
Submitted:
24 June 2025
Posted:
25 June 2025
You are already at the latest version
Abstract
Background/Objectives: Advances in understanding immune checkpoint pathways and tumor immune biology have enabled the development of immune checkpoint inhibitors (ICIs), particularly targeting the PD-1/PD-L1 axis, which has transformed cancer immu-notherapy. Despite their success across multiple tumor types, including melanoma, non-small-cell lung cancer, and gastrointestinal malignancies, variability in patient re-sponse, immune-related adverse events (irAEs), and resistance mechanisms remain sig-nificant. This review aims to evaluate clinical pharmacology, mechanisms of action, re-sistance pathways, and pharmacogenomic influences shaping interindividual responses to ICIs. Methods: This comprehensive review synthesizes current literature on FDA-approved ICIs, exploring their clinical use, underlying biological mechanisms, and emerging pharmacogenomic data. It also assesses key biomarkers such as tumor muta-tional burden (TMB), microsatellite instability (MSI), HLA diversity, and epigenetic factors influencing ICI efficacy and safety. Results: We identify key mechanisms contributing to ICI resistance, including T cell dysfunction, altered antigen presentation, and immuno-suppressive tumor microenvironment components. Furthermore, we highlight promising pharmacogenomic findings, including single-nucleotide polymorphisms (SNPs) in PD-1/PD-L1 and immune-regulatory genes, offering predictive and prognostic utility. Variability in PD-L1 expression and the role of epigenetic modifications are also ad-dressed as challenges in treatment optimization. Conclusions: Interindividual variability in ICI response underscores the need for biomarker-driven strategies. By integrating pharmacogenomic insights with clinical pharmacology, future approaches may support more personalized and effective use of ICIs. Combination therapies and novel modalities hold promise for overcoming resistance, enhancing therapeutic efficacy, and enabling precision oncology.
Keywords:
PD-1/PD-L1 inhibitors
; cancer immunotherapy
; immune checkpoint blockade
; resistance mechanisms
; biomarkers
; precision oncology
1. Introduction
The introduction should briefly place the study in a broad context and highlight why it is important. It should define the purpose of the work and its significance. The current state of the research field should be carefully reviewed and key publications cited. Please highlight controversial and diverging hypotheses when necessary. Finally, briefly mention the main aim of the work and highlight the principal conclusions. As far as possible, please keep the introduction comprehensible to scientists outside your particular field of research. References should be numbered in order of appearance and indicated by a numeral or numerals in square brackets—e.g., [1] or [2,3], or [4,5,6]. See the end of the document for further details on references.
The understanding of the tumours’ immune biology has led to the development of innovative treatments based on immune stimulation, known as cancer immunotherapy. Immunotherapy comprises new drugs that have been proven very promising in treating various malignancies either haemotological or of solid tumors. These therapies are designed to enhance a patient's immune system [1]. Among other treatments, this is achieved by the administration of monoclonal antibodies (mAbs) to proteins known as immune-checkpoint inhibitors (ICIs). The main protein targets are the programmed cell death protein-1 (PD1), and programmed death ligand 1 and 2 (PD-L1 and PD-L2), and the cytotoxic T-lymphocyte-associated antigen-4 (CTLA4); the administration of these agents result in a derepression and/or reactivation of cytotoxic T-cell function enabling patient's immune system to attack cancer cells. That means that immune checkpoint blockade, releases the brake of the immune system to enhance anticancer immune response and their use in various malignancies may have a remarkable and long-lasting effect.
Several ICIs have been approved by the United States Food and Drug Administration (FDA) since 2011 for the treatment of a broad spectrum of advanced cancers and currently various ICIs are available as 1st or 2nd line therapy for several malignancies. The first drugs that have been approved are the anti-PD1 Pembrolizumab and Nivolumab, as well as the anti-CTLA4 ipilimumab for the treatment of metastatic melanoma.
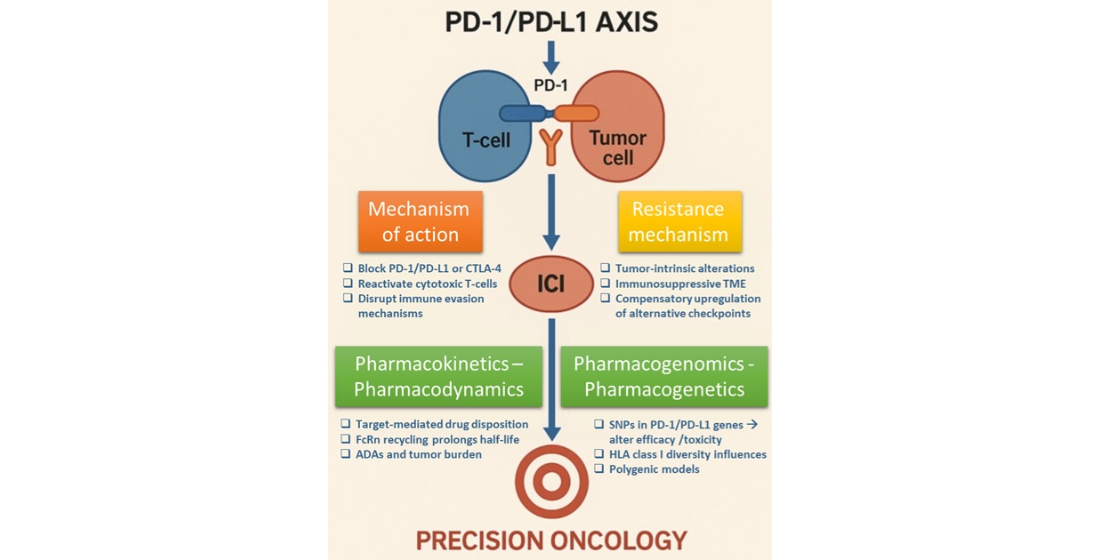
In this review, we focus on the treatment with immune checkpoint inhibitors, in the era of precision medicine.
2. Evolution of Immunotherapy in Cancer Treatment
The use of immune checkpoint inhibitors on clinical cancer care has changed dramatically our knowledge on personalized medicine [2]. Since 2011, US FDA has approved, based on preclinical and clinical data, various agents that demonstrated a substantial survival benefit to cancer patients. The development of immunotherapy in cancer treatment advanced when crucial discoveries were made, showing that T-cell immune responses are controlled through immune checkpoints that function like on/off switches via the cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) pathway or the programmed cell death protein 1 (PD-1)/programmed cell death protein 1 ligand (PD-L1) pathway [3].
The first step towards ICI discovery was understanding its biological components and mechanisms of action. The PD-1 gene was cloned as a new immunoglobulin gene superfamily from stimulated mouse T cell hybridoma by Tasuku Honjo in 1992 [4]. Further studies until 2001 demonstrated that PD-1 negatively regulates the process of T cell activation. The next discovery by several groups was to find out the biological PD-1 ligand showing physical interaction and inhibitory function. This led to the determination of the physical interaction between receptor and ligand (PD-1/PD-L1 and PD-1/PD-L2) contributing to the negative regulation in T cell response [5,6]. In 2002, it was reported by Honjo and Lieping Chen groups that PD-L1, expressed by tumor cells, could inhibit tumor-reactive T cells as one of the immune evolution mechanisms. Thus, targeting the PD-1/PD-L1 axis could be a possible cancer treatment [7]. In 2006, Ono and Medarex developed antibodies targeting human PD-1 and human PD-L1, and started a phase I/II clinical study in cancer patients [8]. The Nobel Prize in Physiology or Medicine 2018 was awarded to James P. Allison and Tasuku Honjo for their discovery of cancer therapy by inhibition of negative immune regulation [9].
CTLA-4 is an immune checkpoint molecule negatively regulates the initiation of T cell activation. It competes with CD28 to bind to CD80 and CD86, thereby downregulating T-cell activation [10]. The first drug which has been approved in 2011 was ipilimumab, a CTLA-4 inhibitor for metastatic and nonresectable melanoma. PD-1 mainly mediates the proliferation of T cells following the activation. PD-1 is expressed on various immune cell types limiting the immune response and the T-cell activity in peripheral tissues. Concerning PD-L1 and PD-L2, these are important mediators of the PD-1 pathway. PD-1/PD-L1 inhibitors have been developed since 2014 and currently they are the cornerstone in immunotherapy. More than 2000 trials of PD-1/PD-L1 inhibitors or their-based combinations have been conducted targeting a wide range of malignancies. The first agents that have been approved were: in 2014 the PD-1 inhibitors for advanced melanoma pembrolizumab and nivolumab, in 2015 nivolumab for advanced non-small-cell lung cancer (NSCLC) and renal cell carcinoma (RCC), as well as pembrolizumab for advanced NSCLC; in 2016 the PD-L1 inhibitors, atezolizumab, for the treatment of locally advanced or metastatic urothelial carcinoma [11], durvalumab for advanced urothelial bladder cancer [12] and in 2017 avelumab for refractory metastatic Merkel cell carcinoma [13]. Currently, common immune checkpoint inhibitors mainly include drugs targeting PD-1(nivolumab, pembrolizumab, cemiplimab, dostarlimab, tislelizumab, retifanlimab, toripalimab and camrelizumab ), PD-L1(atezolizumab, avelumab, durvalumab and cosibelimab), CTLA-4 (Ipilimumab and tremelimumab) and LAG3 (relatlimab, fianlimab and favezelimab) [1]. A timetable with the FDA ICIs approved is shown in Figure 1.
Due to the different way of regulation of T cell function through CTLA-4 and PD-1, the combination of anti-PD-1 antibody (which mainly leads to the expansion and recruitment of existing anti-tumor T cells) and anti-CTLA-4 antibody (which induces new T cell clones) has been used for the simultaneous blocking of the immune escape of tumor cells, enhancing the anti-tumor activity of T cells at different stages. Thus, the ICI combination therapy is currently FDA-approved for several malignancies [14].
Furthermore, over the last decade, other immunotherapies such as the adoptive T cell therapy has been developed. Firstly, Chimeric antigen receptor T (CAR-T) cell therapy, a class of immunotherapy that combines the antigen-binding site of a monoclonal antibody with the signal activated machinery of a T-cell, enabling major histocompatibility complex (MHC)-independent antigen recognition by T cells [15]. CAR-T has been used in the treatment of hematological malignancies, including B-cell leukemia, lymphoma, and multiple myeloma; these drugs have significantly improved patient outcomes and survival rates [16]. Another category of immune cells which are implicated in cancer immunology are the T-cell receptor-engineered T cells (TCR-T). They act in a similar way as the natural T-cells in the human body by recognizing MHC-presented antigens through affinity-optimized or purely natural TCRs. Therefore, TCR-T is able to bind antigens intracellularly, intra-nuclearly and on the surface of the tumor cells [17]. Another promising tumor-infiltrating lymphocyte (TIL) therapy involves isolating TILs from the patient’s own tumor tissue and, following ex vivo expansion, reinfusing them back into the patients [18]. An emerging therapy concerns the Bispecific T cell engager (BiTE) which is a bispecific antibody construct with a unique function, simultaneously binding an antigen on tumor cells and a surface molecule on T cells to induce tumor lysis [19]. These agents have been recently used for the treatment of solid tumors and hematopoietic malignancies may they may offer clinical benefits as an emerging therapy [20]. Another innovative category for cancer treatment are therapies with vaccines which have shown promise by eliciting T cell responses targeting tumor antigens [21]. Finally, oncolytic virus (OV) therapy is also a fairly novel class of immunotherapy that could treat cancers not only by infecting tumor cells specifically and lysing them directly, but also by activating the innate and adaptive immune response [22]. These therapies will not be included in this review.
3. Mechanisms of ICIs Action
The most studied negative regulatory immune checkpoint-related axis in recent years, which plays a prominent role in tumor immune escape is that of PD-1/PD-L1 axis. PD-1 is a type I transmembrane protein and belongs to the B7/CD28 receptor superfamily. It consists of 288 amino acid residues, also known as PDCD1 and CD279. PD-L1 (CD274) and PD-L2 (CD273) are also type I transmembrane proteins and belong to the B7/CD28 family. They are ligands of PD-1 consisting of 290 and 270 amino acid residues, respectively, with a 37% sequence homology [23,24].
The structure of PD-1 consists of four parts: an immunoglobulin variable region (IgV), a transmembrane region, immunoreceptor tyrosine-based inhibitory motifs and immunoreceptor tyrosine-based switch motifs [25]. The interaction of PD-1 and PD-L1 induces those immunoreceptor motifs to be phosphorylated in the intracellular domain of PD-1, which recruits tyrosine acid phosphatase Src homology phosphatase 1 (SHP-1) and Src homology phosphatase 2 (SHP-2) [26]. These phosphatases dephosphorylate several key proteins in the TCR signaling pathway and repress signaling pathways downstream of the TCR, such as phosphoinositide 3-kinase (PI3K), protein kinase B (PKB/AKT), mammalian target of rapamycin (mTOR), rat sarcoma (RAS), mitogen-activated protein kinase (MAPK/MEK), extracellular regulated protein kinase (ERK), etc. In turn, it inhibits the transcription of related genes, hinders the progression of T cell cycle and the expression of related proteins, and ultimately blunts the production of cytokines and the proliferation and differentiation of T cells, causing their immune function to be lost [27].
Concerning the mechanism of anti-PD-1 or anti-PD-L1 antibodies binding (Figure 2), this is based on blocking the interaction of PD-1 with its ligands, PD-L1 and PD-L2 (programmed cell death 1 ligand 2). PD-L1 is more highly expressed than PD-L2 but has a lower affinity for PD-1. Ligands are found at the surface of tumor cells, where their expressions can be induced by type I and II interferons, and at the surface of immune cells, such as macrophages and dendritic cells. PD-1 receptor is mainly expressed by lymphocytes secondarily to their activation. The interaction between PD-1 and PD-L1 or PD-L2 leads to a negative regulation of lymphocytes by inhibiting the signals generated by the TCR and the co-stimulation of molecules as previously described. Thus, the PD-1/PD-L1 axis is a tumor immune escape mechanism. Using anti-PD-1 or anti-PD-L1 antibodies can thus reactivate tumor-specific lymphocytes within the tumor and allows tumor-specific immune cell death. Other closely related immune checkpoints and their ligands control, negatively or positively, lymphocyte activation, such as the lymphocyte activation gene 3 protein (LAG-3), which binds to the MHC-II proteins and to lectins, the T cell immunoglobulin mucin receptor 3 (TIM-3/HAVCR2) and galectin-9, the B and T lymphocyte attenuator (BTLA) and herpesvirus entry mediator (HVEM), the T cell immunoreceptor with Ig and ITIM domains (TIGIT) that binds CD155 and the V-type immunoglobulin domain-containing suppressor of T cell activation (VISTA), for which the ligand is not known [28,29].
4. The Role of ICIs in the Treatment of Solid Tumors
Immunotherapy has been used in different types of malignancies which are mentioned bellow. The PD-1 and PD-L1 inhibitors that have been already approved by FDA are shown on Table 1.
1. Skin Cancers
Melanoma is a skin cancer which develops as the consequence of malignant transformation and proliferation of melanocytes. The main therapeutic procedure is surgery. In several cases metastatic disease occurs and the survival outcome is poor (5-year overall survival (OS) 29.8 %). The development of ICIs has revolutionized the treatment of advanced/metastatic disease [30]. The FDA approved ipilimumab as a first-line therapy for unresectable stage III/IV melanoma and this marked the beginning of the use of ICIs to treat various cancers and ultimately led to an important transformation in the oncologic therapeutic strategy.
In 2016, FDA approved the first combination for immune checkpoint blockade of PD-1 (nivolumab) and CTLA-4 (ipilimumab) for patients with BRAFV600 wild-type unresectable/metastatic melanoma as this combination demonstrated advantage in progression free survival (PFS) and OS compared to monotherapy with ipilimumab [31]. Very recently for patients with resectable, stage III melanoma, neoadjuvant ipilimumab plus nivolumab followed by surgery and response-driven adjuvant therapy resulted in longer event-free survival than surgery followed by adjuvant nivolumab [32]. Another study showed that nivolumab is a proven adjuvant treatment for resected melanoma at high risk of recurrence, with sustained, long-term improvement in recurrence free survival with ipilimumab and high OS rates. Identification of additional biomarkers is needed to better predict treatment outcome [33]. Furthermore, pembrolizumab continued to prolong recurrence-free survival within >4 years follow-up [34].
For advanced/ metastatic non-melanoma skin cancers (such as Merkel cell carcinoma, and basal cell carcinoma) the ICIs avelumab, pembrolizumab, cemiplimab and retifanlimab have also received FDA approval [13,35,36,37]. Finally another anti PD-L1 inhibitor cosibelimab has been very recently approved by FDA for metastatic or locally advanced cutaneous squamous cell carcinoma [38].
2. Lung Cancer
Lung cancer, which is the leading cause of cancer mortality worldwide, is frequently diagnosed in advanced stage. Immunotherapy has provided increases in overall survival during the past decade in patients with non-small cell lung cancer (NSCLC) [39]. Nivolumab, atezolizumab and pembrolizumab have been evaluated as potential first-line therapies especially in cases with PD-L1 expression (determined by Tumor Proportion Score, TPS); it has been shown that patients with TPS ≥50% had a tumor response rate of 50% [40]. Atezolizumab and cemiplimab have also been approved as first-line monotherapy agents for advanced NSCLC patients with PD-L1 tumor expression as they demonstrated improvement in OS compared to platinum-based chemotherapy. Thus, anti-PD-L1 is considered a standard first line monotherapy for PD-L1≥50 % advanced NSCLC [41,42]. Furthermore, a recently published meta-analysis of three phase III clinical trials in 559 advanced non-squamous NSCLC patients reported that patients with KRAS mutations showed improved response, though wildtype KRAS status alone is insufficient to predict lack of benefit [43,44]. Perioperative treatment with nivolumab resulted in significantly longer event-free survival than chemotherapy in patients with resectable NSCLC. No new serious adverse events were observed in this study [45]. FDA has also approved pembrolizumab followed by resection in early stage NSLC as this treatment has benefits in survival [46].
Combination therapies with or without classical chemotherapy have also been approved [46,47,48,49,50]. The most recent breakthrough in the clinical application of immunotherapy is the application of ICIs in treating early-stage NSCLC. Atezolizumab, as a single agent, is indicated as adjuvant treatment following resection and platinum-based chemotherapy for adult patients with stage II-IIIA NSCLC whose tumors have PD-L1 expression on ≥1% of tumor cells [51]. Similar approvals have taken place for pembrolizumab and durvalumab as neoadjuvant treatment for resectable NSCLC as well [46,50].
Furthermore, for small cell lung cancer, a cancer with one of the poorest survival rates of all solid tumors (5-year survival rates less than 5%), ICIs have been evaluated as treatment options however no significant response was recorded. Atezolizumab and durvalumab have been proposed as first-line therapies in combination with etoposide and a platinum-based chemotherapy regimen. The improvement in median OS was 12–13 months [52,53]. In a very recent study adjuvant therapy with durvalumab led to significantly longer overall survival and PFS than placebo among patients with limited-stage small-cell lung cancer [54].
3. Gastrointestinal Malignancies
They are also very frequent malignancies. Specifically, for gastric cancer the outcome is relatively poor mainly for advanced tumors receiving classical chemotherapy. The addition of ICIS may be promising. Nivolumab and pembrolizumab have been administered in combination with classical chemotherapy or trastuzumab for HER2+ tumors and have been approved as second-line treatments for advanced or metastatic gastric and esophageal tumors [55,56,57,58,59]. On March 2025, FDA granted traditional approval to pembrolizumab in combination with trastuzumab and classical chemotherapy for HER2+ tumors gastric or gastroesophageal junction adenocarcinoma expressing PD-L1 (CPS ≥1) [60]. A new PD-1 inhibitor, named tislelizumab has been promising in combination with chemotherapy in advanced HER-2 negative gastric cancer [61]. Durvalumab plus chemotherapy is also a promising treatment for patients with resectable gastric cancer [62].
For metastatic colorectal cancer patients, the median 5-year OS is approximately 14.7 %. Currently, all ICIs approved for the treatment of colorectal cancers have been limited to the subset of patients with microsatellite instability-high/mismatch repair deficient (MSI-H/dMMR) metastatic colorectal cancer (mCRC) [63]. Ipilimumab in combination with nivolumab as well as pembrolizumab were subsequently approved for unresectable/metastatic MSI-H/dMMR metastatic colorectal cancers [63,64]. Recently, in patients with locally advanced dMMR colon cancer, neoadjuvant nivolumab plus ipilimumab showed an acceptable safety profile and led to a significant response in a high proportion of these patients [65].
Concerning patients with advanced hepatocellular carcinoma both nivolumab and pembrolizumab have been approved for patients with advanced hepatocellular carcinoma (HCC); pembrolizumab has been administered in patients previously treated with sorafenib [66]. Other drugs that have been implicated in disease treatment include nivolumab plus ipilimumab as second line treatment [67], more recently atezolizumab in combination with bevacizumab (approved as first-line therapy in 2020), as well as tremelimumab in combination with durvalumab; these combination have been approved due to the significant improvement in OS compared to previous treatments which included sorafenib [68,69]. Recently, camrelizumab in combination with rivoceranib showed a significant and clinically meaningful benefit in PFS and OS compared with sorafenib for patients with unresectable hepatocellular carcinoma, presenting as a new and effective first-line treatment option [70].
4. Breast Cancer
Among breast cancers the triple-negative breast cancer (TNBC) is an aggressive tumor difficult to treat due to lack of targeted agents. Chemotherapy is the the standard-of-care for systemic treatment and includes taxanes or platinum-based agents. However, these tumors can become rapidly resistant to chemotherapy. In 2019, atezolizumab and more recently pembrolizumab have been approved for the treatment of these tumors in combination to chemotherapy showing improvement in PFS [71,72].
5. Gynaecological Malignancies
Although the incidence of cervical cancer has reduced in recent years due to cancer screening programs and vaccination against the human papillomavirus (HPV), cervical cancer remains a significant cause of deaths particularly in developing nations. Several studies have reported relatively high PD-1/PD-L1 expression in cervical tumors, providing potential targets for ICI. Currently, pembrolizumab has been approved since 2018, as monotherapy or in combination therapy for recurrent/metastatic cervical cancer expressing PD-L1 (combined positive score, CPS≥1). Its combination with platinum-based chemotherapy ±bevacizumab, demonstrated a significant survival benefit [73].
Concerning advanced endometrial cancer in 2021, the FDA granted accelerated approval for dostarlimab (anti-PD-1) in the second-line setting, following standard platinum-based chemotherapy, for patients with dMMR/MSI-H endometrial cancers due to a significant overall response rate in these cases [74]. Indeed, improvements in survival and the manageable safety profile support the favorable benefit-risk profile for dostarlimab plus carboplatin-paclitaxel in patients with dMMR/MSI-H primary advanced or recurrent endometrial cancer [75]. Pembrolizumab has also been approved as monotherapy or in combination with the oral TKI lenvatinib as this agent has also improved the objective response rate (ORR) [76]. Carboplatin/paclitaxel plus durvalumab followed by maintenance durvalumab with or without olaparib demonstrated a statistically significant and clinically meaningful PFS benefit in patients with advanced or recurrent endometrial cancer [77].
5. Genitourinary Malignancies
The renal cell carcinoma comprises 90% of all kidney cancers, with clear cell being the most common subtype. Approximately 33% of patients have advanced or metastatic disease at diagnosis. Although RCC is notably resistant to chemotherapy, it is relatively sensitive to immunotherapy and antiangiogenic treatment compared to other tumor types. Except for nivolumab which was the first FDA agent approved, three ICIs have also been approved as first-line therapies for the advanced clear-cell RCC, in combination with either another ICI or with antiangiogenic inhibitors. These combinations comprise nivolumab +ipilimumab, pembrolizumab +axitinib, avelumab +axitinib, nivolumab +cabozantinib and pembrolizumab +lenvatinib [78,79,80,81,82]. Because no direct head-to-head trial comparing dual checkpoint inhibition and checkpoint blockade plus anti-angiogenic TKI regimens exist, first-line therapy selection remains a topic of active debate.
Furthermore, for advanced urothelial carcinoma, the current standard, first-line therapy is platinum-based chemotherapy, while pembrolizumab has been also approved as first-line for platinum-ineligible advanced/metastatic cases [83]. Along with pembrolizumab, avelumab and nivolumab have also been implicated as possible first or second line therapy with/or without chemotherapy as treatment modalities for advanced disease cases [84,85].
6. Head and Neck Squamous Cell Carcinomas (HNSCC)
HNSCC includes cancers of the oral cavity, oropharynx, hypopharynx, and larynx; for these cancers the initial treatment is local intervention. For patients who develop recurrent/metastatic HNSCC the prognosis after treatment with classical chemotherapy is poor. ICIs have not only demonstrated manageable safety in HNSCC but have also been shown to improve OS compared to previous standard-of-care. Pembrolizumab is currently approved as first-line therapy for unresectable recurrent/metastatic HNSCC in combination with chemotherapy with a benefit in OS for the subgroups of patients with PD-L1 positive (CPS ≥1) HNSCC [86]. A randomized, open-label, multicentred, phase III clinical trial showed that adjuvant PD-1 blockade with camrelizumab significantly improved event-free survival with manageable toxicities, highlighting its potential role in the management of locoregionally advanced NPC [87]. A novel PD-1 inhibitor, tislelizumab demonstrated promising efficacy and tolerability in patients with head and neck cancers in both clinical trials and real-world studies [88]. Another PD-1 inhibitor toripalimab may be a highly promising therapy for the treatment of locoregionally advanced nasopharyngeal carcinoma and has been recently approved by FDA as first line therapy [89].
7. Haematological Malignancies
5. ICIs Pharmacokinetics - Pharmacodynamics
Intravenous ICIs are distributed and metabolized by various routes. Extensive binding to target antigens in the plasma or on tissues, reduces the amount of free ICI and increases the volume of distribution. Transvascular movement of free ICIs is principally governed by means of convection, the magnitude of which is limited by factors such as organ perfusion and endothelial permeability. Within tissues, ICIs become distributed by means of diffusion and convection. The neonatal Fc receptor (FcRn) is responsible for the transport of ICIs back into the vascular system, preventing the intracellular lysosomal degradation of these drugs and hence prolonging their half-life (t½ 6–27 days). Prolonged tissue exposure may therefore increase treatment effects without the necessity for frequent drug administration [92]. The distribution of mAbs through the blood, due to their high polarity, is impaired when it comes to peripheral tissues. Their high affinity also affects distribution, by interactions between them and antigens, whereas reduction in tumor size may change the blood flow. Therefore, the volume distribution (Vss) of mAbs is usually very small and approximately equal to the plasma volume [93].
Furthermore, it has been shown that the FcRn is subject to genetic influence based on a variable number of tandem repeats in the promoter region of the FcRn gene (FCGRT). An increased number of tandem repeats has been shown to increase its expression. As FcRn is responsible for salvaging IgG, reduced expression is thought to result in lower serum concentration and increased clearance via alternative mechanisms [94]. Fc isoforms are important determinants of Fcγ receptors (FcγRs)-mediated interactions, wherein the IgG1 subtype displays a higher affinity for FcγR than the IgG4 subtype. Although the IgG1 Fc component of the PD-L1 inhibitor has been tailored to be less susceptible to a specific interaction, the absence of this modification may also explain the relatively short half-life of avelumab compared with other ICIs [93].
