Submitted:
05 June 2025
Posted:
06 June 2025
You are already at the latest version
Abstract
Human Papillomavirus (HPV), particularly high-risk strains like HPV16 and HPV18, is a leading cause of cervical and other epithelial cancers. While the oncogenic roles of viral proteins E6 and E7 are well established, the broader impact of HPV on host transcriptional regulation remains incompletely understood. This study investigates the hypothesis that conserved HPV genomic motifs may sequester human transcription factors (TFs), contributing to oncogenesis through altered gene regulation. Methods: We conducted a computational analysis of the genomes of high-risk HPV types using MEME-ChIP for de novo motif discovery, followed by Tomtom for identifying matching human TFs. Protein–protein interactions among the predicted TFs were examined using STRING, and biological pathway enrichment was performed with Enrichr. Results: The analysis revealed conserved viral motifs that potentially bind host TFs across several functional families, including FOX, HOX, NFAT, and zinc finger proteins. Key regulators such as SMARCA1, DUX4, and CDX1 were identified as central nodes of HPV-induced disruption. Enriched pathways included Wnt signaling, transcriptional misregulation in cancer, immune evasion, and chromatin remodeling. Conclusions: HPV exerts multifaceted control over host transcriptional and epigenetic networks. This study highlights the utility of in silico approaches in mapping viral–host interactions and identifies potential therapeutic targets in disrupted regulatory networks.
Keywords:
HPV (Human Papillomavirus)
; tissue factor binding sites (TFBSs)
; transcription factors (TFs)
Introduction
Human Papillomavirus (HPV) is a double-stranded DNA virus belonging to the Papillomaviridae family. It includes more than 200 types, some of which are known for their role in the development of cervical cancer, other anogenital tumors, and oropharyngeal carcinoma [1]. First discovered in the 1980s, HPV was later classified into high- and low-risk strains, with a global distribution that makes it one of the most common sexually transmitted infections [1].
HPV infection is often asymptomatic, but in symptomatic cases, it can cause benign lesions (such as condylomas) or, in the case of high-risk strains, lead to neoplasia. In particular, HPV16 and HPV18 are responsible for the majority of cervical cancer cases. Scientific interest in this virus has grown significantly due to its etiological role in several tumor pathologies [2]. HPV is primarily transmitted through sexual contact but can also spread through skin-to-skin contact or contaminated surfaces. Persistent infection with high-risk strains is associated with an increased risk of developing dysplasia and carcinomas. Screening programs, such as the Pap smear and the HPV-DNA test, are essential for early diagnosis and prevention [2].
The viral genome consists of approximately 8,000 base pairs and includes early (E) and late (L) genes. The early genes (E1, E2, E4, E5, E6, and E7) are involved in viral replication and oncogenesis, while the late genes (L1 and L2) encode the structural proteins of the virus [2]. The interaction between HPV and host cellular mechanisms is crucial for the virus life cycle and its pathogenesis. HPV hijacks the host transcriptional machinery to promote its own replication and persistence. For example, the viral oncoproteins E6 and E7 can inactivate the tumor suppressor proteins p53 and Rb, respectively, disrupting cell cycle regulation and promoting uncontrolled cell proliferation [3].
In this article, we propose a computational analysis to identify potential interactions between human transcription factors (TFs) and the tissue factor binding sites (TFBSs) shared among the main HPV strains associated with tumor development and classified as high-risk oncogenic types. We hypothesize that the sequestration of these factors could alter host gene expression, producing effects similar to a loss of function. This approach, which has already been applied to other viruses such as SARS-CoV-2, could help clarify the mechanisms underlying HPV pathogenesis and its complications.
Materials and Methods
The analysis pipeline, composed of four main stages, was carried out using various publicly available bioinformatics tools. The in silico approach was applied to the genome sequences of the major HPV strains associated with cancer (Table 1), in order to evaluate the presence of conserved motifs capable of interacting with host proteins (https://pave.niaid.nih.gov/explore/reference_genomes/human_genomes).
MEME-ChIP, a tool from the MEME Suite (http://meme-suite.org/tools/meme-chip, accessed on 11 February 2025), was used to analyze the genomic sequences of the main HPV strains. MEME-ChIP performs comprehensive motif analysis, including de novo motif discovery, on large sets of DNA sequences identified in ChIP-seq or CLIP-seq experiments. It integrates multiple tools, such as MEME, DREME, CentriMo, and FIMO, to identify enriched DNA-binding motifs (TFBSs) and their positional bias, making it suitable for uncovering regulatory elements in viral genomes [4].
