
Submitted:
13 June 2025
Posted:
17 June 2025
You are already at the latest version
Abstract
Gaucher disease (GD) is an autosomal recessive disorder caused by the deficient activity of the lysosomal enzyme glucocerebrosidase (GCase). Although enzyme replacement therapy (ERT) remains the most effective treatment for GD patients, its high cost significantly limits accessibility. To enhance production efficiency, we developed a lentiviral system encoding a codon-optimized GCase gene driven by the human elongation factor 1a (hEF1) promoter for stable production in human cell lines. A functional lentiviral vector, LV_EF1_GBA_Opt, was generated at a titer of 7.88 x 108 LV particles/mL as determined by qPCR. Six transduction cycles were performed at a multiplicity of infection of 30-50. The transduced heterogeneous human cell population showed GCase-specific activity of 307.5 ± 53.49 nmol/ mg protein/ h, which was 3.21-fold increase compared to virgin 293FT cells (95.58 ± 16.5 nmol/ mg protein/ h). Following single-cell cloning, two clones showed specific activity of 763.8 ± 135.1 and 752.0 ± 152.1 nmol/mg/h (clones 15 and 16, respectively). These results show that codon optimization, a lentiviral delivery system, and clonal selection together enable the establishment of stable human cell lines capable of producing high levels of biologically active, synthetic recombinant GCase in vitro. Further studies are warranted for the functional validation in GD patient-derived fibroblasts and animal models.
Keywords:
Gaucher disease
; glucocerebrosidase
; 293FT cells
; codon optimiation
; hEF1a promoter
; lentiviral vector
1. Introduction
Gaucher disease (GD) is an autosomal recessive genetic disorder caused by the deficient activity of the lysosomal enzyme glucosylceramidase (GCase), which catalyzes the hydrolysis of glucosylceramide (GlcCer) into ceramide and glucose [1,2,3]. This enzymatic deficiency leads to the pathological accumulation of GlcCer in cells, primarily in macrophages, which induce progressive damage to multiple organs [4,5,6,7]. The GD is the most common lysosomal storage disease [8] and presents heterogeneous clinical symptomatology ranging from non-neuropathic (Type 1) or presence and severity of neurological pathology (GD Type 2 and 3) [9,10,11,12,13].
Currently, enzyme replacement therapy (ERT) is the most effective treatment for GD patients. ERT significantly improves patient’s quality of life and effectively reverses visceral and hematological manifestations, including hepatosplenomegaly and cytopenia [3,14,15,16]. GD was the first lysosomal storage disease to receive FDA-approved ERT, initially using alglucerase, a placental tissue-derived glucocerebrosidase [17]. To monitor therapeutic outcomes and disease progression, the International Collaborative Gaucher Group (ICGG) established the Gaucher Registry (ClinicalTrials.gov ID NCT00358943, 1991) which collects real-world data on ERT efficacy, safety, and the natural history of GD [3,18,19,20,21].
In 1995, the imiglucerase, produced in Chinese hamster ovary (CHO) cells, was the first recombinant human glucocerebrosidase developed [22,23]. Subsequently, the velaglucerase alfa, an enzyme expressed in human cell culture systems received approval [24,25]. Later, taliglucerase alfa, a plant-derived recombinant enzyme, was introduced as an emergency treatment to ensure therapy continuity for patients, following viral contamination issues in imiglucerase production [26,27].
In Brazil, these therapies are accessible through the Unified Health System (SUS) under Ordinance No. 1.266 (November 14, 2014) [21,28].
The high production cost of recombinant GCase continues to limit the accessibility of enzyme replacement therapy (ERT), driving the need for optimized expression platforms. To address this issue, alternative production strategies have been developed to enhance yield, stability, and tissue-specific delivery. These include viral vector-based approaches using retroviral [29], lentiviral [30] and AAV-mediated targeting of the CCR5 locus [31]. Also, mammalian cell systems make use of methotrexate (MTX) for clone selection and amplification [32] and plant-based platforms [33,34], among other innovative approaches.
Using synonymous codons is a promising strategy to improve human mRNA stability [35] and enhance recombinant protein production [36,37]. This approach has been successfully implemented to increase expression in various genetic diseases, including Hemophilia A and B [38,39], X-linked Severe Combined Immunodeficiency [40], as well as in lysosomal storage disease such as aspartylglucosaminidase [41], Fabry disease [42], Gaucher disease [43,44], globoid cell leukodystrophy [45], mucopolysaccharidosis II [46,47], Krabbe disease [48], Pompe disease [49,50,51], Tay-Sachs and Sandhoff disease [52], among others.
Lentiviral vectors (LVs) have been successfully investigated in ex vivo gene therapy for multiple genetic diseases, including applications using patient-derived induced pluripotent stem cells (iPSCs). Clinical and preclinical studies demonstrate their efficacy in Hemophilia A [39], X-linked Severe Combined Immunodeficiency [53], Fabry disease [54], globoid cell leukodystrophy [55], mucopolysaccharidosis II [56], Pompe disease [57], among others. For GCase production, LV systems offer distinct advantages such as efficient transduction of both dividing and non-dividing cells [58,59,60,61], unlike oncoretroviral vectors. Also, unlike oncoretroviral vectors, LV integration sites are preferably in euchromatin regions, which increases the chances to obtain active transgene expression [62,63,64].
In our previous study [44], we demonstrated the feasibility of transient GCase production using codon-optimized GBA-1 cDNA under the hEF1α promoter in 293FT cells. The engineered variant (GBA-Opt) exhibited a 5.2-fold in mRNA expression and 6.1-fold higher enzymatic activity compared to controls validating the potential of synthetic biology approaches for ERT [44]. Here, we extend this work by developing a scalable producer cell line through lentiviral genomic integration and clonal selection. Compared to virgin 293FT controls (7.037 ± 0.4 nmol hydrolyzed substrate/mL/h), our optimized system achieved 97-fold higher secreted activity (683.9 nmol/mL/h) in clone 16, along with high specific activity (752.0 ± 152.1 nmol hydrolyzed substrate/ mg/ h).
2. Results
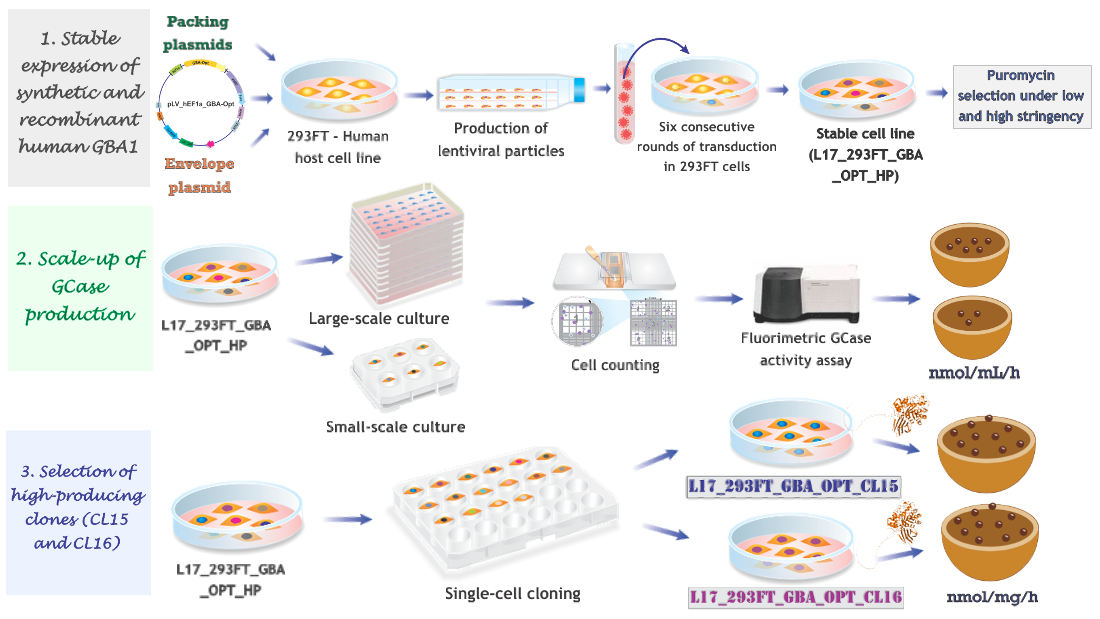
2.1. Production of Stable Lentiviral-Transduced Human Cell Lines
Lentiviral particles encoding the codon-optimized human GBA1 gene (GBA-Opt), under the control of the hEF1α promoter (LV-EF1α_GBA-Opt), were produced and concentrated. Particle formation was confirmed by ELISA for the p24 capsid antigen, and the viral titer was determined by quantitative PCR (qPCR), yielding 7.88 x 108 viral particles per mL.
293FT cells were transduced in six successive cycles using different volumes (2, 5 and 10 μL) of the viral preparation, corresponding to multiplicity of infection (MOI) of 30-50. Following transduction, cells were cultured in 2 μg/mL puromycin for 10 days to select a stable heterogeneous puromycin-resistant population (L17_293FT_GBA_OPT_HP). This population was subsequently expanded under low (1 μg/mL) and high (5 μg/mL) puromycin concentrations to enrich for subpopulations with increased expression, prior to GCase activity analysis.
2.2. GCase Activity in Lentiviral Transduced and Puromycin-Selected L17_293FT_GBA_OPT_HP Cells
The GCase activity was quantified in the supernatants of L17_293FT_GBA_OPT_HP cells following puromycin selection at low (1 μg/mL) and high (5 μg/mL) stringency concentrations. The control 293FT/Virgin cell line demonstrated baseline activity of 7.037 ± 0.4 nmol hydrolyzed substrate/mL/h. Puromycin selection significantly enhanced enzymatic production in the engineered cells. Treatment with 1 µg/mL puromycin resulted in 329.7 ± 31.02 nmol/mL/h of GCase activity, a 47-fold increase compared to control (p = 0.0002). Increasing the puromycin concentration to 5 μg/mL resulted in 459.5 ± 24.82 nmol/mL/h (a 62-fold increase compared to the virgin cell line, p < 0.0001). The higher puromycin concentration induced a 1.4-fold increase compared to the low-dose condition (p = 0.0007) (Figure 1A).
When normalized per million cells, the pattern of enzymatic secretion was maintained. The L17_293FT_GBA_OPT_HP cell line treated with 1 and 5 μg /mL puromycin exhibited enzymatic activities of 215.4 ± 39.46 U GCase/106 cells and 346.1 ± 26.27 U GCase/106 cells, respectively. These values represent a 48.5-fold increase with low stringency (p = 0.0017) and a 78-fold increase with high stringency (p = 0.0001) relative to control, corroborating the effectiveness of the puromycin selection on enzymatic productivity (Figure 1B).
To evaluate scalability of the system, GCase production was assessed in a 10-layer cell factory and compared to that in small-scale cultures. The L17_293FT_GBA_OPT_HP cell line maintained consistent activity levels, producing 209.52 nmol/mL/h in a 6-well plate and 201,931 nmol/mL/h in the 10-layer system. This corresponded to a 280-fold increase in culture volume (from 2 mL to 560 mL), while preserving 96.4% of the volumetric enzymatic efficiency, resulting in a 963-fold increase in total GCase secretion (Figure 2).
