
Submitted:
24 June 2025
Posted:
24 June 2025
You are already at the latest version
Abstract
SNX11, a sorting nexin protein localized on the endosomal membrane, is an important protein closely related to protein sorting and endosomal trafficking. Previously, through a genome-wide CRISPR screening, we identified SNX11 as a critical protein for the entry of Dabie bandavirus. SNX11-deletion significantly inhibits the replication of Dabie bandavirus. We further discovered that the loss of SNX11 alters endosomal pH, potentially affecting the release process of Dabie bandavirus from endosomes to the cytoplasm. However, the mechanism by which SNX11 modulates endosomal pH and whether SNX11-deletion similarly inhibits other viruses remain to be elucidated. This study reveals that SNX11 can interact with the V1 subunit of the endosomal proton pump V-ATPase, affecting the expression level of this subunit on the endosomal membrane and thereby disrupting the assembly of V-ATPase. Additionally, we found that SNX11-deletion significantly inhibits the replication of dengue virus, hantavirus, and influenza virus. These findings suggest that SNX11 may be a key protein in the process of viral infection and could serve as a broad-spectrum antiviral target.
Keywords:
dabie bandavirus
; SNX11
; V-ATPase
Introduction
Dabie bandavirus, belonging to the order Bunyavirales, family Phenuiviridae, and genus Bandavirus [1], was first discovered in 2010 in regions such as Henan and Hubei province, China [2]. It is transmitted through tick bites and can cause a newly emerging infectious disease known as Severe Fever with Thrombocytopenia Syndrome (SFTS) [3]. Since its discovery, cases have been reported in at least 28 provinces in China, including Hubei, Anhui, Jiangsu, and Liaoning province, with multiple localized outbreaks occurring [4,5]. In high-incidence provinces, the infection rate has shown a gradual upward trend in recent years [6]. Beyond mainland China, Dabie bandavirus infections have also been reported in several other countries and regions, such as Taiwan (China), South Korea, Japan, and Vietnam [4,7,8]. As a result, the Dabie bandavirus has become a significant challenge to global public health [9,10].
To date, due to the fact that the Dabie Bandavirus does not induce significant cytopathic effects [11,12], there has been limited research on the screening of key factors involved in its infection [13]. In previous study, we conducted an array-based CRISPR screening to identify host factors associated with Dabie bandavirus infection. Ultimately, we identified eight host proteins, whose absence could inhibit Dabie bandavirus infection to varying degrees, and notably the inhibitory effect was most pronounced in the absence of SNX11. At 96 hours post-infection with Dabie bandavirus, the positive rate of viral nucleoprotein NP in SNX11-deficient cells decreased by 95% compared to the normal group [14].
Furthermore, we found that SNX11-deletion blocks the release of the virus from late endosomes into the cytoplasm, a process that hinges on the fusion of the viral envelope with the endosomal membrane. Subsequently, we discovered that the endosomal pH in SNX11-deficient HeLa cells (5.56) was 0.23 units higher than that in normal cells (5.33), suggesting that this pH alteration affects Dabie bandavirus membrane fusion. Given the previously mentioned role of SNX11 in the formation of acidic endosomal vesicles, we preliminarily hypothesize that SNX11 deficiency may block Dabie Bandavirus release by altering endosomal pH [14]. However, the specific mechanism by which SNX11 regulates endosomal pH remains unclear. and to identify the proteins and mechanisms through which SNX11 modulates endosomal pH will provide important insights for discovering key targets of Dabie bandavirus and developing new antiviral drugs.
Material and Methods
Generation of SNX11-Knockout 293T Cell Line Using CRISPR/Cas9
SNX11 knockout in human embryonic kidney 293T cells was achieved via CRISPR/Cas9 genome editing. Single-guide RNAs (sgRNAs), designed using the CHOPCHOP online tool to target distinct exons within the SNX11 gene (sgRNA sequence:5’-CACCGCCGCTACCGTGAGTTCGTG-3’, was cloned into the pSpCas9(BB)-2A-Puro (PX459) V2.0 vector (Addgene #62988). Cells were cultured in DMEM supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C with 5% CO₂. Approximately 5 × 10⁵ cells were transfected with 2 µg of sgRNA plasmid or a control empty vector using Lipofectamine 3000, according to the manufacturer’s protocol. Transfected cells underwent puromycin selection (2 µg/mL) for 48 hours post-transfection. Surviving cells were diluted and seeded for single-cell cloning. Genomic DNA from expanded monoclonal populations was extracted and the targeted regions amplified by PCR. Successful knockout was confirmed by Sanger sequencing analysis. Absence of SNX11 protein expression was further validated by western blotting.
Quantification of Dabie Bandavirus Viral RNA by RT-qPCR
To assess viral replication kinetics, wild-type (WT) and SNX11-KO 293T cells were infected with Dabie Bandavirus at a multiplicity of infection (MOI) of 0.1. Following a 1-hour adsorption period at 37°C, the inoculum was removed, cells were washed twice with PBS, and fresh complete medium was added. Cell culture supernatants were harvested at defined post-infection time points. Supernatants were clarified by centrifugation (500 x g, 5 min) to remove cellular debris. Viral RNA was extracted from 140 µL of clarified supernatant using the QIAamp Viral RNA Mini Kit (Qiagen, #52906), according to the manufacturer’s instructions. Reverse transcription-quantitative PCR (RT-qPCR) targeting a conserved region of the Dabie bandavirus L segment was performed.
RNA Sequencing and Bioinformatic Analysis
Total RNA from SNX11-KO and wild-type 293T cells (three biological replicates) was extracted. Libraries were prepared with Illumina TruSeq Stranded mRNA Kit and sequenced (150-bp paired-end) on NovaSeq 6000 (20M reads/sample). Differentially expressed genes (DEGs) were identified with DESeq2 (v1.38.3; |log₂FC|>1, adj. p<0.05, Benjamini-Hochberg). Pathway enrichment (KEGG 2023) and protein-protein interaction networks (STRING v12.0; confidence score>0.7) were analyzed.
