
Submitted:
12 June 2025
Posted:
13 June 2025
You are already at the latest version
Abstract
Two germplasm collections, comprising 1026 peach accessions located in Italy, were analyzed with 12 simple sequence repeat (SSR) markers. SSR reactions were performed using the multiplex-ready PCR protocol and 147 alleles were amplified with an average of 12 alleles per locus. BPPCT001 was the most informative marker displaying the highest discrimination power (0.734). The observed heterozygosity showed an average of 0.45 alleles per locus, lower than expected (0.61). The fixation index (F) values were positive in all loci, with an average of 0.27 alleles per locus, suggesting the presence of endogamy. The DNA fingerprinting data allowed the discrimination of 80.95% of the analyzed accessions. If we exclude known sport mutations, known synonymies and cultivars with the same pedigree, 161 accessions are mislabeled , with an error rate of 16.56% within or between collections. Population structure analysis showed three sub-populations: modern peach cultivars, modern nectarine cultivars and a third group mainly composed of traditional peach cultivars. The results obtained in this work will be useful to efficiently manage GenBanks, reducing unwanted redundancy, synonyms and homonyms, mislabeling and spelling errors as well as identifying parents in controlled crosses.
Keywords:
genetic resources
; cultivar identification
; genetic relationship
; Prunus persica (L.) Batsch
; Simple Sequence Repeats (SSR)
1. Introduction
The peach, Prunus persica (L.) Batsch, is a species belonging to the family of the Rosaceae, Spiraeoideae subfamily [1]. The Prunus genus encompasses more than 400 species of trees and shrubs and is divided into six subgenera: (1) Amygdalus (almonds and peaches); (2) Prunus (plums and apricots); (3) Cerasus (cherries); (4) Lithocerasus (one of the representatives is Prunus pumila—sand cherry); (5) Padus (Prunus padus—bird cherry); and (6) Laurocerasus (Prunus laurocerasus—cherry laurel). Prunus species are sources of nuts, oil, wood, ornamental plants and fruits such as apricot (Prunus armeniaca L.), cherry (Prunus avium L.), plum (Prunus domestica L.) and peach (Prunus persica L.), which cater to different tastes and demands on the market [2,3,4,5].
Peach is cultivated world-wide for the high commercial value of its fruits, commonly consumed raw or canned, or processed as jam, jelly and juice, and it is appreciated by consumers for its organoleptic and nutritional properties, providing vitamins, minerals, fiber and antioxidant compounds for healthy diets. According to data from FAOSTAT (http://faostat.fao.org), global peach production in 2023 exceeded 27.08 million tons; 76.33% was produced in Asia, 12.82% in Europe, 6.81% in America, 3.79% in Africa, and 0.23% in Oceania. Italy is the fourth largest producer with an annual production of 1.03 million tons, after People’s Republic of China with 17.50, Spain 1.38 and Turkey 1.07 million tons.
Peach is a diploid self-compatible species and is one of the best genetically characterized deciduous trees; for its characteristics, such as small genome (227 Mb) and absence of recent Genome Wide Duplications [6] it was used as the model species of the Rosaceae.
Peach has a long history of domestication, started in northwest China 4000-5000 years ago [7]. Verde et al. [6] published the high-quality draft genome of peach in 2013 and analyzed Prunus diversity by resequencing 12 peach cultivars of Eastern or Occidental origin and three close relatives, highlighting two genetic bottlenecks compatible with the domestication events and the dissemination path of the species.
In the last 40 years, the problem of diversity reduction in cultivated plants has become evident in many countries and germplasm conservation programs have been established to preserve crop diversity. The entry into force of the International Treaty for Plant Genetic Resources for Food and Agriculture (ITPGRFA) raised awareness and gave an important input on this issue worldwide as well as in Italy [8]. At first, local and commercial cultivars and crop wild relatives were collected resulting, often, in large, unbalanced collections of unknown value. At the beginning of this millennium the main goal shifted from collecting to conserving, managing, characterizing and exploiting genetic resources [9]. Fruit tree germplasm collections, mainly conserved ex-situ and in-vivo, require high financial inputs such as manpower, natural resources, energy and chemicals; USDA has estimated that maintaining each tree costs about 75-100 US dollars per year (https://www.ars.usda.gov/northeast-area/geneva-ny/plant-genetic-resources-unit-pgru/docs/about-pgru/clonal-propagated-crops/). Cryopreservation would be much less costly (approximately 1 US dollar per plant annually) but its routine implementation is hampered by lack of technology transfer and protocol validation across laboratories [10].
Germplasm collections are prone to errors such as mislabeling and landraces renaming during their local diffusion, resulting in synonymies (same genotype with different names) and homonymies (different genotypes with the same name). The level of duplication and errors within and among collections is known to be high, and the elimination of redundancy and errors is economically substantial [9,11]. Incorporating genetic characterization into Genbank management can solve these common issues and lead to more informed decisions and sustainable solutions.
Emanuelli et al. [12], working with 22 SSRs and 384 SNPs in a collection of Vitis vinifera, including interspecific hybrids and Vitis related species, found that approximately half of the collection was composed of putative redundant genotypes. A proportion of the redundancy found in this collection was expected since clonal selections are not generally distinguished by a few molecular markers, but in other cases, mislabeling due to curation errors is the main reason.
Similar results were found in apple using SSRs to investigate germplasm collections and part of the redundancy was due to the difficulty of individuating clonal variants using a low number of markers. Van Treunen et al [13] fingerprinting 695 samples from eight Dutch apple collections found that the redundancy within the total sample was 32%, with the majority between collections, each collection containing only about 50% of unique accessions. The authors proposed that a network of co-operating collection holders would be less costly for a more efficient management. Both Liang et al [14], working in a single Italian collection, and Lassois et al [15], in a French one, detected about 34 % of redundancy. Lower values of duplication were found by Larsen et al. [16] in 448 apple accession, mainly of Danish origins (23%) and by Pina et al. [17] studying a set of 183 accessions coming from mountainous areas in Northeastern Spain (29%). Recently, in apple, Muranty et al [15,18] and Denance et al [19] analyzed a large set of accessions with a set of SSRs and the 20K SNP array and developed the “Malus UNiQue genotype code,” to generate unique codes for each apple accession. The same approach was recently applied in the Danish “Pometum” collection [16]. To our knowledge, no such system has been so far reported for peach.
Molecular characterization has been the way of choice to explore the extent of variability in germplasm. In fruit trees, the use of molecular markers for identification purposes and repository management began at the end of the nineties. Nowadays the markers of choice are Simple Sequence Repeats (SSRs) and Single Nucleotide Polymorphisms (SNPs). SNPs are the most abundant in the genomes and the best choice for high resolution genome-wide assessment in genetic studies. Nevertheless, SSRs still are the marker of choice for collection management due to their high polymorphism with multi-allelic patterns and their use through in-house facilities.
While the first studies were limited to a low number of cultivars, in more recent years faster molecular protocols and/or automation allowed the screening of a higher number of samples and markers.
In peach, several studies have been conducted using both SSR and SNP markers. Aranzana et al [20,21] analyzed a collection of 212 Western accessions with a total of 48 SSRs. These researchers aimed at studying genetic diversity in peach germplasm, solving some cases of homonymies and synonymies. Li et al [22] with the same set of 48 primers analyzed a collection of 434 oriental accessions. They were able to merge their dataset with that of Aranzana et al [21] summing up a total of 653 accessions allowing the identification of 3 main sub-populations Oriental, Occidental, and Landraces, highlighting the higher diversity (higher heterozygosis) of the oriental germplasm. The modern Occidental breeding subgroup was further divided into two subpopulations: nectarines and peaches. Other studies, with limited number of accessions and markers, were conducted using SSRs [23,24,25,26].