On the other hand, the generation of antibodies against ICIs increases clearance. The dominant mechanism of ICI clearance remains through proteolytic catabolism, which occurs in both plasma and peripheral tissues. Lastly, the high-affinity interaction between ICIs and surface receptors precipitates an additional clearance route, i.e. that of receptor-mediated endocytosis. Linear or Nonlinear Clearance of ICIs is governed by numerous physiological mechanisms, the predominant part of which is deemed to occur by nonspecific degradation within plasma and tissues which is not actually impaired by conditions such as age, hepatic impairment and renal failure [95].
An alternative route of elimination consists of receptor-mediated endocytosis which depends on the number of ICI ligands and is the main process to nonlinear clearance. Differences in target proteins can create differences in clearance patterns and degradation [95,96]. Durvalumab and pembrolizumab at doses of 3 and 0.3 mg/kg, respectively present nonlinear clearance due to saturation of receptors, while an absence of nonlinear clearance among other ICIs may indicate another route of clearance or saturation kinetics. Furthermore, clearance can also occur through humoral and cell-mediated degradation pathways of the immune system. Formation of antidrug antibodies (ADAs) facilitates the uptake and endocytic degradation of ICIs, which increases clearance. Under combinational therapies, ADAs might have more significant consequences to the pharmacokinetics of ICIs in comparison to monotherapies; for example, ADAs against nivolumab increased substantially (10–21.9%) in patients receiving concomitant ipilimumab therapy with a 24% increase in nivolumab clearance [97]. An additional route of endocytotic degradation might be facilitated by direct interaction between the Fc component of ICIs and FcγRs on phagocytic cells of the immune system [95].
With the exception of ipilimumab, ICIs exhibit time-varying clearance. This phenomenon has largely been attributed to disease status: clearance decreases when tumor burden declines [98,99]. Cachexia, causes rapid degradation of proteins, including, potentially, ICIs, and ameliorates with improved disease status e.x Durvalumab and pembrolizumab [99,100,101].
Whereas ICIs were originally considered to act in a purely antagonistic manner, more recent advances have demonstrated that several compounds might directly give rise to cytotoxic reactions [102]. Antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) arise by the interaction between the Fc region of ICIs and components of the immune system, which might cause depletion of target cells [103]. The capacity to evoke such an immune response is highly dependent on the isotype involved, where members of the IgG1 group are able to induce ADCC and CDC [103]. IgG1 ICIs mainly serve as ‘classical deleters’ of intratumoral regulatory T cells (Treg cells) because of the capacity to induce cellular and humoral cytotoxicity, while IgG4 ICIs function as true receptor blockers that antagonize the inhibition of T cells [102]. In clinical practice, unmodified IgG1 compounds give rise to a higher degree of infusion-related reactions, as seen in avelumab. In addition, Treg depletion did not occur in the tumor microenvironments of patients treated with ipilimumab or tremelimumab [104], indicating that on a clinical level, ADCC might not be as relevant as preclinical studies suggest [92].
Ameliorating the way of evaluating the pharmacokinetics of ICIs may offer advantage in alterations concerning the dose and the dose interval. This has an important impact in a better management of drug toxicities as well as in the cost effectiveness. Little to no dose-limiting toxicities have been reported for ICIs. The establishment of a therapeutic window within the efficacy range can avert dispensable expenses. A potential candidate for this application is nivolumab, for which the exposure - response curve reaches saturation below marketed doses [105]. This saturation theoretically permits dose minimization, or prolongation of the dose interval, thus potentiating the role of therapeutic drug monitoring in cost reduction [106]. The ICIs (PD-1, PD-L1) pharmacokinetics [107,108,109,110,111,112,113,114] are shown on Table 2 and can be found in FDA database (www.fda.gov).
6. Pharmacogenomics -Pharmacogenetics
Although ICs have changed the treatment strategy for various types of cancer there is a significant interpatient variability in the response to treatment, the overall survival as well as the drug toxicities and the appearance of immune-related adverse events (irAEs) [115]. It is well known that germline genetic variations may be implicated. These genetic variations can either be single-nucleotide polymorphisms (SNPs) or structural variants, such as gene rearrangements, deletions, amplifications. Thus, it is challenging to predict which patients will benefit from ICI treatment and which patients will suffer from severe adverse events [116,117,118]. Many studies have focused on identifying predictive biomarkers for ICI treatment [118]. Pharmacogenetic (or pharmacogenomic) are genetic markers which could potently be used in the evaluation of interpatient variability [119,120].

Table 2.
The ICIs (PD-1, PD-L1) pharmacokinetics. t½: elimination half-life (days); CL: clearance (L/day); Vc: volume of central compartment (L); Vp: volume of peripheral compartment (L); Q: inter-compartmental clearance (L/day); IIV (CV%): inter-individual variability expressed as coefficient of variation (%); CLlinear: clearance of linear elimination (in drugs with linear kinetics).
Table 2.
The ICIs (PD-1, PD-L1) pharmacokinetics. t½: elimination half-life (days); CL: clearance (L/day); Vc: volume of central compartment (L); Vp: volume of peripheral compartment (L); Q: inter-compartmental clearance (L/day); IIV (CV%): inter-individual variability expressed as coefficient of variation (%); CLlinear: clearance of linear elimination (in drugs with linear kinetics).
| Generic name | Dose range (mg/kg ) | t½ (days) | CL (L/day) | Vc (L) | Vp (L) | Q (L/day) | IIV (CV%) | Ref. |
| Atezolizumab | 1–20 | 27 | 0.20 | 3.28 | 3.63 | 0.546 | CL: 29%,Vc: 18%, Vp: 34% | [107] |
| Avelumab | 1–20 | 6,1 | 0.59 | 2.83 | 1.17 | CL: 25.2%, Vc: 18.3%, Vp: 1.05% | * | |
| Durvalumab | 0.1–20 | 21 | 0.232 | 3.51 | 3.45 | 0.476 | CL: 27.2%, Vc: 22.1% | ** |
| Nivolumab | 0.1–20 | 25 | 0.23 | 3.63 | 2.78 | 0.770 | CL: 35%, Vc: 35.1% | [108] |
| Pembrolizumab | 1–10 | 27,3 | 0.22 | 3.48 | 4.06 | 0.795 | CL: 38%,Vc: 21% | [109] |
| Cemiplimab | 1–10 | 28,9 | 0.290 | 3.32 | 1.65 | 0.638 | CL, Q: 8.70% | [110] |
| Dostarlimab | 500 mg or 1000 mg | 23,5 | 0.179 | 2.98 | 2.10 | 0.547 | CL: 23.5%, Vc: 16.1% | [111] |
| Tislelizumab | 0.5-10 | 23.8 | 0.15 | 3.05 | 1.27 | 0.74 | CL:26.3%, Vc:16.7%, Vp:74.7% | [112] |
| Retifanlimab | 1–10 | 18.7 | 0.2928 | 3.76 | 2.64 | 0.684 | CL: 31.4%, Vc:17.9%, Vp:35.5% | *** |
| Toripalimab | 0.3 -10 | 10 ± 1,5 | 0.3576 | 3.7 | … | …. | …. | **** |
| Camrelizumab | 1–10 | 3–11 | 0.231 | 3.07 | 2.90 | 0.414 | CLline: 50.8%, Vc: 49.5% | [113] |
| Cosibelimab | 800 mg or 1200 mg | 17,4 | 0.238 | 3.58 | 2.31 | … | CL: 30.8% ,Vc: 16.8%, Vp: 53.0% | ***** |
* Center for Drug Evaluation and Research [CDER], US FDA . Clinical multi-discipline review: avelumab. Silver Spring: US FDA; 2017. ** US Food and Drug Administration. "Clinical pharmacology and biopharmaceutics review." Silver Spring: US FDA (2007). ***https://www.accessdata.fda.gov/drugsatfda_docs/nda/2023/761334Orig1s000MultidisciplineR.pdf ****https://loqtorzihcp.com/pdf/prescribing-information.pdf *****https://checkpointtx.com/wp-content/uploads/2023/07/Cosibelimab-poster-PAGE-v1-final-3.pdf.
SNPs are germline variations in the DNA occurring in ≥1% of the population. Several SNPs associated with ICI therapy are shown on Table 3. Several genome wide association studies (GWAS) have been performed in order to generate clusters of SNPs, which could be relevant for ICI outcome [121,122,123]. Whereas GWAS offer a great tool for generating biomarker-based hypotheses, they also require large groups of patients and hence have difficulty validating results in other cohorts. Therefore, prospective validation is often missing. In a case-control cohort of 89 ICI-treated patients with melanoma, a GWAS identified 30 SNPs which were significantly associated with the occurrence of immune-related adverse events (irAEs) [122]. Several of these SNPs were located in genes associated with auto-inflammatory diseases, such as SEMA5A for rheumatoid arthritis [122]. Several GWAS and validation studies identified a SNP located on an IL7 intron (rs16906115)[121] which was significantly associated with the occurrence of all grade irAEs [124].
For ICI treatment specifically, genetic variation is studied within i) ICI targeted receptors, ii) pathways related to autoimmunity and iii) variations within the human leukocyte antigen (HLA).
SNPs Within the PD-1 Pathway
Three SNPs for PDCD1, which encodes PD-1, (SNPs 804C>T (rs2227981), 889G>A (rs10204525) and 7146A>G (rs11568821)) are the most investigated, although none is deemed suitable for application in routine clinical care. The first one, 804C>T (rs2227981), is considered to affect gene transcription and PD-1 receptor expression on T cells. Although in a cohort of 119 melanoma patients treated with anti-PD-1, T allele carriers had a shorter OS than wild types, this result was not confirmed in other cohorts of cancer patients [125,126,127,128]. Nonetheless, a trend was seen towards shorter PFS in homozygous variant carriers (TT genotype) although non-significant [129]. Moreover, T carriers may have lower expression of PD-1 in CD4 + T cells [130].
Considering the location of 804C>T within the promotor region of PDCD1, it seems plausible that this SNP alters PDCD1 transcription and thus leads to decreased PD-1 expression [127,131].
For the SNP 889G>A (rs10204525), wild type patients were found to experience more and more severe (≥3) irAEs compared to homozygous variant genotype in a retrospective study from Japan in RCC patients [128]; this finding was not confirmed in other Caucasian populations [120,128,131].
Finally, the third SNP, 7146A>G (rs11568821), located in an enhancer region of intron 4 of PDCD1 regulating gene transcription, was associated with an improved PFS making it an interesting target for validation in upcoming prospective studies [129].
SNPs Within the PD-L1 Receptor Gene
For the PD-L1 receptor, the two most studied SNPs (rs2282055 and rs2890658) were not associated with either toxicity or survival [125,126,127,132], while two other SNPs might predict response to anti-PD-L1 treatment. First, in a cohort of 108 patients with NSCLC treated with nivolumab, both rs1411262 and mainly rs822339 were associated with longer PFS and OS [133] as well as increased frequency of immune-related hypothyroidism [125,133]. This is consistent with previous data suggesting that patients experiencing irAEs might have better treatment outcomes than those without irAEs [134]. These results were further confirmed in the previously mentioned Japanese study in 222 patients with advanced RCC [127], where both SNPs were significantly associated with PFS. While the G/G genotype of CD274 rs4143815 has been associated with elevated PD-L1 expression and favorable ICI responses in some studies, other cohorts have reported inconsistent or null associations, indicating the need for further validation in diverse populations [126]. It has been also suggested in several studies that PD-L1 rs4143815, located in the 3’ untranslated region (UTR), can influence the expression of PD-L1, thus driving tumor cell immune escape [135]. The C allele has been shown to increase production of PD-L1 by attenuating miR-570 [136]. Consequently C/C genotype has an inferior clinical response to paclitaxel - cisplatin chemotherapy but contradicting results and lack of research specificity require further insight in the effects in anti-PD-1 therapy [137]. Also, rs822336 significantly correlates with treatment response and survival outcomes. Specifically, C/C genotype is associated with better ORR, PFS and OS compared to G/C and G/G. In the presence of allele C, PD-L1 transcription is regulated by the binding of both C/EBPβ and NFIC. In contrast, only C/EBPβ regulates PD-L1 transcription in the presence of allele G. Second, a significant correlation between the presence of allele C in rs822336 and that of allele T in rs2282055 was observed. rs822336 and rs2282055 are localized on PD-L1 promoter/enhancer and intron region, respectively. Presence of allele G in rs2282055 was associated with better ORR and PFS as compared to allele T [138]. However, another study has shown better PFS outcome for T/T rs2282055 in the TPS negative population [139]. Thus, those SNPs could be promising predictive biomarkers for ICI treatment, although prospective validation is required.
Germline variants which are known to predispose for autoimmune diseases might also affect the occurrence of irAEs [140,141]. Multiple SNPs were identified in GWAS and whole exome sequencing (WES) but a significant clinical impact could only be replicated for rs16906115, an IL7 SNP, two highly linked FARP1 SNPs – rs685736 and rs643869 – and finally rs4988956, situated within the IL1RL1 gene [121,127]. Some studies focus on genes related with autoimmunity. For instance, in a cohort of 436 patients with metastatic melanoma tested for 25 different autoimmunity related SNPs, the SNP rs17388568, located in a locus containing both IL2 and IL21, was significantly associated with improved response to anti-PD-1 treatment [142].
Another example that autoimmunity is often linked to ICI outcome is found in GZMB gene encoding the granzyme B (a serine protease), an apoptotic effector of T cells. A linkage between the levels of granzyme B and cutaneous autoimmunic activity has been shown [143]; the SNP rs8192917, (128T>C) was associated with development of vitiligo in nivolumab treated NSCLC patients with shorter PFS [144]. One could speculate that these variants may lead to production of less effective granzyme B, thereby impairing the cytotoxic capabilities of T cells [144]. Further SNPs may influence ICI response by inhibiting immune signaling. For CD47 which may alter the macrophage response in ICI treatment, a SNP rs3804639, was associated with longer PFS and OS in patients with NSCLC treated with nivolumab [145].
Finally, polygenic multivariate modelling has been attempted to estimate the large inter-patient variability for a specific germline biomarker expression. Based on testing 166 different SNPs originating in 86 auto-immunity genes, multivariable models for tumor response and irAEs were able to reasonably predict both outcomes [146]. Another study tested 16.751 SNPs to calculate a polygenic risk score, originally developed as a predictor for hypothyroidism in non-oncological patients. Interestingly, the score was also able to predict thyroid irAEs [147]. These results underline the importance of assessing multiple SNPs in polygenic models in relation to each other and provide pharmacogenetic predictive tools for ICI treatment.
HLA and Response to ICI Treatment
HLA consists of a highly polymorphic gene cluster located on the short arm of chromosome 6 (6p21.3). HLA is divided in three subclasses, class I, II and III, which all play an important role in the immune system. Variations of HLA molecules result in different peptide-binding preferences as variations are mainly concentrated in exons encoding for the peptide-binding groove and the interaction with the TCR. Consequently, variations in HLA genes lead to a very diverse group of peptides being presented to both CD4 + and CD8 + T cells (Figure 3) [148].
Variations within the different HLA class I molecules determine the repertoire of peptides which can be presented to CD8+ T cells, directly affecting the diversity of cytotoxic T lymphocytes for an individual patient (i.e., the immunopeptidomes) [149]. The diversity of the HLA classes has been suggested as a predictive biomarker for response to ICI treatment, while an expression of a broader neo-antigen selection could provide a better tumor response to ICIs [150]. Individuals with homozygosity of at least one of the HLA class I alleles are hypothesized to have a poorer survival compared to individuals who are heterozygous for (one of) the HLA class I alleles, as the range in cytotoxic T lymphocytes is much smaller in homozygous patients.
Two studies have shown that homozygosity for one of loci A, B and C could already negatively impact OS after ICI treatment [151,152]. Interestingly, when specifying HLA heterozygosity per loci in the largest study of 1535 patients, those having just one homozygous locus had shorter survival [151]. This might indicate that homozygosity of a single HLA-I locus could already be clinically relevant, which is seen in approximately 18% of all patients [151]. However, this has not always been confirmed [153,154].
HLA-I evolutionary divergence (HED) may also influence the response to treatment. HED is the difference in sequence divergence between the alleles’ peptide binding properties of the corresponding HLA class I molecules [155]. High HED was associated with longer OS for patients with melanoma and NSCLC despite the fact that all included patients were already heterozygous for the HLA class I alleles [155]. Similarly, high HED was associated with more favorable PFS in RCC as well as better PFS and OS in gastrointestinal cancer patients [156,157]. Thus, evaluating the diversity of the HLA class I molecules may play a role in ICI treatment efficacy and may be used as predictive marker, if confirmed prospectively, in different tumor types.
The impact of heterozygosity of the HLA class II alleles on survival after ICI treatment has also been investigated. However, although some trends towards associations between heterozygosity of the HLA-DRB1 locus on OS have been reported, no clinical effect has been proved till now [158].
HLA supertypes are groups of HLA class I alleles classified according to their similar binding affinities. They are combinations of HLA molecules which have binding preferences for certain amino acids, resulting in an overlapping peptide binding specificity [159]. HLA-A*01 supertype was associated with prolonged PFS in a study of metastatic NSCLC patients [158], not confirmed in other studies [151,152,160,161]. A combined HLA-A*01-HLA-A*2 haplotype was associated with prolonged PFS [158]. In large cohorts of patients treated with ICIs, carriage of an HLA-A*03 allele was significantly associated with shortened survival, irrespective of the tumor type. Intriguingly, no effect of HLA-A*03 was seen in patients receiving alternative treatments [159]. All other trials, which studied the impact of HLA-A*03 on survival, did not find a significant correlation [151,152,160,161].
Importantly, the allele frequency of HLA-A*03 differs considerably across different ancestries and this could elucidate the conflicting results [151,152,153,154,161]. It seems that only HLA-A*26 and HLA-B*27 have shown promising results as biomarkers for ICI treatment in small studies and therefore merit further validation in future research [161,162]. Finally, for HLA-C supertypes, no relation with presence of the allele and survival after ICI treatment was found [158]. Thus, based on these results in general, the absence or presence of specific HLA alleles, including zygosity of HLA class I and II, cannot yet be used as predictive biomarkers in patients treated with ICIs.
Concerning the impact of HLA alleles on occurrence of immune-related toxicity a possible association between HLA molecules and occurrence of irAEs was studied in general cohorts of patients treated with ICIs. Most of the results were borderline significant, without adjusting for multiple testing and should thus be interpreted with caution. In addition, most studies were conducted in Japan, which has specific incidence of HLA alleles and consequently, prevents the results from being directly translatable to the non-Asian population.
The role of HLA class II alleles has been investigated more thoroughly as this class is associated with occurrence of autoimmune diseases. Different HLA-DRB1 molecules were found more frequently in patients with ICI-induced inflammatory rheumatoid arthritis [163]. This could indicate a causative mechanism of ICI-induced inflammatory arthritis and should be investigated further in a larger cohort of patients [163]. A significant association was reported concerning the risk of developing specific irAEs with various DR specific HLA class II alleles, such as between HLA-DR4 and the appearance of diabetes mellitus type I, HLA-DR15 with hypophysitis and HLA-DR8 with hypothyroidism [164]. However conflicting results have been reported [165]. When interpreting studies investigating the relationship between HLA alleles and the occurrence of specific irAEs, it is of great importance to take into account the different methods that have been applied. In particular, HLA class II, and more specifically the HLA-DRB1 alleles, could play a role in the onset of irAEs [120].
7. Drug Resistance
A considerable number of patients develop resistance to anti-PD-1/PD-L1 immunotherapy, rendering it ineffective during follow-up. Immunotherapy resistance can be categorized into primary resistance and acquired resistance, depending on the molecular processes behind them [166].
Primary resistance may be caused by impaired tumor-associated antigen presentation as well as alterations in intracellular molecular pathways in tumors affecting immune cell infiltration into tumor microenvironment (TME). The TME is a complex component, consisting of cancer cells, cancer-associated fibroblasts, immunosuppressive cells, cells that activate the immune system and a variety of signaling molecules. TME dynamically influences the progression of tumors and impacts the treatment outcomes. An immunosuppressive TME obstructs T-cell growth and activation, and, ultimately leads to tumor evasion. Interestingly, an irregular activation of the HGF/c-MET signaling pathway may facilitate the communication between cancer-associated fibroblasts and various immune elements, controlling PD-L1 protein expression through chemiotaxis with immunosuppressive cells. Abnormal MET activation reduces the effectiveness of anti-PD-1/PD-L1 immunotherapy [167].
Epigenetic modifications that affect antigen processing and presentation, persistent activation of the WNT/β-catenin signaling pathway, reduced T cell infiltration, and cells promoting immunosuppression are examples of primary resistance [166,168]. Immunosuppression includes deficiencies in interferon signaling and presentation of antigens, T cell immunological dysfunction, upregulated expression of immunosuppressive molecules, as well as modifications in the level of PD-L1.
Acquired resistance occurs when a tumor is initially treated with some inhibitory effect, but the tumor later progresses or reappears. A possible explanation for this is new tumor-derived resistant mutant strains. Immune checkpoints including T cell immunoglobulin mucin 3 (TIM-3) and lymphocyte activation gene protein 3 (LAG-3) show a compensatory effect after treatment, leading to an increase in the expression of other immune surveillance pathways and consequent drug resistance [166,169]. The main resistance mechanisms in ICI treatment are immunosuppression, epigenetic alterations, microbiota alterations and metabolic abnormalities.
Tumor Antigen Deletion
HLA-I is indispensable for the acknowledgment of tumor cells by CD8+ T cells [170]. Tumor cells can cause reduced or even complete down-regulation of HLA-I expression levels through beta-2-microglobulin (B2M) deficiency or mutation leading to failure in presenting neoantigens to tumor-infiltrating T cells (TILs), failure to activate CD8+ T cells, promotion of immune escape from the tumor, and resistance to blockade of immune checkpoint therapy [151,171]. Limited clinical data describe a small group of PD-L1 inhibitor-resistant patients exhibited B2M deficiency in contrast to those improving with no B2M changes detected [172,173]. Additionally, in multifocal HCC with intrahepatic metastases HLA allele heterozygous deletions are associated with a higher recurrence rate in advanced cancers [174].
Immunohistology and RNA sequencing of MHC-II in tumors showed that its expression promotes increased infiltration of CD4 T cells. Tumors adapt to this change either after PD-1 immunotherapy or after tumor progression by expressing the MHC-II inhibitory receptor LAG3 (which competes with CD4 T cells for antigen presentation) or the Fc receptor-like 6 (which binds to MHC-II, directly inhibiting NK and T effector function), leading to anti-PD-1 therapy resistance [175]. It is possible for LAG-3/FCRL6 to be a target for immunotherapy.
T Cell Dysfunction
The PD-1/PD-L1 blockade may leave tumor-specific T cells activated but also causes the overexpression of other immune checkpoints such as TIM-3 which leads to inhibition of cytotoxic T-lymphocyte and Th1 cell function reducing immunotherapeutic response [176]. In addition, PTEN, which is negatively regulated by the pathway mediated by PI3K/AKT and upregulates PD-L1 expression, is absent in a number of malignancies and may result in the development of primary immunotherapy resistance [177]. It also initiates Signal Transducer and Activator of Transcription 3 (STAT3)-mediated immunosuppression, increasing cytokines like IL-10, IL-12, IL-16, and VEGF which suppresses T cell activation and self-phagocytosis in preclinical models [178]. Finally, PD-1/PD-L1 inhibition influences TME, where overexpression of CD38 on T cell surfaces may lead tumor cells to produce adenosine which inhibits proliferation and function of CD8 + T cells (ineffective T cell penetration to TME) [166,179]. PTEN mRNA injection treatment in mouse tumor models lacking PTEN or PTEN mutations revealed that a substantial rise in CD8+ T cells could reverse the immunosuppressive TME [180].
Cucchiara et al., in an in silico analysis concerning genetic profiles of 644 advanced NSCLCs according to the immunotherapy response found at least two mutations in the coding sequence of genes belonging to the chromatin remodeling pathway, and/or at least two mutations of genes involved in cell-to-cell signaling pathways. The study suggested a dependency between mutated genes and a peculiar profile of mutations in late-stage NSCLCs with immune sensitivity. The hypothesis is that somatic loss-of-function mutations in SWI/SNF-related genes and impaired cell-to-cell crosstalk may result in dysfunctional immune evasion and persistent buffering of pro-inflammatory cytokines across the TME. This inflamed state could affect TILs activity once immune checkpoint blockers are inhibited [181].
The activation of WNT-β-collagen pathway is also linked with reduced infiltration of tumor-specific T lymphocytes possibly due to up-regulated TANK-binding kinase 1 (TBK1) and thus drug resistance [182].
Increase in Immunosuppressive Cells
An increase in immunosuppressive cells like Tregs, tumor-associated macrophages (TAMs) and other subtypes may also be implicated in drug resistance. PD-1/PD-L1 inhibitors can have an impact on Treg proliferation and function. They enhance TCR and CD28 signaling in Tregs, promoting the development of Treg-rich TMEs enhancing their inhibitory function [183]. Tregs potentially contribute to resistance against PD-L1 immunotherapy, as indicated by PD-L1 inhibitors' capacity to restore immunity against tumors following Treg depletion.
Subtypes of Myeloid-derived suppressor cells (MDSCs) can boost Arginase-1 (Arg-1) activity, which renders immune cells insensitive or tolerant and impairs their capacity to quickly eliminate tumor cells, leading to tumor immune escape [184]. MDSCs create an immunosuppressive microenvironment by promoting tumor-derived exosomes (Exo) and Hypoxia-inducible factor-1 (HIF-1) and thereby inhibit T cell activity [185] which induces apoptosis in CD8+ T cells and enhances the inhibitory activity of T-regs [186]. Multiple preclinical studies have provided evidence that targeting MDSCs can enhance immunotherapy, and current clinical trials look into how they can be paired with PD-1/PD-L1 blockers for overcoming anti-PD-1/PD-L1 resistance [187].
M2-type TAM macrophages are prompted to enhance the anti-inflammatory molecules like TGF-β and PGE-2, which inhibit normal antigen-presentation-induced T cell activation [188]. Vascular growth-associated factors (VEGF, IGF) and matrix metalloproteinases (MMPs) are produced by M2-type macrophages during the inflammatory response to help promote angiogenesis and tumor growth [189]. Reprogramming myeloid cells in TME has been shown in preclinical models to help overcome resistance [190].
Drug Resistance due to Changes in PD-L1 Expression
Oncogenic signaling pathway KRAS-ERK induces PD-L1 expression. KRAS mutations can induce the release of PD-L1 and promote apoptosis of CD3+ T cells in lung adenocarcinoma through p-ERK signaling significantly contributing to the initial resistance to PD-1 blocking [191]. Another pathway, the JAK/STAT, may be a key pathway for the synthesis of PD-L1, the production of tumor antigens [192]. Mutations in these genes may result in an absence of PD-L1 production and drug resistance [193].
