Submitted:
11 June 2025
Posted:
13 June 2025
You are already at the latest version
Abstract
Simultaneous determination of multiple organic compounds in complex sample matrices remains a significant analytical challenge due to the diverse chemical properties and low concentration levels of target analytes. This review comprehensively evaluates current sample preparation techniques applied in multi-residue analysis coupled with chromatographic methods such as GC-MS/MS and LC-HRMS. Techniques including QuEChERS, solid-phase extraction (SPE), dispersive SPE, and solid-phase microextraction (SPME) demonstrate extraction recoveries ranging from 70% to 95% for various compound classes, with detection limits often reaching low ng/L or ng/g levels. The choice of sample preparation method is critically influenced by analyte polarity, solubility, and matrix complexity, with over 50% of reported studies focusing on food and environmental water samples. Furthermore, this review highlights the growing potential of artificial intelligence (AI) in enhancing chromatographic data interpretation and compound identification, though emphasizes that effective sample preparation remains foundational for AI-driven analysis. The integration of optimized sample preparation workflows with AI-assisted data processing promises to improve throughput, accuracy, and robustness in multi-residue analysis, supporting regulatory compliance and environmental monitoring efforts worldwide.
Keywords:
multi-residue analysis
; sample preparation
; QuEChERS
; solid-phase extraction
1. Introduction
In recent decades, the global chemical landscape has undergone unprecedented expansion, both in scale and complexity. It is now estimated that over 350,000 commercial chemical substances are registered for use worldwide, with approximately 2,000 new compounds entering the market each year [1]. Traditionally, environmental pollutants have been classified either by name (e.g., PFAS, phenolics, non-steroidal compounds) or by function (e.g., antibiotics, persistent organic pollutants (POPs), pesticides). However, the growing structural and functional diversity of these substances has increasingly challenged the practicality and relevance of such conventional classification systems. To address this complexity, scientists have introduced the umbrella term Contaminants of Emerging Concern (CECs) to describe chemicals whose environmental occurrence has been linked to newly identified or evolving risks to human health and ecosystems. Notably, recent studies suggest that CECs may account for up to 15.4% of the known chemical space. In parallel, an even larger subset—estimated at 28.4%—comprises chemicals that remain poorly characterized or unclassified, highlighting a significant knowledge gap in current chemical inventories and risk assessment frameworks [2].
Simultaneous chemical analysis methods for qualitative and quantitative determination of organic compounds are clearly essential to provide comprehensive information from samples quickly and at low cost [3]. Consequently, an increasing number of simultaneous analytical methods have been developed. Figure 1 shows the annual growth in the number of simultaneous analysis studies. These methods are often optimized for specific compound groups such as pharmaceuticals [4], pesticides [5,6], PFAS [7], flame retardants [8], PAHs [9], and plant growth regulators [10].
Table 1 summarizes simultaneous analysis methods conducted with at least 15 peaks of detection in recent studies, demonstrating that simultaneous analytical methods have been applied across various sample matrices and compound groups. These include targeted analyses [11,12], non-targeted analyses [13,14,15], or both [16,17].
While conventional chromatographic methods typically can identify up to around 1,500 peaks [18], multidimensional chromatography methods can separate tens of thousands of peaks simultaneously [19,20].

Table 1.
Selected previous studies on the simultaneous analysis of dozens of compounds using chromatographic techniques.
Table 1.
Selected previous studies on the simultaneous analysis of dozens of compounds using chromatographic techniques.
| Compounds | Matrix | Extraction methods | Instrument | Number detection | Ref. |
|---|---|---|---|---|---|
| PFAS | human serum and semen | QuEChERS | UPLC-MS/MS | 17 | [21] |
| Bromophenols | environment | GC-MS | 19 | [22] | |
| Antibiotic | Plasma | Oasis HLB μEluting Plate | HPLC–MS/MS | 50 | [23] |
| Endocrine disrupting chemicals | human amniotic fluid | SPE | LC-MS/MS | 59 | [24] |
| Pesticides and pharmaceuticals | soil, orange leaves and fruits | QuEChERS | LC-MS/MS | 33 | [25] |
| Lipid-soluble pesticides and metabolites | chicken liver and pork | QuEChERS | HPLC-MS/MS | 24 | [26] |
| Alkaloids | cereal-based food | QuEChERS | LC-MS/MS | 42 | [27] |
| Illegal drugs | Sewage | SPE | SPE-ISTD-UHPLC-MS/MS | 28 | [28] |
| Sulfonamide | Livestock | QuEChERS | LC/MS | 31 | [29] |
| Micro-pollutants | Surface water | LLE | GC-MS and GC-MS-MS | 950 | [15] |
| Semi-volatile organic compounds | Floodwater | LLE | GC-MS | 940 | [30] |
| Micro-pollutants | Surface water | SPE | LC-TOF-MS and GC-MS | 1153 | [31] |
| Pesticides | Medicines | QuEChERS or SPE | GC-MS-MS | 147 | [32] |
| Solvents | Drug | SLE | GC-MS | 50 | [33] |
| SVOCs | indoor air | SLE | GC-MS | 73 | [34] |
Sample preparation methods are employed to remove impurities and matrix interferences. A suitable sample preparation method for the simultaneous analysis of multiple organic compounds must ensure the efficient extraction of all target analytes (which may belong to one or several chemical groups), with recovery rates typically ranging from 70–120%, depending on the requirements of specific analytical guidelines [35,36,37,38]. Additionally, the extracted analytes often need to be concentrated to ensure they can be accurately detected by the analytical instrument. Therefore, as the number of target compounds increases, the sample preparation process must be carried out with greater precision to prevent analyte loss during processing. The objective of this review is to provide a comprehensive overview of current sample preparation strategies that enable the simultaneous determination of multiple organic compounds using chromatographic techniques. Special attention is given to the challenges associated with multi-analyte analysis in complex matrices and how sample preparation directly affects analytical selectivity, sensitivity, and reproducibility.
2. Classification of Organic Compounds in Multi-Residue Analysis
Multi-residue analysis (MRA) aims to detect and quantify a broad spectrum of organic compounds simultaneously in a single run, which presents significant challenges due to the wide range of physicochemical properties exhibited by these compounds. Therefore, a clear classification is essential for designing effective sample preparation strategies and optimizing chromatographic conditions.
2.1. Based on Functional Use or Source
In multi-residue analysis, classifying organic compounds based on their functional use or source of origin helps guide the selection of appropriate sample preparation procedures and analytical methods. One of the most common groups is pesticides [5], which include compounds with highly diverse structures and properties such as organochlorines [39], organophosphates [6,40], carbamates [41], neonicotinoids [42], and pyrethroids [43]. The significant differences in polarity, stability, and volatility among these substances necessitate optimized extraction methods like QuEChERS and analysis via LC-MS/MS [5,44,45,46] or GC-MS/MS [47] to enable simultaneous quantification of multiple analytes.
In addition, antibiotic and veterinary drug residues are frequently analyzed in aquatic and animal-derived food samples [48]. Antibiotic classes such as tetracyclines, sulfonamides, macrolides, and fluoroquinolones typically require selective cleanup methods like SPE and analysis via LC-ESI-MS/MS to achieve high sensitivity due to their low concentrations and complex matrices. Mycotoxins, such as aflatoxins, ochratoxin A, and fumonisins, commonly found in grains and peanuts, demand purification steps using immunoaffinity columns or SPE prior to analysis by LC-FLD or LC-MS.
Personal care products and pharmaceuticals (PPCPs), including diclofenac, estradiol, and triclosan, have gained increasing attention in environmental studies due to their persistence and biological toxicity, with LC-MS/MS being the primary analytical tool [49]. Endocrine-disrupting compounds like bisphenol A, phthalates, and parabens can significantly impact the endocrine system even at very low concentrations, requiring stringent contamination control protocols and highly sensitive instrumentation [50,51].
Finally, persistent environmental and industrial pollutants such as PAHs [52,53], PCBs [54], dioxins [55,56], brominated flame retardants [57], and perfluoroalkyl substances (PFASs) [58] are frequently detected in wastewater, sediments, and food samples. These substances often necessitate multi-step extraction procedures and analysis via gas or liquid chromatography coupled with high-resolution mass spectrometry (HRMS) to ensure optimal resolution and analytical reliability.
Classifying compounds by their functional use or origin not only facilitates effective sample preparation but also reflects their practical applications in agriculture, healthcare, industry, and daily life—thereby informing targeted monitoring and risk management strategies.
2.2. Based on Polarity and Solubility
Classifying organic compounds based on polarity and solubility is one of the fundamental principles guiding the selection of appropriate sample preparation methods, extraction solvents, and analytical conditions in multi-residue analysis. Highly polar compounds such as small organic acids (e.g., glyphosate, formic acid), sugars, amino acids, or certain antibiotics like streptomycin are often difficult to extract using conventional organic solvents [59]. These compounds typically require the use of polar solvents such as methanol, water, acetonitrile, or buffered solutions with adjusted pH. In many cases, cleanup techniques such as ion exchange, polar solid-phase extraction (SPE), or hydrophilic-lipophilic balance (HLB) cartridges are employed to ensure high recovery rates and reduce matrix interferences.
Conversely, non-polar compounds such as polycyclic aromatic hydrocarbons (PAHs), organochlorine pesticides (OCPs), phthalates, and polychlorinated biphenyls (PCBs) are readily extracted using less polar solvents like hexane, dichloromethane, or toluene and are typically analyzed effectively using gas chromatography (GC) coupled with mass spectrometry (MS) or electron capture detectors (ECD).
A group of compounds with intermediate polarity—including carbamate pesticides, triazine herbicides, sulfonamides, or parabens—usually requires a balance between polar and non-polar solvents for optimal extraction [12]. In such cases, mixtures of acetonitrile–water or methanol–water in suitable ratios are employed to enhance recovery efficiency. Additionally, the solubility of compounds in water or fat is another critical factor in matrix selection: hydrophilic compounds like beta-lactam antibiotics or phenoxyacetic acid herbicides are primarily found in plasma or tissue fluids, whereas lipophilic substances such as dioxins, PBDEs, or DDT tend to accumulate in fatty tissues and liver.
Polarity and solubility classification also informs chromatographic conditions, such as using C18 columns for moderately to non-polar compounds, or hydrophilic interaction liquid chromatography (HILIC) columns for highly polar substances. Control over solvent pH, salt type, and the proportion of organic solvents in the mobile phase plays a crucial role in achieving resolution and method repeatability. Therefore, a clear understanding of each compound’s polarity and solubility is essential for developing robust, reliable, and scalable multi-residue analytical methods across diverse matrices, including water, soil, food, biological, and environmental samples.
3. Overview of Sample Matrices
In multi-residue analysis of organic compounds, the sample matrix plays a crucial role in determining the sample preparation approach, the selection of cleanup techniques, and analytical conditions. Common matrices include food (such as vegetables, fruits, meat, fish, milk, and cereals) [5,60,61,62,63], water (drinking water, surface water, wastewater) [11], soil [64,65], sediment [7,15,18,55], air [66,67,68], and biological samples (plasma, tissues, urine) [69,70]. Each matrix possesses distinct physicochemical characteristics that directly affect extraction efficiency, analytical sensitivity, and the risk of matrix interferences.
In the field of food safety, fruits and vegetables are among the most frequently analyzed matrices for determining residues of pesticides [5], plant growth regulators [10,71,72,73,74,75], or environmental contaminants [76,77,78]. These plant matrices are typically rich in water, sugars, organic acids, and pigments, posing significant challenges for removing interfering substances that can affect mass spectrometric signals. In meat and seafood—particularly fish raised in contaminated environments—lipophilic compounds such as chlorinated pesticides, polychlorinated biphenyls (PCBs), and industrial dyes (e.g., malachite green) readily accumulate in fatty tissues and liver, requiring selective extraction methods and thorough cleanup procedures (often involving protein precipitation, solid-phase extraction (SPE), or gel permeation chromatography (GPC)) [79,80,81,82,83].
Water samples—including river water, lake water, and municipal or industrial wastewater—have simpler matrices but usually require large-volume sample processing due to the typically low concentrations of pollutants. Liquid-liquid extraction (LLE), solid-phase extraction (SPE), and solid-phase microextraction (SPME) are commonly employed techniques. In contrast, soil and sediment possess complex solid structures with high levels of natural organic matter, necessitating the use of ultrasonic extraction, pressurized liquid extraction (PLE), or microwave-assisted extraction (MAE) to improve recovery efficiency.
Air and dust samples are important matrices for assessing environmental exposure risks. They are typically collected using glass fiber filters or polyurethane foam (PUF) and processed with strong organic solvents such as toluene or dichloromethane [84]. In biomedical and toxicological studies, biological samples like blood, urine, liver, or animal tissues reflect the actual processes of absorption, metabolism, and excretion of organic compounds. However, these matrices often contain proteins, enzymes, and endogenous substances that can interfere with analysis, thereby requiring protein precipitation, selective extraction, and matrix cleanup using SPE or freeze filtration [85,86].
Understanding the physicochemical characteristics of each matrix is therefore essential not only for selecting the optimal sample preparation method but also for ensuring the accuracy, reproducibility, and reliability of the entire analytical process. Furthermore, the diversity of matrices underscores the need to develop flexible, multi-matrix applicable methods—especially in the context of modern multi-residue analysis.
4. Technical Requirements in Sample Preparation for the Simultaneous Determination of Organic Compounds
Sample preparation is a critical step to ensure the quality and reliability of chromatographic analysis, especially when the goal is the simultaneous determination of multiple organic compounds with diverse chemical characteristics in a complex matrix. To meet this objective, the sample preparation technique must fulfill the following technical criteria:
4.1. High and Uniform Recovery Efficiency Across Compound Groups
One of the most important criteria in sample preparation is the ability to recover the target compounds with high and consistent efficiency, particularly when analyzing multiple substances with varying physicochemical properties [87,88]. In chromatographic analysis, recovery reflects the extent to which the analytes are extracted, cleaned, and preserved intact from the original matrix to the final analytical solution. For quantitative analysis, high recovery not only ensures the accuracy of results but also improves the limits of detection (LOD) and quantification (LOQ).
