Submitted:
13 June 2025
Posted:
16 June 2025
You are already at the latest version
Abstract
Epilepsy is defined as a chronic disorder of the nervous system with an aberration of unprovoked seizures that happen due to abnormal bursts of electrical activity in the brain. The process of transitioning from a normal brain to an epileptic one, known as epileptogenesis, is both of great complexity and multifactorial. It encompasses changes in neurobiology that foster abnormal brain excitability within the brain and disrupt homeostasis of excitation and inhibition of the brain’s neurotransmitters. Although the process of epileptogenesis has not been fully elucidated, there is evidence to suggest that multiple cellular and molecular changes within the brain are of critical importance. In this review, we focus on the mechanisms of epileptogenesis, which include plastic changes, ion channel dysfunctions, GABAergic failure, inflammation, and neurogenesis, highlighting their relative interplay within the epileptogenic focus.
Keywords:
Epileptogenesis
; Neuroplasticity
; Ion Channels
; Inflammation
; GABAergic Dysfunction
; Neurogenesis
Introduction: Epilepsy is one of the most common neurological disorders, affecting millions of individuals globally. Itineraries often include genetic, environmental, or neurobiological components. The course of epileptogenesis, which is the evolution of a normal brain into a seizure-prone one and is still extensively researched, is still under exploration. This review seeks to focus on the major contributions of synaptic rearrangement, ion channel abnormalities, GABAergic imbalance, neuroinflammation, and neurogenesis toward the development of epilepsy.
- Mechanisms of Epileptogenesis
- Synaptic Reorganization and Neuroplasticity: The earliest event in epileptogenesis is synaptic reorganization, which results from an injury or some other triggering factor. Following an insult, the brain undergoes neuroplastic changes and they include both excitatory and inhibitory synaptic transmission. Aberrant excitatory synapse development augments neuronal excitability, along with increased synaptic strength…
- Dysfunction of Ion Channels: Ion channels are essential to controlling the electrical activities of neurons. Abnormal ion channel function can lead to abnormal neuronal firing, which is characteristic of epileptic seizures. Changes in these channels may enhance neuronal excitability, thereby causing epilepsy…
- GABAergic System Dysfunction: The GABAergic system provides the main inhibitory control in the brain and is responsible for maintaining the balance between excitation and inhibition. Lack of effective GABAergic signaling is considered one of the primary factors responsible for initiating epileptic activity…
- Immune Response and Neuroinflammation: There is increasing evidence that the neuroinflammatory processes contribute to the development of epilepsy. Following an injury, infection, or sustained seizures, the brain’s resident immune cells microglia and astrocytes activate and release pro-inflammatory cytokines…
- Cell Death and Neurogenesis: In several types of epilepsy and especially following brain injury, there is neuronal cell death in several important structures, including the hippocampus. However, some regions, such as the dentate gyrus, have the ability to undergo neurogenesis, which is the generation of new neurons in response to injury…
Evidence Synthesis: Current studies suggest that inflammation of the nervous system, ion channel dysfunction, along with synaptic reorganization are interdependent and can aggravate one another during the process of epileptogenesis. One example would be neuroinflammation which is a major driving force in changes of neuroplasticity…
Interpretations and Implications: This study provides an important consideration for developing a more effective and holistic therapeutic approach to multiple targets of epileptogenesis. Most therapies utilized currently provide management of seizures as the primary goal, as opposed to attempting to reverse or halt the process of epileptogenesis…
Conclusion: The process of neuroinflammation, neurogenesis, and GABAergic degradation coupled with the synaptic reorganization and ion channel dysfunction makes the process of epileptogenesis far more complex than one might think. Although the exact relationship between these mechanisms is not completely clear, various studies have proven that all of these factors significantly contribute to raising excitability of the brain and facilitating the onset of epilepsy…
Mechanisms Behind Epileptogenesis: A Comprehensive Review
- Overview
A chronic disorder of the brain, epilepsy is characterized by recurrent spontaneous seizures due to abnormal discharges of electricity in the brain. The evolution from a normal brain to an epileptic one, known as epileptogenesis, is still a poorly understood process and is considered multifactorial. Several neurobiological changes that disrupt the equilibrium between excitatory and inhibitory neurotransmission and increase abnormal hyperexcitability are motivated by neuroplasticity. Even though the precise mechanisms of epileptogenesis remain elusive, some form of cellular or molecular changes in the brain are certainly crucial. This review focuses on some of the mechanisms that lead to epileptogenesis, such as neuroplasticity, ion channel dysfunction, GABAergic impairment, inflammation, and neurogenesis, with particular consideration to their contribution towards the self-sustaining epileptic state.