The GCase-specific activity was quantified in cell lysates from the L17_293FT_GBA_OPT_HP heterogeneous population. Compared to unmodified 293FT cells, the L17 population exhibited significantly higher enzymatic activity. The specific activity reached 307.5 ± 53.49 nmol/mg/h, which represents a 3.2-fold increase relative to control cells (95.58 ± 16.50 nmol/mg/h, p = 0.0026, Figure 3A). When normalized per million cells, the L17 population also demonstrated enhanced intracellular GCase production, reaching 159.3 ± 44.97 U GCase/106 cells, compared to 60.2 ± 19.35 U GCase/106 cells in control cells, a 2.64-fold increase relative to the control (p < 0.05; t = 4.048; df = 4.074, Figure 3B). These findings confirm that the selected population supports increased intracellular GCase secretion following lentiviral transduction and selection.
2.3. High-Producer Clone Selection from Puromycin-Selected Population
Eleven single-cell clones were isolated from the L17_293FT_GBA_OPT_HP heterogeneous population previously selected with 2 μg/mL puromycin. GCase secretion analysis revealed a broad range of enzymatic activities among clones, indicating variable transgene expression and productivity (Table 1). Secreted GCase levels ranged from 89.9 to 683.9 nmol/mL/h, reflecting the inherent heterogeneity of lentiviral transduced human cell populations.
Approximately 27% of the clones (3/11: CL13, CL15, and CL16) exhibited high GCase secretion levels (> 400 nmol/mL/h), 45% (5/11: CL5, CL8, CL9, CL11, CL17) showed moderate production (200–400 nmol/mL/h), and 28% (3/11: CL7, CL10, CL18) demonstrated low secretion (< 200 nmol/mL/h).
Among them, clones 15 and 16 showed the highest enzymatic activities, with 585.4 and 683.9 nmol hydrolyzed substrate/mL/h, respectively, corresponding to 1.8-fold and 2.1-fold increases relative to the heterogeneous parental population. When normalized to cell number, these clones maintained their superior productivity, reaching 390.3 and 455.9 U/106 cells, respectively (Table 1). These results confirm the successful enrichment of high GCase-producing cells through puromycin selection and single-cell cloning.
Morphological analysis by light microscopy revealed that a heterogeneous population (Figure 4B) and clones 15 and 16 (Figure 4C,D) retained a fibroblast-like morphology similar to virgin cells (Figure 4A). Minor differences were observed and appeared to be related to cell density and culture confluence rather than to the transgene expression.
To further characterize these two clones with the highest levels of secreted GCase (clones 15 and 16), we evaluated their intracellular GCase-specific activity. Clone 15 exhibited a specific activity of 763.8 ± 135.1 nmol of hydrolyzed substrate per mg of protein per hour, while clone 16 displayed a comparable activity of 752.0 ± 152.1 nmol/mg/h (Figure 5A). Compared to the L17_293FT_GBA_OPT_HP heterogeneous population, these values represent 2.5-fold (p = 0.01) and 2.4-fold (p = 0.006) increases, respectively. No statistically significant difference was observed between the two clones (p = 0.92). When compared with the 293FT/Virgin control cells, clone 15 exhibited a 7.9-fold increase (p = 0.01), and clone 16 a 7.8-fold increase (p = 0.003), corroborating with the high intracellular production of GCase in both clones. We also normalized GCase production per million cells. Clone 15 reached 313.4 ± 13.4 U GCase/106 cells, and clone 16 reached 293.3 ± 4.9 U GCase/106 cells (Figure 5B). These correspond to 1.96-fold and 1.84-fold increases compared to the L17 heterogeneous population (p = 0.0026 and p = 0.0078, respectively). Compared to the 293FT/Virgin control cells, clone 15 showed 5.2-fold increase (p = 0.0001), and clone 16 showed a 4.8-fold increase (p < 0.0001).
3. Discussion
In this study, we established a stable human cell line capable of producing high levels of recombinant and synthetic human β-glucocerebrosidase (GCase). By integrating a codon-optimized GBA1 gene (GBA-Opt) under the hEF1α promoter into 293FT cells via lentiviral vectors (LVs) we achieved sustained GCase specific activity up to 763.8 ± 135.1 nmol of hydrolyzed substrate per mg of protein per hour in a clonal population. This approach not only addresses the cost and scalability limitations of current ERT production systems but also demonstrates the potential applicability of synthetic biology to optimized therapeutic enzyme manufacture.
Lentiviral vectors (LVs) were selected for their ability to transduce both dividing and non-dividing cells and their preferential integration into transcriptionally active euchromatin regions, thereby enhancing transgene stability [65]. These key features were comprehensively described by Naldini in 2016 [65], and more recently reviewed in 2025, highlighting the continued evolution of LV platforms and their central role in advancing gene therapy for multiple genetic diseases [66]. The clinical relevance of LVs has been demonstrated by recent clinical trials and preclinical studies demonstrating efficacy in several monogenic and lysosomal storage disorders. Srivastava et al [39] showed that CD34+ hematopoietic stem cells transduced with LVs carrying a codon-optimized FVIII gene yielded sustained expression in patients with Hemophilia A. Similarly, Hu et al. [53] reported the successful preclinical application of SIV-LVs encoding IL2RG for X-linked severe combined immunodeficiency (X-SCID), confirming both efficacy and biosafety.
In the context of lysosomal diseases, Ellison et al [56] developed and validated a GMP-grade LV manufacturing platform for hematopoietic stem cell gene therapy targeting MPS II, demonstrating process scalability, regulatory compliance, and preserved vector efficacy. Dogan et al [57] used lentiviral delivery of engineered alpha-glucosidase (GAA) transgenes to improve secretion and tissue targeting in Pompe disease. These vectors conferred GAA enzymatic activity in both hematopoietic and target tissues, including muscle and CNS, highlighting the impact of vector engineering and transduction strategy. Mangiameli et al [55] utilized LV-modified iPSC-derived neurons to model globoid cell leukodystrophy, illustrating the utility of LVs in disease modeling and potential autologous gene therapy. Finally, Saleh et al [54] conducted a phase I trial in five patients with Fabry disease and demonstrated that lentiviral gene therapy resulted in persistent α-galactosidase secretion from transduced cells. Notably, three patients discontinued enzyme replacement therapy and maintained clinical stability, suggesting that LV-based delivery may enable long-term therapeutic enzyme production [54]. Collectively, these studies support broad therapeutic applicability of LV platforms and reinforce their suitability for GCase production.
It is worth noting that, our third-generation SIN LV efficiently delivered a large dual-cassette construct (13.5 kb) encoding GBA-Opt and puromycin resistance, achieving a high titer of 7.88 × 10⁸ VP/ mL despite the near-maximal packaging capacity [67,68]. This performance likely reflects optimized 293FT packing cells, which express the SV40 large T antigen to enhance transfection efficiency [69,70,71,72]. Recent advances have further expanded the utility of this cell line enabling innovative delivery strategies such as dual-pseudotyped LVs (VSV-G/SeV-HN) for enhanced tropism and transduction efficiency in hematopoietic stem cells [73] and for non-integrating lentivirus-likely particles (Gag-Only LVLPs) for safer delivery of base editors in cancer immunotherapy [74].
In our previous study [44], we demonstrated the feasibility of transient expression of codon-optimized GBA1 (GBA-Opt) under the hEF1α promoter in 293FT cells. This construct led to statistically significant 1.89-fold increase in relative mRNA expression compared to the CMV promoter (p = 0.001). Importantly, this transcriptional enhancement was accompanied by increased enzymatic activity: cells expressing GBA-Opt under hEF1α exhibited a mean specific activity of 426.2 + 25.1 nmol/mg/ h, compared to 277.9 + 17.5 nmol/mg/h with CMV, representing a ~1.5-fold increase [44]. Building on these observations and based on its consistent efficacy and stability across multiple human cell lines [75,76], we selected the hEF1α promoter. Kim et al [30] demonstrated hEF1α-driven lentiviral vectors expressing GCase achieved higher expression levels than CMV-driven vector in HEK293, SH-SY5Y, and HeLa cells. In our system, lentiviral transduction with an hEF1α-driven GBA-Opt construct, followed by puromycin selection, led to stable integration into the host genome and resulted in high and sustained recombinant and synthetic GCase.
In parallel, codon-optimization was applied to further enhance transgene expression. Our construct was designed with a codon-optimized GBA1 sequence, aiming to improve mRNA stability and protein expression, as supported by prior studies showing that GC3 codons increase mRNA stability and protein levels in human cells [35]. This strategy has been widely used to optimize therapeutic gene expression in genetic diseases as demonstrated in Hemophilia A trials [39] and SCID-X1 preclinical models [40]. The same strategy has also been successfully applied in several lysosomal storage diseases. Codon optimization of aspartylglucosaminidase (AGA) led to a 2.5 – to 5-fold increase in expression in HEK293 and HeLa cells, supporting its utility for enzyme replacement therapy and gene therapy in aspartylglucosaminuria [41]. In globoid cell leukodystrophy, codon-optimized of galactocerebrosidase (GALC) enabled safe and effective HSPC gene therapy in murine models [45]. The same group later reported long-term benefit in clinical trials using a similar lentiviral strategy for metachromatic leukodystrophy, reinforcing codon optimization as a key component in lysosomal gene therapy [77]. In Mucopolysaccharidosis Type II, a codon-optimized iduronate-2-sulfatase (IDS) construct driven by MNDU3 promoter achieved supraphysiological enzyme levels across tissues and partial restoration in the brain, normalizing glycosaminoglycan (GAG) accumulation and preventing the emergence of cognitive deficits in vivo [47]. Expanding on this strategy, Liang et al fused IGF2 to GAAco to enhance uptake via the CI-M6P/IGF2 receptor, enabling full correction of cardiac, skeletal, and CNS pathology at lower vector doses, thus identifying IGF2.GAA as a promising candidate for clinical translation [51]. A follow-up study by the same group confirmed that LV-IGF2.GAAco nearly normalized the skeletal muscle proteome in preclinical Pompe disease model without inducing off-target effects, reinforcing the therapeutic potential of codon-optimized LVs for lysosomal disorders [78].
While these studies underscore the translational relevance of codon-optimized constructs, we also evaluated the scalability potential of our production system. In cultures of 500 mL (6 x 108 cells), the heterogeneous L17_293FT_GBA-Opt_HP population produced 201,931 nmol/mL/h of GCase, approximately 963-fold more than 2 mL cultures. The 10-layer cell factory run further confirmed this scalability, preserving over 96% of enzymatic efficiency observed in small-scale wells. The observed productivity likely reflects the combined effects of codon optimization, stable lentiviral, and puromycin selection of high-producing cells. In a previous study from our group, Rosa et al. [79] similarly reported enhanced recombinant factor VIII activity using SK-HEP cells cultured on microcarriers in spinner flasks, compared to static conditions. Although, codon optimization was not applied in that study, their findings highlight how optimized microcarrier-based systems can substantially enhance recombinant protein yields, serving as a complement to molecular engineering strategies such as those used in our work. While our current system utilized 10% FBS, the high levels of GCase production levels suggest promising potential for adaptation to serum-free conditions, an important consideration for future GMP-complant production.
To further improve productivity, we tested different concentrations of puromycin under increased stringency. The L17_293FT_GBA_OPT_HP cell line, maintained in medium with 10% FBS, exhibited GCase activity levels of 329.7 ± 5.51 nmol/mL/h and 439.5±12.03 nmol/mL/h after treatment with 1 and 5 μg/mL of puromycin, respectively. Increasing puromycin concentration from 1 µg/mL (low stringency) to 5 µg/mL (high stringency) significantly increased GCase secretion, reaching up to 62-fold higher levels compared to virgin control cells. These results are consistent with the hypothesis that puromycin selection under increasing stringency enriches integration events and transcriptional contexts permissive to high transgene expression, as previously observed in MGMT-based selection systems [60].
