
Submitted:
09 June 2025
Posted:
10 June 2025
You are already at the latest version
Abstract
In this study, we investigate the impact of incorporating energetic substituents such as –NO₂, –NH₂, –NH₃, –N₂ (both with perchlorate anion), and –N₃ into 4,6-dinitrobenzimidazol-2-one on its detonation performance and stability. DFT B3LYP/cc-pVTZ method was employed to evaluate key molecular properties: the HOMO–LUMO gap, cohesive energy, chemical hardness, and electronegativity. Based on these parameters, we assessed the resulting changes in chemical and thermal stability. Our results highlight the significant role of ionic bonding in enhancing both the stability and density of the compounds. Our results indicate that the benzimidazoles enriched by energetic groups possess energetic properties better than TNT, with some variants surpassing HMX. The analysis of the stability and sensitivity based on oxygen balance investigation suggests that by varying the incorporated substituents, it is possible to design both primary and secondary explosives from a common molecular scaffold.
Keywords:
high energy materials (Ex)
; benzimidazoles
; energetic groups
; detonation properties
; density
; stability
1. Introduction
Derivatives of benzimidazoles, heterocyclic aromatic organic compounds consisting of benzene and imidazole rings, are widely used in many approved drugs. For example, animal parasitic infections are treated by fenbendazole, mebendazole, and albendazole, which are used as antiparasitic agents; liarozole and pracinostat are consumed as anticancer agents, omeprazole is a potent proton pump inhibitor, oxfendazole is employed as an anthelmintic, enviroxime exhibits antiviral activity, etc. [1,2]. However, benzimidazole and its polynitro derivatives remain relatively underexplored as high-energy materials. Currently, Klapötke et al. demonstrated that 2-aminobenzimidazole can serve as a valuable and cost-effective precursor for the synthesis of energetic materials [3]. Additionally, Szala et al. reported a novel energetic compound, 5,5′,6,6′-tetranitro-2,2′-bibenzimidazole, characterized as a secondary explosive with high thermal stability [4]. Notably, the contributions of Politzer et al. and Pang et al. also provided significant insights into the development of innovative energetic materials [5,6,7]. We also demonstrated that benzimidazole compounds are energetic materials whose high thermal and chemical stability, low toxicity and sensitivity, and good explosive properties could be achieved by the precise combination of nitro, -CH3, and triazole ring substituents [8]. Naturally, this raises the question of whether even greater improvements in energetic performance and stability can be achieved if other substitutions and/or their combinations are used, i.e., more economical, environmentally friendly, and safer energetic materials will be proposed to be synthesized.
For our continued investigation of the energetic and stability properties of the benzimidazoles, we selected -NO2, -NH2, -NH3, -N2, and -N3 substitutions known as energetic groups, along with energetic anions ClO4 [9,10,11,12,13]. In many reported cases, the incorporation of -NH2 leads to an improvement in the thermal stability of the high-energy materials, while -NO2 enhances energetic properties [14,15,16,17,18,19,20,21]. However, there are a few reports on the influence of the -NH3, -N2, ( both along with energetic anions ClO4 ) and -N3 substitutions on the energetic properties of derivatives, although –N₃ substitution is mentioned as the one increasing density and detonation parameters [22]. The compounds consisting of -NH2, -NH3, -N2, and -N3 substitutions could form hydrogen-bonded organic frameworks offering a viable approach to balance energetic performance with operational safety and design high-energy, ionic, and polymeric materials [23,24,25,26]. It happens not only because of higher density, but also the possibility to form a layer-by-layer structure, which can stabilize the resulting materials towards mechanical stimuli. [27]. Perchlorate (ClO4¯) is used as an extra-anion in the energetic cationic metal-organic frameworks to form hydrogen bonding, which significantly affects the molecular structure and, as a consequence, its properties [28,29]. Hence, we decided to investigate the influence of nitrogen-rich substituents and their combinations on the energetic properties of benzimidazole, aiming to demonstrate that they can function as either primary or secondary explosives, depending on the nature and combination of their chemical substitutions. Additionally, we predict the formation of hydrogen bonds in benzimidazole compounds enriched with perchlorate anions, enabling the design of advanced hydrogen-bonded organic frameworks. This prediction is based on the study by Ying Liang et al., which introduced 4,6-diamino-5,7-dinitro-1H-benzo[d]imidazole as a layered crystal with high thermal stability and low sensitivity, exhibiting energetic properties superior to those of TNT [30].
2. Materials and Methods
The Becke three-parameter hybrid functional method with non-local correlation by Lee, Yang, and Parr (B3LYP), combined with the cc-pVTZ basis set as implemented in the GAUSSIAN package, was employed in our study [31,32,33]. The molecular structures corresponding to the most stable conformations were obtained using the Berny geometry optimization. This procedure was carried out so that all bond lengths, bond angles, and dihedral angles were varied to locate the equilibrium structure. To confirm that a true minimum was found, vibrational frequency analysis was performed. This computational approach, integrated with our research plan, yielded results that agree with experimentally determined data [34,35,36,37,38,39,40,41,42].
To predict the stability and sensitivity of the materials, and to evaluate how these properties are affected by various structural modifications, we calculated and compared several key descriptors: cohesive energy per atom, chemical hardness, chemical softness, electronegativity, HOMO-LUMO gap, and hardness index [43]. The oxygen balance was also determined, considering the presence of chlorine in some of the derivatives under study.
The key methodological criteria used in this study are based on a set of well-established molecular descriptors. Thermal stability was assessed by evaluating cohesive energy per atom, with higher values indicating greater resilience to thermal decomposition. Chemical stability was inferred from the magnitude of the HOMO–LUMO energy gap and chemical hardness; larger values of both parameters suggest a more stable electronic structure. Conversely, chemical softness, inversely related to hardness, serves as an indicator of molecular reactivity, where lower softness implies a higher propensity for degradation. Subsequently, we focused on chemical hardness, as it exhibited a strong inverse correlation with chemical softness (regression coefficient R² = 0.93). This suggests that chemical hardness serves as a reliable indicator of chemical reactivity for these particular compounds. Electronegativity was used to evaluate the likelihood of bond formation, with higher values signifying a greater tendency to attract bonding electrons.
The oxygen balance, particularly in the context of chlorine-containing derivatives, was calculated to assess shock sensitivity of the materials—a more negative oxygen balance generally indicates lower sensitivity and contributes to understanding the brisance and strength of the energetic compounds. The hardness index was evaluated as a measure of the compound’s resistance to indentation and mechanical deformation, where higher values correlate with improved structural durability. The obtained values are not presented separately, but are discussed along with thermal stability and resistance to stimuli.
The density of the compounds was predicted by the equations developed by Politzer et al. and calculated as the division of molecular weight by molar volume obtained by B3LP/cc-pVTZ [44]. These equations differ in the incorporation of factors such as the 0.001 electrons/bohr3 counter of the electronic density of a molecule, the degree of balance between positive potential and negative potential on the surface, and their sum as proposed in Politzer’s model. However, the density values obtained from these equations show excellent correlation, with a regression coefficient of 1.0 (Figure 1).
The densities obtained by the Politizer’s equation are 2-6 % lower than those evaluated by the other model. Thus, for the evaluation of the detonation velocities and pressure, we used the lower density so that these parameters would not be overestimated. It is worth mentioning that the calculated density of C7H6N6O4 is equal to 1.73 g/cm3, while that measured is 1.65 g/cm-3. The difference in the above densities occurred not only due to deviations, but also due to the presence of the solvent molecules [30]. On the other hand, this compound is similar to C7H6N6O5, one of the compounds investigated by us. The difference between them lies in the placement of -H and =O substitutions. The calculated density of C7H6N6O5 varies from 1.53 to 1.63 g/cm-3. The differences in molecular weight and packing of these benzimidazoles result in slight variations in their densities. Nevertheless, our comparison of the predicted values supports the decision to evaluate energetic properties using the lower density values.
The energetic properties of the advanced materials were predicted based on the analysis of the obtained values of detonation pressure and velocity, which were calculated by applying several approaches, adopted for the calculation of these parameters for compounds consisting of Cl [45,46,47]. The analysis of the results revealed that the detonation pressure and velocity values predicted using the method described in [48] are significantly higher than those obtained from the equations reported in [45,46]. Furthermore, the latter method provides predictions for the compound CaHbOcNd comparable to those produced by the well-established Kamlet-Jacobs approach. Hence, the detonation velocity and pressure were evaluated by equations presented in [45,46].
The compounds’ sensitivity to shock stimuli and their classification as either primary or secondary explosives were predicted based on their oxygen balance. In this case, we applied this formula [48,49]:
Here, M is the molar weight in g/mol, a, b, c, and e are the number of carbon, hydrogen, oxygen, and chlorine atoms, respectively, in a compound.
3. Results
The perspective of 4,6-dinitrobenzimidazol-2-one considered as a primary compound, is depicted in Figure 2.
As previously noted, we focus on the influence of the -NH2, -NH3 with perchlorate, -N2 , along with energetic anions ClO4, and -N3 substitutions or their combinations, including with -NO2; Accordingly, based on the key substitutions described above, the compounds are categorized into four groups: (1) ExNH₂, (2) ExNH₃, (3) ExN₂, and (4) ExN₃. Appendix 1 presents the groupings, structural representations, compositions, and the abbreviations used throughout the manuscript.
To predict thermal stability, the cohesive energy per atom (BDE) was evaluated. This parameter reflects the energy needed to dissociate the compound into atoms and can be used to estimate its thermal stability qualitatively. Higher cohesive energy per atom values suggest greater thermal stability and higher decomposition temperatures. Importantly, this approach does not require prior knowledge of the decomposition products, which are often difficult to predict. The computed BDE values and their variation across different substitution groups are presented in Figure 3. Going ahead, there is no general dependency of the parameters described below on the substitution of the primary-group-compound. It indicates that a combination of substitutions could lead to unpredictable changes in the developed properties.
Chemical stability, defined as a compound’s resistance to reacting with environmental species or degrading during normal use while maintaining its functional properties over time, was estimated based on both the HOMO (Highest Occupied Molecular Orbital) – LUMO (Lowest Unoccupied Molecular Orbital) gap and chemical hardness. A decrease in the HOM-LUMO gap indicates an increase in the reactivity of the compounds investigated due to a reduction in energy excitation (Figure 4).
On the other hand, reduced chemical hardness typically correlates with an increased tendency of a compound to undergo chemical transformations and, as mentioned above, represents compound reactivity. So, the obtained values of chemical hardness are given in Figure 5.
In Figure 6, the electronegativity of the compounds, indicating a tendency to attract electrons, a property that may correlate with their aging behavior (degradation), is presented.
