
Submitted:
11 June 2025
Posted:
12 June 2025
You are already at the latest version
Abstract
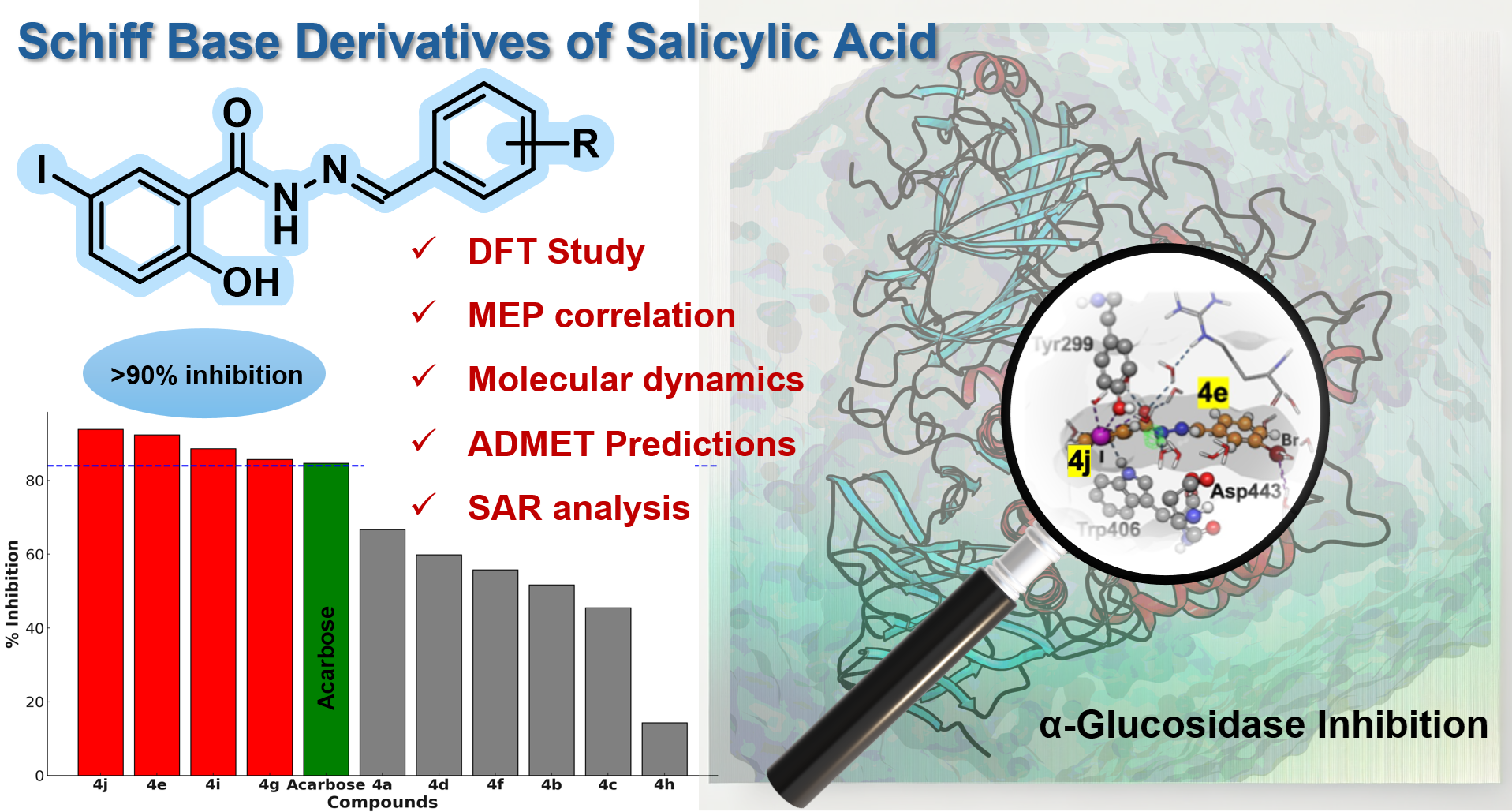
Type 2 diabetes mellitus (T2DM) demands safer and more effective therapies to control postprandial hyperglycemia. Here, we report the synthesis and in vitro evaluation of ten salicylic acid-derived Schiff base derivatives (4a–4j) as α-glucosidase inhibitors. Compounds 4e, 4g, 4i, and 4j exhibited potent enzyme inhibition, with IC50 values ranging from 14.86 to 18.05 µM—substantially better than Acarbose (IC50 = 45.78 µM). Molecular docking and 500 ns molecular dynamics simulations revealed stable enzyme–ligand complexes driven by π–π stacking, halogen bonding, and hydrophobic interactions. Density Functional Theory (DFT) calculations and Molecular Electrostatic Potential (MEP) maps highlighted key electronic factors, while ADMET analysis confirmed favorable drug-like properties and reduced nephrotoxicity. Structure–activity relationship (SAR) analysis emphasized the importance of halogenation and aromaticity in enhancing bioactivity.
Keywords:
α-glucosidase inhibition
; Schiff base derivatives
; Salicylhydrazone
; CNN-based docking
; molecular dynamics
; ADMET profiling
; type 2 diabetes mellitus
1. Introduction
The increasing prevalence of type 2 diabetes mellitus (T2DM) represents a critical global health challenge, with recent reports from the International Diabetes Federation (IDF) estimating that one in ten adults worldwide will have diabetes by 2030 [1,2,3]. Characterized by chronic hyperglycemia due to insulin resistance or impaired insulin secretion, T2DM is a major risk factor for severe complications, including retinopathy, nephropathy, cardiovascular diseases, and neuropathy [2,4]. According to the World Health Organization (WHO), over 830 million people worldwide were affected by diabetes as of 2022, with numbers projected to rise significantly [1]. A key strategy in T2DM management is controlling postprandial blood glucose levels, which mitigates long-term complications [4,5]. α-Glucosidase inhibitors (AGIs) have emerged as effective therapeutic agents, as they delay the digestion and absorption of dietary carbohydrates, thereby preventing rapid glucose spikes [6,7,8,9].
Despite their clinical utility, conventional AGIs such as acarbose, voglibose, and miglitol are often associated with gastrointestinal side effects, including flatulence, diarrhea, and abdominal discomfort due to undigested carbohydrates fermenting in the colon. Studies, such as those by Hanefeld 1998 [10] and Kageyama 1997 [11], have extensively reviewed these limitations, emphasizing the need for novel AGIs with improved safety profiles. These challenges have driven efforts to develop synthetic derivatives and bioactive scaffolds with enhanced selectivity, potency, and reduced adverse effects [12,13].
Among various structural classes, salicylic acid and its derivatives hold significant medicinal importance. As the precursor to aspirin, salicylic acid exhibits unique reactivity due to its ortho-positioned hydroxyl and carboxylic acid groups, which facilitate intramolecular hydrogen bonding and electronic stabilization [14,15]. Recent studies, such as those by Chen 2019 [16] and Aminu 2022 [17], have demonstrated that salicylic acid derivatives exhibit potent α-glucosidase inhibitory activity, highlighting their potential as promising AGI candidates.
Given their versatility, incorporating salicylic acid into hybrid scaffolds has been an effective strategy for enhancing bioactivity. Among various structural classes, Schiff bases have garnered attention for their broad spectrum of biological activities, including antimicrobial, antioxidant, anticancer, anti-inflammatory, and enzyme inhibitory properties [18,19]. Studies, such as those by Afzal 2021 [20] and Sarfaraz 2024 [21], have reported the relevance of Schiff bases in enzyme inhibition, particularly in targeting metabolic enzymes. Schiff bases, formed via the condensation of primary amines with aldehydes or ketones, feature a characteristic imine (–C=N–) linkage, enabling interactions with biological macromolecules, including enzymes and receptors. Their structural simplicity and tunable diversity make Schiff bases attractive candidates for drug discovery [22].
Previous work on Schiff bases derived from salicylic acid scaffolds (Figure 1, top) has demonstrated their biological potential, including antibacterial and enzyme inhibition activities. For instance, compound A [23,24] and compound B [25] exhibit potent antibacterial activity against Gram-positive and Gram-negative bacteria, with MIC values as low as 1.8 µg/mL. Similarly, compound C [26] shows significant antitubercular activity (MIC = 0.39 µg/mL), while compound D [27] effectively inhibits ecKAS III, a key enzyme in bacterial fatty acid biosynthesis. Additionally, compound E [28] targets DNA gyrase and topoisomerase IV, demonstrating dual-inhibitory effects. These reports shows the versatility of Schiff base derivatives of salicylic acid in medicinal chemistry.
Building on this foundation, Khan 2024 [29] (Figure 1, F) demonstrated the potential of Schiff base derivatives of 3,4-dihydroxyphenylacetic acid as α-glucosidase inhibitors. Several of their compounds exhibited superior activity compared to standard drugs like acarbose, underscoring the therapeutic relevance of Schiff bases in T2DM management. These findings support the rationale for further exploration of Schiff base linkages in structurally related scaffolds, such as salicylic acid derivatives, to identify novel AGIs with enhanced pharmacological profiles [30].
Considering the structural diversity and promising bioactivity of Schiff bases, we aimed to design, synthesize, and evaluate a series of salicylic acid-based Schiff base derivatives as α-glucosidase inhibitors. The salicylic acid core was selected due to its ortho-hydroxyl and carboxyl functional groups, which promote intramolecular hydrogen bonding and electronic stabilization. Additionally, iodine substitution at the 5-position of the aromatic ring was introduced to enhance polarizability and facilitate halogen bonding, which can strengthen enzyme interactions. The Schiff base derivatives were synthesized via condensation reactions, where 2-hydroxy-5-iodobenzoic acid hydrazide was coupled with various substituted benzaldehydes. This hydrazone linkage offers hydrogen-bonding and electron-donating capabilities, which are critical for enzyme binding and selectivity.
