Submitted:
06 June 2025
Posted:
09 June 2025
Read the latest preprint version here
Abstract
Whole Genome Sequencing (WGS) has boosted our ability to explore microbial diversity by enabling the recovery of Metagenome-Assembled Genomes (MAGs) directly from environmental DNA. As a result, the vast availability of sequencing data has prompted the development of numerous bioinformatics pipelines for MAG reconstruction, along with challenges to identify the most suitable pipeline to perform the analysis according to the user needs. This report briefly discusses the computational requirements of these pipelines, presents the variety of interfaces, workflow managers and package managers they feature, and describes the typical modular structure. Also, it provides a compacted technical overview of 31 publicly available pipelines or platforms to build MAGs starting from short and/or long sequences. Moreover, recognizing the overwhelming number of factors to consider when selecting an appropriate pipeline, we introduce an interactive decision-support web application, 2Pipe, that helps users to identify a suitable workflow based on their input data characteristics, desired outcomes, and computational constraints. The tool presents a question-driven interface to customize the recommendation, a pipeline gallery to offer a summarized description, and a pipeline comparison based on key factors used for the questionnaire. Beyond this and foreseeing the release of novel pipelines in the near future, we include a quick form and detailed instructions for developers to append their workflow in the application. Altogether, this review and the application equip the researchers with a general outlook of the growing metagenomics pipeline landscape and guide the users towards deciding the workflow that best fits their expectations and infrastructure.
Keywords:
metagenomics
; metagenome-assembled genome
; pipeline
; workflow manager
Introduction
Metagenome-assembled genomes (MAGs) are reconstructed genomes obtained by assembling and binning sequences directly from metagenomic data captured from a broad range of environments, including the human body, soil or oceans. Strictly, the Minimum Information about MAGs (MIMAG) guidelines establish that MAGs can be classified into three quality tiers: high-quality drafts (HQ, ≥90% completeness and ≤5% contamination, presence of rRNA genes and tRNAs), medium-quality drafts (MQ, ≥50% completeness and ≤10% contamination), and low-quality drafts (below medium-quality thresholds) [1]. MAGs can also be divided into SMAGs (MAGs for which a species can be assigned) and HMAGs (MAGs that are supposedly genomes of novel species) according to the genome heterogeneity spectrum proposed by Setubal (2021) [2]. Therefore, MAG study enables the genomic characterization of uncultured microorganisms with consequent community structure, functional potential, and evolutionary relationships inferences [3], [4].
Pipelines for MAG recovery are essential for extracting meaningful information about the structure and function of microbial communities. Through their orchestrated workflow, they simplify and standardize the common tasks that are required to achieve HQ MAGs such as quality control, assembly, binning, and annotation, and therefore they reduce the occurrence of manual errors by improving reproducibility [5]. Nonetheless, pipeline choice may not be a trivial decision given that it should be based on the alignment between of user needs and workflow key factors such as the type of sequencing data they handle (short, long, or hybrid strategy), analytical functions (i.e., co-assembly, sequential co-assembly, taxonomic profiling, eukaryotic recovery), and computational environment (e.g., availability of local resources, HPC, or web-based tools). Therefore, pipeline selection can quickly become an overwhelming process and challenge researchers with a vast landscape of options, delaying the start of the analysis or even in some cases not obtaining the expected results since the incorrect workflow was chosen.
Here, we succinctly highlight important considerations regarding pipeline execution, storage needs and computational. Likewise, we provide a compact overview of 31 publicly available pipelines designed to build and annotate MAGs starting from short and/or long sequences. Finally, considering the main practical features of each pipeline and aiming at aiding researchers in navigating the ecosystem of workflows, we also introduce 2Pipe, a decision-support web application designed to match metagenomics community users with the most suitable MAG pipeline based on their input data, technical requirements, bioinformatics experience and preferred interface.
1. Practical and Technical Considerations for Pipeline Execution
As high-throughput sequencing technologies have grown in the past years, the availability of MAG-centered pipelines has been quickly expanded to handle and integrate different data types and computational strategies. Specifically, recent pipelines have been designed or have evolved to assemble and bin short reads (normally Illumina), long reads (mainly Oxford Nanopore and/or PacBio) or a blend of both technologies to maximize high base accuracy, depth, contiguity and structural information. Eventually, pipelines can also handle MGI reads as this company offers short reads through Nanoball sequencing (DNB) or long reads as a product of CycloneSEQ, a method that relies on a protein nanopore setting similar to Oxford Nanopore. Differences or similarities among these MAG-reconstruction approaches based on the type of sequence used as input have been studied by Goussarov et al. (2024) [6], and Kim et al. (2021) [7] analyzed the variations in terms of genome recovery between Illumina and MGI platforms.
On the other hand, the workflow execution varies in terms of computational demands, where small-scale datasets can be processed on high-end workstations, whilst large or complex metagenomes often require access to high-performance computing (HPC) clusters or cloud-based environments (Azure, AWS, Google Cloud, Terra, among others). Assembly and binning software are the main responsible for the scaling up in these hardware demands, especially when handling datasets with several samples encompassing millions of short-read sequences [8]. Also, the advantages of performing co-assembly and/or co-binning to increase overall recovery rate and quality have been highlighted by Vosloo et al. (2021) [9] and Han et al. (2025) [10]. These strategies increase substantially the computational resources required to achieve these quality standards [8], although sequential co-assembly has emerged recently as an efficient alternative that enhances both time and memory requirements by the assembler [11].
On the other hand, many authors have highlighted the benefits of applying refinement and replication techniques to the bin/MAG set obtained before and/or after quality assessment [12,13,14] (see a detailed explanation about MAG building in the following section and in Figure 1), albeit the inclusion of these tools, as well as their parameters should be always chosen carefully. This is particularly discussed by Evans & Denef (2020) [15] who suggest elements of analysis when choosing the proper strategy for bin refinement and dereplication.
Beyond the computational demands previously mentioned, most metagenomics pipelines rely on external reference databases to perform taxonomic classification, functional annotation, and quality assessment of MAGs. Commonly used databases include RefSeq [16], GTDB [17], UniProtKB [18], KEGG [19], eggNOG [20], among others, which are frequently large and require substantial local storage that ranges from tens to hundreds of gigabytes. For instance, the latest GTDB release (R226) exceeds 140 GB, while comprehensive functional annotation pipelines like DRAM [21] can demand up to 500 GB to exploit its main potential. Being so, MAG building is a demanding process that needs adequate disk space, CPU capacity and memory availability.
For researchers without access to HPC resources, web-based platforms such as KBase [22], MGnify [23], Galaxy [24], BV-BRC [25] (formerly PATRIC), among others, can assist them by carrying out analysis execution in their servers. In addition, these platforms aid users without a strong experience in command line interface (CLI) interaction since they provide user-friendly interfaces where users can upload raw reads and run predefined workflows. Being so, these platforms eliminate the need for command-line interaction and offer built-in visualization applications and databases for downstream interpretation; a complete landscape of web-based applications is compiled by Achudhan et al. (2024) [26].
Furthermore, given the MAG pipeline evolution in complexity, involving multiple tools, dependencies and steps, the use of workflow managers has become the standard to ensure reproducibility, scalability, and portability [27]. Specifically, workflow managers ease pipeline step definition in a modular and automated architecture to orchestrate entire analyses, tracking software versions, managing intermediate files, restarting the process if interrupted, handling multi-sample input and enabling parallel processing in a reproducible manner. The main representatives of these helpful orchestrators are Snakemake [28], Nextflow [29]/nf-core [30], Workflow Description Language (WDL) [31] and Galaxy [32] whose design, implementation, benefits and scope have been reviewed in some reports [28]– [30], [32]; also, important guidelines for pipeline design based on workflow managers have been published by Roach et al. (2022) [33], Reiter et al. (2020) [34] and Ahmed et al. (2021) [5]. Advantageously, containerization platforms such as Docker, Singularity and Seqera Containers, or package managers like Conda or PyPI complement workflow orchestrators by offering a flexible and reproducible solution for software and dependency management [35]. As a result, this combination allows users to run the analysis, without system conflicts, specific versions of the software and libraries.
In contrast, beyond the MAG assembly and annotation, some pipelines feature interesting options that complement the analysis and provide a wider understanding about the microbial community. The range of these special options is wide, and therefore they must be carefully selected. In this sense, read-based taxonomic profiling [27] is one of the most common offerings by the pipelines, as this process does not rely on the main workflow and can be executed in parallel. Furthermore, some pipelines can incorporate tools or modules to recover viral or eukaryotic MAGs [36], and it is even possible to find pipelines mostly focused on this type of MAGs [37]. Another popular extra option is represented by the possibility of establishing genome-scale metabolic models (GEMs) among the built MAGs [38], [39]. However, in many cases some workflows can be considered as unique since they include options that no other pipeline encompasses. Examples of these rare features are the possibility to assemble plasmids [39], genotype recovery [40], RNA-seq transcriptome analysis [41], inverted assembly/binning [42] and controlled resource allocation [39].