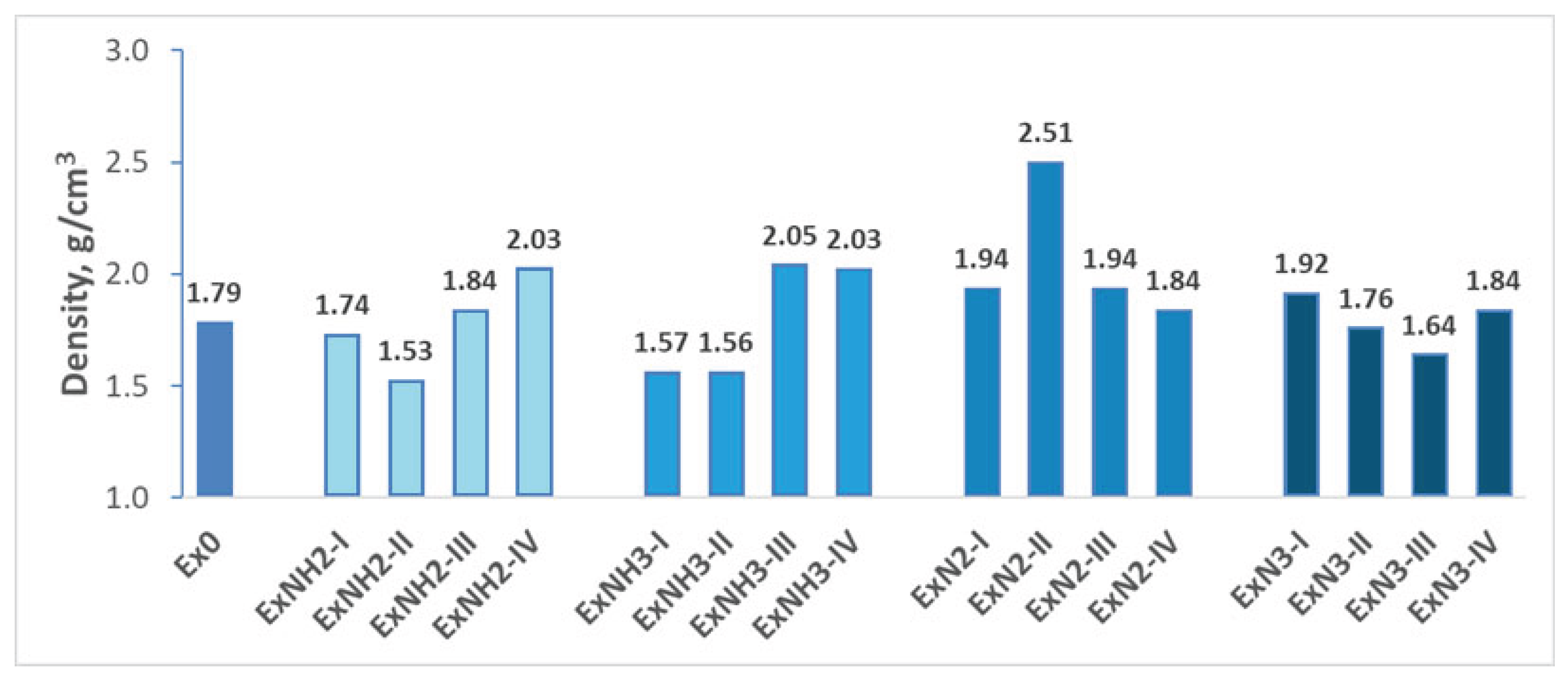
To demonstrate the impact of various substitutions on the density of 4,6-dinitrobenzimidazol-2-one, we present the results in Figure 8.
Figure 7.
Density variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Figure 7.
Density variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Detonation pressure and velocity variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one are depicted in Figure 8 and Figure 9.
The oxygen balance (OB) was calculated to predict the sensitivity to external stimuli such as impact or friction. Generally, compounds with highly negative oxygen balances tend to exhibit decreased sensitivity, likely due to the presence of unbalanced reactive fragments during decomposition. Oxygen balance variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one are presented in Figure 10. In light of the obtained OB and other parameters, we classify the compounds as potential primary or secondary explosives.
4. Discussion
4.1. Stability
It is important to note that BDE and hardness index of 4,5,6-trinitro-1,3-dihydro-2H-benzimidazol-2-one, a closely related compound to 4,6-dinitrobenzimidazol-2-one, are higher than 5,0 eV and 0.80, respectively. It onset at approximately 315 °C, with complete decomposition occurring around 339 °C [50]. Similarly, 5-methyl-4,6,7-trinitro-1,3-dihydro-2H-benzimidazol-2-one, with similar BDE and hardness index, decomposes at a lower temperature range of 270–272 °C. So, considering the cohesive energy of 4,6-dinitrobenzimidazol-2-one (Ex0) of 5.99 eV, and the hardness index of 0.79, we not only state that this compound is thermally stable, but may predict its decomposition temperature in a comparable range, potentially exceeding 270 °C. BDE of compounds of group 2 and 4 (exception ExN3-IV) is higher than 5,00 eV, and their hardness index is larger than 0.8 (Figure 3). So, these compounds are thermally stable, and their decomposition temperature could also be higher than 270 °C. However, only ExN3-I, ExN3-II, and ExN3-III (group 1) possess BDEs higher than that of Ex0, while that in the case of ExNH2-I, ExNH2-II, ExNH2-III, and ExNH2-IV ( group 4) is lower. These results imply that substitution with –N₃, either alone or in combination with –NH₂, –NO₂, or an additional –N₃ group, tends to enhance thermal stability. In contrast, –NH₂ substitution, alone or in its pattern with the above substitutions, appears to slightly raise thermal stability. Consistently, the compounds from groups 1 and 4 exhibit higher hardness indices than Ex0, suggesting enhanced overall stability.
BDE of the compounds consisting of group 2, whose values fluctuate from 4.47 eV to 5.19 eV, is significantly lower than Ex0 (5.99 eV), despite their hardness indexes varying from 0.80 to 0.83. Thus, the thermal stability of the compounds belonging to this group could be lower. This finding is consistent with the understanding that organic compounds containing ionic perchlorate groups are generally less thermally stable than their fully covalent analogues [51].
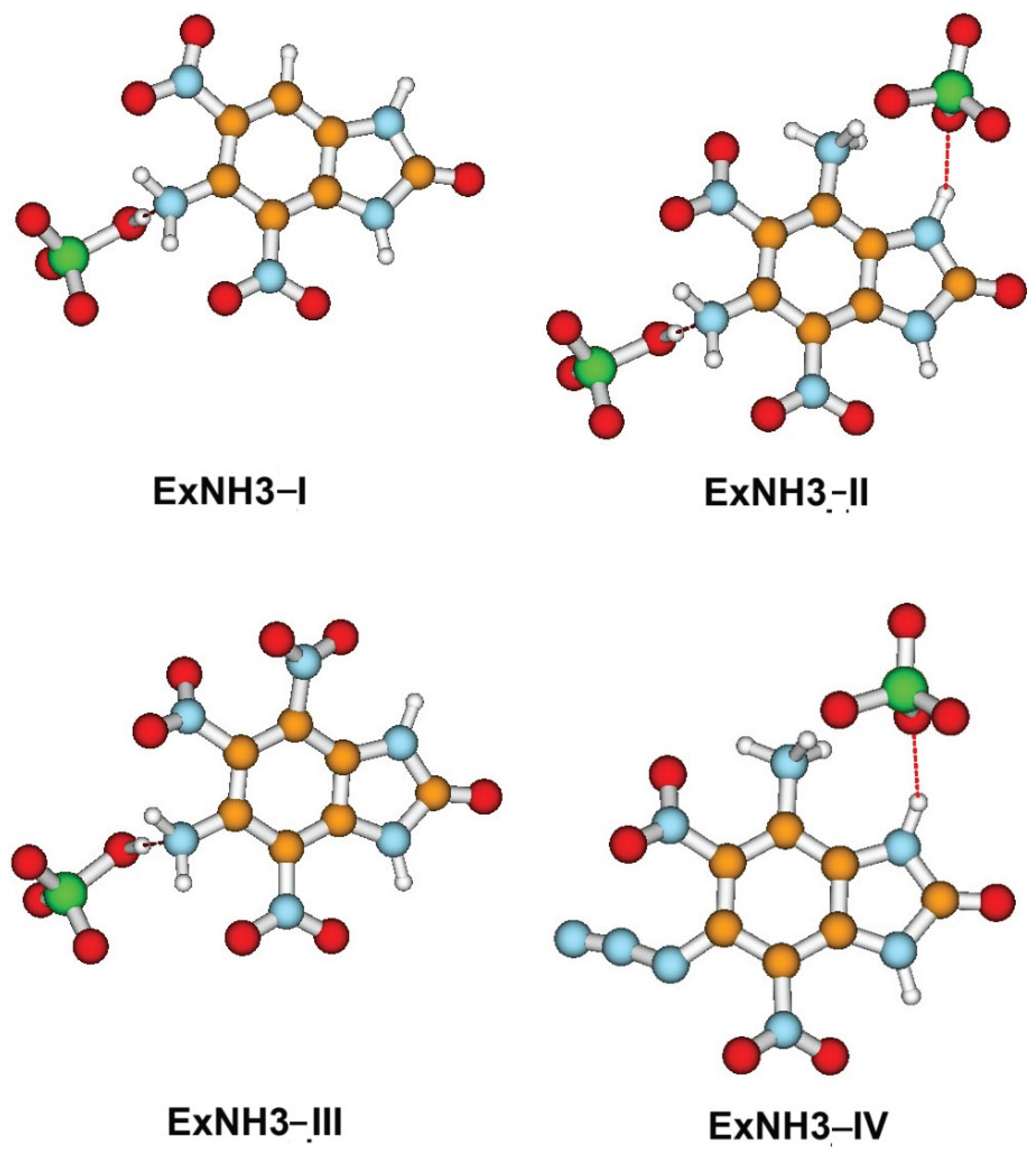
The above reduction in thermal stability is likely due to protonation of the cationic moiety, resulting in the formation of neutral HClO₄, which may facilitate bond cleavage. The protonation is confirmed by the results of our investigation (Figure 11).
Moreover, the performed analysis of BDE indicates that ExNH3-IV, which retains an ionic bond, is more thermally stable than the other group 2 compounds, in which this bond is disrupted by protonation, leading to the formation of a neutral hydrogen-bonded species.