Co-Immunoprecipitation (Co-IP) Analysis of SNX11 and V-ATPase V1 Subunit Interaction
To investigate the interaction between SNX11 and the V-ATPase V1 subunit, HEK293T cells in 10 cm dishes were co-transfected with plasmids encoding HA-tagged SNX11 and FLAG-tagged V1 using Lipofectamine 3000. 48 hours post-transfection, cells were washed with cold PBS and lysed on ice for 30 min in IP lysis buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, protease inhibitors). Clarified lysates (14,000g, 15 min, 4°C) were incubated overnight at 4°C with rotation with an anti-HA rabbit polyclonal antibody. Protein A/G agarose beads were added for 2 hours. After five washes with lysis buffer, bound proteins were eluted by boiling in SDS-PAGE buffer. Eluates were subjected to SDS-PAGE, transferred to PVDF membranes, and probed with anti-FLAG (for V1) and anti-HA (for SNX11) mouse monoclonal antibodies, followed by HRP-conjugated secondary antibodies and ECL detection.
Pharmacological Inhibition of V-ATPase and Viral Replication Analysis
Wild-type (WT) and SNX11-knockout (SNX11-KO) HEK293T cells were pretreated with 100 nM Bafilomycin A1 (BafA1; V-ATPase inhibitor, Abcam, #ab120497) or vehicle control (DMSO) for 1 h at 37°C prior to infection with Dabie Bandavirus (MOI=0.5). Cells were then infected for 1 h in the continuous presence of BafA1/DMSO, followed by removal of inoculum and maintenance in fresh medium containing corresponding treatments. At designated post-infection time points (0, 24, 48, 72 hpi), total RNA was extracted. RNA in supernatants of Dabie bandavirus was quantified using AgPath ID one-step RT-PCR kit (ABI, #4387424).
Subcellular Distribution Analysis of V-ATPase V1 Subunit
Plasma membrane and cytosolic fractions from SNX11-knockout (SNX11-KO) and wild-type (WT) HEK293T cells were isolated using the Minute™ Plasma Membrane Protein Isolation Kit (Invent Biotechnologies, #SM-005). Cells were lysed in hypotonic buffer, followed by differential centrifugation: 700g for 10 min (nuclear removal) and 100,000g for 1 h at 4°C to separate cytosolic (supernatant) and total membrane (pellet) fractions. Membrane pellets were solubilized in RIPA buffer with protease inhibitors. Equal protein amounts (20 μg) from each fraction were resolved by SDS-PAGE and transferred to PVDF membranes. Blots were probed with: Mouse anti-V-ATPase V1 subunit antibody (1:1000; Proteintech, #14920-1-AP), Rabbit β-actin (1:2000; cell signaling technology, #4967S). HRP-conjugated secondary antibodies and ECL were used for detection.
Validation of SNX11 Deficiency on Infection by Diverse Viruses
To determine whether the inhibitory effect of SNX11 deficiency extends beyond Dabie bandavirus, SNX11-knockout (SNX11-KO) and wild-type (WT) 293T cells were infected with dengue virus serotype 2 (DENV-2). Viral infection was assessed at 48 hours post-infection (hpi) by measuring the levels of DENV-2 NS1 antigen released into the culture supernatant using a commercial ELISA kit (MEIMIAN, #MM-2001H1). Additionally, infection with Hantaan virus strain 76-118 (HTNV-76-118) and influenza A/Puerto Rico/8/1934 (H1N1; PR8) was assessed using the newly constructed SNX11-KO A549 cell line alongside their corresponding WT A549 controls. Similarly, at 48 hpi, viral infection for both HTNV-76-118 and PR8 was quantified by ELISA, targeting the viral nucleoprotein (N protein) present in the infected cell supernatants.
Results
In our preliminary investigations, HeLa cells were initially employed for CRISPR screen and validation procedures owing to their experimental robustness and operational convenience. Although 293T cells demonstrate reduced compatibility with CRISPR screening platforms compared to HeLa cells, their superior susceptibility to Dabie bandavirus infection coupled with enhanced transfection efficiency made them more appropriate for subsequent functional characterization. Based on these considerations, we generated SNX11-knockout 293T cell lines using CRISPR/Cas9 system in the current study, The efficiency of protein depletion was confirmed by Western blotting (Figure 1A and 1B).
Quantitative assessment of cellular proliferation kinetics through CCK-8 assays revealed comparable growth profiles between wild-type and SNX11-depleted cells (p > 0.05), confirming that SNX11 ablation does not compromise 293T cell proliferative capacity (Figure 1C). To delineate the antiviral mechanism, isogenic cell models including SNX11 knockout cells and their SNX11-reconstituted counterparts were challenged with Dabie bandavirus. Longitudinal monitoring of viral RNA dynamics in culture supernatants via RT-PCR demonstrated a reduction in viral RNA load in knockout cells relative to controls. This phenotype was effectively rescued upon SNX11 re-expression, providing orthogonal validation that SNX11 depletion exerts significant inhibitory effects on Dabie bandavirus replication (Figure 1D).
To investigate the intracellular function of SNX11, we performed transcriptome sequencing to analyze transcriptomic alterations in cells before and after SNX11 knockout. The results revealed that SNX11 deficiency resulted in the downregulation of 1,862 proteins, including key regulators of intraluminal vesicle formation and endosomal pH (ATP6V1D, MVB12A, VPS37A). Conversely, it induced the upregulation of 1,932 proteins, such as essential endosomal trafficking factors RAB5A and LAMP1(Figure 2A). SNX11 depletion predominantly perturbed signaling pathways associated with retromer complex signaling and PI3P signaling (Figure 2B).
PPI network analysis revealed that among the differentially expressed proteins resulting from SNX11 deficiency, vesicular trafficking proteins such as ATP6V1D, VPS16, VPS26A, VPS26B, and VPS35 were positioned within the core module of the interaction network. This finding further underscores the functional association of SNX11 with vesicular acidification regulation (exemplified by ATP6V1D) and intracellular trafficking (Figure 2C). Notably, the close functional interplay between SNX11 and V-ATPase was further corroborated by this network.
