol. Cell Res. 1833 (2013) 69–79. [CrossRef]
- A.-K. Khimji, D.C. Rockey, Endothelin--biology and disease., Cell. Signal. 22 (2010) 1615–25. [CrossRef]
- J. Kristianto, M.G. Johnson, R. Afzal, R.D. Blank, Endothelin Signaling in Bone, Endocrinol. Metab. Clin. North Am. 46 (2016) 51. [CrossRef]
- A. Sehgal, T. Behl, S. Singh, N. Sharma, M. Albratty, H.A. Alhazmi, A.M. Meraya, L. Aleya, A. Sharma, S. Bungau, Exploring the pivotal role of endothelin in rheumatoid arthritis, Inflammopharmacology 30 (2022) 1555–1567. [CrossRef]
- D. Esibizione, C.-Y. Cui, D. Schlessinger, Candidate EDA targets revealed by expression profiling of primary keratinocytes from Tabby mutant mice, Gene 427 (2008) 42–46. [CrossRef]
- M.T. Cobourne, P.T. Sharpe, Tooth and jaw: molecular mechanisms of patterning in the first branchial arch, Arch. Oral Biol. 48 (2003) 1–14. [CrossRef]
- L. Barske, P. Rataud, K. Behizad, L. Del Rio, S.G. Cox, J.G. Crump, Essential role of Nr2f nuclear receptors in patterning the vertebrate upper jaw, Dev. Cell 44 (2018) 337. [CrossRef]
- J. Che, X. Yang, Z. Jin, C. Xu, Nrf2: A promising therapeutic target in bone-related diseases, Biomed. Pharmacother. 168 (2023) 115748. [CrossRef]
- P. Vogel, J. Liu, K.A. Platt, R.W. Read, M. Thiel, R.B. Vance, R. Brommage, Malformation of Incisor Teeth in Grem2-/- Mice, Vet. Pathol. 52 (2015) 224–229. [CrossRef]
- P. Ducy, G. Karsenty, The two faces of serotonin in bone biology, J. Cell Biol. 191 (2010) 7–13. [CrossRef]
- A. Hori, T. Nishida, S. Takashiba, S. Kubota, M. Takigawa, Regulatory mechanism of CCN2 production by serotonin (5-HT) via 5-HT2A and 5-HT2B receptors in chondrocytes, PLoS One 12 (2017) e0188014. [CrossRef]
- J.R.D. Moiseiwitsch, The role of serotonin and neurotransmitters during craniofacial development, Crit. Rev. Oral Biol. Med. 11 (2000) 230–239. [CrossRef]
- T.L. Brown, E.C. Horton, E.W. Craig, C.E.A. Goo, E.C. Black, M.N. Hewitt, N.G. Yee, E.T. Fan, D.W. Raible, J.P. Rasmussen, Dermal appendage-dependent patterning of zebrafish atoh1a+ Merkel cells, Elife 12 (2023). [CrossRef]
- Y. Chabbi-Achengli, A.E. Coudert, J. Callebert, V. Geoffroy, F. Côté, C. Collet, M.C. De Vernejoul, Decreased osteoclastogenesis in serotonin-deficient mice, Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 2567–2572. [CrossRef]
- E.M. Tsapakis, Z. Gamie, G.T. Tran, S. Adshead, A. Lampard, A. Mantalaris, E. Tsiridis, The adverse skeletal effects of selective serotonin reuptake inhibitors, Eur. Psychiatry 27 (2012) 156–169. [CrossRef]
- C. Xue, G. Li, Q. Zheng, X. Gu, Q. Shi, Y. Su, Q. Chu, X. Yuan, Z. Bao, J. Lu, L. Li, Tryptophan metabolism in health and disease, Cell Metab. 35 (2023) 1304–1326. [CrossRef]
- S. Murab, S. Chameettachal, M. Bhattacharjee, S. Das, D.L. Kaplan, S. Ghosh, Matrix-Embedded Cytokines to Simulate Osteoarthritis-Like Cartilage Microenvironments, (2013) 1733–1753. Available online: https://Home.Liebertpub.Com/Tea 19. [CrossRef]
- D. Docheva, C. Popov, P. Alberton, A. Aszodi, Integrin signaling in skeletal development and function, Birth Defects Res. Part C Embryo Today Rev. 102 (2014) 13–36. [CrossRef]
- D. Kronenberg, M. Brand, J. Everding, L. Wendler, E. Kieselhorst, M. Timmen, M.D. Hülskamp, R. Stange, Integrin α2β1 deficiency enhances osteogenesis via BMP-2 signaling for accelerated fracture repair, Bone 190 (2025) 117318. [CrossRef]