The importance of the ionic bond presence to thermal stability also follows from the BDE analysis of the compounds belonging to group 3. The reduction of these parameters is also obtained, but it (4.82 - 5.47 eV) is smaller than that of group 2. However, the overall stability of the compounds with ionic bonds is significantly lower than that of Ex0. The conclusion follows from the analysis of the hardness indexes that, in the case of the compounds belonging to group 3, does not exceed 0.70, and is especially low (0.34 and below) in the case of ExN2-I and ExN2-II.
Overall, the thermal stability of 4,6-dinitrobenzimidazol-2-one (Ex0) increases with the introduction of one or two -N3 substitutions to the hexagonal ring (Figure 3). However, when -N₃ substitution is combined with –NO₂ or –N₂ along with energetic anions ClO4, groups, a decrease in thermal stability is observed. The formation of perchlorate compounds from Ex0 derivatives reduces the thermal stability of the benzimidazole compounds, although the inclusion of –NO₂ or –N₃ substituents in these compounds can slightly enhance this stability.
The significant decrease of HOMO-LUMO gap and chemical hardness leads to the prediction that -N2, along with energetic anions ClO4, substitution increases the tendency of a compound to undergo chemical transformations (Figure 4 and Figure 5). Consequently, the compounds consisting of group 3 are less chemically stable than the Ex0. In the other cases examined, the HOMO-LUMO gap and chemical hardness are larger than those of Ex0, which indicates that the reactivity of the rest compounds investigated decreases along with the increase in chemical resistance due to -NH2, -NH3, with energetic anions ClO4, and -N3 substitutions. The exception is the ExNH2-III compound, consisting of -NH2 and -NO2 substitutions. The reduction in HOMO-LUMO gap and chemical hardness indicates that its reactivity is larger than that of Ex0 (Figure 4 and Figure 5). The incorporation of -NO2 as a second substitution to compounds consisting of N3 and NH3, along with energetic anions ClO4, reduces their chemical resistance, which becomes comparable to that of Ex0. Interestingly, the incorporation of the above-mentioned substituent into ExN2-I improves its chemical stability.
The chemical stability of Ex0 increases mostly due to -N3, although the incorporation of a second –N₃ substituent appears to reduce it. A similar tendency is obtained in the case of NH3 and N2 (both along with energetic anions ClO4), where additional substituents lead to decreased stability. In contrast, the addition of a second NH₂ group to the hexagonal ring of Ex0 enhances chemical resistance.
In addition to chemical stability, the analysis of the electronegativity of the compounds was performed to predict the influence of substitutions on Ex0 aging behavior. As expected, compounds consisting of perchlorate are highly electronegative. This is illustrated in Figure 6 by the variations of the above compounds from 5.45 eV to 6.60 eV, which is larger than that of Ex0 (5.24 eV). Hence, incorporation of –NH₃ and –N₂ groups, both associated with the energetic anion ClO₄, increases the predicted aging rate of Ex0. In contrast, substitution with –NH₂ or –N₃ reduces the electronegativity ( from 4.58 eV to 5.16 eV) of Ex0 and, as a consequence, the aging tendency. The results also reveal an increase in electronegativity for compounds containing key –NO₂ substitutions. This suggests that NO₂ may contribute to an accelerated aging process.
In summary, compounds belonging to group 4 demonstrate the highest thermal and chemical stability among those studied, along with a slower aging rate. In contrast, the stability of the other compounds varies depending on the combination of key substituents, and no consistent trend is observed as clearly as in the case of group 4.
4.2. Energetic Properties
Notably, the density of a compound is influenced by its chemical composition and molecular geometry. It is therefore a critical factor affecting the detonation products, which in turn directly determine both detonation pressure and velocity [52]. The observed reduction of the density of Ex0 (1.79 g/cm3) could be caused by several factors. Considering the density of ExNH2-I (1.74 g/cm3), similar to that of Ex0, and ExNH2-II (1.53 g/cm3), consisting of NH2, we may predict that incorporation of these two groups increases volume faster than mass, leading to the density decrease. In the case of group 2, the formation of hydrogen bonds instead of ionic bonds in the ExNH3-I and ExNH3-II compounds resulted in density decrease (Figure 7). This is supported by the observation that a compound containing one –NH₃ group combined with energetic ClO₄⁻ anion exhibits a density of 1.57 g/cm³, which is nearly identical to the 1.56 g/cm³ found in compounds containing a double substitution with these groups. This finding is further supported by the significantly higher densities ( 1.84 g/cm³ - 2.51 g/cm³ ) observed in the group 3 compounds in which at least one ionic bond is observed. It is important to emphasize that only the group 3 compound containing both key substitutions (–N₂ along with anion ClO₄⁻ ) exhibits a density significantly higher than that of Ex0. In all other cases studied, compounds with two key substitutions display densities that are either lower than or comparable to that of Ex0. Thus, the combination of the substitutions is crucial for the compound density and, as a consequence, for their energetic properties. It also follows from the analysis of -NO2 substitution influence on the density of the compounds, that is:
- In it combination with –NH₂, –NH₃, or –N₂ (the last two along with anion ClO₄⁻ ), the density of Ex0 increases.
- In it combination with –N₃, the density of Ex0 decreases.
- Incorporation of it in compound with –NH₂, or –NH₃ (along with anion ClO₄⁻ ) leads to their densities increase.
- Incorporation of it in the compound with –N₂ (along with anion ClO₄⁻) does not influence their densities.
- Incorporation of it in the compound with –N3 reduces its density.
In addition, it is necessary to mention that the combination of -N3 and with –N₂ (along with anion ClO₄⁻ ) reduces the density of the compound ExN3-I, which consists of only one N3 group (Figure 7).
As it is predicted, similar correlations are evident between the energetic properties of the compounds under study and substitutions and their combinations. This relationship follows from the similarity in the plot shapes presented in Figure 7, Figure 8 and Figure 9.
Let us recall that high-energy materials typically exhibit detonation velocities ranging from 1.01 km/s to 9.89 km/s. Trinitrotoluene (TNT), commonly used as a standard reference, has a measured detonation velocity of approximately 6.9 km/s. In comparison, RDX and HMX demonstrate higher detonation velocities of 8.7 km/s and 9.1 km/s, respectively. The corresponding detonation pressures are approximately 210 kbar for TNT, 338 kbar for RDX, and 393 kbar for HMX [53,54]. The calculated detonation velocity and pressure of Ex0 are 8.38 km/s and 312.41 kbar, respectively, identifying the compound as a high-energy material with energetic properties comparable to those of RDX (Figure 8 and Figure 9). Moreover, the energetic properties of ExNH3-III, ExNH3-IV, and ExN3-II exceed those of HMX, while only ExNH2-II, ExN2-II, and ExN2-III exhibit detonation density and velocity values comparable to those of TNT. We also found that the energetic properties of the ExNH2-I, ExNH2-IV, ExNH3-II, ExN2-I, and ExN2-III are similar to those of RDX. Hence, the incorporation of various substituents and their combinations into Ex0 enables the tuning of energetic properties across a broad range, from high to very highly energetic performances.
4.3. Oxygen Balance
As it is mentioned above, oxygen balance (OB) was calculated and compared with that of TNT (–73.97 %) and RDX (-21.61%) to predict the sensitivity to external stimuli such as impact or friction. Generally, compounds with highly negative oxygen balances tend to exhibit decreased sensitivity, likely due to the presence of unbalanced reactive fragments during decomposition [55,56,57,58]. So, less sensitive to stimuli than TNT are ExNH2-I, ExNH2-II compounds (Figure 10). These compounds, and Ex0, ExNH2-IV, EXNH3-I, ExN2-II, ExN3-II, EXN3-III, can be classified as insensitive materials due to a highly negative oxygen balance (below -30%), meaning they lack sufficient oxygen for complete combustion and require external oxidizers to function effectively. The moderately sensitive materials OB generally range from -30% to -20%. Hence, ExNH2-III, ExNH3-III, ExNH3-IV, ExN2-I and ExN2-IV(or ExN3-IV) are moderately sensitive materials. The OB of ExNH3-II and ExN2-III are sensitive to stimuli.
Considering the following findings: (i) the detonation pressure and velocity of these sensitive compounds match or exceed those of TNT; (ii) they exhibit the lowest thermal stability and moderate chemical stability among the compounds studied, we propose ExNH₃-II and ExN2-III as candidates for advanced detonators, which can detonate with minimal external stimuli, such as heat, or impact. In contrast, the most thermally and chemically stable insensitive compounds Ex0, ExNH2-I, ExNH2-II, ExNH2-IV, EXNH3-I, ExN3-II, and EXN3-III can be considered as advanced secondary explosives. The insensitive ExN2-II compound is not mentioned among them due to its high chemical reactivity. We also propose that moderately sensitive compounds such as ExNH2-III, ExNH3-III, and ExNH3-IV could be primary explosives, while ExN2-I and ExN2-IV(or ExN3-IV) are secondary ones due to their thermal and chemical stability. The above highlights that, by modifying the functional groups on a common molecular scaffold, it is possible to obtain both primary and secondary energetic materials.
5. Conclusions
This study was conducted to demonstrate that benzimidazoles with incorporated substituents such as –NH₂, –NH₃, –N₂ ( both along with perchlorate ion), –N₃, –NO₂, and their combinations can serve as either primary or secondary explosives. Additionally, the formation of hydrogen bonds in benzimidazole compounds enriched with perchlorate anions was investigated to assess potential improvements in stability and energetic performance.
The results indicate that different combinations of substituents can lead to unpredictable variations in the resulting properties. For example, substitution of of 4,6-dinitrobenzimidazol-2-one (Ex0) with –N₃, either alone or in combination with –NH₂, –NO₂, or an additional –N₃ group, tends to enhance thermal stability. In contrast, the incorporation of –NH₃ and –N₂, whether individually or together with –NH₂, –NO₂, –NH₃, or –N₂ ( both along with perchlorate ion), results in a reduction of chemical stability. The –NH₂ group alone has no significant effect on this parameter. We also found that the protonation process occurred in the compounds containing –NH₃ substituent (s) (along with perchlorate anions) causes a decrease in thermal stability; however, when combined with –NO₂ or –N₃, a slight enhancement in stability can be observed.
The analysis of the HOMO–LUMO gap, chemical hardness, and electronegativity revealed that, in general, the investigated benzimidazoles containing –N₂ substituents (along with perchlorate anions) are less reactive than the other studied compounds and exhibit a slower aging rate. Moreover, the stability of the other compounds varies depending on the combination of key substituents, and no consistent trend is observed.
No consistent trends were observed between the key substitutions (or their combinations and -NO2) and the resulting detonation pressure or velocity. However, based on the obtained results, we conclude that the energetic properties of all studied compounds exceed those of TNT. Moreover, several compounds—ExNH₂-I, ExNH₂-IV, ExNH₃-II, ExN₂-I, and ExN₂-III—exhibit performance comparable to that of RDX.