To determine whether Dabie bandavirus infection relies on V-ATPase activity, we treated both SNX11-knockout (SNX11-KO) and wild-type 293T cells with Bafilomycin A1, a V-ATPase inhibitor, prior to Dabie bandavirus infection. Viral supernatants were collected at various post-infection time points and subjected to quantitative RT-PCR (qRT-PCR) analysis. Notably, viral RNA levels exhibited a significant reduction in both cell types treated with the inhibitor compared to untreated controls, and the viral RNA level were lower in SNX11-KO cells, demonstrating a critical role of V-ATPase in Dabie bandavirus infection, and this role is closely linked to SNX11 (Figure 3A).
To investigate whether SNX11 depletion alters V-ATPase localization on endosomal membranes, thereby impairing endosomal pH regulation, we performed plasma membrane isolation and Western blotting to detect changes in V-ATPase subunit expression before and after SNX11 knockout. Our results revealed that SNX11 deficiency significantly reduced the expression of the V1 subunit on endosomal membranes (Figure 3B and 3C). To explore potential interactions between SNX11 and V-ATPase, we conducted co-immunoprecipitation (co-IP) experiments, which demonstrated that SNX11 specifically interacts with the V1 subunit of V-ATPase (Figure 3D). Supporting these findings, laser confocal microscopy analysis revealed substantial spatial colocalization between the two molecules (Figure 3E). These findings provide compelling evidence that SNX11 regulates endosomal pH through its interaction with V-ATPase.
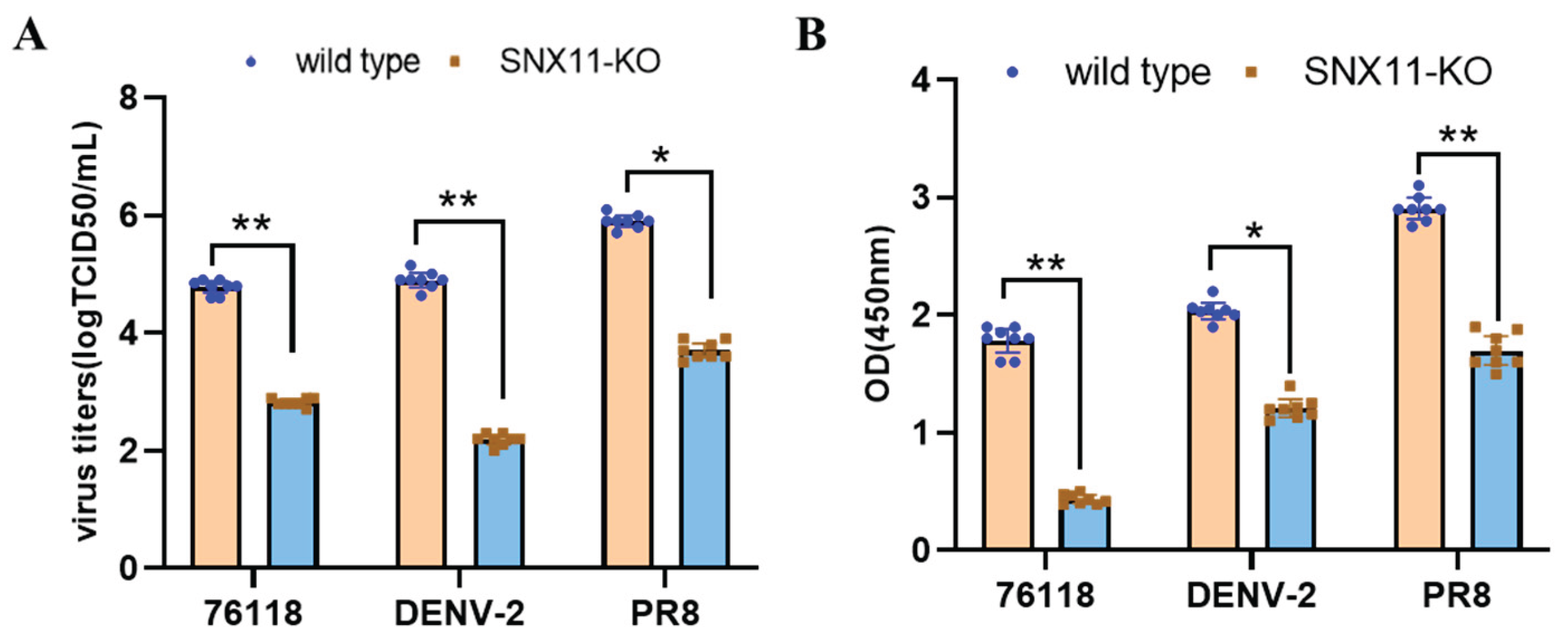
To validate the broad-spectrum nature of SNX11 deficiency in suppressing viral replication, we infected the aforementioned SNX11-deficient 293T cells and wildtype cells with dengue virus and utilized previously generated SNX11-deficient A549 cells [15], and wildtype cells to infect Hantaan virus strain 76-118 (HTNV 76-118) and influenza virus PR8 strain. At 48 hours post-infection, quantification of viral RNA levels and viral antigen load in cell supernatants, including the detection of Dengue virus NS1 antigen, PR8 strain NP antigen, and HTNV 76-118 NP antigen, revealed that SNX11 deficiency significantly suppressed the replication of dengue virus, hantavirus, and influenza virus, as shown in Figure 4A and 4B. Furthermore, flow cytometric analysis of influenza PR8-infected SNX11-KO and wildtype A549 cells demonstrated that SNX11 deficiency markedly reduced the viral antigen-positive cell population from 74.1% to 25.5% (Figure S1). The quantification of these specific viral antigens further supports the conclusion that SNX11 depletion exerts a potent inhibitory effect against diverse viral families.