However, a major challenge in multi-analyte analysis lies in the chemical diversity of organic compounds:
- Some compounds are highly polar (e.g., organic acids, carbamate pesticides, hydroxylated metabolites), while others are non-polar (e.g., polycyclic aromatic hydrocarbons – PAHs, pyrethroid pesticides).
- Some are volatile and thermally stable (suitable for GC analysis), while others decompose at high temperatures and are better suited for LC techniques.
This diversity requires the extraction technique to be flexible and not overly selective toward any specific group of compounds. If the solid-phase sorbents (used in SPE or dSPE) or extraction solvents (used in LLE, DLLME) are too selective, there is a risk of biased extraction—efficiently recovering certain compounds while omitting others [88,89,90]. This significantly compromises the ability to comprehensively and simultaneously assess all target analytes in the sample.
Several strategies are applied to improve uniform recovery across compound groups:
- Optimizing the extraction solvent composition (e.g., acetonitrile can be acidified or basified to extract both neutral and ionizable compounds).
- Combining multiple sorbents in dSPE (e.g., a mixture of PSA, C18, and GCB can simultaneously address matrices rich in organic acids, lipids, and pigments).
- Using internal standards or isotopically labeled standards to correct for losses during sample processing.
- Testing and verifying recovery for each representative compound group, followed by adjustments in extraction and cleanup conditions as needed.
In modern methods such as QuEChERS, the use of salt-induced phase separation and dSPE sorbents has demonstrated good recovery performance for hundreds of different compounds within the same food or environmental sample. However, careful calibration and method validation are still essential to ensure uniform recovery across different compound groups.
4.2. Matrix Effects
Matrix effects are among the most significant challenges in the simultaneous analysis of organic compounds using modern chromatographic techniques, particularly when employing highly sensitive detectors such as tandem mass spectrometry (MS/MS) [91,92]. Matrix effects occur when non-analyte components in the sample—such as matrix constituents or impurities—interfere with the ionization process or detection signal of target analytes. This leads to signal distortion, reducing the accuracy and reliability of analytical results.
In complex samples such as food, environmental, or biological matrices, the background composition is often highly diverse, including proteins, lipids, inorganic salts, pigments, and non-target organic compounds. These components can:
- Suppress or enhance ionization in LC-MS/MS (ion suppression or ion enhancement).
- Clog or damage chromatographic columns, affecting separation efficiency.
- Generate interfering peaks, complicating the identification and quantification of analytes.
Therefore, minimizing matrix effects is a critical requirement during sample preparation to:
- Enhance sensitivity and lower detection limits.
- Improve the accuracy and repeatability of the analysis.
- Protect analytical instruments and extend the lifespan of chromatographic columns and ion sources.
Common strategies for matrix effect reduction in sample preparation include:
-
Selective sorbent-based cleanup:The use of specific sorbents in solid-phase extraction (SPE) or dispersive SPE (dSPE), such as PSA (for removal of organic acids and some pigments), C18 (for lipid adsorption), and GCB (for removing pigments and chlorophyll), helps eliminate many interfering substances.
-
Selective phase extraction:Choosing suitable extraction solvents and pH conditions allows for better separation of target compounds from unwanted matrix components.
-
Standardization and internal standards:Internal standards or isotopically labeled standards not only correct for losses during sample processing but also compensate for matrix effects, improving quantification accuracy.
-
Optimized centrifugation and filtration:Thorough centrifugation and filtration steps remove particulate matter, proteins, and large molecules, reducing the risk of clogging and mechanical interferences.
-
Advanced matrix separation techniques:Emerging techniques such as sample preparation using nanomaterials, functionalized materials, or dual-phase separation methods offer improved efficiency in matrix cleanup while maintaining high recovery of target analytes.
Effectively minimizing matrix effects not only enhances data quality but also reduces instrument maintenance efforts and the need for re-analysis, contributing to improved laboratory efficiency and cost savings.
4.3. High Repeatability and Accuracy
In the simultaneous analysis of multiple organic compounds, the repeatability and accuracy of the sample preparation step play a decisive role in determining the quality and reliability of the final results [93,94]. Repeatability refers to the ability to obtain consistent results when the same method is applied multiple times under identical conditions, while accuracy represents the closeness between the measured value and the true value of the target compound in the sample.
Why are repeatability and accuracy important?
- In applications such as pesticide residue testing, pharmaceutical analysis, or environmental pollution monitoring, high accuracy is essential to ensure that results comply with international regulations and standards.
- Inconsistent or biased results may lead to incorrect decisions, potentially impacting human health, the environment, or industrial processes.
Factors affecting repeatability and accuracy in sample preparation:
- Consistency of procedures: Sample handling steps must be strictly standardized in terms of timing, temperature, solvent volumes, and mixing techniques to minimize human-induced variability.
- Stability of sorbents and solvents: High-purity solvents and reusable sorbents should be used to prevent chemical changes or degradation during extraction.
- Sample storage conditions: Extracted and cleaned samples must be stored under proper conditions to avoid degradation or chemical transformation.
- Contamination and matrix effect control: Minimizing background interference enhances measurement accuracy.
- Use of internal standards and calibration: Isotopically labeled internal standards help correct for errors arising during sample preparation and analysis.
Approaches to improve repeatability and accuracy:
- Automation of sample preparation: Automated systems reduce manual handling errors and improve consistency across analyses.
- Training of laboratory personnel: The skill and experience of the operator significantly influence the stability and reproducibility of the procedure.
- Process validation and quality control: Implementing repeated measurements, using quality control (QC) samples, and regularly checking the system help detect and correct deviations promptly.
Ensuring high repeatability and accuracy in the sample preparation stage is a prerequisite for chromatographic results to truly reflect the actual composition of the sample. It also gives analysts and decision-makers confidence in the reliability of the data obtained.
4.4. Selectivity
Selectivity in the sample preparation process plays a crucial role in ensuring the accurate isolation of target organic compounds from complex sample matrices. Especially in the simultaneous analysis of multiple compounds with diverse chemical characteristics, the selection of sample preparation methods and materials must be optimized to remove unwanted interferences while preserving as much of the target analytes as possible [95,96].
The significance of selectivity:
- Enhances the purity of the analytical extract, reduces background noise, and minimizes unwanted interactions during chromatographic analysis.
- Prevents unnecessary loss of target compounds due to non-specific adsorption or reactions with incompatible sorbents.
- Improves detection and quantification accuracy, especially for low-concentration compounds or those in complex matrices.
Factors influencing selectivity:
- Type of sorbent material: For example, PSA is effective for removing organic acids and some pigments; C18 is suitable for retaining lipids and non-polar compounds; while GCB is selective for pigments and chlorophyll. Combining these sorbents in dSPE techniques enhances multi-dimensional selectivity for complex matrices.
- Extraction and sample handling conditions: Parameters such as pH, solvent composition, and contact time also influence the selective separation between analytes and matrix components.
- Adsorption mechanisms and chemical interactions: Understanding the interaction mechanisms between sorbents and compounds in the sample helps select appropriate materials and avoid non-specific binding or analyte loss.
- Balancing selectivity and recovery: Excessive selectivity may result in the removal of some target compounds, while insufficient selectivity may fail to eliminate interfering substances. Therefore, optimal conditions must be established to achieve the best compromise.
Approaches to enhance selectivity:
- Use dispersive solid-phase extraction (dSPE) with a combination of sorbents possessing different functionalities to target a broad range of matrix interferences.
- Apply additional pre-treatment steps such as filtration, centrifugation, or pH adjustment to improve analyte-matrix separation.
- Develop and select novel functionalized sorbents, such as nanomaterials or specialized polymers, which offer high selectivity toward specific classes of target compounds.
4.5. Integration Capability with Analytical Systems
In the simultaneous analysis of organic compounds, sample preparation not only serves to clean and enrich the sample but must also ensure effective integration with downstream analytical systems—typically chromatographic systems coupled with mass spectrometric detectors (GC-MS/MS, LC-MS/MS) [97,98,99]. Compatibility between the processed sample phase and the analytical instrumentation is essential for maintaining sensitivity, system robustness, and overall data quality.
Importance of integration capability:
- Ensures chemical compatibility between the sample solvent and the mobile phase, preventing issues such as phase separation, syringe clogging, or peak distortion during chromatography.
- Minimizes manual sample transfer steps such as solvent evaporation, solvent phase switching, or additional filtration—saving time and reducing the risk of analyte loss.
- Enhances automation compatibility, aligning with the trend toward integrated, online, or at-line analytical workflows.
Technical requirements for effective integration:
-
Use of extraction solvents compatible with analytical systems:
- ○
- For LC-MS/MS, solvents like acetonitrile or methanol are preferred due to their miscibility with the mobile phase and rapid evaporation at the ion source.
- ○
- For GC-MS/MS, samples must be highly volatile and water-free; thus, solvents like hexane or ethyl acetate are used, and sometimes a solvent evaporation–reconstitution step is required.
- Complete removal of residual solids, proteins, or lipids: These components can clog syringes, affect system pressure, and cause severe background noise in detectors. Strong centrifugation, membrane filtration (0.22–0.45 µm), or lipid removal using C18 sorbents is critical.
- Optimization of sample volume and concentration: The injection volume must meet the requirements of the chromatographic system (typically 1–10 µL for LC, <1 µL for GC), and sample concentration should be adjusted to fall within the detector’s linear range to avoid signal saturation.
- Stability of the processed sample: Samples should remain stable without degradation or transformation during the waiting period before analysis—especially important in automated, chained systems where there may be a delay between sample preparation and analysis.
Modern integration trends:
- Automated QuEChERS systems allow full sample preparation—from extraction to dSPE—and direct injection into LC-MS/MS without manual handling.
- Directly coupled microextraction techniques, such as solid-phase microextraction (SPME) linked directly to GC-MS, eliminate intermediate processing steps entirely.
- On-line SPE–LC-MS/MS systems, where the sample is cleaned directly on an in-line SPE cartridge and transferred to the LC-MS system without manual withdrawal or filtration.
5. Common Sample Preparation Techniques in Simultaneous Analytical Methods
Simultaneous determination of multiple organic compounds presents unique challenges in terms of matrix complexity, compound polarity, volatility, and stability. An effective sample preparation method must be compatible with these diverse chemical properties while minimizing sample loss, interference, and processing time [100,101,102]. Below are several widely used techniques that have demonstrated efficiency in multi-analyte sample preparation for chromatographic analysis.
Table 2.
Common Sample Preparation Techniques in Simultaneous Analysis Methods.
| Method | Advantages | Applications |
|---|---|---|
| QuEChERS | Fast, low-cost, suitable for multi-residue analysis (pesticides, pharmaceuticals) | Food samples, water, plasma |
| SPE (Solid Phase Extraction) | Good cleanup, flexible with separation phases | Environmental samples, wastewater, biological samples |
| SPME (Solid Phase Microextraction) | Solvent-free, ideal for volatile compound analysis | Air, water, food |
| LLE (Liquid-Liquid Extraction) | Widely used, easy to implement | Water samples, biological samples |
| dSPE (Dispersive SPE) | Enhanced matrix cleanup, commonly used in QuEChERS | Combined with complex sample matrices |
5.1. QuEChERS
The QuEChERS method (an acronym for Quick, Easy, Cheap, Effective, Rugged, and Safe) is one of the most widely used sample preparation procedures today for the simultaneous determination of multiple trace organic compounds in complex matrices, especially in food, environmental, and biological samples [103,104]. Originally developed for pesticide residue analysis, QuEChERS has demonstrated high versatility and efficiency in extracting and cleaning a broad range of organic compounds, including pesticides, veterinary drugs, semi-volatile compounds, food additives, antibiotics, and endocrine-disrupting chemicals.
The QuEChERS procedure consists of four main steps (Figure 1):
-
Extraction:Approximately 5–15 g of solid sample (or an equivalent volume of liquid sample) is placed into a centrifuge tube. The most common extraction solvent is acetonitrile due to its excellent ability to extract polar to moderately non-polar compounds while having low miscibility with water. After adding the solvent, the sample is vigorously shaken to extract the target organic compounds into the organic phase.
-
Partitioning:mixture of anhydrous salts, typically magnesium sulfate (MgSO₄) to remove water, combined with sodium chloride (NaCl) or buffering salts such as sodium citrate or sodium acetate, is added. These salts facilitate a clear phase separation between the organic solvent and water while adjusting the pH to stabilize the analytes. The result is a separated acetonitrile phase containing the analytes, isolated from the aqueous matrix.
-
Clean-up (dispersive Solid Phase Extraction, dSPE):The acetonitrile phase obtained after partitioning is transferred to a tube containing sorbents such as PSA (to remove organic acids and sugars), C18 (to remove lipids), and GCB (to eliminate pigments and chlorophyll). The choice of sorbents or their combinations depends on the sample matrix and the analyte groups. This step significantly reduces matrix interference, improving the sensitivity and accuracy of the measurements.
-
Instrumental Analysis:The cleaned supernatant after centrifugation is collected for analysis by instrumentation such as GC, GC-MS, LC, LC-MS/MS, or GC-FID. QuEChERS allows sample preparation in small volumes, suitable for injection requirements in modern chromatographic techniques. Furthermore, the resulting extract generally has good cleanliness and stability, prolonging column lifetime and reducing ion source contamination in mass spectrometry.
Figure 2.
QuEChERS sample preparation process.