Considering sensitivity to stimuli, as inferred from the oxygen balance analysis, and stability of the investigated benzimidazoles, we propose ExNH₃-II, ExN2-III, ExNH2-III, ExNH3-III, and ExNH3-IV as potential candidates for advanced detonators. sEx0, ExNH2-I, ExNH2-II, ExNH2-IV, EXNH3-I, ExN3-II, EXN3-III ExN2-I, and ExN2-IV(or ExN3-IV) compounds are identified as promising advanced secondary explosives. These findings highlight the potential of substituted benzimidazoles as a versatile molecular platform for designing both safer and more efficient primary and secondary explosives.
Author Contributions
Conceptualization, J.T. and J.S.; methodology, J.T., and J.S; formal analysis, J.T, and J.S.; investigation, J.T. and J.S.; writing—original draft preparation, J.T. and J.S.; writing—review and editing, J.T. and J.S. All authors have read and agreed to the published version of the manuscript
Funding
This research received no external funding.
Data Availability Statement
The data presented in this study are available on request from the corresponding author due to access restrictions.
Acknowledgments
The numerical calculations with the GAUSSIAN09 package were performed using the resources of the Information Technology Research Center of Vilnius University and the supercomputer “VU HPC” of Vilnius University in the Faculty of Physics location.
Conflicts of Interest
The funders had no role in the design of the study, in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
The following abbreviations are used in this manuscript:
| BDE | Cohesive energy per atom |
| HOMO-LUMO gap | Difference between the highest occupied and the lowest unoccupied orbitals |
| OB | Oxygen Balance |
| Ex0 | C7H3N5O5 |
| ExNH2-I | C7H5N5O5 |
| ExNH3-I | C7H6ClN5O9 |
| ExN2-I | C7H3ClN6O9 |
| ExN3-I | C7H3N7O5 |
| ExNH2-II | C7H6N6O5 |
| ExNH3-II | C7H8Cl2N6O13 |
| ExN2-II | C7H2Cl2N8O13 |
| ExN3-II | C7H2N10O5 |
| ExNH2-III | C7H2ClN9O9 |
| ExNH3-III | C7H5ClN6O11 |
| ExN2-III | C7H2ClN7O11 |
| ExN3-III | C7H2N8O7 |
| ExNH2-IV | C7H4N8O5 |
| ExNH3-IV | C7H5ClN8O9 |
| ExN2-IV | C7H2ClN9O9 |
| ExN3-IV | C7H2ClN9O9 |
Appendix A
Appendix A.1

Table A1.
Structural formulas of designed benzimidazole energetic materials, their chemical formula and abbreviation (Mol. formula), molecular weights (MW), and calculated elemental composition data .
Table A1.
Structural formulas of designed benzimidazole energetic materials, their chemical formula and abbreviation (Mol. formula), molecular weights (MW), and calculated elemental composition data .
| Substitution | Structure | Mol. formula, Abbreviation | MW | Calculated elemental composition data | ||||
| C, % | H, % | Cl, % | N, % | O, % | ||||
| -NH2 | 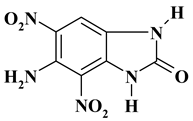 |
C7H5N5O5 ExNH2-I |
239.15 | 35.16 | 2.11 | 0 | 29.28 | 33.45 |
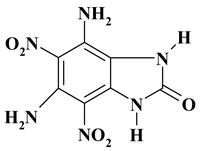 |
C7H6N6O5 ExNH2-II |
254.16 | 33.08 | 2.38 | 0 | 33.07 | 31.47 | |
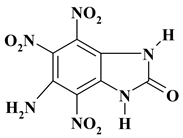 |
C7H4N6O7 ExNH2-III |
284.15 | 29.59 | 1.42 | 0 | 29.58 | 39.41 | |
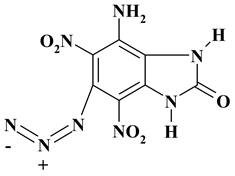 |
C7H4N8O5 ExNH2-IV |
280.16 | 30.01 | 1.44 | 0 | 40.00 | 28.55 | |
| -NH3 with perchlorate | 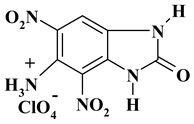 |
C7H5ClN5O9 ExNH3-I |
338.60 | 24.83 | 1.49 | 10.47 | 20.68 | 42.53 |
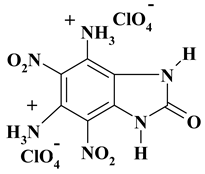 |
C7H8Cl2N6O13 ExNH3-II |
455.08 | 18.48 | 1.77 | 15.58 | 18.47 | 45.70 | |
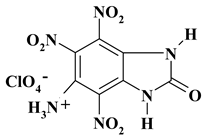 |
C7H5ClN6O11 ExNH3-III |
384.60 | 21.86 | 1.31 | 9.22 | 21.85 | 45.76 | |
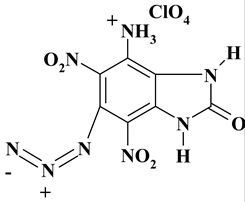 |
C7H5ClN8O9 ExNH3-IV |
380.62 | 22.09 | 1.32 | 9.31 | 29.44 | 37.83 | |
| -N2 with perchlorate | 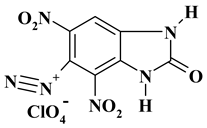 |
C7H3ClN6O9 ExN2-I |
350.59 | 23.98 | 0.86 | 10.11 | 23.97 | 41.07 |
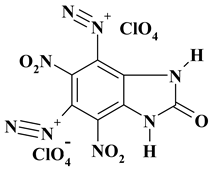 |
C7H2Cl2N8O13 ExN2-II |
477.05 | 17.62 | 0.42 | 14.86 | 23.49 | 43.60 | |
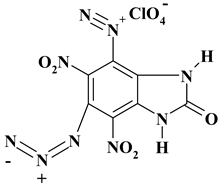 |
C7H2ClN7O11 ExN2-III |
395.59 | 21.20 | 0.76 | 8.94 | 24.72 | 44.38 | |
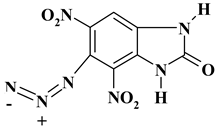 |
C7H2ClN9O9 ExN2-IV |
391.60 | 21.47 | 0.51 | 9.05 | 32.19 | 36.77 | |
| -N3 | 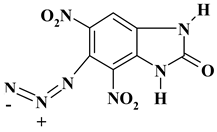 |
C7H3N7O5 ExN3-I |
265.15 | 31.71 | 1.14 | 0 | 36.98 | 30.17 |
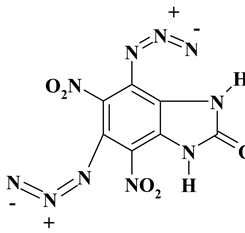 |
C7H2N10O5 ExN3-II |
306.16 | 27.46 | 0.66 | 0 | 45.75 | 26.13 | |
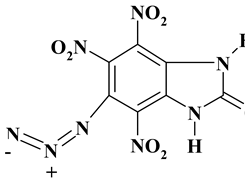 |
C7H2N8O7 ExN3-III |
310.14 | 21.25 | 0.51 | 8.96 | 24.79 | 44.49 | |
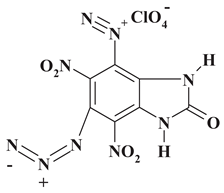 |
C7H2ClN9O9 ExN2-IV |
391.60 | 21.47 | 0.51 | 9.05 | 32.19 | 36.77 | |
References
- Nguyen, J.; Nguyen, T.Q.; Han, B.; Hoang, B.X. Oral Fenbendazole for Cancer Therapy in Humans and Animals. Anticancer Res. 2024, 44, 3725–3735. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, O.; Oyetunde-Joshua, F.; Omotoso, O.D.; Shapi, M. Benzimidazole and its derivatives: Recent Advances (2020–2022). Results Chem. 2023, 5, 100925. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Preimesser, A.; Stierstorfer, J. Energetic Derivatives of 2-Nitrimino-5,6-dinitro-benzimidazole. Propellants Explos. Pyrotech. 2015, 40, 60–66. [Google Scholar] [CrossRef]
- Szala, M.; Gutowski, Ł.; Trzcinski, W. New thermally stable and insensitive energetic compound: 5,5’,6,6’-tetranitro-2,2’-bibenzimidazole. ChemPlusChem 2018, 83, 87–91. [Google Scholar]
- Politzer, P.A.; Murray, J.S. (Eds.) Theoretical and Computational Chemistry, Energetic Materials. Part 1; Decomposition, Crystal and Molecular Properties. Elsevier: Academic Press, Amsterdam, The Netherlands, 2003; Volume 121. [Google Scholar]
- Politzer, P.A.; Murray, J.S. (Eds.) Theoretical and Computational Chemistry, Energetic Materials. Part 2; Detonation, Combustion. Elsevier: Academic Press, Amsterdam, The Netherlands, 2003; Volume 13, pp. 1–474. [Google Scholar]
- Pang, W.; DeLuca, L.T.; Gromov, A.A.; Cumming, A.S. (Eds.) Innovative Energetic Materials. Properties, Combustion Performance and Application. Springer: Nature, Singapore, 2020; pp. 1–569.