To elucidate enzyme-ligand interactions, molecular docking and molecular dynamics simulations were performed, identifying key binding interactions at the α-glucosidase active site. Additionally, Density Functional Theory (DFT) calculations provided insights into molecular orbitals (HOMO-LUMO), charge distribution, and electronic properties, supporting SAR-driven optimization. Finally, ADMET analysis was conducted to assess the pharmacokinetic profiles of the designed inhibitors. By integrating experimental and computational methodologies, this study seeks to develop salicylic acid-based Schiff base derivatives as potent α-glucosidase inhibitors with potential applications in T2DM therapy.
2. Materials and Methods
2.1. General Method
All chemicals and reagents were purchased from Sigma-Aldrich or TCI Chemicals and were used without further purification, unless otherwise stated. Solvents, including ethanol (99.5%), were of analytical grade. Reactions were conducted under a dry nitrogen atmosphere using oven-dried glassware. Heating reactions were performed in a paraffin oil bath to ensure uniform temperature control.
Thin-layer chromatography (TLC) was performed on pre-coated silica gel 60 F254 plates (0.25 mm thickness, E. Merck), and spots were visualized under UV light (254 nm and 365 nm) or by heating after dipping in a ninhydrin solution. Column chromatography was performed using silica gel (100–200 mesh or 230–400 mesh), with eluent selection guided by TLC mobility.
2.2. Synthesis and Characterization
2.2.1. 5-Iodo-2-Salicylic Ester (2)
5-Iodo-salicylic acid (2 g, 7.57 mmol) was dissolved in ethanol (40 mL) and stirred at room temperature for 10 min. To this solution, 1 M H2SO4 (0.56 mL) was added dropwise, and the reaction mixture was refluxed at 90°C for 18 hours. The mixture was then diluted with ethyl acetate (40 mL) and washed successively with aqueous sodium bicarbonate. The organic layer was dried over anhydrous sodium sulfate, concentrated under reduced pressure, and purified by column chromatography (silica gel, 20% ethyl acetate in petroleum ether) to yield 5-iodo-2-salicylic ester as a white solid (1.64 g, 74.14%). 1H NMR (600 MHz, DMSO-d6): δ 10.56 (s, 1H), 7.99 (d, J = 2.3 Hz, 1H), 7.77 (dd, J = 8.7, 2.3 Hz, 1H), 6.82 (d, J = 8.7 Hz, 1H), 4.34 (q, J = 7.1 Hz, 2H), 1.33 (t, J = 7.1 Hz, 3H). 13C NMR (151 MHz, DMSO-d6): δ 167.72, 159.99, 143.86, 138.21, 120.59, 116.44, 81.35, 62.08, 14.42. HRMS (TOF) (m/z): [M + H]+ calculated for C9H9IO3 291.9596; found 293.9572.
2.2.2. 5-Iodo-2-Hydroxybenzo-hydrazide (3)
A solution of 5-iodo-2-salicylic ester (10 mmol) and hydrazine hydrate (20 mmol) in ethanol (50 mL) was heated under reflux for 15 hours. The mixture was concentrated and poured onto crushed ice. The resulting solid was filtered, washed with water, dried, and recrystallized from ethanol to yield 5-iodo-2-hydroxybenzo-hydrazide as colorless crystals. 1H NMR (600 MHz, DMSO-d6): δ 10.07 (s, 1H), 8.12 (d, J = 2.2 Hz, 1H), 7.65 (dd, J = 8.6, 2.2 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 166.62, 159.39, 141.79, 135.99, 120.39, 116.44, 80.98. HRMS (TOF) (m/z): [M + H]+ calculated for C7H7IN2O2 277.9552; found 277.9557.
2.2.3. General Procedure for the Synthesis
5-Iodo-2-hydroxybenzo-hydrazide (10 mmol) and the respective substituted benzaldehydes (10 mmol) were dissolved in ethanol (10 mL) containing a few drops of acetic acid. The reaction mixture was refluxed for 5–9 hours. Upon cooling to room temperature, the precipitate was filtered and recrystallized from ethanol, yielding the corresponding derivatives (4a–4j) as crystalline solids [29,31].
(E)-2-hydroxy-5-iodo-N'-(thiazol-2-ylmethylene)benzohydrazide (4a). White solid (68 %); 1H NMR (600 MHz, DMSO-d6): δ 12.06 (s, 1H), 11.60 (s, 1H), 8.66 (s, 1H), 8.08 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 3.2 Hz, 1H), 7.89 (d, J = 3.2 Hz, 1H), 7.73 (dd, J = 8.6, 2.3 Hz, 1H), 6.84 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 164.40, 163.48, 158.33, 144.64, 143.41, 142.28, 137.32, 122.85, 120.29, 120.08, 81.45. HRMS (TOF) (m/z) : [M + H]+ calculated for C11H8IN3O2S 372.9382; found 372.9389.
(E)-2-hydroxy-5-iodo-N'-(pyridin-3-ylmethylene)benzohydrazide (4b). White solid (78%); 1H NMR (600 MHz, DMSO-d6): δ 11.96 (s, 1H), 11.80 (s, 1H), 8.88 (d, J = 2.2 Hz, 1H), 8.64 (dd, J = 4.8, 1.7 Hz, 1H), 8.50 (s, 1H), 8.20 – 8.12 (m, 2H), 7.73 (dd, J = 8.7, 2.2 Hz, 1H), 7.51 (dd, J = 8.0, 4.8 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.60, 158.68, 151.45, 149.40, 146.70, 142.24, 137.21, 134.09, 130.44, 124.55, 120.36, 119.64, 81.42. HRMS (TOF) (m/z) : [M + H]+ calculated for C13H10IN3O2 366.9818; found 366.9836.
(E)-2-hydroxy-N'-(4-hydroxy-3-nitrobenzylidene)-5-iodobenzohydrazide (4c). yellow solid (75%); 1H NMR (600 MHz, DMSO-d6): δ 11.86 (s, 2H), 11.59 (s, 1H), 8.40 (s, 1H), 8.22 (d, J = 2.3 Hz, 1H), 8.15 (d, J = 2.3 Hz, 1H), 7.99 – 7.90 (m, 1H), 7.76 – 7.67 (m, 1H), 7.23 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 8.5 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.53, 158.78, 154.04, 147.41, 142.19, 137.55, 137.10, 133.49, 125.95, 124.80, 120.35, 120.23, 119.48, 81.39. HRMS (TOF) (m/z) : [M + H]+ calculated for C14H10IN3O5 426.9665; found 426.9654.
(E)-2-hydroxy-5-iodo-N'-(4-nitrobenzylidene)benzohydrazide (4d). Yellow solid (78%); 1H NMR (600 MHz, DMSO-d6): δ 12.04 (s, 1H), 11.73 (s, 1H), 8.54 (s, 1H), 8.31 (d, J = 8.6 Hz, 2H), 8.13 (d, J = 2.3 Hz, 1H), 8.00 (d, J = 8.5 Hz, 2H), 7.73 (dd, J = 8.6, 2.3 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.64, 158.50, 148.47, 146.80, 142.29, 140.80, 137.38, 128.66, 124.58, 120.32, 119.83, 81.47. HRMS (TOF) (m/z): [M + H]+ calculated for C14H10IN3O4 410.9716; found 410.9721.
(E)-N'-(3-bromobenzylidene)-2-hydroxy-5-iodobenzohydrazide (4e). White solid (82%); 1H NMR (600 MHz, DMSO-d6): δ 11.94 (s, 1H), 11.83 (s, 1H), 8.41 (s, 1H), 8.15 (d, J = 2.3 Hz, 1H), 7.94 (t, J = 1.8 Hz, 1H), 7.76 – 7.70 (m, 2H), 7.68 – 7.63 (m, 1H), 7.44 (t, J = 7.9 Hz, 1H), 6.84 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.65, 158.71, 147.60, 142.24, 137.20, 136.96, 133.33, 131.53, 129.81, 126.85, 122.67, 120.35, 119.58, 81.41. HRMS (TOF) (m/z): [M + H]+ calculated for C14H10IN2O2 443.8970; found 443.8959.
(E)-2-hydroxy-N'-(4-hydroxybenzylidene)-5-iodobenzohydrazide (4f). White solid (76%); 1H NMR (600 MHz, DMSO-d6): δ 12.07 (s, 1H), 11.70 (s, 1H), 10.00 (s, 1H), 8.34 (s, 1H), 8.18 (d, J = 2.2 Hz, 1H), 7.71 (dd, J = 8.6, 2.2 Hz, 1H), 7.59 (d, J = 8.3 Hz, 2H), 6.84 (dd, J = 24.2, 8.5 Hz, 3H). 13C NMR (151 MHz, DMSO-d6): δ 163.57, 160.21, 159.22, 149.95, 142.14, 136.77, 129.62, 125.40, 120.42, 119.08, 116.25, 81.26. HRMS (TOF) (m/z): [M + H]+ calculated for C14H11IN2O3 381.9814; found 381.9822.
(E)-N'-(2-fluorobenzylidene)-2-hydroxy-5-iodobenzohydrazide (4g). White solid (80%); 1H NMR (600 MHz, DMSO-d6): δ 11.98 (s, 1H), 11.85 (s, 1H), 8.69 (s, 1H), 8.16 (d, J = 2.3 Hz, 1H), 7.96 (td, J = 7.6, 1.7 Hz, 1H), 7.73 (dd, J = 8.7, 2.2 Hz, 1H), 7.56 – 7.51 (m, 1H), 7.37 – 7.30 (m, 2H), 6.84 (d, J = 8.7 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.92, 162.22, 160.56, 159.09, 142.32, 142.24, 142.21, 136.91, 132.88, 132.82, 126.94, 125.51, 122.09, 122.02, 120.42, 119.28, 116.63, 116.49, 81.31. HRMS (TOF) (m/z) [M + H]+ calculated for C14H10FIN2O2 383.9771; found 383.9770.