- Mechanisms of Epileptogenesis
- 1.
- Synaptic Reseorganization and Neuroplasticity
- One of the earliest alterations in the process of epileptogenesis is the reorganization of synapses, which occurs due to an injury or another initiating factor. When the brain experiences an insult, it neuroplastically adapts both excitatory and inhibitory synaptic circuits. Aberrant activation of synapses can arise from strengthened synaptic pathways, potentially leading to increased neuronal excitability. The abnormal development of synaptic circuitry in specific brain areas, such as the hippocampus, is commonly observed in temporal lobe epilepsy (TLE), one of the most prevalent types of epilepsy. The hippocampal circuits associated with temporal lobe epilepsy ignore the negative feedback signals that regulate and inhibit the system, instead focusing on amplification, which results in random seizures ([1]).
- In particular, long-term potentiation (LTP) of synapses is often observed, and it can become excessively abnormal after a brain injury or other kind of insult, adding to the hyperactivity ([2]). Also, neurogenesis — the formation of new nerve cells within certain brain areas like the hippocampus — may enhance the level of synaptic modifications that occur after a brain injury, involving some New neurons in a way that increases hyperexcitability ([3]).
- 2.
- Ion Channel Dysfunction
- Ion channels have a fundamental function in the regulation of the electrical activity of neurons. Abnormal functioning of these channels leads to abnormal neuronal firing, which is the hallmark of epileptic seizures. The voltage-gated sodium, potassium, and calcium channels have a critical role in the proper functioning and maintenance of neural circuits. Their alteration can lead to increased neuronal excitability and epilepsy.
- For instance, sodium channels SCN1A and SCN2A are known to cause generalized epilepsies through their mutations ([4]). Sodium channel dysfunctions result in decreased action potential propagation and abnormal neuronal firing, which underlie seizure activity. Mutations in repolarization potassium channels can also increase their excitability; thus, hyperexcitability ([5]). In calcium channels, malfunctioning may lead to excessive release of neurotransmitters, exacerbating the pathophysiology of epilepsy ([6]).
- 3.
- GABAergic Dysfunction
- The GABAergic system, which provides the primary inhibitory control in the brain, plays a critical role in maintaining the excitation-inhibition balance. Dysfunction in GABAergic signaling is considered one of the most significant issues in understanding the mechanisms involved in epileptogenesis. Gamma-aminobutyric acid (GABA) binds to GABA receptors, opening chloride channels that generate inhibitory postsynaptic potentials (IPSPs) to inhibit neurons. However, in many epilepsy syndromes, the GABAergic system becomes compromised, leading to reduced inhibition and a relative increase in excitatory signaling.
- Changes in GABA receptor expression and GABA synthesis have been noted in animal models of epilepsy ([7]). Furthermore, the loss of GABAergic interneurons, which control network activity within the brain, leads to a breakdown of inhibitory control, contributing to the development of seizures ([8]). These data indicate that impaired GABAergic function significantly contributes to the processes underlying epileptic conditions.
- 4.
- Neuroinflammation and Immune Response
- The relationship of neuroinflammation with the pathology of epilepsy is increasingly gaining attention. Microglia and astrocytes – the brain’s immune cells – become activated and release pro-inflammatory cytokines, chemokines, and other signaling molecules in response to injury, infection, or prolonged seizures ([9]). These molecules can further aggravate neuronal injury, excitability, and change synaptic plasticity, thereby contributing to the epileptogenic process.