- Sarlauskas, J.; Tamuliene, J.; Bekesiene, S.; Kravcov, A. Benzimidazole Derivatives as Energetic Materials: A Theoretical Study. Materials 2021, 14, 4112. [Google Scholar] [CrossRef]
- Drake, G.W.; Bova, S.J; Shreeve, J.M. Energetic, low-melting salts of simple heterocycles. Propellants Explos. Pyrotech. 2003, 28, 174–180. [Google Scholar] [CrossRef]
- Drake, G.W; Hawkins, T.W.; Boatz, J.; Hall, L.; Vij, A. Experimental and theoretical study of 1,5-diamino-4-H-1,2,3,4-tetrazolium perchlorate. Propellants Explos. Pyrotech. 2005, 30, 156–16. [Google Scholar] [CrossRef]
- Darwich, C.; Klapötke, T.M.; Sabaté, C.M. 1,2,4-Triazolium-cation-based energetic salts. Chem. Eur. J. 2008, 14, 5756–5771. [Google Scholar] [CrossRef]
- Smiglak, M.; Rogers, R.D.; Holbrey, J.D.; et al. Synthesis, limitations, and thermal properties of energetically-substituted, protonated imidazolium picrate and nitrate salts and further comparison with their methylated analogs. New J. Chem. 2012, 36, 702–72. [Google Scholar] [CrossRef]
- Tao, G.H.; He, L.; Yu, Y.; Zheng, S.; et al. Synthesis, structure and property of 5-aminotetrazolate room-temperature ionic liquids. Eur. J. Inorg. Chem. 2012, 3070–3078. [Google Scholar] [CrossRef]
- Liu, Q.; Yuan, M.; He, J.; Yu, P.; Guo, X.; Liu, Y.; Gao, H.; Yin, P. Exchanging of NH₂/NHNH₂/NHOH groups: An effective strategy for balancing the energy and safety of fused-ring energetic materials. Chem. Eng. J. 2023, 466, 143333. [Google Scholar] [CrossRef]
- Man, T.; Wang, K.; Zhang, J.; Niu, X.; Zhang, T. Theoretical Studies of High-nitrogen-containing Energetic Compounds Based on the s-Tetrazine Unit. Cent. Eur. J. Energ. Mater. 2013, 10, 171–1. [Google Scholar]
- Zhang, Y.; Parrish, D.; Shreeve, J. Derivatives of 5-nitro-1,2,3-2H-triazole—High performance energetic materials. J. Mater. Chem. A 2013, 1, 585–5. [Google Scholar] [CrossRef]
- Cao, W.; Qin, J.; Zhang, J.; Sinditskii, V. 4,5-Dicyano-1,2,3-Triazole—A Promising Precursor for a New Family of Energetic Compounds and Its Nitrogen-Rich Derivatives: Synthesis and Crystal Structures. Molecules 2021, 26, 6735. [Google Scholar] [CrossRef] [PubMed]
- Gu, H.; Xiong, H.; Yang, H.; Cheng, G. Tricyclic nitrogen-rich cation salts based on 1,2,3-triazole: Chemically stable and insensitive candidates for novel gas generant. Chem. Eng. J. 2021, 408, 128021. [Google Scholar] [CrossRef]
- Feng, S.; Yin, P.; He, C.; Pang, S.; Shreeve, J. Tunable Dimroth rearrangement of versatile 1,2,3-triazoles towards high performance energetic materials. J. Mater. Chem. A 2021, 9, 12291–12298. [Google Scholar] [CrossRef]
- Lai, Q.; Fei, T.; Yin, P.; Shreeve, J. 1,2,3-Triazole with linear and branched catenated nitrogen chains—The role of regiochemistry in Energetic Materials. Chem. Eng. J. 2020, 410, 128148. [Google Scholar] [CrossRef]
- Yao, W.; Xue, Y.; Qian, L.; Yang, H.; Cheng, G. Combination of 1,2,3-triazole and 1,2,4-triazole frameworks for new high-energy and low-sensitivity compounds. Energ. Mater. Front. 2021, 2, 131–138. [Google Scholar] [CrossRef]
- Li, X.; Zhang, R. Computational studies on energetic properties of nitrogen-rich energetic materials with ditetrazoles. J. Chem. Sci. 2014, 126, 1753–1762. [Google Scholar]
- Wang, Z.; Lai, Q.; Yin, P.; Pang, S. Construction of adaptive deformation block: rational molecular editing of the N-rich host molecule to remove water from the energetic hydrogen-bonded organic frameworks. ACS Appl. Mater. Interfaces 2024, 16, 21849–21856. [Google Scholar] [CrossRef]
- Hisaki, I.; Suzuki, Y.; Gomez, E.; Ji, Q.; Tohnai, N.; Nakamura, T.; Douhal, A. Acid responsive hydrogen-bonded organic frameworks. J. Am. Chem. Soc. 2019, 141, 2111–2121. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Wang, H.; Wang, C.; Wu, H.; Zhou, W.; Chen, B.; Jiang, J. Postsynthetic metalation of a robust hydrogen-bonded organic framework for heterogeneous catalysis. J. Am. Chem. Soc. 2019, 141, 8737–8740. [Google Scholar] [CrossRef] [PubMed]
- Sukhanov, G.; Filippova, Y.; Gatilov, Y.; Sukhanova, A.; Krupnova, I.; Bosov, K.; Pivovarova, E.; Krasnov, V. Energetic Materials Based on N-substituted 4-nitro-1,2,3-triazoles. Materials 2022, 15, 1119. [Google Scholar] [CrossRef] [PubMed]
- Lai, Q.; Pei, L.; Fei, T.; Yin, P.; Pang, S.; Shreeve, J. Size-matched hydrogen bonded hydroxylammonium frameworks for regulation of energetic materials. Nat. Commun. 2022, 13, 6937. [Google Scholar] [CrossRef]
- Du, Y.; Su, H.; Fei, T.; Hu, B.; Zhang, J.; Li, S.; Pang, S.; Nie, F. Structure-Property Relationship in Energetic Cationic Metal–Organic Frameworks: New Insight for Design of Advanced Energetic Materials. Cryst. Growth Des. 2018, 18. [Google Scholar] [CrossRef]
- Mahmoudi, G.; Garcia-Santos, I.; Fernández-Vazquez, R.; Gargari, M.S.; Gomila, R.M.; Castiñeiras, A.; Frontera, A.; Safin, D.A. Anion driven tetrel bonding dictated supramolecular architectures of lead(II) with a zwitterionic form of polydentate N′-(piperidine-1 carbonothioyl)picolinohydrazonamide. CrystEngComm 2024, 26, 435. [Google Scholar] [CrossRef]
- Liang, Y.; Hu, X.; Yang, Z.; Liu, M.; Zhang, Y.; Wu, J.; Zhang, J.; Zhao, T.; Sun, S.; Wang, S. Benzimidazole-based low-sensitivity and heat-resistant energetic materials: design and synthesis. New J. Chem. 2025, 49, 257. [Google Scholar] [CrossRef]
- Becke, A. Density functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 56. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.2; Gaussian, Inc. 2009. [Google Scholar]
- Cardia, R.; Malloci, G.; Mattoni, A.; Cappellini, G. Effects of TIPS-Functionalization and Perhalogenation on the Electronic, Optical, and Transport Properties of Angular and Compact Dibenzochrysene. J. Phys. Chem. A 2014, 118, 5170–5177. [Google Scholar] [CrossRef]
- Cardia, R.; Malloci, G.; Rignanese, G.; Blasé, X.; Molteni, E.; Cappellini, G. Electronic and optical properties of hexathiapentacene in the gas and crystal phases. Phys. Rev. B 2016, 93, 235132. [Google Scholar] [CrossRef]
- Dardenne, N.; Cardia, R.; Li, J.; Malloci, G.; Cappellini, G.; Blasé, X.; Charlier, J.; Rignanese, G. Tuning Optical Properties of Dibenzochrysenes by Functionalization: A Many-Body Perturbation Theory Study. J. Phys. Chem. C 2017, 121, 24480–24488. [Google Scholar] [CrossRef]
- Antidormi, A.; Aprile, G.; Cappellini, G.; Cara, E.; Cardia, R.; Colombo, L.; Farris, R.; d’Ischia, M.; Mehrabanian, M.; Melis, C. Physical and Chemical Control of Interface Stability in Porous Si–Eumelanin Hybrids. J. Phys. Chem. C 2018, 122, 28405–28415. [Google Scholar] [CrossRef]
- Mocci, P.; Cardia, R.; Cappellini, G. Inclusions of Si-atoms in Graphene nanostructures: A computational study on the ground-state electronic properties of Coronene and Ovalene. J. Phys. Conf. Ser. 2018, 956, 012020. [Google Scholar] [CrossRef]
- Mocci, P.; Cardia, R.; Cappellini, G. Si-atoms substitutions effects on the electronic and optical properties of coronene and ovalene. New J. Phys. 2018, 20, 113008. [Google Scholar] [CrossRef]
- Kumar, A.; Cardia, R.; Cappellini, G. Electronic and optical properties of chromophores from bacterial cellulose. Cellulose 2018, 25, 2191–2203. [Google Scholar] [CrossRef]
- Szafran, M.; Koput, J. Ab initio and DFT calculations of structure and vibrational spectra of pyridine and its isotopomers. J. Mol. Struct. 2001, 565, 439–448. [Google Scholar] [CrossRef]
- Begue, D.; Carbonniere, P.; Pouchan, C. Calculations of Vibrational Energy Levels by Using a Hybrid ab Initio and DFT Quartic Force Field: Application to Acetonitrile. J. Phys. Chem. A 2005, 109, 4611–4616. [Google Scholar] [CrossRef]
- Kaya, S.; Kaya, C. New equation based on ionization energies and electron affinities of atoms for calculating of group electronegativity. Comput. Theor. Chem. 2015, 1052, 42–46. [Google Scholar] [CrossRef]
- Politzer, P.; Martinez, J.; Murray, J.; Concha, M.; Toro-Labbé, A. An electrostatic interaction correction for improved crystal density prediction. Mol. Phys. 2009, 107, 2095–2101. [Google Scholar] [CrossRef]
- Keshavarz, M.; Zamani, A. A simple and reliable method for predicting the detonation velocity of CHNOFCl and aluminized explosives. Cent. Eur. Energ. Mater. 2015, 12, 13–33. [Google Scholar]
- Keshavarz, M.; Pouretedal, H. An empirical method for predicting detonation pressure of CHNOFCl explosives. Thermochim. Acta 2004, 414, 203–208. [Google Scholar] [CrossRef]
- Zahari, N.; Montazeri, M.; Hoseini, S. Estimation of the Detonation Pressure of Co-crystal Explosives through a Novel, Simple and Reliable Model. Cent. Eur. J. Energ. Mater. 2020, 17, 492–505. [Google Scholar] [CrossRef]
- Meyer, R.; Köhler, J.; Homburg, A. Explosives, 6th ed.; Wiley-VCH: Weinheim, Germany, 2007; ISBN 978-3-527-31656-4. [Google Scholar]
- Rajakumar, B.; Arathala, P.; Muthiah, B. Thermal Decomposition of 2-Methyltetrahydrofuran behind Reflected Shock Waves over the Temperature Range of 1179–1361 K. J. Phys. Chem. A 2021, 125, 5406–5423. [Google Scholar] [CrossRef]
- Šarlauskas, J.; Stankevičiūtė, J.; Tamulienė, J. An Efficient Synthesis and Preliminary Investigation of Novel 1,3-Dihydro-2H-benzimidazol-2-one Nitro and Nitramino Derivatives. Materials 2022, 15, 8330. [Google Scholar] [CrossRef]
- Christe, K.O.; Wilson, W.W.; Sheehy, J.A.; Boatz, J.A. Thermal Stability of Energetic Ionic Liquids. Inorg. Chem. 2007, 46, 6299–6303. [Google Scholar]
- Džingalašević, V.; Antić, G.; Mlađenović, D. Ratio of detonation pressure and critical pressure of high explosives with different compounds. Sci. Tech. Rev. 2004, 5, 72–76. [Google Scholar]
- Kumar, P. , Ghule, V.D., Dharavath, S. Advancing energetic chemistry: the first synthesis of sulfur-based C–C bonded thiadiazole-pyrazine compounds with a nitrimino moiety. Dalton Trans. 2024, 53, 19112–19115. [Google Scholar] [CrossRef]
- Chandler, J.; Ferguson, R.E.; Forbes, J.; Kuhl, A.L.; Oppenheim, A.K.; Spektor, R. Confined combustion of TNT explosion products in air. In Conference: 8th international Colloquium on Dust Explosions, Schaumburg, IL, September 21-25, 1998. Available online: https://www.osti.gov/biblio/3648 (accessed on day month year).