All identified motifs were used as queries for Tomtom (http://meme-suite.org/doc/tomtom.html, accessed on 20 February 2025), another component of the MEME Suite that compares discovered motifs to a curated, non-redundant database of known DNA-binding protein motifs experimentally validated in the human genome. Tomtom uses a statistical framework based on the Benjamini-Hochberg method to calculate q-values, which represent the minimum false discovery rate (FDR) at which the observed similarity would be deemed significant. This allowed us to generate a list of TFs recognizing the conserved domains found across all viral genome motifs (Figure 1). [5]
Next, we used STRING v11.5 (https://string-db.org/, accessed on February 2025), a database and web resource dedicated to protein–protein interaction (PPI) networks. STRING aggregates known and predicted associations derived from multiple sources, including experimental data, computational prediction methods, and public text collections. The tool was used to identify the strongest associations among the set of predicted TFs using a guilt-by-association approach, providing insight into their functional relationships within the host. [6]
Finally, the entire list of TFs was used as input for Enrichr (https://maayanlab.cloud/Enrichr/, accessed on February 2025), a web-based application that integrates multiple enrichment analysis methods and offers interactive visualizations of the results using the Data Driven Documents (D3) JavaScript library. Enrichr supports a wide range of biological databases and provides detailed graphical summaries of enriched terms, helping to interpret the collective functions of the input TF list and to explore their roles in host gene regulatory networks (Figure 1). [7]
Results
The oncogenesis induced by HPV infection, particularly by the high-risk types HPV16 and HPV18, is a multistep process involving both direct and indirect interactions of the viral oncoproteins E6 and E7 with transcriptional and epigenetic regulatory circuits that are essential for cellular homeostasis. The synergistic action of E6 and E7 leads to the functional inactivation of the tumor suppressors p53 and pRb, through E6-AP–mediated ubiquitination and degradation, and aberrant E2F release, respectively. This imbalance triggers uncontrolled proliferative responses, accompanied by genomic instability, impaired immune responses, and remodeling of the epigenetic landscape, which involves numerous transcription factors (TFs) from various gene families [8].
Our analysis identified several protein families whose dysregulation, caused simply by the presence of the virus in the cell, may drive tumor-related processes. Among the most affected families is the Forkhead Box (FOX): FOXO4, which is crucial in apoptosis and DNA damage response, is rendered functionally inactive due to the degradation of its primary modulator, p53 [9]. FOXC1 and FOXQ1, typically involved in embryonic development and cell migration, become deregulated as a result of chromatin instability induced by E7, promoting mesenchymal and invasive traits [10]. Similarly, FOXJ3, which regulates phenotypic plasticity, may undergo epigenetic alterations that impact epithelial-to-mesenchymal transition (EMT) [11].
The Homeobox (HOX) family represents another key target. HOXA1, HOXA9, HOXB7, HOXB8, and HOXB13, responsible for cellular identity and differentiation along the anteroposterior axis, are frequently disrupted following the integration of viral DNA into the host genome, leading to aggressive and de-differentiated phenotypes [12]. This effect is amplified by the dysregulation of co-associated factors such as MEIS1, PBX2, and LHX3, which are involved in HOX transcriptional complex cooperation [13].
In the immune and inflammatory context, the NFAT family (NFATC2, NFATC3, NFATC4) experiences functional suppression due to impaired intracellular calcium signaling, reducing T cell activation and enabling immune evasion [14]. Concurrently, E7 can interact with histone deacetylase (HDAC) complexes, altering the transcriptional regulation of MEF2 (MEF2A, MEF2C), resulting in the repression of differentiation genes and the activation of oncogenic programs [15].
Among the TFs with direct interactions, NFE2L2 (Nrf2) is activated in response to chronic oxidative stress induced by E7, promoting a pro-survival environment [16]. GLI3, a key node in the Hedgehog pathway, is negatively affected by the coordinated action of E6/E7, impairing epithelial differentiation [17]. The SRY gene, although primarily involved in sex determination, may be indirectly impacted by viral integration that alters local chromatin architecture. CLOCK, a central regulator of circadian rhythm and cellular metabolism, reflects metabolic dysregulation typical of HPV-positive tumor cells, further exacerbated by the suppression of other bZIP proteins such as DBP [18].
A further layer of complexity arises from secondary mutagenesis caused by viral-induced genomic instability, affecting families such as Zinc Finger (ZNF250, ZNF394, ZNF816, ZFP42, ZFP82), compromising DNA-binding capacity and enhancing transcriptional dysregulation [19]. Likewise, the downregulation of MAF, SOX17, NR1H3, NKX3.2, NKX6.1, ALX1, OSR2, and AIRE indicates a broad repression of morphogenetic and immuno-regulatory factors, contributing to tissue identity loss and tumor mimicry [20].
Finally, key targets of viral-induced deregulation identified in our analysis include SMARCA1, DUX4, and CDX1, whose downregulation is critical to tumor progression. SMARCA1, a component of the SWI/SNF chromatin remodeling complex, is essential for DNA accessibility and the regulation of gene expression, including tumor suppressors like TP53. E7-mediated inhibition of SMARCA1 impairs genomic stability and promotes an aggressive tumor phenotype [21]. DUX4, typically active during early development and involved in regeneration and immune response, is silenced in HPV-positive tumors; its loss reduces control over differentiation and lymphocyte activation, enabling immune escape [22]. CDX1, known for its role in epithelial differentiation and EMT suppression, is also downregulated in HPV-associated cervical carcinomas, resulting in loss of cell polarity and activation of oncogenic pathways such as Wnt/β-catenin [23]. In summary, HPV operates across multiple regulatory levels: it interferes with chromatin remodeling complexes (e.g., SWI/SNF via SMARCA1), suppresses differentiation genes (DUX4, CDX1), and impairs immune surveillance, establishing a permissive environment for viral persistence and malignant transformation (Figure 2).
