
Submitted:
11 June 2025
Posted:
12 June 2025
You are already at the latest version
Abstract
Despite significant advances in cancer therapy, the prognosis for patients with advanced, disseminated disease remains poor. This underscores the urgent need for novel treatments that not only eliminate tumor cells effectively but also stimulate a strong, durable anti-cancer immune response. Among emerging strategies, oncolytic viruses have shown exceptional promise due to their selective cytotoxicity and their ability to activate T-cell-mediated immune responses. In this review, we focus on the vaccinia virus (VACV), a member of the Poxviridae family, which has emerged as a leading candidate in modern oncolytic immunotherapy. We examine the virus's properties that enable it to evade antiviral defenses and serve as a versatile, potent oncolytic agent. Furthermore, we explore its interactions with various components of the immune system and how these contribute to the induction of a robust T-cell driven response. Finally, we assess current efforts to harness VACV for the treatment of various cancer types and highlight future directions where its application is most likely to succeed. Overall, our goal is to present VACV as a powerful and broadly applicable platform with the potential to transform the landscape of oncology
Keywords:
immunotherapy
; Vaccinia
; oncolytic virus
; virotherapy
; oncology
1. Introduction
Vaccinia virus (VACV) is an Orthopoxvirus, and a prototypic member of large Poxviridae family. Due to its genomic and antigenic similarities to variola virus, along with high immunogenicity and low virulence, VACV was extensively used as the smallpox vaccine, ultimately contributing to the successful eradication of the disease. Like other Orthopoxviruses, it replicates exclusively in the cytoplasm and does not integrate into the host genome [1,2,3]. The exact origin of VACV remains unclear: One theory suggests that it evolved from either smallpox or cowpox virus; another posits that all three viruses share a common ancestor, while a third proposes that it may have been derived from horsepox virus [4]. In the wild, VACV has been reported to cause sporadic infections in cattle and water buffalos, but its natural host and reservoir remain unknown [5].
VACV is an enveloped, double-stranded DNA virus with a relatively large genome size of approximately 190 to 200kb. Both DNA strands are connected at the termini by partially complementary AT-rich loops, forming one continuous polynucleotide chain [6]. Interestingly, despite many years of genomic and transcriptomic research, the exact number of genes and proteins encoded by the VACV genome remains unknown. Most sources estimate the number of VACV genes to be between 200 and 250 [7,8,9].
Over the years, several strains of VACV have been developed. The most studied strain is the Western Reserve (WR) strain, which was created in the United States through repeated passages of the New York City Board of Health (NYCBH) strain in rabbits, mice and various mammalian cell cultures. Due to its adaptation to multiple hosts, it produces high titer in vitro and causes neuropathogenic effects in vivo, making it unsuitable for use as a vaccine [10].
In contrast to the WR strain, The Lister strain developed at the Lister Institute in the United Kingdom, and the NYCBH strain are much less virulent while still retaining the ability to replicate in humans and other mammals. The Lister and Wyeth strains (with Wyeth being the commercial name for the NYCBH strain) were the most commonly used for vaccinations during the smallpox eradication campaign [11]. They were known to induce strong immunity accompanied by high antibody titers, although they occasionally caused undesired side effects, including severe eczema and encephalitis [12].
Other VACV strains include Copenhagen and IHD (International Health Department), the latter being particularly notable for high production of extracellular enveloped viruses (EEVs) [13]. Finally, the modified vaccinia virus Ankara (MVA) strain represents a more attenuated version of the VACV Ankara strain. MVA cannot replicate in vivo and shows only limited infectivity and replication in vitro. It was developed through over 570 serial passages of the original virus in primary chicken embryo fibroblasts, resulting in the deletion of over 30kb fragment of its genome [11].
Today, most oncolytic VACV strains developed for therapy are based on various modifications of strains such as Lister, Wyeth, Copenhagen, or IHD.
2. Infection and Replication
2.1. Viral Entry
A schematic illustration of the VACV’s life cycle is shown in Figure 1. In contrast to the majority of viruses such as polio or HIV, VACV does not rely on binding to a specific cellular receptor for entry. Instead, it attaches to host cells using glycosaminoglycans (GAGs), specifically heparan sulfate and chondroitin sulfate for attachment [14,15]. Since GAGs are widely expressed on a variety of cell types, this broadens VACV’s host range, allowing it to infect multiple cell types across different species.
Additionally, a scavenger receptor MARCO (Macrophage receptor with collagenous structure) has been reported to facilitate VACV binding and enhance its interaction with GAGs [16].
The primary mechanism of VACV entry involves direct fusion of the viral and cell membranes. This multistep process includes close apposition of both membranes, lipid mixing of the outer membrane leaflets to form a hemifusion intermediate, followed by the formation and expansion of a fusion pore, which permits allowing the viral core or nucleoprotein to enter the cytoplasm [17]. Notably, this fusion can occur at neutral pH allowing VACV to bypass the endocytic pathway. As a result, the virus avoids pathogen recognition receptors (PRRs) localized in endosomes and can initiate replication rapidly.
Alternatively, VACV can enter cells via macropinocytosis, wherein membrane fusion occurs within endosomal vesicles and is triggered by lower pH [18]. This pathway is complex and involves Rho GTPases and the tyrosine kinase activity of the epidermal growth factor receptor (EGFR) in the host cell [19]. Depending on the strain, VACV may induce the formation of blebs or filopodia in the target cell to facilitate the internalization of viral particles.
Overall, at least 16 different viral proteins are involved in VACV entry: four in attachment and twelve in penetration. However, the precise molecular details of these processes remain incompletely understood [17].
2.2. Replication
The VACV virion consists of a compact protein core tightly wrapped around the viral DNA, along with two lateral bodies that contain proteins involved in counteracting host antiviral defenses [20]. Almost immediately after VACV enters a host cell, the lateral bodies dissociate from the core, leading to the release of H1 phosphatase, which inactivates signal transducer and activator of transcription 1 (STAT1) protein and thereby blocks interferon signaling [21].
Within 20 minutes of entry, components of the viral core initiate transcription of approximately 50% of the viral genome, producing a set of early mRNAs [22]. These early transcripts encode proteins required for DNA replication, which begins roughly 2 hours post-infection [23]. DNA replication then triggers the expression of intermediate genes, followed by late genes essential for the assembly of mature virions [22]. The first complete virions are typically produced around 6 hours after initial infection, and cell lysis generally begins within 15-48 hours post-entry [24].
A hallmark of VACV-infected cells is the presence of cytoplasmic structures known as virosomes or virus factories. These complexes, composed of viral proteins and protrusions of endoplasmic reticulum (ER) and Golgi-derived membranes, are the sites of viral DNA replication, transcription, and virion assembly [22,25].
To carry out replication independently of the host cell nucleus, VACV encodes a broad array of viral replication machinery, including enzymes that substitute for nuclear counterparts. These include DNA polymerase (E9), uracil DNA glycosylase (D4), and D5 protein, which possesses helicase and primase activity [26]. Additional proteins such as single-strand DNA-binding protein (I3) [27], Holliday junction resolvase (A22) [28], and ligase (A50) [29] further support autonomous replication, thereby minimizing the risk of non-specific integration into the host genome.
VACV also synthesizes its own nucleotide precursors via enzymes such as thymidine kinase (TK; encoded by J2R) [30], thymidylate kinase (encoded by A48R) [31], and ribonucleotide reductase (RR; encoded by F4L and I4L ) [32].
During its replication cycle, VACV produces four distinct types of virions: intracellular mature virus (IMV), intracellular enveloped virus (IEV), cell-associated enveloped virus (CEV), and extracellular enveloped virus (EEV) [33]. IMV is the most abundant and stable form of the virus, remaining within the cell until released by lysis. It is well-suited for host-to-host transmission. IEV is formed when IMV is wrapped in a secondary membrane, typically derived from the Golgi apparatus, and is transported to the cell surface along microtubules for dissemination. After fusing with the plasma membrane, the IEV loses two of its envelope proteins and becomes a CEV, a form that remains attached to the cell surface [34]. CEV induces the formation of actin tails, which propel the virus away from the cell, facilitating cell-to-cell spread. EEV is the fully released form, enabling long-range dissemination of the virus [34].
It is estimated that approximately 2,500-5,000 new viral particles are produced per infected cells [35].
3. Strategies to Avoid Anti-Viral Mechanisms
Mammalian cells have evolved numerous strategies to detect viruses, block their replication, and inhibit their spread. The general antiviral mechanism involves recognition of the pathogen with specific receptors, signal transduction leading to activation of transcription factors, and expression of target genes that help resolve the infection and alert the neighboring cells.
Most antiviral genes are regulated by two key transcription factors: Interferon Regulatory Factor 3 (IRF3) (along with its relatives IRF5 and IRF7) and Nuclear Factor-kappa B, (NF-κB). IRF3 is essential for the induction of type-I interferons (IFNs): IFN-α and IFN-β — cytokines secreted by infected cells that activate the expression of interferon-stimulated genes (ISGs) in surrounding tissue. ISGs function to block viral replication, promote antigen presentation, and eliminate infected cells through apoptosis [36].
NF-κB, on the other hand, triggers the transcription of multiple pro-inflammatory cytokines (e.g., Tumor necrosis factor (TNF) α, Interleukins (IL) IL-1β, IL-6, IL-8) and chemokines (e.g., chemokine (C-X-C motif) ligand1 (CXCL1), CCL5, CCL9), which recruit and stimulate immune cells, induce fever, and enable a rapid and tailored response to the specific pathogen [37].
To ensure effective replication, VACV has evolved a range of mechanisms to inhibit the IRF3 and NF-kB pathways at multiple levels, including pathogen recognition, signal transduction, transcription factor activation, and cytokine production (see Figure 2). Below, we briefly discuss how various VACV-encoded proteins effectively disarm the host anti-viral response.
3.1. DNA-PK and Viral DNA Detection
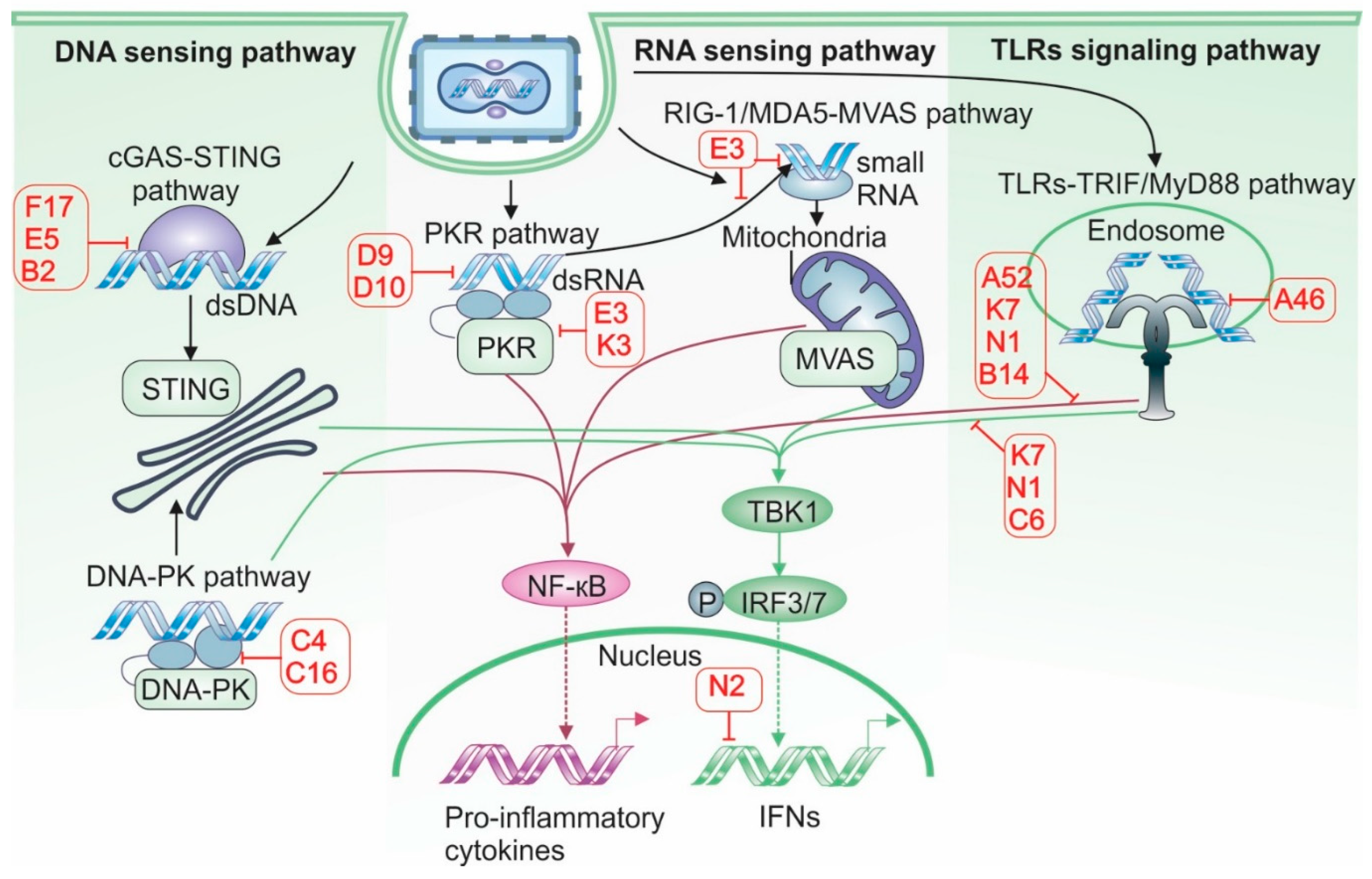
As a DNA virus that replicates in the cytosol, VACV is particularly vulnerable to detection by PRRs, which activate signaling pathways involving IRFs, NF-κB, and the adaptor proteins such as stimulator of interferon genes (STING). To avoid being recognized by PRRs, VACV developed a number of sophisticated mechanisms that are briefly summarized in Figure 3.
One of VACV’s evasion strategies targets the DNA-dependent protein kinase (DNA-PK) complex, which senses cytosolic double-stranded DNA and activates IRF3-mediated IFN production through both STING-dependent and -independent pathways [38]. Two proteins C16, encoded by two copies of the C16L gene, and C4, encoded by the C4L gene bind to the Ku70/Ku80 heterodimer within the DNA-PK complex via their C-terminal domains. This interaction inhibits DNA-PK’s ability to detect cytosolic DNA [39,40]. Interestingly, C16 and C4 appear to function redundantly, as both bind DNA-PK in a similar manner. This redundancy is believed to have evolved to counteract the high levels of DNA-PK present in certain cell types, such as fibroblasts, which are primary targets of VACV infection [38].
Another DNA-sensing mechanism involves DNA-dependent RNA polymerase III (Pol III). Although predominantly nuclear, Pol III is also detected in the cytosol, where it detects AT-rich DNA sequences. Upon recognizing viral DNA, Pol III synthesizes 5’-triphosphate-containing double-stranded (ds)RNA, which activates the RNA sensor Retinoic acid-inducible gene I (RIG-I), leading to downstream activation of IRF3 and NF-kB [41]. The VACV E3 protein inhibits Pol III activity, thereby blocking NF-κB activation. Although E3 contains both RNA- and DNA- binding domains, it is the RNA-binding domain that is responsible for inhibiting Pol III function [42]. VACV’s DNA can also be detected by IFN-γ-inducible protein 16 (IFI16), which, like Pol III, resides mainly in the nucleus but can also be found in the cytosol. IFI16 has been shown to activate both IRF3 and NF-kB in response to viral infections[43]. However, the VACV WR strain can inhibit IFI16-dependent expression of CCL5 and IFN-stimulated genes, although the precise mechanisms underlying this inhibition remain unclear [44].
3.2. cGAS, STING and IRF3
A distinct set of VACV proteins have evolved to counteract the cGAS-STING pathway, one of the most critical antiviral defense systems against cytosolic DNA viruses. Cyclic GMP-AMP synthase (cGAS) senses double-stranded DNA and produces 2'3'-cyclic GMP-AMP (cGAMP), a second messenger that activates the STING adaptor protein. This activation triggers TANK-binding kinase 1 (TBK1)-mediated phosphorylation of IRF3, leading to the induction of type I IFN responses.
Figure 3.
Antiviral sensing pathways and Vaccinia virus antagonists. VACV encodes a wide array of proteins that antagonize host antiviral sensing mechanisms, including cytosolic DNA sensing, RNA sensing, and Toll-Like Receptors (TLRs) signaling pathways. In the cytosolic DNA sensing pathway, the Stimulator of Interferon Genes (STING) axis is inhibited by VACV proteins F17 and E5, which promote degradation of the DNA sensor Cyclic GMP-AMP Synthase (cGAS). Additionally, B2 degrades Cyclic GMP-AMP (cGAMP), thereby preventing the transmission of antiviral signals to neighboring cells. The DNA sensor DNA-dependent protein kinase (DNA-PK) is also targeted by C4 and C16, further impairing DNA-triggered immune responses. In the RNA sensing pathway, VACV proteins D9, D10, E3, and K3 inhibit activation of Protein Kinase R (PKR). E3 also binds and sequesters viral RNA, shielding it from recognition by Retinoic Acid-Inducible Gene I/ Melanoma Differentiation-Associated Protein 5 (RIG-I/MDA5). TLR-mediated signaling is disrupted by multiple viral proteins: A46 blocks interactions between TLRs and adaptor proteins, preventing IRF3 activation; A52 inhibits NF-κB activation; and N1 impairs both IRF3 and NF-κB pathways. Key downstream signaling hubs are also targeted. The serine/threonine kinase TBK1, essential for IRF3 activation downstream of STING, RNA sensors, and TLRs, is inhibited by C6 and K7. In the nucleus, N2 functions as a direct IRF3 inhibitor, preventing transcription of interferon-stimulated genes.
Figure 3.
Antiviral sensing pathways and Vaccinia virus antagonists. VACV encodes a wide array of proteins that antagonize host antiviral sensing mechanisms, including cytosolic DNA sensing, RNA sensing, and Toll-Like Receptors (TLRs) signaling pathways. In the cytosolic DNA sensing pathway, the Stimulator of Interferon Genes (STING) axis is inhibited by VACV proteins F17 and E5, which promote degradation of the DNA sensor Cyclic GMP-AMP Synthase (cGAS). Additionally, B2 degrades Cyclic GMP-AMP (cGAMP), thereby preventing the transmission of antiviral signals to neighboring cells. The DNA sensor DNA-dependent protein kinase (DNA-PK) is also targeted by C4 and C16, further impairing DNA-triggered immune responses. In the RNA sensing pathway, VACV proteins D9, D10, E3, and K3 inhibit activation of Protein Kinase R (PKR). E3 also binds and sequesters viral RNA, shielding it from recognition by Retinoic Acid-Inducible Gene I/ Melanoma Differentiation-Associated Protein 5 (RIG-I/MDA5). TLR-mediated signaling is disrupted by multiple viral proteins: A46 blocks interactions between TLRs and adaptor proteins, preventing IRF3 activation; A52 inhibits NF-κB activation; and N1 impairs both IRF3 and NF-κB pathways. Key downstream signaling hubs are also targeted. The serine/threonine kinase TBK1, essential for IRF3 activation downstream of STING, RNA sensors, and TLRs, is inhibited by C6 and K7. In the nucleus, N2 functions as a direct IRF3 inhibitor, preventing transcription of interferon-stimulated genes.
Importantly, cGAMP’s activity is not confined to the infected cell. It can diffuse into neighboring cells through gap junctions and membrane fusion, or be actively imported from the extracellular milieu [45,46]. As a result, cGAMP produced in even a single cell can initiate a broader antiviral response in surrounding uninfected tissue, thereby limiting viral spread. To overcome this barrier, VACV has evolved multiple proteins that inhibit the cGAS-STING pathway at various levels.
One such protein is F17, which suppresses cGAS activity by hyperactivating the mammalian target of rapamycin (mTOR) pathway. cGAS is known to be inhibited by Akt kinase, which phosphorylates specific serine residues, thereby reducing its activity [47]. F17 promotes this effect by sequestering two key regulatory subunits of the mTOR complexes Raptor and Rictor leading to excessive mTOR activation. This, in turn, stimulates Akt signaling and enhances cGAS degradation [48]. In addition, hyperactive mTOR promotes increased protein synthesis, supporting efficient viral protein production and virion assembly [48].
Another key VACV protein, E5 (encoded by the E5R gene), binds directly to cGAS, inducing its polyubiquitination and subsequent proteasomal degradation. In the presence of E5, intracellular cGAS levels are reduced, resulting in strong inhibition of type I IFN induction [49]. E5 is supported by B2—also known as poxin and coded by the B2R gene—which enzymatically cleaves cGAMP into an inactive linear form. Although B2 has little effect on VACV replication in vitro, its absence significantly reduces viral titers in mouse models, highlighting its importance for in vivo viral spread [50].
VACV also interferes with signaling downstream of STING by targeting the TBK1 complex. The viral protein C6 binds with the cellular TBK1 adaptor proteins TRAF family member-associated NF-kappa-B activator (TANK), similar to NAP1 TBK1 adaptor (SINTBAD), and NAK-associated protein 1 (NAP1), thereby preventing effective IRF3 phosphorylation [51]. Another protein, K7, blocks TBK1 activity by interacting with DEAD-box RNA helicase DDX3, an essential adaptor and substrate for optimal IRF3-mediated responses [52]. Finally, N2—a member of the VACV Bcl-2 family—acts in the nucleus where it inhibits IRF3’s ability to activate gene expression under IFN-β promoter [53]. While deletion of any of these viral genes does not impair VACV replication in in vitro, all significantly affect virulence in mouse models [51,52,53].
3.3. Double Stranded RNA Sensors
Although VACV is a dsDNA virus, it generates significant amounts of dsRNA during its replication cycle due to the synthesis of overlapping transcripts that often anneal into duplexes [54]. As a result, VACV becomes a target to several cellular RNA sensors.
One such sensor is protein kinase R (PKR), which recognizes dsRNA and phosphorylates the eukaryotic translation initiation factor 2 (eIF2). This phosphorylation inhibits cap-dependent translation, ultimately leading to a shutdown of protein synthesis and induction of cell death [55]. To evade PKR-mediated antiviral responses, VACV expresses several proteins— encoded by E3L, K3L, D9R, and D10R—that directly or indirectly inhibit PKR function. The E3 protein binds directly to viral dsRNA, shielding it from PKR recognition and preventing its dimerization and activation [56]. The K3 protein mimics cellular eIF2 and acts as a decoy substrate for PKR, diverting phosphorylation away from the host’s eIF2 [57]. In addition, D9 and D10 function as decapping enzymes for dsRNA, reducing the stability and concentration of viral dsRNA, thereby lowering its visibility to PKR [58].