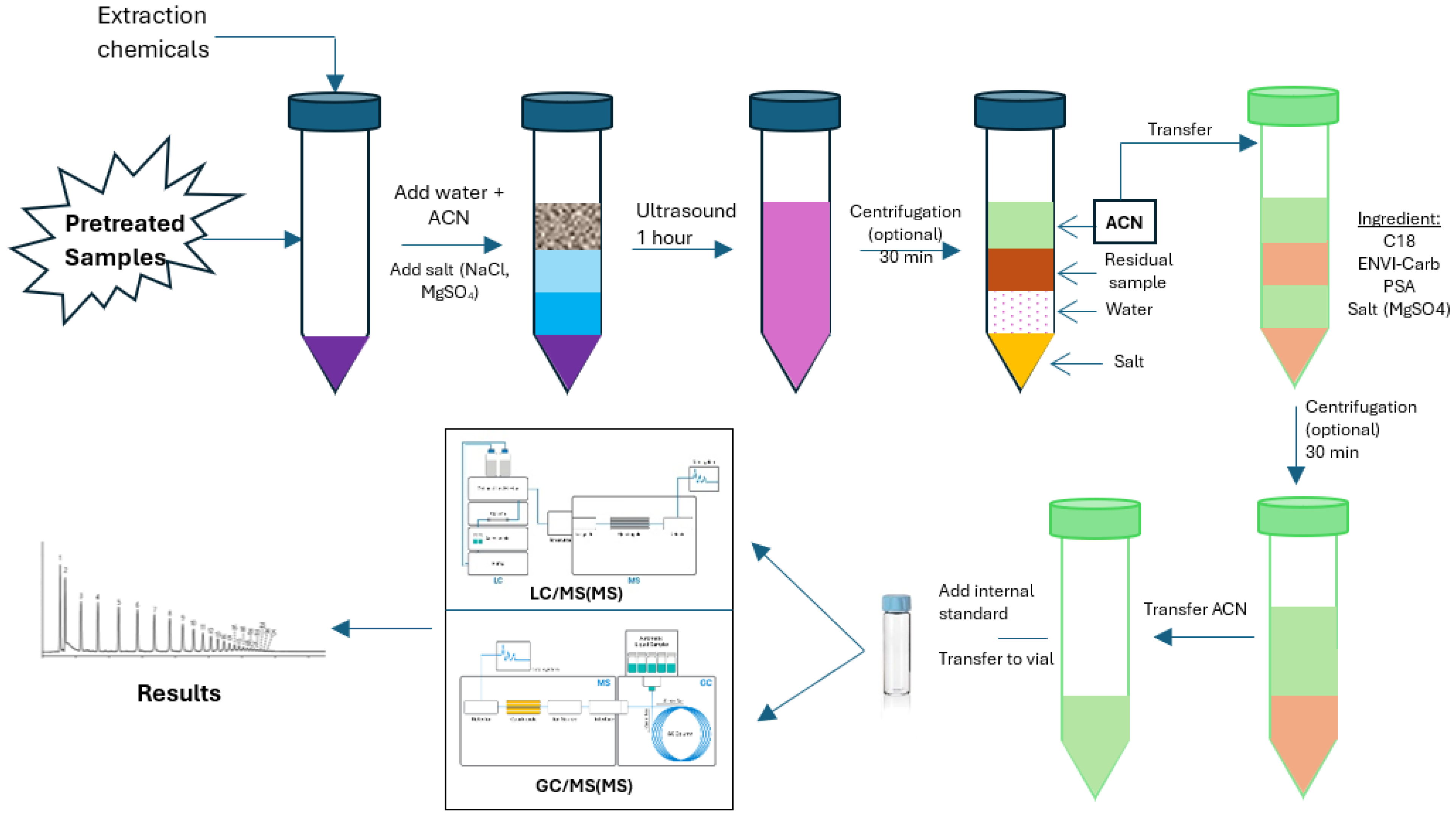
QuEChERS offers several notable advantages in the simultaneous analysis of organic compounds. Primarily, it provides high recovery efficiency across various compound classes due to effective extraction combined with flexible pH adjustment to stabilize analytes. At the same time, it minimizes matrix effects, thereby enhancing analytical accuracy and repeatability [105,106]. This method is particularly suited for multi-target analyses, including pesticides, environmental pollutants, and food additives. Additionally, the QuEChERS extract is readily compatible with modern analytical systems like LC-MS/MS and GC-MS without requiring complex enrichment steps or solvent exchange. Finally, QuEChERS is highly flexible and user-friendly, making it suitable for laboratories of all sizes thanks to its customizable procedure tailored to specific sample characteristics and analytical goals.
5.2. Solid Phase Extraction
Solid Phase Extraction (SPE) is a common and effective sample preparation technique, particularly suitable for applications requiring the cleanup and enrichment of trace organic compounds in complex sample matrices. In the context of simultaneous analysis of multiple compounds from different functional groups, SPE allows flexible process design by selecting specialized solid phases to optimize recovery efficiency and minimize background interference. This technique has been widely applied in environmental, food, pharmaceutical, and biological analyses.
The standard SPE procedure consists of four main steps: conditioning the solid phase, sample loading, column washing, and analyte elution. First, the solid phase inside the SPE cartridge — which may be modified silica (C18), porous polymer (HLB), or ion exchange phases (SCX, SAX) — is conditioned using an organic solvent such as methanol, followed by water or a solvent compatible with the sample matrix [107,108,109]. Then, the liquid sample (or extract from solid samples) is loaded onto the column. Target compounds are retained through physicochemical interactions such as hydrophobic adsorption, ion exchange, or specific molecular interactions.
Next is the column washing step with an intermediate solvent to remove impurities that are not strongly bound to the solid phase, while ensuring that the necessary analytes are not washed away. The final step is elution, where a stronger solvent — such as methanol, acetonitrile, or a pH-adjusted solvent mixture — is used to disrupt interactions between the analytes and the sorbent, recovering the organic compounds in a clean solution ready for chromatographic analysis.
Figure 3.
SPE process workflow for selective multi-compound analysis.
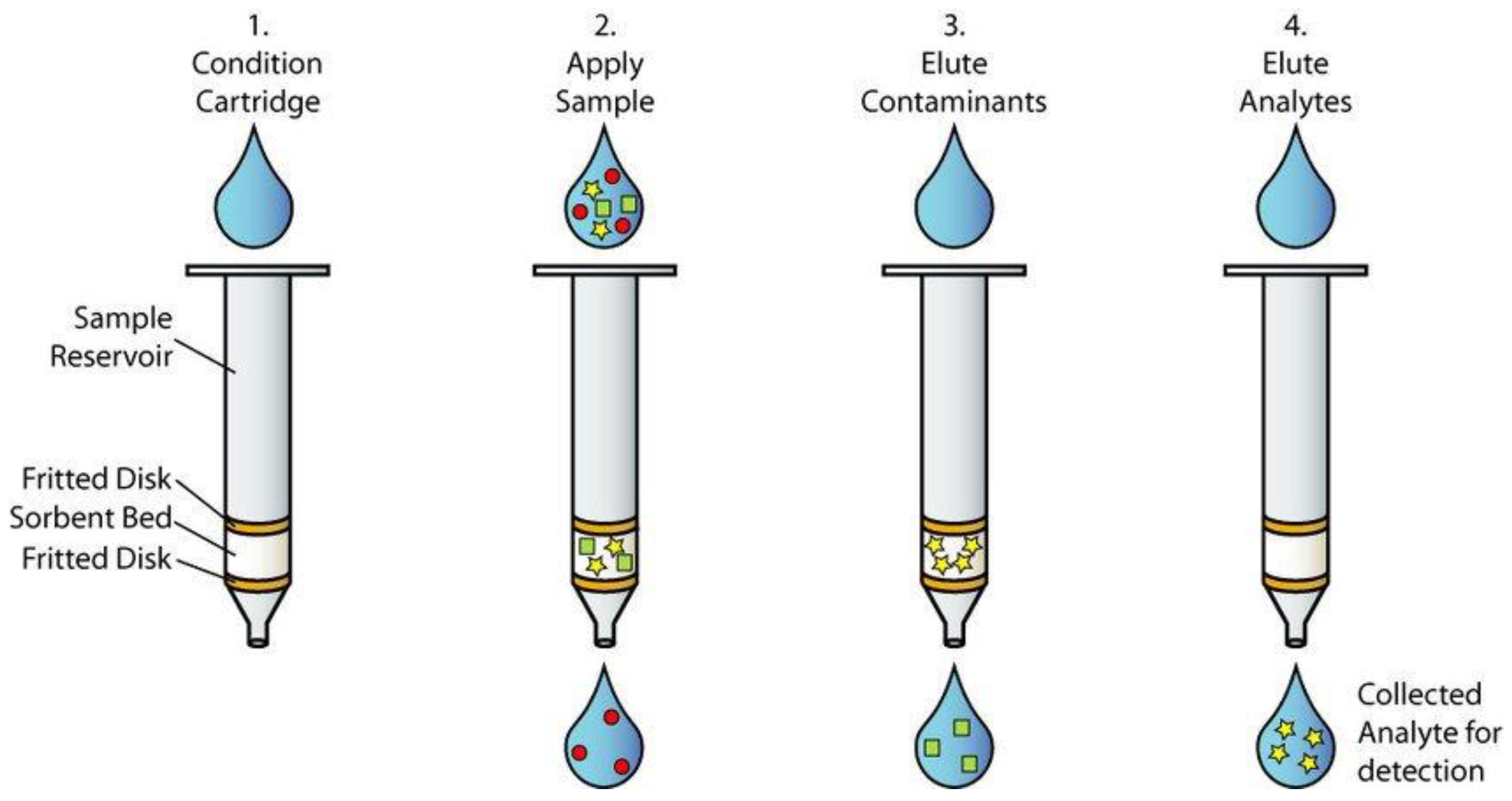
In simultaneous analysis, SPE enables selective sample processing, effectively removing complex matrices while simultaneously retaining multiple target compounds. Depending on the analytical goals, SPE can be configured to prioritize the recovery of polar, semi-polar, or non-polar compounds — or to use sequential combinations of different solid phases to optimize overall recovery efficiency. Additionally, SPE offers the capability to concentrate analytes by reducing the volume of the elution solvent, thereby improving detection limits and sensitivity.
Another significant advantage of SPE is its compatibility with automated analytical systems, which enhances repeatability and throughput in laboratories handling large sample volumes. Thanks to its high flexibility in selecting sorbent materials, solvents, and operating conditions, SPE remains a key choice in modern sample preparation, especially for studies requiring the simultaneous determination of multiple organic compounds with diverse chemical properties.
5.3. Solid Phase MicroExtraction
Solid Phase Microextraction (SPME) is an advanced sample preparation technique that integrates extraction and analyte enrichment in a single step without the use of solvents. With its minimalist and environmentally friendly design, SPME is especially suitable for the analysis of volatile organic compounds (VOCs), semi-volatile organic compounds (SVOCs), and some polar compounds, thereby expanding its potential applications in simultaneous analysis of compounds in air, water, food, biological, and environmental samples.
The SPME procedure consists of four basic steps: fiber conditioning, extraction, desorption, and analysis. Before use, the extraction fiber is conditioned by heating inside the injection port of a GC instrument or a specialized device to remove residual contaminants. During extraction, the fiber is exposed to the sample in one of three modes: direct immersion in the liquid phase, exposure to the gas phase, or in the headspace above the sample. Organic compounds are adsorbed or absorbed into the specially coated fiber surface according to equilibrium or kinetic mechanisms, depending on exposure time, temperature, and the coating material (e.g., PDMS, DVB, CAR).
After extraction, the fiber is transferred directly to the analytical instrument—typically GC or GC-MS—for thermal desorption. The analytes are released from the fiber and introduced into the chromatographic column for separation and identification. SPME can be coupled with gas chromatography systems or, in some cases, liquid chromatography through suitable desorption devices, allowing its application to a wide range of compounds with varying polarity and volatility.
Figure 4.
Schematic diagram of the Solid Phase Microextraction (SPME) process.
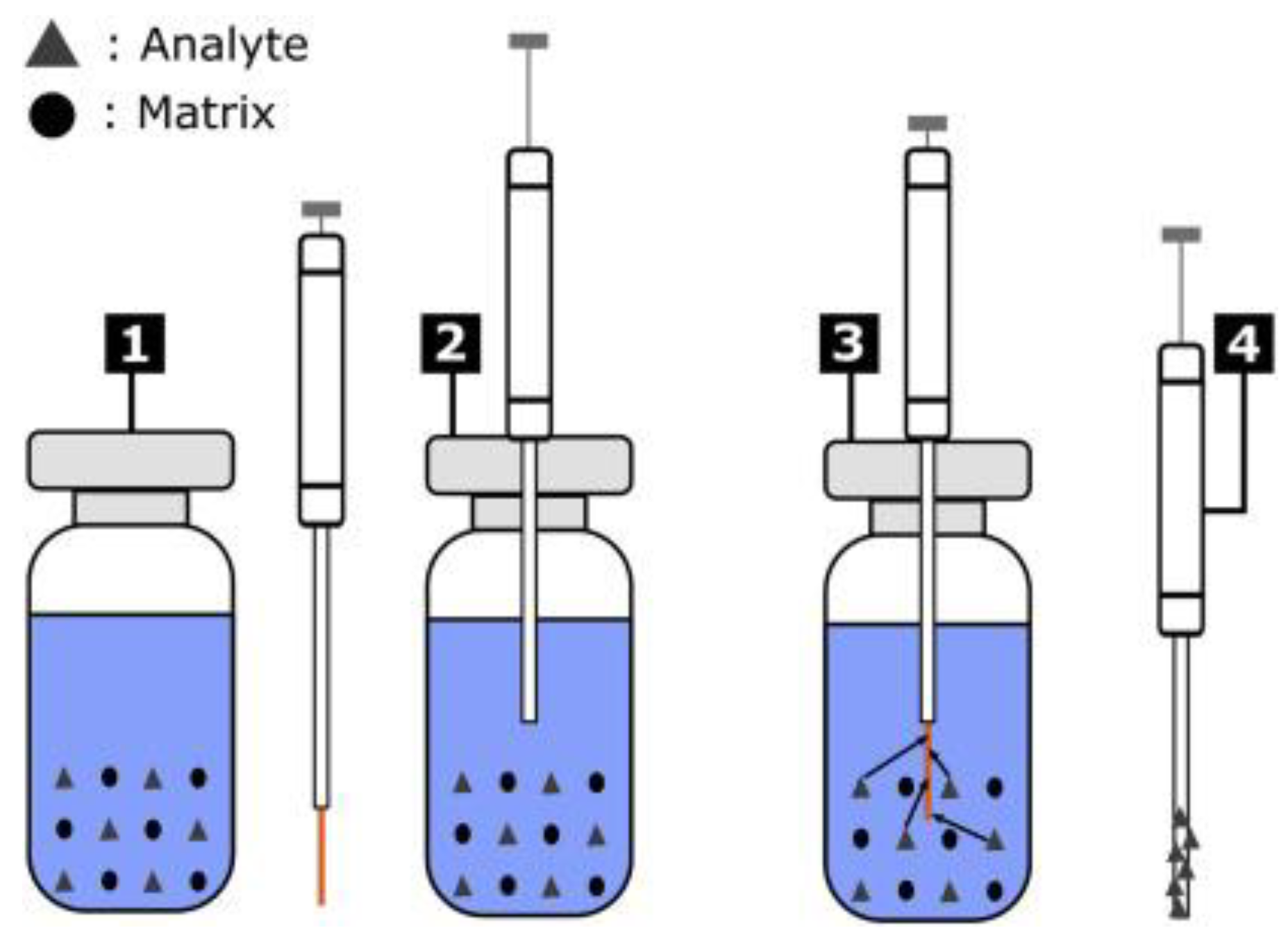
In simultaneous analysis, SPME offers several key advantages: it requires no complex pretreatment, minimizes matrix effects, enhances sensitivity via on-fiber enrichment, and reduces solvent consumption and processing time. Moreover, this technique allows for the processing of multiple compound classes in a single sample without separate fractionation, as long as the analytes can be adsorbed or diffused onto the same type of fiber. Variants such as SPME Arrow, HiSorb, or dual coatings are also being developed to expand the scope of simultaneous analysis, especially in complex matrices.