- E.K. Song, T.J. Park, Integrin signaling in cartilage development, Animal Cells Syst. (Seoul). 18 (2014) 365–371. [CrossRef]
- M.L. Mikkola, J. Pispa, M. Pekkanen, L. Paulin, P. Nieminen, J. Kere, I. Thesleff, Ectodysplasin, a protein required for epithelial morphogenesis, is a novel TNF homologue and promotes cell-matrix adhesion, Mech. Dev. 88 (1999) 133–146. [CrossRef]
- C.Y. Cui, M. Durmowicz, T.S. Tanaka, A.J. Hartung, T. Tezuka, K. Hashimoto, M.S.H. Ko, A.K. Srivastava, D. Schlessinger, EDA targets revealed by skin gene expression profiles of wild-type, Tabby and Tabby EDA-A1 transgenic mice, Hum. Mol. Genet. 11 (2002) 1763–1773. [CrossRef]
- Z. Wang, Y. Jia, X. Huang, D. Zhu, H. Liu, W. Wang, Transcriptome profiling towards understanding of the morphogenesis in the scale development of blunt snout bream (Megalobrama amblycephala), Genomics 113 (2021) 983–991. [CrossRef]
- R. Li, Y. Shi, S. Zhao, T. Shi, G. Zhang, NF-κB signaling and integrin-β1 inhibition attenuates osteosarcoma metastasis via increased cell apoptosis, Int. J. Biol. Macromol. 123 (2019) 1035–1043. [CrossRef]
- S.R.L. Young, R. Gerard-O’Riley, M. Harrington, F.M. Pavalko, Activation of NF-κB by fluid shear stress, but not TNF-α, requires focal adhesion kinase in osteoblasts, Bone 47 (2010) 74–82. [CrossRef]
- C.C. Teixeira, H. Ischiropoulos, P.S. Leboy, S.L. Adams, I.M. Shapiro, Nitric oxide–nitric oxide synthase regulates key maturational events during chondrocyte terminal differentiation, Bone 37 (2005) 37–45. [CrossRef]
- S.J. Wimalawansa, Nitric oxide and bone, Ann. N. Y. Acad. Sci. 1192 (2010) 391–403. [CrossRef]
- J. Klein-Nulend, R.F.M. Van Oers, A.D. Bakker, R.G. Bacabac, Nitric oxide signaling in mechanical adaptation of bone, Osteoporos. Int. 25 (2014) 1427–1437. [CrossRef]
- C.Y. Tsai, F.C.H. Li, C.H.Y. Wu, A.Y.W. Chang, S.H.H. Chan, Sumoylation of IkB attenuates NF-kB-induced nitrosative stress at rostral ventrolateral medulla and cardiovascular depression in experimental brain death, J. Biomed. Sci. 23 (2016) 1–10. [CrossRef]
- M. Sisto, D. Ribatti, S. Lisi, Understanding the Complexity of Sjögren’s Syndrome: Remarkable Progress in Elucidating NF-κB Mechanisms, J. Clin. Med. 2020, Vol. 9, Page 2821 9 (2020) 2821. [CrossRef]
- M. Ostojic, V. Soljic, K. Vukojevic, T. Dapic, Immunohistochemical characterization of early and advanced knee osteoarthritis by NF-κB and iNOS expression, J. Orthop. Res. 35 (2017) 1990–1997. [CrossRef]
- T. Fang, X. Zhou, M. Jin, J. Nie, Xi.I. Li, Molecular mechanisms of mechanical load-induced osteoarthritis, Int. Orthop. 45 (2021) 1125–1136. [CrossRef]
- Y. Zhou, J. Ming, M. Deng, Y. Li, B. Li, J. Li, Y. Ma, Z. Chen, S. Liu, Berberine-mediated up-regulation of surfactant protein D facilitates cartilage repair by modulating immune responses via the inhibition of TLR4/NF-ĸB signaling, Pharmacol. Res. 155 (2020) 104690. [CrossRef]
- V. Veeriah, A. Zanniti, R. Paone, S. Chatterjee, N. Rucci, A. Teti, M. Capulli, Interleukin-1β, lipocalin 2 and nitric oxide synthase 2 are mechano-responsive mediators of mouse and human endothelial cell-osteoblast crosstalk, Sci. Reports 2016 61 6 (2016) 1–14. [CrossRef]
Figure 1.