VACV also inhibits another key dsRNA-sensing pathway involving oligoadenylate synthases (OAS1, OAS2 and OAS3). Upon dsRNA detection, these enzymes produce 2’-5’-oligoadenylates (2-5As), which activate RNAase L, leading to the degradation of cellular and viral mRNAs, shutdown of protein synthesis, and induction of apoptosis. The E3 protein sequesters dsRNA, preventing its recognition by OAS proteins and thereby blocking RNAase L activation [59]. Similarly, D9 and D10 proteins reduce dsRNA levels further limiting activation of the OAS/ RNAase L pathway [58].
Two additional cytosolic sensors critical for type-I IFN induction are RIG-I and melanoma differentiation-associated protein 5 (MDA-5) proteins. They both have helicase activity and can detect dsRNA in the cytosol of infected cells. RIG-I is more specific for shorter RNAs with triphosphate groups while MDA-5 shows higher affinity for longer dsRNA [60]. Upon activation, both sensors interact with the mitochondrial adapter protein Mitochondrial Antiviral Signaling (MAVS), which activates TBK1, leading to IRF3 phosphorylation and induction of type I IFN genes. MAVS can also recruit TNF receptor (TNFR)–associated factor 6 (TRAF6), triggering activation of NF-kB pathway [60].
VACV’s E3 protein has a potent inhibitory effect on the RIG-I/ MDA5 activation. Studies have shown that expression of the E3 RNA-binding domain alone is sufficient to completely block cytokine induction downstream of RIG-I/MDA5 activation [61]. E3 masks viral dsRNA from recognition by these sensors and also inhibits RNA polymerase III, which generates 5’-triphosphate-containing dsRNA species that activate RIG-I [62]. Through these combined actions, VACV effectively suppresses both RNA- and DNA-dependent IFN activation pathways.
3.4. Toll-Like Receptor System
Toll-like receptors (TLRs) are a family of 13 transmembrane PRRs localized on the plasma membrane and within intracellular compartments such as endosomes, ER and, lysosomes. TLRs are characterized by an N-terminal extracellular leucine-rich repeat-containing ectodomain, a single transmembrane helix, and a C-terminal cytoplasmic Toll/interleukin-1 receptor (TIR) domain. These receptors specialize in detecting pathogen-associated molecular pattern (PAMPs), such as viral nucleic acids, lipopolysaccharide (LPS) or flagellin [63].
Upon recognition of PAMPs, TLRs can signal through either the Myeloid differentiation primary response 88 (MyD88) adapter protein—activating NF-κB (e.g., TLRs 2, 6, 7, 8, 9)—or through the TIR-domain-containing adaptor-inducing interferon-β (TRIF), used by TLR3 and TLR4, leading to IRF3 activation and the expression of type-I IFN genes [64]. Studies have shown that VACV can interfere with signaling via both endosomal (TLRs 3, 8, and 9) and plasma membrane-bound (TLRs 2 and 4) receptors.
TLR3, an endosomal sensor specific for viral dsRNA, is capable of inducing both IRF3 and NF-kB activation. VACV proteins A52 and A46 are known to effectively interfere with different branches of TLR3 signaling. A52 blocks TLR3-mediated NF-kB activation induced by polyinosinic:polycytidylic acid (poly(I:C)), a synthetic dsRNA analog, by interacting with downstream adapter proteins such as IL-1 receptor-associated kinase2 (IRAK2) and tumor necrosis factor receptor-associated factor 6 (TRAF6) [65]. A46, on the other hand, inhibits IRF3 activation by binding to the adapter TIR domain containing adaptor molecule 1(TICAM1), thereby preventing it from transmitting signals from TLR3 [66]. Together, A52 and A46 effectively shut down both arms of the TLR3 signaling pathway.
Interestingly, in vivo studies have shown that TLR3-deficient mice display reduced morbidity following VACV infection and exhibit lower viral replication in the respiratory tract. This suggests that VACV may exploit functional TLR3 to its advantage.
Other endosomal TLRs, such as TLR8 and TLR9, recognize viral nucleic acids—single- stranded (ss)RNA and DNA, respectively. The VACV E3 protein has been shown to inhibit cytokines expression induced by TLR8 [67] and TLR9 ligands [68]. Notably, the DNA-binding domain of E3 alone is sufficient to block TLR9 signaling [68]. Since E3 also contains RNA-binding domain that inhibits dsRNA-sensing receptors, it likely serves as a broad-spectrum shield that protects both VACV RNA and DNA from antiviral detection mechanisms.
VACV can also interfere with signaling from cell membrane-bound TLRs, including TLR2 and TLR4, although it remains unclear whether the virus can be directly recognized by these receptors [58].
3.5. NF-kB, IFNs and Other Cytokines
In addition to inhibiting foreign nuclear acid-recognition systems, VACV can also downregulate NF-kB signaling, a critical pathway for inducing many pro-inflammatory cytokines essential for combating viral infections. The N1 protein targets the IκB kinase (IKK) complex, inhibits NF-kB signaling via the TNF superfamily of receptors, and blocks NF-kB activation through toll-like receptors [69]. The F14 protein binds to the transcriptional co-activator CREB-binding protein (CBP) and disrupts its ability to acetylate the p65 subunit of NF-kB, thereby impairing NF-kB’s capacity to recruit the transcription regulator bromodomain-containing protein 4 (BRD4). This leads to reduced expression of chemokine genes such as CCL2 and CXCL10 [70].
Another protein, B14, binds to the dimerization and kinase domain of IKKβ, preventing it from phosphorylating IκBα [40]. Additionally, the A49 protein mimics IκBα and is phosphorylated by IKKβ, but it then blocks the E3 ubiquitin ligase responsible for IκBα degradation [71]. Both mechanisms result in the accumulation of unphosphorylated IκBα, which sequesters NF-κB in the cytoplasm and prevents its activation and nuclear translocation. Furthermore, two other VACV proteins, K1 and A55, also contribute to NF-κB suppression, either by interfering with its activation or by blocking the translocation of the NF-κB heterodimer [38].
Another strategy employed by VACV to subvert host immune responses is the use of soluble cytokine decoys that inhibit host cytokine signaling [72]. The B18 protein, coded by the B18R gene, acts as a broad-spectrum decoy receptor for type-I IFNs (IFN-α and IFN-β). Secreted from infected cells, B18 binds IFNs from a wide range of species, preventing their interaction with cell surface receptors. B18 can also attach to the external membranes of both infected and uninfected neighboring cells via glycosaminoglycans, creating a barrier that intercepts interferons before they can engage their receptor [73].
Even if IFN receptors are activated, VACV can still block signaling through the viral phosphatase H1, which dephosphorylates the STAT1 transcription factor, thereby preventing its nuclear translocation and inhibiting the expression of IFN-stimulated genes [74].
In addition, VACV produces a pair of secreted proteins known as cytokine response modifiers (CrmC and CrmE) that target soluble TNF-α. These proteins possess N-terminal domain homologous to TNFRs, enabling them to sequester TNF-α from extracellular milieu. While CrmC functions primarily as a free soluble molecule, CrmE can also associate with the cell membrane, blocking TNF-α from binding its receptor in a manner similar to B18’s inhibition of IFNs [75].
Other VACV-encoded cytokine decoy proteins include B8, which neutralizes IFN-γ [76]; viral IL-18 binding protein (vIL-18BP), which blocks IL-18 [77], and B15, which captures IL-1β [78]. Collectively, these immune evasion strategies allow VACV to evade early detection by the innate immune system and delay the onset of the adaptive response, that finally is responsible for clearing the infection.
3.6. Complement Evasion
The complement system is a network of cell associated and soluble proteins that react with the surface of pathogens, leading to their lysis and inactivation [79]. Complement activation can be initiated through three pathways: the classical pathway, triggered by immunoglobulin-bound pathogens; the lectin pathway, activated by mannose-binding lectin (MBL); and the alternative pathway, which involves direct recognition by complement components. This activation is a multi-step cascade process that culminates in the formation of membrane attack complex (MAC), a structure composed of enzymes that effectively lyse pathogens such as viruses, bacteria, and parasites [79].
To evade complement-mediated destruction, VACV has evolved a protein called VCP (VACV complement control protein), encoded by the gene C21L. VCP interferes with key complement components C3b and C4b, which are crucial for the function of C3 convertase, the central complex responsible for amplifying the complement signal—and the formation of C5 convertase, which drives MAC assembly. Additionally, C3b acts as an opsonin, coating viral particles to target them for phagocytosis.
Since all three complement pathways converge at the stages of C3 and C5 convertases, VACV can efficiently disrupt the entire system with a single protein. VCP also serves as a cofactor for the serine protease factor I, promoting the cleavage and inactivation of C3b and C4b and accelerating the decay of the C3 convertase complex [80]. By neutralizing complement activity, VACV effectively modulates both innate and adaptive immune responses.
3.7. Antigen Processing and Presentation
The adaptive immune response, driven by the activity of T and B lymphocytes, depends on the precise recognition of specific antigens associated with pathogens. To evade this response, VACV has evolved mechanisms to interfere with the processing and presentation of its antigens to the immune system.
Studies have shown that VACV infection impairs the ability of antigen-presenting cells (APCs) to display peptide antigens in the context of Major Histocompatibility Complex class II (MHCII) molecules. This effect is observed in infections with both replication-competent and replication-defective forms of the virus. Interestingly, the expression and cell surface levels of MHCII molecules remained unchanged, suggesting that VACV disrupts antigen loading rather than MHCII expression [81].
Further research identified the VACV protein A35, encoded by the A35R gene, as a key factor in this immune evasion. A35 localizes to endosomes and reduces the amount of peptide presented on MHC II molecules [82]. Its expression inhibits the activation of CD4+ T lymphocytes by APCs and diminishes their production of nitric oxide (NO) and cytokines in response to antigen stimulation. Although A35 is not essential for viral replication in vitro, its absence significantly attenuates viral virulence in vivo, underscoring the critical role of CD4+ T cell–mediated responses in viral clearance [82].
4. Strategies to Avoid Cell Death Mechanisms
Programmed cell death is a fundamental defense mechanism that helps prevent viral spread within the host. If an infected cell undergoes death before the virus can complete replication, it fails to produce progeny virions and is eliminated through phagocytosis by immune cells such as dendritic cells and macrophages [83,84]. Consequently, VACV evolved multiple tools to control mechanisms of programmed cell death and utilize them to its advantage (Figure 4).
Apoptosis is the most common form of programmed cell death. It can be triggered by internal or external signals and relies on caspases—a family of cysteine proteases that cleave target proteins at aspartic acid residues. Two major apoptotic pathways exist: extrinsic and intrinsic.
The extrinsic pathway is initiated by the activation of specific cell surface receptors, such as the TNFR and the Fas-ligand (FASL) receptor. Activated receptors induce the formation of a protein complex composed of death receptor adaptor proteins—TNFR-associated death domain (TRADD) and/or Fas-associated death domain (FADD)—which in turn activate the intracellular protease caspase-8. Activated caspase-8 then triggers downstream caspases 3 and 7, which cleave several cellular substrates, such as nuclear lamins and gelsolin, leading to DNA fragmentation and cell death [83].
The intrinsic pathway is activated by cellular stress such as DNA damage, oxidative stress, or nutrient deprivation, resulting in the permeabilization of the outer mitochondrial membrane. This process is mediated by Bax and Bak proteins, which form pores in the membrane, allowing cytochrome c to escape into the cytosol. Cytochrome c binds to apoptotic protease activating factor-1 (APAF-1), forming a complex that activates caspase-9. Caspase-9, in turn, activates caspases 3 and 7, culminating in apoptosis [83].
Both apoptotic pathways can be triggered during viral infection, and VACV has evolved mechanisms to counteract them. The VACV protein B13 binds and inhibits both caspase-8 and caspase-9, effectively blocking the extrinsic and intrinsic pathways simultaneously [85]. Another viral protein, F1, inhibits both the precursor and active forms of caspase-9 and
prevents the activation of caspases 3 and 7. F1 also interferes with Bak’s ability to bind Bax and form pores in the mitochondrial membrane. In addition, F1 blocks the pro-apoptotic protein Bim, a key antagonist of the anti-apoptotic factor Bcl2. These actions are reinforced by the N1 protein, which neutralizes other Bcl-2 antagonists, Bid and Bad [86]. As a result, the mitochondrial pathway of apoptosis is effectively dismantled in VACV-infected cells. Other VACV proteins, such as B22, Golgi anti-apoptotic protein (GAAP), and E-3, have also been implicated in blocking various stages of apoptosis [85].
In addition to apoptosis, VACV can inhibit pyroptosis, another form of programmed cell death that is dependent on the activation of caspase-1 via the inflammasome—a multiprotein complex. Pyroptosis is a highly proinflammatory process, typically triggered when intracellular PRRs such as NOD-like receptors (NLRs), especially NLRP1 (nucleotide-binding oligomerization domain, Leucine rich Repeat and Pyrin domain containing 1), detect PAMPs. This recognition initiates the assembly of the adaptor protein ASC (Apoptosis-associated speck-like protein containing a CARD), which activates caspase-1. Activated caspase-1 processes pro-interleukin-1β (pro-IL-1β) into its active form and also activates gasdermin D (GSDMD), which forms pores in the cellular membrane, releasing pro-inflammatory contents [87].
Although commonly associated with bacterial infections, pyroptosis also plays a role in VACV infection [88]. VACV’s F1 protein binds the NLRP1 subunit, preventing inflammasome assembly and caspase-1 activation [89]. Additionally, B13 can directly inhibit caspase-1 activity [85], while B15 acts as a soluble decoy receptor for IL-1β, neutralizing pyroptosis-induced inflammatory signals and suppressing immune cell activation [78].
Interestingly, when apoptosis is inhibited in VACV-infected cells, necroptosis, another form of programmed cell death, may be triggered. Necroptosis occurs when caspase-8 is inhibited and cannot cleave two of its downstream targets, Receptor-interacting serine/threonine kinase 1 (RIP1) and its relative RIP3. In this context, RIP1 and RIP3, along with inactive caspase-8, form the ripoptosome complex, which activates Mixed Lineage Kinase Domain-like pseudokinase (MLKL), a protein that oligomerizes and forms pores in the cell membrane, leading to cell lysis and the release of intracellular contents [90]. Because B13 strongly inhibits caspase-8, it can induce inadvertently promote ripoptosome formation and necroptosis [91].
This strategy offers dual benefits to VACV: by delaying apoptosis, VACV gains time to complete replication and produce progeny virions; and by inducing necroptosis, it facilitates viral release and dissemination to neighboring cells.
5. Interactions with Immune Cells
5.1. Dendritic Cells
As a pathogen, VACV interacts with various immune cell types, either directly by infecting them or indirectly altering immune system function. Studies have shown that VACV exhibits a preference for infecting myeloid cells such as monocytes and dendritic cells (DCs), rather than B or T lymphocytes, although both lymphocyte types can still be infected [92].
Dendritic cells play a central role in the immune response by presenting viral antigens and initiating T cell mediated immunity. In one study, VACV infection of immature DCs derived from peripheral blood mononuclear cells (PBMCs) impaired their maturation, as evidenced by reduced expression of key maturation markers including CD83, CD86, Human Leukocyte Antigen – DR isotype, (HLA-DR), and CD25 [93]. These infected DCs also showed a diminished capacity to stimulate T cell proliferation and exhibited increased levels of apoptosis. Notably, immature DCs were found to be significantly more susceptible to VACV infection than their mature counterparts [93].
Similar findings were observed in murine models: bone marrow-derived DCs failed to mature following VACV infection, as indicated by reduced expression of CD40, CD80, and CD86, diminished production of pro-inflammatory cytokines, and impaired activation of CD8+ T lymphocytes [94]. In contrast, mature DCs displayed greater resistance to VACV infection, with a lower proportion of infected cells and preserved antigen presentation and CD8+ T cell activation capabilities [94].
Interestingly, studies in mice infected with VACV reported that viral infection may actually promote DC maturation. Ex vivo analysis of splenic dendritic cells from VAVCV-infected mice showed increased expression of MHC I and co-stimulatory molecules CD40 and CD86 on their cell surface [95]. These DCs also exhibited elevated levels of IFN-β and demonstrated enhanced capacity to produce IL-10 and IL-12 upon LPS stimulation [95]. While these findings appear to contradict earlier results, they may be reconciled by the observation that fewer than 1% of splenic DCs were directly infected by VACV. This suggests that the increased maturation may have occurred in uninfected DCs in response to cytokine signaling, particularly type I IFNs.
Conversely, the same study found that VACV infection reduced expression of MHCII on DCs and impaired their ability to present antigens to CD4+ T lymphocytes [95], a disruption potentially attributable to the viral A35 protein. Collectively, these findings suggest that DCs respond to VACV infection by preferentially activating CD8+ cell responses over CD4+ T cell responses, which aligns with the immune system’s typical strategy for combating viral infections.
5.2. Macrophages
Macrophages are another type of immune cell that can be infected by VACV. Studies have demonstrated that VACV is capable of infecting and replicating in both human M1 and M2 macrophages in vitro, with the M2 subset producing more than twice the viral titer per cell compared to M1 macrophages [96]. Infected macrophages exhibited cytoplasmic viral factories, while levels of apoptosis and necrosis remained low even 48 hours post-infection. Notably, activation of macrophages with LPS and IFN-γ did not affect viral replication, whereas stimulation with IL-10 or a combination of LPS and IL-1β significantly inhibited viral production.
Interestingly, the majority of virions generated in macrophages were EEVs, which are primarily involved in long-range viral dissemination. Given the migratory capacity of macrophages between tissues, these findings suggest that macrophages may play a critical role in the systemic spread of the virus within the host [96]. In contrast, monocytes examined in the same study supported only minimal VACV replication and exhibited high levels of apoptosis, indicating a less permissive environment for viral propagation [96].
5.3. Neutrophils and NK Cells
While the role of neutrophils in VACV infection has not been extensively characterized, studies suggest they contribute significantly to early virus clearance and the initiation of adaptive T-cell responses. Experiments using oncolytic VACV demonstrated that neutrophils internalize more virus than any other cell subset accounting for over 80% of viral uptake and subsequently degrade the virus within phagocytic vesicles [97]. Interestingly, depletion of neutrophils using anti-Ly6G antibodies significantly enhanced the efficacy of oncolytic VACV in treating B16F10 tumors [97]. Additionally, neutrophils have been shown to transport VACV antigens to the bone marrow, where they deliver them to antigen-presenting cells, thereby promoting the development of CD8+ T cell responses [98].
Natural killer (NK) cells also play an important role in the immune response to VACV. Although VACV can directly infect NK cells, the functional consequences of this infection remain poorly understood [97]. Nonetheless, studies have shown that NK cells are recruited to sites of VACV infection, where they become activated, proliferate and lyse infected cells [99,100,101]. Notably, VACV infection does not reduce MHC I expression or the expression of ligands for the NK Group 2 Member D (NKG2D) receptor on infected cells. Instead, it induces the expression of ligands for other activating NK cell receptors, including NKp46, NKp44, and NKp30 [101]. In a mouse model of pulmonary VACV infection, NK cells were found to produce high levels of IFN-γ prior to the infiltration of CD8+ T cells, helping to limit viral replication during the early stages of infection [99]. Furthermore, NK cells were observed to form a memory-like Thy+ subset capable of protecting naïve immunodeficient mice from lethal VACV challenge [102].
5.4. B and T Lymphocytes
VACV also interacts with components of the adaptive immune system. B cells, which produce virus-neutralizing antibodies, can also function as APCs to stimulate T cell responses. Although VACV can bind to multiple B cell subpopulations, it primarily infects and replicates in memory B cells [103]. However, successful viral replication requires B cell activation; in the absence of stimulation, infection is abortive and late gene expression is not induced [103].
A murine model of respiratory VACV infection revealed that virus-specific antibodies begin to appear around day 15 post-infection, peak at day 148, and play only a minor role in controlling primary infection [104]. In contrast, serum from mice with a fully developed B cell response was highly effective in mitigating the effects of subsequent VACV exposure [104]. Although the B cell response to VACV develops slowly, it results in durable immune memory. In humans, memory B cells and protective antibodies specific to VACV-based smallpox vaccine have been detected up to 50 and 59 years post-vaccination, respectively [105]. Similarly, in mice, VACV induces a robust B cell memory response, though its protective role appears secondary to that of memory T cells [106].
T cells, while only weakly susceptible to VACV infection [107], play a central role in viral clearance. Experimental data show that CD8+ T lymphocytes are both necessary and sufficient to protect mice against VACV respiratory infection [104]. Depletion of CD8+ cells renders immunocompetent mice susceptible to VACV-induced mortality, whereas adoptive transfer of naïve CD8+ T cells rescues immunodeficient RAG-/- mice from lethal infection [104]. Although CD4+ T cells are essential for the development of a functional antibody response, their depletion does not impair survival or virus clearance in the respiratory model [104]. Interestingly, in an intraperitoneal model of VACV infection, CD4+ T cells played a more substantial role by supporting proper CD8+ T cell priming [108].
Additional evidence points to a role for γδ T cells in CD8+ T cell activation. These cells can present VACV antigens via MHC I and secrete cytokines such as IL-1 and IFN-α, thereby contributing to antiviral defense [109].
Like B cells, T cells also generate robust and long-lasting memory. In humans, VACV-specific CD8+ memory T cells have been detected up to 50 years post-vaccination, maintaining cytotoxic activity against infected cells [110]. Remarkably, CD4+ memory T cells isolated form these individuals also exhibited cytotoxic, rather than regulatory, properties [110].
6. VACV as a Tool in Cancer Therapy
6.1. Advantages of VACV as an Anti-Cancer Agent
VACV possesses several characteristics that make it an exceptionally valuable tool in cancer therapy. First, it can infect a wide range of cell types. While many viruses are restricted to infecting cells that express a specific receptor, VACV can infect tumors originating from various tissues [1,111,112,113]. Moreover, VACV binds to MARCO, a protein commonly found on tumor-associated macrophages and myeloid derived suppressor cells (MDSCs). This property makes it a promising candidate for modifying the tumor microenvironment, for example, by delivering genes that reverse the immunosuppressive phenotype of myeloid cells.
Second, the large genome of VACV allows for the insertion of sizable therapeutic transgene constructs of at least 25 kilobases [114,115]. Because the virus replicates entirely in the cytosol, there is no risk of transgene integration into the hosts genome, thereby minimizing the chance of transformation if normal cells are infected.
Third, VACV has a rapid replication cycle. It produces new virions within 6-8 hours and causes lysis of infected cells within 48 hours, enabling it to quickly infect and destroy tumor cells before the immune system can neutralize the virus. Notably, VACV replicates at comparable rates under both normoxic and hypoxic conditions, making it particularly effective against tumors in low oxygen environments such as pancreatic cancer [116].
Additionally, VACV suppresses proteins involved in apoptosis, allowing it to replicate efficiently in cancer cells with defective apoptosis pathways. While resistance to apoptosis undermines many conventional therapies, it actually enhances VACV replication by prolonging host cell survival. Instead of apoptosis, VACV induces necroptosis and cell lysis, resulting in the release of damage-associated molecular pattern molecules (DAMPs) such as high mobility group box 1 protein (HMGB1), adenosine triphosphate (ATP), calreticulin (CRT), and heat shock protein 90 (HSP90) [117]. The release of these DAMPS characterizes immunogenic cell death (ICD), which promotes leukocyte recruitment, dendritic cell maturation, and priming CD8+ T lymphocytes against tumor antigens [118].