Discussion
Structurally, V-ATPase consists of two functional domains: the membrane-embedded V0 complex, which mediates proton transport, and the cytoplasmic V1 complex, which hydrolyzes ATP to energize this process [16,17]. This enzyme is indispensable for maintaining the acidic luminal pH of lysosomes and endosomes, as well as modulating the pericellular microenvironment in specialized cells [18,19]. V-ATPase mechanistically regulates endosomal pH through two interconnected pathways: Primarily by modulating the dynamic equilibrium between V0 and V1 subunit dissociation and assembly, which directly governs its proton-pumping activity [20,21]. Additionally, through controlling the membrane abundance of V0 and V1 subunits, it determines the density of functional proton-transporting complexes on endosomal membranes [22]. These coordinated processes collectively establish the acidic endosomal environment essential for intracellular trafficking [23].
SNX11, a 264-amino acid member of the SNX family, features an N-terminal lipid-binding motif and two C-terminal α-helical domains [24]. We previously identified SNX11 as a critical host factor required for the cellular entry of Dabie bandavirus, with initial evidence suggesting its involvement in the viral endocytic pathway. However, the precise mechanism by which SNX11 facilitates viral entry remained unclear. In our previous attempts to investigate potential interactions between SNX11 and Dabie bandavirus proteins using co-immunoprecipitation (co-IP) assays, we did not detect any interaction between SNX11 and viral proteins. This suggests that SNX11 likely facilitates infection indirectly via host pathway modulation rather than through direct virion binding. This study demonstrates for the first time that SNX11 interacts with the V1 subunit of V-ATPase, regulating its trafficking to endosomal membranes. This interaction modulates endosomal pH, ultimately affecting the endocytic entry of Dabie bandavirus. This conclusion provides a new perspective for investigating viral endocytosis and expands the functional repertoire of the SNX protein family.
While our study establishes SNX11's role in v-ATPase assembly and endosomal acidification, broader functions of SNXs in viral infection warrant consideration. SNXs typically regulate viral entry through three primary mechanisms: SNXs can bind viral envelope proteins, mediating viral trafficking from early to late endosomes [25,26]; SNXs interact with viral nucleoproteins to facilitate membrane fusion and uncoating processes [27]; SNXs regulate actin cytoskeleton dynamics by binding microfilament-associated proteins, thereby modulating intracellular viral transport through actin depolymerization/polymerization [28]. Notably, prior characterization of SNX11 has been limited to TRPV3 degradation [29] and functional interplay with SNX10 in endosomal acidification. Our findings now position SNX11 as a central coordinator of endosomal maturation required for diverse viral life cycles.
To validate the broad impact of SNX11 deficiency on viral entry, we selected phylogenetically diverse viruses: Hantaan virus (segmented negative-sense RNA virus like Dabie bandavirus [30,31]), Influenza virus (segmented negative-sense RNA virus [32]), and Dengue virus (positive-sense single-stranded RNA virus [33,34]). SNX11 knockout consistently inhibited infection by all tested viruses. This pan-viral suppression can be interpreted through dual mechanisms: Primarily, as a sorting nexin (SNX) family member that intrinsically regulates endosomal trafficking of cellular cargoes [35,36,37], SNX11 ablation would be expected to disrupt endosomal transport of viruses. However, our earlier genome-wide CRISPR screen uniquely identified SNX11 among all SNX family members as an essential host factor for viral entry, suggesting its distinctive role as a specific regulator of viral endocytosis. Future investigations will focus on delineating the precise mechanisms underlying SNX11-virus interactions.
Author Contributions
Conceptualization, S.W. and J.L.; Data curation, T.L., Y.F., X.W.; Funding acquisition, S.W. and T.L.; Investigation, T.L., X.W., Q.S. and P.Z.; Methodology, T.L., J.L, Supervision, S.W; Writing—review & editing, T.L., All authors have read and agreed to the published version of the manuscript.
Funding
This study received funding from the Young Scientist Fund, IVDC, Grant No. IVDC-202301, and National Key R&D Program of China, grant number(2022YFC2303402).
Conflicts of Interest
The authors declare that they have no conflict of interest.
References
- Tu Y, Deng M, Zhang X, et al. Dabie bandavirus and Mycoplasma pneumoniae co-infection: a case report. BMC Infect Dis 2025, 25, 47. [Google Scholar] [CrossRef]
- Yu X J, Liang M F, Zhang S Y, et al. Fever with thrombocytopenia associated with a novel bunyavirus in China. N Engl J Med 2011, 364, 1523–1532. [Google Scholar] [CrossRef] [PubMed]
- Sheng R, Cheng T, Wang Y, et al. Molecular evolution and geographic migration of severe fever with thrombocytopenia syndrome virus in Asia. PLoS Pathog 2025, 21, e1012970. [Google Scholar] [CrossRef]
- Fang Y, Shen S, Zhang J, et al. Thrombocytopenia in Severe Fever with Thrombocytopenia Syndrome Due to Platelets With Altered Function Undergoing Cell Death Pathways. The Journal of infectious diseases 2025, 231, e183–e194. [Google Scholar] [CrossRef]
- Casel M A, Park S J, Choi Y K. Severe fever with thrombocytopenia syndrome virus: emerging novel phlebovirus and their control strategy. Exp. Mol. Med. 2021, 53, 713–722. [Google Scholar] [CrossRef]