Predicted regulatory crosstalk of the EDA pathway components with major skeletal development pathways. (A–D) Molecular and cellular details of EDA signaling crosstalk with the BMP, Hedgehog, Notch, and Wnt pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. In addition, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
Figure 1.
Predicted regulatory crosstalk of the EDA pathway components with major skeletal development pathways. (A–D) Molecular and cellular details of EDA signaling crosstalk with the BMP, Hedgehog, Notch, and Wnt pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. In addition, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
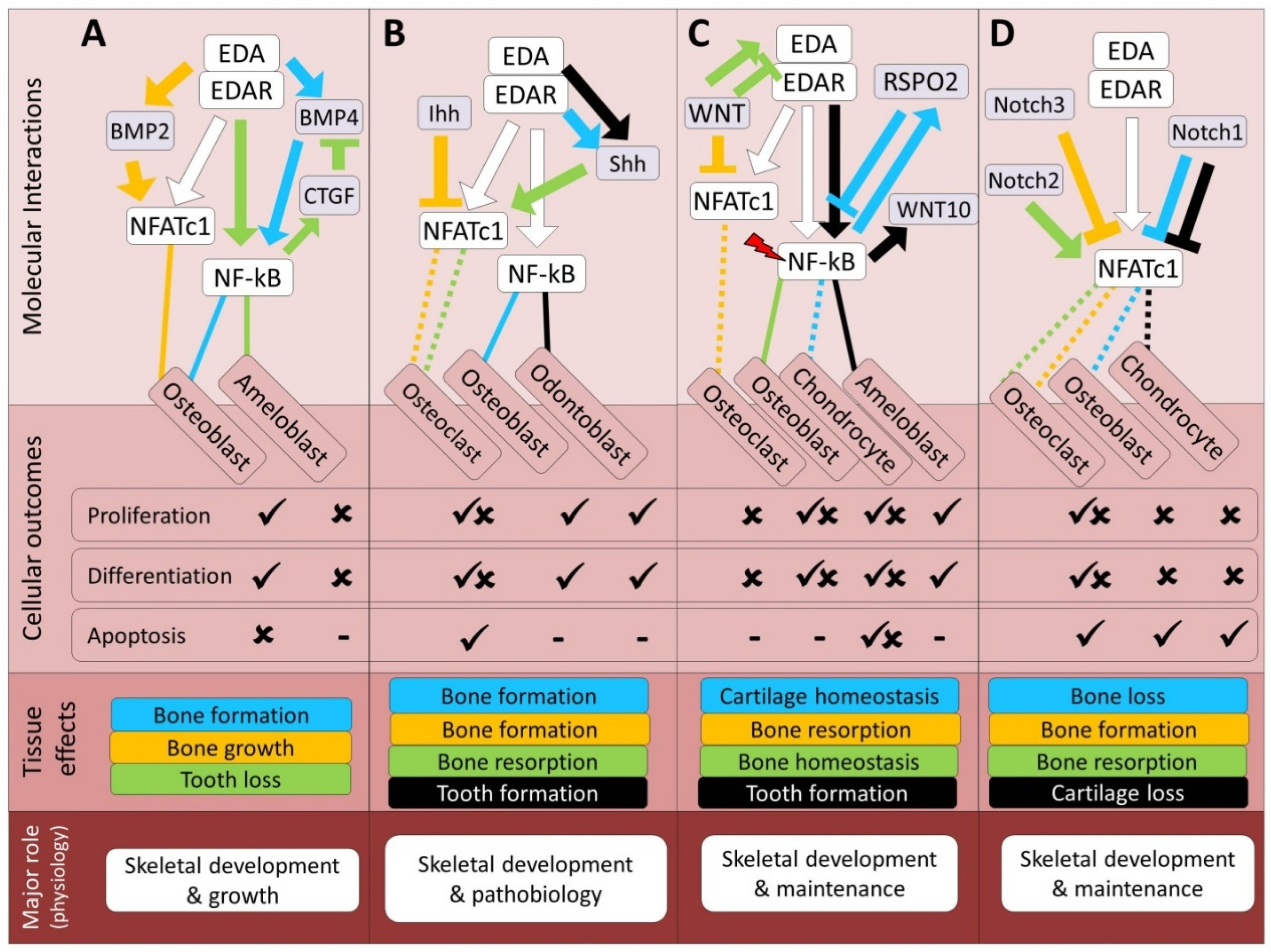
Figure 2.