Consequently, VACV is especially promising for treating “cold” tumors—those with poor immune cell infiltration—by converting them into “hot” tumors actively infiltrated by NK and T cells. Finally, VACV induces a predominantly CD8+ cytotoxic T cell response, which is particularly beneficial in cancer therapy, as CD4+ T cells and B cells can sometimes exert immunosuppressive effects.
6.2. Chimeric VACV: CF33
To further enhance the oncolytic properties of VACV, researchers have employed several strategies that are summarized in Figure 5. One such approach is chimerization, which involves combining genetic elements from different, closely related poxviruses to generate chimeric viruses with optimal oncolytic potential. In a study conducted at City of Hope, researchers co-infected cells with cowpox, raccoonpox, rabbitpox, and six different strains of VACV to generate a diverse pool of chimeric orthopoxviruses. From this pool, 100 chimeric viruses were isolated and screened using high-throughput methods for their efficacy against the NCI-60 panel of human cancer cell lines [119,120].
Among these candidates, the chimeric virus CF33 demonstrated superior oncolytic activity compared to its parental strains, including robust cytotoxicity against six different human pancreatic cancer cell lines. It also exhibited a strong ability to induce immunogenic cell death and significantly inhibited the growth of pancreatic and triple-negative breast cancer xenografts in mouse models [119,120]. Sequence analysis of the CF33 genome revealed that it is primarily derived from three VACV strains—IHD, Lister, and WR—which together account for approximately 60% of its genome. No genetic material from raccoonpox or cowpox viruses was detected, indicating that CF33 is in fact a chimeric VACV, rather than a broader orthopoxvirus hybrid [121].
Building on CF33, several derivative viruses have been developed. One such derivative, CF33-hNIS (also known as VAXINIA), incorporates the human sodium iodide symporter (hNIS) gene, enabling non-invasive imaging of viral distribution via using PET scans. This modification allows researchers to track the virus's localization and replication within the body [122].
Another derivative, CF33-hNIS-antiPDL1, expresses both hNIS and a single-chain variable fragment targeting PD-L1, an immune checkpoint protein. Preclinical studies in models of triple-negative breast cancer and gastric cancer peritoneal metastases have shown that this dual-function virus not only enhances direct tumor cell killing but also reprograms the tumor microenvironment to improve immune recognition and activation[123,124].
A third variant, CF33-CD19, is engineered to express a truncated CD19 on the surface of infected cancer cells. This strategy enables targeting of solid tumors with CD19-specific CAR-T cells, which typically show limited efficacy against solid tumors due to the lack of appropriate surface antigens [125].
All three CF33-based viruses are currently under clinical evaluation:
CF33-hNIS is being tested as a monotherapy or in combination with pembrolizumab in adults with metastatic or advanced solid tumors (NCT05346484),
CF33-hNIS-antiPDL1 is in clinical trials for the treatment of metastatic triple-negative breast cancer (NCT05081492),
CF33-CD19 is being evaluated as a monotherapy or in combination with blinatumomab for metastatic solid tumors (NCT06063317).
6.3. Genetic Engineering of VACV
Creating novel oncolytic variants of VACV typically involves precise insertions and deletions within the viral genome. However, due to its large genome size (~195kb), VACV is not amenable to standard molecular cloning techniques commonly used for smaller viral vectors, such as lentiviral systems. Consequently, genetic engineering approaches are required.
The most traditional and widely used method is based on homologous recombination. In this approach, a plasmid carrying the desired genetic construct is introduced into VACV-infected cells. Recombination occurs between homologous sequences on the plasmid and the viral genome, enabling the exchange of genetic material [126]. A typical construct includes a desired transgene (e.g., a cytokine) accompanied by a selection marker (e.g., GFP or β-galactosidase) each driven by separate promoters. The expression cassette is flanked by sequences homologous to regions of the viral genome—commonly the termini of the TK gene—to allow for targeted insertion. Recombinant viruses are isolated using marker-based selection or screening, followed by multiple rounds of plaque purification.
While conceptually straightforward and technically accessible, this method is labor-intensive and inefficient: recombination events occur at low frequency, yielding approximately one recombinant per 1,000 wild-type virions [127].
To enhance efficiency, several alternative strategies have been developed, including the use of bacterial artificial chromosomes (BACs) [128] and genome editing techniques based on CRISPR-Cas9 [129]. A newest method, known as MAVERICC (marker-free vaccinia virus engineering of recombinants through in vitro CRISPR/Cas9 cleavage) integrates the strengths of previous approaches [127].
In the MAVERICC method, the VACV genome is first cleaved in vitro at specific sites using Cas9 guided by sequence-specific RNAs. This cleaved genome is then co-transfected with an amplicon containing the desired transgene flanked by sequences homologous to the targeted genomic region. Recombination occurs within the host cell, which is also infected with a replication-defective helper poxvirus. The helper virus supplies the necessary proteins to facilitate homologous recombination but cannot replicate itself. As a result, only recombinant VACV that have repaired the Cas9-induced cleavage through homologous recombination with the amplicon can be propagated. This method enables the generation of engineered viruses with >90% efficiency—without the need for selection markers, serial passaging, or extensive screening [127].
6.4. Commonly Targeted Viral Genes for Modification in VACV-Based Oncolytic Viruses
Over the years, numerous strategies have been developed to enhance the suitability of VACV for cancer therapy [130]. Cancer cells possess several characteristics that facilitate enhanced replication of VACV, ultimately leading to cell lysis. These include deficient apoptotic pathways [131] , reduced expression of tumor suppressor genes (such p53), and impaired antiviral and interferon signaling mechanisms [132]. In addition, due to their high proliferative rates and intense DNA synthesis demands, cancer cells typically exhibit elevated levels of cytosolic TK and RR [133,134].
To exploit this metabolic environment, researchers have engineered VACV strains with deletions in the viral genes encoding TK and/or RR. These modified viruses rely on host cell-derived enzymes to synthesize DNA precursors, thereby restricting viral replication to cancer cells where these enzymes are abundant [135,136]. Interestingly, endothelial cells in tumor-associated vasculature also exhibit elevated TK expression, largely due to stimulation by vascular endothelial growth factor (VEGF). This makes VACV a potential anti-angiogenic agent. For example, the oncolytic strain JX-594—engineered to express human granulocyte-monocyte colony stimulating factor (hGM-CSF) and β-galactosidase (β-gal )—has been shown to selectively replicate within tumor-associated blood vessels, causing their collapse while sparing normal vasculature [137].
Another modification involves deletion of the C11R, which encodes the viral growth factor (VGF). VGF stimulates the EGFR-Ras signaling pathway to promote cell proliferation. In the absence of VGF, VACV replication is impaired unless the host cell has an active EGFR pathway—a common feature of many cancer cells [138].
Additional deletions used to enhance oncolytic specificity and safety include A56R and F14.5L. The A56R gene encodes a hemagglutinin-like protein (A56) that prevents infected cells from forming syncytia, protects against superinfection, and inhibits complement-mediated lysis [139]. Despite these roles, A56 is not essential for viral replication and is often replaced with therapeutic transgenes. The F14.5L gene is involved in regulating cell adhesion and contributes to viral virulence in vivo, though it is not required for replication [140]. Its deletion is commonly employed as a safety measure, especially important for treating potentially immunocompromised cancer patients [141]. Other deletions introduced to increase safety and tumor specificity include genes such as: SPI-1 (B22) and -2 (B13), B18R, N1L, A41L, A49L and F1L. A list of these deletions and their associated viruses is provided in Table 1.
6.5. Expression of Chemokines and Cytokines
Many VACV-based oncolytic viruses are engineered to express immunostimulatory genes, primarily chemokines and cytokines, to enhance anti-tumor immunity. These modifications generally pursue two main objectives: (1) to recruit antigen presenting cells, particularly DCs, promote their maturation, and improve their capacity to present antigens to T cells; (2) to modulate existing T cell responses by promoting T cell survival, sustaining activation, and preventing exhaustion.
One of the most commonly used transgene is GM-CSF, which is known to promote tumor infiltration by NK and DCs, and to accelerates DC maturation [142]. GM-CSF is expressed by the only FDA-approved oncolytic virus in the U.S., the herpesvirus-based T-VEC. VACV vectors engineered to express GM-CSF have shown promising results in preclinical studies [137,143,144].
An alternative approach strategy involves incorporating genes encoding chemokines such as CXCL11 and CCL5 (RANTES). A VACV strain expressing CCL5 induced significant infiltration of DCs, CD4+ T cells, and NK cells, effectively suppressing tumor growth in the MC38 murine model [145]. Similarly, another VACV expressing CXCL11 was highly effective in the same model, attracting large numbers of CD8+ lymphocytes and inducing strong IFN-γ expression [146].

Table 1.
Examples of viral genes deleted from Vaccinia virus to enhance tumor selectivity and promote immune system activation.
Table 1.
Examples of viral genes deleted from Vaccinia virus to enhance tumor selectivity and promote immune system activation.
| Deleted viral genes | Functions of deleted viral genes | Example of virus | Reference |
|---|---|---|---|
| TK (Thymidine kinase) | Provides material for viral DNA replication | vCB2 (vvLuc) | [136] |
| TK and VGF (Viral growth factor) | Supports viral replication, accelerates cell growth and metabolism | vvDD-GFP | [147] |
| SPI-1 (B22) and -2 (B13) | Inhibit apoptosis | vSP | [148] |
| TK, SPI-1 and -2 | Supports viral replication, inhibit apoptosis | vSPT | [149] |
| Soluble type I IFN receptor (B18R) | Inhibits mobilization of the immune cells | WR-delB18 | [150] |
| Soluble type I IFN receptor and TK | Inhibits mobilization of the immune cell, supports viral replication | ΔB18RΔTK | [150] |
| F14.5L, TK and HA | Promotes virulence in vivo, supports viral replication, inhibits complement-mediated lysis of infected cell | GLV-1h68 | [151] |
| TK, RR (ribonucleotide reductase) | Support viral replication by providing material for DNA synthesis | TG6002 | [152] |
| TK, N1L, and A41L | Supports viral replication, inhibits apoptosis, interferes with chemokine signaling | VVLΔTKΔN1LΔA41L | [153] |
| A49L | Inhibits NF-kB activation | vΔA49L | [154] |
| TK, F1L | Supports viral replication, Inhibits apoptosis | ΔTK/F1L | [155] |
| TK, B2R | Supports viral replication, Inhibits the cGAS/STING pathway | WR/TK−/ΔB2 | [156] |
To further enhance T cell-mediated responses, pro-inflammatory cytokine genes such as IL-12 [157], Il-15 [158], and IL-23 [159] have been inserted into the VACV genome. These modifications led to improved anti-tumor effects with increased CD8⁺ T cell infiltration—consistent with the roles of these cytokines in supporting cytotoxic T lymphocyte (CTL) proliferation, survival, and effector function.
Interestingly, the anti-inflammatory cytokine IL-10 also enhanced anti-tumor effects when expressed by oncolytic VACV in a mouse model of pancreatic cancer [160]. This finding is counterintuitive, given that IL-10 is generally associated with suppression of immune responses, including inhibition of IL-12 production. The study revealed that IL-10 expressing VACV persisted longer within tumors, led to fewer VACV-specific CD8⁺ T cells, and resulted in reduced infiltration by macrophages [160]. This suggests that IL-10 may prolong viral replication within tumor tissue, thereby enhancing direct oncolysis and potentially facilitating a more robust anti-tumor immune response. Additionally, IL10 is known to impair macrophage antigen presentation to CD4+ cells [161], potentially skewing the immune response toward a CD8⁺ T cell-driven cytotoxic pathway.
6.6. Induction of the IFN System
Robust activation of the type I IFN response is critical for the development of effective cytotoxic immunity, which is a key objective in cancer immunotherapy [162]. However, VACV encodes multiple genes that inhibit the host IFN response, posing a challenge for its use as an oncolytic agent. To address this, researchers have developed strategies to prevent VACV from suppressing IFN production. One such strategy involves deletion of the B2R gene, which encodes a nuclease that degrades cGAMP—a key activator of the cGAS-STING pathway [163]. Another targeted gene is E5R, a gene that promotes degradation of cGAS itself [49]. VACV strains lacking B2R exhibit elevated IFN expression and enhanced anti-tumor activity in vivo [163]. Notably, B2R deletion also reduces viral virulence, making the modified virus potentially safer for use in immunocompromised patients.
Alternative strategy involves the insertion of the gene encoding DNA-dependent activator of IFN-regulatory factors (DAI), which can activate IRF3 through a cGAS independent pathway [164]. In one study, DAI-expressing VACV demonstrated significantly improved inhibition of melanoma tumor growth in both syngeneic mouse models and humanized mice, even when the B2R gene remained intact [164].
A more novel approach utilized the gene encoding white-spotted charr lectin (WCL), a plant-derived protein previously linked to strong anti-tumor effects. VACV engineered to express WCL gene was shown to robustly activate IRF3 and induce high levels of type I IFNs, leading to effective suppression of hepatocellular carcinoma in a mouse model [165].
6.7. Expression of Costimulatory Molecules
To boost the activity of tumor-primed T cells, various research groups have developed VACVs encoding co-stimulatory molecules such as CD40L, 4-1BBL, and OX40L. Signals derived from these molecules are known to enhance T cell activity, reduce apoptosis, and increase the production of proinflammatory cytokines. When delivered intratumorally, these engineered viruses successfully inhibited the growth of B16 melanoma tumors in immunocompetent mice and extended the survival of treated animals [166,167,168].
Additionally, a CD40L-expressing VACV effectively slowed the progression of bladder cancer in a mouse xenograft model by activating the NF-κB pathway in T cells and promoting the secretion of TNF-α, IL-1α and RANTES [167]. In another study, a VACV encoding a tandem of Fms-related tyrosine kinase 3 ligand (Flt3l) and OX40L genes completely eliminated A20 lymphoma tumors and significantly delayed the development of spontaneous tumors in the MMTV-PyMT mouse model of triple negative breast cancer (TNBS) [168]. The use of the OX40L/FLt3l-expressing virus also led to a marked depletion of regulatory T cells (Tregs), reprogramming them into more cytotoxic-like phenotype [168].
6.8. Other Strategies
Researchers have also explored several innovative strategies to enhance the anti-tumor properties of VACV. In one notable study, a VACV engineered to express a secretory bispecific T-cell engager composed of single chain variable antibody fragments specific for CD3 and the tumor antigen EphA. This virus effectively recruited T lymphocytes to EphA-expressing cancer cells and induced complete clearance of A549 tumors in SCID mice infused with human PBMCs [169].
Another approach targeted the transforming growth factor beta (TGF-β) pathway, which is frequently upregulated in immune-resistant tumors. A VACV expressing a soluble TGF-β inhibitor was able to eliminate head and neck squamous cell carcinoma (HNSCC) tumors that were resistant to treatment with a control VACV containing B2R and TK deletions. The engineered virus reduced the number of Tregs and increased their sensitive to IFN-γ signaling [170].
Finally, to target tumor vasculature, scientists engineered a VACV to express the anti-VEGF single-chain antibody GLAF1. Treatment with this virus dramatically decreased blood vessel density within tumors and inhibited disease progression in xenograft models [171].
Overall, as our understanding of immune stimulation continues to grow, we can expect an increasing number of innovative genetic constructs to be tested as transgenes in the new generation of oncolytic VACVs.
7. Vaccinia Virus in Combination with Other Therapeutic Strategies
7.1. Combination with CAR-T Therapies
Although various forms of oncolytic VACV have demonstrated promising results as monotherapies in cancer treatment, many researchers are now exploring their use in combination with other therapeutic approaches. One notable strategy involves combining VACV with chimeric antigen receptor T-cell (CAR-T) therapy.
CAR-T based therapies are highly effective against CD19-expressing lymphomas; however, their efficacy in treating solid tumors remains limited due to the lack of tumor-specific antigens [172]. To overcome this challenge, researchers engineered VACV to express the CD19 gene. Because VACV preferentially replicates in tumor tissues, it can induce CD19 expression on the surface of cancer cells, rendering them susceptible to CAR-T cell-mediated cytotoxicity. This combination demonstrated promising results in the B16 melanoma model [173], as well as in the MC-38 colorectal cancer model and human tumor xenograft models [174].
In another approach, a CXCL11-expressing VACV was used to attract mesothelin-specific CAR-T cells toward mesothelin-positive TC-1 tumors. While both the virus and CAR-T cells individually inhibited tumor progression following intravenous injections, their combination produced a significantly more potent effect [175].
These findings suggest that VACV can serve as a valuable adjunct to enhance the efficacy of CAR-T therapies, particularly in the treatment of solid tumors.
7.2. Combination with Checkpoint Inhibitors
Since many oncolytic VACVs can activate immune responses in immunologically “cold” tumors, a logical strategy is to combine them with immune checkpoint inhibitors, which can sustain and amplify T cell activity once it has been initiated [176,177]. For example, the combination of IL-21-expressing VACV with an anti-PD-1 antibody produced a significantly stronger therapeutic effect in a mouse glioma model than either treatment alone [178]. Similar synergistic effects were observed in A20 and EL4 lymphoma models when combining a VACV expressing manganese superoxide dismutase (MnSOD) with anti-PD-L1 antibodies [179]. In the MC-38 colon cancer model, the efficacy of a CXCL11-expressing VACV was also substantially enhanced by co-administration of anti-PD-L1 therapy [180].
Because systemic administration of checkpoint inhibitors is often associated with immune-related toxicities, researchers have also explored engineering VACVs to express checkpoint- inhibitory molecules directly within tumors. Leveraging the virus’s tumor-specific replication, this approach allows for localized delivery of checkpoint inhibitors, potentially reducing systemic side effects. A VACV co-expressing a soluble PD-L1 inhibitor and GM-CSF demonstrated high efficacy in treating B16 melanoma tumors [181]. Another engineered virus encoding a cell-depleting anti-CTLA4 antibody and GM-CSF successfully eradicated tumors in multiple models, including breast (EMT6), colon (MC-38), and melanoma (B16) models, following intratumoral administration. Tumor regression in these models was associated with a marked reduction in CD4+ Treg cells and exhausted CD8+ T cells, alongside expansion of activated cytotoxic CD8+ T cells. Combining this therapy with anti-PD -1 antibodies further improved its therapeutic efficacy [182].
Another checkpoint target explored with VACV is TIGIT (T cell immunoreceptor with Ig and ITIM domains), a common marker of exhausted and regulatory T cells. Researchers engineered a VACV to express the variable domains of heavy and light chains of an anti-TIGIT antibody. This virus effectively slowed the progression of several subcutaneously implanted tumor models, including EMT6 (breast), CT26, MC-38 (colon), and H22 (liver) tumors. Once again, combining the treatment with anti-PD-1 therapy further enhanced the anti-tumor effects [183].
Collectively, these findings support the idea that immune checkpoint inhibitors, whether co-administered or encoded directly within the virus, are likely to play a critical role in the future development of VACV-based cancer therapies.
7.3. Combination with Radio- and Chemotherapy
Chemotherapy and radiotherapy remain among the most widely used cancer treatments, and the potential of combining these with VACV has been explored in several studies. Radiotherapy has been shown to enhance the efficacy of oncolytic VACV in preclinical models of glioblastoma [184,185] and pancreatic cancer [186]. In the TC-1 lung cancer model, combining VACV with radiotherapy increased tumor cells necroptosis and stimulated the release of DAMP molecules. This, in turn, activated T cells and reduced the populations of Tregs and M2 macrophages [187]. Owing to its tumor selectivity, VACV is a promising agent for sensitizing tumors to radiation.
One commonly employed strategy involved engineering VACV to express the sodium iodide symporter (NIS) [188,189,190]. Infection with this virus significantly increased the uptake of radioactive Iodine-131 (131I) in prostate cancer xenografts, leading to tumor growth inhibition and prolonged survival in treated mice [191]. In another strategy, researchers developed a radiotherapy system using VACV expressing the somatotropin receptor, in combination with a radioisotope labeled somatotropin analogue. In both subcutaneous and disseminated mouse models of colorectal cancer, intraperitoneal administration of the virus resulted in tumor-specific localization and targeted uptake of the radiolabeled compound. This specific accumulation of radioisotope within tumor tissue significantly inhibited tumor growth, improved survival, and did not cause systemic toxicity [192].
VACV has also been studied in combination with conventional chemotherapy. Paclitaxel, for example, significantly enhanced the anti-tumor efficacy of VACV in HCT116 tumors grown in athymic mice. This effect was associated with type-I IFN production and the release of HMGB1, a key DAMP molecule [193]. Furthermore, both cisplatin and gemcitabine improved the efficacy of oncolytic VACV in pancreatic tumor xenografts in nude mice [194]. Similarly, treatment with cyclophosphamide (CPA) or rapamycin increased VACV’s ability to inhibit the growth of malignant gliomas in rat models [195]. CPA also enhanced the efficacy of intravenously administered VACV in lung cancer xenografts by increasing viral spread within the tumor and reducing tumor vasculature [196].
Another commonly employed strategy involves arming VACV with genes that convert inactive prodrugs into active chemotherapeutic agents. Given VACV’s selectivity for tumor tissue, this approach allows high local concentrations of the active drug while minimizing systemic toxicity. For example, a VACV engineered to express super cytosine deaminase (SCD) effectively converted the prodrug 5-fluorocytosine (5-FC) into the chemotherapeutic compound 5-fluorouracil (5-FU). In the presence of prodrug, the virus induced death in cancer cell lines that were otherwise resistant to VACV-mediated lysis [197]. Another engineered VACV expressed β-galactosidase to activate a prodrug containing a β-galactosidase-specific cleavage site. In a breast cancer xenograft model, this virus-prodrug system led to accelerated tumor shrinkage and induced apoptosis in multiple cancer cell lines when the prodrug was present [198].
7.4. Combination with Small-Molecule Inhibitors
The oncolytic potential of VACV has also been evaluated in combination with various small-molecule inhibitors used in cancer therapy. Trametinib, a clinically approved inhibitor of mitogen-activated protein kinase MEK, was found to enhance VACV replication in vitro and improve its ability to inhibit the growth of ovarian cancer xenografts [199]. Idelalisib, an FDA-approved selective inhibitor of phosphoinositide 3-kinase delta (PI3Kδ) , significantly increased viral delivery to tumors following intravenous injection and substantially enhanced the therapeutic effect [200]. Other notable inhibitors that have been shown to improve the anti-cancer activity of oncolytic VACVs include trichastatin A, a histone deacetylase inhibitor, and sunitinib, a multitargeted receptor tyrosine kinase inhibitor. Trichastatin A markedly increased viral replication and spread within tumor tissues [201], while sunitinib robustly enhanced CD8+ T cell infiltration, suppressed Tregs, and increased tumor cell apoptosis by more than threefold [202]. Inhibitors targeting the VEGF pathway have also been found to potentiate the oncolytic effects of VACV [171,203]. As the number of novel inhibitors targeting tumor-specific pathways continues to grow, combining them with VACV holds great promise for achieving even more effective cancer therapies.