The network illustrates functional and physical associations among selected TFs, constructed using data derived from integrated bioinformatics tools. Each node represents a TF, with colors indicating functional or gene family groupings while colors and outlined clusters group proteins according to their main biological functions: immune regulation (red), genomic instability (orange), tumor progression (purple), inflammation and immune response (green), transcriptional regulation (light green), viral DNA integration (yellow), direct interactions (light blue), and apoptosis and DNA damage response (dark blue). The lines connecting the nodes indicate known functional or physical interactions between the factors. This map highlights the interconnection between different pathways, emphasizing the key role of certain factors such as MEIS1, MEF2C, and NFATC2 as central hubs in the transcriptional network.
2.4. Enriched Analysis
Subsequently, the same group of transcription factors (TFs) was analyzed using Enrichr (https://maayanlab.cloud/Enrichr/, accessed on march 2025), a platform that allows simultaneous interrogation of numerous biological databases. As expected, the results confirmed the association of the identified TFs with key regulatory processes in tumor development, as highlighted by the analysis of the Human KEGG database [24]. Several molecular pathways are directly or indirectly involved in this dynamic.
The cGMP-PKG signaling pathway, known for its role in regulating proliferation, apoptosis, and oxidative stress response, can, when altered, promote the survival of HPV-infected cells by preventing their normal elimination through apoptosis [25]. Similarly, the oxytocin signaling pathway, which typically has antiproliferative effects in certain tissues, may lose this protective function due to HPV-induced dysfunctions, thereby contributing to tumor progression [26].
Particularly relevant is the involvement of the Wnt signaling pathway, essential for development and control of cell growth. Aberrant activation of this pathway by HPV can lead to uncontrolled proliferation and malignant transformation of epithelial cells [27]. This is further exacerbated by transcriptional dysregulation caused by the viral oncoproteins, resulting in massive oncogene expression and silencing of tumor suppressor genes, as observed in the transcriptional misregulation in cancer pathway [28].
The immune system is also profoundly affected: alteration in the Th1/Th2 balance impairs effective antiviral responses, favoring viral persistence and increasing the risk of neoplastic transformation [29]. In parallel, HPV can negatively modulate the C-type lectin receptor signaling pathway, reducing the immune cells’ ability to recognize and eliminate infected cells [30].
Even pathways seemingly unrelated to carcinogenesis, such as fluid shear stress and atherosclerosis, may play an indirect role. The mechanotransduction response, influenced by the infection, can alter epithelial cell behavior, contributing to a tumor-promoting microenvironment [31].
Another key element is cellular senescence. HPV is capable of evading this crucial defense mechanism, which prevents the replication of damaged cells, thus promoting the unlimited replication of infected cells [32]. Although hepatitis B virus (HBV) differs in tropism and pathogenesis, it shares notable similarities with HPV in terms of viral persistence, immune evasion, and oncogenic potential, offering a useful comparative model to understand viral carcinogenesis [33].
Finally, HPV infection can also alter the axon guidance pathway, typically associated with neuronal development but increasingly recognized for its role in regulating cell migration, adhesion, and invasiveness, key characteristics in metastatic spread [34].
Collectively, these pathways highlight the extreme complexity of HPV’s role in tumorigenesis (Figure 3).
Discussion
High-risk HPV infection, such as HPV-16 and HPV-18, is much more than a biological event. It marks the beginning of a conflicting dialogue between two evolutionary intelligences: the viral and the cellular, both competing for control over the circuits of life. In this context, carcinogenesis does not arise merely as a consequence of the presence of two viral oncoproteins (E6 and E7), but rather as the product of a multilayered adaptive network, where each move by the virus triggers a response or a defeat of the host [35].
HPV acts as a sophisticated “molecular hacker,” capable of deconstructing the host eukaryote’s epigenetic and transcriptional programs: it degrades p53 and pRb, captures Tfs such as those from the FOX, HOX, and NFAT families, and interferes with chromatin remodeling by inhibiting SWI/SNF complexes (e.g., SMARCA1) [36]. However, each intrusion leaves a signature, every alteration generates an observable perturbation. This is where in silico models become not just technical tools but epistemic necessities: they allow for the controlled exploration of emerging disorder by artificially reproducing hidden patterns of pathogenesis [37].