However, to achieve high analytical performance and good reproducibility in simultaneous analysis, appropriate fiber coatings must be selected, extraction time and desorption conditions optimized, and competitive interactions among analytes on the fiber surface considered. With recent technical advancements, SPME is increasingly becoming a reliable choice for modern laboratories aiming for non-invasive, rapid, and efficient sample preparation for multi-organic compound analysis.
5.4. Liquid-Liquid Extraction
Liquid-Liquid Extraction (LLE) is one of the most traditional and widely used sample preparation techniques in chemical analysis, based on the different partitioning of analytes between two immiscible liquid phases—typically an organic solvent and an aqueous solvent (or aqueous extract). This method is commonly applied to separate, clean up, and concentrate organic compounds in biological, food, environmental, and pharmaceutical samples.
In simultaneous analysis, LLE can be effectively used to recover groups of organic compounds with varying polarity and solubility, especially when multiple consecutive extraction steps or mixed solvent systems are employed. A typical LLE procedure includes: (1) mixing the sample with an appropriate extraction solvent, (2) vigorous shaking to reach partition equilibrium, (3) phase separation by centrifugation or separatory funnel, and (4) collection of the organic phase containing the analytes.
Figure 5.
Liquid-Liquid Extraction (LLE) procedure overview.
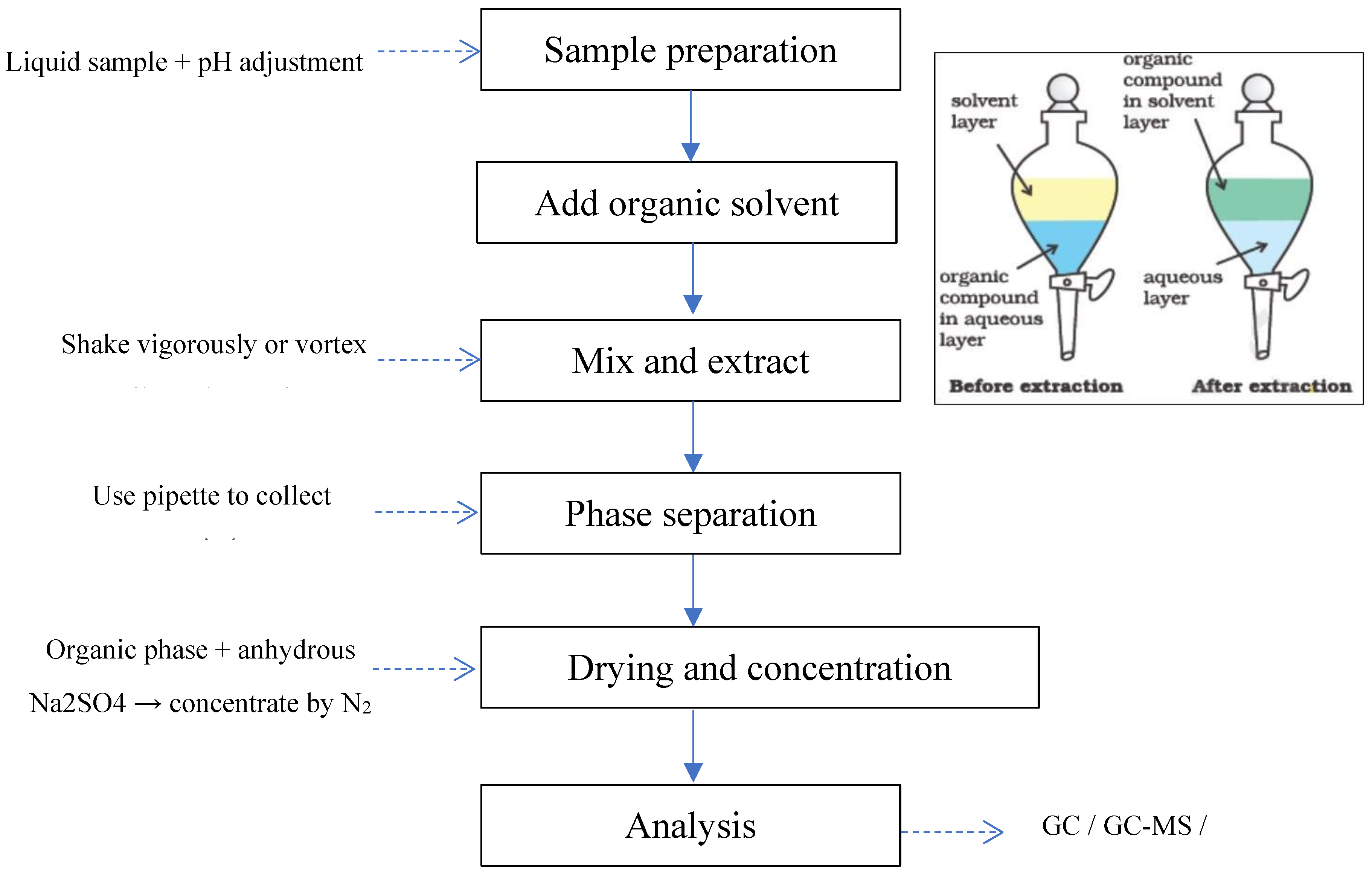
One of LLE’s strengths in simultaneous analysis is its simple and flexible sample handling with many solvents (ethyl acetate, dichloromethane, hexane, acetonitrile, etc.) whose polarity can be adjusted to match the chemical range of target compounds. Additionally, this method allows processing large sample volumes, significantly improving detection limits (LOD) through volume enrichment.
However, LLE also faces several challenges in simultaneous analysis:
- Low selectivity when compounds with similar physicochemical properties partition into the organic phase, causing high background interference.
- Uneven recovery efficiency since some analytes may remain in the aqueous phase or be lost during solvent evaporation.
- High solvent consumption, impacting sustainability and analysis costs.
To address these issues, improved strategies such as micro-liquid-liquid extraction (micro-LLE), ultrasound-assisted extraction (UAE-LLE), or dispersive liquid-liquid microextraction (DLLME) have been developed to enhance extraction efficiency, reduce solvent use, and improve reproducibility.
In the context of simultaneous analysis of trace organic compounds with high diversity, LLE remains a useful choice when fast, flexible preliminary sample processing is needed without requiring specialized equipment. However, to achieve optimal performance, careful consideration of the analyte chemistry, appropriate solvent selection, and strict control of operating parameters are necessary throughout the procedure.
5.5. Dispersive Solid Phase Extraction
Dispersive Solid Phase Extraction (dSPE) is a simple and effective sample clean-up technique, commonly integrated into modern procedures such as QuEChERS for rapid treatment of extracts containing trace organic compounds. The main advantage of dSPE lies in its ability to remove unwanted matrix components such as organic acids, sugars, pigments, and lipids without the need for traditional SPE columns, thereby reducing processing time and increasing flexibility in sample preparation.
The dSPE process begins after the initial extraction step, which typically uses acetonitrile as the solvent in the presence of salts (e.g., MgSO₄ and NaCl) to induce phase separation. The resulting extract is transferred into centrifuge tubes pre-filled with a selected mixture of sorbents suited to the sample matrix. Commonly used sorbents include:
- PSA (Primary Secondary Amine): removes organic acids, certain sugars, and fatty acids.
- C18 (Octadecylsilane): adsorbs lipids and non-polar compounds.
- GCB (Graphitized Carbon Black): targets pigments and aromatic ring-containing compounds.
- Anhydrous MgSO₄: helps remove residual water from the organic phase.
After the extract is added to the dSPE tube, the mixture is vigorously vortexed or manually shaken to ensure thorough dispersion of the sorbent particles and effective contact with the entire solution. The adsorption process occurs quickly, usually within a few minutes. The tube is then centrifuged to separate the sorbents—now bound to the interfering substances—leaving behind a clean supernatant containing the target analytes.
Figure 6.
Step-by-Step Illustration of dSPE Sample Clean-Up Process.
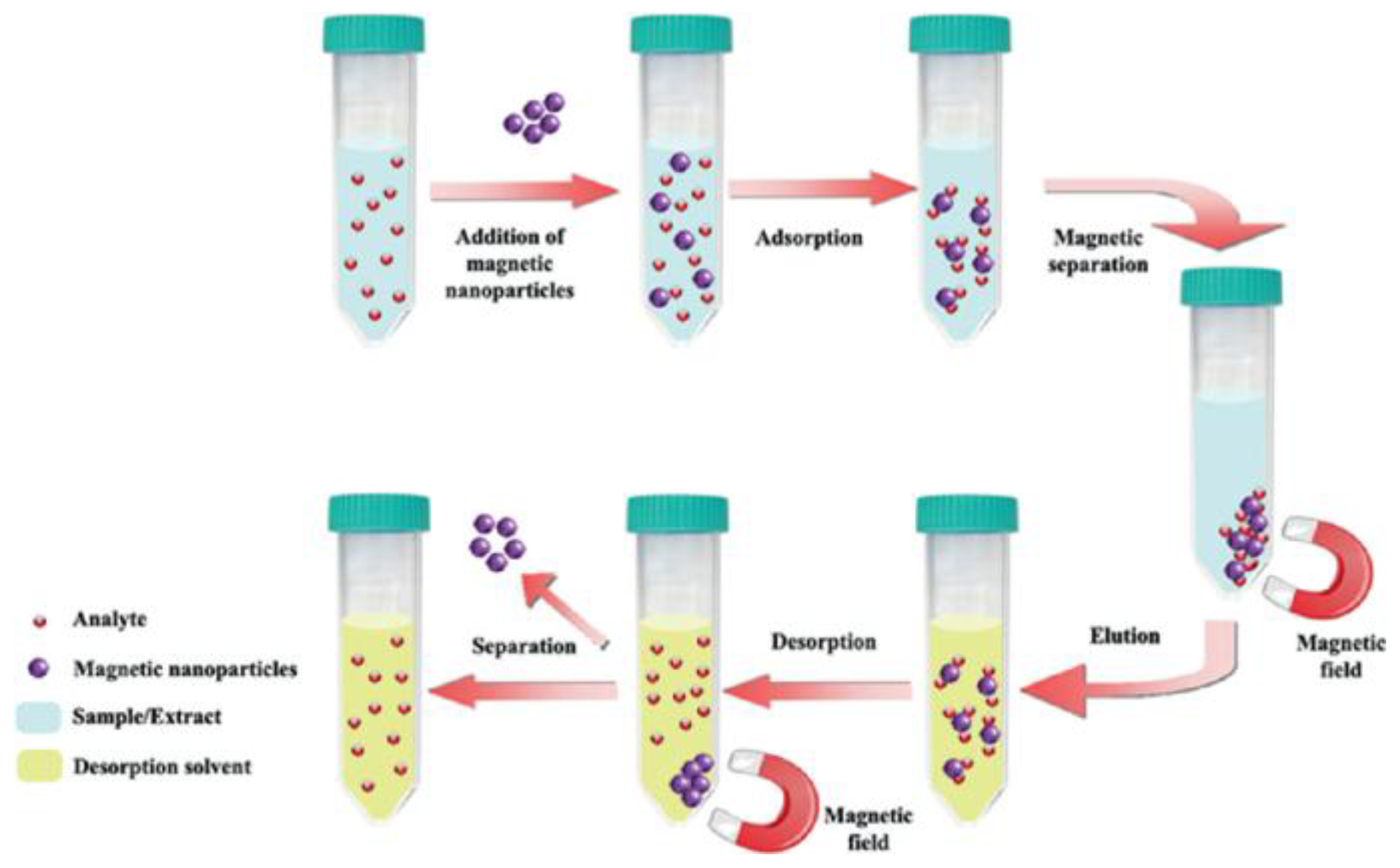
dSPE offers several advantages in simultaneous multi-compound analysis:
- Fast and efficient clean-up, minimizing matrix effects on analytical performance.
- No requirement for specialized or vacuum equipment, making it suitable for small-scale laboratories.
- Easily customizable based on sample type by selecting or combining appropriate sorbents.
- Highly compatible with modern chromatographic systems such as LC-MS/MS and GC-MS, enhancing sensitivity and accuracy.
However, to optimize dSPE performance in multi-residue analysis, careful consideration must be given to factors such as matrix type, chemical nature of the analytes, and their interactions with each sorbent. Inappropriate sorbent selection can lead to analyte loss or inadequate matrix removal.
Thanks to its simplicity, efficiency, and strong integration into modern sample preparation workflows, dSPE is increasingly employed for the simultaneous quantification of trace organic compounds in fields such as food safety, environmental monitoring, agriculture, and biomedical research.
6. Comparison and Selection of Appropriate Sample Preparation Techniques
Selecting the appropriate sample preparation technique plays a crucial role in the efficiency of simultaneous analysis of multiple organic compounds. Each technique offers distinct advantages and limitations depending on the chemical nature of the target analytes, the sample matrix, sensitivity and accuracy requirements, and the laboratory's capabilities [110,111,112,113]. The comparison table below summarizes the key characteristics of several commonly used techniques.