8. Clinical Trials and Potential Obstacles
The specificity of VACV for cancer cells and the promising results from pre-clinical models have spurred the development of multiple VACV-based oncolytic viruses, many of which have progressed to clinical trials. A key consideration in patient treatment is the choice of viral delivery route. Systemic administration, such as intravenous or intraperitoneal injection, is the most convenient; however, it requires the virus to have high tumor-targeting specificity. Moreover, repeated systemic dosing can be challenging due to the development of neutralizing antibodies, which reduce the efficacy of subsequent treatments.
In contrast, intratumoral injection allows for direct delivery of the virus into the tumor, enabling the use of lower doses and minimizing the impact of circulating neutralizing antibodies. However, this method has its limitations—it is not feasible when tumors are inaccessible or numerous, and it often requires complex medical procedures. These procedures can be burdensome for patients and may disrupt their daily lives.
Current clinical trials involving VACV utilize both systemic and intratumoral delivery approaches. Table 2 summarizes clinical studies completed between 2009 and 2025, while a comprehensive list of earlier clinical trials has been published elsewhere [204]. An up-to-date list of ongoing clinical trials involving VACV can be found at https://clinicaltrials.gov/.
As of April 2025, only two trials involving oncolytic VACV have reached phase III. The first is a study evaluating intraperitoneal delivery of Genelux’s Olvi-Vec (GL-ONC1) in combination with a platinum-based chemotherapy regimen for the treatment of platinum-resistant refractory ovarian cancer (NCT05281471). Olvi-Vec was engineered by inserting three expression cassettes—encoding a Renilla luciferase-Aequorea green fluorescent protein fusion, β-galactosidase, and β-glucuronidase —into the F14.5L, J2R (encoding TK) and A56R (encoding hemagglutinin) loci of the parental VACV genome. In addition to this phase-III trial, Olvi-Vec is currently in phase I trials for the treatment of small-cell and non-small lung cancer.
Table 02562755. is Pexa-Vac (JX-594), developed by SillaJen. JX-594 is engineered with a deletion of the TK gene and insertion of transgenes encoding human GM-CSF and β-galactosidase. It is administered via intratumoral injection. Pexa-Vac has been evaluated in clinical trials for renal, colorectal, and liver cancers, and advanced to a phase III trial for hepatocellular carcinoma in combination with the targeted therapy sorafenib. Unfortunately, this trial failed to demonstrate a survival benefit compared to the control group [130].
Nevertheless, additional trials exploring JX-594 in combination with other therapeutic agents and across different cancer types are still ongoing. As newer generations of oncolytic VACV are continuously being developed, more candidates are expected to enter clinical trials, potentially yielding more promising results.
Despite its many advantages, VACV also presents certain challenges that may limit its therapeutic potential. As a replicating virus, it poses a theoretical risk to immunocompromised patients, particularly when administered systemically (e.g., via intravenous injection). However, this risk is significantly mitigated by the presence of multiple attenuating mutations engineered into oncolytic VACV strains, many of which are based on the Lister strain, which has a long history of safe use as a smallpox vaccine.
Another major challenge is the strong immune memory elicited by VACV, at both the B and T cell levels. This response can limit the efficacy of repeated administrations, especially through intravenous delivery. High titers of neutralizing antibodies may block the virus from binding to target cells, while cytotoxic memory T cells can destroy infected cancer cells before sufficient viral replication and spread occurs.
Strategies to overcome these immune barriers have shown promise. For example, treatment with the cyclooxygenase-2 (COX-2) inhibitor celecoxib prior to a second viral dose significantly reduced the generation of neutralization antibodies and restored viral titers in pre-immunized models [226]. Similarly, inhibiting the complement system with the synthetic compound CP40 or depleting it with cobra venom factor (CVF) markedly increased viral titers in both blood and tumor tissue of pre-immunized animals [227].
To further protect VACV from complement-mediated neutralization, researchers have engineered a modified that expresses the N-terminal fragment of the complement regulatory protein CD55 fused to six membrane proteins found on intracellular mature virions (IMV). This engineered virus maintained its replication efficiency and infectivity while successfully evading neutralization by VACV-specific antibodies generated after multiple systemic administrations [228].
In conclusion, although pre-existing immunity to VACV poses a significant challenge for repeated dosing, recent advances offer practical strategies to overcome these challenges and enhance the clinical efficacy of oncolytic VACV.
9. Conclusions
The limited efficacy of current treatments for disseminated cancer underscores the urgent need for more innovative and potent treatment strategies. VACV has emerged as a highly promising platform for viro-immunotherapy due to its safety, genetic flexibility, and ability to deliver multiple therapeutic genes directly to tumor tissue. It has shown significant potential both as monotherapy and in combination with other treatment modalities.
Although the phase III clinical trial of JX-594 did not yield the desired outcome, it is important to note that this virus represents an earlier generation construct—far less advanced than the more sophisticated candidates currently under development. As our understanding of tumor biology, immunology, and viral engineering continues to evolve, we are poised to design increasingly sophisticated VACV derivatives with improved efficacy and tumor specificity. Moreover, the strategic integration of VACV into combination therapies is expected to become more refined and effective.
With ongoing research and technological advancements, the future of cancer treatment involving VACV appears highly promising and is likely to play a pivotal role in the next generation of oncologic therapies.
Acknowledgments
All authors are employees of ViroMissile, Inc.
Disclosure/Conflict of Interest
M.K.S, Y.Y. and N.G.C are all employees of ViroMissile, Inc.
References
- Xu, L.; Sun, H.; Lemoine, N.R.; Xuan, Y.; Wang, P. Oncolytic Vaccinia Virus and Cancer Immunotherapy. Front. Immunol. 2024, 14. [CrossRef]
- Kaynarcalidan, O.; Moreno Mascaraque, S.; Drexler, I. Vaccinia Virus: From Crude Smallpox Vaccines to Elaborate Viral Vector Vaccine Design. Biomedicines 2021, 9, 1780. [CrossRef]
- Greseth, M.D.; Czarnecki, M.W.; Bluma, M.S.; Traktman, P. Isolation and Characterization of vΔI3 Confirm That Vaccinia Virus SSB Plays an Essential Role in Viral Replication. Journal of Virology 2018, 92, 10.1128/jvi.01719-17. [CrossRef]
- Wittek, R. VACCINIA VIRUS (POXVIRIDAE). In Encyclopedia of Virology (Second Edition); Granoff, A., Webster, R.G., Eds.; Elsevier: Oxford, 1999; pp. 1865–1872 ISBN 978-0-12-227030-7.
- Molteni, C.; Forni, D.; Cagliani, R.; Clerici, M.; Sironi, M. Genetic Ancestry and Population Structure of Vaccinia Virus. npj Vaccines 2022, 7, 1–9. [CrossRef]
- Moss, B. Poxvirus DNA Replication. Cold Spring Harb Perspect Biol 2013, 5, a010199. [CrossRef]
- Yang, Z.; Maruri-Avidal, L.; Sisler, J.; Stuart, C.A.; Moss, B. Cascade Regulation of Vaccinia Virus Gene Expression Is Modulated by Multistage Promoters. Virology 2013, 447, 10.1016/j.virol.2013.09.007. [CrossRef]
- Chou, W.; Ngo, T.; Gershon, P.D. An Overview of the Vaccinia Virus Infectome: A Survey of the Proteins of the Poxvirus-Infected Cell. J Virol 2012, 86, 1487–1499. [CrossRef]
- Laudermilch, E.; Chandran, K. MAVERICC: Marker-Free Vaccinia Virus Engineering of Recombinants through in Vitro CRISPR/Cas9 Cleavage. J Mol Biol 2021, 433, 166896. [CrossRef]
- Jacobs, B.L.; Langland, J.O.; Kibler, K.V.; Denzler, K.L.; White, S.D.; Holechek, S.A.; Wong, S.; Huynh, T.; Baskin, C.R. Vaccinia Virus Vaccines: Past, Present and Future. Antiviral Res 2009, 84, 1–13. [CrossRef]
- de Freitas, L.F.D.; Oliveira, R.P.; Miranda, M.C.G.; Rocha, R.P.; Barbosa-Stancioli, E.F.; Faria, A.M.C.; da Fonseca, F.G. The Virulence of Different Vaccinia Virus Strains Is Directly Proportional to Their Ability To Downmodulate Specific Cell-Mediated Immune Compartments In Vivo. J Virol 2019, 93, e02191-18. [CrossRef]
- Belongia, E.A.; Naleway, A.L. Smallpox Vaccine: The Good, the Bad, and the Ugly. Clin Med Res 2003, 1, 87–92.
- Meiser, A.; Boulanger, D.; Sutter, G.; Krijnse Locker, J. Comparison of Virus Production in Chicken Embryo Fibroblasts Infected with the WR, IHD-J and MVA Strains of Vaccinia Virus: IHD-J Is Most Efficient in Trans-Golgi Network Wrapping and Extracellular Enveloped Virus Release. J Gen Virol 2003, 84, 1383–1392. [CrossRef]
- Bengali, Z.; Townsley, A.C.; Moss, B. Vaccinia Virus Strain Differences in Cell Attachment and Entry. Virology 2009, 389, 132–140. [CrossRef]
- Carter, G.C.; Law, M.; Hollinshead, M.; Smith, G.L. Entry of the Vaccinia Virus Intracellular Mature Virion and Its Interactions with Glycosaminoglycans. Journal of General Virology 2005, 86, 1279–1290. [CrossRef]
- MacLeod, D.T.; Nakatsuji, T.; Wang, Z.; di Nardo, A.; Gallo, R.L. Vaccinia Virus Binds to the Scavenger Receptor MARCO on the Surface of Keratinocytes. J Invest Dermatol 2015, 135, 142–150. [CrossRef]
- Laliberte, J.P.; Weisberg, A.S.; Moss, B. The Membrane Fusion Step of Vaccinia Virus Entry Is Cooperatively Mediated by Multiple Viral Proteins and Host Cell Components. PLoS Pathog 2011, 7, e1002446. [CrossRef]
- Townsley, A.C.; Weisberg, A.S.; Wagenaar, T.R.; Moss, B. Vaccinia Virus Entry into Cells via a Low-pH-Dependent Endosomal Pathway. J Virol 2006, 80, 8899–8908. [CrossRef]
- Mercer, J.; Knébel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia Virus Strains Use Distinct Forms of Macropinocytosis for Host-Cell Entry. Proceedings of the National Academy of Sciences 2010, 107, 9346–9351. [CrossRef]
- Greseth, M.D.; Traktman, P. The Life Cycle of the Vaccinia Virus Genome. Annual Review of Virology 2022, 9, 239–259. [CrossRef]
- Schmidt, F.I.; Bleck, C.K.E.; Reh, L.; Novy, K.; Wollscheid, B.; Helenius, A.; Stahlberg, H.; Mercer, J. Vaccinia Virus Entry Is Followed by Core Activation and Proteasome-Mediated Release of the Immunomodulatory Effector VH1 from Lateral Bodies. Cell Reports 2013, 4, 464–476. [CrossRef]
- Tolonen, N.; Doglio, L.; Schleich, S.; Locker, J.K. Vaccinia Virus DNA Replication Occurs in Endoplasmic Reticulum-Enclosed Cytoplasmic Mini-Nuclei. MBoC 2001, 12, 2031–2046. [CrossRef]
- Moss, B. Cascade Regulation of Vaccinia Virus Gene Expression. In Regulation of Gene Expression in Animal Viruses; Carrasco, L., Sonenberg, N., Wimmer, E., Eds.; Springer US: Boston, MA, 1993; pp. 13–24 ISBN 978-1-4615-2928-6.
- Howell, L.M.; Gracie, N.P.; Newsome, T.P. Single-Cell Analysis of VACV Infection Reveals Pathogen-Driven Timing of Early and Late Phases and Host-Limited Dynamics of Virus Production. PLOS Pathogens 2024, 20, e1012423. [CrossRef]
- Liu, L.; Cooper, T.; Howley, P.M.; Hayball, J.D. From Crescent to Mature Virion: Vaccinia Virus Assembly and Maturation. Viruses 2014, 6, 3787–3808. [CrossRef]
- Boyle, K.A.; Stanitsa, E.S.; Greseth, M.D.; Lindgren, J.K.; Traktman, P. Evaluation of the Role of the Vaccinia Virus Uracil DNA Glycosylase and A20 Proteins as Intrinsic Components of the DNA Polymerase Holoenzyme *. Journal of Biological Chemistry 2011, 286, 24702–24713. [CrossRef]
- Rochester, S.C.; Traktman, P. Characterization of the Single-Stranded DNA Binding Protein Encoded by the Vaccinia Virus I3 Gene. J Virol 1998, 72, 2917–2926. [CrossRef]
- Garcia, A.D.; Moss, B. Repression of Vaccinia Virus Holliday Junction Resolvase Inhibits Processing of Viral DNA into Unit-Length Genomes. J Virol 2001, 75, 6460–6471. [CrossRef]
- Paran, N.; De Silva, F.S.; Senkevich, T.G.; Moss, B. Cellular DNA Ligase I Is Recruited to Cytoplasmic Vaccinia Virus Factories and Masks the Role of the Vaccinia Ligase in Viral DNA Replication. Cell Host Microbe 2009, 6, 563–569. [CrossRef]
- El Omari, K.; Solaroli, N.; Karlsson, A.; Balzarini, J.; Stammers, D.K. Structure of Vaccinia Virus Thymidine Kinase in Complex with dTTP: Insights for Drug Design. BMC Struct Biol 2006, 6, 22. [CrossRef]
- Topalis, D.; Collinet, B.; Gasse, C.; Dugué, L.; Balzarini, J.; Pochet, S.; Deville-Bonne, D. Substrate Specificity of Vaccinia Virus Thymidylate Kinase. The FEBS Journal 2005, 272, 6254–6265. [CrossRef]
- Gammon, D.B.; Gowrishankar, B.; Duraffour, S.; Andrei, G.; Upton, C.; Evans, D.H. Vaccinia Virus–Encoded Ribonucleotide Reductase Subunits Are Differentially Required for Replication and Pathogenesis. PLoS Pathog 2010, 6, e1000984. [CrossRef]
- Smith, G.L.; Vanderplasschen, A.; Law, M. The Formation and Function of Extracellular Enveloped Vaccinia Virus. Journal of General Virology 2002, 83, 2915–2931. [CrossRef]
- Smith, G.L.; Law, M. The Exit of Vaccinia Virus from Infected Cells. Virus Research 2004, 106, 189–197. [CrossRef]
- Zeh, H.J.; Downs-Canner, S.; McCart, J.A.; Guo, Z.S.; Rao, U.N.M.; Ramalingam, L.; Thorne, S.H.; Jones, H.L.; Kalinski, P.; Wieckowski, E.; et al. First-in-Man Study of Western Reserve Strain Oncolytic Vaccinia Virus: Safety, Systemic Spread, and Antitumor Activity. Molecular Therapy 2015, 23, 202–214. [CrossRef]
- Schwanke, H.; Stempel, M.; Brinkmann, M.M. Of Keeping and Tipping the Balance: Host Regulation and Viral Modulation of IRF3-Dependent IFNB1 Expression. Viruses 2020, 12, 733. [CrossRef]
- Cell Fate in Antiviral Response Arises in the Crosstalk of IRF, NF-κB and JAK/STAT Pathways | Nature Communications Available online: https://www.nature.com/articles/s41467-017-02640-8 (accessed on 18 April 2025).
- El-Jesr, M.; Teir, M.; Maluquer de Motes, C. Vaccinia Virus Activation and Antagonism of Cytosolic DNA Sensing. Front Immunol 2020, 11, 568412. [CrossRef]
- Peters, N.E.; Ferguson, B.J.; Mazzon, M.; Fahy, A.S.; Krysztofinska, E.; Arribas-Bosacoma, R.; Pearl, L.H.; Ren, H.; Smith, G.L. A Mechanism for the Inhibition of DNA-PK-Mediated DNA Sensing by a Virus. PLoS Pathog 2013, 9, e1003649. [CrossRef]
- Scutts, S.R.; Ember, S.W.; Ren, H.; Ye, C.; Lovejoy, C.A.; Mazzon, M.; Veyer, D.L.; Sumner, R.P.; Smith, G.L. DNA-PK Is Targeted by Multiple Vaccinia Virus Proteins to Inhibit DNA Sensing. Cell Rep 2018, 25, 1953-1965.e4. [CrossRef]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [CrossRef]
- Valentine, R.; Smith, G.L. Inhibition of the RNA Polymerase III-Mediated dsDNA-Sensing Pathway of Innate Immunity by Vaccinia Virus Protein E3. Journal of General Virology 2010, 91, 2221–2229. [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 Is an Innate Immune Sensor for Intracellular DNA. Nat Immunol 2010, 11, 997–1004. [CrossRef]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS Cooperate in the Activation of STING during DNA Sensing in Human Keratinocytes. Nat Commun 2017, 8, 14392. [CrossRef]
- Ablasser, A.; Schmid-Burgk, J.L.; Hemmerling, I.; Horvath, G.L.; Schmidt, T.; Latz, E.; Hornung, V. Cell Intrinsic Immunity Spreads to Bystander Cells via the Intercellular Transfer of cGAMP. Nature 2013, 503, 530–534. [CrossRef]
- Luteijn, R.D.; Zaver, S.A.; Gowen, B.G.; Wyman, S.K.; Garelis, N.E.; Onia, L.; McWhirter, S.M.; Katibah, G.E.; Corn, J.E.; Woodward, J.J.; et al. SLC19A1 Transports Immunoreactive Cyclic Dinucleotides. Nature 2019, 573, 434–438. [CrossRef]
- Seo, G.J.; Yang, A.; Tan, B.; Kim, S.; Liang, Q.; Choi, Y.; Yuan, W.; Feng, P.; Park, H.-S.; Jung, J.U. Akt Kinase-Mediated Checkpoint of cGAS DNA Sensing Pathway. Cell Rep 2015, 13, 440–449. [CrossRef]
- Meade, N.; Furey, C.; Li, H.; Verma, R.; Chai, Q.; Rollins, M.G.; DiGiuseppe, S.; Naghavi, M.H.; Walsh, D. Poxviruses Evade Cytosolic Sensing through Disruption of an mTORC1-mTORC2 Regulatory Circuit. Cell 2018, 174, 1143-1157.e17. [CrossRef]
- Yang, N.; Wang, Y.; Dai, P.; Li, T.; Zierhut, C.; Tan, A.; Zhang, T.; Xiang, J.Z.; Ordureau, A.; Funabiki, H.; et al. Vaccinia E5 Is a Major Inhibitor of the DNA Sensor cGAS. Nat Commun 2023, 14, 2898. [CrossRef]
- Eaglesham, J.B.; Pan, Y.; Kupper, T.S.; Kranzusch, P.J. Viral and Metazoan Poxins Are cGAMP-Specific Nucleases That Restrict cGAS-STING Signaling. Nature 2019, 566, 259–263. [CrossRef]
- Unterholzner, L.; Sumner, R.P.; Baran, M.; Ren, H.; Mansur, D.S.; Bourke, N.M.; Randow, F.; Smith, G.L.; Bowie, A.G. Vaccinia Virus Protein C6 Is a Virulence Factor That Binds TBK-1 Adaptor Proteins and Inhibits Activation of IRF3 and IRF7. PLoS Pathog 2011, 7, e1002247. [CrossRef]
- Schröder, M.; Baran, M.; Bowie, A.G. Viral Targeting of DEAD Box Protein 3 Reveals Its Role in TBK1/IKKɛ-Mediated IRF Activation. EMBO J 2008, 27, 2147–2157. [CrossRef]
- Ferguson, B.J.; Benfield, C.T.O.; Ren, H.; Lee, V.H.; Frazer, G.L.; Strnadova, P.; Sumner, R.P.; Smith, G.L. Vaccinia Virus Protein N2 Is a Nuclear IRF3 Inhibitor That Promotes Virulence. J Gen Virol 2013, 94, 2070–2081. [CrossRef]
- Willis, K.L.; Langland, J.O.; Shisler, J.L. Viral Double-Stranded RNAs from Vaccinia Virus Early or Intermediate Gene Transcripts Possess PKR Activating Function, Resulting in NF-κB Activation, When the K1 Protein Is Absent or Mutated *. Journal of Biological Chemistry 2011, 286, 7765–7778. [CrossRef]
- García, M.A.; Gil, J.; Ventoso, I.; Guerra, S.; Domingo, E.; Rivas, C.; Esteban, M. Impact of Protein Kinase PKR in Cell Biology: From Antiviral to Antiproliferative Action. Microbiol Mol Biol Rev 2006, 70, 1032–1060. [CrossRef]
- Szczerba, M.; Subramanian, S.; Trainor, K.; McCaughan, M.; Kibler, K.V.; Jacobs, B.L. Small Hero with Great Powers: Vaccinia Virus E3 Protein and Evasion of the Type I IFN Response. Biomedicines 2022, 10, 235. [CrossRef]
- Cao, J.; Varga, J.; Deschambault, Y. Poxvirus Encoded eIF2α Homolog, K3 Family Proteins, Is a Key Determinant of Poxvirus Host Species Specificity. Virology 2020, 541, 101–112. [CrossRef]
- Yu, H.; Bruneau, R.C.; Brennan, G.; Rothenburg, S. Battle Royale: Innate Recognition of Poxviruses and Viral Immune Evasion. Biomedicines 2021, 9, 765. [CrossRef]
- Rivas, C.; Gil, J.; Mělková, Z.; Esteban, M.; Díaz-Guerra, M. Vaccinia Virus E3L Protein Is an Inhibitor of the Interferon (IFN)-Induced 2-5A Synthetase Enzyme. Virology 1998, 243, 406–414. [CrossRef]
- Chan, Y.K.; Gack, M.U. RIG-I-like Receptor Regulation in Virus Infection and Immunity. Curr Opin Virol 2015, 12, 7–14. [CrossRef]
- Deng, L.; Dai, P.; Parikh, T.; Cao, H.; Bhoj, V.; Sun, Q.; Chen, Z.; Merghoub, T.; Houghton, A.; Shuman, S. Vaccinia Virus Subverts a Mitochondrial Antiviral Signaling Protein-Dependent Innate Immune Response in Keratinocytes through Its Double-Stranded RNA Binding Protein, E3. Journal of Virology 2008, 82, 10735–10746. [CrossRef]
- Valentine, R.; Smith, G.L. Inhibition of the RNA Polymerase III-Mediated dsDNA-Sensing Pathway of Innate Immunity by Vaccinia Virus Protein E3. Journal of General Virology 2010, 91, 2221–2229. [CrossRef]
- Botos, I.; Segal, D.M.; Davies, D.R. The Structural Biology of Toll-like Receptors. Structure 2011, 19, 447–459. [CrossRef]
- El-Zayat, S.R.; Sibaii, H.; Mannaa, F.A. Toll-like Receptors Activation, Signaling, and Targeting: An Overview. Bulletin of the National Research Centre 2019, 43, 187. [CrossRef]
- Harte, M.T.; Haga, I.R.; Maloney, G.; Gray, P.; Reading, P.C.; Bartlett, N.W.; Smith, G.L.; Bowie, A.; O’Neill, L.A.J. The Poxvirus Protein A52R Targets Toll-like Receptor Signaling Complexes to Suppress Host Defense. J Exp Med 2003, 197, 343–351. [CrossRef]
- Stack, J.; Haga, I.R.; Schröder, M.; Bartlett, N.W.; Maloney, G.; Reading, P.C.; Fitzgerald, K.A.; Smith, G.L.; Bowie, A.G. Vaccinia Virus Protein A46R Targets Multiple Toll-like–Interleukin-1 Receptor Adaptors and Contributes to Virulence. Journal of Experimental Medicine 2005, 201, 1007–1018. [CrossRef]
- Dempsey, A.; Keating, S.E.; Carty, M.; Bowie, A.G. Poxviral Protein E3–Altered Cytokine Production Reveals That DExD/H-Box Helicase 9 Controls Toll-like Receptor–Stimulated Immune Responses. J Biol Chem 2018, 293, 14989–15001. [CrossRef]
- Dai, P.; Cao, H.; Merghoub, T.; Avogadri, F.; Wang, W.; Parikh, T.; Fang, C.-M.; Pitha, P.M.; Fitzgerald, K.A.; Rahman, M.M.; et al. Myxoma Virus Induces Type I Interferon Production in Murine Plasmacytoid Dendritic Cells via a TLR9/MyD88-, IRF5/IRF7-, and IFNAR-Dependent Pathway. Journal of Virology 2011, 85, 10814–10825. [CrossRef]
- DiPerna, G.; Stack, J.; Bowie, A.G.; Boyd, A.; Kotwal, G.; Zhang, Z.; Arvikar, S.; Latz, E.; Fitzgerald, K.A.; Marshall, W.L. Poxvirus Protein N1L Targets the I-κB Kinase Complex, Inhibits Signaling to NF-κB by the Tumor Necrosis Factor Superfamily of Receptors, and Inhibits NF-κB and IRF3 Signaling by Toll-like Receptors *. Journal of Biological Chemistry 2004, 279, 36570–36578. [CrossRef]
- Albarnaz, J.D.; Ren, H.; Torres, A.A.; Shmeleva, E.V.; Melo, C.A.; Bannister, A.J.; Brember, M.P.; Chung, B.Y.-W.; Smith, G.L. Molecular Mimicry of NF-κB by Vaccinia Virus Protein Enables Selective Inhibition of Antiviral Responses. Nat Microbiol 2022, 7, 154–168. [CrossRef]
- Mansur, D.S.; Motes, C.M. de; Unterholzner, L.; Sumner, R.P.; Ferguson, B.J.; Ren, H.; Strnadova, P.; Bowie, A.G.; Smith, G.L. Poxvirus Targeting of E3 Ligase β-TrCP by Molecular Mimicry: A Mechanism to Inhibit NF-κB Activation and Promote Immune Evasion and Virulence. PLOS Pathogens 2013, 9, e1003183. [CrossRef]
- Smith, G.L.; Benfield, C.T.O.; Maluquer de Motes, C.; Mazzon, M.; Ember, S.W.J.; Ferguson, B.J.; Sumner, R.P. Vaccinia Virus Immune Evasion: Mechanisms, Virulence and Immunogenicity. Journal of General Virology 2013, 94, 2367–2392. [CrossRef]
- Symons, J.A.; Alcamí, A.; Smith, G.L. Vaccinia Virus Encodes a Soluble Type I Interferon Receptor of Novel Structure and Broad Species Specificity. Cell 1995, 81, 551–560. [CrossRef]
- Mann, B.A.; Huang, J.H.; Li, P.; Chang, H.-C.; Slee, R.B.; O’Sullivan, A.; Mathur, A.; Yeh, N.; Klemsz, M.J.; Brutkiewicz, R.R.; et al. Vaccinia Virus Blocks Stat1-Dependent and Stat1-Independent Gene Expression Induced by Type I and Type II Interferons. J Interferon Cytokine Res 2008, 28, 367–379. [CrossRef]
- Alvarez-de Miranda, F.J.; Alonso-Sánchez, I.; Alcamí, A.; Hernaez, B. TNF Decoy Receptors Encoded by Poxviruses. Pathogens 2021, 10, 1065. [CrossRef]
- Symons, J.A.; Tscharke, D.C.; Price, N.; Smith, G.L. A Study of the Vaccinia Virus Interferon-Gamma Receptor and Its Contribution to Virus Virulence. J Gen Virol 2002, 83, 1953–1964. [CrossRef]
- Reading, P.C.; Smith, G.L. Vaccinia Virus Interleukin-18-Binding Protein Promotes Virulence by Reducing Gamma Interferon Production and Natural Killer and T-Cell Activity. J Virol 2003, 77, 9960–9968. [CrossRef]
- Alcami, A.; Smith, G.L. A Soluble Receptor for Interleukin-1β Encoded by Vaccinia Virus: A Novel Mechanism of Virus Modulation of the Host Response to Infection. Cell 1992, 71, 153–167. [CrossRef]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. The Complement System and Innate Immunity. In Immunobiology: The Immune System in Health and Disease. 5th edition; Garland Science, 2001.