2. Pipelines
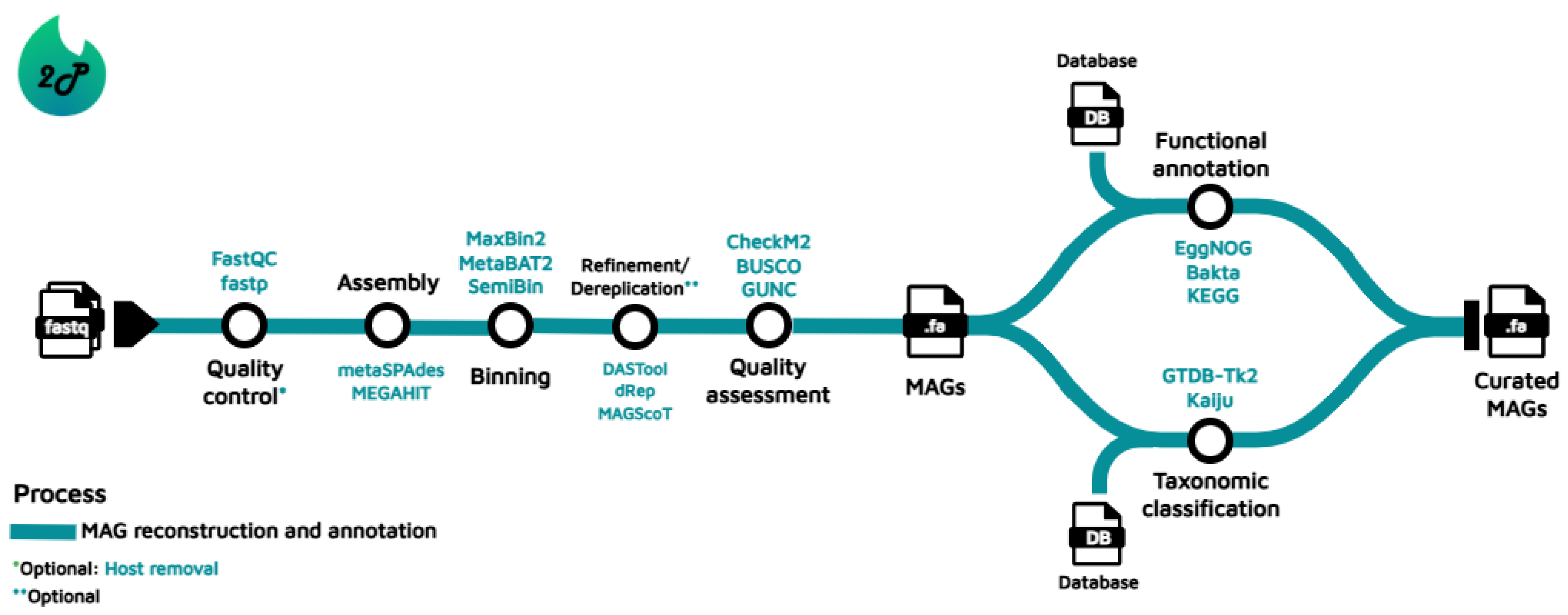
The traditional computational workflow to build and annotate MAGs involves several steps [8]; Figure 1 introduces the general series of steps to potentially achieve MQ or HQ MAGs, along with some common software integrated by the pipelines. It begins with quality control, where low-quality reads and contaminants are removed; when required, some pipelines include the option to discard host organism sequences. This is followed by the assembly step, where reads are extended to create contiguous sequences, also called contigs. The contigs are then grouped into bins that potentially represent individual genomes, based on sequence composition, coverage patterns, among other genomic features. Optionally, the bins are subjected to a process of refinement or dereplication when researchers consider it necessary. Afterwards, these bins are evaluated for common metrics such as completeness and contamination to assess their quality, and hence determine whether they constitute MAGs or not, using the criteria previously mentioned. In some cases, the quality of the bins/MAGs is measured before and/or after refinement/dereplication. To conclude with the workflow, the MAGs are then taxonomically affiliated and functionally annotated to assign biological meaning, enabling insights into the identity and potential roles of the recovered genomes within their microbial communities. A detailed description of the tools for each step of the workflow is provided by Yang et al. (2021) [8], and Wajid et al. (2022) [43] present an overview of the typical analysis pipeline and software using an interesting music analogy.
At the moment of writing this report, we have documented 31 pipelines, resources or platforms that enable MAG reconstruction and annotation. These pipelines vary in their ability to process different data types, scale across sample sizes, and integrate multiple software for quality control, assembly, binning, refinement, taxonomic classification, and functional annotation. On Table 1, we present a summarized overview of the technical features and methodological factors each workflow presents, and hence these same pipeline aspects are also the basis for the design of 2Pipe. Methodological factors include the ability to assemble short reads, long sequences or both in a hybrid approach, the possibility to request a co-assembly/co-binning natively, whether the user can use a multi-sample input or not, if the pipeline includes a bin refinement or dereplication module, and special functionalities they may incorporate. In the same sense, technical features are described by which kind of resources the user is planning to use for the pipeline execution, the interface they feel more comfortable working with, the workflow manager they expect to orchestrate the data flow, and the software/package technology management available within each workflow.
Figure 1.
Traditional bioinformatics workflow followed to perform MAG recovery, classification and annotation. Some common tools incorporated by the pipelines are highlighted.
Figure 1.
Traditional bioinformatics workflow followed to perform MAG recovery, classification and annotation. Some common tools incorporated by the pipelines are highlighted.
2.1. Descriptive Pipeline Overview
Below we introduce a descriptive overview of the main workflow for each pipeline, where important technical considerations such as the type of input (short reads, long reads or both), key tools employed at each step, advantages, limitations and/or special features they depict are documented.
2.1.1. Short-Read Centered Pipelines
2.1.1.1. Anvi’o [45]
Anvi’o is a comprehensive modular platform for the analysis and visualization of microbial omics including, but not restricted to, metagenomics, metatranscriptomics and metapangenomics. Anvi’o is developed to be highly customizable through exchangeable programs (tools) that perform specific tasks, empowering the user with a wide range of tools to explore. Being so, a metagenomics workflow is proposed by the developers of the platforms that begins with short-read quality cleaning, proceeds to read assembly to be used for read recruitment (mapping), and finalizes contig annotation (functions, Hidden Markov Models, and taxonomy). Optionally, the user can achieve read taxonomic profiling with KrakenUniq [65], and more recently binning tools have been made available such as MetaBAT2 [66], CONCOCT [67], MaxBin2 [68], BinSanity [69], as well as DASTool [14] as a refinement alternative. Nonetheless, the user must run the analysis manually, requiring them to account with some experience regarding software installation, execution and debugging. Moreover, although Anvi’o is in principle a command line tool, it incorporates a user-friendly graphical interface for data inspection and visualization that is commonly used for contig visualization.
2.1.1.2. DATMA [42]
DATMA (Distributed AuTomatic Metagenomic Assembly and annotation framework) is a pipeline focused on speed and automation, leveraging distributed computing for efficiency. As a starting point, DATMA applies a quality filter with RAPPIFILT (customized tool developed for this pipeline), Trimmomatic [70] and FastQC [71], and if the input sequences are paired-end, it merges them using FLASH [72] and ForceMerge. Following this procedure, this pipeline identifies and removes 16S rDNA sequences based on RFAM [73] (RNA sequence families), NCBI [16], RDP (Ribosomal Database Project) [74] and SILVA [75] to cluster the remaining sequences with CLAME [76]. The clusters (or bins in definition of the traditional workflow) generated then are assembled in batches by metaSPAdes [77], Velvet [78], and MEGAHIT [79] for a subsequent taxonomic annotation relying on BLAST [80] and Kaiju [81], as well as ORF prediction with Prodigal [82] and GeneMark [83]. To conclude with the analysis a detailed HTML report is generated with interactive Krona [84] plots for taxonomic visualization; this report integrates the 16S rDNA annotation (RDP Classified) along with the annotated bins. As inferred from the described workflow, DATMA performs an inverted approach to generate bins by first grouping the reads using CLAME and attempting to assemble only these groups individually afterwards. Further, this pipeline is wrapped by COMP Superscalar which facilitates the development and execution of parallel applications for distributed infrastructures such as clusters, cloud services and containerized platforms.
2.1.1.3. EasyMetagenome [46]
EasyMetagenome integrates a classical workflow starting with short reads to provide a de-replicated (dRep [13]) set of bins and pangenome analysis that relies on an Anvi’o module. The assembly is performed with MEGAHIT [79], a MetaWRAP [57] module is in charge of the binning task, CheckM2 [85] controls the quality of the bins, and GTDB-Tk2 [17] finalizes the execution by taxonomically annotating them. Notably, this pipeline performs functional annotation (GhostKOALA [86], eggNOG [20], dbCAN3 [87]) and taxonomy assignment on the contigs after a pre-filtering step that generates a non-redundant gene set. EasyMetagenome uses Conda environments to assure reproducibility, the user can input multi-sample data, although it is not orchestrated by any workflow manager. As special remarks, it carries out a taxonomic profiling (MetaPhlAn [88], HUMAnN [89], Kraken2 [90]) of the post-filtered (KneadData) reads, and the functional annotation of the gene set is expanded to identify virulence factors (VFDB [91]) and antibiotic resistant genes (CARD [92]).
| Pipeline/Platform | Short reads | Long reads* | Hybrid Assembly | Multi-sample | Assembly/Binning strategies | Bin refinement | Computational resources** | Interface*** | Workflow manager | Software execution | Special features | |
| 1 | aDNA [44] | Yes | No | No | No | Single | Yes | Local, HPC | CLI | None | Local | Designed for ancient DNA |
| 2 | Anvi’o [45] | Yes | No | No | Yes | Single/Co | Yes | Local | CLI/GUI | None | Conda | Eukaryotic/Viral MAGs |
| 3 | Aviary [40] | Yes | Yes | Yes | Yes | Single | Yes | Local, HPC, CC | CLI | Snakemake | Conda | Genotype recovery |
| 4 | BV-BRC [25] | Yes | No | No | Yes | Single | No | External | GUI | None | None | Taxonomic profiling, Viral MAGs |
| 5 | DATMA [42] | Yes | No | No | No | Single | No | Local/HPC | CLI | COMP Superscalar | Local | Inverted binning and assembly |
| 6 | EasyMetagenome [46] | Yes | No | No | Yes | Single/Co | Yes | Local, HPC | CLI | None | Conda | Taxonomic profiling |
| 7 | EasyNanoMeta [47] | No | Yes (ONT) | Yes | Yes | Single | No | Local, HPC | CLI | None | Conda, Singularity | Taxonomic profiling |
| 8 | Eukfinder [37] | Yes | Yes | No | No | Single | No | Local, HPC | CLI | None | Conda | Eukaryotic MAGs |
| 9 | Galaxy [48] | Yes | Yes | Yes | No | Single | Yes | External | GUI | None | None | Taxonomic profiling |
| 10 | GEN-ERA [49] | Yes | Yes (ONT) | No | Yes | Single | No | Local, HPC, CC | CLI | Nextflow | Singularity | Metabolic modeling |
| 11 | HiFi-MAG [50] | No | Yes (PacBio) | No | Yes | Single | Yes | Local, HPC, CC | CLI | Snakemake | Conda | |
| 12 | IDseq [51] | Yes | Yes (ONT) | No | No | Single | No | External | GUI | None | None | Viral MAGs |
| 13 | KBase [22] | Yes | Yes | Yes | Yes | Single/Co | Yes | External | GUI | None | None | Taxonomic profiling, metabolic modeling |
| 14 | MAGNETO [52] | Yes | No | No | Yes | Single/Co | No | Local, HPC, CC | CLI | Snakemake | Conda | Taxonomic profiling |
| 15 | MetaGEM [38] | Yes | No | No | Yes | Single | Yes | Local, HPC, CC | CLI | Snakemake | Conda | Eukaryotic MAGs, Metabolic modeling |
| 16 | MetaGenePipe [53] | Yes | No | No | Yes | Single/Co | No | Local, HPC, CC | CLI | WDL | Singularity | |
| 17 | Metagenome-Atlas [54] | Yes | No | No | Yes | Single | Yes | Local, HPC, CC | CLI | Snakemake | Conda | |
| 18 | Metagenomics-Toolkit [39] | Yes | Yes (ONT) | No | Yes | Single | Yes | Local, HPC, CC | CLI | Nextflow | Docker |
Plasmid assembly, Metabolic modeling, Controlled resource allocation |
| 19 | Metaphor [55] | Yes | No | No | Yes | Single/Co | Yes | Local, HPC, CC | CLI | Snakemake | Conda | |
| 20 | metaWGS [56] | Yes | Yes (PacBio) | No | Yes | Single/Co | Yes | Local, HPC, CC | CLI | Nextflow | Singularity | |
| 21 | MetaWRAP [57] | Yes | No | No | Yes | Single/Co | Yes | Local/HPC | CLI | None | Conda, Docker | Taxonomic profiling |
| 22 | MGnify [23] | Yes | Yes | Yes | Yes | Single/co | No | External | GUI | None | None | Taxonomic profiling |
| 23 | MOSHPIT [58] | Yes | No | No | Yes | Single | Yes | Local, HPC | CLI | None | Conda | Taxonomic profiling |
| 24 | MUFFIN [41] | No | Yes (ONT) | Yes | Yes | Single | Yes | Local, HPC, CC | CLI | Nextflow |