The L17 heterogeneous population exhibited a specific activity of 307.5 nmol/mg/h, representing 3.21 times higher than the non-transduced 293FT cell line. This enhancement was achieved through six cycles of lentiviral transduction at MOIs of 30-50. Our findings align with Spencer et al [80], corroborating that repeated transduction cycles allow for high-level production of recombinant and synthetic enzymes.
To isolate the best producers, we performed clonal selection from the L17 population. Among 11 clones, two clones of L17 showed specific activity of 763.8 ± 135.1 nmol/mg/h (clone 15) and 752.0 ± 152.1 nmol/mg/h (clone 16), which were 6 and 8 times higher compared to the virgin cell line. While our results are promising, certain limitations warrant future study. For example, the investigation of long-term expression in fibroblasts from Gaucher disease patients as well to evaluate in vivo efficacy in GD models will be essential to validate therapeutic potential.
In conclusion, our integrated platform combining codon optimization, hEF1α-driven expression, and LV-mediated delivery provides an effective strategy for generating biologically active recombinant and synthetic human GCase. The selected clones showed sustained enzyme production and high specific activity highlighting its potential to reduce ERT costs and improve accessibility for GD patients. Beyond Gaucher, this production system could be readily adapted to the lysosomal enzymes, supporting future application across a broad spectrum of lysosomal storage disorders.
4. Materials and Methods
4.1. Plasmid Constructs
The lentiviral expression vector used in this study were derived from pDEST_R4_R2 and contains a codon-optimized human GBA1 cDNA, including the R534H missense mutation previously described [44]. The production of the lentiviral particles was performed using two accessory plasmids: pCMV-VSV-G (5,824 bp) and pCMV△R8.91 (12,120 bp), kindly provided by Dr. Lucas Eduardo Botelho de Souza (Laboratory of Gene Transfer, Blood Center of Ribeirão Preto, SP, Brazil).
4.2. Cell Culture
The 293FT human cell line was purchased from Thermo, USA (R700-07). Cells were maintained in DMEM (Dulbecco modified Eagle’s medium) supplemented with 10% fetal bovine serum (FBS) (HyClone, USA), 1% Penicillin/Streptomycin/L-glutamine (Sigma-Aldrich), and 1% non-essential amino acids (HyClone). The serum was heat-inactivated at 56 °C for 30 minutes prior to use. Cell cultures were maintained at 37 oC in a humidified incubator with a 5% CO2 atmosphere. Cell growth was monitored every two days by phase-contrast microscopy, and the medium was refreshed as needed.
4.3. Production of Lentiviral Particles
To generate lentiviral particles, the construct DNA (pLV-hEF1a-GBA-Opt [44]) was transiently introduced into 293FT cells by triple co-transfection using lipofectamine (Life Technologies) following the manufacturer’s instructions. The transfection mix included 6.5 μg of plasmid pCMV△R8.9, 3.2 μg of pCMV-VSV-G and 12 μg of the expression vector coding the GBA-Opt cDNA. Lentiviral supernatants were collected 48 h and 72 h post-transfection, filtered through a 0.22 μm Millex-GV filter (Millipore), and concentrated by ultracentrifugation (1.40 h at 31,000 x g) using an OptimatTMXL-100K ultracentrifuge (Beckman Coulter) with a SW28 rotor, as previously described by [81,82]. The concentrated virus was stored at -80oC. The viral titration was calculated by absolute quantification using real time PCR with the TaqMan system (Applied Biosystems). The endogenous control was human β-actin gene (Hs03023880_g1), and lentiviral genome copies were quantified using primers and a probe targeting the LTR sequence: forward primer (5’- GCCCGAACAGGGACTTGA-3'), reverse primer (5’-CGAGTCCTGCGTCGAGAGA-3') and the probe (5’-FAM-AGCGAAAGGGAAACC-MGB-3'). The viral titer (VP/mL) was calculated as described by [83], using the following formula: {[(Average LTR copy number * 2 / Average β-actin copy number) * cell plated number] * dilution factor}. The lentiviral transductions were subsequently carried out at a multiplicity of infection (MOI) ranging from 30 to 50.
4.4. Lentiviral Transduction and Establishment of Stable Transduced Cell Lines
Stable cell populations expressing the codon-optimized GBA1 (GBA-Opt) were generated by six consecutive rounds of lentiviral transduction in 293FT cells, using MOIs ranging from 30 to 50. For each round, 2 x 105 cells were seeded in 6-well plates (2 mL DMEM per well). After 8 h, the medium was replaced with Opti-MEM (Gibco) and lentiviral particles were added in volume calculated based on viral tier (VP/mL) and desired MOI. Polybrene (6 μg/ mL; Sigma-Aldrich) was added to enhance transduction efficiency. Cells were subjected to centrifugation (spinoculation) at 1,200 rpm for 40 min. at 22oC. The following day, the medium was replaced with DMEM containing 10% FBS and 1% penicillin/streptomycin. Ater recovery and expansion, cells were trypsinized and replated for the next round of transduction. The interval between transduction cycles ranged from 2 to 7 days. Aliquots of transduced cells were cryopreserved between cycles to ensure experimental reproducibility.
4.5. Puromycin Treatment of L17_293FT_GBA_OPT_HP Heterogeneous Population
After the generation of the L17_293FT_GBA_OPT_HP heterogeneous population through 10 days of selection with 2 μg/mL of puromycin, cells were further exposed to 1 µg/mL or 5 μg/mL puromycin for 5 additional days to investigate the effect of different selection stringencies on GCase production. Following treatment, cells were expanded in T-75 flasks for enzymatic assays.
To ensure consistent selection pressure and maintain the expression of the integrated transgene, all experimental procedures involving this population, including scale-up studies, were preceded by a standardized 5-day puromycin treatment at 2 μg/mL. This approach was adopted across all assays involving L17_293FT_GBA_OPT_HP cells.
4.6. GCase Activity Analysis: Secreted and Intracellular (GCase-Specific Activity)
For enzymatic activity of secreted GCase activity, 3 × 105 cells were seeded per well in 6-well plates and cultured for 48 h in DMEM supplemented with 10% FBS. The medium was then replaced for serum-free DMEM, and cells were incubated for an additional 48 h. At the end of this period, supernatants were collected, centrifuged at 1,500 rpm for 5 minutes at 10oC to remove cellular debris, and stored at -80oC until enzymatic analysis.
For intracellular GCase-specific activity, 2 × 105 cells were seeded in 6-well plates and cultured in DMEM with 10% FBS and 1% antibiotics. After 48 h, the medium was replaced with fresh DMEM containing 10% FBS and 1% antibiotics. Following additional 48 h, cell pellets were collected in 245 μL of Mammalian Protein Extraction Buffer (GE Healthcare Life Sciences), supplemented with 5 μL of protease inhibitor cocktail (Sigma-Aldrich). Lysates were homogenized and stored at -80 oC for subsequent analysis.
4.7. Scaling of GCase Production in L17_293FT_GBA_OPT_HP Cell Supernatants
For scale-up, 2 x 107 cells from L17_293FT_GBA_OPT_HP population were seeded in 10-layer Cell Culture Multi-Flask – BD Biosciences) and cultured for 72 h in 560 mL of DMEM containing 10% FBS and 1% antibiotic. After this period, the medium was replaced with serum-free DMEM, and cells were maintained for an additional 48 h. The supernatants were then harvested for enzymatic activity analysis. Cell pellets were collected by centrifugation at 2000 rpm, for 10 minutes at 10oC and used for total cell count.
4.8. Clone Cell Selection (Isolation)
Following the selection of the heterogeneous populations with 2 μg/mL puromycin, cells were expanded and 1x103 transduced cells were seeded in 100 mm culture dishes to allow for single-cell clonal expansion. After approximately one-week, discrete clonal colonies became visible and were individually harvested by trypsinization and transferred to 6-well plates for expansion. Each clonal population was then treated with puromycin (2 μg/mL) for five additional days to ensure transgene expression stability. Expanded clones were cryopreserved in aliquots of 3 × 106 cells per cryotube and stored at -80 oC for further analyses.
4.9. Biological Activity by Fluorimetric Assay
Lysosomal GCase activities were measured using the synthetic fluorescent substrate 4-methylumbelliferyl-β-D-glucopyranoside (4-MUG) in the presence of sodium taurodeoxycholate according to the method described by [84], adapted from [85], and previously described by our group [44].
Before the enzymatic assay, total protein was quantified using the Lowry method [86] with the DCTM Protein Assay Kit (Bio-Rad), following the manufacturer’s instructions. All samples, including leukocytes lysates, virgin cells, and transduced cell lines, were homogenized by sonication (3x 10s at 60 W, on ice) to guarantee complete cell lysis and protein extraction. The protein concentration was determined using a standard curve generated with serial dilutions of bovine serum albumin (BSA; 1.4 mg/mL to 0.175 mg/mL).
Each enzymatic reaction was prepared in amber tubes with 30μL of the sample (containing 40-60 μg of total protein), 50 μL of 20 mM 4-MUG substrate, and 20 μL of 2% sodium taurodeoxycholate in 1M citrate-phosphate buffer (pH=5.0). Reactions were incubated at 37 oC for 2 h, with gentle agitation, then cooled on ice for 5 minutes and stopped with the addition of 2 mL of glycine-KOH buffer (0.25 M, pH 10.3). All reactions were performed in triplicate.
A 200 μL aliquot of each reaction was transferred to black 96-well microplates, and relative fluorescence was measured using a Cary Eclipse Fluorescence Spectrophotometer (Agilent) with excitation at 360 nm and emission at 450 nm. Fluorescence readings were corrected using blanks and quantified by interpolation against a 4-methylumbelliferone (4-MU) standard curve. The enzymatic activity, as described in the literature [84], was expressed as nmol of hydrolyzed substrate per mg of protein per 1h. As reference, leukocyte lysates from healthy donors were included, with expected values between 8.68 and 11.57 nmol/ mg/ h as previously established [84].
The biological activity of secreted GCase present in the cell culture supernatant was also evaluated. The reaction conditions were identical to those described for intracellular lysates, except that 30 μL of supernatant was directly added to the reaction mix without adjustment for protein concentration. As with intracellular activity, all reactions were performed in triplicate, and fluorescence values were corrected using blanks and interpolated from the 4-MU standard curve.
4.10. Statistical Analysis
Statistical analyzes were performed using the non-parametric t-test with Welch correction, with p-value set at 0.05. Graphs were generated using GraphPad Prism software version 8.00 (GraphPad Software, San Diego California USA).
Author Contributions
A.C.C.: Methodology: Investigation, Writing - original draft. C.E.V.W.: Methodology, Visualization, Writing - original draft. L.F., G.S., J.B., V.F.: Methodology, Investigation. D.T., C.C-J.: Conceptualization, Resources, Visualization. A.C., K.A.: Data curation, Writing - review and editing, Visualization. V.F.: Methodology, Investigation, Writing – review and editing. S.G., V.S.: Conceptualization, Resources, Writing—review and editing. R.W.: Conceptualization, Resources, Funding acquisition, A.M.F.: Conceptualization, Formal analysis, Supervision, Project administration, Writing - review and editing, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
Funding
This study was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) in collaboration with Agilent Technologies (Grant No. 2013/50450-2), the FAPESP-MIT program (Grant No. 2013/50764-7), and a FAPESP doctoral fellowship (Grant No. 2014/15846-5). Additional support was provided by Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES – Finance Code 001), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) and Foundation for Support to Teaching, Research, and Assistance of the HC-FMRP-USP (FAEPA).