Predicted regulatory crosstalk of the EDA pathway components with growth factors mediated signals during skeletogenesis. (A–C) Molecular and cellular details of EDA signaling crosstalk with the FGF, IGF, and MAPK pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. In addition, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
Figure 2.
Predicted regulatory crosstalk of the EDA pathway components with growth factors mediated signals during skeletogenesis. (A–C) Molecular and cellular details of EDA signaling crosstalk with the FGF, IGF, and MAPK pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. In addition, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
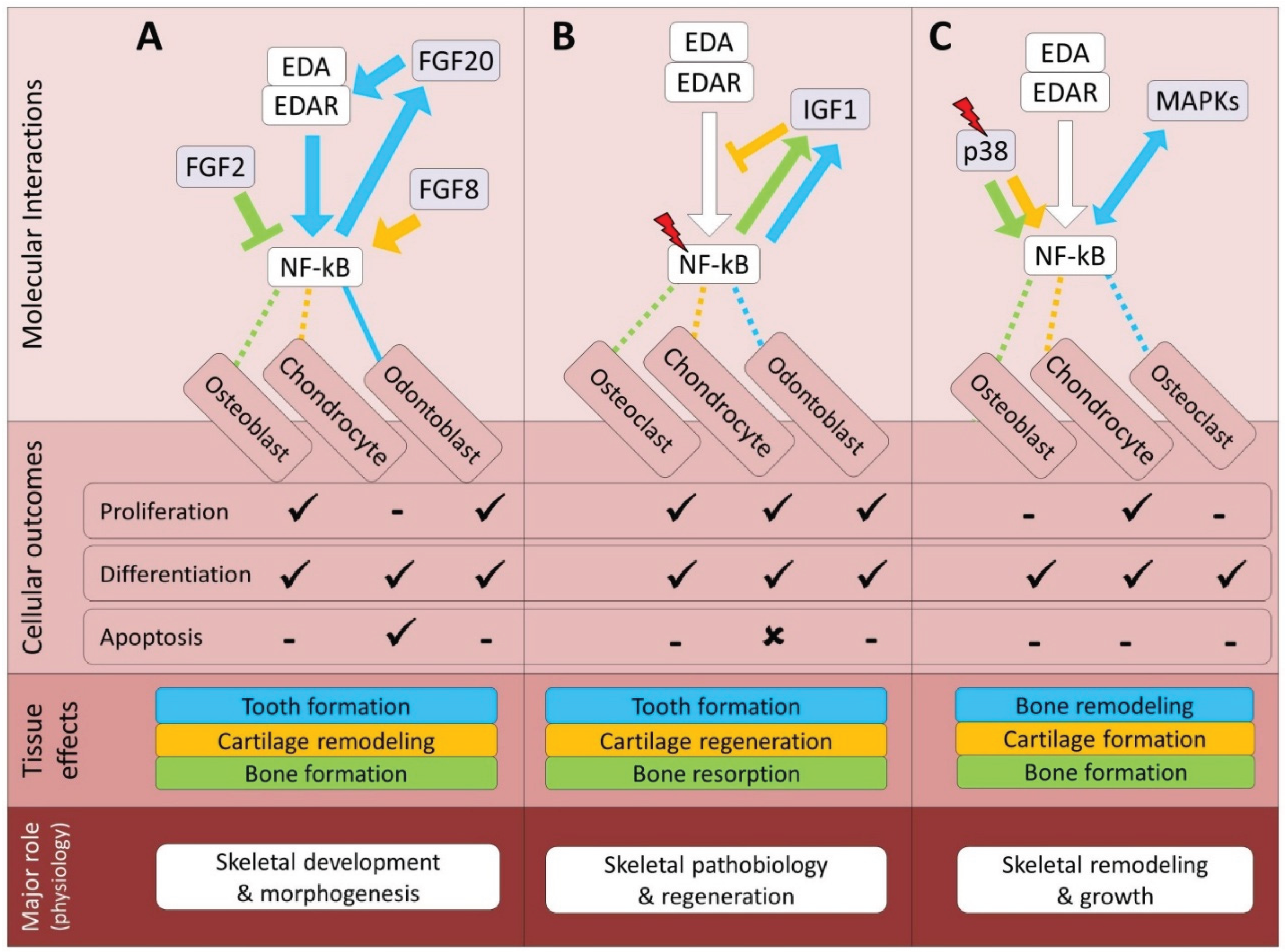
Figure 3.
Predicted regulatory crosstalk of the EDA pathway components with nuclear receptor signals during skeletogenesis. (A–D) Molecular and cellular details of EDA signaling crosstalk with the RA, AHR, GR and estrogen pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
Figure 3.