Conda, Docker, Singularity |
RNA-seq transcriptome analysis |
| 25 | NanoPhase [59] | No | Yes (ONT) | Yes | No | Single | Yes | Local, HPC | CLI | None | Conda | |
| 26 | nf-core/mag [60] | Yes | No | Yes | Yes | Single/Co | Yes | Local, HPC, CC | CLI | Nextflow |
Conda, Docker, Singularity, Other |
Taxonomic profiling |
| 27 |
ngs-preprocess- MpGAp-Bacannot [61] |
Yes | Yes | Yes | Yes | Single | No | Local, HPC, CC | CLI | Nextflow | Conda, Docker, Singularity | |
| 28 | SnakeMAGs [62] | Yes | No | No | Yes | Single | No | Local, HPC, CC | CLI | Snakemake | Conda | |
| 29 | SqueezeMeta [63] | Yes | Yes | Yes | Yes | Single/co | Yes | Local, HPC | CLI | None | Conda | |
| 30 | Sunbeam [64] | Yes | No | No | Yes | Single | No | Local, HPC | CLI | Snakemake | Conda, Docker | Taxonomic profiling |
| 31 | VEBA [36] | Yes | No | No | Yes | Pseudo-coassembly | No | Local, HPC | CLI | None | Conda | Eukaryotic/Viral MAGs |
| *Long reads: ONT: Oxford Nanopore Technology. PacBio: Pacific Biosciences. **Computational resources: HPC: High Performance Cluster. CC: Cloud Computing. ***Interface: CLI: Command Line Interface. GUI: Graphical User Interface. | ||||||||||||
2.1.1.4. MAGNETO [52]
MAGNETO is an automated, modularized and scalable pipeline wrapped with Snakemake [28] and executed with Conda. It is focused on allowing the user the selection of different assembly and/or binning strategies, involving several steps from read pre-processing until MAG annotation and gene catalog generation. The Pre-processing module leverages fastp [93], Bowtie2 [94] and FastQ Screen [95], whilst the Assembly mode uses Simka [96] and hierarchical agglomerative clustering to cluster the samples if the users pre-defines a co-assembly strategy; the reads are assembled using MEGAHIT [79]. Furthermore, contig abundances are computed by alignment against the raw reads to be bin by MetaBAT2 [66] afterwards. Quality estimation and dereplication are carried out with CheckM [97] v1.0 and dRep [13], respectively. To end the workflow, a gene catalog is produced for both the contigs and the MAGs by running Prodigal [82], Linclust [98] and CD-HIT [99], and the MAGs are annotated with GTDB-Tk2 [17], Mummer [100] and EggNOGmapper [101]. As a special feature, MAGNETO can provide a read-based taxonomy abundance with mOTU [102] profiler. MAGNETO exhibits all the advantages Snakemake wrapping, and executed with Conda, represents such as multi-sample handling, scalability across different computing infrastructures and checkpoint control for workflow restarting.
2.1.1.5. metaGEM [38]
metaGEM represents a traditional end-to-end pipeline designed to reconstruct MAGs from metagenomics raw reads; however, its main feature relies on an integrated module that provides genome scale metabolic models (GEMS). The workflow starts with the read quality cleaning using fastp [93] for a subsequent assembly with MEGAHIT [79] and a contig coverage estimation with BWA [103]. The bins are then obtained via three different tools (MetaBAT2 [66], MaxBin2 [68] and CONCOCT [67]) along a posterior refining by the metaWRAP [57] refinement module. As a result, the bins or MAGs are used as input for CarveMe [104] (Genome Scale Metabolic Models), and SMETANA [105] is called for metabolic interaction predictions and MEMOTE [106] is in charge of generating quality reports. The resulting GEMs can then be used for various downstream analyses, such as predicting metabolic interactions within the community, simulating growth under different conditions, and identifying key metabolic pathways. The pipeline ends with MAG characterization through Prokka [107] and Roary [108] (functional annotation and pangenome analysis), GRiD [109] (growth rate estimation), GTDB-Tk2 [17] (taxonomic annotation) and BWA [103] (genome abundance). As additional features, metaGEM identifies eukaryotic MAGs via EukRep [110] and evaluates contamination with EukCC [111]. Also, this pipeline produces taxonomic abundance profiles from the filtered reads using mOTUS2 [112]. Naturally, this pipeline exhibits the benefits Snakemake [28] orchestration provides, as mentioned previously.
2.1.1.6. MetaGenePipe [53]
MetaGenePipe is a pipeline developed with Workflow Definition Language (WDL), self-executed within a Singularity container, whose primary goal is performing a contig-based functional and taxonomic analysis from short read sequences. It is composed of 4 subworkflows, where the operation starts with the quality control workflow, the subsequent one assembles the reads with MEGAHIT [79] to map them back against the short reads within the third subworkflow. Meanwhile, the last subworkflow is in charge of gene prediction and functional annotation based on two main strategies: alignment with the Swiss-Prot database and Hidden Markov Models search in KOfam database [113]. Although MetaGenePipe does not include binning software to provide MAGs as main output, its versatility that allows an analysis adapted for eukaryotic and viral analyses with minimal modifications, and its uncommon workflow manager within the pipelines considered in this review, makes MetaGenePipe an interesting alternative for users with advanced computational infrastructures. Additionally, MetaGenePipe is designed to handle a co-assembly strategy in case the user requires this feature.
2.1.1.7. Metagenome-Atlas [54]
Metagenome-Atlas is an end-to-end, Snakemake [28]-based and Conda-executed pipeline supporting Illumina short reads and providing a modular workflow. It is divided into four modules, namely Quality Control, Assembly, Genomic Binning and Annotation. The initial module removes host, common contaminants and PCR duplicates, and if necessary, trims low-quality sequences according to user pre-specified parameters. The Assembly module corrects sequence errors based on k-mer coverage, merges paired-end sequences, assembles them using MEGAHIT [79] and/or metaSPAdes [77] along with a contig-length filtering. The following module uses MetaBAT2 [66], MaxBin2 [68], and optionally VAMB [114] and SemiBin2 [115] to bin the contigs; CheckM2 [85], BUSCO [116] and GUNC [117] are run to measure the bin quality, as well as DASTool [14] and dRep [13] for bin refinement and dereplication. For the last module, Metagenome-Atlas taxonomically and functionally annotates the MAGs using GTDB-Tk2 [17] and DRAM [21], respectively, and it finally produces a gene catalog through mapping the predicted coding sequences using EggNOG mapper [101]. Among the main advantages of Metagenome-Atlas, it is possible to describe the possibility of running individual modules and its energetic supporting community and developers. Moreover, the Snakemake wrapper allows for flexibility, multi-sample handling, and adaptability to medium to large projects running on local servers or High-Performance Cluster (HPC) environments.
2.1.1.8. Metagenomics-Toolkit [39]
Metagenomics-Toolkit is a workflow designed to increase scalability of task execution, enabling optimal resource allocation from its machine learning-optimized assembly step. This optimized assembly tailors the peak RAM value requested by a metagenome assembler to match actual requirements, thereby minimizing the dependency on dedicated high-memory hardware. Metagenomics-Toolkit is wrapped by Nextflow [29] and powered with Docker containerization technology, and it can take either short or Oxford Nanopore (ONT) long reads as input. As a result, this pipeline is highly scalable and adaptable across computational infrastructures with a backbone workflow that relies on the traditional MAG-aimed steps such as quality control, assembly, binning, and annotation, plus an aggregation module that captures the output from each sample to “polish” the final MAGs. Regarding special features offered by Metagenomics-Toolkit, it offers plasmid identification based on various tools, the recovery of unassembled microbial community members, and the discovery of microbial interdependencies through a combination of dereplication, co-occurrence, and genome-scale metabolic modeling.
2.1.1.9. Metaphor [55]
Metaphor is a classic metagenomics pipeline aiming at MAG reconstruction and annotation wrapped by Snakemake [28] and leveraging Conda as package manager. The pipeline is triggered by the user with a .csv file pointing to the sequence directories and a .yaml file with the pipeline configuration. A quality control will be carried out then with FastQC [71] and fastp [93], with a posterior assembly with MEGAHIT [79], contig evaluation with MetaQUAST [118] and mapping against the input sequences using Minimap2 [119] and Samtools; the contigs are binned (VAMB [114], MetaBAT2 [66], CONCOCT [67]) and refined (DASTool [14]). Metaphor execution finalizes with bin annotation through Prodigal, Diamond, and the NCBI COG database. Complementary to Snakemake orchestration capabilities, Metaphor provides a series of plots depicting runtime and memory with the goal of identifying computational bottlenecks during the analyses.
2.1.1.10. MetaWRAP [57]
MetaWRAP is a popular and customizable pipeline built primarily as a command-line framework with a focus on flexibility and user control. MetaWRAP consists of individual modules that can be run independently or combined into custom workflows. Its core functionalities encompasses read QC and cleaning (FastQC [71], Trim Galore and BMTagger), assembly (MEGAHIT [79], metaSPAdes [77], BWA [103] and MetaQUAST [118]), and a binning suite that incorporates MetaBAT2 [66], MaxBin2 [68], and CONCOCT [67]. MetaWRAP also includes a native refinement module that produces hybrid bin sets to explore over the different variants of each bin (original and hybridized bin sets) to determine the “best bin” according to the user pre-specified quality values based on completeness and contamination (CheckM [97] v1.0). This module is frequently executed in independent metagenomics analysis, and even some pipelines described in this review incorporate it within their workflows. If decided by the user, MetaWRAP offers the possibility of bin re-assembling guided by their previous versions, improving the overall bin quality. For MAG taxonomic and functional analysis, MetaWRAP relies on Prokka [107] and Taxator-tk [120] (combined with NCBI [16] databases), and it provides visualization modules for summarizing results. Analogous to MAGNETO [52], MetaWRAP can produce read-based taxonomic profiles in parallel. Although MetaWRAP does not integrate full pipeline automation, its high modularity and straightforward design have promoted a wide supporting community. Nonetheless, at the moment of writing this report, MetaWRAP is not maintained by the developers, with the subsequent lack of tool updates.