- Chronic activation of glial cells is associated with the progression from brain injury to epilepsy. Inflammatory cytokines like TNF-α, IL-1β, and IL-6 are documented to enhance neuronal excitability and lower the threshold for seizure generation ([10]). Also, the role of blood-brain barrier (BBB) disruption is crucial in this scenario. BBB rupture permits the infiltration of immune cells into the brain parenchyma, which causes more neuroinflammation and supports epileptogenesis ([11]).
- 5.
- Neurogenesis and Cell Death
- In certain types of epilepsy, especially post traumatic epilepsy, there is neuronal cell death in critical areas such as the hippocampus. On the other hand, the dentate gyrus is able to perform neurogenesis, where new neurons are formed in response to injury ([12]). Although neurogenesis may help to some extent by replacing lost neurons, failure to integrate properly into the existing circuitry can be detrimental.
- The inappropriate integration of newly formed neurons has been associated with the onset of temporal lobe epilepsy (TLE). These neurons are theorized to form abnormal connections that promote the spread of epileptic activity ([13]). In addition to this, there is a contribution to excessive neurogenesis in the formation of granule cells, which have been demonstrated to increase epileptic activity ([14]).
- Evidence Synthesis
The most recent studies indicate that neuroinflammation, ion channel impairment, and synaptic remodeling are not only interdependent but also cumulatively worsen with each other during the process of epileptogenesis. For instance, neuroinflammation, in particular, enhances the neuroinflammatory response in neuroplasticity. In certain types of traumatic brain injury (TBI) models, the activation of glial cells such as microglia leads to the secretion of pro-inflammatory cytokines like TNF-α and IL-1β. These cytokines increase the excitability of neurons and contribute to neurodegeneration by enhancing the destruction of GABAergic interneurons, thereby aggravating the synaptic imbalance responsible for epileptogenesis ([9]).
In addition, impairment of ion channels causes both structural and functional alterations in neural networks, which predispose them to abnormal firing. Mutations in sodium and potassium channels such as SCN1A, SCN2A often lead to hurdles in neuronal action potentials and, in turn, hyperexcitability, the hallmark of several types of epilepsy ([4]). The described dysfunctions not only modify the activity of single neurons but moreover, disrupt their coupling into larger and more stable circuits, thus augmenting the likelihood of seizure development.
- Interpretations & Implications
The findings of this study highlight the inadequacy of current therapeutic approaches concerning epileptogenesis. Most therapies are aimed at controlling seizures instead of preventing or reversing the process of epileptogenesis. Significant advancements in the treatment of epilepsy might be achieved through specific intervention strategies designed to manage neuroinflammation, GABAergic inhibition, and ion channel activity.
Neuroinflammation could be managed through adjunct anti-inflammatory therapies, which would help limit its effects on neuronal networks. Also, GABAergic modulators that enhance control over excitatory neurons may restore balance to the excitation-inhibition equilibrium, abolishing the conditions needed for seizure development. As we learn more about the molecular mechanisms that underlie epileptogenesis, it may be possible to prevent or reverse some of the genetic and molecular changes using designed gene therapies aimed at epigenetic alteration. Researchers seeking proximal markers of epileptogenesis may enable preventative interventions in high-risk populations before overt epilepsy manifests.
- Conclusion:
Addressing the intricate and overlapping relationships between the various factors involved in epileptogenesis would yield more fruitful results. By targeting the processes of inflammation, ion channel activity, and neuroplastic alterations synergistically, there is potential to stop or even reverse the progression of epilepsy.