Another oncogenic factor which alters the expression of PD-L1 on tumor cells, is the oncogenic transcription factor Yin Yang 1 (YY1), a known factor overexpressed in many cancers [194]. Emerging data suggest that the transcription factor YY1 may modulate PD-L1 expression through promoter binding and chromatin remodeling, although additional mechanistic studies are warranted to establish its role across different tumor types. Multiple studies have implicated the YY1/PD-L1 axis in immune escape in melanoma, NSCLC, liver cancer, and lymphoma [195]. It has been determined that YY1 is an important regulator of PD-L1 during T-cell exhaustion and promotes immune escape in prostate cancer. Additionally, YY1 is linked to tumor immune evasion through the upregulation of PD-L1 involving the p53/miR-34/PD-L1 pathway [194]. Targeting YY1 may result in a significant inhibition of cancer oncogenic activities. Various strategies are proposed to selectively target YY1 in human cancers and present a promising novel therapeutic approach for treating unresponsive cancer phenotypes. Finally, interferon gamma (IFNγ) produced by activated T cells and NK cells may strongly trigger PD-L1 expression in the tumor microenvironment. It has been shown that mutations in IFNGR1/2 or JAK1/2 components of the IFNγ signaling cascade, are a common cause of acquired and primary resistance to ICI blockade therapies [196].
Epigenetic Mechanisms of Drug Resistance
Epigenetic marks, such as DNA methylation and histone post-translational modifications (histone PTMs), participate in the regulation of gene expression and chromatin structures allowing or not allowing transcriptional machinery to access DNA. Several epigenetic mechanisms are involved in resistance to the immune checkpoint inhibitors: the main ones are the modifications of histone marks and chromatin structures, alteration of DNA methylation and changes in miRNA expression levels [197].
i. Histone Deacetylases (HDACs)
HDACs are important epigenetic regulators. Due to epigenetic silencing, they could diminish the expression of cell surface molecules essential to tumor recognition by the immune system. HDAC inhibitors may enhance the response to immunotherapy by increasing levels of tumor antigens and the reactivation of proapoptotic genes and are tested in clinical trial with ICIs [198].
ii. Histone Methyltransferases (HMT/EZH2)
EZH2 (enhancer of zeste homolog 2) may play an important role in the differentiation of Treg cells that suppress immune responses. The expression of EZH2 is linked to tumor immunogenicity and could be targeted in order to modulate the response to ICIs [199].
iii. miRNAs in Cancers and in Resistance to ICIs
MicroRNAs (miRNAs) are single-stranded, noncoding small RNA that negatively regulate gene expression at the posttranscriptional level. Their pairing with an mRNA target can lead to the inhibition of its translation or to its degradation. Many studies have linked various miRNAs with PD-L1 or PD-1 expression with the resistance to immunotherapy by modulating T-cell functions [200,201]. Altered miR expressions also act on the tumor immune response through epithelial-mesenchymal transition (EMT) induction. Furthermore, embryonic transcription factors (such as the ZEB family SNAIL, SLUG1 and TWIST1) are inducers of EMT and may be reactivated in cancer cells. MiRs like miR-200s are well-characterized inhibitors of EMT through transcription factor upregulations. The EMT and PD-L1 are linked by dysregulation of the miR-200s/ZEB1 axis, a central regulator of the EMT. These findings suggest that a subgroup of patients in whom malignant progression is driven by EMT activators may respond to treatments with PD-L1 inhibitors [201].
iv. Alteration of Tumor Immunogenicity
Epigenetic alterations participate in the remodeling of the TME and, thus, facilitate its growth and its escape from the immune system. The activation and differentiation of CD8+ T cells are associated with epigenetic changes. It has been demonstrated that chromatin remodeling is involved in the resistance to ICIs through mutations in the chromatin remodeling complex SWI/SNF (SWItch/Sucrose Non-Fermentable) complexes. PBAF, a chromatin regulatory complex (PBRM1, ARID2 and BRD7), regulates chromatin accessibility for the IFN γ pathway within tumor cells, resulting in an increased resistance to T cell–mediated cytotoxicity. PBRM1 and possibly ARID1A inactivation restores the response to immunotherapy by increasing the tumor immunogenicity [202,203].
v. DNA Methylation and Anti-PD-1/PD-L1 Treatment Resistance
DNA methylation is crucial to the development of tumors. This modification of DNA is associated with gene silencing and is carried out by specific enzymes called DNMTs for “DNA methyltransferase”. DNA hypomethylation results from the regulation of DNMT1 by PD-L1 across the STAT3 signaling pathway, which induces the derivation of new drug-resistant mutant strains in vivo [204]. The overall hypomethylation of DNA may also contribute to the constitutive upregulation of cytokines such as VEGF and IL-6, which could contribute to the resistance to immunotherapy [205]. On the other hand, the overall hypermethylation of DNA associates with low levels of PD-L1 and correlates with a poor prognosis in melanoma patients [206].
Further Difficulties of ICIs Treatments
Clinical trials have shown that not all patients are sensitive to monoclonal antibody therapy. In melanoma treatment, PD-1 antibodies exhibit a 50% response rate, but the overall response rate for other solid tumors is generally low ~15%–20% [207]. Furthermore, the timing of initiating treatment should also be investigated taking into consideration the mutational evolution within the tumor.
Another concern for the failure of ICIs treatment is that they may not effectively penetrate tumor tissues and reach all regions of the tumor to accumulate at a sufficient concentration[208]. Furthermore, the immunogenicity of antibody drugs may induce the production of anti-antibodies and lead to the loss of efficacy in some patients [209]. Moreover, immune imbalance caused by ICIs may lead to immune intolerance clinically manifested as autoimmune side effects that cause collateral damage to normal organ systems and tissues, including liver, gastrointestinal tract, lung, skin, and endocrine system [210]. As an example, in melanoma patients, skin toxicity was observed in 34% of patients treated with nivolumab and 39% of patients treated with pembrolizumab [207].
8. Biomarkers Participating in the Immune Inhibition Process
ICIs have revolutionized cancer treatment but aside from the very expensive therapeutic agents, only a limited percentage of patients benefit from them while severe immune-related adverse events may occur. A more personalized use is of outmost importance, people and waste-wise. A step towards that is distinguishing between responders and non-responders and predicting patients who are likely to develop serious side effects by identifying biomarkers [211].
Cancer - immune interaction follows a dynamic relationship characterized by the presence of checkpoints, the immune-activation mechanism and the immune-inhibitory mechanism [212], while other parameters may be also implicated. Recently, the gut microbiome has attracted attention from many clinical researches as a biomarker for ICIs and strategy to enhance ICIs efficacy. Additionally, it has been suggested that signaling pathways that affect DNA repair or antigen presentation and other immune checkpoint molecules are potential biomarkers.
The investigation of possible biomarkers is therefore possible through understanding the complexity of this relationship adding further to the precision medicine era. TME immunogenicity and response to therapy is heavily influenced by the presence or the lack of intratumoral T-cell infiltration. However, it should be noted that the character of this dynamic relationship during immunotherapy is redefined constantly. For instance, PD-L1 expressing tumor cells will be eliminated by PD-1 inhibitors, however, in turn this leaves more room for non-PD-L1 expressing clones to progress. To guide immunotherapy, it is important to dynamically monitor combinations of biomarker assays and of the cancer–immune status.
Checkpoints
The most important biomarker is the PD-L1 expression in cancer cells. Various clinical studies show an association between pretreatment PD-L1 expression and PD-1/PD-L1 blockade treatment outcomes [40,213,214]. Other biomarkers could possibly guide more precisely the decision for treatment with ICIs in addition to PD-L1 expression.
Moreover, other regulating factors of immune checkpoints candidate as potential complementary biomarkers. A study in NSCLC demonstrated that EGFR mutations could upregulate PD-L1 through the ERK–c–Jun pathway, suggesting that the adoption of anti PD-1/PD-L1 treatment in EGFR–TKIs resistant NSCLC patients with EGFR mutation is of importance [215]. Similarly, ALK rearrangements are associated with low ORRs in PD-1/PD-L1 inhibition. Low rates of concurrent PD-L1 expression and CD8+ TILs within the TME may underlie these clinical observations [216].
Immune-Activation Mechanism
The immune activation mechanism consists of tumor neoantigens known as tumor-specific novel immunogenic peptides. These are recognized by the MHC and subsequently induce specific adaptive immune response. They are diverse and include differentiation antigens, mutational antigens, over-expressed/amplified antigens, viral antigens and cancer-testis antigens with their expression determining tumor immunogenicity[217].
Gene mutational load expressed as tumor mutation burden (TMB) has been accepted as a surrogate marker for tumor neoantigen load, which varies between and within tumor types. Higher TMB is correlated with better sensitivity to PD-1 blockade of pembrolizumab in NSCLC and thus with improved objective response and durable clinical benefit in these patients [218].
Furthermore, DNA repair deficiency caused by mutations in DNA repair genes (mainly epigenetic alterations) could increase the odds of tumor immunogenicity, thus activating human immune response [219]. The status of MMR genes has been shown to be predictive of clinical benefit from anti PD-1 therapy [220]. MMR-deficient colorectal cancer compared with MMR-proficient tumors has better treatment outcome from pembrolizumab. MMR deficiency may develop through an inherited germline mutation in an MMR gene such as MLH1, MSH2, MSH6 or PMS2, which results in microsatellite instability (genetic hypermutability) [221].
When it comes to the role of immune cells the pool of circulating lymphocytes in peripheral circulation are indispensable for immune cell-mediated tumor control. A systemic dysfunction of general immune response will inevitably lead to intra-tumoral immune inhibition. Neutrophil-to-lymphocyte ratio pretherapy may be an independent marker for evaluating ipilimumab’s benefit in metastatic melanoma patients [222]. Immunotherapy potential could be enhanced by therapies that stimulate general immune function
Immune Inhibitory Mechanisms
Tumor metabolites may be implicated in alteration of the TME. Most cancer cells depend on energy by converting pyruvate to lactate via lactate dehydrogenase. Large amounts of lactic acid are pumped out of cells and cause low pH in tumor microenvironment, which can impair local T-cell functions including cytolytic activity and cytokine secretion. Particularly, lactic acid uptake in Treg cells promotes PD-1 expression which dampens the efficacy of anti-PD-1 immunotherapy [223]. Furthermore, inhibitory tumor metabolites and tumor proinflammatory factors such as interleukins (IL-1, IL-6, IL-17) as well as the expression of interferon may too, alter the response to ICI treatment. Tumor inflammatory factors could be a potential biomarker to evaluate the immune status in TME and ICI responsiveness [212].
Potential Biomarkers
i. Biomarkers Related to DNA Damage/Antigen Presentation/Interferon Signaling
Currently undergoing evaluation various tumor-derived elements with the potential to function as predictors and/or used in prognostic stratification (including DNA damage response, DDR) are: DDR gene alterations, MHC-I genotypes, beta-2 microglobulin (B2M) deficiency, POLE mutations, and JAK1/2 mutations.
ii. Tumor-Infiltrated T Cells (TILs)
TILs and peripheral T cells, particularly tumor-reactive cytotoxic CD8+ T cells, are associated with improved clinical outcomes in a wide range of tumor types with ICI efficacy depending on their sustained activation and proliferation [224]. Based on their differentiation status, CD8+ T cells are broadly classified into naive T cells, effector T cells, memory T cells, and exhausted T cells [225]. However, which of these populations is activated by ICI therapy is still not fully understood. Tissue-resident memory T cells (TRMs), a population of CD103+CD69+ cells have recently been identified to be excluded from the circulation and present in various tissues, including tumors [226]. Interestingly, increased intra-tumoral TRMs after anti-PD-1 therapy were associated with significantly improved outcomes in clinical cases of NSCLC and oral cancer patients shows that [226]. Thus, TRMs have potential as a biomarker for ICIs.
T-cell subsets may also serve as biomarkers in immunotherapy. T cells play a vital role in the development of immune tolerance to self, auto immunity and antitumor activities. The infiltration of different T-cell subsets (such as CD3, CD4, CD8, CD45RO or FoxP3 T cells, etc.) and their proportion are responsible for an effective immune response [227]. The CD8+ to PD-L1+ ratio or the CD8+ to CD3+FoxP3+ regulatory T-cell ratio may serve as a more significant predictor for immune checkpoint blockade than purely population numbers [228]. Other biomarkers in T cells are currently under investigation.
Tumor-reactive cytotoxic CD8+ T cells transform to terminal exhausted T cells (Tex) after chronic antigen stimulation, which are also candidates as biomarkers. Exhausted T cells are characterized by increased expression of the immunosuppressive receptors PD-1, CTLA-4. Recent studies have shown that Tex cells are composed of a highly heterogeneous cell population, including precursors of exhausted T cells (Tpex cells) [229]. Analysis of tumors from melanoma patients shows that higher percentages of Tpex cells are associated with longer duration of response to anti-PD-1 treatment [230]. Similarly, analysis of NSCLC patients treated with anti-PD-1 antibodies shows that Tpex cells are increased in responsive tumors [229]. Therefore, Tpex cells may be along with a possible biomarker, a target for anti-PD-1 therapy. Recently, more markers are identified to define Tpex subsets. Tsui et al. reported that a small subset of TCF-1+CD62L+ Tpex cells are the stem-like population essential for long-term self-renewal, maintenance of Tex lineage and responsiveness to immunotherapy [231]. The PD-1(-) TIGIT(-) progenitors are committed to a functional Tex differentiation, whereas PD-1(+)TIGIT(+) progenitors are differentiated into a dysfunctional and exhausted state [232].
Interestingly, epigenetic landscape analysis demonstrates that the phenotypic changes of Tex cell development coincide with the chromatin accessibility of key genes [233]. Long-term antigen stimulation leads to epigenetic reprogram which enforces the terminal exhaustion of T cells marked by high expression of checkpoint receptors, diminished effector-related molecules (IFN-γ, TNF, granzymes, and T-bet) and loss of stemness and proliferation potential (TCF-1, MYB, MYC, and Ki67) [233]. Moreover, it has been intra-tumoral tertiary lymphoid structures (TLSs), composed of T cells and B cells, can be detected by IHC staining for T cell and B cell markers, may also be used as biomarkers. TLS have been associated with improved survival in melanoma patients treated with anti-CTLA-4 or anti-PD-1 antibodies [234]. Thus, TLS may function as a biomarker in certain tumors.
iii. Peripheral T Cells
Peripheral T cells are also involved in the response to ICI treatment. The expansion of effector T cells from the periphery is highly correlated with the efficacy of anti-PD-1 therapy as is shown by T- cell receptors and scRNA sequencing analyses of tumors and peripheral blood of patients with various types of cancer [235]. In neoadjuvant anti-PD-1 treatment of NSCLC patients, T cell clones specific for mutation-associated neoantigens have been shown to rapidly proliferate in peripheral blood in patients with major pathological responses as well as Tpex populations [229,236]. Most importantly, peripheral T cells can be monitored regularly during ICI treatment which is advantageous for biomarker use.
Gut Microbiome
Gut microbiome, is a community of microorganisms that coexist in the human digestive system and, play an important role in human health and disease, having effects on important biological functions such as: nutrient absorption, regulation of the immune system, and resistance to pathogens within the digestive tract. It is associated with various diseases such as obesity, diabetes, autoimmune diseases, and cancer [237]. Because both innate and adaptive immunity are significantly influenced by the gut microbiome and its metabolites it may influence the effectiveness of immunotherapy primarily through modulation of the immune system [238]. In patients with hepatobiliary carcinoma, the amount of gut microbiota correlates with the clinical outcome of anti-PD-1 immunotherapy. In a trial of patients with hepatobiliary carcinoma treated with anti-PD-1 treatment patients who obtained longer PFS and OS showed substantially enriched microbiotaAlthough studies suggest that the gut microbiome plays a critical role in response to ICIs, it has not yet been established as a biomarker.
Other Immune Check Points
Upregulation of other co-inhibitory receptors, including LAG-3, TIM-3, and T cell immunoreceptor with immunoglobulin and immunoreceptor tyrosine-based inhibition motif domain (TIGIT), as well as their ligands, may act as a compensatory mechanism of resistance to ICIs, indicating a need for novel treatment strategies [239]. In a phase II/III clinical trial, the combination of relatlimab (the first anti-LAG-3 antibody) and nivolumab resulted in superior PFS, in comparison to nivolumab monotherapy, regardless of LAG-3 expression [240].Thus, use of LAG-3 expression as a biomarker requires further investigation. LAG-3’s importance is further underlined by the fact that in 2022, FDA approved the combination of relatlimab with nivolumab to treat unresectable or metastatic melanoma, making LAG-3 the third checkpoint target for cancer immunotherapy. Co-inhibitory receptors are expressed in various immune cells and bind to multiple ligands, making it important to determine which ligand/interaction is dominant for their immuno- suppressive functions, understanding further their biological background and designing drugs and biomarkers [118].
Other Potential Peripheral Blood Biomarkers
Given the limitations for the tumor tissue-based biomarkers, such as the difficulty of obtaining biopsies and the spatial heterogeneity within tumors, certain plasma biomarkers are considered to be easier to be implemented in the clinic. Circulating tumor DNA (ctDNA), neutrophil-to-lymphocyte ratio, and soluble forms of immune checkpoint and costimulatory molecules have been detected in the blood of cancer patients and could serve as potential biomarkers [118]. A recent metanalyses, based on 63 studies, showed that a high monocyte/lymphocyte ratio is a prognostic biomarker for short PFS and OS. The increased percentage of classical monocytes was an unfavorable predictor of survival, while low baseline rates of monocytic myeloid-derived suppressor cells were favorable. So, baseline monocyte phenotyping may serve as a composite biomarker of response to ICI [241]. Another recent study showed the implication of IL-6 as a possible biomarker for ICI responsiveness, depending on PD-L1 status. It may predict a poor response and outcome after ICI therapy, particularly in patients with PD-L1-high NSCLC via the participation of the IL-6/Jak/Stat3 pathway which in turn drives immunosuppression by MDSCs [242]
New indexes and signatures based on genomic data have been a promising new insight on ICI treatment prognosis. Further clinical research is of importance considering LATPS in lung adenocarcinoma, immune-related gene prognostic index (IRGPI), NAD+ metabolism-related gene signature (NMRGS), a novel immune signature (IMS) in HNSCC and the Lung Immune Prognostic Index (LIPI) [243,244].
9. Biomarkers in ICI Treatments in Clinical Practice
FDA-approved biomarkers used to predict ICI efficacy include PD-L1 expression, TMB, and DNA repair defects; including deficient MMR and microsatellite instability high (MSI-H)[245]. TILs are also widely recognized as a potential biomarker of ICIs [211].
PD-L1 Expression
PD-L1 as mentioned above, is a crucial biomarker for anti-PD-1/PD-L1 therapy across various cancers. Τhe dominant method for assessing PD-L1 in patients is immunohistochemical staining (IHC). Four PD-L1 IHC antibodies; SP142, 22C3, 28-8, and SP263 have been approved by the US FDA and European Medicines Agency (EMA), along with their associated staining protocols. Each antibody is approved for specific PD-1/PD-L1 inhibitors and cancer types. Scoring of PD-L1 detection involves algorithms based on the cell type, location, frequency, and intensity of PD-L1 expression, nevertheless clinically three scoring algorithms are used: TPS (tumor proportion score, also known as TC for tumor cells), CPS (combined positive score), and ICs (immune cells).
TPS is the percentage of viable tumor cells exhibiting partial or complete membrane staining, relative to all viable tumor cells (scored by positive or negative staining of PD-L1). TC is the percentage of tumor cells stained with PD-L1. CPS is the percentage of PD L1- positive cells (tumor cells, lymphocytes, and macrophages) relative to the total number of viable tumor cells. IC represents the portion of the tumor occupied by PD-L1-positive immune cells at any intensity.
Despite its detection variability among different types of cancer, PD-L1’ s expression affects the efficacy of PD-1/PD-L1 inhibitors, influencing response rates (RR), PFS and OS. Retrospective studies and meta-analyses indicate that PD-L1 expression should guide the initial choice between PD-1 blockade and chemotherapy in treating naive patients [246,247]. For previously treated patients, PD-1 inhibitors generally provide better outcomes compared to chemotherapy across all PD-L1 subgroups [246] with an increased PD-L1 expression being associated with better OS. There is also a correlation between PD-L1 expression with survival outcomes in advanced/metastatic NSCLC. Topalian et al reported that patients with PD-L1 positive tumors correlated significantly with objective response (9 of 25 patients, p=0.006) to nivolumab, while none of the 17 PD-L1 negative tumors showed response [191]. Another study on various tumors including melanoma, NSCLC, renal cell cancer has shown significant objective response of nivolumab in PD-L1 positive (≥ 5% on tumor cells) tumors [248]. Although the aforementioned studies indicated the necessity for the detection of PD-L1 expression it should be noted that half of the PD-L1 positive patients did not respond to PD-1/PD-L1 blockade while, some PD-L1 negative patients also demonstrated drug response and clinical benefit [248].
The relevance of PD-L1 staining diverges among cancer types [249]. In order to administer Pembrolizumab in NSCLC (either at stage III NSCLC localized to the chest, unsuitable for local therapy, or metastatic NSCLC) the tumor must be PD-L1 positive and lack EGFR or ALK mutations or it can be prescribed if other treatment have failed. Interestingly, in metastatic NSCLC, a beneficial effect has been recorded in combination with chemotherapy even in PD-L1 scores<1% although the greatest improvement is observed in patients with a PD-L1 score >50% [250]. Furthermore, pembrolizumab’s treatment for melanoma, renal cell carcinoma, Hodgkin lymphoma, hepatocellular carcinoma and other types of cancer is not significantly influenced by PD-L1 staining.
Considering atezolizumab, PD-L1 staining is obligatory for first-line NSCLC treatment qualification. However, the drug can be administered in combination with bevacizumab, paclitaxel, and carboplatin, when no PD-L1 has been found, for first-line treatment of metastatic non-squamous NSCLC without EGFR or ALK mutations or for metastatic NSCLC post-platinum chemotherapy. Atezolizumab is recommended in combination therapy for small-cell lung cancer, hepatocarcinoma, and melanoma which are BRAF V600 positive, regardless of PD-L1 expression.
An issue that has to be considered is the possibility of glycosylation of PD-L1 and other confounding factors leading to false-negative detection of PD-L1. Glycosylation is a post-translational modification in the extracellular domain of PD-L1 that significantly modulates its stability and interaction with the immune system [251]. This modification influences the biological functions of PD-L1 and its detection in tumor tissues, especially with traditional IHC staining further complicating diagnosis and treatment decision. Unfortunately, PD-L1 expression is thought to be underestimated in up to 40% of patient tissues, varying with therapeutic antibodies, staining antibodies, and scoring systems. Thus, the implications of glycosylation-induced, false-negative results are a rising concern when it comes to depriving patients of effective ICI therapy. An inclusive protocol incorporating pre-treatment of glycosylation removal before IHC staining has been developed [251]. Interestingly, a study involving patients with advanced TNBC, previously deemed PD-L1 negative, showed unexpected positive responses to atezolizumab in patients positive to PD-L1 post de-glycosylation. This suggests the potential of de-glycosylated PD-L1 as a more accurate biomarker for predicting treatment responses [252].
Several confounding factors that are correlated to PD-L1 expression as a biomarker are being investigated. An important issue is the heterogeneity of PD-L1 expression within and between tumors. This heterogeneity can lead to sampling errors if the biopsy fails to accurately represent the overall PD-L1 status within tumors. PD-L1 expression varies not only across different regions of the same tumor (intra-tumoral heterogeneity) but also among different tumors, such as the primary tumor and metastatic lesions in the same patient (intertumoral heterogeneity). Additionally, PD-L1 expression can change over time due to factors such as tumor progression, further mutations, and the influence of various therapeutic treatments. To conclude, it is of outmost importance to gain a better understanding of the biology of PD-L1 regulation at the transcriptional and post-translational levels, applying more comprehensive approaches to selecting cancer immunotherapies. Moving beyond PD-L1 staining to include a broader panel of biomarkers, genetic, and molecular profiling as well as considering clinical factors will pave the way of personalized ICI treatment [253].
Tumor Mutational Burden (TMB)
TMB, which serves as a metric of the genetic mutations, is present in the genome of cancer cells, reflecting the mutagenic processes induced by environmental and intracellular factors. It is suggested that a higher TMB leads to an increased presence of neoantigens, increasing the probability of immune recognition and subsequent tumor cell elimination by T cells. Whilst this concept revolves around the total number of mutations in a tumor specimen, the actual criteria for genetic alterations in TMB vary across different methodologies.
Initial TMB measurements, conducted through whole-exome sequencing (WES), included non-synonymous mutations in coding regions and omitted germline alterations by subtracting matched normal samples [254]. In clinical practice, comprehensive large cancer gene-targeted sequencing panels utilize next-generation sequencing (NGS) [254]. The FDA has approved four tests for measuring TMB, including FoundationOne CDx (F1CDx), Me- morial Sloan Kettering-Integrated Mutation Profiling of Action- able Cancer Targets (MSK-IMPACT), Guardant360 CDx, and FoundationOne Liquid CDx (F1 Liquid CDx). Another advantage of NGS liquid biopsy test is that it may identify gene mutations in circulating cell-free DNA (cfDNA) from blood samples. In practice the assessment of TMB is important. It has been reported that high TBM patients receiving pembrolizumab have more favourable ORR compared to those with low TMB (29% vs 6% respectively) [255].
The combination of the two biomarkers, the PD-L1 expression with the TMB may identify patients most likely to respond to anti-PD-1/PD-L1 therapy, with those having high TMB and PD-L1 levels above 50% experiencing a 57% ORR and the longest PFS [256]. Recent literature has showed a possible amelioration of the use of the TMB biomarker combining it with other parameters. As an example, the landscape of frameshift mutations in combination with TMB, especially in tumors with low TMB. They have been found in a high proportion of patients with low TMB who might benefit from ICIs when T-cell immunity is already present, without the need to distinguish between high- and low-TMB tumors [257,258].
MSI-H/dMMR
Microsatellites are repetitive DNA sequences containing 1–6 nucleotides, spread in tandem across the genome. Under normal conditions, DNA integrity and the length of microsatellites are maintained by the DNA MMR system, which rectifies base mismatches generated by DNA polymerases during replication or due to DNA damage. The presence of dMMR results in variations in microsatellite length, causing microsatellite instability (MSI) compared to matched normal DNA.
The phenotypic evidence for dMMR is the presence of MSI [259]. Although high-MSI (MSI-H) can manifest in various cancer types, Lynch syndrome (a familial syndrome with an increased risk of various cancer developement) stands out as the most well-known inherited condition responsible for MSI-H or dMMR tumors. It is thought that in tumors characterized as MSI-H and/or high-dMMR, faulty DNA repair mechanisms lead to a significant accumulation of mutations, rendering the tumor cells more recognizable by the immune system [259]. Thus, several studies have highlighted dMMR and MSI-H as predictive biomarkers for ICIs.