Acknowledgments
The authors thank Profa. Dra. Elza Tiemi Sakamoto Hojo (FFCLRP-USP) for providing access to the spectrophotometer used in the Lowry protein quantification assays. We are also grateful to Elisabete Maria S. Barreto-Beira and Rosana Márcia Silva Cruz for their technical support. Lysosome illustration were adapted from BioRender.com and integrated into a custom-designed graphical abstract.
Conflicts of Interest
The authors declare no conflicts of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| Abbreviation | Meaning |
| CHO | Chinese hamster ovary cell |
| DMEM | Dulbecco's Modified Eagle's Medium |
| ERT | Enzyme replacement therapy |
| ELISA | Enzyme-linked immunosorbent assay |
| FBS | Fetal bovine serum |
| FDA | Food and Drug Administration |
| GBA1 | Glucocerebrosidase, Glucosylceramidase beta 1 |
| GCase | Glucocerebrosidase, Glucosylceramidase beta 1 |
| GD | Gaucher disease |
| GlcCer | Glucosylceramide |
| 293_FT | Human embryonic kidney 293 cells |
| HP | Heterogeneous population |
| HS | High stringency |
| ICGG | International Collaborative Gaucher Group |
| L17 | Lineage 17 |
| LS | Low stringency |
| LV | Lentiviral vector |
| LTR | Long terminal repeat |
| MOI | Multiplicity of infection |
| MTX | Methotrexate |
| 4MU | 4-methylumbiliferone |
| 4MUG | 4-methylumbiliferon-β-D-glucopyranoside |
| qPCR | Real-time quantitative PCR |
| SUS | Unified Health System |
| TDC | Sodium taurodeoxycholate hydrate |
References
- Hughes DA, Pastores GM: Gaucher Disease. In GeneReviews((R)). Edited by Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A. Seattle (WA); 1993.
- Brady RO, Kanfer JN, Shapiro D: Metabolism of Glucocerebrosides. Ii. Evidence of an Enzymatic Deficiency in Gaucher's Disease. Biochem Biophys Res Commun 1965, 18:221-225. [CrossRef]
- Elstein D, Belmatoug N, Bembi B, Deegan P, Fernandez-Sasso D, Giraldo P, Goker-Alpan O, Hughes D, Lau H, Lukina E, et al: Twelve Years of the Gaucher Outcomes Survey (GOS): Insights, Achievements, and Lessons Learned from a Global Patient Registry. J Clin Med 2024, 13. [CrossRef]
- Belinsky G, Ruan J, Fattahi N, Mehta S, Boddupalli CS, Mistry PK, Nair S: Modeling bone marrow microenvironment and hematopoietic dysregulation in Gaucher disease through VavCre mediated Gba deletion. Hum Mol Genet 2025. [CrossRef]
- Ducatez F, Berger MG, Pilon C, Plichet T, Lesueur C, Berger J, Belmatoug N, Marret S, Bekri S, Tebani A: Deciphering metabolic shifts in Gaucher disease type 1: a multi-omics study. J Mol Med (Berl) 2025, 103:187-203. [CrossRef]
- Grabowski GA: Phenotype, diagnosis, and treatment of Gaucher's disease. Lancet 2008, 372:1263-1271. [CrossRef]
- Cohen D, Levy Y, Bar-Ziv Y, Revel-Vilk S, Zimran A, Lebel E: Simultaneous Bilateral Femoral Osteonecrosis in Gaucher Disease. Life (Basel) 2023, 13. [CrossRef]
- Grabowski GA, Antommaria AHM, Kolodny EH, Mistry PK: Gaucher disease: Basic and translational science needs for more complete therapy and management. Mol Genet Metab 2021, 132:59-75. [CrossRef]
- Grabowski GA, Kishnani PS, Alcalay RN, Prakalapakorn SG, Rosenbloom BE, Tuason DA, Weinreb NJ: Challenges in Gaucher disease: Perspectives from an expert panel. Mol Genet Metab 2025, 145:109074. [CrossRef]
- Furderer ML, Hertz E, Lopez GJ, Sidransky E: Neuropathological Features of Gaucher Disease and Gaucher Disease with Parkinsonism. Int J Mol Sci 2022, 23. [CrossRef]
- Hertz E, Chen Y, Sidransky E: Gaucher disease provides a unique window into Parkinson disease pathogenesis. Nat Rev Neurol 2024, 20:526-540. [CrossRef]
- Imbalzano G, Ledda C, Romagnolo A, Covolo A, Lopiano L, Artusi CA: Neurological symptoms in adults with Gaucher disease: a systematic review. J Neurol 2024, 271:3897-3907. [CrossRef]
- Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER, Bar-Shira A, Berg D, Bras J, Brice A, et al: Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009, 361:1651-1661. [CrossRef]
- Motta I, Delbini P, Scaramellini N, Ghiandai V, Duca L, Nava I, Nascimbeni F, Lugari S, Consonni D, Trombetta E, et al: Enzyme replacement therapy improves erythropoiesis and iron dysregulation in Gaucher disease. Ann Hematol 2024, 103:5113-5121. [CrossRef]
- Deegan P, Lau H, Elstein D, Fernandez-Sasso D, Giraldo P, Hughes D, Zimran A, Istaiti M, Gadir N, Botha J, et al: Long-Term Treatment of Gaucher Disease with Velaglucerase Alfa in ERT-Naive Patients from the Gaucher Outcome Survey (GOS) Registry. J Clin Med 2024, 13. [CrossRef]
- Revel-Vilk S, Mansfield R, Feder-Krengel N, Machtiger-Azoulay N, Kuter D, Szer J, Rosenbaum H, Ferreira DC, Ruhrman-Shahar N, Wajnrajch M, Zimran A: Real-World Experiences with Taliglucerase Alfa Home Infusions for Patients with Gaucher Disease: A Global Cohort Study. J Clin Med 2023, 12. [CrossRef]
- Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI, Argoff CE, et al.: Replacement therapy for inherited enzyme deficiency--macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med 1991, 324:1464-1470. [CrossRef]
- Weinreb NJ, Camelo JS, Jr., Charrow J, McClain MR, Mistry P, Belmatoug N, International Collaborative Gaucher Group Gaucher Registry i: Gaucher disease type 1 patients from the ICGG Gaucher Registry sustain initial clinical improvements during twenty years of imiglucerase treatment. Mol Genet Metab 2021, 132:100-111. [CrossRef]
- Mistry PK, Belmatoug N, vom Dahl S, Giugliani R: Understanding the natural history of Gaucher disease. Am J Hematol 2015, 90 Suppl 1:S6-11. [CrossRef]
- Stepien KM, Kiec-Wilk B, Lampe C, Tangeraas T, Cefalo G, Belmatoug N, Francisco R, Del Toro M, Wagner L, Lauridsen AG, et al: Challenges in Transition From Childhood to Adulthood Care in Rare Metabolic Diseases: Results From the First Multi-Center European Survey. Front Med (Lausanne) 2021, 8:652358. [CrossRef]
- Borin MC, Alvares-Teodoro J, Acurcio FA, Guerra AA, Jr.: Gaucher disease in Brazil: a comprehensive 16 year retrospective study on survival, cost, and treatment insights. Front Pharmacol 2024, 15:1433970. [CrossRef]
- Grabowski GA, Barton NW, Pastores G, Dambrosia JM, Banerjee TK, McKee MA, Parker C, Schiffmann R, Hill SC, Brady RO: Enzyme therapy in type 1 Gaucher disease: comparative efficacy of mannose-terminated glucocerebrosidase from natural and recombinant sources. Ann Intern Med 1995, 122:33-39. [CrossRef]
- Zimran A, Elstein D, Levy-Lahad E, Zevin S, Hadas-Halpern I, Bar-Ziv Y, Foldes J, Schwartz AJ, Abrahamov A: Replacement therapy with imiglucerase for type 1 Gaucher's disease. Lancet 1995, 345:1479-1480. [CrossRef]
- Zimran A, Altarescu G, Philips M, Attias D, Jmoudiak M, Deeb M, Wang N, Bhirangi K, Cohn GM, Elstein D: Phase 1/2 and extension study of velaglucerase alfa replacement therapy in adults with type 1 Gaucher disease: 48-month experience. Blood 2010, 115:4651-4656. [CrossRef]
- Zimran A: Velaglucerase alfa: a new option for Gaucher disease treatment. Drugs Today (Barc) 2011, 47:515-529. [CrossRef]
- Zimran A, Brill-Almon E, Chertkoff R, Petakov M, Blanco-Favela F, Munoz ET, Solorio-Meza SE, Amato D, Duran G, Giona F, et al: Pivotal trial with plant cell-expressed recombinant glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement therapy for Gaucher disease. Blood 2011, 118:5767-5773. [CrossRef]
- van Dussen L, Zimran A, Akkerman EM, Aerts JM, Petakov M, Elstein D, Rosenbaum H, Aviezer D, Brill-Almon E, Chertkoff R, et al: Taliglucerase alfa leads to favorable bone marrow responses in patients with type I Gaucher disease. Blood Cells Mol Dis 2013, 50:206-211. [CrossRef]
- BRASIL: Ministério da Saúde. Secretaria de Atenção à Saúde. Protocolo Clínico e Diretrizes Terapêuticas da Doença de Gaucher. Brasília, 2014 (atualizado em 2017). 2017.