Complexity, often seen as a barrier to interpretation, becomes an epistemological opportunity. The virus does not act linearly: it is not E6 or E7 alone that induces cancer, but the systemic synergy among E6, E7, downregulation of DUX4, SMARCA1, CDX1, and the sequestration of regulators such as FOXM1 or CLOCK [38]. This requires a paradigm shift: from molecular reductionism to computational holism. Computational models such as protein–protein interaction networks, dynamic simulations, and machine learning algorithms trace the invisible lines of the host–virus war. They quantify the likelihood that the loss of a transcription factor leads to genomic instability or simulate the systemic impact of reactivating a repressor gene [39]. Thus, simulation becomes not just a “model” but a virtual laboratory: a place to test hypotheses.
In the phenomenon of host shutoff, where the virus suppresses host translation despite depending on it, lies the paradox of how a lifeform survives by sabotaging its own replication vehicle [40]. Models suggest a response: HPV creates a precarious balance, selectively silencing immune and tumor suppressor genes while preserving circuits essential for its propagation. The downregulation of DUX4 or CDX1 is not accidental, but an evolutionary compromise that maximizes viral fitness [41].
This leads us to the therapeutic frontier. In silico models not only show us where to strike, but how to think. Drugs targeting multiple nodes (epigenetic inhibitors, immune activators) may prove more effective than unidirectional therapies. Instead of seeking a drug against E6 alone, we might aim at vulnerable networks, such as the E7–SMARCA1 interaction, or exploit the instability of the viral system within the framework of pathway-oriented targeted therapy [42].
Conclusions
The study of HPV in its complexity demands a holistic view of events related to the virus’s mere presence within the host—even before clinical manifestations of the infection appear. We are no longer looking at a list of genes and mutations but at a complex system, a constellation of probabilistic events converging into various pathological phenotypes. In this new framework, HPV is not merely a biological adversary but an epistemological interlocutor. Its challenge is not limited to understanding a symptom or the evolution of disease, but to our ability to comprehend the complexity of the entire event network.
References
- Schiffman, M.; Castle, P.E.; Jeronimo, J.; Rodriguez, A.C.; Wacholder, S. Human papillomavirus and cervical cancer. Lancet 2007, 370, 890–907. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Egawa, N.; Griffin, H.; Kranjec, C.; Murakami, I. Human papillomavirus molecular biology and disease association. Rev. Med. Virol. 2015, 25, 2–23. [Google Scholar] [CrossRef]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Gupta, S.; Stamatoyannopoulos, J.A.; Bailey, T.L.; Noble, W.S. Quantifying similarity between motifs. Genome Biol. 2007, 8, R24. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; Jensen, L.J.; von Mering, C. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Calnan, D.R.; Brunet, A. The FoxO code. Oncogene 2008, 27, 2276–2288. [Google Scholar] [CrossRef]
- Qiao, Y.; He, H.; Jonsson, P.; Sinha, I.; Zhao, C.; Dahlman-Wright, K. AP-1 is a key regulator of proinflammatory cytokine TNFα-mediated triple-negative breast cancer progression. J. Biol. Chem. 2016, 291, 5068–5079. [Google Scholar] [CrossRef]
- Gomis, R.R.; Alarcón, C.; He, W.; Wang, Q.; Seoane, J.; Lash, A.; Massagué, J. A FoxO–Smad synexpression group in human keratinocytes. Proc. Natl. Acad. Sci. USA 2006, 103, 12747–12752. [Google Scholar] [CrossRef] [PubMed]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Moens, C.B.; Selleri, L. Hox cofactors in vertebrate development. Dev. Biol. 2006, 291, 193–206. [Google Scholar] [CrossRef]
- Macián, F. NFAT proteins: Key regulators of T-cell development and function. Nat. Rev. Immunol. 2005, 5, 472–484. [Google Scholar] [CrossRef]
- Lu, J.; McKinsey, T.A.; Nicol, R.L.; Olson, E.N. Signal-dependent activation of MEF2 transcription factors by dissociation from histone deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 4070–4075. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- Regl, G.; Neill, G.W.; Eichberger, T.; Kasper, M.; Ikram, M.S.; Koller, J.; Hintner, H.; Quinn, A.G.; Moyes, D.L.; Aberger, F. Human GLI2 and GLI1 are part of a positive feedback mechanism in basal cell carcinoma. Oncogene 2002, 21, 5529–5539. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Kettner, N.M. The circadian clock in cancer development and therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar]
- Cassandri, M.; Smirnov, A.; Novelli, F.; Pitolli, C.; Agostini, M.; Malewicz, M.; Melino, G.; Raschellà, G. Zinc-finger proteins in health and disease. Cell Death Discov. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Zaret, K.S.; Carroll, J.S. Pioneer transcription factors: Establishing competence for gene expression. Genes Dev. 2011, 25, 2227–2241. [Google Scholar] [CrossRef]