Nonetheless, given the popularity of MetaWRAP, a Snakemake [28] wrapper was developed to automate the metagenomics analysis known as SnakeWRAP [121]. Therefore, SnakeWRAP can carry out the MetaWRAP end-to-end read processing to generate MAGs in a single run, retaining the flexibility of MetaWRAP while reducing the burden of manual execution and dependency handling. Additionally, SnakeWRAP’s integrated environment management via Conda and support for HPC environments enables seamless execution of multiple MetaWRAP modules and samples in parallel, being particularly useful for multi-sample execution.
2.1.1.11. MOSHPIT [58]
According to its documentation, MOSHPIT (MOdular SHotgun metagenome Pipelines with Integrated provenance Tracking) is a toolkit of plugins for whole metagenome assembly, annotation, and analysis built on the microbiome multi-omics data science framework QIIME 2 [122]. MOSHPIT enables flexible, modular, fully reproducible workflows for read-based or assembly-based analysis of metagenome data. The core components of MOSHPIT include q2-assembly, which provides functionalities for genome assembly and quality control, and q2-annotate, which supports contig binning, taxonomic classification, and functional annotation. Additional plugins, such as q2-viromics and q2-amrfinderplus, extend capabilities to viral sequence detection and antimicrobial resistance gene annotation, respectively. In technical terms, MOSHPIT must be run locally or on an HPC environment with the possibility to execute the processes in parallel by the explicit declaration of partitions, a native QIIME2 functionality. Further, the entire QIIME2 ecosystem relies on Conda, and hence this a sine-qua-non requisite to perform MAG reconstruction with MOSHPIT.
2.1.1.12. SnakeMAGs [62]
SnakeMAGs is a simple yet useful pipeline that as its name indicates is controlled by a Snakemake [28] wrapper with Conda as software administrator. It integrates basic modules starting with quality control with Illumina-utils [123] and Trimmomatic [70], and if required, host removal with Bowtie2 [94]. Afterwards, the reads are assembled through MEGAHIT [79], the contigs are binned by MetaBAT2 [66], a quality assessment is carried out with CheckM [97] v1.1 and GUNC [117], MAG abundances are obtained using CoverM [124], and finally the taxonomic classification is performed using GTDB-Tk2 [17]. Similar to the previous pipelines governed by Snakemake, SnakeMAGs eases automation, reproducibility, scalability and workflow management.
2.1.1.13. Sunbeam [64]
Sunbeam is a modular pipeline orchestrated by Snakemake [28] with Conda as dependency manager; this configuration makes Sunbeam analysis reliable, reproducible and scalable. The main feature Sunbeam depicts is its modularized and extensible design that allows users to build off the core functionality. The execution backbone of Sunbeam is represented by an initial quality control that encloses adapter trimming, host read removal and low-complexity filtering (Trimmomatic [70], FastQC [71], BWA [103] and Komplexity), followed the assembly of reads into contigs with MEGAHIT [79] along with their corresponding annotation with Prodigal [82], BLAST [80] and Diamond [125] (with nucleotide or protein databases). As complementary procedures, Sunbeam maps the reads to reference genomes (user pre-specified) and delivers a taxonomic assignment of the clean reads using Kraken [126] v1.0. As previously stated, its modularization and ready-to-use templates to create new modules have enabled the development of additional extensions for assigning metagenomic reads to a full bacterial phylogeny, single genome assembly, among others.
2.1.1.14. VEBA [36]
VEBA (Viral Eukaryotic Bacterial Archaeal) is a Conda-executed pipeline designed that enables the recovery and classification of genomes from all domains of life including archaeas, prokaryotes, microeukaryotes, and viruses. It starts with a common short read-preprocessing and assembly from which the process is bifurcated for prokaryotic and viral binning; unbinned contigs from the viral module are reincorporated into the prokaryotic contig set. Residual contigs from the prokaryotic module are then considered for eukaryotic MAG generation to proceed with the annotation and classification covering the genomes obtained in each module. Hence, several databases are considered at this step such as KOfam [113], Pfam [127] and NCBI [16] non-redundant. Also, a joint phylogeny is obtained based on MAG-gene models and lineage marker detection. An interesting approach VEBA follows is represented by the module coverage.py that collects all the unbinned contigs, from viral, eukaryotic and prokaryotic steps, to pursue a pseudo-coassembly, where iteratively the reference fasta (built from the contigs) and the sorted BAM files used as a final pass through prokaryotic and eukaryotic binning modules. Notably, it automates the detection of candidate phyla radiation (CPR) bacteria and integrates a consensus microeukaryotic database to optimize gene modeling and taxonomic classification.
2.1.2. Long-Read Focused Pipelines
2.1.2.1. EasyNanoMeta [128]
EasyNanoMeta is a specialized pipeline designed to process ONT long reads either solely or in combination with short reads (hybrid assembly). This pipeline relies on a dual approach that uses both assembly-based and assembly-free strategies. Particularly, EasyNanoMeta incorporates four assemblers (MetaFlye [129], OPERA-MS [130], MetaSPAdes [77], MetaPlatanus [131]), five binners (SemiBin2 [115], MetaBAT2 [66], MaxBin2 [68], CONCOCT [67], VAMB [114]) and a polishing tool (NextPolish [132]) to assure the best possible outcome. Additionally, once the bins are obtained, it performs the common tasks such as functional annotation with Prokka [107], quality control with CheckM2 [85], phylogeny inference with PhyloPhlan [133] and taxonomic classification with GTDB-Tk2 [17]. For the assembly-free methodology, EasyNanoMeta provides a full report containing composition, diversity and correlation among the identified species with Kraken2 [90] and Centrifuge [134]. Regarding operational characteristics, this pipeline can be run automatically on a Singularity/Apptainer image that streamlines the setup process and minimizes dependency issues or experienced users can execute individual modules through shell scripts that rely on Conda environments.
2.1.2.2. Hi-Fi-MAG-Pipeline [50]
Hi-Fi-MAG is a simple, yet time-saving pipeline developed and maintained by Pacific Biosciences specially designed to build MAGs from Hi-Fi reads (long PacBio reads). It encompasses different binning tools (MetaBAT2 [66] and SemiBin2 [115]) along with DASTool [14] as refinement software; CheckM2 [85] serves a quality control tool, where contigs above 500 kb are kept as single bins if they show a completeness above 93%, otherwise they are sent back to the binning module. This approach enhances the recovery of high-quality and single-contig MAGs, outperforming traditional binning methods. After MAG de-replication, taxonomic annotation is achieved with GTDB-Tk2 [17], and a complete graphical report is compiled automatically. One important caveat about this workflow is represented by its lack of assembly step, and hence the user must prepare the assembly of the PacBio sequences beforehand using tools such as hifiasm [135] in its meta version, metaFlye [129], OPERA-MS [130], among others. Hi-Fi-MAG-Pipeline requires Conda as software manager, and it is orchestrated by Snakemake [28].
2.1.2.3. metaWGS [136]
metaWGS is one of the most recently released pipelines whose main differential is related with the possibility to assemble either short reads or long sequences (PacBio). This Nextflow [29] pipeline is built off Singularity with consequent benefits this kind of setup brings as discussed previously. It incorporates a wide variety of tools as it must ensure a proper workflow for both types of sequencing technologies in a traditional end-to-end framework divided into 8 steps. The first step aims at cleaning and performing quality control with proper tools according to the input, while the second step allows the assembly of the sequences using either metaSPAdes [77]/MEGAHIT [79] for short sequences and hifiasm [135]/metaFlye [129] for PacBio reads. Following with the process, this pipeline filters the contigs and performs structural annotation during steps 3 and 4, respectively; step 5 is designed to estimate contig abundance by mapping them against the reads. Afterwards, a complete subworkflow for functional annotation is undergone with EggNOG mapper [101] at its core (step 6), and contig taxonomic affiliation is achieved through home-made scripts (step 7) to conclude with step 8, where the contigs are binned with MaxBin2 [68], MetaBAT2 [66] and CONCOCT [67]. Remarkably, metaWGS utilizes BINETTE [137], a state-of-the-art binning refinement tool designed to construct high-quality MAGs from the output of multiple binning tools.
2.1.2.4. NanoPhase [59]
NanoPhase is a pipeline that enables building high-quality MAGs from ONT long reads, optionally enhanced with short read-based MAG polishing. The backbone of the pipeline is represented by an assembly with metaFlye [129] followed by contig binning with MetaBAT2 [66] and MaxBin2 [68], and bin refinement with a MetaWRAP [57] module. To estimate abundance and coverage, the contigs are mapped against the reads, and several polishing rounds with Racon [138] and medaka, complete the workflow to generate high-accuracy final bins; If the user decides to include short reads in the analysis, these are used for polishing with Pilon [139]. Complementary, MetaQuast [118] and CheckM [97] v1.0 are in charge of MAG quality control, IDEEL [140] evaluates the fraction of predicted full-length proteins in each MAG, full-length proteins are detected via alignment with UniProtKB [18], and Prokka [107] serves as functional annotation software. Remarkably, NanoPhase allows prophage and active prophage identification within the reconstructed MAGs with VIBRANT [141] and PropagAtE [142]. Among pipeline technical specifications, this pipeline requires Conda as package manager and it offers parallelized execution with GNU Parallel to speed up the analysis.
2.1.3. Hybrid Pipelines
2.1.3.1. Aviary [40]
Aviary is a modular, Snakemake [28]-based pipeline, with Conda as package manager, designed for single or hybrid metagenomic assembly and MAG recovery, supporting both short and long-read input sequences. The workflow is distributed in 8 modules following a traditional workflow starting with quality and diversity assessment of the reads, followed by a discriminated assembly according to the type of input, MEGAHIT [79] or metaSPAdes [77] for short reads only or metaFlye [129] in case of long reads solely. For hybrid assembly the process is divided into four stages: polishing with Racon [138] and Pilon [139], metrics-based filtering, assembly and discard of low-quality bins and re-assembly with Unicycler [143]. The pipeline proceeds with a subsequent assembly evaluation in terms of fragmentation, misassembly detection and diversity quantification, and a complementary module moves forward with a read mapping of the assembly and abundance statistics calculation. To continue with the workflow, the contigs are binned using up to 6 tools (MetaBAT2 [66], Rosella, MetaBAT1 [144], VAMB [114], MaxBin2 [68] and CONCOCT [67]) and refined afterwards with 5-time loop that includes CheckM2 [85], Rosella Refine and DASTool [14]. The pipeline ends with MAG recovery assessment via CoverM, CheckM2 and SingleM to proceed with MAG annotation through GTDB-Tk2 [17], Prodigal [82] and EggNOG [20]. Variant calling, ANI analysis and genotype recovery with Lorikeet are interesting attributes offered by Aviary as a complement to the traditional genomic feature detection. Aviary’s design presents a series of advantages that include the possibility of running modules, multi-sample handling and scalability across different computational infrastructures.