MSI-H has been approved by the FDA as a biomarker for applying ICIs to different cancer types [260]. In 2017 pembrolizumab was first approved for pre-treated MSI-H colorectal cancer and in 2020 for first-line MSI-H colorectal cancer treatment [255]. FDA granted full approval to pembrolizumab for patients with pre-treated MSI-H solid tumors in 2023. Furthermore, EMA approves pembrolizumab for patients with MSI-H or dMMR advanced colorectal, endometrial, gastric, small intestine, or biliary cancers and PD- L1-positive metastatic cervical cancer. Pembrolizumab showed an ORR of 33.3% across a wide range of advanced MSI-H/dMMR solid cancers[260]. While these results suggest that TMB-H and MSI-H are indicative of pembrolizumab outcomes across various tumor types, they also underline the limitations of relying on these markers in isolation.
Considering dostarlimab, in 2021, it gained accelerated FDA approval for pre-treated MSI-H endometrial cancer and solid tumors and in 2023, full approval for first-line MSI-H endometrial cancer treatment. Additionally, nivolumab monotherapy and ipilimumab plus nivolumab were approved by the FDA for pre-treated MSI-H colorectal cancer in 2017 and 2018, respectively [261].
To enhance predictive precision, a composite predictor that encompasses critical variables such as MHC, T cell receptor repertoire, and TIL status is suggested [262].
Cost Effectiveness of Biomarkers
Firstly, monoclonal antibodies are difficult to produce, expensive and inconvenient to store and transport, which might limit the clinical application of PD-1/PD-L1 antibody drugs. A recent systematic review and metanalysis investigated the health economic evaluations of predictive biomarker testing in solid tumors treated with ICIs [263]. In this metanalysis 58 relevant articles were reviewed out of the 730 screened. The focus was predominantly on NSCLC (60%) and other solid tumours (40%). Among the NSCLC studies, 21 out of 35 demonstrated cost-effectiveness, notably for pembrolizumab as first-line treatment when preceded by PD-L1 assessment, cost-effective at a threshold of $100,000/QALY compared to the standard of care. However, for bladder, cervical, and triple-negative breast cancers (TNBCs), no economic evaluations met the affordability threshold of $100,000/QALY. Overall, the review highlights a certain degree of uncertainty about the cost- effectiveness of ICI.
In this study the PD-L1 expression associated with ICI treatment is a cost-effective strategy, particularly in NSCLC, urothelial, and renal cell carcinoma. The findings suggest the potential value of predictive biomarker testing, specifically with pembrolizumab in NSCLC, while indicating challenges in achieving cost-effectiveness for certain other solid tumours. Therefore, the economic impact of biomarkers upstream of the choice of the specific therapy represents an imperative to validate its effectiveness, the eventual relationship with the quality of life, and economic sustainability. A biomarker testing approach is therefore a virtuous model to invest in, providing the patient with a greater chance of receiving increasingly effective therapy and minimizing adverse events due to the administration of untargeted therapies, resulting in an undoubted improvement
in quality of life, while also optimizing the management of health care resources [263].
10. Discussion
Prospectives – Novel Therapies – Precision Medicine
Cancer immunotherapy has revolutionized the clinical landscape of oncology and brought renewed hope to the treatment of previously difficult-to-treat malignancies. Yet, more questions and challenges continue to lie ahead. Future directions of cancer immunotherapy should include the investigation of immune-oncologic agents earlier in the cancer disease progression, such as in the context of neoadjuvant therapy across solid tumors. It should be recognized that the application of these therapies has difficulties mainly related to drug resistance [264]. This resistance arises from a complex interplay of diverse dynamic mechanisms within the TME, include few T cell infiltration, T cell exhaustion and immunosuppressive network [265].
Therefore, combination immunotherapies, or immunotherapies in combination with targeted therapies, chemotherapies, and radiation therapy, are promising treatment strategies to be investigated. Combination immunotherapy by counteracting several immunosuppressive elements in the TME and activating multiple steps of the cancer-immunity cycle is an intriguing strategy although the toxicity of combination therapy may also be increased [266,267]. As previously mentioned, MET inhibitors may alter the TME by neutralizing the irregular expression of PD-L1 caused by MET irregularities, thereby stimulating the cancer-fighting abilities of T and NK cells, reducing the gathering and penetration of immunosuppressive cells. Ultimately MET inhibitors may be used therapeutically in ICI combination [167].
The combination of ICI and tyrosine kinase inhibitors (TKI) have gained therapeutical significance in cancer therapy recently, due to the high efficacy of each substance group individually or combined. The majority of available data results from clinical phase I and II trials. So far, ICI-TKI combinations with nivolumab + cabozantinib, pembrolizumab + axitinib or lenvatinib, avelumab + axitinib, have been approved in advanced renal cell (RCC), with pembrolizumab + lenvatinib in endometrial carcinoma and with camrelizumab + rivoceranib in hepatocellular carcinoma (HCC). Further issues to be delt with are the optimal sequence of ICI-TKI, cross-resistances and higher toxicities [268].
Many recent studies on the mechanisms of resistance to ICI therapy and its associated biomarkers show promising prospects. However, there are still challenges in implementing these in clinical practice, and further clinical research is required.
New molecules have been tested recently as possible alternative treatment options. Such agents are small molecule inhibitors. They are intervening in the PD-1/PD-L1 signaling pathway presenting different mechanisms of action. They have significant advantages such as controllable pharmacokinetic characteristics suitability for oral administration avoidance of immune-related SAEs and favorable biosafety, thus enabling them as a novel monotherapy or complementary therapy [269,270,271]. Novel small molecule inhibitors considering the PD-1/PDL-1 axis include a variety of targets. Some block the binding of PD-1 and PD-L1 utilizing the physicochemical properties of the amino acid residues such as AUNP-12, cyclic peptide compounds DPPA-1, TPP-1, nonpeptide-based small molecule CA-170, BMS-202 and its derivatives and INCB086550, INCB099280, INCB099318 in combination with axitinib. Other targets may include suppressing the expression level of PD-L1 like JQ1 inhibitor, eFT508 (tomivosertib), Osimertinib (AZD9291), platycodin D and promoting the degradation of PD-L1 like PD-LYSO, curcumin and Metformin. All of these are currently in clinical or preclinical stages of research [269,270,272,273,274,275,276,277,278,279,280]. Furthermore, development of small-molecule inhibitors of YY1 presents a promising method of YY1 inhibition. It should be noted that YY1 inhibition novel therapies include a variety of other possible treatments [194]. These molecules are shown on Table 4. There is massive research in order to find effective small molecule inhibitors with hundreds of candidates like the BMS or the INCB families.
Another factor is long non-coding RNAs. Studies have shown that lncRNAs are abnormally expressed in tumors and play an important role in tumor proliferation, angiogenesis, apoptosis and metastasis. Lots of lncRNAs have been demonstrated to regulate the expression of key immune checkpoints, PD-1/PD-L1, CTLA-4, TIGIT, and TIM-3. One possible post-transcriptional regulatory mechanism is the lncRNA-miRNA network in PD-1/PD-L1 pathway. LncRNAs can sponge miRNAs as competing endogenous-RNA to upregulate downstream target gene PD-L1, thus contributing to immune evasion. Several lncRNAs exert their roles in immunotherapy through modulating T cells. It was found that lncRNAs inhibit the antitumor activity, decreased proportion and induced exhaustion of CD8+ T cells and they play an important role in modulating oncogenic signaling pathways involved in tumor proliferation and invasion, such as NF-κB, PI3K/AKT, Wnt/β-catenin pathways. These are preclinical data and clinical data are few. One of the greatest challenges is developing a delivery system to deliver lncRNAs efficiently and with lasting effects to specific targets [281]. Several lncRNAs are thought to be of prognostic value by some clinical studies in advanced tumors; recently, the association of the lncRNA NEAT with a favorable response to PD1/PD-L1 therapy in melanoma and glioblastoma patients was uncovered [282,283].
The impact of race, gender, and age on the outcome of immune-oncologic therapies should also be also considered. As an example, women generally exhibit stronger adaptive and innate immune responses in the immune response, as estrogen can affect the immune response by regulating the function and distribution of immune cells [284]. In addition, elderly patients often have a poorer response to immunotherapy, which may be related to the decline of the immune system [285].
The variability in patient responses to immunotherapy poses a significant challenge, creating a pressing need to identify effective biomarkers that can predict immunotherapeutic efficacy. Currently, the main biomarkers used in the clinical applications are limited to PD-L1 expression, TMB, and MSI status [286]. Although several biomarkers for ICIs have been used in clinic, their effectiveness as biomarkers to predict responders for ICIs is not sufficient, as evidenced by the low response rate of ICIs. Because individual tumor types are diverse, and each patient’s health status and immune status may also play a role in the sensitivity or resistance to ICIs, it is difficult to develop a single universal biomarker for ICIs. Therefore, combining multiple bio- markers may be necessary for more accurate prediction of the efficacy of ICI therapy. This requires developing machine learning on larger datasets and more optimized algorithms.
PD-L1 can be used to guide the selection of patients to receive immune-oncologic therapy in certain types of malignancies. However, temporal and spatial dynamics of PD-L1 has increased the complexity of using it as a biomarker in the clinic [287]. Nevertheless, components in the TME are emerging as new biomarkers although these biomarkers are often prognostic, not predictive for the response to immunotherapy [288].
Thus, more efforts to discover and validate new biomarkers are needed. Moreover, non-invasive biomarkers must be developed, and detection costs must be minimized for economical and widespread implementation in clinical applications.
Promising Ongoing Clinical Trials
The field of inhibiting the PD-1/PD-L1 axis is rapidly evolving. New antibodies, innovative combination therapies, biomarkers, and methods of administration are currently under investigation. In this dynamic research environment, it is essential to showcase clinical trials that could advance and transform this area. The latest ongoing phase III clinical trials (referred in www.clinicaltrials.gov) are summarized in the Table 5.
11. Conclusions
Future research based on the characterization of immune checkpoint mechanisms will revolutionize the field and lead to the development of promising immunotherapies for previously refractory cancers.
Polygenic models may more clearly indicate the differences in the interpatient variability and better predict those cases who could have increased chance for clinical benefit of ICIs treatment and simultaneously limit those patients at risk for the development of (severe) irAEs. The discovery and validation of effective biomarkers for ICI therapy are critical for advancing personalized cancer treatment. While significant novelties have been made, the knowledge towards precision immunotherapy is ongoing. Future research should focus on addressing the current limitations, standardizing biomarker assessment, and exploring innovative approaches to biomarker discovery in order to optimize treatment outcomes, and improve patient’s quality of life. New drugs more effective and with a safer profile are under investigation in the new era of immunotherapy.
Author Contributions
Conceptualization, I.S.V., D.-I.Z., E.G., and A.M.; methodology, D.-I.Z. and A.M.; formal analysis, D.-I.Z., A.M., and E.G.; investigation, A.M. and N.F.T.; resources, I.S.V., D.-I.Z. and A.M.; data curation, A.M. and D.-I.Z.; writing—original draft preparation, D.-I.Z., E.G., A.M., and N.F.T.; writing—review and editing, I.S.V., D.-I.Z., E.G., A.M., and N.G.; visualization, D.-I.Z.; supervision, I.S.V.; project administration, I.S.V., D.-I.Z. and A.M.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Wang, J.; Chen, Q.; Shan, Q.; Liang, T.; Forde, P.; Zheng, L. Clinical development of immuno-oncology therapeutics. Cancer letters 2025, 617, 217616. [CrossRef]
- Sharma, P.; Allison, J.P. The future of immune checkpoint therapy. Science (New York, N.Y.) 2015, 348, 56-61. [CrossRef]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews. Cancer 2012, 12, 252-264. [CrossRef]
- Ishida, Y.; Agata, Y.; Shibahara, K.; Honjo, T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. The EMBO journal 1992, 11, 3887-3895. [CrossRef]
- Freeman, G.J.; Long, A.J.; Iwai, Y.; Bourque, K.; Chernova, T.; Nishimura, H.; Fitz, L.J.; Malenkovich, N.; Okazaki, T.; Byrne, M.C.; et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. The Journal of experimental medicine 2000, 192, 1027-1034. [CrossRef]
- Latchman, Y.; Wood, C.R.; Chernova, T.; Chaudhary, D.; Borde, M.; Chernova, I.; Iwai, Y.; Long, A.J.; Brown, J.A.; Nunes, R.; et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nature immunology 2001, 2, 261-268. [CrossRef]
- Hirano, F.; Kaneko, K.; Tamura, H.; Dong, H.; Wang, S.; Ichikawa, M.; Rietz, C.; Flies, D.B.; Lau, J.S.; Zhu, G.; et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer research 2005, 65, 1089-1096.
- Brahmer, J.R.; Drake, C.G.; Wollner, I.; Powderly, J.D.; Picus, J.; Sharfman, W.H.; Stankevich, E.; Pons, A.; Salay, T.M.; McMiller, T.L.; et al. Phase I study of single-agent anti-programmed death-1 (MDX-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2010, 28, 3167-3175. [CrossRef]
- Smyth, M.J.; Teng, M.W. 2018 Nobel Prize in physiology or medicine. Clinical & translational immunology 2018, 7, e1041. [CrossRef]
- Schwartz, R.H. Costimulation of T lymphocytes: the role of CD28, CTLA-4, and B7/BB1 in interleukin-2 production and immunotherapy. Cell 1992, 71, 1065-1068. [CrossRef]
- Rosenberg, J.E.; Hoffman-Censits, J.; Powles, T.; van der Heijden, M.S.; Balar, A.V.; Necchi, A.; Dawson, N.; O'Donnell, P.H.; Balmanoukian, A.; Loriot, Y.; et al. Atezolizumab in patients with locally advanced and metastatic urothelial carcinoma who have progressed following treatment with platinum-based chemotherapy: a single-arm, multicentre, phase 2 trial. Lancet (London, England) 2016, 387, 1909-1920. [CrossRef]
- Massard, C.; Gordon, M.S.; Sharma, S.; Rafii, S.; Wainberg, Z.A.; Luke, J.; Curiel, T.J.; Colon-Otero, G.; Hamid, O.; Sanborn, R.E.; et al. Safety and Efficacy of Durvalumab (MEDI4736), an Anti-Programmed Cell Death Ligand-1 Immune Checkpoint Inhibitor, in Patients With Advanced Urothelial Bladder Cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2016, 34, 3119-3125. [CrossRef]
- Kaufman, H.L.; Russell, J.; Hamid, O.; Bhatia, S.; Terheyden, P.; D'Angelo, S.P.; Shih, K.C.; Lebbé, C.; Linette, G.P.; Milella, M.; et al. Avelumab in patients with chemotherapy-refractory metastatic Merkel cell carcinoma: a multicentre, single-group, open-label, phase 2 trial. The Lancet. Oncology 2016, 17, 1374-1385. [CrossRef]
- Migden, M.R.; Rischin, D.; Schmults, C.D.; Guminski, A.; Hauschild, A.; Lewis, K.D.; Chung, C.H.; Hernandez-Aya, L.; Lim, A.M.; Chang, A.L.S.; et al. PD-1 Blockade with Cemiplimab in Advanced Cutaneous Squamous-Cell Carcinoma. The New England journal of medicine 2018, 379, 341-351. [CrossRef]
- Wang, L.; Zhang, L.; Dunmall, L.C.; Wang, Y.Y.; Fan, Z.; Cheng, Z.; Wang, Y. The dilemmas and possible solutions for CAR-T cell therapy application in solid tumors. Cancer letters 2024, 591, 216871. [CrossRef]
- Al Hadidi, S.; Heslop, H.E.; Brenner, M.K.; Suzuki, M. Bispecific antibodies and autologous chimeric antigen receptor T cell therapies for treatment of hematological malignancies. Molecular therapy : the journal of the American Society of Gene Therapy 2024, 32, 2444-2460. [CrossRef]
- Lin, P.; Lin, Y.; Mai, Z.; Zheng, Y.; Zheng, J.; Zhou, Z.; Zhao, X.; Cui, L. Targeting cancer with precision: strategical insights into TCR-engineered T cell therapies. Theranostics 2025, 15, 300-323. [CrossRef]
- Lopez de Rodas, M.; Villalba-Esparza, M.; Sanmamed, M.F.; Chen, L.; Rimm, D.L.; Schalper, K.A. Biological and clinical significance of tumour-infiltrating lymphocytes in the era of immunotherapy: a multidimensional approach. Nature reviews. Clinical oncology 2025, 22, 163-181. [CrossRef]
- Zhou, S.; Liu, M.; Ren, F.; Meng, X.; Yu, J. The landscape of bispecific T cell engager in cancer treatment. Biomarker research 2021, 9, 38. [CrossRef]
- Tian, Z.; Liu, M.; Zhang, Y.; Wang, X. Bispecific T cell engagers: an emerging therapy for management of hematologic malignancies. Journal of hematology & oncology 2021, 14, 75. [CrossRef]
- Fan, T.; Zhang, M.; Yang, J.; Zhu, Z.; Cao, W.; Dong, C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal transduction and targeted therapy 2023, 8, 450. [CrossRef]
- Bommareddy, P.K.; Shettigar, M.; Kaufman, H.L. Integrating oncolytic viruses in combination cancer immunotherapy. Nature reviews. Immunology 2018, 18, 498-513. [CrossRef]
- Chen, L. Co-inhibitory molecules of the B7-CD28 family in the control of T-cell immunity. Nature reviews. Immunology 2004, 4, 336-347. [CrossRef]
- Intlekofer, A.M.; Thompson, C.B. At the bench: preclinical rationale for CTLA-4 and PD-1 blockade as cancer immunotherapy. Journal of leukocyte biology 2013, 94, 25-39. [CrossRef]
- Boussiotis, V.A. Molecular and Biochemical Aspects of the PD-1 Checkpoint Pathway. The New England journal of medicine 2016, 375, 1767-1778. [CrossRef]
- Lázár-Molnár, E.; Yan, Q.; Cao, E.; Ramagopal, U.; Nathenson, S.G.; Almo, S.C. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proceedings of the National Academy of Sciences of the United States of America 2008, 105, 10483-10488. [CrossRef]
- Sharpe, A.H.; Pauken, K.E. The diverse functions of the PD1 inhibitory pathway. Nature reviews. Immunology 2018, 18, 153-167. [CrossRef]
- Laderach, D.; Movassagh, M.; Johnson, A.; Mittler, R.S.; Galy, A. 4-1BB co-stimulation enhances human CD8(+) T cell priming by augmenting the proliferation and survival of effector CD8(+) T cells. International immunology 2002, 14, 1155-1167. [CrossRef]
- Liu, K.; Cui, J.J.; Zhan, Y.; Ouyang, Q.Y.; Lu, Q.S.; Yang, D.H.; Li, X.P.; Yin, J.Y. Reprogramming the tumor microenvironment by genome editing for precision cancer therapy. Molecular cancer 2022, 21, 98. [CrossRef]
- Barrios, D.M.; Do, M.H.; Phillips, G.S.; Postow, M.A.; Akaike, T.; Nghiem, P.; Lacouture, M.E. Immune checkpoint inhibitors to treat cutaneous malignancies. Journal of the American Academy of Dermatology 2020, 83, 1239-1253. [CrossRef]
- Hodi, F.S.; Chesney, J.; Pavlick, A.C.; Robert, C.; Grossmann, K.F.; McDermott, D.F.; Linette, G.P.; Meyer, N.; Giguere, J.K.; Agarwala, S.S.; et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. The Lancet. Oncology 2016, 17, 1558-1568. [CrossRef]
- Blank, C.U.; Lucas, M.W.; Scolyer, R.A.; van de Wiel, B.A.; Menzies, A.M.; Lopez-Yurda, M.; Hoeijmakers, L.L.; Saw, R.P.M.; Lijnsvelt, J.M.; Maher, N.G.; et al. Neoadjuvant Nivolumab and Ipilimumab in Resectable Stage III Melanoma. The New England journal of medicine 2024, 391, 1696-1708. [CrossRef]
- Larkin, J.; Del Vecchio, M.; Mandalá, M.; Gogas, H.; Arance Fernandez, A.M.; Dalle, S.; Cowey, C.L.; Schenker, M.; Grob, J.J.; Chiarion-Sileni, V.; et al. Adjuvant Nivolumab versus Ipilimumab in Resected Stage III/IV Melanoma: 5-Year Efficacy and Biomarker Results from CheckMate 238. Clinical cancer research : an official journal of the American Association for Cancer Research 2023, 29, 3352-3361. [CrossRef]
- Luke, J.J.; Ascierto, P.A.; Khattak, M.A.; Rutkowski, P.; Del Vecchio, M.; Spagnolo, F.; Mackiewicz, J.; Merino, L.C.; Chiarion-Sileni, V.; Kirkwood, J.M.; et al. Pembrolizumab versus placebo as adjuvant therapy in resected stage IIB or IIC melanoma: Long-term follow-up, crossover, and rechallenge with pembrolizumab in the phase III KEYNOTE-716 study. European journal of cancer (Oxford, England : 1990) 2025, 220, 115381. [CrossRef]