- Mathur, R. ARID1A loss in cancer: Towards a mechanistic understanding. Pharmacol. Ther. 2018, 190, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tapscott, S.J. DUX4 activates germline genes, retroelements, and immune mediators: Implications for facioscapulohumeral dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef]
- Guo, R.J.; Suh, E.R.; Lynch, J.P. The role of Cdx proteins in intestinal development and cancer. Cancer Biol. Ther. 2004, 3, 593–601. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; McDermott, M.G.; Monteiro, C.D.; Gundersen, G.W.; Ma’ayan, A. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Pilz, R.B.; Casteel, D.E. Regulation of gene expression by cyclic GMP. Circ. Res. 2003, 93, 1034–1046. [Google Scholar] [CrossRef] [PubMed]
- Szeto, A.; Nation, D.A.; Mendez, A.J.; Dominguez-Bendala, J.; Brooks, L.G.; Schneiderman, N.; McCabe, P.M. Oxytocin attenuates NADPH-dependent superoxide activity and IL-6 secretion in macrophages and vascular cells. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E1495–E1501. [Google Scholar] [CrossRef]
- Nusse, R.; Clevers, H. Wnt/β-catenin signaling, disease, and emerging therapeutic modalities. Cell 2017, 169, 985–999. [Google Scholar] [CrossRef]
- Bradner, J.E.; Hnisz, D.; Young, R.A. Transcriptional addiction in cancer. Cell 2017, 168, 629–643. [Google Scholar] [CrossRef]
- Romagnani, S. Th1/Th2 cells. Inflamm. Bowel Dis. 1999, 5, 285–294. [Google Scholar] [CrossRef]
- Dambuza, I.M.; Brown, G.D. C-type lectins in immunity: Recent developments. Curr. Opin. Immunol. 2015, 32, 21–27. [Google Scholar] [CrossRef]
- Li, Y.S.; Haga, J.H.; Chien, S. Molecular basis of the effects of shear stress on vascular endothelial cells. J. Biomech. 2005, 38, 1949–1971. [Google Scholar] [CrossRef] [PubMed]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular senescence: Defining a path forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Revill, P.A.; Chisari, F.V.; Block, J.M.; Gehring, A.J.; Guo, H.; Cooper, S.; Locarnini, S.A. A global scientific strategy to cure hepatitis B. Lancet Gastroenterol. Hepatol. 2019, 4, 545–558. [Google Scholar] [CrossRef]
- Mehlen, P.; Delloye-Bourgeois, C.; Chedotal, A. Novel roles for Slits and netrins: Axon guidance cues as anticancer targets? Nat. Rev. Cancer 2011, 11, 188–197. [Google Scholar] [CrossRef] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nat. Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Vázquez-Arreguín, K.; Teyra, J.; Weisburd, B.; Serra, R.W.; Yip, K.Y.; Fernandez-Zapico, M.E.; Uetz, P. HPV oncoproteins target the SWI/SNF chromatin remodeling complex to repress tumor suppressor gene expression. Cell Rep. 2021, 35, 109103. [Google Scholar]
- Ma, J.; Yu, M.K.; Fong, S.; Ono, K.; Sage, E.; Demchak, B.; Sharan, R.; Ideker, T. Using deep learning to model the hierarchical structure and function of a cell. Nat. Methods 2018, 15, 290–298. [Google Scholar] [CrossRef]
- Zhang, C.; Richon, V.M.; Ni, X.; Talpur, R.; Duvic, M. Selective inhibition of HDAC1 and HDAC2 contributes to transcriptional activation of DUX4 and apoptosis in cancer cells. J. Invest. Dermatol. 2019, 139, 1281–1284. [Google Scholar]
- Zitnik, M.; Agrawal, M.; Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 2018, 34, i457–i466. [Google Scholar] [CrossRef]
- Gaglia, M.M.; Rycroft, C.H.; Glaunsinger, B.A. Transcript degradation by the herpesvirus host shutoff factor accelerates global changes in gene expression and mRNA turnover. Cell Host Microbe 2012, 12, 559–571. [Google Scholar]
- Liao, X.; Li, Y.; Wang, X.; Deng, Z.; Zhang, Y. DUX4 modulates stem cell pluripotency and endows human embryonic stem cells with a higher capacity to form teratomas. Stem Cell Rep. 2020, 14, 345–358. [Google Scholar]
- Tamborero, D.; Rubio-Perez, C.; Deu-Pons, J.; Schroeder, M.P.; Vivancos, A.; Rovira, A.; Tusquets, I.; Albanell, J.; Rodon, J.; Tabernero, J.; López-Bigas, N. Cancer Genome Interpreter annotates the biological and clinical relevance of tumor alterations. Genome Med. 2018, 10, 25. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Bioinformatics pipeline used to detect TFs from high-risk HPV strains. Spep1, 15 HPV strand’s viral are examined to see whether any common TFBSs with the human genome can be found. Step 2: Known transcription factor motifs are found using Tomtom. Step 3: Enrichr and STRING are used to investigate functional enrichment and protein-protein interactions.
Figure 1.
Bioinformatics pipeline used to detect TFs from high-risk HPV strains. Spep1, 15 HPV strand’s viral are examined to see whether any common TFBSs with the human genome can be found. Step 2: Known transcription factor motifs are found using Tomtom. Step 3: Enrichr and STRING are used to investigate functional enrichment and protein-protein interactions.