- Bernet, J.; Mullick, J.; Panse, Y.; Parab, P.B.; Sahu, A. Kinetic Analysis of the Interactions between Vaccinia Virus Complement Control Protein and Human Complement Proteins C3b and C4b. Journal of Virology 2004, 78, 9446–9457. [CrossRef]
- Li, P.; Wang, N.; Zhou, D.; Yee, C.S.K.; Chang, C.-H.; Brutkiewicz, R.R.; Blum, J.S. Disruption of MHC Class II-Restricted Antigen Presentation by Vaccinia Virus1. The Journal of Immunology 2005, 175, 6481–6488. [CrossRef]
- Rehm, K.E.; Connor, R.F.; Jones, G.J.B.; Yimbu, K.; Roper, R.L. Vaccinia Virus A35R Inhibits MHC Class II Antigen Presentation. Virology 2010, 397, 176. [CrossRef]
- Orzalli, M.H.; Kagan, J.C. Apoptosis and Necroptosis as Host Defense Strategies to Prevent Viral Infection. Trends Cell Biol 2017, 27, 800–809. [CrossRef]
- Gregory, C.D.; Devitt, A. The Macrophage and the Apoptotic Cell: An Innate Immune Interaction Viewed Simplistically? Immunology 2004, 113, 1–14. [CrossRef]
- Veyer, D.L.; Carrara, G.; Maluquer de Motes, C.; Smith, G.L. Vaccinia Virus Evasion of Regulated Cell Death. Immunology Letters 2017, 186, 68–80. [CrossRef]
- Maluquer de Motes, C.; Cooray, S.; Ren, H.; Almeida, G.M.F.; McGourty, K.; Bahar, M.W.; Stuart, D.I.; Grimes, J.M.; Graham, S.C.; Smith, G.L. Inhibition of Apoptosis and NF-κB Activation by Vaccinia Protein N1 Occur via Distinct Binding Surfaces and Make Different Contributions to Virulence. PLoS Pathog 2011, 7, e1002430. [CrossRef]
- Liu, Y.; Pan, R.; Ouyang, Y.; Gu, W.; Xiao, T.; Yang, H.; Tang, L.; Wang, H.; Xiang, B.; Chen, P. Pyroptosis in Health and Disease: Mechanisms, Regulation and Clinical Perspective. Sig Transduct Target Ther 2024, 9, 1–28. [CrossRef]
- Gerlic, M.; Faustin, B.; Postigo, A.; Yu, E.C.-W.; Proell, M.; Gombosuren, N.; Krajewska, M.; Flynn, R.; Croft, M.; Way, M.; et al. Vaccinia Virus F1L Protein Promotes Virulence by Inhibiting Inflammasome Activation. Proceedings of the National Academy of Sciences 2013, 110, 7808–7813. [CrossRef]
- Gerlic, M.; Faustin, B.; Postigo, A.; Yu, E.C.-W.; Proell, M.; Gombosuren, N.; Krajewska, M.; Flynn, R.; Croft, M.; Way, M.; et al. Vaccinia Virus F1L Protein Promotes Virulence by Inhibiting Inflammasome Activation. Proceedings of the National Academy of Sciences 2013, 110, 7808–7813. [CrossRef]
- Liu, Y.; Liu, T.; Lei, T.; Zhang, D.; Du, S.; Girani, L.; Qi, D.; Lin, C.; Tong, R.; Wang, Y. RIP1/RIP3-Regulated Necroptosis as a Target for Multifaceted Disease Therapy (Review). Int J Mol Med 2019, 44, 771–786. [CrossRef]
- Koehler, H.S.; Jacobs, B.L. Subversion of Programed Cell Death by Poxviruses. Curr Top Microbiol Immunol 2023, 442, 105–131. [CrossRef]
- Chahroudi, A.; Chavan, R.; Koyzr, N.; Waller, E.K.; Silvestri, G.; Feinberg, M.B. Vaccinia Virus Tropism for Primary Hematolymphoid Cells Is Determined by Restricted Expression of a Unique Virus Receptor. J Virol 2005, 79, 10397–10407. [CrossRef]
- Engelmayer, J.; Larsson, M.; Subklewe, M.; Chahroudi, A.; Cox, W.I.; Steinman, R.M.; Bhardwaj, N. Vaccinia Virus Inhibits the Maturation of Human Dendritic Cells: A Novel Mechanism of Immune Evasion1. The Journal of Immunology 1999, 163, 6762–6768. [CrossRef]
- Object, object Vaccinia Virus Infection of Mature Dendritic Cells Results in Activation of Virus-Specific Naïve CD8+ T Cells: A Potential Mechanism for Direct Presentation.
- Yao, Y.; Li, P.; Singh, P.; Thiele, A.T.; Wilkes, D.S.; Renukaradhya, G.J.; Brutkiewicz, R.R.; Travers, J.B.; Luker, G.D.; Hong, S.-C.; et al. Vaccinia Virus Infection Induces Dendritic Cell Maturation but Inhibits Antigen Presentation by MHC Class II. Cell Immunol 2007, 246, 92–102. [CrossRef]
- Byrd, D.; Shepherd, N.; Lan, J.; Hu, N.; Amet, T.; Yang, K.; Desai, M.; Yu, Q. Primary Human Macrophages Serve as Vehicles for Vaccinia Virus Replication and Dissemination. J Virol 2014, 88, 6819–6831. [CrossRef]
- Zhou, D.; Xu, W.; Ding, X.; Guo, H.; Wang, J.; Zhao, G.; Zhang, C.; Zhang, Z.; Wang, Z.; Wang, P.; et al. Transient Inhibition of Neutrophil Functions Enhances the Antitumor Effect of Intravenously Delivered Oncolytic Vaccinia Virus. Cancer Sci 2024, 115, 1129–1140. [CrossRef]
- Duffy, D.; Perrin, H.; Abadie, V.; Benhabiles, N.; Boissonnas, A.; Liard, C.; Descours, B.; Reboulleau, D.; Bonduelle, O.; Verrier, B.; et al. Neutrophils Transport Antigen from the Dermis to the Bone Marrow, Initiating a Source of Memory CD8+ T Cells. Immunity 2012, 37, 917–929. [CrossRef]
- Abboud, G.; Tahiliani, V.; Desai, P.; Varkoly, K.; Driver, J.; Hutchinson, T.E.; Salek-Ardakani, S. Natural Killer Cells and Innate Interferon Gamma Participate in the Host Defense against Respiratory Vaccinia Virus Infection. Journal of Virology 2015, 90, 129–141. [CrossRef]
- Dokun, A.O.; Kim, S.; Smith, H.R.; Kang, H.S.; Chu, D.T.; Yokoyama, W.M. Specific and Nonspecific NK Cell Activation during Virus Infection. Nat Immunol 2001, 2, 951–956. [CrossRef]
- Chisholm, S.E.; Reyburn, H.T. Recognition of Vaccinia Virus-Infected Cells by Human Natural Killer Cells Depends on Natural Cytotoxicity Receptors. J Virol 2006, 80, 2225–2233. [CrossRef]
- Gillard, G.O.; Bivas-Benita, M.; Hovav, A.-H.; Grandpre, L.E.; Panas, M.W.; Seaman, M.S.; Haynes, B.F.; Letvin, N.L. Thy1+ Nk Cells from Vaccinia Virus-Primed Mice Confer Protection against Vaccinia Virus Challenge in the Absence of Adaptive Lymphocytes. PLOS Pathogens 2011, 7, e1002141. [CrossRef]
- Shepherd, N.; Lan, J.; Li, W.; Rane, S.; Yu, Q. Primary Human B Cells at Different Differentiation and Maturation Stages Exhibit Distinct Susceptibilities to Vaccinia Virus Binding and Infection. J Virol 2019, 93, e00973-19. [CrossRef]
- Goulding, J.; Bouge, R.; Tahiliani, V.; Croft, M.; Salek-Ardakani, S. CD8 T Cells Are Essential for Recovery from a Respiratory Vaccinia Virus Infection. J Immunol 2012, 189, 2432–2440. [CrossRef]
- Bernasconi, N.L.; Traggiai, E.; Lanzavecchia, A. Maintenance of Serological Memory by Polyclonal Activation of Human Memory B Cells. Science 2002, 298, 2199–2202. [CrossRef]
- Liu, L.; Zhong, Q.; Tian, T.; Dubin, K.; Athale, S.K.; Kupper, T.S. Epidermal Injury and Infection during Poxvirus Immunization Is Crucial for the Generation of Highly Protective T Cell-Mediated Immunity. Nat Med 2010, 16, 224–227. [CrossRef]
- Sánchez-Puig, J.M.; Sánchez, L.; Roy, G.; Blasco, R. Susceptibility of Different Leukocyte Cell Types to Vaccinia Virus Infection. Virol J 2004, 1, 10. [CrossRef]
- Hu, Z.; Molloy, M.J.; Usherwood, E.J. CD4+ T-Cell Dependence of Primary CD8+ T-Cell Response against Vaccinia Virus Depends upon Route of Infection and Viral Dose. Cell Mol Immunol 2016, 13, 82–93. [CrossRef]
- Dai, R.; Huang, X.; Yang, Y. γδT Cells Are Required for CD8+ T Cell Response to Vaccinia Viral Infection. Front. Immunol. 2021, 12. [CrossRef]
- Demkowicz, W.E.; Littaua, R.A.; Wang, J.; Ennis, F.A. Human Cytotoxic T-Cell Memory: Long-Lived Responses to Vaccinia Virus. J Virol 1996, 70, 2627–2631.
- Mercer, J.; Knébel, S.; Schmidt, F.I.; Crouse, J.; Burkard, C.; Helenius, A. Vaccinia Virus Strains Use Distinct Forms of Macropinocytosis for Host-Cell Entry. Proceedings of the National Academy of Sciences 2010, 107, 9346–9351. [CrossRef]
- Yaghchi, C.A.; Zhang, Z.; Alusi, G.; Lemoine, N.R.; Wang, Y. Vaccinia Virus, a Promising New Therapeutic Agent for Pancreatic Cancer. Immunotherapy 2015, 7, 1249–1258. [CrossRef]
- Lei, W.; Ye, Q.; Hao, Y.; Chen, J.; Huang, Y.; Yang, L.; Wang, S.; Qian, W. CD19-Targeted BiTE Expression by an Oncolytic Vaccinia Virus Significantly Augments Therapeutic Efficacy against B-Cell Lymphoma. Blood Cancer J. 2022, 12, 1–10. [CrossRef]
- Smith, G.L.; Moss, B. Infectious Poxvirus Vectors Have Capacity for at Least 25 000 Base Pairs of Foreign DNA. Gene 1983, 25, 21–28. [CrossRef]
- Gallardo, F.; Schmitt, D.; Brandely, R.; Brua, C.; Silvestre, N.; Findeli, A.; Foloppe, J.; Top, S.; Kappler-Gratias, S.; Quentin-Froignant, C.; et al. Fluorescent Tagged Vaccinia Virus Genome Allows Rapid and Efficient Measurement of Oncolytic Potential and Discovery of Oncolytic Modulators. Biomedicines 2020, 8, 543. [CrossRef]
- Hiley, C.T.; Yuan, M.; Lemoine, N.R.; Wang, Y. Lister Strain Vaccinia Virus, a Potential Therapeutic Vector Targeting Hypoxic Tumours. Gene Ther 2010, 17, 281–287. [CrossRef]
- Ma, J.; Ramachandran, M.; Jin, C.; Quijano-Rubio, C.; Martikainen, M.; Yu, D.; Essand, M. Characterization of Virus-Mediated Immunogenic Cancer Cell Death and the Consequences for Oncolytic Virus-Based Immunotherapy of Cancer. Cell Death Dis 2020, 11, 1–15. [CrossRef]
- Fucikova, J.; Kepp, O.; Kasikova, L.; Petroni, G.; Yamazaki, T.; Liu, P.; Zhao, L.; Spisek, R.; Kroemer, G.; Galluzzi, L. Detection of Immunogenic Cell Death and Its Relevance for Cancer Therapy. Cell Death Dis 2020, 11, 1–13. [CrossRef]
- O’Leary, M.P.; Choi, A.H.; Kim, S.-I.; Chaurasiya, S.; Lu, J.; Park, A.K.; Woo, Y.; Warner, S.G.; Fong, Y.; Chen, N.G. Novel Oncolytic Chimeric Orthopoxvirus Causes Regression of Pancreatic Cancer Xenografts and Exhibits Abscopal Effect at a Single Low Dose. J Transl Med 2018, 16, 110. [CrossRef]
- Choi, A.H.; O’Leary, M.P.; Lu, J.; Kim, S.-I.; Fong, Y.; Chen, N.G. Endogenous Akt Activity Promotes Virus Entry and Predicts Efficacy of Novel Chimeric Orthopoxvirus in Triple-Negative Breast Cancer. Mol Ther Oncolytics 2018, 9, 22–29. [CrossRef]
- Chaurasiya, S.; Yang, A.; Zhang, Z.; Lu, J.; Valencia, H.; Kim, S.-I.; Woo, Y.; Warner, S.G.; Olafsen, T.; Zhao, Y.; et al. A Comprehensive Preclinical Study Supporting Clinical Trial of Oncolytic Chimeric Poxvirus CF33-hNIS-Anti-PD-L1 to Treat Breast Cancer. Mol Ther Methods Clin Dev 2021, 24, 102–116. [CrossRef]
- Warner, S.G.; Kim, S.-I.; Chaurasiya, S.; O’Leary, M.P.; Lu, J.; Sivanandam, V.; Woo, Y.; Chen, N.G.; Fong, Y. A Novel Chimeric Poxvirus Encoding hNIS Is Tumor-Tropic, Imageable, and Synergistic with Radioiodine to Sustain Colon Cancer Regression. Mol Ther Oncolytics 2019, 13, 82–92. [CrossRef]
- Yang, A.; Zhang, Z.; Chaurasiya, S.; Park, A.K.; Jung, A.; Lu, J.; Kim, S.-I.; Priceman, S.; Fong, Y.; Woo, Y. Development of the Oncolytic Virus, CF33, and Its Derivatives for Peritoneal-Directed Treatment of Gastric Cancer Peritoneal Metastases. J Immunother Cancer 2023, 11, e006280. [CrossRef]
- Chaurasiya, S.; Yang, A.; Zhang, Z.; Lu, J.; Valencia, H.; Kim, S.-I.; Woo, Y.; Warner, S.G.; Olafsen, T.; Zhao, Y.; et al. A Comprehensive Preclinical Study Supporting Clinical Trial of Oncolytic Chimeric Poxvirus CF33-hNIS-Anti-PD-L1 to Treat Breast Cancer. Mol Ther Methods Clin Dev 2021, 24, 102–116. [CrossRef]
- Chen, C.; Park, A.K.; Monroy, I.; Ren, Y.; Kim, S.-I.; Chaurasiya, S.; Priceman, S.J.; Fong, Y. Using Oncolytic Virus to Retask CD19-Chimeric Antigen Receptor T Cells for Treatment of Pancreatic Cancer: Toward a Universal Chimeric Antigen Receptor T-Cell Strategy for Solid Tumor. J Am Coll Surg 2024, 238, 436–447. [CrossRef]
- Earl, P.L.; Moss, B.; Wyatt, L.S. Generation of Recombinant Vaccinia Viruses. Curr Protoc Protein Sci 2017, 89, 5.13.1-5.13.18. [CrossRef]
- Laudermilch, E.; Chandran, K. MAVERICC: Marker-Free Vaccinia Virus Engineering of Recombinants through in Vitro CRISPR/Cas9 Cleavage. J Mol Biol 2021, 433, 166896. [CrossRef]
- Domi, A.; Moss, B. Cloning the Vaccinia Virus Genome as a Bacterial Artificial Chromosome in Escherichia Coli and Recovery of Infectious Virus in Mammalian Cells. Proc Natl Acad Sci U S A 2002, 99, 12415–12420. [CrossRef]
- Yuan, M.; Zhang, W.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.R.; Wang, Y. Efficiently Editing the Vaccinia Virus Genome by Using the CRISPR-Cas9 System. J Virol 2015, 89, 5176–5179. [CrossRef]
- Xu, L.; Sun, H.; Lemoine, N.R.; Xuan, Y.; Wang, P. Oncolytic Vaccinia Virus and Cancer Immunotherapy. Front. Immunol. 2024, 14. [CrossRef]
- Pfeffer, C.M.; Singh, A.T.K. Apoptosis: A Target for Anticancer Therapy. Int J Mol Sci 2018, 19, 448. [CrossRef]
- de Queiroz, N.M.G.P.; Xia, T.; Konno, H.; Barber, G.N. Ovarian Cancer Cells Commonly Exhibit Defective STING Signaling Which Affects Sensitivity to Viral Oncolysis. Molecular Cancer Research 2019, 17, 974–986. [CrossRef]
- Aye, Y.; Li, M.; Long, M.J.C.; Weiss, R.S. Ribonucleotide Reductase and Cancer: Biological Mechanisms and Targeted Therapies. Oncogene 2015, 34, 2011–2021. [CrossRef]
- Bitter, E.E.; Townsend, M.H.; Erickson, R.; Allen, C.; O’Neill, K.L. Thymidine Kinase 1 through the Ages: A Comprehensive Review. Cell & Bioscience 2020, 10, 138. [CrossRef]
- Potts, K.G.; Irwin, C.R.; Favis, N.A.; Pink, D.B.; Vincent, K.M.; Lewis, J.D.; Moore, R.B.; Hitt, M.M.; Evans, D.H. Deletion of F4L (Ribonucleotide Reductase) in Vaccinia Virus Produces a Selective Oncolytic Virus and Promotes Anti-Tumor Immunity with Superior Safety in Bladder Cancer Models. EMBO Mol Med 2017, 9, 638–654. [CrossRef]
- Puhlmann, M.; Brown, C.K.; Gnant, M.; Huang, J.; Libutti, S.K.; Alexander, H.R.; Bartlett, D.L. Vaccinia as a Vector for Tumor-Directed Gene Therapy: Biodistribution of a Thymidine Kinase-Deleted Mutant. Cancer Gene Ther 2000, 7, 66–73. [CrossRef]
- Breitbach, C.J.; Arulanandam, R.; De Silva, N.; Thorne, S.H.; Patt, R.; Daneshmand, M.; Moon, A.; Ilkow, C.; Burke, J.; Hwang, T.-H.; et al. Oncolytic Vaccinia Virus Disrupts Tumor-Associated Vasculature in Humans. Cancer Research 2013, 73, 1265–1275. [CrossRef]
- Lai, A.C.-K.; Pogo, B.G.-T. Attenuated Deletion Mutants of Vaccinia Virus Lacking the Vaccinia Growth Factor Are Defective in Replication in Vivo. Microbial Pathogenesis 1989, 6, 219–226. [CrossRef]
- DeHaven, B.C.; Gupta, K.; Isaacs, S.N. The Vaccinia Virus A56 Protein: A Multifunctional Transmembrane Glycoprotein That Anchors Two Secreted Viral Proteins. Journal of General Virology 2011, 92, 1971–1980. [CrossRef]
- Izmailyan, R.; Chang, W. Vaccinia Virus WR53.5/F14.5 Protein Is a New Component of Intracellular Mature Virus and Is Important for Calcium-Independent Cell Adhesion and Vaccinia Virus Virulence in Mice. J Virol 2008, 82, 10079–10087. [CrossRef]
- Zhang, Q.; Liang, C.; Yu, Y.A.; Chen, N.; Dandekar, T.; Szalay, A.A. The Highly Attenuated Oncolytic Recombinant Vaccinia Virus GLV-1h68: Comparative Genomic Features and the Contribution of F14.5L Inactivation. Mol Genet Genomics 2009, 282, 417–435. [CrossRef]
- Papatriantafyllou, M. GM-CSF in Focus. Nat Rev Immunol 2011, 11, 370–371. [CrossRef]
- Deng, L.; Fan, J.; Guo, M.; Huang, B. Oncolytic and Immunologic Cancer Therapy with GM-CSF-Armed Vaccinia Virus of Tian Tan Strain Guang9. Cancer Letters 2016, 372, 251–257. [CrossRef]
- Xuan, Y.; Yan, W.; Wang, R.; Wang, X.; Guo, Y.; Dun, H.; Huan, Z.; Xu, L.; Han, R.; Sun, X.; et al. GM-CSF and IL-21-Armed Oncolytic Vaccinia Virus Significantly Enhances Anti-Tumor Activity and Synergizes with Anti-PD1 Immunotherapy in Pancreatic Cancer. Front Immunol 2025, 15, 1506632. [CrossRef]
- Li, J.; O’Malley, M.; Urban, J.; Sampath, P.; Guo, Z.S.; Kalinski, P.; Thorne, S.H.; Bartlett, D.L. Chemokine Expression From Oncolytic Vaccinia Virus Enhances Vaccine Therapies of Cancer. Molecular Therapy 2011, 19, 650–657. [CrossRef]
- Liu, Z.; Ravindranathan ,Roshni; Li ,Jun; Kalinski ,Pawel; Guo ,Z. Sheng; and Bartlett, D.L. CXCL11-Armed Oncolytic Poxvirus Elicits Potent Antitumor Immunity and Shows Enhanced Therapeutic Efficacy. OncoImmunology 2016, 5, e1091554. [CrossRef]
- McCart, J.A.; Ward, J.M.; Lee, J.; Hu, Y.; Alexander, H.R.; Libutti, S.K.; Moss, B.; Bartlett, D.L. Systemic Cancer Therapy with a Tumor-Selective Vaccinia Virus Mutant Lacking Thymidine Kinase and Vaccinia Growth Factor Genes. Cancer Res 2001, 61, 8751–8757.