Micheletti et al [27] analyzed 1.540 peach accessions from several repositories around the world, including Western and oriental germplasm, with the 9k SNP Peach array [28]. Using about four thousand informative SNPs, a detailed understanding of the genetic diversity and population structure in peach germplasm was proposed. In particular, the study indicates the presence of 3 distinct genetic groups (namely Occidental breeding, Occidental traditional and Oriental) with the structure influenced by both the geographic origin and pedigrees. They confirmed the stratification obtained by Li et al. [22] and the further division of the Occidental breeding group (K=4) into two distinct subpopulations: peaches and nectarines. More recently, genome wide association studies (GWAS) and structure analyses were performed using Genotyping by Sequencing (GBS), [29,30,31,32].
In this work, 1026 peach accessions were evaluated using a set of 12 SSR markers. The samples investigated came from two peach collections mostly composed of local and commercial peaches. One is held in Rome and belongs to the National Fruit Germplasm Collection (NFGC) of the Consiglio per la Ricerca in Agricoltura e l’analisi dell’Economia agraria - Centro Olivicoltura, Frutticoltura e Agrumicoltura (CREA-OFA), the other is located in Imola and results from the public-private program MAS.PES (Breeding apricot and peach through Marker-Assisted Selection) [33]. The aim was the assessment of the genetic diversity in the two Italian peach collections and to support their management.
2. Results & Discussion
2.1. Genetic Diversity Analysis and Collections Characterization
All the 1026 accessions were subjected to the diversity analysis. Among them, 62 names were represented twice each and 6 three times each, within or between the two collections: the accessions bearing a distinct name were 952 (Table S1). After genotyping, all the triplicated accessions and 53 couples out of 62 were found identical, thus confirming as replicates of the same genotype. Only one for each group of replicates was retained for further analysis for a total of 961 samples, the genetic profiles for all the accessions are reported in Table S2. Compared with previous studies that analyzed large peach datasets for genetic diversity [21,22,23,25,26,27], a total of 377 peach accessions were characterized for the first time in this study, while 495 accessions, belonging to the same germplasm collections that had been previously genotyped using SNPs markers [27]. The dendrogram (Figure S1) detected 93 groups containing from 2 to 10 accessions for a total of 223 undistinguished genotypes.
European and North American germplasm represented 89.9% of the total with known geographical origin (426 and 325 accessions, respectively). Within this germplasm there were several “traditional varieties”, namely landraces and local varieties not coming from breeding activities.
A total of 147 alleles were amplified with 12 SSR loci, the complete list of alleles and their frequency is reported in Table 1 with an average of alleles (Table 2). The average number of alleles is 12 per locus, higher than those observed in other works: almost double than in Aranzana et al. [21] and Bouhadida et al. [24] and triple than in Paula et al. [34], but similar to Li et al. [22], Chavez et al. [25] and Shen et al. [26]. The allele number ranged from a minimum of 8 (UDP-022) to a maximum of 19 (for BPPCT017). A high proportion (72%) of the total were rare alleles (frequency <0.05%) and the allele distributions deviated from Hardy-Weinberg equilibrium (Table 3). The observed heterozygosity varied between 0.32 and 0.61 (UDP-409 and EPPCU5176, respectively) with an average of 0.45 per locus. This value was lower than the expected heterozygosity in all loci with the exception of EPPCU5176, which is the only marker used in this paper deriving from Expressed Sequence Tags (ESTs). EPPCU5176 is located on chromosome 7 of the peach genome v2.0, partially overlapping the Prupe.7G192600 gene coding for an auxin-responsive protein (SAUR50).
A deviation from Hardy-Weinberg equilibrium with an excess of homozygotes was reported by many authors [20,34,35,36]. This excess is consistent with the presence of inbreeding, which naturally occurs in an autogamous species such as peach.
According to Botstein et al. [37], informativeness is limited for loci showing PIC values lower than 0.50. In this study, PIC values were equal or above 0.50 for all loci, except UDP-409. Similarly to Bouhadida et al. [24], the most informative marker was BPPCT001 with a PIC value of 0.73, while the least informative was UDP-409 (PIC value of 0.40). The discrimination power (PD) showed an average of 0.79. In line with previous studies [24,38], marker BPPCT001 showed the highest discrimination power (PD=0.92). The inbreeding coefficients were positive for all loci showing an average of 0.27, suggesting the presence of endogamy. A total of 107 rare alleles (MAF < 0.05; Table 3) and 18 private alleles were detected (Table 3). The peach x plum rootstock Citation and the peach x P. webbii selection IF817023 carry three private alleles each, while twelve accessions, including a peach x almond hybrid (AB/6) and four oriental genotypes (‘Chui Huang Tao’, Ferganensis, ‘Shan Dong’, ‘Xiaguang’), carry one each.
Nine couples of accessions sharing the name were shown to be different and were renamed adding a letter code (Azurite, Harvester, Limonini, Lizbeth, Norman, Poppa di Venere, Romagna Red, Ross and Xia Guang - Table S1), thus reducing the total number of samples with unique labels to 961. Thirty-nine couples were replicated between collections, the remaining 24 were duplications inside a single collection.
The dendrogram representing the similarity among accessions is presented in Supplementary Figure S1. An accession belonging to P. davidiana, an outcrossing wild species close to peach, but whose variability was not modified by human selection, was inserted in the analyses as outgroup.
It stands out that most nectarines (71%) are grouped in a single cluster (node 9). However, there are a few peaches (26) scattered among them. The remaining nectarines are dispersed among the peaches, individually or in small groups. Consistent with multiple independent origins of the yellow flesh trait [39,40] no clear pattern was detected for flesh color except for the group of peaches at the top of the dendrogram (node 10), mainly white fleshed cultivars. A subgroup containing about half of them comes from Asia (node 10.b) and is a result of breeding programs, often involving American founders, as Beijing 3 (Elberta) and Hakuto and its descendants. Hakuto is a Japanese cultivar dating about 1900 and possibly a Chinese Cling mutant, but Li [22] classifies it as coming from breeding with structure analysis placing it in the ‘Hakuho population’. Most of the other samples are European landraces, which as such could well have an Asian or old American origin, or groups of modern cultivars with landraces in the pedigree, as the Cesarini cluster. All flat peaches not coming from modern breeding (9 out of 17) are clustered in subcluster 10.a including the traditional Sicilian flat peaches and a Chinese one (‘Shao Hong Pantao’). Similarly, Micheletti et al. [27] analyzing the same Chinese flat cultivar together with traditional Sicilian (‘Tabacchiera’) and Spanish flat peaches (‘Paraguayo’) grouped them all in the same cluster together with other Chinese flat peaches and in the Oriental sub population, suggesting a common origin of these varietal groups and related to the oriental one. Both Spanish and Sicilian groups are cultivated since the XIX century, likely derived from the same source from China. Six flat cultivars, coming from modern breeding, are also grouped together in cluster 8. Aranzana and coworkers [20,21] found a clear pattern based on flesh melting/non melting character: both peaches and nectarines were grouped according to fruit flesh texture with only six exceptions. In our dendrogram a pattern emerges only for the melting flesh type, while the non-melting types are scattered individually or in small groups, often according to their pedigree. The cluster 11 contains mainly melting peaches (279), with the exception of 13 non-melting nectarines and 5 non-melting peaches.
The small group closer to P. davidiana (cluster 1), contains three accessions all coming from China: ‘Pesco Fiore Rosa’ (in English ‘Pink Flower Peach’), and in a sub-cluster ‘Yu Bai’ and ‘Chui Huang Tao’). ‘Pesco Fiore Rosa’ was collected in China (unknown origins) and is an ornamental peach, showing double light pink flowers. ‘Yu Bai’ was obtained in China by cross and one of its parents is a landrace according to Cao [41]. The last one is ‘Chui Huang Tao’ (unknown origins).