2.1.3.2. GEN-ERA [49]
GEN-ERA suite is a collection of Nextflow [29] pipelines aiming at supporting MAG reconstruction and annotation with as many methodologies as possible starting from either short or long reads. Specifically, this toolbox counts with more that 10 workflows specifically designed for tasks ranging from assembly and binning, quality assessment and decontamination, orthologous inference and maximum likelihood phylogenomic analyses, SSU rRNA phylogeny (constrained by ribosomal phylogenomic), Average Nucleotide Identity (ANI) clustering, taxonomic identification and metabolic modelling. Moreover, GEN-ERA incorporates specific tools designed to handle eukaryotic assembly annotation such as BRAKER2 [145] and AMAW [146]. Thus, GEN-ERA suits almost all requirements any user might demand given the variety of goals that can be achieved within a single software suite. From a technical point of view, operational GEN-ERA features, Nextflow-managed and Singularity-executed, ensures portability and reproducibility across environments.
2.1.3.3. MUFFIN [41]
MUFFIN is a reproducible pipeline built with Nextflow [29] designed for hybrid assembly by integrating short-read (Illumina) and long-read (nanopore) sequencing data. MUFFIN begins its workflow with a quality control of the reads (fastp [93] and Filtlong) to progress through hybrid assembly (metaSPAdes [77] or metaFlye [129] with polishing) and differential binning (CONCOCT [67], MetaBAT2 [66], and MaxBin2 [68]). After bin refining with the MetaWRAP [57] refinement module, a hybrid reassembly is pursued with Unicycler [143]. The pipeline ends with bin classification through CheckM [97] v1.1 and sourmash [147] (combined with GTDB [148]), and with bin annotation with EggNOG [20] and a KEGG [19] parser, providing high-quality, annotated MAGs and insights into the metabolic potential of the microbial community. Optionally, the user can provide metatranscriptomics data to perform a de novo transcript assembly (Trinity [149]), quantification (Salmon [150]) and annotation (EggNOG). Additionally, given its modularity design, the workflow can start as well with user-provided bins, differential reads or only RNA-seq data. MUFFIN can be executed with either Conda or Docker, and its native Nextflow features confer to it the possibility to restart the pipeline in case of failing, run on different computing infrastructures, multi-sample handling, among others.
2.1.3.4. nf-core/mag [60]
nf-core/mag is a Nextflow [29] pipeline developed following the nf-core guidelines that ensures robustness and reproducibility. It supports both short-read and hybrid sequences, and it leverages a modular design, containerization (Docker, Singularity, among others) and package managers (Conda) to confer portability across different computing environments, including HPC and cloud systems. Beyond these important features, as part of the workflow orchestration, nf-core/mag can handle multi-sample input, it can be restarted if it is interrupted at any point thanks to its native checkpoint control and different assembly/binning modes can be selected. This pipeline encompasses tools for quality control of the reads (fastp [93]), host removal (Bowtie2 [94]) and adapter trimming (AdapterRemoval [151]), as well as two assemblers (MEGAHIT [79] and metaSPAdes [77]). In addition, it offers three binning software options (MetaBAT2 [66], MaxBin2 [68] and CONCOCT [67]) along with an optional refinement tool (DASTool [14]). nf-core/mag checks assembly and bin quality through several tools that include CheckM2 [85], MetaQUAST [118], BUSCO [116] and GUNC [117], and for genome annotation, it uses GTDB-Tk2 [17] or CAT [152] (taxonomic) and Prokka [107] or MetaEuk [153] (functional). As special features, this pipeline can carry out a taxonomic annotation of the sequences (Kraken2 [90] and Centrifuge [134]), validates the presence of typical ancient DNA damages (PyDamage [154]), attempts MAG domain classification with Tiara [155] and identifies viruses after assembly with geNomad [156]. After workflow execution, nf-core/mag generates detailed multi-sample summaries through MultiQC [157], and it creates HTML reports to track resource usage. Finally, the nf-core framework is actively maintained and updated as it relies on a numerous and enthusiastic developing community.
2.1.3.5. ngs-preprocess-MpGAp-Bacannot [61]
Ngs-preprocess, MpGAP and Bacannot are a series of Nextflow [29]-based and container-powered pipelines designed to achieve a wide variety of specific tasks. ngs-preprocess performs several quality-control steps required for Next-Generation Sequencing (NGS) data assessment, while MPGAP supports de novo genome assembly from Illumina, PacBio, and ONT reads, enabling short-read, long-read, and hybrid assemblies using tools like metaSPAdes [77], metaFlye [129], Canu [158], and Unicycler [143], followed by polishing and quality assessment. Meanwhile, Bacannot provides an annotation workflow that incorporates gene prediction, rRNA detection, sequence typing, KEGG-based metabolic reconstruction, and secondary metabolite identification, integrating tools such as Prokka [107], Bakta [159], Barrnap, MLST [160], KofamScan [113], KEGGDecoder, and antiSMASH [161]. As an additional analytical procedure, Bacannot incorporates additional support for methylation analysis via Nanopolish [162]. Noticeably, this set of pipelines do not include at any point neither contig binning nor bin quality assessment; however, the smooth interconnection among the pipelines makes them an interesting option for metagenome assembly and annotation, boosted by the native benefits conferred by Nextflow and container technology.
2.1.3.6. SqueezeMeta [63]
SqueezeMeta is a fully automatic pipeline written in Perl scripts that relies on Conda for software execution. As special features, this pipeline can handle short and long reads (ONT and Hi-Fi) in both single or hybrid approaches, supports for de-novo metatranscriptome assembly and hybrid metagenomics/metatranscriptomics analysis, carries out taxonomic annotation of unassembled reads, and empowers the user with a GUI application for downstream analysis. Also, SqueezeMeta’s flexibility enables different assembly modes such as sequential (samples assembled individually), co-assembly (samples assembled ensemble), merged (samples assembled individually with a posterior pooling) and seqmerge (similar to merged with a guided pooling based on assembly similarity). This pipeline follows the traditional workflow by applying quality filtering and trimming with Trimmomatic [70], then the reads are assembled by MEGAHIT [79] and SPAdes (rnaSPAdes [163], Canu [158] and metaFlye [129] are run if transcriptomics or long read data are provided) to be binned afterwards with MaxBin2 [68], MetaBAT2 [66] and CONCOCT [67]; DASTool [14] is in charge of bin refinement. MAG Quality checks are established through CheckM2 [85], and optionally taxonomic classification is achieved by GTDB-Tk2 [17]. To complement MAG annotation with KEGG [19] and MetaCyc [164], SqueezeMeta analyzes the assembly by performing an homology searching against taxonomic and functional databases, an Hmmer search against Pfam [127] database, and an estimation of taxa and function abundances. An important remark of this pipeline is its numerous and helpful developing and maintaining community.
2.1.4. Web-Based Pipelines with External Computational Resource Support
2.1.4.1. BV-BRC [25]
BV-BRC (Bacterial and Viral Bioinformatics Resource Center) is web-based platform that supports a broad spectrum of microbial genomics analyses, including genome-resolved metagenomics. This platform offers an intuitive interface to perform tailored quality control, assembly, binning, annotation, and downstream comparative analyses. For MAG building, BV-BRC has developed a specific metagenomic binning service, which offers genome assembly with metaSPAdes [77] and MEGAHIT [79] and a customized approach for genome binning based on kmer distribution and multi-genome functionality. Moreover, BV-BRC leverages PATRIC [165] genomes to create reference bins as a starting point for annotation with RASTtk [166] and/or VIGOR [167]. Regarding technical features, BV-BRC runs entirely on a remote infrastructure, allowing users to execute workflows without local installations or advanced computational setups. Aside from the features already mentioned, customizable analysis jobs, visualization tools and integrated comparative genomics tools are available, making BV-BRC a valuable resource for users seeking an accessible, reproducible, and data-rich environment for metagenomic studies.
2.1.4.2. Galaxy [48]
Galaxy is a web-based platform and open-source project that empowers scientists all over the world to conduct bioinformatics analysis in an user-friendly and intuitive graphical interface that requires no programming skills. Galaxy offers a broad range of tools covering genomics, transcriptomics, metagenomics, among many others, where the user is free to select the software that best suits their needs. In addition, the users can share their workflows in the platform, and therefore users can just follow pre-established methodologies validated by a world-wide community. As a result, there are multiple pipelines designed for MAG reconstruction that feature common tools like MEGAHIT [79] for assembly, MetaBAT2 [66] or MaxBin2 [68] for binning, and Prokka [107] or GTDB-Tk2 [17] for annotation and classification. Also, given Galaxy’s flexibility the traditional workflow can be expanded to include long reads, accomplish read-based taxonomic profiling or detect and classify viral sequences. Being so, Galaxy ensures reproducibility through automatic tracking of parameters and tool versions, and supports HPC and cloud deployment, making it scalable for projects of various sizes. Notwithstanding, the users may experience limitations in performance for large datasets and/or delays in result processing as Galaxy’s community of users grows every day with the subsequent demand for more computational resources.
2.1.4.3. IDseq [51]
IDseq is an open-source, cloud-based platform developed for metagenomic next-generation sequencing (mNGS) analysis. IDseq has a specific scope focused on pathogen detection, antibiotic resistance detection and infection control. IDseq supports short-reads or long reads (ONT) to provide analyses that encompass host read removal, quality control, alignment, and taxonomic classification using a curated reference database based on NCBI [16] nt and nr databases. Although IDseq is not primarily focused on MAG reconstruction, it is highly valuable in the initial stages of metagenomics data analysis projects. As interesting remarks, IDseq’s results are visualized through interactive dashboards that provide taxonomic trees, abundance plots, and detailed sample metrics thanks to its web-based interface that requires minimal bioinformatics expertise. Also, the users can find alternative pipelines for viral consensus genome recovery and antimicrobial resistance gene detection.
2.1.4.4. KBase [22]
KBase (the Department of Energy Systems Biology Knowledgebase) is a collaborative, web-based platform that enables researchers to perform comprehensive metagenomics analyses through its customized interactive Narrative Interface. This platform allows users to build and share workflows (narratives) for genome assembly, comparative genomics, metagenomics, among others. Specifically, the metagenomics narrative offers running MAG-centered pipeline steps such as quality control, assembly (e.g., metaSPAdes [77], MEGAHIT [79]), binning (i.e., MetaBAT2 [66]), annotation (e.g., RASTtk [166], DRAM [21]), and metabolic modeling using ModelSEED [168]. KBase platform offers automated data provenance, seamless integration with public databases, and interactive visualizations to interpret MAG quality, taxonomy, and metabolic pathways. The possibility of running analyses using external resources makes KBase a powerful and accessible environment for genome-resolved metagenomics, particularly valuable for users lacking access to HPC systems.