- Charoensakulchai S, Matsuno K, Nakayama E E, et al. Epidemiological Characteristics of Severe Fever with Thrombocytopenia Syndrome. The American journal of tropical medicine and hygiene 2025, 112, 956–962. [Google Scholar] [CrossRef]
- Yu S, Zhang Q, Su L, et al. Dabie bandavirus infection induces macrophagic pyroptosis and this process is attenuated by platelets. PLoS neglected tropical diseases 2023, 17, e0011488. [Google Scholar] [CrossRef]
- Lam T T, Liu W, Bowden T A, et al. Evolutionary and molecular analysis of the emergent severe fever with thrombocytopenia syndrome virus. Epidemics 2013, 5, 1–10. [Google Scholar] [CrossRef]
- Woo D, Michelow I C, Choi Y, et al. Transmission of Severe Fever with Thrombocytopenia Syndrome (SFTS) to humans: A systematic review of individual participant data and meta-analysis. J Infect Public Health 2025, 18, 102685. [Google Scholar] [CrossRef]
- Xu D, Ji L, Wu X, et al. Whole-genome sequence analysis of SFTS bunyavirus in Huzhou, China. PLoS One 2025, 20, e0318742. [Google Scholar] [CrossRef]
- Liu J, Xu M, Tang B, et al. Single-Particle Tracking Reveals the Sequential Entry Process of the Bunyavirus Severe Fever with Thrombocytopenia Syndrome Virus. Small 2019, 15, e1803788. [Google Scholar] [CrossRef] [PubMed]
- Plegge T, Hofmann-Winkler H, Spiegel M, et al. Evidence that Processing of the Severe Fever with Thrombocytopenia Syndrome Virus Gn/Gc Polyprotein Is Critical for Viral Infectivity and Requires an Internal Gc Signal Peptide. PLoS One 2016, 11, e0166013. [Google Scholar] [CrossRef]
- Xing C, Zhang C, Xu Z, et al. Genome-wide CRISPR screening identifies LRP1 as an entry factor for SFTSV. Nat Commun 2025, 16, 4036. [Google Scholar] [CrossRef] [PubMed]
- Liu T, Li J, Liu Y, et al. SNX11 Identified as an Essential Host Factor for SFTS Virus Infection by CRISPR Knockout Screening. Virol Sin 2019, 34, 508–520. [Google Scholar] [CrossRef]
- Liu T, Li A, Li C, et al. Construction of A SNX11-knockout A549 Cell Line Based on the CRISPR/Cas9 System. Chin. J. Virol. Vol. 2022, 38, 652–657. [Google Scholar] [CrossRef]
- Eaton A F, Danielson E C, Tu L J, et al. Knockout of the V-ATPase interacting protein Tldc2 in B-type kidney intercalated cells impairs urine alkalinization. Am. J. Physiol. Renal Physiol. 2025, 328, F890–F906. [Google Scholar] [CrossRef]
- Wang C, Jiang W, Leitz J, et al. Structure and topography of the synaptic V-ATPase-synaptophysin complex. Nature 2024, 631, 899–904. [Google Scholar] [CrossRef]
- Sekiya M, Nakanishi-Matsui M, Matsumoto N, et al. The a subunit isoforms of V-ATPase are involved in glucose-dependent trafficking of insulin granules. Sci. Rep. 2025, 15, 18982. [Google Scholar] [CrossRef]
- De Luca M, Bucci C. A new V-ATPase regulatory mechanism mediated by the Rab interacting lysosomal protein (RILP). Commun. Integr. Biol. [CrossRef]
- Huang T, Sun C, Du F, et al. STING-induced noncanonical autophagy regulates endolysosomal homeostasis. Proc. Natl. Acad. Sci. U. S. A. 2025, 122, e2415422122. [Google Scholar] [CrossRef]
- Baars T L, Petri S, Peters C, et al. Role of the V-ATPase in regulation of the vacuolar fission-fusion equilibrium. Mol. Biol. Cell 2007, 18, 3873–3882. [Google Scholar] [CrossRef]
- Chen Y Y, Liu C X, Liu H X, et al. The Emerging Roles of Vacuolar-Type ATPase-Dependent Lysosomal Acidification in Cardiovascular Disease. Biomolecules. [CrossRef]
- Peng S Q, Wu Q Q, Wang W Y, et al. A gain-of-function mutation in ATP6V0A4 drives primary distal renal tubular alkalosis with enhanced V-ATPase activity. J Clin Invest. [CrossRef]
- Xu J, Xu T, Wu B, et al. Structure of sorting nexin 11 (SNX11) reveals a novel extended phox homology (PX) domain critical for inhibition of SNX10-induced vacuolation. J. Biol. Chem. 2013, 288, 16598–16605. [Google Scholar] [CrossRef] [PubMed]
- Bergant Marusic M, Ozbun M A, Campos S K, et al. Human papillomavirus L2 facilitates viral escape from late endosomes via sorting nexin 17. Traffic 2012, 13, 455–467. [Google Scholar] [CrossRef] [PubMed]
- Nanbo A, Imai M, Watanabe S, et al. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog 2010, 6, e1001121. [Google Scholar] [CrossRef]
- Ganti K, Massimi P, Manzo-Merino J, et al. Interaction of the Human Papillomavirus E6 Oncoprotein with Sorting Nexin 27 Modulates Endocytic Cargo Transport Pathways. PLoS Pathog 2016, 12, e1005854. [Google Scholar] [CrossRef]
- Lim J P, Wang J T, Kerr M C, et al. A role for SNX5 in the regulation of macropinocytosis. BMC Cell Biol. 2008, 9, 58. [Google Scholar] [CrossRef]
- Li C, Ma W, Yin S, et al. Sorting Nexin 11 Regulates Lysosomal Degradation of Plasma Membrane TRPV3. Traffic 2016, 17, 500–514. [Google Scholar] [CrossRef]
- Lu S, Chen S, Zhang Y, et al. Hantaan virus glycoprotein Gc induces NEDD4-dependent PTEN ubiquitination and degradation to escape the restriction of autophagosomes and facilitate viral propagation. FASEB J.
- Chang N, Huang W, Niu Y, et al. Risk of hemorrhagic fever with renal syndrome associated with meteorological factors in diverse epidemic regions: a nationwide longitudinal study in China. Infect Dis Poverty 2025, 14, 3. [Google Scholar] [CrossRef]
- Varghese J N, McKimm-Breschkin J L, Caldwell J B, et al. The structure of the complex between influenza virus neuraminidase and sialic acid, the viral receptor. Proteins Structure Function & Bioinformatics 2010, 14, 327–332. [Google Scholar]
- de Araújo L P, Weisshahn S K, Do Carmo E T, et al. Oral manifestations of dengue virus infection: a scoping review for clinical dental practice. BMC Oral Health.