At a short distance follows a single branch with a selection obtained at CREA-OFA Rome (IF817023), a putative open pollination hybrid of P. webbii, a Mediterranean wild almond maintained at CNGF, with an accession of P. persica. The selection has small leaves and short internodes. The next cluster 3 contains four apparently heterogeneous accessions: the Chinese peach Ferganensis, formerly classified as a distinct species [6](Verde et al 2013), shares a sub-cluster with an old local Italian peach belonging to a varietal population (Poppa di Venere); the other sub-cluster contains ‘Nettarina pendula’ and ‘Souvenir Nikitsky’, two nectarines of uncertain origin; the first is an ornamental tree with weeping habit, the latter is a tree with no evident ornamental features coming from the Nikita botanical garden in Crimea. This institution actively collected ornamental and edible Prunus persica specimens coming from China, and from other gardens of the former USSR [3,42]. There are only five selections known to be cultivated as ornamental in this analysis, the one in cluster 1 (Pesco Fiore Rosa), ‘Nettarina pendula’ in cluster 3, ‘Flordahome, which is also a relative of P. davidiana, ‘Double Crimson’ and ‘Zansetsushidare’ at a short distance (branch 5 and cluster 6). In cluster 6 are grouped many accessions carrying ornamental characters, such as S2678, ‘NJ Weeping’, ‘Zansetsushidare’ (weeping habit, white double flower), ‘Double Crimson’ (crimson double flower). According to Cao and coworkers [41] the ornamental peaches originated in China from an ancient edible group. This happened about 2000 years after the domestication event which separated peach from its wild progenitor, well before the second bottleneck related to the introduction of peach from Europe to the United States. This suggests that clusters 3-4-5-6 contain the more diverse accessions of oriental origin among those sampled in this study. This is also supported by some accessions carrying resistance to diseases: for example, in cluster 3 Ferganensis carries resistance to Sphaerotheca pannosa, S2678 to green aphids, ‘Rancho Resistant’ and ‘Higama’ in cluster 6 to nematodes. Clusters 4 and 6 also contain three out of the four peach x almond hybrids.
Next cluster (cluster 7 - 27 accessions) is also composed by a great majority of peaches, which have in common the origin from warm climate regions (Brasil, South Italy, Florida, Mexico, Texas) and low chilling requirements.
Using the 12 SSR markers 227 accessions cannot be separated (Table S3). The probability of identity, which is the likelihood that two unrelated individuals by chance share the same multilocus genotype, ranges from 2.98x10-20 to 3.79x10-5. Twenty-nine accessions refer to known mutations, 17 refer to known synonymies, 22 have a shared parent while the remaining 161 refer to accessions that should be different, thus have to be considered as mislabeling errors. The estimated error rate present in the two collections is equal to 16.56% considering both the accessions (nine) with the same name and different genetic profile and the genotypes with different labels and the same genetic fingerprint.
Using the 12 SSR markers from the present work, it was possible to distinguish closely related accessions such as (Stark) ‘Redgold’ and ‘Maria Aurelia’ (derived from selfing of ‘Stark Redgold’), the couples ‘Orion’ and ‘Venus’ (‘Stark Redgold’ x ‘Flamekist’), ‘Caldesi 2000’ and ‘Caldesi 2010’ (‘Stark Redgold’ x ‘Snow Queen’) the three flat peaches ‘Ufo 1’, ‘Ufo 2’ and ‘Ufo 4’ (‘Maybelle’ x ‘Stark Saturn’) sharing the same parents. ‘Sweet Lady’ (derived from open pollination of (Stark) ‘Redgold’, ‘Borgia’ (open pollination of ‘Maycrest’) and ‘Capucci 18’ (open pollination of ‘S. Anna Balducci’) showed different fingerprints from their parents and ‘Villa_Ada’, ‘Villa_Doria’, ‘Villa_Giulia’, ‘Villa Adriana’ and ‘Romea’ (all derived from ‘Catherina’ open pollinated) with genetic profile different from their parent and among them (Figure S1). Different genetic patterns were observed between the accessions ‘Loadel’ and ‘Loadel mutata’. In contrast to two previous studies [21,27], ‘Spring Lady’ and ‘Queen Crest’ showed different genotypes, highlighting a putative mislabelling error in the NFCG collection. However, considering both collections, some accessions could not be distinguished (Figure S1), probably because they are highly genetically related. ‘Bella di Cesena’ and ‘Bella di Cesena Precoce’ showed the same profile for all the markers analyzed, as well as ‘Maycrest’ and ‘Spring Lady’, both known to be sport mutations of ‘Springcrest’ [27]. This was expected for sports that are identical at all loci except for a few mutations [27,43,44].
The cultivar ‘Queen Crest’ showed a genetic profile identical to ‘Springbelle’. Although pedigree information is not available for ‘Springbelle’, this result is confirmed in two different genetic diversity studies using SSR markers and SNPs markers [20,27].
‘Betty’ and ‘Lamone’ were not discriminated, although they stem from different pedigrees: Redwing’ x ‘W6-120’ and ‘Babygold 6’ x ‘Shasta’, respectively. A similar situation was observed with the accessions ‘Appia’ and ‘Brighton’: ‘Appia’ is an open pollination of ‘Southland’ x ‘Pesco Noce 1’, while ‘Brighton’ was selected from a progeny of ‘Sunhigh’ x ‘Redhaven’. Similarly, ‘Ambergold’ and ‘Morsiani 51’ could not be distinguished. Inconsistencies between pedigree information and genetic profiles were also observed in other studies [20,27], and may be explained by labelling errors.
According to pedigree information, the accessions ‘Vivian’ and ‘Fortuna’ have ‘Leader’ as a common parent [45] which could justify the same genetic profile and indicate that a higher number of (or different) markers may be necessary to distinguish accessions sharing common parentage. Other accessions like ‘Armking Precoce’ and ‘Escaline’ could not be distinguished, probably because both are derived from an ‘Armking’ mutation.
In the collections from MAS.PES and NCFG-CREA, 38 out of the 72 accessions in duplicate showed the same profile, confirming their identity. One of the duplications of the accessions ‘Babygold 9’, ‘Flaminia’, ‘Palazzina’, ‘Alitop’, ‘Big Top’, ‘Pieri 81’, ‘Royal Time’, ‘Angelo Marzocchela’, ‘Bella di Cesena’, ‘Botto’, ‘Glohaven’, ‘Grenat’, ‘Magique’, ‘Maria Bianca’, ‘Merril Gem Free’, ‘Nectagrand 1’, ‘Oro A’, ‘Pillar’, ‘Redhaven’; ‘Romagna Bright’, ‘Romagna Gold’, ‘Romagna Red’, ‘Romagna Star’, ‘Romagna Top’, ‘Rosa Dardi, ‘Splendor’, ‘Summer Rich’, ‘Turquoise’ and ‘Zee Glo’ showed missing data to at least one marker. However, the same fingerprint profile was observed in the markers with no missing data. In both collections, the confirmation of their identity can be very useful for tracing plant material and for clarifying and/or confirming a possible parentage of the accessions.
2.2. Parentage Analysis
Overall, 53.0% of the comparisons confirm the full matching of declared parentage varying from a minimum of 34% (two known parents) to a maximum of 71.2% (one known parent) (Table 4). This could be ascribed to the controlled crossing method that consists only of the emasculation of the flowers, which doesn’t prevent the access of the bees and unwanted pollinations. The parentage analysis of 42 accessions declared open pollinated, revealed that 57% could be a result of self-pollination. On the other hand, out of 14 declared self-pollinations checked, only five (35.7 %) matched. Those numbers are susceptible to slight change if taking possible null alleles into consideration.
As regards accessions declared as mutants, we observed 55.2% of matching accessions (not including the accessions having a name suggesting a clonal origin, but not described in literature to our best knowledge).