(E)-2-hydroxy-N'-(2-hydroxy-4-methoxybenzylidene)-5-iodobenzohydrazide (4h). White solid (70%); 1H NMR (600 MHz, DMSO-d6): δ 11.94 (s, 2H), 11.42 (s, 1H), 8.58 (s, 1H), 8.17 (d, J = 2.2 Hz, 1H), 7.72 (dd, J = 8.7, 2.2 Hz, 1H), 7.46 (d, J = 8.6 Hz, 1H), 6.83 (d, J = 8.7 Hz, 1H), 6.54 (dd, J = 8.6, 2.4 Hz, 1H), 6.51 (d, J = 2.5 Hz, 1H), 3.79 (s, 3H). 13C NMR (151 MHz, DMSO-d6): δ 163.29, 162.83, 159.94, 159.10, 150.14, 142.29, 136.85, 131.48, 120.42, 118.88, 112.16, 107.11, 101.62, 81.32, 55.82. HRMS (TOF) (m/z) : [M + H]+ calculated for C15H13IN2O4 411.9920; found 411.9912.
(E)-2-hydroxy-5-iodo-N'-(naphthalen-2-ylmethylene)benzohydrazide (4i). White solid (71%); 1H NMR (600 MHz, DMSO-d6): δ 11.93 (d, J = 14.5 Hz, 2H), 8.61 (s, 1H), 8.19 (d, J = 2.1 Hz, 2H), 8.05 – 8.02 (m, 1H), 8.00 (d, J = 1.2 Hz, 2H), 7.98 – 7.95 (m, 1H), 7.74 (dd, J = 8.6, 2.2 Hz, 1H), 7.60 – 7.57 (m, 2H), 6.85 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.70, 158.91, 149.42, 142.21, 137.05, 134.35, 133.30, 132.24, 129.59, 129.06, 128.90, 128.29, 127.79, 127.31, 123.18, 120.39, 119.55, 81.38. HRMS (TOF) (m/z): [M + H]+ calculated for C18H13IN2O2 416.0022; found 416.0022.
(E)-N'-benzylidene-2-hydroxy-5-iodobenzohydrazide (4j). White solid (76%); 1H NMR (600 MHz, DMSO-d6): δ 11.91 (s, 1H), 11.85 (s, 1H), 8.45 (s, 1H), 8.17 (d, J = 2.2 Hz, 1H), 7.82 – 7.67 (m, 3H), 7.48 (td, J = 5.1, 4.7, 3.0 Hz, 3H), 6.84 (d, J = 8.6 Hz, 1H). 13C NMR (151 MHz, DMSO-d6): δ 163.67, 158.93, 149.51, 142.21, 137.04, 134.48, 130.88, 129.37, 127.77, 120.39, 119.41, 81.36. HRMS (TOF) (m/z) : [M + H]+ calculated for C14H11IN2O2 366.9865; found 366.9856.
2.3. Biological Assay
2.3.1. Screening for α-Glucosidase Inhibitory Activity
The α-glucosidase inhibitory activity of 4a–4j was evaluated using p-nitrophenyl-α-D-glucopyranoside (pNPG) as the substrate. Test samples were prepared as 0.1 mM solutions in dimethyl sulfoxide (DMSO) (Khan 2024). In a 96-well microplate, 10 µL of each sample was mixed with 70 µL of 100 mM phosphate buffer (pH 6.8) and 10 µL of α-glucosidase enzyme solution (0.05 U/mL final concentration). The reaction mixture was incubated at room temperature for 15 minutes, after which 10 µL of the substrate solution (5 mM pNPG in phosphate buffer) was added to each well.
After an additional 1-hour incubation, the release of p-nitrophenol was monitored spectrophotometrically at 450 nm using a microplate reader. Individual blanks were prepared by replacing the substrate with phosphate buffer to correct for background absorbance. Acarbose, a commercially available α-glucosidase inhibitor, was used as a positive control, while DMSO served as the vehicle control [31].
2.3.2. Determination of IC50 Values
For the most active compounds, the IC50 values (concentration required to inhibit 50% of enzyme activity) were determined. Samples were prepared at varying concentrations (100, 50, 25, 12.5, 6.25, 3.125, and 1.5625 µM). The assay was performed as described above, and the percentage inhibition at each concentration was calculated using the following formula:
The percentage inhibition values were plotted against the log-transformed concentrations of the compounds. Nonlinear regression analysis using the four-parameter logistic model was performed to determine the IC50 values.
2.4. Theoretical Study
To complement the experimental findings, comprehensive theoretical investigations were carried out to elucidate the electronic, structural, and pharmacokinetic properties of 4a–4j, as well as their interactions with the α-glucosidase enzyme. The following approaches were employed:
2.4.1. Density Functional Theory (DFT) and Molecular Electrostatic Potential (MEP)
The electronic properties of 4a–4j were evaluated using DFT calculations. Geometry optimizations were carried out using the Gaussian09 software package at the ωB97X-D/def2-tzvpp level of theory [32,33]. No symmetry constraints were applied during the optimization. ΔE were calculated to assess the electronic reactivity of the compounds. The MEP maps were generated to visualize the charge distribution over the molecule's surface, highlighting the electrophilic and nucleophilic regions.
2.4.2. Molecular Docking (MD) and Molecular Dynamics Simulations (MDS)
Molecular docking studies were performed to identify the binding interactions between the Schiff base derivatives and the active site of α-glucosidase. An ensemble docking-virtual screening pipeline was employed using 10 different conformations of α-glucosidase obtained from 500 ns of molecular dynamics simulations. The binding free energies (ΔG) were calculated using GNINA’s convolutional neural network (CNN)-based scoring method [34,35,36].
For the top-ranked compounds, 4e and 4j, molecular dynamics simulations were conducted for 500 ns in triplicate to evaluate the stability and intermolecular interactions of the enzyme-ligand complexes. Key parameters, including root mean square deviations (RMSD), root mean square fluctuations (RMSF), and ligand residence time, were analyzed to confirm the stability and binding efficiency of the compounds.
2.4.3. ADMET Profiling
The pharmacokinetic and toxicity profiles of the synthesized Schiff base derivatives were evaluated using ADMETLab v3.0 (https://admetlab3.scbdd.com/server/evaluationCal) [37]. Parameters such as human intestinal absorption, aqueous solubility, plasma protein binding, cytochrome P450 interactions, and nephrotoxicity potential were calculated to assess their drug-likeness and suitability for therapeutic use.
2.4.4. SAR and Statistical Analysis
The SAR analysis was conducted to identify the structural features of the Schiff base derivatives that contribute to their α-glucosidase inhibitory activity. Key descriptors, including electronic properties, hydrophobicity, and steric factors, were evaluated to correlate structural modifications with biological activity.
All statistical analyses, including analysis of variance (ANOVA) and post-hoc Tukey's HSD tests, were performed using R programming. Nonlinear regression models were utilized to determine IC50 values and their confidence intervals.
All graphs and plots, including dose-response curves, molecular property distributions, and correlation analyses, were generated using Python. Libraries such as Matplotlib and Seaborn were employed to ensure high-quality visual representations of the data.
3. Results and Discussion
3.1. Synthesis and Characterization
The synthesis of Schiff base derivatives 4a–4j was accomplished via a three-step synthetic strategy, as depicted in Scheme 1. The starting material, 5-iodo-salicylic acid 1, underwent esterification using ethanol and catalytic sulfuric acid, yielding 5-iodo-2-salicylic ester 2 in high yield. Subsequent hydrazinolysis of compound 2 with hydrazine hydrate under reflux conditions afforded the key intermediate, 5-iodo-2-hydroxybenzo-hydrazide 3 [38].
Scheme 1.
Synthesis of Schiff base derivatives 4a–4j starting from 5-iodo-salicylic acid.
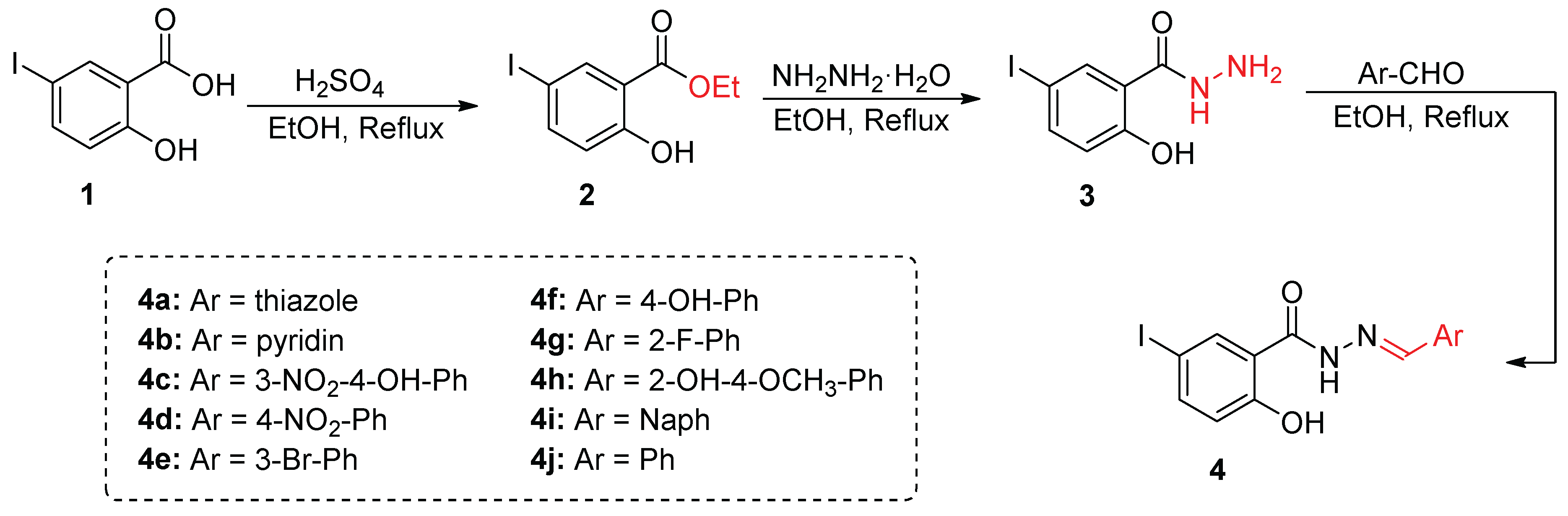
The final step involved the condensation of the hydrazide derivative 3 with various substituted benzaldehydes in the presence of catalytic acetic acid in refluxing ethanol, leading to the formation of 4a–4j. The reaction proceeded smoothly with excellent yields ranging from 68% to 82%, confirming the efficiency and reproducibility of the synthetic route [31].