- Liu C, Bahnson AB, Dunigan JT, Watkins SC, Barranger JA: Long-term expression and secretion of human glucocerebrosidase by primary murine and human myoblasts and differentiated myotubes. J Mol Med (Berl) 1998, 76:773-781. [CrossRef]
- Kim EY, Hong YB, Lai Z, Kim HJ, Cho YH, Brady RO, Jung SC: Expression and secretion of human glucocerebrosidase mediated by recombinant lentivirus vectors in vitro and in vivo: implications for gene therapy of Gaucher disease. Biochem Biophys Res Commun 2004, 318:381-390. [CrossRef]
- Scharenberg SG, Poletto E, Lucot KL, Colella P, Sheikali A, Montine TJ, Porteus MH, Gomez-Ospina N: Engineering monocyte/macrophage-specific glucocerebrosidase expression in human hematopoietic stem cells using genome editing. Nat Commun 2020, 11:3327. [CrossRef]
- Novo JB, Morganti L, Moro AM, Paes Leme AF, Serrano SM, Raw I, Ho PL: Generation of a Chinese hamster ovary cell line producing recombinant human glucocerebrosidase. J Biomed Biotechnol 2012, 2012:875383. [CrossRef]
- Naphatsamon U, Ohashi T, Misaki R, Fujiyama K: The Production of Human beta-Glucocerebrosidase in Nicotiana benthamiana Root Culture. Int J Mol Sci 2018, 19. [CrossRef]
- Uthailak N, Kajiura H, Misaki R, Fujiyama K: Production of recombinant beta-glucocerebrosidase in wild-type and glycoengineered transgenic Nicotiana benthamiana root cultures with different N-glycan profiles. J Biosci Bioeng 2022, 133:481-488. [CrossRef]
- Hia F, Yang SF, Shichino Y, Yoshinaga M, Murakawa Y, Vandenbon A, Fukao A, Fujiwara T, Landthaler M, Natsume T, et al: Codon bias confers stability to human mRNAs. EMBO Rep 2019, 20:e48220. [CrossRef]
- Wu G, Zheng Y, Qureshi I, Zin HT, Beck T, Bulka B, Freeland SJ: SGDB: a database of synthetic genes re-designed for optimizing protein over-expression. Nucleic Acids Res 2007, 35:D76-79. [CrossRef]
- Plotkin JB, Kudla G: Synonymous but not the same: the causes and consequences of codon bias. Nat Rev Genet 2011, 12:32-42. [CrossRef]
- VandenDriessche T, Chuah MK: Hemophilia Gene Therapy: Ready for Prime Time? Hum Gene Ther 2017, 28:1013-1023. [CrossRef]
- Srivastava A, Abraham A, Aboobacker F, Singh G, Geevar T, Kulkarni U, Selvarajan S, Korula A, Dave RG, Shankar M, et al: Lentiviral Gene Therapy with CD34+ Hematopoietic Cells for Hemophilia A. N Engl J Med 2025, 392:450-457. [CrossRef]
- Poletti V, Charrier S, Corre G, Gjata B, Vignaud A, Zhang F, Rothe M, Schambach A, Gaspar HB, Thrasher AJ, Mavilio F: Preclinical Development of a Lentiviral Vector for Gene Therapy of X-Linked Severe Combined Immunodeficiency. Mol Ther Methods Clin Dev 2018, 9:257-269. [CrossRef]
- Banning A, Konig JF, Gray SJ, Tikkanen R: Functional Analysis of the Ser149/Thr149 Variants of Human Aspartylglucosaminidase and Optimization of the Coding Sequence for Protein Production. Int J Mol Sci 2017, 18. [CrossRef]
- Jeyakumar JM, Kia A, Tam LCS, McIntosh J, Spiewak J, Mills K, Heywood W, Chisari E, Castaldo N, Verhoef D, et al: Preclinical evaluation of FLT190, a liver-directed AAV gene therapy for Fabry disease. Gene Ther 2023, 30:487-502. [CrossRef]
- Sinclair G, Choy FY: Synonymous codon usage bias and the expression of human glucocerebrosidase in the methylotrophic yeast, Pichia pastoris. Protein Expr Purif 2002, 26:96-105. [CrossRef]
- Figueiredo LLS, Lau Jr W, Goncalves VWS, Ramos ES, D’Almeida V, Souza LEB, Orellana MD, Abraham KJ, Lichtenstein F, Bleicher L, et al: Engineering Synthetic and Recombinant Human Lysosomal β-Glucocerebrosidase for Enzyme Replacement Therapy for Gaucher Disease. Discover Applied Sciences 2024, 6. [CrossRef]
- Ungari S, Montepeloso A, Morena F, Cocchiarella F, Recchia A, Martino S, Gentner B, Naldini L, Biffi A: Design of a regulated lentiviral vector for hematopoietic stem cell gene therapy of globoid cell leukodystrophy. Mol Ther Methods Clin Dev 2015, 2:15038. [CrossRef]
- Pimentel N, Rodriguez-Lopez A, Diaz S, Losada JC, Diaz-Rincon DJ, Cardona C, Espejo-Mojica AJ, Ramirez AM, Ruiz F, Landazuri P, et al: Production and characterization of a human lysosomal recombinant iduronate-2-sulfatase produced in Pichia pastoris. Biotechnol Appl Biochem 2018, 65:655-664. [CrossRef]
- Smith MC, Belur LR, Karlen AD, Erlanson O, Podetz-Pedersen KM, McKenzie J, Detellis J, Gagnidze K, Parsons G, Robinson N, et al: Phenotypic Correction of Murine Mucopolysaccharidosis Type II by Engraftment of Ex Vivo Lentiviral Vector-Transduced Hematopoietic Stem and Progenitor Cells. Hum Gene Ther 2022, 33:1279-1292. [CrossRef]
- Pan X, Sands SA, Yue Y, Zhang K, LeVine SM, Duan D: An Engineered Galactosylceramidase Construct Improves AAV Gene Therapy for Krabbe Disease in Twitcher Mice. Hum Gene Ther 2019, 30:1039-1051. [CrossRef]
- Doerfler PA, Todd AG, Clement N, Falk DJ, Nayak S, Herzog RW, Byrne BJ: Copackaged AAV9 Vectors Promote Simultaneous Immune Tolerance and Phenotypic Correction of Pompe Disease. Hum Gene Ther 2016, 27:43-59. [CrossRef]
- Stok M, de Boer H, Huston MW, Jacobs EH, Roovers O, Visser TP, Jahr H, Duncker DJ, van Deel ED, Reuser AJJ, et al: Lentiviral Hematopoietic Stem Cell Gene Therapy Corrects Murine Pompe Disease. Mol Ther Methods Clin Dev 2020, 17:1014-1025. [CrossRef]
- Liang Q, Catalano F, Vlaar EC, Pijnenburg JM, Stok M, van Helsdingen Y, Vulto AG, van der Ploeg AT, van Til NP, Pijnappel W: IGF2-tagging of GAA promotes full correction of murine Pompe disease at a clinically relevant dosage of lentiviral gene therapy. Mol Ther Methods Clin Dev 2022, 27:109-130. [CrossRef]
- Ornaghi F, Sala D, Tedeschi F, Maffia MC, Bazzucchi M, Morena F, Valsecchi M, Aureli M, Martino S, Gritti A: Novel bicistronic lentiviral vectors correct beta-Hexosaminidase deficiency in neural and hematopoietic stem cells and progeny: implications for in vivo and ex vivo gene therapy of GM2 gangliosidosis. Neurobiol Dis 2020, 134:104667. [CrossRef]
- Hu M, Xu Q, Zhang F, Buckland KF, Gao Y, Du W, Ding Y, Zhou L, Sun X, Ma L, et al: Preclinical ex vivo IL2RG gene therapy using autologous hematopoietic stem cells as an effective and safe treatment for X-linked severe combined immunodeficiency disease. Genes Dis 2025, 12:101445. [CrossRef]
- Saleh AH, Rothe M, Barber DL, McKillop WM, Fraser G, Morel CF, Schambach A, Auray-Blais C, West ML, Khan A, et al: Persistent hematopoietic polyclonality after lentivirus-mediated gene therapy for Fabry disease. Mol Ther Methods Clin Dev 2023, 28:262-271. [CrossRef]
- Mangiameli E, Cecchele A, Morena F, Sanvito F, Matafora V, Cattaneo A, Della Volpe L, Gnani D, Paulis M, Susani L, et al: Human iPSC-based neurodevelopmental models of globoid cell leukodystrophy uncover patient- and cell type-specific disease phenotypes. Stem Cell Reports 2021, 16:1478-1495. [CrossRef]
- Ellison S, Buckland K, Learmonth Y, Day V, Kalra S, Howe L, Roman-Rodriguez FJ, Bonafont J, Booth L, Holley R, et al: Design and validation of a GMP stem cell manufacturing protocol for MPSII hematopoietic stem cell gene therapy. Mol Ther Methods Clin Dev 2024, 32:101271. [CrossRef]
- Dogan Y, Barese CN, Schindler JW, Yoon JK, Unnisa Z, Guda S, Jacobs ME, Oborski C, Maiwald T, Clarke DL, et al: Screening chimeric GAA variants in preclinical study results in hematopoietic stem cell gene therapy candidate vectors for Pompe disease. Mol Ther Methods Clin Dev 2022, 27:464-487. [CrossRef]
- Yilmaz BS, Gurung S, Perocheau D, Counsell J, Baruteau J: Gene therapy for inherited metabolic diseases. J Mother Child 2020, 24:53-64. [CrossRef]
- Zielske SP, Gerson SL: Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther 2003, 7:325-333. [CrossRef]
- Fontes AM, Davis BM, Encell LP, Lingas K, Covas DT, Zago MA, Loeb LA, Pegg AE, Gerson SL: Differential competitive resistance to methylating versus chloroethylating agents among five O6-alkylguanine DNA alkyltransferases in human hematopoietic cells. Mol Cancer Ther 2006, 5:121-128. [CrossRef]
- Fontes AM, Melo FU, Greene LJ, Faca VM, Lin Y, Gerson SL, Covas DT: Production of human factor VIII-FL in 293T cells using the bicistronic MGMT(P140K)-retroviral vector. Genet Mol Res 2012, 11:775-789. [CrossRef]
- Castilho-Fernandes A, Fontes AM, Abraham KJ, de Freitas MC, da Rosa NG, Picanco-Castro V, de Sousa Russo-Carbolante EM, Covas DT: Significant differences in integration sites of Moloney murine leukemia virus/Moloney murine sarcoma virus retroviral vector carrying recombinant coagulation factor IX in two human cell lines. Biotechnol Lett 2015, 37:991-1001. [CrossRef]
- Correa de Freitas MC, Fontes AM, de Castilho Fernandes A, Picanco-Castro V, de Sousa Russo EM, Covas DT: Murine leukemia virus-derived retroviral vector has differential integration patterns in human cell lines used to produce recombinant factor VIII. Rev Bras Hematol Hemoter 2014, 36:213-218. [CrossRef]
- Milone MC, O'Doherty U: Clinical use of lentiviral vectors. Leukemia 2018, 32:1529-1541. [CrossRef]
- Naldini L, Trono D, Verma IM: Lentiviral vectors, two decades later. Science 2016, 353:1101-1102. [CrossRef]
- Scotti C, Aiuti A, Naldini L: Challenges and solutions to the sustainability of gene and cell therapies. Nat Rev Genet 2025. [CrossRef]
- al Yacoub N, Romanowska M, Haritonova N, Foerster J: Optimized production and concentration of lentiviral vectors containing large inserts. J Gene Med 2007, 9:579-584. [CrossRef]
- Hirch T, Brander N, Schenk F, Pollmann SJ, Reichenbach J, Schubert R, Modlich U: Expression of a large coding sequence: Gene therapy vectors for Ataxia Telangiectasia. Sci Rep 2023, 13:19386. [CrossRef]
- Wang Z, Chen C, Ge X: Large T antigen mediated target gene replication improves site-specific recombination efficiency. Front Bioeng Biotechnol 2024, 12:1377167. [CrossRef]
- Abaandou L, Quan D, Shiloach J: Affecting HEK293 Cell Growth and Production Performance by Modifying the Expression of Specific Genes. Cells 2021, 10. [CrossRef]
- Thomas P, Smart TG: HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods 2005, 51:187-200. [CrossRef]
- Perry C, Rayat A: Lentiviral Vector Bioprocessing. Viruses 2021, 13. [CrossRef]
- Jargalsaikhan BE, Muto M, Been Y, Matsumoto S, Okamura E, Takahashi T, Narimichi Y, Kurebayashi Y, Takeuchi H, Shinohara T, et al: The Dual-Pseudotyped Lentiviral Vector with VSV-G and Sendai Virus HN Enhances Infection Efficiency through the Synergistic Effect of the Envelope Proteins. Viruses 2024, 16. [CrossRef]
- Jia J, Hao Y, Zhang L, Cao X, An L, Wang H, Ma Q, Jin X, Ma X: Development and validation of optimized lentivirus-like particles for gene editing tool delivery with Gag-Only strategy. Eur J Med Res 2025, 30:242. [CrossRef]
- Cabrera A, Edelstein HI, Glykofrydis F, Love KS, Palacios S, Tycko J, Zhang M, Lensch S, Shields CE, Livingston M, et al: The sound of silence: Transgene silencing in mammalian cell engineering. Cell Syst 2022, 13:950-973. [CrossRef]
- Xu ZJ, Jia YL, Wang M, Yi DD, Zhang WL, Wang XY, Zhang JH: Effect of promoter, promoter mutation and enhancer on transgene expression mediated by episomal vectors in transfected HEK293, Chang liver and primary cells. Bioengineered 2019, 10:548-560. [CrossRef]
- Fumagalli F, Calbi V, Natali Sora MG, Sessa M, Baldoli C, Rancoita PMV, Ciotti F, Sarzana M, Fraschini M, Zambon AA, et al: Lentiviral haematopoietic stem-cell gene therapy for early-onset metachromatic leukodystrophy: long-term results from a non-randomised, open-label, phase 1/2 trial and expanded access. Lancet 2022, 399:372-383. [CrossRef]
- Liang Q, Vlaar EC, Pijnenburg JM, Rijkers E, Demmers JAA, Vulto AG, van der Ploeg AT, van Til NP, Pijnappel W: Lentiviral gene therapy with IGF2-tagged GAA normalizes the skeletal muscle proteome in murine Pompe disease. J Proteomics 2024, 291:105037. [CrossRef]
- da Rosa NG, Swiech K, Picanco-Castro V, Russo-Carbolante EM, Soares Neto MA, de Castilho-Fernandes A, Faca VM, Fontes AM, Covas DT: SK-HEP cells and lentiviral vector for production of human recombinant factor VIII. Biotechnol Lett 2012, 34:1435-1443. [CrossRef]
- Spencer HT, Denning G, Gautney RE, Dropulic B, Roy AJ, Baranyi L, Gangadharan B, Parker ET, Lollar P, Doering CB: Lentiviral vector platform for production of bioengineered recombinant coagulation factor VIII. Mol Ther 2011, 19:302-309. [CrossRef]
- Kutner RH, Zhang XY, Reiser J: Production, concentration and titration of pseudotyped HIV-1-based lentiviral vectors. Nat Protoc 2009, 4:495-505. [CrossRef]
- Fantacini DM, Fontes AM, de Abreu Neto MS, Covas DT, Picanco-Castro V: The F309S mutation increases factor VIII secretion in human cell line. Rev Bras Hematol Hemoter 2016, 38:135-140. [CrossRef]
- Matsui H, Shibata M, Brown B, Labelle A, Hegadorn C, Andrews C, Hebbel RP, Galipeau J, Hough C, Lillicrap D: Ex vivo gene therapy for hemophilia A that enhances safe delivery and sustained in vivo factor VIII expression from lentivirally engineered endothelial progenitors. Stem Cells 2007, 25:2660-2669. [CrossRef]
- Peters SP, Coyle P, Glew RH: Differentiation of beta-glucocerebrosidase from beta-glucosidase in human tissues using sodium taurocholate. Arch Biochem Biophys 1976, 175:569-582. [CrossRef]
- Muller KB, Rodrigues MD, Pereira VG, Martins AM, D'Almeida V: Reference values for lysosomal enzymes activities using dried blood spots samples - a Brazilian experience. Diagn Pathol 2010, 5:65. [CrossRef]
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193:265-275.