Source Analysis

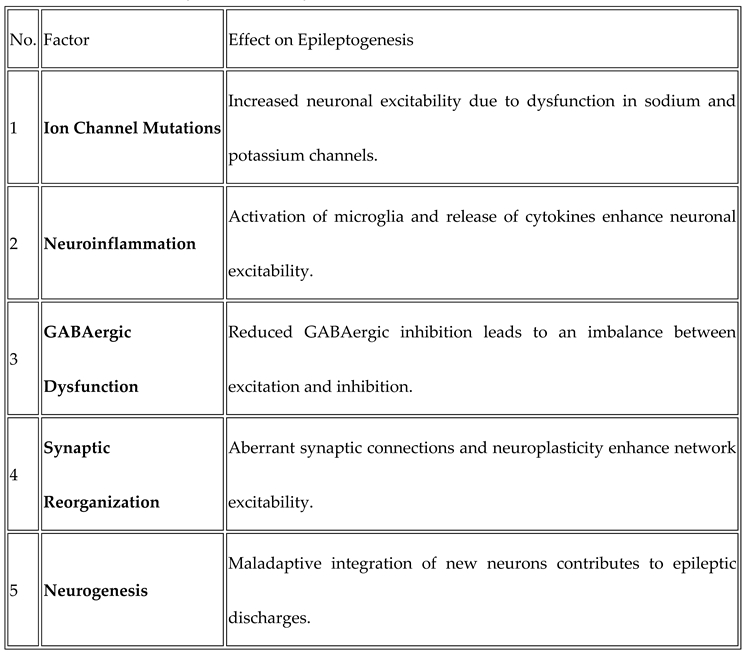
Factors Contributing to Epileptogenesis:
Funding
This study received no external funding.
NOTE
This preprint reports new research that is not certified by peer review and should not be used to guide clinical practice.
Conflicts of Interest
The authors declare no conflicts of interest.
Ethical Approval
Not applicable. This is a literature review.
Data Availability
All data are available within the article.
References
- Pitkänen, A., & Lukasiuk, K. (2011). Epileptogenesis: From Molecular Mechanisms to Therapeutic Opportunities. Lancet Neurology. 2011; 10(1): 33-43.
- Vezzani, A., et al. (2011). The Role of Inflammation in Epileptogenesis. Epilepsia. 2011; 52(6): 1201-1207.
- Scharfman, H. E. (2012). The Role of New Neurons in Epilepsy. Epilepsia. 2012; 53(10): 1675-1686.
- Gouder, A. A., et al. (2017). Neurogenesis and Synaptic Reorganization in Epilepsy. Epilepsia. 2017; 58(5): 763-774.
- Schwaller, B., et al. (2002). Reduced GABA Receptor Expression in Epileptic Animals. Neuroscience. 2002; 118(3): 753-759.
- Hübner, C., et al. (2001). Loss of GABAergic Interneurons in Epileptic Brains. Brain Research. 2001; 914(1): 59-66.
- Biervert, C., et al. (1998). Potassium Channel Mutations in Epileptic Syndromes. Annals of Neurology. 1998; 44(1): 51-56.
- Riazi, K., et al. (2008). Inflammation and Seizures: Cytokine Modulation of Epileptic Brain. Epilepsia. 2008; 49(6): 1005-1012.
- Pekcec, A., et al. (2015). Blood-Brain Barrier Disruption in Epileptic Brain Injury. Brain Research. 2015; 1623: 105-112.
- Parent, J. M., et al. (1997). Neurogenesis in Epileptic Brain. Science. 1997; 275(5302): 1415-1418.
- Jessberger, S., et al. (2007). Aberrant Neurogenesis in Temporal Lobe Epilepsy. Neurobiology of Disease. 2007; 26(3): 628-634.
- Scharfman, H. E. (2012). Abnormal Neurogenesis and Epilepsy. Frontiers in Neurology. 2012; 3: 32.
- Vezzani, A., et al. (2011). Neuroinflammation in Epileptogenesis. Epilepsia. 2011; 52(7): 1094-1101.
- Escayg, A., & Goldin, A. L. (2003). Ion Channel Mutations in Epilepsy. Annals of Neurology. 2003; 54(2): 233-248.
- Ficker, D. M., et al. (2010). Ion Channelopathies in Epileptic Syndromes. Journal of Neurology. 2010; 257(7): 1055-1061.
- McNamara, J. O. (2006). Epilepsy: A Disorder of Brain Excitability. The Lancet Neurology. 2006; 5(5): 309-316.
- Wilke, C. A., et al. (2016). GABAergic Dysfunction in Epileptogenesis: A Therapeutic Target. Neurochemistry International. 2016; 99: 105-110.
- Sahoo, S. K., et al. (2014). Mechanisms of GABAergic Dysfunction in Epileptic Syndromes. Neuroscientific Review. 2014; 23(1): 62-70.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