Predicted regulatory crosstalk of the EDA pathway components with nuclear receptor signals during skeletogenesis. (A–D) Molecular and cellular details of EDA signaling crosstalk with the RA, AHR, GR and estrogen pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed and solid lines respectively represent indirect and direct regulatory connections between the upstream components of EDA and other pathways. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
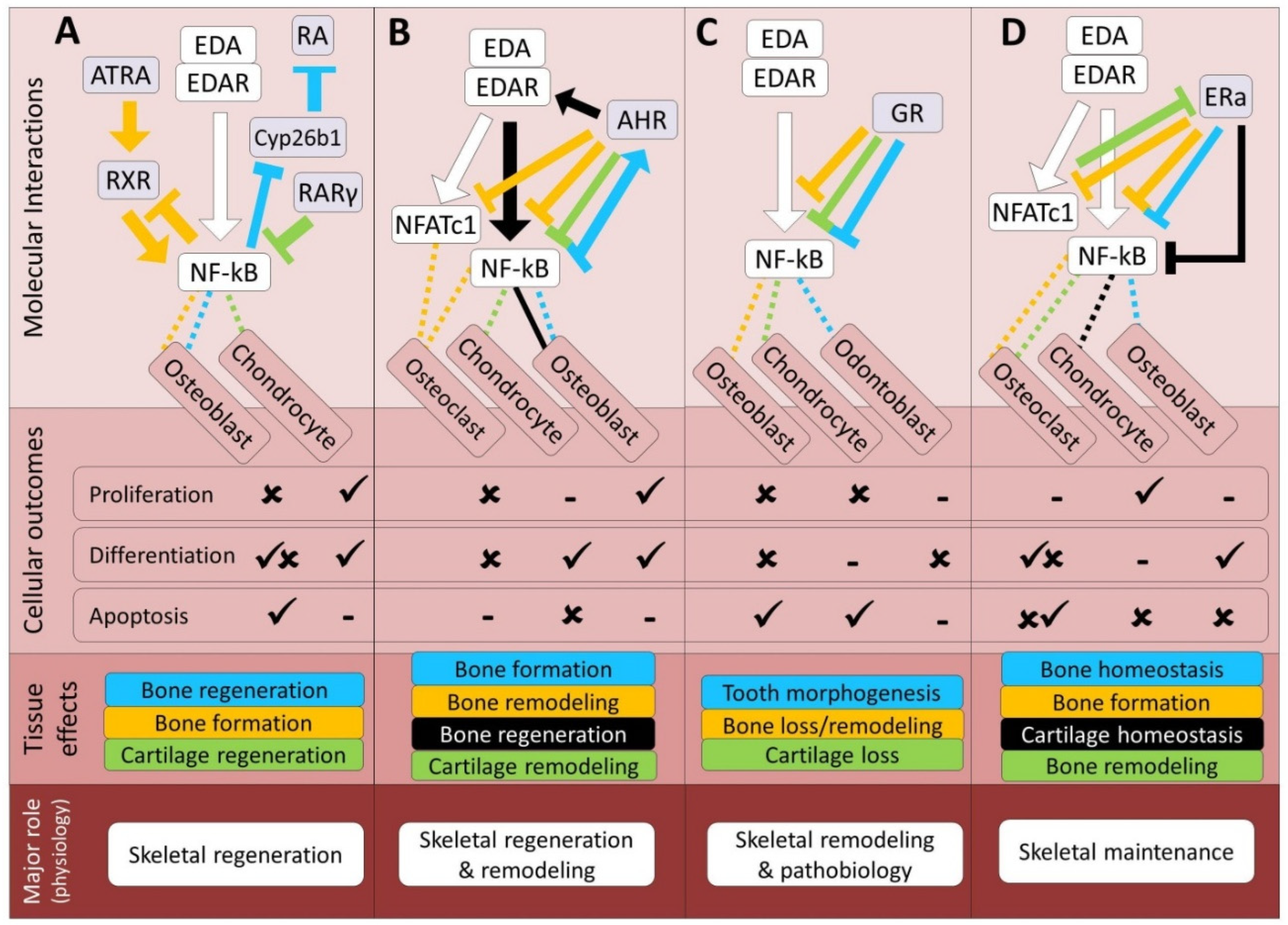
Figure 4.