Figure 1.
GCase activity in L17_293FT_GBA_OPT_HP cell supernatants following puromycin selection. (A) Enzymatic activity (nmol hydrolyzed substrate per mL per h) significantly increased upon puromycin treatment. Treatment with 1 μg/mL puromycin (low stringency, ls) 329.7 ± 31.02 nmol/mL/h of GCase activity, while 5 μg/mL (high stringency, hs) increased activity to 459.7 ± 24.82 nmol/mL/h. Compared to the untreated 293FT/virgin control, GCase activity increased by 47-fold in ls (p = 0.0002; t = 20.81, df = 3.001; n = 4) and by 62.4-fold in hs (p < 0.0001; t = 36.48, df = 3.002; n = 4). The comparison between hs and ls showed a 1.4-fold increase in activity (p = 0.0007; t = 6.547, df = 5.725; n = 4). (B) GCase activity normalized per million cells (U GCase/10⁶ cells) was also enhanced by puromycin selection. The hp_ls population exhibited 215.4 ± 39.46 U/10⁶ cells, while hp_hs reached 346.1 ± 26.27 U/10⁶ cells. Compared to 293FT/Virgin cells (4.44 ± 0.21 U/10⁶ cells), this corresponds to 48.5-fold (p = 0.0017; t = 10.69, df = 3.000; n = 4) and 78-fold (p = 0.0001; t = 26.01, df = 3.000; n = 4) increases, respectively. The comparison between hp_hs and hp_ls showed a 1.6-fold increase (p = 0.0023; t = 5.515, df = 5.223; n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. ** indicates p ≤ 0.01, *** indicates p ≤ 0.001, and **** indicates p ≤ 0.0001. [293FT: human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α: human elongation factor 1-alpha promoter]; [GBA-Opt: codon-optimized human GBA (glucocerebrosidase) cDNA sequence]; [hp_ls: heterogeneous population under low-stringency puromycin selection (1 µg/mL)]; [hp_hs: heterogeneous population under high-stringency puromycin selection (5 µg/mL)]; [GCase: β-glucocerebrosidase enzyme].
Figure 1.
GCase activity in L17_293FT_GBA_OPT_HP cell supernatants following puromycin selection. (A) Enzymatic activity (nmol hydrolyzed substrate per mL per h) significantly increased upon puromycin treatment. Treatment with 1 μg/mL puromycin (low stringency, ls) 329.7 ± 31.02 nmol/mL/h of GCase activity, while 5 μg/mL (high stringency, hs) increased activity to 459.7 ± 24.82 nmol/mL/h. Compared to the untreated 293FT/virgin control, GCase activity increased by 47-fold in ls (p = 0.0002; t = 20.81, df = 3.001; n = 4) and by 62.4-fold in hs (p < 0.0001; t = 36.48, df = 3.002; n = 4). The comparison between hs and ls showed a 1.4-fold increase in activity (p = 0.0007; t = 6.547, df = 5.725; n = 4). (B) GCase activity normalized per million cells (U GCase/10⁶ cells) was also enhanced by puromycin selection. The hp_ls population exhibited 215.4 ± 39.46 U/10⁶ cells, while hp_hs reached 346.1 ± 26.27 U/10⁶ cells. Compared to 293FT/Virgin cells (4.44 ± 0.21 U/10⁶ cells), this corresponds to 48.5-fold (p = 0.0017; t = 10.69, df = 3.000; n = 4) and 78-fold (p = 0.0001; t = 26.01, df = 3.000; n = 4) increases, respectively. The comparison between hp_hs and hp_ls showed a 1.6-fold increase (p = 0.0023; t = 5.515, df = 5.223; n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. ** indicates p ≤ 0.01, *** indicates p ≤ 0.001, and **** indicates p ≤ 0.0001. [293FT: human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α: human elongation factor 1-alpha promoter]; [GBA-Opt: codon-optimized human GBA (glucocerebrosidase) cDNA sequence]; [hp_ls: heterogeneous population under low-stringency puromycin selection (1 µg/mL)]; [hp_hs: heterogeneous population under high-stringency puromycin selection (5 µg/mL)]; [GCase: β-glucocerebrosidase enzyme].
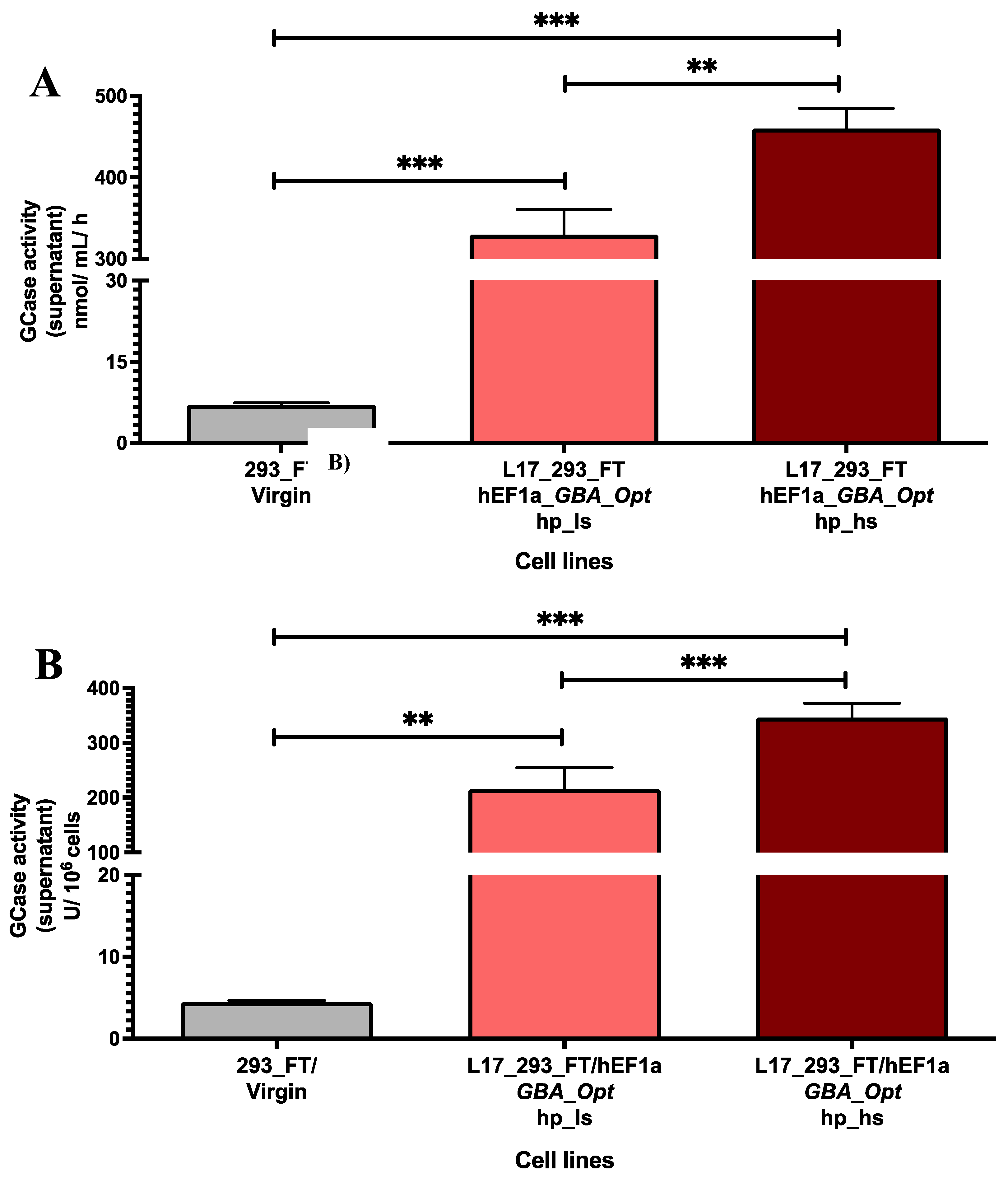
Figure 2.
Total GCase production at different culture scales. Total β-glucocerebrosidase (GCase) activity (nmol/h) obtained from 6-well plate (2 mL) and from a 10-layer Cell Factory system (560 mL) for the L17_293FT_GBA_OPT_HP cell line. A 963-fold increase in total enzyme production was observed upon scale-up, with preserved volumetric activity. Values are presented on a base-10 logarithmic scale. [293_FT = human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α = human elongation factor 1-alpha promoter]; [GBA-Opt = codon-optimized human GBA (glucocerebrosidase) cDNA]; [GCase = β-glucocerebrosidase].