In particular the SSR profile of ‘Gage Elberta’ is identical to that of ‘Elberta’, thus confirming Okie’s hypothesis of ‘Gage Elberta’ being a mutant of ‘Elberta’ [45].
Aranzana et al [20] analyzed 38 declared pedigrees and found that 100% of SSR data from open-pollinated seedlings matched their known parent, but this decreased to 47.8 with genotypes with both known parents. This is almost half of the matches, similarly to what we observed in our study on a larger sample.
2.3. Population Structure
According to the Evanno method, the population structure analysis revealed three subpopulations (K = 3) (Figure 1). Considering the membership coefficient Q ≥ 0.80, 477 samples were clustered into three main subpopulations (Table S4). The subpopulation Q2 was mainly composed of nectarines (143 out of 153 accessions, Supplementary Table S4). The other two subpopulations, Q1 and Q3, (134 and 190 accessions, Table S4) were largely composed of fuzzy peaches (301 out of 324 accessions). The remaining accessions (350) could not be assigned under the 80% membership coefficient criterion to any of the three subgroups and remained admixed. The two main subpopulations (peaches and nectarines) match the two major varietal groups in P. persica, namely pubescent (fuzzy) peaches (PE) and glabrous peaches (nectarines: NE). The main reason for this separation is related to the breeding history until a few decades ago: the breeding programs of nectarines and peaches were mainly kept distinct, and this may have created different allele frequencies between the two groups. Indeed, the breeding of modern Western nectarines can be traced back to three main cultivars: ‘Quetta’, a local Pakistani cultivar, introduced in Europe and USA at the beginning of the last century, ‘Goldmine’ and ‘Lippiat’, both discovered in New Zealand in the early XX century. The origin of the nectarine trait/gene (namely a retrotransposon insertion disrupting the function of the causal gene PpeMYB025) is common to all the Western nectarines known so far [46]. In accordance with our results Micheletti et al [27] and Li et al [22] divided modern Western cultivars into peaches and nectarines.
The other two groups were mainly composed of fuzzy peaches. The biggest group, Q3, i is predominantly composed of cultivars obtained during the modern breeding activity in Western countries (129 accessions). This started in the middle of XIX century in US and can be traced back to a limited number of accessions as founders (‘Chinese cling’, imported from China in the middle of XIX century, and other few European cultivars brought in US by the early settlers, [21,22,47]). Most breeding programs in US and Europe (and partly in Japan and Korea) in the last century rely on this reduced number of genotypes, explaining the difference in allele frequencies observed in this group. The other subgroup, Q1, is equally composed of traditional Western cultivars (56) and modern breeding cultivars (56). The results of this study are in agreement with those obtained by Micheletti et al. [27] that described three main groups namely Occidental varieties from breeding programs, Occidental landraces and Oriental accessions. In our study the number of Oriental accessions was limited (40), thus preventing the detection of a separate group. Most of the cultivars (16 out of 31) obtained by modern breeding activities in South America (Argentina, Brazil and Mexico) were included in the third group (traditional Western varieties), highlighting the possible origin of these cultivars from a gene pool related to the traditional Western one.
2.4. SNP and SSR Markers for GenBank Management
The main difference between SNPs and SSRs is their rate of polymorphism. SSR markers are multi-allelic with more than 10 alleles at a single locus (up to 19 in this study) making them useful for fingerprinting and diversity studies, while SNPs are biallelic and the most abundant class of markers in the genome. The high throughput low-cost technologies and their abundance make SNPs the markers of choice for whole genome analyses such as GWAS. On the other hand SSRs analysis is more flexible and can be carried out by small laboratories.
From a GenBank perspective SSRs are more suitable and affordable to efficiently manage germplasm collections, reducing unwanted redundancy, mislabeling and spelling errors.
3. Materials and Methods
3.1. Plant Materials and DNA Extraction
A panel of 1025 peaches (Prunus persica) and a Prunus davidiana accession (a species closely related to P. persica) were analyzed. The samples came from two collections: 872 from the NFGC held at CREA-OFA in Rome (41°47’42”N, 12°33’46”E) and 154 from the MAS.PES collection in Imola (44°20′12″N, 11°45′26″E). The accessions are listed in Supplementary Table S1. The geographical origin (22 countries representing all the continents), the pedigree and the most important fruit traits evaluated in the fields and/or retrieved from literature [45,48], are also reported when available.
DNA was extracted from young leaves, using the DNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions. Quant-iTPicogreen (Invitrogen) was used for DNA quantification and the DNA concentration of each sample was estimated based on a standard concentration curve. The final concentrations of all DNA samples were adjusted to 20 ng/µl.
3.2. SSR Analysis
DNA samples were genotyped by 12 SSR markers (Table 1). Markers were selected based on an earlier study by Jouy et al. [49], whose objective was to choose a set of markers for characterization of large peach collections. SSR reactions were performed using the multiplex-ready PCR protocol [50] with some modifications by Eduardo et al [51]. After amplification, PCR products labelled with different dyes, were diluted with 10 μl of distilled water and pooled in a 1:1:1:1 proportion for capillary electrophoresis on an ABI Prism 3730 DNA Analyzer (Applied Biosystems, MA, USA): the loading mix consisted of 2 μl of PCR pool, 10 μl of HI-DI formamide and 0.15 μl of GeneScan500 LIZ-250 size standard (ThermoFisher).
Allele size was determined with the Gene-Marker demo version 1.70 (SoftGenetics). Using the multiplex-ready approach with “tagged” SSR primers, the expected size of the alleles corresponded to30 base pairs more than the reported allele sizes (Pacheco et al., 2014). To improve accuracy and allele scoring reliability two independent readings of the microsatellite sample profiles were done.
Genetic diversity analysis
NTSYSpc version 2.11Q [52] was employed to estimate and graphically represent the genetic similarity among cultivars. The SimGend procedure with the Lynch Shared band similarity index [53] was used to calculate the similarity; cultivars were then clustered using the SAHN procedure with the unweighted pair-group method (UPGMA).
The number of alleles per locus (Na), the effective number of alleles per locus (Ne), the number of rare (frequency <0.05) and unique alleles (specific to a genotype), the observed and expected heterozygosity (Ho and He), the polymorphic information content (PIC), the probability of identity (PI) and the combined non-exclusion probability were estimated by the software packages Cervus 3.0.7 [54] and GenAlEx 6.5.0.3 [55]. The discrimination power (DP) of each marker was calculated from the probability of identity (PI) as in the following formula: PD=1-PI.
3.3. Population Structure Analysis
Population structure analysis was conducted with the software Structure 2.3.4 [56], based on Bayesian statistics using 12 SSR markers. The admixture model of ancestry and correlated allele frequencies was adopted to analyze the data set, with no preliminary subpopulation information. The proportion of the ancestry of each individual was tested considering a K number from 1 to 10, with 10 iterations for each value of K. The settings for burning-in and MCMC (Markov Chain Monte Carlo) were 100,000 and 500,000, respectively. To determine the K number, the model established by Evanno et al., 2005, was adopted using the structure selector website [57]. CLUMPP v 1.1.2 [58] was used to find the optimal alignment of the ten independent replications and compute the Q-matrix. The R package ‘pophelper’ v. 2.3.1 [59] was used to graphically display the population structure.
4. Conclusions
The Molecular genetic characterization of 1026 peach accessions was carried out in order to estimate the genetic diversity and to build a fingerprint database of the germplasm from MAS.PES and NGFC CREA collections. In accordance with previous studies, the marker with the highest value of effective number of alleles, PIC and discrimination power was ‘BPPCT001’. A prevalence of homozygosity was observed for all loci of the evaluated accessions, as expected in the case of an autogamous species. The chosen markers also allowed us to confirm the identity of most duplicated peach accessions in both collections. Some accessions, in a few cases, even if not genetically related, showed the same fingerprinting pattern, and additional SSRs markers are needed to discriminate them. The cultivars analyzed have mostly Occidental origin and represent both traditional and modern germplasm. The present study reports novel fingerprinting and genetic diversity information for 377 peach accessions, while allowing comparison with the cited previous studies.