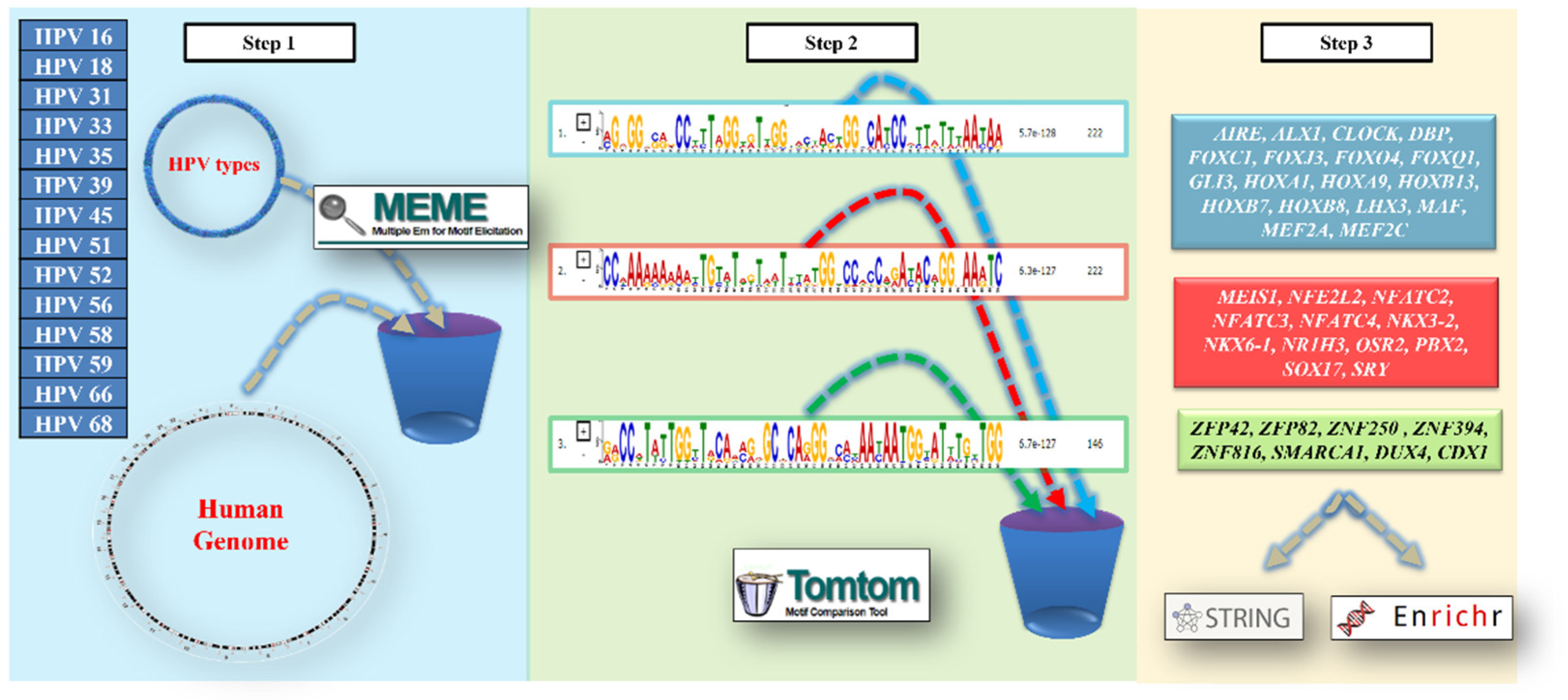
Figure 2.
Protein–Protein Interaction (PPI) Network of Transcription Factors.