- Nghiem, P.; Bhatia, S.; Lipson, E.J.; Sharfman, W.H.; Kudchadkar, R.R.; Brohl, A.S.; Friedlander, P.A.; Daud, A.; Kluger, H.M.; Reddy, S.A.; et al. Durable Tumor Regression and Overall Survival in Patients With Advanced Merkel Cell Carcinoma Receiving Pembrolizumab as First-Line Therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019, 37, 693-702. [CrossRef]
- Damsin, T.; Lebas, E.; Marchal, N.; Rorive, A.; Nikkels, A.F. Cemiplimab for locally advanced and metastatic basal cell carcinoma. Expert review of anticancer therapy 2022, 22, 243-248. [CrossRef]
- Yan, F.; Schmalbach, C.E. Updates in the Management of Advanced Nonmelanoma Skin Cancer. Surgical oncology clinics of North America 2024, 33, 723-733. [CrossRef]
- Lee, A. Cosibelimab: First Approval. Drugs 2025, 85, 695-698. [CrossRef]
- Patel, S.A.; Weiss, J. Advances in the Treatment of Non-Small Cell Lung Cancer: Immunotherapy. Clinics in chest medicine 2020, 41, 237-247. [CrossRef]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the treatment of non-small-cell lung cancer. The New England journal of medicine 2015, 372, 2018-2028. [CrossRef]
- Herbst, R.S.; Giaccone, G.; de Marinis, F.; Reinmuth, N.; Vergnenegre, A.; Barrios, C.H.; Morise, M.; Felip, E.; Andric, Z.; Geater, S.; et al. Atezolizumab for First-Line Treatment of PD-L1-Selected Patients with NSCLC. The New England journal of medicine 2020, 383, 1328-1339. [CrossRef]
- Sezer, A.; Kilickap, S.; Gümüş, M.; Bondarenko, I.; Özgüroğlu, M.; Gogishvili, M.; Turk, H.M.; Cicin, I.; Bentsion, D.; Gladkov, O.; et al. Cemiplimab monotherapy for first-line treatment of advanced non-small-cell lung cancer with PD-L1 of at least 50%: a multicentre, open-label, global, phase 3, randomised, controlled trial. Lancet (London, England) 2021, 397, 592-604. [CrossRef]
- Landre, T.; Justeau, G.; Assié, J.B.; Chouahnia, K.; Davoine, C.; Taleb, C.; Chouaïd, C.; Duchemann, B. Anti-PD-(L)1 for KRAS-mutant advanced non-small-cell lung cancers: a meta-analysis of randomized-controlled trials. Cancer immunology, immunotherapy : CII 2022, 71, 719-726. [CrossRef]
- Chen, W.; Li, L.; Cheng, S.; Yu, J. The Efficacy of Immune Checkpoint Inhibitors vs. Chemotherapy for KRAS-Mutant or EGFR-Mutant Non-Small-Cell Lung Cancers: A Meta-Analysis Based on Randomized Controlled Trials. Disease markers 2022, 2022, 2631852. [CrossRef]
- Cascone, T.; Awad, M.M.; Spicer, J.D.; He, J.; Lu, S.; Sepesi, B.; Tanaka, F.; Taube, J.M.; Cornelissen, R.; Havel, L.; et al. Perioperative Nivolumab in Resectable Lung Cancer. The New England journal of medicine 2024, 390, 1756-1769. [CrossRef]
- Wakelee, H.; Liberman, M.; Kato, T.; Tsuboi, M.; Lee, S.H.; Gao, S.; Chen, K.N.; Dooms, C.; Majem, M.; Eigendorff, E.; et al. Perioperative Pembrolizumab for Early-Stage Non-Small-Cell Lung Cancer. The New England journal of medicine 2023, 389, 491-503. [CrossRef]
- Hellmann, M.D.; Paz-Ares, L.; Bernabe Caro, R.; Zurawski, B.; Kim, S.W.; Carcereny Costa, E.; Park, K.; Alexandru, A.; Lupinacci, L.; de la Mora Jimenez, E.; et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. The New England journal of medicine 2019, 381, 2020-2031. [CrossRef]
- West, H.; McCleod, M.; Hussein, M.; Morabito, A.; Rittmeyer, A.; Conter, H.J.; Kopp, H.G.; Daniel, D.; McCune, S.; Mekhail, T.; et al. Atezolizumab in combination with carboplatin plus nab-paclitaxel chemotherapy compared with chemotherapy alone as first-line treatment for metastatic non-squamous non-small-cell lung cancer (IMpower130): a multicentre, randomised, open-label, phase 3 trial. The Lancet. Oncology 2019, 20, 924-937. [CrossRef]
- Antonia, S.J.; Villegas, A.; Daniel, D.; Vicente, D.; Murakami, S.; Hui, R.; Kurata, T.; Chiappori, A.; Lee, K.H.; de Wit, M.; et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. The New England journal of medicine 2018, 379, 2342-2350. [CrossRef]
- Heymach, J.V.; Harpole, D.; Mitsudomi, T.; Taube, J.M.; Galffy, G.; Hochmair, M.; Winder, T.; Zukov, R.; Garbaos, G.; Gao, S.; et al. Perioperative Durvalumab for Resectable Non-Small-Cell Lung Cancer. The New England journal of medicine 2023, 389, 1672-1684. [CrossRef]
- Felip, E.; Altorki, N.; Zhou, C.; Csőszi, T.; Vynnychenko, I.; Goloborodko, O.; Luft, A.; Akopov, A.; Martinez-Marti, A.; Kenmotsu, H.; et al. Adjuvant atezolizumab after adjuvant chemotherapy in resected stage IB-IIIA non-small-cell lung cancer (IMpower010): a randomised, multicentre, open-label, phase 3 trial. Lancet (London, England) 2021, 398, 1344-1357. [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. The New England journal of medicine 2018, 379, 2220-2229. [CrossRef]
- Goldman, J.W.; Dvorkin, M.; Chen, Y.; Reinmuth, N.; Hotta, K.; Trukhin, D.; Statsenko, G.; Hochmair, M.J.; Özgüroğlu, M.; Ji, J.H.; et al. Durvalumab, with or without tremelimumab, plus platinum-etoposide versus platinum-etoposide alone in first-line treatment of extensive-stage small-cell lung cancer (CASPIAN): updated results from a randomised, controlled, open-label, phase 3 trial. The Lancet. Oncology 2021, 22, 51-65. [CrossRef]
- Cheng, Y.; Spigel, D.R.; Cho, B.C.; Laktionov, K.K.; Fang, J.; Chen, Y.; Zenke, Y.; Lee, K.H.; Wang, Q.; Navarro, A.; et al. Durvalumab after Chemoradiotherapy in Limited-Stage Small-Cell Lung Cancer. The New England journal of medicine 2024, 391, 1313-1327. [CrossRef]
- Janjigian, Y.Y.; Shitara, K.; Moehler, M.; Garrido, M.; Salman, P.; Shen, L.; Wyrwicz, L.; Yamaguchi, K.; Skoczylas, T.; Campos Bragagnoli, A.; et al. First-line nivolumab plus chemotherapy versus chemotherapy alone for advanced gastric, gastro-oesophageal junction, and oesophageal adenocarcinoma (CheckMate 649): a randomised, open-label, phase 3 trial. Lancet (London, England) 2021, 398, 27-40. [CrossRef]
- Shah, M.A.; Kojima, T.; Hochhauser, D.; Enzinger, P.; Raimbourg, J.; Hollebecque, A.; Lordick, F.; Kim, S.B.; Tajika, M.; Kim, H.T.; et al. Efficacy and Safety of Pembrolizumab for Heavily Pretreated Patients With Advanced, Metastatic Adenocarcinoma or Squamous Cell Carcinoma of the Esophagus: The Phase 2 KEYNOTE-180 Study. JAMA oncology 2019, 5, 546-550. [CrossRef]
- Kelly, R.J.; Ajani, J.A.; Kuzdzal, J.; Zander, T.; Van Cutsem, E.; Piessen, G.; Mendez, G.; Feliciano, J.; Motoyama, S.; Lièvre, A.; et al. Adjuvant Nivolumab in Resected Esophageal or Gastroesophageal Junction Cancer. The New England journal of medicine 2021, 384, 1191-1203. [CrossRef]
- Rha, S.Y.; Oh, D.Y.; Yañez, P.; Bai, Y.; Ryu, M.H.; Lee, J.; Rivera, F.; Alves, G.V.; Garrido, M.; Shiu, K.K.; et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for HER2-negative advanced gastric cancer (KEYNOTE-859): a multicentre, randomised, double-blind, phase 3 trial. The Lancet. Oncology 2023, 24, 1181-1195. [CrossRef]
- Janjigian, Y.Y.; Kawazoe, A.; Bai, Y.; Xu, J.; Lonardi, S.; Metges, J.P.; Yanez, P.; Wyrwicz, L.S.; Shen, L.; Ostapenko, Y.; et al. Pembrolizumab plus trastuzumab and chemotherapy for HER2-positive gastric or gastro-oesophageal junction adenocarcinoma: interim analyses from the phase 3 KEYNOTE-811 randomised placebo-controlled trial. Lancet (London, England) 2023, 402, 2197-2208. [CrossRef]
- Grieb, B.C.; Agarwal, R. HER2-Directed Therapy in Advanced Gastric and Gastroesophageal Adenocarcinoma: Triumphs and Troubles. Current treatment options in oncology 2021, 22, 88. [CrossRef]
- Moehler, M.; Oh, D.Y.; Kato, K.; Arkenau, T.; Tabernero, J.; Lee, K.W.; Rha, S.Y.; Hirano, H.; Spigel, D.; Yamaguchi, K.; et al. First-Line Tislelizumab Plus Chemotherapy for Advanced Gastric Cancer with Programmed Death-Ligand 1 Expression ≥ 1%: A Retrospective Analysis of RATIONALE-305. Advances in therapy 2025, 42, 2248-2268. [CrossRef]
- Yoshinami, Y.; Shoji, H. Recent advances in immunotherapy and molecular targeted therapy for gastric cancer. Future science OA 2023, 9, Fso842. [CrossRef]
- Ganesh, K.; Stadler, Z.K.; Cercek, A.; Mendelsohn, R.B.; Shia, J.; Segal, N.H.; Diaz, L.A., Jr. Immunotherapy in colorectal cancer: rationale, challenges and potential. Nature reviews. Gastroenterology & hepatology 2019, 16, 361-375. [CrossRef]
- Overman, M.J.; McDermott, R.; Leach, J.L.; Lonardi, S.; Lenz, H.J.; Morse, M.A.; Desai, J.; Hill, A.; Axelson, M.; Moss, R.A.; et al. Nivolumab in patients with metastatic DNA mismatch repair-deficient or microsatellite instability-high colorectal cancer (CheckMate 142): an open-label, multicentre, phase 2 study. The Lancet. Oncology 2017, 18, 1182-1191. [CrossRef]
- Chalabi, M.; Verschoor, Y.L.; Tan, P.B.; Balduzzi, S.; Van Lent, A.U.; Grootscholten, C.; Dokter, S.; Büller, N.V.; Grotenhuis, B.A.; Kuhlmann, K.; et al. Neoadjuvant Immunotherapy in Locally Advanced Mismatch Repair-Deficient Colon Cancer. The New England journal of medicine 2024, 390, 1949-1958. [CrossRef]
- Zhu, A.X.; Finn, R.S.; Edeline, J.; Cattan, S.; Ogasawara, S.; Palmer, D.; Verslype, C.; Zagonel, V.; Fartoux, L.; Vogel, A.; et al. Pembrolizumab in patients with advanced hepatocellular carcinoma previously treated with sorafenib (KEYNOTE-224): a non-randomised, open-label phase 2 trial. The Lancet. Oncology 2018, 19, 940-952. [CrossRef]
- El-Khoueiry, A.B.; Sangro, B.; Yau, T.; Crocenzi, T.S.; Kudo, M.; Hsu, C.; Kim, T.Y.; Choo, S.P.; Trojan, J.; Welling, T.H.R.; et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet (London, England) 2017, 389, 2492-2502. [CrossRef]
- Finn, R.S.; Qin, S.; Ikeda, M.; Galle, P.R.; Ducreux, M.; Kim, T.Y.; Kudo, M.; Breder, V.; Merle, P.; Kaseb, A.O.; et al. Atezolizumab plus Bevacizumab in Unresectable Hepatocellular Carcinoma. The New England journal of medicine 2020, 382, 1894-1905. [CrossRef]
- Sangro, B.; Chan, S.L.; Kelley, R.K.; Lau, G.; Kudo, M.; Sukeepaisarnjaroen, W.; Yarchoan, M.; De Toni, E.N.; Furuse, J.; Kang, Y.K.; et al. Four-year overall survival update from the phase III HIMALAYA study of tremelimumab plus durvalumab in unresectable hepatocellular carcinoma. Annals of oncology : official journal of the European Society for Medical Oncology 2024, 35, 448-457. [CrossRef]
- Qin, S.; Chan, S.L.; Gu, S.; Bai, Y.; Ren, Z.; Lin, X.; Chen, Z.; Jia, W.; Jin, Y.; Guo, Y.; et al. Camrelizumab plus rivoceranib versus sorafenib as first-line therapy for unresectable hepatocellular carcinoma (CARES-310): a randomised, open-label, international phase 3 study. Lancet (London, England) 2023, 402, 1133-1146. [CrossRef]
- Schmid, P.; Rugo, H.S.; Adams, S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Henschel, V.; Molinero, L.; Chui, S.Y.; et al. Atezolizumab plus nab-paclitaxel as first-line treatment for unresectable, locally advanced or metastatic triple-negative breast cancer (IMpassion130): updated efficacy results from a randomised, double-blind, placebo-controlled, phase 3 trial. The Lancet. Oncology 2020, 21, 44-59. [CrossRef]
- Cetin, B.; Gumusay, O. Pembrolizumab for Early Triple-Negative Breast Cancer. The New England journal of medicine 2020, 382, e108. [CrossRef]
- Colombo, N.; Dubot, C.; Lorusso, D.; Caceres, M.V.; Hasegawa, K.; Shapira-Frommer, R.; Tewari, K.S.; Salman, P.; Hoyos Usta, E.; Yañez, E.; et al. Pembrolizumab for Persistent, Recurrent, or Metastatic Cervical Cancer. The New England journal of medicine 2021, 385, 1856-1867. [CrossRef]
- Oaknin, A.; Tinker, A.V.; Gilbert, L.; Samouëlian, V.; Mathews, C.; Brown, J.; Barretina-Ginesta, M.P.; Moreno, V.; Gravina, A.; Abdeddaim, C.; et al. Clinical Activity and Safety of the Anti-Programmed Death 1 Monoclonal Antibody Dostarlimab for Patients With Recurrent or Advanced Mismatch Repair-Deficient Endometrial Cancer: A Nonrandomized Phase 1 Clinical Trial. JAMA oncology 2020, 6, 1766-1772. [CrossRef]
- Powell, M.A.; Cibula, D.; O'Malley, D.M.; Boere, I.; Shahin, M.S.; Savarese, A.; Chase, D.M.; Gilbert, L.; Black, D.; Herrstedt, J.; et al. Efficacy and safety of dostarlimab in combination with chemotherapy in patients with dMMR/MSI-H primary advanced or recurrent endometrial cancer in a phase 3, randomized, placebo-controlled trial (ENGOT-EN6-NSGO/GOG-3031/RUBY). Gynecologic oncology 2025, 192, 40-49. [CrossRef]
- Di Dio, C.; Bogani, G.; Di Donato, V.; Cuccu, I.; Muzii, L.; Musacchio, L.; Scambia, G.; Lorusso, D. The role of immunotherapy in advanced and recurrent MMR deficient and proficient endometrial carcinoma. Gynecologic oncology 2023, 169, 27-33. [CrossRef]
- Westin, S.N.; Moore, K.; Chon, H.S.; Lee, J.Y.; Thomes Pepin, J.; Sundborg, M.; Shai, A.; de la Garza, J.; Nishio, S.; Gold, M.A.; et al. Durvalumab Plus Carboplatin/Paclitaxel Followed by Maintenance Durvalumab With or Without Olaparib as First-Line Treatment for Advanced Endometrial Cancer: The Phase III DUO-E Trial. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2024, 42, 283-299. [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. The New England journal of medicine 2018, 378, 1277-1290. [CrossRef]
- Rini, B.I.; Plimack, E.R.; Stus, V.; Gafanov, R.; Hawkins, R.; Nosov, D.; Pouliot, F.; Alekseev, B.; Soulières, D.; Melichar, B.; et al. Pembrolizumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. The New England journal of medicine 2019, 380, 1116-1127. [CrossRef]
- Motzer, R.J.; Penkov, K.; Haanen, J.; Rini, B.; Albiges, L.; Campbell, M.T.; Venugopal, B.; Kollmannsberger, C.; Negrier, S.; Uemura, M.; et al. Avelumab plus Axitinib versus Sunitinib for Advanced Renal-Cell Carcinoma. The New England journal of medicine 2019, 380, 1103-1115. [CrossRef]
- Choueiri, T.K.; Powles, T.; Burotto, M.; Escudier, B.; Bourlon, M.T.; Zurawski, B.; Oyervides Juárez, V.M.; Hsieh, J.J.; Basso, U.; Shah, A.Y.; et al. Nivolumab plus Cabozantinib versus Sunitinib for Advanced Renal-Cell Carcinoma. The New England journal of medicine 2021, 384, 829-841. [CrossRef]
- Motzer, R.; Alekseev, B.; Rha, S.Y.; Porta, C.; Eto, M.; Powles, T.; Grünwald, V.; Hutson, T.E.; Kopyltsov, E.; Méndez-Vidal, M.J.; et al. Lenvatinib plus Pembrolizumab or Everolimus for Advanced Renal Cell Carcinoma. The New England journal of medicine 2021, 384, 1289-1300. [CrossRef]
- Powles, T.; Csőszi, T.; Özgüroğlu, M.; Matsubara, N.; Géczi, L.; Cheng, S.Y.; Fradet, Y.; Oudard, S.; Vulsteke, C.; Morales Barrera, R.; et al. Pembrolizumab alone or combined with chemotherapy versus chemotherapy as first-line therapy for advanced urothelial carcinoma (KEYNOTE-361): a randomised, open-label, phase 3 trial. The Lancet. Oncology 2021, 22, 931-945. [CrossRef]
- Powles, T.; Park, S.H.; Voog, E.; Caserta, C.; Valderrama, B.P.; Gurney, H.; Kalofonos, H.; Radulović, S.; Demey, W.; Ullén, A.; et al. Avelumab Maintenance Therapy for Advanced or Metastatic Urothelial Carcinoma. The New England journal of medicine 2020, 383, 1218-1230. [CrossRef]
- van der Heijden, M.S.; Sonpavde, G.; Powles, T.; Necchi, A.; Burotto, M.; Schenker, M.; Sade, J.P.; Bamias, A.; Beuzeboc, P.; Bedke, J.; et al. Nivolumab plus Gemcitabine-Cisplatin in Advanced Urothelial Carcinoma. The New England journal of medicine 2023, 389, 1778-1789. [CrossRef]
- Oridate, N.; Takahashi, S.; Tanaka, K.; Shimizu, Y.; Fujimoto, Y.; Matsumoto, K.; Yokota, T.; Yamazaki, T.; Takahashi, M.; Ueda, T.; et al. First-line pembrolizumab with or without chemotherapy for recurrent or metastatic head and neck squamous cell carcinoma: 5-year follow-up of the Japanese population of KEYNOTE-048. International journal of clinical oncology 2024, 29, 1825-1839. [CrossRef]
- Liang, Y.L.; Liu, X.; Shen, L.F.; Hu, G.Y.; Zou, G.R.; Zhang, N.; Chen, C.B.; Chen, X.Z.; Zhu, X.D.; Yuan, Y.W.; et al. Adjuvant PD-1 Blockade With Camrelizumab for Nasopharyngeal Carcinoma: The DIPPER Randomized Clinical Trial. Jama 2025. [CrossRef]
- Zheng, B.; Huang, Z.; Liu, W.; Zhao, D.; Xu, X.; Xiao, S.; Sun, Y.; Wang, W. A real-world evaluation of tislelizumab in patients with head and neck cancer. Translational cancer research 2024, 13, 808-818. [CrossRef]
- Liu, S.L.; Li, X.Y.; Yang, J.H.; Wen, D.X.; Guo, S.S.; Liu, L.T.; Li, Y.F.; Luo, M.J.; Xie, S.Y.; Liang, Y.J.; et al. Neoadjuvant and adjuvant toripalimab for locoregionally advanced nasopharyngeal carcinoma: a randomised, single-centre, double-blind, placebo-controlled, phase 2 trial. The Lancet. Oncology 2024, 25, 1563-1575. [CrossRef]
- Younes, A.; Santoro, A.; Shipp, M.; Zinzani, P.L.; Timmerman, J.M.; Ansell, S.; Armand, P.; Fanale, M.; Ratanatharathorn, V.; Kuruvilla, J.; et al. Nivolumab for classical Hodgkin's lymphoma after failure of both autologous stem-cell transplantation and brentuximab vedotin: a multicentre, multicohort, single-arm phase 2 trial. The Lancet. Oncology 2016, 17, 1283-1294. [CrossRef]
- Armand, P.; Rodig, S.; Melnichenko, V.; Thieblemont, C.; Bouabdallah, K.; Tumyan, G.; Özcan, M.; Portino, S.; Fogliatto, L.; Caballero, M.D.; et al. Pembrolizumab in Relapsed or Refractory Primary Mediastinal Large B-Cell Lymphoma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2019, 37, 3291-3299. [CrossRef]
- Centanni, M.; Moes, D.; Trocóniz, I.F.; Ciccolini, J.; van Hasselt, J.G.C. Clinical Pharmacokinetics and Pharmacodynamics of Immune Checkpoint Inhibitors. Clinical pharmacokinetics 2019, 58, 835-857. [CrossRef]
- Yan, T.; Yu, L.; Shangguan, D.; Li, W.; Liu, N.; Chen, Y.; Fu, Y.; Tang, J.; Liao, D. Advances in pharmacokinetics and pharmacodynamics of PD-1/PD-L1 inhibitors. International immunopharmacology 2023, 115, 109638. [CrossRef]
- Sachs, U.J.; Socher, I.; Braeunlich, C.G.; Kroll, H.; Bein, G.; Santoso, S. A variable number of tandem repeats polymorphism influences the transcriptional activity of the neonatal Fc receptor alpha-chain promoter. Immunology 2006, 119, 83-89. [CrossRef]
- Keizer, R.J.; Huitema, A.D.; Schellens, J.H.; Beijnen, J.H. Clinical pharmacokinetics of therapeutic monoclonal antibodies. Clinical pharmacokinetics 2010, 49, 493-507. [CrossRef]
- Wang, W.; Wang, E.Q.; Balthasar, J.P. Monoclonal antibody pharmacokinetics and pharmacodynamics. Clinical pharmacology and therapeutics 2008, 84, 548-558. [CrossRef]
- Zhao, X.; Suryawanshi, S.; Hruska, M.; Feng, Y.; Wang, X.; Shen, J.; Vezina, H.E.; McHenry, M.B.; Waxman, I.M.; Achanta, A.; et al. Assessment of nivolumab benefit-risk profile of a 240-mg flat dose relative to a 3-mg/kg dosing regimen in patients with advanced tumors. Annals of oncology : official journal of the European Society for Medical Oncology 2017, 28, 2002-2008. [CrossRef]
- Liu, C.; Yu, J.; Li, H.; Liu, J.; Xu, Y.; Song, P.; Liu, Q.; Zhao, H.; Xu, J.; Maher, V.E.; et al. Association of time-varying clearance of nivolumab with disease dynamics and its implications on exposure response analysis. Clinical pharmacology and therapeutics 2017, 101, 657-666. [CrossRef]
- Li, H.; Yu, J.; Liu, C.; Liu, J.; Subramaniam, S.; Zhao, H.; Blumenthal, G.M.; Turner, D.C.; Li, C.; Ahamadi, M.; et al. Time dependent pharmacokinetics of pembrolizumab in patients with solid tumor and its correlation with best overall response. Journal of pharmacokinetics and pharmacodynamics 2017, 44, 403-414. [CrossRef]
- Porporato, P.E. Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 2016, 5, e200. [CrossRef]
- Baverel, P.G.; Dubois, V.F.S.; Jin, C.Y.; Zheng, Y.; Song, X.; Jin, X.; Mukhopadhyay, P.; Gupta, A.; Dennis, P.A.; Ben, Y.; et al. Population Pharmacokinetics of Durvalumab in Cancer Patients and Association With Longitudinal Biomarkers of Disease Status. Clinical pharmacology and therapeutics 2018, 103, 631-642. [CrossRef]
- Beers, S.A.; Glennie, M.J.; White, A.L. Influence of immunoglobulin isotype on therapeutic antibody function. Blood 2016, 127, 1097-1101. [CrossRef]
- Kretschmer, A.; Schwanbeck, R.; Valerius, T.; Rösner, T. Antibody Isotypes for Tumor Immunotherapy. Transfusion medicine and hemotherapy : offizielles Organ der Deutschen Gesellschaft fur Transfusionsmedizin und Immunhamatologie 2017, 44, 320-326. [CrossRef]
- Sharma, A.; Subudhi, S.K.; Blando, J.; Scutti, J.; Vence, L.; Wargo, J.; Allison, J.P.; Ribas, A.; Sharma, P. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3(+) Regulatory T Cells (Tregs) in Human Cancers. Clinical cancer research : an official journal of the American Association for Cancer Research 2019, 25, 1233-1238. [CrossRef]
- Agrawal, S.; Feng, Y.; Roy, A.; Kollia, G.; Lestini, B. Nivolumab dose selection: challenges, opportunities, and lessons learned for cancer immunotherapy. Journal for immunotherapy of cancer 2016, 4, 72. [CrossRef]
- Oude Munnink, T.H.; Henstra, M.J.; Segerink, L.I.; Movig, K.L.; Brummelhuis-Visser, P. Therapeutic drug monitoring of monoclonal antibodies in inflammatory and malignant disease: Translating TNF-α experience to oncology. Clinical pharmacology and therapeutics 2016, 99, 419-431. [CrossRef]
- Stroh, M., H. Winter, M. Marchand, L. Claret, S. Eppler, J. Ruppel, O. Abidoye, S. L. Teng, W. T. Lin, S. Dayog, R. Bruno, J. Jin, and S. Girish. "Clinical Pharmacokinetics and Pharmacodynamics of Atezolizumab in Metastatic Urothelial Carcinoma." Clin Pharmacol Ther 102, no. 2 (2017): 305-12.