- Maillard O, Belot J, Adenis T, et al. Early diagnosis of dengue: Diagnostic utility of the SD BIOLINE Dengue Duo rapid test in Reunion Island. PLoS neglected tropical diseases 2023, 17, e0011253. [Google Scholar] [CrossRef]
- Cullen P J, Korswagen H C. Sorting nexins provide diversity for retromer-dependent trafficking events. Nat. Cell Biol. 2011, 14, 29–37. [Google Scholar] [CrossRef]
- Teasdale R D, Collins B M. Insights into the PX (phox-homology) domain and SNX (sorting nexin) protein families: structures, functions and roles in disease. Biochem. J. 2012, 441, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Mas C, Norwood S J, Bugarcic A, et al. Structural basis for different phosphoinositide specificities of the PX domains of sorting nexins regulating G-protein signaling. J. Biol. Chem. 2014, 289, 28554–28568. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Generation and validation of SNX11-knockout (SNX11-KO) 293T cells and their resistance to Dabie bandavirus infection. (A) Sanger sequencing of the SNX11 locus in wild-type (WT) and SNX11-KO 293T cells. Arrows indicate deletion (indel) mutations causing frameshifts. (B) Western blot analysis of SNX11 protein expression in wild-type (WT) and SNX11-KO 293T cells. β-actin served as a loading control. (C) Cell proliferation assay (CCK-8) showing no significant difference in viability between WT and SNX11-KO cells over 72 hours. (D) Dabie bandavirus infection efficiency in WT vs. SNX11-KO cells. Viral RNA in supernatants was quantified by RT-qPCR.
Figure 1.
Generation and validation of SNX11-knockout (SNX11-KO) 293T cells and their resistance to Dabie bandavirus infection. (A) Sanger sequencing of the SNX11 locus in wild-type (WT) and SNX11-KO 293T cells. Arrows indicate deletion (indel) mutations causing frameshifts. (B) Western blot analysis of SNX11 protein expression in wild-type (WT) and SNX11-KO 293T cells. β-actin served as a loading control. (C) Cell proliferation assay (CCK-8) showing no significant difference in viability between WT and SNX11-KO cells over 72 hours. (D) Dabie bandavirus infection efficiency in WT vs. SNX11-KO cells. Viral RNA in supernatants was quantified by RT-qPCR.
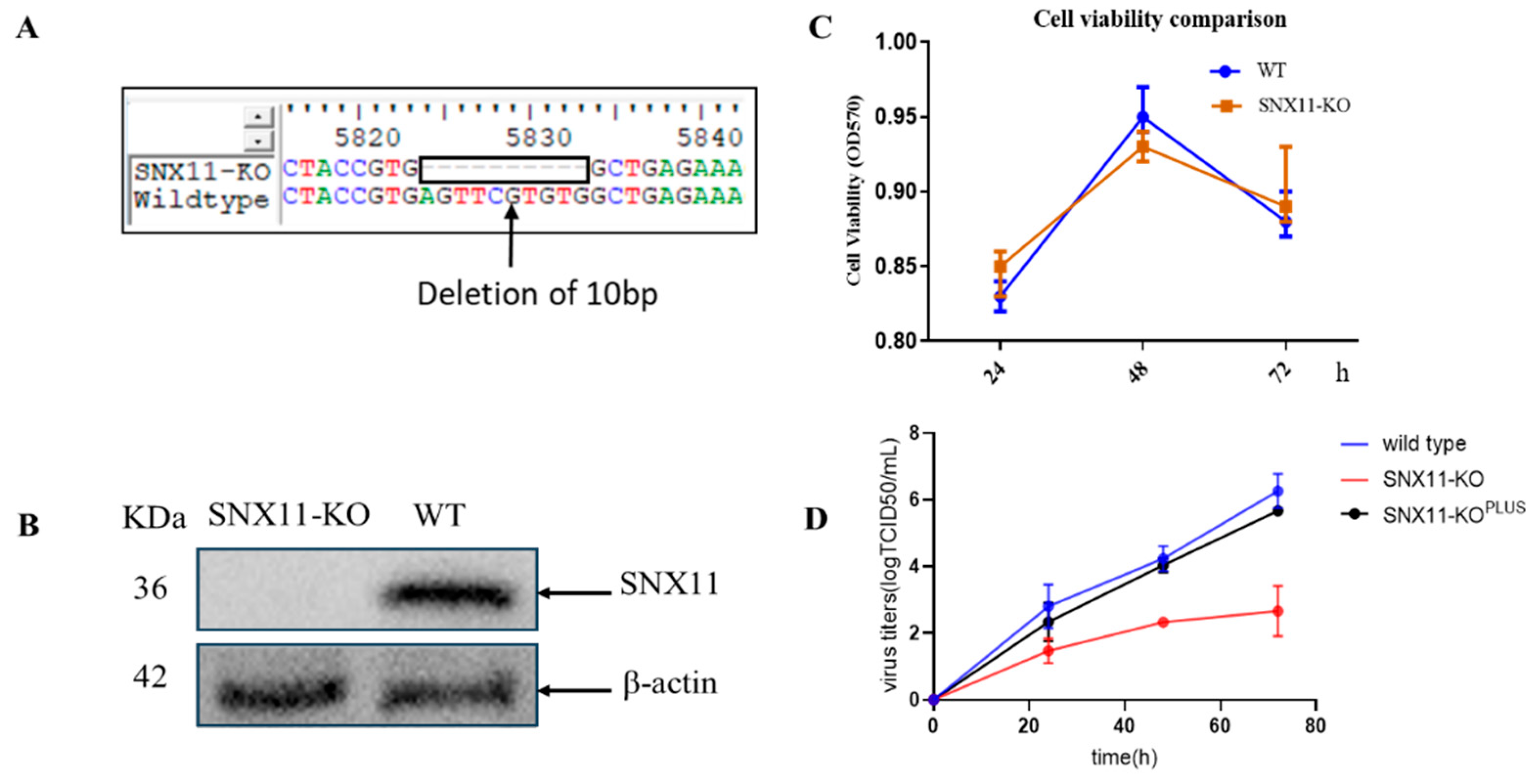
Figure 2.