2.1.4.5. MGnify [23]
MGnify is a web-based platform hosted by EMBL-EBI with an automatized service for submitting and annotating microbiome-derived sequence data. It counts with a standardized pipeline that receives raw reads to perform functional and taxonomic annotation with an extensive series of tools encompassing mOTUs2 [112], InterProScan [169], KEGG annotation (hmmscan [170]), EggNOG mapper [101] and/or antiSMASH [161]. Optionally, MGnify offers the possibility for read assembly through metaSPAdes with a prior contamination removal to continue with the annotation. In the recent years, MGnify has evolved to accept and process long reads from PacBio and ONT with the pipeline MGnify-lr that carries out read pre-filtering, assembly with Flye and re-mapping against the initial sequences. Furthermore, users can contribute to the resource MGnify Genomes which stores a genome catalogues each user can create with their own MAGs. Once the MAGs are submitted to this space, they are automatically analyzed with a pipeline that establishes overall quality and annotates them. Given that MGnify is a service controlled by EMBL-EBI, the user is only requested to submit the data and make it publicly available before the analysis to ENA. As a result, MGnify is a powerful computational resource and user-friendly as the user interacts with the platform to upload the data through its web interface, taking the burden off the user. However, MGnify’s reliance on predefined workflows may limit flexibility for users seeking to customize specific steps or parameters in the analysis, while at the same time heavy use by multiple users may delay result delivery.
2.1.5. Special Pipelines
2.1.5.1. Pipeline for ancient DNA [171]
MAG recovery from ancient DNA can be challenging due to DNA intrinsic properties such as degradation, fragmentation, chemical damage, low-abundance and contamination. Nonetheless, a validated pipeline to manage this type of data is proposed by Standeven et al. (2024) [171], where the MAGs are obtained by following the classic steps involving quality check, decontamination, assembly, binning, bin quality assessment and refinement, and taxonomic annotation. The main advantage of this pipeline is the integration of different bin software, and it can also authenticate the sequence provenance by estimating damage authentication of the host DNA (mainly human) via mapDamage2 [172]. Despite its validation to recover high-quality MAGs, this pipeline is only proposed and it has not been properly compiled in a single repository or container, and hence users should run the tools manually or leverage any of the other available pipelines in this suite.
2.1.5.2. Eukfinder [37]
Eukfinder is a specialized pipeline designed to recover microbial eukaryotic genomes, including both nuclear and mitochondrial DNA. Considering the inherent complexity and underrepresentation of eukaryotic genomes in metagenomics, this tool is composed by two workflows: the first one for Illumina short reads (Eukfinder_short) and another one for assembled contigs or long-read data (Eukfinder_long). In the workflow for short reads, they are first classified into five major taxonomic groups using Centrifuge [134] and PLAST [173], and afterwards ‘Eukaryotic’ and ‘Unknown’ reads are subsequently assembled and reclassified to refine candidate eukaryotic sequences. On the other hand, the long-read version focuses on classifying pre-assembled contigs before proceeding to genome binning and downstream analysis. The binning procedure is common to both approaches and it relies on MyCC [174] output, Centrifuge, and PLAST results in customized and tailored integration of kmer analysis and contigs mapping to eukaryotic genomes. Given its specificity, Eukfinder represents a flexible solution for studying eukaryotic microbial communities in environmental metagenomics.
3. 2Pipe: It Starts with a Question
Considering the pipeline landscape identified in this review, we have developed a decision-support application that concatenates most of the features described for each workflow. 2Pipe is an interactive web application designed to help researchers to identify the most suitable metagenomics pipeline for reconstructing and annotating MAGs. 2Pipe can be used by users with different expertise levels and computational access, simplifying the often complex selection process by mapping user needs to a curated database of available pipelines.
At the core of 2Pipe is a dynamic, question-driven interface that guides users through a questionnaire. This adaptive form collects information related to the methodological factors and technical features detailed on Table 1. Therefore, every response is used to assign a score to each pipeline based on the presence or absence of specific features that align with the user’s input. It is worthy to mention that the scoring is weighted, and some features have prevalence as they are definitive for the pipeline suggestion. For instance, computational resources, type of sequences and the incorporation of a GUI are prioritized towards ensuring realistic guidance. Furthermore, the recommendation system also shows the second “best hit” among the available pipelines for the user to check in case that the first option does not fulfill their requirements; these suggestions can be as well the starting point for the user to dig into the other sections of 2Pipe. In case that the users decide to promptly benchmark the recommended pipelines in terms of performance, tools such as MAGFlow/BIgMAG [175] and MAGqual [176] can accelerate the process; similarly, Wood et al. (2021) [177] and Meyer et al. (2021) [178] have proposed interesting methodologies to evaluate and compare the outcomes from metagenomics software and pipelines.
Aside from the accession to the questionnaire and the response-based recommendation at the end of this, 2Pipe as well encompasses a pipeline gallery, where a visual catalog is displayed offering individual summaries of each pipeline, describing their main characteristics, supporting technology and a direct access to the source code or publication documenting the pipeline. Additionally, 2Pipe makes available an interactive view of Table 1 that includes the possibility of filtering by each category or by a combination of them, allowing users to directly tailor the search for the pipeline that best suits their needs; the displayed categories are the same key attributes the question-based suggestion system relies on.
The source code for 2Pipe is available at the repository https://github.com/jeffe107/2pipe, and foreseeing the possibility of new pipelines being released in the near future, we provide a quick form for developers to include their workflow into 2Pipe’s recommendation system, pipeline gallery and table comparison. Complementary, at the GitHub repository, developers can find a simple template and detailed instructions for the inclusion of their pipeline through a pull request.
Conclusions
The rapid evolution of sequencing technologies has boosted the availability of metagenomics datasets that demand bioinformatics tools adjusted to the user requirements to achieve cutting-edge analysis, including MAG reconstruction. As a result, in the past 10 years a rise in the number of MAG reconstruction pipelines available has been observed, and the selection of the proper pipeline for the analysis has become an essential step during the execution of metagenomics projects. This review offers a compacted description of 31 publicly available pipelines or platforms, with special focus on their capabilities and distinctive features to serve as a valuable resource for researchers navigating this overwhelming landscape. Expanding the scope of a classical review, we streamlined the selection process by introducing 2Pipe, an interactive decision-support web application that aligns the user needs with the most suitable workflow for their analysis and allows a general overview of the pipeline landscape with its gallery and pipeline-comparison sections. Finally, this review and its accompanying application provide a unified framework that simplifies the decision-making process, releasing part of the burden and uncertainty when setting a metagenomics data analysis project.
Availability of data and materials
2Pipe is hosted under the domain https://2pipe.app/. The source code is available at https://github.com/jeffe107/2pipe, along with a template to include new pipelines. The quick form to add a new pipeline can be found at https://form.jotform.com/jeffe10789/2pipe-form. For version tracking, 2Pipe v.1.1 release has been deposited at Zenodo, and it can be followed with the identifier https://doi.org/10.5281/zenodo.15608773.
Acknowledgments
JYG specially thanks the Federal Commission for Scholarships for Foreign Students (FCS) for their support through the Swiss Government Excellence Scholarship.
References
- Bowers, R.M.; et al. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat. Biotechnol. 2017, 35, 725–731. [Google Scholar] [CrossRef] [PubMed]
- Setubal, J.C. Metagenome-assembled genomes: concepts, analogies, and challenges. Biophys. Rev. 2021, 13, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; et al. Genome-resolved metagenomics: a game changer for microbiome medicine. Exp. Mol. Med. 2024, 56, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Lemos, L.N.; Mendes, L.W.; Baldrian, P.; Pylro, V.S. Genome-Resolved Metagenomics Is Essential for Unlocking the Microbial Black Box of the Soil. Trends Microbiol. 2021, 29, 279–282. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, A.E.; et al. Design considerations for workflow management systems use in production genomics research and the clinic. Sci. Rep. 2021, 11, 1–18. [Google Scholar] [CrossRef]
- Goussarov, G.; et al. Benchmarking short-, long- and hybrid-read assemblers for metagenome sequencing of complex microbial communities. Microbiology 2024, 170, 001469. [Google Scholar] [CrossRef]
- Kim, H.-M.; et al. Comparative analysis of 7 short-read sequencing platforms using the Korean Reference Genome: MGI and Illumina sequencing benchmark for whole-genome sequencing. GigaScience, 2021; 10, giab014. [Google Scholar]
- Yang, C.; et al. A review of computational tools for generating metagenome-assembled genomes from metagenomic sequencing data. Computational and Structural Biotechnology Journal, 2021; 19, 6301–6314 . [Google Scholar]
- Vosloo, S.; et al. Evaluating de Novo Assembly and Binning Strategies for Time Series Drinking Water Metagenomes. Microbiol. Spectr. 2021; 9. [Google Scholar]
- Han, H.; Wang, Z.; Zhu, S. Benchmarking metagenomic binning tools on real datasets across sequencing platforms and binning modes. Nat. Commun. 2025, 16, 2865. [Google Scholar] [CrossRef]
- Lynn, H.M.; Gordon, J.I. Sequential co-assembly reduces computational resources and errors in metagenome-assembled genomes. Cell Rep. Methods, 2025; 5. [Google Scholar]
- Christoph, M.; Hlemann, R.; Wacker, E.M.; Ellinghaus, D.; Franke, A. MAGScoT: a fast, lightweight and accurate bin-refinement tool. Bioinformatics 2022, 38, 5430–5433. [Google Scholar]
- Olm, M.R.; Brown, C.T.; Brooks, B.; Banfield, J.F. dRep: a tool for fast and accurate genomic comparisons that enables improved genome recovery from metagenomes through de-replication. ISME J. 2017, 11, 2864–2868. [Google Scholar] [CrossRef]
- Sieber, C.M.K.; et al. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 2018, 3, 836–843. [Google Scholar] [CrossRef]
- Evans, J.T.; Denef, V.J. To Dereplicate or Not To Dereplicate? mSphere, 2020; 5. [Google Scholar]
- Goldfarb, T.; et al. NCBI RefSeq: reference sequence standards through 25 years of curation and annotation. Nucleic Acids Res. 2025, 53, D243–D257. [Google Scholar] [CrossRef]
- Chaumeil, P.-A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk v2: memory friendly classification with the genome taxonomy database. Bioinformatics 2022, 38, 5315–5316. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res. 2025, 53, D609–D617. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019; 47, D309–D314 . [Google Scholar]
- Shaffer, M.; et al. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res. 2020, 48, 8883–8900. [Google Scholar] [CrossRef]
- Arkin, A.P.; et al. KBase: The United States Department of Energy Systems Biology Knowledgebase. Nat. Biotechnol. 2018, 36, 566–569. [Google Scholar] [CrossRef]
- Richardson, L.; et al. MGnify: the microbiome sequence data analysis resource in 2023. Nucleic Acids Res. 2023, 51, D753–D759. [Google Scholar] [CrossRef]
- Sloggett, C.; Goonasekera, N.; Afgan, E. BioBlend: automating pipeline analyses within Galaxy and CloudMan. Bioinformatics 2013, 29, 1685–1686. [Google Scholar] [CrossRef]
- Olson, R.D.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): a resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Achudhan, A.B.; Kannan, P.; Gupta, A.; Saleena, L.M. A Review of Web-Based Metagenomics Platforms for Analysing Next-Generation Sequence Data. Biochem. Genet. 2024, 62, 621–632. [Google Scholar] [CrossRef]
- Navgire, G.S.; et al. Analysis and Interpretation of metagenomics data: an approach. Biol. Proced. Online 2022, 24, 1–22. [Google Scholar] [CrossRef]
- Köster, J.; et al. Sustainable data analysis with Snakemake. F1000Research 2021, 10, 33. [Google Scholar]
- Tommaso, P.D.; et al. Nextflow enables reproducible computational workflows. Nat. Biotechnol. 2017, 35, 316–319. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.A.; et al. The nf-core framework for community-curated bioinformatics pipelines. Nat. Biotechnol. 2020, 38, 276–278. [Google Scholar] [CrossRef] [PubMed]
- Voss, K.; Auwera, G.V.d.; Gentry, J. Full-stack genomics pipelining with GATK4 + WDL + Cromwell. F1000Research, 2017; 6. [Google Scholar]
- Wratten, L.; Wilm, A.; Göke, J. Reproducible, scalable, and shareable analysis pipelines with bioinformatics workflow managers. Nat. Methods 2021, 18, 1161–1168. [Google Scholar] [CrossRef]
- Roach, M.J.; et al. Ten simple rules and a template for creating workflows-as-applications. PLOS Comput. Biol. 2022, 18, e1010705. [Google Scholar] [CrossRef]
- Reiter, T.; et al. Streamlining data-intensive biology with workflow systems. GigaScience, 2021; 10. [Google Scholar]
- Kadri, S.; Sboner, A.; Sigaras, A.; Roy, S. Containers in Bioinformatics: Applications, Practical Considerations, and Best Practices in Molecular Pathology. J. Mol. Diagn. 2022, 24, 442–454. [Google Scholar] [CrossRef]
- Espinoza, J.L.; Dupont, C.L. VEBA: a modular end-to-end suite for in silico recovery, clustering, and analysis of prokaryotic, microeukaryotic, and viral genomes from metagenomes. BMC Bioinformatics 2022, 23, 1–36. [Google Scholar] [CrossRef]
- Zhao, D.; et al. Eukfinder: a pipeline to retrieve microbial eukaryote genome sequences from metagenomic data. mBio 2025, 16, e00699–25. [Google Scholar] [CrossRef]
- Zorrilla, F.; Buric, F.; Patil, K.R.; Zelezniak, A. metaGEM: reconstruction of genome scale metabolic models directly from metagenomes. Nucleic Acids Res. 2021, 49, e126. [Google Scholar] [CrossRef]
- Belmann, P.; et al. Metagenomics-Toolkit: The Flexible and Efficient Cloud-Based Metagenomics Workflow featuring Machine Learning-Enabled Resource Allocation. ( 2024.