Table 3.
Comparison of main features, advantages, and limitations of common sample preparation techniques for multi-residue analysis.
Table 3.
Comparison of main features, advantages, and limitations of common sample preparation techniques for multi-residue analysis.
| Technique | Advantages | Limitations | Typical Applications |
|---|---|---|---|
| QuEChERS | – Fast, simple, low-cost – Suitable for various analyte groups – Integrates extraction and clean-up | – May not effectively clean complex matrices – Unsuitable for highly polar compounds | Pesticide residues, contaminants in food and environmental samples |
| SPE | – Excellent clean-up, high reproducibility – Allows sample enrichment – Highly customizable by analyte | – Multi-step procedure requiring equipment – Costly when multiple cartridges are used | Pharmaceuticals, pollutants in water and biological matrices |
| dSPE | – Simple and time-saving – Easily integrated with QuEChERS – No need for specialized equipment | – Strongly dependent on sorbent selection – Unsuitable for strongly adsorptive analytes | Rapid clean-up of food extracts, environmental matrices |
| SPME | – Solvent-free – Combines extraction and preconcentration – Well-suited for GC and GC-MS | – Limited to analytes that can be extracted – Requires specialized fibers and equipment; high cost | VOCs and SVOCs in air, water, food headspace |
| LLE | – Effective for non-polar compounds – Easy to perform, no complex equipment needed | – High solvent consumption, not environmentally friendly – Poor phase separation with emulsions or complex matrices | Organic compounds in water, serum, biological samples |
Based on the above comparison, choosing an appropriate sample preparation technique should consider the following factors:
- Chemical diversity of target analytes: For multi-residue analysis, techniques like QuEChERS or SPE are often preferred due to their flexibility and effective matrix removal.
- Sample matrix type: For complex matrices such as food and environmental samples, techniques with strong matrix removal capabilities like SPE or QuEChERS-dSPE are recommended.
- Required detection limits: For high sensitivity requirements, SPE or SPME techniques can preconcentrate analytes prior to analysis.
- Compatibility with analytical instrumentation: Techniques such as QuEChERS, dSPE, and SPME are easily compatible with LC-MS/MS and GC-MS without requiring intermediate processing.
- Automation potential and sample throughput: For labs handling high sample volumes, cartridge-based or 96-well plate SPE and autosampler-compatible SPME are ideal choices.
In many cases, combining techniques is a viable strategy to optimize sample preparation efficiency, such as QuEChERS + dSPE or liquid–liquid extraction followed by SPE.
Table 4.
Summary comparison of key operational criteria among five sample preparation techniques.
| Criterion | LLE | SPE | dSPE/QuEChERS | SPME | DLLME |
|---|---|---|---|---|---|
| Selectivity | Low | High | Medium | High | High |
| Automation | Limited | Possible | Possible | Possible | Difficult |
| Solvent saving | No | Moderate | Yes | Yes | Very high |
| Processing time | Moderate | Moderate | Fast | Moderate | Very fast |
7. Coupling Sample Preparation with Chromatographic Techniques
In multi-residue analysis of organic compounds, the effective integration of sample preparation and chromatographic techniques plays a pivotal role in ensuring the accuracy, sensitivity, and reliability of analytical results. Sample preparation not only removes interfering matrix components but also concentrates and preserves the target analytes, thereby optimizing the separation and detection performance of the chromatographic system.
Depending on the chemical properties of the compounds and the matrix type, common sample preparation techniques such as solid-phase extraction (SPE), liquid-liquid extraction (LLE), solid-phase microextraction (SPME), and ultrasound- or microwave-assisted extraction (UAE, MAE) are selected to efficiently recover a wide range of polar and non-polar compounds. After sample preparation, organic compounds are primarily analyzed using two major chromatographic techniques: high-performance (or ultra-high performance) liquid chromatography (HPLC/UHPLC) and gas chromatography (GC), each of which can be coupled with various detectors, particularly mass spectrometry (MS or MS/MS), to enhance selectivity and sensitivity.
The coordination between sample processing and chromatography must be carefully designed to avoid analyte loss or the formation of analytical artifacts. For instance, when simultaneously analyzing compounds with a wide polarity range—such as chlorinated pesticides (non-polar), dyes like malachite green (moderately polar), and polar metabolites—the sample preparation method must be flexible. This often involves multi-step procedures such as modified QuEChERS or mixed-mode SPE. These samples are then introduced into UHPLC-MS/MS systems using dynamic gradient elution to effectively separate target compounds within a short run time while maintaining resolution.
For volatile and semi-volatile compounds—especially hydrocarbons, light organic solvents, phosphorus-based pesticides, or phthalate esters—GC-MS/MS is considered optimal due to its superior separation capacity and high sensitivity. However, the sample must be thoroughly cleaned beforehand to remove water and non-volatile matrix components. This typically involves drying with anhydrous Na₂SO₄ or concentration using rotary evaporation.
The close integration of sample pre-treatment and chromatographic analysis is critical in designing multi-residue analytical methods, particularly when processing dozens or even hundreds of compounds in a single run. Developing sample preparation protocols that are fully compatible with modern chromatographic systems not only enhances analytical performance but also helps minimize errors, reduce time and cost, and ultimately promotes widespread application in food safety control, environmental monitoring, and biomonitoring of human exposure.
8. Method Validation in Multi-Residue Analysis
Method validation is an indispensable step to ensure that a multi-residue analytical procedure can provide accurate, reliable, and reproducible results under varying experimental conditions. Due to the complexity of simultaneously analyzing multiple compounds in diverse and challenging matrices, the validation process typically requires rigorous and comprehensive evaluation of numerous technical parameters.
The key parameters commonly assessed include accuracy, trueness, repeatability, reproducibility, limit of detection (LOD), limit of quantification (LOQ), selectivity, linearity, and matrix effects.
Accuracy and trueness reflect the method's ability to recover the actual concentration of the analytes. In multi-residue analysis, validation is typically performed at several concentration levels (e.g., low, medium, and high) to ensure method applicability across the relevant analytical range. According to international guidelines such as SANTE/11312/2021 or ICH Q2(R1), acceptable recovery typically ranges from 70% to 120%, with relative standard deviation (RSD) below 20%.
LOD and LOQ are critical parameters, especially for applications in food safety or environmental monitoring, where trace-level detection is essential. These limits are often determined based on signal-to-noise ratios or calculated from the standard deviation of the regression line in calibration curves.
Selectivity and linearity ensure that the method can accurately distinguish the analytes from matrix interferences and structurally similar compounds. Multi-residue analysis often requires a wide linear range (typically from a few µg/kg to mg/kg) and a correlation coefficient (R²) greater than 0.99.
Matrix effects represent a major challenge in the simultaneous analysis of organic compounds, as co-extracted matrix components can affect the mass spectrometric signal, causing ion suppression or ion enhancement. These effects can significantly compromise accuracy if not properly compensated, typically through the use of internal standards or matrix-matched calibration.
Stability of the analytes throughout sample processing, storage, and analysis must also be evaluated, especially for labile or easily degradable compounds.
Validating methods for multi-residue analysis requires a combination of robust statistical strategies and deep understanding of analytical chemistry. The advancement of high-resolution mass spectrometry (HRMS) platforms not only improves sensitivity but also facilitates structural confirmation and non-targeted screening, thus expanding the application scope of validated methods. Standardizing and harmonizing validation protocols according to international guidelines is a critical step toward achieving global acceptance of analytical results, supporting regulatory oversight in health, environment, and international trade.
9. Perspectives
Simultaneous analysis of multiple organic compounds in complex matrices continues to be one of the greatest challenges in modern analytical chemistry. Advances in chromatographic and mass spectrometric technologies—particularly the integration of ultra-high-performance and high-resolution systems—have significantly expanded the range of detectable compounds, while enhancing both sensitivity and selectivity.
Simultaneous analytical methods are increasingly expected to be applicable across diverse sample types such as environmental samples, food products, and plant-based medicines. These methods aim to quantify a broad spectrum of compound classes, including nutrients, contaminants, and bioaccumulative substances. The resulting analytical data have wide-ranging applications—not only in contaminant detection but also in fields such as food traceability and human health risk assessment.
In this context of increasing data volume and complexity, artificial intelligence (AI)—particularly machine learning and deep learning models—has emerged as a promising tool for data processing, classification, and compound identification. AI has shown considerable effectiveness in interpreting chromatographic and mass spectral data, detecting anomalies, and identifying unknown compounds [114]. Trained on large spectral databases, AI systems can accelerate compound identification, reduce reliance on costly reference standards, and even aid in the discovery of novel trace-level contaminants. Additionally, AI can assist in optimizing chromatographic conditions and recommending sample preparation steps based on matrix type and analyte chemistry.
However, the success of AI-powered analysis depends critically on the quality and reliability of input data, which places robust sample preparation at the core of the workflow. In analytical practice, any inconsistencies or errors in sample preparation may lead to incorrect data interpretation, thereby undermining the entire analysis—even when supported by advanced algorithms. As a result, modern sample preparation techniques must be designed to handle a wide range of compounds simultaneously, while maintaining high selectivity, reproducibility, and compatibility with downstream AI-assisted analysis.
Furthermore, the integration of AI into analytical instrumentation—such as automated chromatographic-mass spectrometric systems capable of learning and adapting to specific sample matrices—is ushering in a new era of non-targeted analysis, in which the challenge shifts from compound detection to data interpretation. In such a paradigm, sample preparation is no longer merely a preprocessing step, but rather a strategic component that underpins accuracy, speed, and adaptability in real-world analytical scenarios.
In conclusion, the future of multi-residue analysis lies not in replacing traditional processes but in harmonizing advanced sample preparation techniques with cutting-edge analytical technologies and artificial intelligence—toward building an intelligent, accurate, and sustainable analytical ecosystem.
10. Conclusions
Simultaneous analysis of multiple organic compounds in complex matrices is becoming increasingly essential for food quality control, environmental risk assessment, and public health surveillance. Achieving high accuracy, reproducibility, and appropriate detection limits requires robust and well-optimized sample preparation workflows as the critical foundation of the entire analytical process. Modern preparation strategies such as QuEChERS, SPE, d-SPE, and SPME have demonstrated strong performance in simultaneously extracting chemically diverse compound classes, thus enabling the effective application of advanced chromatographic platforms such as GC-MS/MS and LC-HRMS.
Classifying analytes based on their origin, function, or physicochemical properties—such as polarity or solvent solubility—serves as a guiding principle for selecting extraction solvents and cleanup protocols. At the same time, the diversity of sample matrices—including food, water, soil, and biological tissues—demands increasingly flexible and adaptable preparation methods. Efficient integration between sample preparation and chromatographic analysis is therefore key to enabling accurate, high-throughput detection of a wide range of target compounds in a single analytical run.
Simultaneous analytical methods are now expected to support a broad spectrum of applications beyond contaminant monitoring. These include analysis of environmental samples, food products, and plant-based medicines, aiming to detect and quantify various compound groups—such as essential nutrients, pollutants, and bioaccumulative substances. The resulting comprehensive datasets can inform diverse research and regulatory fields, including food traceability, health risk assessment, and origin verification.
In the era of large and complex datasets, artificial intelligence (AI) opens new opportunities for spectral data processing, compound identification, and automation of analytical workflows. AI models—particularly those based on machine learning and deep learning—can accelerate the detection of unknown or emerging compounds, optimize chromatographic conditions, and reduce dependence on expensive reference standards. However, the effectiveness of AI remains highly dependent on the reliability and consistency of input data, which in turn hinges on rigorous sample preparation. Rather than replacing traditional workflows, AI should be viewed as a powerful enabler that enhances the value of existing analytical and sample preparation techniques.
In conclusion, the future of multi-residue analysis lies in the convergence of modern chromatographic instrumentation, optimized sample preparation strategies, and intelligent AI platforms. Together, these components can deliver analytical systems that are faster, more accurate, cost-effective, and better suited to the real-world demands of chemical monitoring and environmental protection.