Predicted regulatory crosstalk of the EDA pathway components with signals mediated by calcium-dependent pathways during skeletogenesis. (A–D) Molecular and cellular details of EDA signaling crosstalk with the NFAT, PTH/PTHrP, Ca2+/CaM and Edn/Ednr pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed lines represent indirect regulatory connections between the upstream components of EDA and other pathways. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
Figure 4.
Predicted regulatory crosstalk of the EDA pathway components with signals mediated by calcium-dependent pathways during skeletogenesis. (A–D) Molecular and cellular details of EDA signaling crosstalk with the NFAT, PTH/PTHrP, Ca2+/CaM and Edn/Ednr pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed lines represent indirect regulatory connections between the upstream components of EDA and other pathways. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
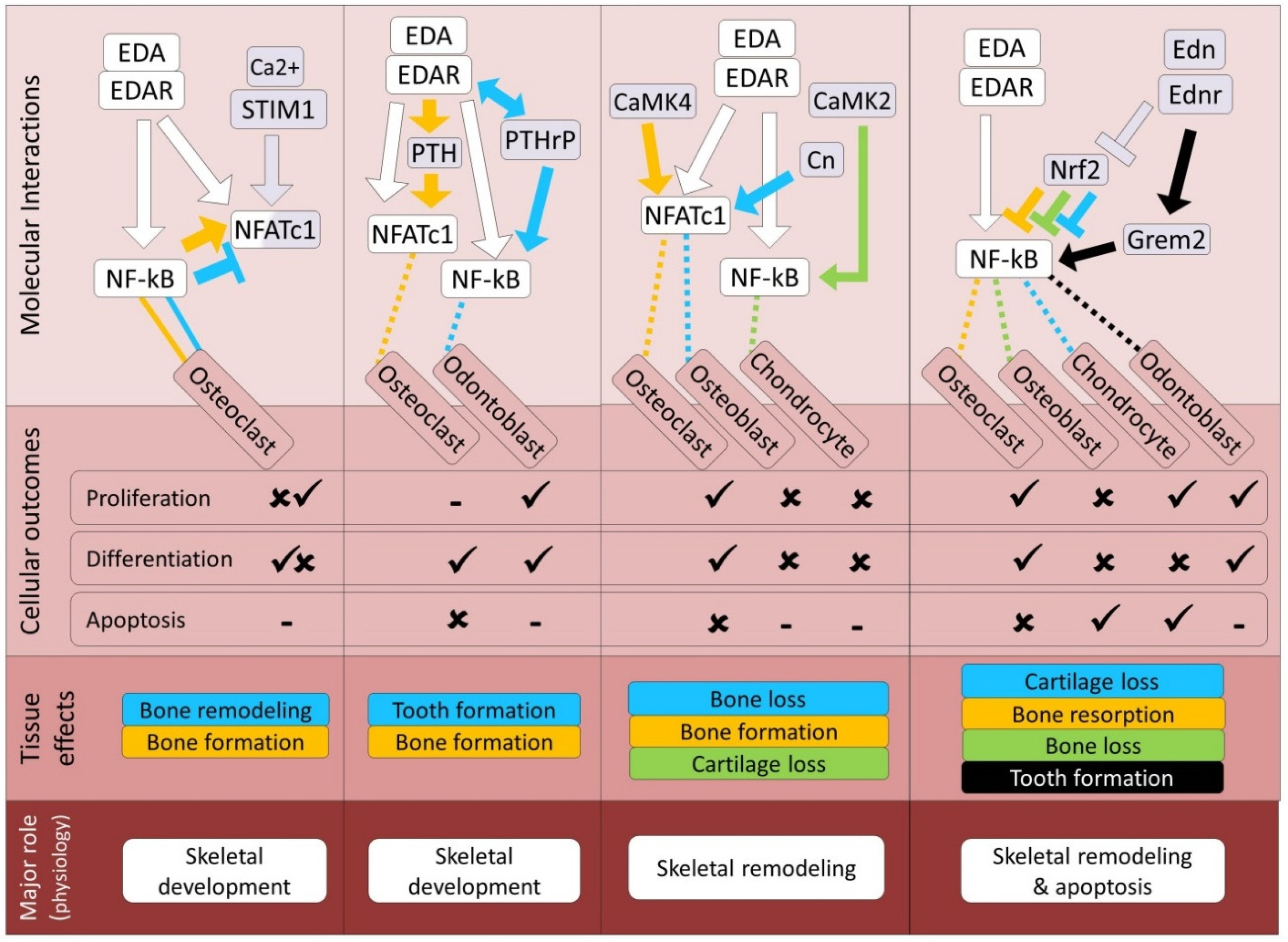
Figure 5.