- Guo, Z.S.; Naik, A.; O’Malley, M.E.; Popovic, P.; Demarco, R.; Hu, Y.; Yin, X.; Yang, S.; Zeh, H.J.; Moss, B.; et al. The Enhanced Tumor Selectivity of an Oncolytic Vaccinia Lacking the Host Range and Antiapoptosis Genes SPI-1 and SPI-2. Cancer Res 2005, 65, 9991–9998. [CrossRef]
- Yang, S.; Guo, Z.S.; O’Malley, M.E.; Yin, X.; Zeh, H.J.; Bartlett, D.L. A New Recombinant Vaccinia with Targeted Deletion of Three Viral Genes: Its Safety and Efficacy as an Oncolytic Virus. Gene Ther 2007, 14, 638–647. [CrossRef]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of Interferon-Beta to Produce a Specific, Multi-Mechanistic Oncolytic Vaccinia Virus. PLoS Med 2007, 4, e353. [CrossRef]
- Eradication of Solid Human Breast Tumors in Nude Mice with an Intravenously Injected Light-Emitting Oncolytic Vaccinia Virus | Cancer Research | American Association for Cancer Research Available online: https://aacrjournals.org/cancerres/article/67/20/10038/533730/Eradication-of-Solid-Human-Breast-Tumors-in-Nude (accessed on 28 May 2025).
- Foloppe, J.; Kempf, J.; Futin, N.; Kintz, J.; Cordier, P.; Pichon, C.; Findeli, A.; Vorburger, F.; Quemeneur, E.; Erbs, P. The Enhanced Tumor Specificity of TG6002, an Armed Oncolytic Vaccinia Virus Deleted in Two Genes Involved in Nucleotide Metabolism. Mol Ther Oncolytics 2019, 14, 1–14. [CrossRef]
- Jia, Y.; Wang, Y.; Zhao, G.; Yang, Y.; Yan, W.; Wang, R.; Han, B.; Wang, L.; Zhang, Z.; Chen, L.; et al. Novel Oncolytic Vaccinia Virus Armed with Interleukin-27 Is a Potential Therapeutic Agent for the Treatment of Murine Pancreatic Cancer. J Immunother Cancer 2025, 13, e010341. [CrossRef]
- Neidel, S.; Torres, A.A.; Ren, H.; Smith, G.L. Leaky Scanning Translation Generates a Second A49 Protein That Contributes to Vaccinia Virus Virulence. J Gen Virol 2020, 101, 533–541. [CrossRef]
- Pelin, A.; Foloppe, J.; Petryk, J.; Singaravelu, R.; Hussein, M.; Gossart, F.; Jennings, V.A.; Stubbert, L.J.; Foster, M.; Storbeck, C.; et al. Deletion of Apoptosis Inhibitor F1L in Vaccinia Virus Increases Safety and Oncolysis for Cancer Therapy. Mol Ther Oncolytics 2019, 14, 246–252. [CrossRef]
- Riederer, S.; del Canizo, A.; Navas, J.; Peter, M.G.; Link, E.K.; Sutter, G.; Rojas, J.J. Improving Poxvirus-Mediated Antitumor Immune Responses by Deleting Viral cGAMP-Specific Nuclease. Cancer Gene Ther 2023, 30, 1029–1039. [CrossRef]
- Ge, Y.; Wang, H.; Ren, J.; Liu, W.; Chen, L.; Chen, H.; Ye, J.; Dai, E.; Ma, C.; Ju, S.; et al. Oncolytic Vaccinia Virus Delivering Tethered IL-12 Enhances Antitumor Effects with Improved Safety. J Immunother Cancer 2020, 8, e000710. [CrossRef]
- Shakiba, Y.; Vorobyev, P.O.; Yusubalieva, G.M.; Kochetkov, D.V.; Zajtseva, K.V.; Valikhov, M.P.; Kalsin, V.A.; Zabozlaev, F.G.; Semkina, A.S.; Troitskiy, A.V.; et al. Oncolytic Therapy with Recombinant Vaccinia Viruses Targeting the Interleukin-15 Pathway Elicits a Synergistic Response. Molecular Therapy - Oncolytics 2023, 29, 158–168. [CrossRef]
- Chen, L.; Chen, H.; Ye, J.; Ge, Y.; Wang, H.; Dai, E.; Ren, J.; Liu, W.; Ma, C.; Ju, S.; et al. Intratumoral Expression of Interleukin 23 Variants Using Oncolytic Vaccinia Virus Elicit Potent Antitumor Effects on Multiple Tumor Models via Tumor Microenvironment Modulation. Theranostics 2021, 11, 6668–6681. [CrossRef]
- Chard, L.S.; Maniati, E.; Wang, P.; Zhang, Z.; Gao, D.; Wang, J.; Cao, F.; Ahmed, J.; El Khouri, M.; Hughes, J.; et al. A Vaccinia Virus Armed with Interleukin-10 Is a Promising Therapeutic Agent for Treatment of Murine Pancreatic Cancer. Clinical Cancer Research 2015, 21, 405–416. [CrossRef]
- Mittal, S.K.; Cho, K.-J.; Ishido, S.; Roche, P.A. Interleukin 10 (IL-10)-Mediated Immunosuppression. J Biol Chem 2015, 290, 27158–27167. [CrossRef]
- Holicek, P.; Guilbaud, E.; Klapp, V.; Truxova, I.; Spisek, R.; Galluzzi, L.; Fucikova, J. Type I Interferon and Cancer. Immunol Rev 2024, 321, 115–127. [CrossRef]
- Riederer, S.; del Canizo, A.; Navas, J.; Peter, M.G.; Link, E.K.; Sutter, G.; Rojas, J.J. Improving Poxvirus-Mediated Antitumor Immune Responses by Deleting Viral cGAMP-Specific Nuclease. Cancer Gene Ther 2023, 30, 1029–1039. [CrossRef]
- Hirvinen, M.; Capasso, C.; Guse, K.; Garofalo, M.; Vitale, A.; Ahonen, M.; Kuryk, L.; Vähä-Koskela, M.; Hemminki, A.; Fortino, V.; et al. Expression of DAI by an Oncolytic Vaccinia Virus Boosts the Immunogenicity of the Virus and Enhances Antitumor Immunity. Molecular Therapy - Oncolytics 2016, 3. [CrossRef]
- Wang, X.; Zhou, N.; Liu, T.; Jia, X.; Ye, T.; Chen, K.; Li, G. Oncolytic Vaccinia Virus Expressing White-Spotted Charr Lectin Regulates Antiviral Response in Tumor Cells and Inhibits Tumor Growth In Vitro and In Vivo. Marine Drugs 2021, 19, 292. [CrossRef]
- Hinterberger, M.; Giessel, R.; Fiore, G.; Graebnitz, F.; Bathke, B.; Wennier, S.; Chaplin, P.; Melero, I.; Suter, M.; Lauterbach, H.; et al. Intratumoral Virotherapy with 4-1BBL Armed Modified Vaccinia Ankara Eradicates Solid Tumors and Promotes Protective Immune Memory. J Immunother Cancer 2021, 9, e001586. [CrossRef]
- Parviainen, S.; Ahonen, M.; Diaconu, I.; Hirvinen, M.; Karttunen, Å.; Vähä-Koskela, M.; Hemminki, A.; Cerullo, V. CD40 Ligand and tdTomato-Armed Vaccinia Virus for Induction of Antitumor Immune Response and Tumor Imaging. Gene Ther 2014, 21, 195–204. [CrossRef]
- Yang, N.; Wang, Y.; Liu, S.; Tariq, S.B.; Luna, J.M.; Mazo, G.; Tan, A.; Zhang, T.; Wang, J.; Yan, W.; et al. OX40L-Expressing Recombinant Modified Vaccinia Virus Ankara Induces Potent Antitumor Immunity via Reprogramming Tregs. Journal of Experimental Medicine 2023, 220, e20221166. [CrossRef]
- Yu, F.; Wang, X.; Guo, Z.S.; Bartlett, D.L.; Gottschalk, S.M.; Song, X.-T. T-Cell Engager-Armed Oncolytic Vaccinia Virus Significantly Enhances Antitumor Therapy. Molecular Therapy 2014, 22, 102–111. [CrossRef]
- DePeaux, K.; Rivadeneira, D.B.; Lontos, K.; Dean, V.G.; Gunn, W.G.; Watson, M.J.; Yao, T.; Wilfahrt, D.; Hinck, C.; Wieteska, L.; et al. An Oncolytic Virus–Delivered TGFβ Inhibitor Overcomes the Immunosuppressive Tumor Microenvironment. Journal of Experimental Medicine 2023, 220, e20230053. [CrossRef]
- Frentzen, A.; Yu, Y.A.; Chen, N.; Zhang, Q.; Weibel, S.; Raab, V.; Szalay, A.A. Anti-VEGF Single-Chain Antibody GLAF-1 Encoded by Oncolytic Vaccinia Virus Significantly Enhances Antitumor Therapy. Proceedings of the National Academy of Sciences 2009, 106, 12915–12920. [CrossRef]
- Brown, C.E.; Mackall, C.L. CAR T Cell Therapy: Inroads to Response and Resistance. Nat Rev Immunol 2019, 19, 73–74. [CrossRef]
- Aalipour, A.; Boeuf, F.L.; Tang, M.; Murty, S.; Simonetta, F.; Lozano, A.X.; Shaffer, T.M.; Bell, J.C.; Gambhir, S.S. Viral Delivery of CAR Targets to Solid Tumors Enables Effective Cell Therapy. Molecular Therapy - Oncolytics 2020, 17, 232–240. [CrossRef]
- Park, A.K.; Fong, Y.; Kim, S.-I.; Yang, J.; Murad, J.P.; Lu, J.; Jeang, B.; Chang, W.-C.; Chen, N.G.; Thomas, S.H.; et al. Effective Combination Immunotherapy Using Oncolytic Viruses to Deliver CAR Targets to Solid Tumors. Sci Transl Med 2020, 12, eaaz1863. [CrossRef]
- Moon, E.K.; Wang ,Liang-Chuan S.; Bekdache ,Kheng; Lynn ,Rachel C.; Lo ,Albert; Thorne ,Stephen H.; and Albelda, S.M. Intra-Tumoral Delivery of CXCL11 via a Vaccinia Virus, but Not by Modified T Cells, Enhances the Efficacy of Adoptive T Cell Therapy and Vaccines. OncoImmunology 2018, 7, e1395997. [CrossRef]
- Sivanandam, V.; LaRocca, C.J.; Chen, N.G.; Fong, Y.; Warner, S.G. Oncolytic Viruses and Immune Checkpoint Inhibition: The Best of Both Worlds. Mol Ther Oncolytics 2019, 13, 93–106. [CrossRef]
- Chaurasiya, S.; Chen, N.G.; Fong, Y. Oncolytic Viruses and Immunity. Curr Opin Immunol 2018, 51, 83–90. [CrossRef]
- Sun, Y.; Zhang, Z.; Zhang, C.; Zhang, N.; Wang, P.; Chu, Y.; Dunmall, L.S.C.; Lemoine, N.R.; Wang, Y. An Effective Therapeutic Regime for Treatment of Glioma Using Oncolytic Vaccinia Virus Expressing IL-21 in Combination with Immune Checkpoint Inhibition. Molecular Therapy - Oncolytics 2022, 26, 105–119. [CrossRef]
- Lou, J.; Dong, J.; Xu, R.; Zeng, H.; Fang, L.; Wu, Y.; Liu, Y.; Wang, S. Remodeling of the Tumor Microenvironment Using an Engineered Oncolytic Vaccinia Virus Improves PD-L1 Inhibition Outcomes. Bioscience Reports 2021, 41, BSR20204186. [CrossRef]
- Liu, Z.; Ravindranathan, R.; Kalinski, P.; Guo, Z.S.; Bartlett, D.L. Rational Combination of Oncolytic Vaccinia Virus and PD-L1 Blockade Works Synergistically to Enhance Therapeutic Efficacy. Nat Commun 2017, 8, 14754. [CrossRef]
- Wang, G.; Kang, X.; Chen, K.S.; Jehng, T.; Jones, L.; Chen, J.; Huang, X.F.; Chen, S.-Y. An Engineered Oncolytic Virus Expressing PD-L1 Inhibitors Activates Tumor Neoantigen-Specific T Cell Responses. Nat Commun 2020, 11, 1395. [CrossRef]
- Semmrich, M.; Marchand, J.-B.; Fend, L.; Rehn, M.; Remy, C.; Holmkvist, P.; Silvestre, N.; Svensson, C.; Kleinpeter, P.; Deforges, J.; et al. Vectorized Treg-Depleting αCTLA-4 Elicits Antigen Cross-Presentation and CD8+ T Cell Immunity to Reject ‘Cold’ Tumors. J Immunother Cancer 2022, 10, e003488. [CrossRef]
- Zuo, S.; Wei, M.; Xu, T.; Kong, L.; He, B.; Wang, S.; Wang, S.; Wu, J.; Dong, J.; Wei, J. An Engineered Oncolytic Vaccinia Virus Encoding a Single-Chain Variable Fragment against TIGIT Induces Effective Antitumor Immunity and Synergizes with PD-1 or LAG-3 Blockade. J Immunother Cancer 2021, 9, e002843. [CrossRef]
- Advani, S.J.; Buckel, L.; Chen, N.G.; Scanderbeg, D.J.; Geissinger, U.; Zhang, Q.; Yu, Y.A.; Aguilar, R.J.; Mundt, A.J.; Szalay, A.A. Preferential Replication of Systemically Delivered Oncolytic Vaccinia Virus in Focally Irradiated Glioma Xenografts. Clinical Cancer Research 2012, 18, 2579–2590. [CrossRef]
- Storozynsky, Q.T.; Agopsowicz, K.C.; Noyce, R.S.; Bukhari, A.B.; Han, X.; Snyder, N.; Umer, B.A.; Gamper, A.M.; Godbout, R.; Evans, D.H.; et al. Radiation Combined with Oncolytic Vaccinia Virus Provides Pronounced Antitumor Efficacy and Induces Immune Protection in an Aggressive Glioblastoma Model. Cancer Letters 2023, 562, 216169. [CrossRef]
- Dai, M.H.; Liu, S.L.; Chen, N.G.; Zhang, T.P.; You, L.; Q. Zhang, F.; Chou, T.C.; Szalay, A.A.; Fong, Y.; Zhao, Y.P. Oncolytic Vaccinia Virus in Combination with Radiation Shows Synergistic Antitumor Efficacy in Pancreatic Cancer. Cancer Letters 2014, 344, 282–290. [CrossRef]
- Chen, W.-Y.; Chen, Y.-L.; Lin, H.-W.; Chang, C.-F.; Huang, B.-S.; Sun, W.-Z.; Cheng, W.-F. Stereotactic Body Radiation Combined with Oncolytic Vaccinia Virus Induces Potent Anti-Tumor Effect by Triggering Tumor Cell Necroptosis and DAMPs. Cancer Letters 2021, 523, 149–161. [CrossRef]
- Gholami, S.; Haddad, D.; Chen, C.-H.; Chen, N.G.; Zhang, Q.; Zanzonico, P.B.; Szalay, A.A.; Fong, Y. Novel Therapy for Anaplastic Thyroid Carcinoma Cells Using an Oncolytic Vaccinia Virus Carrying the Human Sodium Iodide Symporter. Surgery 2011, 150, 1040–1047. [CrossRef]
- Gholami, S.; Chen, C.-H.; Lou, E.; De Brot, M.; Fujisawa, S.; Chen, N.G.; Szalay, A.A.; Fong, Y. Vaccinia Virus GLV-1h153 Is Effective in Treating and Preventing Metastatic Triple-Negative Breast Cancer. Annals of Surgery 2012, 256, 437. [CrossRef]
- Haddad, D.; Chen, N.G.; Zhang, Q.; Chen, C.-H.; Yu, Y.A.; Gonzalez, L.; Carpenter, S.G.; Carson, J.; Au, J.; Mittra, A.; et al. Insertion of the Human Sodium Iodide Symporter to Facilitate Deep Tissue Imaging Does Not Alter Oncolytic or Replication Capability of a Novel Vaccinia Virus. J Transl Med 2011, 9, 36. [CrossRef]
- Mansfield, D.C.; Kyula, J.N.; Rosenfelder, N.; Chao-Chu, J.; Kramer-Marek, G.; Khan, A.A.; Roulstone, V.; McLaughlin, M.; Melcher, A.A.; Vile, R.G.; et al. Oncolytic Vaccinia Virus as a Vector for Therapeutic Sodium Iodide Symporter Gene Therapy in Prostate Cancer. Gene Ther 2016, 23, 357–368. [CrossRef]
- Ottolino-Perry, K.; Mealiea, D.; Sellers, C.; Acuna, S.A.; Angarita, F.A.; Okamoto, L.; Scollard, D.; Ginj, M.; Reilly, R.; McCart, J.A. Vaccinia Virus and Peptide-Receptor Radiotherapy Synergize to Improve Treatment of Peritoneal Carcinomatosis. Molecular Therapy - Oncolytics 2023, 29, 44–58. [CrossRef]
- Huang, B.; Sikorski, R.; Kirn, D.H.; Thorne, S.H. Synergistic Anti-Tumor Effects between Oncolytic Vaccinia Virus and Paclitaxel Are Mediated by the IFN Response and HMGB1. Gene Ther 2011, 18, 164–172. [CrossRef]
- Yu, Y.A.; Galanis, C.; Woo, Y.; Chen, N.; Zhang, Q.; Fong, Y.; Szalay, A.A. Regression of Human Pancreatic Tumor Xenografts in Mice after a Single Systemic Injection of Recombinant Vaccinia Virus GLV-1h68. Molecular Cancer Therapeutics 2009, 8, 141–151. [CrossRef]
- Lun, X.Q.; Jang, J.-H.; Tang, N.; Deng, H.; Head, R.; Bell, J.C.; Stojdl, D.F.; Nutt, C.L.; Senger, D.L.; Forsyth, P.A.; et al. Efficacy of Systemically Administered Oncolytic Vaccinia Virotherapy for Malignant Gliomas Is Enhanced by Combination Therapy with Rapamycin or Cyclophosphamide. Clinical Cancer Research 2009, 15, 2777–2788. [CrossRef]
- Hofmann, E.; Weibel, S.; Szalay, A.A. Combination Treatment with Oncolytic Vaccinia Virus and Cyclophosphamide Results in Synergistic Antitumor Effects in Human Lung Adenocarcinoma Bearing Mice. Journal of Translational Medicine 2014, 12, 197. [CrossRef]
- Berchtold, S.; Beil, J.; Raff, C.; Smirnow, I.; Schell, M.; D’Alvise, J.; Gross, S.; Lauer, U.M. Assessing and Overcoming Resistance Phenomena against a Genetically Modified Vaccinia Virus in Selected Cancer Cell Lines. International Journal of Molecular Sciences 2020, 21, 7618. [CrossRef]
- Seubert, C.M.; Stritzker, J.; Hess, M.; Donat, U.; Sturm, J.B.; Chen, N.; Hof, J.M. von; Krewer, B.; Tietze, L.F.; Gentschev, I.; et al. Enhanced Tumor Therapy Using Vaccinia Virus Strain GLV-1h68 in Combination with a β-Galactosidase-Activatable Prodrug Seco-Analog of Duocarmycin SA. Cancer Gene Ther 2011, 18, 42–52. [CrossRef]
- Lee, S.; Yang, W.; Kim, D.K.; Kim, H.; Shin, M.; Choi, K.U.; Suh, D.S.; Kim, Y.H.; Hwang, T.-H.; Kim, J.H. Inhibition of MEK-ERK Pathway Enhances Oncolytic Vaccinia Virus Replication in Doxorubicin-Resistant Ovarian Cancer. Molecular Therapy - Oncolytics 2022, 25, 211–224. [CrossRef]
- Ferguson, M.S.; Dunmall, L.S.C.; Gangeswaran, R.; Marelli, G.; Tysome, J.R.; Burns, E.; Whitehead, M.A.; Aksoy, E.; Alusi, G.; Hiley, C.; et al. Transient Inhibition of PI3Kδ Enhances the Therapeutic Effect of Intravenous Delivery of Oncolytic Vaccinia Virus. Molecular Therapy 2020, 28, 1263–1275. [CrossRef]
- MacTavish, H.; Diallo, J.-S.; Huang, B.; Stanford, M.; Boeuf, F.L.; Silva, N.D.; Cox, J.; Simmons, J.G.; Guimond, T.; Falls, T.; et al. Enhancement of Vaccinia Virus Based Oncolysis with Histone Deacetylase Inhibitors. PLOS ONE 2010, 5, e14462. [CrossRef]
- Kim, M.; Nitschké, M.; Sennino, B.; Murer, P.; Schriver, B.J.; Bell, A.; Subramanian, A.; McDonald, C.E.; Wang, J.; Cha, H.; et al. Amplification of Oncolytic Vaccinia Virus Widespread Tumor Cell Killing by Sunitinib through Multiple Mechanisms. Cancer Research 2018, 78, 922–937. [CrossRef]
- Heo, J.; Breitbach, C.J.; Moon, A.; Kim, C.W.; Patt, R.; Kim, M.K.; Lee, Y.K.; Oh, S.Y.; Woo, H.Y.; Parato, K.; et al. Sequential Therapy With JX-594, A Targeted Oncolytic Poxvirus, Followed by Sorafenib in Hepatocellular Carcinoma: Preclinical and Clinical Demonstration of Combination Efficacy. Molecular Therapy 2011, 19, 1170–1179. [CrossRef]
- Chen, N.G.; and Szalay, A.A. Oncolytic Vaccinia Virus: A Theranostic Agent for Cancer. Future Virology 2010, 5, 763–784. [CrossRef]
- Heo, J.; Reid, T.; Ruo, L.; Breitbach, C.J.; Rose, S.; Bloomston, M.; Cho, M.; Lim, H.Y.; Chung, H.C.; Kim, C.W.; et al. Randomized Dose-Finding Clinical Trial of Oncolytic Immunotherapeutic Vaccinia JX-594 in Liver Cancer. Nat Med 2013, 19, 329–336. [CrossRef]
- Moehler, M.; Heo, J.; Lee, H.C.; Tak, W.Y.; Chao, Y.; Paik, S.W.; Yim, H.J.; Byun, K.S.; Baron, A.; Ungerechts, G.; et al. Vaccinia-Based Oncolytic Immunotherapy Pexastimogene Devacirepvec in Patients with Advanced Hepatocellular Carcinoma after Sorafenib Failure: A Randomized Multicenter Phase IIb Trial (TRAVERSE). Oncoimmunology 2019, 8, 1615817. [CrossRef]
- Abou-Alfa, G.K.; Galle, P.R.; Chao, Y.; Erinjeri, J.; Heo, J.; Borad, M.J.; Luca, A.; Burke, J.; Pelusio, A.; Agathon, D.; et al. PHOCUS: A Phase 3, Randomized, Open-Label Study of Sequential Treatment with Pexa-Vec (JX-594) and Sorafenib in Patients with Advanced Hepatocellular Carcinoma. Liver Cancer 2023, 13, 256–272. [CrossRef]
- Toulmonde, M.; Cousin, S.; Kind, M.; Guegan, J.-P.; Bessede, A.; Le Loarer, F.; Perret, R.; Cantarel, C.; Bellera, C.; Italiano, A. Randomized Phase 2 Trial of Intravenous Oncolytic Virus JX-594 Combined with Low-Dose Cyclophosphamide in Patients with Advanced Soft-Tissue Sarcoma. J Hematol Oncol 2022, 15, 149. [CrossRef]
- Heo, J.; Breitbach, C.; Cho, M.; Hwang, T.-H.; Kim, C.W.; Jeon, U.B.; Woo, H.Y.; Yoon, K.T.; Lee, J.W.; Burke, J.; et al. Phase II Trial of Pexa-Vec (Pexastimogene Devacirepvec; JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus, Followed by Sorafenib in Patients with Advanced Hepatocellular Carcinoma (HCC). JCO 2013, 31, 4122–4122. [CrossRef]
- Kim, S.-G.; Ha, H.K.; Lim, S.; De Silva, N.S.; Pelusio, A.; Mun, J.H.; Patt, R.H.; Breitbach, C.J.; Burke, J.M. Phase II Trial of Pexa-Vec (Pexastimogene Devacirepvec; JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus, in Patients with Metastatic, Refractory Renal Cell Carcinoma (RCC). JCO 2018, 36, 671–671. [CrossRef]
- Immunization Strategy With Intra-Tumoral Injections of Pexa-Vec With Ipilimumab in Metastatic / Advanced Solid Tumors. (ISI-JX) Available Online: Https://Clinicaltrials.Gov/Study/NCT02977156?term=NCT02977156&rank=1 (Accessed on 8/5/2025).