The structures of the synthesized compounds 4a–4j were confirmed using 1H NMR, 13C NMR, and high-resolution mass spectrometry (HRMS). The 1H NMR spectra confirmed Schiff base formation by the presence of characteristic imine proton signals (–CH=N–) at δ 8.34–8.88 ppm, along with downfield shifts of the hydrazide NH and aromatic protons, indicative of imine formation. The 13C NMR spectra exhibited characteristic signals for the imine carbon (C=N) in the range of δ 158–164 ppm, further supporting the formation of the Schiff base linkage. Additionally, HRMS analysis provided molecular ion peaks consistent with the expected molecular formulas, confirming the structural integrity of the synthesized compounds.
3.2. α-Glucosidase Inhibition Assay
The α-glucosidase inhibitory activity of compounds 4a–4j was evaluated to identify potent enzyme inhibitors [29,31]. The percentage inhibition at a fixed concentration of 100 µM was determined, and the results are summarized in Table 1 and illustrated in Figure 1. Among the tested compounds, 4e, 4j, 4i, and 4g exhibited the highest inhibitory activities, with % inhibition values of 92.4 ± 2.52%, 93.8 ± 1.49%, 88.6 ± 0.78%, and 85.7 ± 1.59%, respectively. These values exceeded the inhibition observed for the standard drug Acarbose (84.7 ± 0.71%), indicating superior inhibitory potential. In contrast, some compounds, such as 4h, demonstrated minimal activity (14.3 ± 7.47%), while others displayed moderate activity, with % inhibition values ranging from 45.5 ± 7.45% (4c) to 66.7 ± 4.27% (4a).
To further quantify the inhibitory potency, IC50 values were determined for the most active compounds (4e, 4g, 4i, and 4j) using a concentration-response assay[20,23]. The IC50 value of 4e was found to be 14.86 ± 0.24 µM, while 4g, 4i, and 4j exhibited IC50 values of 15.58 ± 0.30 µM, 18.05 ± 0.92 µM, and 17.56 ± 0.39 µM, respectively (Figure 2). These values were significantly lower than that of Acarbose (45.78 ± 1.95 µM), confirming the superior potency of these derivatives.
Statistical analysis of the % inhibition data, including one-way ANOVA followed by Tukey’s HSD test, confirmed that the differences between inhibitory activities of the compounds were statistically significant (p < 0.05). The statistical validation indicated that compounds 4e, 4j, and 4i performed significantly better than weaker inhibitors like 4h, reinforcing the observed trends in inhibition.
3.3. Density Functional Theory (DFT) Study
To investigate the electronic properties influencing the reactivity and potential inhibitory activity of compounds 4a–4j, DFT calculations were performed using the ωB97X-D/def2-tzvpp level of theory [32,33]. The HOMO-LUMO energy gaps (ΔE) for the compounds ranged from 7.24 to 8.09 eV, reflecting differences in their electronic reactivity. Compounds with lower ΔE values, such as 4c (7.24 eV) and 4h (7.50 eV), exhibited higher theoretical reactivity, while higher ΔE values, such as in 4b (8.09 eV), indicated reduced electronic reactivity.
Despite these variations, no direct correlation was observed between ΔE values and the IC50 values. For instance, 4e, which exhibited the lowest IC50 value (14.86 ± 0.24 µM), had a relatively high ΔE (8.03 eV), while 4g and 4j, with IC50 values of 15.58 ± 0.30 µM and 17.56 ± 0.39 µM, respectively, also had higher ΔE values (8.04 eV and 8.01 eV, respectively). Meanwhile, 4h, which displayed minimal % inhibition (14.33 ± 7.47%) and had one of the smallest ΔE values (7.50 eV), was not tested for IC50 due to its weak inhibition. These results implies that factors beyond electronic properties, such as steric effects, solvation, and binding dynamics, play a dominant role in determining biological activity.
To complement these findings, molecular electrostatic potential (MEP) maps were generated to visualize charge distribution and identify regions of electrophilicity and nucleophilicity. The MEP analysis revealed that compounds with balanced charge distributions, such as 4e, 4i, and 4j, demonstrated higher inhibition, highlighting the critical role of well-positioned electrophilic and nucleophilic regions in enzyme binding. Conversely, uneven or poorly localized charge distributions, as seen in compounds like 4h, resulted in significantly lower inhibitory activity which proves that optimized charge balance is essential for effective binding interactions (Detailed MEP data is presented in Section 4.3 and 7.9 of the Supporting Information).
3.4. Molecular Dynamics Simulations
An in silico virtual screening campaign consisting of molecular docking, binding free energy calculations, conventional molecular dynamics simulations (cMDS), and drug-likeness analysis was conducted to evaluate the binding potential of Schiff base derivatives toward human α-glucosidase (α-GLU) and to identify potential drug candidates [34].
To improve the accuracy of molecular docking predictions, an ensemble docking-virtual screening pipeline was implemented. This approach utilized ten distinct α-GLU conformations, extracted from 500 ns of cMDS, to account for conformational flexibility and enhance the robustness of molecular docking evaluations [35,36]. The binding free energies and docking scores (kcal mol⁻1) were averaged and further corrected using GNINA’s Convolutional Neural Network (CNN) method for more reliable affinity predictions [39,40].
As shown in Figure 3, Schiff base derivatives exhibited favorable binding free energies, comparable to or better than Acarbose, a well-known commercial α-GLU inhibitor. Among the tested compounds, 4j displayed the lowest energy value (−7.38 kcal mol⁻1), followed by 4e (−7.23 kcal mol⁻1), and 4i and 4g (both ~−7.10 kcal mol⁻1). In comparison, Acarbose exhibited a binding free energy of −7.41 kcal mol⁻1, suggesting that some Schiff base derivatives have comparable or stronger binding potential than the reference inhibitor. The observed differences in binding free energies may be attributed to variations in binding modes, functional group interactions, and molecular flexibility.
3.5. Molecular Dynamics Simulation for 4e and 4j
To further assess the binding stability and interaction dynamics of the most potent inhibitors, cMDS were performed in triplicate for 500 ns for the top-ranked Schiff base derivatives, 4e and 4j. These simulations aimed to evaluate intermolecular interactions, conformational stability, and binding mode persistence within the α-glucosidase (α-GLU) active site. The root mean square deviation (RMSD), root mean square fluctuation (RMSF), and key intermolecular interactions were analyzed to quantify the stability and molecular adaptability of the enzyme-inhibitor complexes.
As illustrated in Figure 4A, the overall conformational stability of the 4e/α-GLU and 4j/α-GLU complexes exhibited minimal structural deviations (<0.5 Å), comparable to the control Acarbose-bound enzyme simulation. Notably, 4e induced fewer atomic motions than Acarbose, suggesting higher stability in the binding pocket. Both compounds also exhibited similar fluctuations in α-GLU side chains, particularly around catalytic residues, further confirming binding site accommodation and retention (Figure 4B).
Analysis of active site flexibility revealed that general acid-base residues within the α-GLU catalytic region underwent restricted motion (<1.4 Å RMSF values) in the presence of 4e and 4j, implying that these inhibitors successfully stabilized the enzyme’s active conformation. The presence of π-stacking interactions between the iodine-modified phenyl moiety of Schiff bases and Trp406 significantly contributed to binding affinity and stability. Additionally, halogen moieties facilitated hydrogen bond formation with adjacent water molecules, reinforcing ligand stability in the enzyme binding pocket.
Interestingly, center-to-edge π-stacking interactions with Tyr299 played a pivotal role in enhancing ligand residence time, measured at 88% for 4e and 92% for 4j during each simulation replica (Figure 4C and 4D). This interaction promoted a single, dominant binding mode, maintaining proximity to catalytically relevant residues. However, despite these stabilizing forces, the formation of hydrogen bonds with surrounding water molecules may increase the ΔG of desolvation, potentially reducing the efficiency of intermolecular interactions and affecting the overall drugability of these Schiff base derivatives.
3.6. ADMET Profiling of Acarbose, 4e, and 4j
The Schiff base derivatives 4e and 4j were selected for further ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) profiling based on their strong enzyme-inhibitory activity, favorable docking scores, and stable ligand-enzyme interactions observed during cMDS [37]. Their pharmacokinetic properties were compared with Acarbose, the reference α-glucosidase inhibitor, to evaluate their potential as oral drug candidates (Table 2).