- Rice, B. M.; Byrd, E.F.C. Relationships with oxygen balance and bond dissociation energies. Theoretical and Computational Chemistry, 2022; 22, 67–75. [Google Scholar] [CrossRef]
- Zeman, S.; Jungov, M. ; Sensitivity and Performance of Energetic Materials. Propellants Explos. Pyrotech, 2016; 41, 426–451. [Google Scholar] [CrossRef]
- Lia, G.; Zhang, Ch. ; Review of the molecular and crystal correlations on sensitivities of energetic materials. Journal of Hazardous Materials 2020, 398, 122910. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. High Performance, Low Sensitivity: Conflicting or Compatible? Propellants Explos. Pyrotech. 2016, 41, 414–425. [Google Scholar] [CrossRef]
Figure 1.
Comparison of densities: Density I, calculated by dividing the molecular weight by the molar volume obtained from B3LYP/cc-pVTZ calculations; and Density II, derived using the equation proposed by Politzer et al.
Figure 1.
Comparison of densities: Density I, calculated by dividing the molecular weight by the molar volume obtained from B3LYP/cc-pVTZ calculations; and Density II, derived using the equation proposed by Politzer et al.
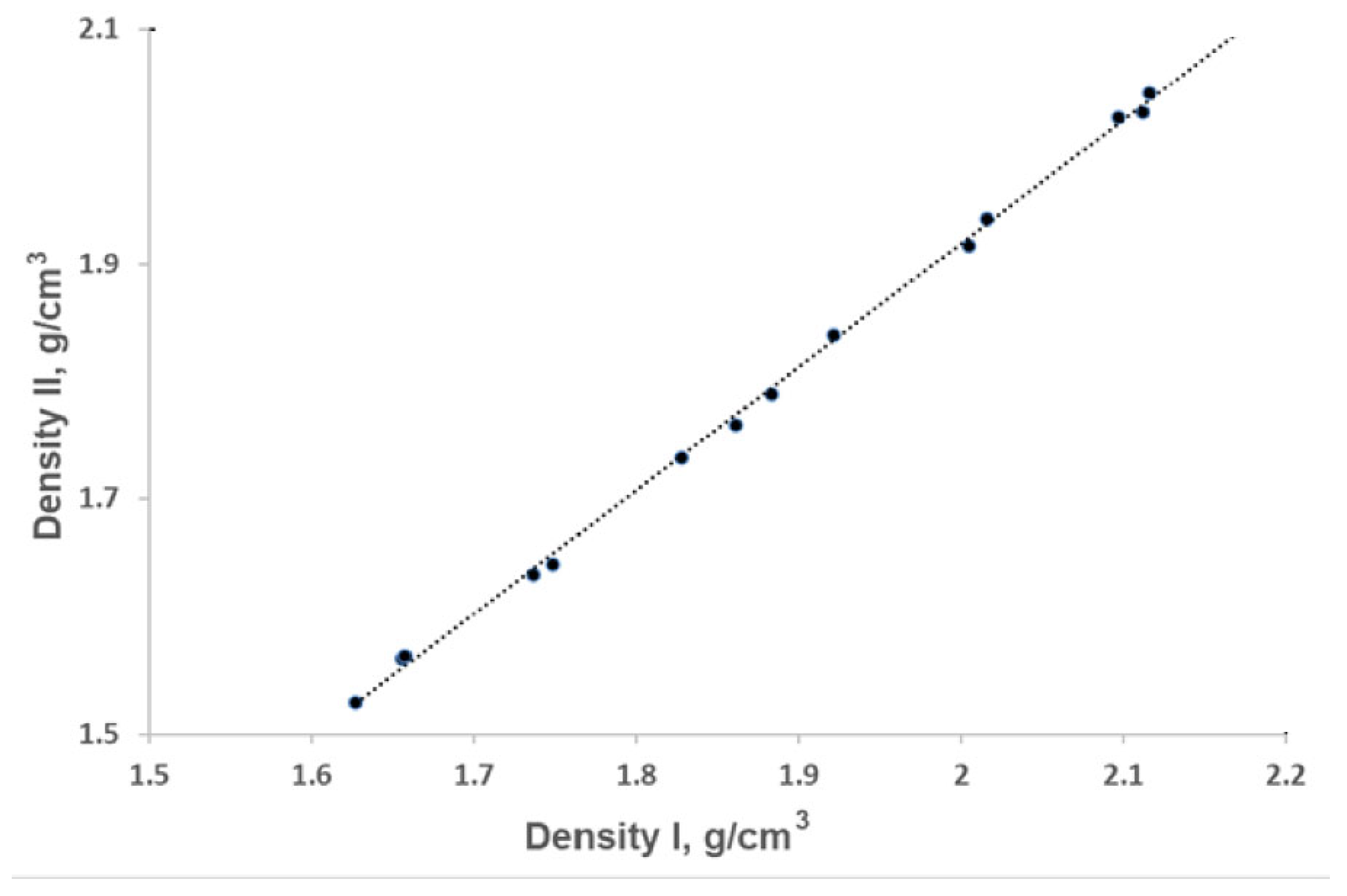
Figure 2.
The perspective of 4,6-dinitrobenzimidazol-2-one. The compound comprises 37.51% carbon, 1.80% hydrogen, 25.00% nitrogen, and 35.69% oxygen by weight. The abbreviation of this compound in the manuscript is Ex0.
Figure 2.
The perspective of 4,6-dinitrobenzimidazol-2-one. The compound comprises 37.51% carbon, 1.80% hydrogen, 25.00% nitrogen, and 35.69% oxygen by weight. The abbreviation of this compound in the manuscript is Ex0.
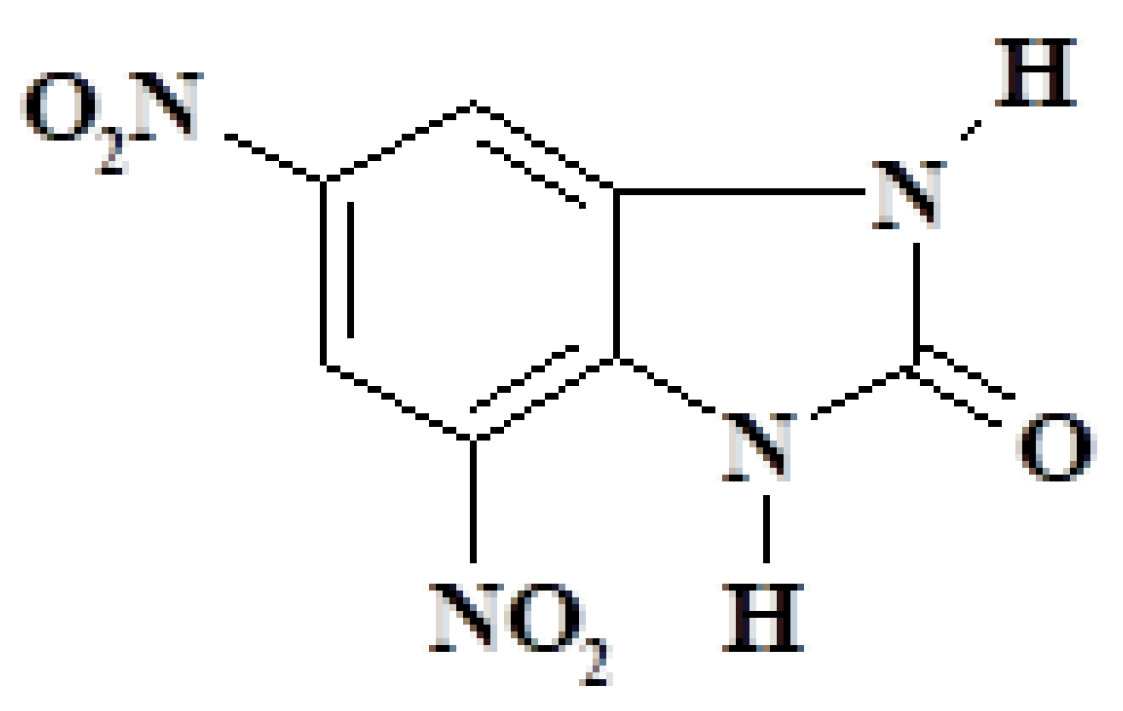
Figure 3.
Cohesive energy per atom for 4,6-dinitrobenzimidazol-2-one with various functional group substitutions on the hexagonal ring.
Figure 3.
Cohesive energy per atom for 4,6-dinitrobenzimidazol-2-one with various functional group substitutions on the hexagonal ring.