- Shek, D.; Read, S.A.; Ahlenstiel, G.; Piatkov, I. Pharmacogenetics of anticancer monoclonal antibodies. Cancer drug resistance (Alhambra, Calif.) 2019, 2, 69-81. [CrossRef]
- Michot, J.M.; Bigenwald, C.; Champiat, S.; Collins, M.; Carbonnel, F.; Postel-Vinay, S.; Berdelou, A.; Varga, A.; Bahleda, R.; Hollebecque, A.; et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. European journal of cancer (Oxford, England : 1990) 2016, 54, 139-148. [CrossRef]
- Blank, C.U.; Haanen, J.B.; Ribas, A.; Schumacher, T.N. CANCER IMMUNOLOGY. The "cancer immunogram". Science (New York, N.Y.) 2016, 352, 658-660. [CrossRef]
- Yamaguchi, H.; Hsu, J.M.; Sun, L.; Wang, S.C.; Hung, M.C. Advances and prospects of biomarkers for immune checkpoint inhibitors. Cell reports. Medicine 2024, 5, 101621. [CrossRef]
- Heersche, N.; Veerman, G.D.M.; de With, M.; Bins, S.; Assaraf, Y.G.; Dingemans, A.C.; van Schaik, R.H.N.; Mathijssen, R.H.J.; Jansman, F.G.A. Clinical implications of germline variations for treatment outcome and drug resistance for small molecule kinase inhibitors in patients with non-small cell lung cancer. Drug resistance updates : reviews and commentaries in antimicrobial and anticancer chemotherapy 2022, 62, 100832. [CrossRef]
- de Joode, K.; Heersche, N.; Basak, E.A.; Bins, S.; van der Veldt, A.A.M.; van Schaik, R.H.N.; Mathijssen, R.H.J. Review - The impact of pharmacogenetics on the outcome of immune checkpoint inhibitors. Cancer treatment reviews 2024, 122, 102662. [CrossRef]
- Stroh, M.; Winter, H.; Marchand, M.; Claret, L.; Eppler, S.; Ruppel, J.; Abidoye, O.; Teng, S.L.; Lin, W.T.; Dayog, S.; et al. Clinical Pharmacokinetics and Pharmacodynamics of Atezolizumab in Metastatic Urothelial Carcinoma. Clinical pharmacology and therapeutics 2017, 102, 305-312. [CrossRef]
- Bajaj, G.; Wang, X.; Agrawal, S.; Gupta, M.; Roy, A.; Feng, Y. Model-Based Population Pharmacokinetic Analysis of Nivolumab in Patients With Solid Tumors. CPT: pharmacometrics & systems pharmacology 2017, 6, 58-66. [CrossRef]
- Ahamadi, M.; Freshwater, T.; Prohn, M.; Li, C.H.; de Alwis, D.P.; de Greef, R.; Elassaiss-Schaap, J.; Kondic, A.; Stone, J.A. Model-Based Characterization of the Pharmacokinetics of Pembrolizumab: A Humanized Anti-PD-1 Monoclonal Antibody in Advanced Solid Tumors. CPT: pharmacometrics & systems pharmacology 2017, 6, 49-57. [CrossRef]
- Yang, F.; Paccaly, A.J.; Rippley, R.K.; Davis, J.D.; DiCioccio, A.T. Population pharmacokinetic characteristics of cemiplimab in patients with advanced malignancies. Journal of pharmacokinetics and pharmacodynamics 2021, 48, 479-494. [CrossRef]
- Melhem, M.; Hanze, E.; Lu, S.; Alskär, O.; Visser, S.; Gandhi, Y. Population pharmacokinetics and exposure-response of anti-programmed cell death protein-1 monoclonal antibody dostarlimab in advanced solid tumours. British journal of clinical pharmacology 2022, 88, 4142-4154. [CrossRef]
- Budha, N.; Wu, C.Y.; Tang, Z.; Yu, T.; Liu, L.; Xu, F.; Gao, Y.; Li, R.; Zhang, Q.; Wan, Y.; et al. Model-based population pharmacokinetic analysis of tislelizumab in patients with advanced tumors. CPT: pharmacometrics & systems pharmacology 2023, 12, 95-109. [CrossRef]
- Wang, C.Y.; Sheng, C.C.; Ma, G.L.; Xu, D.; Liu, X.Q.; Wang, Y.Y.; Zhang, L.; Cui, C.L.; Xu, B.H.; Song, Y.Q.; et al. Population pharmacokinetics of the anti-PD-1 antibody camrelizumab in patients with multiple tumor types and model-informed dosing strategy. Acta pharmacologica Sinica 2021, 42, 1368-1375. [CrossRef]
- Groha, S.; Alaiwi, S.A.; Xu, W.; Naranbhai, V.; Nassar, A.H.; Bakouny, Z.; El Zarif, T.; Saliby, R.M.; Wan, G.; Rajeh, A.; et al. Germline variants associated with toxicity to immune checkpoint blockade. Nature medicine 2022, 28, 2584-2591. [CrossRef]
- Abdel-Wahab, N.; Diab, A.; Yu, R.K.; Futreal, A.; Criswell, L.A.; Tayar, J.H.; Dadu, R.; Shannon, V.; Shete, S.S.; Suarez-Almazor, M.E. Genetic determinants of immune-related adverse events in patients with melanoma receiving immune checkpoint inhibitors. Cancer immunology, immunotherapy : CII 2021, 70, 1939-1949. [CrossRef]
- Montaudié, H.; Beranger, G.E.; Reinier, F.; Nottet, N.; Martin, H.; Picard-Gauci, A.; Troin, L.; Ballotti, R.; Passeron, T. Germline variants in exonic regions have limited impact on immune checkpoint blockade clinical outcomes in advanced melanoma. Pigment cell & melanoma research 2021, 34, 978-983. [CrossRef]
- Taylor, C.A.; Watson, R.A.; Tong, O.; Ye, W.; Nassiri, I.; Gilchrist, J.J.; de Los Aires, A.V.; Sharma, P.K.; Koturan, S.; Cooper, R.A.; et al. IL7 genetic variation and toxicity to immune checkpoint blockade in patients with melanoma. Nature medicine 2022, 28, 2592-2600. [CrossRef]
- Parakh, S.; Musafer, A.; Paessler, S.; Witkowski, T.; Suen, C.; Tutuka, C.S.A.; Carlino, M.S.; Menzies, A.M.; Scolyer, R.A.; Cebon, J.; et al. PDCD1 Polymorphisms May Predict Response to Anti-PD-1 Blockade in Patients With Metastatic Melanoma. Frontiers in immunology 2021, 12, 672521. [CrossRef]
- Refae, S.; Gal, J.; Ebran, N.; Otto, J.; Borchiellini, D.; Peyrade, F.; Chamorey, E.; Brest, P.; Milano, G.; Saada-Bouzid, E. Germinal Immunogenetics predict treatment outcome for PD-1/PD-L1 checkpoint inhibitors. Investigational new drugs 2020, 38, 160-171. [CrossRef]
- de With, M.; Hurkmans, D.P.; Oomen-de Hoop, E.; Lalouti, A.; Bins, S.; El Bouazzaoui, S.; van Brakel, M.; Debets, R.; Aerts, J.; van Schaik, R.H.N.; et al. Germline Variation in PDCD1 Is Associated with Overall Survival in Patients with Metastatic Melanoma Treated with Anti-PD-1 Monotherapy. Cancers 2021, 13. [CrossRef]
- Kobayashi, M.; Numakura, K.; Hatakeyama, S.; Muto, Y.; Sekine, Y.; Sasagawa, H.; Kashima, S.; Yamamoto, R.; Koizumi, A.; Nara, T.; et al. Severe Immune-Related Adverse Events in Patients Treated with Nivolumab for Metastatic Renal Cell Carcinoma Are Associated with PDCD1 Polymorphism. Genes 2022, 13. [CrossRef]
- Suminaga, K.; Nomizo, T.; Yoshida, H.; Ozasa, H. The impact of PD-L1 polymorphisms on the efficacy of immune checkpoint inhibitors depends on the tumor proportion score: a retrospective study. Journal of cancer research and clinical oncology 2025, 151, 61. [CrossRef]
- Nomizo, T.; Ozasa, H.; Tsuji, T.; Funazo, T.; Yasuda, Y.; Yoshida, H.; Yagi, Y.; Sakamori, Y.; Nagai, H.; Hirai, T.; et al. Clinical Impact of Single Nucleotide Polymorphism in PD-L1 on Response to Nivolumab for Advanced Non-Small-Cell Lung Cancer Patients. Scientific reports 2017, 7, 45124. [CrossRef]
- Del Re, M.; Cucchiara, F.; Rofi, E.; Fontanelli, L.; Petrini, I.; Gri, N.; Pasquini, G.; Rizzo, M.; Gabelloni, M.; Belluomini, L.; et al. A multiparametric approach to improve the prediction of response to immunotherapy in patients with metastatic NSCLC. Cancer immunology, immunotherapy : CII 2021, 70, 1667-1678. [CrossRef]
- Shiota, M.; Miyake, H.; Takahashi, M.; Oya, M.; Tsuchiya, N.; Masumori, N.; Matsuyama, H.; Obara, W.; Shinohara, N.; Fujimoto, K.; et al. Effect of genetic polymorphisms on outcomes following nivolumab for advanced renal cell carcinoma in the SNiP-RCC trial. Cancer immunology, immunotherapy : CII 2023, 72, 1903-1915. [CrossRef]
- Funazo, T.Y.; Nomizo, T.; Ozasa, H.; Tsuji, T.; Yasuda, Y.; Yoshida, H.; Sakamori, Y.; Nagai, H.; Hirai, T.; Kim, Y.H. Clinical impact of low serum free T4 in patients with non-small cell lung cancer treated with nivolumab. Scientific reports 2019, 9, 17085. [CrossRef]
- Yoshida, H.; Nomizo, T.; Ozasa, H.; Tsuji, T.; Funazo, T.; Yasuda, Y.; Ajimizu, H.; Yamazoe, M.; Kuninaga, K.; Ogimoto, T.; et al. PD-L1 polymorphisms predict survival outcomes in advanced non-small-cell lung cancer patients treated with PD-1 blockade. European journal of cancer (Oxford, England : 1990) 2021, 144, 317-325. [CrossRef]
- Polcaro, G.; Liguori, L.; Manzo, V.; Chianese, A.; Donadio, G.; Caputo, A.; Scognamiglio, G.; Dell'Annunziata, F.; Langella, M.; Corbi, G.; et al. rs822336 binding to C/EBPβ and NFIC modulates induction of PD-L1 expression and predicts anti-PD-1/PD-L1 therapy in advanced NSCLC. Molecular cancer 2024, 23, 63. [CrossRef]
- Chat, V.; Ferguson, R.; Simpson, D.; Kazlow, E.; Lax, R.; Moran, U.; Pavlick, A.; Frederick, D.; Boland, G.; Sullivan, R.; et al. Autoimmune genetic risk variants as germline biomarkers of response to melanoma immune-checkpoint inhibition. Cancer immunology, immunotherapy : CII 2019, 68, 897-905. [CrossRef]
- Hurkmans, D.P.; Basak, E.A.; Schepers, N.; Oomen-De Hoop, E.; Van der Leest, C.H.; El Bouazzaoui, S.; Bins, S.; Koolen, S.L.W.; Sleijfer, S.; Van der Veldt, A.A.M.; et al. Granzyme B is correlated with clinical outcome after PD-1 blockade in patients with stage IV non-small-cell lung cancer. Journal for immunotherapy of cancer 2020, 8. [CrossRef]
- Ogimoto, T.; Ozasa, H.; Yoshida, H.; Nomizo, T.; Funazo, T.; Yoshida, H.; Hashimoto, K.; Hosoya, K.; Yamazoe, M.; Ajimizu, H.; et al. CD47 polymorphism for predicting nivolumab benefit in patients with advanced non-small-cell lung cancer. Oncology letters 2023, 26, 364. [CrossRef]
- Bins, S.; Basak, E.A.; El Bouazzaoui, S.; Koolen, S.L.W.; Oomen-de Hoop, E.; van der Leest, C.H.; van der Veldt, A.A.M.; Sleijfer, S.; Debets, R.; van Schaik, R.H.N.; et al. Association between single-nucleotide polymorphisms and adverse events in nivolumab-treated non-small cell lung cancer patients. British journal of cancer 2018, 118, 1296-1301. [CrossRef]
- Haratani, K.; Hayashi, H.; Chiba, Y.; Kudo, K.; Yonesaka, K.; Kato, R.; Kaneda, H.; Hasegawa, Y.; Tanaka, K.; Takeda, M.; et al. Association of Immune-Related Adverse Events With Nivolumab Efficacy in Non-Small-Cell Lung Cancer. JAMA oncology 2018, 4, 374-378. [CrossRef]
- Lee, S.Y.; Jung, D.K.; Choi, J.E.; Jin, C.C.; Hong, M.J.; Do, S.K.; Kang, H.G.; Lee, W.K.; Seok, Y.; Lee, E.B.; et al. Functional polymorphisms in PD-L1 gene are associated with the prognosis of patients with early stage non-small cell lung cancer. Gene 2017, 599, 28-35. [CrossRef]
- Wang, W.; Li, F.; Mao, Y.; Zhou, H.; Sun, J.; Li, R.; Liu, C.; Chen, W.; Hua, D.; Zhang, X. A miR-570 binding site polymorphism in the B7-H1 gene is associated with the risk of gastric adenocarcinoma. Human genetics 2013, 132, 641-648. [CrossRef]
- Lee, S.Y.; Jung, D.K.; Choi, J.E.; Jin, C.C.; Hong, M.J.; Do, S.K.; Kang, H.G.; Lee, W.K.; Seok, Y.; Lee, E.B.; et al. PD-L1 polymorphism can predict clinical outcomes of non-small cell lung cancer patients treated with first-line paclitaxel-cisplatin chemotherapy. Scientific reports 2016, 6, 25952. [CrossRef]
- Khan, Z.; Hammer, C.; Carroll, J.; Di Nucci, F.; Acosta, S.L.; Maiya, V.; Bhangale, T.; Hunkapiller, J.; Mellman, I.; Albert, M.L.; et al. Genetic variation associated with thyroid autoimmunity shapes the systemic immune response to PD-1 checkpoint blockade. Nature communications 2021, 12, 3355. [CrossRef]
- Postow, M.A.; Sidlow, R.; Hellmann, M.D. Immune-Related Adverse Events Associated with Immune Checkpoint Blockade. The New England journal of medicine 2018, 378, 158-168. [CrossRef]
- Russo, V.; Klein, T.; Lim, D.J.; Solis, N.; Machado, Y.; Hiroyasu, S.; Nabai, L.; Shen, Y.; Zeglinski, M.R.; Zhao, H.; et al. Granzyme B is elevated in autoimmune blistering diseases and cleaves key anchoring proteins of the dermal-epidermal junction. Scientific reports 2018, 8, 9690. [CrossRef]
- Luo, J.; Martucci, V.L.; Quandt, Z.; Groha, S.; Murray, M.H.; Lovly, C.M.; Rizvi, H.; Egger, J.V.; Plodkowski, A.J.; Abu-Akeel, M.; et al. Immunotherapy-Mediated Thyroid Dysfunction: Genetic Risk and Impact on Outcomes with PD-1 Blockade in Non-Small Cell Lung Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2021, 27, 5131-5140. [CrossRef]
- Pierini, F.; Lenz, T.L. Divergent Allele Advantage at Human MHC Genes: Signatures of Past and Ongoing Selection. Molecular biology and evolution 2018, 35, 2145-2158. [CrossRef]
- Paul, S.; Weiskopf, D.; Angelo, M.A.; Sidney, J.; Peters, B.; Sette, A. HLA class I alleles are associated with peptide-binding repertoires of different size, affinity, and immunogenicity. Journal of immunology (Baltimore, Md. : 1950) 2013, 191, 5831-5839. [CrossRef]
- Cuppens, K.; Baas, P.; Geerdens, E.; Cruys, B.; Froyen, G.; Decoster, L.; Thomeer, M.; Maes, B. HLA-I diversity and tumor mutational burden by comprehensive next-generation sequencing as predictive biomarkers for the treatment of non-small cell lung cancer with PD-(L)1 inhibitors. Lung cancer (Amsterdam, Netherlands) 2022, 170, 1-10. [CrossRef]
- Chowell, D.; Morris, L.G.T.; Grigg, C.M.; Weber, J.K.; Samstein, R.M.; Makarov, V.; Kuo, F.; Kendall, S.M.; Requena, D.; Riaz, N.; et al. Patient HLA class I genotype influences cancer response to checkpoint blockade immunotherapy. Science (New York, N.Y.) 2018, 359, 582-587. [CrossRef]
- Abed, A.; Calapre, L.; Lo, J.; Correia, S.; Bowyer, S.; Chopra, A.; Watson, M.; Khattak, M.A.; Millward, M.; Gray, E.S. Prognostic value of HLA-I homozygosity in patients with non-small cell lung cancer treated with single agent immunotherapy. Journal for immunotherapy of cancer 2020, 8. [CrossRef]
- Chhibber, A.; Huang, L.; Zhang, H.; Xu, J.; Cristescu, R.; Liu, X.; Mehrotra, D.V.; Shen, J.; Shaw, P.M.; Hellmann, M.D.; et al. Germline HLA landscape does not predict efficacy of pembrolizumab monotherapy across solid tumor types. Immunity 2022, 55, 56-64.e54. [CrossRef]
- Negrao, M.V.; Lam, V.K.; Reuben, A.; Rubin, M.L.; Landry, L.L.; Roarty, E.B.; Rinsurongkawong, W.; Lewis, J.; Roth, J.A.; Swisher, S.G.; et al. PD-L1 Expression, Tumor Mutational Burden, and Cancer Gene Mutations Are Stronger Predictors of Benefit from Immune Checkpoint Blockade than HLA Class I Genotype in Non-Small Cell Lung Cancer. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2019, 14, 1021-1031. [CrossRef]
- Chowell, D.; Krishna, C.; Pierini, F.; Makarov, V.; Rizvi, N.A.; Kuo, F.; Morris, L.G.T.; Riaz, N.; Lenz, T.L.; Chan, T.A. Evolutionary divergence of HLA class I genotype impacts efficacy of cancer immunotherapy. Nature medicine 2019, 25, 1715-1720. [CrossRef]
- Lee, C.H.; DiNatale, R.G.; Chowell, D.; Krishna, C.; Makarov, V.; Valero, C.; Vuong, L.; Lee, M.; Weiss, K.; Hoen, D.; et al. High Response Rate and Durability Driven by HLA Genetic Diversity in Patients with Kidney Cancer Treated with Lenvatinib and Pembrolizumab. Molecular cancer research : MCR 2021, 19, 1510-1521. [CrossRef]
- Lu, Z.; Chen, H.; Jiao, X.; Wang, Y.; Wu, L.; Sun, H.; Li, S.; Gong, J.; Li, J.; Zou, J.; et al. Germline HLA-B evolutionary divergence influences the efficacy of immune checkpoint blockade therapy in gastrointestinal cancer. Genome medicine 2021, 13, 175. [CrossRef]
- Correale, P.; Saladino, R.E.; Giannarelli, D.; Giannicola, R.; Agostino, R.; Staropoli, N.; Strangio, A.; Del Giudice, T.; Nardone, V.; Altomonte, M.; et al. Distinctive germline expression of class I human leukocyte antigen (HLA) alleles and DRB1 heterozygosis predict the outcome of patients with non-small cell lung cancer receiving PD-1/PD-L1 immune checkpoint blockade. Journal for immunotherapy of cancer 2020, 8. [CrossRef]
- Naranbhai, V.; Viard, M.; Dean, M.; Groha, S.; Braun, D.A.; Labaki, C.; Shukla, S.A.; Yuki, Y.; Shah, P.; Chin, K.; et al. HLA-A*03 and response to immune checkpoint blockade in cancer: an epidemiological biomarker study. The Lancet. Oncology 2022, 23, 172-184. [CrossRef]
- Takahashi, S.; Narita, S.; Fujiyama, N.; Hatakeyama, S.; Kobayashi, T.; Kato, R.; Naito, S.; Sakatani, T.; Kashima, S.; Koizumi, A.; et al. Impact of germline HLA genotypes on clinical outcomes in patients with urothelial cancer treated with pembrolizumab. Cancer science 2022, 113, 4059-4069. [CrossRef]
- Wang, L.; Zhu, Y.; Zhang, B.; Wang, X.; Mo, H.; Jiao, Y.; Xu, J.; Huang, J. Prognostic and predictive impact of neutrophil-to-lymphocyte ratio and HLA-I genotyping in advanced esophageal squamous cell carcinoma patients receiving immune checkpoint inhibitor monotherapy. Thoracic cancer 2022, 13, 1631-1641. [CrossRef]
- Ishida, Y.; Otsuka, A.; Tanaka, H.; Levesque, M.P.; Dummer, R.; Kabashima, K. HLA-A*26 Is Correlated With Response to Nivolumab in Japanese Melanoma Patients. The Journal of investigative dermatology 2017, 137, 2443-2444. [CrossRef]
- Cappelli, L.C.; Dorak, M.T.; Bettinotti, M.P.; Bingham, C.O.; Shah, A.A. Association of HLA-DRB1 shared epitope alleles and immune checkpoint inhibitor-induced inflammatory arthritis. Rheumatology (Oxford, England) 2019, 58, 476-480. [CrossRef]
- Akturk, H.K.; Couts, K.L.; Baschal, E.E.; Karakus, K.E.; Van Gulick, R.J.; Turner, J.A.; Pyle, L.; Robinson, W.A.; Michels, A.W. Analysis of Human Leukocyte Antigen DR Alleles, Immune-Related Adverse Events, and Survival Associated With Immune Checkpoint Inhibitor Use Among Patients With Advanced Malignant Melanoma. JAMA network open 2022, 5, e2246400. [CrossRef]
- Hasan Ali, O.; Berner, F.; Bomze, D.; Fässler, M.; Diem, S.; Cozzio, A.; Jörger, M.; Früh, M.; Driessen, C.; Lenz, T.L.; et al. Human leukocyte antigen variation is associated with adverse events of checkpoint inhibitors. European journal of cancer (Oxford, England : 1990) 2019, 107, 8-14. [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer discovery 2018, 8, 1156-1175. [CrossRef]
- Xia, Y.; Huang, C.; Zhong, M.; Zhong, H.; Ruan, R.; Xiong, J.; Yao, Y.; Zhou, J.; Deng, J. Targeting HGF/c-MET signaling to regulate the tumor microenvironment: Implications for counteracting tumor immune evasion. Cell communication and signaling : CCS 2025, 23, 46. [CrossRef]
- Zhang, G. Regulatory T-cells-related signature for identifying a prognostic subtype of hepatocellular carcinoma with an exhausted tumor microenvironment. Frontiers in immunology 2022, 13, 975762. [CrossRef]
- Morad, G.; Helmink, B.A.; Sharma, P.; Wargo, J.A. Hallmarks of response, resistance, and toxicity to immune checkpoint blockade. Cell 2021, 184, 5309-5337. [CrossRef]
- McGranahan, N.; Rosenthal, R.; Hiley, C.T.; Rowan, A.J.; Watkins, T.B.K.; Wilson, G.A.; Birkbak, N.J.; Veeriah, S.; Van Loo, P.; Herrero, J.; et al. Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell 2017, 171, 1259-1271.e1211. [CrossRef]
- Jhunjhunwala, S.; Hammer, C.; Delamarre, L. Antigen presentation in cancer: insights into tumour immunogenicity and immune evasion. Nature reviews. Cancer 2021, 21, 298-312. [CrossRef]
- Sade-Feldman, M.; Jiao, Y.J.; Chen, J.H.; Rooney, M.S.; Barzily-Rokni, M.; Eliane, J.P.; Bjorgaard, S.L.; Hammond, M.R.; Vitzthum, H.; Blackmon, S.M.; et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nature communications 2017, 8, 1136. [CrossRef]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (New York, N.Y.) 2017, 357, 409-413. [CrossRef]
- Dong, L.Q.; Peng, L.H.; Ma, L.J.; Liu, D.B.; Zhang, S.; Luo, S.Z.; Rao, J.H.; Zhu, H.W.; Yang, S.X.; Xi, S.J.; et al. Heterogeneous immunogenomic features and distinct escape mechanisms in multifocal hepatocellular carcinoma. Journal of hepatology 2020, 72, 896-908. [CrossRef]
- Johnson, D.B.; Nixon, M.J.; Wang, Y.; Wang, D.Y.; Castellanos, E.; Estrada, M.V.; Ericsson-Gonzalez, P.I.; Cote, C.H.; Salgado, R.; Sanchez, V.; et al. Tumor-specific MHC-II expression drives a unique pattern of resistance to immunotherapy via LAG-3/FCRL6 engagement. JCI insight 2018, 3. [CrossRef]
- Koyama, S.; Akbay, E.A.; Li, Y.Y.; Herter-Sprie, G.S.; Buczkowski, K.A.; Richards, W.G.; Gandhi, L.; Redig, A.J.; Rodig, S.J.; Asahina, H.; et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nature communications 2016, 7, 10501. [CrossRef]
- Chow, J.T.; Salmena, L. Recent advances in PTEN signalling axes in cancer. Faculty reviews 2020, 9, 31. [CrossRef]
- Dong, Y.; Richards, J.A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2014, 33, 4632-4642. [CrossRef]
- Sepúlveda, C.; Palomo, I.; Fuentes, E. Role of adenosine A2b receptor overexpression in tumor progression. Life sciences 2016, 166, 92-99. [CrossRef]
- Crunkhorn, S. Reactivating PTEN promotes antitumour immunity. Nature reviews. Drug discovery 2021, 20, 588. [CrossRef]
- Cucchiara, F.; Crucitta, S.; Petrini, I.; de Miguel Perez, D.; Ruglioni, M.; Pardini, E.; Rolfo, C.; Danesi, R.; Del Re, M. Gene-network analysis predicts clinical response to immunotherapy in patients affected by NSCLC. Lung cancer (Amsterdam, Netherlands) 2023, 183, 107308. [CrossRef]
- Trujillo, J.A.; Luke, J.J.; Zha, Y.; Segal, J.P.; Ritterhouse, L.L.; Spranger, S.; Matijevich, K.; Gajewski, T.F. Secondary resistance to immunotherapy associated with β-catenin pathway activation or PTEN loss in metastatic melanoma. Journal for immunotherapy of cancer 2019, 7, 295. [CrossRef]
- de Visser, K.E.; Joyce, J.A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer cell 2023, 41, 374-403. [CrossRef]
- Veglia, F.; Sanseviero, E.; Gabrilovich, D.I. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nature reviews. Immunology 2021, 21, 485-498. [CrossRef]
- Poggio, M.; Hu, T.; Pai, C.C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L.; et al. Suppression of Exosomal PD-L1 Induces Systemic Anti-tumor Immunity and Memory. Cell 2019, 177, 414-427.e413. [CrossRef]
- Yang, Y.; Li, C.W.; Chan, L.C.; Wei, Y.; Hsu, J.M.; Xia, W.; Cha, J.H.; Hou, J.; Hsu, J.L.; Sun, L.; et al. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell research 2018, 28, 862-864. [CrossRef]
- Hou, A.; Hou, K.; Huang, Q.; Lei, Y.; Chen, W. Targeting Myeloid-Derived Suppressor Cell, a Promising Strategy to Overcome Resistance to Immune Checkpoint Inhibitors. Frontiers in immunology 2020, 11, 783. [CrossRef]
- Kuang, D.M.; Zhao, Q.; Peng, C.; Xu, J.; Zhang, J.P.; Wu, C.; Zheng, L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma foster immune privilege and disease progression through PD-L1. The Journal of experimental medicine 2009, 206, 1327-1337. [CrossRef]
- Sprinzl, M.F.; Galle, P.R. Immune control in hepatocellular carcinoma development and progression: role of stromal cells. Seminars in liver disease 2014, 34, 376-388. [CrossRef]
- De Henau, O.; Rausch, M.; Winkler, D.; Campesato, L.F.; Liu, C.; Cymerman, D.H.; Budhu, S.; Ghosh, A.; Pink, M.; Tchaicha, J.; et al. Overcoming resistance to checkpoint blockade therapy by targeting PI3Kγ in myeloid cells. Nature 2016, 539, 443-447. [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. The New England journal of medicine 2012, 366, 2443-2454. [CrossRef]
- Song, T.L.; Nairismägi, M.L.; Laurensia, Y.; Lim, J.Q.; Tan, J.; Li, Z.M.; Pang, W.L.; Kizhakeyil, A.; Wijaya, G.C.; Huang, D.C.; et al. Oncogenic activation of the STAT3 pathway drives PD-L1 expression in natural killer/T-cell lymphoma. Blood 2018, 132, 1146-1158. [CrossRef]
- Luo, N.; Formisano, L.; Gonzalez-Ericsson, P.I.; Sanchez, V.; Dean, P.T.; Opalenik, S.R.; Sanders, M.E.; Cook, R.S.; Arteaga, C.L.; Johnson, D.B.; et al. Melanoma response to anti-PD-L1 immunotherapy requires JAK1 signaling, but not JAK2. Oncoimmunology 2018, 7, e1438106. [CrossRef]
- Dillen, A.; Bui, I.; Jung, M.; Agioti, S.; Zaravinos, A.; Bonavida, B. Regulation of PD-L1 Expression by YY1 in Cancer: Therapeutic Efficacy of Targeting YY1. Cancers 2024, 16. [CrossRef]
- Li, M.; Wei, J.; Xue, C.; Zhou, X.; Chen, S.; Zheng, L.; Duan, Y.; Deng, H.; Xiong, W.; Tang, F.; et al. Dissecting the roles and clinical potential of YY1 in the tumor microenvironment. Frontiers in oncology 2023, 13, 1122110. [CrossRef]
- Shin, D.S.; Zaretsky, J.M.; Escuin-Ordinas, H.; Garcia-Diaz, A.; Hu-Lieskovan, S.; Kalbasi, A.; Grasso, C.S.; Hugo, W.; Sandoval, S.; Torrejon, D.Y.; et al. Primary Resistance to PD-1 Blockade Mediated by JAK1/2 Mutations. Cancer discovery 2017, 7, 188-201. [CrossRef]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: from mechanism to therapy. Cell 2012, 150, 12-27. [CrossRef]