- Monge, C.; Xie, C.; Myojin, Y.; Coffman, K.; Hrones, D.M.; Wang, S.; Hernandez, J.M.; Wood, B.J.; Levy, E.B.; Juburi, I.; et al. Phase I/II Study of PexaVec in Combination with Immune Checkpoint Inhibition in Refractory Metastatic Colorectal Cancer. J Immunother Cancer 2023, 11, e005640. [CrossRef]
- Lauer, U.M.; Schell, M.; Beil, J.; Berchtold, S.; Koppenhöfer, U.; Glatzle, J.; Königsrainer, A.; Möhle, R.; Nann, D.; Fend, F.; et al. Phase I Study of Oncolytic Vaccinia Virus GL-ONC1 in Patients with Peritoneal Carcinomatosis. Clinical Cancer Research 2018, 24, 4388–4398. [CrossRef]
- Pedersen, J.V.; Karapanagiotou, E.M.; Biondo, A.; Tunariu, N.; Puglisi, M.; Denholm, K.A.; Sassi, S.; Mansfield, D.; Yap, T.A.; De Bono, J.S.; et al. A Phase I Clinical Trial of a Genetically Modified and Imageable Oncolytic Vaccinia Virus GL-ONC1 with Clinical Green Fluorescent Protein (GFP) Imaging. JCO 2011, 29, 2577–2577. [CrossRef]
- Mell, L.K.; Brumund, K.T.; Daniels, G.A.; Advani, S.J.; Zakeri, K.; Wright, M.E.; Onyeama, S.-J.; Weisman, R.A.; Sanghvi, P.R.; Martin, P.J.; et al. Phase I Trial of Intravenous Oncolytic Vaccinia Virus (GL-ONC1) with Cisplatin and Radiotherapy in Patients with Locoregionally Advanced Head and Neck Carcinoma. Clinical Cancer Research 2017, 23, 5696–5702. [CrossRef]
- Krug, L.M.; Zauderer, M.G.; Adusumili, P.S.; McGee, E.; Sepkowitz, K.; Klang, M.; Yu, Y.A.; Scigalla, P.; Rusch, V.W. Phase I Study of Intra-Pleural Administration of GL-ONC1, an Oncolytic Vaccinia Virus, in Patients with Malignant Pleural Effusion. JCO 2015, 33, 7559–7559. [CrossRef]
- Chintala, N.K.; Choe, J.K.; McGee, E.; Bellis, R.; Saini, J.K.; Banerjee, S.; Moreira, A.L.; Zauderer, M.G.; Adusumilli, P.S.; Rusch, V.W. Correlative Analysis from a Phase I Clinical Trial of Intrapleural Administration of Oncolytic Vaccinia Virus (Olvi-Vec) in Patients with Malignant Pleural Mesothelioma. Front Immunol 2023, 14, 1112960. [CrossRef]
- Holloway, R.W.; Mendivil, A.A.; Kendrick, J.E.; Abaid, L.N.; Brown, J.V.; LeBlanc, J.; McKenzie, N.D.; Mori, K.M.; Ahmad, S. Clinical Activity of Olvimulogene Nanivacirepvec–Primed Immunochemotherapy in Heavily Pretreated Patients With Platinum-Resistant or Platinum-Refractory Ovarian Cancer. JAMA Oncol 2023, 9, 903–908. [CrossRef]
- Genelux Clinical Trial Summary. Available Online: Https://Genelux.Com/Clinical-Trials-Summary/ (Accessed on May 8, 2025).
- GL-ONC1 Administered Intravenously Prior to Surgery to Patients With Solid Organ Cancers. Available Online: Https://Clinicaltrials.Gov/Study/NCT02714374?intr=NCT02714374&rank=1 (Accessed on May 8, 2025).
- Expanded Access to Provide GL-ONC1 for the Treatment of Advanced Cancers With No Standard of Care. Available Online: Https://Clinicaltrials.Gov/Study/NCT03420430?term=NCT03420430&rank=1 (Accessed on May 8, 2025).
- Safety and Efficacy of the ONCOlytic VIRus Armed for Local Chemotherapy, TG6002/5-FC, in Recurrent Glioblastoma Patients (ONCOVIRAC). Available Online: Https://Clinicaltrials.Gov/Study/NCT03294486?intr=NCT03294486&rank=1 (Accessed on May 8, 2025).
- Bendjama, K.; Cassier, P.; Moreno, V.; Doger, B.; Calvo, E.; de Miguel, M.; Jungels, C.; Erbs, P.; Carpentier, D.; Sadoun, A. Abstract LB179: Oncolytic Virus TG6002 Locates to Tumors after Intravenous Infusion and Induces Tumor-Specific Expression of a Functional pro-Drug Activating Enzyme in Patients with Advanced Gastrointestinal Carcinomas. Cancer Research 2021, 81, LB179. [CrossRef]
- West, E.J.; Sadoun, A.; Bendjama, K.; Erbs, P.; Smolenschi, C.; Cassier, P.A.; de Baere, T.; Sainte-Croix, S.; Brandely, M.; Melcher, A.A.; et al. A Phase I Clinical Trial of Intrahepatic Artery Delivery of TG6002 in Combination with Oral 5-Fluorocytosine in Patients with Liver-Dominant Metastatic Colorectal Cancer. Clin Cancer Res 2025, 31, 1243–1256. [CrossRef]
- Study of TBio-6517 Given Alone or in Combination With Pembrolizumab in Solid Tumors (RAPTOR). Available Online: Https://Clinicaltrials.Gov/Study/NCT04301011?term=NCT04301011&rank=1 (Accessed on May 8, 2025).
- Chang, C.-L.; Ma, B.; Pang, X.; Wu, T.-C.; Hung, C.-F. Treatment With Cyclooxygenase-2 Inhibitors Enables Repeated Administration of Vaccinia Virus for Control of Ovarian Cancer. Molecular Therapy 2009, 17, 1365–1372. [CrossRef]
- Evgin, L.; Acuna, S.A.; Souza, C.T. de; Marguerie, M.; Lemay, C.G.; Ilkow, C.S.; Findlay, C.S.; Falls, T.; Parato, K.A.; Hanwell, D.; et al. Complement Inhibition Prevents Oncolytic Vaccinia Virus Neutralization in Immune Humans and Cynomolgus Macaques. Molecular Therapy 2015, 23, 1066–1076. [CrossRef]
- Lee, N.; Jeon, Y.-H.; Yoo, J.; Shin, S.; Lee, S.; Park, M.-J.; Jung, B.-J.; Hong, Y.-K.; Lee, D.-S.; Oh, K. Generation of Novel Oncolytic Vaccinia Virus with Improved Intravenous Efficacy through Protection against Complement-Mediated Lysis and Evasion of Neutralization by Vaccinia Virus-Specific Antibodies. J Immunother Cancer 2023, 11, e006024. [CrossRef]
Figure 1.
Life cycle of Vaccinia virus. VACV enters host cells either through direct membrane fusion or via micropinocytosis after binding to the cell surface. Following entry, the viral core is released into the cytoplasm, where core-associated enzymes initiate transcription of early genes. These early genes promote viral DNA replication and suppress host antiviral responses. DNA replication then triggers the expression of intermediate genes, which in turn activates the transcription of late genes responsible for virion assembly. VACV produces four distinct forms of virions: (1) Intracellular Mature Virions (IMVs): accumulate within the cytoplasm and are released only upon cell lysis; (2) Intracellular Enveloped Virions (IEVs): acquire additional membranes from the trans-Golgi network or endosomes and are transported the cell surface via microtubules; (3) Cell-associated Enveloped Virions (CEVs): formed when IEVs fuse with the plasma membrane, enabling direct cell-to-cell spread; (4) Extracellular Enveloped Virions (EEVs): a subset of CEVs that are released from the cell and facilitate long-range viral dissemination to distant cells.
Figure 1.
Life cycle of Vaccinia virus. VACV enters host cells either through direct membrane fusion or via micropinocytosis after binding to the cell surface. Following entry, the viral core is released into the cytoplasm, where core-associated enzymes initiate transcription of early genes. These early genes promote viral DNA replication and suppress host antiviral responses. DNA replication then triggers the expression of intermediate genes, which in turn activates the transcription of late genes responsible for virion assembly. VACV produces four distinct forms of virions: (1) Intracellular Mature Virions (IMVs): accumulate within the cytoplasm and are released only upon cell lysis; (2) Intracellular Enveloped Virions (IEVs): acquire additional membranes from the trans-Golgi network or endosomes and are transported the cell surface via microtubules; (3) Cell-associated Enveloped Virions (CEVs): formed when IEVs fuse with the plasma membrane, enabling direct cell-to-cell spread; (4) Extracellular Enveloped Virions (EEVs): a subset of CEVs that are released from the cell and facilitate long-range viral dissemination to distant cells.
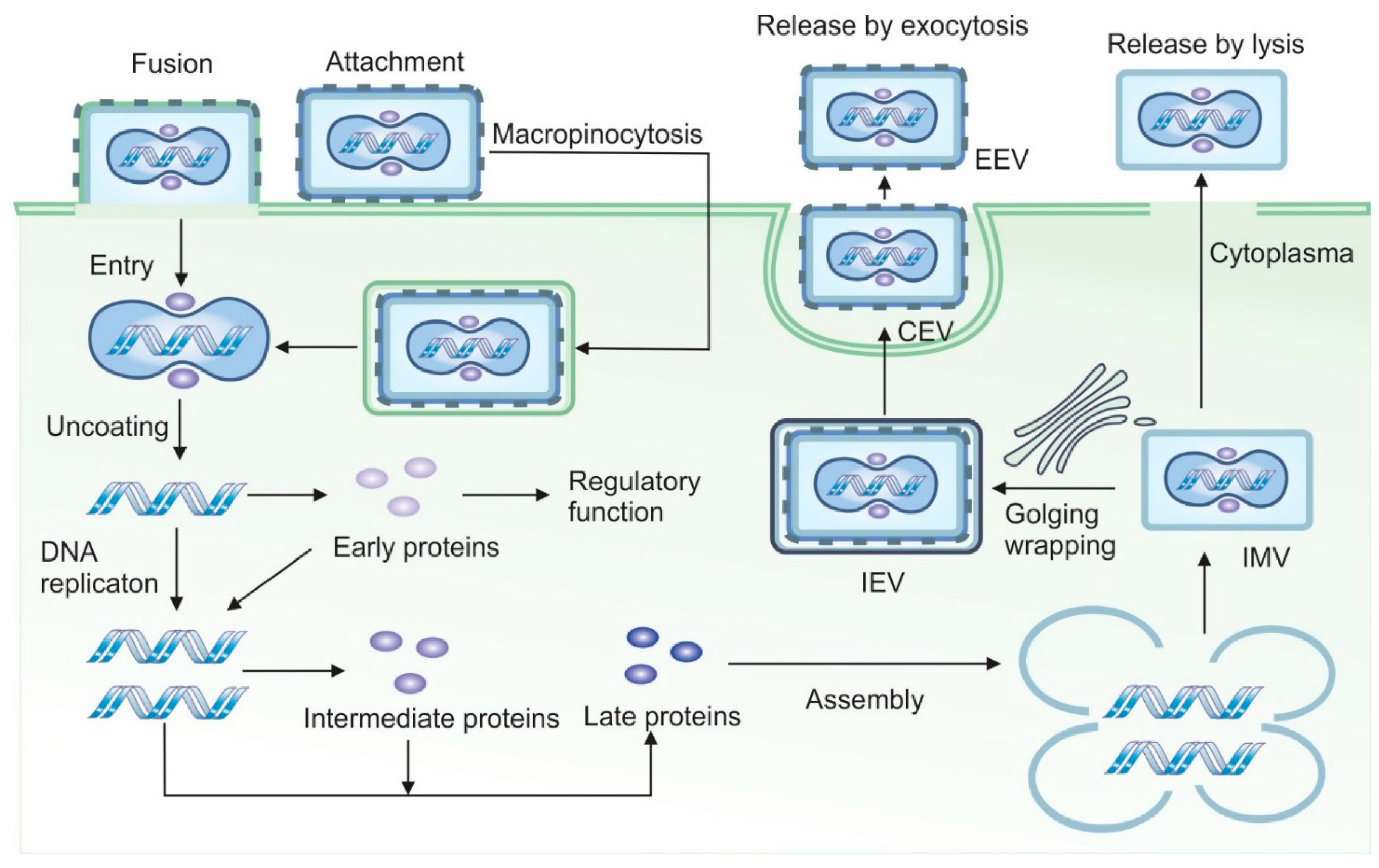
Figure 2.
Inhibition of antiviral mechanisms by Vaccinia virus. VACV has evolved multiple strategies to evade host immune responses at both molecular and cellular levels. It encodes multiple viral proteins that disrupt pathogen recognition receptor (PRR) signaling pathways, including those mediated by cGAS, DNA-PK, and Toll-like receptors (TLRs). These viral proteins inhibit key transcription factors such as interferon regulatory factor 3 (IRF3) and NF-κB, thereby blocking the expression of pro-inflammatory and antiviral genes. VACV also impairs interferon (IFN) signaling by suppressing IFN production, scavenging soluble IFNs, and blocking downstream signaling from IFN receptors. Additionally, the virus secretes immunomodulatory proteins that neutralize pro-inflammatory cytokines. To promote efficient viral replication, VACV encodes multiple proteins that inhibit apoptosis by targeting key components of apoptotic pathways. It also interferes with antigen presentation by antigen-presenting cells (APCs) and suppress the activation of other immune cells, including natural killer (NK) cells and T cells. Finally, VACV expresses a distinct set of proteins that inhibit the complement cascade, further promoting immune evasion.
Figure 2.
Inhibition of antiviral mechanisms by Vaccinia virus. VACV has evolved multiple strategies to evade host immune responses at both molecular and cellular levels. It encodes multiple viral proteins that disrupt pathogen recognition receptor (PRR) signaling pathways, including those mediated by cGAS, DNA-PK, and Toll-like receptors (TLRs). These viral proteins inhibit key transcription factors such as interferon regulatory factor 3 (IRF3) and NF-κB, thereby blocking the expression of pro-inflammatory and antiviral genes. VACV also impairs interferon (IFN) signaling by suppressing IFN production, scavenging soluble IFNs, and blocking downstream signaling from IFN receptors. Additionally, the virus secretes immunomodulatory proteins that neutralize pro-inflammatory cytokines. To promote efficient viral replication, VACV encodes multiple proteins that inhibit apoptosis by targeting key components of apoptotic pathways. It also interferes with antigen presentation by antigen-presenting cells (APCs) and suppress the activation of other immune cells, including natural killer (NK) cells and T cells. Finally, VACV expresses a distinct set of proteins that inhibit the complement cascade, further promoting immune evasion.
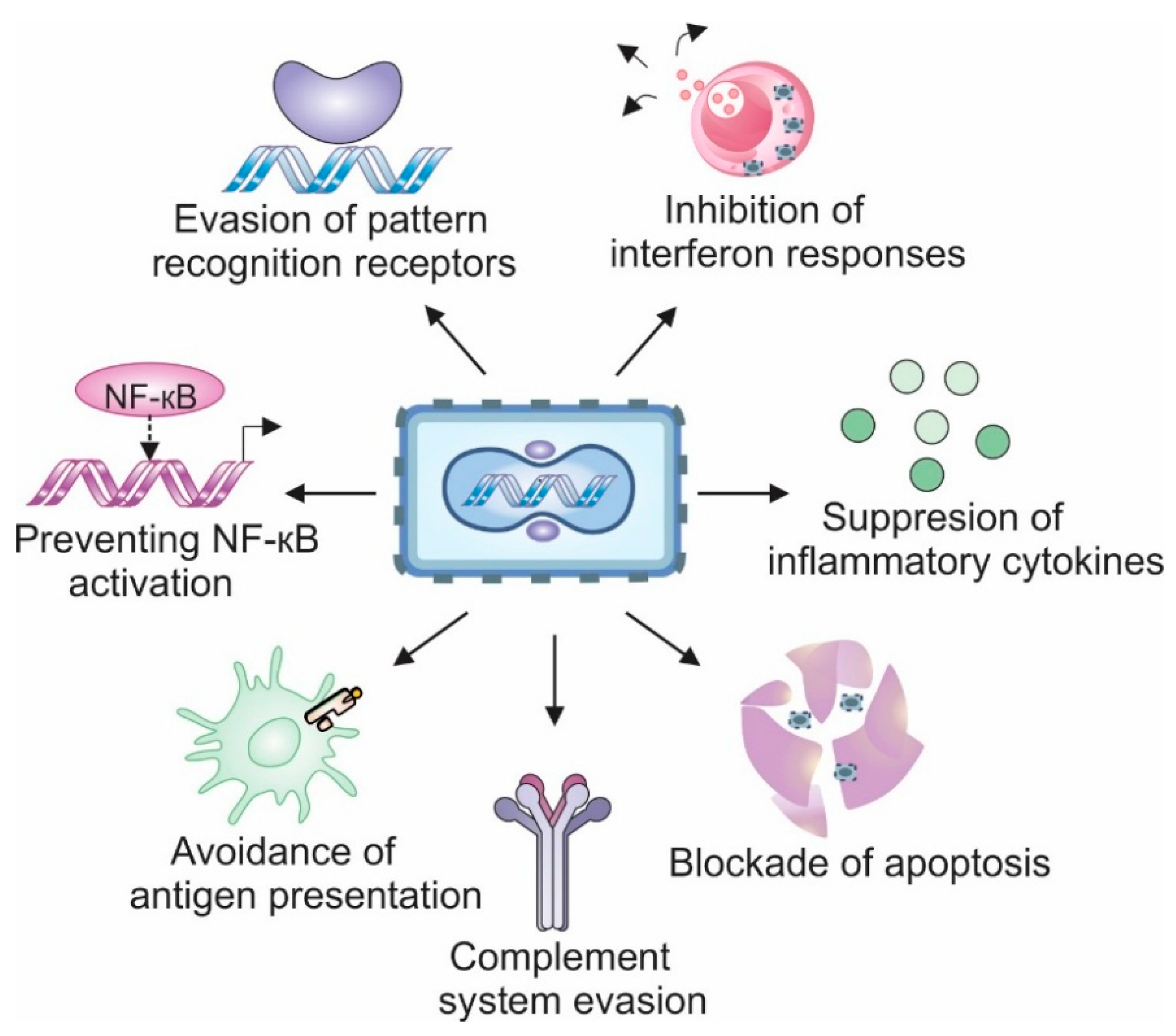
Figure 4.