Figure 2.
Total GCase production at different culture scales. Total β-glucocerebrosidase (GCase) activity (nmol/h) obtained from 6-well plate (2 mL) and from a 10-layer Cell Factory system (560 mL) for the L17_293FT_GBA_OPT_HP cell line. A 963-fold increase in total enzyme production was observed upon scale-up, with preserved volumetric activity. Values are presented on a base-10 logarithmic scale. [293_FT = human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α = human elongation factor 1-alpha promoter]; [GBA-Opt = codon-optimized human GBA (glucocerebrosidase) cDNA]; [GCase = β-glucocerebrosidase].
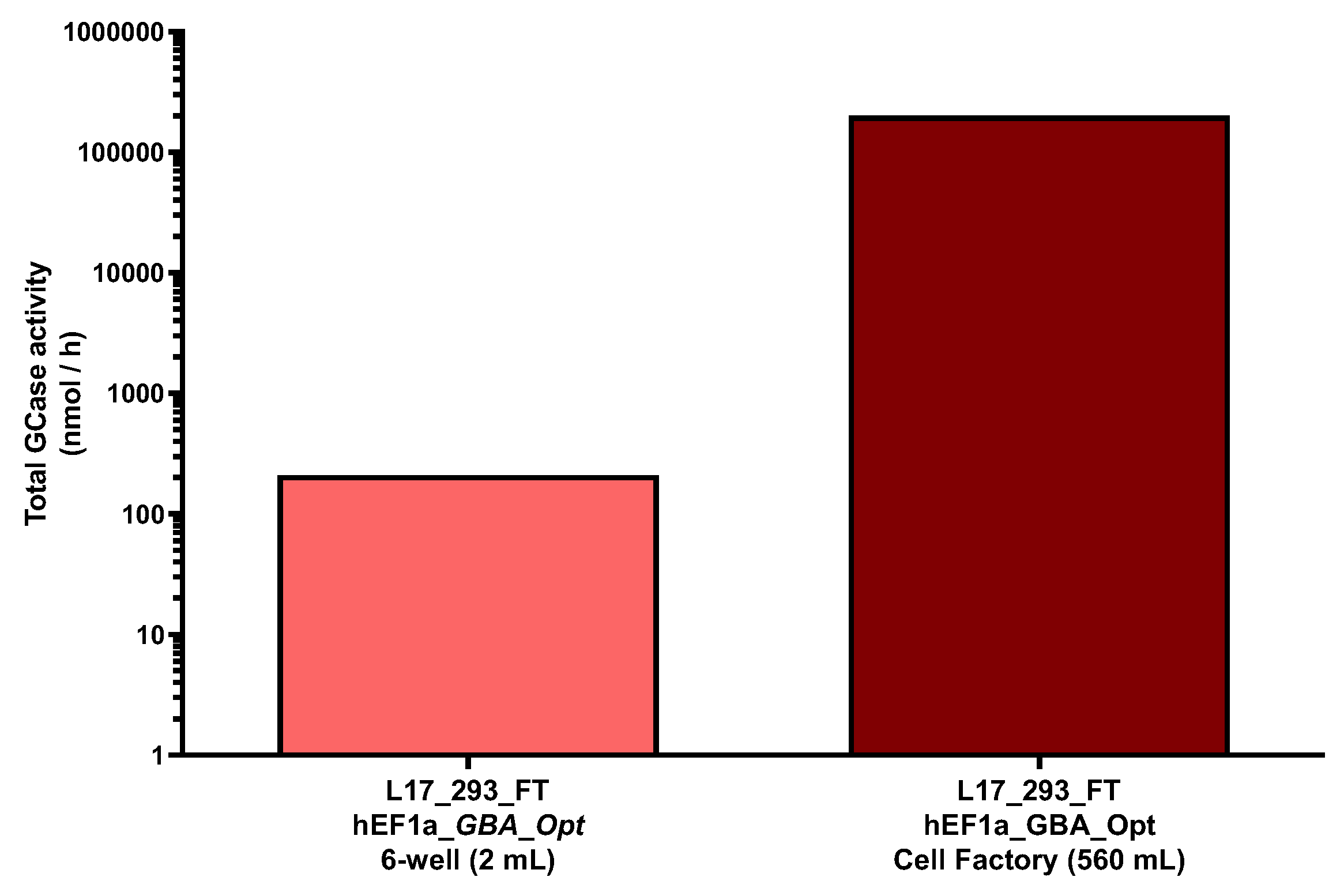
Figure 3.
Intracellular GCase-specific activity in L17_293FT_GBA_OPT_HP cells. (A) GCase-specific activity in cell lysates (nmol of hydrolyzed substrate per mg of total protein per hour) was significantly higher in the L17_293FT_GBA_OPT_HP population (307.5 ± 53.49 nmol/mg/h) compared to 293FT/virgin controls (95.58 ± 16.5 nmol/mg/h; p = 0.0026; t =7.572, df = 3.566; n = 4). (B) GCase activity normalized per million cells (U GCase/106 cells) also showed enhanced production in the L17 population (159.3 ± 44.97 U/106 cells) versus controls (60.2 ± 19.35 U/106 cells; p = 0.0149; t = 4.048, df = 4.074; n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. * indicates p ≤ 0.05; ** indicates p ≤ 0.01. [293_FT = human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α = human elongation factor 1-alpha promoter]; [GBA-Opt = codon-optimized human GBA (glucocerebrosidase) cDNA]; [GCase = β-glucocerebrosidase]; [hp = puromycin-selected heterogeneous population].
Figure 3.
Intracellular GCase-specific activity in L17_293FT_GBA_OPT_HP cells. (A) GCase-specific activity in cell lysates (nmol of hydrolyzed substrate per mg of total protein per hour) was significantly higher in the L17_293FT_GBA_OPT_HP population (307.5 ± 53.49 nmol/mg/h) compared to 293FT/virgin controls (95.58 ± 16.5 nmol/mg/h; p = 0.0026; t =7.572, df = 3.566; n = 4). (B) GCase activity normalized per million cells (U GCase/106 cells) also showed enhanced production in the L17 population (159.3 ± 44.97 U/106 cells) versus controls (60.2 ± 19.35 U/106 cells; p = 0.0149; t = 4.048, df = 4.074; n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. * indicates p ≤ 0.05; ** indicates p ≤ 0.01. [293_FT = human embryonic kidney cells expressing SV40 large T antigen]; [hEF1α = human elongation factor 1-alpha promoter]; [GBA-Opt = codon-optimized human GBA (glucocerebrosidase) cDNA]; [GCase = β-glucocerebrosidase]; [hp = puromycin-selected heterogeneous population].
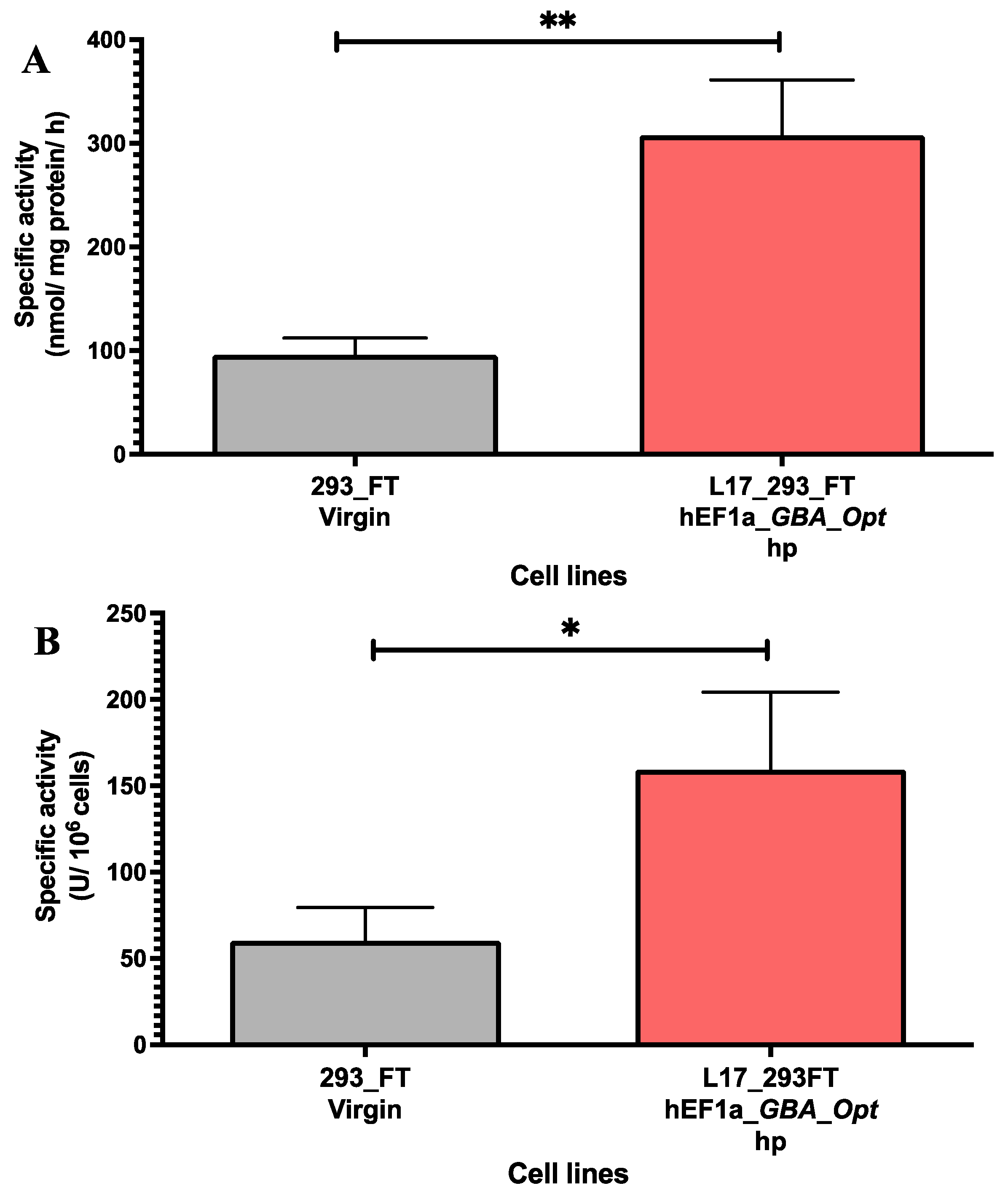
Figure 4.
Morphological analysis of 293FT/Virgin and transgenic cell lines. (A) 293FT/Virgin cells showing characteristic fibroblast-like morphology; (B) Heterogeneous population L17_293FT_GBA_OPT_HP after 96 h in culture (3x105 cells plated in a 25 cm2 flask). (C) Clone 15 (L17_293FT_GBA_OPT_CL15) after 120 h in cell culture (5x104 cells plated in a 9.6 cm2 well); (D) Clone 16 (L17_293FT_GBA_OPT_CL16) after 24 h in culture (3x105 cells plated in a 25 cm2 flask). Microscopy images were acquired using a ZEISS Axio inverted microscope with 10x objective, and a 60N-C 1" 1,0x Axiocam 503 color digital camera (ZEISS, Germany). Scale bar = 100 μm.
Figure 4.