- Conte, M.; De Palma, R.; Altucci, L. HDAC inhibitors as epigenetic regulators for cancer immunotherapy. The international journal of biochemistry & cell biology 2018, 98, 65-74. [CrossRef]
- Wang, D.; Quiros, J.; Mahuron, K.; Pai, C.C.; Ranzani, V.; Young, A.; Silveria, S.; Harwin, T.; Abnousian, A.; Pagani, M.; et al. Targeting EZH2 Reprograms Intratumoral Regulatory T Cells to Enhance Cancer Immunity. Cell reports 2018, 23, 3262-3274. [CrossRef]
- Keir, M.E.; Butte, M.J.; Freeman, G.J.; Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annual review of immunology 2008, 26, 677-704. [CrossRef]
- Perrier, A.; Didelot, A.; Laurent-Puig, P.; Blons, H.; Garinet, S. Epigenetic Mechanisms of Resistance to Immune Checkpoint Inhibitors. Biomolecules 2020, 10. [CrossRef]
- Pan, D.; Kobayashi, A.; Jiang, P.; Ferrari de Andrade, L.; Tay, R.E.; Luoma, A.M.; Tsoucas, D.; Qiu, X.; Lim, K.; Rao, P.; et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (New York, N.Y.) 2018, 359, 770-775. [CrossRef]
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nature medicine 2018, 24, 556-562. [CrossRef]
- Yuan, Y.; Adam, A.; Zhao, C.; Chen, H. Recent Advancements in the Mechanisms Underlying Resistance to PD-1/PD-L1 Blockade Immunotherapy. Cancers 2021, 13. [CrossRef]
- Emran, A.A.; Chatterjee, A.; Rodger, E.J.; Tiffen, J.C.; Gallagher, S.J.; Eccles, M.R.; Hersey, P. Targeting DNA Methylation and EZH2 Activity to Overcome Melanoma Resistance to Immunotherapy. Trends in immunology 2019, 40, 328-344. [CrossRef]
- Madore, J.; Strbenac, D.; Vilain, R.; Menzies, A.M.; Yang, J.Y.; Thompson, J.F.; Long, G.V.; Mann, G.J.; Scolyer, R.A.; Wilmott, J.S. PD-L1 Negative Status is Associated with Lower Mutation Burden, Differential Expression of Immune-Related Genes, and Worse Survival in Stage III Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 3915-3923. [CrossRef]
- Robert, C.; Schachter, J.; Long, G.V.; Arance, A.; Grob, J.J.; Mortier, L.; Daud, A.; Carlino, M.S.; McNeil, C.; Lotem, M.; et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. The New England journal of medicine 2015, 372, 2521-2532. [CrossRef]
- Perez, H.L.; Cardarelli, P.M.; Deshpande, S.; Gangwar, S.; Schroeder, G.M.; Vite, G.D.; Borzilleri, R.M. Antibody-drug conjugates: current status and future directions. Drug discovery today 2014, 19, 869-881. [CrossRef]
- Naidoo, J.; Page, D.B.; Li, B.T.; Connell, L.C.; Schindler, K.; Lacouture, M.E.; Postow, M.A.; Wolchok, J.D. Toxicities of the anti-PD-1 and anti-PD-L1 immune checkpoint antibodies. Annals of oncology : official journal of the European Society for Medical Oncology 2015, 26, 2375-2391. [CrossRef]
- Spain, L.; Diem, S.; Larkin, J. Management of toxicities of immune checkpoint inhibitors. Cancer treatment reviews 2016, 44, 51-60. [CrossRef]
- Havel, J.J.; Chowell, D.; Chan, T.A. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nature reviews. Cancer 2019, 19, 133-150. [CrossRef]
- Wang, W.; Liu, J.; He, Y.; McLeod, H.L. Prospect for immune checkpoint blockade: dynamic and comprehensive monitorings pave the way. Pharmacogenomics 2017, 18, 1299-1304. [CrossRef]
- Yi, M.; Jiao, D.; Xu, H.; Liu, Q.; Zhao, W.; Han, X.; Wu, K. Biomarkers for predicting efficacy of PD-1/PD-L1 inhibitors. Molecular cancer 2018, 17, 129. [CrossRef]
- Vilain, R.E.; Menzies, A.M.; Wilmott, J.S.; Kakavand, H.; Madore, J.; Guminski, A.; Liniker, E.; Kong, B.Y.; Cooper, A.J.; Howle, J.R.; et al. Dynamic Changes in PD-L1 Expression and Immune Infiltrates Early During Treatment Predict Response to PD-1 Blockade in Melanoma. Clinical cancer research : an official journal of the American Association for Cancer Research 2017, 23, 5024-5033. [CrossRef]
- Chen, N.; Fang, W.; Zhan, J.; Hong, S.; Tang, Y.; Kang, S.; Zhang, Y.; He, X.; Zhou, T.; Qin, T.; et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer 2015, 10, 910-923. [CrossRef]
- Gainor, J.F.; Shaw, A.T.; Sequist, L.V.; Fu, X.; Azzoli, C.G.; Piotrowska, Z.; Huynh, T.G.; Zhao, L.; Fulton, L.; Schultz, K.R.; et al. EGFR Mutations and ALK Rearrangements Are Associated with Low Response Rates to PD-1 Pathway Blockade in Non-Small Cell Lung Cancer: A Retrospective Analysis. Clinical cancer research : an official journal of the American Association for Cancer Research 2016, 22, 4585-4593. [CrossRef]
- Kaufman, H.L. Precision immunology: the promise of immunotherapy for the treatment of cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 2015, 33, 1315-1317. [CrossRef]
- Rizvi, N.A.; Hellmann, M.D.; Snyder, A.; Kvistborg, P.; Makarov, V.; Havel, J.J.; Lee, W.; Yuan, J.; Wong, P.; Ho, T.S.; et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science (New York, N.Y.) 2015, 348, 124-128. [CrossRef]
- Scarpa, M.; Ruffolo, C.; Canal, F.; Scarpa, M.; Basato, S.; Erroi, F.; Fiorot, A.; Dall'Agnese, L.; Pozza, A.; Porzionato, A.; et al. Mismatch repair gene defects in sporadic colorectal cancer enhance immune surveillance. Oncotarget 2015, 6, 43472-43482. [CrossRef]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. The New England journal of medicine 2015, 372, 2509-2520. [CrossRef]
- Kawakami, H.; Zaanan, A.; Sinicrope, F.A. Microsatellite instability testing and its role in the management of colorectal cancer. Current treatment options in oncology 2015, 16, 30. [CrossRef]
- Delyon, J.; Mateus, C.; Lefeuvre, D.; Lanoy, E.; Zitvogel, L.; Chaput, N.; Roy, S.; Eggermont, A.M.; Routier, E.; Robert, C. Experience in daily practice with ipilimumab for the treatment of patients with metastatic melanoma: an early increase in lymphocyte and eosinophil counts is associated with improved survival. Annals of oncology : official journal of the European Society for Medical Oncology 2013, 24, 1697-1703. [CrossRef]
- Kumagai, S.; Koyama, S.; Itahashi, K.; Tanegashima, T.; Lin, Y.T.; Togashi, Y.; Kamada, T.; Irie, T.; Okumura, G.; Kono, H.; et al. Lactic acid promotes PD-1 expression in regulatory T cells in highly glycolytic tumor microenvironments. Cancer cell 2022, 40, 201-218.e209. [CrossRef]
- Barnes, T.A.; Amir, E. HYPE or HOPE: the prognostic value of infiltrating immune cells in cancer. British journal of cancer 2018, 118, e5. [CrossRef]
- Oliveira, G.; Stromhaug, K.; Klaeger, S.; Kula, T.; Frederick, D.T.; Le, P.M.; Forman, J.; Huang, T.; Li, S.; Zhang, W.; et al. Phenotype, specificity and avidity of antitumour CD8(+) T cells in melanoma. Nature 2021, 596, 119-125. [CrossRef]
- Luoma, A.M.; Suo, S.; Wang, Y.; Gunasti, L.; Porter, C.B.M.; Nabilsi, N.; Tadros, J.; Ferretti, A.P.; Liao, S.; Gurer, C.; et al. Tissue-resident memory and circulating T cells are early responders to pre-surgical cancer immunotherapy. Cell 2022, 185, 2918-2935.e2929. [CrossRef]
- Joyce, J.A.; Fearon, D.T. T cell exclusion, immune privilege, and the tumor microenvironment. Science (New York, N.Y.) 2015, 348, 74-80. [CrossRef]
- Feng, Z.; Puri, S.; Moudgil, T.; Wood, W.; Hoyt, C.C.; Wang, C.; Urba, W.J.; Curti, B.D.; Bifulco, C.B.; Fox, B.A. Multispectral imaging of formalin-fixed tissue predicts ability to generate tumor-infiltrating lymphocytes from melanoma. Journal for immunotherapy of cancer 2015, 3, 47. [CrossRef]
- Liu, B.; Hu, X.; Feng, K.; Gao, R.; Xue, Z.; Zhang, S.; Zhang, Y.; Corse, E.; Hu, Y.; Han, W.; et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-PD-1 therapy in lung cancer. Nature cancer 2022, 3, 108-121. [CrossRef]
- Miller, B.C.; Sen, D.R.; Al Abosy, R.; Bi, K.; Virkud, Y.V.; LaFleur, M.W.; Yates, K.B.; Lako, A.; Felt, K.; Naik, G.S.; et al. Subsets of exhausted CD8(+) T cells differentially mediate tumor control and respond to checkpoint blockade. Nature immunology 2019, 20, 326-336. [CrossRef]
- Tsui, C.; Kretschmer, L.; Rapelius, S.; Gabriel, S.S.; Chisanga, D.; Knöpper, K.; Utzschneider, D.T.; Nüssing, S.; Liao, Y.; Mason, T.; et al. MYB orchestrates T cell exhaustion and response to checkpoint inhibition. Nature 2022, 609, 354-360. [CrossRef]
- Galletti, G.; De Simone, G.; Mazza, E.M.C.; Puccio, S.; Mezzanotte, C.; Bi, T.M.; Davydov, A.N.; Metsger, M.; Scamardella, E.; Alvisi, G.; et al. Two subsets of stem-like CD8(+) memory T cell progenitors with distinct fate commitments in humans. Nature immunology 2020, 21, 1552-1562. [CrossRef]
- Beltra, J.C.; Manne, S.; Abdel-Hakeem, M.S.; Kurachi, M.; Giles, J.R.; Chen, Z.; Casella, V.; Ngiow, S.F.; Khan, O.; Huang, Y.J.; et al. Developmental Relationships of Four Exhausted CD8(+) T Cell Subsets Reveals Underlying Transcriptional and Epigenetic Landscape Control Mechanisms. Immunity 2020, 52, 825-841.e828. [CrossRef]
- Cabrita, R.; Lauss, M.; Sanna, A.; Donia, M.; Skaarup Larsen, M.; Mitra, S.; Johansson, I.; Phung, B.; Harbst, K.; Vallon-Christersson, J.; et al. Tertiary lymphoid structures improve immunotherapy and survival in melanoma. Nature 2020, 577, 561-565. [CrossRef]
- Wu, T.D.; Madireddi, S.; de Almeida, P.E.; Banchereau, R.; Chen, Y.J.; Chitre, A.S.; Chiang, E.Y.; Iftikhar, H.; O'Gorman, W.E.; Au-Yeung, A.; et al. Peripheral T cell expansion predicts tumour infiltration and clinical response. Nature 2020, 579, 274-278. [CrossRef]
- Forde, P.M.; Chaft, J.E.; Smith, K.N.; Anagnostou, V.; Cottrell, T.R.; Hellmann, M.D.; Zahurak, M.; Yang, S.C.; Jones, D.R.; Broderick, S.; et al. Neoadjuvant PD-1 Blockade in Resectable Lung Cancer. The New England journal of medicine 2018, 378, 1976-1986. [CrossRef]
- Gilbert, J.A.; Quinn, R.A.; Debelius, J.; Xu, Z.Z.; Morton, J.; Garg, N.; Jansson, J.K.; Dorrestein, P.C.; Knight, R. Microbiome-wide association studies link dynamic microbial consortia to disease. Nature 2016, 535, 94-103. [CrossRef]
- Sepich-Poore, G.D.; Zitvogel, L.; Straussman, R.; Hasty, J.; Wargo, J.A.; Knight, R. The microbiome and human cancer. Science (New York, N.Y.) 2021, 371. [CrossRef]
- Kraehenbuehl, L.; Weng, C.H.; Eghbali, S.; Wolchok, J.D.; Merghoub, T. Enhancing immunotherapy in cancer by targeting emerging immunomodulatory pathways. Nature reviews. Clinical oncology 2022, 19, 37-50. [CrossRef]
- Tawbi, H.A.; Schadendorf, D.; Lipson, E.J.; Ascierto, P.A.; Matamala, L.; Castillo Gutiérrez, E.; Rutkowski, P.; Gogas, H.J.; Lao, C.D.; De Menezes, J.J.; et al. Relatlimab and Nivolumab versus Nivolumab in Untreated Advanced Melanoma. The New England journal of medicine 2022, 386, 24-34. [CrossRef]
- Ezdoglian, A.; Tsang, A.S.M.; Khodadust, F.; Burchell, G.; Jansen, G.; de Gruijl, T.; Labots, M.; van der Laken, C.J. Monocyte-related markers as predictors of immune checkpoint inhibitor efficacy and immune-related adverse events: a systematic review and meta-analysis. Cancer metastasis reviews 2025, 44, 35. [CrossRef]
- Jeong, H.; Koh, J.; Kim, S.; Yim, J.; Song, S.G.; Kim, H.; Li, Y.; Lee, S.H.; Chung, Y.K.; Kim, H.; et al. Cell-intrinsic PD-L1 signaling drives immunosuppression by myeloid-derived suppressor cells through IL-6/Jak/Stat3 in PD-L1-high lung cancer. Journal for immunotherapy of cancer 2025, 13. [CrossRef]
- Strati, A.; Adamopoulos, C.; Kotsantis, I.; Psyrri, A.; Lianidou, E.; Papavassiliou, A.G. Targeting the PD-1/PD-L1 Signaling Pathway for Cancer Therapy: Focus on Biomarkers. International journal of molecular sciences 2025, 26. [CrossRef]
- Lu, W.; Su, J. Predictive Value of the Lung Immune Prognostic Index for Immune Checkpoint Inhibitor Therapy Outcomes in Non-Small Cell Lung Cancer: A Systematic Review and Meta-Analysis. Iranian journal of allergy, asthma, and immunology 2025, 24, 132-142. [CrossRef]
- Wang, Y.; Tong, Z.; Zhang, W.; Zhang, W.; Buzdin, A.; Mu, X.; Yan, Q.; Zhao, X.; Chang, H.H.; Duhon, M.; et al. FDA-Approved and Emerging Next Generation Predictive Biomarkers for Immune Checkpoint Inhibitors in Cancer Patients. Frontiers in oncology 2021, 11, 683419. [CrossRef]
- Man, J.; Millican, J.; Mulvey, A.; Gebski, V.; Hui, R. Response Rate and Survival at Key Timepoints With PD-1 Blockade vs Chemotherapy in PD-L1 Subgroups: Meta-Analysis of Metastatic NSCLC Trials. JNCI cancer spectrum 2021, 5. [CrossRef]
- Vallejo, J.; Singh, H.; Larkins, E.; Drezner, N.; Ricciuti, B.; Mishra-Kalyani, P.; Tang, S.; Beaver, J.A.; Awad, M.M. Impact of Increasing PD-L1 Levels on Outcomes to PD-1/PD-L1 Inhibition in Patients With NSCLC: A Pooled Analysis of 11 Prospective Clinical Trials. The oncologist 2024, 29, 422-430. [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clinical cancer research : an official journal of the American Association for Cancer Research 2014, 20, 5064-5074. [CrossRef]
- Zdrenka, M.; Kowalewski, A.; Ahmadi, N.; Sadiqi, R.U.; Chmura, Ł.; Borowczak, J.; Maniewski, M.; Szylberg, Ł. Refining PD-1/PD-L1 assessment for biomarker-guided immunotherapy: A review. Biomolecules & biomedicine 2024, 24, 14-29. [CrossRef]
- Gandhi, L.; Rodríguez-Abreu, D.; Gadgeel, S.; Esteban, E.; Felip, E.; De Angelis, F.; Domine, M.; Clingan, P.; Hochmair, M.J.; Powell, S.F.; et al. Pembrolizumab plus Chemotherapy in Metastatic Non-Small-Cell Lung Cancer. The New England journal of medicine 2018, 378, 2078-2092. [CrossRef]
- Lee, H.H.; Wang, Y.N.; Xia, W.; Chen, C.H.; Rau, K.M.; Ye, L.; Wei, Y.; Chou, C.K.; Wang, S.C.; Yan, M.; et al. Removal of N-Linked Glycosylation Enhances PD-L1 Detection and Predicts Anti-PD-1/PD-L1 Therapeutic Efficacy. Cancer cell 2019, 36, 168-178.e164. [CrossRef]
- Mei, J.; Xu, J.; Yang, X.; Gu, D.; Zhou, W.; Wang, H.; Liu, C. A comparability study of natural and deglycosylated PD-L1 levels in lung cancer: evidence from immunohistochemical analysis. Molecular cancer 2021, 20, 11. [CrossRef]
- Yin, X.; Song, Y.; Deng, W.; Blake, N.; Luo, X.; Meng, J. Potential predictive biomarkers in antitumor immunotherapy: navigating the future of antitumor treatment and immune checkpoint inhibitor efficacy. Frontiers in oncology 2024, 14, 1483454. [CrossRef]
- Meléndez, B.; Van Campenhout, C.; Rorive, S.; Remmelink, M.; Salmon, I.; D'Haene, N. Methods of measurement for tumor mutational burden in tumor tissue. Translational lung cancer research 2018, 7, 661-667. [CrossRef]
- Marcus, L.; Fashoyin-Aje, L.A.; Donoghue, M.; Yuan, M.; Rodriguez, L.; Gallagher, P.S.; Philip, R.; Ghosh, S.; Theoret, M.R.; Beaver, J.A.; et al. FDA Approval Summary: Pembrolizumab for the Treatment of Tumor Mutational Burden-High Solid Tumors. Clinical cancer research : an official journal of the American Association for Cancer Research 2021, 27, 4685-4689. [CrossRef]
- Ricciuti, B.; Wang, X.; Alessi, J.V.; Rizvi, H.; Mahadevan, N.R.; Li, Y.Y.; Polio, A.; Lindsay, J.; Umeton, R.; Sinha, R.; et al. Association of High Tumor Mutation Burden in Non-Small Cell Lung Cancers With Increased Immune Infiltration and Improved Clinical Outcomes of PD-L1 Blockade Across PD-L1 Expression Levels. JAMA oncology 2022, 8, 1160-1168. [CrossRef]
- Florou, V.; Floudas, C.S.; Maoz, A.; Naqash, A.R.; Norton, C.; Tan, A.C.; Sokol, E.S.; Frampton, G.; Soares, H.P.; Puri, S.; et al. Real-world pan-cancer landscape of frameshift mutations and their role in predicting responses to immune checkpoint inhibitors in cancers with low tumor mutational burden. Journal for immunotherapy of cancer 2023, 11. [CrossRef]
- Georgoulias, G.; Zaravinos, A. Genomic landscape of the immunogenicity regulation in skin melanomas with diverse tumor mutation burden. Frontiers in immunology 2022, 13, 1006665. [CrossRef]
- Bonneville, R.; Krook, M.A.; Kautto, E.A.; Miya, J.; Wing, M.R.; Chen, H.Z.; Reeser, J.W.; Yu, L.; Roychowdhury, S. Landscape of Microsatellite Instability Across 39 Cancer Types. JCO precision oncology 2017, 2017. [CrossRef]
- Marabelle, A.; O'Malley, D.M.; Hendifar, A.E.; Ascierto, P.A.; Motola-Kuba, D.; Penel, N.; Cassier, P.A.; Bariani, G.; De Jesus-Acosta, A.; Doi, T.; et al. Pembrolizumab in microsatellite-instability-high and mismatch-repair-deficient advanced solid tumors: updated results of the KEYNOTE-158 trial. Nature cancer 2025, 6, 253-258. [CrossRef]
- Fan, A.; Wang, B.; Wang, X.; Nie, Y.; Fan, D.; Zhao, X.; Lu, Y. Immunotherapy in colorectal cancer: current achievements and future perspective. International journal of biological sciences 2021, 17, 3837-3849. [CrossRef]
- McGrail, D.J.; Pilié, P.G.; Rashid, N.U.; Voorwerk, L.; Slagter, M.; Kok, M.; Jonasch, E.; Khasraw, M.; Heimberger, A.B.; Ueno, N.T.; et al. Validation of cancer-type-dependent benefit from immune checkpoint blockade in TMB-H tumors identified by the FoundationOne CDx assay. Annals of oncology : official journal of the European Society for Medical Oncology 2022, 33, 1204-1206. [CrossRef]
- Mucherino, S.; Lorenzoni, V.; Triulzi, I.; Del Re, M.; Orlando, V.; Capuano, A.; Danesi, R.; Turchetti, G.; Menditto, E. Cost-Effectiveness of Treatment Optimisation with Biomarkers for Immunotherapy in Solid Tumours: A Systematic Review. Cancers 2024, 16. [CrossRef]
- Zhang, C.; Zhang, C.; Wang, H. Immune-checkpoint inhibitor resistance in cancer treatment: Current progress and future directions. Cancer letters 2023, 562, 216182. [CrossRef]
- Bell, H.N.; Zou, W. Beyond the Barrier: Unraveling the Mechanisms of Immunotherapy Resistance. Annual review of immunology 2024, 42, 521-550. [CrossRef]
- Heinhuis, K.M.; Ros, W.; Kok, M.; Steeghs, N.; Beijnen, J.H.; Schellens, J.H.M. Enhancing antitumor response by combining immune checkpoint inhibitors with chemotherapy in solid tumors. Annals of oncology : official journal of the European Society for Medical Oncology 2019, 30, 219-235. [CrossRef]
- Moya-Horno, I.; Viteri, S.; Karachaliou, N.; Rosell, R. Combination of immunotherapy with targeted therapies in advanced non-small cell lung cancer (NSCLC). Therapeutic advances in medical oncology 2018, 10, 1758834017745012. [CrossRef]
- Starzer, A.M.; Wolff, L.; Popov, P.; Kiesewetter, B.; Preusser, M.; Berghoff, A.S. The more the merrier? Evidence and efficacy of immune checkpoint- and tyrosine kinase inhibitor combinations in advanced solid cancers. Cancer treatment reviews 2024, 125, 102718. [CrossRef]
- Wu, Q.; Jiang, L.; Li, S.C.; He, Q.J.; Yang, B.; Cao, J. Small molecule inhibitors targeting the PD-1/PD-L1 signaling pathway. Acta pharmacologica Sinica 2021, 42, 1-9. [CrossRef]
- Uzar, W.; Kaminska, B.; Rybka, H.; Skalniak, L.; Magiera-Mularz, K.; Kitel, R. An updated patent review on PD-1/PD-L1 antagonists (2022-present). Expert opinion on therapeutic patents 2024, 34, 627-650. [CrossRef]
- Hao, X.; Chen, Z.; Li, H.; Wei, M.; Zuo, Z.; Su, Q. Small-Molecule Drugs in Immunotherapy. Mini reviews in medicinal chemistry 2023, 23, 1341-1359. [CrossRef]
- Li, C.; Zhang, N.; Zhou, J.; Ding, C.; Jin, Y.; Cui, X.; Pu, K.; Zhu, Y. Peptide Blocking of PD-1/PD-L1 Interaction for Cancer Immunotherapy. Cancer immunology research 2018, 6, 178-188. [CrossRef]
- Kamijo, H.; Sugaya, M.; Takahashi, N.; Oka, T.; Miyagaki, T.; Asano, Y.; Sato, S. BET bromodomain inhibitor JQ1 decreases CD30 and CCR4 expression and proliferation of cutaneous T-cell lymphoma cell lines. Archives of dermatological research 2017, 309, 491-497. [CrossRef]
- Liu, M.; Cao, Z.; Zhang, R.; Chen, Y.; Yang, X. Injectable Supramolecular Hydrogel for Locoregional Immune Checkpoint Blockade and Enhanced Cancer Chemo-Immunotherapy. ACS applied materials & interfaces 2021, 13, 33874-33884. [CrossRef]
- Shafi, H.; Lora, A.J.; Donow, H.M.; Dickinson, S.E.; Wondrak, G.T.; Chow, H.S.; Curiel-Lewandrowski, C.; Mansour, H.M. Comprehensive Advanced Physicochemical Characterization and In Vitro Human Cell Culture Assessment of BMS-202: A Novel Inhibitor of Programmed Cell Death Ligand. Pharmaceutics 2024, 16. [CrossRef]
- Sasikumar, P.G.; Sudarshan, N.S.; Adurthi, S.; Ramachandra, R.K.; Samiulla, D.S.; Lakshminarasimhan, A.; Ramanathan, A.; Chandrasekhar, T.; Dhudashiya, A.A.; Talapati, S.R.; et al. PD-1 derived CA-170 is an oral immune checkpoint inhibitor that exhibits preclinical anti-tumor efficacy. Communications biology 2021, 4, 699. [CrossRef]
- Chen, S.; Wang, Q.; Ming, S.; Zheng, H.; Hua, B.; Yang, H.S. Platycodin D induces apoptosis through JNK1/AP-1/PUMA pathway in non-small cell lung cancer cells: A new mechanism for an old compound. Frontiers in pharmacology 2022, 13, 1045375. [CrossRef]
- Wang, H.; Yao, H.; Li, C.; Shi, H.; Lan, J.; Li, Z.; Zhang, Y.; Liang, L.; Fang, J.Y.; Xu, J. HIP1R targets PD-L1 to lysosomal degradation to alter T cell-mediated cytotoxicity. Nature chemical biology 2019, 15, 42-50. [CrossRef]
- Salih, D.J.; Barsoom, S.H.; Ahmed, G.F.; Hussien, S.Q.; Al Ismaeel, Q.; Alasady, A.A.B.; Alsalim, T.A.; Salih, A.M. Curcumin inhibits IFN-γ induced PD-L1 expression via reduction of STAT1 Phosphorylation in A549 non-small cell lung cancer cells. Saudi pharmaceutical journal : SPJ : the official publication of the Saudi Pharmaceutical Society 2025, 33, 16. [CrossRef]
- Amato, L.; De Rosa, C.; Di Guida, G.; Sepe, F.; Ariano, A.; Capaldo, S.; Ul Haq, F.; Di Liello, A.; Tuccillo, C.; Lucà, S.; et al. Addition of metformin to anti-PD-1/PD-L1 drugs activates anti-tumor immune response in peripheral immune cells of NSCLC patients. Cell death & disease 2025, 16, 286. [CrossRef]
- Ferrario, C.; Mackey, J.; Gelmon, K.A.; Levasseur, N.; Sorensen, P.H.; Oo, H.Z.; Negri, G.L.; Tse, V.W.L.; Spencer, S.E.; Cheng, G.; et al. Phase Ib Pharmacodynamic Study of the MNK Inhibitor Tomivosertib (eFT508) Combined With Paclitaxel in Patients With Refractory Metastatic Breast Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2025, 31, 491-502. [CrossRef]
- Riess, J.W.; de Langen, A.J.; Ponce, S.; Goldberg, S.B.; Piotrowska, Z.; Goldman, J.W.; Le, X.; Cho, B.C.; Yoneshima, Y.; Ambrose, H.; et al. ORCHARD: Osimertinib Plus Necitumumab in Patients With Epidermal Growth Factor Receptor-Mutated Advanced Non-Small Cell Lung Cancer With a Secondary Epidermal Growth Factor Receptor Alteration Whose Disease Had Progressed on First-Line Osimertinib. JCO precision oncology 2025, 9, e2400818. [CrossRef]
- Pan, X.; Li, C.; Feng, J. The role of LncRNAs in tumor immunotherapy. Cancer cell international 2023, 23, 30. [CrossRef]
- Zhou, J.G.; Liang, B.; Liu, J.G.; Jin, S.H.; He, S.S.; Frey, B.; Gu, N.; Fietkau, R.; Hecht, M.; Ma, H.; et al. Identification of 15 lncRNAs Signature for Predicting Survival Benefit of Advanced Melanoma Patients Treated with Anti-PD-1 Monotherapy. Cells 2021, 10. [CrossRef]
- Toker, J.; Iorgulescu, J.B.; Ling, A.L.; Villa, G.R.; Gadet, J.; Parida, L.; Getz, G.; Wu, C.J.; Reardon, D.A.; Chiocca, E.A.; et al. Clinical Importance of the lncRNA NEAT1 in Cancer Patients Treated with Immune Checkpoint Inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research 2023, 29, 2226-2238. [CrossRef]
- Diao, M.; Wang, Y.; Wu, S.; He, S.; Liao, Y. Estrogen, estrogen receptor and the tumor microenvironment of NSCLC. International journal of cancer 2025, 156, 1501-1508. [CrossRef]
- Ontiveros, C.O.; Murray, C.E.; Crossland, G.; Curiel, T.J. Considerations and Approaches for Cancer Immunotherapy in the Aging Host. Cancer immunology research 2023, 11, 1449-1461. [CrossRef]
- Duffy, M.J.; Crown, J. Biomarkers for Predicting Response to Immunotherapy with Immune Checkpoint Inhibitors in Cancer Patients. Clinical chemistry 2019, 65, 1228-1238. [CrossRef]
- Doroshow, D.B.; Bhalla, S.; Beasley, M.B.; Sholl, L.M.; Kerr, K.M.; Gnjatic, S.; Wistuba, II; Rimm, D.L.; Tsao, M.S.; Hirsch, F.R. PD-L1 as a biomarker of response to immune-checkpoint inhibitors. Nature reviews. Clinical oncology 2021, 18, 345-362. [CrossRef]
- Chennamadhavuni, A.; Abushahin, L.; Jin, N.; Presley, C.J.; Manne, A. Risk Factors and Biomarkers for Immune-Related Adverse Events: A Practical Guide to Identifying High-Risk Patients and Rechallenging Immune Checkpoint Inhibitors. Frontiers in immunology 2022, 13, 779691. [CrossRef]
Figure 1.
A timetable with the FDA ICIs approved.