References
- Wang, Z., Walker, G. W., Muir, D. C. G., & Nagatani-Yoshida, K. (2020). Toward a Global Understanding of Chemical Pollution: A First Comprehensive Analysis of National and Regional Chemical Inventories. Environmental Science & Technology, 54(5), 2575–2584. [CrossRef]
- Muir DCG, Getzinger GJ, McBride M, Ferguson PL. How Many Chemicals in Commerce Have Been Analyzed in Environmental Media? A 50 Year Bibliometric Analysis. Environ Sci Technol. 2023 Jun 27;57(25):9119-9129. [CrossRef]
- Lin, Y., Liu, R., Hu, F., Liu, R., Ruan, T., & Jiang, G. (2016). Simultaneous qualitative and quantitative analysis of fluoroalkyl sulfonates in riverine water by liquid chromatography coupled with Orbitrap high resolution mass spectrometry. Journal of Chromatography A, 1435, 66–74. [CrossRef]
- Kalogeropoulou AG, Kosma CI, Albanis TA. Simultaneous determination of pharmaceuticals and metabolites in fish tissue by QuEChERS extraction and UHPLC Q/Orbitrap MS analysis. Anal Bioanal Chem. 2021 Nov;413(28):7129-7140. Epub 2021 Oct 1. [CrossRef] [PubMed]
- Vu-Duc, N.; Nguyen-Quang, T.; Le-Minh, T.; Nguyen-Thi, X.; Tran, T.M.; Vu, H.A.; Nguyen, L.A.; Doan-Duy, T.; Van Hoi, B.; Vu, C.T.; et al. Multiresidue pesticides analysis of vegetables in Vietnam by ultrahigh-performance liquid chromatography in combination with high-resolution mass spectrometry (UPLC-Orbitrap MS). J. Anal. Methods Chem. 2019, 2019, 3489634. [CrossRef]
- D.A. Truong, H.T. Trinh, G.T. Le, T.Q. Phan, H.T. Duong, T.T.L. Tran, T.Q. Nguyen, M.T.T. Hoang, T.V. Nguyen, Occurrence and ecological risk assessment of organophosphate esters in surface water from rivers and lakes in urban Hanoi, Vietnam, Chemosphere, 331 (2023), Article 138805. [CrossRef]
- H.T. Duong, K. Kadokami, H. Shirasaka, R. Hidaka, H.T.C. Chau, L. Kong, T.Q. Nguyen, T.T. Nguyen, Occurrence of perfluoroalkyl acids in environmental waters in Vietnam, Chemosphere, 122 (2015), pp. 115-124. [CrossRef]
- Hoang, T. T. M.; Truong, G.; Kiwao, K.; Thi, H. Chemosphere Occurrence and Risk of Human Exposure to Organophosphate Flame Retardants in Indoor Air and Dust in Hanoi, Vietnam. Chemosphere, 2023, 328, 138597. [CrossRef]
- Thang, P.Q., Taniguchi, T., Nabeshima, Y. et al. Distribution of polycyclic aromatic hydrocarbons concentrations simultaneously obtained in gas, rainwater and particles. Air Qual Atmos Health 7, 273–281 (2014). [CrossRef]
- V. N. Le, Q. T. Nguyen, T. D. Nguyen, N. T. Nguyen, T. Janda, G. Szalai, and T. G. Le, “The potential health risks and environmental pollution associated with the application of plant growth regulators in vegetable production in several suburban areas of hanoi, Vietnam,” Biologia Futura, vol. 71, no. 3, pp. 323–331, Sep. 2020. [CrossRef]
- Nguyen HMN, Khieu HT, Ta NA, Le HQ, Nguyen TQ, Do TQ, Hoang AQ, Kannan K, Tran TM, Distribution of cyclic volatile methylsiloxanes in drinking water, tap water, surface water, and wastewater in Hanoi, Vietnam. Environmental Pollution, 2021, 285:117260. [CrossRef]
- T.M. Le, P.T. Pham, Truong Quang Nguyen, Trung Quang Nguyen, M.Q. Bui, H.Q. Nguyen, N.D. Vu, K. Kannan, T.M. Tran, A survey of parabens in aquatic environments in Hanoi, Vietnam and its implications for human exposure and ecological risk, Environ. Sci. Pollut. Res., 29 (2022), pp. 46767-46777. [CrossRef]
- Alexa Canchola, Lillian N. Tran, Wonsik Woo, Linhui Tian, Ying-Hsuan Lin, Wei-Chun Chou, Advancing non-target analysis of emerging environmental contaminants with machine learning: Current status and future implications, Environment International, 2025, 198, 109404. [CrossRef]
- Rebryk A, Haglund P. Comprehensive non-target screening of biomagnifying organic contaminants in the Baltic Sea food web. Sci Total Environ. 2022 Dec 10;851(Pt 1):158280. Epub 2022 Aug 24. [CrossRef] [PubMed]
- D.T. Hanh, K. Kadokami, N. Matsuura, N.Q. Trung, Screening analysis of a thousand micro-pollutants in Vietnamese rivers, Southeast Asian Water Environ., 5 (2013), pp. 195-202.
- Yang Gao, Yanhua Chen, Xiaofei Yue, Jiuming He, Ruiping Zhang, Jing Xu, Zhi Zhou, Zhonghua Wang, Rui Zhang, Zeper Abliz, Development of simultaneous targeted metabolite quantification and untargeted metabolomics strategy using dual-column liquid chromatography coupled with tandem mass spectrometry, Analytica Chimica Acta, 2018, 1037, 369-379. [CrossRef]
- Pengwei Guan, Yuting Wang, Tiantian Chen, Jun Yang, Xiaolin Wang, Guowang Xu, and Xinyu Liu, Novel Method for Simultaneously Untargeted Metabolome and Targeted Exposome Analysis in One Injection, Analytical Chemistry, 2025 97 (7), 3996-4004. [CrossRef]
- H.T. Duong, K. Kadokami, S. Pan, N. Matsuura, T.Q. Nguyen, Screening and analysis of 940 organic micro-pollutants in river sediments in Vietnam using an automated identification and quantification database system for GC–MS, Chemosphere, 107 (2014), pp. 462-472. [CrossRef]
- Winnike JH, Wei X, Knagge KJ, Colman SD, Gregory SG, Zhang X. Comparison of GC-MS and GC×GC-MS in the analysis of human serum samples for biomarker discovery. J Proteome Res. 2015 Apr 3;14(4):1810-7. Epub 2015 Mar 16. [CrossRef] [PubMed] [PubMed Central]
- Luiz Antonio Fonseca de Godoy, Ronei Jesus Poppi, Márcio Pozzobon Pedroso, Fabio Augusto, Leandro Wang Hantao, GCxGC-FID for Qualitative and Quantitative Analysis of Perfumes, LCGC Europe, 2010, 23, 8, 430 – 438.
- Alessandro Di Giorgi, Giuseppe Basile, Francesco Bertola, Francesco Tavoletta, Francesco Paolo Busardò, Anastasio Tini, A green analytical method for the simultaneous determination of 17 perfluoroalkyl substances (PFAS) in human serum and semen by ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS), Journal of Pharmaceutical and Biomedical Analysis, 2024, 246, 116203. [CrossRef]
- Yanwei Liu, Xingwang Hou, Xiaoying Li, Jiyan Liu, Guibin Jiang, Simultaneous determination of 19 bromophenols by gas chromatography–mass spectrometry (GC-MS) after derivatization, Talanta, 2024, 274, 126015. [CrossRef]
- Jun Hu, Yina Ba, Zhifeng Pan, Xiaogang Li, Simultaneous determination of 50 antibiotic residues in plasma by HPLC–MS/MS, Heliyon, 2024, 10, 24, e40629. [CrossRef]
- Lei Zhao, Zi Lin, Duan Ju, Jiayan Ni, Yuxuan Ma, Bin Chen, Xiaozhou Li, Congcong Sun, Jianqiong Zheng, Hongping Zhang, Shike Hou, Penghui Li, Shanjun Song, Liqiong Guo, Simultaneous determination of multiple endocrine disrupting chemicals in human amniotic fluid samples by solid phase extraction coupled with liquid chromatography tandem mass spectrometry (LC-MS/MS), Talanta, 2025, 293, 128088. [CrossRef]
- Mahdiyeh Otoukesh, Claudia Simarro-Gimeno, Félix Hernández, Elena Pitarch, Simultaneous LC-MS/MS determination of multi-class emerging contaminants in an orange plant system, Environmental Nanotechnology, Monitoring & Management, 2025, 23, 101077. [CrossRef]
- Qianqian Deng, Yang Liu, Dan Liu, Ziwei Meng, Xianghong Hao, Development of a design of experiments (DOE) assistant modified QuEChERS method coupled with HPLC-MS/MS simultaneous determination of twelve lipid-soluble pesticides and four metabolites in chicken liver and pork, Journal of Food Composition and Analysis, 2024, 133, 106379. [CrossRef]
- Alejandro García-Juan, Nuria León, Sergio Armenta, Olga Pardo, Development and validation of an analytical method for the simultaneous determination of 12 ergot, 2 tropane, and 28 pyrrolizidine alkaloids in cereal-based food by LC-MS/MS, Food Research International, 2023, 174, 1, 113614. [CrossRef]
- Shunqin Chen, Han Yang, Shan Zhang, Faze Zhu, Shan Liu, Huan Gao, Qing Diao, Wenbo Ding, Yuemeng Chen, Peng Luo, Yubo Liu, Simultaneous determination of 28 illegal drugs in sewage by high throughput online SPE-ISTD-UHPLC-MS/MS, Heliyon, 2024, 10, 6, e27897. [CrossRef]
- Kim, Y.R., Park, S., Kim, J.Y. et al. Simultaneous determination of 31 Sulfonamide residues in various livestock matrices using liquid chromatography-tandem mass spectrometry. Appl Biol Chem 67, 13 (2024). [CrossRef]
- Trinh, H. T.; Marcussen, H.; Hansen, H. C. B.Screening of inorganic and organic contaminants in floodwater in paddy fields of Hue and Thanh Hoa in Vietnam. Environ. Sci. Pollut Res. 2017, 24 (8), 7348– 7358,. [CrossRef]
- Chau, H. T. C.; Kadokami, K.; Duong, H. T.; Kong, L.; Nguyen, T. T.; Nguyen, T. Q.; Ito, Y. Occurrence of 1153 organic micropollutants in the aquatic environment of Vietnam. Environ. Sci. Pollut. Res. 2018, 25, 7147– 7156,. [CrossRef]
- Chao Ji, Li Xiao, Xingyu Wang, Marti Z. Hua, Yifeng Wu, Yifan Wang, Zhiqiang Wu, Xiahong He, Dunming Xu, Wenjie Zheng, and Xiaonan Lu, Simultaneous Determination of 147 Pesticide Residues in Traditional Chinese Medicines by GC–MS/MS, ACS Omega 2023 8 (31), 28663-28673. [CrossRef]
- WU Zhao-wei,CHEN An-dong,WANG Tie-song,TONG Yan-hua,CONG Luo-luo* ,CHE Bao-quan. Simultaneous Determination of 50 Residual Solvents in Solid Drug Preparations by GC-MS[J]. Chinese Pharmaceutical Journal, 2014, 49(9): 764-768. [CrossRef]
- Yoshida T, Matsunaga I, Oda H. Simultaneous determination of semivolatile organic compounds in indoor air by gas chromatography-mass spectrometry after solid-phase extraction. J Chromatogr A. 2004 Jan 16;1023(2):255-69. [CrossRef] [PubMed]
- Xu, M.-L., Gao, Y., Wang, X., Han, X. X., & Zhao, B. (2021). Comprehensive Strategy for Sample Preparation for the Analysis of Food Contaminants and Residues by GC–MS/MS: A Review of Recent Research Trends. Foods, 10(10), 2473. [CrossRef]
- Veloo, K. V., & Ibrahim, N. A. S. (2021). Analytical Extraction Methods and Sorbents’ Development for Simultaneous Determination of Organophosphorus Pesticides’ Residues in Food and Water Samples: A Review. Molecules, 26(18), 5495. [CrossRef]
- Duong, T. T., Nguyen, T. T. L., Dinh, T. H. V., Hoang, T. Q., Vu, T. N., Doan, T. O., Dang, T. M. A., Le, T. P. Q., Tran, D. T., Le, V. N., Nguyen, Q. T., Le, P. T., Nguyen, T. K., Pham, T. D., & Bui, H. M. (2021). Auxin production of the filamentous cyanobacterial Planktothricoides strain isolated from a polluted river in Vietnam. Chemosphere, 284, 131242. [CrossRef]
- Alahmad, W., Kaya, S. I., Cetinkaya, A., Varanusupakul, P., & Ozkan, S. A. (2023). Green chemistry methods for food analysis: Overview of sample preparation and determination. Advances in Sample Preparation, 5, 100053. [CrossRef]
- Jayaraj, R., Megha, P., & Sreedev, P. (2016). Review Article. Organochlorine pesticides, their toxic effects on living organisms and their fate in the environment. Interdisciplinary Toxicology, 9(3–4), 90–100. [CrossRef]
- Shekhar, C., Khosya, R., Thakur, K., Mahajan, D., Kumar, R., Kumar, S., & Sharma, A. K. (2024). A systematic review of pesticide exposure, associated risks, and long-term human health impacts. Toxicology Reports, 13, 101840. [CrossRef]
- Mdeni, N. L., Adeniji, A. O., Okoh, A. I., & Okoh, O. O. (2022). Analytical Evaluation of Carbamate and Organophosphate Pesticides in Human and Environmental Matrices: A Review. Molecules, 27(3), 618. [CrossRef]
- Araújo, M. F., Castanheira, E. M. S., & Sousa, S. F. (2023). The Buzz on Insecticides: A Review of Uses, Molecular Structures, Targets, Adverse Effects, and Alternatives. Molecules, 28(8), 3641. [CrossRef]
- Hodoșan, C., Gîrd, C. E., Ghica, M. V., Dinu-Pîrvu, C.-E., Nistor, L., Bărbuică, I. S., Marin, Ștefan-C., Mihalache, A., & Popa, L. (2023). Pyrethrins and Pyrethroids: A Comprehensive Review of Natural Occurring Compounds and Their Synthetic Derivatives. Plants, 12(23), 4022. [CrossRef]
- Nguyen Thi, K.-O., Do, H.-G., Duong, N.-T., Nguyen, T. D., & Nguyen, Q.-T. (2021). Geographical Discrimination of Curcuma longa L. in Vietnam Based on LC-HRMS Metabolomics. Natural Product Communications, 16(10). [CrossRef]
- Ngoc, P. H., An, T. C., Hiep, N. T., Nhu, T. P. H., Hung, L. N., Trung, N. Q., Minh, B. Q., & Van Trung, P. (2023). UHPLC-Q-TOF-MS/MS-guided dereplication to study chemical constituents of Hedera nepalensis leaves in northern Vietnam. Journal of Analytical Science and Technology, 14(1). [CrossRef]
- Le, V. N., Nguyen, Q. T., Nguyen, N. T., Le, T. G., Janda, T., Szalai, G., & RUI, Y.-K. (2021). Simultaneous determination of plant endogenous hormones in green mustard by liquid chromatography – Tandem mass spectrometry. Chinese Journal of Analytical Chemistry, 49(12), 111–117. [CrossRef]
- Hoang, A. Q., Trinh, H. T., Nguyen, H. M. N., Nguyen, T. Q., Nguyen, T. X., Duc, T. V., Nguyen, T. T., Do, T. Q., Minh, T. B., & Tran, T. M. (2022). Assessment of cyclic volatile methyl siloxanes (CVMSs) in indoor dust from different micro-environments in northern and central Vietnam. Environmental Geochemistry and Health, 45(5), 1711–1722. [CrossRef]
- Minh, T. N., Minh, B. Q., Duc, T. H. M., Thinh, P. V., Anh, L. V., Dat, N. T., Nhan, L. V., & Trung, N. Q. (2022). Potential Use of Moringa oleifera Twigs Extracts as an Anti-Hyperuricemic and Anti-Microbial Source. Processes, 10(3), 563. [CrossRef]
- Hai, C. T., Luyen, N. T., Giang, D. H., Minh, B. Q., Trung, N. Q., Chinh, P. T., Hau, D. V., & Dat, N. T. (2023). <i>Atractylodes macrocephala</i> Rhizomes Contain Anti-inflammatory Sesquiterpenes. Chemical and Pharmaceutical Bulletin, 71(6), 451–453. [CrossRef]
- Metcalfe, C. D., Bayen, S., Desrosiers, M., Muñoz, G., Sauvé, S., & Yargeau, V. (2022). Methods for the analysis of endocrine disrupting chemicals in selected environmental matrixes. Environmental Research, 206, 112616. [CrossRef]
- Manh Tri Tran, Thuy Le Minh, Thi Ngoc Anh Nguyen, Trinh Le Thi, Huong Le Quang, Thi Phuong Thao Pham, Quang Trung Nguyen, Determination and distribution of phthalate diesters in plastic bottled beverages collected in Hanoi, Vietnam, VNU Journal of Science: Natural Sciences and Technology, 2018, 34, 4.