The population structure analysis showed three clusters, with two main subpopulations matching the major varietal groups in pubescent (PE) and glabrous (NE) peaches. Among the pubescent group, two main groups were identified: one included genotypes obtained from breeding activities and one with most of the traditional landraces. Considering the high values of PIC and PD, the SSR markers used in this work proved to be very efficient for estimating the genetic diversity present in both collections.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org, Table S1: List of Peach accessions genotyped by the 12 SSR markers, pedigree if known; Table S2: Fingerprint profiles of 961peache samples from CREA and MAS.PES collections; Table S3: List of accessions with pairwise exact match and related probability of identity (pID and pIDsib) as calculated by CERVUS; Table S4: Results of the structure analysis with membership coefficients for each cultivar. Figure S1: Dendrogram of 961 peach accessions by 12 SSR markers. Characters are coded by colors in the diagram presented across the dendrogram: nectarines are represented by yellow and peaches by orange ones (first column), yellow flesh by green and white flesh by light pink rectangles (second column).
Author Contributions
Conceptualization, Daniele Bassi, Laura Rossini and Ignazio Verde; Data curation, Elisa Vendramin and Cássia Da Silva Linge; Funding acquisition, Daniele Bassi and Ignazio Verde; Investigation, Elisa Vendramin, Cássia Da Silva Linge, Sabrina Micali, Giorgiana Chietera, Maria Teresa Dettori, Valeria Aramini, Jessica Giovinazzi, Igor Pacheco and Ignazio Verde; Methodology, Elisa Vendramin, Cássia Da Silva Linge, Sabrina Micali, Giorgiana Chietera, Maria Teresa Dettori, Jessica Giovinazzi, Laura Rossini and Ignazio Verde; Resources, Daniele Bassi and Ignazio Verde; Software, Sabrina Micali, Maria Teresa Dettori and Valeria Aramini; Supervision, Daniele Bassi, Laura Rossini and Ignazio Verde; Validation, Elisa Vendramin, Cássia Da Silva Linge, Valeria Aramini; Writing – original draft, Elisa Vendramin, Cássia Da Silva Linge, Maria Teresa Dettori, Laura Rossini and Ignazio Verde; Writing – review & editing, Daniele Bassi, Sabrina Micali, Giorgiana Chietera, Valeria Aramini, Jessica Giovinazzi and Igor Pacheco. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Ministero delle Politiche Agricole Alimentari e Forestali (MiPAAF, Italy) through the projects DRUPOMICS (grant DM14999/7303/08), and MAS.PES apricot and peach breeding by molecular-assisted selection, a project funded by private and public agencies; IP was supported by a post-doctoral fellowship ‘Becas Chile’, managed by Conicyt and INTA, Universidad de Chile. CDSL was supported by a ‘Dote Ricerca Applicata’ fellowship co-funded by Regione Lombardia and Parco Tecnologico Padano (Italy) and by a post doc fellowship by CNPq, Conselho Nacional de Desenvolvimento Científico e Tecnologico (Brazil).
Acknowledgments
We thank S. Foschi, M. Lama, and E. Quattrini for their valuable contribution in field tree management and sample harvest.
Conflicts of Interest
The authors declare no conflicts of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| NGFC CREA | National Fruit Germplasm Collection |
| MAS.PES | Breeding apricot and peach through Marker-Assisted Selection |
| ITPGRFA | International Treaty for Plant Genetic Resources for Food and Agriculture |
| GWAS | Genome Wide Association Studies |
| GBS | Genotyping by Sequencing |
| SSRs | Simple Sequence Repeats |
| SNPs | Single Nucleotide Polymorphisms |
| PD | Discrimination Power |
| Ho | observed Heterozygosity |
| He | expected Heterozygosity |
| PIC | Polymorphic Information content |
| PI | Probability of Identity |
| MAF | Minor Allele Frequency |
References
- D. Potter et al., “Phylogeny and classification of Rosaceae,” Plant Systematics and Evolution, vol. 266, no. 1–2, pp. 5–43, Jul. 2007. [CrossRef]
- S. Lee and J. Wen, “A phylogenetic analysis of Prunus and the Amygdaloideae (Rosaceae) using ITS sequences of nuclear ribosomal DNA,” Am J Bot, vol. 88, no. 1, pp. 150–160, Jan. 2001. [CrossRef]
- V. Shulaev et al., “Multiple Models for Rosaceae Genomics,” Plant Physiol, vol. 147, no. 3, pp. 985–1003, Jul. 2008. [CrossRef]
- K. Boudehri, A. Bendahmane, G. Cardinet, C. Troadec, A. Moing, and E. Dirlewanger, “Phenotypic and fine genetic characterization of the D locus controlling fruit acidity in peach,” BMC Plant Biol, vol. 9, no. 1, pp. 1–14, May 2009. [CrossRef]
- S. A. Gilani, R. A. Qureshi, A. M. Khan, and D. Potter, “A molecular phylogeny of selected species of genus Prunus L. (Rosaceae) from Pakistan using the internal transcribed spacer (ITS) spacer DNA,” Afr J Biotechnol, vol. 9, no. 31, pp. 4867–4872, 2010, Accessed: Jun. 10, 2025. [Online]. Available online: https://www.ajol.info/index.php/ajb/article/view/92035.
- I. Verde et al., “The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution,” Nature Genetics 2013 45:5, vol. 45, no. 5, pp. 487–494, Mar. 2013. [CrossRef]
- M. Faust and B. Timon, “Origin and Dissemination of Peach,” Hortic Rev (Am Soc Hortic Sci), pp. 331–379, Nov. 1995. [CrossRef]
- P. Vaccino et al., “Plant Genetic Resources for Food and Agriculture: The Role and Contribution of CREA (Italy) within the National Program RGV-FAO,” Agronomy, vol. 14, no. 6, p. 1263, Jun. 2024. [CrossRef]
- R. Van Treuren and T. J. L. Van Hintum, “Marker-assisted reduction of redundancy in germplasm collections: Genetic and economic aspects,” Acta Hortic, vol. 623, pp. 139–149, Jul. 2003. [CrossRef]
- J. C. Bettoni, R. Bonnart, and G. M. Volk, “Challenges in implementing plant shoot tip cryopreservation technologies,” Plant Cell Tissue Organ Cult, vol. 144, no. 1, pp. 21–34, Jan. 2021. [CrossRef]
- P. Tripodi et al., “Global range expansion history of pepper (Capsicum spp.) revealed by over 10,000 genebank accessions,” Proc Natl Acad Sci U S A, vol. 118, no. 34, p. e2104315118, Aug. 2021. [CrossRef]
- F. Emanuelli et al., “Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape,” BMC Plant Biol, vol. 13, no. 1, pp. 1–17, Mar. 2013. [CrossRef]
- R. van Treuren, H. Kemp, G. Ernsting, B. Jongejans, H. Houtman, and L. Visser, “Microsatellite genotyping of apple (Malus × domestica Borkh.) genetic resources in the Netherlands: Application in collection management and variety identification,” Genet Resour Crop Evol, vol. 57, no. 6, pp. 853–865, Jan. 2010. [CrossRef]
- W. Liang, L. Dondini, P. De Franceschi, R. Paris, S. Sansavini, and S. Tartarini, “Genetic Diversity, Population Structure and Construction of a Core Collection of Apple Cultivars from Italian Germplasm,” Plant Mol Biol Report, vol. 33, no. 3, pp. 458–473, Jun. 2015. [CrossRef]
- L. Lassois et al., “Genetic Diversity, Population Structure, Parentage Analysis, and Construction of Core Collections in the French Apple Germplasm Based on SSR Markers,” Plant Mol Biol Report, vol. 34, no. 4, pp. 827–844, Aug. 2016. [CrossRef]
- B. Larsen, T. B. Toldam-Andersen, C. Pedersen, and M. Ørgaard, “Unravelling genetic diversity and cultivar parentage in the Danish apple gene bank collection,” Tree Genet Genomes, vol. 13, no. 1, pp. 1–12, Feb. 2017. [CrossRef]
- A. Pina, J. Urrestarazu, and P. Errea, “Analysis of the genetic diversity of local apple cultivars from mountainous areas from Aragon (Northeastern Spain),” Sci Hortic, vol. 174, no. 1, pp. 1–9, Jul. 2014. [CrossRef]
- C. M. H. D. C.-E. Denancé, “MUNQ - Malus UNiQue genotype code for grouping apple accessions corresponding to a unique genotypic profile,” 2020.