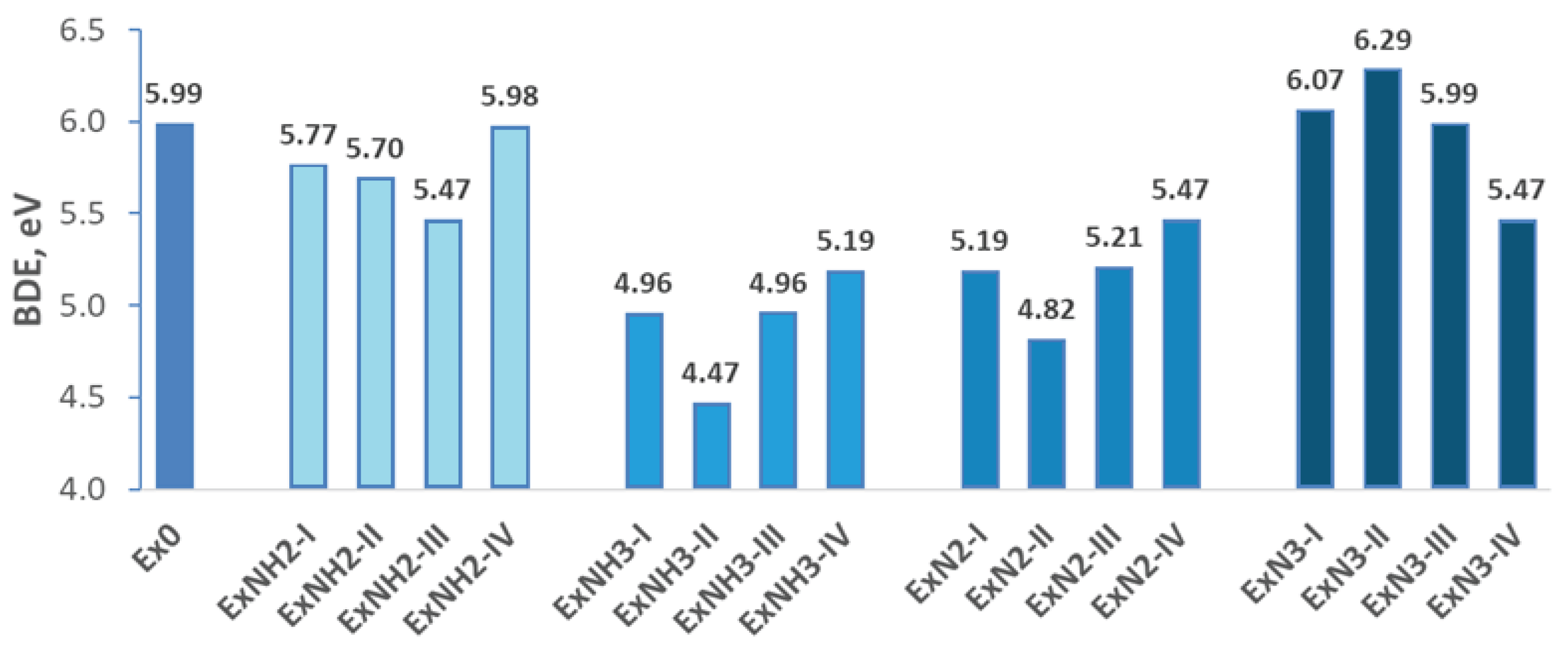
Figure 4.
HOMO–LUMO energy gap variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Figure 4.
HOMO–LUMO energy gap variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
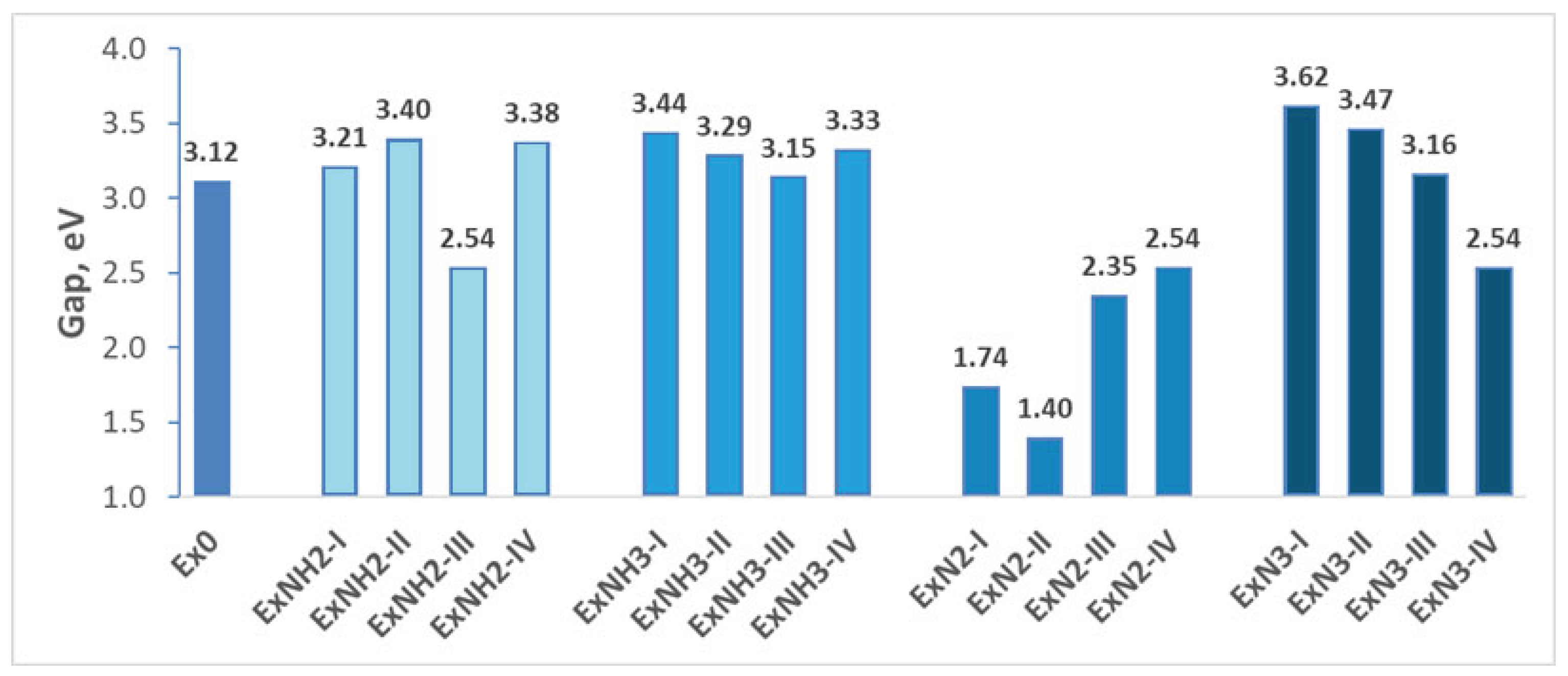
Figure 5.
Chemical harness variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Figure 5.
Chemical harness variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
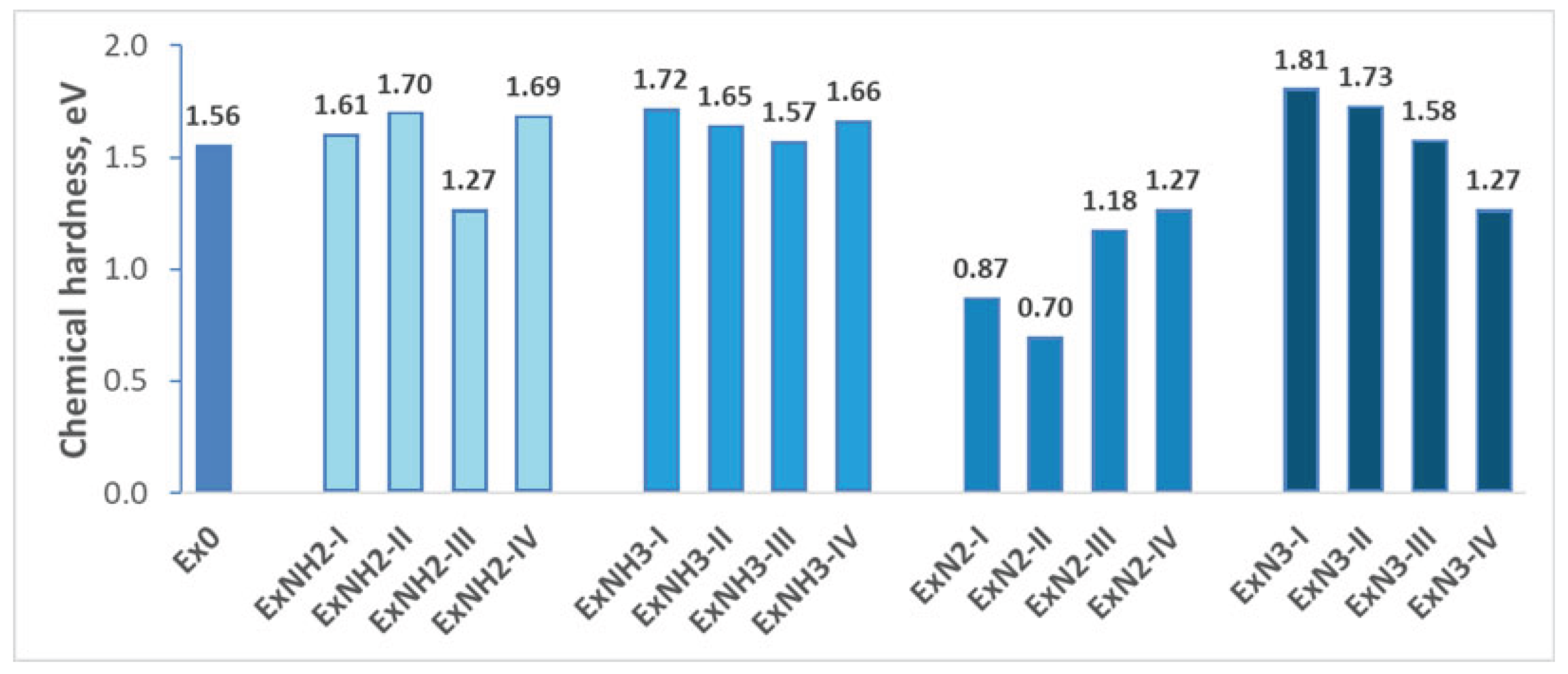
Figure 6.
Electronegativity variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Figure 6.