Predicted regulatory crosstalk of the EDA pathway components with non-canonical microenvironment-responsive pathways. (A–C) Molecular and cellular details of EDA signaling crosstalk with the serotonin, integrin and nitric oxide pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed lines represent indirect regulatory connections between the upstream components of EDA and other pathways. Additionally, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
Figure 5.
Predicted regulatory crosstalk of the EDA pathway components with non-canonical microenvironment-responsive pathways. (A–C) Molecular and cellular details of EDA signaling crosstalk with the serotonin, integrin and nitric oxide pathways, respectively, along with their associated skeletal outcomes at the tissue and physiological levels. In the molecular interactions section, arrows and block heads represent regulatory induction and inhibition, respectively. The color of lines connecting EDA signal to each cell type follows the color of the specified interaction, and dashed lines represent indirect regulatory connections between the upstream components of EDA and other pathways. Additionally, red slash symbols above certain genes indicate upstream components that detect mechanical signals within each molecular signaling. The color coding of skeletal effects corresponds to the colors of related regulatory interactions in the molecular section. In the cellular section, ticks and crosses denote the promotion and impairment of processes, based on the crosstalk studied. The lowest section presents the primary expected physiological role of EDA in the context of its crosstalk with each pathway.
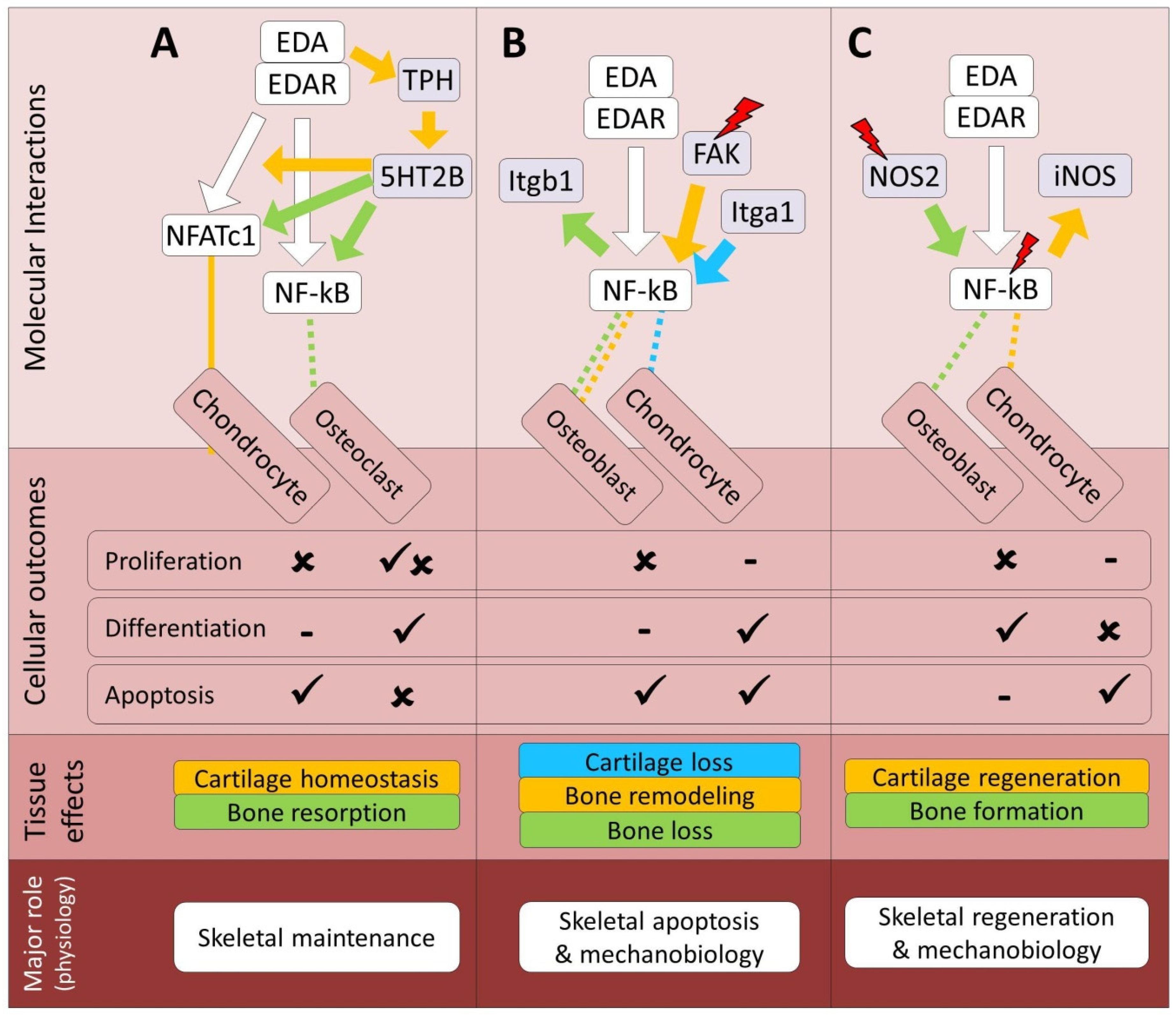

Table 1.