- B. Larsen et al., “Cultivar fingerprinting and SNP-based pedigree reconstruction in Danish heritage apple cultivars utilizing genotypic data from multiple germplasm collections in the world,” Genet Resour Crop Evol, vol. 72, no. 2, pp. 2397–2411, Feb. 2024. [CrossRef]
- M. J. Aranzana, J. Carbó, and P. Arús, “Microsatellite variability in peach [Prunus persica (L.) Batsch]: Cultivar identification, marker mutation, pedigree inferences and population structure,” Theoretical and Applied Genetics, vol. 106, no. 8, pp. 1341–1352, May 2003. [CrossRef]
- M. J. Aranzana, E. K. Abbassi, W. Howad, and P. Arús, “Genetic variation, population structure and linkage disequilibrium in peach commercial varieties,” BMC Genet, vol. 11, no. 1, pp. 1–11, Jul. 2010. [CrossRef]
- X. wei Li et al., “Peach genetic resources: Diversity, population structure and linkage disequilibrium,” BMC Genet, vol. 14, no. 1, pp. 1–16, Sep. 2013. [CrossRef]
- M. Méndez, G. Rojas, C. Muñoz, G. Lemus, and P. Hinrichsen, “Identification of a minimal microsatellite marker panel for the fingerprinting of peach and nectarine cultivars,” 2008.
- M. Bouhadida, M. Á. Moreno, M. J. Gonzalo, J. M. Alonso, and Y. Gogorcena, “Genetic variability of introduced and local Spanish peach cultivars determined by SSR markers,” Tree Genet Genomes, vol. 7, no. 2, pp. 257–270, Apr. 2011. [CrossRef]
- D. J. Chavez, T. G. Beckman, D. J. Werner, and J. X. Chaparro, “Genetic diversity in peach [prunus persica (l.) batsch] at the university of florida: past, present and future,” Tree Genet Genomes, vol. 10, no. 5, pp. 1399–1417, Oct. 2014. [CrossRef]
- Z. J. Shen, R. J. Ma, Z. X. Cai, and M. L. Yu, “Diversity, Population structure, And evolution of local peach cultivars in China identified by simple sequence repeats,” Genetics and Molecular Research, vol. 14, no. 1, pp. 101–117, Jan. 2015. [CrossRef]
- D. Micheletti et al., “Whole-Genome Analysis of Diversity and SNP-Major Gene Association in Peach Germplasm,” PLoS One, vol. 10, no. 9, p. e0136803, Sep. 2015. [CrossRef]
- I. Verde et al., “Development and evaluation of a 9K SNP array for peach by internationally coordinated SNP detection and validation in breeding germplasm.,” PLoS One, vol. 7, no. 4, p. e35668, 2012. [CrossRef]
- K. Cao et al., “Genome-wide association study of 12 agronomic traits in peach,” Nat Commun, vol. 7, no. 1, pp. 1–10, Nov. 2016. [CrossRef]
- J. Guo et al., “An integrated peach genome structural variation map uncovers genes associated with fruit traits,” Genome Biol, vol. 21, no. 1, pp. 1–19, Oct. 2020. [CrossRef]
- Y. Li et al., “Genomic analyses of an extensive collection of wild and cultivated accessions provide new insights into peach breeding history,” Genome Biol, vol. 20, no. 1, pp. 1–18, Feb. 2019. [CrossRef]
- Y. Yu et al., “Population-scale peach genome analyses unravel selection patterns and biochemical basis underlying fruit flavor,” Nature Communications 2021 12:1, vol. 12, no. 1, pp. 1–13, Jun. 2021. [CrossRef]
- F. S. R. M. and C. L. Bassi D, “Progetto ‘MAS.PES‘ per il miglioramento genetico del pesco: criteri di selezione e individuazione ideotipi di riferimento,” Italus Hortus 17 (5), pp. 60–62, 2010.
- B. V. F. J. Paula LA, “Caracterização molecular variabilidade genética entre porta-enxertos de pessegueiro com base em marcadores codominantes,” Pesquisa Agropecuária Brasileira, Brasília., vol. 47, no. 2, pp. 193–199, 2012.
- C. A. E. A. C. P. Z. A. and D. E. Chalak L, “Morphological and molecular characterization of peach accessions (Prunus persica L.) cultivated in Lebanon,” Lebanese Sci. J., vol. 7, no. 2, pp. 23–31, 2003.
- D. J. Chavez, T. G. Beckman, D. J. Werner, and J. X. Chaparro, “Genetic diversity in peach [Prunus persica (L.) Batsch] at the University of Florida: past, present and future,” Tree Genet Genomes, 2014. [CrossRef]
- D. Botstein, R. L. White, M. Skolnick, and R. W. Davis, “Construction of a genetic linkage map in man using restriction fragment length polymorphisms,” Am J Hum Genet, vol. 32, no. 3, p. 314, 1980, Accessed: Jun. 10, 2025. [Online]. Available online: https://pmc.ncbi.nlm.nih.gov/articles/PMC1686077/.
- D. Giovannini et al., “Assessment of genetic variability in Italian heritage peach resources from Emilia-Romagna using microsatellite markers,” J Hortic Sci Biotechnol, vol. 87, no. 5, pp. 435–440, 2012. [CrossRef]
- R. Falchi et al., “Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach,” Plant Journal, vol. 76, no. 2, pp. 175–187, 2013. [CrossRef]
- M. Adami et al., “Identifying a Carotenoid Cleavage Dioxygenase (ccd4) Gene Controlling Yellow/White Fruit Flesh Color of Peach,” Plant Mol Biol Report, vol. 31, no. 5, pp. 1166–1175, 2013. [CrossRef]
- K. Cao et al., “Comparative population genomics reveals the domestication history of the peach, Prunus persica, and human influences on perennial fruit crops,” Genome Biol, vol. 15, no. 7, pp. 1–15, Jul. 2014. [CrossRef]
- A. Smykov, O. Fedorova, T. Shishova, and I. Ivashchenko, “Introduction and use of the peach gene pool from China in Nikita Botanical Garden,” Acta Hortic, vol. 1208, pp. 1–6, Aug. 2018. [CrossRef]
- R. Testolin et al., “Microsatellite DNA in peach (Prunus persica L. Batsch) and its use in fingerprinting and testing the genetic origin of cultivars,” https://doi.org/10.1139/g00-010, vol. 43, no. 3, pp. 512–520, Jun. 2011. [CrossRef]
- R. Falchi et al., “Three distinct mutational mechanisms acting on a single gene underpin the origin of yellow flesh in peach,” The Plant Journal, vol. 76, no. 2, pp. 175–187, Oct. 2013. [CrossRef]
- W. R. Okie, Handbook of peach and nectarine varieties. National TechnicaU.S. Dept. of Agriculture, Agricultural Research Service; National Technical Information Service, distributor, 1998.