- Thang, P.Q., Taniguchi, T., Nabeshima, Y. et al. Distribution of polycyclic aromatic hydrocarbons concentrations simultaneously obtained in gas, rainwater and particles. Air Qual Atmos Health 7, 273–281 (2014). [CrossRef]
- Thang, P. Q., Taniguchi, T., Nabeshima, Y., Bandow, H., Trung, N. Q., & Takenaka, N. (2014). Distribution of polycyclic aromatic hydrocarbons concentrations simultaneously obtained in gas, rainwater and particles. Air Quality, Atmosphere & Health, 7(3), 273–281. [CrossRef]
- Sikiti, P., Msagati, T. A. M., Mishra, A. K., & Mamba, B. B. (2012). Simultaneous determination of tetrachloro dibenzo-p-dioxin and poly-aromatic chlorinated biphenyls in aqueous environment using liquid phase microextraction. Physics and Chemistry of the Earth, Parts A/B/C, 50–52, 98–103. [CrossRef]
- Nguyen Xuan, H., Phan Dinh, Q., Nguyen Thi, X., Mai Thi Hong, H., Nguyen Phuc, A., Le Van, N., Bui Quang, M., Nguyen Quang, T., Chu Dinh, B., Nguyen Tien, D., Anh Le Hoang, T., & Vu Duc, N. (2024). Occurrence and Contamination Levels of Polychlorinated Dibenzo- P -Dioxins (PCDDs) and Polychlorinated Dibenzofurans (PCDFs) in Soil and Sediment in the Vicinity of Recycled Metal Casting Villages: A Case Study in Vietnam. Soil and Sediment Contamination: An International Journal, 1–26. [CrossRef]
- Nguyen, H. X., Nguyen, X. T., Mai, H. T. H., Nguyen, H. T., Vu, N. D., Pham, T. T. P., Nguyen, T. Q., Nguyen, D. T., Duong, N. T., Hoang, A. L. T., Nguyen, T. N., Le, N. V., Dao, H. V., Ngoc, M. T., & Bui, M. Q. (2024). A Comprehensive Evaluation of Dioxins and Furans Occurrence in River Sediments from a Secondary Steel Recycling Craft Village in Northern Vietnam. Molecules, 29(8), 1788. [CrossRef]
- Tran, L. T., Kieu, T. C., Bui, H. M., Nguyen, N. T., Nguyen, T. T. T., Nguyen, D. T., Nguyen, T. Q., Nguyen, H. T. A., Le, T. H., Takahashi, S., Tu, M. B., & Hoang, A. Q. (2021). Polybrominated diphenyl ethers in indoor dusts from industrial factories, offices, and houses in northern Vietnam: Contamination characteristics and human exposure. Environmental Geochemistry and Health, 44(8), 2375–2388. [CrossRef]
- Duong, H. T., Kadokami, K., Shirasaka, H., Hidaka, R., Chau, H. T. C., Kong, L., Nguyen, T. Q., & Nguyen, T. T. (2015). Occurrence of perfluoroalkyl acids in environmental waters in Vietnam. Chemosphere, 122, 115–124. [CrossRef]
- Silvestro, L., Tarcomnicu, I., & Rizea, S. (2012). HPLC-MS/MS of Highly Polar Compounds. In Tandem Mass Spectrometry - Applications and Principles. InTech. [CrossRef]
- Nguyen, T. P. L., Nguyen, V. T. A., Do, T. T. T., Nguyen Quang, T., Pham, Q. L., & Le, T. T. (2020). Fatty Acid Composition, Phospholipid Molecules, and Bioactivities of Lipids of the Mud Crab Scylla paramamosain. Journal of Chemistry, 2020, 1–9. [CrossRef]
- Le, L. H. T., Tran-Lam, T.-T., Nguyen, H. Q., Quan, T. C., Nguyen, T. Q., Nguyen, D. T., & Dao, Y. H. (2021). A study on multi-mycotoxin contamination of commercial cashew nuts in Vietnam. Journal of Food Composition and Analysis, 102, 104066. [CrossRef]
- Nguyen-Quang, T., Bui-Quang, M., & Truong-Ngoc, M. (2021). Rapid Identification of Geographical Origin of Commercial Soybean Marketed in Vietnam by ICP-MS. Journal of Analytical Methods in Chemistry, 2021, 1–9. [CrossRef]
- Nguyen, T. Q., Tran-Lam, T.-T., Nguyen, H. Q., Dao, Y. H., & Le, G. T. (2021). Assessment of organic and inorganic arsenic species in Sengcu rice from terraced paddies and commercial rice from lowland paddies in Vietnam. Journal of Cereal Science, 102, 103346. [CrossRef]
- Anh, B. T. K., Minh, N. N., Ha, N. T. H., Kim, D. D., Kien, N. T., Trung, N. Q., Cuong, T. T., & Danh, L. T. (2018). Field Survey and Comparative Study of Pteris Vittata and Pityrogramma Calomelanos Grown on Arsenic Contaminated Lands with Different Soil pH. Bulletin of Environmental Contamination and Toxicology, 100(5), 720–726. [CrossRef]
- Bui, T. K. A., Dang, D. K., Nguyen, T. K., Nguyen, N. M., Nguyen, Q. T., & Nguyen, H. C. (2014). Phytoremediation of heavy metal polluted soil and water in Vietnam. Journal of Vietnamese Environment, 6(1), 47–51. [CrossRef]
- Markus Amann, Zbigniew Klimont, T An Ha, Peter Rafaj, Gregor Kiesewetter, Adriana Gomez Sanabria, Binh Nguyen, TN Thi Thu, Kimminh Thuy, Wolfgang Schöpp, Jens Borken-Kleefeld, L Höglund-Isaksson, Fabian Wagner, Robert Sander, Chris Heyes, Janusz Cofala, Nguyen Quang Trung, Nguyen Tien Dat, Nguyen Ngoc Tung, Future Air Quality in Ha Noi and Northern Vietnam. http://pure.iiasa.ac.at/15803 (2019).
- Truong, A. H., Kim, M. T., Nguyen, T. T., Nguyen, N. T., & Nguyen, Q. T. (2018). Methane, Nitrous Oxide and Ammonia Emissions from Livestock Farming in the Red River Delta, Vietnam: An Inventory and Projection for 2000–2030. Sustainability, 10(10), 3826. [CrossRef]
- Thang, P. Q., Taniguchi, T., Nabeshima, Y., Bandow, H., Trung, N. Q., & Takenaka, N. (2014). Distribution of polycyclic aromatic hydrocarbons concentrations simultaneously obtained in gas, rainwater and particles. Air Quality, Atmosphere & Health, 7(3), 273–281. [CrossRef]
- Duong, T. T., Nguyen, T. T. L., Dinh, T. H. V., Hoang, T. Q., Vu, T. N., Doan, T. O., Dang, T. M. A., Le, T. P. Q., Tran, D. T., Le, V. N., Nguyen, Q. T., Le, P. T., Nguyen, T. K., Pham, T. D., & Bui, H. M. (2021). Auxin production of the filamentous cyanobacterial Planktothricoides strain isolated from a polluted river in Vietnam. Chemosphere, 284, 131242. [CrossRef]
- Hanh, T. T. H., Anh, D. H., Huong, P. T. T., Thanh, N. V., Trung, N. Q., Cuong, T. V., Mai, N. T., Cuong, N. T., Cuong, N. X., Nam, N. H., & Minh, C. V. (2018). Crinane, augustamine, and β -carboline alkaloids from Crinum latifolium. Phytochemistry Letters, 24, 27–30. [CrossRef]
- Van Cong, P., Anh, H. L. T., Trung, N. Q., Quang Minh, B., Viet Duc, N., Van Dan, N., Trang, N. M., Phong, N. V., Vinh, L. B., Anh, L. T., & Lee, K. Y. (2022). Isolation, structural elucidation and molecular docking studies against SARS-CoV-2 main protease of new stigmastane-type steroidal glucosides isolated from the whole plants of Vernonia gratiosa. Natural Product Research, 37(14), 2342–2350. [CrossRef]
- Nguyen, Q., Nguyen, T., Le, V., Nguyen, N., Truong, N., Hoang, M., Pham, T., & Bui, Q. (2023). Towards a Standardized Approach for the Geographical Traceability of Plant Foods Using Inductively Coupled Plasma Mass Spectrometry (ICP-MS) and Principal Component Analysis (PCA). Foods, 12(9), 1848. [CrossRef]
- Darkó, E., Khalil, R., Elsayed, N., Pál, M., Hamow, K. A., Szalai, G., Tajti, J., Nguyen, Q. T., Nguyen, N. T., Le, V. N., & Janda, T. (2019). Factors playing role in heat acclimation processes in barley and oat plants. Photosynthetica, 57(4), 1035–1043. [CrossRef]
- Minh, T. N., Anh, L. V., Trung, N. Q., Minh, B. Q., & Xuan, T. D. (2022). Efficacy of Green Extracting Solvents on Antioxidant, Xanthine Oxidase, and Plant Inhibitory Potentials of Solid-Based Residues (SBRs) of Cordyceps militaris. Stresses, 3(1), 11–21. [CrossRef]
- Le, V. N., Nguyen, Q. T., Nguyen, N. T., Janda, T., Szalai, G., Nguyen, T. D., & Le, T. G. (2021). FOLIAGE APLLIED GIBBERELIC ACID INFLUENCES GROWTH, NUTRIENT CONTENTS AND QUALITY OF LETTUCES (LACTUCA SATIVA L.). The Journal of Animal and Plant Sciences, 31(6), 1720–1727. [CrossRef]
- Dang, T. T., Vo, T. A., Duong, M. T., Pham, T. M., Van Nguyen, Q., Nguyen, T. Q., Bui, M. Q., Syrbu, N. N., & Van Do, M. (2022). Heavy metals in cultured oysters (Saccostrea glomerata) and clams (Meretrix lyrata) from the northern coastal area of Vietnam. Marine Pollution Bulletin, 184, 114140. [CrossRef]
- Nguyen, T. P. L., Nguyen, V. T. A., Do, T. T. T., Nguyen Quang, T., Pham, Q. L., & Le, T. T. (2020). Fatty Acid Composition, Phospholipid Molecules, and Bioactivities of Lipids of the Mud Crab Scylla paramamosain. Journal of Chemistry, 2020, 1–9. [CrossRef]
- Le, L. H. T., Tran-Lam, T.-T., Nguyen, H. Q., Quan, T. C., Nguyen, T. Q., Nguyen, D. T., & Dao, Y. H. (2021). A study on multi-mycotoxin contamination of commercial cashew nuts in Vietnam. Journal of Food Composition and Analysis, 102, 104066. [CrossRef]
- Quang, T. H., Phong, N. V., Anh, L. N., Hanh, T. T. H., Cuong, N. X., Ngan, N. T. T., Trung, N. Q., Nam, N. H., & Minh, C. V. (2020). Secondary metabolites from a peanut-associated fungus Aspergillus niger IMBC-NMTP01 with cytotoxic, anti-inflammatory, and antimicrobial activities. Natural Product Research, 36(5), 1215–1223. [CrossRef]
- Hanh, T. T. H., Hang, L. T. T., Huong Giang, V., Trung, N. Q., Thanh, N. V., Quang, T. H., & Cuong, N. X. (2021). Chemical constituents of Blumea balsamifera. Phytochemistry Letters, 43, 35–39. [CrossRef]
- Janda, T., Lejmel, M. A., Molnár, A. B., Majláth, I., Pál, M., Nguyen, Q. T., Nguyen, N. T., Le, V. N., & Szalai, G. (2020). Interaction between elevated temperature and different types of Na-salicylate treatment in Brachypodium dystachion. PLOS ONE, 15(1), e0227608. [CrossRef]
- Nguyen-Quang, T., Bui-Quang, M., & Truong-Ngoc, M. (2021). Rapid Identification of Geographical Origin of Commercial Soybean Marketed in Vietnam by ICP-MS. Journal of Analytical Methods in Chemistry, 2021, 1–9. [CrossRef]