The predicted human intestinal absorption (HIA) model indicated low absorption for Acarbose, 4e, and 4j, with values ranging from 36.76% to 41.19%, suggesting limited bioavailability. The logP (partition coefficient between octanol/water) values for 4e and 4j fell within the acceptable range for lipophilicity, which is critical for membrane permeability and drug absorption. Calculated aqueous solubility (logS) values for both Schiff base derivatives were lower than Acarbose, indicating reduced solubility, which may require formulation strategies to enhance their bioavailability.
The Caco-2 cell permeability assay, a model for intestinal absorption, predicted values less than -6.0, indicating potential for oral bioavailability despite moderate permeability. In contrast, plasma protein binding (PPB) predictions suggested that 4e and 4j exhibited high protein binding affinity (>98%), which may prolong their half-life and systemic circulation time but could also reduce free drug availability.
In terms of metabolism and clearance, cytochrome P450 inhibition profiling suggested that both Schiff base derivatives strongly inhibit CYP1A2 and CYP2C8, which may lead to potential drug-drug interactions and reduced metabolic clearance. Additionally, nephrotoxicity probability values indicated lower toxicity risks for 4e (0.374) and 4j (0.493) compared to Acarbose (0.981), suggesting that these derivatives pose a reduced risk of kidney toxicity. This indicate that the pharmacokinetic properties of Schiff base derivatives 4e and 4j, highlighting their potential as therapeutic candidates based on their ADMET profiles (Table 2).
3.7. Structure-Activity Relationship (SAR) Analysis
The SAR analysis of derivatives 4a–4j reveals critical structural features that influence α-glucosidase inhibitory activity. The most potent inhibitors, 4j, 4e, 4i, and 4g, exhibited the highest % inhibition values (93.8%, 92.4%, 88.6%, and 85.5%, respectively), suggesting that specific substituents and molecular architectures significantly impact enzyme binding and activity. The planar aromatic systems in 4i (naphthalene) and 4j (phenyl) likely enhance π–π stacking interactions within the enzyme's active site, thereby contributing to their superior inhibitory effects. Similarly, halogenated derivatives such as 4e (3-Br) and 4g (2-F) demonstrated strong inhibition, suggesting that electron-withdrawing groups (EWGs) optimize electronic distribution and hydrophobic interactions, facilitating stable enzyme-ligand interactions.
Conversely, compounds bearing electron-donating groups (EDGs), such as hydroxyl (-OH) and methoxy (-OCH₃) substituents, exhibited reduced activity. For example, 4c (3-NO₂-4-OH) and 4h (2-OH-4-OCH₃) demonstrated only 45.5% and 14.3% inhibition, respectively, indicating that steric hindrance and uneven charge distribution may disrupt the optimal binding conformation at the enzyme’s active site. These findings emphasize that substituent effects, particularly the balance between EWGs and EDGs, are pivotal in modulating inhibitory activity.
The analysis of HOMO-LUMO energy gaps (ΔE values, Table 1) further highlights the complexity of inhibitory activity predictions. While lower ΔE values are typically associated with higher reactivity, no clear correlation was observed in this series. For instance, 4e, which exhibited a relatively high ΔE (8.03 eV), showed exceptional inhibitory activity, whereas 4h, with a lower ΔE (7.50 eV), displayed minimal inhibition. This discrepancy suggests that electronic properties alone cannot fully predict bioactivity, and that structural and steric effects must also be considered.
MEP maps further confirmed that balanced charge distributions in 4e, 4i, and 4j contribute to favorable enzyme binding, reinforcing their high inhibitory potential. This indicate that optimal electronic properties, combined with steric accessibility and favorable binding interactions, are essential for enhancing α-glucosidase inhibition.
The SAR analysis underscores the multifaceted nature of enzyme inhibition, where electronic, structural, and steric factors collectively determine activity. Among the synthesized Schiff base derivatives, 4e and 4j emerge as the most promising candidates, demonstrating high inhibitory activity and favorable molecular properties. (Detailed SAR analysis is provided in Section 7 of the Supporting Information).
4. Conclusions
This study highlights the therapeutic potential of salicylic acid-based Schiff base derivatives as α-glucosidase inhibitors, offering a promising approach for managing postprandial hyperglycemia in Type 2 diabetes mellitus (T2DM). Among the synthesized derivatives, 4e, 4g, 4j, and 4i exhibited significant inhibitory activity, with IC50 values notably lower than Acarbose, indicating their potential as lead compounds for further development. Molecular dynamics simulations revealed stable ligand-enzyme interactions, including π–π stacking and halogen bonding, which likely contribute to their enhanced potency. Additionally, DFT studies provided insights into their electronic properties, while SAR analysis identified key structural features, such as resonance stabilization and halogen substitution, that promote effective enzyme binding.
While these results are promising, the study is limited to in vitro assays, necessitating further validation through in vivo experiments. Future studies should focus on structural optimization to improve potency and selectivity, along with comprehensive pharmacokinetic and toxicological evaluations to assess drug-likeness and safety. These results provide a strong foundation for advancing Schiff base derivatives toward preclinical evaluation, contributing to the development of innovative therapeutic strategies for T2DM management.
Supplementary Materials
The following supporting information can be downloaded at the website of this paper posted on Preprints.org.
Author Contributions
Conceptualization: T.J.P. and S.V.P.; Data curation: S.K.B., F.H.R., A.V.L., B.K.G., V.D.B., T.J.P. and S.V.P.; Formal analysis: A.V.L., F.H.R., J.O.C.J.H., E.D.A., and T.J.P.; Funding acquisition: F.H.R. A.V.L., E.D.A., T.J.P. and S.V.P.; Investigation: S.K.B., A.V.L., B.K.G., V.D.B. and F.H.R.; Methodology: S.K.B., A.V.L., T.J.P. and S.V.P.; Project administration: J.O.C.J.H., E.D.A., F.H.R., T.J.P. and S.V.P.; Software: A.V.L. and T.J.P.; Resources: F.H.R., E.D.A., T.J.P. and S.V.P.; Supervision: T.J.P. and S.V.P.; Validation: A.V.L., J.O.C.J.H., E.D.A., T.J.P. and S.V.P.; Visualization: A.V.L. and T.J.P.; Writing – Original Draft: F.H.R., A.V.L., J.O.C.J.H., T.J.P. and S.V.P.; Writing – Review & Editing: T.J.P. and S.V.P. All authors have read and agreed to the published version of the manuscript.
Funding
National Supercomputing Center—IPICYT (Instituto Potosino de Investigación Científica y Tecnológica, A.C.) for the computational research [TKII-AMVL001] and supercomputing resources at Miztli [LANCAD-UNAM-DGTIC-347].
Data Availability Statement
No new data was created.
Acknowledgments
Authors thank Dr. Pradeep Chopra and Dr. Geert-Jan Boons (complex carbohydrate research center, University of Georgia, USA) for their assistance with glucosidase inhibition assay. Authors also acknowledge the technical support of Emanuel Villafán in Huitzilin High-Performance Computing at INECOL and Patricia Romero-Arellano for technical support.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- World Health Organization. Diabetes: Fact Sheet; World Health Organization: Geneva, 2024; Available online: https://www.who.int/news-room/fact-sheets/detail/diabetes (accessed on 31 December 2024).
- International Diabetes Federation. IDF Diabetes Atlas, 10th ed.; International Diabetes Federation: Brussels, 2021; Available online: https://diabetesatlas.org (accessed on 31 December 2024).
- International Diabetes Federation. Diabetes Facts and Figures; International Diabetes Federation: Brussels, 2024; Available online: https://idf.org (accessed on 31 December 2024).