- E. Vendramin et al., “A Unique Mutation in a MYB Gene Cosegregates with the Nectarine Phenotype in Peach.,” PLoS One, vol. 9, no. 3, p. e90574, 2014. [CrossRef]
- R. Scorza, S. A. Mehlenbacher, and G. W. Lightner, “Inbreeding and Coancestry of Freestone Peach Cultivars of the Eastern United States and Implications for Peach Germplasm Improvement,” Journal of the American Society for Horticultural Science, vol. 110, no. 4, pp. 547–552, Jul. 1985. [CrossRef]
- AA.VV., Atlante dei fruttiferi autoctoni italiani, 2016th ed., vol. I-II–III. Roma: CREA Olivicoltura, Frutticoltura e Agrumicoltura & MIPAAF, 2016.
- C. Jouy et al., “Management of peach tree reference collections: Ongoing research & development program relevant to the community plant variety rights protection system,” Acta Hortic, vol. 962, pp. 51–56, Oct. 2012. [CrossRef]
- M. J. Hayden, T. M. Nguyen, A. Waterman, and K. J. Chalmers, “Multiplex-Ready PCR: A new method for multiplexed SSR and SNP genotyping,” BMC Genomics, vol. 9, no. 1, pp. 1–12, Feb. 2008. [CrossRef]
- I. Eduardo et al., “QTL analysis of fruit quality traits in two peach intraspecific populations and importance of maturity date pleiotropic effect,” Tree Genet Genomes, vol. 7, no. 2, pp. 323–335, Apr. 2011. [CrossRef]
- F. J. Rohlf, “NTSYS-pc, Numerical Taxonomy and Multivariate Analysis System. Ver. 1. 80,” 1994, Exeter Software, New York.
- M. Lynch, “The similarity index and DNA fingerprinting.,” Mol Biol Evol, vol. 7, no. 5, pp. 478–484, Sep. 1990. [CrossRef]
- S. T. Kalinowski, M. L. Taper, and T. C. Marshall, “Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment,” Mol Ecol, vol. 16, no. 5, pp. 1099–1106, Mar. 2007. [CrossRef]
- R. Peakall and P. E. Smouse, “GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research—an update,” Bioinformatics, vol. 28, no. 19, pp. 2537–2539, Oct. 2012. [CrossRef]
- J. K. Pritchard, M. Stephens, and P. Donnelly, “Inference of Population Structure Using Multilocus Genotype Data,” Genetics, vol. 155, no. 2, pp. 945–959, Jun. 2000. [CrossRef]
- Y. L. Li and J. X. Liu, “StructureSelector: A web-based software to select and visualize the optimal number of clusters using multiple methods,” Mol Ecol Resour, vol. 18, no. 1, pp. 176–177, Jan. 2018. [CrossRef]
- M. Jakobsson and N. A. Rosenberg, “CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure,” Bioinformatics, vol. 23, no. 14, pp. 1801–1806, Jul. 2007. [CrossRef]
- R. M. Francis, “pophelper: an R package and web app to analyse and visualize population structure,” Mol Ecol Resour, vol. 17, no. 1, pp. 27–32, Jan. 2017. [CrossRef]
Figure 1.
Dendrogram of 961 peach accessions by 12 SSR markers. Characters are coded by colors in the diagram presented across the dendrogram: nectarines are represented by yellow and peaches by orange ones (first column), yellow flesh by green and white flesh by light pink rectangles (second column).
Figure 1.
Dendrogram of 961 peach accessions by 12 SSR markers. Characters are coded by colors in the diagram presented across the dendrogram: nectarines are represented by yellow and peaches by orange ones (first column), yellow flesh by green and white flesh by light pink rectangles (second column).
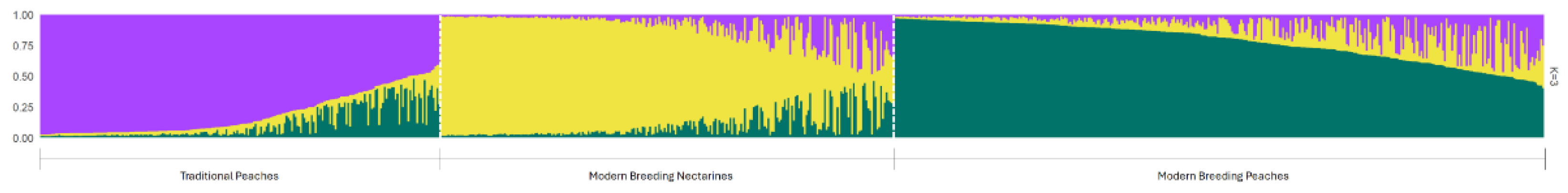

Table 1.
Allele size (bp), and frequencies (f) of the 12 SSR markers analyzed.
| BPPCT017 | BPPCT001 | UDP-005 | BPPCT007 | BPPCT038 | UDP-412 | EPPCU5176 | BPPCT015 | CPDCT045 | UDP-409 | CPPCT006 | UDP-022 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| size | 134 | 162 | 149 | 157 | 119 | 128 | 183 | 171 | 161 | 152 | 208 | 147 |
| f | 0,001 | 0,009 | 0,001 | 0,001 | 0,001 | 0,001 | 0,001 | 0,001 | 0,001 | 0,12 | 0,291 | 0,001 |
| size | 149 | 166 | 157 | 161 | 121 | 133 | 185 | 178 | 167 | 156 | 210 | 157 |
| f | 0,012 | 0,001 | 0,001 | 0,025 | 0,002 | 0,001 | 0,001 | 0,001 | 0,001 | 0,001 | 0,001 | 0,173 |
| size | 151 | 170 | 162 | 163 | 123 | 143 | 191 | 180 | 172 | 158 | 216 | 159 |
| f | 0,026 | 0,004 | 0,001 | 0,001 | 0,594 | 0,003 | 0,001 | 0,016 | 0,003 | 0,742 | 0,001 | 0,002 |
| size | 155 | 176 | 166 | 165 | 125 | 145 | 193 | 182 | 174 | 160 | 218 | 165 |
| f | 0,001 | 0,076 | 0,002 | 0,001 | 0,038 | 0,003 | 0,001 | 0,022 | 0,024 | 0,101 | 0,157 | 0,143 |
| size | 157 | 178 | 168 | 167 | 127 | 150 | 197 | 186 | 176 | 162 | 220 | 167 |
| f | 0,002 | 0,007 | 0,004 | 0,001 | 0,018 | 0,004 | 0,505 | 0,257 | 0,438 | 0,004 | 0,544 | 0,15 |
| size | 159 | 182 | 170 | 169 | 129 | 152 | 199 | 188 | 178 | 168 | 222 | 169 |
| f | 0,017 | 0,102 | 0,001 | 0,001 | 0,061 | 0,262 | 0,026 | 0,006 | 0,124 | 0,001 | 0,002 | 0,021 |
| size | 161 | 184 | 172 | 171 | 131 | 154 | 201 | 190 | 180 | 170 | 224 | 171 |
| f | 0,539 | 0,152 | 0,001 | 0,001 | 0,006 | 0,09 | 0,229 | 0,02 | 0,406 | 0,001 | 0,002 | 0,509 |
| size | 163 | 186 | 174 | 175 | 133 | 156 | 203 | 198 | 182 | 179 | 226 | 173 |
| f | 0,007 | 0,072 | 0,185 | 0,044 | 0,021 | 0,142 | 0,001 | 0,614 | 0,002 | 0,005 | 0,001 | 0,002 |
| size | 165 | 188 | 176 | 177 | 135 | 158 | 205 | 200 | 184 | 181 | 228 | |
| f | 0,011 | 0,439 | 0,066 | 0,437 | 0,25 | 0,448 | 0,022 | 0,034 | 0,001 | 0,021 | 0,002 | |
| size | 167 | 190 | 178 | 179 | 137 | 160 | 207 | 202 | 186 | 183 | ||
| f | 0,003 | 0,058 | 0,004 | 0,067 | 0,004 | 0,038 | 0,209 | 0,026 | 0,001 | 0,004 | ||
| size | 169 | 192 | 187 | 181 | 139 | 162 | 209 | 206 | ||||
| f | 0,009 | 0,06 | 0,004 | 0,01 | 0,005 | 0,005 | 0,004 | 0,003 | ||||
| size | 171 | 194 | 189 | 183 | 141 | 164 | 211 | |||||
| f | 0,004 | 0,01 | 0,104 | 0,408 | 0,001 | 0,004 | 0,001 | |||||
| size | 173 | 196 | 191 | 186 | 149 | |||||||
| f | 0,001 | 0,002 | 0,611 | 0,002 | 0,001 | |||||||
| size | 175 | 198 | 193 | 198 | ||||||||
| f | 0,001 | 0,006 | 0,015 | 0,001 | ||||||||
| size | 177 | 200 | 195 | |||||||||
| f | 0,013 | 0,003 | 0,001 | |||||||||
| size | 179 | |||||||||||
| f | 0,343 | |||||||||||
| size | 181 | |||||||||||
| f | 0,009 | |||||||||||
| size | 183 | |||||||||||
| f | 0,001 | |||||||||||
| size | 185 | |||||||||||
| f | 0,001 |
Table 2.