Transcriptomic profiling reveals SNX11 knockout-induced signaling rewiring in 293T cells. (A) Volcano plot of differentially expressed genes (DEGs) in SNX11-knockout (SNX11-KO) vs. wild-type (WT) 293T cells. (B) KEGG pathway enrichment of DEGs. Top 12 significantly altered pathways (ranked by enrichment p value < 0.05) are shown. Dot size reflects gene count; color scale indicates -log10(p value). (C) Protein-protein interaction (PPI) network of high-confidence DEG-encoded protein.
Figure 2.
Transcriptomic profiling reveals SNX11 knockout-induced signaling rewiring in 293T cells. (A) Volcano plot of differentially expressed genes (DEGs) in SNX11-knockout (SNX11-KO) vs. wild-type (WT) 293T cells. (B) KEGG pathway enrichment of DEGs. Top 12 significantly altered pathways (ranked by enrichment p value < 0.05) are shown. Dot size reflects gene count; color scale indicates -log10(p value). (C) Protein-protein interaction (PPI) network of high-confidence DEG-encoded protein.
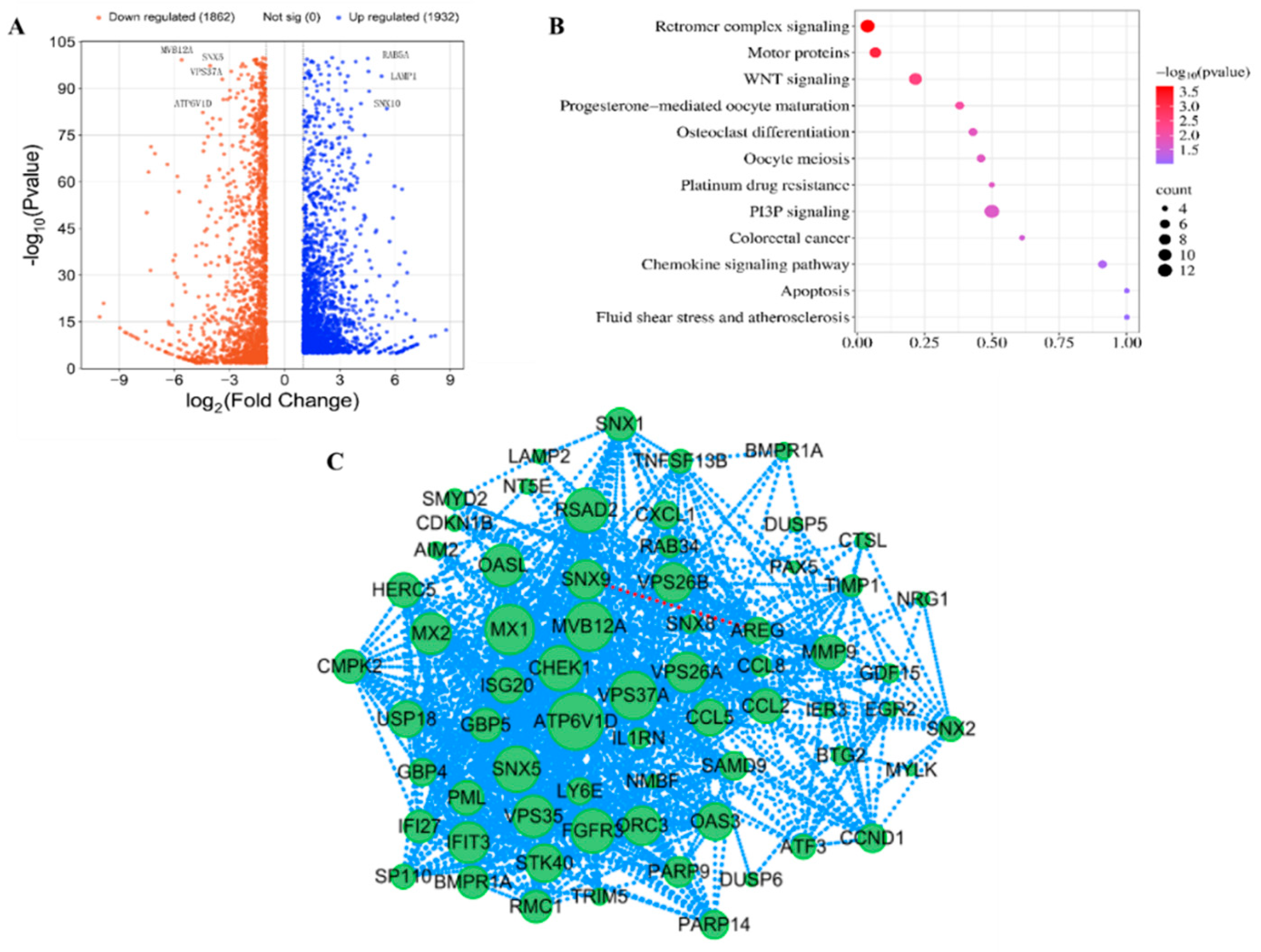
Figure 3.
SNX11 regulates V-ATPase assembly and promotes Dabie bandavirus entry via V1 subunit interaction. (A) Bafilomycin A1 (Baf A1) inhibition assay: Viral RNA in supernatants after Dabie bandavirus infection. (B) Plasma membrane fractionation: Immunoblot of V1 subunits in membrane fractions and cytosol of WT vs. SNX11-KO cells. (C) Quantification of V1 subunit expression: Reduced V1A levels in SNX11-KO membrane fractions (n=3; **p<0.01, unpaired t-test). (D) Co-immunoprecipitation (Co-IP): SNX11-HA immunoprecipitates with V1A in 293T cells. Lanes: Input (10%). (E) Confocal microscopy: Colocalization of SNX11 (red) and V1 (green) in perinuclear regions, scale bars: 20 μm.