Inhibition of cell death pathways by Vaccinia virus proteins. VACV employs multiple strategies to inhibit host cell death pathways and promote viral replication. The VACV F1 disrupts the intrinsic apoptosis pathway by blocking caspase-9 activation and suppressing pro-apoptotic factors BAX and BAK. Additionally, B13 functions as a pan-caspase inhibitor, blocking the death receptor-mediated extrinsic apoptosis pathway, whereas N1 blocks this signaling cascade by targeting the pro-apoptotic protein tBid. B13 also inhibits caspase-8 activation, and in its inactive form, caspase-8 allows the stabilization of RIP1 and RIP3, which interact via the RIP homotypic interaction motif (RHIM). This complex promotes the phosphorylation and oligomerization of MLKL (mixed lineage kinase domain-like protein), ultimately leading to plasma membrane rupture and necrotic cell death. VACV can also counteract pyroptosis using F1, which blocks the NLRP1 inflammasome. In addition, B13 inhibits caspase-1 intracellularly, and B15 acts extracellularly as a soluble IL-1β receptor.
Figure 4.
Inhibition of cell death pathways by Vaccinia virus proteins. VACV employs multiple strategies to inhibit host cell death pathways and promote viral replication. The VACV F1 disrupts the intrinsic apoptosis pathway by blocking caspase-9 activation and suppressing pro-apoptotic factors BAX and BAK. Additionally, B13 functions as a pan-caspase inhibitor, blocking the death receptor-mediated extrinsic apoptosis pathway, whereas N1 blocks this signaling cascade by targeting the pro-apoptotic protein tBid. B13 also inhibits caspase-8 activation, and in its inactive form, caspase-8 allows the stabilization of RIP1 and RIP3, which interact via the RIP homotypic interaction motif (RHIM). This complex promotes the phosphorylation and oligomerization of MLKL (mixed lineage kinase domain-like protein), ultimately leading to plasma membrane rupture and necrotic cell death. VACV can also counteract pyroptosis using F1, which blocks the NLRP1 inflammasome. In addition, B13 inhibits caspase-1 intracellularly, and B15 acts extracellularly as a soluble IL-1β receptor.
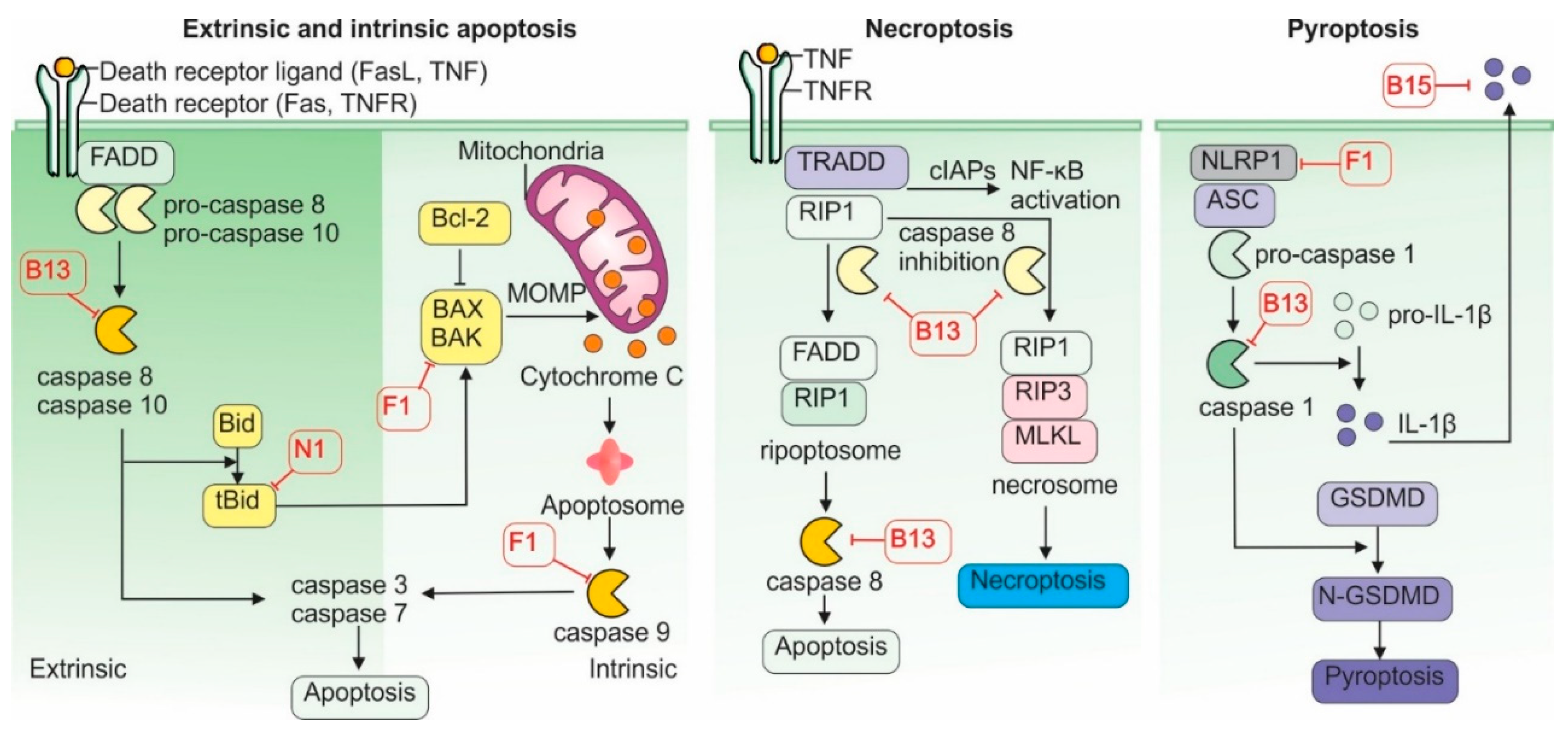
Figure 5.
Vaccinia virus in cancer therapy. Wild-type or chimeric strains of VACV can be genetically engineered in multiple ways to improve both safety and therapeutic efficacy for cancer treatment. Deletion of the thymidine kinase (TK) gene restricts viral replication to rapidly dividing tumor cells, improving tumor selectivity. Removal of the B2R gene enhances host interferon responses further supporting antitumor immunity. Therapeutic transgenes can be inserted into the VACV genome to modulate the tumor microenvironment. For example, expression of cytokines and chemokines such as GM-CSF (Granulocyte-Macrophage Colony-Stimulating Factor) or CCL5 (RANTES) promotes the recruitment and activation of immune cells within tumors. Additionally, the expression of costimulatory molecules such as CD40L or OX40L enhances T cell activation and survival. As an oncolytic agent, VACV can be employed either as a monotherapy or in combination with other treatments to improve therapeutic outcomes. Combination strategies include conventional approaches such as radio- and chemotherapy, as well as emerging modalities such as CAR T cells and other adoptive cell therapies, immune checkpoint inhibitors, and targeted therapies involving antibody-drug conjugates and small molecules inhibitors.
Figure 5.
Vaccinia virus in cancer therapy. Wild-type or chimeric strains of VACV can be genetically engineered in multiple ways to improve both safety and therapeutic efficacy for cancer treatment. Deletion of the thymidine kinase (TK) gene restricts viral replication to rapidly dividing tumor cells, improving tumor selectivity. Removal of the B2R gene enhances host interferon responses further supporting antitumor immunity. Therapeutic transgenes can be inserted into the VACV genome to modulate the tumor microenvironment. For example, expression of cytokines and chemokines such as GM-CSF (Granulocyte-Macrophage Colony-Stimulating Factor) or CCL5 (RANTES) promotes the recruitment and activation of immune cells within tumors. Additionally, the expression of costimulatory molecules such as CD40L or OX40L enhances T cell activation and survival. As an oncolytic agent, VACV can be employed either as a monotherapy or in combination with other treatments to improve therapeutic outcomes. Combination strategies include conventional approaches such as radio- and chemotherapy, as well as emerging modalities such as CAR T cells and other adoptive cell therapies, immune checkpoint inhibitors, and targeted therapies involving antibody-drug conjugates and small molecules inhibitors.
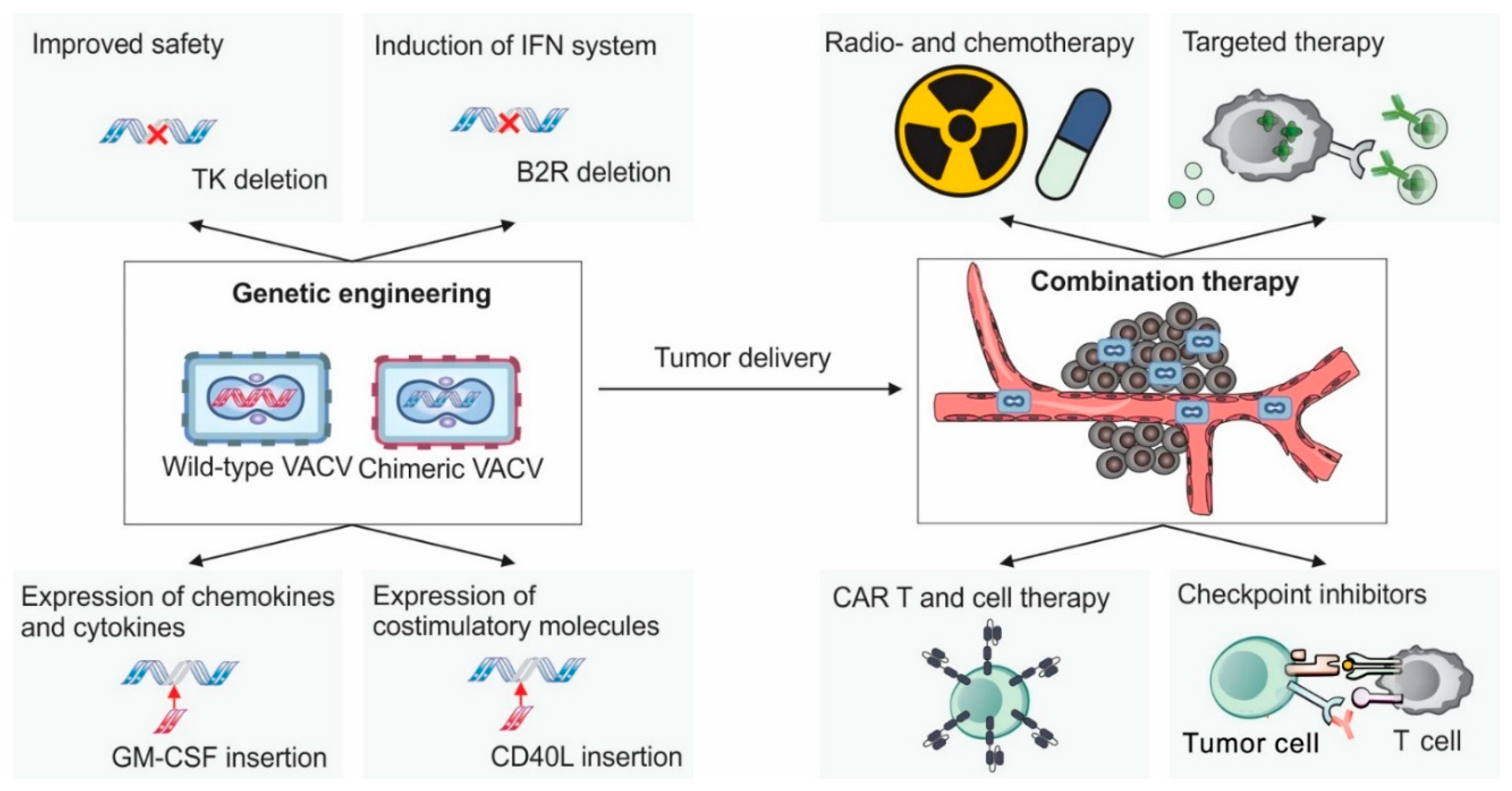
Table 2.
Clinical studies of Vaccinia virus as an oncolytic agent completed between 2009 and 2025.
| Trial# Name, strain and sponsor |
Gene Inactivation | Transgene | Phase (route) | Number of patients (cancer type) | Design | Toxicity | Response | Ref. |
|---|---|---|---|---|---|---|---|---|
|
NCT00554372 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase II (intratumoral) | 30 (Advanced Hepatocellular Carcinoma, HCC) |
Two groups treated with high dose 1x109 PFU and low dose 1x108 PFU, respectively | Fever, chills, fatigue, nausea. Also: hypertension in 10% of patients and thrombocytopenia in 7% |
62.5% of high dose group showed tumor necrosis on MRI vs 35.7% of low-dose group. Median overall survival (OS): 14.1 months for high dose group and 6.7 months for low dose group. Historical controls: 6-8 months | [205] |
|
NCT01387555 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase IIb (Intravenous infusion + later Intratumoral injections) | 129 (Advanced HCC, refractory or ineligible to sorafenib) |
Two groups: Virus + Best Supportive Care (BSC) vs BSC alone. Pexa-Vec was given as a single intravenous (IV) infusion followed by up to 5 IT injections. at 1x109 PFU each |
Fever, chills, fatigue, nausea. Also: hypertension in 8% of patients and thrombocytopenia in 6% |
No statistically significant difference between two groups. Median OS: ~4.2 months (Virus+BSC group) vs. 4.4 months (BSC only) Study terminated early as it did not meet its primary survival endpoint and lacked clear benefit |
[206] |
|
NCT02562755 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase III (intratumoral) |
459 patients (Advanced HCC) |
Three intratumoral doses at 1x109 PFU plus sorafenib orally (400mg BID) vs sorafenib alone | Pyrexia, diarrhea, decreased appetite, nausea, fatigue | No survival benefit, trial was stopped for futility after interim analysis. Median OS: ~ 12.7 months (virus+sorafenib) vs 14.0 months (sorafenib) |
[207] |
|
NCT02630368 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase II (intravenous) |
20 patients (Soft tissue sarcoma) | Dose of 1x109 PFU every 2 weeks for the first 3 injections and then every 3 weeks combined with low-dose cyclophosphamide, compared to the patients treated with low-dose phosphamide only |
Fever and fatigue, single case of grade 3 lymphopenia and grade 3 fever. | Both progression free survival (PFS) and OS were lower in the JX-594+cyclophosphamide group vs the cyclophosphamide-only group. | [208] |
|
NCT01171651 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase II (Intravenous and Intratumoral) |
25 patients with unresectable primary HCC |
One intravenous dose followed by two intratumoral doses each once a week followed by sorafenib at Day 25 | Fever, chills, headache and nausea | 47% of patients displayed responses after treatment with virus only, after including sorafenib, responses were observed in 75% of patients | [209] |
|
Unspecified JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase II (Intravenous) |
17 patients with with Metastatic, Refractory Renal Cell Carcinoma (RCC). |
5 weekly intravenous infusions, followed by extra doses if response is observed | Transient flu-like illness, vomiting, chills, nausea | 76% of patients displayed disease control including one complete response | [210] |
|
NCT02977156 JX-594 (Pexa-Vec/TG6006) Wyeth Transgene |
TK | GM-CSF lacZ |
Phase I (Intravenous and Intratumoral) |
22 patients with Metastatic / Advanced Solid Tumors |
Single intravenous injection at 1x109 PFU followed by up to 4 intratumoral injections every week followed by treatment with ipilimumab |
No data available | The trial is marked as complete but no information about efficacy has been revealed. Since the asset is no longer in the company’s pipeline, it’s likely that efficacy was not satisfactory | [211] |
|
NCT03206073 JX-594 (Pexa-Vec/TG6006) Wyeth SillaJen, Inc. |
TK | GM-CSF lacZ |
Phase I/II (Intravenous and Intratumoral) |
34 patients with refractory metastatic colorectal cancer |
Four doses every 2 weeks at 3x108 or 1x109 PFU followed by treatment with durvalumab. 18 patients received additional single dose of tremelimumab on day 1 | Fever and decreased lymphocyte count | Disease control rate was 12.5% in the group without tremelimumab and 16.7% in the group with tremelimumab. OS was 5.2 months with tremelimumab and 7.5 months without tremelimumab. | [212] |
|
NCT01443260 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase I (intraperitoneal) | 9 patients (7 with peritoneal carcinomatosis and 2 with peritoneal mesothelioma) |
Three doses ranging from 107 to 109 PFU per cycle via intraperitoneal infusion | Decrease in lymphocyte count, increase in C-reactive protein (CRP), pyrexia, and abdominal pain | Stable disease observed in ~30% of evaluable patients (no complete/partial responses). Median OS ~5 months vs 3-6 months in historical controls |
[213] |
|
NCT00794131 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase I (Intravenous) |
24 patients with advanced solid tumors (including colorectal, head/neck, and lung tumors) | IIV infusion (dose-escalation study) 3 infusions at doses ranging from 105 to 109 PFU |
Pyrexia, musculoskeletal pain, fatigue, nausea. | Stable disease observed in 33% patients, but no partial or complete response | [214] |
|
NCT01584284 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase I (Intravenous) |
19 patients with Advanced Head and Neck Carcinoma |
3 cohorts of patients treated with single dose at 3x108, 1x109 and 3x109 PFU. 1 cohort treated with 2 doses at 3x109 PFU and 1 cohort treated with 4 doses at 3x109 PFU every 5-7 days. Virus treatment combined with cis-platin and radiotherapy |
Nausea, fatigue, mucositis, dysphagia | Favorable results in terms of PFS and OS were observed in the virus treated group compared with historical data. | [215] |
|
NCT01766739 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase I (Intraperitoneal) | 18 patients with malignant pleural effusions from mesothelioma, breast or non-small cell lung cancer | Single doses of 1x107, 1x108, 1x109, or 3x109 PFU |
Fever, chills and flu-like symptoms | Virus was detected in tumor tissues, tumor cell density decreased, while density of immune cells increased. Following completion of the trial, patients received subsequent other therapies so it’s hard to precisely determine the effect of virus alone | [216,217] |
|
NCT02759588 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase II (Intraperitoneal) |
27 patients with ovarian cancer | 3 × 109 PFU administered on 2 consecutive days followed by platinum doublet chemotherapy with or without bevacizumab | Pyrexia, abdominal pain and nausea | 54% evaluable by RECIST 1.1 had an objective response, with a median PFS of 11.0 months. | [218] |
|
NCT02714374 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Phase I (Intravenous) |
5 patients with solid organ cancers | 1 × 109 PFU administered intravenously prior to surgery | Limited data, according to the company’s website, it was well tolerated | Study terminated due to insufficient funding. According to company’s website, virus replication and increased infiltration by immune cells were detected in treated patients | [219,220] |
|
NCT03420430 GL-ONC1 (GLV-1h68) Lister Genelux, Inc. |
TK, hemagglutinin, F14.5L | Ruc-GFP, β-glucuronidase, and β-galactosidase | Expanded access (Intravenous) |
4 patients with solid tumors and 6 patients with blood tumors with no standard of care treatment available | IV administration, dose not officially revealed. | Limited data, according to the company’s website, it was well tolerated | No official results posted. | [219,221] |
|
NCT03294486 TG6002 Copenhagen Transgene |
TK, RR | cytosine deaminase and uracil phosphoribosyl transferase | Phase I/IIa (Intravenous) |
78 patients with recurrent glioblastoma | 3 weekly IV infusions at the dose of 1x105 PFU combined with fluorocytosine (5-FC) | Limited data. According to the company, toxicity was low and included usual mild symptoms (fever, fatigue, etc.) | Limited data. According to the official company website, the results were favorable. However, the asset was dropped from the pipeline, suggesting that efficacy was not high enough | [222] |
|
NCT03724071 TG6002 Copenhagen Transgene |
TK, RR | cytosine deaminase and uracil phosphoribosyl transferase | Phase I (Intravenous) |
15 patients with gastric carcinoma | 3 infusions once a week at different doses 3x108, 1x109 and 3x109 PFU combined with 5-Fluorocytosine | None of the patients displayed signs of vaccinia induced disease. | Virus and transgene expression were detected in tumors. No other data available probably due to low efficacy. TG6002 is no longer in the company’s pipeline | [223] |
|
Eudra-CT 2018-004103-39 TG6002 Copenhagen Transgene |
TK, RR | cytosine deaminase and uracil phosphoribosyl transferase | Phase I (Intrahepatic Artery Delivery) |
15 patients with Liver-Dominant Metastatic Colorectal Cancer |
Two infusions 43 days apart with the dose range from 1x106 to 1x109 PFU combined with 5-fluorocytosine | Gastrointestinal disorders, pyrexia, fatigue. | Virus and transgene expression were detected in tumors No patients had an objective response based on a 10-week disease control rate | [224] |
|
Unspecified vvDD Western Reserve NCI |
TK, VGF | NA | Phase I (Intratumoral) | 17 patients with advanced solid tumors | Single injections of the virus with the dose range from 3x107 to 3x109 PFU | Fever, nausea, fatigue | Virus replication detected in tumors, but true clinical benefit was not achieved in any of the patients | [35] |
|
NCT04301011 Tbio-6517 Copenhagen Turnstone Biologics |
25 kb deletion from the virus genome including 32 genes | anti-CTLA-4 antibody, FLT3 ligand (fms7 -like tyrosine kinase 3 ligand; FLT3L), and membrane-bound IL-12 (p35 subunit) | Phase I/IIa (Intratumorally or Intravenously) |
27 Patients with advanced solid tumors | Administered Intratumorally or Intravenously Alone and in Combination with Pembrolizumab, No info on dose revealed |
Lack of information | Study was terminated and the asset was withdrawn from pipeline. Official reason not revealed | [225] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