Summary of the crosstalk between EDA and skeletogenic signaling pathways. This table summarizes the nature of molecular crosstalk between Ectodysplasin-A (EDA) signaling and major skeletogenic signaling pathways in bone, cartilage, and tooth tissues. Each cell indicates the overall interaction type between EDA and the respective pathway as interpreted from studies discussed in this article: ‘Synergistic’, ‘Antagonistic’, ‘Both’ (if dual effects are reported), or ‘Unknown’ (if no relevant data exists). The type of regulatory link is provided in parentheses: D = direct interaction (through EDA or EDAR); In1 = indirect interaction via NFATc1; In2 = indirect interaction via NF-κB. Multiple terms (e.g., D, In1, In2) reflect convergence of multiple regulatory inputs.
Table 1.
Summary of the crosstalk between EDA and skeletogenic signaling pathways. This table summarizes the nature of molecular crosstalk between Ectodysplasin-A (EDA) signaling and major skeletogenic signaling pathways in bone, cartilage, and tooth tissues. Each cell indicates the overall interaction type between EDA and the respective pathway as interpreted from studies discussed in this article: ‘Synergistic’, ‘Antagonistic’, ‘Both’ (if dual effects are reported), or ‘Unknown’ (if no relevant data exists). The type of regulatory link is provided in parentheses: D = direct interaction (through EDA or EDAR); In1 = indirect interaction via NFATc1; In2 = indirect interaction via NF-κB. Multiple terms (e.g., D, In1, In2) reflect convergence of multiple regulatory inputs.
| Pathway | Bone | Cartilage | Tooth | References |
|---|---|---|---|---|
| TGF-β | Unknown | Unknown | Antagonistic (D, In2) | 20, 24, 42-44 |
| BMP | Synergistic (D, In1) | Unknown | Antagonistic (D, In1) | 19-21, 52-54 |
| Hh | Both (D) | Synergistic (D) | Synergistic (D) | 2, 20-21, 33, 52, 62-65 |
| Wnt | Both (D, In1, In2) | Both (D, In1, In2) | Synergistic (D, In1, In2) | 23, 31, 33, 54, 62, 73-82 |
| Notch | Both (In1, In2) | Antagonistic (In1, In2) | Unknown | 90-97 |
| FGF | Synergistic (D, In2) | Synergistic (D, In2) | Synergistic (D, In2) | 6, 33, 62, 107-112 |
| IGF | Both (In1, In2) | Both (In1, In2) | Both (In1, In2) | 119-125 |
| MAPK | Synergistic (In2) | Synergistic (In2) | Unknown | 131-137 |
| RA | Both (In2) | Both (In2) | Synergistic (In2) | 6, 144-149 |
| Ahr | Both (In2) | Both (In2) | Unknown | 153-158 |
| GR | Antagonistic (In2) | Antagonistic (In2) | Antagonistic (In2) | 169-173 |
| ER | Antagonistic (In1, In2) | Antagonistic (In1, In2) | Unknown | 185-192 |
| NFAT | Both (D, In1, In2) | Both (D, In1, In2) | Synergistic (D, In1, In2) | 17, 201-204 |
| PTH/PTHrP | Synergistic (D, In2) | Synergistic (D, In2) | Synergistic (D, In2) | 23, 208-210 |
| Ca2+/CaM | Both (In1, In2) | Antagonistic (In1, In2) | Unknown | 212, 216-219 |
| Edn/Ednr | Synergistic (D, In2) | Both (D, In2) | Synergistic (D, In2) | 223-227 |
| Serotonin | Both (In1, In2) | Both (In1, In2) | Unknown | 17, 231-235 |
| Integrin | Both (D, In2) | Both (D, In2) | Unknown | 239-243 |
| NO | Synergistic (In2) | Both (In2) | Unknown | 244-252 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