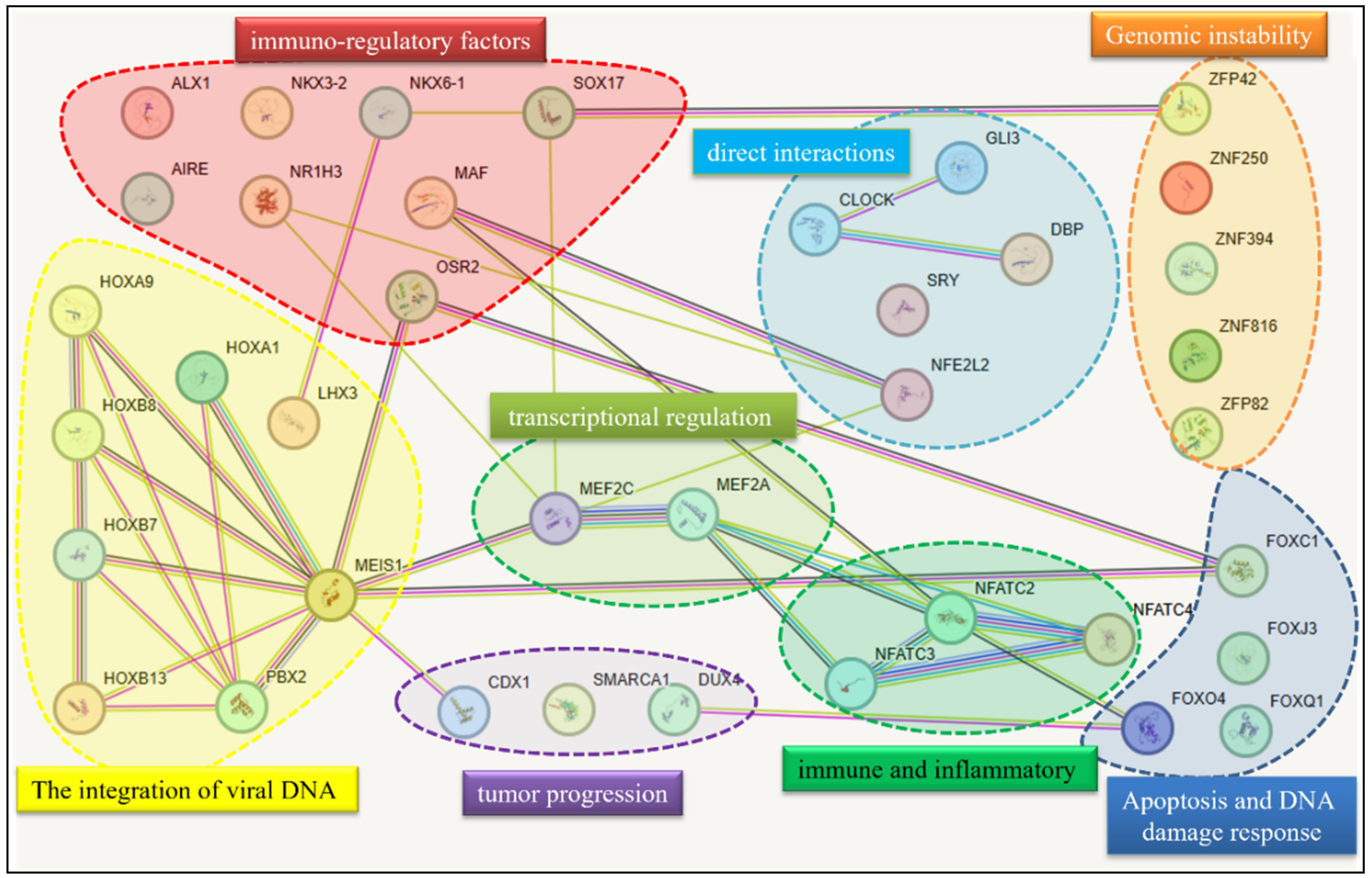
Figure 3.
Functional enrichment analysis of transcription factor-associated genes using KEGG 2021 Human pathways. Panel A: UMAP (Uniform Manifold Approximation and Projection) clustering shows the spatial distribution of significantly enriched pathways, each labeled and color-coded, including Wnt signaling, cellular senescence, axon guidance, and transcriptional misregulation in cancer. Panel B: Bar plot representing the top 10 most significantly enriched pathways ranked by –log₁₀(p-value), highlighting the involvement of key biological processes such as cGMP-PKG and oxytocin signaling. Together, the panels illustrate the diverse functional implications of transcription factor networks across various disease and signaling contexts.
Figure 3.
Functional enrichment analysis of transcription factor-associated genes using KEGG 2021 Human pathways. Panel A: UMAP (Uniform Manifold Approximation and Projection) clustering shows the spatial distribution of significantly enriched pathways, each labeled and color-coded, including Wnt signaling, cellular senescence, axon guidance, and transcriptional misregulation in cancer. Panel B: Bar plot representing the top 10 most significantly enriched pathways ranked by –log₁₀(p-value), highlighting the involvement of key biological processes such as cGMP-PKG and oxytocin signaling. Together, the panels illustrate the diverse functional implications of transcription factor networks across various disease and signaling contexts.