Figure 3.
SNX11 regulates V-ATPase assembly and promotes Dabie bandavirus entry via V1 subunit interaction. (A) Bafilomycin A1 (Baf A1) inhibition assay: Viral RNA in supernatants after Dabie bandavirus infection. (B) Plasma membrane fractionation: Immunoblot of V1 subunits in membrane fractions and cytosol of WT vs. SNX11-KO cells. (C) Quantification of V1 subunit expression: Reduced V1A levels in SNX11-KO membrane fractions (n=3; **p<0.01, unpaired t-test). (D) Co-immunoprecipitation (Co-IP): SNX11-HA immunoprecipitates with V1A in 293T cells. Lanes: Input (10%). (E) Confocal microscopy: Colocalization of SNX11 (red) and V1 (green) in perinuclear regions, scale bars: 20 μm.
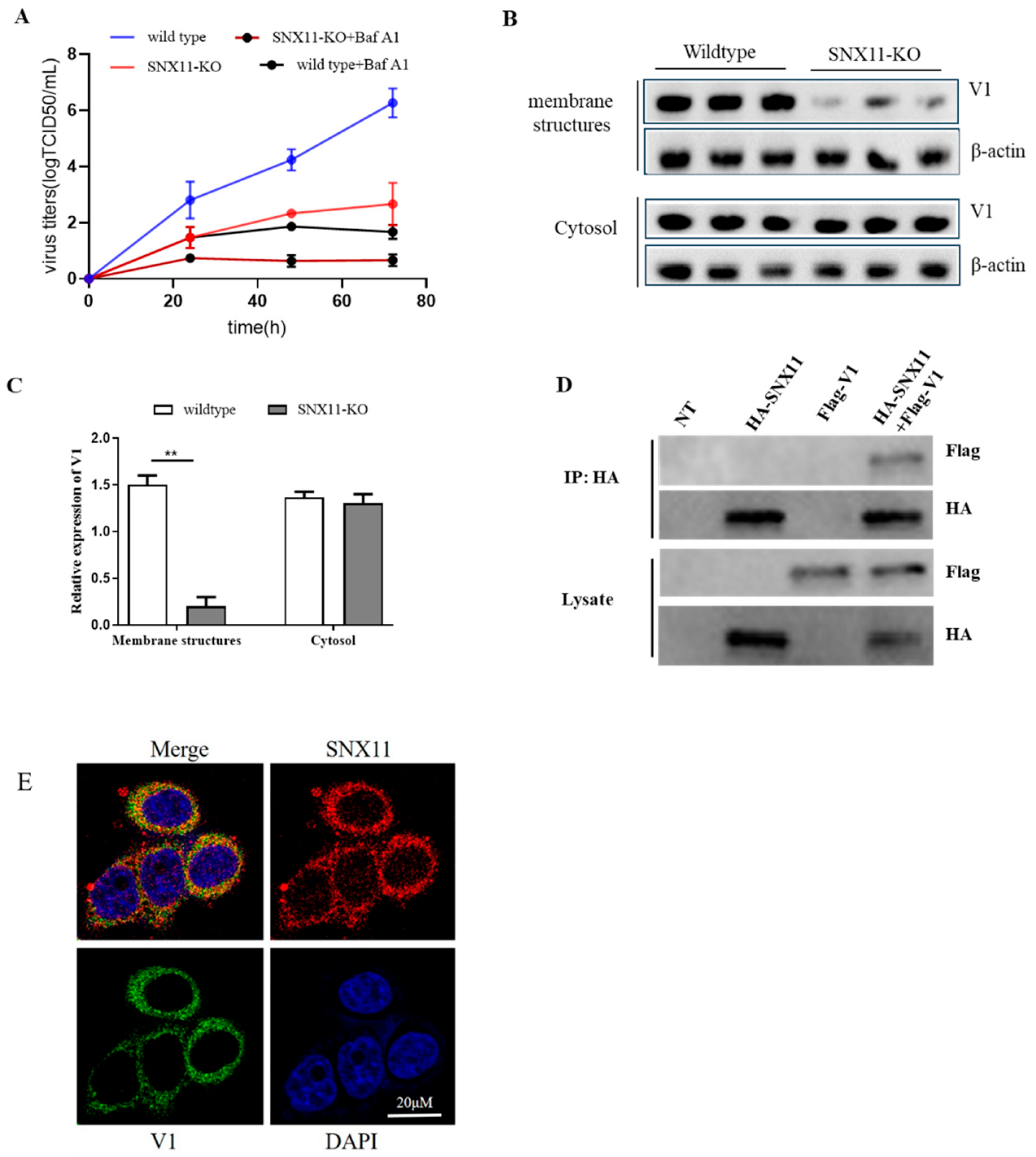
Figure 4.
SNX11 deficiency broadly inhibits viral infection. (A) Viral RNA levels in supernatants of SNX11-knockout (SNX11-KO) and control cells infected with Dengue virus (DENV; 293T cells), Hantaan virus (HTNV 76-118; A549 cells), or Influenza virus (PR8; A549 cells), quantified by qRT-PCR at 48 hours post-infection (hpi). (B) Viral antigen load in supernatants of infected SNX11-KO and control cells, measured by ELISA at 48 hpi. *Data represent mean ± SD (n=8 independent experiments). **p<0.01, SNX11-KO: SNX11-knockout.
Figure 4.
SNX11 deficiency broadly inhibits viral infection. (A) Viral RNA levels in supernatants of SNX11-knockout (SNX11-KO) and control cells infected with Dengue virus (DENV; 293T cells), Hantaan virus (HTNV 76-118; A549 cells), or Influenza virus (PR8; A549 cells), quantified by qRT-PCR at 48 hours post-infection (hpi). (B) Viral antigen load in supernatants of infected SNX11-KO and control cells, measured by ELISA at 48 hpi. *Data represent mean ± SD (n=8 independent experiments). **p<0.01, SNX11-KO: SNX11-knockout.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