- American Diabetes Association Professional Practice Committee. Retinopathy, Neuropathy, and Foot Care: Standards of Medical Care in Diabetes—2022. Diabetes Care 2022, 45, S185–S194. [Google Scholar] [CrossRef]
- Ratner, R. E. Controlling Postprandial Hyperglycemia. Am. J. Cardiol. 2001, 88, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Citarella, A.; Cavinato, M.; Rosini, E.; Shehi, H.; Ballabio, F.; Camilloni, C.; Fasano, V.; Silvani, A.; Passarella, D.; Pollegioni, L.; Nardini, M. Nicotinic Acid Derivatives as Novel Noncompetitive α-Amylase and α-Glucosidase Inhibitors for Type 2 Diabetes Treatment. ACS Med. Chem. Lett. 2024, 15, 1474–1481. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, C.; Hu, L. Fragment-Based Dynamic Combinatorial Chemistry for Identification of Selective α-Glucosidase Inhibitors. ACS Med. Chem. Lett. 2022, 13, 1791–1796. [Google Scholar] [CrossRef] [PubMed]
- Mwakalukwa, R.; Amen, Y.; Nagata, M.; Shimizu, K. Postprandial Hyperglycemia Lowering Effect of the Isolated Compounds from Olive Mill Wastes – An Inhibitory Activity and Kinetics Studies on α-Glucosidase and α-Amylase Enzymes. ACS Omega 2020, 5, 20070–20079. [Google Scholar] [CrossRef]
- Xu, Y.; Xie, L.; Xie, J.; Liu, Y.; Chen, W. Pelargonidin-3-O-Rutinoside as a Novel α-Glucosidase Inhibitor for Improving Postprandial Hyperglycemia. Chem. Commun. 2019, 55, 39–42. [Google Scholar] [CrossRef]
- Hanefeld, M. The Role of Acarbose in the Treatment of Non-Insulin-Dependent Diabetes Mellitus. J. Diabetes Complicat. 1998, 12, 228–237. [Google Scholar] [CrossRef]
- Kageyama, S.; Nakamichi, N.; Sekino, H.; Nakano, S. Comparison of the Effects of Acarbose and Voglibose in Healthy Subjects. Clin. Ther. 1997, 19, 720–729. [Google Scholar] [CrossRef]
- Kashtoh, H.; Baek, K.-H. Recent Updates on Phytoconstituent Alpha-Glucosidase Inhibitors: An Approach towards the Treatment of Type Two Diabetes. Plants 2022, 11, 2722. [Google Scholar] [CrossRef]
- Cai, Y.-S.; Xie, H.-X.; Zhang, J.-H.; Li, Y.; Zhang, J.; Wang, K.-M.; Jiang, C.-S. An Updated Overview of Synthetic α-Glucosidase Inhibitors: Chemistry and Bioactivities. Curr. Med. Chem. 2023, 23, 2488–2526. [Google Scholar] [CrossRef]
- Moya-Garzón, M. D.; Martín Higueras, C.; Peñalver, P.; Romera, M.; Fernandes, M. X.; Franco-Montalbán, F.; Gómez-Vidal, J. A.; Salido, E.; Díaz-Gavilán, M. Salicylic Acid Derivatives Inhibit Oxalate Production in Mouse Hepatocytes with Primary Hyperoxaluria Type 1. J. Med. Chem. 2018, 61, 7144–7167. [Google Scholar] [CrossRef]
- Yang, X.; Forster, E. R.; Darabedian, N.; Kim, A. T.; Pratt, M. R.; Shen, A.; Hang, H. C. Translation of Microbiota Short-Chain Fatty Acid Mechanisms Affords Anti-Infective Acyl-Salicylic Acid Derivatives. ACS Chem. Biol. 2020, 15, 1141–1147. [Google Scholar] [CrossRef]
- Chen, J.; Lu, W.; Chen, H.; Bian, X.; Yang, G. A New Series of Salicylic Acid Derivatives as Non-Saccharide α-Glucosidase Inhibitors and Antioxidants. Biol. Pharm. Bull. 2019, 42, 231–246. [Google Scholar] [CrossRef] [PubMed]
- Aminu, K. S.; Uzairu, A.; Umar, A. B.; Ibrahim, M. T. Salicylic Acid Derivatives as Potential α-Glucosidase Inhibitors: Drug Design, Molecular Docking, and Pharmacokinetic Studies. Bull. Natl. Res. Cent. 2022, 46, 162. [Google Scholar] [CrossRef]
- Harohally, N.V.; Cherita, C.; Bhatt, P.; Appaiah, K.A. Antiaflatoxigenic and Antimicrobial Activities of Schiff Bases of 2-Hydroxy-4-methoxybenzaldehyde, Cinnamaldehyde, and Similar Aldehydes. J. Agric. Food Chem. 2017, 65, 8773–8778. [Google Scholar] [CrossRef] [PubMed]
- Thakor, P.M.; Patel, J.D.; Patel, R.J.; Chaki, S.H.; Khimani, A.J.; Vaidya, Y.H.; Chauhan, A.P.; Dholakia, A.B.; Patel, V.C.; Patel, A.J.; Bhavsar, N.H.; Patel, H.V. Exploring New Schiff Bases: Synthesis, Characterization, and Multifaceted Analysis for Biomedical Applications. ACS Omega 2024, 9, 35431–35448. [Google Scholar] [CrossRef]
- Afzal, H.R.; Khan, N.H.; Sultana, K.; Mobashar, A.; Lareb, A.; Khan, A.; Gull, A.; Afzaal, H.; Khan, M.T.; Rizwan, M.; Imran, M. Schiff Bases of Pioglitazone Provide Better Antidiabetic and Potent Antioxidant Effect in a Streptozotocin–Nicotinamide-Induced Diabetic Rodent Model. ACS Omega 2021, 6, 4470–4479. [Google Scholar] [CrossRef]
- Sarfraz, M.; Ayyaz, M.; Rauf, A.; Yaqoob, A.; Tooba, *!!! REPLACE !!!*; Ali, M.A.; Siddique, S.A.; Qureshi, A.M.; Sarfraz, M.H.; Aljowaie, R.M.; Almutairi, S.M.; Arshad, M. New Pyrimidinone Bearing Aminomethylenes and Schiff Bases as Potent Antioxidant, Antibacterial, SARS-CoV-2, and COVID-19 Main Protease MPro Inhibitors: Design, Synthesis, Bioactivities, and Computational Studies. ACS Omega 2024, 9, 25730–25747. [Google Scholar] [CrossRef]
- Mushtaq, I.; Ahmad, M.; Saleem, M.; Ahmed, A. Pharmaceutical Significance of Schiff Bases: An Overview. Futur. J. Pharm. Sci. 2024, 10, 16. [Google Scholar] [CrossRef]
- Souza, A. O.; Galetti, F.; Silva, C. L.; Bicalho, B.; Parma, M. M.; Fonseca, S. F.; et al. Synthesis and Antimicrobial Activity of Novel Schiff Bases. Quim. Nova 2007, 30, 1563–1566. [Google Scholar] [CrossRef]
- Hejchman, E.; Kruszewska, H.; Maciejewska, D.; Sowirka-Taciak, B.; Tomczyk, M.; Sztokfisz-Ignasiak, A.; Jankowski, J.; Młynarczuk-Biały, I. Design, Synthesis, and Biological Activity of Schiff Bases Bearing Salicyl and 7-Hydroxycoumarinyl Moieties. Monatsh. Chem. 2019, 150, 255–266. [Google Scholar] [CrossRef]
- Shi, L.; Ge, H.-M.; Tan, S.-H.; Li, H.-Q.; Song, Y.-C.; Zhu, H.-L. Antibacterial Activity of Schiff Bases Derived from 3-Hydroxyquinoxaline-2-carboxaldehyde. Eur. J. Med. Chem. 2007, 42, 558–564. [Google Scholar] [CrossRef]
- Abdu-Allah, H. H.; Youssif, B. G.; Abdelrahman, M. H.; Abdel-Hamid, M. K.; Reshma, R. S.; Yogeeswari, P.; et al. Synthesis and Biological Activity of Hydrazones of o- and p-Hydroxybenzoic Acids. Russ. J. Gen. Chem. 2017, 87, 2299–2306. [Google Scholar] [CrossRef]
- Cheng, K.; Zheng, Q.-Z.; Hou, J.; Zhou, Y.; Liu, C.-H.; Zhao, J.; et al. Design, Synthesis, and Biological Evaluation of Novel Schiff Bases as Enzyme Inhibitors. Bioorg. Med. Chem. 2010, 18, 2447–2455. [Google Scholar] [CrossRef]
- Al-Wahaibi, L. H.; Mahmoud, M. A.; Alzahrani, H. A.; Abou-Zied, H. A.; Gomaa, H. A. M.; Youssif, B. G. M.; Bräse, S.; Rabea, S. M. Investigating Novel Chemical Scaffolds in Medicinal Chemistry. Front. Chem. 2024, 12, Article–1419242. [Google Scholar] [CrossRef]
- Khan, H.; Jan, F.; Shakoor, A.; Khan, A.; AlAsmari, A. F.; Alasmari, F.; et al. Design, Synthesis, Molecular Docking Study, and α-Glucosidase Inhibitory Evaluation of Novel Hydrazide–Hydrazone Derivatives of 3,4-Dihydroxyphenylacetic Acid. Sci. Rep. 2024, 14, 11410. [Google Scholar] [CrossRef]
- Guha, R. On Exploring Structure–Activity Relationships. In Methods in Molecular Biology; Ekins, S., Ed.; Humana Press: Totowa, NJ, 2013. [Google Scholar] [CrossRef]
- Waziri, I.; Yusuf, T. L.; Kelani, M. T.; Akintemi, E. O.; Olofinsan, K. A.; Muller, A. J. Exploring the Potential of N-Benzylidenebenzohydrazide Derivatives as Antidiabetic and Antioxidant Agents: Design, Synthesis, Spectroscopic, Crystal Structure, DFT, and Molecular Docking Study. ChemistrySelect 2024, 9, e202401631. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-Range Corrected Hybrid Density Functionals with Damped Atom–Atom Dispersion Corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef]
- Sim, L.; Quezada-Calvillo, R.; Sterchi, E.E.; Nichols, B.L.; Rose, D.R. Human Intestinal Maltase–Glucoamylase: Crystal Structure of the N-Terminal Catalytic Subunit and Basis of Inhibition and Substrate Specificity. J. Mol. Biol. 2008, 375, 782–792. [Google Scholar] [CrossRef]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent Treatment of Internal and Surface Residues in Empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Maxwell, D.S.; Tirado-Rives, J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. [Google Scholar] [CrossRef]
- Xiong, G.; Wu, Z.; Yi, J.; Fu, L.; Yang, Z.; Hsieh, C.; Yin, M.; Zeng, X.; Wu, C.; Chen, X.; Hou, T.; Cao, D. ADMETlab 2.0: An Integrated Online Platform for Accurate and Comprehensive Predictions of ADMET Properties. Nucleic Acids Res. 2021, 49, W5–W14. [Google Scholar] [CrossRef] [PubMed]
- Nurkenov, O.A.; Satpaeva, Zh.B.; Schepetkin, I.A.; Khlebnikov, A.I.; Turdybekov, K.M.; Seilkhanov, T.M.; Fazylov, S.D. Synthesis and Biological Activity of Hydrazones of o- and p-Hydroxybenzoic Acids. Russ. J. Gen. Chem. 2017, 87, 2299–2306. [Google Scholar] [CrossRef]
- McNutt, A.T.; Francoeur, P.; Aggarwal, R.; Masuda, T.; Meli, R.; Ragoza, M.; Sunseri, J.; Koes, D.R. GNINA 1.0: Molecular Docking with Deep Learning. J. Cheminf. 2021, 13, 43. [Google Scholar] [CrossRef]
- Quiroga, R.; Villarreal, M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS One 2016, 11, e0155183. [Google Scholar] [CrossRef]
Figure 1.