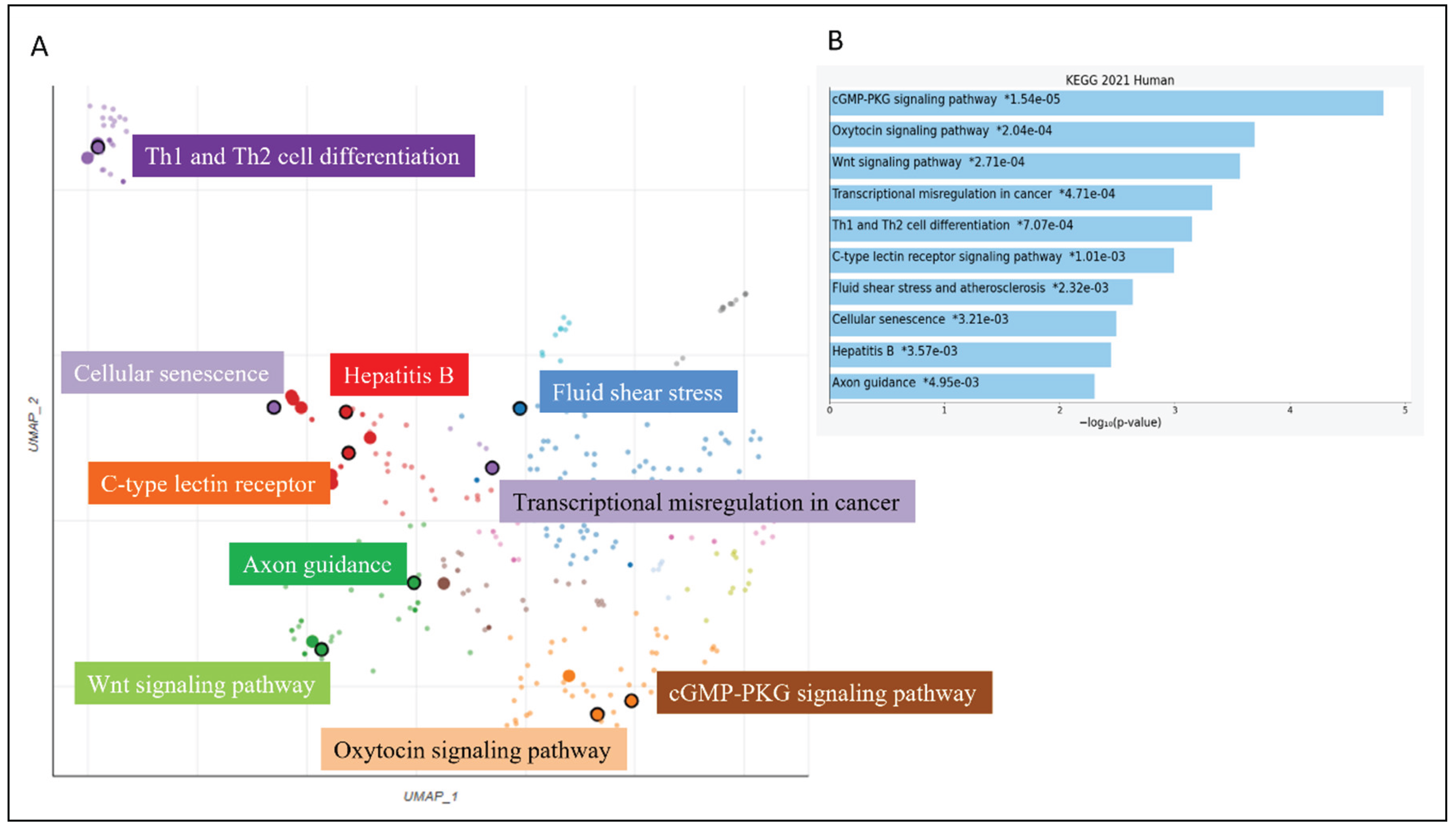

Table 1.
High-risk HPV types included in the analysis, along with their prevalence in tumors, tumor localization, clinical notes, and genome access information (https://pave.niaid.nih.gov/explore/reference_genomes/human_genomes).
Table 1.
High-risk HPV types included in the analysis, along with their prevalence in tumors, tumor localization, clinical notes, and genome access information (https://pave.niaid.nih.gov/explore/reference_genomes/human_genomes).
| HPV Type | Prevalence in Tumors | Tumor Localization | Clinical Notes |
| HPV 16 | ~60% of cervical cancers~70% of oropharyngeal cancers | Cervix, oropharynx, anus, penis, vulva, vagina | Highly oncogenicPersistent infection linked to integration into the human genome |
| HPV 18 | ~10–15% of cervical cancers | Cervix, vagina, anus | Often associated with glandular carcinomas |
| HPV 31 | 2–5% | Cervix, anus, penis | Common in CIN2/CIN3 precancerous lesions |
| HPV 33 | 2–4% | Cervix, vulva | Frequent precancerous lesions |
| HPV 35 | <2% | Cervix, vagina | Detected in advanced cervical neoplasms |
| HPV 39 | <2% | Cervix, anus | Moderate oncogenic potential |
| HPV 45 | ~5% | Cervix, vagina | Strong association with adenocarcinomas |
| HPV 51 | ~1–2% | Cervix | Rare but oncogenic |
| HPV 52 | ~2–3% | Cervix, anus | Included in Gardasil 9 vaccine |
| HPV 56 | <1% | Cervix | Lower relative risk |
| HPV 58 | ~2–4% | Cervix, vulva | Common in East Asia |
| HPV 59 | <1% | Cervix | Rare but classified as high-risk |
| HPV 66 | Rarely isolated | Cervix | Often found in co-infections |
| HPV 68 | ~1% | Cervix, oropharynx | May integrate into host genome |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Copyright: This open access article is published under a Creative Commons CC BY 4.0 license, which permit the free download, distribution, and reuse, provided that the author and preprint are cited in any reuse.