- Newell, R.J.P.; et al. Aviary: Hybrid assembly and genome recovery from metagenomes. ( 2025.
- Damme, R. van et al. Metagenomics workflow for hybrid assembly, differential coverage binning, metatranscriptomics and pathway analysis (MUFFIN). PLOS Comput. Biol. 2021, 17, 1–13. [Google Scholar]
- Benavides, A.; Sanchez, F.; Alzate, J.F.; Cabarcas, F. DATMA: Distributed Automatic Metagenomic Assembly and annotation framework. PeerJ, 2020; 8. [Google Scholar]
- Wajid, B.; et al. Music of metagenomics—a review of its applications, analysis pipeline, and associated tools. Funct. Integr. Genomics 2022, 22, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Standeven, F.J.; Dahlquist-Axe, G.; Speller, C.F.; Meehan, C.J.; Tedder, A. An efficient pipeline for creating metagenomic-assembled genomes from ancient oral microbiomes. ( 2024.
- Eren, A.M.; et al. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 2020, 6, 3–6. [Google Scholar] [CrossRef]
- Bai, D.; et al. EasyMetagenome: A user-friendly and flexible pipeline for shotgun metagenomic analysis in microbiome research. iMeta 2025, 4, e70001. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; et al. Benchmarking of analysis tools and pipeline development for nanopore long-read metagenomics. Sci. Bull. 2025, 70, 1591–1595. [Google Scholar] [CrossRef]
- The Galaxy, Community; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 2022, 50, W345–W351. [Google Scholar] [CrossRef]
- Cornet, L.; et al. The GEN-ERA toolbox: unified and reproducible workflows for research in microbial genomics. GigaScience 2022, 12, 1–10. [Google Scholar] [CrossRef]
- Pacific Biosciences. HiFi-MAG-Pipeline. (2025).
- Kalantar, K.L.; et al. IDseq—An open source cloud-based pipeline and analysis service for metagenomic pathogen detection and monitoring. GigaScience 2020, 9, giaa111. [Google Scholar] [CrossRef] [PubMed]
- Churcheward, B.; Millet, M.; Bihouée, A.; Fertin, G.; Chaffron, S. MAGNETO: An Automated Workflow for Genome-Resolved Metagenomics. mSystems, 2022; 7. [Google Scholar]
- Shaban, B.; et al. MetaGenePipe: An Automated, Portable Pipeline for Contig-based Functional and Taxonomic Analysis. ( 2022. [CrossRef]
- Kieser, S.; Brown, J.; Zdobnov, E.M.; Trajkovski, M.; McCue, L.A. ATLAS: A Snakemake workflow for assembly, annotation, and genomic binning of metagenome sequence data. BMC Bioinformatics 2020, 21, 1–8. [Google Scholar] [CrossRef]
- Salazar, V.W.; et al. Metaphor—A workflow for streamlined assembly and binning of metagenomes. GigaScience 2023, 12, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Mainguy, J.; et al. metagWGS, a comprehensive workflow to analyze metagenomic data using Illumina or PacBio HiFi reads. ( 2024.
- Uritskiy, G.V.; Diruggiero, J.; Taylor, J. MetaWRAP - A flexible pipeline for genome-resolved metagenomic data analysis. Microbiome 2018, 6, 1–13. [Google Scholar] [CrossRef]
- Ziemski, M.; et al. MOSHPIT: accessible, reproducible metagenome data science on the QIIME 2 framework. ( 2025.
- Liu, L.; Yang, Y.; Deng, Y.; Zhang, T. Nanopore long-read-only metagenomics enables complete and high-quality genome reconstruction from mock and complex metagenomes. Microbiome 2022, 10, 209. [Google Scholar] [CrossRef]
- Krakau, S.; Straub, D.; Gourlé, H.; Gabernet, G.; Nahnsen, S. nf-core/mag: a best-practice pipeline for metagenome hybrid assembly and binning. NAR Genomics Bioinforma. 2022; 4. [Google Scholar]
- Almeida, F.M.d.; Campos, T.A.d.; Pappas, G.J. Scalable and versatile container-based pipelines for de novo genome assembly and bacterial annotation. F1000Research 2023, 12, 1205. [Google Scholar] [CrossRef]
- Tadrent, N.; et al. SnakeMAGs: a simple, efficient, flexible and scalable workflow to reconstruct prokaryotic genomes from metagenomes. F1000Research 2023, 11, 1522. [Google Scholar] [CrossRef]
- Tamames, J.; Puente-Sánchez, F. SqueezeMeta, a highly portable, fully automatic metagenomic analysis pipeline. Front. Microbiol. 2019, 10, 3349. [Google Scholar] [CrossRef] [PubMed]
- Clarke, E.L.; et al. Sunbeam: An extensible pipeline for analyzing metagenomic sequencing experiments. Microbiome 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Breitwieser, F.P.; Baker, D.N.; Salzberg, S.L. KrakenUniq: confident and fast metagenomics classification using unique k-mer counts. Genome Biol. 2018, 19, 198. [Google Scholar] [CrossRef]
- Kang, D.D.; et al. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ, 2019; 2019. [Google Scholar]
- Alneberg, J.; et al. Binning metagenomic contigs by coverage and composition. Nat. Methods 2014, 11, 1144–1146. [Google Scholar] [CrossRef]
- Wu, Y.W.; Simmons, B.A.; Singer, S.W. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 2016, 32, 605–607. [Google Scholar] [CrossRef]
- Graham, E.D.; Heidelberg, J.F.; Tully, B.J. BinSanity: unsupervised clustering of environmental microbial assemblies using coverage and affinity propagation. PeerJ 2017, 5, e3035. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Simon, A. FastQC A Quality Control tool for High Throughput Sequence Data. ( 2010.
- Magoč, T.; Salzberg, S.L. FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Ontiveros-Palacios, N.; et al. Rfam 15: RNA families database in 2025. Nucleic Acids Res. 2025, 53, D258–D267. [Google Scholar] [CrossRef]
- Maidak, B.L.; et al. The RDP (Ribosomal Database Project). Nucleic Acids Res. 1997, 25, 109–110. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Benavides, A.; Isaza, J.P.; Niño-García, J.P.; Alzate, J.F.; Cabarcas, F. CLAME: a new alignment-based binning algorithm allows the genomic description of a novel Xanthomonadaceae from the Colombian Andes. BMC Genomics, 2018; 19. [Google Scholar]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; et al. MEGAHIT v1.0: A fast and scalable metagenome assembler driven by advanced methodologies and community practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef]
- Morgulis, A.; et al. Database indexing for production MegaBLAST searches. Bioinformatics 2008, 24, 1757–1764. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef]
- Hyatt, D.; et al. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 1–11. [Google Scholar] [CrossRef]
- Besemer, J.; Borodovsky, M. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 2005, 33, W451–W454. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics 2011, 12, 1–10. [Google Scholar] [CrossRef]
- Chklovski, A.; Parks, D.H.; Woodcroft, B.J.; Tyson, G.W. CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 2023, 20, 1203–1212. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; et al. dbCAN3: automated carbohydrate-active enzyme and substrate annotation. Nucleic Acids Res. 2023, 51, W115–W121. [Google Scholar] [CrossRef] [PubMed]
- Blanco-Míguez, A.; et al. Extending and improving metagenomic taxonomic profiling with uncharacterized species using MetaPhlAn 4. Nat. Biotechnol. 2021; 1–12. [Google Scholar]
- Beghini, F.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. eLife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 1–13. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Alcock, B.P.; et al. CARD 2023: expanded curation, support for machine learning, and resistome prediction at the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2023, 51, D690–D699. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. ( 2018.