Comparison of previous work on Schiff base derivatives (A–F) with this study. Previous studies explored antibacterial, antitubercular, and α-glucosidase inhibition activities (top). Structural features of Schiff base derivatives. Key functional groups and substitutions enhance electronic balance, molecular stability, and enzyme binding efficiency (bottom).
Figure 1.
Comparison of previous work on Schiff base derivatives (A–F) with this study. Previous studies explored antibacterial, antitubercular, and α-glucosidase inhibition activities (top). Structural features of Schiff base derivatives. Key functional groups and substitutions enhance electronic balance, molecular stability, and enzyme binding efficiency (bottom).
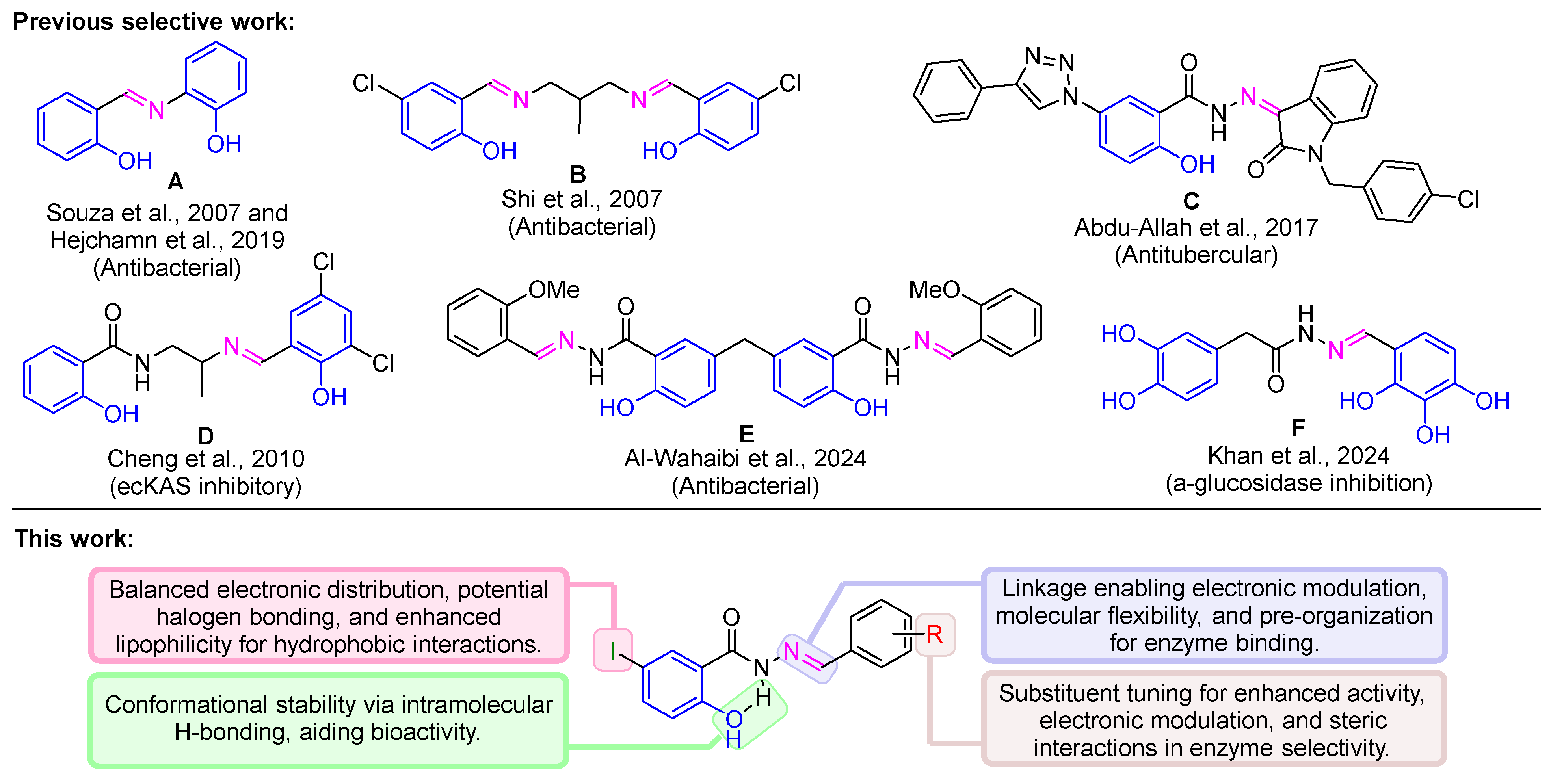
Figure 2.
The % inhibition of α-glucosidase activity for compounds 4a–4j and the standard control Acarbose at 100 µM.
Figure 2.
The % inhibition of α-glucosidase activity for compounds 4a–4j and the standard control Acarbose at 100 µM.
Figure 3.
Binding free energies of Schiff base derivatives of salicylic acid. Average values were calculated from derivatives interacting with the active site of ten different α-GLU conformations.
Figure 3.
Binding free energies of Schiff base derivatives of salicylic acid. Average values were calculated from derivatives interacting with the active site of ten different α-GLU conformations.
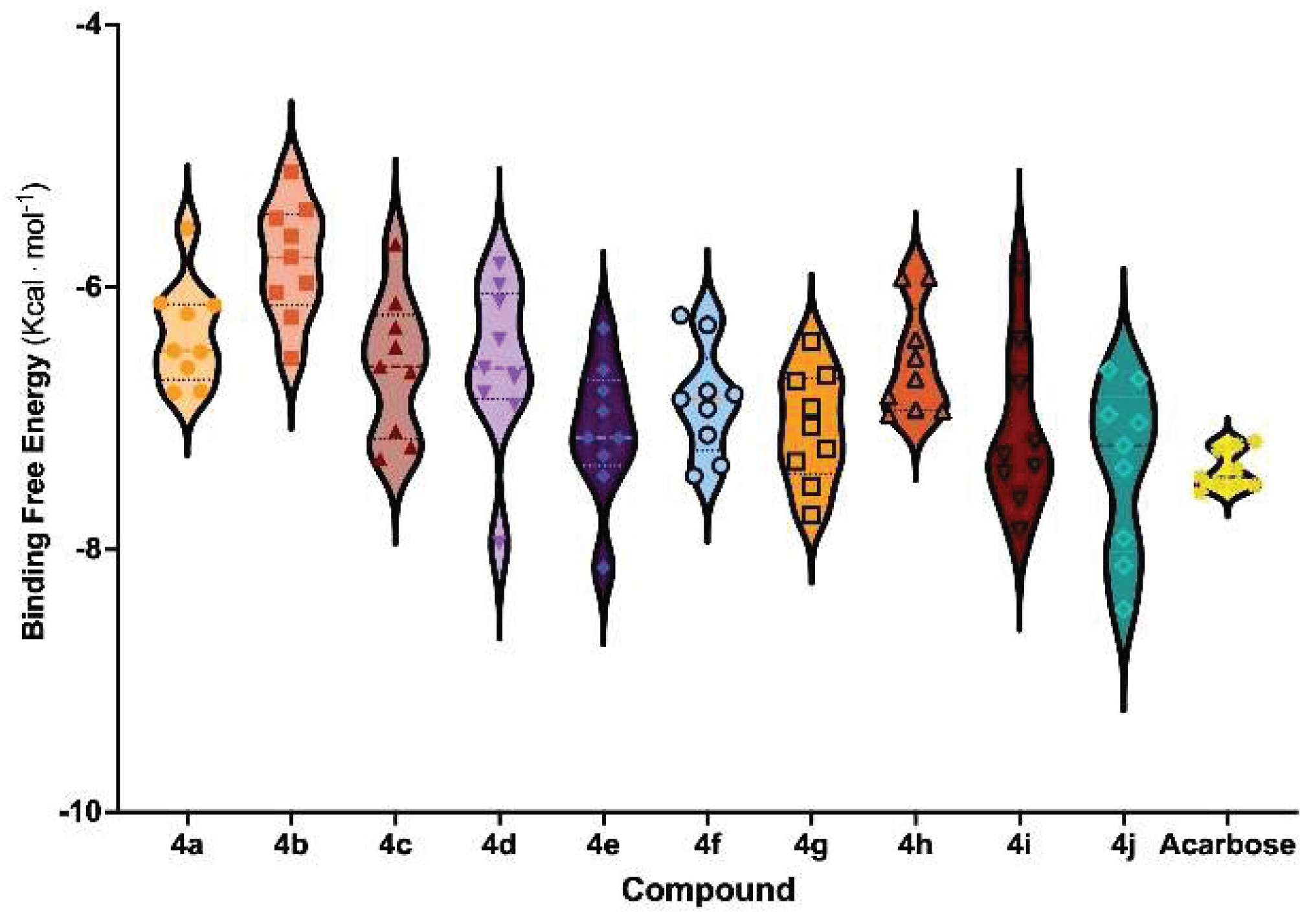
Figure 4.
Molecular Dynamics Simulations summary of Schiff base derivatives complexed to α-GLU. (A) Root Mean Square Deviations (RMSD) of α-carbon stereogenic centers of the enzyme, assessing overall conformational stability; (B) Root Mean Square Fluctuations (RMSF) of α-GLU sidechains, indicating local residue flexibility; (C) 3D Ligand interaction diagram of 4j, highlighting key binding interactions; (D) 3D Ligand interaction diagram of 4e, showing molecular recognition patterns. Solvent-Accessible Surface Areas (SASA) of α-GLU’s binding site are shown as grey surfaces, while intermolecular interactions (hydrogen bonds and π-stacking interactions) are represented as yellow and blue dotted lines, respectively.
Figure 4.