- Hanh, T. T. H., Hang, L. T. T., Huong, P. T. T., Trung, N. Q., Cuong, T. V., Thanh, N. V., Cuong, N. X., Nam, N. H., & Minh, C. V. (2017). Two new guaiane sesquiterpene lactones from the aerial parts of Artemisia vulgaris. Journal of Asian Natural Products Research, 20(8), 752–756. [CrossRef]
- Le-Quang, H., Phuong, T. P. T., Bui-Quang, M., Nguyen-Tien, D., Nguyen-Thanh, T., Nguyen-Ha, M., Shimadera, H., Kondo, A., Luong-Viet, M., & Nguyen-Quang, T. (2022). Comprehensive Analysis of Organic Micropollutants in Fine Particulate Matter in Hanoi Metropolitan Area, Vietnam. Atmosphere, 13(12), 2088. [CrossRef]
- Hanh, T. T. H., Hang, L. T. T., Huong Giang, V., Trung, N. Q., Thanh, N. V., Quang, T. H., & Cuong, N. X. (2021). Chemical constituents of Blumea balsamifera. Phytochemistry Letters, 43, 35–39. [CrossRef]
- Hanh, T. T. H., Anh, D. H., Quang, T. H., Trung, N. Q., Thao, D. T., Cuong, N. T., An, N. T., Cuong, N. X., Nam, N. H., Kiem, P. V., & Minh, C. V. (2019). Scutebarbatolides A-C, new neo-clerodane diterpenoids from Scutellaria barbata D. Don with cytotoxic activity. Phytochemistry Letters, 29, 65–69. [CrossRef]
- Quang Trung, N., Thi Luyen, N., Duc Nam, V., & Tien Dat, N. (2018). Chemical Composition and in Vitro Biological Activities of White Mulberry Syrup during Processing and Storage. Journal of Food and Nutrition Research, 6(10), 660–664. [CrossRef]
- Hanh, T. T. H., Cham, P. T., Anh, D. H., Cuong, N. T., Trung, N. Q., Quang, T. H., Cuong, N. X., Nam, N. H., & Minh, C. V. (2021). Dammarane-type triterpenoid saponins from the flower buds of Panax pseudoginseng with cytotoxic activity. Natural Product Research, 36(17), 4343–4351. [CrossRef]
- Nguyen-Quang, T., Do-Hoang, G., & Truong-Ngoc, M. (2021). Multielement Analysis of Pakchoi (Brassica rapa L. ssp. chinensis) by ICP-MS and Their Classification according to Different Small Geographical Origins. Journal of Analytical Methods in Chemistry, 2021, 1–11. [CrossRef]
- Trung, N. Q., Van Nhan, L., Thao, P. T. P., & Giang, L. T. (2017). Novel draw solutes of iron complexes easier recovery in forward osmosis process. Journal of Water Reuse and Desalination, 8(2), 244–250. [CrossRef]
- Van, Pc. P., Ngo Van, H., Quang, M. B., Duong Thanh, N., Nguyen Van, D., Thanh, T. D., Tran Minh, N., Thi Thu, H. N., Quang, T. N., Thao Do, T., Thanh, L. P., Do Thi Thu, H., & Le Tuan, A. H. (2023). Stigmastane-type steroid saponins from the leaves of Vernonia amygdalina and their α -glucosidase and xanthine oxidase inhibitory activities. Natural Product Research, 38(4), 601–606. [CrossRef]
- Minh, T. N., Minh, B. Q., Duc, T. H. M., Thinh, P. V., Anh, L. V., Dat, N. T., Nhan, L. V., & Trung, N. Q. (2022). Potential Use of Moringa oleifera Twigs Extracts as an Anti-Hyperuricemic and Anti-Microbial Source. Processes, 10(3), 563. [CrossRef]
- Quang, T. H., Phong, N. V., Anh, D. V., Hanh, T. T. H., Cuong, N. X., Ngan, N. T. T., Trung, N. Q., Oh, H., Nam, N. H., & Minh, C. V. (2021). Bioactive secondary metabolites from a soybean-derived fungus Aspergillus versicolor IMBC-NMTP02. Phytochemistry Letters, 45, 93–99. [CrossRef]
- Bui, M. Q., Quan, T. C., Nguyen, Q. T., Tran-Lam, T.-T., & Dao, Y. H. (2022). Geographical origin traceability of Sengcu rice using elemental markers and multivariate analysis. Food Additives & Contaminants: Part B, 15(3), 177–190. [CrossRef]
- Thang, P. Q., Muto, Y., Maeda, Y., Trung, N. Q., Itano, Y., & Takenaka, N. (2016). Increase in ozone due to the use of biodiesel fuel rather than diesel fuel. Environmental Pollution, 216, 400–407. [CrossRef]
- Hanh, T. T. H., Anh, L. N., Trung, N. Q., Quang, T. H., Anh, D. H., Cuong, N. X., Nam, N. H., & Van Minh, C. (2021). Cytotoxic phenolic glycosides from the seeds of Senna tora. Phytochemistry Letters, 45, 190–194. [CrossRef]
- Trung, N. Q., Dat, N. T., Anh, H. N., Tung, Q. N., Nguyen, V. T. H., Van, H. N. B., Van, N. M. N., & Minh, T. N. (2024). Substrate Influence on Enzymatic Activity in Cordyceps militaris for Health Applications. Chemistry, 6(4), 517–530. [CrossRef]
- Bach, M. X., Minh, T. N., Anh, D. T. N., Anh, H. N., Anh, L. V., Trung, N. Q., Minh, B. Q., & Xuan, T. D. (2022). Protection and Rehabilitation Effects of Cordyceps militaris Fruit Body Extract and Possible Roles of Cordycepin and Adenosine. Compounds, 2(4), 388–403. [CrossRef]
- Tung, N. N., Hung, T. T., Trung, N. Q., & Giang, L. T. (2019). Symmetrical Fatty Dialkyl Carbonates as Potential Green Phase Change Materials: Synthesis and Characterisation. Russian Journal of General Chemistry, 89(7), 1513–1518. [CrossRef]
- Thang, P. Q., Maeda, Y., Trung, N. Q., & Takenaka, N. (2014). Low molecular weight methyl ester in diesel/waste cooking oil biodiesel blend exhausted gas. Fuel, 117, 1170–1171. [CrossRef]
- Minh, T. N., Anh, L. V., Trung, N. Q., Minh, B. Q., & Xuan, T. D. (2022). Efficacy of Green Extracting Solvents on Antioxidant, Xanthine Oxidase, and Plant Inhibitory Potentials of Solid-Based Residues (SBRs) of Cordyceps militaris. Stresses, 3(1), 11–21. [CrossRef]
- Trung, N. Q., Quyen, P. D. T., Ngoc, N. T. T., & Minh, T. N. (2024). Diversity of Host Species and Optimized Cultivation Practices for Enhanced Bioactive Compound Production in Cordyceps militaris. Applied Sciences, 14(18), 8418. [CrossRef]
- Ngoc, T., Thinh, P., Mui, D., Uyen, L., Ngan, N., Tran, N., Khang, P., Huy, L., Minh, T., & Trung, N. (2024). Influences of Fermentation Conditions on the Chemical Composition of Red Dragon Fruit (Hylocereus polyrhizus) Wine. Beverages, 10(3), 61. [CrossRef]
- Duong Thanh, N., Tran Thi, H., Nguyen Quang, T., Nguyen Van, H., Hoang Nguyen, G., Nguyen Huu, Q., & Tran Son, T. (2023). Performance evaluation of multiple particulate matter monitoring instruments under higher temperatures and relative humidity in Southeast Asia and design of an affordable monitoring instrument (ManPMS). Instrumentation Science & Technology, 51(6), 660–680. [CrossRef]
- Cong, P. V., Anh, H. L. T., Vinh, L. B., Han, Y. K., Trung, N. Q., Minh, B. Q., Duc, N. V., Ngoc, T. M., Hien, N. T. T., Manh, H. D., Lien, L. T., & Lee, K. Y. (2023). Alpha-Glucosidase Inhibitory Activity of Saponins Isolated from Vernonia gratiosa Hance. Journal of Microbiology and Biotechnology, 33(6), 797–805. [CrossRef]
- Nguyen, T. N., Trinh, H. T., Hoang, T. M., Van Le, N., Bui, M. Q., & Nguyen, T. Q. (2022). Experimental investigation into the room thermoregulation efficiency in tropical climate of novel green phase change material eutectic mixture. Journal of Thermal Analysis and Calorimetry, 147(20), 11039–11048. [CrossRef]
- Doncheva, T., Kostova, N., Toshkovska, R., Philipov, S., Vu, N., Nguyen, D., Nguyen, T., Do, G., & Dang, H. (2022). Alkaloids from Pandanus amaryllifolius and Pandanus tectorius from Vietnam and Their Anti-inflammatory Properties. Proceedings of the Bulgarian Academy of Sciences, 75(6), 812–820. [CrossRef]
- Quang, T. H., Anh, L. N., Hanh, T. T. H., Cuong, N. X., Ngan, N. T. T., Trung, N. Q., & Nam, N. H. (2021). Cytotoxic and antimicrobial benzodiazepine and phenolic metabolites from Aspergillus ostianus<scp>IMBC-NMTP03</scp>. Vietnam Journal of Chemistry, 59(5), 660–666. [CrossRef]
- Bui, Q. M., Nguyen, Q. T., Nguyen, T. T., Nguyen, H. M., Phung, T. T., Le, V. A., Truong, N. M., Mac, T. V., Nguyen, T. D., Hoang, L. T. A., Tran, H. M. D., Le, V. N., & Nguyen, M. D. (2024). Multivariate Statistical Analysis for the Classification of Sausages Based on Physicochemical Attributes, Using Attenuated Total Reflectance-Fourier Transform Infrared (ATR-FTIR) and Inductively Coupled Plasma-Mass Spectrometry (ICP-MS). Journal of Analytical Methods in Chemistry, 2024, 1–13. [CrossRef]
- Wieczorek, M. N., Zhou, W., & Pawliszyn, J. (2024). Perspective on sample preparation fundamentals. Advances in Sample Preparation, 10, 100114. [CrossRef]
- Ingle, R. G., Zeng, S., Jiang, H., & Fang, W.-J. (2022). Current developments of bioanalytical sample preparation techniques in pharmaceuticals. Journal of Pharmaceutical Analysis, 12(4), 517–529. [CrossRef]
- Lou, Y., Xu, Q., Chen, J., Yang, S., Zhu, Z., & Chen, D. (2023). Advancements in Sample Preparation Methods for the Chromatographic and Mass Spectrometric Determination of Zearalenone and Its Metabolites in Food: An Overview. Foods, 12(19), 3558. [CrossRef]
- Almeida, C. M. M. (2021). Overview of Sample Preparation and Chromatographic Methods to Analysis Pharmaceutical Active Compounds in Waters Matrices. Separations, 8(2), 16. [CrossRef]
- Kuppusamy, S., Meivelu, M., Praburaman, L., Mujahid Alam, M., Al-Sehemi, A. G., & K, A. (2024). Integrating AI in food contaminant analysis: Enhancing quality and environmental protection. Journal of Hazardous Materials Advances, 16, 100509. [CrossRef]
Figure 1.
Increase in simultaneous analysis studies over time (Data collected from Elsevier’s platform using the keywords “simultaneous analysis” and “simultaneous determination”).
Figure 1.
Increase in simultaneous analysis studies over time (Data collected from Elsevier’s platform using the keywords “simultaneous analysis” and “simultaneous determination”).
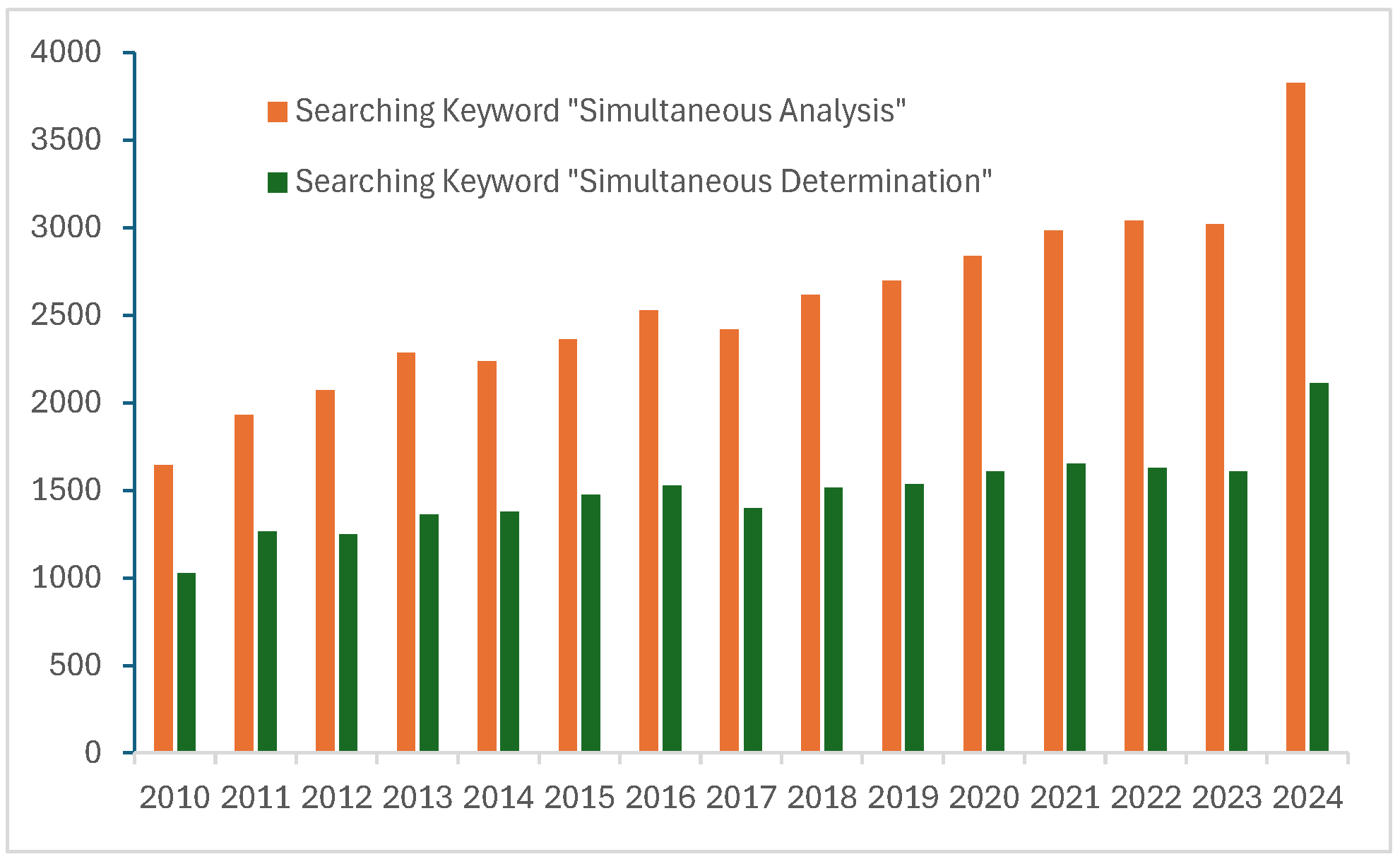
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