Genetic diversity of 961 peach accessions analyzed by 12 SSRs markers. Locus name, number of alleles per locus (Na), effective number of alleles per locus (Ne), observed heterozygosity (Ho), expected heterozygosity (He), polymorphic information.
Table 2.
Genetic diversity of 961 peach accessions analyzed by 12 SSRs markers. Locus name, number of alleles per locus (Na), effective number of alleles per locus (Ne), observed heterozygosity (Ho), expected heterozygosity (He), polymorphic information.
| Locus | N | Na | Ne | I | Ho | He | F | PIC | PD | Nra |
|---|---|---|---|---|---|---|---|---|---|---|
| BPPCT001 | 885 | 15 | 4,1 | 1,81 | 0,5 | 0,76 | 0,34 | 0,73 | 0,92 | 8 |
| BPPCT007 | 935 | 14 | 2,74 | 1,24 | 0,5 | 0,64 | 0,21 | 0,57 | 0,8 | 11 |
| BPPCT015 | 871 | 11 | 2,24 | 1,15 | 0,37 | 0,55 | 0,34 | 0,5 | 0,75 | 9 |
| BPPCT017 | 939 | 19 | 2,44 | 1,23 | 0,46 | 0,59 | 0,22 | 0,52 | 0,76 | 17 |
| BPPCT038 | 949 | 13 | 2,38 | 1,21 | 0,44 | 0,58 | 0,24 | 0,53 | 0,77 | 10 |
| CPDCT045 | 909 | 10 | 2,69 | 1,13 | 0,49 | 0,63 | 0,22 | 0,55 | 0,78 | 7 |
| CPPCT006 | 921 | 9 | 2,47 | 1,03 | 0,44 | 0,6 | 0,26 | 0,53 | 0,77 | 6 |
| EPPCU5176 | 948 | 12 | 2,84 | 1,24 | 0,5 | 0,65 | 0,23 | 0,59 | 0,82 | 9 |
| UDP-005 | 841 | 15 | 2,36 | 1,21 | 0,4 | 0,58 | 0,3 | 0,54 | 0,78 | 11 |
| UDP-022 | 894 | 8 | 3,01 | 1,32 | 0,42 | 0,67 | 0,38 | 0,63 | 0,85 | 4 |
| UDP-409 | 926 | 10 | 1,74 | 0,88 | 0,32 | 0,42 | 0,24 | 0,4 | 0,64 | 7 |
| UDP-412 | 932 | 12 | 3,35 | 1,45 | 0,53 | 0,7 | 0,24 | 0,66 | 0,87 | 8 |
Table 3.
Genetic profiles of the accessions bearing private alleles (in bold).
| Sample/SSR | BPPCT001 | BPPCT007 | BPPCT015 | BPPCT017 | BPPCT038 | CPDCT045 | CPPCT006 | EPPCU5176 | UDP-005 | UDP-022 | UDP-409 | UDP-412 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AB/6 | 170 190 | 169 177 | - - | 157 161 | 125 129 | 178 184 | 220 224 | 205 205 | 162 174 | 167 167 | 158 168 | 154 154 |
| Chui_Huang_Tao | 182 182 | 177 177 | 186 186 | 151 151 | 131 149 | 176 176 | - - | 193 193 | 157 157 | 165 169 | 181 181 | 152 152 |
| Citation | 162 166 | 171 177 | 198 198 | 161 161 | 123 131 | 176 178 | 220 226 | 185 197 | 191 191 | 165 165 | 158 158 | 152 160 |
| Dourado | 186 188 | 161 177 | 180 180 | 151 165 | 123 127 | 176 178 | 208 220 | 197 207 | 172 191 | 165 167 | 158 158 | 152 158 |
| Ferganensis | 182 182 | 183 183 | - - | 161 161 | 123 123 | 178 178 | 220 220 | 209 209 | 193 195 | 165 165 | 183 183 | 162 162 |
| Fiorenza | 188 188 | 177 183 | 198 198 | 161 185 | 123 123 | 180 180 | 220 220 | 197 205 | 191 191 | 165 171 | 152 158 | 158 160 |
| Glowin_Star | 170 190 | 179 179 | 198 198 | 134 169 | 123 123 | 176 186 | 220 224 | 207 207 | 174 189 | 165 165 | 160 183 | 156 156 |
| IF_817023 | 162 162 | - - | 186 186 | 155 161 | 123 123 | 167 180 | 216 228 | 191 191 | 174 174 | - - | 183 183 | 128 133 |
| P1/12 | 186 186 | 177 198 | 198 198 | 161 161 | 123 123 | 176 176 | 220 220 | 197 207 | 176 176 | 157 157 | 158 158 | 156 158 |
| P5/645 | 182 188 | 161 177 | 186 186 | 177 179 | 135 135 | 176 180 | 208 208 | 203 207 | 189 191 | 165 171 | 158 158 | 158 158 |
| Queen_Ruby | 176 184 | 179 183 | 198 200 | 161 161 | 123 123 | 178 180 | 208 220 | 201 211 | 189 191 | 165 171 | 158 158 | 152 156 |
| Red_Robin | 182 182 | 165 175 | 198 198 | 149 179 | 129 135 | 176 180 | 208 208 | 197 201 | 191 191 | 165 171 | 152 158 | 152 158 |
| Shan_Dong | 184 184 | 161 177 | 198 198 | 149 179 | 123 141 | 176 180 | - - | 197 199 | 174 191 | - - | 158 181 | 152 158 |
| XIAGUANG_a | 186 186 | 167 183 | 198 198 | 159 161 | 123 123 | 176 176 | 208 208 | 201 201 | 174 176 | 171 171 | 158 160 | - - |
Table 4.
Matching of accessions with known pedigree with their putative parents in different type of pedigree.
Table 4.
Matching of accessions with known pedigree with their putative parents in different type of pedigree.
| Type of Pedigree | N° | N Match | % Match |
|---|---|---|---|
| P1xP2 | 47 | 16 | 34.0 |
| PxSelf | 14 | 5 | 35.7 |
| P op | 59 | 42 | 71.2 |
| Clones | 29 | 17 | 55.2 |
| N° TOT | 149 | 80 | 53.0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