- Benoit, G.; et al. Multiple comparative metagenomics using multiset k-mer counting. PeerJ Comput. Sci. 2016, 2, e94. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Steinegger, M.; Söding, J. Clustering huge protein sequence sets in linear time. Nat. Commun. 2018, 9, 2542. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Marçais, G.; et al. MUMmer4: A fast and versatile genome alignment system. PLOS Comput. Biol. 2018, 14, e1005944. [Google Scholar] [CrossRef] [PubMed]
- Cantalapiedra, C.P.; Hernández-Plaza, A.; Letunic, I.; Bork, P.; Huerta-Cepas, J. eggNOG-mapper v2: Functional Annotation, Orthology Assignments, and Domain Prediction at the Metagenomic Scale. Mol. Biol. Evol. 2021, 38, 5825–5829. [Google Scholar] [CrossRef]
- Ruscheweyh, H.-J.; et al. Cultivation-independent genomes greatly expand taxonomic-profiling capabilities of mOTUs across various environments. Microbiome 2022, 10, 212. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Machado, D.; Andrejev, S.; Tramontano, M.; Patil, K.R. Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 2018, 46, 7542–7553. [Google Scholar] [CrossRef]
- Zelezniak, A.; et al. Metabolic dependencies drive species co-occurrence in diverse microbial communities. Proc. Natl. Acad. Sci. 2015, 112, 6449–6454. [Google Scholar] [CrossRef]
- Lieven, C.; et al. MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 2020, 38, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Page, A.J.; et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef]
- Emiola, A.; Oh, J. High throughput in situ metagenomic measurement of bacterial replication at ultra-low sequencing coverage. Nat. Commun. 2018, 9, 4956. [Google Scholar] [CrossRef]
- West, P.T.; Probst, A.J.; Grigoriev, I.V.; Thomas, B.C.; Banfield, J.F. Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res. 2018, 28, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Saary, P.; Mitchell, A.L.; Finn, R.D. Estimating the quality of eukaryotic genomes recovered from metagenomic analysis with EukCC. Genome Biol. 2020, 21, 244. [Google Scholar] [CrossRef]
- Milanese, A.; et al. Microbial abundance, activity and population genomic profiling with mOTUs2. Nat. Commun. 2019, 10, 1014. [Google Scholar] [CrossRef] [PubMed]
- Aramaki, T.; et al. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef]
- Líndez, P.P.; et al. Adversarial and variational autoencoders improve metagenomic binning. Commun. Biol. 2023, 6, 1–10. [Google Scholar] [CrossRef]
- Pan, S.; Zhao, X.M.; Coelho, L.P. SemiBin2: self-supervised contrastive learning leads to better MAGs for short- and long-read sequencing. Bioinformatics 2023, 39, i21–i29. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Zdobnov, E.M. BUSCO: Assessing Genomic Data Quality and Beyond. Curr. Protoc. 2021, 1, e323. [Google Scholar] [CrossRef] [PubMed]
- Orakov, A.; et al. GUNC: detection of chimerism and contamination in prokaryotic genomes. Genome Biol. 2021, 22, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Mikheenko, A.; Saveliev, V.; Gurevich, A. MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 2016, 32, 1088–1090. [Google Scholar] [CrossRef]
- Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Dröge, J.; Gregor, I.; McHardy, A.C. Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics 2015, 31, 817–824. [Google Scholar] [CrossRef]
- Krapohl, J.; Pickett, B.E. SnakeWRAP: a Snakemake workflow to facilitate automated processing of metagenomic data through the metaWRAP pipeline. F1000Research, 2022; 11. [Google Scholar]
- Bolyen, E.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Eren, A.M.; Vineis, J.H.; Morrison, H.G.; Sogin, M.L. A Filtering Method to Generate High Quality Short Reads Using Illumina Paired-End Technology. PLOS ONE 2013, 8, e66643. [Google Scholar] [CrossRef]
- Aroney, S.T.N.; et al. CoverM: read alignment statistics for metagenomics. Bioinformatics 2025, 41, btaf147. [Google Scholar] [CrossRef]
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive protein alignments at tree-of-life scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Mistry, J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; et al. Benchmarking of analysis tools and pipeline development for nanopore long-read metagenomics. Sci. Bull. 2025, 70, 1591–1595. [Google Scholar] [CrossRef]
- Kolmogorov, M.; et al. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat. Methods 2020, 17, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, D.; et al. Hybrid metagenomic assembly enables high-resolution analysis of resistance determinants and mobile elements in human microbiomes. Nat. Biotechnol. 2019, 37, 937–944. [Google Scholar] [CrossRef]
- Kajitani, R.; et al. MetaPlatanus: a metagenome assembler that combines long-range sequence links and species-specific features. Nucleic Acids Res. 2021, 49, e130. [Google Scholar] [CrossRef]
- Hu, J.; Fan, J.; Sun, Z.; Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 2020, 36, 2253–2255. [Google Scholar] [CrossRef]
- Asnicar, F.; et al. Precise phylogenetic analysis of microbial isolates and genomes from metagenomes using PhyloPhlAn 3.0. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.; Concepcion, G.T.; Feng, X.; Zhang, H.; Li, H. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat. Methods 2021, 18, 170–175. [Google Scholar] [CrossRef]
- Mainguy, J.; et al. metagWGS, a comprehensive workflow to analyze metagenomic data using Illumina or PacBio HiFi reads. ( 2024.
- Mainguy, J.; Hoede, C. Binette: a fast and accurate bin refinement tool to construct high quality Metagenome Assembled Genomes. J. Open Source Softw. 2024, 9, 6782. [Google Scholar] [CrossRef]
- Vaser, R.; Sovic, I.; Nagarajan, N.; Sikic, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. gr.214270.116 ( 2017. [CrossRef] [PubMed]
- Walker, B.J.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLOS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.D.; et al. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat. Biotechnol. 2019, 37, 953–961. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Kieft, K.; Anantharaman, K. Deciphering Active Prophages from Metagenomes. mSystems 2022, 7, e00084–22. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLOS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Kang, D.D.; Froula, J.; Egan, R.; Wang, Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 2015, 3, e1165. [Google Scholar] [CrossRef]
- Brůna, T.; Hoff, K.J.; Lomsadze, A.; Stanke, M.; Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genomics Bioinforma. 2021, 3, lqaa108. [Google Scholar] [CrossRef]
- Meunier, L.; Baurain, D.; Cornet, L. AMAW: automated gene annotation for non-model eukaryotic genomes. ( 2023. [CrossRef]
- Irber, L.; et al. sourmash v4: A multitool to quickly search, compare, and analyze genomic and metagenomic data sets. J. Open Source Softw. 2024, 9, 6830. [Google Scholar] [CrossRef]
- Parks, D.H.; et al. GTDB: an ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef]
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- von Meijenfeldt, F.A.B.; Arkhipova, K.; Cambuy, D.D.; Coutinho, F.H.; Dutilh, B.E. Robust taxonomic classification of uncharted microbial sequences and bins with CAT and BAT. Genome Biol. 2019, 20, 217. [Google Scholar] [CrossRef]
- Levy Karin, E.; Mirdita, M.; Söding, J. MetaEuk—sensitive, high-throughput gene discovery, and annotation for large-scale eukaryotic metagenomics. Microbiome 2020, 8, 48. [Google Scholar] [CrossRef]
- Borry, M.; Hübner, A.; Rohrlach, A.B.; Warinner, C. PyDamage: automated ancient damage identification and estimation for contigs in ancient DNA de novo assembly. PeerJ 2021, 9, e11845. [Google Scholar] [CrossRef]
- Karlicki, M.; Antonowicz, S.; Karnkowska, A. Tiara: deep learning-based classification system for eukaryotic sequences. Bioinformatics 2022, 38, 344–350. [Google Scholar] [CrossRef]
- Camargo, A.P.; et al. Identification of mobile genetic elements with geNomad. Nat. Biotechnol. 2024, 42, 1303–1312. [Google Scholar] [CrossRef]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Schwengers, O.; et al. Bakta: Rapid and standardized annotation of bacterial genomes via alignment-free sequence identification. Microb. Genomics 2021, 7, 000685. [Google Scholar] [CrossRef] [PubMed]
- Jolley, K.A.; Maiden, M.C. BIGSdb: Scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 2010, 11, 595. [Google Scholar] [CrossRef]
- Blin, K.; et al. antiSMASH 7.0: new and improved predictions for detection, regulation, chemical structures and visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Huang, N.; Zou, Y.; Liao, X.; Wang, J. MultiNanopolish: refined grouping method for reducing redundant calculations in Nanopolish. Bioinformatics 2021, 37, 2757–2760. [Google Scholar] [CrossRef]
- Bushmanova, E.; Antipov, D.; Lapidus, A.; Prjibelski, A.D. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 2019, 8, giz100. [Google Scholar] [CrossRef]
- Caspi, R.; et al. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef]
- Gillespie, J.J.; et al. PATRIC: the Comprehensive Bacterial Bioinformatics Resource with a Focus on Human Pathogenic Species. Infect. Immun. 2011, 79, 4286–4298. [Google Scholar] [CrossRef]
- Brettin, T.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef]
- Wang, S.; Sundaram, J.P.; Spiro, D. VIGOR, an annotation program for small viral genomes. BMC Bioinformatics 2010, 11, 451. [Google Scholar] [CrossRef] [PubMed]
- Seaver, S.M.D.; et al. The ModelSEED Biochemistry Database for the integration of metabolic annotations and the reconstruction, comparison and analysis of metabolic models for plants, fungi and microbes. Nucleic Acids Res. 2021; 49, D575–D588 . [Google Scholar]
- Zdobnov, E.M.; Apweiler, R. InterProScan – an integration platform for the signature-recognition methods in InterPro. Bioinformatics 2001, 17, 847–848. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Standeven, F.J.; Dahlquist-Axe, G.; Speller, C.F.; Meehan, C.J.; Tedder, A. An efficient pipeline for creating metagenomic-assembled genomes from ancient oral microbiomes. ( 2024.
- Jónsson, H.; Ginolhac, A.; Schubert, M.; Johnson, P.L.F.; Orlando, L. mapDamage2.0: fast approximate Bayesian estimates of ancient DNA damage parameters. Bioinformatics 2013, 29, 1682–1684. [Google Scholar] [CrossRef]
- Van Nguyen, H.; Lavenier, D. PLAST: parallel local alignment search tool for database comparison. BMC Bioinformatics 2009, 10, 329. [Google Scholar] [CrossRef]
- Lin, H.-H.; Liao, Y.-C. Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 2016, 6, 24175. [Google Scholar] [CrossRef]
- Yepes-García, J.; Falquet, L. Metagenome quality metrics and taxonomical annotation visualization through the integration of MAGFlow and BIgMAG. F1000Research, 2024; 13, 640. [Google Scholar]
- Cansdale, A.; Chong, J.P.J. MAGqual: a stand-alone pipeline to assess the quality of metagenome-assembled genomes. Microbiome 2024, 12, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.M.; et al. Performance of Multiple Metagenomics Pipelines in Understanding Microbial Diversity of a Low-Biomass Spacecraft Assembly Facility. Front. Microbiol. 2021, 12. [Google Scholar] [CrossRef]
- Meyer, F.; et al. Tutorial: assessing metagenomics software with the CAMI benchmarking toolkit. Nat. Protoc. 2021, 16, 1785–1801. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