Molecular Dynamics Simulations summary of Schiff base derivatives complexed to α-GLU. (A) Root Mean Square Deviations (RMSD) of α-carbon stereogenic centers of the enzyme, assessing overall conformational stability; (B) Root Mean Square Fluctuations (RMSF) of α-GLU sidechains, indicating local residue flexibility; (C) 3D Ligand interaction diagram of 4j, highlighting key binding interactions; (D) 3D Ligand interaction diagram of 4e, showing molecular recognition patterns. Solvent-Accessible Surface Areas (SASA) of α-GLU’s binding site are shown as grey surfaces, while intermolecular interactions (hydrogen bonds and π-stacking interactions) are represented as yellow and blue dotted lines, respectively.
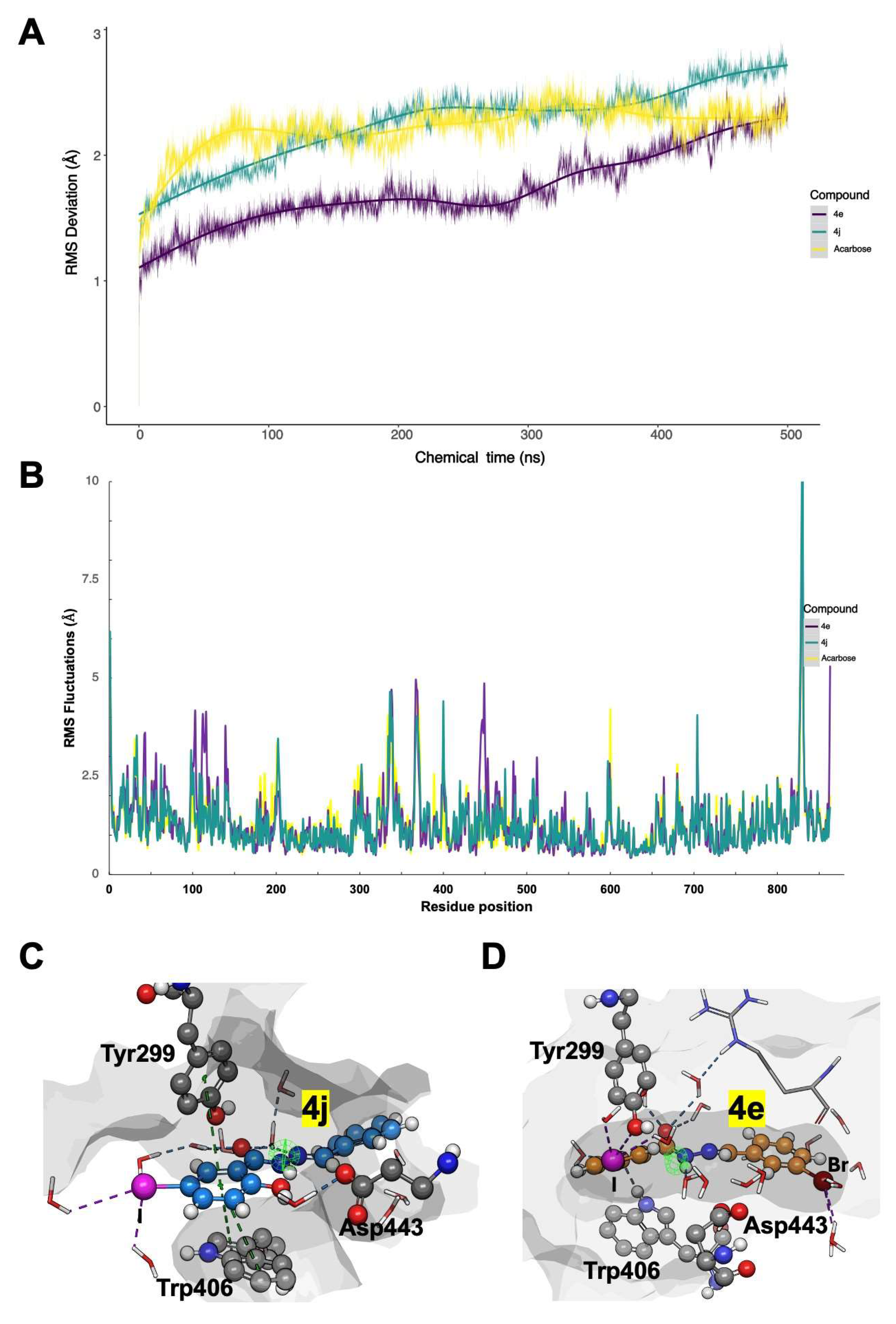

Table 1.
α-Glucosidase inhibitory activity of compounds 4a–4j at 100 µM concentration, IC50 values, HOMO-LUMO energy gap, binding free energy (BFE), and CNN-based binding affinity (AFF).
Table 1.
α-Glucosidase inhibitory activity of compounds 4a–4j at 100 µM concentration, IC50 values, HOMO-LUMO energy gap, binding free energy (BFE), and CNN-based binding affinity (AFF).
| Entry | Compounds | Ar | % inhibition 1 | IC50 (µM) | ΔE 2 (eV) |
BFE 3 (Kcal mol-1) |
Affinity 4 (pK units) |
|---|---|---|---|---|---|---|---|
| 1 | 4a | thiazole | 66.71 ± 4.27 | nd5 | 7.89 | -6.46 ± 0.62 | 4.74 ± 0.33 |
| 2 | 4b | pyridine | 51.65 ± 4.72 | nd | 8.09 | -5.83 ± 0.58 | 4.78 ± 0.35 |
| 3 | 4c | 3-NO2-4-OH-Ph | 45.51 ± 7.45 | nd | 7.24 | -6.62 ± 0.66 | 4.60 ± 0.27 |
| 4 | 4d | 4-NO2-Ph | 59.86 ± 2.60 | nd | 7.60 | -6.61 ± 0.78 | 4.69 ± 0.27 |
| 5 | 4e | 3-Br-Ph | 92.35 ± 2.52 | 14.86 ± 0.24 | 8.03 | -7.23 ± 0.64 | 4.63 ± 0.26 |
| 6 | 4f | 4-OH-Ph | 55.75 ± 1.91 | nd | 7.73 | -6.87 ± 0.60 | 4.77 ± 0.26 |
| 7 | 4g | 2-F-Ph | 85.69 ± 1.59 | 15.58 ± 0.30 | 8.04 | -7.09 ± 0.63 | 4.71 ± 0.23 |
| 8 | 4h | 2-OH-4-OCH3-Ph | 14.33 ± 7.47 | nd | 7.50 | -6.59 ± 0.59 | 4.64 ± 0.24 |
| 9 | 4i | Naph | 88.64 ± 0.78 | 18.05 ± 0.92 | 7.62 | -7.10 ± 0.88 | 4.67 ± 0.20 |
| 10 | 4j | Ph | 93.84 ± 1.49 | 17.56 ± 0.39 | 8.01 | -7.38 ± 0.82 | 4.78 ± 0.31 |
| 11 | Acarbose | - | 84.66 ± 0.71 | 45.78 ± 1.95 | - | -7.41 ± 0.79 | 5.00 ± 0.22 |
1 % Inhibition values were calculated based on absorbance at 450 nm at a fixed concentration of 100 µM and converted to % inhibition using the standard formula; 2 ΔE (eV): HOMO-LUMO energy gap calculated at the ωB97X-D/def2-tzvpp level, indicating electronic reactivity. 3 Binding Free Energy and 4 CNN-based calculated affinity in pK units obtained from molecular dynamic simulations; 5 nd: IC50 not determined.
Table 2.
Evaluated drug-likeness parameters for Acarbose, 4e, and 4j.
| Parameters | Acarbose | 4e | 4j |
|---|---|---|---|
| MW (g/mol) | 645.2 | 443.9 | 365.99 |
| H-bond donors | 14.0 | 2.0 | 2.0 |
| H-bond acceptors | 19.0 | 4.0 | 4.0 |
| logPo/w1 | −4.064 | 4.559 | 4.253 |
| logSwat2 | 0.591 | −5.999 | −5.466 |
| Lipinski’s Rule | Rejected | Accepted | Accepted |
| Pfizer Rule | Accepted | Rejected | Rejected |
| Apparent Caco-2 Permeability log(nm/s)3 | −6.472 | −4.928 | −4.750 |
| Apparent MDCK Permeability log(nm/s)4 | 0.0 | −4.704 | −4.837 |
| Skin sensitization5 | 1.0 | 0.944 | 0.906 |
| Human Intestinal Absorption | Low | Low | Low |
| % Plasma Protein Binding6 | 13.03 | 99.09 | 98.82 |
| CYP1A2 inhibitors7 | 0.0 | 0.979 | 0.999 |
| CYP2C8 inhibitors8 | 0.942 | 0.999 | 0.998 |
| Nephrotoxicity9 | 0.981 | 0.374 | 0.493 |
1 Predicted logarithm of partitioning coefficient for octanol/water phases (range for 95% of drugs: −2.0 to 6.0). 2 Predicted logarithm of aqueous solubility in mol/dm3 (range for 95% of drugs: −6.0 to 0.5). 3 Predicted apparent Caco-2 cell rate permeability in log nm/s (range for 95% of drugs: <25 and >500). 4 Predicted apparent MDCK cells rate permeability in log nm/s (range for 95% of drugs: <25 and >500). 5 Predicted apparent for skin permeability rate permeability Kp in cm/h). 6 Predicted logarithm of serum protein binding (range for 95% of drugs: −1.5 to 1.5). 7 Metabolism profile associated with inhibition of cytochrome P450 isoforms. 8 Nephotoxicity probability (range for 95% of drugs: 0 to 1). 9 Toxicophore prediction of water non-degradable functional groups.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