Morphological analysis of 293FT/Virgin and transgenic cell lines. (A) 293FT/Virgin cells showing characteristic fibroblast-like morphology; (B) Heterogeneous population L17_293FT_GBA_OPT_HP after 96 h in culture (3x105 cells plated in a 25 cm2 flask). (C) Clone 15 (L17_293FT_GBA_OPT_CL15) after 120 h in cell culture (5x104 cells plated in a 9.6 cm2 well); (D) Clone 16 (L17_293FT_GBA_OPT_CL16) after 24 h in culture (3x105 cells plated in a 25 cm2 flask). Microscopy images were acquired using a ZEISS Axio inverted microscope with 10x objective, and a 60N-C 1" 1,0x Axiocam 503 color digital camera (ZEISS, Germany). Scale bar = 100 μm.
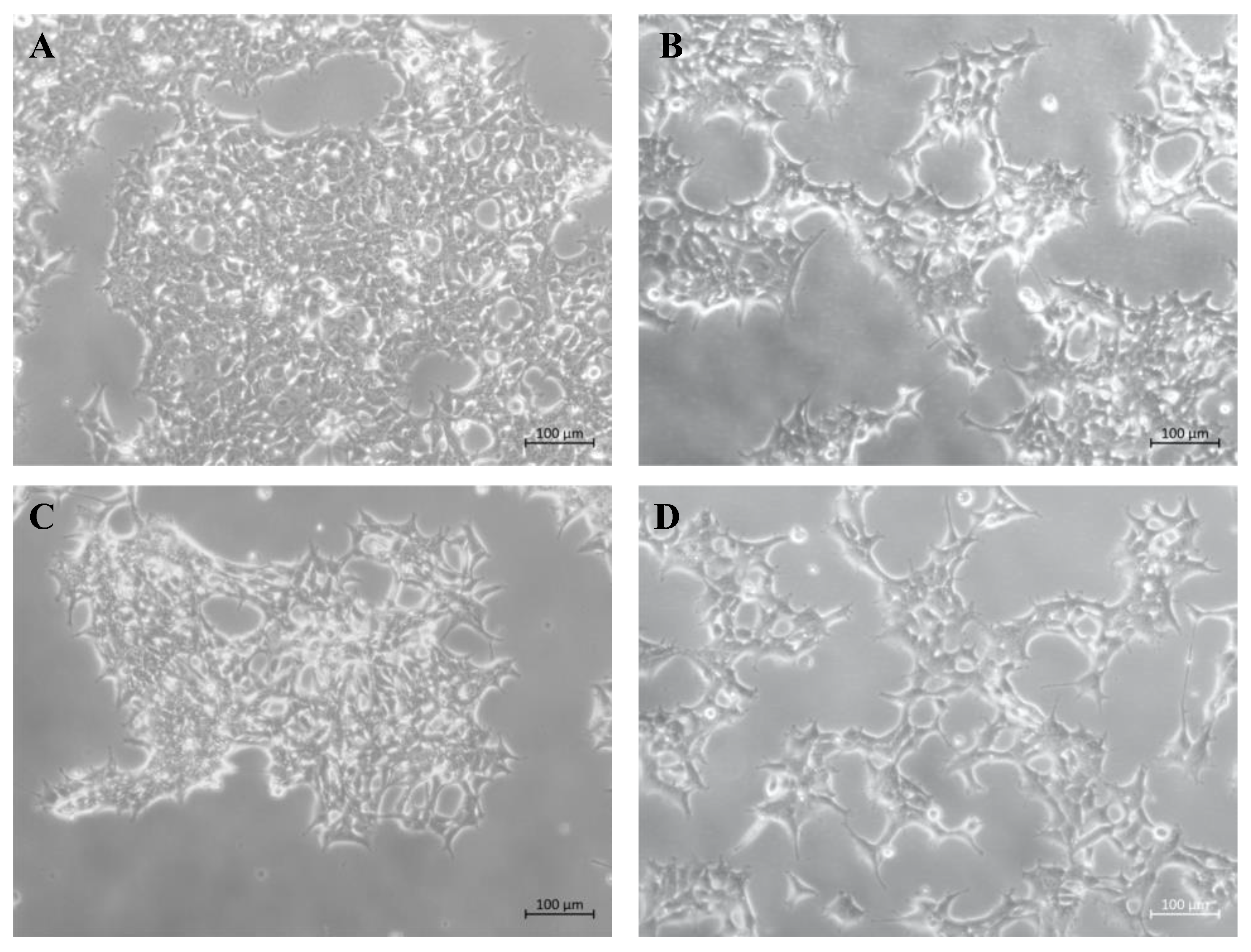
Figure 5.
Intracellular GCase-specific activity in L17-derived clones 15 and 16. (A) GCase-specific activity in cell lysates (nmol of hydrolyzed substrate per mg of total protein per hour) was comparable between clone 15 (763.8 ± 135.1, n=3) and clone 16 (752.0 ± 152.1, n=4), with no significant difference (p = 0.9292; t = 0.106, df = 4.749; n= 3 and n= 4). (B) GCase activity normalized per million cells (U GCase/106 cells) was also similar between clone 15 (313.4 ± 13.4, n = 3) and clone 16 (293.3 ± 4.9, n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. ns = not significant. [GCase = β-glucocerebrosidase]; [GBA_Opt_CL15 and CL16 = codon-optimized GBA-expressing single-cell clones derived from the L17_293FT_GBA_OPT line]. The number of replicates (n) for each clone is indicated in the figure legend.
Figure 5.
Intracellular GCase-specific activity in L17-derived clones 15 and 16. (A) GCase-specific activity in cell lysates (nmol of hydrolyzed substrate per mg of total protein per hour) was comparable between clone 15 (763.8 ± 135.1, n=3) and clone 16 (752.0 ± 152.1, n=4), with no significant difference (p = 0.9292; t = 0.106, df = 4.749; n= 3 and n= 4). (B) GCase activity normalized per million cells (U GCase/106 cells) was also similar between clone 15 (313.4 ± 13.4, n = 3) and clone 16 (293.3 ± 4.9, n = 4). Statistical analysis was performed using a two-tailed unpaired t-test with Welch’s correction. ns = not significant. [GCase = β-glucocerebrosidase]; [GBA_Opt_CL15 and CL16 = codon-optimized GBA-expressing single-cell clones derived from the L17_293FT_GBA_OPT line]. The number of replicates (n) for each clone is indicated in the figure legend.
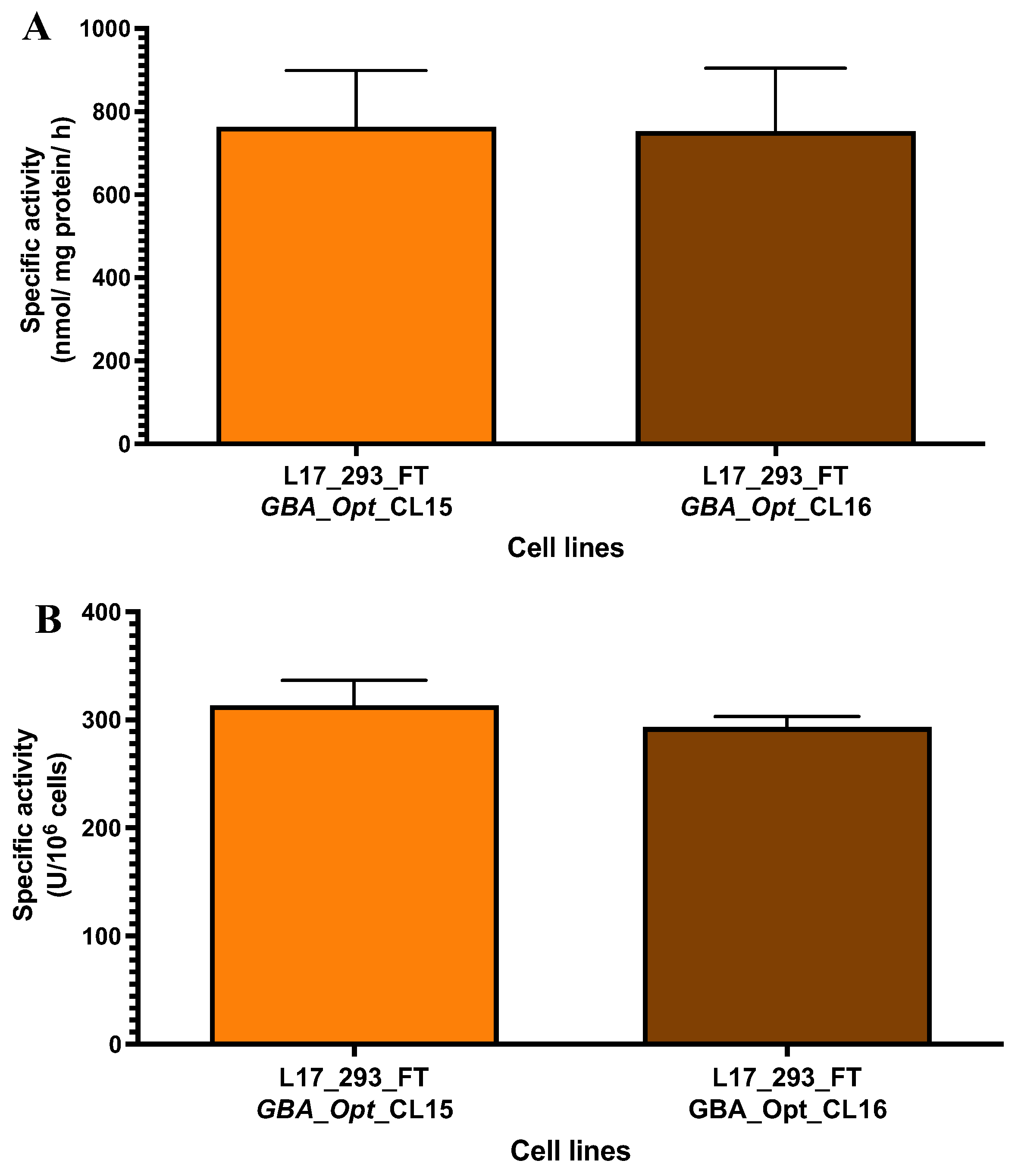

Table 1.
GCase activity in the supernatant of single-cell clones derived from L17_293FT_GBA_OPT cell line.
Table 1.
GCase activity in the supernatant of single-cell clones derived from L17_293FT_GBA_OPT cell line.
| Sample | GCase activity (nmol/mL/h) |
GCase activity (U/106cells) |
|---|---|---|
| L17_293FT_GBA_OPT_CL5 | 265.087 | 252.464 |
| L17_293FT_GBA_OPT_CL7 | 89.911 | 128.444 |
| L17_293FT_GBA_OPT_CL8 | 230.045 | 135.320 |
| L17_293FT_GBA_OPT_CL9 | 301.160 | 200.773 |
| L17_293FT_GBA_OPT_CL10 | 118.907 | 72.065 |
| L17_293FT_GBA_OPT_CL11 | 285.561 | 219.662 |
| L17_293FT_GBA_OPT_CL13 | 440.955 | 275.597 |
| L17_293FT_GBA_OPT_CL15 | 585.464 | 390.310 |
| L17_293FT_GBA_OPT_CL16 | 683.952 | 455.968 |
| L17_293FT_GBA_OPT_CL17 | 207.610 | 143.180 |
| L17_293FT_GBA_OPT_CL18 | 167.931 | 108.342 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