Electronegativity variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
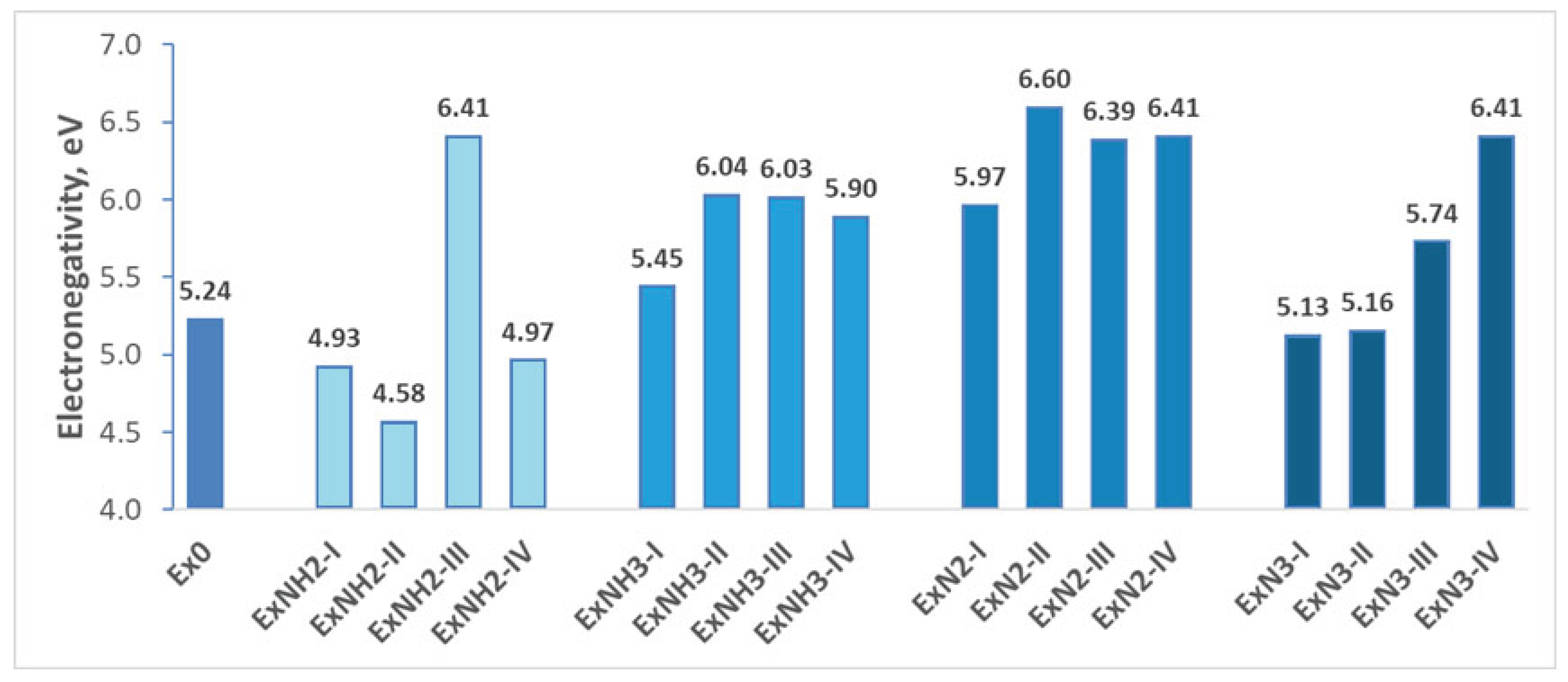
Figure 8.
Detonation pressure variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.The obtained values are compared with those of TNT, RDX, and HMX, whose parameters are indicated by red lines.
Figure 8.
Detonation pressure variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.The obtained values are compared with those of TNT, RDX, and HMX, whose parameters are indicated by red lines.
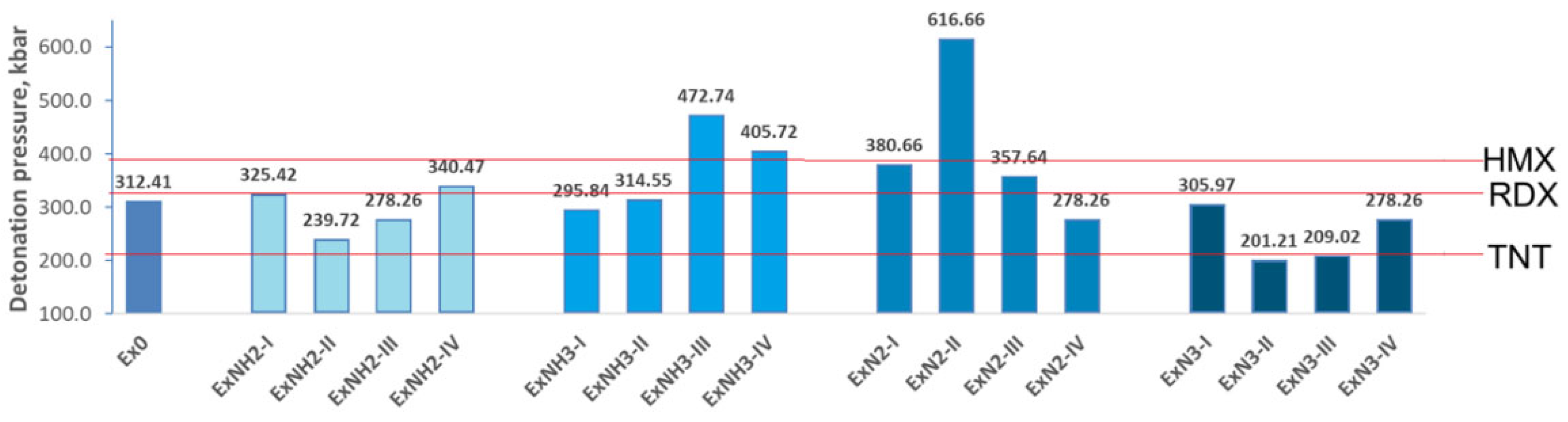
Figure 9.
Detonation velocity variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one. The obtained values are compared with those of TNT, RDX, and HMX, whose parameters are indicated by red lines.
Figure 9.
Detonation velocity variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one. The obtained values are compared with those of TNT, RDX, and HMX, whose parameters are indicated by red lines.
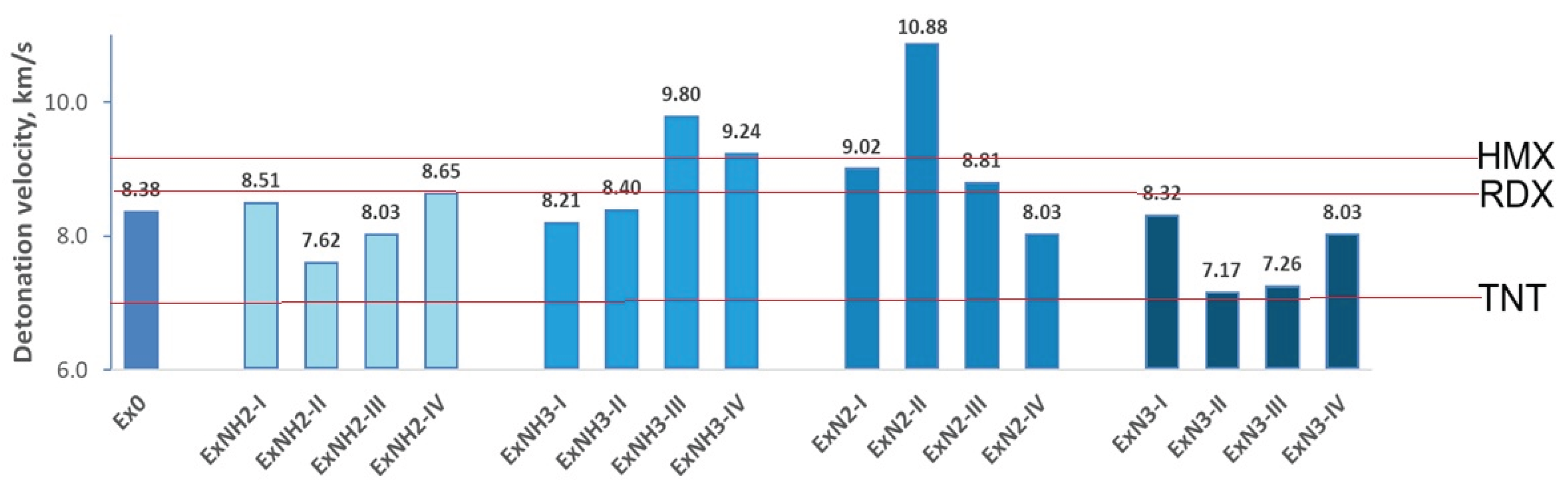
Figure 10.
Oxygen balance variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
Figure 10.
Oxygen balance variations for various substituents introduced on the hexagonal ring of 4,6-dinitrobenzimidazol-2-one.
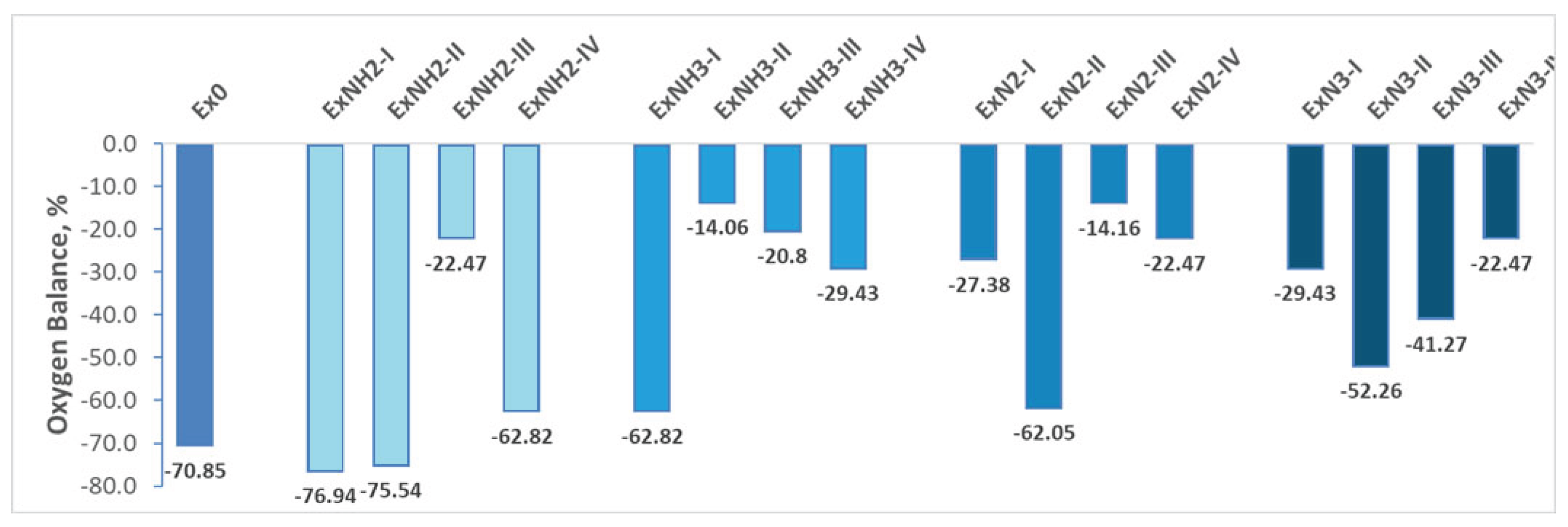
Figure 11.
Equilibrium structures of the compounds containing ExNH3 groups, optimized using the B3LYP/cc-pVTZ method. Carbon atoms are shown in brown, nitrogen in light blue, oxygen in red, chlorine in green, and hydrogen in grey.
Figure 11.
Equilibrium structures of the compounds containing ExNH3 groups, optimized using the B3LYP/cc-pVTZ method. Carbon atoms are shown in brown, nitrogen in light blue, oxygen in red, chlorine in green, and hydrogen in grey.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