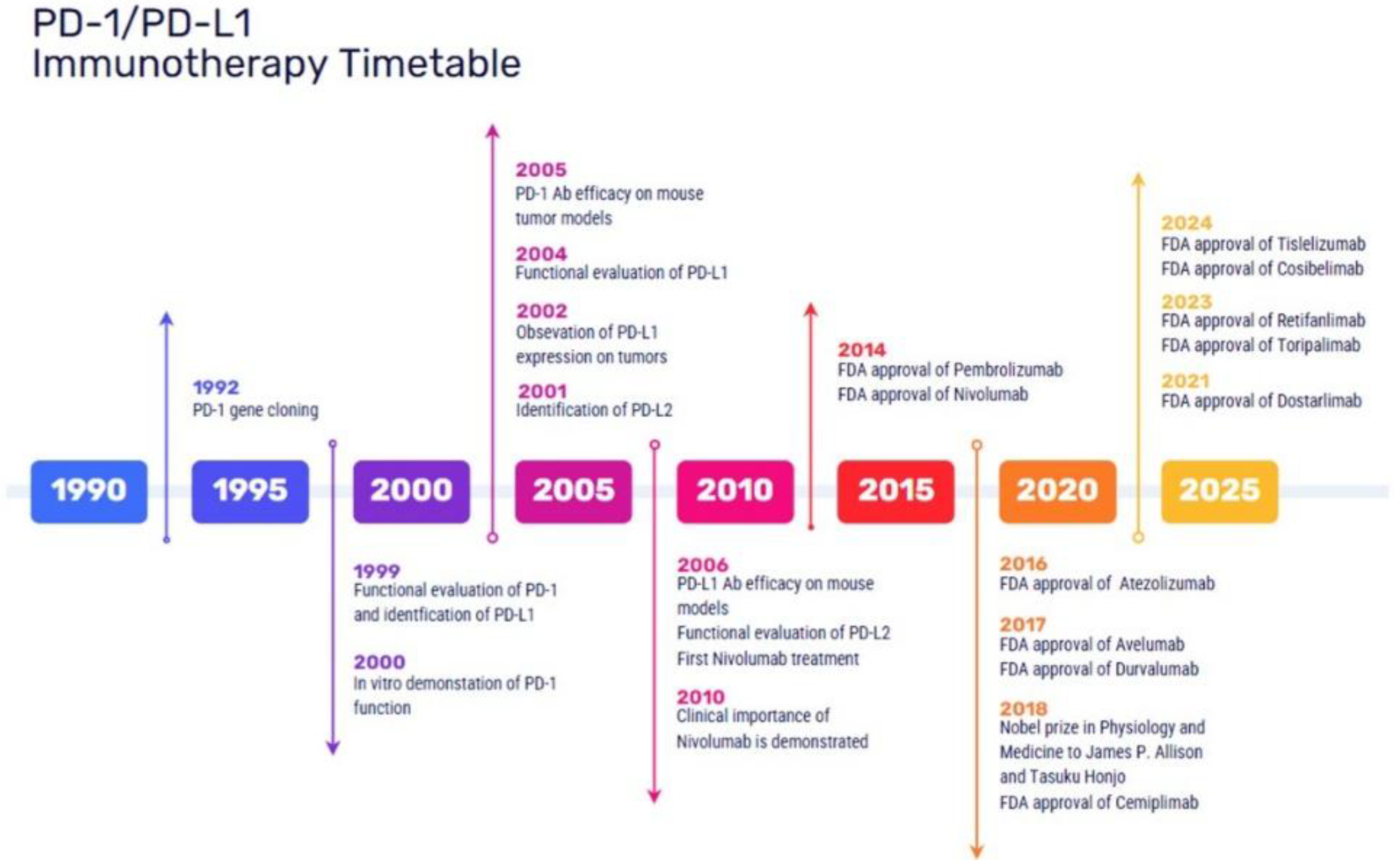
Figure 2.
Mechanism of action of PD-1 PD-L1 inhibitors against cancer cells. TCR: T cell receptor; MHC: major histocompatibility complex; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; anti-PD-1: monoclonal antibody against PD-1; anti-PD-L1: monoclonal antibody against PD-L1.
Figure 2.
Mechanism of action of PD-1 PD-L1 inhibitors against cancer cells. TCR: T cell receptor; MHC: major histocompatibility complex; PD-1: programmed cell death protein 1; PD-L1: programmed death-ligand 1; anti-PD-1: monoclonal antibody against PD-1; anti-PD-L1: monoclonal antibody against PD-L1.
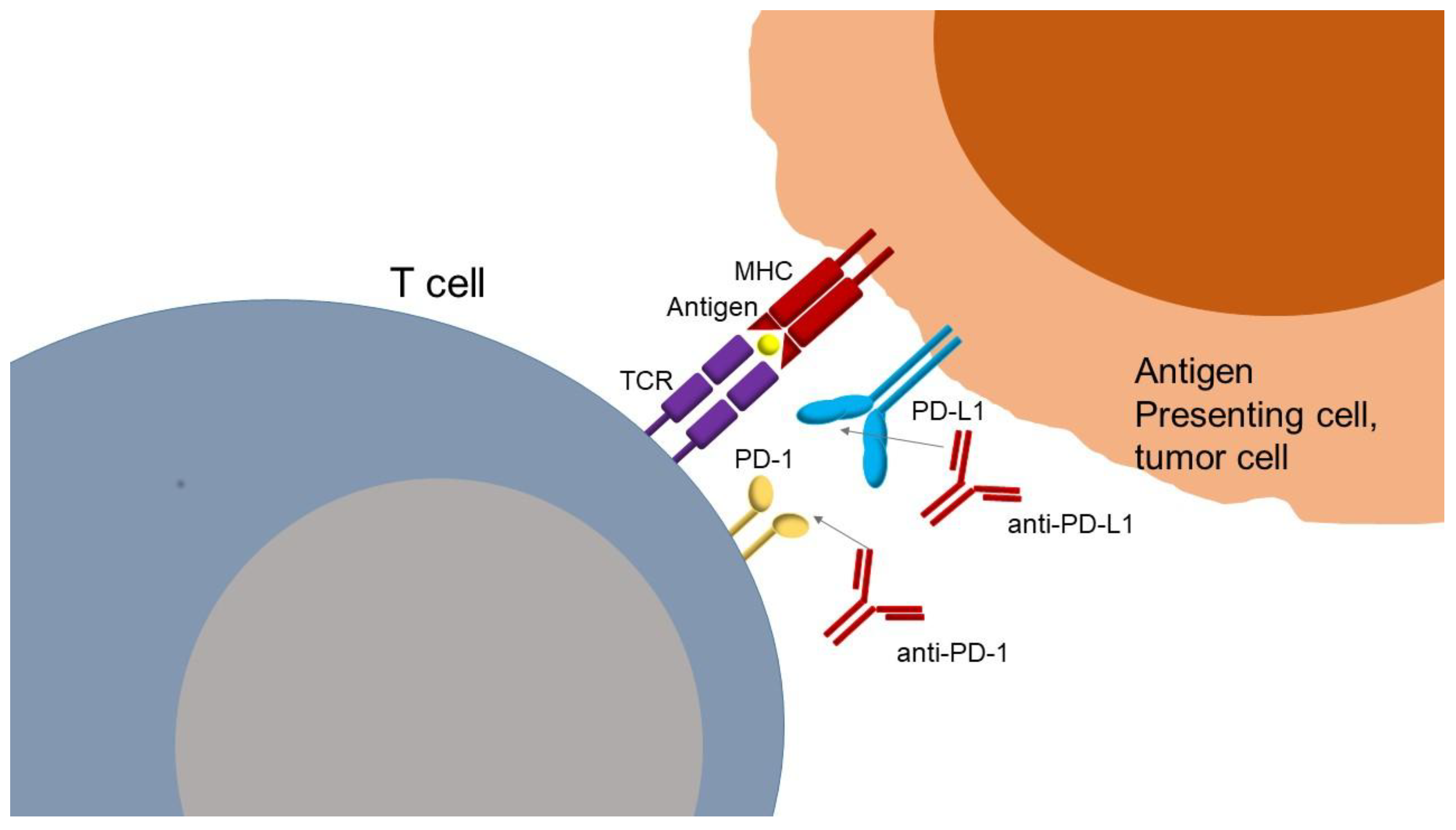
Figure 3.
HLA and response to ICI treatment. HLA: human leukocyte antigen; DP, DQ, DR: Class II HLA molecules; HLA-A, HLA-B, HLA-C: Class I HLA molecules; Class III: region encoding complement and other immune-related proteins.
Figure 3.
HLA and response to ICI treatment. HLA: human leukocyte antigen; DP, DQ, DR: Class II HLA molecules; HLA-A, HLA-B, HLA-C: Class I HLA molecules; Class III: region encoding complement and other immune-related proteins.
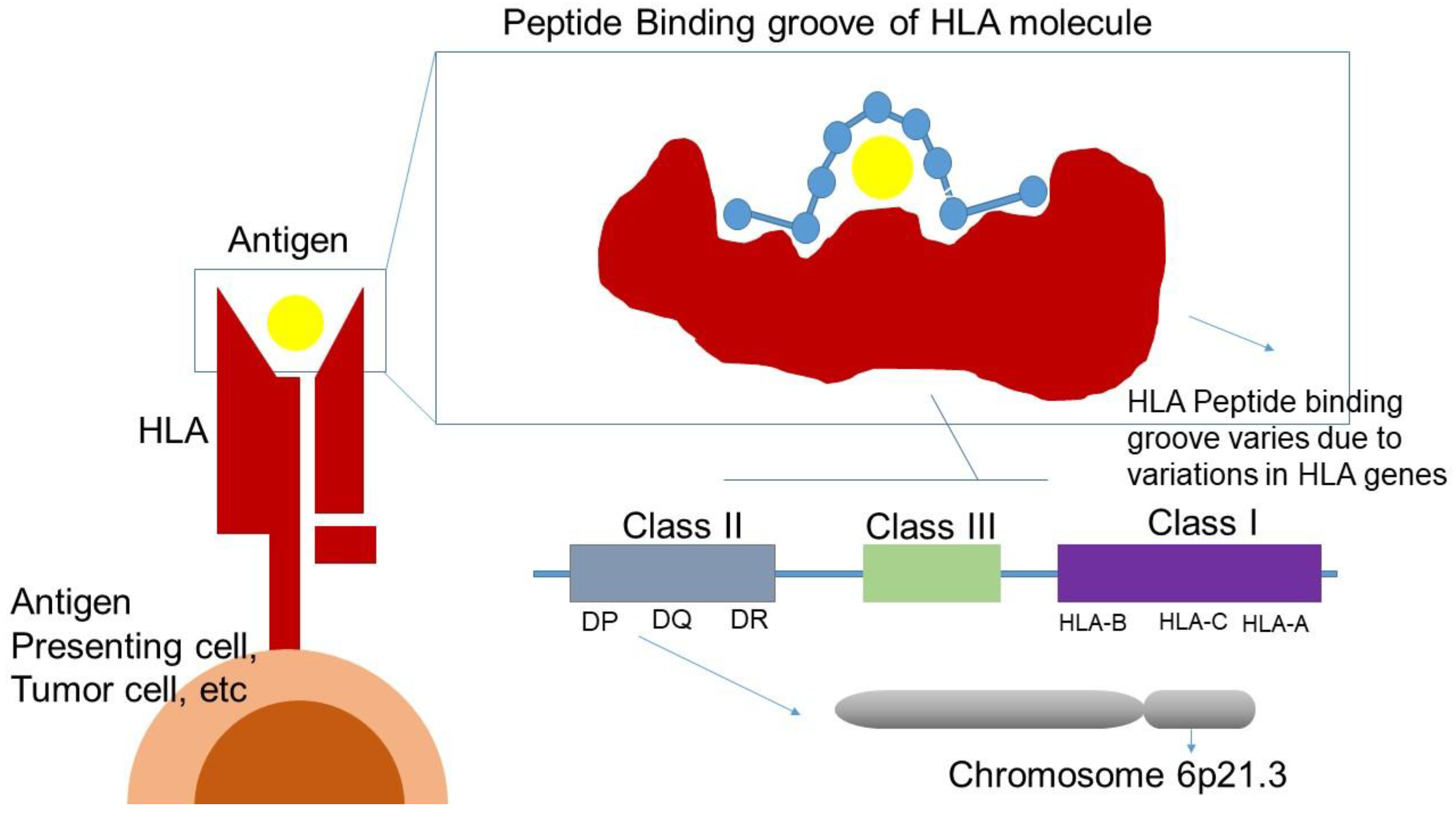
Table 1.
The PD-1 and PD-L1 inhibitors that have been already approved by FDA for the treatment of various types of cancer.
Table 1.
The PD-1 and PD-L1 inhibitors that have been already approved by FDA for the treatment of various types of cancer.
| PD-1 inhbitors FDA approved | Cancer type | Clinical trial |
| Nivolumab (OPDIVO) |
Melanoma, NSCLC, gastric, squamous-cell esophageal/gastroesophageal junction adenocarcinoma MSI-H/dMMMR or TMB-H Solid tumors, hepatocelllular, renal cell, urothelial, head & necksquamous cell, lymphomas |
CheckMate trials |
| Pembrolizumab (KEYTRUDA) |
Melanoma, Merckel-cell cutaneous squamous cell, NSCLC, triple-negative breast cancer, gastric, esophageal/gastroesophageal junction adenocarcinoma, MSI-H/dMMMR or TMB-H Solid tumors, hepatocellular, cervical, endometrial, renal, urothelial, head & necksquamous cell, |
KEYNOTE trials |
| Cemiplimab (LIBTAYO) |
Cutaneous squamous cell, basal cell NSLCC, | Studies 1540, 1620, EMPOWER-lung 1 |
| Dostarlimab (JEMPERLI) |
MSI-H/dMMMR or TMB-H Solid tumors, endometrial | GARNET trials |
| Tislelizumab (TEVIMBRA) |
Gastric Cancer; Gastro-Oesophageal Junction Cancer; Oesophageal Cancer, NSCLC, Oesophageal squamous cell carcinoma |
RATIONALE TRIAL |
| Retifanlimab (ZYNYZ) |
Merkel cell carcinoma | POD1UM-203 |
| Toripalimab (LOQTORZI) |
nasopharyngeal carcinoma | JUPITER-02 |
| PD-L1 inhbitors FDA approved | Cancer type | Clinical trial |
| Atezolizumab (TECENTRIQ) |
Melanoma, NSCLC, triple-negative breast, hepatocellular cancers |
IMspire, ImPower trials, POPLAR, OAK, IMBrave |
| Avelumab (BAVENCIO) |
Merkel-cell, renal cell, urothelial cancers | JAVELIN trials |
| Durvalumab (IMFINZI) |
NSCLC | PACIFIC, CASPIAN |
| Cosibelimab (UNLOXCYT) |
cutaneous squamous cell carcinoma | Study CK-301-101 |
Table 3.
SNPs associated with ICI therapy.
| Gene | Variant | Treatment | Observable result | No. of patients/Ethnicity/Cancer Type | Ref. |
| PDCD1 |
7146A>G (rs11568821) | Nivolumab or Pembrolizumab | BOR: GG vs AG CR vs other response rate (16.5 % vs. 2.6 %), PFS:GG longer PFS 0.05 (95 %CI 0.003–0.87; p = 0.040) | 115 Australian patients with metastatic melanoma | [129] |
| anti-PD-1, anti-PD-L1 | BOR: No relationship with hyperprogressive disease | Cohort of 98 French patients with various types of cancer | [146] | ||
| 804C>T (rs2227981) | anti-PD-1 monotherapy (i.e., PemBrolizumab or Nivolumab) | OS: poorer OS with a 3-year OS rate of 51.8%, as compared to 71% in wild type patients, HR: 2.37; 95% CI: 1.11–5.04; p = 0.026) | 119 patients of Caucasian decent with metastatic melanoma | [130] | |
| anti-PD-1, anti-PD-L1 | BOR: No relationship with hyperprogressive disease | Cohort of 98 French patients with various types of cancer | [146] | ||
| 889G>A (rs10204525) | Nivolumab or Nivolumab+iPilimumab | G allele carriers had more AE (OR: 3.712 CI95% (1.591 – 8.658); p = 0.002) | 106 patients from Japan with metastatic RCC | [128] | |
| anti-PD-1, anti-PD-L1 | BOR: No relationship with hyperprogressive disease | Cohort of 98 French patients with various types of cancer | [146] | ||
| CD274 | rs2282055 T/T | Nivolumab, Atezolizumab, Pembrolizumab | PFS: better PFS for TPS-negative population, T/T vs G/T and G/G p=0.008 | 104 Japanese patients with NSCLC | [139] |
| 395G>C (rs4143815) | Nivolumab | BOR: better clinical response of G vs C allele (p = 0.0319), PFS: CC + CG vs GG: 2.6 vs 2.1 m (HR: 0.46 (95 %-CI: 0.22–1.04); p = 0.0438) | 50 patients from Japan with NSCLC | [126] | |
| Nivolumab or Pembrolizumab | PFS: CC + CG vs GG: not reached vs 2.3 m (p = 0.41; n = 20, nivolumab only) | 32 Italian patients with NSCLC | [132] | ||
| anti-PD-1, anti-PD-L1 | No relationship with hyperprogressive disease | Cohort of 98 French patients with various types of cancer | [146] | ||
| Nivolumab | PFS: GG: HR: 1.69, 95 % CI 1.09–2.59; p = 0.018 | 222 Japanese patients with advanced RCC | [127] | ||
| rs1411262 (T>C) | Nivolumab | PFS: HR 1.65 (p = 0.040), rAEs: Low fT4 or liver dysfunction or rash or fever, p = 0.0013 | 111 Japanese patients with NSCLC | [133] | |
| Nivolumab | OS: T/T vs C/T or C/C HR 0.40 (0.21–0.70) p = 0.017 | 133 Japanese patients with NSCLC | [125] | ||
| Nivolumab | PFS: TT + TC: HR: 0.58, 95 % CI 0.37–0.89; P = 0.014 | 222 Japanese patients with advanced RCC | [127] | ||
| rs822339 (A>G) | Nivolumab | PFS: HR 1.76 (p = 0.025), rAEs:low fT4 or liver dysfunction or rash or fever, p = 0.0204 | 111 Japanese patients with NSCLC | [133] | |
| Nivolumab | OS: A/A vs A/G or G/G HR 0.38 CI 95%(0.19–0.69) p < 0.001 | 133 Japanese patients with NSCLC | [125] | ||
| Nivolumab | PFS: GG: HR: 1.59, 95 % CI 1.04–2.45; P = 0.034 | 222 Japanese patients with advanced RCC | [127] | ||
| rs822336 | Nivolumab, Pembrolizumab or Atezolizumab following platinum-based chemotherapy, Pembrolizumab + platinum-based chemotherapy |
a= C/C vs C/G and G/G better ORR (P = 0.004), PFS (P = 0.003) and OS (P = 0.002), ss in multivariate models:PFS HR 0.5163762 95% CI (0.3241144–0.8226861) p=0.005, OS: HR 0.5424147 95% CI (0.3408916–0.8630712)p= 0.010, b= C/C vs C/G and G/G better PFS (P = 0.0258) and OS (P = 0.0455) | 2 cohorts (a=n44, b=n19) of Caucasian populations with advanced non-oncogene addicted NSCLC | [138] | |
| FARP1 |
rs685736 | Nivolumab | BOR: OR, 3.82; 95 % CI 2.17–6.70; P < 0.0001, PFS:GA + AA: HR, 1.67; 95 % CI 1.18–2.38; P = 0.0041 | 222 Japanese patients with advanced RCC | [127] |
| rs643869 | Nivolumab | BOR: OR, 0.23; 95 % CI 0.13–0.41; P < 0.0001, PFS: CC: HR, 0.57; 95 % CI 0.40–0.80; P = 0.0013 | 222 Japanese patients with advanced RCC | [127] | |
| IL2, ADAD1, IL21 | rs17388568 | anti-PD-1 (Nivolumab, Pembrolizumab; N = 176) | BOR: Responders (CR, PR, SD) vs. non responders (PD) OR: 0.26, 95%CI(0.12 - 0.53) (GA or AA vs GG 74% less likely to resist anti-PD-1 treatment | 169 patients from America with metastatic melanoma | [142] |
| IL1RL1 | rs4988956 | Pembrolizumab, Nivolumab, Ipilimumab, Nivolumab +Ipilimumab |
BOR: Better response (p = 5.4E-02) in A/A and A/R vs R/R genotypes | GWAS study in 57 patients in France with metastatic melanoma, Validation occured in another cohort of 57 patients. | [123] |
| IL7 | rs16906115 | Immune checkpoint inhibitors | Increased all grade toxicity (HR: 2.1; p = 36E-11) | GWAS study in 1751 patients, 12 different tumour types | [121] |
| GZMB | c.128C>A (rs8192917) | Nivolumab | BOR: (CR/PR vs SD vs PD) OR 1.60 95%CI (1.01–2.52) p=0.044, PFS:HR: 1.38; 95% CI:1.02 to 1.87; p=0.036 | 322 mostly Caucasian patients with NSCLC | [144] |
| CD47 | rs3804639 | Nivolumab | PFS: GG longer PFS (HR 0.70; p = 0.026), OS:GG longer OS (HR 0.64; p = 0.021) | 164 Japanese patients with NSCLC | [145] |
Table 4.
Small molecule inhibitors of PD-1, PD-L1 as possible cancer treatment.
| Small Molecule Inhibitors | Site of action | Research phase | Cancer type | Ref. |
| AUNP-12 | Binding of PD-1 to PD-L1 | preclinical | Melanoma | [269] |
| DPPA-1 | preclinical | Solid Tumors | [274] | |
| TPP-1 | preclinical | LCLC | [272] | |
| BMS-202 | preclinical | Cancer | [275] | |
| CA-170 | clinical | NSCLC | [276] | |
| INCB086550 | clinical | NSCLC, UC, RCC, HCC, Melanoma | [270] | |
| INCB099280 | clinical | cSCC | [270] | |
| INCB099318 + axitinib |
clinical | Advanced Solid Tumors | [270] | |
| JQ1 | Expression of PD-L1 | clinical | Lymphoma | [273] |
| eFT508 Tomivosertib |
clinical | Breast cancer | [289] | |
| Osimertinib | clinical | NSCLC | [290] | |
| Platycodin D | preclinical | NSCLC | [277] | |
| PD-LYSO | Degradation of PD-L1 | preclinical | Cancer | [278] |
| Curcumin | preclinical | Cancer | [279] | |
| Metformin | clinical | NSCLC | [280] |
Table 5.
Recent ongoing clinical trials phase III (2024 -2025) for ICIs cancer therapy (www.clinicaltrials.gov).
Table 5.
Recent ongoing clinical trials phase III (2024 -2025) for ICIs cancer therapy (www.clinicaltrials.gov).
| Immunotherapy | Cancer type | Identifier | Clinical trial |
| V940 + Pembrolizumab | Cutaneous Squamous Cell Carcinoma | NCT06295809 | V940-007 |
| V940 + Pembrolizumab | High-Risk Melanoma | NCT05933577 | V940-001 |
| Pembrolizumab + chemoradiation therapy followed by pembrolizumab with or without olaparib | Stage III Non-Small Cell Lung Cancer | NCT04380636 | MK-7339-012/KEYLYNK-012 |
| Carboplatin-paclitaxel + retifanlimab or placebo | Advanced or Metastatic Squamous Cell Anal Carcinoma | NCT04472429 | POD1UM-303/InterAACT 2 |
| Safety and Efficacy of Lenvatinib (E7080/MK-7902) + Pembrolizumab (MK-3475) in Combination with Transarterial Chemoembolization (TACE) | Incurable/Non-metastatic Hepatocellular Carcinoma | NCT04246177 | MK-7902-012/E7080-G000-318/LEAP-012 |
| Rucaparib + nivolumab following response to front-line platinum-based chemotherapy | Ovarian Cancer | NCT03522246 | ATHENA |
| Regorafenib + nivolumab (RegoNivo) vs Standard of Care Chemotherapy | Advanced gastro-oesophageal cancer | NCT04879368 | INTEGRATEIIb |
| Sitravatinib ± nivolumab, pembrolizumab, enfortumab vedotin-ejfv, ipilimumab | Advanced or Metastatic Solid Malignancies | NCT04887870 | CA248-0003 |
| Nivolumab-relatlimab vs regorafenib or TAS-102 | Metastatic Colorectal Cancer |
NCT05328908 | RELATIVITY-123 |
| IO102-IO103 + Pembrolizumab vs Pembrolizumab | Advanced Melanoma | NCT05155254 | IOB-013 / KN-D18 |
| Nemvaleukin Alfa + Pembrolizumab | Platinum-Resistant Epithelial Ovarian Cancer | NCT05092360 | ARTISTRY-7 |
| Nivolumab + Relatlimab vs Nivolumab | After Complete Resection of Stage III-IV Melanoma | NCT05002569 | RELATIVITY-098 |
| Ociperlimab + Tislelizumab vs Pembrolizumab |
Untreated Lung Cancer | NCT04746924 | AdvanTIG-302 |
| Cemiplimab vs Placebo After Surgery and Radiation Therapy | High Risk Cutaneous Squamous Cell Carcinoma | NCT03969004 | R2810-ONC-1788 |
| Cabozantinib + Nivolumab + Ipilimumab | Untreated Advanced or Metastatic Renal Cell Carcinoma | NCT03937219 | COSMIC-313 |
| Chemotherapy vs Chemotherapy + Nivolumab vs Nivolumab + BMS-986205 | Muscle Invasive Bladder Cancer | NCT03661320 | CA017-078 |
| PD-L1 inhbitors | Cancer type | Clinical trial | |
| Durvalumab and Tremelimumab | Advanced Hepatocellular Carcinoma | NCT05883644 | SIERRA |
| XL092 + Atezolizumab vs Regorafenib | Metastatic Colorectal Cancer | NCT05425940 | STELLAR-303 |
| Patient-Specific Neoantigen Vaccine + ICI | Metastatic Colorectal Cancer | NCT05141721 | GO-010 |
| Lurbinectedin + Atezolizumab vs Atezolizumab |
Extensive-Stage Small-Cell Lung Cancer | NCT05091567 | IMforte |
| Durvalumab +Tremelimumab + Enfortumab Vedotin or Durvalumab + Enfortumab Vedotin | Muscle Invasive Bladder Cancer ineligible to Cisplatin | NCT04960709 | VOLGA |
| Atezolizumab + Tiragolumab vs Durvalumab |
Locally Advanced, Unresectable Stage III Non-Small Cell Lung Cancer | NCT04513925 | SKYSCRAPER-03 |
| Durvalumab+ Gemcitabine/Cisplatin and Durvalumab With MIBC | Muscle-Invasive Bladder Cancer. | NCT03732677 | NIAGARA |
| Durvalumab + Chemotherapy vs Durvalumab + Tremelimumab + Chemotherapy vs Chemotherapy | Unresectable Urothelial Cancer | NCT03682068 | NILE |
| Both | |||
| Pembrolizumab/Vibostolimab (MK-7684A) or Atezolizumab + Chemotherapy | Extensive-Stage Small Cell Lung Cancer | NCT05224141 | MK-7684A-008, KEYVIBE-008 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



